Embed Size (px)
Citation preview

| André Filipe Almeida da Cunha
qwertyuiopasdfghjklçzxcvbnmq
wertyuiopasdfghjklçzxcvbnmqwe
rtyuiopasdfghjklçzxcvbnmqwerty
uiopasdfghjklçzxcvbnmqwertyui
opasdfghjklçzxcvbnmqwertyuiop
asdfghjklçzxcvbnmqwertyuiopas
dfghjklçzxcvbnmqwertyuiopasdf
ghjklçzxcvbnmqwertyuiopasdfgh
jklçzxcvbnmqwertyuiopasdfghjkl
çzxcvbnmqwertyuiopasdfghjklçz
xcvbnmqwertyuiopasdfghjklçzxc
vbnmqwertyuiopasdfghjklçzxcvb
nmqwertyuiopasdfghjklçzxcvbn
mqwertyuiopasdfghjklçzxcvbnm
qwertyuiopasdfghjklçzxcvbnmq
wertyuiopasdfghjklçzxcvbnmqwe
rtyuiopasdfghjklçzxcvbnmrtyuio
CASE
REPORT
Doença de Charcot-Marie-Tooth Nefropatia de IgA
Instituto de Ciências Biomédicas Abel Salazar
André Filipe Almeida da Cunha
Mestrado Integrado de Medicina
Ano lectivo 2009/ 2010

CASE REPORT ________________________________________________________________ CMT & NIgA
2
Índice
Resumo/ Summary --------------------------------------------------------------------------------------- 3
Introdução -------------------------------------------------------------------------------------------------- 4
o Doença de Charcot-Marie-Tooth ----------------------------------------------------------- 4
o Nefropatia IgA ---------------------------------------------------------------------------------- 5
Epidemiologia ------------------------------------------------------------------------ 5
Fisiopatologia ------------------------------------------------------------------------- 5
Morfologia Renal -------------------------------------------------------------------- 6
Clínica ---------------------------------------------------------------------------------- 7
Tratamento --------------------------------------------------------------------------- 8
Caso Clínico ------------------------------------------------------------------------------------------------ 9
o Ecografia Abdominal ------------------------------------------------------------------------- 10
o Ecografia Renal -------------------------------------------------------------------------------- 10
o Estudo Genético ------------------------------------------------------------------------------ 10
o Biopsia Renal ---------------------------------------------------------------------------------- 10
o Curso Clínico ----------------------------------------------------------------------------------- 11
Discussão ------------------------------------------------------------------------------------------------- 11
Abreviaturas ---------------------------------------------------------------------------------------------- 13
Agradecimentos ----------------------------------------------------------------------------------------- 14
Bibliografia ----------------------------------------------------------------------------------------------- 14

CASE REPORT ________________________________________________________________ CMT & NIgA
3
---------------------------------------------------------------------------------------------------------- Case Report
Doença de Charcot-Marie-Tooth e Nefropatia de IgA
Charcot-Marie-Tooth disease and IgA Nephropathy
-------------------------------------------------------------------------------------------------------------------
Cunha, André Filipe Almeida da
Instituto de Ciências Biomédicas Abel Salazar - Mestrado Integrado de Medicina
Junho de 2010
Resumo
A doença de Charcot-Marie-Tooth (CMT) engloba um conjunto de neuropatias hereditárias
causadas por mutações dos genes que codificam as proteínas da mielina periférica provocando
defeitos na sua estrutura, manutenção e formação. As características clínicas e patológicas
destas variantes são semelhantes, e incluem uma atrofia muscular lenta e progressiva, com
alterações da sensibilidade dos membros.
A nefropatia por IgA (NIgA) é a forma mais comum de glomerulonefrite primária. Tem
um pico de incidência na terceira década de vida, é mais comum nos homens, nos Caucasianos
e nos Asiáticos. Geralmente é idiopática, contudo pode associar-se a patologia sistémica ou
apresentar carácter heredo-familiar. Esta doença é mediada por complexos imunes e definida
pela presença de depósitos glomerulares de IgA aos quais geralmente se associam depósitos
de C3 e por vezes de IgG. Apresenta grande variedade de lesões histopatológicas, havendo
sempre expansão mesangial. Nos casos graves pode haver proliferação difusa, crescentes
celulares, inflamação intersticial e áreas de glomeruloesclerose focal. Normalmente os doentes
apresentam-se com hipertensão, hematúria e proteinúria, podendo evoluir para insuficiência
renal crónica terminal.
Apresenta-se um homem com doença de CMT desde os 20 anos, portador de mutação
não amiloidogénica da transtirretina (TTR), microhematúria, proteinúria de 300 mg/dL e
função renal normal há 10 anos. A biopsia renal revelou nefropatia de IgA com padrão
mesangiopático, sem depósitos de amilóide.
A associação de CMT e nefropatia é rara estando descritos casos de
glomeruloesclerose focal e nefropatia fibrilar. A associação entre esta neuropatia e NIgA nunca
foi relatada. Será abordada esta relação e seus mecanismos fisiopatológicos.
Palavras-chave: Charcot-Marie-Tooth. Nefropatia IgA. Gene.
Summary
The Charcot-Marie-Tooth´s (CMT) disease comprises a group of hereditary
neuropathies caused by mutations of genes encoding proteins of peripheral myelin causing

