Embed Size (px)
Citation preview

Phasenübergänge in Festkörpern
Phänomenologie und Thermodynamik
Skriptum zu einer Vorlesung im Wintersemester 2010
Wolfgang Püschl
Fakultät für Physik
Universität Wien

Phasenübergänge in Festkörpern- Phänomenologie und Thermodynamik
1. Einleitung ..................................................................................................................... 1
1.1. Motivation ........................................................................................................ 1 1.2. Historische Perspektive .................................................................................... 1
2. Klassifizierung der Phasenübergänge ........................................................................... 6
3. Phasengleichgewichte .................................................................................................. 11
3.1. Thermodynamische Grundlagen 3.2. Thermodynamik von Lösungen (Mischkristallen) .......................................... 13 3.3. Gibbs’sche Phasenregel .................................................................................... 16 3.4. Klassifizierung der Typen von Phasendiagrammen ....................................... 17 3.5. Ternäre Phasendiagramme .............................................................................. 23
4. Diffusion………………………………………………………………………………24
4.1. Mechanismen………………………………………………………..………...24 4.2. Statistische Betrachtung der Diffusion………………………………………..24 4.3. Korrelation…………………………………………………………………….29 4.4. Atomare Zufallsbewegung mit chemischer treibender Kraft………………... 32 4.5. Nicht-gegengleiche Diffusion, Kirkendall-Effekt…………………………….33
5. (Kontinuierliche)Ausscheidung ……………… ........................................................... 36 5.1. Keimbildung……………................................................................................ 39
5.1.1. Die klassische Keimbildungstheorie .................................................. 42 5.1.2. Einschränkungen und Modifikationen der K.K.T. ............................. 44 5.1.3. Nichtklassische Keimbildung .............................................................. 47 5.1.4. Clusterdynamik ................................................................................... 48
5.2. Spinodale Entmischung ................................................................................... 50 5.2.1. Die klassische Theorie (Cahn-Hilliard) ............................................... 48 5.2.2. Vergleich mit dem Experiment und verbesserte Theorien .................. 53
5.3. Wachstum und Vergröberung........................................................................... 57 5.3.1. Die LSW (Lifshitz-Slyozov-Wagner)-Theorie .................................... 60 5.3.2. Verfeinerungen der Vergröberungstheorie .......................................... 62
6. Erweiterte Betrachtung thermisch aktivierter Phasenübergänge ................................ 64
6.1. Allgemeine Beschreibung der Transformationskinetik .................................. 64 6.2. Diskontinuierliche Ausscheidung .................................................................... 67 6.3. Eutektoide Reaktion ......................................................................................... 68 6.4. Polymorphe Umwandlungen ........................................................................... 75 6.5. Massive Transformation .................................................................................. 76
7. Martensitische Umwandlungen .................................................................................. 79 7.1. Charakter der Martensitischen Umwandlung ................................................. 79 7.2. Die Kristallographie des Martensits ................................................................ 81 7.3. Bildung von Martensitkeimen ......................................................................... 86 7.4. Kinetik der Martensitumwandlung ................................................................. 87 7.5. Transformations-induzierte Plastizität (TRIP) ................................................ 89 7.6. Mechanische Effekte bei thermoelastischen Martensitumwandlungen ........... 91

7.6.1. Der Formgedächtniseffekt ................................................................... 91 7.6.2. Superelastizität ..................................................................................... 94
Literatur……………………………………………………………………………………96

1
1. Einleitung
1.1. Motivation Phasenumwandlungen im festen Aggregatzustand haben eine große Bedeutung für die technologische Herstellung von Werkstoffen, insbesondere von Metallen. Im Herstellungsprozess werden die Metalle legiert, gegossen bzw. gesintert, Temperaturwechseln und mechanischer Deformation wie Schmieden, Walzen, Tiefziehen etc. unterworfen. Dabei kommt es zu einer Fülle von Umwandlungsprozessen, deren Folge die gewünschte Mikrostruktur ist, in der Regel ein mehrphasiges Gefüge. Bei den besonders wichtigen Ausscheidungsreaktionen (precipitation reactions) ist eine Legierungskomponente bei hohen Temperaturen löslich und fällt bei tieferen Temperaturen als zweite Phase aus (wichtig v.a. bei Al-, Cu- und Ni- Legierungen). Die fein verteilten Partikel der Ausscheidungsphase wirken als Hindernisse (Verankerungspunkte) für Versetzungen (dislocations), das sind lineare Gitterbaufehler, über die die Verformung der Metalle abläuft. Dadurch wird Verfestigung (Härtung) erzielt. Ordnungsübergänge sind für die Entstehung und Stabilität (nah- oder fern-) geordneter Phasen verantwortlich. In diesen herrschen besondere Bedingungen und Einschränkungen für die Bewegung von Versetzungen, mit deren Hilfe gewünschte mechanische Eigenschaften wie etwa Hochtemperaturfestigkeit (Beispiel: Superlegierungen auf Basis intermetallischer Verbindungen, etwa Ni3Al) gewonnen werden. Durch gezielte Kombination mehrerer Phasen können gute Eigenschaften kombiniert werden wie etwa Festigkeit und Bruchzähigkeit. Die mehrphasigen Gefüge haben also Auswirkungen auf die mechanischen Eigenschaften), aber auch auf den elektrischen Widerstand (Streuung der Leitungselektronen an Ausscheidungsteilchen und Phasengrenzflächen, bessere Leitung in ferngeordneten Phasen) und magnetische Eigenschaften. So ist z.B. die Ausbildung ferromagnetischer Weissscher Bezirke durch das Gefüge eingeschränkt.
1.2. Historische Perspektive Metallbearbeitung und damit Phasenumwandlungen und die Herstellung mehrphasiger Gefüge im Zuge technologischer Herstellungsprozesse haben bereits prähistorisch große Rolle gespielt. Fortschritte darin waren bekanntlich Auslöser kultureller Umwälzungen (Bronzezeit, Eisenzeit etc.). Auf rein empirischer Grundlage wurden dabei z. T. hervorragende Ergebnisse erzielt. Als Beispiel ist das durch kunstvolle Schmiedebearbeitung hergestellte Gefüge einer Fe-Legierung (Stahl einer Damaszenerklinge) in Abb. 1-1 dargestellt.

2
Abb. 1-1
Frühe Behandlung metallurgischer Themen sind schon bei Plinius, Biringuccio (1540), Agricola („De Re Metallica 1556, Abb. 1-2) sowie bei Naturphilosophen des 17. und 18. Jahrhunderts (Francis Bacon, Hooke, Newton, Réaumur) zu finden. Elastizitätstheorie Cauchy, Poisson ca. 1820.
Abb. 1-3
Abb. 1-2 Der Österreicher Aloys von Widmannstätten zerschnitt 1808 einen Eisenmeteor, polierte und ätzte die Schnittfläche und machte dadurch ein Figurenmuster sichtbar (Abb. 1-3).
Damals hat man allerdings darin noch nicht ein mehrphasiges Gefüge erkannt.

3
Wichtige frühe Meilensteine auf dem Weg zu einem wissenschaftlichen Verständnis der Phasen im Festkörper waren: ~1864 ff Sorby Beobachtung von Kristallschliffen in polarisiertem Licht 1876 Gibbssche Phasenregel 1890 Van ’t Hoff 1890 Analogie zwischen festen und flüssigen Lösungen 1896 William Roberts-Austen Diffusion von Gold in festem Blei.
Eine systematische Untersuchung auf quantitativer wissenschaftlicher Grundlage setzte im Rahmen der physikalischen Metallkunde (physical metallurgy) etwa vor 100 Jahren ein: Die thermodynamische Grundlage für das Verständnis der Phasengleichgewichte lieferten die theoretischen Arbeiten des bedeutenden amerikanischen Physikers Josiah Willard Gibbs (1839-1903, Abb. 1-4) ~1884 Heycock und Neville erstellen das erste präzise Phasendiagramm des Cu-Sn-Systems.
Abb. 1-5
Abb. 1-4 Beginn des 20. Jahrhunderts: Gustav Tammann (Göttingen) (Abb. 1-5): Erstellung einer großen Anzahl metallischer Phasendiagramme, Erforschung von Keimbildung und Wachstum.
Ausscheidungshärtung: Adolf Wilm: Entwicklung von Duralumin 1903-1909 Al - (3-4,5%) Cu (Gewichtsprozente) - (0,4-1%) Mg
- (0-0,7%) Mn - (0,4-1%) Fe - (0-0,7%) Si
4
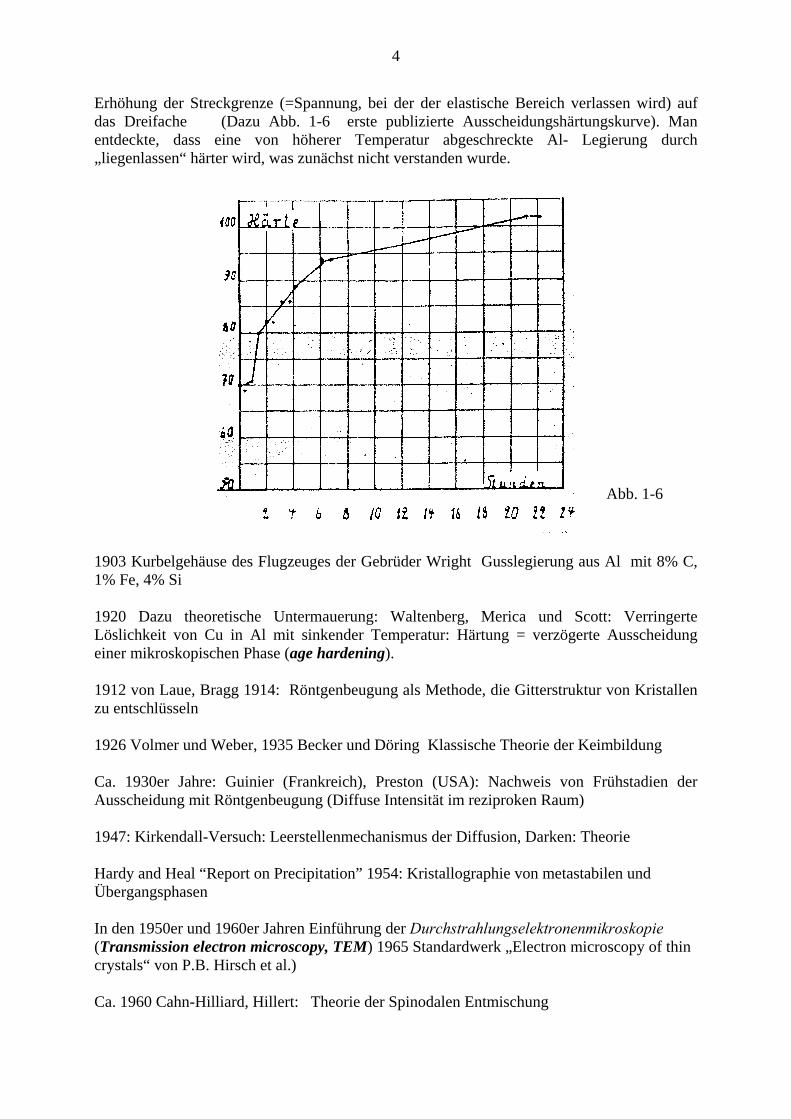
Erhöhung der Streckgrenze (=Spannung, bei der der elastische Bereich verlassen wird) auf das Dreifache (Dazu Abb. 1-6 erste publizierte Ausscheidungshärtungskurve). Man entdeckte, dass eine von höherer Temperatur abgeschreckte Al- Legierung durch „liegenlassen“ härter wird, was zunächst nicht verstanden wurde.
Abb. 1-6
1903 Kurbelgehäuse des Flugzeuges der Gebrüder Wright Gusslegierung aus Al mit 8% C, 1% Fe, 4% Si 1920 Dazu theoretische Untermauerung: Waltenberg, Merica und Scott: Verringerte Löslichkeit von Cu in Al mit sinkender Temperatur: Härtung = verzögerte Ausscheidung einer mikroskopischen Phase (age hardening). 1912 von Laue, Bragg 1914: Röntgenbeugung als Methode, die Gitterstruktur von Kristallen zu entschlüsseln 1926 Volmer und Weber, 1935 Becker und Döring Klassische Theorie der Keimbildung Ca. 1930er Jahre: Guinier (Frankreich), Preston (USA): Nachweis von Frühstadien der Ausscheidung mit Röntgenbeugung (Diffuse Intensität im reziproken Raum) 1947: Kirkendall-Versuch: Leerstellenmechanismus der Diffusion, Darken: Theorie Hardy and Heal “Report on Precipitation” 1954: Kristallographie von metastabilen und Übergangsphasen In den 1950er und 1960er Jahren Einführung der Durchstrahlungselektronenmikroskopie (Transmission electron microscopy, TEM) 1965 Standardwerk „Electron microscopy of thin crystals“ von P.B. Hirsch et al.) Ca. 1960 Cahn-Hilliard, Hillert: Theorie der Spinodalen Entmischung

5
Ab ca. 1970- zunehmendes Interesse an Formgedächtnis-Legierungen (Shape Memory Effect ) unter martensitischer Umwandlung. ~1970 ff. Hochauflösende Elektronenmikroskopie: „Atome sehen“. Ab 1980er-1990er Jahre: Steigende Computerleistung ermöglicht theoretische Fortschritte bei quantenmechanischen „voraussetzungslosen“ (first principles) Berechnungen auf Basis von Dichtefunktionalmethoden. Berechnung der Entwicklung von Mikrostrukturen mit kinetischen Monte-Carlo-Simulationen. 21. Jahrhundert: Zunehmende Bedeutung von dimensional beschränkten Strukturen: Dünnschichten, Oberflächen, Nanostrukuren (auch im Zusammenhang mit der Technologie extrem miniaturisierter elektronischer Schaltungen).

6
2. Klassifizierung der Phasenübergänge Ein Aggregat von Atomen oder Molekülen, das unter vorgegebenen äußeren physikalischen Bedingungen (Temperatur, Druck, Magnetfeld, Gravitation...) einen thermodynamischen Gleichgewichtszustand erreicht hat, besteht aus einer oder mehreren Phasen. Wie ist eine Phase definiert? Es handelt sich um ein homogenes (bis auf Gitterbaufehler (defects)) und eindeutig unterscheidbares Gebiet, in dem intensive Parameter wie Dichte, Zusammensetzung, Gitterstruktur etc. gleich sind. Wenn die äußeren Parameter verändert werden, dann kann es zu Phasenumwandlungen kommen, wobei folgende Fragen interessant sind:
1) Warum kommt es zu einer bestimmten Phasenumwandlung? 2) Was ist der Mechanismus, mit dem dies geschieht?
Die erste Frage ist eng verknüpft mit der Frage nach der Stabilität und damit nach dem Energieinhalt einer bestimmten Phase. Früher musste man mit verschiedenen näherungsweisen Ansätzen die Wechselwirkung der Atome beschreiben. (Paarpotenziale, „Embedded Atom“-Methode, “Tight Binding”-Approximation. Seit Einführung der Dichtefunktionaltheorie (Hohenberg und Kohn 1964, Kohn und Sham 1965) und der Leistungsexplosion moderner Elektronenrechner (1980er-1990er Jahre) ist es möglich, die Grundzustandsenergie einer Phase mit einer Genauigkeit zu berechnen, die deutlich kleiner als die Phasenübergangsenergie ist, und so die Frage zu beantworten, welche von zwei Phasen stabiler ist (Ein strenger Beweis, dass eine bestimmte Phase diejenige mit der absolut kleinsten Grundzustandsenergie ist, lässt sich damit allerdings noch immer nicht führen, einen weiteren Schritt dazu liefert die Clusterentwicklungsmethode (cluster expansion method, Sanchez et al. 1984). Wir werden uns allerdings hauptsächlich mit den Mechanismen der Phasenumwandlung beschäftigen. Die Klassifizierung der Phasenübergänge kann nach verschiedenen Kriterien vorgenommen werden, wobei die Zuordnung nicht immer eindeutig ist: So bedingt eine große Anzahl von Komponenten bzw. Freiheitsgraden einen entsprechenden Reichtum des Verhaltens. Es kann kombinierte Übergänge geben (z.B. gleichzeitiges Auftreten von Rekristallisation, Phasentrennung, Ordnungsübergängen etc.) und Mischformen, zwischen denen experimentell nicht leicht zu unterscheiden ist. Eine thermodynamische Unterteilung geht zurück auf Ehrenfest (1933). Dabei wird die Ordnung des Phasenüberganges benannt nach dem Grad der niedrigsten Ableitung der Freien Enthalpie nach einem geeigneten intensiven Parameter wie Temperatur oder Druck, die beim Übergang unstetig wird. Demnach ist z.B. der klassische Übergang gasförmig-flüssig (z.B. Wasserdampf - flüssiges Wasser) ein Übergang 1. Ordnung, denn das spezifische Volumen
pGv∂∂
−= (2-1)
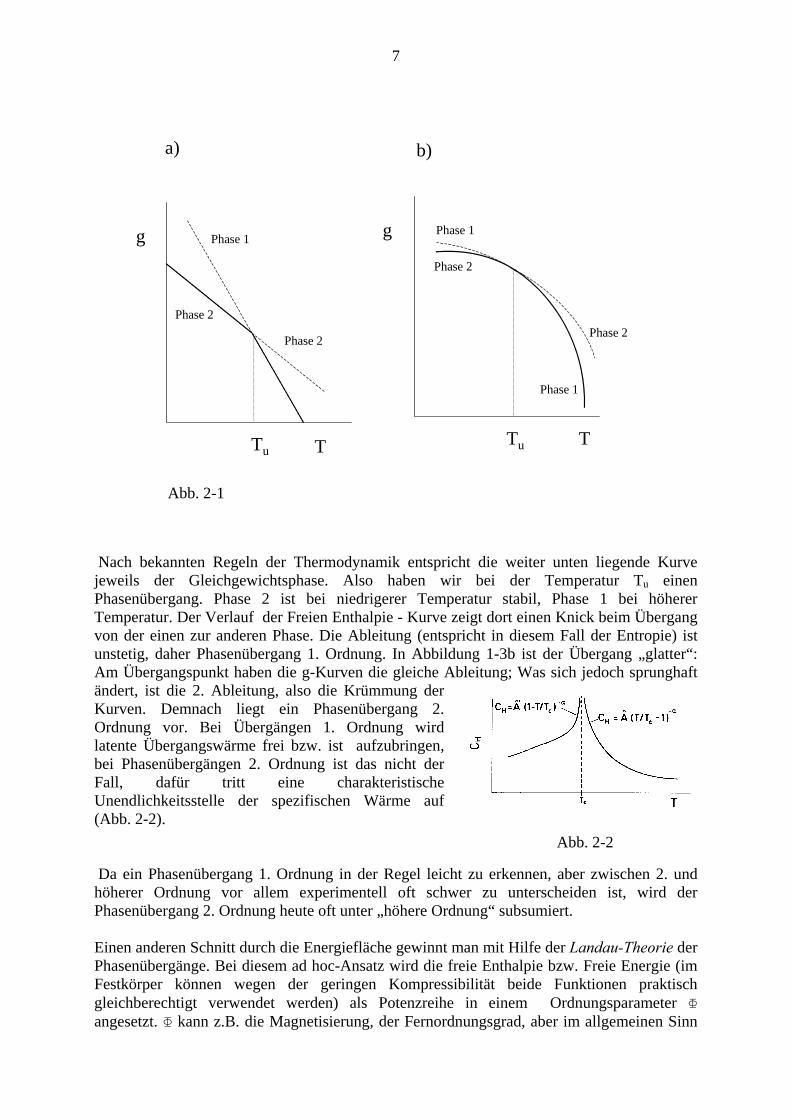
macht einen „Sprung“ von gasförmig zu flüssig. Um dies zu verdeutlichen, wollen wir schematisch den Verlauf der Freien Enthalpie pro Volumseinheit gegen die Temperatur für ein 1-komponentiges System (bei konstantem Druck) skizzieren (Abb. 2-1a).
7
g
T
g
T
Phase 1
Phase 2
Tu
Phase 1
Phase 1
Phase 2
Phase 2Phase 2
Tu
a) b)
Abb. 1-3
Abb. 2-1
Nach bekannten Regeln der Thermodynamik entspricht die weiter unten liegende Kurve jeweils der Gleichgewichtsphase. Also haben wir bei der Temperatur Tu einen Phasenübergang. Phase 2 ist bei niedrigerer Temperatur stabil, Phase 1 bei höherer Temperatur. Der Verlauf der Freien Enthalpie - Kurve zeigt dort einen Knick beim Übergang von der einen zur anderen Phase. Die Ableitung (entspricht in diesem Fall der Entropie) ist unstetig, daher Phasenübergang 1. Ordnung. In Abbildung 1-3b ist der Übergang „glatter“: Am Übergangspunkt haben die g-Kurven die gleiche Ableitung; Was sich jedoch sprunghaft ändert, ist die 2. Ableitung, also die Krümmung der Kurven. Demnach liegt ein Phasenübergang 2. Ordnung vor. Bei Übergängen 1. Ordnung wird latente Übergangswärme frei bzw. ist aufzubringen, bei Phasenübergängen 2. Ordnung ist das nicht der Fall, dafür tritt eine charakteristische Unendlichkeitsstelle der spezifischen Wärme auf (Abb. 2-2). Abb. 2-2 Da ein Phasenübergang 1. Ordnung in der Regel leicht zu erkennen, aber zwischen 2. und höherer Ordnung vor allem experimentell oft schwer zu unterscheiden ist, wird der Phasenübergang 2. Ordnung heute oft unter „höhere Ordnung“ subsumiert. Einen anderen Schnitt durch die Energiefläche gewinnt man mit Hilfe der Landau-Theorie der Phasenübergänge. Bei diesem ad hoc-Ansatz wird die freie Enthalpie bzw. Freie Energie (im Festkörper können wegen der geringen Kompressibilität beide Funktionen praktisch gleichberechtigt verwendet werden) als Potenzreihe in einem Ordnungsparameter Φ angesetzt. Φ kann z.B. die Magnetisierung, der Fernordnungsgrad, aber im allgemeinen Sinn
8
durchaus auch Zusammensetzung oder spezifisches Volumen sein. Die Entwicklungskoeffizienten sind Funktionen des Abstandes von der kritischen Temperatur Tc
( ) ( ) ....420 +−+−+= ΦΦ cc TTBTTAFF (2-2)
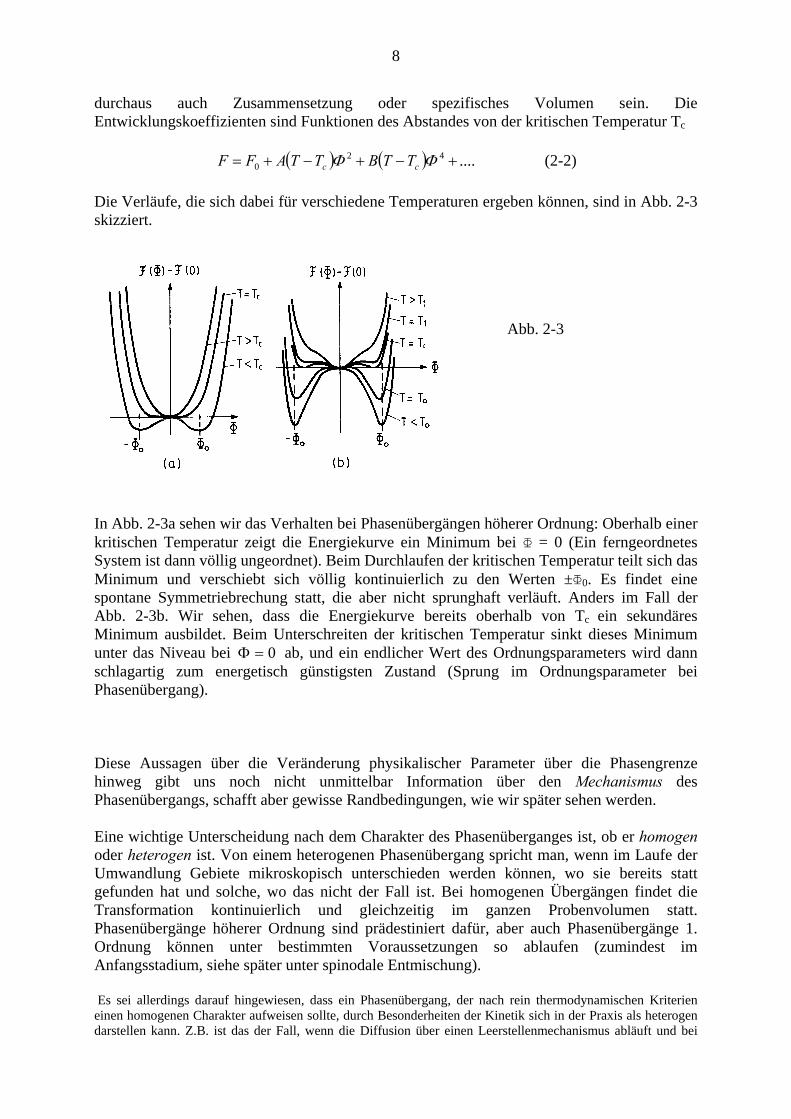
Die Verläufe, die sich dabei für verschiedene Temperaturen ergeben können, sind in Abb. 2-3 skizziert.
Abb. 2-3
In Abb. 2-3a sehen wir das Verhalten bei Phasenübergängen höherer Ordnung: Oberhalb einer kritischen Temperatur zeigt die Energiekurve ein Minimum bei Φ = 0 (Ein ferngeordnetes System ist dann völlig ungeordnet). Beim Durchlaufen der kritischen Temperatur teilt sich das Minimum und verschiebt sich völlig kontinuierlich zu den Werten ±Φ0. Es findet eine spontane Symmetriebrechung statt, die aber nicht sprunghaft verläuft. Anders im Fall der Abb. 2-3b. Wir sehen, dass die Energiekurve bereits oberhalb von Tc ein sekundäres Minimum ausbildet. Beim Unterschreiten der kritischen Temperatur sinkt dieses Minimum unter das Niveau bei 0=Φ ab, und ein endlicher Wert des Ordnungsparameters wird dann schlagartig zum energetisch günstigsten Zustand (Sprung im Ordnungsparameter bei Phasenübergang). Diese Aussagen über die Veränderung physikalischer Parameter über die Phasengrenze hinweg gibt uns noch nicht unmittelbar Information über den Mechanismus des Phasenübergangs, schafft aber gewisse Randbedingungen, wie wir später sehen werden. Eine wichtige Unterscheidung nach dem Charakter des Phasenüberganges ist, ob er homogen oder heterogen ist. Von einem heterogenen Phasenübergang spricht man, wenn im Laufe der Umwandlung Gebiete mikroskopisch unterschieden werden können, wo sie bereits statt gefunden hat und solche, wo das nicht der Fall ist. Bei homogenen Übergängen findet die Transformation kontinuierlich und gleichzeitig im ganzen Probenvolumen statt. Phasenübergänge höherer Ordnung sind prädestiniert dafür, aber auch Phasenübergänge 1. Ordnung können unter bestimmten Voraussetzungen so ablaufen (zumindest im Anfangsstadium, siehe später unter spinodale Entmischung). Es sei allerdings darauf hingewiesen, dass ein Phasenübergang, der nach rein thermodynamischen Kriterien einen homogenen Charakter aufweisen sollte, durch Besonderheiten der Kinetik sich in der Praxis als heterogen darstellen kann. Z.B. ist das der Fall, wenn die Diffusion über einen Leerstellenmechanismus abläuft und bei

9
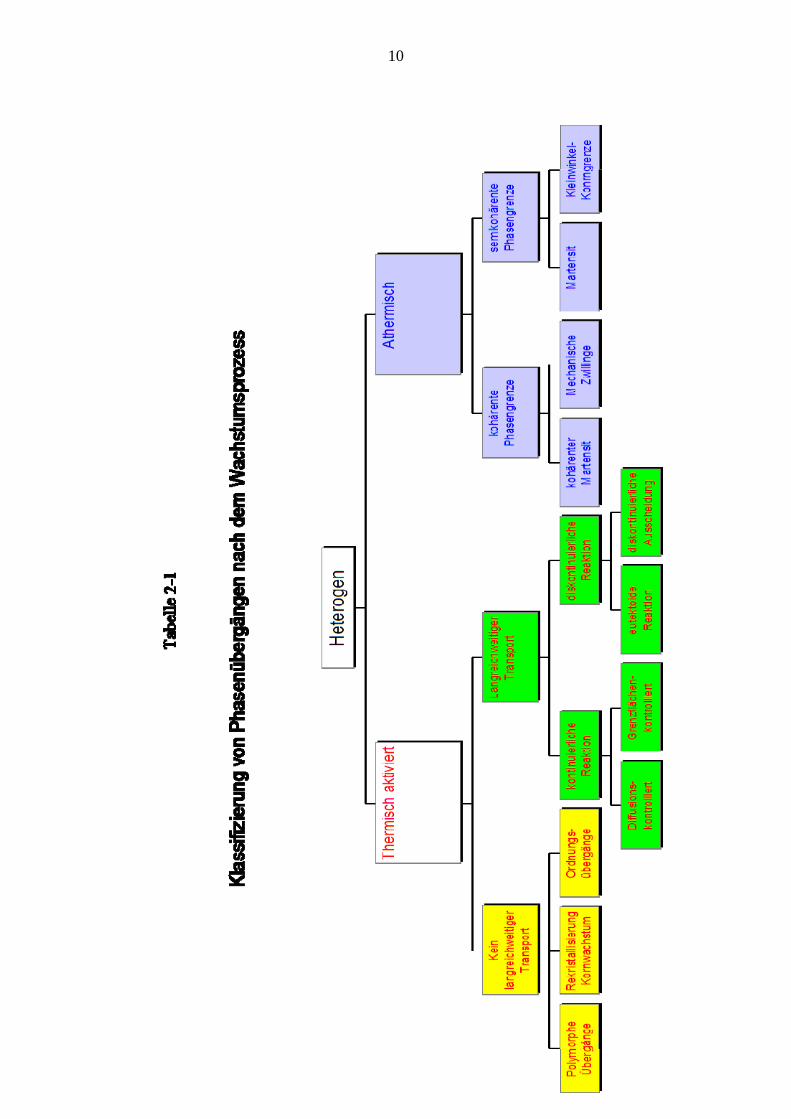
niedriger Temperatur und geringer Leerstellenbeweglichkeit die Kinetik auf Teilgebiete der Probe beschränkt bleibt. Die meisten Phasenübergänge sind heterogener Natur. Sie können nach der Abhängigkeit der Reaktionsgeschwindigkeit und des Anteils der transformierten Phase von der Temperatur unterschieden werden (siehe Tabelle 2-1, beiliegend) In der einen großen Klasse heterogener Übergänge muss ein Zustand erhöhter Energie durch thermische Aktivierung überwunden werden. In diesen ist die Bewegung der Phasengrenzfläche von der Temperatur abhängig. Gerade im Festkörper besonders wichtig ist das Auftreten metastabiler Phasen, wenn die Energiebarriere bei bestimmten Temperaturverhältnissen nur mit einer so geringen Rate überwunden wird, dass für die Praxis diese Phase als stabil angesehen werden kann. Des weiteren kann unterschieden werden, ob ein langreichweitiger Transport von Atomen zur Reaktionsfront stattfindet oder nicht. Besteht zwischen der Ausgangsphase und der neu gebildeten Phase ein Unterschied in der Zusammensetzung (Konzentration), dann kann die Geschwindigkeit der Umwandlung von der Rate abhängen, mit der Atome mittels Diffusion an die Phasengrenzfläche gebracht werden können (diffusionskontrollierte Phasenübergangskinetik). Dieser Fall liegt sehr oft bei einer Ausscheidungsreaktion vor. Hängt die Geschwindigkeit nur von der Rate ab, mit der Atome über die Grenzfläche treten, dann spricht man von einer reaktionskontrollierten oder grenzflächenkontrollierten (interface controlled) Kinetik. Mit letzterer hat man es auf jeden Fall dann zu tun, wenn kein Konzentrationsunterschied zwischen den beiden Phasen herrscht, wie etwa in polymorphen Umwandlungen, in der Regel die Umwandlung eines Reinelementes in eine andere Modifikation, Ordnungsübergängen oder Rekristallisierung, die im erweiterten Sinne auch zu den Phasenübergängen gerechnet wird. Völlig anders liegen die Verhältnisse bei athermischem Wachstum. Hier ist die Phasengrenzfläche unter Spannung frei gleitfähig (glissile). Diese Art von Phasenübergängen treten nur in Festkörpern auf. Die Reaktionsgeschwindigkeit ist von der Temperatur praktisch unabhängig und in der Regel sehr hoch. Nur der Anteil der umgewandelten Phase hängt von der Temperatur ab. In diese Klasse gehören Martensitbildung, die damit verwandte Bildung mechanischer Deformationszwillinge und die Bewegung von Kleinwinkelkorngrenzen. Bei martensitischen Umwandlungen nehmen die Atome mittels kooperativer Bewegung neue Lagen innerhalb der Gitterzelle ein, wobei diese verzerrt wird. Die Nächstnachbar-Verhältnisse bleiben im wesentlichen erhalten, es kommt nicht zu einem Atomaustausch. (In Tabelle 2-1 nicht mehr dargestellt ist eine Klasse von Phasenübergängen, die durch den Wärmetransport gesteuert werden. Dazu gehört Erstarrung aus der Schmelze)
10

11
3. Phasengleichgewichte 3.1. Thermodynamische Grundlagen
Wir wollen hier Phasengleichgewichte mit den Methoden der makroskopischen Thermodynamik beschreiben. Zu den Annahmen dieser Methode gehört, dass die Systeme genügend groß ausgedehnt sind, sodass die Wirkung der Oberfläche auf die Eigenschaften des Systems nicht berücksichtigt werden muss. Oberflächeneigenschaften werden ggf. getrennt eingeführt, wobei auch die Oberfläche bzw. Grenzfläche mit einem makroskopischen Konzept behandelt wird. Oft werden makroskopisch-thermodynamische Methoden auf sehr kleine Teilsysteme angewendet wie etwa kleine Keime einer neuen Phase, was zu Problemen führen kann.
In der klassischen Thermodynamik wird davon ausgegangen, dass mit einer relativ geringen Anzahl von Zustandsvariablen (p, V, T, c) (state properties) der Zustand eines Systems im Gleichgewicht eindeutig beschrieben werden kann. Bei Festkörpern ist dieses einfache Konzept problematisch, denn eine bisweilen sehr komplizierte Defekt- und Mikrostruktur hängt von der Vorgeschichte der thermischen und Deformationsbehandlung ab. Festkörper, z.B. metallische Werkstoffe, werden oft in einem metastabilen Zustand verwendet, der kein echtes Gleichgewicht im thermodynamischen Sinn darstellt. Dennoch lassen sich auf thermodynamischer Basis viele grundsätzliche Aussagen machen. Wo die makroskopisch-thermodynamische Behandlung zu fundamentalen Problemen führt, werden wir gesondert darauf hinweisen. Das thermodynamische Begriffsgebäude geht von den Hauptsätzen der Thermodynamik aus (three laws of thermodynamics)
1. Hauptsatz WQdE δδ −= (3-1)
Hier ist die innere Energie E eine Zustandsfunktion (eindeutige Funktion der Zustandsvariablen, unabhängig davon, auf welchem Weg der Zustand erreicht wurde, daher das vollständige Differenzial), die vom System aufgenommene Wärmemenge Qδ und die vom System geleistete Arbeit Wδ jedoch nicht. (Eine Wärmekraftmaschine kehrt z.B. nach einem Zyklus in den gleichen makroskopischen Zustand mit dem gleichen E zurück, es wurde aber netto Wärme Q aufgenommen und Arbeit W abgegeben.) Für die mechanische Arbeit gegen einen äußeren hydrostatischen Druck lässt sich schreiben
pdVW =δ (3-2)
(analoge Ausdrücke lassen sich angeben, wenn Arbeit gegenüber anderen Feldern geleistet wird, z.B.
ikik dW εσδ = , wenn Arbeit durch elastische Deformationen εt
gegenüber einem Spannungsfeld σt geleistet
wird oder HdBWrr
•=δ , wenn Magnetisierungsarbeit in einem Magnetfeld geleistet wird etc.) Eine große Bedeutung in der Geschichte der Thermodynamik hatte die Untersuchung reversibler Kreisprozesse (Carnot-Prozess und äquivalente, z.B. Stirlingprozess), wobei reversibel bedeutet, dass die Zustandsänderungen so behutsam und langsam vorgenommen werden, dass dabei eine Folge von Gleichgewichtszuständen durchlaufen wird. Ganz allgemein lässt sich für geschlossene Kreisprozesse zeigen
∑ ≤i i
i
TQ 0δ bzw. 0≤∫ T
Qδ (3-3)

12
(Clausius-Theorem), wobei das Gleichheitszeichen genau für die reversiblen Vorgänge gilt.
Bei Rückkehr in den Ausgangszustand ist daher die Summe aller reversibel aufgenommenen Wärmemengen, dividiert durch die jeweilige absolute Temperatur, ist also gleich 0. Daher kann man eine Zustandsfunktion S definieren
TQdS revδ
= . (3-4)
Für abgeschlossene Systeme (closed systems), d.h. solche, die mit ihrer Umgebung weder Wärme noch Arbeit austauschen, gilt dann
2. Hauptsatz (3-5) 0≥dS Das Gleichheitszeichen gilt genau für die reversiblen Veränderungen. Ist der Anfangszustand ein Nicht-Gleichgewichtszustand, dann nimmt die Entropie zu. Ein typischer solcher Vorgang wäre das Fließen von Wärme von einem wärmeren Teilsystem des abgeschlossenen Systems zu einem kälteren Teilsystem. Der umgekehrte Vorgang würde bedeuten, dass sich die Entropie verringert. Darum lautet eine der vielen Formulierungen des zweiten Hauptsatzes auch, dass kein Prozess ausschließlich darin bestehen kann, dass Wärme von einem kälteren zu einem wärmeren Teilsystem fließt. Mit Hilfe von (3-2), (3-3) und (3-4) lässt sich (3-1) schreiben als:
pdVTdSdE −= (3-6) Somit haben wir die Zustandsfunktion E als vollständiges Differenzial in den Zustands-variablen S und V geschrieben (wobei der zweite Hauptsatz seinen Ausdruck darin gefunden hat, dass die Beziehung (3-6) nur für reversible Zustandsänderungen gilt). Die hier vorkommenden Variablen S und V sind für die Praxis denkbar unhandlich, denn S lässt sich in einem Versuch ohne vorherige Kenntnis der Systemeigenschaften nicht konstant halten, und auch die Einstellung eines konstanten Volumens ist bei einem Festkörper nicht ohne weiteres möglich. Man definiert daher als neue Zustandsfunktion die Freie Energie (Helmholtz free energy) F = E – TS. Für das Differenzial ergibt sich dann
( ) pdVSdTTdSSdTpdVTdSTSddEdF −−=−−−=−= (3-7) Als unabhängige Variable erhalten wir T, V. Man kann nun einen Schritt weiter gehen und die Freie Enthalpie (Gibbs free energy) G = F + PV = E – TS + PV einführen. (Man schreibt auch G = H –TS, wobei H = E + PV Enthalpie genannt wird.) Für das Differenzial von G gilt dann
VdpSdTpdVVdPpdVSdTPVddFdG +−=++−−=+= )( (3-8). Die sich jetzt ergebenden unabhängigen Variablen p, T lassen sich im Laufe eines Experimentes gut kontrollieren. Der Wert der beiden thermodynamischen Potenziale (=Zustandsfunktionen) F und G liegt in den Extremalprinzipien, die für sie gelten, und aus denen sich Eigenschaften von Gleichgewichtszuständen ableiten lassen. So gilt für ein

