Embed Size (px)
Citation preview

PRZYGOTOWANO W KATEDRZE ONKOLOGII AKADEMII MEDYCZNEJ, WROCŁAW 2008 r.
WYKONANO W RAMACH NARODOWEGO GRANTU: „MODYFIKACJA I WDROŻENIE PROGRAMU NAUCZANIA ONKOLOGII W POLSKICH UCZELNIACH MEDYCZNYCH”
Nowotwory lite
u młodych dorosłych
Nietypowe i klasyczne objawy,
nietypowych w tym wieku chorób

Miejsce nowotworów wśród przyczyn
zgonów u dzieci
• Nowotwory są drugą, statystyczną przyczyną
zgonów u dzieci - po wypadkach,
urazach,zatruciach
• Nowotwory są pierwszą wśród chorób
przyczyną zgonów u dzieci

Epidemiologia nowotworów
Geny naprawy DNA
- hMSH2, hMLH1, hPMS1, hPMS2
- ich uszkodzenie uniemożliwia rozpoznawanie oraz naprawę
uszkodzeń DNA
- mutacja genu mutatorowego umożliwia powstawanie
kolejnych mutacji genomu – uruchamia kaskadę zdarzeń
prowadzących do transformacji nowotworowej
Geny
- CYP, GST
- fenotyp „zły metabolizer” często jest skojarzony z komórką
nowotoworową – większa podatność na oddziaływanie
szkodliwych czynników środowiska

Epidemiologia nowotworów
Czynniki predestynujące do powstawania nowotworów:
- zespół Downa – 1 % dzieci zachoruje na białaczkę
przed ukończeniem 10 rż (60% ALL, 40%AML)
- zespół Turnera (mozaika) luz zespół niewrażliwości
na androgeny – 25% ryzyko zachorowania na
gonadoblastomę
- zespół WAGR – Tu Wilms + aniridia + zaburzenia
ukłądu moczowo-płciowego + upośledzenie umysłowe –
uszkodzenie genu supresorowego WT1. WT2. 40%
przypadków nerczaka występuje rodzinnie

Epidemiologia nowotworów cechy przenoszone autosomalnie dominująco:
- mutacja jednego allelu genu Rb – retinoblastoma, osteosarcoma, czerniak złośliwy,
najczęściej skojarzona z obustronną lub rozsianą formą nowotworu
- mutacja genu p53 – najczęściej występujące w nowotworach zaburzenie
genetyczne – zespół Li-Fraumeni – zwiększona podatność zachorowania na mięsaki,
białaczki, raka sutka, raka kory nadnerczy, nowotwory OUN
- zespół genów odpowiedzialnych za MEN – multiple endocrine neoplasia –
zachorowania na nowotwory tarczycy, przytarczyc, trzustki, przysadki mózgowej,
nadnerczy
- neurofibromatoza typ I – mutacja protoonkogenu ras - nowotwory OUN, osłonek
nerwów obwodowych, AML
- stwardnienie guzowate – hamartoma, opóźnienie umysłowe, angiofibroma,
rhabdomyoma w sercu, astrocytoma, hemartoma siatkówki
- zespół von Hippel-Lindau – uszkodzony gen VHL odpowiedzialny za procesy
transkrypcji – rek nerki, pheochromocytoma, naczyniaki móżdżku i siatkówki

Epidemiologia nowotworów
cechy przenoszone autosomalnie recesywnie:
-Xeroderma Pigmentosum – zaburzenia genów naprawy DNA i regulujących transkrypcję –
skłonność do nowotworów skóry łącznie z czerniakiem złośliwym
- ataxia-teleangiectasia – uszkodzony gen naprawy DNA – ATM. Zespół nadwrażliwości na
promieniowanie – ataxia, teleangiektazja, skłonność do rozwoju chłoniaków
- anemia Fanconiego – wrodzona łamliwość chromosomów, zespół wad wrodzonych i
niewydoności szpiku, nadmierna wrażliwość na działanie środków alkilujących, w patogenezie
udział ok 12 genów
stany obniżonej odporności
- ciężki złożony niedobór odporności
- zespół Wiskotta-Aldricha – trombocytopenia, egzema, upośledzenie funkcji limfocytów T –
skłonność do rozwoju chłoniaków
- zespół limfoproliferacyjny indukowany wirusem EBV
- infekcja HIV – mięsak Kaposiego, chłoniaki (szczególnie w OUN), leyomyosarcoma

Epidemiologia nowotworów
Czynniki środowiskowe
-promieniowanie jonizujące – bomba atomowa, diagnostyka RTG u
ciężarnych – dane niepewne!
- promieniowanie elektromagnetyczne?
- toksyny:
dioksyny – ziarnica złośliwa, AML, rak tarczycy
trichloroetan – białaczki
pestycydy – guzy OUN
rozpuszczalniki organiczne – guzy OUN, neuroblastoma,
hepatoblastoma
metale ciężkie – hepatoblastoma, guz Wilmsa, białaczka
produkty ropopochodne - hepatoblastoma, guz Wilmsa, białaczka
chemioterapia - białaczka

Epidemiologia nowotworów
• W Polsce na chorobę nowotworową choruje ok. 118 dzieci/1mln osób, w USA – 125 na 1
mln, Nigeria – 155 na 1 mln, Fidżi – 40 na 1 mln
• W rejonie obejmującym 50-75tys. dzieci obserwuje się ok 10-15 zachorowań rocznie
• W ogólnej populacji chorych na nowotwory dzieci stanowią 0,5% - 1.0%
•50% wszystkich zachorowań wśród dzieci stanowią białaczki i chłoniaki
• różnice pomiędzy zapadalnością zależą od rasy (u murzynów częściej chłoniaki, sa. Ewing,
rak tarczycy)
• ... wieku:
- dorośli – 80-88% nowotwory nabłonkowe – raki
- młodzież – osteosarcoma, guz Ewing. HD, guzy germinalne
- dzieci – białaczka, u chłopców guzy germinalne - Pierwszy wierzchołek zachorowań (do 4
roku życia) tłumaczy się przewagą u małych dzieci guzów embrionalnych, rozwijających się w
tkankach, których rozwój nie został całkowicie zakończony.
- niemowlęta- NBL, guz Wilmsa, retinoblastoma
• ... i płci
- przewaga chłopców – chłoniaki, białaczka, NBL, guzy OUN, mięsaki tk miękkich
- przewaga dziewcząt – gonadalne guzy zarodkowe, rak kory nadnerczy, rak tarczycy

ZESPOŁY GENETYCZNE
Z RYZYKIEM NOWOTWORU
zespoły przewodu pokarmowego
Zespół gruczolakowatej polipowatości
rodzinnej – mutacja genu APC – ryzyko raka
jelita grubego i hepatoblastoma
Dziedziczny rak jelita grubego bez
polipowatości – zespól Lych – mutacje w
obrębie genów naprawy

Zaburzenia związane z
dziedziczeniem autosomalnie
dominującym
Retinoblastoma – mutacja genu Rb. U nosicieli
ryzyko czerniaka i osteosarcoma
Zespół Li-Fraumeni – mutacja genu p53 – u
nosicieli wzrost ryzyka : RMS, białaczki, guzów
mózgu, guzów kości, raka piersi

Klasyfikacja nowotworów
dziecięcych
I. białaczki
II chłoniaki
III. nowotwory CUN
IV. nowotwory układu współczulnego
V. retinoblastoma
VI. nowotwory nerek
VII. nowotwory wątroby
VIII. nowotwory kości
IX. mięsaki tkanek miękkich
X. nowotwory zarodkowe
XI. nowotwory nabłonkowe
XII. inne

Polska Europa
• Białaczki 28,7 33,5
• Guzy mózgu 16,3 22,6
• Chłoniaki 14,3 9,9
• Nowotwory układu nerwowego 6,9 6,8
• Nowotwory tkanek miękkich 6,6 6,0
• Nowotwory nerek 6,5 6,9
• Nowotwory kości 8,2 4,2
• Inne 1,3 0,2
nowotwory zarodkowe 5,8 2,3
retinoblastoma 2,7 3,3
nowotwory wątroby 1,5 0.9
nowotwory nabłonkowe 1,3 1,8
J.Kowalczyk
Rocznie w Polsce rozpoznaje się 1000-1200 nowych zachorowań na nowotwory złośliwe u dzieci.
CZĘSTOŚĆ POSZCZEGÓLNYCH TYPÓW NOWOTWORÓW W % W POLSCE I EUROPIE

Przewiduje się, iż w pierwszej dekadzie
bieżącego stulecia jedna osoba
spośród 900 dorosłych między 18 a 44
rokiem życia jest ozdrowieńcem z
choroby nowotworowej przebytej w
dzieciństwie.

