Embed Size (px)
Citation preview

最近の安全対策行政について
日本製薬団体連合会医薬品の安全対策に関する講習会
【大阪会場】平成26年9月25日(木)
エル・おおさか(大阪府立労働センター)
【東京会場】平成26年9月30日(火)
きゅりあん(品川区立総合区民会館)
厚生労働省医薬食品局安全対策課 山本 剛

1
本日の話題1 薬事法改正への対応
・ 一般用医薬品の販売制度改正(平成26年6月施行)
・ 医薬品・医療機器等法(平成26年11月施行)
2 医療情報データベース事業
3 その他

薬事法及び薬剤師法の一部を改正する法律(概要)
2
1.医薬品の販売規制の見直し
(1)一般用医薬品:適切なルールの下、全てネット販売可能○ 第1類医薬品は、これまでどおり薬剤師が販売し、その際は、・年齢、他の医薬品の使用状況等について、薬剤師が確認・適正に使用されると認められる場合を除き、薬剤師が情報提供
○ その他の販売方法に関する遵守事項は、法律に根拠規定を置いて省令等で規定
(2)スイッチ直後品目・劇薬(=要指導医薬品):対面販売○ スイッチ直後品目※・劇薬については、他の一般用医薬品とは性質が異なるため、要指導医薬品(今回新設)に指
定し、薬剤師が対面で情報提供・指導※ 医療用から一般用に移行して間もなく、一般用としてのリスクが確定していない薬
○ スイッチ直後品目については、原則3年で一般用医薬品へ移行させ、ネット販売可能
(3)医療用医薬品(処方薬):引き続き対面販売○ 医療用医薬品については、人体に対する作用が著しく、重篤な副作用が生じるおそれがあるため、これまでどお
り※薬剤師が対面で情報提供・指導※ これまでは、省令で対面販売を規定
2.指定薬物の所持・使用等の禁止
3.施行期日
○ 公布日から半年以内(政令で規定)(1:平成26年6月12日、2:平成26年4月1日)
○ 指定薬物※について、学術研究等を除き、その所持、使用等を禁止し、違反した場合には 罰則※ 精神毒性(幻覚、中枢神経系の興奮・抑制)を有する蓋然性が高く、人に使用された場合に保健衛生上の危害の
おそれがある物質

医薬品の分類と販売方法等について
3
【従来】
【改正後】
医療用医薬品(処方薬)
対面販売(省令)
一般用医薬品
第1類 第2類
対面販売
第3類
ネット販売可
医療用医薬品(処方薬)
対面販売(法律)
一般用医薬品
第1類
ネット販売可
第3類
ネット販売可
第2類
ネット販売可
対面販売
スイッチ直後品目:スイッチOTC(原則3年後)、ダイレクトOTC(原則8年後)
要指導医薬品
対面販売
※ 重篤な副作用の発生状況や製造販売承認の拒否事由に該当する状況にないことを確認する。(安全対策部会の下の安全対策調査会において実施。)要指導医薬品は、問題がなければ、一般用医薬品に移行してから1年間は第1類医薬品となり、
その後、1年間で、安全対策部会において第1類~第3類のいずれに分類するか検討・決定する。
スイッチ直後品目
劇薬