CASE REPORT ________________________________________________________________ CMT & NIgA
4
defects in its structure, maintenance and formation. The clinical and pathological features of
these variants are similar and include a slowly progressive muscular atrophy, with changes in
sensitivity of members.
The IgA nephropathy (NIgA) is the most common form of primary glomerulonephritis.
Has a peak incidence in the third decade of life, is more common in men, Caucasians and
Asians. It is usually idiopathic, but may be associated with systemic pathology or present
Hereditary-familial character. This is a disease mediated by immune complexes, defined by the
presence of glomerular IgA deposits which are usually associated with deposits of C3 and some
times of IgG. It a great variety of histopathological lesions, but there is always mesangial
expansion. In severe cases there may be diffuse proliferation, cellular crescents, interstitial
inflammation and areas of focal glomerulosclerosis. Typically patients present with
hypertension, hematuria and proteinuria and may progress to end-stage renal failure.
The author presents a man with CMT disease since 20 years, suffering from non
amyloidogenic transthyretin (TTR) mutation, microhematuria, proteinuria of 300 mg / dL and
normal renal function 10 years ago. The renal biopsy revealed IgA nephropathy with standard
mesangiophatic without amyloid deposits.
The association of CMT and nephropathy is rare and few cases are described cases
with focal glomerulosclerosis, and fibrillary nephropathy. The association between this
neuropathy and NIgA has not been reported so far. In this presentation it will be addressed
this relationship and its causes.
Key words: Charcot-Marie-Tooth disease. IgA Nephropathy. Gene.
Introdução
A neuropatia de Charcot-Marie-
Tooth (CMT), compreende um grupo
heterogéneo de doenças hereditárias do
sistema nervoso periférico. A transmissão é
frequentemente autossómica dominante,
contudo também pode ser autossómica
recessiva ou ligada ao X. Trata-se da
neuropatia sensitivo-motora primária
hereditária mais comum, com uma
incidência estimada de 1/2500 pessoas1.
As características clínicas e
patológicas incluem uma atrofia muscular
lenta e progressiva, com fraqueza e perda
de sensibilidade dos pés e pernas seguido
pelo envolvimento das mãos, ocorre ainda
uma diminuição dos reflexos tendinosos.
Os sintomas e sinais comuns estão
relacionados com a fraqueza e atrofia
muscular, havendo normalmente uma
história de marcha parética com
“steppage”. As queixas resultantes da
deformidade dos pés (pé cavo ou com
grande curvatura) são devidas à atrofia dos
músculos intrínsecos dos pés. Apesar do
envolvimento sensorial, as queixas
sensitivas são raras2.
O início é mais frequente durante a
primeira ou segunda décadas de vida,
embora possa aparecer mais tarde. A
variação na apresentação clínica é bastante
ampla, desde aqueles que são
praticamente assintomáticos até aqueles
que com atrofia distal grave e com
deformidades marcadas3.

CASE REPORT ________________________________________________________________ CMT & NIgA
5
Actualmente, não existem terapias
que possam prevenir o início ou a
progressão da incapacidade associada a
esta doença.
A neuropatia de CMT é subdividida
em tipo I e em tipo II, tendo por base
critérios electrofisiológicos e
neuropatológicos. Sendo que o tipo I trata-
se de uma neuropatia desmielinizante
caracterizada por uma velocidade de
condução nervosa motora e sensorial baixa
(<38 m/s) e achados patológicos de
neuropatia desmielinizante hipertrófica. O
tipo II trata-se de uma neuropatia axonal
com uma velocidade de condução nervosa
motora e sensorial normal ou ligeiramente
reduzida, com preservação relativa da
bainha de mielina4.
A associação entre nefropatia e
CMT é pontual. Os poucos doentes
documentados com doença renal
apresentaram glomerulosclerose focal e
segmentar e nefropatia fibrilar.
Descrevemos um caso de CMT que cursou
com nefropatia de IgA (NIgA) e revemos a
fisiopatologia a morfologia desta doença.
Os aspectos clínicos relevantes também
serão abordados.
Epidemiologia_______________________
A NIgA é uma das doenças
glomerulares mais comuns em todo o
mundo e responsável por 20 a 40% das
glomerulopatias primárias. Afecta
indivíduos de todas as idades, mas é
diagnosticada mais frequentemente em
crianças e jovens adultos, nomeadamente
do sexo masculino. A frequência desta
doença varia substancialmente nos vários
países do mundo, sendo mais comum nos
Asiáticos e nos Caucasianos5. Porém esta
discrepância pode reflectir apenas uma
diferença de critérios na pertinência de
biópsia renal em casos de microhemtúria6.
Ocasionalmente a NIgA pode afectar vários
membros da mesma família, contudo esta
predisposição familiar é rara. O
envolvimento familiar e diferenças clínicas
entre grupos étnicos sugerem que a NIgA
pode abranger vários subgrupos de
doenças que não são passíveis de distinção
pelas ferramentas clínicas disponíveis. Esta
variabilidade sugere a existência de genes
de susceptibilidade com frequência
variável nessas populações. Os dados
recentes indicam que defeitos de
glicosilação da IgA1 são herdados e
constituem um factor de risco para NIgA
hereditária. O mapeamento dos genes de
susceptibilidade da doença tem-se
revelado difícil não tendo sido até agora
identificada nenhuma mutação específica.
Alguns dos genes candidatos nas doenças
renais potencialmente hereditárias
incluindo a NIgA são aqueles que codificam
a nefrina e a podocina6.
Fisiopatologia_______________________
Apesar da intensa investigação, a
patogénese da NIgA continua obscura.
Numerosas alterações foram descritas,
tanto a nível renal quanto a nível
circulatório com em outros órgãos. Os
achados da imuno-histologia e da
microscopia electrónica suportam a
hipótese de uma doença mediada por
imuno-complexos. Além disso, a
recorrência da nefropatia em
transplantados renais, o aumento dos
níveis de IgA1 e a presença de depósitos de
IgA1, sugerem que estes imuno-complexos
derivam do plasma. Assim vários
mecanismos foram propostos para tentar
esclarecer como se desenvolve a NIgA7.