Klasyfikacja nowotworów
Dzieci - od 0 do końca 17 r.ż.
U dorosłych - narządowa, u dzieci - histologiczna
Różna częstość występowania poszczególnych nowotworów
Różna dynamika wzrastania
Różnice w etiologii?
Wyniki leczenia

Rzadkość pojawiania się typowych raków z nabłonka
gruczołowego błon śluzowych lub nabłonka
wielowarstwowego płaskiego, w przeciwieństwie do
patologii dorosłych:
Około 92 % wszystkich nowotworów dziecięcych –
charakter nienabłonkowy
Około 80 – 88 % nowotworów dorosłych –
nowotwory nabłonkowe
Zmiana proporcji między 15 a 18 rokiem życia
Cechy charakterystyczne histopatologii
nowotworów wieku dziecięcego

Problemy prewencji nowotworów
dziecięcych DOROŚLI DZIECI
Często długi czas ekspozycji na
kancerogeny
Zwykle niewyjaśniona etiologia, często we
wczesnym dzieciństwie
Skłonność rodzinna do określonych
nowotworów – możliwość badań
przesiewowych
Możliwość występowania na podłożu
wrodzonych deficytów immunologicznych
i innych zespołów chorobowych
Częste występowanie i charakterystyczne
dolegliwości – duża czujność onkologiczna
lekarzy rodzinnych
Rzadsze występowanie, niespecyficzne
objawy – niewystarczająca czujność
onkologiczna
Często leczenie lokalne, mały odsetek
wieloletnich obserwacji po wyleczeniu ze
względu na podeszły wiek
Intensywne leczenie systemowe,
skojarzona CHT, RT, BMT – zwiększenie
ryzyka wtórnych nowotworów wiele lat po
wyleczeniu
Ogromna rola prewencji oraz badań
przesiewowych
Zapobieganie nowotworom praktycznie
NIEMOŻLIWE !!!
Istotne wczesne wykrywanie, związane z
lepszym rokowaniem

Nowotwory u dzieci
• Czy zwiększa się liczba zachorowań ?
• Jaka jest przyczyna choroby ?
• Czy można ją wcześniej rozpoznać ?
• Czy można ją wyleczyć ?
• Jakie mogą być następstwa?
• Czyn różnią się od nowotworów dorosłych

Polska populacja w wieku 0 –17 lat
1995 r - 10.644.803
1996 r - 10.417.517
1997 r - 10.165.537
1998 r. - 9.888.636
1999 r. - 9.613.822
2000 r. - 9.305.936
2001 r. - 8.971.182
2002 r. - 8.704.011
2003 r. – 8.474.026
2004 r. - 8.219.185
2005 r. - 7.863.799 2006 r. - 7.761.094 J.Kowalczyk

19
Populacja polska a populacja
standardowa Europy
Gr. wiekowa Polska Standardowa 0 - 4 lat 1.780.631 8% 3.040.000
5 - 9 lat 1.982.614 7% 2.660.000
10 -14 lat 2.425.930 7% 2.660.000
15 - 17 lat 1.674.930 4,5% 1.710.000
0-17 lat 7.863.799 26.5% 10.109.750
J.Kowalczyk

Nowotwory u dzieci
Czy zwiększa się liczba zachorowań ?
Jaka jest przyczyna choroby ?
Czy można je wcześniej rozpoznać ?
Czy można je wyleczyć ?
Jakie mogą być następstwa?

21
Zachorowania na nowotwory
dziecięce
110-130 / 1 mln dzieci
5-10 nowych przypadków rocznie w rejonie
50-75 tys.dzieci
ryzyko zachorowania w ciągu 15 lat życia:
Europa 0.0012 (0.12%) 12/10.000
- 0.0017 (0.17%) 17/10.000
Polska 0.16% (ok.1/625)

22
Zwiększanie się liczby zachorowań:
• guzy mózgu
• nerwiak zarodkowy
• ALL

Polska
Ok. 1100 – 1200 nowych rozpoznań
nowotworów dziecięcych rocznie
Ok. 2200 – 2500 dzieci w czasie
intensywnej terapii
Ok. 12 000 dzieci monitorowanych po
zakończonym leczeniu

24
Największa częstość zachorowań
1 r.ż. -neuroblastoma, retinoblastoma, hepatoblastoma
2 - 4 r.ż. - ALL
wiek młodzieńczy - ch. Hodgkina, mięsak kościopochodny, mięsak Ewinga
wiek niemowlęcy, następnie 10 -14 l. - fibrosarcoma

białaczki; 26,4
chłoniaki; 15,8
inne; 0,3nabłonk.; 2zarodkowe; 5,8
mięsaki; 6,6
kości; 8,2
wątroba; 1,5
nerki ; 6,5
retinobl.; 2,9
ukł.wsp.; 6,9
oun; 22,2
Częstość poszczególnych typów nowotworów

Leczenie onkologiczne
CHEMIOTERAPIA + RADIOTERAPIA +
CHIRURGIA + INTENSYWNA TERAPIA
W Polsce wszystkie dzieci leczone są w ośrodkach
akademickich zrzeszonych w Polskiej Grupie ds Leczenia
Białaczek i Chłoniaków oraz w Polskiej Grupie ds
Leczenia Guzów Litych, które ustalają wspólny standard
postępowania oparty najczęściej na protokołach
europejskich i amerykańskich. Obecnie w Polsce nie ma
zasadniczych różnic w terapii, dostępie do nowoczesnych
leków, metod diagnostycznych itp. w porównaniu z innymi
krajami. Wyniki leczenia również nie odbiegają od danych
spotykanych w literaturze światowej.

Chemioterapia
• leczenie przy użyciu środków hamujących podziały lub zabijające
komórki nowotworowe
• ma działanie systemowe
• podstawowym mechanizmem działania cytostatyków jest
wprowadzenie komórki na drogę programowanej śmierci - apoptozy
Grupy leków przeciwnowotworowych
1.Leki cytotoksyczne (CYTOSTATYKI)
- Leki alkilujące
- Antymetabolity
- Antybiotyki przeciwnowotworowe
- Leki pochodzenia roślinnego
2. Hormony i związki modyfikujące działanie hormonów
3. nasilają przeciwnowotworową odpowiedź immunologiczną
gospodarza - np modulatory odpowiedzi biologicznej.
4. Inne leki przeciwnowotworowe

Chemioterapia 7. Inne leki przeciwnowotworowe - modulatory odpowiedzi biologicznej
na nowotwór oraz najważniejsze nowe i użyteczne starsze leki
przeciwnowotworowe
- interferony
- Il-2
–rituksymab - anty-CD20
- ibritumomab: przeciwciało monoklonalne przeciw CD20 sprzężone z
izotopem 90Y
- tositumomab: przeciwciało monoklonalne przeciw CD20 sprzężone z
izotopem 131I,
- Gleevec - inhibitor kinazy tyrozynowej BCR-ABL patologicznie
nadaktywnej w komórkach nowotworowych, szczególnie białaczkowych,
- L-asparaginaza - enzym rozkładający asparaginę, niszczy komórki
białaczkowe, które w przeciwieństwie do zdrowych, nie mogą same
syntetyzować tego aminokwasu
- retinoidy – stymulują dojrzewanie komórek
- hydroksymocznik – użyteczny w leczeniu CML

Chemioterapia
1. Chemioterapia wskazana jest w wypadku procesów uogólnionych i
obecności przerzutów. W przypadku niektórych guzów litych
chemioterapia jest leczeniem z wyboru (np.ziarnica złośliwa). W
przypadku procesów umiejscowionych głównym leczeniem jest leczenie
lokalne tj. interwencja chirurgiczna i radioterapia.
2. Chemioterapia może być jedyną metodą leczenia choroby nowotworowej
lub może być stosowana jako chemioterapia wspomagająca (adjuvant
chemiotherapy, podawanie leków po zabiegu chirurgicznym w celu
eliminacji mikroprzerzutów) oraz chemioterapia wstępnie wspomagająca
(neoadjuvant chemiotherapy, podawanie leków przed interwencją
chirurgiczną w celu zmniejszenia masy guza.
3. Chemioterapia może być stosowana jako leczenie systemowe oraz
miejscowo (podawanie leków do tętnicy zaopatrującej dany obszar np.
do tętnicy wątrobowej w przypadku przerzutów do wątroby).
Ogólne zasady chemioterapii przeciwnowotworowej

Leczenie chirurgiczne • Wspólnie z chemioterapią i radioterapią część
onkologicznego leczenia skojarzonego
• W wielu nowotworach radykalna resekcja guza jest
istotnym warunkiem pełnego wyleczenia i znacząco
poprawia rokowanie
• Rozwój nowych technik chirurgicznych (laser, nóż
ultradźwiękowy, laparoskopia, torakoskopia itp), materiałów
(endoprotezy kości), metod obrazowania (MR, TK), opieki
przed- i pooperacyjnej, anestezjologii dziecięcej pozwolił z
jednej strony na usunięcie guzów uważanych do tej pory za
nieoperacyjne z maksymalnym oszczędzeniem tkanek
zdrowych i zminimalizowaniem ryzyka powikłań
• niejednokrotnie postępowanie chirurgiczne jest uzależnione od
wyniku oceny histopatologicznej materiału pobranego na drodze
biopsji guza