要指導・一般用医薬品の審議について
【従来】
【改正後】
要指導・一般用医薬品部会承認の可否
(医療用から要指導医薬品・一般用医薬品への移行の可否を判断)
一般用医薬品部会承認の可否
(医療用から一般用医薬品への移行の可否を判断) 区分の移行(第1類~第3類)
医薬品等安全対策部会市販後のリスク評価
市販後調査の結果を踏まえリスク評価を実施
・ リスク区分の移行(第1類~第3類)
対策部会医薬品等安全対策部会市販後のリスク評価
市販後調査の結果を踏まえリスク評価の実施
・ 要指導医薬品から一般用への移行・ リスク区分の移行(第1類~第3類)
※要指導医薬品から一般用医薬品への移行の評価については、安全対策調査会において実施
※薬事分科会規定第3条第10項
※薬事分科会規定第3条第10項
※薬事分科会規定第3条第12項
※薬事分科会規定第3条第12項※平成25年12月20日安全対策部会申合せ
4
安全対策調査会
安全対策調査会※
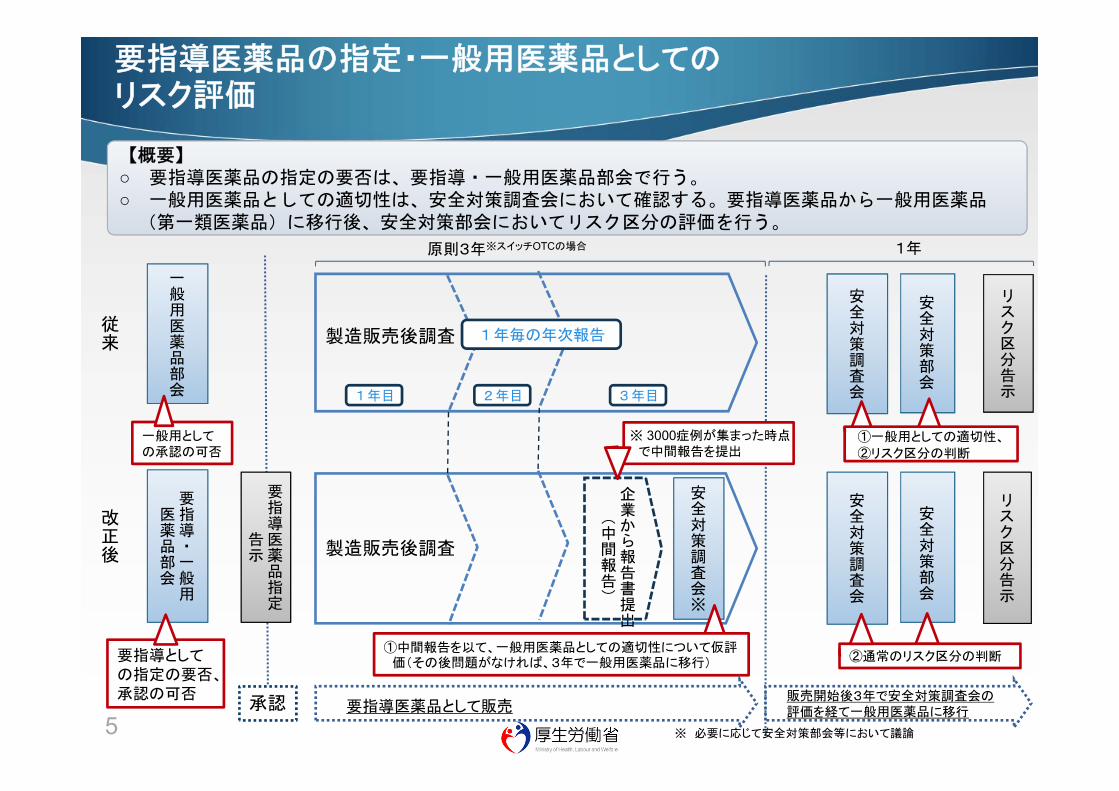
要指導医薬品の指定・一般用医薬品としてのリスク評価
5
リスク区分告示
1年
販売開始後3年で安全対策調査会の評価を経て一般用医薬品に移行
原則3年※スイッチOTCの場合
【概要】○ 要指導医薬品の指定の要否は、要指導・一般用医薬品部会で行う。○ 一般用医薬品としての適切性は、安全対策調査会において確認する。要指導医薬品から一般用医薬品
(第一類医薬品)に移行後、安全対策部会においてリスク区分の評価を行う。
改正後
安全対策部会
製造販売後調査従来
①一般用としての適切性、
②リスク区分の判断
1年毎の年次報告
企業から報告書提出
(中間報告)
①中間報告を以て、一般用医薬品としての適切性について仮評価(その後問題がなければ、3年で一般用医薬品に移行)
製造販売後調査
1年目 2年目 3年目
リスク区分告示
安全対策調査会
②通常のリスク区分の判断
※ 3000症例が集まった時点で中間報告を提出
要指導医薬品として販売
要指導医薬品指定
告示
一般用医薬品部会
要指導・一般用
医薬品部会
一般用としての承認の可否
要指導としての指定の要否、承認の可否
承認
※ 必要に応じて安全対策部会等において議論
安全対策部会

多量・頻回購入の防止
6
過去の通知による指導
昭和62年3月5日企画課長通知により、①リン酸コデイン、②リン酸ジヒドロコデイン、③塩酸メチルエフェドリンを含有する鎮咳去痰薬の内用液剤を規制。
平成22年6月1日総務課長・安対課長通知により、①リン酸コデイン、②リン酸ジヒドロコデイン、④リン酸ヒドロコデインセキサノールを含有する鎮咳去痰薬の内用剤を規制。
平成23年5月13日監麻課長通知により、⑤エフェドリン、⑥プソイドエフェトリンを含む医薬品を規制。
習慣性・依存性のある医薬品や覚醒剤原料になりうる医薬品等について、多量頻回購入を法律に基づき規制
対象薬物:通知で指導していた6成分に加えて、⑦ブロムワレリル尿素を追加
購入数量:原則1包装単位
購入にあたっての確認内容:①若年者の場合、氏名及び年齢、②他店での購入状況、③2包装以上の購入を希望する場合はその理由 等
平成26年2月26日省令公布、6月4日対象品目告示。

