Embed Size (px)
Citation preview
1
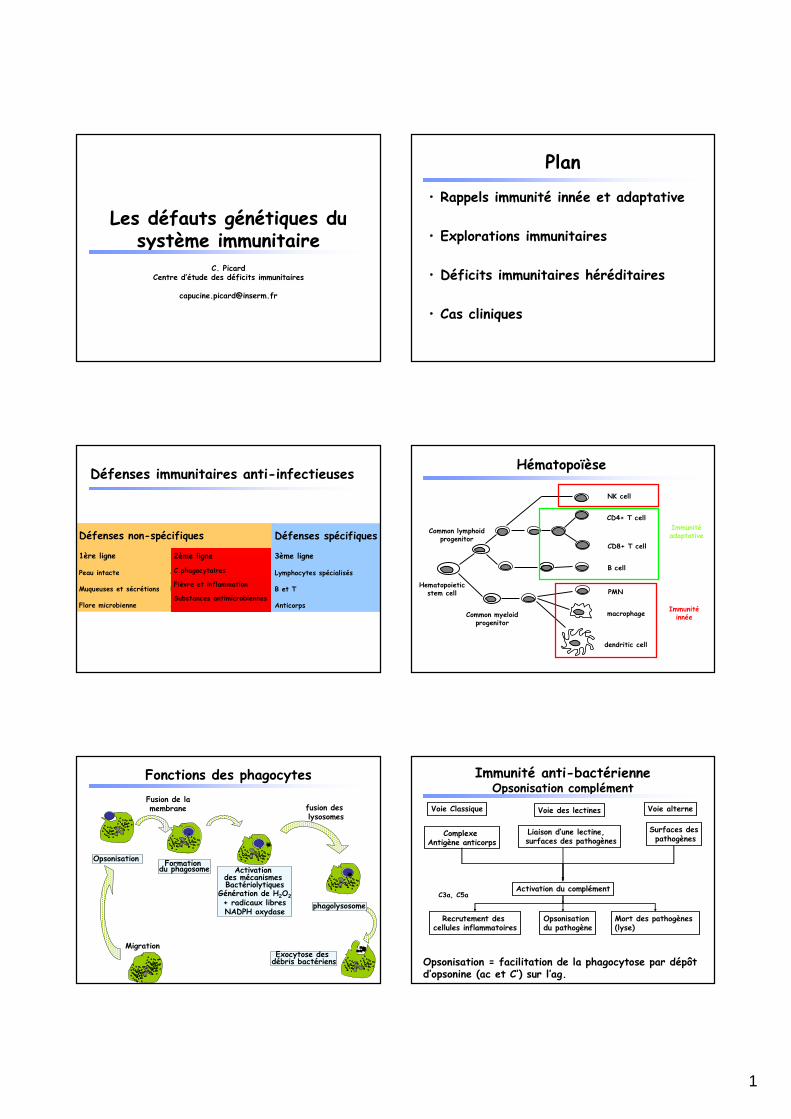
Les défauts génétiques du système immunitaire
C. PicardCentre d’étude des déficits immunitaires
Plan
• Rappels immunité innée et adaptative
• Explorations immunitaires
• Déficits immunitaires héréditaires
• Cas cliniques
Défenses immunitaires anti-infectieuses
Défenses non-spécifiques
1ère ligne 2ème ligne
Peau intacte C. phagocytaires
Muqueuses et sécrétions Fièvre et inflammation
Flore microbienne Substances antimicrobiennes
Défenses spécifiques
3ème ligne
Lymphocytes spécialisés
B et T
Anticorps
2ème ligne
C.phagocytaires
Fièvre et inflammation
Substances antimicrobiennes
CD8+ T cell
CD4+ T cell
NK cell
B cell
Common lymphoidprogenitor
Hematopoieticstem cell
Common myeloidprogenitor
macrophage
dendritic cell
PMN
Hématopoïèse
Immunitéinnée
Immunitéadaptative
Migration
phagolysosome
Exocytose des débris bactériens
Activation des mécanismes Bactériolytiques
Génération de H2O2
+ radicaux libresNADPH oxydase
fusion deslysosomes
OpsonisationFormation
du phagosome
Fusion de la membrane
Fonctions des phagocytes
Voie Classique
Complexe Antigène anticorps
Voie des lectines
Liaison d’une lectine, surfaces des pathogènes
Voie alterne
Surfaces despathogènes
Activation du complément
Recrutement des cellules inflammatoires
Opsonisationdu pathogène
Mort des pathogènes (lyse)
C3a, C5a
Immunité anti-bactérienneOpsonisation complément
Opsonisation = facilitation de la phagocytose par dépôt d’opsonine (ac et C’) sur l’ag.