Leczenie chirurgiczne
I. Diagnostyka
aspiracyjna biopsja cienkoigłowa (BAC) – nakłucie igłą
powierzchniowych guzów pod kontrolą wzroku, USG, RTG. Zaleta: mała
inwazyjność, możliwość wykonania bez znieczulenia ogólnego. Wada:
mała ilość materiału do badania histopatologicznego, przypadkowość
miejsca aspiracji, brak możliwości oceny pełnej architektoniki
histopatologicznej. U dorosłych wykonywana stosunkowo często, w
onkologii dziecięcej niewielkie zastosowanie.
biopsja otwarta – zabieg połączony z częściową (w przypadku
niekorzystnej lokalizacji, dużej rozległości, naciekania życiowo istotnych
struktur) lub całkowitą resekcją guza. Dla diagnostyki histopatologicznej
najlepsze fragmenty obwodowe guza (w centralnych często występuje
martwica). Zabieg zgodnie z zasadami chirurgii onkologicznej tj. bez
przerwania ciągłości tkanek guza, z marginesem zdrowych tkanek. Kanał
biopsji powinien być usunięty podczas zabiegu podstawowego.

Leczenie chirurgiczne
II. Zabieg główny (odroczony)
- resekcja mikroskopowo radykalna – zdrowy brzeg wyciętej tkanki bez
komórek nowotworowej w ocenie histopatologicznej
- resekcja makroskopowo radykalna, mikroskopowo nieradykalna –
obecne komórki npl w marginesie zdrowych tkanek
- resekcja makroskopowo nieradykalna (częściowa)
• dokładna ocena przyległych narządów, tkanek, węzłów chłonnych, w
przypadku podejrzenia zajęcia węzłów – ich usunięcie
• CHT przedoperacyjna pozwala zmniejszyć masę guza i zakres resekcji
chirurgicznej. CHT pooperacyjna i radioterapia lokalna zniszczyć komórki
rezydualne w loży po guzie i tkankach sąsiadujących
III. Zabieg second-look
– usunięcie guza resztkowego, rewizja miejscowa obszaru zajmowanego
przez nowotwór
- przypadki wznowy miejscowej

Leczenie chirurgiczne
Przerzuty odległe – najczęściej płuca, ew. wątroba, kości, OUN
-chirurgia tylko w przypadku opanowania ogniska pierwotnego w wyniku
CHT i/lub radioterapii
- głównie dotyczy zmian w płucach, najczęściej w osteosarcoma, czasami
pojedyncze zmiany w wątrobie, OUN
redukcja masy guza, odbarczenie tchawicy, przełyku, nerwów, naczyń, jelit
itp.
Leczenie powikłań -perforacja jelit, niedrożność jelit
- ropnie
- odma płuc
- martwica tkanek
- zabiegi rekonstrukcyjne, plastyczne, usprawniające
- zakładanie gastrostomii, tracheostomii
- wyłanianie jelita, moczowodów
- usunięcie grzybni

Nowotwory
przegląd systematyczny

Guzy OUN


Guzy OUN
• 2-gi pod względem częstości nowotwór u dzieci /po
białaczkach/, 1-szy z guzów litych
• najczęściej 2-7 rż
• stosunkowo często jako guz wtórny po zakończeniu
leczenia białaczki
• defekty genetyczne /z. Li-Fraumeni/
• złośliwość histopatologiczna i lokalna guzów
CNS – rozsiew drogą płynu m-r, wzrost ciśnienia
śródczaszkowego, ucisk ważnych życiowo
struktur mózgowia

Guzy OUN - Histologia:
- zdecydowana większość): Astrocytoma (I, II st.), Astr.
Anaplasticum (IIIst), Glioblastoma multiforme (IV st.),
Oligodendroglioma, Ependymoma, PNET-
Medulloblastoma
- guzy zwojów nerwowych
- guzy oponowe
- pierwotne chłoniaki mózgu
- craniopharyngeoma
- guzy naczyniowe
- guzy germinalne
- guzy przerzutowe

Guzy OUN
Topografia
- guzy podnamiotowe 50-60% (guzy móżdżku,
ok.. IV komory, pnia mózgu)
- guzy nadnamiotowe 35-40% (guzy półkul
mózgowych, ok. siodła tureckiego, III komory,
skrzyżowanie wzrokowe)
- guzy kanału kręgowego 5% (zwenątrz- i
wewnątrzoponowe)
wzmożone ciśnienie śródczaszkowe!

40

OBJAWY OGÓLNE
WZMOŻONE CIŚNIENIE ŚRÓDCZASZKOWE
1. Zmiana zachowania – apatia i senność → rozdrażnienie i
płaczliwość
2. Wymioty (zwłaszcza rano) i biegunka
3. Wielkie uwypuklone ciemię
4. Obrzęk tarcz nerwu wzrokowego
5. Ból głowy
6. Zwolnienie akcji serca, wzrost RR
7. Objawy wklinowania mózgu – zaburzenia świadomości,
zatrzymanie krążenia i oddychania, odmóżdżeniowe
napady toniczne
Guzy OUN - objawy kliniczne:

Guzy OUN - objawy specyficzne
związane z lokalizacją
1. Móżdżek- zab. równowagi, oczopląs, ataksja, ↓napięcia mięśniowego
2. Pień mózgu – obj. porażenia nn V-XII, niedowłady spastyczne kończyn, zab.
czucia
3. Ok. czuciowo-ruchowa – przeciwstronne niedowłady spastyczne, zab. czucia
4. Płaty czołowe – zab. psychiczne, zmiany zachowania
5. Ok. skroniowo-ciemieniowo-potyliczna – zab. mowy, orientacji schematu
ciała, agrafia, aleksja
6. Płaty skroniowe – omamu słuchowe, wzrokowe, epi, ubytki pola widzenia
7. Płaty potyliczne - ubytki pola widzenia
8. Ok. analizatora wzrokowego – ślepota korowa
9. Ok. analizatora mowy – afazja
10.Guzy struktur głębokich mózgu – obj. piramidowe, oporny na leczenia ból
11.Guzy komór bocznych – krwawienia dokomorowe
12.Ok. podwzgórza i przysadki – zaburzenia endokrynologiczne
13. Rdzeń kręgowy – odcinek szyjny – porażenie kurczowe czterokończynowe
- o. piersiowy – niedowład i porażenie kk dolnych
- o. lędźwiowy – niedowłady kk dolnych, porażenia zwieraczy
- ból
- zaburzenia czucia – zaniki i przeczulica

Guzy OUN
- RTG czaszki
- TK
- MR
- PET
- badanie okulistyczne (dno oka)
- punkcja lędźwiowa – obecność komórek
nowotworowych w płynie m-r, ↑ poziomu białka
przy prawidłowej liczbie elementów komórkowych
- biopsja, biopsja stereotaktyczna

Guzy OUN
Leczenie skojarzone
zabieg operacyjny – podstawa wyleczenia!!!
radioterapia – duże dawki naświetlań do 55 Gy, <3
rż w ostateczności
CHT – medulloblastoma, PNET, astrocytoma III i IV
st, , ependymoma
- bariera krew mózg
- CCNU, cisplatyna, karboplatyna, etopozyd,
ifosfamid, cyklofosfamid, adriamycyna, temozolamid

Guzy kości

Guzy kości
Typ histologiczny Łagodne Złośliwe
Hematopoetyczne
Myeloma
Chłoniaki nieziarnicze
Chrzęstnopochodne
Osteochondroma
Chondroma
Chondroblastoma
Chondromyxoid fibroma
Chondrosarcoma
Dedifferentiated
chondrosarcoma
Kostnopochodne Osteoid osteoma
Osteoblastoma
Osteosarcoma
Pochodzenie nieznane Giant cell tumour Sa Ewing
Giant cell tumour
Histiocytarne
Włóknistopochodne
Naczyniowe
Tłuszczopochodne
Neurogenne
Fibrous histiocytoma
Fibroma
MFH
Fibrosarcoma
Chordoma

• najczęściej występujący złośliwy guz
kości u dzieci
•II dekada życia - skok pokwitaniowy, intensywny
wzrost kości na długość
• przynasada bliższa k. piszczelowej i strzałkowej
przynasada przynasada dalsza k. udowej
przynasada bliższa trzonów k. udowej i ramieniowej
kości płaskie szkieletu osiowego
• przerzuty: → płuca
→ kości
→ szpik kostny
Osteosarcoma /kostniakomięsak, mięsak kościopochodny/

Osteosarcoma /kostniakomięsak, mięsak kościopochodny/

Osteosarcoma:
1.osteoblastic
2.fibroblastic
3.chondroblastic
4.telangiectatic
Lepsze rokowanie, raczej bez meta
-paraosteal
Osteosarcoma
/kostniakomięsak, mięsak kościopochodny/