副作用報告(企業報告)の変更
7
○ネット販売に関する薬事法改正に対応した変更
要指導医薬品と一般用医薬品の別に関する備考欄への記載
○施行日 平成26年6月12日実施
「『市販後副作用等報告及び治験副作用等報告について』の一部改正について」 (平成26年6月5日付け薬食審査発0605第1号・薬食安発0605第1号審査管理課長・安全対策課長連名通知)「『市販後副作用等報告及び治験副作用等報告の留意点について』の一部改正について」 (平成26年6月5日付け薬機審マ発第0605001号・薬機安一発第0605001号・薬機安二発第0605001号医薬品医療機器総合機構審査マネジメント部長・安全第一部長・安全第二部長連名通知)
区分 区切り文字 コード
第一類医薬品 _ (アンダーバー) OTC1第二類医薬品 _ (アンダーバー) OTC2指定第二類医薬品 _ (アンダーバー) OTC2S第三類医薬品 _ (アンダーバー) OTC3要指導医薬品 _ (アンダーバー) YS

安全性情報報告(医療機関報告)様式の変更
8
○ネット販売に関する薬事法改正に対応した変更
要指導医薬品欄の新設
被疑薬と使用状況に関する情報をより正確に把握するため、一般用医薬品の「購入経路」(選択式)を記載する欄を追加
○報告様式の改善
健康被害救済制度に関し、副作用等の被害を受けた患者の今後の申請の意向等を把握するため、「患者が請求予定」か「患者に紹介済み」かどうかに関する選択肢を追加
「化粧品・医薬部外品安全性情報報告書」を新設
従来の報告事項にチェック欄を追加 など
○施行日 平成26年6月12日実施
「『医薬品・医療機器等安全性情報報告制度』の報告様式の変更について」 (平成26年6月12日付薬食発0612第1号医薬食品局長通知)

9
本日の話題1 薬事法改正への対応
・ 一般用医薬品の販売制度改正(平成26年6月施行)
・ 医薬品・医療機器等法(平成26年11月施行)
2 医療情報データベース事業
3 その他

薬事法改正の経緯
10
平成26年7月30日、8月6日 関係政令・省令・告示 公布
平成25年11月27日薬事法等の一部を改正する法律(平成25年法律第84号) 公布
平成26年11月25日より施行予定
・日本発の革新的な医薬品・医療機器の創出により、健康長寿社会の実現と経済成長への貢献・iPS細胞の活用等への国民の関心が高まる中、再生医療の迅速な実用化
等を実現するため、薬事法の改正を検討
「薬害肝炎事件の検証及び再発防止のための医薬品行政のあり方検討会」最終提言(平成22年4月)
厚生科学審議会医薬品等制度改正検討部会とりまとめ(平成24年1月)添付文書の位置付けの見直しをはじめとする医薬品、医療機器等の安全対策の強化や医療機器や再生医療
製品の特性を踏まえた制度の構築を検討すること等が提言

薬事法改正の概要① 安全対策の強化
11
【添付文書の位置付け等の見直し】(1) 医薬品等の製造販売業者は、最新の知見に基づき添付文書を作成し、厚生労働大臣に届け出るものとする。
併せて、迅速な情報提供を行う観点から、届け出た添付文書を直ちにウェブサイトに掲載することとする。
【その他の改正事項】
(2) 薬事法の目的に、保健衛生上の危害の発生・拡大防止のため必要な規制を行うことを明示。(3) 医薬品等の品質、有効性及び安全性の確保等のための関連事業者、医療従事者等の関係者の役割の明確化。(4) 医療機関の副作用等の報告先を、製造販売業者の報告先と一元化して独立行政法人医薬品医療機器総合機構
(PMDA)とし、国はPMDAに情報の整理等を行わせることができることとするほか、必要な市販後安全対策を講じる。
○ 医薬品・医療機器等の実用化を促進するに当たっては、併せて、安全対策を強化することが必要である。
○ 医薬品、医療機器等に添付する添付文書は、使用上の注意等を現場に伝える重要なものであり、薬害肝炎事件の検証において、添付文書の位置付けを改めるべきことが指摘されている。また、添付文書は常に最新の知見が反映されていることが重要であるが、現行の薬事法では、これが明確となっていない。
○ このため、添付文書の位置付け等を見直すこと等により、医薬品、医療機器等に係る安全対策の強化を図ることが必要。
改正の背景