2
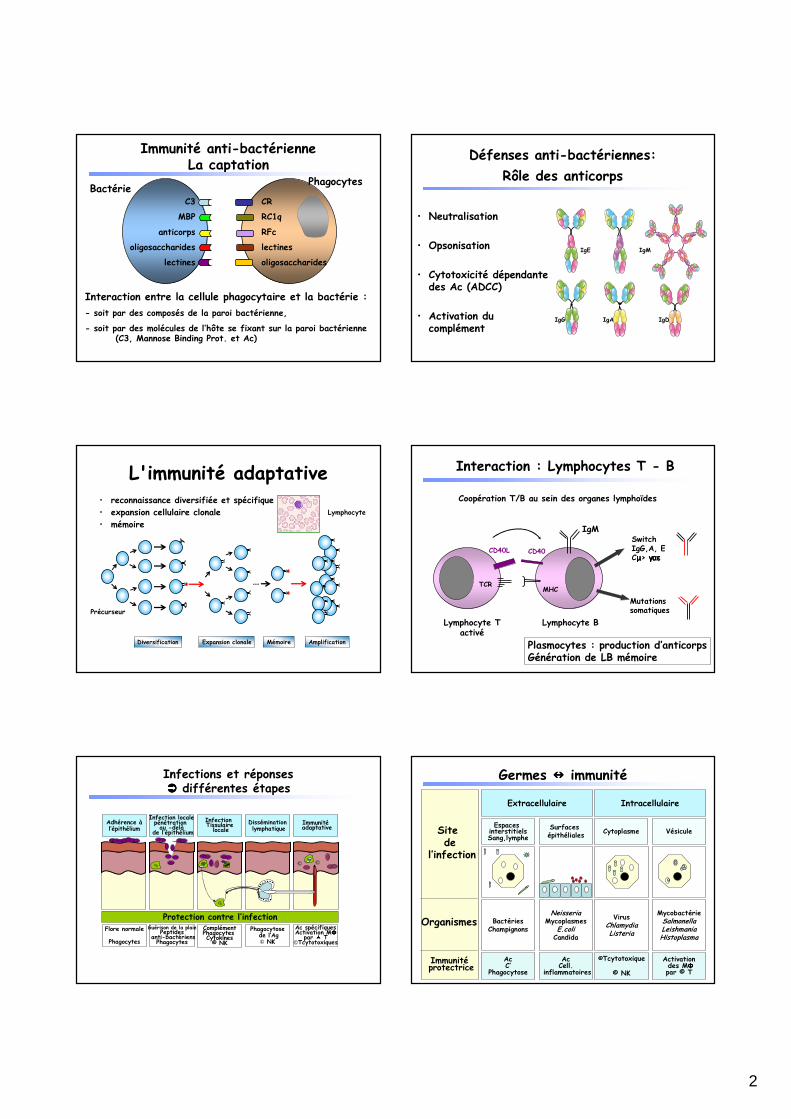
Immunité anti-bactérienneLa captation
BactériePhagocytes
CR
RC1q
RFc
lectines
oligosaccharides
C3
MBP
anticorps
oligosaccharides
lectines
Interaction entre la cellule phagocytaire et la bactérie :
- soit par des composés de la paroi bactérienne,
- soit par des molécules de l’hôte se fixant sur la paroi bactérienne (C3, Mannose Binding Prot. et Ac)
Défenses anti-bactériennes:
Rôle des anticorps
• Neutralisation
• Opsonisation
• Cytotoxicité dépendante des Ac (ADCC)
• Activation du complément
IgMIgE
IgDIgAIgG
Précurseur
Diversification
…
MémoireExpansion clonale Amplification
Lymphocyte
L'immunité adaptative• reconnaissance diversifiée et spécifique
• expansion cellulaire clonale
• mémoire
Coopération T/B au sein des organes lymphoïdes
Lymphocyte Tactivé
Lymphocyte B
CD40L CD40
TCRMHC
SwitchIgG,A, ECµµµµ> γγγγααααεεεε
Mutationssomatiques
IgM
Plasmocytes : production d’anticorpsGénération de LB mémoire
Interaction : Lymphocytes T - B
Infections et réponses ���� différentes étapes
Guérison de la plaiePeptides
anti-bactériensPhagocytes
Infection localepénétration
au -delàde l’épithélium
Flore normale
Phagocytes
Protection contre l’infection
Adhérence àl’épithélium
ComplémentPhagocytes Cytokines
© NK
Infection Tissulaire
locale
Ac spécifiquesActivation MΦΦΦΦ
par ���� T©Tcytotoxiques
© ©
Immunitéadaptative
Phagocytosede l’Ag© NK
Disséminationlymphatique Cytoplasme Vésicule
Intracellulaire
VirusChlamydiaListeria
©Tcytotoxique
© NK
Activationdes MΦΦΦΦpar © T