13
System, das bei konstanter Temperatur und konstantem Druck (und konstanter Masse und Zusammensetzung) gehalten wird:
0,≤
pTdG (3-9)
Das Gleichheitszeichen gilt für den Gleichgewichtsfall, somit erreicht G bei festgehaltenem Druck und Temperatur im Gleichgewicht ein Minimum. Beweis: Mit Hilfe von (3-7) und (3-8) können wir schreiben
VdPpdVSdTTdSdEdG ++−−= (3-10) Betrachten wir das System bei festgehaltenen Werten von p, T, dann verschwindet der dritte und fünfte Term auf der rechten Seite. Mit Hilfe des 1. Hauptsatzes (3-1) und (3-2) können wir zusammenfassen: QpdVdE δ=+ . Wenden wir die Definition des Differenzials der Entropie an (3-4), dann wird (3-10) zu
revQQdG δδ −= (3-10’) Es lässt sich nun leicht einsehen, dass aus dem Clausiusschen Theorem (3-3) für einen beliebigen Übergang zwischen zwei Zuständen A und B folgt
∫∫ =−≤B
A
revB
A TQASBS
TQ δδ )()( (3-3’).
(Um dies zu zeigen, genügt es, das Integral durch Rückkehr von B nach A auf reversiblem
Wege zu einem geschlossenen zu ergänzen: 0≤+∫∫A
B
revB
A TQ
TQ δδ nach Clausius. Ferner gilt
)()( BSASTQ
TQ B
A
revA
B
rev −=−= ∫∫δδ und daraus unmittelbar Gl. (3-3’)). Macht man nun den
Unterschied zwischen A und B beliebig klein, so folgt revQQ δδ ≤ und damit , q.e.d. 0≤dG Analoge Überlegungen zeigen, dass
0,≤
VTdF (3-11)
Für theoretische Überlegungen bei Festkörpern macht es allerdings meistens wenig Unterschied, ob das Kriterium (3-9) oder (3-11) angewendet wird, da die Veränderung des Produkts PV bei Übergängen im Festkörper wegen der vergleichsweise geringen Kompressibilität klein ist gegenüber den sonstigen Veränderungen von F bzw. G. Entscheidend ist, dass durch alle thermodynamischen Potenziale der gleiche Gleichgewichts-Zustand beschrieben wird, vorausgesetzt, man hat die „richtigen“, zusammenpassenden Werte (p, T), (V, T), (S, V) etc. eingestellt. Es wird nur der gleiche Sachverhalt auf verschiedene Art beschrieben. Der Übergang E→F→G ist jeweils eine sogenannte Legendretransformation. Er ist völlig analog zu jenem, wenn man eine Funktion statt durch die Koordinaten (x,y(x)) ihres Graphen durch Achsenabschnitt und Anstieg (a,k) der an diese y
a = y – k x
x

14
Kurve gelegten Tangenten beschreibt. Die Kurve stellt sich dann als Einhüllende dieser Tangentenschar dar.
xdxdyykxya −=−=
(thermodynamisch SdSdEETSEF −=−= ).
Von der Funktion y wird zur Legendre-transformierten Funktion, d.i. der Abschnitt a auf der y-Achse (thermodynamisch: von E zu F), und von der unabhängigen Variablen x wird zur unabhängigen Variablen k = dy/dx übergegangen (im thermodynamischen Fall von S zu T=dE/dS).
3.2. Thermodynamik von Lösungen (Mischkristallen) Während wir bisher die Freie Enthalpie für Systeme mit konstanter Masse bzw. Teilchenzahl betrachtet haben, fassen wir sie jetzt als Funktion der Molzahlen n1, n2 der einzelnen Komponenten auf und schreiben bei einer binären Lösung für das Differenzial der Funktion G = G (n1, n2)
22
11
dnnGdn
nGdG
∂∂
+∂∂
= (3-12)
Die partielle Ableitung ijnTpi
ii nGg
≠∂∂
==,,
μ wird als partielle molare Enthalpie oder
chemisches Potenzial bezeichnet und stellt den Energieaufwand pro Mol für das Hinzufügen der betrachteten Teilchensorte (Atome, Moleküle etc) dar. (Genauso kann man es natürlich auch auf ein Teilchen beziehen, indem man nicht nach der Molzahl, sondern nach der Teilchenzahl differenziert; Der Unterschied zur vorliegenden Definition ist dann ein Faktor NA=6,023x1023) Definitionsgemäß sind zwei verschiedene Phasen dann im thermodynami-schen Gleichgewicht, wenn für jede Teilchensorte die chemischen Potenziale für beide Phasen gleich sind. Eine besonders unter Chemikern gerne gebrauchte Größe ist die Aktivität ai . Definition:
iiii aRTggg ln0 =−=Δ (3-13),
wobei Δgi das auf einen Referenzzustand (z.B. wie in Abb. 3-1 die reine Komponente i) bezogene chemische Potenzial ist. Ohne Beweis: Bei einer idealen Lösung (keine Wechselwirkung zwischen den Komponenten) gilt ai = Xi = ni / (n1 + n2) (Grund: Nur die Mischungsentropie trägt zur freien Enthalpie bei). Für ein monoatomares, ideales Gas ist die Aktivität gleich dem Druck des Gases. Aufintegrieren von (3-12) bei konstanter Zusammensetzung, dh. Hinzufügen von Atomen im richtigen Verhältnis, bis die gesamte Menge des Mischkristalls erreicht ist, ergibt
2211 ngngG += (3-14) Bildet man das Differenzial von G, aufgefasst als Funktion der g’s und der n’s und berücksichtigt man (3-12), so erhält man die sog. Gibbs-Duhem-Gleichung

15
02211 =+ dgndgn (3-15),
die besagt, dass die Änderungen der chemischen Potentiale nicht unabhängig von einander erfolgen dürfen, sondern im umgekehrten Verhältnis der Konzentrationen.
Dividiert man nun Gl. (3-14) durch n1 + n2 und definiert die Molenbrüche Xi = ni / (n1 + n2), so lassen sich mit g = G / (n1 + n2) die Gln. (3-12) und (3-14) folgendermaßen schreiben:
2211 dXgdXgdg += (3-12’)
2211 XgXgg += (3-14’)
Bei (3-12’) handelt es sich allerdings nicht mehr um ein vollständiges Differenzial, da X1 und X2 nicht mehr von einander unabhängig sind. Es gilt vielmehr
211
121 )( ggXgdXggdg −=
∂∂
⇒−= (3-16)
Drücken wir g aus (3-14’) aus:
12121212221 )()1(
XgXgggXgXgXgg
∂∂
−=−−=+−= oder
11
121 )1(
XgXg
XgXgg
∂∂
−+=∂∂
+= (3-17)
Diese Formel begründet eine nützliche Tangentenkonstruktion: Die chemischen Potenziale der beiden Komponenten sind an den Schnittpunkten einer Tangente an die g(X)-Kurve bei der gegebenen Zusammensetzung mit den Ordinaten X = X1 und X = X2 abzulesen (Abbildung 3-1).
Abb. 3-1
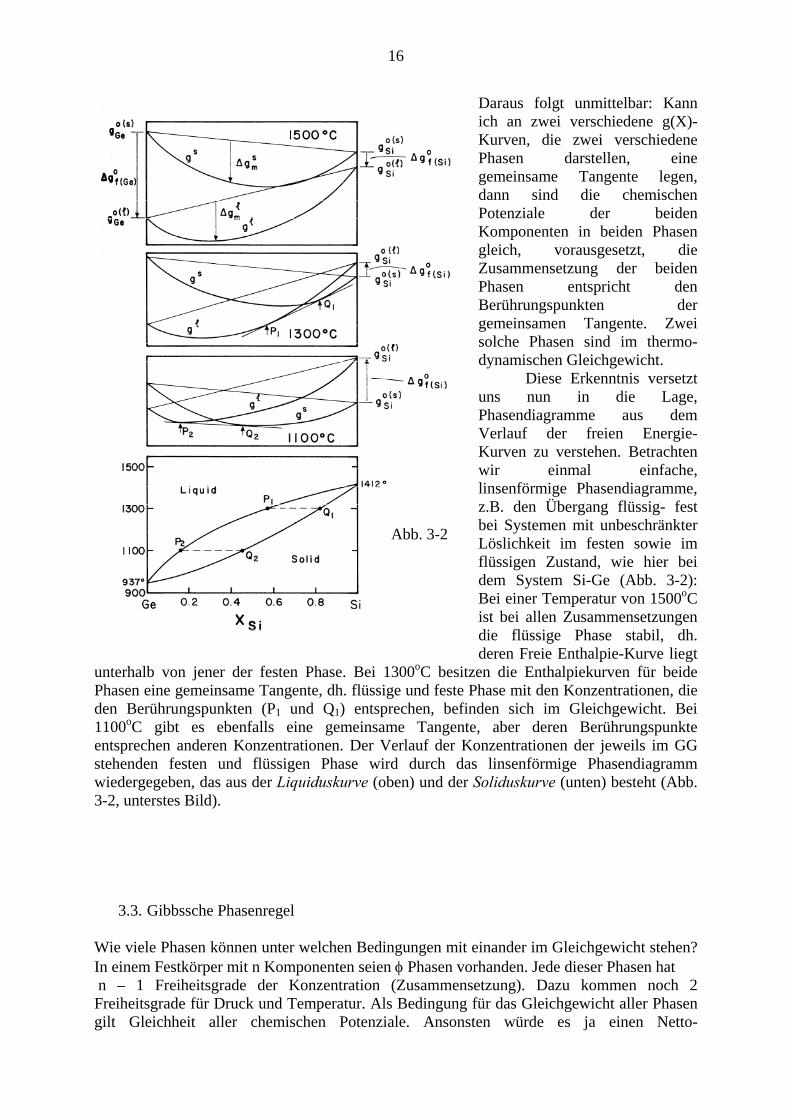
16
Daraus folgt unmittelbar: Kann ich an zwei verschiedene g(X)-Kurven, die zwei verschiedene Phasen darstellen, eine gemeinsame Tangente legen, dann sind die chemischen Potenziale der beiden Komponenten in beiden Phasen gleich, vorausgesetzt, die Zusammensetzung der beiden Phasen entspricht den Berührungspunkten der gemeinsamen Tangente. Zwei solche Phasen sind im thermo-dynamischen Gleichgewicht. Diese Erkenntnis versetzt uns nun in die Lage, Phasendiagramme aus dem Verlauf der freien Energie- Kurven zu verstehen. Betrachten wir einmal einfache, linsenförmige Phasendiagramme, z.B. den Übergang flüssig- fest bei Systemen mit unbeschränkter Löslichkeit im festen sowie im flüssigen Zustand, wie hier bei dem System Si-Ge (Abb. 3-2): Bei einer Temperatur von 1500oC ist bei allen Zusammensetzungen die flüssige Phase stabil, dh. deren Freie Enthalpie-Kurve liegt
unterhalb von jener der festen Phase. Bei 1300oC besitzen die Enthalpiekurven für beide Phasen eine gemeinsame Tangente, dh. flüssige und feste Phase mit den Konzentrationen, die den Berührungspunkten (P1 und Q1) entsprechen, befinden sich im Gleichgewicht. Bei 1100oC gibt es ebenfalls eine gemeinsame Tangente, aber deren Berührungspunkte entsprechen anderen Konzentrationen. Der Verlauf der Konzentrationen der jeweils im GG stehenden festen und flüssigen Phase wird durch das linsenförmige Phasendiagramm wiedergegeben, das aus der Liquiduskurve (oben) und der Soliduskurve (unten) besteht (Abb. 3-2, unterstes Bild).
Abb. 3-2
3.3. Gibbssche Phasenregel
Wie viele Phasen können unter welchen Bedingungen mit einander im Gleichgewicht stehen? In einem Festkörper mit n Komponenten seien φ Phasen vorhanden. Jede dieser Phasen hat n – 1 Freiheitsgrade der Konzentration (Zusammensetzung). Dazu kommen noch 2 Freiheitsgrade für Druck und Temperatur. Als Bedingung für das Gleichgewicht aller Phasen gilt Gleichheit aller chemischen Potenziale. Ansonsten würde es ja einen Netto-

17
Energiegewinn bringen, ein Teilchen von einer zu einer anderen Phase zu transferieren. Also gilt z.B. für die Komponente i :
λβα μμμ iii === ..... (3-18)
mit den Phasen α, β,...λ. Das sind insgesamt (φ - 1) n Bedingungsgleichungen. Für die übrig bleibenden Freiheitsgrade lautet somit die Bilanz:
f = φ (n – 1) + 2 – (φ - 1) n = n - φ + 2 (Gibbssche Phasenregel) (3-19)
Beispiele: Wenn Wasser und Dampf koexistieren, dann bleibt ein Freiheitsgrad übrig. Man kann also z.B. die Temperatur in einem gewissen Breich frei wählen, dann ergibt sich für den Druck p = p (T) (Gleichgewichts-Dampfdruckkurve). Am Tripelpunkt des Wassers koexistieren 3 Phasen (Wasserdampf, fl. Wasser, Eis). Es bleiben also keine (intensiven) Freiheitsgrade übrig, insbesondere sind Druck und Temperatur festgelegt. Man beachte aber, dass die relativen Anteile der einzelnen Phasen verschoben werden können. Es verbleiben daher extensive „Freiheitsgrade“. Deswegen gibt es auch in einem (p, V, T)- Diagramm keinen Tripelpunkt, sondern eine Tripellinie.
Abb. 3-2-1: Phasenkoexistenzflächen für Wasser in einem dreidimensionalen p-V-T- Diagramm und projiziert auf ein p-T- Diagramm. Die Tripellinie ist an der Grenze der Koexistenzflächen flüssig-gasförmig und fest-gasförmig zu sehen.
3.4. Klassifizierung der Typen von Phasendiagrammen
Wir führen den Begriff der Überschusseigenschaften ein. So ist z.B. die Überschussenthalpie (excess enthalpy) der Anteil der molaren Mischungsenthalpie, der über eine ideale Lösung hinausgeht. Bei einer idealen Lösung üben die Atome keine Wechselwirkungskräfte aufeinander aus. besteht daher nur aus der Mischungsentropie, multipliziert mit (-T). Für eine zweikomponentige Legierung haben wir demnach
idealmm
E ggg ΔΔ −=
idealmgΔ
( ))1ln()1(ln XXXXRTg idealm −−+=Δ . Die Überschussenthalpie stellt also nichts
Anderes dar als die Wechselwirkungsenthalpie der Atome, . Anstelle des Verlaufes in Abb (2) können, falls , d.h. die Überschussenthalpie in der flüssigen Phase kleiner als in der festen Phase ist (das bedeutet: Bindung in der flüssigen Phase stärker), die Enthalpiekurven wie folgt aussehen (Abb. 3-3a)
mE hg Δ=
sElE gg ,, <
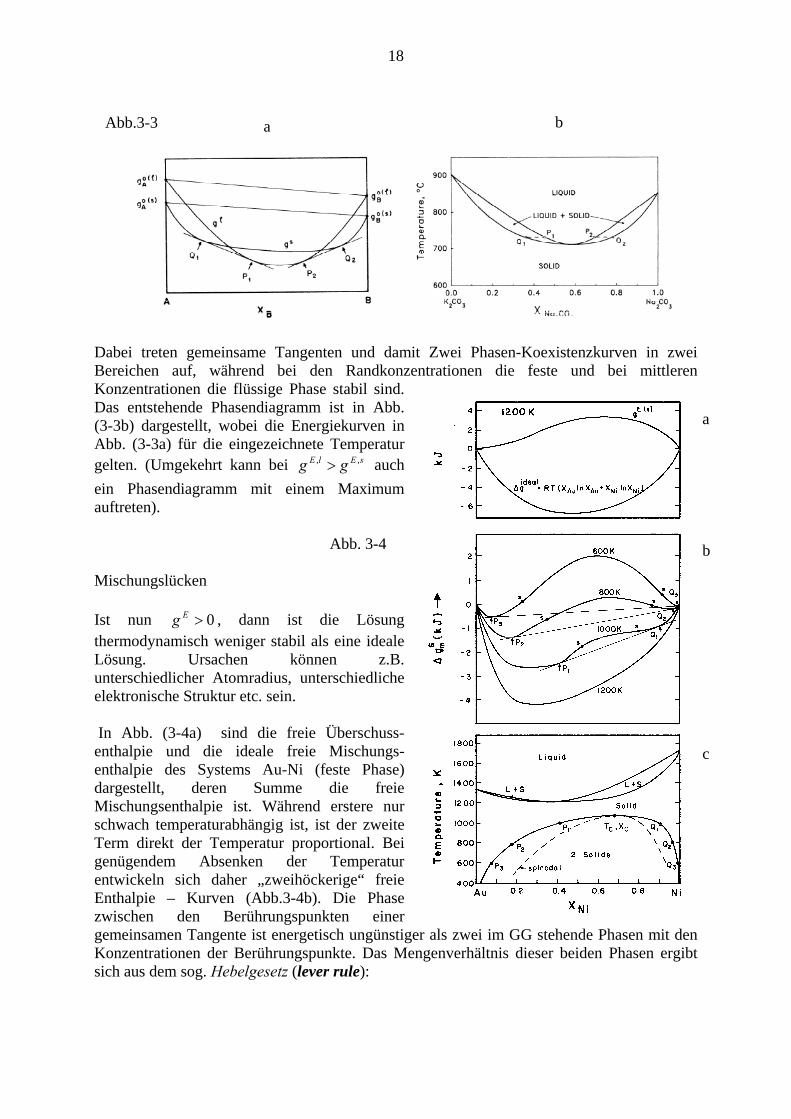
18
b Abb.3-3 a
Dabei treten gemeinsame Tangenten und damit Zwei Phasen-Koexistenzkurven in zwei Bereichen auf, während bei den Randkonzentrationen die feste und bei mittleren Konzentrationen die flüssige Phase stabil sind. Das entstehende Phasendiagramm ist in Abb. (3-3b) dargestellt, wobei die Energiekurven in Abb. (3-3a) für die eingezeichnete Temperatur gelten. (Umgekehrt kann bei auch ein Phasendiagramm mit einem Maximum auftreten).
sElE gg ,, >
Abb. 3-4 Mischungslücken Ist nun , dann ist die Lösung thermodynamisch weniger stabil als eine ideale Lösung. Ursachen können z.B. unterschiedlicher Atomradius, unterschiedliche elektronische Struktur etc. sein.
0>Eg
In Abb. (3-4a) sind die freie Überschuss-enthalpie und die ideale freie Mischungs-enthalpie des Systems Au-Ni (feste Phase) dargestellt, deren Summe die freie Mischungsenthalpie ist. Während erstere nur schwach temperaturabhängig ist, ist der zweite Term direkt der Temperatur proportional. Bei genügendem Absenken der Temperatur entwickeln sich daher „zweihöckerige“ freie Enthalpie – Kurven (Abb.3-4b). Die Phase zwischen den Berührungspunkten einer gemeinsamen Tangente ist energetisch ungünstiger als zwei im GG stehende Phasen mit den Konzentrationen der Berührungspunkte. Das Mengenverhältnis dieser beiden Phasen ergibt sich aus dem sog. Hebelgesetz (lever rule):
c
b
a
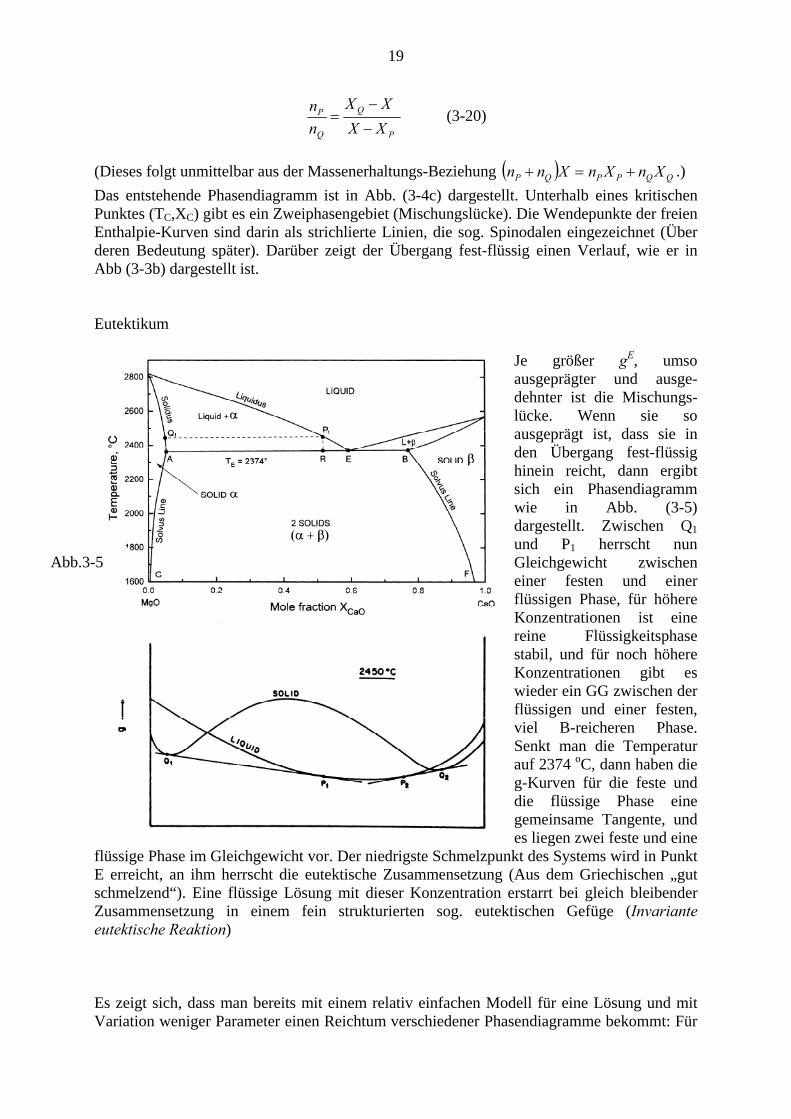
19
P
Q
Q
P
XXXX
nn
−
−= (3-20)
(Dieses folgt unmittelbar aus der Massenerhaltungs-Beziehung ( ) QQPPQP XnXnXnn +=+ .) Das entstehende Phasendiagramm ist in Abb. (3-4c) dargestellt. Unterhalb eines kritischen Punktes (TC,XC) gibt es ein Zweiphasengebiet (Mischungslücke). Die Wendepunkte der freien Enthalpie-Kurven sind darin als strichlierte Linien, die sog. Spinodalen eingezeichnet (Über deren Bedeutung später). Darüber zeigt der Übergang fest-flüssig einen Verlauf, wie er in Abb (3-3b) dargestellt ist. Eutektikum
Je größer gE, umso ausgeprägter und ausge-dehnter ist die Mischungs-lücke. Wenn sie so ausgeprägt ist, dass sie in den Übergang fest-flüssig hinein reicht, dann ergibt sich ein Phasendiagramm wie in Abb. (3-5) dargestellt. Zwischen Q1 und P1 herrscht nun Gleichgewicht zwischen einer festen und einer flüssigen Phase, für höhere Konzentrationen ist eine reine Flüssigkeitsphase stabil, und für noch höhere Konzentrationen gibt es wieder ein GG zwischen der flüssigen und einer festen, viel B-reicheren Phase. Senkt man die Temperatur auf 2374 oC, dann haben die g-Kurven für die feste und die flüssige Phase eine gemeinsame Tangente, und es liegen zwei feste und eine
flüssige Phase im Gleichgewicht vor. Der niedrigste Schmelzpunkt des Systems wird in Punkt E erreicht, an ihm herrscht die eutektische Zusammensetzung (Aus dem Griechischen „gut schmelzend“). Eine flüssige Lösung mit dieser Konzentration erstarrt bei gleich bleibender Zusammensetzung in einem fein strukturierten sog. eutektischen Gefüge (Invariante eutektische Reaktion)
Abb.3-5
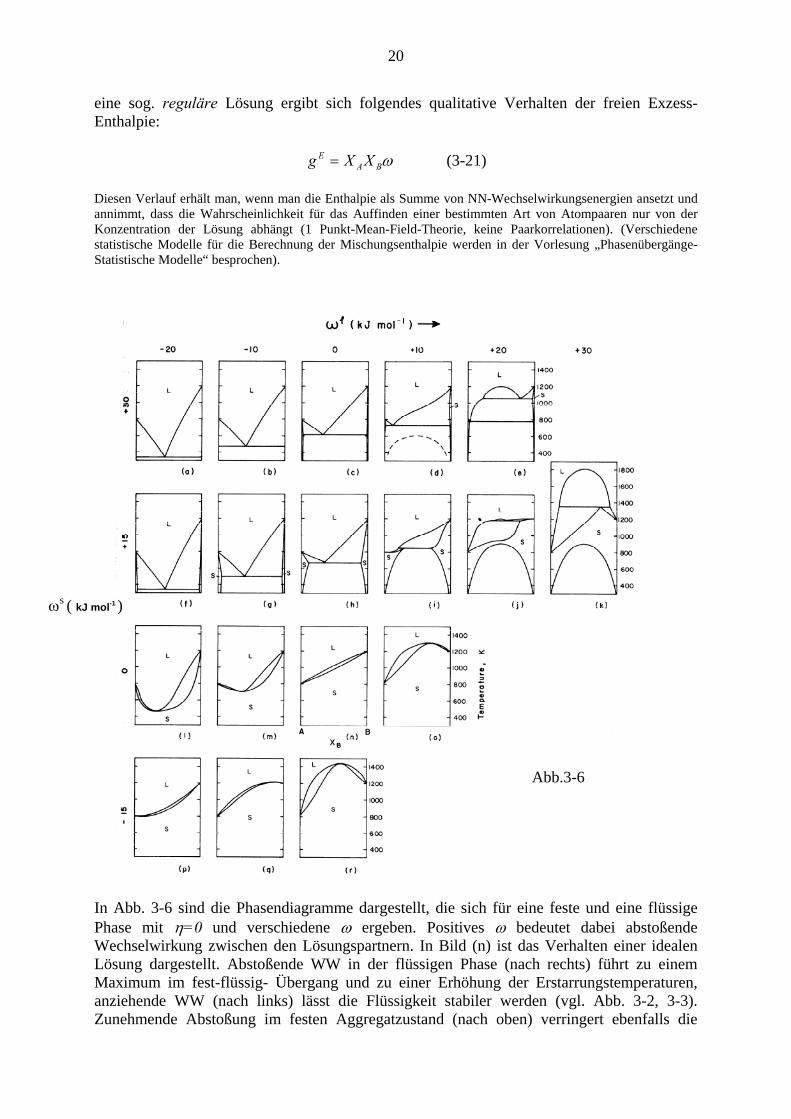
Es zeigt sich, dass man bereits mit einem relativ einfachen Modell für eine Lösung und mit Variation weniger Parameter einen Reichtum verschiedener Phasendiagramme bekommt: Für
20
eine sog. reguläre Lösung ergibt sich folgendes qualitative Verhalten der freien Exzess-Enthalpie:
ωBAE XXg = (3-21)
Diesen Verlauf erhält man, wenn man die Enthalpie als Summe von NN-Wechselwirkungsenergien ansetzt und annimmt, dass die Wahrscheinlichkeit für das Auffinden einer bestimmten Art von Atompaaren nur von der Konzentration der Lösung abhängt (1 Punkt-Mean-Field-Theorie, keine Paarkorrelationen). (Verschiedene statistische Modelle für die Berechnung der Mischungsenthalpie werden in der Vorlesung „Phasenübergänge-Statistische Modelle“ besprochen).
ωs ( kJ mol-1 )
Abb.3-6
In Abb. 3-6 sind die Phasendiagramme dargestellt, die sich für eine feste und eine flüssige Phase mit η=0 und verschiedene ω ergeben. Positives ω bedeutet dabei abstoßende Wechselwirkung zwischen den Lösungspartnern. In Bild (n) ist das Verhalten einer idealen Lösung dargestellt. Abstoßende WW in der flüssigen Phase (nach rechts) führt zu einem Maximum im fest-flüssig- Übergang und zu einer Erhöhung der Erstarrungstemperaturen, anziehende WW (nach links) lässt die Flüssigkeit stabiler werden (vgl. Abb. 3-2, 3-3). Zunehmende Abstoßung im festen Aggregatzustand (nach oben) verringert ebenfalls die
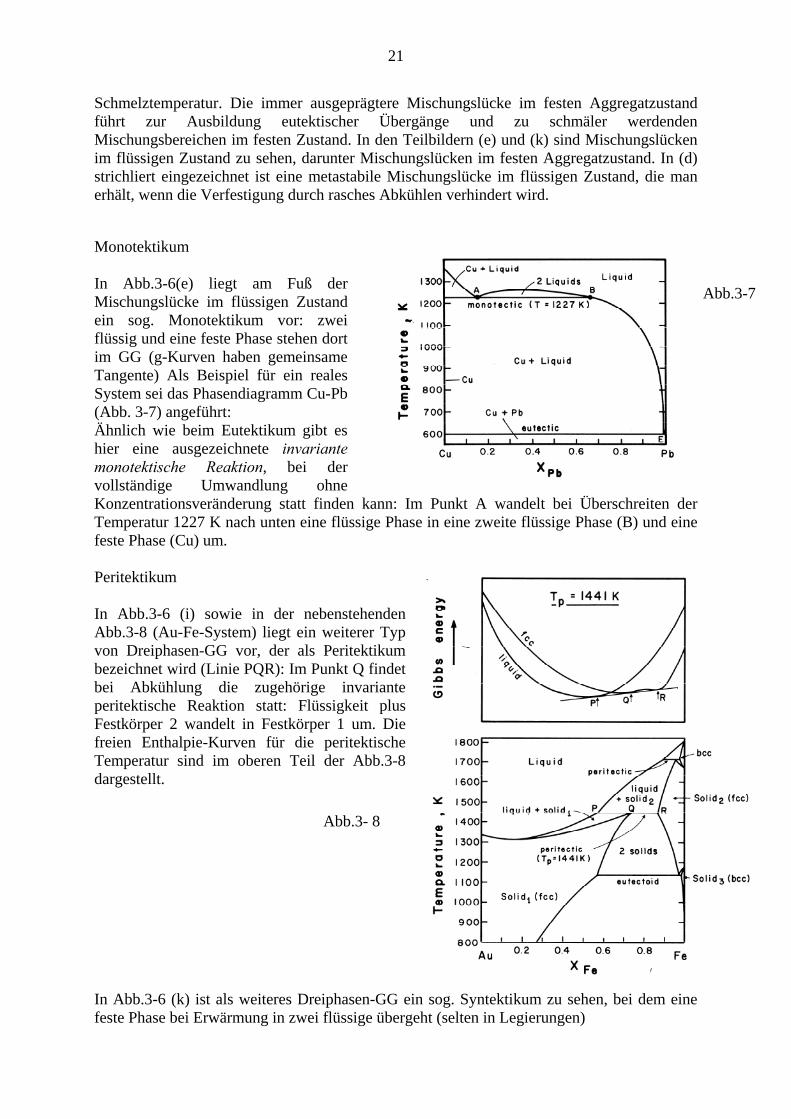
21
Schmelztemperatur. Die immer ausgeprägtere Mischungslücke im festen Aggregatzustand führt zur Ausbildung eutektischer Übergänge und zu schmäler werdenden Mischungsbereichen im festen Zustand. In den Teilbildern (e) und (k) sind Mischungslücken im flüssigen Zustand zu sehen, darunter Mischungslücken im festen Aggregatzustand. In (d) strichliert eingezeichnet ist eine metastabile Mischungslücke im flüssigen Zustand, die man erhält, wenn die Verfestigung durch rasches Abkühlen verhindert wird.
Monotektikum
In Abb.3-6(e) liegt am Fuß der Mischungslücke im flüssigen Zustand ein sog. Monotektikum vor: zwei flüssig und eine feste Phase stehen dort im GG (g-Kurven haben gemeinsame Tangente) Als Beispiel für ein reales System sei das Phasendiagramm Cu-Pb (Abb. 3-7) angeführt: Ähnlich wie beim Eutektikum gibt es hier eine ausgezeichnete invariante monotektische Reaktion, bei der vollständige Umwandlung ohne Konzentrationsveränderung statt finden kann: Im Punkt A wandelt bei Überschreiten der Temperatur 1227 K nach unten eine flüssige Phase in eine zweite flüssige Phase (B) und eine feste Phase (Cu) um.
Abb.3-7
Peritektikum
In Abb.3-6 (i) sowie in der nebenstehenden Abb.3-8 (Au-Fe-System) liegt ein weiterer Typ von Dreiphasen-GG vor, der als Peritektikum bezeichnet wird (Linie PQR): Im Punkt Q findet bei Abkühlung die zugehörige invariante peritektische Reaktion statt: Flüssigkeit plus Festkörper 2 wandelt in Festkörper 1 um. Die freien Enthalpie-Kurven für die peritektische Temperatur sind im oberen Teil der Abb.3-8 dargestellt.
Abb.3- 8
In Abb.3-6 (k) ist als weiteres Dreiphasen-GG ein sog. Syntektikum zu sehen, bei dem eine feste Phase bei Erwärmung in zwei flüssige übergeht (selten in Legierungen)
22
Zusammenfassend die vorgestellten Dreiphasen-GG anhand ihrer invarianten Reaktionen bei Abkühlung:
Eutektikum L -> S1 + S2
Eutektischer Typ Monotektikum L1 -> L2 + S
Peritektikum L + S1 -> S2
Peritektischer Typ Syntektikum L1 + L2 -> S
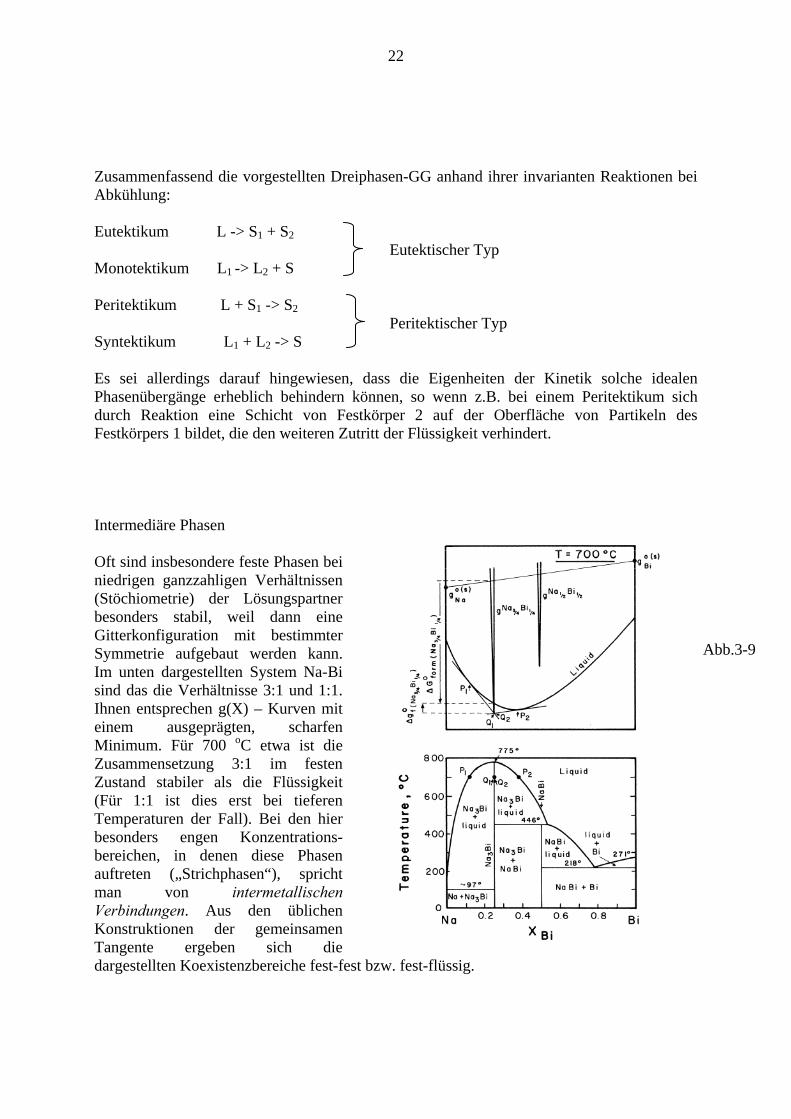
Es sei allerdings darauf hingewiesen, dass die Eigenheiten der Kinetik solche idealen Phasenübergänge erheblich behindern können, so wenn z.B. bei einem Peritektikum sich durch Reaktion eine Schicht von Festkörper 2 auf der Oberfläche von Partikeln des Festkörpers 1 bildet, die den weiteren Zutritt der Flüssigkeit verhindert. Intermediäre Phasen Oft sind insbesondere feste Phasen bei niedrigen ganzzahligen Verhältnissen (Stöchiometrie) der Lösungspartner besonders stabil, weil dann eine Gitterkonfiguration mit bestimmter Symmetrie aufgebaut werden kann. Im unten dargestellten System Na-Bi sind das die Verhältnisse 3:1 und 1:1. Ihnen entsprechen g(X) – Kurven mit einem ausgeprägten, scharfen Minimum. Für 700 oC etwa ist die Zusammensetzung 3:1 im festen Zustand stabiler als die Flüssigkeit (Für 1:1 ist dies erst bei tieferen Temperaturen der Fall). Bei den hier besonders engen Konzentrations-bereichen, in denen diese Phasen auftreten („Strichphasen“), spricht man von intermetallischen Verbindungen. Aus den üblichen Konstruktionen der gemeinsamen Tangente ergeben sich die dargestellten Koexistenzbereiche fest-fest bzw. fest-flüssig.
Abb.3-9

23
3.5. Ternäre Phasendiagramme Gleichgewichtsbedingungen und Gibbs’sche Phasenregel gelten auch dann, wenn die Anzahl der Komponenten größer als 2 ist. Die Darstellung der Phasendiagramme wird natürlich zunehmend schwieriger. Bei ternären Lösungen (3 Komponenten) benützt man oft die Projektion auf ein gleichseitiges Zusammensetzungs-Dreieck, das die gleichberechtigte Darstellung auf der Basis der Bedingung XA + XB + XC = 1 ermöglicht (Abb.3-10, wo auch für einen ausgewählten Punkt die Ablesung der Konzentrationen dargestellt ist)
Abbildung 3-10 Abbildung 3-11 Die Ecken entsprechen den reinen Komponenten, die Seiten den binären Verbindungen ohne die jeweils dritte Komponente. Linien parallel zu den Seiten, die zwei Komponenten repräsentieren, entsprechen konstanten Molenbrüchen (Konzentrationen) der dritten Komponente. In Abb.3-10 ist die Projektion der Liquidusflächen des dreidimensionalen Eutektikums Bi-Sn-Cd eingezeichnet, wobei die Isolinien Temperaturen sind, in Abb.3- 11 eine dreidimensionale Darstellung desselben (qualitativen) Sachverhaltes. Der Punkt E entspricht der Eutektischen Zusammensetzung. Kühlt man eine Flüssigkeit mit dieser Zusammensetzung ab, dann erstarrt sie invariant in ein Gefüge aus 3 festen Phasen. Die Eutektika der jeweiligen binären Systeme (ohne dritte Komponente) sind an den Seitenflächen zu sehen. In Abb. 3-10 ist der kinetische Pfad bei der Abkühlung einer flüssigen Phase mit der Zusammensetzung a dargestellt. Es wird zunächst Cd- reicher Festkörper ausgeschieden, wodurch die Cd- Konzentration der Flüssigkeit sinkt. Schließlich wird Punkt b erreicht. Die weitere Bewegung erfolgt entlang der univarianten Linie, während Cd-reicher und Sn-reicher Festkörper zugleich aus der Flüssigkeit ausgeschieden werden, bis schließlich E erreicht wird und die gleichzeitige Ausscheidung von drei festen Phasen (einschl. der Bi-reichen) aus der Flüssigkeit beginnt.