【医薬品と別個の章を新設・法律の名称にも明示】(1) 医療機器の製造販売業・製造業について、医薬品等と章を区分して規定する。(2) 「薬事法」の名称について、医療機器を明示。
※改正後の名称は「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」(略称:医薬品医療機器等法)とする。
【迅速な実用化に向けた規制・制度の簡素化】(3) 民間の第三者機関を活用した認証制度を、基準を定めて高度管理医療機器にも拡大する。これにより、
PMDAの審査について新医療機器に重点化・迅速化を図る。(例)歯科インプラント、コンタクトレンズなど
※ このほか、製造販売の認証を受けた者の地位の承継、登録認証機関の業務規程の認可、厚生労働大臣による認証取消し等の命令など、認証制度の拡大に合わせた所要の規定の整備を行う。
薬事法改正の概要②医療機器の特性を踏まえた規制の構築(1)
12
○ 医療機器は、パソコン等の他の機械製品と同様に短いサイクルで改善・改良が行われた製品が市場に供給される場合が多いことなど、医薬品と異なる特性を有している。
○ 新医療機器の開発・実用化については、医療の質の向上に寄与するとともに、我が国の経済成長を牽引する産業分野としても期待されているが、承認・上市に時間がかかる等といった課題も指摘。
○ さらに、医療機器の国際展開を進めるためには、国際整合性に配慮する必要がある。
○ このため、医療機器の特性を踏まえた制度改正を行い、医療機器の迅速な実用化と規制の合理化を図ることが必要。
改正の背景

薬事法改正の概要③医療機器の特性を踏まえた規制の構築(2)
13
【単体プログラムの位置付けの明確化】(4) 単体プログラムについて、欧米では既に医療機器として位置付けられていることを踏まえ、これを医療機器
の範囲に加え、製造販売等の対象とする。(例)MRI等で撮影された画像データの処理、保存、表示等を行うプログラム
【その他の改正事項】(5) 医療機器の製造業について、許可制・認定制から登録制に改め、要件を簡素化する。
(6) 承認・認証において、個別製品ごとに行われていたQMS調査(製造管理・品質管理が基準に基づいて行われているかの調査)を合理化し、製品群(医療機器の特性等に応じて種類別に大くくりしたもの)単位で調査を実施することとする。※ 既にQMS調査で基準に適合している製品と同じ製品群に属する製品についてのQMS調査が原則免除されることとなる。
なお、都道府県によるQMS調査は廃止し、認証機関とPMDAに集約する。
(7) 現行の再審査・再評価に代えて、厚生労働大臣が指定する医療機器(※)について、製品の特性に応じて期間を設定し、当該期間中に使用成績に係る調査を行い、有効性や安全性を確認することとする。※人工心臓など長期間に渡って体内に留置される製品を想定。
(8) 高度管理医療機器等の賃貸について、業として対価を得ずに貸与を行う場合についても、許可又は届出の対象とする。
(9) 医療機器を医療機関等に販売する際に、ウェブサイトに情報を掲載すること、医療機関の了解があること等の一定の条件を満たした場合は、添付文書の製品への添付を省略できることとする。

【医薬品・医療機器と別個の定義付け】
(1) 医薬品や医療機器とは別に「再生医療等製品」を新たに定義し、再生医療等製品の「章」を設ける。
<再生医療等製品の範囲>・人の細胞に培養等の加工を施したものであって、①身体の構造・機能の再建・修復・形成や、②疾病の
治療・予防を目的として使用するもの、又は・遺伝子治療を目的として、人の細胞に導入して使用するもの※これらはいずれも人の細胞等を用いることから、品質が不均一であり、有効性の予測が困難な場合があるという特性を
有している。
【条件及び期限付承認制度の導入】(2) 均質でない再生医療等製品については、有効性が推定され、安全性が確認されれば、条件及び期限付きで特
別に早期に承認できる仕組みを導入する。その場合、承認後に有効性・安全性を改めて検証する。※ 条件及び期限については、販売先を専門的な医師や設備を有する医療機関等に限定する条件や、原則として7年を超えな
い範囲内の期限を想定。また、承認を受けた者は、期限内に使用成績に関する資料等を添付して、再度承認申請を行うことが必要。
薬事法改正の概要④再生医療等製品の特性を踏まえた規制の構築(1)
14
○ iPS細胞等による再生医療は、革新的な医療として実用化に向けた国民の期待が高い。一方で、安全面などの課題が存在。
○ このため、再生医療等製品については、安全性を確保しつつ、迅速な実用化が図られるよう、その特性(※)を踏まえた制度等を設けることが必要。
※ 再生医療等製品の主な特性人の細胞等を用いることから個人差などを反映し、品質が不均一となること
改正の背景