CASE REPORT ________________________________________________________________ CMT & NIgA
6
A IgA é uma imunoglobulina
secretada nas superfícies mucosas,
alcançando um nível reduzido na circulação
sistémica. Um aumento nos níveis de IgA
circulantes foi documentado em alguns
doentes, principalmente durante os
períodos de exacerbação. No entanto, as
tentativas de correlacionar os níveis destes
complexos com a actividade da doença não
são consistentes. Sabe-se que o nível
plasmático aumentado de IgA, por si só
não é suficiente para produzir a sua
deposição mesangial, daí esta ser atribuída
a moléculas de IgA com características
anormais8.
Existem evidências crescentes de
que a anomalia molecular nos doentes com
NIgA envolve um defeito na galactosilação
da região flexível (hinge region) da IgA1. A
IgA presente nos depósitos glomerulares
pertence à subclasse IgA19,10. Em alguns
doentes, também tem sido descrita uma
alteração da sialilação da IgA110. Estudos
recentes têm mostrado estas anomalias da
Ο-galactosilação das IgA´s tanto a nível
circulatório, quanto a nível dos depósitos
glomerulares. Uma IgA urinária
anormalmente galactosilada tem sido
descrita nos doentes com NIgA, mas não
naqueles com outras formas de doença
glomerular11.
Outra possível contribuição para o
aumento dos níveis de IgA circulantes e sua
deposição glomerular é a sua deficiente
depuração. A redução no catabolismo de
IgA pelos leucócitos na NIgA foi outra das
hipóteses colocadas12. Assim, a taxa de
deposição pode exceder a capacidade de
depuração da IgA glomerular ou há
resistência à depuração mesangial. No
entanto, sabe-se que os depósitos não são
irreversíveis, uma vez podem desaparecer
quando um rim de um dador com NIgA
subclínica é transplantado num receptor
que desenvolveu insuficiência renal de
outra causa.
Geralmente a NIgA não está
associada a grande infiltração celular dos
glomérulos, sugerindo que grande parte da
lesão glomerular é mediada pela expansão
das células glomerulares residentes. Isso
ocorre principalmente através da activação
induzida pelas IgA´s nas células mesangiais,
que lhes confere um fenótipo de células
pró-inflamatórias. Estudos recentes têm
demonstrado um aumento na ligação dos
complexos de IgA às células mesangiais,
particularmente de complexos que contêm
IgA1 pouco galactosilada. Esta ligação está
associada a um aumento da proliferação
mesangial, apoptose e aumento na síntese
de componentes da matriz extracelular. Os
complexos imunes de IgA e a IgA1
mesangial podem activar o complemento,
desencadear uma cascata inflamatória e
potenciar a lesão glomerular. No entanto, a
contribuição da activação do complemento
na lesão glomerular progressiva ainda não
é conhecida13.
Morfologia renal_____________________
Os depósitos de IgA também são
encontrados no mesângio glomerular
numa grande variedade patologias (tabela
1). Contudo, nestas situações não há
habitualmente uma inflamação glomerular
significativa nem disfunção renal não
devendo receber a designação de NIgA.
A imunohistologia é mandatória
para o diagnóstico da NIgA. Esta nefropatia
é uma glomerulonefrite mediada por
imunocomplexos, em que os depósitos de
IgA estão presentes em todos os
glomérulos, até naqueles que se mostram
normais à microscopia. Normalmente estes
depósitos surgem como massas

CASE REPORT ________________________________________________________________ CMT & NIgA
7
confluentes de alta intensidade. A
extensão para áreas para-mesangiais é
frequente, sendo menos comum nas áreas
periféricas. Outras imunoglobulinas (IgG e
IgM) também podem estar presentes,
nomeadamente em áreas de esclerose. O
C3 também costuma estar presente
assumindo praticamente a mesma
localização dos depósitos de IgA. A
ausência depósitos de C4 e C1q é de
extrema importância, uma vez que nos
permite distinguir a NIgA de outras nefrites
proliferativas. O fibrinogénio também pode
ser observado no mesângio14.
A microscopia electrónica permite-
nos verificar a presença de uma
proliferação com aumento da matriz e a
presença de depósitos electron-densos ao
nível mesangial. Estes depósitos também
podem ser observados ao nível dos
capilares glomerulares, tanto a nível
subendotelial como subepitelial15.
Tabela 1 - Patologias associadas a NIgA
Primária
Nefropatia de IgA (doença de Berger)
Sistémica
Púrpura de Henoch-Schönlein
Secundária
Doença hepática crónica Doença de Crohn Doença Celíaca Dermatite herpetiforme Psoríase Episclerite Sindrome de Sjögren Gamopatia monoclonal de IgA Carcinomas (pulmão, pâncreas, secretor de mucina) Micose fungoide Espondilite Anquilosante Doença de Reiter Lepra Toxoplasmose Infecção HIV Doença da membrana fina
Clínica______________________________
O desenvolvimento de hematúria
macroscópica, imediatamente após um
episódio de infecção respiratória superior
sugeriu que a NIgA se pudesse tratar de
uma complicação de tais infecções.
Contudo, nenhum organismo viral ou
bacteriano tem sido associado
consistentemente ao seu desenvolvimento.
Também foi apontado que a NIgA pudesse
surgir devido a uma hipersensibilidade a
antigénios alimentares, nomeadamente ao
glúten. No entanto, na maioria destes
doentes, não há qualquer tipo de evidência
de hipersensibilidade a alimentos. Neste
momento, há consenso de que a NIgA
geralmente se desenvolve em
consequência a uma resposta imune
aberrante com produção de IgA para uma
variedade de antigénios, quer sejam
microrganismos ou de origem alimentar13.
A biopsia renal continua a técnica
standard para o diagnóstico da nefropatia
IgA primária. Assim uma grande variedade
de lesões histológicas podem ser
observadas, tais como glomerulonefrite
proliferativa difusa, esclerose segmentar e
raramente necrose segmentar com
formação de crescentes celulares, que se
manifesta tipicamente como insuficiência
renal rapidamente progressiva14.
As duas manifestações mais
comuns são a hematúria macroscópica
recorrente durante ou imediatamente após
uma infecção do tracto respiratório
superior em crianças ou hematúria
microscópica assintomática observada
mais frequentemente em adultos. Entre os
episódios, o exame da urina é normal.
Quando a hematúria persiste, os níveis de
proteinúria aumentam, no entanto a
síndrome nefrótica é incomum. Raramente
os doentes podem apresentar-se com