24
4. Diffusion Unter Diffusion versteht man den langreichweitigen Transport von Atomen durch viele einzelne Schritte, die zufällig, aber nach Wahrscheinlichkeiten verteilt erfolgen. Diffusion gibt es in allen Aggregatzuständen. Das Besondere an der Diffusion im Festkörper ist dabei, dass sie auf einem Gitter stattfindet, mit einer kleinen Auswahl von Sprungvektoren, zwischen denen Symmetrierelationen herrschen.
4.1. Mechanismen Leerstellenmechanismus: Der am häufigsten vorkommende Mechanismus, mit dem wir uns hauptsächlich beschäftigen werden, tritt in der Regel bei substitutionellen Mischkristallen auf, bei denen Atome verschiedener Sorten, aber vergleichbarer Größe auf dem gleichen Kristallgitter sitzen. Aus thermodynamischen Gründen ist immer ein Teil dieser Gitterplätze unbesetzt. Atome auf Gitterplätzen, die Nachbarn solchen Leerstellen sind, können in diese springen und daher mit der Leerstelle Platz tauschen. Zwischengitterdiffusion: Sind die Fremdatome viel kleiner als die Atome des Wirtsgitters (z.B. C oder H in Metallen), dann können sie Zwischengitterplätze einnehmen und diffundieren, indem sie von einem Zwischengitterplatz zum nächsten springen. Da in der Regel sehr viele davon unbesetzt sind, ist dieser Diffusionsmechanismus sehr wirksam, da nicht auf gewartet werden muss, bis eine Leerstelle vorbeikommt. Weitere Mechanismen mit untergeordneter Bedeutung sind „Interstitialcy“-Mechanismus: Ein Atom auf einem Zwischengitterplatz verdrängt ein Atom auf einem regulären Gitterplatz und nimmt dessen Platz ein. „Crowdion“-Mechanismus: In eine Atomreihe eines Kristallgitters wird an einem Ende ein zusätzliches Atom hineingequetscht, welches dann am anderen Ende wieder herausspringt.
4.2. Statistische Betrachtung der Diffusion Die Atome vollführen also auf dem Kristallgitter (auf den regulären oder den Zwischengitter-plätzen) eine Zufallsbewegung (random walk). Die Sprungwahrscheinlichkeit kann dabei durch eine treibende Kraft beeinflusst sein, etwa wenn geladene Atome in einem elektrischen Feld diffundieren oder wenn ein Gradient des chemischen Potenzials vorliegt. Verschiedene Einflüsse können dafür sorgen, dass die lokalen Barrieren für den Atomsprung von Ort zu Ort variieren. In der Abbildung 1 sind diese verschiedenen Möglichkeiten schematisch dargestellt.

25
Abbildung 4-1: Diffusions-Sprungprofile: a) Für ideale Lösung im homogenen Gitter, b) mit treibender Kraft, c) bei ortsabhängigem Diffusionskoeffizienten.
Um aus den mikroskopischen Sprüngen das makroskopische Verhalten abzuleiten, kann man zwei Wege gehen: Entweder 1) man betrachte zwei benachbarte Netzebenen und ermittelt den Nettofluss von Atomen einer bestimmten Sorte zwischen ihnen oder 2) man verfolgt die Zufallsbewegung eines Atoms über eine gewisse Distanz und schließt auf den Atomtransport, indem man Mittelwerte dieser Bewegung berechnet, etwa die in einem Zeitintervall zurückgelegte Distanz vom Ursprung. Wir wollen zunächst Weg 1) gehen und der Einfachheit halber ein kubisch-primitives Gitter und die Diffusion normal zu den Würfelebenen {100} betrachten, wodurch die Diffusion auf ein eindimensionales Problem zurückgeführt ist. Sei z.B. die Anzahl der A-Atome pro Flächeneinheit in Ebene 1 mit n1 bezeichnet und die in der benachbarten Ebene 2 mit n2 . Weiters betrachten wir ein Szenario gemäß Abb. 1b und Sprungfrequenzen ( )ε+Γ=Γ 1012 und ( )ε−Γ=Γ 1021 . (4-1) Der Nettofluss pro Flächeneinheit (Flussdichte) beträgt dann
( ) ( ) lCxClnnnnnnj εε 0
20210210212121 2Γ+
∂∂
Γ−=+Γ+−Γ=Γ−Γ= , (4-2)
Wobei wir die Relation ( 212/1 nnlC )+= für die Konzentration C der A-Atome verwendet haben. Eine treibende Kraft F führt im Abstand zweier Netzebenen zu einem

26
Energieunterschied F.l, sodass )2/( TkFl B=ε .1 Definiert man nun den Diffusionskoeffizienten als , so erhält man für die Flussdichte 2
0lD Γ=
kTDFC
xCDj +∂∂
−= . (4-3)
Das ist in einer Dimension das erste Fick’sche Gesetz mit einem Driftterm. Wenn die Sprünge nicht mit der Diffusionsrichtung zusammenfallen, sondern Winkel iα mit ihr einschließen, muss über die Projektionen summiert werden. Für Nächst-Nachbarsprünge bekommt man dann
∑=
=z
iilD
1
220 cos
21 αΓ (4-4)
Für kubische Kristallstrukturen erhält man, unabhängig von der Diffusionsrichtung
Γ=Γ= 20
2
61
61 lzlD (4-5)
mit der totalen Sprungfrequenz z0Γ=Γ , wobei z die Anzahl der nächsten Nachbarn, die sog. Koordinationszahl ist. Mit der Kontinuumsgleichung für eindimensionalen Fluss
xj
tC
∂∂
−=∂∂ (4-6)
Bekommen wir das zweite Fick’sche Gesetz.
xC
kTDF
xCD
tC
∂∂
−∂∂
=∂∂
2
2
. (4-7)
Gehen wir nun den Weg 2) und verfolgen wir ein Atom auf seinem Weg. Sprünge nach links und rechts finden mit den Wahrscheinlichkeiten p und q = (1- p) statt. Nach N Sprüngen kann die Wahrscheinlichkeit, eine Distanz m nach rechts zurückgelegt zu haben (das bedeutet
2mN + Sprünge nach rechts und
2mN − Sprünge nach links) durch eine Binomialverteilung
ausgedrückt werden
2222 )1(2!
2!
2
!),(mNmNmNmN
ppmNN
qpmNmN
NmNP−+−+
−⎟⎟
⎠
⎞
⎜⎜
⎝
⎛+=
⎟⎠⎞
⎜⎝⎛ −
⎟⎠⎞
⎜⎝⎛ +
= (4-8)
Für eine große Anzahl n von Sprüngen kann die Stirling’sche Näherungsformel verwendet werden 1 Diese Beziehung folgt aus der Transition State Theory (Vineyard 1957) der Atomsprünge und ist hier ohne Beweis angeführt.

27
( 12ln21ln
21!ln −++−⎟⎠⎞
⎜⎝⎛ += nOnnnn π ) (4-9)
Nimmt man darüber hinaus an, dass m << N , sodass Terme ⎟⎟⎠
⎞⎜⎜⎝
⎛⎟⎠⎞
⎜⎝⎛
3
NmO oder höher
vernachlässigt werden können, und (wie vorher) dass sich die Links- und Rechts-Sprungwahrscheinlichkeiten nur sehr wenig unterscheiden
)(2
1 2εε Op ++
= (4-10)
)(2
11 2εε Opq +−
=−= , (4-11)
So erhält man für die Wahrscheinlichkeitsverteilung der Zufallsbewegung eine Gauß’sche Normalverteilung
( ) ( )⎟⎟⎠
⎞⎜⎜⎝
⎛ −−=
NNm
NNmP
2exp2,
2επ
. (4-12)
Wir führen nun eine kontinuierliche Ortsvariable mlx = (mit der Sprungdistanz l ) und eine kontinuierliche Zeitvariable ein, wobei angenommen wird, dass das Teilchen im Mittel 02Γ Sprünge pro Zeiteinheit macht. Die differentielle Wahrscheinlichkeit dafür, dass das Atom nach der Zeit t im Intervall [
),( txW]xxx Δ+, angekommen ist, ergibt sich dann zu
( )⎟⎟⎠
⎞⎜⎜⎝
⎛ −−=
Dttux
tDtxW
4exp
21),(
2
π , (4-13)
Wobei wir den Diffusionskoeffizienten und die Driftgeschwindigkeit 20lD Γ= εlu 02Γ= ein-
geführt haben. Mit dieser Funktion haben wir gleichzeitig auch die Green-Funktion für das Anfangswertproblem der Diffusion gewonnen (für die hier gemachten Annahmen, dass weder Diffusionskoeffizient noch treibende Kraft vom Ort abhängen). Da nämlich das zweite Fick’sche Gesetz eine lineare Differentialgleichung ist, können Lösungen durch Superposition konstruiert werden. Die obige Funktion gibt die Lösung an für den Fall, dass sich genau 1 A-Atom zum Zeitpunkt 0 an der Stelle 0 befindet, oder in Kontinuumssprache formuliert, dass dort eine Dirac’sche Deltafunktion sitzt. Die allgemeine Lösung für den Konzentrations-verlauf in Ort und Zeit bei einer gegebenen Anfangsverteilung stellt sich dann als Faltung mit dieser Green-Funktion dar:
),( txc )0,()(0 xcxc =
, (4-14) ∫+∞=
−∞=
−=ξ
ξ
ξξξ dtWxctxc ),()(),( 0
Da die Zufallsbewegung nicht von der Vorgeschichte abhängen soll, muss sich die Verteilung zu einem späteren Zeitpunkt τ+t in derselben Weise aus der Verteilung zum Zeitpunkt t entwickeln:

28
. (4-15) ∫+∞=
−∞=
−′=+′x
x
dxxWtxxCtxC ),(),(),( ττ
Wir entwickeln nun die linke Seite der Gleichung in der Variablen t und den Integranden auf der rechten Seite in x
∫+∞=
−∞=⎥⎦
⎤⎢⎣
⎡+
∂∂
+∂∂
−′=+∂∂
+′x
x
dxxWxCx
xCxtxC
tCtxC ),(....
21),(...),( 2
22 ττ . (4-16)
Definiert man nun das n-te Moment der Wahrscheinlichkeitsverteilung W als
∫+∞=
−∞=
=x
x
nn dxxWxx ),( τ , so kann man (für genügend kurze Zeiten τ) die Gl (4-16) in folgender
Form schreiben
xCx
xCx
tC
∂∂
−∂∂
=∂∂
ττ 2
22
2 . (4-17)
Vergleicht man dies nun mit dem zweiten Fick’schen Gesetz, so bekommen die darin vorkommenden Konstanten eine statistische Bedeutung (Einstein 1905):
ττ 2
lim2
0
xD
→= ,
τx
uTk
DF
B
== . (4-18)
Zu beachten ist dabei, dass bei Berechnung der Momente der Verteilungsfunktion Gl (4-13)
für endliche Zeiten zwar tutkTFDx == , aber Dtx 22 = nur bei Abwesenheit von
treibenden Kräften oder im Grenzfall sehr kurzer Zeiten gilt. Die Definition (4-18) bezieht sich in der Regel auf den Selbstdiffusionskoeffizenten (tracer diffusion coefficient)2, der normalerweise mit bezeichnet wird. Um eine Vorstellung von den mittleren Wanderungsdistanzen zu bekommen, halten wir fest, dass für substitutionelle Legierungen mit kfz Struktur ein typischer Diffusionskoeffizient beträgt.
Daraus folgt eine Wanderungsdistanz nach einem Tag von
*D
12910 −−≈ smD
cmx 3.121
2 ≈ . Eine Entwicklung wie in Gl. (4-16) kann auch im 3-dimensionalen Fall durchgeführt werden und liefert (wiederum für kleine Zeiten τ)
∑∑∑ ∂∂∂
+∂∂
−=∂∂
i j jiij
ii
i
xxCD
xCx
tC 2
τ . (4-19)
2 Dieser Diffusionskoeffizient spiegelt das Wanderungsverhalten von Isotopen (z.B. radioaktive Tracer) wieder, die sich in einer Umgebung von Atomen der gleichen Ordnungszahl bewegen. Bei Diffusionsexperimenten wird am Anfang eine Schicht solcher Tracer auf die Oberfläche aufgetragen und nach einer bestimmten Zeit durch schichtweises Abschleifen und Messung der Aktivität ermittelt, wie viel davon wie weit in den Probekörper eingedrungen ist.

29
Der Diffusionskoeffizient ist nun im allgemeinen Fall ein 2-stufiger Tensor
ττ 2lim
0
jiij
xxD
→= . (4-20)
Man kann ihn durch eine Hauptachsentransformation diagonalisieren, die die Symmetrie-eigenschaften des Kristalls widerspiegelt. Besonders einfach liegen wegen der hohen Symmetrie die Verhältnisse bei kubischen Kristallen, wo die Diffusion isotrop ist und stets
23
22
21 xxx ==
erfüllt ist. Dann gilt für dreidimensionale Wanderungsdistanzen
R=(x1, x2, x3)
ττ 6lim
2Rklein
D =, (4-21)
sodass für den isotropen, dreidimensionalen Fall die Fick’schen Gesetze der Diffusion folgen-dermaßen lauten
Tk
DCCDB
Fj +∇−= (4-22)
( )⎥⎦
⎤⎢⎣
⎡•∇−=
∂∂
TkCCD
tC
B
FΔ . (4-23)
Die 3-dimensionale Verallgemeinerung von Gl. (4-13) ergibt sich nun als Produkt von drei unabhängigen Zufallswanderungen in die drei Hauptdiffusionsrichtungen:
. Ohne treibende Kräfte erhalten wir explizit ),(),(),(),,( 321321 txWtxWtxWxxxW =
( )
⎥⎦
⎤⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛++−=
tDx
tDx
tDx
tDDDxxxW
3
21
2
22
1
21
21
3321
3321 444
exp8
1),,(π
. (4-24)
Die Oberflächen konstanter (Wanderungs-)Wahrscheinlichkeit sind also Ellipsoide, deren Achsen die Hauptdiffusionskoeffizienten sind. Für isotrope Diffusion (z.B. in kubischen Kristallen) werden die Ellipsoide zu Kugeln.
321 ,, DDD
4.3. Korrelation Wir betrachten eine Zufallsbewegung (random walk) ohne treibende Kräfte, wobei wir den Tracer-Diffusionskoeffizienten über Gl. (4-21) definieren. Die gesamte Atomverlagerung R stellt sich als Folge von Sprüngen r1, r2, …..rn dar, die nicht unbedingt gleiche Länge oder Richtung haben müssen. Wir schreiben dann die mittlere quadratische Verlagerung vom Ausgangspunkt weg wie folgt:
∑ ∑∑∑=
−
= +==
•+=⎟⎠
⎞⎜⎝
⎛=
n
i
n
i
n
ijjii
n
ii
1
1
1 1
22
1
2 2 rrrrR . (4-25)

30
Für eine komplett unkorrelierte Zufallsbewegung kürzen sich die Kreuzterme im Mittel weg, denn jeder Sprung ist genauso wahrscheinlich wie einer in die genau entgegengesetzte Richtung. Ist das nicht der Fall, so weist dies auf eine „Erinnerung“ des Atoms an den bisher zurückgelegten Weg hin. Warum dies auftritt, sei am Leerstellenmechanismus beispielhaft erklärt: Ein schwarzes Aufatom (Abbildung 4-2) hat gerade mit einer Leerstelle den Platz getauscht. Von allen weiteren Sprungrichtungen dieses Atoms ist nun die in Richtung der noch vorhandenen Leerstelle am wahrscheinlichsten, womit es an seinen ursprünglichen Platz zurückkehrt. Für jeden anderen Sprung muss es ja warten, bis in dieser Richtung eine Leerstelle auftaucht. Die Bewegung der Leerstelle selbst ist jedoch völlig unkorreliert, denn diese kann ja mit Hilfe jedes beliebigen Atoms ihren Platz verlagern.
Abbildung 4-2: Leerstellenmechanismus Abbildung 4-3: Interstitialcy – Mechanismus Ein anderes Beispiel ist die Korrelation, die sich beim „Interstitialcy“-Mechanismus aus dem Wechselspiel von regulären und Zwischengitterplätzen ergibt (Abbildung 4-3). Das Aufatom (schwarz) liegt ursprünglich auf einem Zwischengitterplatz und springt dann auf einen regulären Gitterplatz; das ursprünglich dort sitzende Atom wird zu einem Zwischengitter-atom. Es gibt nun eine erhöhte Wahrscheinlichkeit dafür, dass der nächste Sprung die Umkehr des gerade stattgefundenen Austauschs ist, eben weil der dafür notwendige Defekt (Zwischen-gitteratom) noch vorhanden ist. Um die Korrelationseffekte bei Atomarer Diffusion quantitativ zu beschreiben, definiert man einen Korrelationsfaktor als Verhältnis von 2R aus Gl. (4-25) mit
random
2R einer völlig
zufälligen Bewegung
⎪⎪⎭
⎪⎪⎬
⎫
⎪⎪⎩
⎪⎪⎨
⎧•
+==
∑
∑∑
=
−
= +=
∞→ n
ii
n
i
n
ijji
nrandom
f
1
2
1
1 12
2 21lim
r
rr
R
R. (4-26)
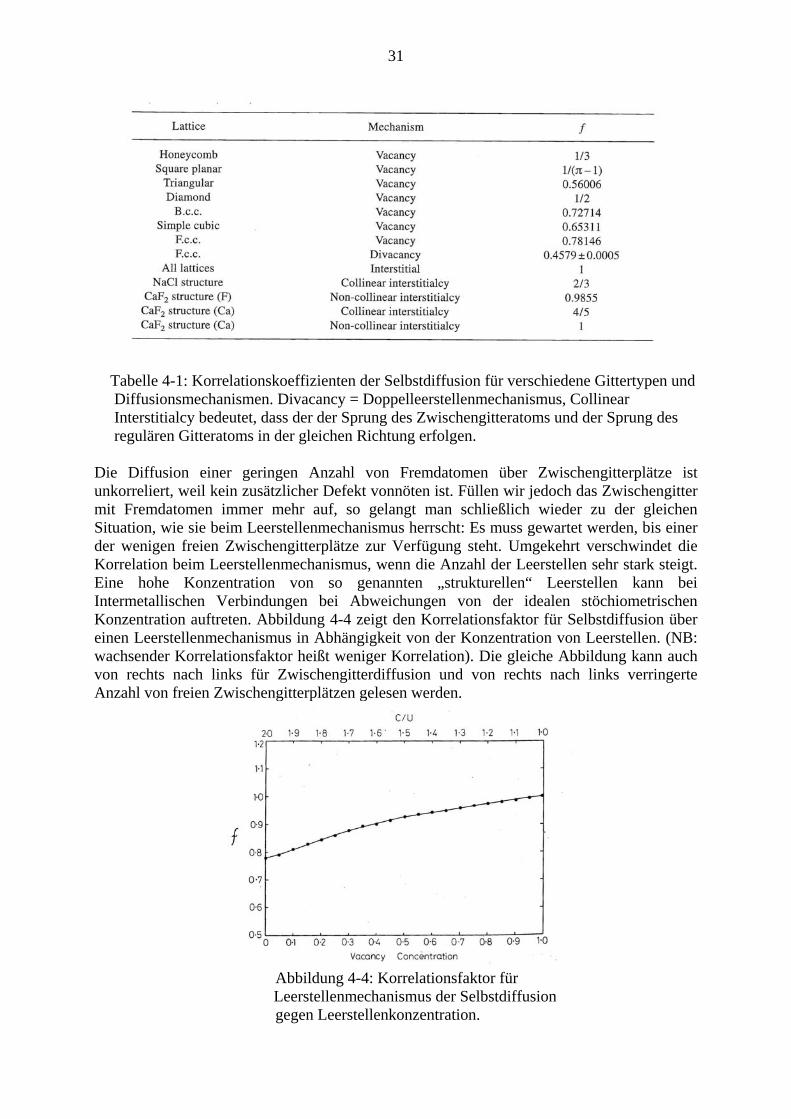
Dieser beträgt 1 für völlig unkorrelierte Bewegung und wird umso kleiner, je mehr Korrelation auftritt, die zu einer Umkehr von Sprüngen führt. Korrelationsfaktoren für verschiedene Gittertypen und Diffusionsmechanismen können mit theoretischen Mitteln oder durch Computersimulationen berechnet werden und sind in einschlägigen Lehrbüchern tabelliert. Tabelle 4-1 gibt ein Beispiel für Selbstdiffusion.
31
Tabelle 4-1: Korrelationskoeffizienten der Selbstdiffusion für verschiedene Gittertypen und Diffusionsmechanismen. Divacancy = Doppelleerstellenmechanismus, Collinear Interstitialcy bedeutet, dass der der Sprung des Zwischengitteratoms und der Sprung des regulären Gitteratoms in der gleichen Richtung erfolgen. Die Diffusion einer geringen Anzahl von Fremdatomen über Zwischengitterplätze ist unkorreliert, weil kein zusätzlicher Defekt vonnöten ist. Füllen wir jedoch das Zwischengitter mit Fremdatomen immer mehr auf, so gelangt man schließlich wieder zu der gleichen Situation, wie sie beim Leerstellenmechanismus herrscht: Es muss gewartet werden, bis einer der wenigen freien Zwischengitterplätze zur Verfügung steht. Umgekehrt verschwindet die Korrelation beim Leerstellenmechanismus, wenn die Anzahl der Leerstellen sehr stark steigt. Eine hohe Konzentration von so genannten „strukturellen“ Leerstellen kann bei Intermetallischen Verbindungen bei Abweichungen von der idealen stöchiometrischen Konzentration auftreten. Abbildung 4-4 zeigt den Korrelationsfaktor für Selbstdiffusion über einen Leerstellenmechanismus in Abhängigkeit von der Konzentration von Leerstellen. (NB: wachsender Korrelationsfaktor heißt weniger Korrelation). Die gleiche Abbildung kann auch von rechts nach links für Zwischengitterdiffusion und von rechts nach links verringerte Anzahl von freien Zwischengitterplätzen gelesen werden.
Abbildung 4-4: Korrelationsfaktor für
Leerstellenmechanismus der Selbstdiffusion gegen Leerstellenkonzentration.

32
Diffusion in ferngeordneten Intermetallischen Verbindungen stellt einen Sonderfall dar, weil Atomsprünge zwischen Untergittern zu einer Änderung der Ordnungsstruktur führen. Wird dabei ein Antistrukturdefekt (antisite) erzeugt, also ein Atom, das auf dem falschen Untergitter sitzt, erhöht sich dadurch die Energie, sodass ein unmittelbar folgender Rücksprung sehr wahrscheinlich und damit die Korrelation sehr stark wird. Bei Diffusion von Fremdatomen über Leerstellenmechanismus lässt sich der Korrelationsfaktor als Funktion von fünf charakteristischen Wechselwirkungen zwischen Atomen bzw. Defekten angeben (five-frequency-model).
4.4. Atomare Zufallsbewegung mit chemischer Treibender Kraft Das 1. Fick’sche Gesetz Gl. (4-3) beschreibt in Abwesenheit einer Treibenden Kraft (Abb.4-1, Szenario a)) einen Diffusionstrom, der nur dadurch zustande kommt, dass die Konzentration der betrachteten Atomsorte an verschiedenen Orten des Kristalls verschieden ist. Formal könnte man in diesem Fall zwar sagen, dass die Mischungsentropie die Treibende Kraft der Diffusion ist. Betrachten wir aber einen einzelnen Atomsprung, dann geht die Mischungsentropie nicht explizit in die Sprungrate ein, denn sie ergibt sich direkt aus der Statistik der Atome, die nach links oder nach rechts springen. Für eine Ideale Lösung (keine Wechselwirkungskräfte zwischen den Atomen) haben daher Sprünge in verschiedene Richtungen gleiche Sprungfrequenzen. Im allgemeinen Fall einer nicht-idealen Lösung erwarten wir jedoch als Folge des Atomsprunges eine Enthalpiedifferenz, etwa weil sich die Anzahl der A-Atome in der Nachbarschaft des diffundierenden Atoms geändert hat. Ursache für die chemische Treibende Kraft ist der Unterschied an Freier Enthalpie bei Hinzufügung eines Atoms an verschiedenen Orten des Kristalls, ausgedrückt durch das chemische Potenzial3. Für die Atomsorte i schreiben wir es als
cexi
ideali
ii N
G μμμμ ++=∂∂
= 0 , (4-28)
Wobei sich μ0 das chemische Potenzial eines (konstanten) Referenzzustandes ist, der zweite Beitrag nur von der Mischungsentropie kommt (und daher nicht als treibende Kraft für einen Atomsprung auftritt). Für die bei einem einzelnen Atomsprung wirksame Treibende Kraft gilt daher
l
xFl
exci
∂∂
−=μ . (4-29)
Mit Hilfe des Aktivitätsfaktors γi ,
ideali
ii a
a=γ (4-30)
(Zur Definition der Aktivität siehe Gl. (3-13)) lässt sich schreiben
, (4-31) iB
exci Tk γμ ln=
3 Anders als in Kapitel 3 ist das chemische Potenzial hier nicht auf ein Mol (6,023x1023 Teilchen), sondern auf ein Teilchen bezogen.

33
und analog zu Gl. (4-3) (2. Fick’sches Gesetz mit Treibender Kraft) können wir schreiben
x
Cc
Dx
Cc
cDx
CDx
CDj i
i
ii
i
i
iii
iii
iii ∂
∂⎥⎦
⎤⎢⎣
⎡∂∂
+−=∂∂
⎥⎦
⎤⎢⎣
⎡∂
∂+−=
∂∂
−∂∂
−=lnln1ln1ln **** γγγ . (4-32)
Hier haben wir die Gitterplatzbesetzung (site fraction) ii Cc Ω= , eingeführt, wobei Ω das Atomvolumen ist (Für homogene Legierungen ci = Xi, Molenbruch). Wir sehen also, dass die Diffusion in einer chemischen Treibenden Kraft statt durch einen additiven Driftterm auch durch einen Thermodynamischen Faktor ϕ zum Diffusionskoeffizienten beschrieben werden kann:
i
i
clnln1
∂∂
+=γϕ (4-33)
Es lässt sich zeigen, dass g ′′∝ϕ (vergleiche Spinodale Entmischung, Kapitel 5.2.). Das bedeutet, dass einer thermodynamisch instabilen Legierung der Diffusionskoeffizient negativ wird, wodurch kleine Konzentrationsschwankungen nicht mehr durch Diffusion ausgeglichen, sondern aufgeschaukelt werden. Mit dem Stern und dem Index i haben wir darauf hingewiesen, dass es sich in (4-33) um den Tracer-Diffusionskoeffizienten für die Atomsorte i handelt4.
Wir sehen also, dass eine Reihe von Einflüssen in die Diffusion mittels Defekten (Leerstellen!) eingehen: Geometrie des Gitters, die Verfügbarkeit von Defekten, Korrelation der Sprünge, Gitterschwingungen, die Höhe der Sprungbarriere, chemische Treibende Kraft.
4.5. Nicht-gegengleiche Diffusion (Non-reciprocal diffusion), Kirkendall-Effekt. Wir haben bei unseren bisherigen Betrachtungen immer nur die Statistik für die Bewegung einer einzigen Atomsorte in Betracht gezogen. Natürlich kommt es bei binären Legierungen zur Durchmischung infolge Diffusion beider Atomsorten, Interdiffusion oder Chemische Diffusion genannt. Diese lässt sich in binären Legierungen trotzdem mit einem einzigen effektiven Parameter beschreiben, dem Interdiffusionskoeffizienten (chemischen Diffusions-koeffzizienten) D~ . Diffundieren beide Atomsorten gleich schnell (gegengleiche Diffusion), dann ist er gleich den jeweiligen Diffusionskoeffizenten (intrinsischen Diffusionskoeffizi-enten) der beiden Atomsorten DA=DB=B D~ . Bewegen sich bei einem Leerstellenmechanismus die beiden Atomsorten verschieden schnell (nicht-gegengleiche Diffusion, non-reciprocal diffusion), dann kommt es zu einem Netto-Fluss von Leerstellen in eine Richtung (Leerstellenwind, vacancy wind). Diese Erscheinung wird als Kirkendall-Effekt bezeichnet. Im Originalversuch von Smigelskas and Kirkendall (1947) wurde ein Cu-Kristall an einen Messing-Kristall (Cu70Zn30) gefügt. In der Mitte befanden sich in periodischen Abständen inerte Mo-Drähte als Markierung. Man beobachtet nun nach genügend langer Auslagerungszeit bei hoher Temperatur, dass die Probe auf der einen Seite der Phasengrenze anschwillt, während ihr auf der anderen Seite Material entzogen wird, bis hin zum Auftreten von Löchern. Die Markierungsdrähte wandern relativ zu den Enden der Probe in Richtung des Gebietes, dem Material entzogen wird.
4 Der gleiche Faktor tritt jedoch auch beim chemischen Diffusionskoeffizienten (= Interdiffusionskoeffizienten) im Falle einer chemischen Treibenden Kraft auf.

34
Abbildung 5: Kirkendall-Effekt. Links schematisch, rechts Cu-Zn-Diffusionspaar. Zn diffundiert schneller, daher entstehen auf der linken Seite Löcher (unregelmäßiger Abbau des Kristallgitters infolge Ansammlung von Leerstellen). Das Diffusionsgeschehen kann in zwei alternativen Bezugssystemen betrachtet werden: 1) dem Laborsystem, in dem die Enden der Probe ruhen, 2) dem Gittersystem, in dem die Gitterebenen der Diffusionszone ruhen, gekennzeichnet durch die Lage der Markierungs-drähte. Diese bewegen sich mit der Geschwindigkeit vM im Laborsystem. Im Gittersystem sind die Flüsse der A- und B-Atome nach dem 1. Fick’schen Gesetz gegeben durch
xCDj A
AA ∂∂
−= und x
CDj BBB ∂∂
−= . (4-34)
Wenn DA ≠ DB, dann gibt es einen Nettofluss an Atomen jB net= jA+jBB. Wo dieser hinfließt, wird Material aufgehäuft, wo er herkommt, wird Material entzogen, und es werden Leerstellen aufgehäuft, denn es gilt 0=++ BAV jjj bzw. netBAV jjjj −=+−= )( (4-35) Entfernen von Material (= Anhäufen von Leerstellen) ist aber gleichbedeutend mit einem Abbau von Gitterzellen. Dabei wird pro Atom das Volumen Ω vernichtet, sodass Ω−= netM jv . (4-36) für den Leerstellenstrom im Gitter-Referenzsystem
x
CDDj B
ABV ∂∂
−= )( . (4-37)
Der Leerstellenwind ist also proportional zur Differenz der intrinsischen Diffusionskoeffi-zienten und bläst entgegen der schneller diffundierenden Atomsorte durch das Gitter. Bei

35
Vorliegen eines Kirkendall-Effektes werden die Fick’schen Gesetze durch die Drift der Gitterebenen (Advektionsbewegung) modifiziert
(4-38) CCD Mvj −∇−= ~
CCDtC
M ∇+∇∇=∂∂ .)~.( v . (4-39)
Gl. (4-42) wird auch Diffusions-Advektionsgleichung genannt. L. Darken lieferte eine phänomenologische Erklärung des Kirkendall-Effektes (Darken 1948). Nach ihm sind folgende Beziehungen für die Markierungsgeschwindigkeit und den Interdiffusionskoeffizienten benannt:
x
cDx
cDv BB
AAM ∂
∂+
∂∂
= (4-40)
(1. Darken-Gleichung), (4-41) BAAB DcDcD +≡~
(2. Darken-Gleichung). cA und cB sind die Gitterplatzkonzentrationen (site fractions, cB i = Ω Ci ). Daraus lassen sich nach Ermittlung von vM und D~ die intrinsischen Diffusionskoeffizienten DA und DB bestimmen. wird dabei aus dem entstehenden Konzentrationsprofil mithilfe der Methode von Boltzmann-Matano gewonnen (Siehe angegebene Literatur).
)(~~ CDD =
Literaturangaben zur Diffusion: W. Püschl, H. Numakura, W. Pfeiler, Chapter 5, Point Defects, Atom Jumps, and Diffusion in Alloy Physics (W. Pfeiler, Ed.), Wiley-VCH 2007 Mehrer, H., Diffusion in Solids, Springer, Berlin Heidelberg 2007. Glicksman, M.E., Diffusion in Solids, John Wiley & Sons, New York, 2000. Murch, G. E., Diffusion Kinetics in Solids, in Phase Transformations in Materials, G. Kostorz, Ed., Wiley-VCH, Weinheim 2001, p. 195. Manning, J. R., Diffusion Kinetics for Atoms in Crystals, Van Nostrand, Princeton, New Jersey 1968 Smigelskas, A.D. and Kirkendall, E.O. et al. Trans. AIME 171, 130 (1947).
36
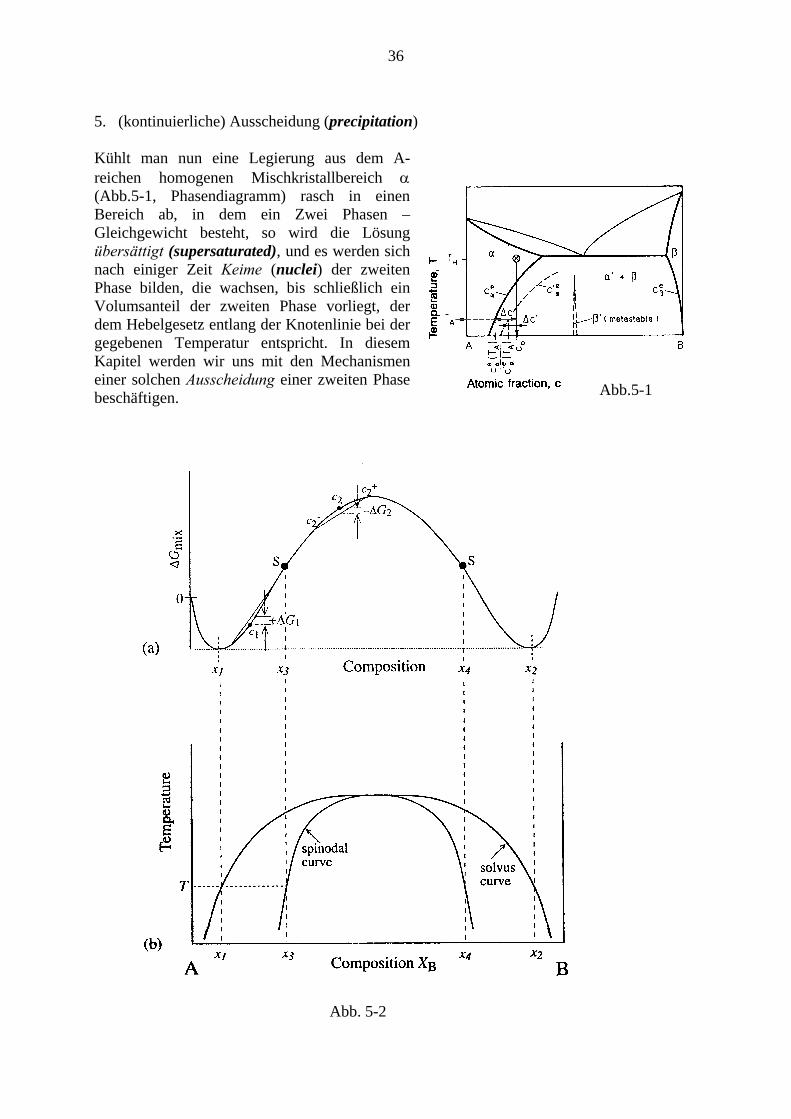
5. (kontinuierliche) Ausscheidung (precipitation) Kühlt man nun eine Legierung aus dem A-reichen homogenen Mischkristallbereich α (Abb.5-1, Phasendiagramm) rasch in einen Bereich ab, in dem ein Zwei Phasen – Gleichgewicht besteht, so wird die Lösung übersättigt (supersaturated), und es werden sich nach einiger Zeit Keime (nuclei) der zweiten Phase bilden, die wachsen, bis schließlich ein Volumsanteil der zweiten Phase vorliegt, der dem Hebelgesetz entlang der Knotenlinie bei der gegebenen Temperatur entspricht. In diesem Kapitel werden wir uns mit den Mechanismen einer solchen Ausscheidung einer zweiten Phase beschäftigen. Abb.5-1
Abb. 5-2