薬事法改正の概要⑤再生医療等製品の特性を踏まえた規制の構築(2)
15
【安全対策等の整備】(3) 医師等は、製品の使用に当たって患者に対して適切な説明を行い、使用の同意を得るよう努めるものとする。(4) 使用成績に関する調査、感染症定期報告や使用の対象者等に係る記録と保存など、市販後の安全対策を講じ
る。※ 厚生労働大臣が指定した再生医療等製品については、製造販売業者は長期に記録を保存するとともに、医療機関は使用の対
象者等について記録・保存しなければならないこととする。
(5) 再生医療等製品による健康被害について、副作用被害救済制度及び感染等被害救済制度の対象とする。(*独立行政法人医薬品医療機器総合機構法関係)
【その他の改正事項】(6) 製造所における製造管理又は品質管理の基準を作成し、品質・安全性等を確保する。(7) 業として人体から採血することは原則禁止されているが、再生医療等製品について、その製造業者や医療機
関が人体から採取した血液を原料として、製品を製造することを可能とする。(*安全な血液製剤の安定供給の確保等に関する法律の改正)
4.施行期日公布の日(平成25年11月27日)から起算して1年を超えない範囲内において政令で定める日とする。(平成26年11月25日施行予定)

16
本日の話題1 薬事法改正への対応
・ 一般用医薬品の販売制度改正(平成26年6月施行)
・ 医薬品・医療機器等法(平成26年11月施行)
2 医療情報データベース事業
3 その他

協力
期待される成果:医薬品等の迅速で的確な安全対策の実施①同種同効薬での副作用の発生頻度などの比較②薬の副作用であるのか、病気自体の症状によるものかの判別③安全対策の措置が副作用の低減に十分な効果があったか検証
医薬品医療機器総合機構(PMDA)
副作用情報等の安全性情報の収集及び分析
研究者・製薬企業
迅速な安全対策
拠点病院データベース
データベース
全国10カ所ネットワークの形成
拠点病院
データの調査分析
医療情報データベースを活用した薬剤疫学的手法による医薬品等の安全対策を推進する。 1,000万人規模のデータを収集するための医療情報データベースを拠点病院に構築するとと
もに、 独立行政法人医薬品医療機器総合機構(PMDA)に情報分析システムを構築する。平成23年度 より5年計画で事業を実施中。
拠点病院
データベース
拠点病院
データベース
拠点病院
データベース
※10拠点病院東北大、東大、千葉大、NTT病院、北里大、 浜松医大、徳洲会、香川大、九大、佐賀大
平成26年度予算 1.1億円(国費分) ※全事業費を国費とPMDAで半分ずつ負担。
データベース構築と活用推進
電子カルテデータ
レセプトデータ
オーダリングデータ
検査データ
医療情報データベース基盤整備事業
17

副作用報告制度の限界と医療情報の活用で期待される安全対策
18
現在の副作用報告の限界その医薬品を投与されている人数を把握できない。(分母が不明のため発生頻度が不明)
他剤との副作用発生頻度の比較、安全対策措置前後での副作用発生頻度の比較等をできない。
原疾患による症状と「副作用」の鑑別が難しい。
医薬関係者が報告しなければ、副作用の存在が分からない。
医療情報の活用により可能となる安全対策の例
症状の発生割合
(症状/正確な使用患者数)
A薬治療群 A薬なしの治療
副作用の発生割合
(副作用/正確な使用患者数)
副作用発生割合(率)を措置の前後で比較できる
他剤との比較
例えば、投与後の異常な行動。A薬なしでも発生する。
緊急安全情報前 緊急安全情報後
副作用発生割合
(副作用/正確な使用患者数)
A薬治療群 B薬治療群
同種同効薬との発現頻度の比較ができる。
原疾患による症状発現との比較 安全対策の効果の検証

1000万人規模の医療情報DBの必要性
19
米国、欧州等では、既に1000万人~数千万人規模のデータベースが存在し、医薬品安全対策に積極的に利用し始めている。
(例)・ダビガトランの重篤出血リスク市販後に重篤な出血リスクが問題となり、日本をはじめとして世界的にも対策が講じられた。FDAのミニセンチネルプロジェ
クトによる、大規模データベースを利用したリスク評価の結果、他剤に比較してリスクが高まらないとの評価。
・オルメサルタンのセリアック病のリスクオルメサルタンについて予想外に多数のセリアック病の報告(AERS)が見られたため、ミニセンチネルプロジェクトにより、本
薬と他のARBの使用者での発現率比較を実施。本薬のセリアック病のリスクは他のARBと大差なかった。オルメサルタンに
ついて予想外に多数のセリアック病の報告(AERS)が見られたため、ミニセンチネルプロジェクトにより、本薬と他のARBの使
用者での発現率比較を実施。本薬のセリアック病のリスクは他のARBと大差なかった。
・ACEI/ARB等の血管浮腫リスクβ遮断薬に比較して、ACEIでは血管浮腫のリスクが約3倍高く、ARBに伴う血管浮腫のリスクはACEIより低かったとの評価
。
日本人患者について、諸外国に匹敵する評価を可能にするためには、数100万~1000
万人規模のデータベースが最低限必要。
(注)年間100万人に用いられている大型医薬品の場合でも、例えば0.01%レベル(10万人に10人)の稀な副作用
について2倍のリスク上昇を検出するには、検討対象医薬品について10万人規模の使用者数が確保できる
データベースが必要であり、このレベルの使用者数を確保するためには、1000万人規模のDBが必要。