CASE REPORT ________________________________________________________________ CMT & NIgA
8
insuficiência renal e um quadro
rapidamente progressivo15.
A NIgA é uma doença benigna para
a maioria dos doentes, ocorrendo
progressão para insuficiência renal em
cerca de 25 a 30% durante um período de
20 a 25 anos. De facto 5 a 30 % dos
doentes entram em remissão completa15.
Os factores de risco para a perda de função
renal incluem a presença de hipertensão,
proteinúria, a ausência de episódios de
hematúria macroscópica, o sexo masculino,
a idade mais avançada por ocasião do
início e alterações graves evidenciadas pela
biopsia renal16,17.
A evidência clínica e laboratorial
sugere grandes semelhanças entre a
púrpura de Henoch-Schönlein e a
nefropatia de IgA. Contudo, a púrpura
pode ser clinicamente diferenciada da
nefropatia, uma vez que esta apresenta
sintomas sistémicos proeminentes, uma
idade mais jovem (<20 anos), infecção
precedente e queixas abdominais18.
Tratamento_________________________
O tratamento ideal da NIgA é um
assunto controverso. O objectivo ideal do
tratamento seria corrigir quaisquer
defeitos da glicosilação da IgA1, modificar a
deposição e remover os depósitos de IgA.
No entanto, as opções actuais de
tratamento são abordagens mais
genéricas, que são comuns para uma
variedade de doenças glomerulares
crónicas. São direccionadas essencialmente
aos processos imunes e inflamatórios
situados tanto a nível glomerular como
tubulointersticial.
Uma vez que a NIgA pode levar a
doença renal terminal, isto tem servido de
estímulo à realização de vários estudos,
nomeadamente utilizando uma dieta sem
glúten, amigdalectomia, fenitoína para
reduzir os níveis séricos de IgA, danazol
para dissolver imunocomplexos,
cromoglicato de sódio para reduzir a
mucosa permeabilidade a antigénios,
antiplaquetários, anticoagulantes e outros.
Com estes tratamentos podemos obter
alguma redução da hematúria, dos níveis
séricos de IgA, e na circulação
imunocomplexos, mas não existem
evidências claras que possam interferir
com o avanço da doença19.
A maioria dos autores recomenda
apenas a observação dos doentes cuja taxa
de filtração glomerular (TGF) ainda não
está reduzida e que apresentam uma
proteinúria <1g/dia20. Em geral
prescrevem-se inibidores de enzima de
conversão da angiotensina (IECA´s) em
virtude do seu efeito protector renal nos
doentes com doença mais grave. Um
estudo clínico, sugeriu uma redução do
risco de insuficiência renal para a
administração prolongada (6meses) de
glicorticoides em altas doses nos doentes
com diminuição da TGF e com proteinúria
>1g21. Estudos clínicos controlados e
randomizados para avaliar a eficácia dos
óleos de peixe e forneceram resultados
divergentes quanto aos seus efeitos na
NIgA22. A eficácia de outros agentes
imunossupressores, como a ciclofosfamida
e o micofenolato de mofetil, ainda não está
comprovada. Em geral reservam-se estes
agentes para aqueles que apresentam uma
síndrome nefrítica ou glomerulonefrite
rapidamente progressiva, nos quais a
biopsia renal revela a formação agressiva
de crescentes e inflamação glomerular
proeminente.
Um esquema simplificado de
abordagem terapêutica para os doentes
com NIgA é apresentado na figura 1.