37
Eine wichtige Fallunterscheidung wurde traditionell danach getroffen, ob der nach der Abkühlung zunächst noch immer homogene Mischkristall stabil gegenüber kleinen Konzentrationsfluktuationen ist oder nicht. Unter der Voraussetzung, dass man die freie Enthalpie pro Volumseinheit der metastabilen Phase durch „höckerartige“ Kurven wie in Abb. 5-2 (siehe auch Abb.3-4b) darstellen kann, ergibt sich: Ist die Kurve konkav („hohl“ gegen die x-Achse), dann bringt eine Aufspaltung der Mischung in Gebiete eng benachbarter Konzentrationen jedenfalls Energiegewinn, denn die freie Enthalpie des zweiphasigen Ergebnisses liegt auf der Sehne, welche die Punkte auf der Kurve für die Konzentrationen der beiden entstehenden Phasen verbindet, somit unter der Kurve.5 Anderenfalls (konvexe Kurve) muss die ausgeschiedene Phase sehr viel höhere Konzentration haben, um insgesamt einen
Energiegewinn zu ermöglichen. Die beiden Fälle werden getrennt durch die Linie der Wendepunkte der „Kamelhöckerkurven“ (S in Abb.3-4b), welche Spinodale („spinodal curve“ in Abb. 5-2) genannt wird. Innerhalb der Spinodalen wächst demnach jede Konzentrationsfluktuation spontan an (spinodale Entmischung, spinodal decomposition). Im Vordergrund stehen hier „Homophasenfluktuationen“ (Abb.5-3) Außerhalb der Spinodalen müssen Keime der zweiten Phase mit einer genügend hohen Konzentration gebildet werden (Keimbildung, nucleation) („Heterophasenfluktuationen“, Abb.5-3). Diese beiden Typen von Umwandlungen werden nach Josiah Willard Gibbs, einem der Väter der Thermodynamik, auch als Gibbs-Typ I (Heterophasen-Fluktuation) und Gibbs-Typ II (Homophasen-Fluktuation) bezeichnet.
Abb.5-3
So klar ist die Trennung der beiden Fälle jedoch in Wirklichkeit nicht. Was tatsächlich experimentell beobachtet wird und aus Simulationen der atomaren Kinetik hervor geht, ist vielmehr ein kontinuierlicher bzw. verwaschener Übergang (siehe dazu auch Diskussion im Kapitel über spinodale Entmischung).
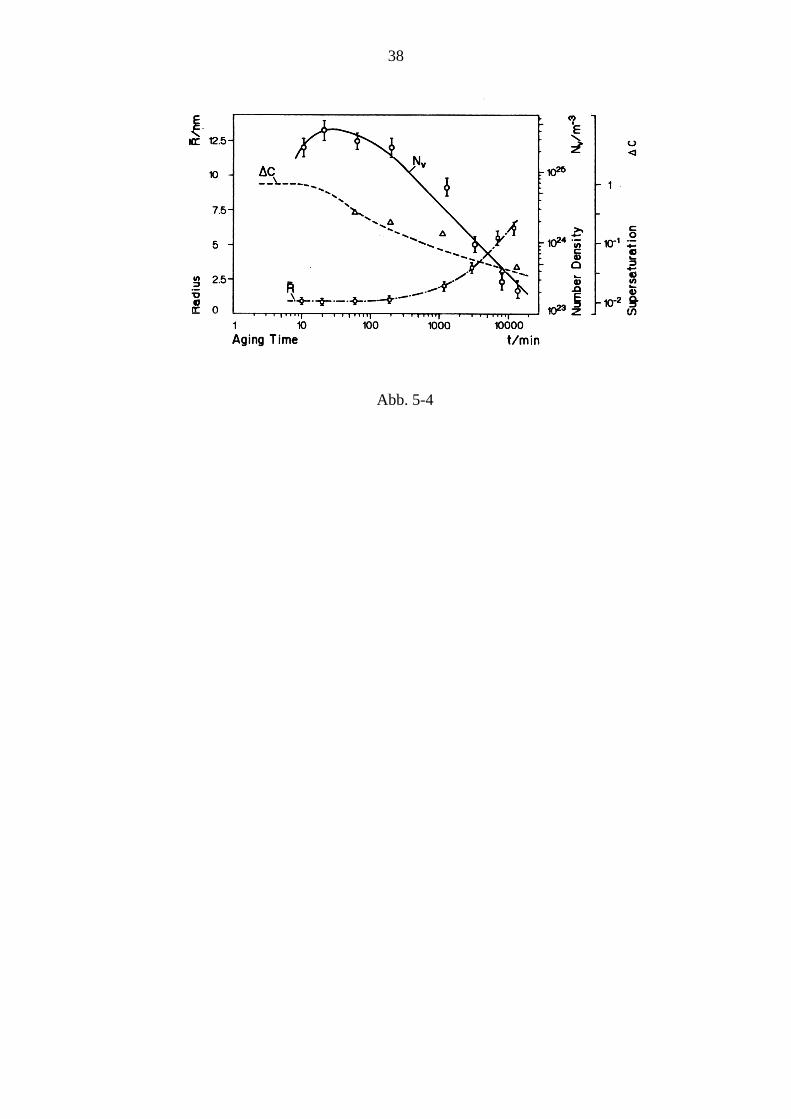
Der gesamte Ausscheidungsvorgang verläuft im Wesentlichen in drei Stufen: Zunächst werden Keime gebildet, diese wachsen dann, wodurch die Konzentration (Übersättigung) in der Matrix relativ rasch bis zum Gleichgewichtswert sinkt. Schon nach relativ geringem Absinken der Übersättigung kommt die Keimbildung zum Erliegen. In der lange dauernden Spätphase kommt es zur Vergröberung (coarsenig), auch Ostwaldreifung genannt, bei der große Ausscheidungsteilchen auf Kosten kleinerer, die verschwinden, wachsen. In Abb. 4-3 ist dieser Verlauf anhand der Kurven für Teilchenzahl NV, mittleren Radius R und Übersättigung Δc bei γ ′ - Ausscheidung bei einer Ni36Cu9Al55 - Legierung dargestellt (Man beachte die logarithmische Darstellung der Zeitachse sowie von NV und Δc)
5 Für die Mengenverhältnisse gilt wieder das Hebelgesetz Gl. (3-20). Addiert man die beiden Teilphasen zur Gesamtenergie, so erhält man Punkte auf der Verbindungsgeraden.
38
Abb. 5-4
39
5.1. Keimbildung (nucleation)
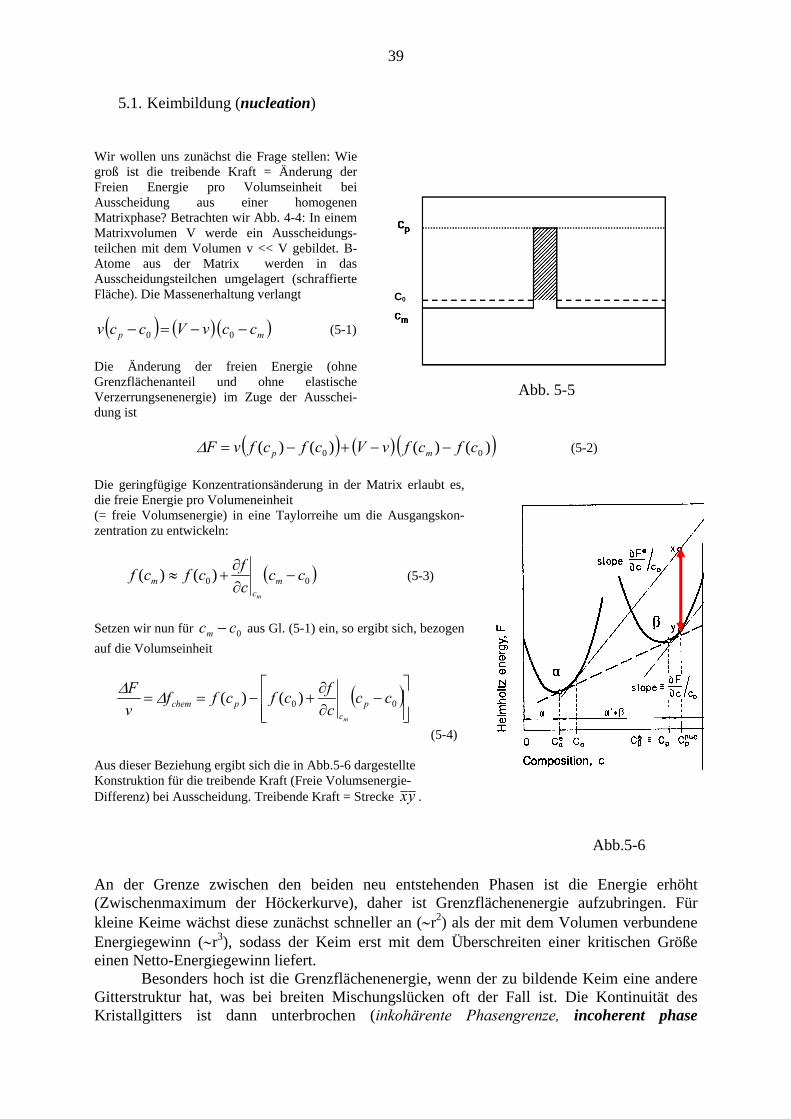
Wir wollen uns zunächst die Frage stellen: Wie groß ist die treibende Kraft = Änderung der Freien Energie pro Volumseinheit bei Ausscheidung aus einer homogenen Matrixphase? Betrachten wir Abb. 4-4: In einem Matrixvolumen V werde ein Ausscheidungs-teilchen mit dem Volumen v << V gebildet. B-Atome aus der Matrix werden in das Ausscheidungsteilchen umgelagert (schraffierte Fläche). Die Massenerhaltung verlangt C0
( ) ( ) ( mp ccvVccv −−=− 00 ) (5-1)
Die Änderung der freien Energie (ohne Grenzflächenanteil und ohne elastische Verzerrungsenenergie) im Zuge der Ausschei-dung ist
Abb. 5-5
( ) ( ) ( ))()()()( 00 cfcfvVcfcfvF mp −−+−=Δ (5-2)
Die geringfügige Konzentrationsänderung in der Matrix erlaubt es, die freie Energie pro Volumeneinheit (= freie Volumsenergie) in eine Taylorreihe um die Ausgangskon-zentration zu entwickeln:
( 00 )()( cccfcfcf m
cm
m
−∂∂
+≈ )
m −
(5-3)
Setzen wir nun für c aus Gl. (5-1) ein, so ergibt sich, bezogen auf die Volumseinheit
0c
( )⎥⎥⎦
⎤
⎢⎢⎣
⎡−
∂∂
+−== 00 )()( cccfcfcff
vF
pc
pchemm
ΔΔ
(5-4)
Aus dieser Beziehung ergibt sich die in Abb.5-6 dargestellte Konstruktion für die treibende Kraft (Freie Volumsenergie-Differenz) bei Ausscheidung. Treibende Kraft = Strecke yx .
Abb.5-6 An der Grenze zwischen den beiden neu entstehenden Phasen ist die Energie erhöht (Zwischenmaximum der Höckerkurve), daher ist Grenzflächenenergie aufzubringen. Für kleine Keime wächst diese zunächst schneller an (∼r2) als der mit dem Volumen verbundene Energiegewinn (∼r3), sodass der Keim erst mit dem Überschreiten einer kritischen Größe einen Netto-Energiegewinn liefert.
Besonders hoch ist die Grenzflächenenergie, wenn der zu bildende Keim eine andere Gitterstruktur hat, was bei breiten Mischungslücken oft der Fall ist. Die Kontinuität des Kristallgitters ist dann unterbrochen (inkohärente Phasengrenze, incoherent phase
40
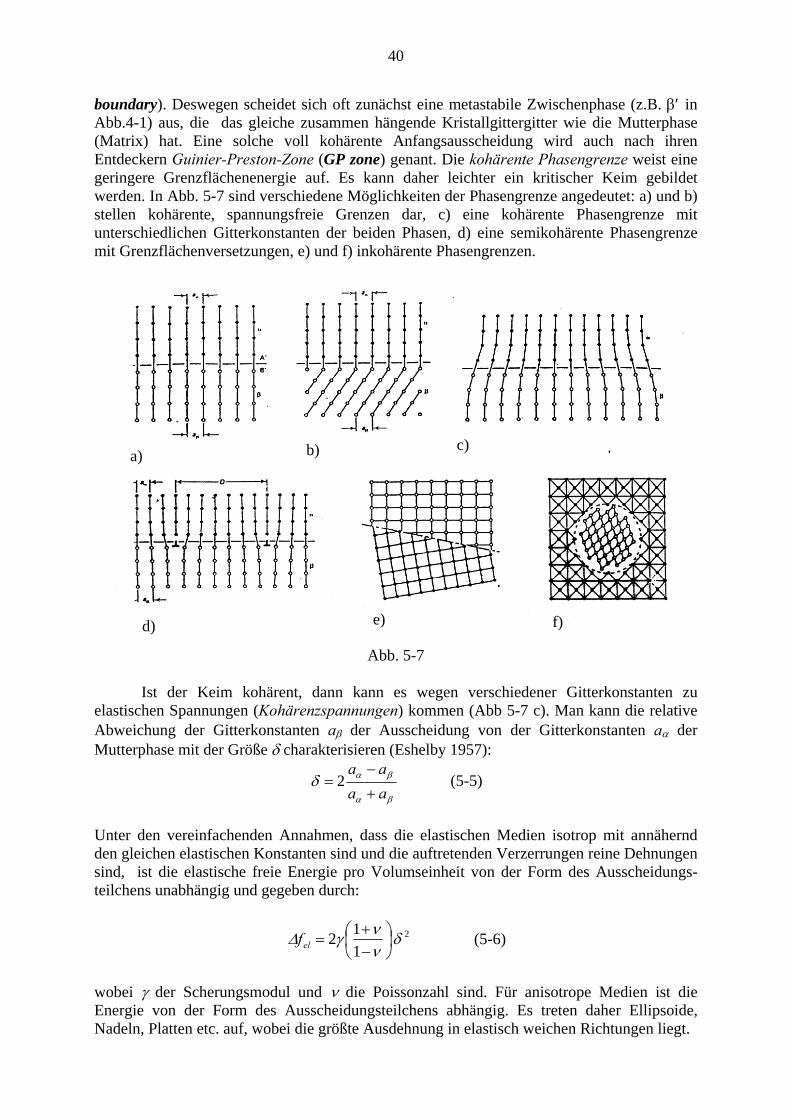
boundary). Deswegen scheidet sich oft zunächst eine metastabile Zwischenphase (z.B. β′ in Abb.4-1) aus, die das gleiche zusammen hängende Kristallgittergitter wie die Mutterphase (Matrix) hat. Eine solche voll kohärente Anfangsausscheidung wird auch nach ihren Entdeckern Guinier-Preston-Zone (GP zone) genant. Die kohärente Phasengrenze weist eine geringere Grenzflächenenergie auf. Es kann daher leichter ein kritischer Keim gebildet werden. In Abb. 5-7 sind verschiedene Möglichkeiten der Phasengrenze angedeutet: a) und b) stellen kohärente, spannungsfreie Grenzen dar, c) eine kohärente Phasengrenze mit unterschiedlichen Gitterkonstanten der beiden Phasen, d) eine semikohärente Phasengrenze mit Grenzflächenversetzungen, e) und f) inkohärente Phasengrenzen.
c) b) a)
e) f) d)
Abb. 5-7
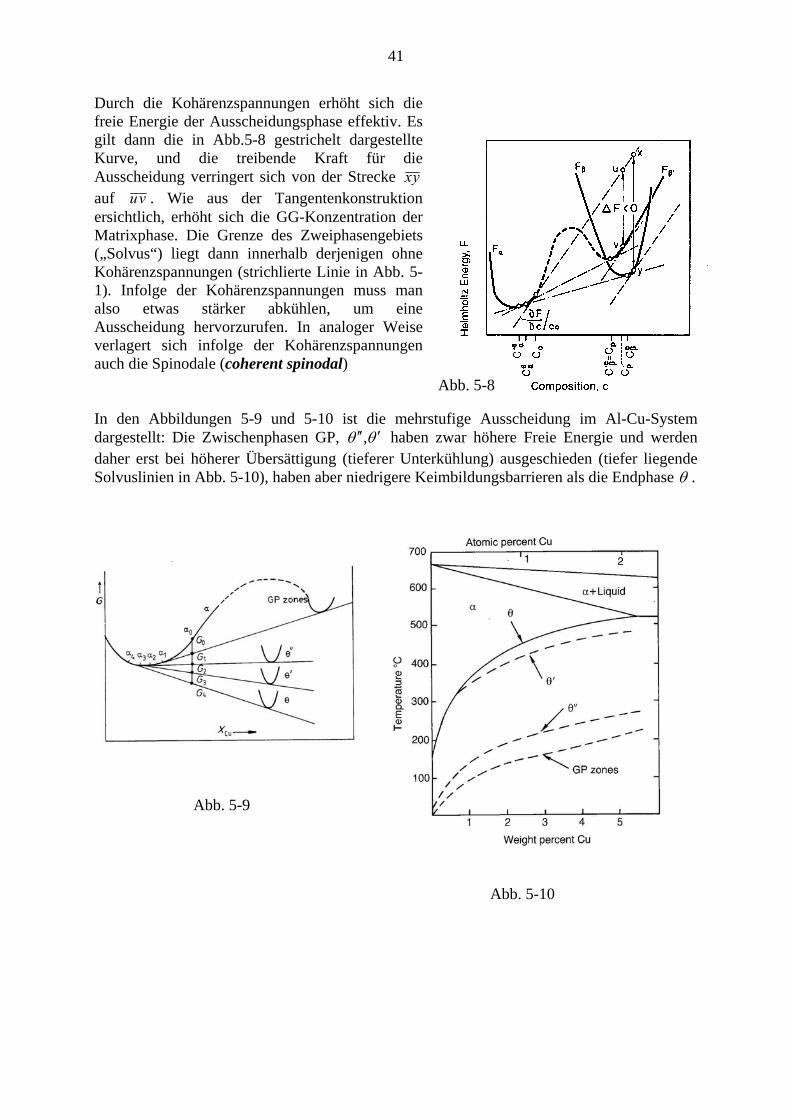
Ist der Keim kohärent, dann kann es wegen verschiedener Gitterkonstanten zu elastischen Spannungen (Kohärenzspannungen) kommen (Abb 5-7 c). Man kann die relative Abweichung der Gitterkonstanten aβ der Ausscheidung von der Gitterkonstanten aα der Mutterphase mit der Größe δ charakterisieren (Eshelby 1957):
βα
βαδaaaa
+
−= 2 (5-5)
Unter den vereinfachenden Annahmen, dass die elastischen Medien isotrop mit annähernd den gleichen elastischen Konstanten sind und die auftretenden Verzerrungen reine Dehnungen sind, ist die elastische freie Energie pro Volumseinheit von der Form des Ausscheidungs-teilchens unabhängig und gegeben durch:
2
112 δ
ννγΔ ⎟⎠⎞
⎜⎝⎛−+
=elf (5-6)
wobei γ der Scherungsmodul und ν die Poissonzahl sind. Für anisotrope Medien ist die Energie von der Form des Ausscheidungsteilchens abhängig. Es treten daher Ellipsoide, Nadeln, Platten etc. auf, wobei die größte Ausdehnung in elastisch weichen Richtungen liegt.
41
Durch die Kohärenzspannungen erhöht sich die freie Energie der Ausscheidungsphase effektiv. Es gilt dann die in Abb.5-8 gestrichelt dargestellte Kurve, und die treibende Kraft für die Ausscheidung verringert sich von der Strecke yx auf vu . Wie aus der Tangentenkonstruktion ersichtlich, erhöht sich die GG-Konzentration der Matrixphase. Die Grenze des Zweiphasengebiets („Solvus“) liegt dann innerhalb derjenigen ohne Kohärenzspannungen (strichlierte Linie in Abb. 5-1). Infolge der Kohärenzspannungen muss man also etwas stärker abkühlen, um eine Ausscheidung hervorzurufen. In analoger Weise verlagert sich infolge der Kohärenzspannungen auch die Spinodale (coherent spinodal)
Abb. 5-8 In den Abbildungen 5-9 und 5-10 ist die mehrstufige Ausscheidung im Al-Cu-System dargestellt: Die Zwischenphasen GP, θθ ′′′ , haben zwar höhere Freie Energie und werden daher erst bei höherer Übersättigung (tieferer Unterkühlung) ausgeschieden (tiefer liegende Solvuslinien in Abb. 5-10), haben aber niedrigere Keimbildungsbarrieren als die Endphase θ .
Abb. 5-9
Abb. 5-10

42
5.1.1. Die klassische Keimbildungstheorie
Energiebilanz eines Keims
Als einfachste Näherung geht man im sog. Tröpfchenmodell von einem kugelförmigen Ausscheidungskeim mit Radius R und scharfer Phasengrenzfläche aus. Diesem kommt dann mit der spezifischen Grenzflächenenergie αβσ folgende freie Bildungsenergie zu:
( ) αβσππ 23 43
4)( RRffRF elchem +Δ+Δ=Δ (5-7)
Der Verlauf dieser Funktion ist schematisch in Abb. 5-11 dargestellt. Sie erreicht für den Radius R* eines kritischen Keims (critical nucleus) ein Maximum. Die Energieschwelle (Keimbildungsenergie) beträgt dann
( )23
*
316)(
elchem ffRF
ΔΔσπΔ αβ
+= (5-8)
Die klassische Keimbildungstheorie (Volmer und Weber 1926, Becker und Döring 1935, Zeldovich 1943) wurde zunächst für den Kondensationsvorgang gasförmig-flüssig entwickelt und erst später für Phasenübergänge im festen Aggregatzustand adaptiert. Sie geht von einer diskreten Verteilungsfunktion N( i , t ) aus, die angibt, wie viele Keime mit i Atomen (bzw. Molekülen) zum Zeitpunkt t existieren. Sie soll sich nur durch den Übergang i → i ± 1 ändern, d.h. das „Kondensieren“ bzw. „Verdampfen“ einzelner Atome (Monomere). Der Netto-Fluss im Phasenraum von Keimen zwischen zwei Größenklassen beträgt dann Abb.5-11
−+
+ +−= 1),1(),( iii tiNtiNJ γγ (5-9)
mit als Kondensationsrate und als Evaporationsrate einzelner Monomere. +iγ
−+1iγ
Die freie Energie der gesamten Anordnung von verschieden großen Keimen in der Matrixphase beträgt
∑ −=n
mixTStiNiFF1
),()(Δ (5-10)
wobei ΔF(i) die freie Energie eines Keimes der Größe i ist und Smix ein Mischungsentropie-Term, der die Anzahl der Möglichkeiten berücksichtigt, die Keime auf die Gitterplätze zu verteilen. Minimierung von (5-10) nach N(i,t) ergibt als Gleichgewichts-Größenverteilung
⎟⎠⎞
⎜⎝⎛−=
kTiFNiN ee)(exp)1()( Δ (5-11)

43
Da bei Vorliegen einer Gleichgewichtsverteilung der Nettostrom zwischen den Größenklassen 0 sein muss (Prinzip des detaillierten Gleichgewichts, detailed balance), folgt
)1()(
1 += +−
+ iNiN
e
eii γγ (5-12)
Der Strom im Phasenraum lässt sich dann folgendermaßen schreiben:
⎥⎥⎥⎥⎥
⎦
⎤
⎢⎢⎢⎢⎢
⎣
⎡
∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂
−≈⎥⎦
⎤⎢⎣
⎡++
−= ++
iiNtiN
iNiN
tiNiNtiNiNJ e
eiee
eii
)(),(
)()1(),1(
)(),()( γγ (5-13)
Die ursprüngliche Theorie von Volmer und Weber macht nun die Annahme, dass Keime nur bis zur kritischen Größe existieren und dann aus dem System „entfernt“ werden. Da aber überkritische Keime durch Abdampfen von Monomeren auch wieder unterkritisch werden können, macht man dabei einen beträchtlichen numerischen Fehler. In der Behandlung von Becker und Döring wird daher angenommen, dass es Keime über die kritische Größe hinaus gibt. Dividiert man in der linken Gleichung von (5-13) beide Seiten durch , so bleibt rechts nur mehr der Ausdruck in eckiger Klammer. Man summiert nun über i von einer Keimgröße i
)(iNei+γ
0 , die deutlich kleiner als der kritische Keim ist, bis zu einer Keimgröße im , die deutlich größer als der kritische Keim ist. In aufeinander folgenden Summanden hebt sich jeweils der rechte Term in eckiger Klammer (für i = k) mit dem linken Term (für i = k+1) weg, sodass nur der erste und der letzte Term übrig bleiben:
101)()(
)()(
)( 0
0
0
=−≈−=∑=
+me
m
e
i
ii ei
i
iNiN
iNiN
iNJm
γ (5-14)
Für genügend kleine Keime wird die Voraussetzung gemacht, dass deren Häufigkeit der Gleichgewichtsverteilung entspricht, die Häufigkeit von Keimen, die deutlich größer als der kritische sind, soll gegen 0 gehen, vor allem relativ zur Gleichgewichtsverteilung, die rein formal in diesem Bereich sehr stark anwächst. Damit ergibt sich (5-14). Geht man zum kontinuierlichen Limes über, dann kann man (5-14) als Integral schreiben
∫ =+
mi
i ei
i diiN
J
0
1)(γ
(5-15)
Von den Termen im Integranden hat Ne in der Nähe des kritischen Keims wegen (5-11) und der Tatsache, dass dort maximal wird, ein sehr scharfes Minimum. Im Vergleich dazu sind und praktisch konstant. Die Vorstellung ist ja, dass ein kleiner, aber konstanter Strom von Keimen über die kritische Größe hinweg besteht. Das Integral (5-15) ist also durch das scharfe Maximum an der Stelle des kritischen Keims dominiert. Man kann dort für den Exponenten in (5-11) nach einer Taylorreihe entwickeln:
)(iFΔSBDi JJ ≈ *γγ ≈+
i
( 2*2
2*
*21 ii
iFFF
ii
−∂Δ∂
−Δ=Δ=
) (5-16)

44
Setzt man diesen Ausdruck in (5-11) und dieses in (5-15) ein, dann ergibt sich nach
Integration mit der Standardformel ∫+∞
∞−
=−a
dxax π)exp( 2 und Definition des sog. Zeldovich-
Faktors Z :
2/1
2
2
*21
⎥⎥⎦
⎤
⎢⎢⎣
⎡
∂Δ∂−
==iii
FkT
Zπ
(5-17)
die stationäre Keimbildungsrate (steady-state nucleation rate) als:
⎟⎟⎠
⎞⎜⎜⎝
⎛−=
kTFNZJ S
BD
*
0* exp Δγ (5-18)
(Dabei wurde formal statt von i0 bis im von -∞ bis +∞ integriert. Das macht praktisch kleinen Unterschied, da nur ein schmaler Bereich rund um i* zur Integration beiträgt.)
Die Kinetik wird bei der klassischen Keimbildung also nur von den Verhältnissen in einem kleinen Bereich um die kritische Keimgröße bestimmt (bis die freie Energie um etwa kT abfällt). Im zugänglichen experimentellen Bereich beträgt der Zeldovich-Faktor zwischen 1/20 und 1/40.
5.1.2. Einschränkungen und Modifikationen der klassischen Keimbildungstheorie
In der Praxis ist dieses idealisierte Bild aus verschiedenen Gründen meistens beeinträchtigt. Zunächst ist zu bedenken, dass eine Quasi-Gleichgewichts-Verteilung der Keimgrößen nicht sofort nach Einstellung der Übersättigung (etwa durch Abkühlen) vorliegt, sondern erst nach Verstreichen einer gewissen Inkubationszeit t, sodass der Verlauf durch folgende Funktion beschrieben werden kann
⎟⎠⎞
⎜⎝⎛−=
tJtJ S τexp)(* (5-19).
t kann identifiziert werden mit der Zeit, die benötigt wird, den entscheidenden Bereich der Breite 1/Z um den kritischen Keim durch Zufallssprünge (random walk) im Phasenraum zu überdecken:
Z*21γ
τ = (5-20)
Es kann nun der Fall eintreten, dass die Übersättigung in der Umgebung der Keime so rasch abgebaut wird, dass die Keimbildung zum Stillstand kommt, bevor ein stationärer Zustand überhaupt erreicht wird. Man spricht in diesem Fall von sog. katastrophischer Keimbildung (catastrophic nucleation). Formal liegt sie dann vor, wenn ct>τ , wobei tc eine Zeit ist, bei der die Diffusionszonen rund um die Keime, über die B-Atome aus der Matrix abgezogen werden, zu überlappen beginnen. Dann aber ist die Übersättigung soweit abgesunken, dass die Keimbildung zum Erliegen kommt. Die stationäre Keimbildungsrate hat also gar keine Zeit, sich einzustellen. Bei den meisten untersuchten Legierungen scheinen die Ausscheidungs-reaktionen so zu verlaufen.

45
Weiters ist zu beachten, dass die Grenzflächenenergie und Kohärenzspannungen die Gestalt kleiner und großer Keime verschieden beeinflussen. Bei kleinen Keimen ist die Grenzflächenenergie dominant, sie tendieren zu sphärischer Form. Größere Ausscheidungsteilchen werden durch Kohärenzspannungen in elastisch weiche Richtungen (<100> bei den meisten kubischen Kristallen, während <111> eine harte Richtung ist) zu Platten oder Nadeln deformiert. Abgesehen von der Gitter-Fehlpassung (misfit) können auch bestimmte Orientierungen der Oberfläche energetisch bevorzugt sein, etwa wenn sie entlang einer dicht gepackten Ebene liegen.
Dominiert die Grenzflächenenergie gegenüber der elastischen Verzerrungsenergie, dann lässt sich das Aussehen des Ausscheidungsteilchens mit Hilfe der sog. Wulffschen Konstruktion ermitteln (Abbildung 5-12: Man trage die spezifische Oberflächen-(bzw. Grenzflächen-) Energie als Polardiagramm auf. Dabei ist die Orientierung der betrachteten Ebene durch die Richtung ihres Normalenvektors repräsentiert. Die einspringenden Ecken (cusps) stellen besonders günstige Orientierungen dar. Durch die Spitze jedes Vektors lege man eine Normalebene
(strichliert in 5-12). Die innere Einhüllende dieser Ebenen (strichpunktiert in 5-12) stellt dann den Verlauf der Oberfläche minimaler Energie dar. Eine solche Facettierung der Oberfläche ist hauptsächlich bei niedrigen Temperaturen von Bedeutung. Bei T > 0,4Tc (Tc ist die kritische Temperatur der Entmischungsreaktion) kann im Wesentlichen sphärische Form angenommen werden, wenn nicht anisotrope elastische Wechselwirkungsenergie eine Rolle spielt.
Abb. 5-12
Eine anschauliche Illustration des Wechselspiels zwischen Phasengrenzflächen-Energie, elastischer Kohärenzenergie und Temperatur bieten die in Abb. 5-13 dargestellten Berechnungen der Form von Zn- Ausscheidungen nach der Clusterentwicklungsmethode durch S. Müller (Erlangen)
Abbildung 5-13: Entwicklung von kohärenten Zn-Keimen bei Ausscheidung aus einer Al-Zn-Legierung (S. Müller 2003). Abszisse: Größe des Keims, Ordinate: Temperatur.

46
Je größer das Ausscheidungsteilchen wird (nach rechts in der Darstellung), umso mehr macht sich der Einfluss der Kohärenzspannungen bemerkbar, und die Form wird immer mehr abgeplattet. Verringert man die Temperatur (nach unten in der Darstellung), dann wird der Einfluss der Grenzflächenenergie spürbar, und es tritt eine deutliche Facettierung des Teilchens auf. Es ist klar, dass die Energie des kritischen Keims im Allgemeinen von seiner Form und diese wiederum von der Größe abhängt. In Festkörpern ist die Monomer-Kondensationsrate über einen Boltzmannfaktor mit der atomaren Wanderungsenergie (energy of migration)
*γ
mFΔ verknüpft. Es gilt also insgesamt
⎟⎟⎠
⎞⎜⎜⎝
⎛ Δ−⎟
⎠⎞
⎜⎝⎛ Δ−∝
kTF
kTFJ ms
*
expexp (5-21)
Mit sinkender Temperatur wird der zweite der beiden Faktoren rasch größer, weil die Übersättigung und damit die treibende Kraft (im Wesentlichen die Volumsenergie-Differenz) für die Keimbildung immer größer wird. Gleichzeitig wird der erste Faktor kleiner, da die Atombeweglichkeit kleiner wird und somit die Kinetik „einschläft“. Man wird also nur in einem gewissen Temperaturbereich Keimbildung beobachten können (Abb. 5-14)
Abb. 5-14
Als die am meisten einschränkende Annahme der klassischen Keimbildungstheorie stellt sich jedoch die freie Entstehung des Keims in der homogenen Matrixphase heraus. In den meisten Fällen bilden sich nämlich die Ausscheidungskeime zuerst an Grenzen einer bereits vorhandenen zweiten Phase, an Korngrenzen oder an Defekten (heterogene Keimbildung). Dadurch kann Grenzflächenenergie eingespart und *FΔ erniedrigt werden. An Korngrenzen wird die Keimbildung dadurch erleichtert, dass gleichzeitig mit der Erzeugung von Grenzflächen Keim-Matrix ein Stück der alten Korngrenze aufgelöst wird. Die Abb. 5-15 zeigt einen solchen linsenförmigen Keim an der Grenze zwischen zwei Körnern. Die Energiebilanz lautet in diesem Fall
αααααβαβ γγ AAfVF −+Δ=Δ (5-22)
Abb. 5-15
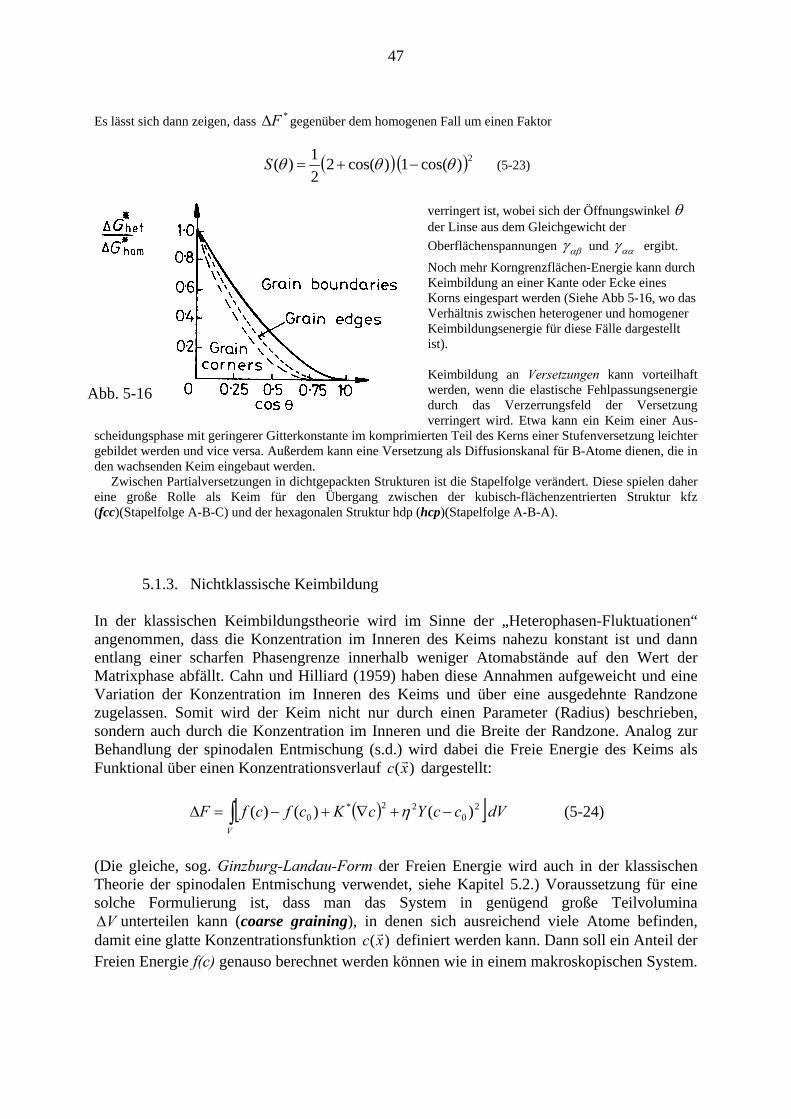
47
Es lässt sich dann zeigen, dass *FΔ gegenüber dem homogenen Fall um einen Faktor
( ) ( 2)cos(1)cos(221)( θθθ −+=S ) (5-23)
verringert ist, wobei sich der Öffnungswinkel θ der Linse aus dem Gleichgewicht der Oberflächenspannungen αβγ und ααγ ergibt. Noch mehr Korngrenzflächen-Energie kann durch Keimbildung an einer Kante oder Ecke eines Korns eingespart werden (Siehe Abb 5-16, wo das Verhältnis zwischen heterogener und homogener Keimbildungsenergie für diese Fälle dargestellt ist). Keimbildung an Versetzungen kann vorteilhaft werden, wenn die elastische Fehlpassungsenergie durch das Verzerrungsfeld der Versetzung verringert wird. Etwa kann ein Keim einer Aus-
scheidungsphase mit geringerer Gitterkonstante im komprimierten Teil des Kerns einer Stufenversetzung leichter gebildet werden und vice versa. Außerdem kann eine Versetzung als Diffusionskanal für B-Atome dienen, die in den wachsenden Keim eingebaut werden.
Abb. 5-16
Zwischen Partialversetzungen in dichtgepackten Strukturen ist die Stapelfolge verändert. Diese spielen daher eine große Rolle als Keim für den Übergang zwischen der kubisch-flächenzentrierten Struktur kfz (fcc)(Stapelfolge A-B-C) und der hexagonalen Struktur hdp (hcp)(Stapelfolge A-B-A).
5.1.3. Nichtklassische Keimbildung In der klassischen Keimbildungstheorie wird im Sinne der „Heterophasen-Fluktuationen“ angenommen, dass die Konzentration im Inneren des Keims nahezu konstant ist und dann entlang einer scharfen Phasengrenze innerhalb weniger Atomabstände auf den Wert der Matrixphase abfällt. Cahn und Hilliard (1959) haben diese Annahmen aufgeweicht und eine Variation der Konzentration im Inneren des Keims und über eine ausgedehnte Randzone zugelassen. Somit wird der Keim nicht nur durch einen Parameter (Radius) beschrieben, sondern auch durch die Konzentration im Inneren und die Breite der Randzone. Analog zur Behandlung der spinodalen Entmischung (s.d.) wird dabei die Freie Energie des Keims als Funktional über einen Konzentrationsverlauf )(xc r dargestellt:
( )[ ]∫ −+∇+−=ΔV
dVccYcKcfcfF 20
22*0 )()()( η (5-24)
(Die gleiche, sog. Ginzburg-Landau-Form der Freien Energie wird auch in der klassischen Theorie der spinodalen Entmischung verwendet, siehe Kapitel 5.2.) Voraussetzung für eine solche Formulierung ist, dass man das System in genügend große Teilvolumina
unterteilen kann (coarse graining), in denen sich ausreichend viele Atome befinden, damit eine glatte Konzentrationsfunktion
VΔ)(xc r definiert werden kann. Dann soll ein Anteil der
Freien Energie f(c) genauso berechnet werden können wie in einem makroskopischen System.
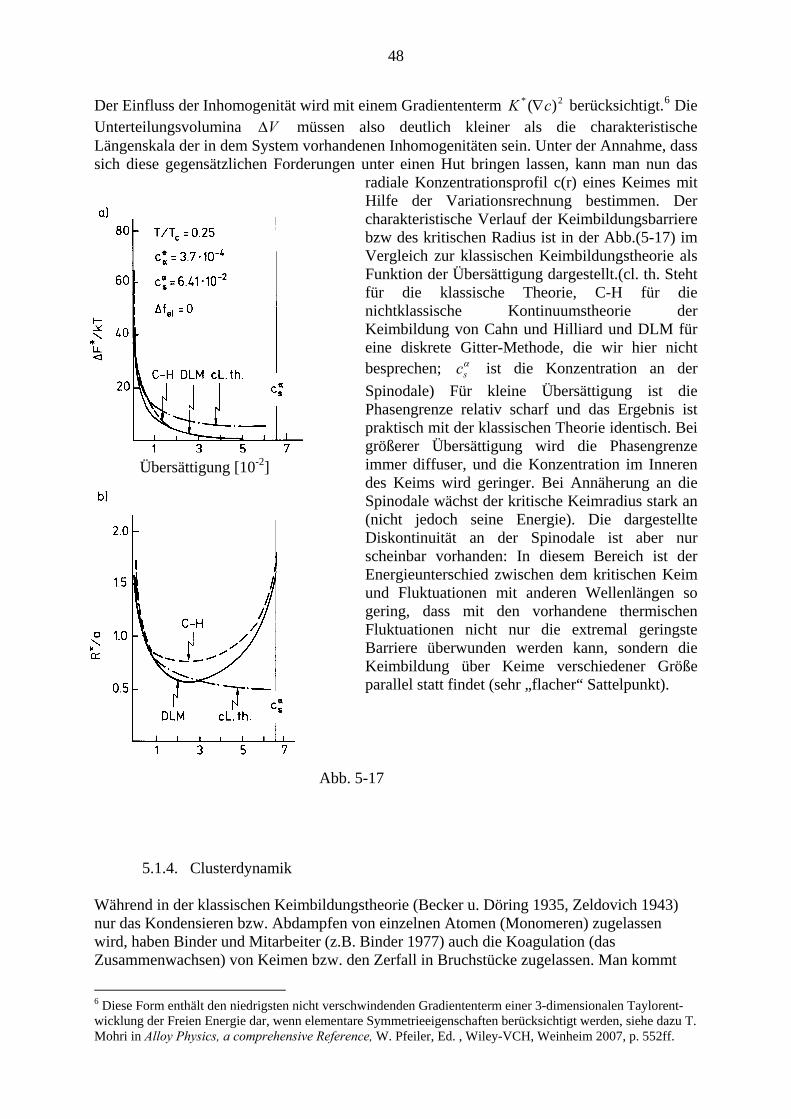
48
Der Einfluss der Inhomogenität wird mit einem Gradiententerm berücksichtigt.2* )( cK ∇ 6 Die Unterteilungsvolumina müssen also deutlich kleiner als die charakteristische Längenskala der in dem System vorhandenen Inhomogenitäten sein. Unter der Annahme, dass sich diese gegensätzlichen Forderungen unter einen Hut bringen lassen, kann man nun das
radiale Konzentrationsprofil c(r) eines Keimes mit Hilfe der Variationsrechnung bestimmen. Der charakteristische Verlauf der Keimbildungsbarriere bzw des kritischen Radius ist in der Abb.(5-17) im Vergleich zur klassischen Keimbildungstheorie als Funktion der Übersättigung dargestellt.(cl. th. Steht für die klassische Theorie, C-H für die nichtklassische Kontinuumstheorie der Keimbildung von Cahn und Hilliard und DLM für eine diskrete Gitter-Methode, die wir hier nicht besprechen; ist die Konzentration an der Spinodale) Für kleine Übersättigung ist die Phasengrenze relativ scharf und das Ergebnis ist praktisch mit der klassischen Theorie identisch. Bei größerer Übersättigung wird die Phasengrenze immer diffuser, und die Konzentration im Inneren des Keims wird geringer. Bei Annäherung an die Spinodale wächst der kritische Keimradius stark an (nicht jedoch seine Energie). Die dargestellte Diskontinuität an der Spinodale ist aber nur scheinbar vorhanden: In diesem Bereich ist der Energieunterschied zwischen dem kritischen Keim und Fluktuationen mit anderen Wellenlängen so gering, dass mit den vorhandene thermischen Fluktuationen nicht nur die extremal geringste Barriere überwunden werden kann, sondern die Keimbildung über Keime verschiedener Größe parallel statt findet (sehr „flacher“ Sattelpunkt).
VΔ
αsc
Übersättigung [10-2]
Abb. 5-17
5.1.4. Clusterdynamik
Während in der klassischen Keimbildungstheorie (Becker u. Döring 1935, Zeldovich 1943) nur das Kondensieren bzw. Abdampfen von einzelnen Atomen (Monomeren) zugelassen wird, haben Binder und Mitarbeiter (z.B. Binder 1977) auch die Koagulation (das Zusammenwachsen) von Keimen bzw. den Zerfall in Bruchstücke zugelassen. Man kommt
6 Diese Form enthält den niedrigsten nicht verschwindenden Gradiententerm einer 3-dimensionalen Taylorent-wicklung der Freien Energie dar, wenn elementare Symmetrieeigenschaften berücksichtigt werden, siehe dazu T. Mohri in Alloy Physics, a comprehensive Reference, W. Pfeiler, Ed. , Wiley-VCH, Weinheim 2007, p. 552ff.