システム開発の現状・今後の予定
20
H23年度 H24年度 H26年度
協力医療機関選定
H25年度 H28年度H27年度
仕
様
策
定
システムのテスト・試行利活用
システムの本格運用
6病院システム導入
バリデーション1病院システム開発・導入・データ蓄積
バリデーション
バリデーション
PMDA分析システム開発・導入
データ蓄積
H23年度導入病院(1病院)東大
H24年度導入病院(6病院)東北大、浜松医大、香川大、九大、佐賀大、徳洲会(グループ)
H25年度導入病院(3病院)千葉大、北里(グループ)、NTT病院(グループ)
データマッピングの精緻化を協力医療機関・PMDA共同で実施
データ
マッピング 引き続き実施
複数医療機関のデータの統合分析
システム改修
↑3病院システム導入
データ蓄積

協力医療機関C
協力医療機関C
協力医療機関C協力医療機関C協力医療機関C協力医療機関C
協力医療機関C
データベース構成概念図
21 21
協力医療機関B
協力医療機関A
10医療機関の統合分析センター(PMDAが管理)
抽出結果
標準化ストレージ
電子カルテ等システム
匿名化
・・・
抽出結果
協力医療機関H抽出結果
統合・標準化 統計処理・
集計結果
統計処理・集計結果
統計処理・集計結果
PMDA
リモートログイン
10医療機関それぞれの了承後送信
各医療機関の集計結果が中心必要時は医療機関の合意のもとで抽出結果も分析

日本再興戦略(平成25年6月14日閣議決定)
22
○医療・介護情報の電子化の促進
・ 医薬品の副作用データベースシステムについて、データ収集の拠点となる病
院の拡充や地域連携の推進を図ることにより、利活用できる十分な情報を確保
し、医薬品の有効性・安全性評価や健康長寿の延伸につなげる。
○地域連携の推進
・地域連携の在り方の検討→本事業の拠点病院を中心とした効果的な地域連携の在り方
・拠点病院の基準の検討 →本事業の主旨に合致した「標準的な病院」の検討
○データ収集の拠点となる病院の拡充
・拠点病院の拡充 →事業参加病院確保のため、協力拠点病院のメリットを提示
・症例集積手法の見直し
○医療情報データベースの本格運用にかかる体制整備
・DB本格運用の体制 →PMDAにおけるインフラと運用の管理体制の整備
・DB本格運用の財源 →データベース管理・事業運用にかかる経費の財源の検討

行政事業レビュー公開プロセス評価(平成25年6月21日)
23
評価結果:[事業全体の抜本的改善]とりまとめコメント:データベースの規模や達成時期等の検証・明
確化、手法の再検討、費用負担の在り方の検証を念頭に更なる見直しを行い、概算要求へ反映させることが必要
(主な指摘)
①1000万人規模のデータとする必要性が明確でない。(数百万人規模のデータであれば意味がないのではないか)
②本事業に参加する医療機関のメリットを明確にし、拡大を図るべき。
③既存事業の10病院は事業目的と合致していない。病院に来る前の治療情報を得るため、レセプトデータとの連携が必要ではないか。
④財源を見直すべき(製薬企業の負担を考慮すべき)
⑤1000万人規模に到達する道筋、将来構想を明確にすべき。 等

医療情報DB基盤整備事業のあり方に関する検討会
24
1.趣旨医療情報データベース基盤整備事業(以下「本事業」という。)における進捗や状況の変化に伴い
対応が必要となる各種の課題を改めて整理し、今後の本事業のあり方について検討し、今後の政策に反映することを目的とする。
2.検討事項(1)本事業のあり方に関する検討(2)本事業の協力医療機関・連携医療機関の拡充等のあり方に関する検討(3)その他、必要に応じて本事業の実施に当たり必要な検討
3.構成員 ○:座長
青木 事成 日本製薬団体連合会赤沢 学 明治薬科大学公衆衛生・疫学教室教授秋山 祐治 川崎医療福祉大学医療情報学科教授石川 広己 日本医師会常任理事井出 健二郎 和光大学経済経営学部教授大江 和彦 東京大学大学院医学系研究科医療情報経済学分野教授川上 純一 浜松医科大学医学部附属病院薬剤部教授土屋 文人 日本薬剤師会副会長冨山 雅史 日本歯科医師会常務理事
○永井 良三 自治医科大学学長松村 泰志 大阪大学大学院医学系研究科医学専攻教授山口 拓洋 東北大学大学院医学系研究科医学統計学分野教授山本 隆一 医療情報システム開発センター理事長