CASE REPORT ________________________________________________________________ CMT & NIgA
9
Caso Clínico
Um homem de 49 anos,
caucasiano, foi referido a Nefrologia por
microhematúria e proteinúria estimada em
300 mg/dL. Na segunda década de vida
apresentava neuropatia sensitivo-motora
periférica sendo estabelecido o diagnóstico
de CMT. Tinha uma história de infecções
cutâneas crónicas, recorrentes, nos
membros inferiores, no contexto da
expressão clínica da sua doença
neurológica. Como outros antecedentes
pessoais, oligoartrite reactiva do punho há
14 anos e osteomielites de ossos do tarso.
Mencionava o consumo esporádico de
anti-inflamatórios não esteróides e álcool.
Negava hipertensão arterial, edemas,
hematúria macroscópica e púrpura
cutânea. Sem história de exposição a
produtos tóxicos, nem consumo de drogas.
Como história familiar, apresenta vários
casos desta neuropatia na família,
nomeadamente o seu pai, tios paternos e
primos paternos. Porém, nos membros
afectados não existe qualquer tipo de
patologia renal conhecida.
Figura 1- Algoritmo com abordagens terapêuticas para doentes com NIgA13
. A terapêutica depende das características clínicas, dos achados patológicos e da idade do doente. Este algoritmo não inclui o tratamento da glomerulonefrite rapidamente progressiva. * Os doentes com características histológicas muito leves e com proteinúria moderada (UP / C <2,0) devem ser monitorizados, e o tratamento para a MCNS deve ser considerado se os níveis de UP / C permanecem elevados (UP / C >1,0). # Ao realizar tratamento imunossupressor, é necessário ter cuidado com os doentes e estadio 4-5 de DRC. Ω3AG, ácidos gordos ômega-3; IECA, inibidor da enzima conversora da angiotensina; ARA, bloqueador do receptor da angiotensina; TA, tensão arterial; DRC, doença renal crónica; DAM, Doença com alteração mínima; UP / C, razão proteína urinária / creatinina.
CASE REPORT ________________________________________________________________ CMT & NIgA
10
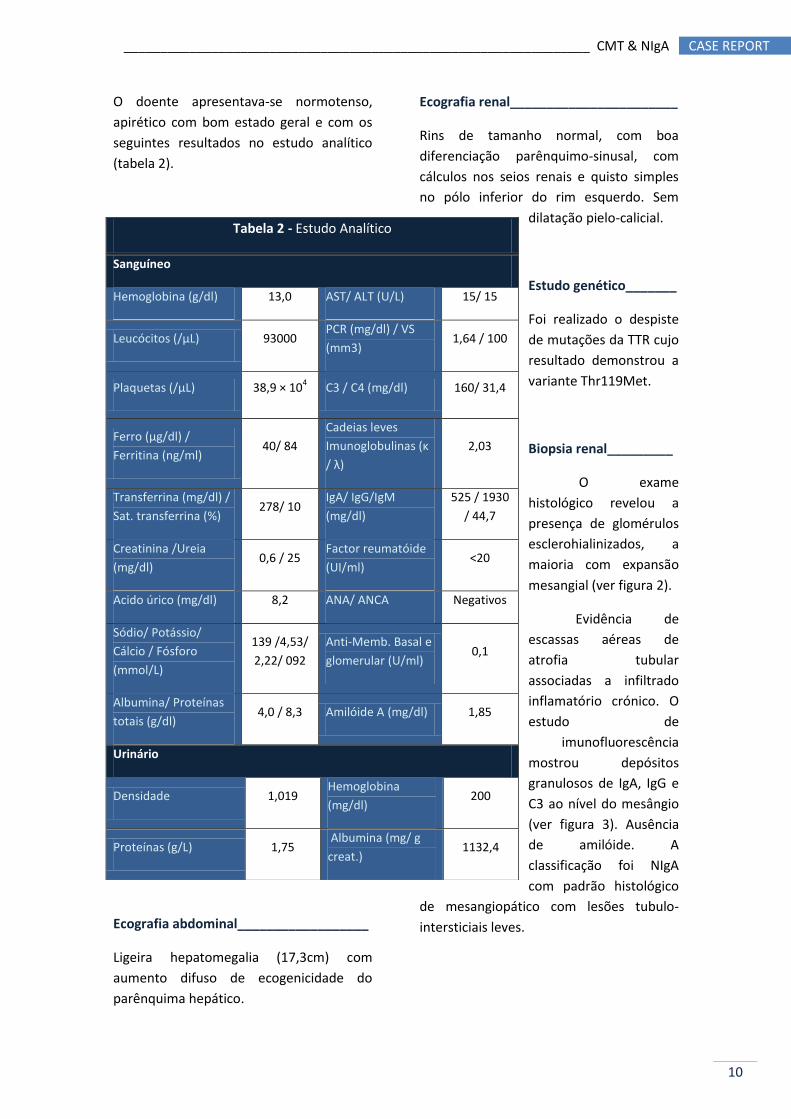
O doente apresentava-se normotenso,
apirético com bom estado geral e com os
seguintes resultados no estudo analítico
(tabela 2).
Ecografia abdominal__________________
Ligeira hepatomegalia (17,3cm) com
aumento difuso de ecogenicidade do
parênquima hepático.
Ecografia renal_______________________
Rins de tamanho normal, com boa
diferenciação parênquimo-sinusal, com
cálculos nos seios renais e quisto simples
no pólo inferior do rim esquerdo. Sem
dilatação pielo-calicial.
Estudo genético_______
Foi realizado o despiste
de mutações da TTR cujo
resultado demonstrou a
variante Thr119Met.
Biopsia renal_________
O exame
histológico revelou a
presença de glomérulos
esclerohialinizados, a
maioria com expansão
mesangial (ver figura 2).
Evidência de
escassas aéreas de
atrofia tubular
associadas a infiltrado
inflamatório crónico. O
estudo de
imunofluorescência
mostrou depósitos
granulosos de IgA, IgG e
C3 ao nível do mesângio
(ver figura 3). Ausência
de amilóide. A
classificação foi NIgA
com padrão histológico
de mesangiopático com lesões tubulo-
intersticiais leves.
Tabela 2 - Estudo Analítico
Sanguíneo
Hemoglobina (g/dl) 13,0 AST/ ALT (U/L) 15/ 15
Leucócitos (/µL) 93000 PCR (mg/dl) / VS
(mm3) 1,64 / 100
Plaquetas (/µL) 38,9 × 104 C3 / C4 (mg/dl) 160/ 31,4
Ferro (µg/dl) /
Ferritina (ng/ml) 40/ 84
Cadeias leves
Imunoglobulinas (κ
/ λ)
2,03
Transferrina (mg/dl) /
Sat. transferrina (%) 278/ 10
IgA/ IgG/IgM
(mg/dl)
525 / 1930
/ 44,7
Creatinina /Ureia
(mg/dl) 0,6 / 25
Factor reumatóide
(UI/ml) <20
Acido úrico (mg/dl) 8,2 ANA/ ANCA Negativos
Sódio/ Potássio/
Cálcio / Fósforo
(mmol/L)
139 /4,53/
2,22/ 092
Anti-Memb. Basal e
glomerular (U/ml) 0,1
Albumina/ Proteínas
totais (g/dl) 4,0 / 8,3 Amilóide A (mg/dl) 1,85
Urinário
Densidade 1,019 Hemoglobina
(mg/dl) 200
Proteínas (g/L) 1,75 Albumina (mg/ g
creat.) 1132,4