49
dann auf eine Gleichung für die zeitliche Veränderung der Clusterpopulation, die den klassischen Fall als Spezialfall enthält:
∑ ∑ ∑ ∑∞
=′
−
=′
−
=′
∞
=′′′′−′′′+ ′−′−′+−′+=
1
1
1
1
1 1,,2
1,2
1, ),(),(),(),(),(),(),(
i
i
i
i
i iiiiiiiiiii tiftiftiiftiftiftiiftif
dtd ββαα
(4- 25)
Die Anzahl der Keime mit Größe i steigt also durch Zerfall von größeren (Term1), Zusammenwachsen von kleineren (Term 3), verringert sich durch Zerfall in kleinere (Term 2) und Zusammenwachsen mit größeren (Term 4). Erhebt man die Forderung nach detailliertem Fließgleichgewicht (detailed balance) und macht man möglichst plausible Annahmen über die Reaktionsraten, die a priori nicht bekannt sind, dann lässt sich Gleichung (5-25) integrieren. Damit können zumindest qualitative Aussagen gewonnen werden. Es zeigt sich, dass Keimbildung und Wachstum nicht als getrennte Stadien auftreten, sondern ineinander greifend stattfinden. Besonders aufschlussreich ist ein Vergleich des Verhaltens charakteristischer Größen der Keimbildung in Abhängigkeit von der Übersättigung in den verschiedenen Modellen (Abb. 4-13): Bei Annäherung an die Spinodale divergiert im nichtklassischen Kontinuumsmodell von Cahn und Hilliard (C-H non-cl.) der kritische Keimradius, während im Clusterdynamikmodell (Binder et al.) ein kontinuierlicher Übergang zur Größe der kritischen Fluktuationen im spinodalen Bereich (siehe Abschnitt (5-2)) erfolgt.
Abb. 5-18

50
5.2. Spinodale Entmischung
5.2.1. Die klassische Theorie (Cahn-Hilliard)
Wie bereits in der Einleitung zu Abschnitt 4 erläutert, ist in einem Bereich des Phasendiagramms, wo die f(c)-Kurve negative Krümmung aufweist, eine homogene Phase formal instabil gegen Fluktuationen der Konzentration. Wir wollen schon an dieser Stelle auf eine gewisse Willkürlichkeit hinweisen, die sich aus dem coarse graining ergibt. Um eine lokale Konzentration überhaupt definieren zu können, muss man ja die Anzahl der Atome über gewisse kleine Volumina ΔV mitteln. Je größer man ΔV wählt, umso größere Inhomogenitäten werden durch die Mittelung „geschluckt“ und daher durch den )(xc r -Verlauf gar nicht mehr wiedergegeben. In Abbildung (4-14) ist dieser Sachverhalt deutlich gemacht. Je größer die Länge L der Mittelungsbereiche, umso mehr verflacht sich die „Kamelhöckerkurve“, bis für völlig flachen Verlauf schließlich die komplette zweiphasige Gleichgewichtsstruktur im Mittelungsvolumen ΔV∼L3 enthalten ist. Eine analoge „Verflachung“ dieser Kurve erhält man, wenn man in der Clustervariationsmethode (R. Kikuchi) die Größe der maximal berücksichtigten Cluster so erhöht, dass sie schließlich den gesamten Kristall umfassen.
Abb. 5-19
Nehmen wir an, es lässt sich dennoch auf geeignete Weise eine lokale Konzentration
)(xc r definieren und die Freie Energie des Systems durch einen Funktionalansatz wie in Gl. (5-24) darstellen. Da die treibende Kraft für das Springen von Atomen im wesentlichen von Unterschieden des chemischen Potenzials her rührt, ist es sinnvoll, den mittleren Diffusionsstrom als proportional zum lokalen Gradienten des effektiven chemischen Potenzials BA μμμ −=~ anzusetzen:
),(~),( txMtxj rrrμ∇−= (5-26)

51
wobei M eine Atom-Mobilität darstellt.7 Für homogene Legierungen berechnet sich das chemische Potenzial durch Ableitung der Freien Energie (bzw. Enthalpie) nach der Atomzahl bzw. Konzentration (siehe die Tangentenkonstruktion in Abschnitt 3). Es würde daher in der Formulierung der Thermodynamik für homogene Systeme folgen
( ) ( )[ ] ( ) ( txccftxc
ctxctx ,, )
~,, 2
2 vvvr∇
∂∂
=∇∂∂
=∇=∇μμμ (5-27), daher
( ) ( txcDtxcc
fMj ,~,2
2 vvv∇−=∇
∂∂
−= ) (5-28).
Hier erkennen wir das 1. Fick’sche Gesetz der Diffusion wieder, allerdings mit einem Diffusionskoeffizienten, der proportional zur zweiten Ableitung der Freien Energie nach der Konzentration ist. Hier sieht man unmittelbar, dass in einem Gebiet, in dem diese negativ ist, wo also die f(c) - Kurve konkav verläuft, die Diffusion spontan entgegen dem Konzentrationsgradienten verläuft. Hier wird nun dieser Verfahren für inhomogene Legierungen, deren Freie Energie durch einen Ausdruck wie in Gl. (5-24) gegeben ist, verallgemeinert, indem die Funktionalableitung (Variationsableitung) gebildet wird8:
{ }( ) )(*2
)()(~ 2 xcK
cf
cf
cf
xcxcf rr ∇−
∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∇∂Δ∂
∇−∂Δ∂
=Δ
=δ
δμ (5-29)
wobei wir für den Integranden aus Gl. (5-24) ohne den elastischen Term eingesetzt haben. Setzen wir dieses Ergebnis in die Gl. (5-26) ein, dann bekommen wir für den Diffusionsstrom
fΔ
( ⎥⎦⎤
⎢⎣⎡ ∇∇−
∂∂
∇−= ),(2),( 2* txcKcfMtxj rrr ) (5-30)
Die Diffusionsgleichung ergibt sich dann wie immer aus der Kontinuitätsbedingung (Massenerhalt)
0),(.),(=∇+
∂∂ txj
ttxc rrr
(5-31)
7 Die Formulierung des Diffusionsstroms mit Hilfe des Konzentrationsgradienten und eines thermodynamischen Faktors Gl. 4-33 drückt den gleichen Sachverhalt in einer anderen Darstellung aus. Es zeigt sich dann, dass der
thermodynamische Faktor gerade proportional zu 2
2
cf
∂∂ ist.
8 Der Grund dafür ist, dass die gesamte Freie Energie durch den einen Ausdruck der allgemeinen Form gegeben ist. Entwickelt man diesen für kleine Änderungen der Konzentration ( )dVccfF ∫ ∇Δ=Δ ,.., cδ , so
ergibt sich ( ) dVccfc
cfcFccF ∫ ⎟
⎠⎞
⎜⎝⎛ ∇
∂∇Δ∂
+∂Δ∂
≈Δ−+Δ δδδ )()( . Da man jedoch die Änderung auf die Variation
cδ und nicht auf ( c∇ )δ zurückführen will, transformiert man den zweiten Term durch eine partielle Integration so, dass der Gradient auf den ersten Faktor wirkt und erhält
dVccf
cfcFccF ∫ ⎟⎟
⎠
⎞⎜⎜⎝
⎛⎥⎦⎤
⎢⎣⎡∂∇Δ∂
∇−∂Δ∂
≈Δ−+Δ δδ )()( . Der Ausdruck in runder Klammer ist dann der Beitrag zur
Änderung der Freien Energie pro lokaler Variation der Konzentration. Dessen Gradient ist die Treibende Kraft für Diffusion.

52
was schließlich zu folgender Gleichung führt:
⎭⎬⎫
⎩⎨⎧ ∇−
∂∂
∇=∂
∂ ),(2)],([),( 2*2 txcKc
txcfMt
txc rrr
(5-32)
In dieser Gleichung ist c implizit und nichtlinear enthalten. Betrachtet man jedoch die Entwicklung für kleine Abweichungen von der gleichförmigen Konzentration des Ausgangszustandes
0),(),( ctxctxc −=rrδ (5-33),
erhält man eine lineare partielle Differenzialgleichung
),(2)(),( 2*2
22
0
txcKc
cfMtxct c
rr δδ⎪⎭
⎪⎬⎫
⎪⎩
⎪⎨⎧
∇−⎟⎟⎠
⎞⎜⎜⎝
⎛∂
∂∇=
∂∂ (5-34)
Diese lässt sich durch einen Produktansatz )()(),( txtxf ϕη vr
= lösen. Die linke Seite der Gleichung wirkt nur auf den zeitabhängigen Teil, die rechte Seite nur auf den ortsabhängigen Teil. Nach Division durch )()( tx ϕη v erhalten wir
ϕ
ϕ
ηη ⎪⎭
⎪⎬⎫
⎪⎩
⎪⎨⎧
∇−⎟⎟⎠
⎞⎜⎜⎝
⎛∂
∂∇
==
2*2
22 2)(
0
Kc
cfM
R c& (5-34’)
Worin sich ausdrückt, dass beide Seiten, wenn sie gleich sein müssen und von verschiedenen Variablen abhängig sind, gleich einer Konstanten R sind. Nach den üblichen Exponentialansätzen für lineare Differentialgleichungen mit konstanten Koeffizienten ergibt sich für den zeitabhängigen Teil )exp()( Rtt =ϕ und für den ortsabhängigen Teil eine harmonische Funktion ).exp()( xkix rrr
=η , somit insgesamt eine Lösungsbasis der Form ])(exp[).exp(),( tkRxkitxk
rrrrr =ς , (5-35)
Wobei die Konstanten R und kk
r= durch folgende Bedingung miteinander verknüpft sind
⎥⎥⎦
⎤
⎢⎢⎣
⎡+⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
−= 2*2
22 2)(
0
kKc
fMkkRc
r (5-36)
Die Zeitkonstante R ist positiv für Wellenvektoren 0<k<kc mit
*2
2
22
0
Kc
fkcc
c ⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−==λπ (5-37)

53
und erreicht ihr Maximum für 2c
mkk = . Man sollte also erwarten, dass eine Modulation der
Konzentration mit dieser Wellenlänge am schnellsten wächst und sich daher durchsetzt, während Konzentrationsschwankungen mit Wellenlänge kc konstant bleiben, da dort R=0 ist.
5.2.2. Vergleich mit dem Experiment und verbesserte Theorien Eine experimentelle Verifikation dieses Verhaltens sowie überhaupt eine Entscheidung, ob spinodale Entmischung vorliegt, erweist sich als grundsätzlich schwierig. Direkte Beobachtungsmethoden wie optische oder Durchstrahlungs-Elektronenmikroskopie (TEM) können das System erst in einem fortgeschrittenen Zustand der Ausscheidung sichtbar machen, zu einem Zeitpunkt also, wo die Abweichung vom homogenen Zustand bereits groß ist und die Linearisierung der Gleichungen nicht mehr gilt. Lange wurde das Vorliegen einer zusammenhängenden, labyrinthartigen Struktur (Abb. 5-21) bzw. einer sog. Tweed –Struktur (Abb. 5-22), wie sie sich bei unter dem Einfluss von elastischen Kohärenzspannungen ausbilden kann, als experimenteller Beweis für vorangegangene spinodale Entmischung angesehen.
Abb. 5-20: FIM-Aufnahme Abb. 5-21

54
Computersimulationen zeigen aber, dass sowohl nach Keimbildung als auch nach spinodaler Entmischung, je nach Volumsanteil der ausgeschiedenen Phase, im Spätstadium eine zusammenhängende modu-lierte Struktur oder aber eine Teilchen-struktur beobachtet werden kann (Abb 5-22, Fratzl u. Penrose 1994, Monte-Carlo Rechnung eines Ising-Modells der spinodalen Entmischung, a) und b) mit Volumsanteil c=0,5, c) und d) mit Volumsanteil c=0,1). Man spricht im ersteren Fall davon, dass eine Perkolationsgrenze (percolation limit) überschritten wurde.
Abb. 5-22 Für die Beobachtung der Anfangsstadien der Ausscheidung sind hingegen Streumethoden (insbesondere Röntgen- oder Neutronenkleinwinkelstreuung, SAXS bzw SANS) prädestiniert. Hier zeigt sich, dass der Gültigkeitsbereich des oben dargestellten, linearen Modells relativ rasch verlassen wird. Aufwändigere Berechnungsmethoden wurden daher entwickelt, um auch spätere Stadien der spinodalen Entmischung beschreiben zu können und auch die Wechselwirkung der Atome realistischer zu erfassen. In Abb. 5-23 sind in a die Ergebnisse einer Neutronen-Kleinwinkelstreuung an einer Au-Pt-Legierung wiedergegeben, in b das Ergebnis einer Computersimulation (Nächst-Nachbar-Isingmodell), in c das Ergebnis einer Rechnung nach einem diskreten Ginzburg-Landau Modell. Dargestellt ist jeweils der Strukturfaktor, der als Fouriertransformierte der Streuintensität den jeweiligen Anteil der Modulation mit Wellenvektor k angibt, für verschiedene Zeiten. In allen Fällen ist das vom linearen Modell geforderte „crossing-over“, also die Konstanz für eine Wellenlänge mit Wachstumsfaktor R=0, nicht erfüllt. Ebenso zeigen die „Cahn plots“ (R(k) / k2 gegen k2 aufgetragen) nicht den idealen linearen Verlauf, wie er aus Gl.(5-36) folgen würde sondern einen gekrümmten Verlauf.
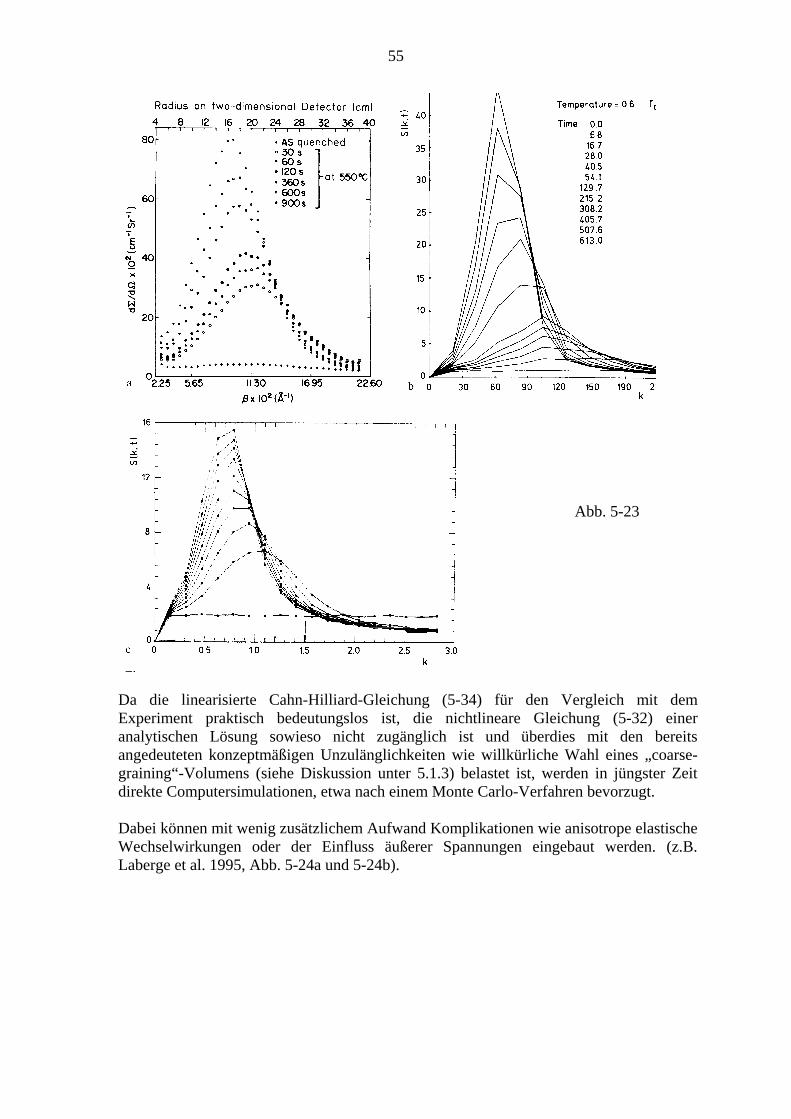
55
Abb. 5-23
Da die linearisierte Cahn-Hilliard-Gleichung (5-34) für den Vergleich mit dem Experiment praktisch bedeutungslos ist, die nichtlineare Gleichung (5-32) einer analytischen Lösung sowieso nicht zugänglich ist und überdies mit den bereits angedeuteten konzeptmäßigen Unzulänglichkeiten wie willkürliche Wahl eines „coarse-graining“-Volumens (siehe Diskussion unter 5.1.3) belastet ist, werden in jüngster Zeit direkte Computersimulationen, etwa nach einem Monte Carlo-Verfahren bevorzugt. Dabei können mit wenig zusätzlichem Aufwand Komplikationen wie anisotrope elastische Wechselwirkungen oder der Einfluss äußerer Spannungen eingebaut werden. (z.B. Laberge et al. 1995, Abb. 5-24a und 5-24b).
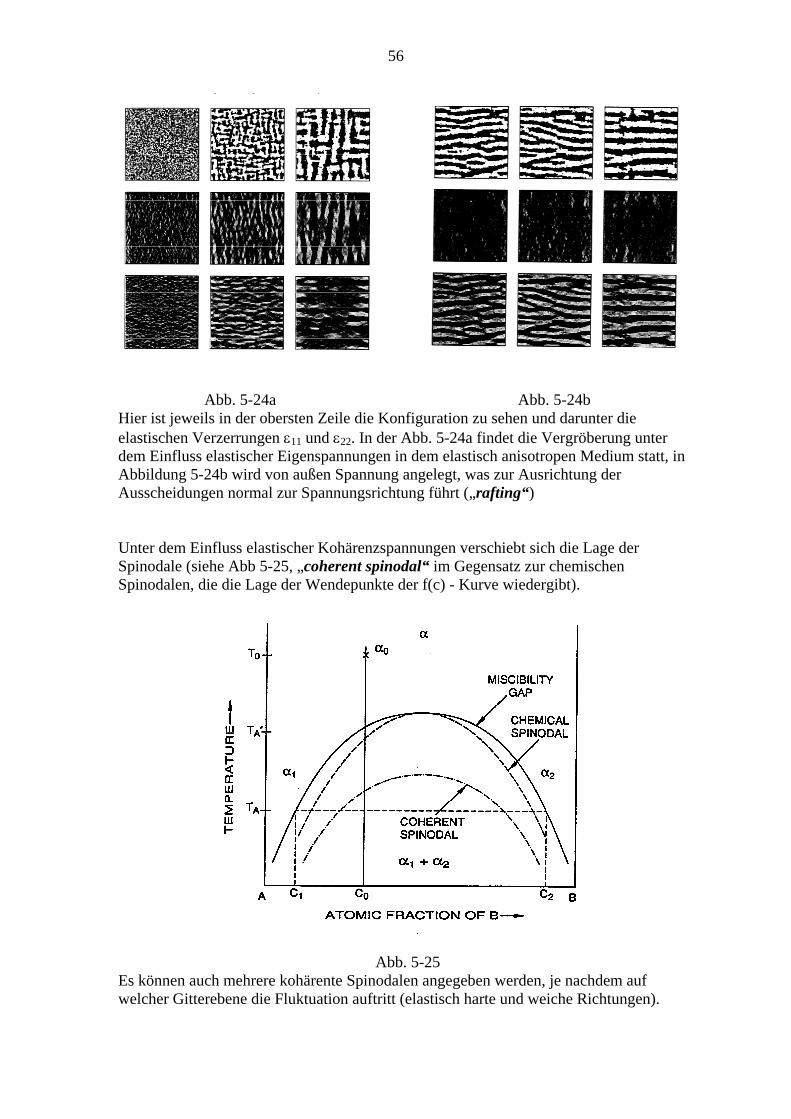
56
Abb. 5-24a Abb. 5-24b Hier ist jeweils in der obersten Zeile die Konfiguration zu sehen und darunter die elastischen Verzerrungen ε11 und ε22. In der Abb. 5-24a findet die Vergröberung unter dem Einfluss elastischer Eigenspannungen in dem elastisch anisotropen Medium statt, in Abbildung 5-24b wird von außen Spannung angelegt, was zur Ausrichtung der Ausscheidungen normal zur Spannungsrichtung führt („rafting“) Unter dem Einfluss elastischer Kohärenzspannungen verschiebt sich die Lage der Spinodale (siehe Abb 5-25, „coherent spinodal“ im Gegensatz zur chemischen Spinodalen, die die Lage der Wendepunkte der f(c) - Kurve wiedergibt).
Abb. 5-25 Es können auch mehrere kohärente Spinodalen angegeben werden, je nachdem auf welcher Gitterebene die Fluktuation auftritt (elastisch harte und weiche Richtungen).
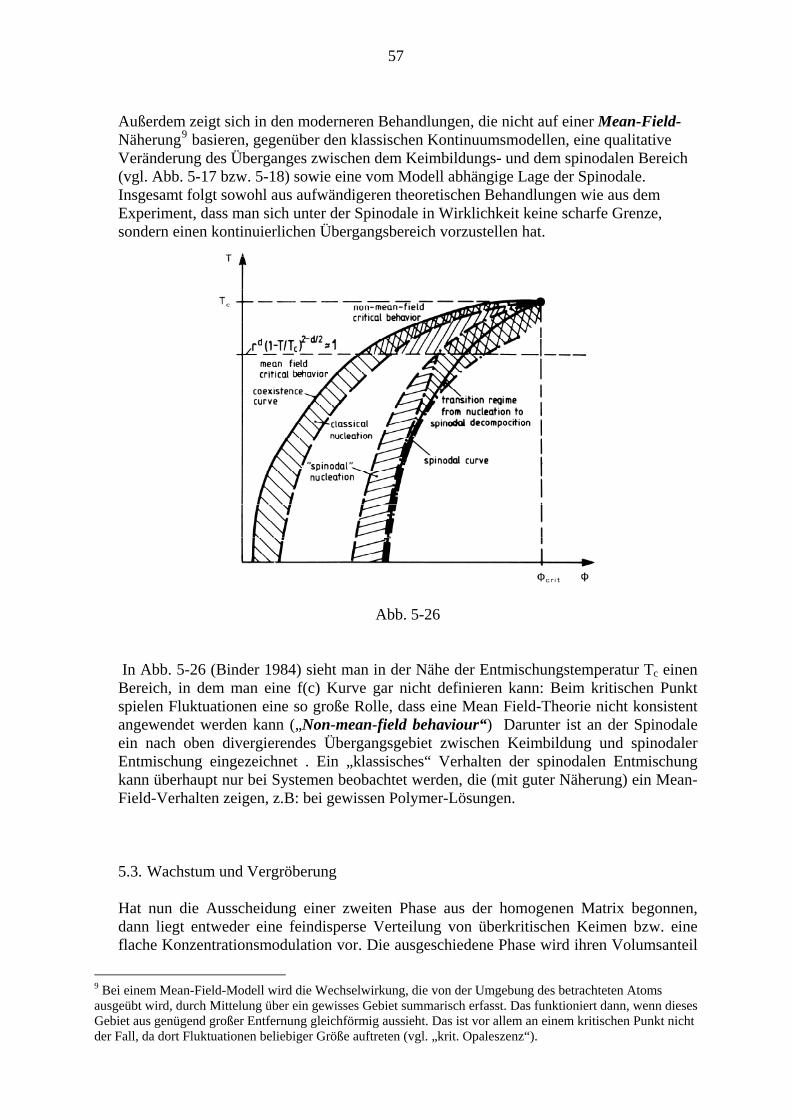
57
Außerdem zeigt sich in den moderneren Behandlungen, die nicht auf einer Mean-Field-Näherung9 basieren, gegenüber den klassischen Kontinuumsmodellen, eine qualitative Veränderung des Überganges zwischen dem Keimbildungs- und dem spinodalen Bereich (vgl. Abb. 5-17 bzw. 5-18) sowie eine vom Modell abhängige Lage der Spinodale. Insgesamt folgt sowohl aus aufwändigeren theoretischen Behandlungen wie aus dem Experiment, dass man sich unter der Spinodale in Wirklichkeit keine scharfe Grenze, sondern einen kontinuierlichen Übergangsbereich vorzustellen hat.
Abb. 5-26
In Abb. 5-26 (Binder 1984) sieht man in der Nähe der Entmischungstemperatur Tc einen Bereich, in dem man eine f(c) Kurve gar nicht definieren kann: Beim kritischen Punkt spielen Fluktuationen eine so große Rolle, dass eine Mean Field-Theorie nicht konsistent angewendet werden kann („Non-mean-field behaviour“) Darunter ist an der Spinodale ein nach oben divergierendes Übergangsgebiet zwischen Keimbildung und spinodaler Entmischung eingezeichnet . Ein „klassisches“ Verhalten der spinodalen Entmischung kann überhaupt nur bei Systemen beobachtet werden, die (mit guter Näherung) ein Mean-Field-Verhalten zeigen, z.B: bei gewissen Polymer-Lösungen. 5.3. Wachstum und Vergröberung Hat nun die Ausscheidung einer zweiten Phase aus der homogenen Matrix begonnen, dann liegt entweder eine feindisperse Verteilung von überkritischen Keimen bzw. eine flache Konzentrationsmodulation vor. Die ausgeschiedene Phase wird ihren Volumsanteil
9 Bei einem Mean-Field-Modell wird die Wechselwirkung, die von der Umgebung des betrachteten Atoms ausgeübt wird, durch Mittelung über ein gewisses Gebiet summarisch erfasst. Das funktioniert dann, wenn dieses Gebiet aus genügend großer Entfernung gleichförmig aussieht. Das ist vor allem an einem kritischen Punkt nicht der Fall, da dort Fluktuationen beliebiger Größe auftreten (vgl. „krit. Opaleszenz“).

58
durch Wachstum vergrößern, solange in der Matrixphase noch eine Übersättigung vorhanden ist. Betrachten wir ein typisches, sagen wir kugelförmiges Ausscheidungsteilchen, das in einer Matrixphase wächst. Wäre die Grenzfläche eben (entspricht einem Teilchen mit unendlichem Radius), dann würde es mit einer Matrixphase der Konzentration im Gleichgewicht stehen, die dem Berührungspunkt der gemeinsamen Tangente an die „Doppelhöckerkurve“ (etwa in Abb. 3-4b) entspricht. Werden hingegen Atome in ein Teilchen endlicher Größe eingebaut, sodass es wächst, muss die Grenzfläche vergrößert und damit zusätzliche Energie aufgebracht werden. Dadurch verschiebt sich die Gleichgewichtskonzentration zu höheren Werten, ähnlich wie der Druck des Dampfes, der mit einem Flüssigkeitströpfchen im Gleichgewicht steht, umso höher ist, je kleiner dessen Radius ist. Eine thermodynamische Analyse liefert für eine reguläre feste Lösung folgende Gibbs-Thomson-Gleichung:
ecα
⎟⎟⎠
⎞⎜⎜⎝
⎛=
rRTV
crc e 12exp)( βαβ
α
σ (5-38),
wobei das molare Volumen der Ausscheidungsphase ist. Für große Teilchenradien kann nach Entwicklung der Exponentialfunktion annähernd geschrieben werden
βV
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
rRTV
crc e 121)( βαβ
α
σ (5-39)
Die Wachstumsgeschwindigkeit eines Teilchens kann entweder von der Rate des Übertritts der Atome über die Phasengrenze bestimmt sein (rate controlled) oder durch die Rate, mit der Atome zum Einbau in das Teilchen aus der Matrix heran geschafft werden (diffusion controlled). Während für sehr kleine Keime die Grenzflächenreaktion in der Regel ratenbestimmend ist, wird das Wachstum großer Teilchen oft vom Diffusionsstrom gesteuert. Ein schematisches Konzentrationsprofil in der Umgebung eines solchen Teilchens ist in Abb. 5-27 dargestellt.
Abb. 5-27
An der Oberfläche des Teilchens stellt sich die der Krümmung entsprechende Gleichgewichtskonzentration c(r) der Matrix ein. Entsprechend einem stationären Diffusionsstrom nimmt der Gradient ab, bis in großer Entfernung die mittlere Matrixkonzentration c(t) erreicht wird. Aus der Lösung der Diffusionsgleichung in

59
Kugelsymmetrie folgt, dass der Konzentrations-Gradient an der Teilchenoberfläche bei stationärem Diffusionsstrom (c(t) – c(r)) / r beträgt. Der Radius würde dann, wenn c(t) als konstant angenommen werden kann, also die Wirkung der anderen Teilchen vernachlässigbar ist, nach folgendem Gesetz anwachsen:
( ) 2/1tDR iλ= (5-40)
Ein solches parabolisches Wachstum ergibt sich ganz allgemein für diffusionsgesteuertes Wachstum, unabhängig von der Morphologie. Dies lässt sich mit einem Skalierungsargument zeigen. Betrachtet man die Diffusionsgleichung (2. Ficksches Gesetz, Gl. 4-23 ohne Treibende Kraft)
),(),( txcDt
txc rr
Δ=∂
∂ (5-41)
mit der Lösung ),(1 txc r unter bestimmten Randbedingungen. Man definiert nun eine neue Funktion . Es ist durch Nachrechnen leicht zu verifizieren, dass auch diese Funktion die Differenzialgleichung erfüllt. Es haben sich alle linearen Dimensionen nach der Zeit um den Faktor
),(:),( 21 txctxc ααα
rr=
t2α α vergrößert. Daraus folgt, dass die Dimensionen der Ausscheidung proportional zu t1/2 wachsen. Dies gilt natürlich nur wenn (5-41) gilt, also bei einem homogenen und isotropen Diffusionsgesetz. Insbesondere muss der Diffusionsmechanismus Volumendiffusion und nicht Korngrenzen- oder Versetzungsdiffusion sein.
In Wirklichkeit jedoch bewirkt die „Konkurrenz“ durch die anderen Teilchen schließlich eine Änderung des Wachstumsgesetzes.
Was nämlich in der Regel nach der Wachstumsphase, aber auch schon übergreifend mit Keimbildung und Wachstum passiert , ist von Ausscheidungen aus wässriger Lösung unter „Ostwaldreifung“ (Ostwald ripening) bzw. „Umlösung“ bekannt: Die Anzahl der Teilchen verringert sich, kleine verschwinden zu Gunsten größerer, die wachsen. Es gibt einen „kritischen“ Teilchenradius, der im Gleichgewicht mit der mittleren Matrixkonzentration steht. Bei größeren Teilchen sieht das umgebende Konzentrationsprofil wie in Abb. 5-27 aus, bei kleinen ist die Gleichgewichts-konzentration an der Teilchenoberfläche gemäß der Gibbs-Thomson-Gleichung (5-38) höher und B-Atome strömen in einer Anreicherungszone vom Teilchen weg. In einer typischen zweiphasigen Legierung wird durch diesen Vorgang die Dichte an Ausscheidungsteilchen von ∼1025 m-3 auf weniger als ∼1019 m-3 verringert. Die treibende Kraft dieses Vorganges ist die Grenzflächenenergie, welche durch diesen Vorgang insgesamt verringert wird. Ein solcher Vergröberungsprozess (coarsening) setzt sowohl nach Keimbildung als auch nach spinodaler Entmischung ein. Während man im letzteren Fall zur Beschreibung auf Computermodelle angewiesen ist, gibt es unter bestimmten vereinfachenden Voraussetzungen eine elegante analytische Theorie für die Vergröberung einer feindispersen Verteilung nach Keimbildung (Lifshitz u. Slyozov 1961, Wagner 1961)

60
5.3.1. Die LSW (Lifshitz-Slyozov-Wagner)-Theorie
In der LSW-Theorie wird angenommen, dass die Keimbildung abgeschlossen ist und die Übersättigung durch Keimwachstum bereits auf eine solche Matrixkonzentration c abgebaut wurde, dass der Volumsanteil fp (volume fraction) der ausgeschiedenen Phase fortan annähernd konstant bleibt. Teilchen mit einem Radius
⎟⎟⎠
⎞⎜⎜⎝
⎛′=
⎟⎟⎠
⎞⎜⎜⎝
⎛=
ee cc
K
ccRT
VR
α
αβ
α
βαβ σσ
ln
1
ln
12* (5-42)
verändern dann ihre Größe nicht, während Teilchen mit einer Größe r < R* schrumpfen und Teilchen mit r > R* wachsen (bei letzteren ist die Situation wie in Abb. 5-27, bei ersteren gilt ja crc >)( , und damit ist der Konzentrationsgradient negativ). Es gilt nun die zeitliche Entwicklung einer anfänglich vorhandenen Teilchen-Größenverteilungsfunktion f(r,t) zu berechnen. Diese folgt aus einer verallgemeinerten Diffusionsgleichung im Raum der Teilchengrößen:
0=⎥⎦⎤
⎢⎣⎡
∂∂
∂∂
+∂∂
trf
rtf (5-43)
Vereinfachend wird bei LSW angenommen, dass der Volumsanteil fp klein ist (verdünnte Lösung) und sich damit die Diffusionsfelder der einzelnen Teilchen nicht gegenseitig beeinflussen. Da die Übersättigung bereits verschwunden ist, befindet man sich in einem Spätstadium der Ausscheidung. Die Teilchen haben also relativ großen Radius, sodass die linearisierte Version der Gibbs-Thomson-Gl. (5-39) verwendet werden kann. Unter all diesen Annahmen wird folgendes Verhalten abgeleitet: Die mittlere Radius wächst mit t1/3 („t1/3-Gesetz“) oder genauer
ttcc
cDKrr LSWep
e
ασα
ααβ =
−′=− 9
430
3 (5-44)
Entsprechend entwickelt sich die Teilchenzahl wie
tfNN LSWp
VV
απ 1
0 3411 −=− (5-45)
Bemerkenswert ist, dass sich im asymptotischen Verlauf eine universelle Form der Verteilungsfunktion einstellt:
230
23
)()(,
>=
<⎟⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛
rrfürf
rrfür
rrh
trtNt
rrf
LSW
vLSW
(5-46)
wobei h eine zeitinvariante Formfunktion ist. In diesem asymptotischen Limes gilt auch
*rr = . Explizit ergibt sich für die Formfunktion
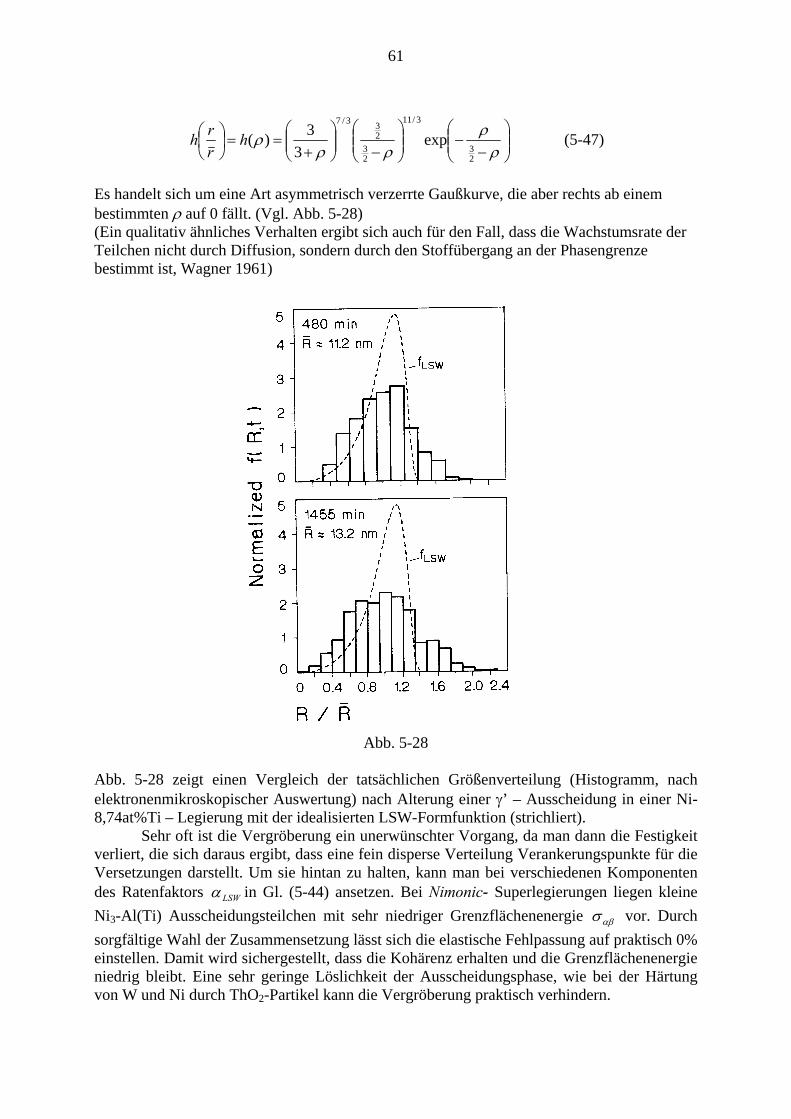
61
⎟⎟⎠
⎞⎜⎜⎝
⎛−
−⎟⎟⎠
⎞⎜⎜⎝
⎛−⎟⎟
⎠
⎞⎜⎜⎝
⎛+
==⎟⎠⎞
⎜⎝⎛
ρρ
ρρρ
23
3/11
23
233/7
exp3
3)(hrrh (5-47)
Es handelt sich um eine Art asymmetrisch verzerrte Gaußkurve, die aber rechts ab einem bestimmten ρ auf 0 fällt. (Vgl. Abb. 5-28) (Ein qualitativ ähnliches Verhalten ergibt sich auch für den Fall, dass die Wachstumsrate der Teilchen nicht durch Diffusion, sondern durch den Stoffübergang an der Phasengrenze bestimmt ist, Wagner 1961)
Abb. 5-28
Abb. 5-28 zeigt einen Vergleich der tatsächlichen Größenverteilung (Histogramm, nach elektronenmikroskopischer Auswertung) nach Alterung einer γ’ – Ausscheidung in einer Ni-8,74at%Ti – Legierung mit der idealisierten LSW-Formfunktion (strichliert). Sehr oft ist die Vergröberung ein unerwünschter Vorgang, da man dann die Festigkeit verliert, die sich daraus ergibt, dass eine fein disperse Verteilung Verankerungspunkte für die Versetzungen darstellt. Um sie hintan zu halten, kann man bei verschiedenen Komponenten des Ratenfaktors LSWα in Gl. (5-44) ansetzen. Bei Nimonic- Superlegierungen liegen kleine Ni3-Al(Ti) Ausscheidungsteilchen mit sehr niedriger Grenzflächenenergie αβσ vor. Durch sorgfältige Wahl der Zusammensetzung lässt sich die elastische Fehlpassung auf praktisch 0% einstellen. Damit wird sichergestellt, dass die Kohärenz erhalten und die Grenzflächenenergie niedrig bleibt. Eine sehr geringe Löslichkeit der Ausscheidungsphase, wie bei der Härtung von W und Ni durch ThO2-Partikel kann die Vergröberung praktisch verhindern.