本検討会の提言の取りまとめ(1)
25 25
医療情報データベース基盤整備事業のあり方に関する検討会報告書(平成26年7月1日)
1. 従来の副作用報告制度では困難であった副作用の定量的な評価や低頻度であるが重大な影響を与えるリスクの迅速な検出等を可能とする医療情報DBを活用した薬剤疫学的手法の平成28年度以降の本格運用に向けて、10拠点における基盤整備を進めるべき。
2. 試行期間における10拠点の患者データ(300万人規模)の利活用実績を評価した上で、より有用性の高い1,000万人規模を(地域連携等も視野に入れ)目指すべき。
3. 本格運用に向けて、実践的な利活用を可能とする体制整備に必要な予算・人員の確保が必要。

本検討会の提言の取りまとめ(1)
26
4. 試行期間における利活用の実績等も踏まえて、本格運用後のルールづくりが必要。
5. 本格運用開始後の運営に必要な費用・人員等の精査とともに、利用者負担も含めた費用負担の枠組み構築に向けて、引き続き検討が必要。
6. 短期的には、DB整備及び薬剤疫学研究等に必要な人材の確保・育成、中長期的には、必要な環境(社会的合意や法令等)が整備され、各種DB間の情報連携が技術的にも可能となった際に、情報の長期追跡性・正確性等の向上を図った形での横断的な利活用の推進に向けて引き続き検討が必要。
7. 今後の拠点病院の拡充にあたっては既存の基盤を活用したコンパクトなシステム導入やデータの標準化等の効率化を図ることが重要。

27
本日の話題1 薬事法改正への対応
・ 一般用医薬品の販売制度改正(平成26年6月施行)
・ 医薬品・医療機器等法(平成26年11月施行)
2 医療情報データベース事業
3 その他

安全確保業務と市販直後調査
28
医薬品、医薬部外品、化粧品及び医療機器の製造販売後安全管理の基準に関する省令(GVP省令)に規定
製造販売業者は、安全管理を適正かつ円滑に行うため、以下の製造販売後安全管理業務手順書を作成しなければならない・情報の収集
・情報の検討・安全確保措置の立案
・安全確保措置の実施
・安責から総責への報告
・安全管理実施責任者から安責への報告
・市販直後調査
・自己点検
・製販後安全管理業務従事者への教育訓練
・製販後安全管理業務の記録保存
・品責、他の業務責任者との相互連携
・他の製販後安全管理業務の適正・円滑実施
SOP作成(5条1項)
・販売を開始した後の6ヶ月間、診療において、医薬品の適正な使用を促し、副作用報告義務の症例等の発生を迅速に把握するために行うもの
・承認条件として付されるもの
市販直後調査(2条3項)
・製販業者は、SOP等に基づき、以下の安全管理情報を安責又は安全管理実施責任者に収集させ、その記録を作成させなければならない。①医療関係者からの情報、②学会報告等研究報告に関する情報④外国政府、外国法人等からの情報
安全管理情報の収集(7条)

市販直後調査の徹底
29
新規性が高く、国内の治験症例が少ない新医薬品若しくは、重点監視医薬品について、市販後一定期間、臨床現場の企業の活動状況、医薬品使用状況等の情報を直接収集・評価の上、
必要な対応を図る。
販売
副作用報告
・納入前説明・市販直後調査協力依頼・情報提供
重要な添付文書改訂指示等
医療機関
医薬品企業
厚生労働省製品情報の提供、副作用等の把握・市販直後調査中間報告・添付文書改訂のお知らせ・適正使用のお願い・イエローレター・ブルーレター
副作用情報6ケ月
副作用報告
承認
・倫理的無償提供・選定療養・臨床使用医薬品
定点観測・全国6カ所程度の医療機関・医師と薬剤師の先生各1名・月1回定期的に報告を受ける

副作用報告に基づく安全対策の実施
30
統合失調症治療薬について、4月17日開催
の安全対策調査会の審議結果に基づき、同日、安全性速報を発出。
副作用と本剤の因果関係は不明であったが、使用状況から、添付文書を改訂し、適正使用を強化。