Osteosarcoma /kostniakomięsak, mięsak kościopochodny/
• Ból, często spoczynkowy!
• Widoczny guz, powiększenie obrysu kości,
kolana
• złamanie patologiczne kości
• ↑ fosf. alk. ↑ LDH
• RTG kości, klatki piersiowej, TK, scyntygrafia
• biopsja otwarta → histopatologia
Leczenie skojarzone:
CHT przedoperacyjna → zabieg operacyjny /endoproteza, amputacja/
→ CHT pooperacyjna

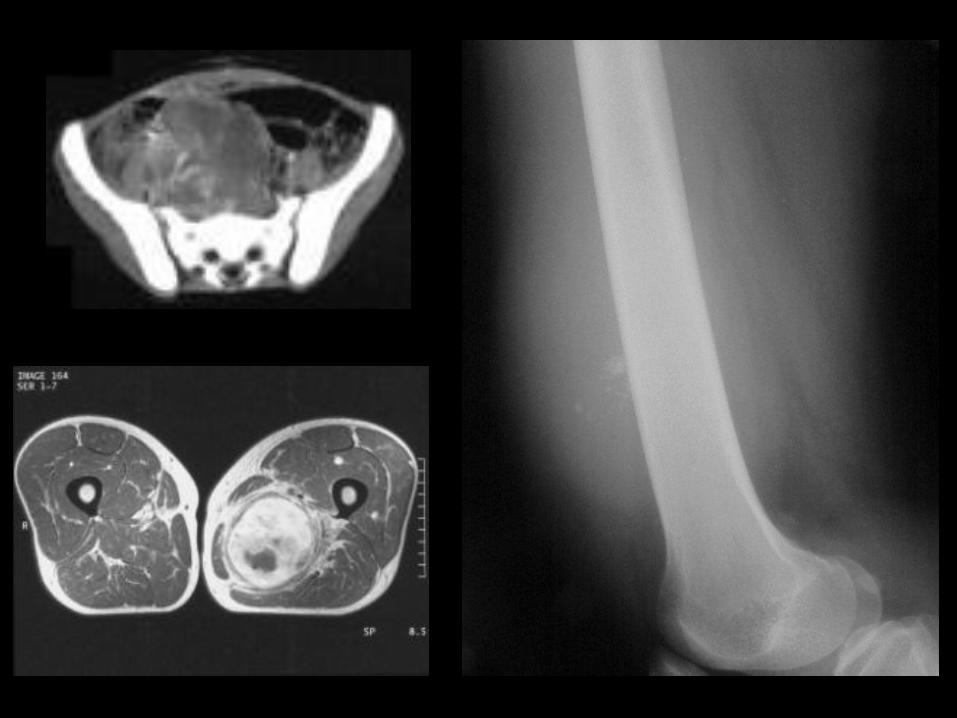
Guzy kości
OS
sunburst pattern

Guzy kości OS

Guzy kości OS
TK
MR

Guzy kości
OS

Guzy kości OS
Guzy kości OS

Guzy kości OS



Guzy kości
Sa Ewing
Sa Ewing family – PNET, Tu Askin
glycoprotein MIC-2 (+) /CD99+/
t(11;22) – 95% - EWS/FLI1 lub EWS/ERG
objawy ogólne – gorączka, spadek masy,
„maska zapalna”
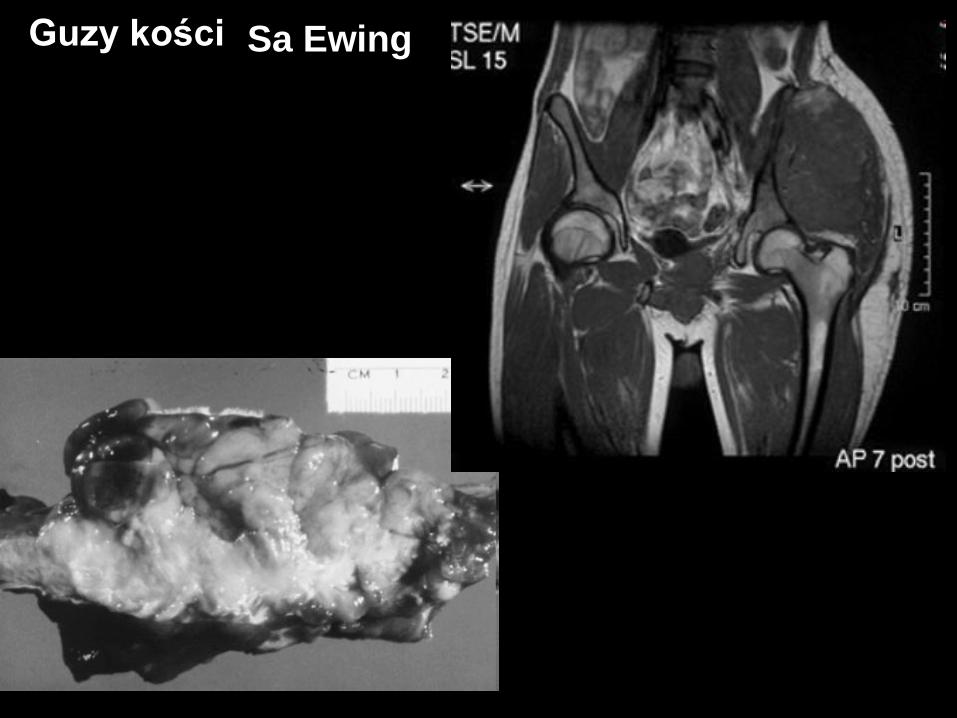
Guzy kości Sa Ewing

Guzy kości Sa Ewing

Guzy kości
Sa Ewing

Guzy kości
Osteosarcoma Sa Ewing
2 dekada 2 dekada
wszystkie rasy białą dominuje
M/Ż 1.5/1 M/Ż 1.5/1
kom. wrzecionowate prod.
osteoid
niezróżnicowany,
drobnokrągłokomórkowy
prawdopodobnie poch. neuronalne
RB, Li-Fraumeni, ch. Pageta,
radioterapia
predyspozycje nieznane
przynasady k. długich nasady k. długich, k. płaskie
ból miejscowy i obrzęk
często uraz w wywiadzie
ból miejscowy i obrzęk
gorączka
zmiany sklerotyczne, rzadziej
lityczne, sunburst pattern
zmiany głównie lityczne, reakcja
okostnowa, onion skinning

Guzy kości
Osteosarcoma Sa Ewing
Sa Ewing, osteomyelitis osteomyelitis, ziarniniak
kwasochłonny, NHL, NBL,
RMS, LCH
płuca, kości, szpik kostny płuca, kości, szpik kostny
CHT
resekcja guza pierwotnego
CHT, radioterapia
resekcja guza pierwotnego
surv – 70% bez meta
- <20% z meta!!!
surv – 60% bez meta
- 20-30% z meta!!!

MIĘSAKI TKANEK
MIĘKKICH U DZIECI

MIĘSAKI TKANEK MIĘKKICH
heterogenna grupa nowotworów
rozwój z embrionalnej tkanki mezenchymalnej i
neuroektodermalnej
wspólne cechy biologiczne, kliniczne, strategia leczenia
pseudorozprężający wzrost
ograniczane przez bariery naturalne
szerzenie się wzdłuż przestrzeni i otworów naturalnych, naciekają
sąsiadującą tkankę
guzy inwazyjne miejscowo, z tendencją do nawrotu w miejscu
ogniska pierwotnego
tworzenie przerzutów głównie drogą krwionośną
w 20% już w chwili rozpoznania rozsiana postać choroby

EPIDEMIOLOGIA
• 6,5% wszystkich nowotworów
dziecięcych
• najczęstsze u dzieci w wieku 2-6 lat oraz u
młodzieży powyżej 12 roku życia
• występują niezależnie od płci
• najczęstszy (70% mięsaków dziecięcych)
- mięsak prążkowanokomórkowy (RMS)