62
5.3.2. Verfeinerungen der Vergröberungstheorie Eine Reihe späterer Arbeiten (z.B. Ardell 1972, Brailsford u. Wynblatt 1979, Davies et al. 1980) haben gegenüber der LSW-Theorie die Annahme verschwindend kleinen Volumsanteils der ausgeschiedenen Phase fallen gelassen und die Wechselwirkung zwischen den Diffusionsfeldern der Teilchen berücksichtigt. Obwohl sie alle zu einem Anwachsen des mittleren Radius 3/1tr ∝ führen, sind die Vorfaktoren andere und die asymptotisch erreichte Verteilungsfuntion breiter und niedriger (vgl. den „realen“ Verlauf in Abb. 5-28). Lässt man aber Diffusion und Koagulation von Clustern zu (Binder und Heermann 1985, Fratzl und Penrose 1997), so verlässt man das -Regime. Die Zeitkonstanten für das Anwachsen des mittleren Radius liegen in diesem Fall zwischen ¼ und
3/1t6
1 . Eine weitere Stoßrichtung geht dahin, Keimbildung und Vergröberung nicht als hinter einander ablaufende, sondern zugleich stattfindende Prozesse zu behandeln und auch höhere Volumsanteile zuzulassen (Langer u. Schwarz 1980, Kampmann und Wagner 1984). Eine völlig einschränkungsfreie Behandlung ist nur numerisch möglich (Wagner u. Kampmann 1984) Natürlich wird nicht nur bei einer dispersen Phase (Teilchen eingebettet in Matrix) die Grenzflächenenergie als treibende Kraft für Vergröberung der Struktur wirksam, sondern auch bei einer „spinodalen“ Struktur, bei der die ausgeschiedene Phase zusammen hängt (Perkolation). Statt einer geschlossenen mathematischen Formulierung ist man hier jedoch auch Computersimulationen angewiesen (Filme!). Siehe dazu die Zeitentwicklung in der Abbildung 5-24.
Kohärenzspannungen verändern nicht nur die Keimbildungsbarriere, sondern auch das Vergröberungsverhalten. Die Morphologie der Ausscheidung wird schließlich bestimmt durch Grenzflächenenergie und elastische Wechselwirkungsenergie der Ausscheidungs-teilchen mit der Matrix und untereinander. Dies kann zu dem paradoxen Phänomen führen, dass Ausscheidungsteilchen bei Überschreiten einer bestimmten Größe in zwei gleiche Teilchen aufspalten. Die Verringerung der elastischen WW-Energie überkompensiert dabei den damit verbundenen Anstieg der Grenzflächenenergie. Es tritt eine Bifurkations-Instabilität auf. Ein solches Verhalten wird z.B. bei den γ′-Ausscheidungen in Ni-Al-Superlegierungen beobachtet (Abbildung 5-29, Doi et al. 1984).
Abbildung 5-29: Aufspaltung von geordneten Ausscheidungsteilchen durch elastische Wechselwirkung. Die elastische Wechselwirkung kann den Vergröberungsprozess nicht nur verlangsamen, sondern unter Umständen sogar gänzlich zum Stillstand bringen.

63
Beispiele für verschiedene Ausscheidungs-Morphologien, die durch anisotrope elastische Wechselwirkung zwischen Ausscheidung und Matrix bzw. zwischen den Ausscheidungsteilchen bedingt werden, sind in den Abb. 5-30 (Ni-Al-Mo Legierungen, Fährmann et al. 1995) und 5-31 dargestellt (sog. Widmannstätten-Struktur plattenförmiger hexagonaler Ausscheidungen entlang kristallographischer Vorzugsrichtungen in einer kubisch-flächenzentrierten Matrix).
Abb. 5-31
Abb. 5-30
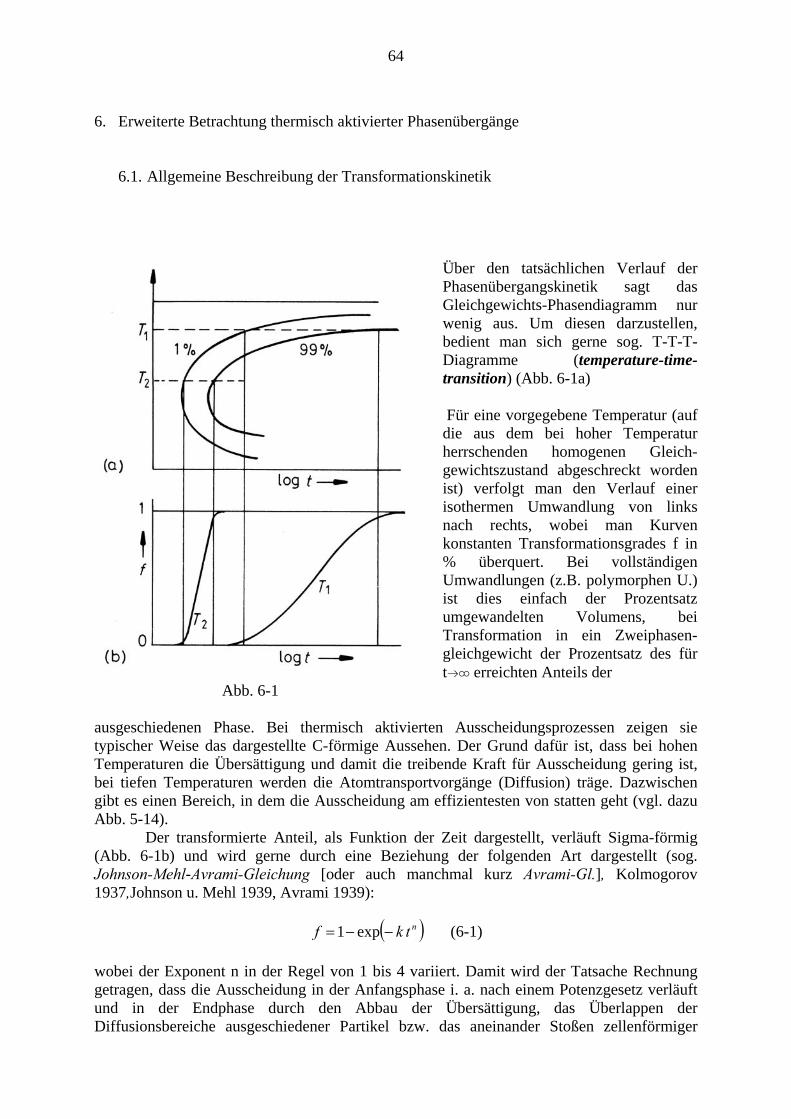
64
6. Erweiterte Betrachtung thermisch aktivierter Phasenübergänge
6.1. Allgemeine Beschreibung der Transformationskinetik
Über den tatsächlichen Verlauf der Phasenübergangskinetik sagt das Gleichgewichts-Phasendiagramm nur wenig aus. Um diesen darzustellen, bedient man sich gerne sog. T-T-T- Diagramme (temperature-time-transition) (Abb. 6-1a) Für eine vorgegebene Temperatur (auf die aus dem bei hoher Temperatur herrschenden homogenen Gleich-gewichtszustand abgeschreckt worden ist) verfolgt man den Verlauf einer isothermen Umwandlung von links nach rechts, wobei man Kurven konstanten Transformationsgrades f in % überquert. Bei vollständigen Umwandlungen (z.B. polymorphen U.) ist dies einfach der Prozentsatz umgewandelten Volumens, bei Transformation in ein Zweiphasen-gleichgewicht der Prozentsatz des für t→∞ erreichten Anteils der
Abb. 6-1 ausgeschiedenen Phase. Bei thermisch aktivierten Ausscheidungsprozessen zeigen sie typischer Weise das dargestellte C-förmige Aussehen. Der Grund dafür ist, dass bei hohen Temperaturen die Übersättigung und damit die treibende Kraft für Ausscheidung gering ist, bei tiefen Temperaturen werden die Atomtransportvorgänge (Diffusion) träge. Dazwischen gibt es einen Bereich, in dem die Ausscheidung am effizientesten von statten geht (vgl. dazu Abb. 5-14). Der transformierte Anteil, als Funktion der Zeit dargestellt, verläuft Sigma-förmig (Abb. 6-1b) und wird gerne durch eine Beziehung der folgenden Art dargestellt (sog. Johnson-Mehl-Avrami-Gleichung [oder auch manchmal kurz Avrami-Gl.], Kolmogorov 1937,Johnson u. Mehl 1939, Avrami 1939):
( )ntkf −−= exp1 (6-1)
wobei der Exponent n in der Regel von 1 bis 4 variiert. Damit wird der Tatsache Rechnung getragen, dass die Ausscheidung in der Anfangsphase i. a. nach einem Potenzgesetz verläuft und in der Endphase durch den Abbau der Übersättigung, das Überlappen der Diffusionsbereiche ausgeschiedener Partikel bzw. das aneinander Stoßen zellenförmiger

65
Ausscheidungsgebiete beschränkt wird und sich asymptotisch dem Gleichgewichtszustand annähert. Um die Form (6-1) abzuleiten, bedient man sich folgenden Kunstgriffs: Man definiert ein erweitertes Volumen (extended volume) der Ausscheidung, das sich ergeben würde, wenn alle Ausscheidungsteilchen ungehindert ohne aneinander Stoßen (impingement) wachsen könnten. Dieses lässt sich relativ einfach mit Hilfe des jeweils zutreffenden Keimbildungs- und Wachstumsgesetzes angeben. Für den einfachen Fall kugelförmiger Ausscheidungen bei homogener Keimbildungsrate I(t) erhält man
eV ,β
∫ −=t
e dtrIVV0
3, )()(
34 τττπ
β (6-2)
Hierbei haben Keime, die zum Zeitpunkt τ entstanden sind, zum Zeitpunkt t den Radius r (t - τ) erreicht. Dieses fiktive Volumen kann ohne Weiteres ein Mehrfaches des gesamten Probenvolumens erreichen. Die Korrektur bringt man jetzt mit folgendem Argument an: Die Vergrößerung von wird nur dort für die Vergrößerung des tatsächlichen Ausscheidungs-volumens wirksam, wo sie auf noch nicht umgewandeltes Volumen fällt. Da die Keimbildung und das Wachstum homogen erfolgen sollen, ist die Wahrscheinlichkeit dafür durch das Verhältnis der entsprechenden Volumina gegeben, sodass
eV ,β
βV
eß dVVV
dV ,1 ββ⎟⎟⎠
⎞⎜⎜⎝
⎛−= (6-3)
Mittels Trennung der Variablen integriert man nun und erhält
⎟⎟⎠
⎞⎜⎜⎝
⎛−−=
VV
VV e,exp1 ββ (6-4),
Was mit den geeigneten Potenzgesetzen für das Wachstum der Ausscheidung der Gestalt (5-1) entspricht. Nimmt man etwa an, dass alle N Keime zum Zeitpunkt t=0 gebildet wurden und danach mit einer Rate ttr ν=)( wachsen, so erhält man
33,
34 tvN
VV
f e πβ == (6-5)
und somit und 3=n 3
34 vNk π
= . (Die Ableitung anderer Fälle ist dem interessierten Leser
als Übung anempfohlen) In der Tabelle 5-1 sind Parameter verschiedener Spezialfälle der Ausscheidungskinetik zusammen gefasst. Das soeben betrachtete Beispiel findet sich unter Kategorie a) eutektoide und diskontinuierliche Ausscheidungen, grenzflächenkontrolliertes Wachstum unter verschwindende Keimbildungsrate (zero nucleation rate). Dieser und ein weiterer wichtiger Spezialfall (konstante Keimbildungsrate) sind in der Tabelle 5-1 durch Pfeile hervor gehoben.

66
Tabelle 6-1
Bei der Auswertung experimentell gewonnener ζ(t) – Kurven versucht man das Verhalten einem der Standardfälle in der Tabelle 6-1 zuzuordnen. Zu diesem Zweck wird ln[-ln(1-ζ)] gegen ln t aufgetragen. Der Anstieg der eventuell dadurch erhaltenen Geraden ist gerade der Exponent n in der Johnson-Mehl-Avrami-Gleichung (6-1). Aus Gl. (6-1) ergibt sich näherungsweise für den Zeitpunkt, an dem die Hälfte ausgeschieden ist,
n
kt
1
5,02ln⎟⎠⎞
⎜⎝⎛= (6-6)
Daraus folgt: Die Umwandlung geht umso schneller vor sich, je größer die Wachstums-(und Keimbildungs-)Konstante k ist. Diese hängt empfindlich von der Temperatur ab, während der Exponent n für den Mechanismus der Umwandlung kennzeichnend und somit weitgehend von der Temperatur unabhängig ist.

67
6.2. Diskontinuierliche Ausscheidung (discontinuous precipitation) Während bei den bisher behandelten kontinuierlichen Ausscheidungsprozessen die Keime gleichmäßig überall im Volumen der Matrixphase entstehen können, wachsen bei den diskontinuierlichen Ausscheidungsvorgängen Duplex-Zellen (Lamellen) der beiden neuen Gleichgewichtsphasen mit einer Reaktionsfront in die Matrixphase, was schematisch in Abb. 6-2 dargestellt ist. Ein klassisches Beispiel sind Sn-Ausscheidungen im Zweistoffsystem Pb-Sn.
Abb. 6-2
Der Phasenübergang geht dabei meistens von einer Korngrenze aus. Ein möglicher Mechanismus wird in einer Theorie von C. Smith (1953) vorgeschlagen: Ein β -Keim wird zunächst in Korn 1 gebildet, kann aber dort schwer wachsen, weil kein bevorzugter Diffusionsweg zur Verfügung steht. Wächst jedoch die neue α -Phase zusammen mit β in das benachbarte Korn 2, so kann Diffusion besonders schnell über die inkohärente Phasengrenze αα ′− erfolgen. Die Ausscheidung nimmt also in Korn 1 ihren Anfang, wächst aber dann aber in das Korn 2, weil ein schneller Diffusionsweg zur Verfügung steht. Wie groß die Bedeutung der Korn- bzw. Zellgrenzendiffusion ist, lässt sich daran erkennen, dass z.B. bei Sn-Pb-Legierungen die Umwandlung bereits bei der Gleichgewichtstemperatur von Trockeneis (∼-78oC) abläuft. Wenn sich die Lamellen strahlenförmig ausbreiten, dann müssen sie verzweigen bzw. neue bilden, um den Lamellenabstand beizubehalten. Der Lamellenabstand ist für eine bestimmte Matrixkonzentration eine Funktion der Temperatur. Oft geht diskontinuierliche Ausscheidung von beweglichen Korngrenzen aus, wobei die Richtung der Ausscheidung mit der Krümmung der Korngrenze korreliert ist (Williams und Butler 1981, Abb. 6-3), Hierbei kann sowohl durch die Korngrenzenbewegung die Ausscheidung hervorgerufen werden als auch umgekehrt.
Abb. 6-3 In manchen Fällen wird eine zweistufige diskontinuierliche Ausscheidungsreaktion beobachtet (Turnbull und Treaftis 1955). Ein Großteil der Übersättigung wird in einer ersten, schnellen Reaktion aufgebraucht, wo bei halbkugelförmigem Wachstum lamellarer Aggregate ein Avrami-Gesetz der Form
( )3exp1 ktf −−= (6-7)

68
mit ( ) 33/2 vNk π= (6-8)
beobachtet wird, wobei f der Anteil an der Transformation in der ersten Phase, N die Anzahl der Zellkeime und v die Wachstumsrate bedeutet. In einer zweiten, wesentlich langsameren Phase wird dann, wiederum diskontinuierlich, eine viel gröbere Struktur eingeführt und der Rest der Übersättigung abgebaut.
6.3. Eutektoide Reaktion
Im Festkörper bedeutet diese Reaktion, dass beim Abkühlen eine feste (statt einer flüssigen wie beim eigentlichen Eutektikum) Phase in zwei andere mit gleich bleibender Zusammensetzung zerfällt. In Abbildung 6-4 ist noch einmal daran erinnert, was zu erwarten ist, wenn man eine Legierung mit der Zusammensetzung x1 aus der γ -Phase heraus abkühlt.
Abb. 6-4 Ab der Temperatur T1 scheidet sich α -Mischkristall aus, bis bei TE die Temperatur
des eutektoiden Gleichgewichts erreicht ist und sich ein fein lamellares eutektoides Gefüge ausscheidet. Die entsprechende T-T-T-Kurvenschar (Abb. 6-5) für unser Beispiel zeigt, dass oberhalb von TE nur α -Mischkristall entsteht, unterhalb von TE bei der hier gewählten hypoeutektoiden Zusammensetzung x1<xγ (Gegenteil: hypereutektoid x2> xγ) zuerst α-Mischkristall, dann das eutektoide Gefüge (ab der strichlierten Linie) ausgeschieden wird, während unterhalb einer Temperatur T2 sofort das eutektoide Gefüge ausgeschieden wird.
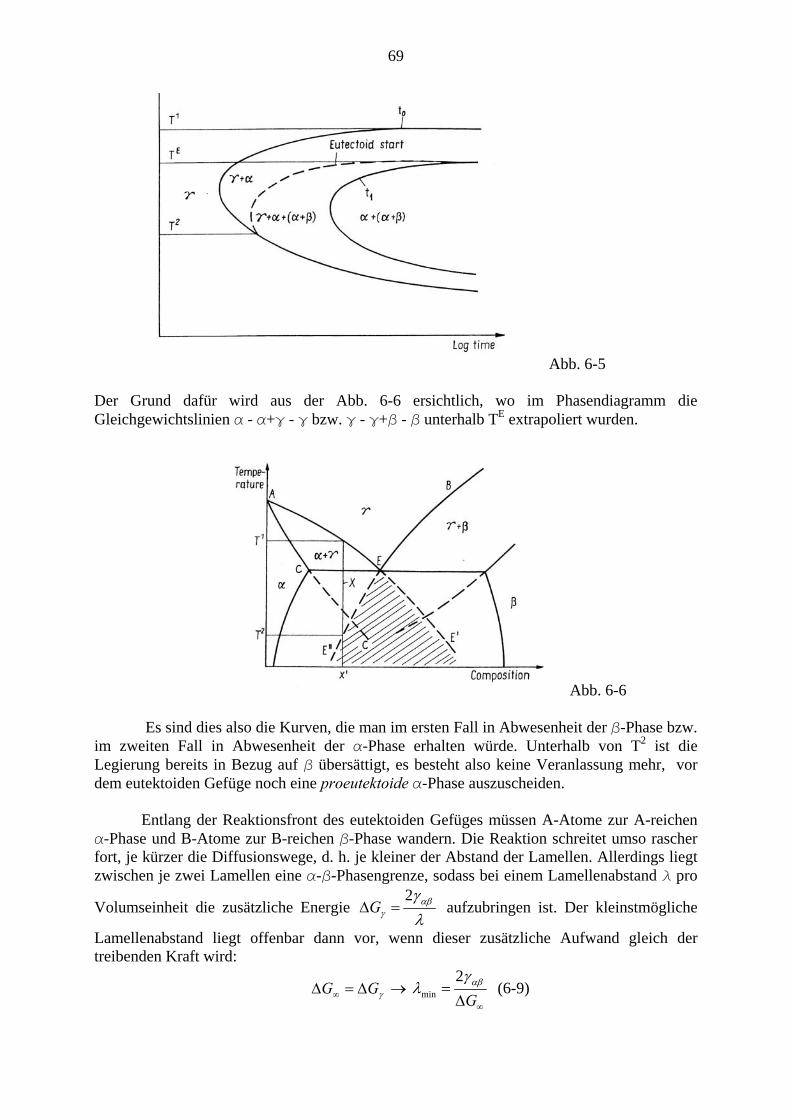
69
Abb. 6-5
Der Grund dafür wird aus der Abb. 6-6 ersichtlich, wo im Phasendiagramm die Gleichgewichtslinien α - α+γ - γ bzw. γ - γ+β - β unterhalb TE extrapoliert wurden.
Abb. 6-6 Es sind dies also die Kurven, die man im ersten Fall in Abwesenheit der β-Phase bzw.
im zweiten Fall in Abwesenheit der α-Phase erhalten würde. Unterhalb von T2 ist die Legierung bereits in Bezug auf β übersättigt, es besteht also keine Veranlassung mehr, vor dem eutektoiden Gefüge noch eine proeutektoide α-Phase auszuscheiden.
Entlang der Reaktionsfront des eutektoiden Gefüges müssen A-Atome zur A-reichen
α-Phase und B-Atome zur B-reichen β-Phase wandern. Die Reaktion schreitet umso rascher fort, je kürzer die Diffusionswege, d. h. je kleiner der Abstand der Lamellen. Allerdings liegt zwischen je zwei Lamellen eine α-β-Phasengrenze, sodass bei einem Lamellenabstand λ pro
Volumseinheit die zusätzliche Energie λγαβ
γ
2=ΔG aufzubringen ist. Der kleinstmögliche
Lamellenabstand liegt offenbar dann vor, wenn dieser zusätzliche Aufwand gleich der treibenden Kraft wird:
γGG Δ=Δ ∞ → ∞Δ
=Gαβγ
λ2
min (6-9)
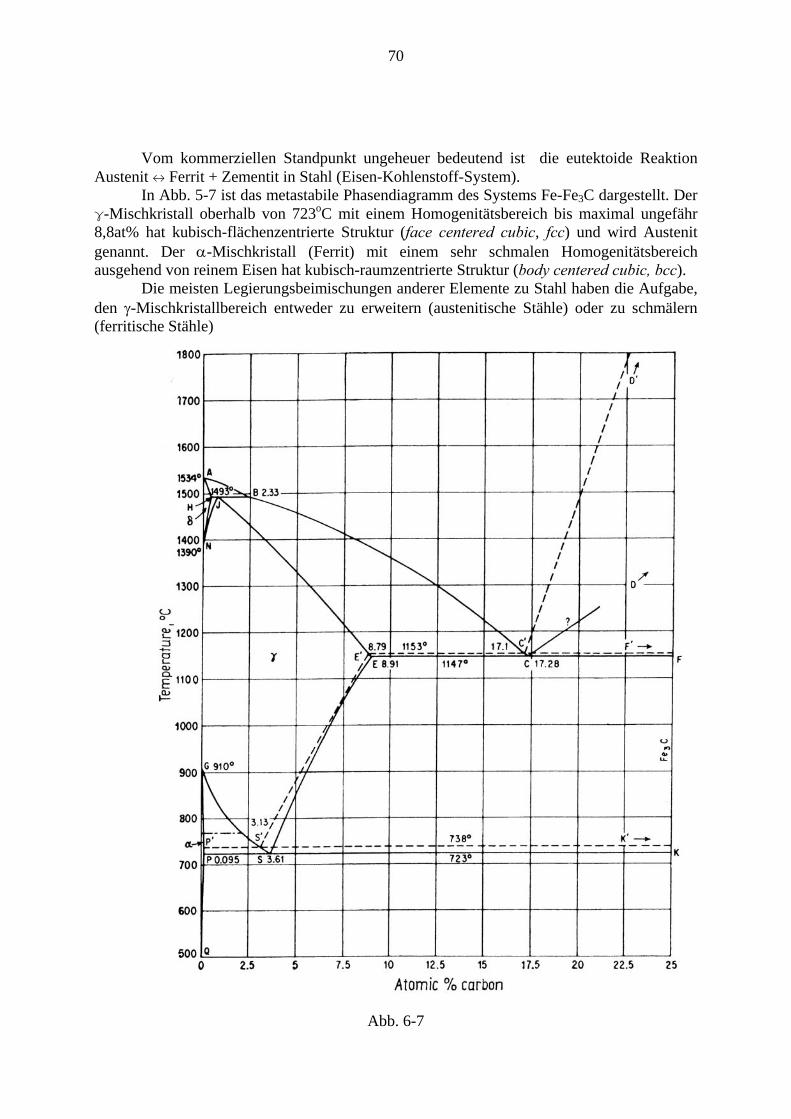
70
Vom kommerziellen Standpunkt ungeheuer bedeutend ist die eutektoide Reaktion
Austenit ↔ Ferrit + Zementit in Stahl (Eisen-Kohlenstoff-System). In Abb. 5-7 ist das metastabile Phasendiagramm des Systems Fe-Fe3C dargestellt. Der
γ-Mischkristall oberhalb von 723oC mit einem Homogenitätsbereich bis maximal ungefähr 8,8at% hat kubisch-flächenzentrierte Struktur (face centered cubic, fcc) und wird Austenit genannt. Der α-Mischkristall (Ferrit) mit einem sehr schmalen Homogenitätsbereich ausgehend von reinem Eisen hat kubisch-raumzentrierte Struktur (body centered cubic, bcc).
Die meisten Legierungsbeimischungen anderer Elemente zu Stahl haben die Aufgabe, den γ-Mischkristallbereich entweder zu erweitern (austenitische Stähle) oder zu schmälern (ferritische Stähle)
Abb. 6-7

71
In der proeutektoiden Phase wird hier kubisch-raumzentrierter Ferrit (α) in kubisch-flächenzentriertem Austenit (γ) ausgeschieden. Dabei treten komplexe semikohärente Phasengrenzen auf, bei denen zwischen den verschiedenen Gitterstrukturen charakteristische, bevorzugten Orientierungsrelationen herrschen. So können etwa die dichtest gepackten Ebenen (110) der bcc- Struktur und (111) der fcc-Struktur annähernd parallel liegen. Bezüglich der Richtungen treten besonders häufig folgende Orientierungsrelationen auf
[ ] [ ] fccbccfccbcc 011//001,)111//()110( Nishiyama-Wassermann (N-W)-Orientierungsrelation,
[ ] [ ]fccbccfccbcc 110//111,)111//()110( Kurdjumov-Sachs (K-S)-Orientierungsrelation (// bedeutet „ungefähr parallel“)
Abb. 6-8