事業参加医療機関の報告より
31
・ 最初の説明会以後、まったく訪問がない。
・ 市販直後調査への協力依頼はなかった。(処方医師)
・ MRが「市販直後調査」について理解していなかった。
・ MRが訪問しても副作用情報の収集・提供は行わず、プロモーションばかりだった。提携販売業者のMRのためか不勉強(質問には迅速な対応があった)。
・ 販売当初、安全なのでたくさん使用してくださいという営業活動があった。禁忌等の説明が不十分だった。
副作用症例を根拠とする使用上の注意の改訂等の指示を期に情報提供活動に力を入れるようになった感がある。
・ 副作用情報や市販直後調査結果の情報提供が遅い。適正使用のお願いの発行日の翌営業日の夕方でもMRが把握していなかった。
・ 未知の副作用報告が製造販売業者に把握されていなかった。

事業参加医療機関の報告より
32
(医師には情報提供等があるが)薬剤部、DI室への情報提供等が少ない。要請したら薬剤部向けに説明会を開いてくれた。適正使用のお願いについて担当MRから薬剤部への情報提供がなかった。
「医療用医薬品の市販直後調査の実施方法等について」(平成18年3月24日付け薬食安発第0324001号厚生労働省医薬食品局安全対策課長通知)2 製造販売業者は、その製造販売した医薬品を使用する医療機関に対し、原則として、納入前に次のような説明及び協力依頼を医薬情報担当者により行うこと。(略)
3 製造販売業者は、当該医薬品を使用する医療機関に対し、原則として、納入開始後2か
月間は、おおむね2週間以内に1回の頻度で、その後も適切な頻度(おおむね1か月以内に1回)で、協力依頼等を行うこと。
市販直後調査の趣旨(適正使用の促進、重篤な副作用等の発生の迅速な把握)について理解し、適切な実施を願う。
薬剤部への協力依頼・情報提供は法令上必ずしも必要ではないが、効果的な情報伝達・収集の実施のためご配慮願いたい。

ノバルティス社における安全管理上の問題事例
3333
ノバルティス社による社内調査や社外調査委員会の調査等から判明した問題点○ 同社が関与した臨床研究及びアンケート調査において、複数のMRが有害事象を把握していたにも関わらず、
安全管理統括部門に伝えてなかった。⇒これらの症例は、当然、重篤性の評価も行われず、副作用の期限内の報告が行われていない。
① 臨床研究やアンケート調査等で知り得た有害事象など、医療関係者等からの自発的、積極的な報告以外の方法により知り得た情報についても、安全管理統括部門に報告すべき対象であることを明確にすること、
② 有害事象等の報告について、全てのMRから安全管理統括部門に確実に報告される体制をとること及びそのための教育訓練を実施すること
等の改善を命令
(ノバルティス社の改善計画の内容(8月29日付))① 情報源にかかわらず、知り得た全ての有害事象が報告対象であることを業務手順書で明記② 業務報告システムの改善、教育研修の充実、安全管理実施責任者への注意喚起、有害事象報告業務を人事評価項目に追加、
社内監査及び第三者による定期的な点検の実施 等
・ 臨床研究やアンケート調査など、通常の医療機関の自発報告と異なる有害事象も安全管理統括部門に報告されることになっているか
・ MR以外に有害事象の情報を把握した者も報告することになっているか
安全管理上の問題点
平成26年7月31日 ノバルティス社に対する行政処分(業務改善命令)製造販売業者におけるGVP省令等の遵守について(平成26年8月4日付薬食安発0804第2号通知)製造販売業者におけるGVP省令等の遵守について(平成26年8月4日付薬食安発0804第2号通知)
製造販売業者における留意事項

「PMDAメディナビ」への登録のお願い
34
PMDAでは、医薬品・医療機器情報提供ホームページで、安全性情報等を提供するとともに、掲載情報をメール配信サービス「PMDAメディナビ」 により、登録者に対し、迅速にメール配信している。
医療関係者、医薬関係者への安全性情報等の迅速な提供のため、一層の登録推進が必要。 平成25年11月よりスマートフォン用のページを開設し、より使い易くしている。
配信される情報厚生労働省緊急安全性情報緊急安全性情報・安全性速報医薬品・医療機器等安全性情報使用上の注意改訂情報医療機器自主点検通知回収情報(クラスⅠ回収)承認情報PMDA医療安全情報DSU(医薬品安全対策情報)医薬品の評価中のリスク等情報関連通知
引き続き、PMDAメディナビの登録及び関係者への周知をお願いします。
http://www.info.pmda.go.jp/info/idx-push.html
スマートフォン用のページ 登録件数の推移
H26年3月31日
102,790件

35
御清聴ありがとうございました。





