ETIOLOGIA
Nieznana powstawanie na bazie mutacji germinalnych (10% dziecięcych MTM) Zwiększona częstość występowania w wielu zespołach chorobowych (Li-Fraumeni, Beckwith-Wiedmann Gardnera, Wernera, Gorlina, neurofibromatoza, wrodzony zespół alkoholowy) częste współwystępowanie RMS z wrodzonymi anomaliami układu moczowo- płciowego lub CNS
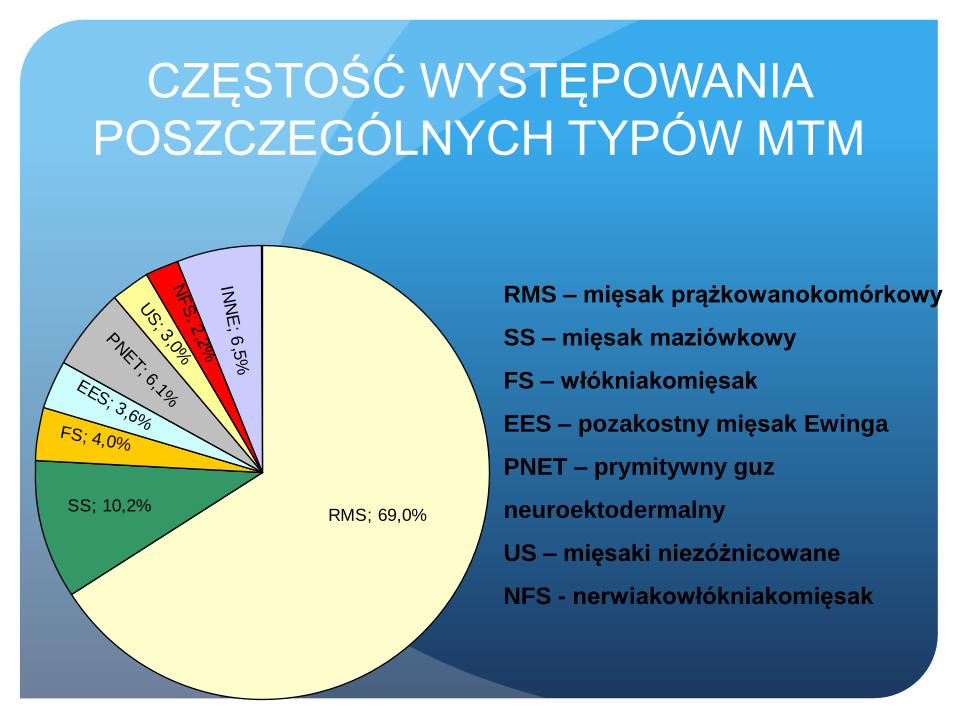
CZĘSTOŚĆ WYSTĘPOWANIA
POSZCZEGÓLNYCH TYPÓW MTM
SS; 10,2%
US; 3
,0%
FS; 4,0%
INN
E; 6
,5%
NF
S; 2
,2%
PNET; 6,1%EES; 3,6%
RMS; 69,0%
RMS – mięsak prążkowanokomórkowy
SS – mięsak maziówkowy
FS – włókniakomięsak
EES – pozakostny mięsak Ewinga
PNET – prymitywny guz
neuroektodermalny
US – mięsaki niezóżnicowane
NFS - nerwiakowłókniakomięsak

TRANSLOKACJE
W NOWOTWORACH
TKANEK MIĘKKICH
aktywacja protoonkogenów przez amplifikację
lub aranżację genów
utrata genów supresorowych procesu
nowotworowego przez delecję lub inaktywację
tworzenie białek fuzyjnych poprzez
translokacje chromosomalne

TRANSLOKACJE
W NOWOTWORACH TKANEK MIĘKKICH
Mięsak prążkowanokomórkowy
pęcherzykowy
PAX3-FKHR
PAX7-FKHR
t(2;13)(q35;q14)
t(1;13)(p36;q14)
Guzy rodziny Ewinga EWS-FLI1
EWS-ERG
t(11;22)(q24;q12)
t(21;22)(q22;q12)
Sarcoma synoviale SYT-SSX1
SYT-SSX2
t(X;18)(p11;q11)
t(X;18)(p11;q11)
Desmoplastyczny guz
drobnookrągłokomórkowy
EWS-WT1
t(11;22)(p13;q12)
Fibrosarcoma wrodzona,
Nerczak mezoblastyczny
ETV6-NTRK3 t(12;15)(p13;q25)
Jasnokomórkowy
mięsak tkanek miękkich
EWS-ATF1 t(12;22)(q13;q12)
Myxoid lipocarcoma FUS-CHOP
EWS-CHOP
t(12;16)(q13;p11)
t(12;22)(q13;q12)
Myxoid chondrosarcoma EWS-CHN t(9;22)(q22;q13)
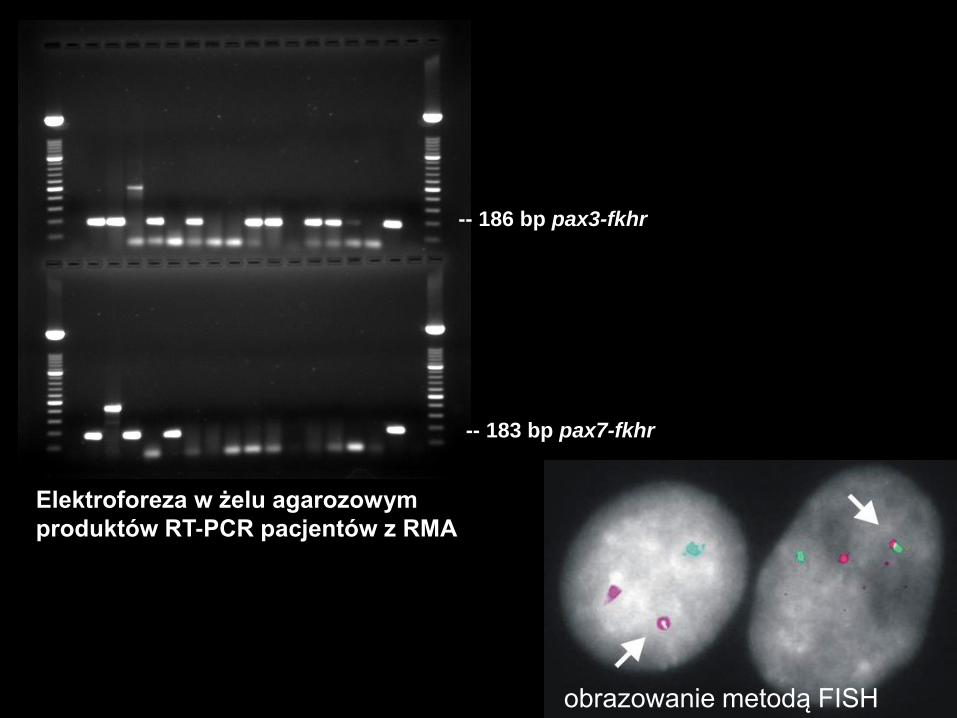
-- 183 bp pax7-fkhr
-- 186 bp pax3-fkhr
Elektroforeza w żelu agarozowym
produktów RT-PCR pacjentów z RMA
obrazowanie metodą FISH