MycobactérieSalmonellaLeishmaniaHistoplasma
Site de
l’infection
Organismes
Immunitéprotectrice
Extracellulaire
Espaces interstitielsSang,lymphe
Surfaces épithéliales
AcC’
Phagocytose
AcCell.
inflammatoires
BactériesChampignons
NeisseriaMycoplasmes
E.coliCandida
Germes ���� immunité

3
QUAND SUSPECTER ?
• 1) infections récurrentes des voies respiratoires (ORL-poumons) :– Fréquence :
• >6-8 /automne-hiver chez < de 4 ans
• >2-4 /automne-hiver chez > de 4 ans
– OMA > 5 ans
– Présence de lésions de dilatations des bronches (bronchiectasies)
• 2) 1 ou > infections bactériennes sévères: méningite, ostéomyélite…
QUAND SUSPECTER ?
3) Une seule infection opportuniste
4) Mycose digestive persistante
5) Abcès récidivants
6) Diarrhée réfractaire
QUAND SUSPECTER ?
PARFOIS DIFFICILE
� OMA
� Angines
� retard de croissance
AVEC RECIDIVES +++
QUAND SUSPECTER ?
CLINIQUE A MODERER QUAND :
� ORL ISOLE
� CROISSANCE NORMALE
� ASTHME; ATOPIE
� COLLECTIVITE
� INFECTIONS VIRALES
� INFECTIONS URINAIRES
• 1) croissance staturo-pondéral
• 2) examen des aires ganglionnaires, foie et rate
• 3) examen ORL:
– tympans et cavité buccale (amygdales, muguet?),
– présence ou non d’une obstruction nasale.
• 4) examen pulmonaires : râles, crépitants ou sibilants ?
• 5) examen cutané : eczéma,
» cicatrices d’infections anciennes,
» BCG (cicatrice ?)
Examen clinique Explorations immunitaires (1)
Age (mois)
Lymph
ocyte
s x 1
0-
3/m
m3
1) Numération de la formule sanguine :– Anémie ?
– Thrombopénie ?
– Neutropénie ?
– Lymphopénie ?