72
Im Gegensatz zu Cu- und Al- Legierungen kommt es hier nicht zur Ausscheidung
metastabiler Zwischenphasen (GP-Zonen u. dgl.), sondern es wird gleich der thermodynamisch stabile Ferrit ausgeschieden. Die Reaktion geht aus Energiegründen von den Korngrenzen aus. Es bilden sich blockartige Strukturen (Korngrenzen-Allotriomorphe, grain boundary allotriomorphs) bzw. bei stärkerer Unterkühlung Aggregate von Ferrit-Platten (Widmanstättensche Seitenplatten,Widmannstaetten side plates). In Abb. 6-8 sind solche typischen Strukturen wiedergegeben. Die weißen Bereiche sind Ferrit-Ausscheidungen. Das Gefüge der Widmannstättenschen Seitenplatten wird umso feiner, je stärker die Unterkühlung (Sequenz b-d) Die eutektoide Reaktion findet bei 3,6 at% C bei 723oC statt. Die bei höherer Temperatur stabile γ-Phase Austenit wandelt sich in ein Lamellen-Gefüge von Zementit (Fe3C) und Ferrit (α) um. Sie werden zusammen als Kolonien mit gemeinsamer Orientierung ausgeschieden. (Abb. 6-9)
Abb. 6-9
Verschiedene Kolonien wachsen als kugelförmige Gebilde von sog. Perlit. Auch hier beginnt die Ausscheidung an einer Korngrenze. Es bildet sich ein Ferrit- oder Zementit-Keim, der mit einem der beiden Kristallite (=Körner) eine semikohärente, mit dem anderen eine inkohärente Phasengrenze hat. Durch Verarmung einer Atomsorte bildet sich anschließend die komplementäre Phase (Zementit bzw. Ferrit). Beide wachsen nun zusammen in jenes Korn, zu dem eine inkohärente Grenze besteht. Grund dafür ist der schnellere Materialtransport durch Diffusion entlang der inkohärenten Phasengrenze (Abb. 5-10). Das Wachstum erfolgt also immer in dasjenige Korn, das nicht die gleiche Orientierung wie das neu entstehende eutektoide Gefüge hat.
Der Vorgang ähnelt also sehr stark dem Phasenwachstum bei diskontinuierlicher Ausscheidung, wobei jedoch zwei neue Phasen entstehen (α → β + γ), während bei der diskontinuierlichen Ausscheidung die Ausscheidungsphase zusammen mit einem anders orientierten Korn der ursprünglichen Matrixphase in dieselbe wächst (α → α′ + β).
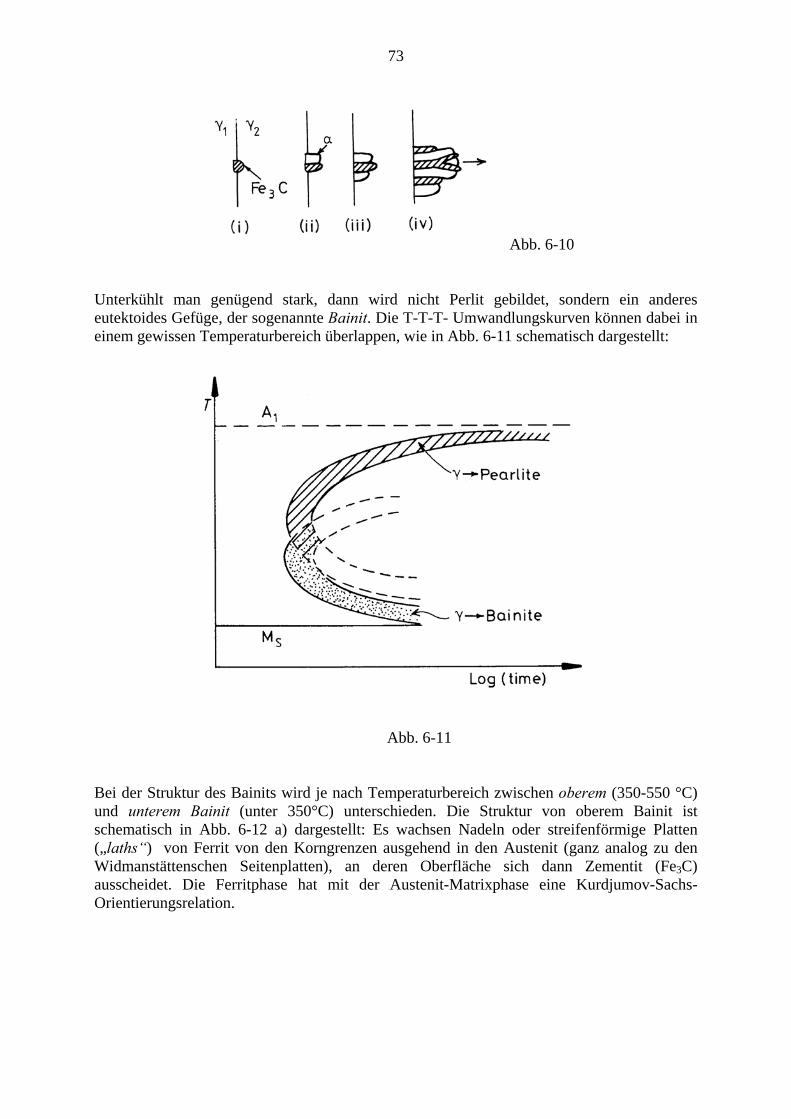
73
Abb. 6-10
Unterkühlt man genügend stark, dann wird nicht Perlit gebildet, sondern ein anderes eutektoides Gefüge, der sogenannte Bainit. Die T-T-T- Umwandlungskurven können dabei in einem gewissen Temperaturbereich überlappen, wie in Abb. 6-11 schematisch dargestellt:
Abb. 6-11
Bei der Struktur des Bainits wird je nach Temperaturbereich zwischen oberem (350-550 °C) und unterem Bainit (unter 350°C) unterschieden. Die Struktur von oberem Bainit ist schematisch in Abb. 6-12 a) dargestellt: Es wachsen Nadeln oder streifenförmige Platten („laths“) von Ferrit von den Korngrenzen ausgehend in den Austenit (ganz analog zu den Widmanstättenschen Seitenplatten), an deren Oberfläche sich dann Zementit (Fe3C) ausscheidet. Die Ferritphase hat mit der Austenit-Matrixphase eine Kurdjumov-Sachs-Orientierungsrelation.
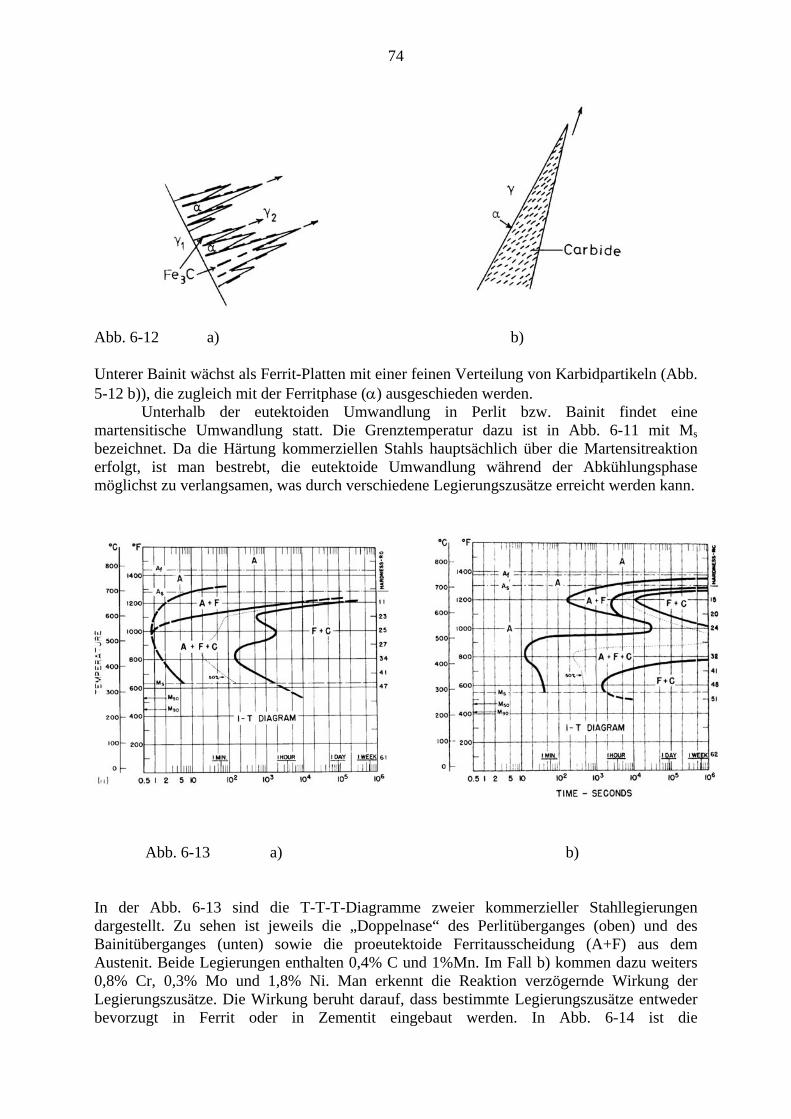
74
Abb. 6-12 a) b) Unterer Bainit wächst als Ferrit-Platten mit einer feinen Verteilung von Karbidpartikeln (Abb. 5-12 b)), die zugleich mit der Ferritphase (α) ausgeschieden werden.
Unterhalb der eutektoiden Umwandlung in Perlit bzw. Bainit findet eine martensitische Umwandlung statt. Die Grenztemperatur dazu ist in Abb. 6-11 mit Ms bezeichnet. Da die Härtung kommerziellen Stahls hauptsächlich über die Martensitreaktion erfolgt, ist man bestrebt, die eutektoide Umwandlung während der Abkühlungsphase möglichst zu verlangsamen, was durch verschiedene Legierungszusätze erreicht werden kann.
Abb. 6-13 a) b)
In der Abb. 6-13 sind die T-T-T-Diagramme zweier kommerzieller Stahllegierungen dargestellt. Zu sehen ist jeweils die „Doppelnase“ des Perlitüberganges (oben) und des Bainitüberganges (unten) sowie die proeutektoide Ferritausscheidung (A+F) aus dem Austenit. Beide Legierungen enthalten 0,4% C und 1%Mn. Im Fall b) kommen dazu weiters 0,8% Cr, 0,3% Mo und 1,8% Ni. Man erkennt die Reaktion verzögernde Wirkung der Legierungszusätze. Die Wirkung beruht darauf, dass bestimmte Legierungszusätze entweder bevorzugt in Ferrit oder in Zementit eingebaut werden. In Abb. 6-14 ist die
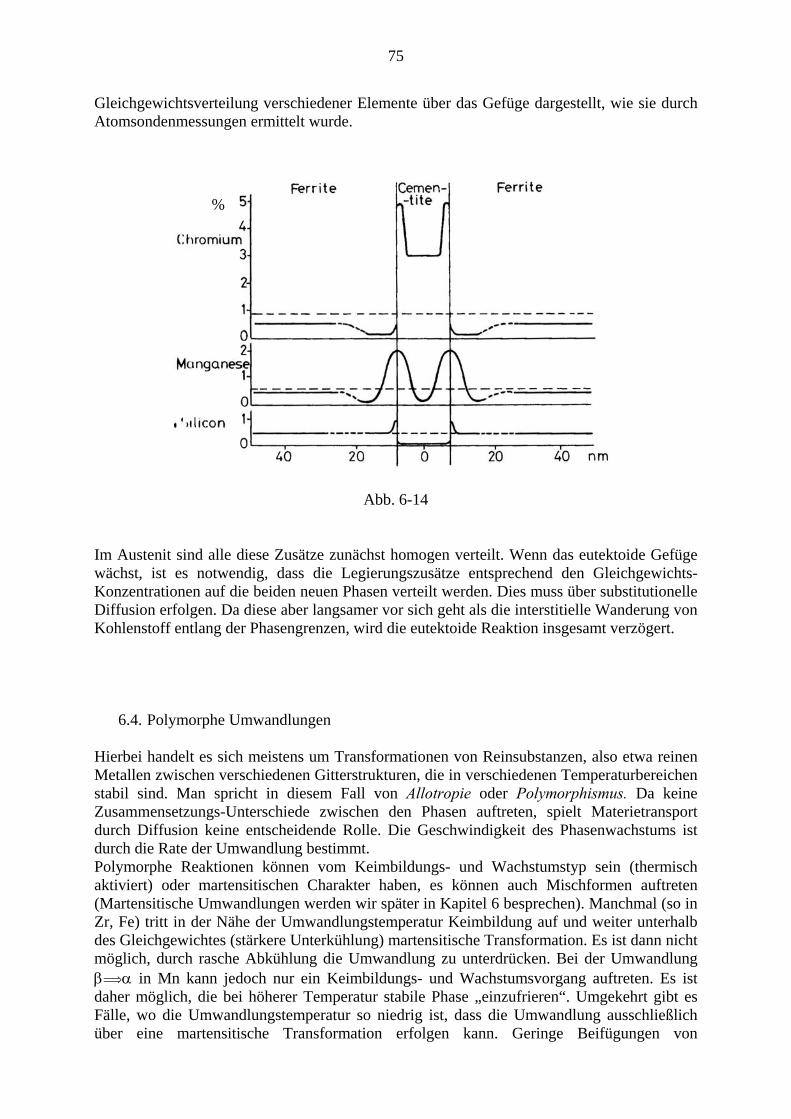
75
Gleichgewichtsverteilung verschiedener Elemente über das Gefüge dargestellt, wie sie durch Atomsondenmessungen ermittelt wurde.
%
Abb. 6-14
Im Austenit sind alle diese Zusätze zunächst homogen verteilt. Wenn das eutektoide Gefüge wächst, ist es notwendig, dass die Legierungszusätze entsprechend den Gleichgewichts-Konzentrationen auf die beiden neuen Phasen verteilt werden. Dies muss über substitutionelle Diffusion erfolgen. Da diese aber langsamer vor sich geht als die interstitielle Wanderung von Kohlenstoff entlang der Phasengrenzen, wird die eutektoide Reaktion insgesamt verzögert.
6.4. Polymorphe Umwandlungen
Hierbei handelt es sich meistens um Transformationen von Reinsubstanzen, also etwa reinen Metallen zwischen verschiedenen Gitterstrukturen, die in verschiedenen Temperaturbereichen stabil sind. Man spricht in diesem Fall von Allotropie oder Polymorphismus. Da keine Zusammensetzungs-Unterschiede zwischen den Phasen auftreten, spielt Materietransport durch Diffusion keine entscheidende Rolle. Die Geschwindigkeit des Phasenwachstums ist durch die Rate der Umwandlung bestimmt. Polymorphe Reaktionen können vom Keimbildungs- und Wachstumstyp sein (thermisch aktiviert) oder martensitischen Charakter haben, es können auch Mischformen auftreten (Martensitische Umwandlungen werden wir später in Kapitel 6 besprechen). Manchmal (so in Zr, Fe) tritt in der Nähe der Umwandlungstemperatur Keimbildung auf und weiter unterhalb des Gleichgewichtes (stärkere Unterkühlung) martensitische Transformation. Es ist dann nicht möglich, durch rasche Abkühlung die Umwandlung zu unterdrücken. Bei der Umwandlung β α in Mn kann jedoch nur ein Keimbildungs- und Wachstumsvorgang auftreten. Es ist daher möglich, die bei höherer Temperatur stabile Phase „einzufrieren“. Umgekehrt gibt es Fälle, wo die Umwandlungstemperatur so niedrig ist, dass die Umwandlung ausschließlich über eine martensitische Transformation erfolgen kann. Geringe Beifügungen von
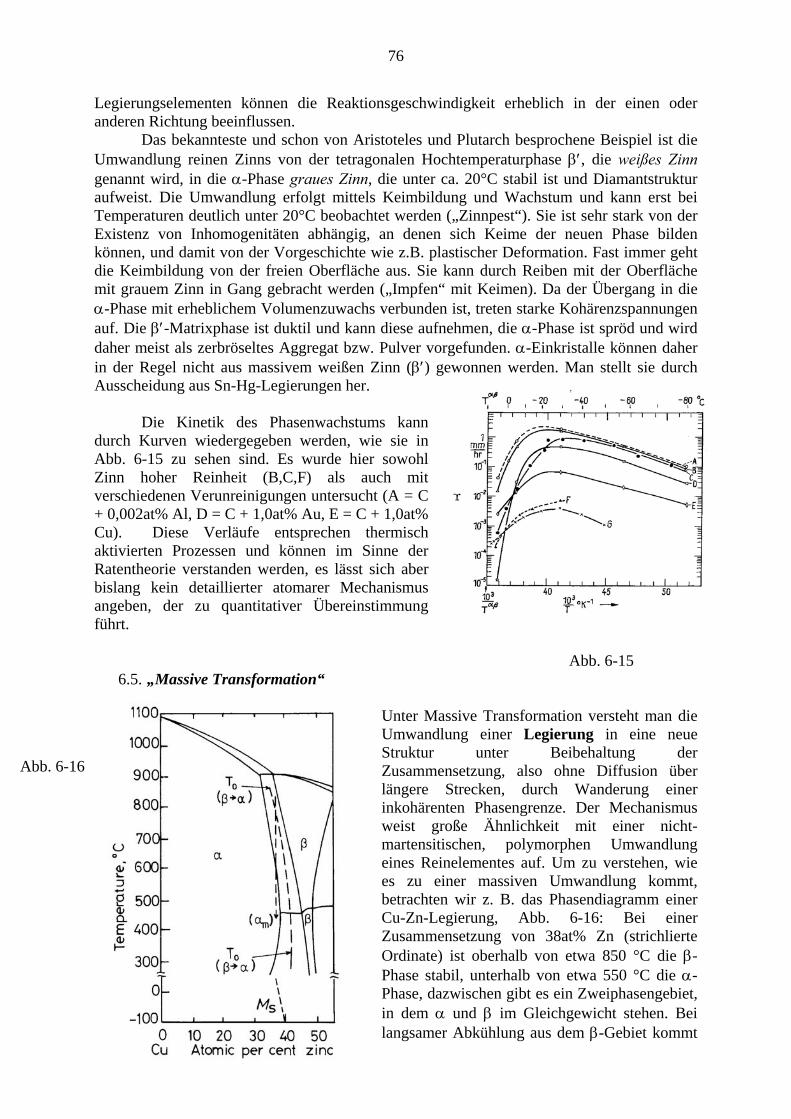
76
Legierungselementen können die Reaktionsgeschwindigkeit erheblich in der einen oder anderen Richtung beeinflussen.
Das bekannteste und schon von Aristoteles und Plutarch besprochene Beispiel ist die Umwandlung reinen Zinns von der tetragonalen Hochtemperaturphase β′, die weißes Zinn genannt wird, in die α-Phase graues Zinn, die unter ca. 20°C stabil ist und Diamantstruktur aufweist. Die Umwandlung erfolgt mittels Keimbildung und Wachstum und kann erst bei Temperaturen deutlich unter 20°C beobachtet werden („Zinnpest“). Sie ist sehr stark von der Existenz von Inhomogenitäten abhängig, an denen sich Keime der neuen Phase bilden können, und damit von der Vorgeschichte wie z.B. plastischer Deformation. Fast immer geht die Keimbildung von der freien Oberfläche aus. Sie kann durch Reiben mit der Oberfläche mit grauem Zinn in Gang gebracht werden („Impfen“ mit Keimen). Da der Übergang in die α-Phase mit erheblichem Volumenzuwachs verbunden ist, treten starke Kohärenzspannungen auf. Die β′-Matrixphase ist duktil und kann diese aufnehmen, die α-Phase ist spröd und wird daher meist als zerbröseltes Aggregat bzw. Pulver vorgefunden. α-Einkristalle können daher in der Regel nicht aus massivem weißen Zinn (β′) gewonnen werden. Man stellt sie durch Ausscheidung aus Sn-Hg-Legierungen her.
Die Kinetik des Phasenwachstums kann
durch Kurven wiedergegeben werden, wie sie in Abb. 6-15 zu sehen sind. Es wurde hier sowohl Zinn hoher Reinheit (B,C,F) als auch mit verschiedenen Verunreinigungen untersucht (A = C + 0,002at% Al, D = C + 1,0at% Au, E = C + 1,0at% Cu). Diese Verläufe entsprechen thermisch aktivierten Prozessen und können im Sinne der Ratentheorie verstanden werden, es lässt sich aber bislang kein detaillierter atomarer Mechanismus angeben, der zu quantitativer Übereinstimmung führt.
Abb. 6-15
6.5. „Massive Transformation“ Unter Massive Transformation versteht man die Umwandlung einer Legierung in eine neue Struktur unter Beibehaltung der Zusammensetzung, also ohne Diffusion über längere Strecken, durch Wanderung einer inkohärenten Phasengrenze. Der Mechanismus weist große Ähnlichkeit mit einer nicht-martensitischen, polymorphen Umwandlung eines Reinelementes auf. Um zu verstehen, wie es zu einer massiven Umwandlung kommt, betrachten wir z. B. das Phasendiagramm einer Cu-Zn-Legierung, Abb. 6-16: Bei einer Zusammensetzung von 38at% Zn (strichlierte Ordinate) ist oberhalb von etwa 850 °C die β-Phase stabil, unterhalb von etwa 550 °C die α-Phase, dazwischen gibt es ein Zweiphasengebiet, in dem α und β im Gleichgewicht stehen. Bei langsamer Abkühlung aus dem β-Gebiet kommt
Abb. 6-16

77
es zu einer Ausscheidung von den Korngrenzen her ähnlich dem Übergang Austenit-Ferrit, bei größerer Abkühlgeschwindigkeit in Form von Widmanstätten-Nadeln. Die ausgeschiedene α-Phase ist Cu-reicher als die Matrix, Zn muss also mittels Diffusion von der Reaktionsfront entfernt werden. Durchfährt man jedoch durch rasche Abkühlung das Zweiphasengebiet, dann wird die Ausscheidung unterdrückt, und es kann sich β in α direkt ohne Konzentrationsverschiebung umwandeln. Es wachsen massive Körner von α von den β-Korngrenzen her. Die Geschwindigkeit des Phasenwachstums ist nicht von der Diffusion begrenzt, sondern nur davon, wie schnell die Atome in die neue Struktur eingebaut werden können. Bei genügend hoher Abkühlungsrate kann schließlich ein martensitischer Übergang stattfinden. Eine massive Umwandlung kann auch innerhalb des Zweiphasengebietes stattfinden, wenn die thermodynamische Bedingung dafür gegeben ist, nämlich dass die freie Energie pro Volumseinheit für die neue Phase kleiner ist als für die alte. Die strichlierte Kurve mit der Bezeichnung T0 in der Mitte des Zweiphasengebietes in Abb. 6-16 gibt die Temperatur an, bei der für eine gegebene Zusammensetzung diese Bedingung erfüllt ist. Das Ergebnis der Umwandlung ist dann keine Gleichgewichtsphase, sondern eine übersättigte Tieftemperaturphase.
Die Verhältnisse können besonders anschaulich durch ein CCT-Diagramm (continuous cooling transformation) dargestellt werden, in dem der Temperaturverlauf entlang verschiedener Abkühlkurven (mit verschiedenen Abkühlgeschwindigkeiten) als eine Kurvenschar und die dabei auftretenden Transformationen mit den entsprechenden Umwandlungsgraden als weitere (kreuzende) Kurvenscharen erscheinen (Abb. 6-17)
Abb. 6-17

78
Abb. 6-18 a) b) In Abb. 6-18 sind mikroskopische Aufnahmen massiver Umwandlungen a) in der besprochenen Cu-38at%Zn – Legierung, b) in einer Cu-20at%Al – Legierung wiedergegeben. Man erkennt die typisch unregelmäßig begrenzten, von den Korngrenzen ausgehenden Umwandlungsgebiete.

79
7. Athermische Phasenübergänge (martensitische Phasenübergänge)
7.1. Charakter der martensitischen Umwandlung Im englischen Sprachraum werden martensitische Phasenübergänge gern als militärisch (military) charakterisiert, im Gegensatz zu den bisher betrachteten Übergängen, die als zivil (civilian) bezeichnet werden. Damit wird ausgesagt, dass die Atome bei ersteren gewissermaßen im Gleichschritt, also mit kooperativen Bewegungen, ihre Plätze ändern, während bei letzteren die Atombewegungen statistisch zufällig, wenn auch mit bestimmten Wahrscheinlichkeiten verteilt, stattfinden. Die kooperativen Bewegungen erfolgen über kurze Strecken, die kleiner als die Abmessungen der Gitterzelle sind. Dabei wird das Gitter der Ausgangsphase durch eine homogene Verzerrung in das Gitter der Martensitphase umgewandelt, die Nächst-Nachbar-Relationen bleiben dabei erhalten. Eine Ebene bleibt bei der Verzerrung unverändert (invariant-plane strain). Daher werden martensitische Umwandlungen auch als „verschiebend“ (displacive), „scherungsartig“ (shear-like) oder „diffusionsfrei“ (diffusion-less) bezeichnet. Die Phasengrenze kann dabei völlig kohärent sein (wie bei der fcc→hcp Umwandlung in Co und seinen Legierungen) oder semikohärent wie üblicherweise bei Fe-Legierungen. In jedem Fall laufen martensitische Umwandlungen, wenn sie erst einmal in Gang gesetzt sind, mit großer Geschwindigkeit ab, wobei die Geschwindigkeit der Reaktionsgrenze nicht von der Temperatur abhängt (wohl aber der Anteil der umgewandelten Phase im Gleichgewicht). Die bisher besprochenen Phasenübergänge sind im Gegensatz dazu thermisch aktiviert: Es muss eine Energiebarriere mit thermischen Fluktuationen überwunden werden (vgl. Tabelle 2-1). Zur experimentellen Beobachtung der martensitischen Umwandlungen: Martensit (nach dem deutschen Metallographen Adolf Martens, 1850-1914) wurde ursprünglich der harte Bestandteil in Abschreckungs-gehärteten Stählen genannt. Dabei bleiben die C-Atome der Ausgangsphase Austenit zunächst in Lösung, sodass nach der Martensit-Transformation eine übersättigte, metastabile Ferritphase vorliegt. Die Martensitumwandlung in Stahl ist ein sehr komplexer Vorgang, der bis heute nicht vollkommen verstanden wird.
Abb. 7-1
Poliert man die Oberfläche der Ausgangsphase und schreckt dann unter Martensitbildung ab, dann entsteht ein charakteristisches Oberflächenrelief (Abbildung 7-1, Fe-24,5%Pt – Legierung, lichtmikroskopische Aufnahme). Auf der Oberfläche geritzte Linien werden geknickt, bleiben aber kontinuierlich. Zumindest auf der Skala der lichtmikroskopischen Beobachtung ist die Phasengrenze kohärent. Dort wo die Phasengrenze zwischen Martensit und untransformiertem Volumen die Oberfläche trifft, tritt keine Stufe auf.

80
(f) (g)
Abb. 7-2 : a) und b) sukzessives Wachstum von Martensitplatten (α′). Martensit-Morphologien: c) Fe mit niedrigem C – Gehalt: Streifen (laths), d) Fe mit mittlerem C-Gehalt: Platten, e) Fe-Ni-Legierung: Platten, f) Dünnplatten-Morphologie (Fe-31%Ni-0,29%C), g) „butterfly“-Morphologie (Fe-20% Ni-0,7%C)

81
In Abb. 7-2 a) und b) ist schematisch dargestellt, wie die linsenförmigen Martensitplatten wachsen und sukzessive Zwischenräume zwischen bereits existierenden Platten ausfüllen. Das Wachstum erfolgt in bestimmten kristallographischen Richtungen und kann fast Schallgeschwindigkeit erreichen, dh. in 10-7s kann ein ganzes Korn des Austenits durchquert werden. Die Martensitbildung setzt bei einer Start-Temperatur Ms ein und ist bei einer Endtemperatur Mf abgeschlossen. Es wird i.a. nicht der gesamte Austenit umgewandelt, sondern es verbleibt ein restliches, nicht transformiertes Volumen. Grund dafür scheinen zunehmende Kohärenzspannungen zu sein.
Die Abb. 7-3 zeigt den Verlauf der Anfangs- und Endtemperatur der Martensitreaktion im Fe-C-Phasendiagramm. T0 markiert jeweils die Temperatur, ab der die freie Enthalpie pro Volumseinheit der Martensitphase geringer ist als die der Matrixphase (Austenit) und somit eine treibende Kraft für die Umwandlung vorhanden ist. Abb. 7-3
7.2. Die Kristallographie des Martensits Aus Abb. 6-1 geht hervor, dass die transformierten Bereiche des Kristalls eine Verzerrung erfahren, die auf den ersten Blick einer plastischen Scherungsdeformation ähnelt. Eine genauere Untersuchung zeigt aber, dass die Transformation komplexer und mehrstufig ist. Aus der Lage verschiedener (nicht paralleler und auf verschiedenen Oberflächen befindlicher) geritzter Linien vor und nach der Deformation lässt sich eine Matrix der Deformation ableiten. Eine Ebene, die die ursprüngliche Phase und der Martensit gemeinsam besitzen (habit plane), bleibt dabei unverändert und unverzerrt (invariant-plane strain deformation). Ansonsten erfolgt die Verschiebung jedes Punktes während der Umwandlung in die gleiche Richtung und ist der Größe nach proportional dem Abstand von der invarianten Ebene.
In Abb. 7-4 ist dieser Sachverhalt schematisch dargestellt, wobei ABC die invariante Ebene ist. Abb. 7-4

82
Abb. 7-5 Eine Verzerrung dieser Art kann erzeugt werden, wenn eine anfängliche Kugel in ein Ellipsoid deformiert wird, wobei eine Hauptachsendehnung (principal strain) 1 ist und die beiden anderen entgegengesetztes Vorzeichen haben. Dann könne der ursprüngliche und der verzerrte Bereich entlang der unverzerrten Ebenen AOX oder BOX zusammen gefügt werden. (Abb. 7-5). Eine Strukturuntersuchung mittels Röntgenbeugung zeigt, dass die neue Phase eine andere Gitterstruktur hat (Beim „klassischen“ Martensit wandelt sich fcc in bcc bzw. bct -bodycentered tetragonal- um). Eine Tatsache, die ursprünglich das Verständnis der Martensitbildung sehr erschwert hat, ist nun, dass die Anwendung der oben erwähnten Deformationsmatrix auf die Austenitstruktur nicht zur Kristallstruktur des Martensits führt. Die Habitebene entspricht meistens keinen einfachen, ganzzahligen Millerindices. In der Tabelle 7-1 sind Eigenschaften der Martensitgeometrie zusammen gefasst, wie sie in verschiedenen Legierungen beobachtet wurden:
Tab. 7-1
Abb. 7-6 Die Orientierungen der tatsächlichen Habitebenen streuen um die angegebenen Werte (ein Beispiel ist in Abb. 7-6 dargestellt, eine Fe – 7%Al – 1,5%C – Legierung; Orientierungsdreieck in stereographischer Projektion)

83
Indessen wurde relativ frühzeitig erkannt (Bain 1924), dass eine einfache homogene Verzerrung die fcc- Gitterstruktur (Austenit) in die bcc-Gitterstruktur (Martensit) überführt. Die von Bain vorgeschlagene Transformation bzw. Korrespondenz zwischen den beiden Gittertypen (Bain transformation) ist in Abb. 6-7 dargestellt.
Abb. 7-7 Abb. 7-8 Sie geht mit einer ∼20% Kompression in z-Richtung und einer ∼12% Expansion normal dazu einher. Es entsprechen dann die Richtungen bzw. Ebenen
( ) ( )bccfcc 001001 ≈ (Basisebene) (7-1) [ ] [ ]bccfcc 001001 ≈ (z – Richtung)
[ ] [ ]bccfcc 111011 ≈ (Flächendiagonale fcc – Raumdiagonale bcc)
bccfcc )011()111( ≈ (Pyramidenfläche fcc- Prismenfläche bcc). Grundsätzlich ist eine solche Zuordnung nicht eindeutig und könnte auf verschiedene Weisen hergestellt werden. Eine alternative Möglichkeit ist in Abb. 6-8 zu sehen. Jedoch ist die von Bain vorgeschlagene Transformation diejenige, die mit den geringsten Hauptachsendehnungen verbunden ist. Dass die beiden Gitter tatsächlich in dieser Weise einander zuordenbar sind, wurde mittels Elektronenbeugung durch kontinuierliche Beobachtung der Übergitterreflexe (superlattice reflections) während der Transformation in einer Fe3Pt – Legierung gezeigt (Tadaki und Shimizu 1970). Die Bain-Transformation erzeugt also die richtige Gitterstruktur. Sie stellt aber nicht die makroskopische bzw. im Lichtmikroskop festgestellte Ebenen-invariante Transformation zum Martensit dar. Die Auflösung dieses Widerspruchs ergibt sich aus der Feinstruktur des Martensits: Es findet zusätzlich zur Bain-Transformation eine Gitter-invariante Transformation statt, wobei entweder eine plastische Scherung oder eine Zwillingsdeformation stattfinden kann.
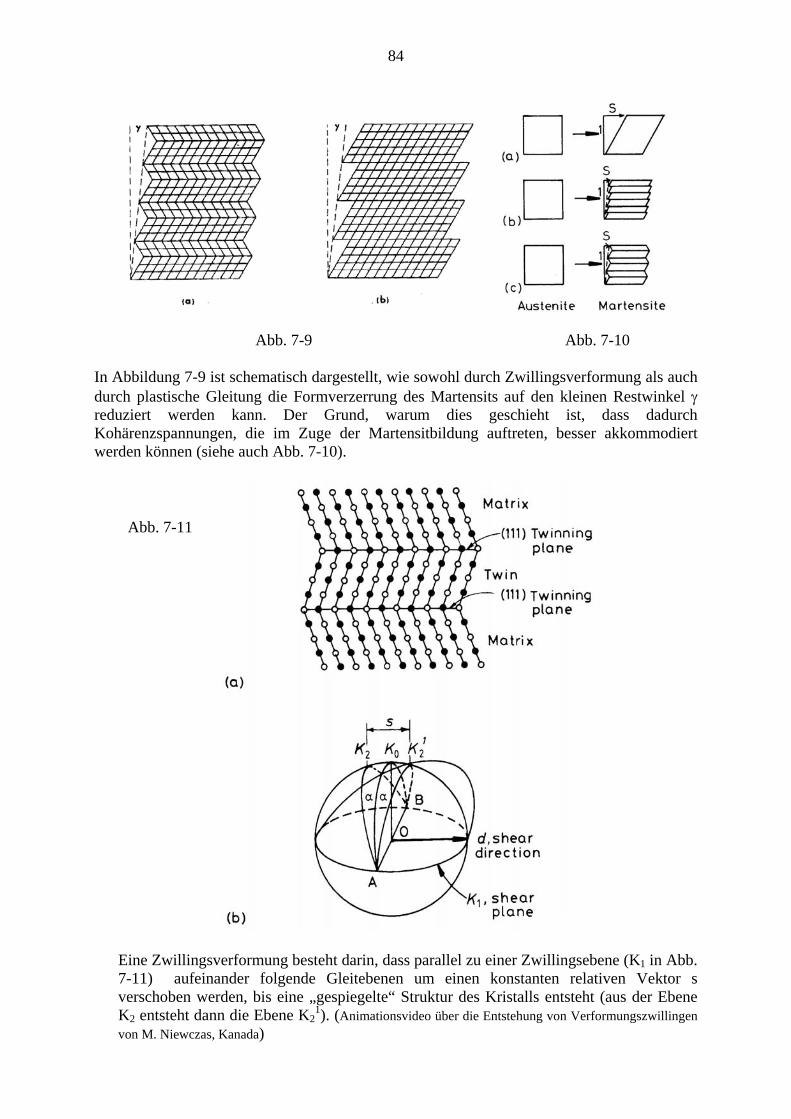
84
Abb. 7-9 Abb. 7-10 In Abbildung 7-9 ist schematisch dargestellt, wie sowohl durch Zwillingsverformung als auch durch plastische Gleitung die Formverzerrung des Martensits auf den kleinen Restwinkel γ reduziert werden kann. Der Grund, warum dies geschieht ist, dass dadurch Kohärenzspannungen, die im Zuge der Martensitbildung auftreten, besser akkommodiert werden können (siehe auch Abb. 7-10).
Abb. 7-11
Eine Zwillingsverformung besteht darin, dass parallel zu einer Zwillingsebene (K1 in Abb. 7-11) aufeinander folgende Gleitebenen um einen konstanten relativen Vektor s verschoben werden, bis eine „gespiegelte“ Struktur des Kristalls entsteht (aus der Ebene K2 entsteht dann die Ebene K2
1). (Animationsvideo über die Entstehung von Verformungszwillingen von M. Niewczas, Kanada)

85
In Abb. 7-12 ist zu sehen, wie sich die verzwillingte bzw scherungsdeformierte Feinstruktur in die linsenförmigen Martensitgebiete einfügt.
Abb. 7-12
Diese zusätzliche Deformation (Gleitung oder Zwillingsbildung) wird auch inhomogene oder komplementäre Deformation genannt. Sie macht zwar eine Hauptdehnung zu eins und stellt invariante Ebenen her. Diese befinden sich aber noch nicht in der richtigen Lage, und es bedarf aber noch einer kleinen, starren Drehung, um die vollständige Martensit-Deformation zu erzeugen. Diese stellt sich dann formal durch folgende kombinierte Matrixoperation dar:
BPRP =1 (7-2)
mit B Bain-Transformation, P inhomogener Scherung oder Zwillingsbildung und R starrer Rotation. Alle beteiligten Größen sind 3x3-Matrizen. Die Bain-Transformation etwa lässt sich schreiben als
⎟⎟⎟
⎠
⎞
⎜⎜⎜
⎝
⎛=
⎟⎟⎟⎟
⎠
⎞
⎜⎜⎜⎜
⎝
⎛
=8,000
012,100012,1
/000/2000/2
0
0
0
acaa
aaB (7-3)
mit den typischen Werten für Stahl. In der Analyse der Martensitgeometrie erweist es sich als rechentechnisch einfacher, die komplementäre Deformation formal nicht im Martensit, sondern vorher in der Ausgangsphase stattfinden zu lassen, also
PBRP =1 (7-4)
Eine Deformation mit invarianter Ebene lässt sich immer in folgender Form darstellen (in Komponenten geschrieben):
( ) ′+= jiijij pmdP δ1 (7-5), wobei also eine Verzerrung durch ein Tensorprodukt (dyadisches Produkt) aus einem Richtungsvektor d und einem Ebenen-Normalvektor p’ ausgedrückt werden kann. Hat man also m1 , d1 und p1’ bestimmt, dann ist die Geometrie des Martensits festgelegt. Es sei darauf hingewiesen, dass das soeben geschilderte Bild typisch für die Umwandlung in Stählen ist, aber in einigen rostfreien Stählen und in Fe-Mn-C – Legierungen eine martensitische Transformation fcc → hcp (anstelle von bcc bzw. bct) stattfindet. Dabei wird die Deformation vollständig durch Partialversetzungen erzeugt, die parallel zur