MIĘSAKI TKANEK MIĘKKICH
RHABDOMYOSARCOMA NON-RHABDO
SARCOMAS

75
RHABDOMYOSARCOMA (RMS)
• najczęstszy spośród dziecięcych MTM (do 69%)
• pochodzenie – pierwotne komórki
mezenchymalne, prekursory mięśni poprzecznie prążkowanych
• występowanie 70% dziecięcych RMS przed 10 r.ż.
• szczyty zachorowalności:
– 2 – 5 lat
– okres dojrzewania
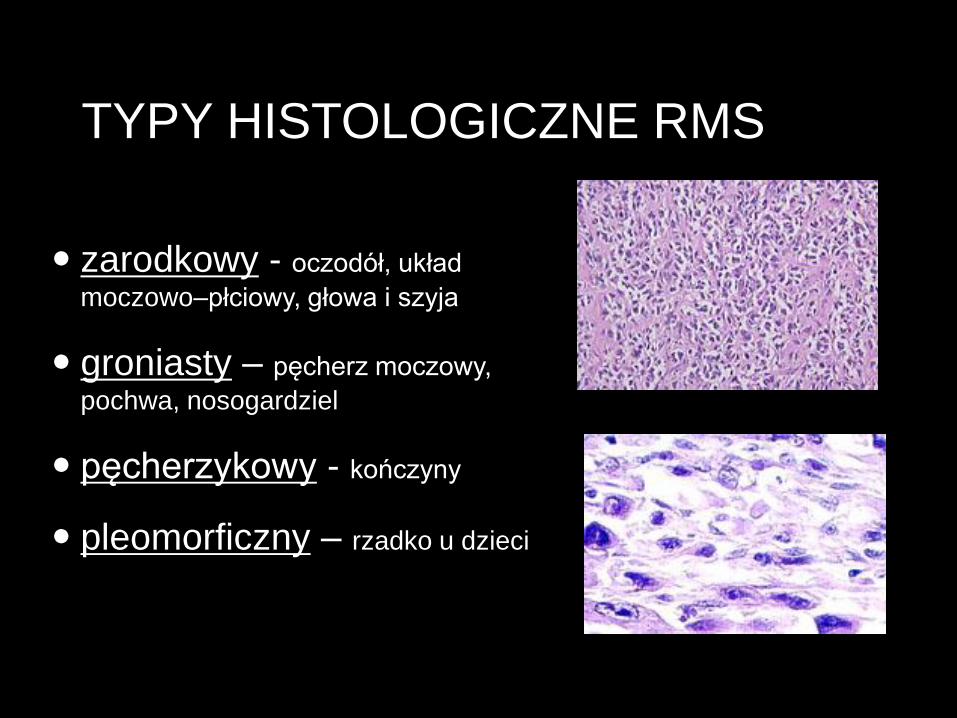
TYPY HISTOLOGICZNE RMS
zarodkowy - oczodół, układ
moczowo–płciowy, głowa i szyja
groniasty – pęcherz moczowy,
pochwa, nosogardziel
pęcherzykowy - kończyny
pleomorficzny – rzadko u dzieci
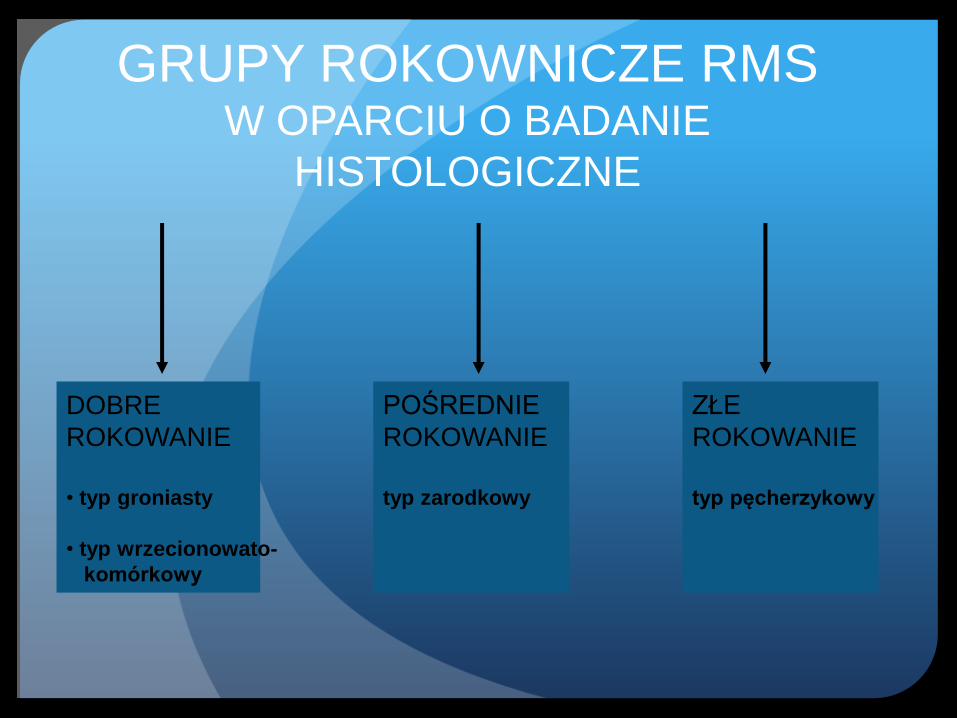
GRUPY ROKOWNICZE RMS W OPARCIU O BADANIE
HISTOLOGICZNE
DOBRE
ROKOWANIE
• typ groniasty
• typ wrzecionowato-
komórkowy
POŚREDNIE
ROKOWANIE
typ zarodkowy
ZŁE
ROKOWANIE
typ pęcherzykowy

LOKALIZACJA GUZA PIERWOTNEGO
gałka oczna głowa i szyja bez manifestacji okołooponowej głowa i szyja z zajęciem okołooponowym układ moczowo – płciowy
z zajęciem pęcherza moczowego lub gruczołu krokowego z zajęciem pochwy, sromu, macicy, okolicy okołojądrowej
kończyny inne

AKTYWNOŚĆ BIOLOGICZNA RMS
ogromna agresywność guza
naciekanie okolicy wzdłuż powięzi
tworzenie przerzutów •droga chłonna i krwionośna
•u 20% dzieci przerzuty obecne w momencie rozpoznania
•najczęstsza lokalizacja: płuca, węzły chłonne, kości, szpik
•rzadziej: wątroba, mózg, sutek

OBJAWY KLINICZNE
W ZALEŻNOŚCI OD LOKALIZACJI -
GŁOWA I SZYJA
gałka oczna - zez, zwężenie szpary powiekowej, wytrzeszcz gałki ocznej, obrzęk spojówki, zaczerwienienie powiek
nosogardziel – obstrukcja dróg oddechowych, mowa nosowa, lokalny ból, dysfagia, porażenie nerwów czaszkowych
zatoki przynosowe – obrzęk, ból, sinusitis, obstrukcja, porażenie nerwów czaszkowych
ucho środkowe – polipowaty twór, przewlekłe zapalenie ucha, wyciek krwi, porażenie nerwów czaszkowych
krtań – chrypka, męczący kaszel
szyja – dysfagia, dusznośc, zespół Hornera







OBJAWY KLINICZNE
W ZALEŻNOŚCI OD LOKALIZACJI –
UKŁAD MOCZOWO - PŁCIOWY
pochwa – gronowate masy, „wypadające” z jej światła
trzon macicy – śluzowokrwista wydzielina z pochwy
pęcherz moczowy - polipowaty guz pobliża trójkąta, nawracające ZUM, krwiomocz, krwinkomocz, uniemożliwienie odpływu moczu
prostata – rozległy guz miednicy małej, zaparcia, dysuria
jądro – guz długo bezobjawowy



OBJAWY KLINICZNE
W ZALEŻNOŚCI OD LOKALIZACJI –
KOŃCZYNY
głęboko wewnątrz masy mięśniowej
długo bezobjawowe
zmianie może towarzyszyć siniaczenie
szybkie zajęcie węzłów chłonnych i rozsiew


OBJAWY KLINICZNE
W ZALEŻNOŚCI OD LOKALIZACJI –
INNE LOKALIZACJE
okolica przykręgosłupowa
– jednostronne, asymetryczne
zgrubienie, objawy korzeniowe
przestrzeń zaotrzewnowa
drogi żółciowe, powłoki
brzusznej i ściany klatki
piersiowej – objawy zależne
zniszczenia/ucisku pobliskich
struktur
śródpiersie – duszność,
zespół żyły czczej górnej


DIAGNOSTYKA STADIUM dokładne badanie fizykalne:
ocena umiejscowienia i rozmiarów guza
ocena węzłów chłonnych
badania laboratoryjne: morfologia,
biochemia (ocena wątroby i nerek)
poziom elektolitów
badania obrazowe: NMR, TK, USG
punkcja szpiku kostnego (+ dwie trepanobiopsje w przypadku przerzutów i w typie
pęcherzykowym)
scyntygrafia kośćca z użyciem technetu
RTG / TK klatki piersiowej
biopsja podejrzanych węzłów chłonnych








ROZSZERZONA DIAGNOSTYKA
ZALEŻNA OD LOKALIZACJI
guzy okołooponowe: TK/NMR głowy badanie PMR
układ moczowo-płciowy TK/NMR +/- USG przestrzeni pozaotrzewnowej cystouretroskopia z biopsją pęcherza moczowego
kończyny TK +/- USG przestrzeni pozaotrzewnowej i węzłów chłonnych (przy zajęciu kończyny dolnej) TK głowy (częste przerzuty do mózgu) biopsja regionalnych węzłów chłonnych
tułów TK rdzenia kręgowego mielografia (przy obecności objawów kompresji rdzenia)
jama brzuszna TK +/- USG wątroby

OCENA KLINICZNEGO
STADIUM ZAAWANSOWANIA
• systemy IRS oraz TNM (z oceną przed i pooperacyjną)
• rozwój CHT mniejsza liczba ograniczenie
pierwotnych resekcji znaczenia w/w systemów
• próba klasyfikacji pacjentów do leczenia w oparciu o nowe
czynniki prognostyczne

STADIUM KLINICZNE W OCENIE
POOPERACYJNEJ
STADIUM
IRS OKREŚLENIE
STADIUM
pT
I
I A
I B
Guz usunięty doszczętnie makroskopowo i mikroskopowo,
bez zajęcia węzłów chłonnych
- ograniczony do narządu
- wychodzący poza narząd
pT1
pT2
II
II A
II B
II C
Guz usunięty makroskopowo, lecz niedoszczętnie
mikroskopowo i
- regionalne węzły chłonne nie zajęte
- regionalne węzły chłonne zajęte, ale usunięte
- regionalne węzły chłonne zajęte i nie usunięte
pT3a
III Niedoszczętna resekcja z pozostawieniem resztek guza
lub tylko biopsja z rozsiewem komórek nowotworowych do
okolicznych tkanek i jam ciała
pT3b
pT3c
IV Wyjściowo przerzuty odległe lub wyjściowe zajęcie
pozaregionalnych węzłów chłonnych
pT4