4
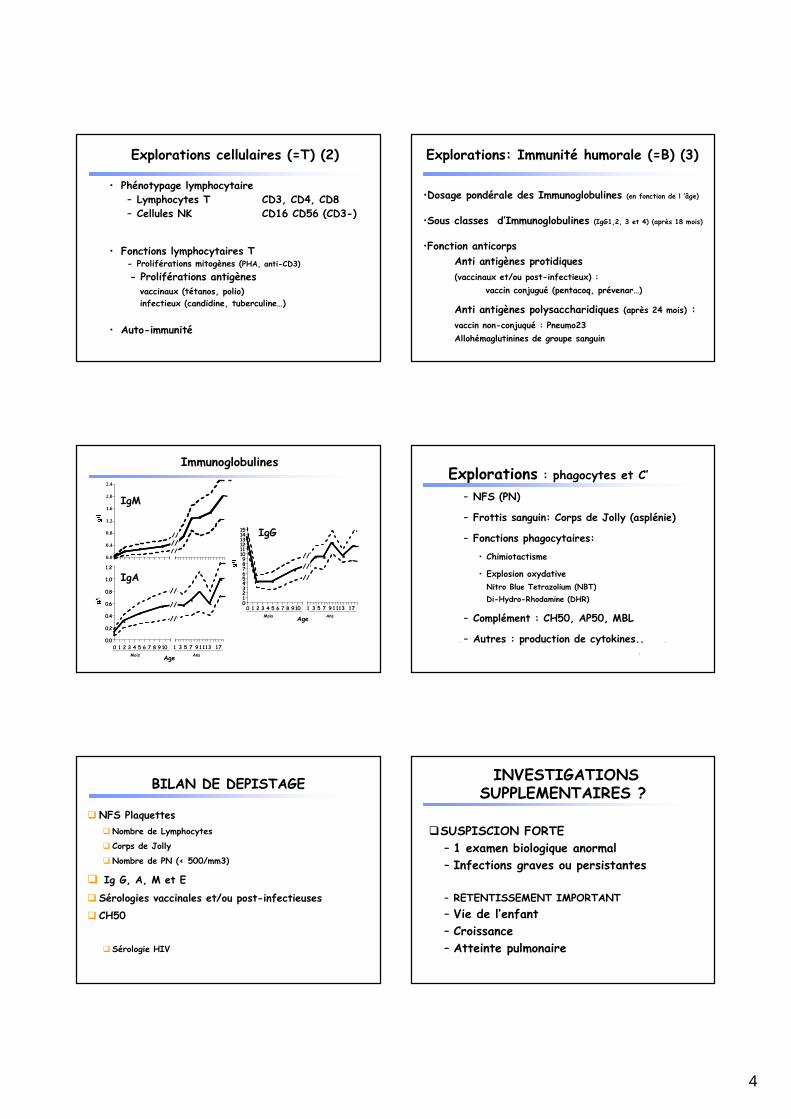
• Phénotypage lymphocytaire– Lymphocytes T CD3, CD4, CD8– Cellules NK CD16 CD56 (CD3-)
• Fonctions lymphocytaires T - Proliférations mitogènes (PHA, anti-CD3)
- Proliférations antigènes vaccinaux (tétanos, polio)
infectieux (candidine, tuberculine…)
• Auto-immunité
Explorations cellulaires (=T) (2)
•Dosage pondérale des Immunoglobulines (en fonction de l ’âge)
•Sous classes d’Immunoglobulines (IgG1,2, 3 et 4) (après 18 mois)
•Fonction anticorps
Anti antigènes protidiques
(vaccinaux et/ou post-infectieux) :
vaccin conjugué (pentacoq, prévenar…)
Anti antigènes polysaccharidiques (après 24 mois) :
vaccin non-conjuqué : Pneumo23
Allohémaglutinines de groupe sanguin
Explorations: Immunité humorale (=B) (3)
Immunoglobulines
0123456789
101112131415
0 1 2 3 4 5 6 7 8 910 1 3 5 7 91113 17
AgeMois Ans
0.0
0.2
0.4
0.6
0.8
1.0
1.2
0 1 2 3 4 5 6 7 8 910 1 3 5 7 91113 17
AgeMois Ans
//
//
//
0.0
0.4
0.8
1.2
1.6
2.0
2.4
//
////
//
//
//
IgM
IgA
IgG
Explorations : phagocytes et C’
– NFS (PN)
– Frottis sanguin: Corps de Jolly (asplénie)
– Fonctions phagocytaires:
• Chimiotactisme
• Explosion oxydative
Nitro Blue Tetrazolium (NBT)
Di-Hydro-Rhodamine (DHR)
– Complément : CH50, AP50, MBL
– Autres : production de cytokines..