CASE REPORT ________________________________________________________________ CMT & NIgA
11
Curso clínico_________________________
Doente mantém estado clínico,
com uma função renal normal. Apresenta
uma proteinúria que oscila entre 0,3 e 1g,
mantendo microhematúria recorrente.
Discussão
As questões que se levantam é se a
coexistência de doença de CMT e NIgA é
apenas fortuita ou se trata de uma
associação nunca descrita. Algumas das
hipóteses que serão colocadas podem
corroborar esta relação.
Como já foi referido anteriormente
a associação da doença de CMT a NIgA
nunca foi documentada. No entanto
existem vários relatos que associam esta
doença e nefropatia, nomeadamente a
casos de glomeruloesclerose focal e
nefropatia fibrilar.
Já em 1967 tinham sido descritos
casos de CMT e doença glomerular. Nesse
artigo foram apresentados 3 doentes com
esta associação. Dois irmãos, sexo
masculino, com proteinúria, hematúria
microscópica e cilindrúria. O terceiro
tratava-se de uma mulher que apresentava
apenas proteinúria persistente. Todos os 3
doentes tinham uma função renal normal.
As suas biopsias revelavam focos de atrofia
tubular com infiltrado linfocitário ou
fibrose, um dos casos além destas
alterações apresentava um espessamento
da membrana glomerular23.
No único caso reportado sobre a
nefropatia fibrilar e a CMT é apresentado
um indivíduo que desenvolveu proteinúria,
hipertensão arterial e insuficiência renal
por volta dos 15anos, em que a sua biopsia
renal mostrava o depósito de microfibrilas
não-amiloidóticas24.
A presença de glomeruloesclerose
focal e a doença de CMT, já foi
documentada em 6 doentes. Num desses
casos é apresentado um jovem admitido
por síndrome nefrótico, em que
rapidamente se desenvolveu uma falência
renal terminal. A biopsia renal revelou a
fusão dos processos podocitários,
espessamento e enrugamento da lâmina
da membrana basal, podócitos
vacuolizados e a presença de inclusões
intracelulares25.
Ambas doenças podem ser
mediadas imunologicamente, logo existe
uma forte possibilidade de existir uma
Figura 2 – Lesões de Esclerose Segmentar. Tricrómio
de Masson (400x)
Figura 3 - Microscopia de imunofluorescência
demonstrando depósitos mesangiais de IgA.

CASE REPORT ________________________________________________________________ CMT & NIgA
12
ligação entre elas. Anticorpos contra a
mielina do nervo periférico têm sido
demonstrados em soros de doentes com
neuropatia periférica (incluindo CMT) e
paraproteinemia IgM. Nestes doentes, a
maior parte apresenta anticorpos
essencialmente da classe IgM, no entanto
alguns apresentam essencialmente
anticorpos IgA e outros apresentam
anticorpos das duas classes. Esta situação
poderia condicionar NIgA por aumento de
produção de IgA, este excesso ir-se-ia
depositar ao nível do mesângio26.
Na tentativa de explicar o
surgimento da NIgA, foi pesquisada a
presença de todas as doenças enunciadas
na tabela 1, as quais se vieram a revelar
negativas neste doente. Nos antecedentes
o doente apresentava uma história de
infecções cutâneas crónicas, recorrentes,
nas áreas de deformidades dos membros
inferiores. Numa tentativa de relacionar
este achado com a nefropatia,
averiguamos que apenas estão descritos
casos em que é associada a Epidermólise
bulhosa (EB) com a NIgA.
A EB é constituída por um grupo de
doenças autossómicas, caracterizadas pela
fragilidade da pele e mucosas. A síndrome
inflamatória crónica, secundária às
infecções cutâneas recorrentes, pode dar
origem a diversas complicações, incluindo
hidroureteronefrose geniturinária,
infecções recorrentes do trato urinário,
amiloidose renal, nefropatia IgA e
glomerulonefrite pós-infecciosa27.
A variante Hallopeau-Siemens da
epidermólise bolhosa distrófica recessiva
(HS-EBDR) é uma dermatose hereditária
grave, associada a uma deficiência de
colagénio VII. Já foram relatados vários
casos de associação desta doença com
NIgA. Um dos quais é m menino de 6 anos
de idade com diagnóstico de HS-EBDR, que
apresentava inicialmente uma
microhematúria que evoluiu para
hematúria macroscópica. A biopsia renal
revelou um aumento das células da matriz
extracelular e do mesângio e
glomeruloesclerose focal e segmentar com
lesões crónicas graves a nível tubulo-
intersticial. A imunofluorescência mostrou
depósitos de IgA no mesângio28.
Na pesquisa da variante Val30Met
da TTR, que condiciona o depósito de
amilóide a nível tecidular, verificou-se que
esta não estava presente neste doente.
Contudo detectou-se a variante Thr119Met
da TTR, não amiloidogénica. Esta
manifestou-se como não patogénica tanto
no sistema nervoso como a nível renal,
uma vez que não foram detectados
depósitos de amilóide nestes dois sistemas.
Pensa-se que a variante Thr119Met119
possa ser protectora em relação aos efeitos
deletérios da variante Val30Met, uma vez
que a evolução da doença parece ser mais
benigna. Pensa-se que este efeito pode
ocorrer, porque a Thr119Met estabiliza a
estrutura da TTR29.
A doença de CMT tipo I pode ser
causada por mutações no gene peripheral
myelin protein, 22 kDa (PMP22), no gene
da protein zero (P0), no gene 2 early
growth response (EGR-2), no gene da
connexin-32 e no gene lipopolysaccharide-
induced TNF- alpha factor (LITAF)30. Muitas
destas mutações podem condicionar
patologia renal, nomeadamente NIgA,
assim vamos tentar elucidar sobre as
consequências destas alterações e tentar
arranjar correlações entre estas duas
doenças.
Expressão de TM4SF10, um
constituinte de uma família de proteínas de
junção celular Claudin/EMP/PMP22,
também está presente na mielina. Foi
estudada a sua presença durante o