86
{111}-Habitebene laufen, und es tritt keine zusätzliche Scherung bzw. Zwillingsdeformation auf. Dasselbe gilt für Co und seine Legierungen. In allen diesen Fällen ist die Phasengrenze voll kohärent. Die Morphologie des Martensits entspricht dann geraden Streifen parallel zur Habitebene anstatt linsenförmigen Strukturen. 7.3. Bildung von Martensitkeimen Das Phasenwachstum des Martensits ist zwar inhärent athermisch, aber zuvor müssen thermisch aktivert Keime gebildet werden. Die zur Bildung eines Martensitkeimes notwendige freie Enthalpie kann geschrieben werden als
σπΔπΔ 22 23
4 aacAgcaG ch +⎟⎠⎞
⎜⎝⎛ += (7-6)
Das Teilchen wird dabei als abgeplattetes Ellipsoid mit Radius a und Höhe c angenommen. ist die freie Enthalpie der Umwandlung pro Volumseinheit, A c/a die elastische Verzerrungsenergie pro Volumseinheit. Der Verzerrungsenergiefaktor A ist dabei gegeben durch
chgΔ
( )( )
20
20 418
2 εμπγμννπ
+⎥⎦
⎤⎢⎣
⎡−−
=A (7-7),
wobei angenommen wurde, dass die Poissonzahl ν und der Schubmodul μ für beide Phasen gleich sind. Der letzte Term in (7-6) stellt die Grenzflächenenergie dar. Die Martensit-Deformation habe eine Scherungskomponente 0γ parallel und eine Dehnungskomponente 0ε normal auf die invariante Habitebene. A beträgt typischerweise 2,4.103 MJm-3. Die Energie der für die Eisenlegierungen typischen semikohärenten Grenzfläche beträgt etwa 100-200mJm-2. chgΔ beträgt bei Fe-Legierungen typischerweise -170 MJm-3 bei der Martensit-Starttemperatur. In die obigen Formeln (Gln. 7-6 u. 7-7) eingesetzt, erhält man als Keimbildungsbarriere etwa 8.10-16 J, das entspricht ca. 105 kT bei 580K, einer Temperatur, bei der bereits spontane Keimbildung beobachtet wird. Homogene Keimbildung ist daher auszuschließen. Aus Versuchen der Martensitkeimbildung an Einkristallpartikeln verschiedener Größe ergibt sich, dass die Anzahl der Keime nicht von der Korngröße abhängt. Daher ist die Oberfläche bzw. Korngrenze für die Keimbildung nicht ausschlaggebend. Man nimmt daher an, dass die Keimbildung vorzugsweise an Defekten wie Versetzungsanordnungen erfolgt.
Dass Versetzungen eine wichtige Rolle bei der Martensitbildung spielen können, wird schon anhand von Transformationen wie der Umwandlung fcc → hcp in Co – Legierungen klar: eine Partialversetzung des Typs a/6 <112> bringt eine Verschiebung ein, die die Stapelung zweier auf einander folgender Ebenen ändert. Nach Durchlaufen mehrerer solcher Partialversetzungen in parallelen Ebenen wird aus der Stapelfolge A-B-C-A-B-C des kubisch-flächenzentrierten Gitters die Stapelfolge A-B-A-B-A-B des hexagonalen Gitters. Findet die Verschiebung, die einer solchen Shockley- Partialversetzung entspricht, nur zur Hälfte statt, dann kommt man zu einem möglichen
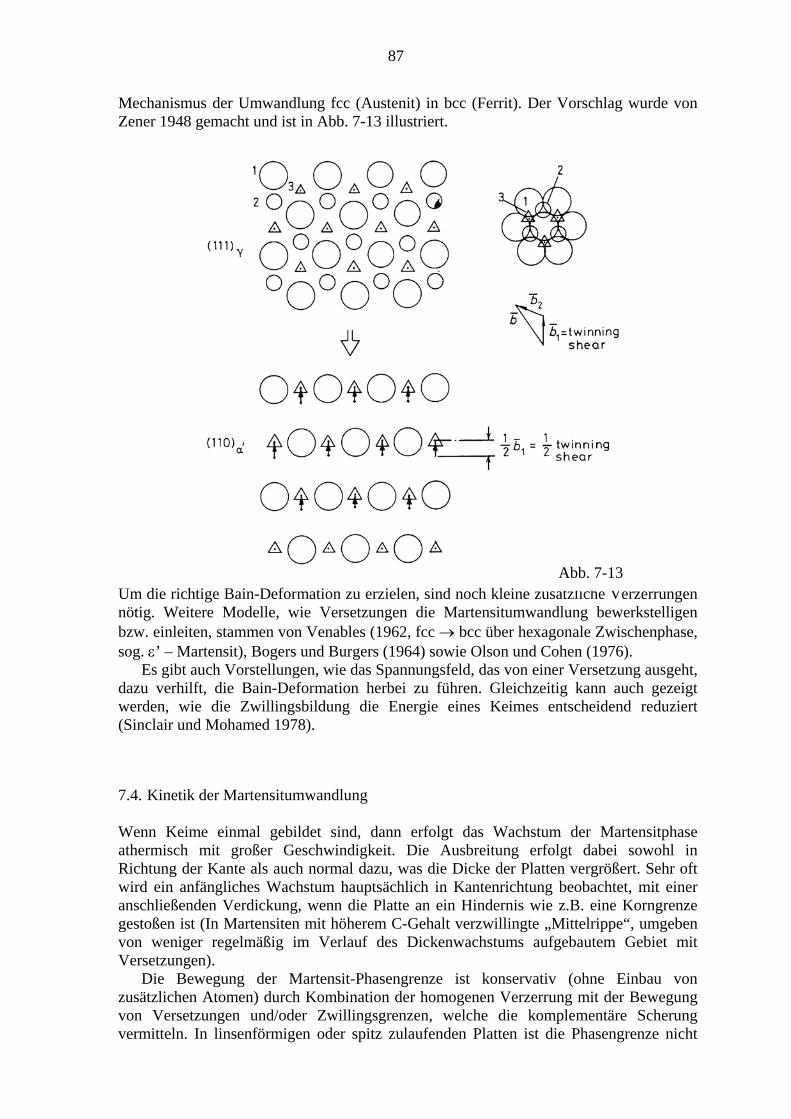
87
Mechanismus der Umwandlung fcc (Austenit) in bcc (Ferrit). Der Vorschlag wurde von Zener 1948 gemacht und ist in Abb. 7-13 illustriert.
Um die richtige Bain-Deformation zu erzielen, sind noch kleine zusätzliche Verzerrungen nötig. Weitere Modelle, wie Versetzungen die Martensitumwandlung bewerkstelligen bzw. einleiten, stammen von Venables (1962, fcc → bcc über hexagonale Zwischenphase, sog. ε’ – Martensit), Bogers und Burgers (1964) sowie Olson und Cohen (1976).
Abb. 7-13
Es gibt auch Vorstellungen, wie das Spannungsfeld, das von einer Versetzung ausgeht, dazu verhilft, die Bain-Deformation herbei zu führen. Gleichzeitig kann auch gezeigt werden, wie die Zwillingsbildung die Energie eines Keimes entscheidend reduziert (Sinclair und Mohamed 1978). 7.4. Kinetik der Martensitumwandlung Wenn Keime einmal gebildet sind, dann erfolgt das Wachstum der Martensitphase athermisch mit großer Geschwindigkeit. Die Ausbreitung erfolgt dabei sowohl in Richtung der Kante als auch normal dazu, was die Dicke der Platten vergrößert. Sehr oft wird ein anfängliches Wachstum hauptsächlich in Kantenrichtung beobachtet, mit einer anschließenden Verdickung, wenn die Platte an ein Hindernis wie z.B. eine Korngrenze gestoßen ist (In Martensiten mit höherem C-Gehalt verzwillingte „Mittelrippe“, umgeben von weniger regelmäßig im Verlauf des Dickenwachstums aufgebautem Gebiet mit Versetzungen).
Die Bewegung der Martensit-Phasengrenze ist konservativ (ohne Einbau von zusätzlichen Atomen) durch Kombination der homogenen Verzerrung mit der Bewegung von Versetzungen und/oder Zwillingsgrenzen, welche die komplementäre Scherung vermitteln. In linsenförmigen oder spitz zulaufenden Platten ist die Phasengrenze nicht
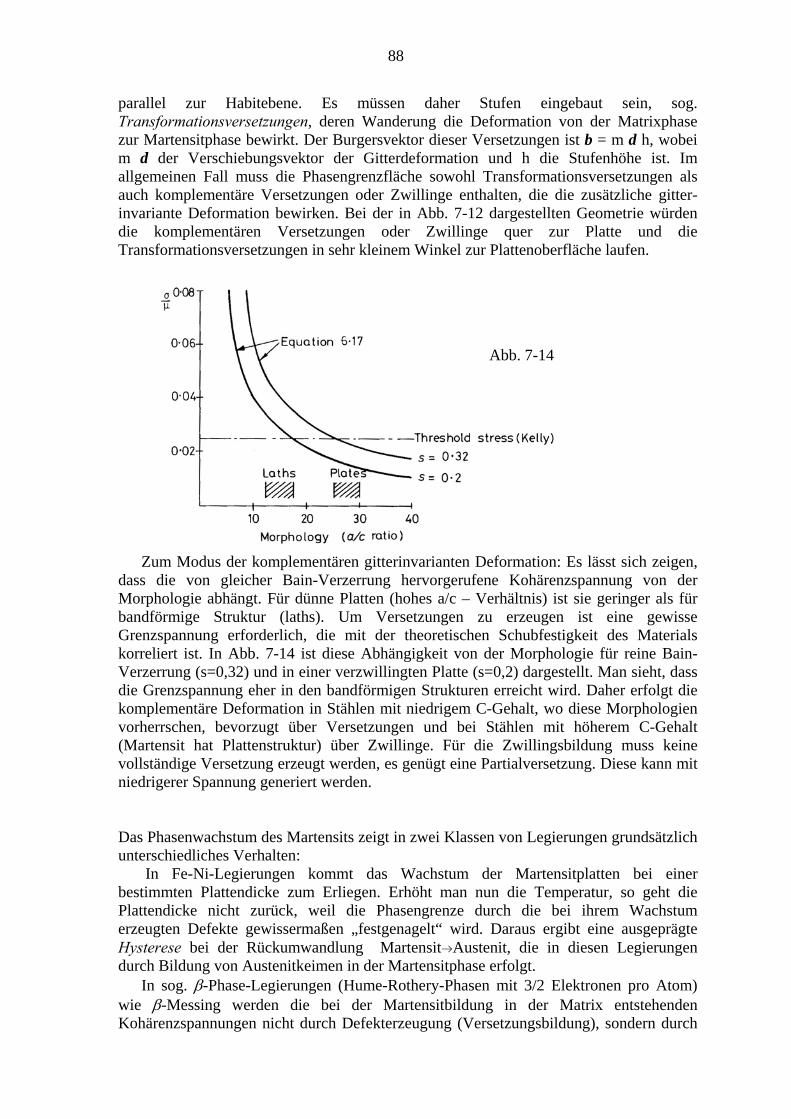
88
parallel zur Habitebene. Es müssen daher Stufen eingebaut sein, sog. Transformationsversetzungen, deren Wanderung die Deformation von der Matrixphase zur Martensitphase bewirkt. Der Burgersvektor dieser Versetzungen ist b = m d h, wobei m d der Verschiebungsvektor der Gitterdeformation und h die Stufenhöhe ist. Im allgemeinen Fall muss die Phasengrenzfläche sowohl Transformationsversetzungen als auch komplementäre Versetzungen oder Zwillinge enthalten, die die zusätzliche gitter-invariante Deformation bewirken. Bei der in Abb. 7-12 dargestellten Geometrie würden die komplementären Versetzungen oder Zwillinge quer zur Platte und die Transformationsversetzungen in sehr kleinem Winkel zur Plattenoberfläche laufen.
Abb. 7-14
Zum Modus der komplementären gitterinvarianten Deformation: Es lässt sich zeigen, dass die von gleicher Bain-Verzerrung hervorgerufene Kohärenzspannung von der Morphologie abhängt. Für dünne Platten (hohes a/c – Verhältnis) ist sie geringer als für bandförmige Struktur (laths). Um Versetzungen zu erzeugen ist eine gewisse Grenzspannung erforderlich, die mit der theoretischen Schubfestigkeit des Materials korreliert ist. In Abb. 7-14 ist diese Abhängigkeit von der Morphologie für reine Bain-Verzerrung (s=0,32) und in einer verzwillingten Platte (s=0,2) dargestellt. Man sieht, dass die Grenzspannung eher in den bandförmigen Strukturen erreicht wird. Daher erfolgt die komplementäre Deformation in Stählen mit niedrigem C-Gehalt, wo diese Morphologien vorherrschen, bevorzugt über Versetzungen und bei Stählen mit höherem C-Gehalt (Martensit hat Plattenstruktur) über Zwillinge. Für die Zwillingsbildung muss keine vollständige Versetzung erzeugt werden, es genügt eine Partialversetzung. Diese kann mit niedrigerer Spannung generiert werden. Das Phasenwachstum des Martensits zeigt in zwei Klassen von Legierungen grundsätzlich unterschiedliches Verhalten:
In Fe-Ni-Legierungen kommt das Wachstum der Martensitplatten bei einer bestimmten Plattendicke zum Erliegen. Erhöht man nun die Temperatur, so geht die Plattendicke nicht zurück, weil die Phasengrenze durch die bei ihrem Wachstum erzeugten Defekte gewissermaßen „festgenagelt“ wird. Daraus ergibt eine ausgeprägte Hysterese bei der Rückumwandlung Martensit→Austenit, die in diesen Legierungen durch Bildung von Austenitkeimen in der Martensitphase erfolgt. In sog. β-Phase-Legierungen (Hume-Rothery-Phasen mit 3/2 Elektronen pro Atom) wie β-Messing werden die bei der Martensitbildung in der Matrix entstehenden Kohärenzspannungen nicht durch Defekterzeugung (Versetzungsbildung), sondern durch
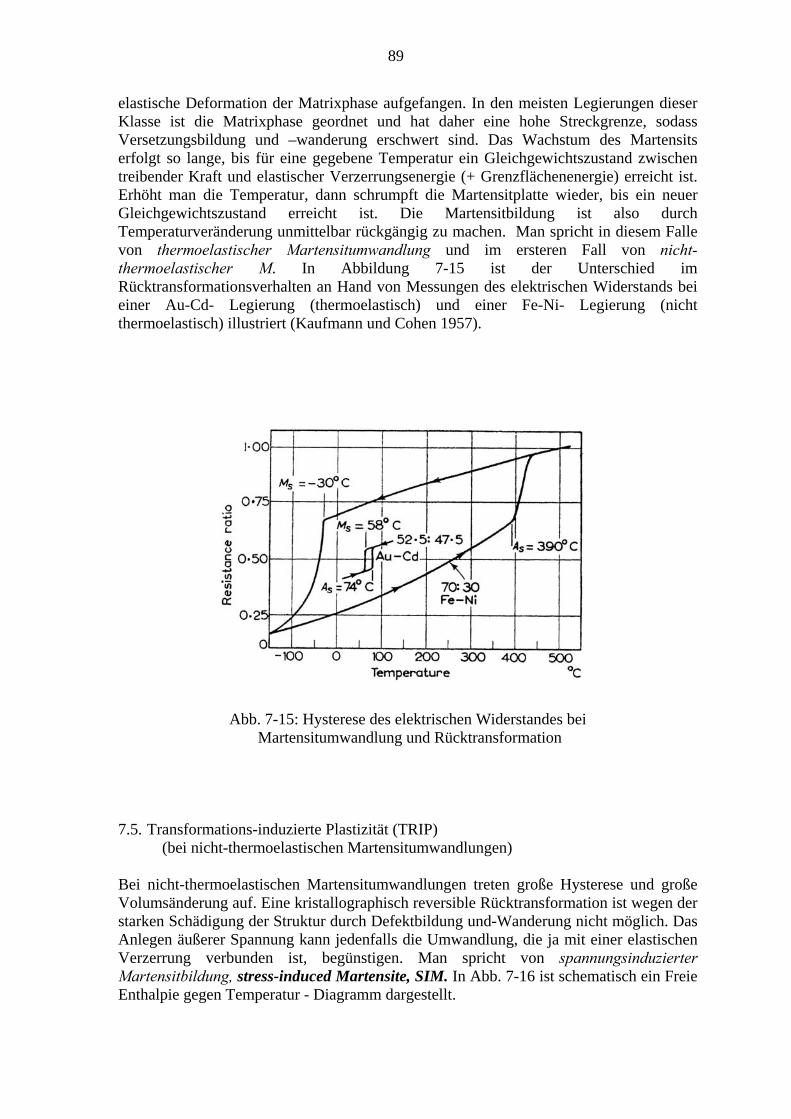
89
elastische Deformation der Matrixphase aufgefangen. In den meisten Legierungen dieser Klasse ist die Matrixphase geordnet und hat daher eine hohe Streckgrenze, sodass Versetzungsbildung und –wanderung erschwert sind. Das Wachstum des Martensits erfolgt so lange, bis für eine gegebene Temperatur ein Gleichgewichtszustand zwischen treibender Kraft und elastischer Verzerrungsenergie (+ Grenzflächenenergie) erreicht ist. Erhöht man die Temperatur, dann schrumpft die Martensitplatte wieder, bis ein neuer Gleichgewichtszustand erreicht ist. Die Martensitbildung ist also durch Temperaturveränderung unmittelbar rückgängig zu machen. Man spricht in diesem Falle von thermoelastischer Martensitumwandlung und im ersteren Fall von nicht-thermoelastischer M. In Abbildung 7-15 ist der Unterschied im Rücktransformationsverhalten an Hand von Messungen des elektrischen Widerstands bei einer Au-Cd- Legierung (thermoelastisch) und einer Fe-Ni- Legierung (nicht thermoelastisch) illustriert (Kaufmann und Cohen 1957).
Abb. 7-15: Hysterese des elektrischen Widerstandes bei Martensitumwandlung und Rücktransformation
7.5. Transformations-induzierte Plastizität (TRIP) (bei nicht-thermoelastischen Martensitumwandlungen) Bei nicht-thermoelastischen Martensitumwandlungen treten große Hysterese und große Volumsänderung auf. Eine kristallographisch reversible Rücktransformation ist wegen der starken Schädigung der Struktur durch Defektbildung und-Wanderung nicht möglich. Das Anlegen äußerer Spannung kann jedenfalls die Umwandlung, die ja mit einer elastischen Verzerrung verbunden ist, begünstigen. Man spricht von spannungsinduzierter Martensitbildung, stress-induced Martensite, SIM. In Abb. 7-16 ist schematisch ein Freie Enthalpie gegen Temperatur - Diagramm dargestellt.
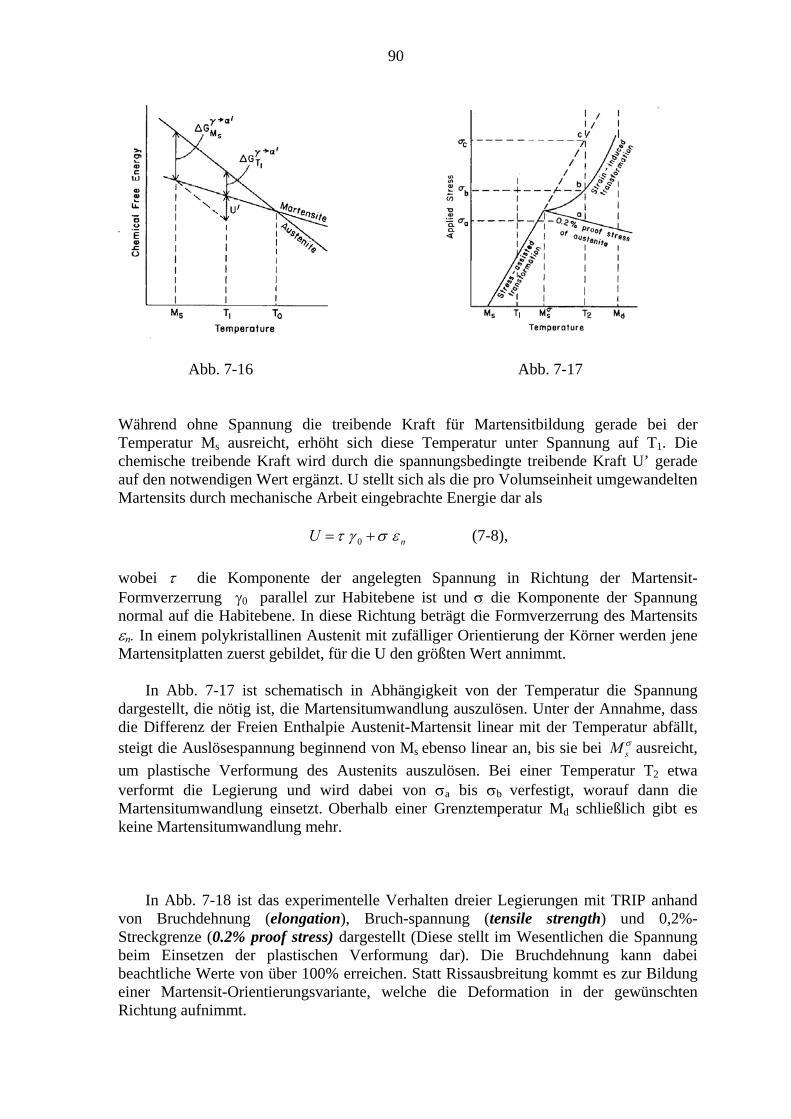
90
Abb. 7-16 Abb. 7-17 Während ohne Spannung die treibende Kraft für Martensitbildung gerade bei der Temperatur Ms ausreicht, erhöht sich diese Temperatur unter Spannung auf T1. Die chemische treibende Kraft wird durch die spannungsbedingte treibende Kraft U’ gerade auf den notwendigen Wert ergänzt. U stellt sich als die pro Volumseinheit umgewandelten Martensits durch mechanische Arbeit eingebrachte Energie dar als
nU εσγτ += 0 (7-8), wobei τ die Komponente der angelegten Spannung in Richtung der Martensit-Formverzerrung γ0 parallel zur Habitebene ist und σ die Komponente der Spannung normal auf die Habitebene. In diese Richtung beträgt die Formverzerrung des Martensits εn. In einem polykristallinen Austenit mit zufälliger Orientierung der Körner werden jene Martensitplatten zuerst gebildet, für die U den größten Wert annimmt.
In Abb. 7-17 ist schematisch in Abhängigkeit von der Temperatur die Spannung dargestellt, die nötig ist, die Martensitumwandlung auszulösen. Unter der Annahme, dass die Differenz der Freien Enthalpie Austenit-Martensit linear mit der Temperatur abfällt, steigt die Auslösespannung beginnend von Ms ebenso linear an, bis sie bei ausreicht, um plastische Verformung des Austenits auszulösen. Bei einer Temperatur T
σsM
2 etwa verformt die Legierung und wird dabei von σa bis σb verfestigt, worauf dann die Martensitumwandlung einsetzt. Oberhalb einer Grenztemperatur Md schließlich gibt es keine Martensitumwandlung mehr.
In Abb. 7-18 ist das experimentelle Verhalten dreier Legierungen mit TRIP anhand
von Bruchdehnung (elongation), Bruch-spannung (tensile strength) und 0,2%-Streckgrenze (0.2% proof stress) dargestellt (Diese stellt im Wesentlichen die Spannung beim Einsetzen der plastischen Verformung dar). Die Bruchdehnung kann dabei beachtliche Werte von über 100% erreichen. Statt Rissausbreitung kommt es zur Bildung einer Martensit-Orientierungsvariante, welche die Deformation in der gewünschten Richtung aufnimmt.
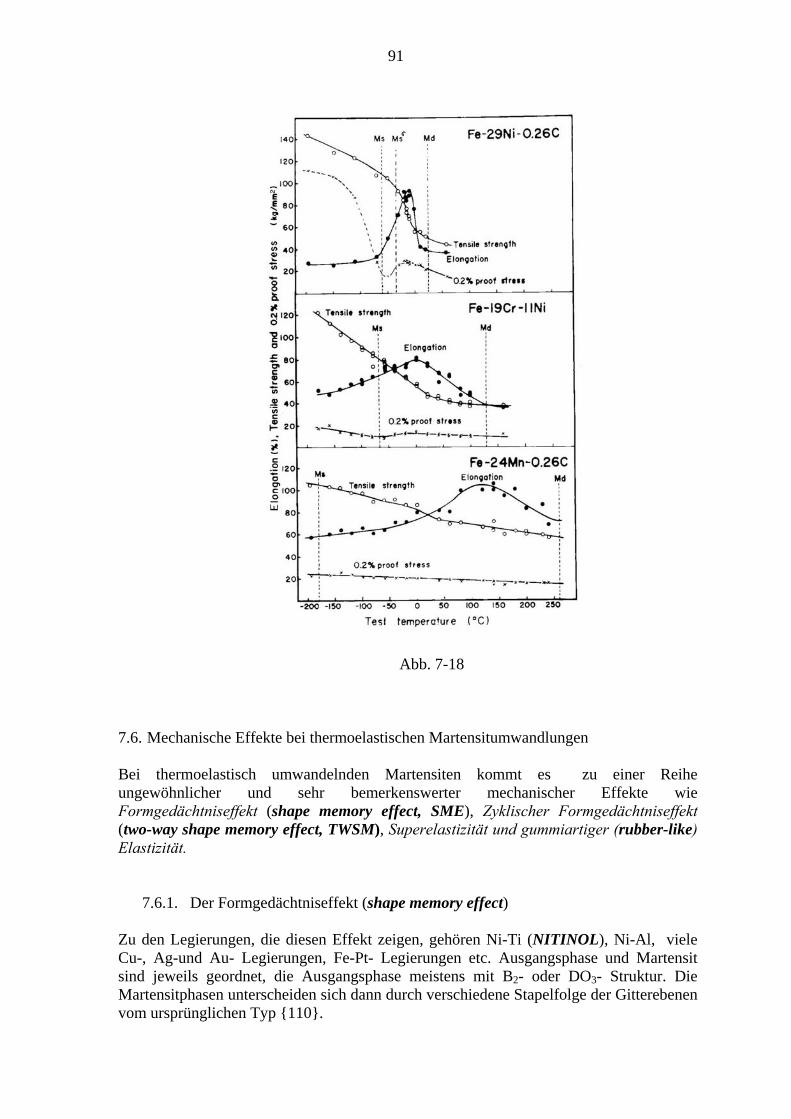
91
Abb. 7-18
7.6. Mechanische Effekte bei thermoelastischen Martensitumwandlungen Bei thermoelastisch umwandelnden Martensiten kommt es zu einer Reihe ungewöhnlicher und sehr bemerkenswerter mechanischer Effekte wie Formgedächtniseffekt (shape memory effect, SME), Zyklischer Formgedächtniseffekt (two-way shape memory effect, TWSM), Superelastizität und gummiartiger (rubber-like) Elastizität.
7.6.1. Der Formgedächtniseffekt (shape memory effect) Zu den Legierungen, die diesen Effekt zeigen, gehören Ni-Ti (NITINOL), Ni-Al, viele Cu-, Ag-und Au- Legierungen, Fe-Pt- Legierungen etc. Ausgangsphase und Martensit sind jeweils geordnet, die Ausgangsphase meistens mit B2- oder DO3- Struktur. Die Martensitphasen unterscheiden sich dann durch verschiedene Stapelfolge der Gitterebenen vom ursprünglichen Typ {110}.
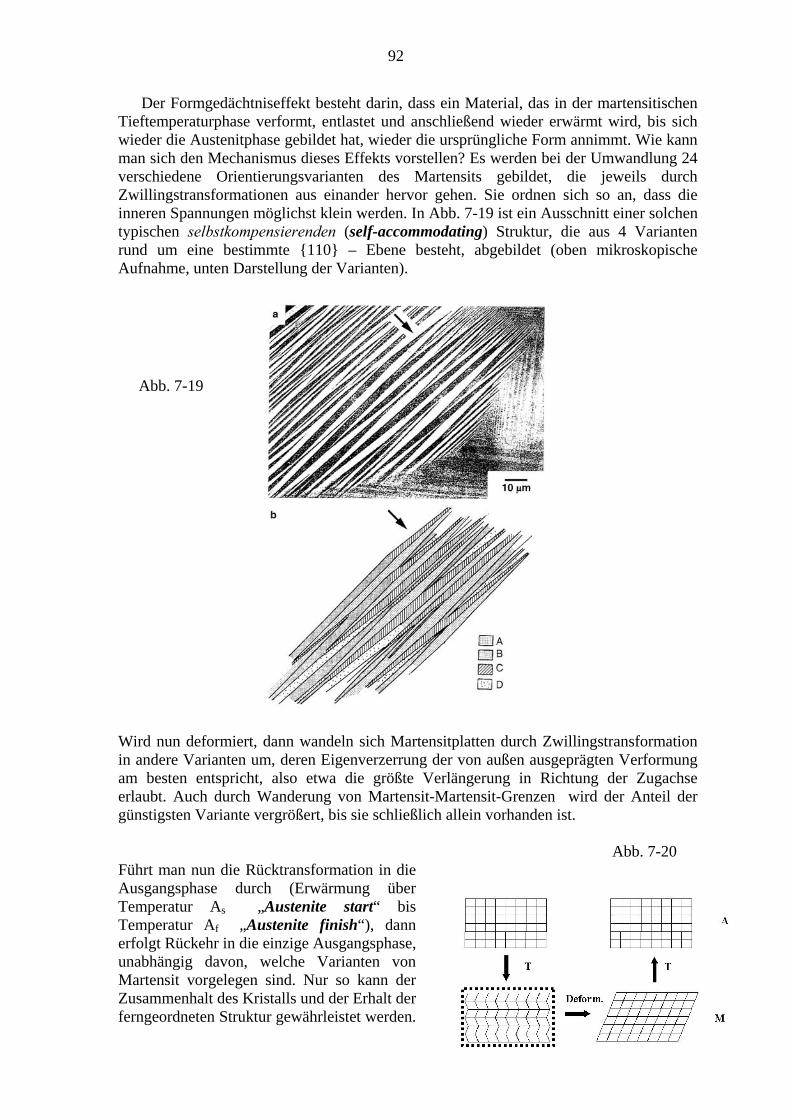
92
Der Formgedächtniseffekt besteht darin, dass ein Material, das in der martensitischen Tieftemperaturphase verformt, entlastet und anschließend wieder erwärmt wird, bis sich wieder die Austenitphase gebildet hat, wieder die ursprüngliche Form annimmt. Wie kann man sich den Mechanismus dieses Effekts vorstellen? Es werden bei der Umwandlung 24 verschiedene Orientierungsvarianten des Martensits gebildet, die jeweils durch Zwillingstransformationen aus einander hervor gehen. Sie ordnen sich so an, dass die inneren Spannungen möglichst klein werden. In Abb. 7-19 ist ein Ausschnitt einer solchen typischen selbstkompensierenden (self-accommodating) Struktur, die aus 4 Varianten rund um eine bestimmte {110} – Ebene besteht, abgebildet (oben mikroskopische Aufnahme, unten Darstellung der Varianten).
Abb. 7-19
Wird nun deformiert, dann wandeln sich Martensitplatten durch Zwillingstransformation in andere Varianten um, deren Eigenverzerrung der von außen ausgeprägten Verformung am besten entspricht, also etwa die größte Verlängerung in Richtung der Zugachse erlaubt. Auch durch Wanderung von Martensit-Martensit-Grenzen wird der Anteil der günstigsten Variante vergrößert, bis sie schließlich allein vorhanden ist. Abb. 7-20 Führt man nun die Rücktransformation in die Ausgangsphase durch (Erwärmung über Temperatur As „Austenite start“ bis Temperatur Af „Austenite finish“), dann erfolgt Rückehr in die einzige Ausgangsphase, unabhängig davon, welche Varianten von Martensit vorgelegen sind. Nur so kann der Zusammenhalt des Kristalls und der Erhalt der ferngeordneten Struktur gewährleistet werden.
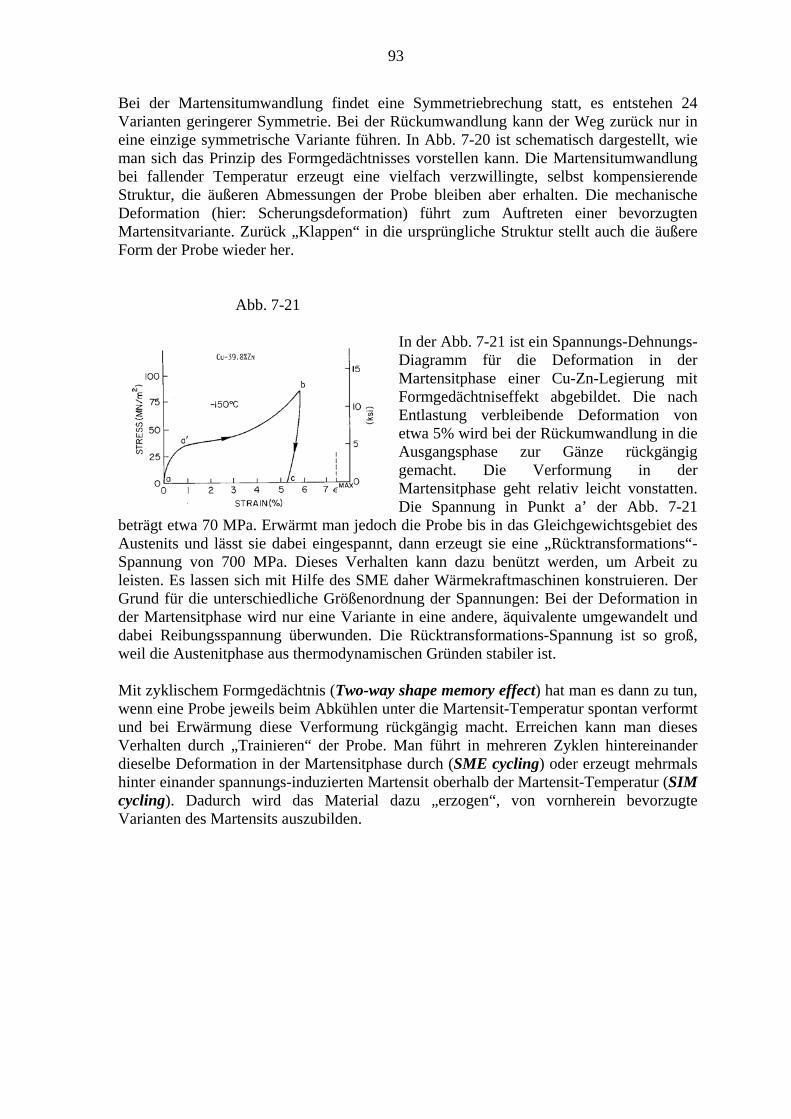
93
Bei der Martensitumwandlung findet eine Symmetriebrechung statt, es entstehen 24 Varianten geringerer Symmetrie. Bei der Rückumwandlung kann der Weg zurück nur in eine einzige symmetrische Variante führen. In Abb. 7-20 ist schematisch dargestellt, wie man sich das Prinzip des Formgedächtnisses vorstellen kann. Die Martensitumwandlung bei fallender Temperatur erzeugt eine vielfach verzwillingte, selbst kompensierende Struktur, die äußeren Abmessungen der Probe bleiben aber erhalten. Die mechanische Deformation (hier: Scherungsdeformation) führt zum Auftreten einer bevorzugten Martensitvariante. Zurück „Klappen“ in die ursprüngliche Struktur stellt auch die äußere Form der Probe wieder her. Abb. 7-21
In der Abb. 7-21 ist ein Spannungs-Dehnungs-Diagramm für die Deformation in der Martensitphase einer Cu-Zn-Legierung mit Formgedächtniseffekt abgebildet. Die nach Entlastung verbleibende Deformation von etwa 5% wird bei der Rückumwandlung in die Ausgangsphase zur Gänze rückgängig gemacht. Die Verformung in der Martensitphase geht relativ leicht vonstatten. Die Spannung in Punkt a’ der Abb. 7-21
beträgt etwa 70 MPa. Erwärmt man jedoch die Probe bis in das Gleichgewichtsgebiet des Austenits und lässt sie dabei eingespannt, dann erzeugt sie eine „Rücktransformations“-Spannung von 700 MPa. Dieses Verhalten kann dazu benützt werden, um Arbeit zu leisten. Es lassen sich mit Hilfe des SME daher Wärmekraftmaschinen konstruieren. Der Grund für die unterschiedliche Größenordnung der Spannungen: Bei der Deformation in der Martensitphase wird nur eine Variante in eine andere, äquivalente umgewandelt und dabei Reibungsspannung überwunden. Die Rücktransformations-Spannung ist so groß, weil die Austenitphase aus thermodynamischen Gründen stabiler ist. Mit zyklischem Formgedächtnis (Two-way shape memory effect) hat man es dann zu tun, wenn eine Probe jeweils beim Abkühlen unter die Martensit-Temperatur spontan verformt und bei Erwärmung diese Verformung rückgängig macht. Erreichen kann man dieses Verhalten durch „Trainieren“ der Probe. Man führt in mehreren Zyklen hintereinander dieselbe Deformation in der Martensitphase durch (SME cycling) oder erzeugt mehrmals hinter einander spannungs-induzierten Martensit oberhalb der Martensit-Temperatur (SIM cycling). Dadurch wird das Material dazu „erzogen“, von vornherein bevorzugte Varianten des Martensits auszubilden.
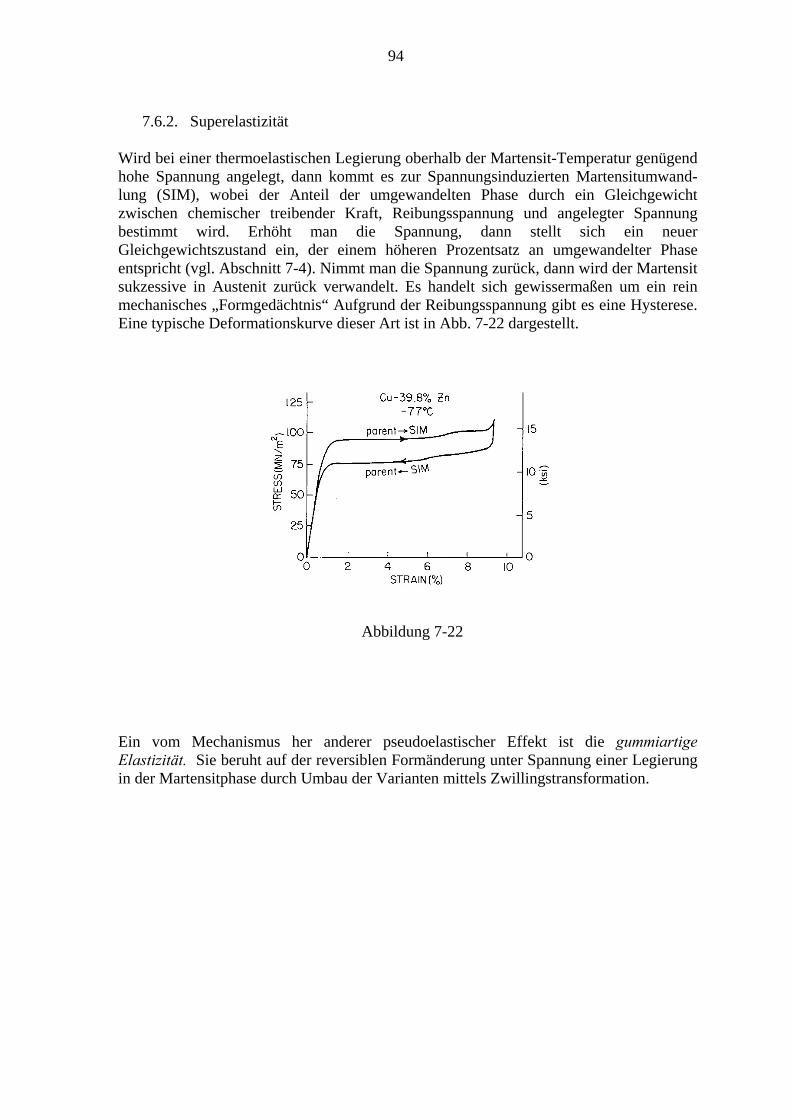
94
7.6.2. Superelastizität
Wird bei einer thermoelastischen Legierung oberhalb der Martensit-Temperatur genügend hohe Spannung angelegt, dann kommt es zur Spannungsinduzierten Martensitumwand-lung (SIM), wobei der Anteil der umgewandelten Phase durch ein Gleichgewicht zwischen chemischer treibender Kraft, Reibungsspannung und angelegter Spannung bestimmt wird. Erhöht man die Spannung, dann stellt sich ein neuer Gleichgewichtszustand ein, der einem höheren Prozentsatz an umgewandelter Phase entspricht (vgl. Abschnitt 7-4). Nimmt man die Spannung zurück, dann wird der Martensit sukzessive in Austenit zurück verwandelt. Es handelt sich gewissermaßen um ein rein mechanisches „Formgedächtnis“ Aufgrund der Reibungsspannung gibt es eine Hysterese. Eine typische Deformationskurve dieser Art ist in Abb. 7-22 dargestellt.
Abbildung 7-22 Ein vom Mechanismus her anderer pseudoelastischer Effekt ist die gummiartige Elastizität. Sie beruht auf der reversiblen Formänderung unter Spannung einer Legierung in der Martensitphase durch Umbau der Varianten mittels Zwillingstransformation.
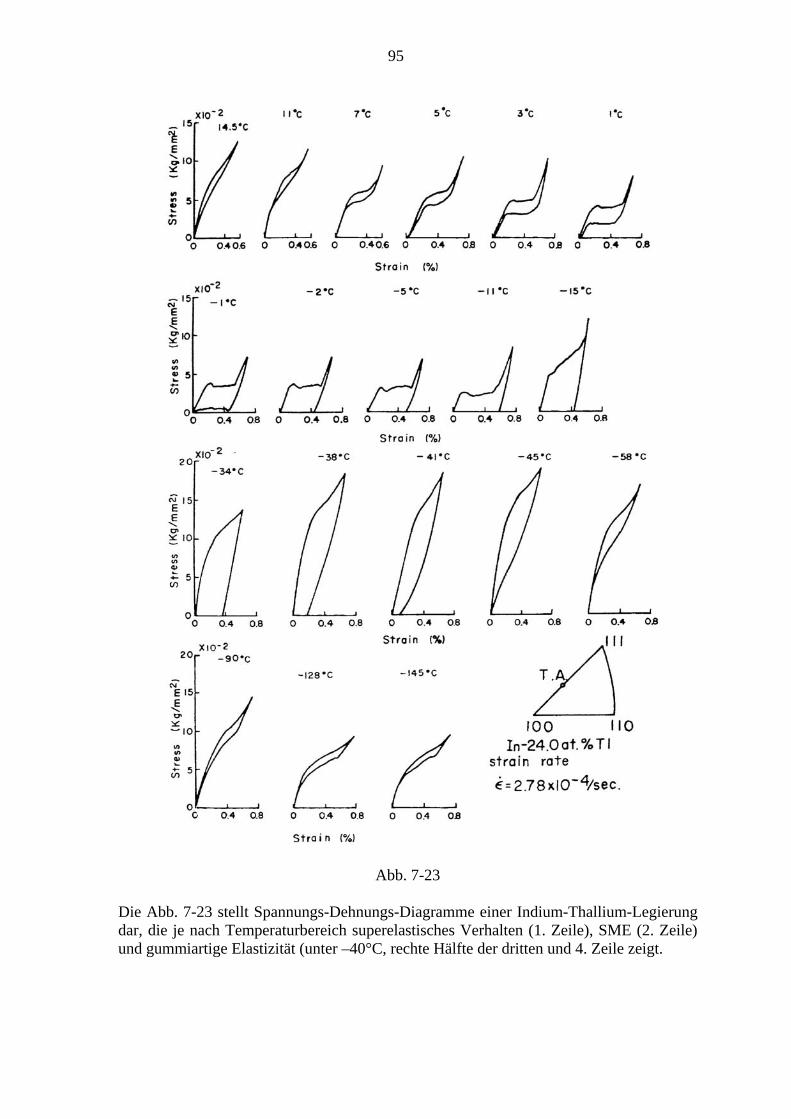
95
Abb. 7-23
Die Abb. 7-23 stellt Spannungs-Dehnungs-Diagramme einer Indium-Thallium-Legierung dar, die je nach Temperaturbereich superelastisches Verhalten (1. Zeile), SME (2. Zeile) und gummiartige Elastizität (unter –40°C, rechte Hälfte der dritten und 4. Zeile zeigt.

Literatur zur Vorlesung „Phasenübergänge in Festkörpern – Phänomenologie und Thermodynamik H. Böhm, „Einführung in die Metallkunde“, BI-Hochschultaschenbuch 196, BI-Wissenschaftsverlag, Mannheim 1992 Enthält leichtfassliche Einführungen in einige Themen der Vorlesung. Als Bettlektüre geeignet R. W. Cahn, P. Haaasen (Eds.), „Physical Metallurgy“ , Part I/II (diverse Kapitel), North-Holland, Amsterdam 1983 Umfangreiche Monographiensammlung über physikalische Metallkunde mit diversen Beiträgen über Phasenübergänge J.W. Christian, „The Theory of Transformations in Metals and Alloys“ Part I/II, Pergamon, Amsterdam 2002 Umfangreiches, auf Vollständigkeit bedachtes Kompendium, Lebenswerk von J.W.C., 2. Teil vor kurzem posthum erschienen, als Bettlektüre nicht geeignet P. Haasen (Ed.) Series „Materials Science and Technology“, Vol. 5 „Phase Transformations in Materials“, VCH, Weinheim 1991 Sehr wertvolle Artikelsammlung von führenden Autoritäten der jeweiligen Themen G. Kostorz (Ed.) “Phase Transformations in Materials”., Wiley-VCH, Weinheim 2001 Im wesentlichen auf neuen Stand gebrachte Ausgabe des obigen Werkes, tw. inhaltsgleich D.A. Porter, K. Easterling „Phase Transformations in Metals and Alloys“, Chapman and Hall, London 1991 Viel verwendetes, gut lesbares und praxisorientiertes Lehrbuch W. Pfeiler (Ed.), „Alloy Physics. A Comprehensive Reference“, Alloy Physics, Wiley-VCH, Weinheim-New York, 2007. Ca. 1000-seitiges Handbuch der Legierungsphysik mit Behandlung aller relevanten Themen. Enthält auch Kapitel über Phasenübergänge und Diffusion. Weiters: M. Hillert, „Phase Equilibria, Phase Diagrams and Phase Transformations”, Cambridge University Press 1998 Detaillierte Behandlung der Thermodynamik der Phasengleichgewichte Diverse Tagungsberichte, z.B. J. M. Howe, D. E. Laughlin, J. K. Lee, U. Dahmen and W. A. Soffa (Eds.) „Solid-solid phase transformations in inorganic materials 2005“, Volume I/II, Proceedings of a Conference at Phoenix, Ariz.,TMS Publications, Warrendale, Pennsylvania 2005, “Stand der Technik” M. Koiwa, K. Otsuka, T. Miyazaki (Eds.), „Solid-Solid Phase Transformations“, Kyoto 1999, The Japan Institute of Metals Proceedings Vol. 12, Part I/II





