M
N
T
T0: nie stwierdza się obecności guza pierwotnego
T1: guz ograniczony wyjściowo do jednego narządu lub tkanki :
T1a : największa średnica guza =< 5 cm
T1b : największa średnica guza > 5 cm
T2: guz wyjściowo wychodzący poza narząd lub tkankę:
T2a : największa średnica guza =< 5 cm
T2b : największa średnica guza > 5 cm
TX: brak adekwatnych danych o wyjściowych rozmiarach guza
(oceniać jak T2).
N0: nie stwierdza się zajęcia regionalnych węzłów chłonnych
N1: zajęcie regionalnych węzłów chłonnych
NX: brak adekwatnych danych o stanie węzłów chłonnych
(oceniać jak N0).
REGIONALNE WĘZŁY CHŁONNE :
głowa/szyja - szyjne i nadobojczykowe;
brzuch/miednica - podprzeponowe i biodrowo-
pachwinowe;
kończyna górna - pachowe i łokciowe;
kończyna dolna - podkolanowe i pachwinowe.
M0: nie stwierdza się przerzutów odległych ani zajęcia
pozaregionalnych węzłów chłonnych
M1: przerzuty odległe lub zajęcie pozaregionalnych węzłów
chłonnych
MX: brak adekwatnych danych o obecności przerzutów
(oceniać jak M0).
PRZERZUTY ODLEGŁE
- zajęcie węzłów chłonnych pozaregionalnych,
(czyli drugiego piętra po stronie guza)
- zajęcia węzłów chłonnych po stronie
przeciwnej, nawet jeśli odpowiadały węzłom
regionalnym
STADIUM KLINICZNE WG SYSTEMU TNM

ISTOTNE CZYNNIKI PROGNOSTYCZNE
status T – istotny we wszystkich lokalizacjach poza zajęciem gałki ocznej
typ histopatologiczny
zarodkowy – lepsze rokowanie
pęcherzykowy – złe rokowanie
wiek w momencie diagnozy
(najlepsze rokowanie 1-9 lat)
stadium zaawansowania
odpowiedź na leczenie
czynniki biologiczne

OKALECZAJĄCE:
•Enukleacja
•Operacje twarzoczaszki
•prowadzące do kosmetycznych defektów
•Amputacje kończyn
•Wyłonienie jelita
•Totalna cystektomia
•Trwałe nadpęcherzowe
•odprowadzenie moczu
•Usunięcie pochwy / macicy
•Wycięcie gruczołu krokowego
•pneumonektomia
•Amputacja kończyny
•Lokalna resekcja powodująca
•poważne uposledzenie funkcji kończyny
ZABIEGI CHIRURGICZNE
NIEOKALECZAJĄCE:
lobektomia płuca
częściowa resekcja jelita / wątroby
nefrektomia
częściowa cystektomia
jednostronna
orchidektomia/ovariektomia
amputacja palca

RADYKALNOŚĆ
ZABIEGU
PIERWOTNEGO
resekcja R0 – resekcja całkowita,
margines mikroskopowo wolny
od komórek nowotworowych
resekcja R1 – resekcja
makroskopowo całkowita
z pozostawieniem marginesu
z obecnością komórek
nowotworowych
resekcja R2 - resekcja z
pozostawieniem
makroskopowych resztek guza

RADIOTERAPIA
niezbędna metoda miejscowej kontroli nowotworu u
pacjentów z mikro/makroskopową chorobą resztkową
po resekcji chirurgicznej lub CHT wstępnej
wskazania do napromieniania i wybór dawki zależą
od:
pierwotnej lokalizacji i wielkości guza
wieku chorego
rozpoznania histopatologicznego
rodzaju resekcji pierwotnej
ogólnie rekomendowana dawka – 32 – 54 Gy
różny czas napromieniania (między 7 a 20 tygodniem
terapii)
unikanie RT u dzieci < 3 roku życia

NOWE TECHNIKI RADIOTERAPII FRAKCJONOWANIE (zwiększenie całkowitej dawki napromieniania podawanej w krótszym czasie, działanie protekcyjne względem zdrowych tkanek)
•konwencjonalne – 1.8 Gy 1 x dziennie (CFRT) •hiperfrakcjonowanie – 1.2 Gy 2x dziennie (HFRT)
AKCELERACJA (HART) (napromienianie z użyciem przyspieszaczy) BRACHYTERAPIA (LDR, HDR)
•u pacjentów po niedoszczętnej resekcji z lokalizacją umożliwiającą założenie aplikatora (otrzewna, pęcherz, prostata, układ rozrodczy) •zmniejszenie ryzyka późnych powikłań

CHEMIOTERAPIA CELE CHEMIOTERAPII:
eliminacja makro- lub mikroprzerzutów zmniejszenie masy guza pierwotnego w celu umożliwienia lokalnej radioterapii i radykalnej operacji
POSZUKIWANIA OPTYMALNEJ STRATEGII TERAPEUTYCZNEJ:
zastosowanie CHT z udziałem: winkrystyny, doxorubicyny, aktynomycyny i cyklofosfamidu badania nad rolą ifosfamidu w CHT etoposid – wzmocnienie CHT w grupie wysokiego ryzyka
LECZENIE ZALEŻNE OD CZYNNIKÓW RYZYKA: charakter naciekania i wielkość guza kliniczne stadium zaawansowania choroby rodzaj resekcji pierwotnej podtyp histopatologiczny

NAJCZĘSTSZE
PROTOKOŁY CHT


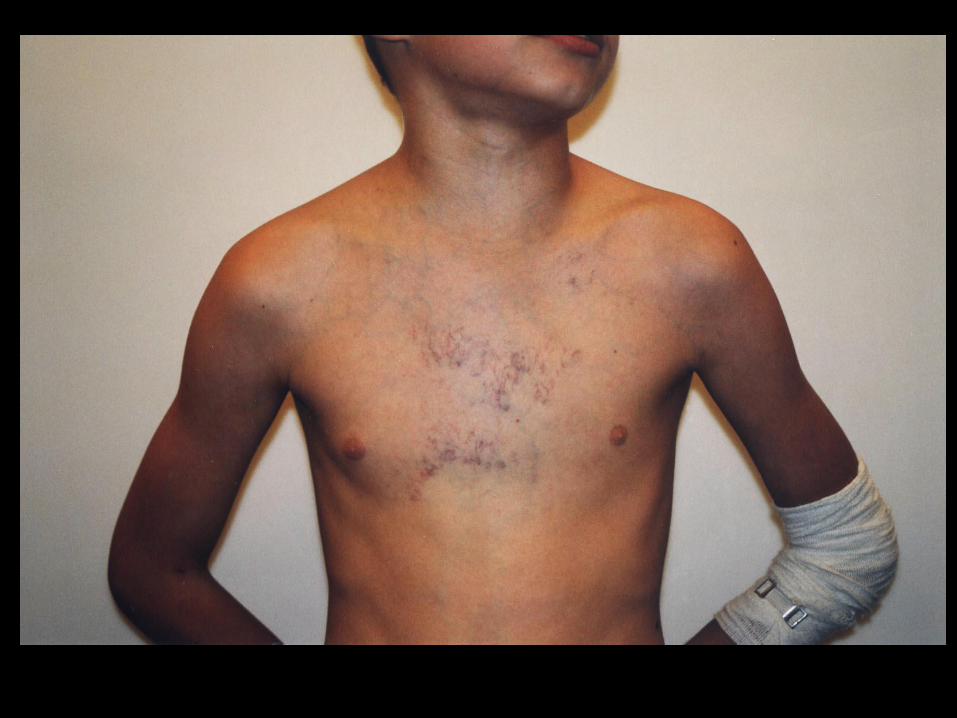



Wyniki
Stadium III, grupa RMS
Zabieg radykalny
n=27
Zabieg nieradykalny
n=10
EFS w zależności od radykalności zabiegu wtórnego
Kompletne Ucięte
Log rank test: p=0,02
Czas (miesiące)
Ku
mu
low
an
a p
rop
. prz
eży
wa
jąc
yc
h
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
0 20 40 60 80 100 120 140

Ki-67<20%
n=62
Ki-67 >20%
n=51
Przeżycie całkowite
Kompletne Ucięte
Test log rank: p=0,03
Czas obserwacji (miesiące)
Ku
mu
low
an
a p
rop
. p
rze
ży
wa
jąc
yc
h
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
0 20 40 60 80 100 120 140 160
Aktywność proliferacyjna

STRATEGIA LECZENIA NIEKTÓRYCH
LOKALIZACJI RMS
• GAŁKA OCZNA » CHT (VA/IVA) +RT
» chirurgia – ograniczona do biopsji i oceny węzłów chłonnych
» z powodu bardzo dobrej prognozy dążymy do zredukowania RT
• LOKALIZACJA OKOŁOOPONOWA » bez pierwotnej a zwykle również wtórnej resekcji
» RT stosuje się najszybciej jak to możliwe
» naświetlanie całego mózgu ograniczone do lokalizacji wewnątrzczaszkowych
» w innej sytuacji – naświetlanie podstawy czaszki i okolicy guza z marginesem
• ZAJĘCIE PĘCHERZA / PROSTATY » unikanie cystektomii
» radioterapia ograniczona do pęcherza moczowego
» dalsze postępowanie zależy od odpowiedzi na leczenie
• LOKALIZACJA OKOŁOJĄDROWA » bardzo dobra prognoza
» podstawa to orchidektomia, laparotomię wykonujemy jedynie w przypadku stwierdzenia „niepewnych” węzłów chłonnych
» zwykle wystarcza mało intensywna CHT