CASE REPORT ________________________________________________________________ CMT & NIgA
13
desenvolvimento do rim e na diferenciação
podócitos do rato e descobriu-se que
TM4SF10 se localiza na região mais basal
dos precursores dos podócitos. A
expressão desta proteína atingiu um pico
aos 4 dias pós-parto e foi praticamente
ausente no rim adulto. Estes resultados
sugerem que o TM4SF10 tem funções
durante a diferenciação dos podócitos e
poderá participar na maturação das
junções celulares a partir de cruzamentos
simples aderências para elaborar as fendas
do diafragma. Este tipo de alterações
poderia condicionar uma filtração
deficiente, com diminuição da clearance
das IgA, levando ao seu depósito31.
A P0 é uma glicoproteína
transmembranar que representa o
componente mais abundante da mielina.
Como já foi referido, a doença de CMT
pode estar associada com envolvimento
renal, assim podíamos supor que P0
poderia ser expressa no rim. Um estudo
veio demonstrar isto mesmo, que P0 é
expressa durante a nefrogênese e no rim
maduro, principalmente a nível dos
podócitos. Eles sugerem que as mutações
do gene P0 podem estar envolvidas em
diversas doenças renais. Os podócitos, são
células altamente especializadas que
participam na barreira de filtração
glomerular e na síntese de componentes
da membrana basal glomerular32.
A manutenção da tolerância de
células T aos auto-antigénios é
fundamental para evitar as doenças auto-
imunes. A maneira como as células T auto-
reactivas são mantidas funcionalmente
inactivas, é desconhecida. Foi mostrado
num estudo, que o gene EGR-2 é expresso
em células T CD44 (high) e controla a sua
proliferação e activação. Na ausência de
EGR-2, as células T CD44 (high), mas não as
CD44 (low), tornam-se hiperreactivas e
hiperproliferativas. O aumento das células
T CD4 CD44 (high) activadas, leva ao
desenvolvimento de um síndrome lupuslike
de início tardio caracterizado pelo acúmulo
de interferon-gama e interleucina-17,
perda da tolerância a antígenos nucleares,
infiltração maciça de células T em vários
órgãos e glomerulonefrite33.
Foi documentado que na NIgA, a
lesão dos podócitos pode ser induzida por
citocinas derivadas do mesângio. O factor
de necrose tumoral alfa (TNF-alfa) é um
mediador importante envolvido na
comunicação glomérulo-tubular e no
desenvolvimento da lesão intersticial na
NIgA. Os resultados demonstraram que os
marcadores dos podócitos estão reduzidos
na NIgA. Um estudo in vitro utilizou estes
factores humorais (predominantemente
TNF-alfa e TGF-beta) obtidos a partir de
células mesangiais, e conseguiu alterar a
permeabilidade glomerular provocando
proteinúria e lesões tubulointersticiais
características da IgAN34,35.
Para além das possibilidades aqui
mencionadas, podem haver outros
processos envolvidos. Isto é viável uma vez
que a própria patiofisiologia da NIgA ainda
é um processo obscuro.
Uma vez que o doente se
apresenta normotenso, a função renal
ainda não está diminuída e tem
actualmente uma proteinúria entre 0,3 e 1
g/L, devemos ter uma atitude expectante.
No entanto, podemos prescrever um IECA
em virtude do seu efeito protector renal.
Em conclusão, a doença de CMT
deverá ser mais uma das patologias a
associar à NIgA.
Abreviaturas
CMT - Charcot-Marie-Tooth
NIgA - Nefropatia por IgA
TTR - Transtirretina
EB - Epidermólise bulhosa

CASE REPORT ________________________________________________________________ CMT & NIgA
14
IECA - Inibidor da enzima conversora da
angiotensina
HS-EBDR - Hallopeau-Siemens da
epidermólise bolhosa distrófica recessiva
Agradecimentos
Este espaço é dedicado àqueles
que deram a sua contribuição para que
esta dissertação fosse realizada. A todos
eles deixo aqui o meu agradecimento
sincero.
Agradeço à Prof. Doutora Luísa
Lobato a forma como orientou o meu
trabalho. As notas dominantes da sua
orientação foram a utilidade das suas
recomendações e a cordialidade com que
sempre me recebeu. Estou grato por
ambas e também pela liberdade de acção
que me permitiu, que foi decisiva para que
este trabalho contribuísse para o meu
desenvolvimento pessoal.
Bibliografia
1. Chance PF, Shapiro BE: Charcot-Marie-Tooth disease and related disorders, in B Katirji et al (eds): Neuromuscular Disorders in Clinical Pratice, Philadelphia, Butterworth-Heinemann, pp 513-525, 2002.
2. Online Mendelian Inheritance in Man, OMIM (TM). McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), {17-03-2010}. World Wide Web URL: http://www.ncbi.nlm.nih.gov/omim/
3. Fauci, Anthony; Kasper, Dennis; Longo, Dan ; Braunwald, Eugene; . Hauser, Stephen; Jameson, Larry & Loscalzo, Joseph. Harrison's - PRINCIPLES OF INTERNAL MEDICINE, Seventeenth Edition (2008). The McGraw-Hill Companies.
4. Salisachs, P. Wide spectrum of motor conduction velocity in Charcot-Marie-Tooth disease: an anatomico-physiological interpretation. J. Neurol. Sci. 23: 25-31, 1974
5. Donadio, JV, Grande, JP. IgA nephropathy. N Engl J Med 2002; 347:738
6. de Borst MH, Benigni A, Remuzzi G. Primer: strategies for identifying genes involved in renal disease. Nat Clin Pract Nephrol. 2008 May;4(5):265-76. Epub 2008 Mar 25.
7. H.H. Malluche; B.P. Sawaya; R.M. Hakim; M.H. Sayegh. Clinical Nephrology, Dialysis and Transplantation . Second Edition (2004). Dustri-Verlag
8. Barratt J., Feehally J., Smith A.C.: Pathogenesis of IgA nephropathy. Semin Nephrol 2004; 24:197-217
9. Murakami T, Furuse A, Hattori S, Kobayashi K, Matsuda I 1983 Glomerular IgA1 and IgA2 deposits in IgA nephropathies. Nephron 35: 120-123
10. Tomana M., Novak J., Julian B.A., et al: Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest 1999; 104:73-81.
11. Barratt J., Smith A.C., Feehally J.: The pathogenic role of IgA1 O-linked glycosylation in the pathogenesis of IgA nephropathy [review]. Nephrology (Carlton) 2007; 12:275-284.
12. Sato M; Kinugasa E ; Ideura T; Koshikawa S . Phagocytic activity of polymorphonuclear leucocytes in patients with IgA nephropathy. Clinical Nephrology. 1983 Apr ; 19(4):166-71.
13. Greenberg , Arthur ; Cheung , Alfred K. ; Coffman , Thomas M. ; FalkJ , Ronald J. & Jennette , Charles. Primer on Kidney Diseases, 5th Edition (2009) .Elsevier
14. Hall, CL, Bradley, R, Kerr, A, et al. Clinical value of renal biopsy in