NON-RHABDO SARCOMA
• różne pochodzenie
• indywidualna charakterystyka histologiczna
• częstsze występowanie u dorosłych niż u dzieci
• szczyty zachorowalności: • < 5 roku życia
• wczesny okres dojrzewania

SZCZEGÓLNA BIOLOGIA NIEKTÓRYCH MTM
• TENDENCJA DO WZNOWY LOKALNEJ • giant cell fibrosarcoma
• fibrous plexiform histiocytoma
• fibrous angiomatoid histiocytoma
• AGRESYWNE, PRZERZUTUJĄCE GUZY,
ZŁA PROGNOZA • extrarenal rhabdoid tumour
• intraabdominal desmoplastic small-cell tumour

OBJAWY KLINICZNE
• niebolesny guz, najczęściej szybko rosnący
• objawy wynikające z ucisku nerwów, naczyń
• mechaniczna niedrożność jelit
• powiększenie regionalnych węzłów chłonnych
• odległe przerzuty przy rozpoznaniu (10-12%)








WRAŻLIWOŚĆ NA CHT
GRUPA A
(guzy chemiowrażliwe)
leczone jak RMS
• pPNET
• EOE
• undiferentiated
sarcoma
GRUPA B
(guzy o możliwej
chemiowrażliwości)
leczenie w zależności
od oceny po
zabiegu chirurgicznym
• SS
• MFH
• infantile FS
GRUPA C
(niewykazujące
chemiowrażliwości)
leczenie chirurgiczne –
podstawa terapii
•juvenile FS
•MPNST

NAJCZĘSTSZE MTM
TYP MTM WIEK UMIEJSCOWIENIE LECZENIE/PROGNOZA
pPNET / EOE młodzież klatka piersiowa
głowa
kończyny
Agresywne leczenie systemowe.
Prognoza zależna od stadium
choroby.
Infantile
fibrosarcoma
< 2 lat kończyny
(dystalnie)
Bardzo dobre rokowanie.
W leczeniu – tylko doszczętna
resekcja.
Adolescent
fibrosarcoma
10 -15 lat kończyny
(proksymalnie)
Agresywny wzrost.
Zła odpowiedź na CHT / RT.
Złe rokowanie.
MPNST dzieci,
młodzież
kończyny
lokalizacja
pozaotrzewnowa
Agresywne leczenie chirurgiczne.
Zła odpowiedź na CHT / RT
Sarcoma synoviale młodzi
dorośli
kończyny dolne
(kolano, stopa)
Resekcja. Niepewna odpowiedź
na CHT / RT.
Rokowanie zależne od stadium.
Haemangio-
-sarcoma
rzadko u
dzieci
wątroba,
głowa i szyja
Leczenie systemowe. Wysoka
złośliwość.
Niekorzystne rokowanie.

pPNET / EOE
słabo zróżnicowana klasa guzów pozaczaszkowych i pozardzeniowych, z okrągłych, charakterystycznych komórek
dzieci i młodzi dorośli (mediana 12-15 lat)
przerzutuje do węzłów chłonnych, płuc i kości
czynniki złego rokowania: rozsiana choroba przy rozpoznaniu
w zlokalizowanych postaciach choroby : wielkość guza, lokalizacja (najgorsza – miednica, najlepsza – dystalny odcinek kończyn)
duży guz, często zajmuje sąsiadujące kości
po leczeniu chirurgicznym pacjenci często wymagają rekonstrukcji, protez

FIBROSARCOMA CONGENITAL (INFANTILE) FIBROSARCOMA
JUVENILE („ADULT TYPE”) FIBROSARCOMA
TYP DOROSŁYCH TYP DZIECIĘCY
• proksymalne odcinki kończyn,
tułów
• złe rokowanie
• radykalna resekcja
• w razie dużego stopnia choroby
resztkowej - dodatkowo RT
•dystalne odcinki kończyn
• szybko rosnący guz
• tylko leczenie chirurgiczne
• w guzach nieoperacyjnych CHT
• wznowy lokalne mogą wystąpić,
• odległe przerzuty – bardzo rzadko

SARCOMA SYNOWIALE (SS)
MONOFAZOWY
DWUFAZOWY (z domieszką tkanki nabłonkowo –
gruczołowej)
duże stawy – kolana, inne stawy kończyny dolnej
istotna jest radykalna resekcja
w nieoperacyjnych guzach – CHT + kontrola lokalna
CHT/RT
obecność odległych przerzutów – złe rokowanie

ODLEGŁE NASTĘPSTWA TERAPII
• leczenie chirurgiczne poważne ubytki funkcji
lub defekty kosmetyczne
• naświetlanie głowy zaćma, zespół suchego oka,
hipoplazja gałki ocznej,
niedoczynność przysadki
• lokalizacja pęcherzowa całkowity brak pęcherza,
upośledzenie funkcji
• następstwa CHT zaburzenia wzrostu i dojrzewania,
bezpłodność, kardiomiopatia, defekty
neurologiczne, uszkodzenie słuchu,
zwłóknienie płuc
• wzrost ryzyka rozwoju wtórnych nowotworów





GUZ JAMY BRZUSZNEJ Pomyśl o nim! Ból Powiększenie obwodu brzucha Objawy z ucisku (niedrożność mechaniczna, zanerkowa niewydolność nerek, zespół żyły czczej dolnej) Naprzemienne zaparcia i biegunki Nietrzymanie moczu
Inne objawy Krwiomocz (guz Wilmsa) Objawy wyrzutu katecholamin, nadciśnienie (Neuroblastoma) Sekrecyjna biegunka (Neuroblastoma)

GUZY JAMY BRZUSZNEJ
NEUROBLASTOMA
GUZ WILMSA
MIĘSAKI
HEPATOBLASTOMA
GUZY GERMINALNE
POZAKOSTNY MIĘSAK EWINGA




GUZ ŚRÓDPIERSIA Pomyśl o nim!
Duszność, kaszel, niewydolność oddechowa
Zespół żyły głównej górnej poszerzenie żył szyjnych
cechy krążenia obocznego
obrzęk i sinica twarzy i przedniej części klatki piersiowej
osłabienie
Nawracające zapalenia oskrzeli i płuc
Inne objawy: Zespół Hornera i ubytki neurologiczne
Objawy zespołu lizy guza (szybko proliferujące NHL)

GUZ ŚRÓDPIERSIA • Badania dodatkowe
– RTG klatki piersiowej (AP i boczne)!
– TK klatki piersiowej
– USG (płyn w jamie opłucnowej)
– Morfologia krwi obwodowej
• Skierowanie do specjalisty
– Onkolog dziecięcy
– Bez pierwotnych interwencji chirurgicznych!



GUZY KLATKI
PIERSIOWEJ
ŚRÓDPIERSIE
PRZEDNIE ŚRODKOWE TYLNE
chłoniaki
grasiczaki
potworniaki
białaczki
chłoniaki
mięsaki
przerzuty
neuroblastoma
PNET
neurofibro -
sarcoma

Wyleczalność nowotworów
u dzieci jest wysoka
i wynosi ok. 70 – 80%
ALL - 80-%
AML - 60%
HD - 98%
Guz Wilmsa - 80%
Osteosarcoma - 60%

Prz
eżyc
ie
Czas przeżycia
Nieziarniczy chłoniak złośliwy B leczony wg LMB 89 EFS w
zależności od stadium zaawansowania

1
2
3
4
Stadium I
Stadium
II
StadiumIII
Stadium IV
RMS. Prawdopodobieństwo przeżycia zależne
od stadium
Prz
eżycie
Lata

Guz rozległy
nieresekowalny całkowicie
Guz całkowicie usunięty
Lata
Prz
eżyc
ie
Osteosarcoma (COSS) EFS w zależności od zaawansowania i
możliwości wykonania pierwotnego radykalnego zabiegu
chirurgicznego

Wczesne podejrzenie
sformułowane przez
lekarza I-go kontaktu jest
najistotniejszym
czynnikiem rokowniczym
w chorobie nowotworowej













![I - [ NIKODEM - STRONA GŁÓWNA ] · Web viewEpidemiologia nowotworów u dzieci Nowotwory wieku rozwojowego stanowią ok. 0.5-2 % wszystkich nowotworów występujących u ludzi. Rodzaj](https://img.pdfslide.tips/doc/110x75/5c77e5f209d3f21d538c5b90/i-nikodem-strona-glowna-web-viewepidemiologia-nowotworow-u-dzieci.jpg)