CASE REPORT ________________________________________________________________ CMT & NIgA
15
patients with asymptomatic microscopic hematuria with and without low-grade proteinuria. Clin Nephrol 2004; 62:267.
15. Galla, JH. IgA nephropathy. Kidney Int 1995; 47:377.
16. Coppo R., D’Amico G.: Factors predicting progression of IgA nephropathies. J Nephrol 2005; 18:503-512.
17. D'Amico, G. Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin Nephrol 2004; 24:179.
18. Pillebout E., Thervet E., Hill G., et al: Henoch-Schönlein purpura in adults: Outcome and prognostic factors. J Am Soc Nephrol 2002; 13:1271-1278.
19. Lee G, Glassock RJ 1997 Immunoglobulin A nephropathy. In: Ponticelli C, Glassock JG (eds): Treatment of Primary Glomerulonephritis. Oxford University Press, Oxford, pp 186-217
20. Barratt J., Feehally J.: Treatment of IgA Nephropathy. Kidney Int 2006; 69:1934.
21. Southwest Pediatric Nephrology Study Group 1985 Association of IgA nephropathywith steroidresponsive nephrotic syndrome. Am J Kidney Dis 3: 157-164
22. Hogg R.J., Lee J., Nardelli N., et al: Placebo-controlled clinical trial evaluating omega-3 fatty acids and alternate day prednisone in patients with IgA nephropathy. Clin J Am Soc Nephrol 2006; 1:467-474.
23. G. Lemieux and J. A. Neemeh. Charcot-Marie-Tooth disease and nephritis.Can Med Assoc J. 1967 November 11; 97(20): 1193–1198.
24. Nadal MA; Lago NR, Oliveri LE, de Rosa G, Pierri T. Fribilary glomerulonephritis and Charcot-Marie-Tooth disease. Am J Kidney Dis. 1998 Nov; 32(5): E3.
25. Gherardi R, Belghiti-Deprez D, Hirbec G, Bouche P, WeilB, Lagrue G. Focal glomerulosclerosis associated with Charcot-Marie- Tooth disease. Nephron. 1985;40(3):357-61.
26. Cruz M; Ernerudh J; Olsson T; Höjeberg B; Link H. Occurrence and isotype of antibodies against peripheral nerve myelin in serum from patients with peripheral neuropathy and healthy controls. J Neurol Neurosurg Psychiatry. 1988 Jun;51(6):820-5.
27. Farhi D; Ingen-Housz-Oro S; Ducret F; Rioux-Leclercq N; Cam G; Simon P; Martinez F; Fumeron C; Dubertret L; Blanchet-Bardon C.Recessive dystrophic epidermolysis bullosa (Hallopeau-Siemens) with IgA nephropathy: 4 cases. Ann Dermatol Venereol. 2004 Nov;131(11):963-7.
28. Tammaro F; Calabrese R; Aceto G; Lospalluti L; Garofalo L; Bonifazi E; Piccolo T; Pannarale G; Penza R. End-stage renal disease secondary to IgA nephropathy in recessive dystrophic epidermolysis bullosa: a case report. Pediatr Nephrol. 2008 Jan; 23(1):141-4. Epub 2007 Oct 23.
29. Longo Alves I, Hays MT, Saraiva MJ. Comparative stability and clearance of [Met30]transthyretin and [Met119]transthyretin. Eur J Biochem. 1997 Nov 1; 249(3):662-8
30. Kamholz J; Menichella D; Jani A; Garben J; Lewis RA; Krajewski KM; Lilien J; Scherer SS; Shy ME. Hereditary primary motor sensory neuropathies, including Charcot-Marie-Tooth disease. Brain 2000 Feb; 123 (Pt2): 222-33.
31. Bruggeman LA; Martinka S; Simske JS. Expression of TM4SF10, a Claudin/EMP/PMP22 family cell junction protein, during mouse kidney development and podocyte differentiation. Dev Dyn. 2007 Feb;236(2):596-605.
32. Plaisier ,Emmanuelle; Mougenot , Béatrice; Verpont, Marie; Jouanneau , Chantal; Archelos, Joan; Martini , Rudolf; Kerjaschki, Dontscho; Ronco,

CASE REPORT ________________________________________________________________ CMT & NIgA
16
Pierre.Glomerular Permeability Is Altered by Loss of P0, a Myelin Protein Expressed in Glomerular Epithelial Cells. J Am Soc Nephrol 16: 3350-3356, 2005
33. Zhu B; Symonds AL; Martin JE; Kioussis D; Wraith DC; Li S; Wang P. Early growth response gene 2 (Egr-2) controls the self-tolerance of T cells and prevents the development of lupuslike autoimmune disease. J Exp Med. 2008 Sep 29;205(10):2295-307. Epub 2008 Sep 8
34. Lai KN; Leung JC; Chan LY; Saleem MA; Mathieson PW; Tam KY; Xiao J; Lai FM; Tang SC. Podocyte injury induced by mesangial-derived cytokines in IgA nephropathy. Nephrol Dial Transplant. 2009 Jan;24(1):62-72. Epub 2008 Aug 6.
35. Lee EY, Yang DH, Hwang KY, Hong SY. Is tumor necrosis factor genotype (TNFA2/TNFA2)a genetic prognostic factor of an unfavorable outcome in IgA nephropathy? J Korean Med Sci. 2001 Dec;16(6):751-5.





