BILAN DE DEPISTAGE
� NFS Plaquettes
�Nombre de Lymphocytes
� Corps de Jolly
�Nombre de PN (< 500/mm3)
� Ig G, A, M et E
� Sérologies vaccinales et/ou post-infectieuses
� CH50
� Sérologie HIV
INVESTIGATIONS SUPPLEMENTAIRES ?
�SUSPISCION FORTE
– 1 examen biologique anormal
– Infections graves ou persistantes
– RETENTISSEMENT IMPORTANT
– Vie de l’enfant
– Croissance
– Atteinte pulmonaire

5
Déficits immunitaires héréditaires
• Déficits immunitaires « classiques » :
� > 150 déficits décrits
� ~ 120 gènes
� 1/5000 naissances
� Infections sévères
à germes « opportunistes »
à germes « communs » multiples
(Notarangelo et al. J allergy clin Immunol,2006)
Classification des DIH
• Déficits immunitaires humoraux (B)
• Déficits immunitaires combinés (T et B)
• Déficits de la phagocytose et de l’opsonisation
• Déficits de l’immunité innée
• Déficits de l’homéostasie du système immunitaire
• Pathologies auto-inflammatoires
Déficits de l’immunité humorale
Les PLUS fréquents
� Premiers signes > 6 mois
� Germes : Bactéries PYOGENES
� Signes associés : PULMONAIRES, ORL,
articulaire, retard de croissance +/-
→→→→ Diagnostic :Ig G, A et M; Sérologies vaccinales et/ou post-infectieuses +/- sous classes IgG (>2 ans)
• Agammaglobulinémie liée à l’X (M. Bruton)
• Syndrome hyper IgM (CD40L, autres)
• Déficit en sous-classes d’IgG
• Défaut de production d’ac dirigés contre ag
polysaccharidiques (après 2 ans)
• Déficit en IgA (1/600) : isolé ou associé
Déficits Humoraux
Agammaglobulinémie
• 1/ 380 000
• X- Récessif (BRUTON) , AR (rare)
• Age de révélation 12 mois
• Otites 70%
• Sinusites 59%
• Pneumopathies 62%
• Inf. gastro-intestinales 23%
• Abcès 18%
• Méningo-encéphalites 12%
• Arthrites, Ostéomyélites 10%
Agammaglobulinémie
• Absence de cellule B au phénotypagelymphocytaire (CD19~0)
• Agammaglobulinémie :
– IgG< 2-3g/l, IgA et IgM basses
– Sérologies vaccinales et infectieuses négatives
ProB PréBLymphoïde
T, NK
CD19+CD34+
CD19+CD34-Cµµµµ cyto BTK

6
Syndromes d’hyper IgM
UNGHIGM4<5%AR
CD40HIGM3<5%AR
AIDHIGM250%?AR
CD40LHIGM130%X-linked
1/500 000 naissances
Autre(s) gène(s) impliqués
DDééficits en CD40L et CD40 ficits en CD40L et CD40
• Mode de transmission• Liée au chromosome X (CD40-ligand) : 95 %• Transmission autosomique récessive (CD40) : 5 %
• Défaut de coopération T-B/monocytes– Défaut de l’immunité cellulaire : infections à germes
opportunistes (pneumocystose et cryptosporidies)
– Infections bactériennes
– Défaut humoral : – taux élevé ou normal d’IgM,
– absence d’IgG et d’IgA
– neutropénie
Déficit en CD40L (Syndrome hyperIgM lié à l’X)
Absence d’expression de CD40L sur les Blastes T J7 (conA+IL-2) activés par la PMA+Ionomycine
CD69 CD40 CD40L
CD69 CD40LCD40
Patient
Contrôle
HIGM2: défaut d’AID
• 35 mutations différentes chez 73 patients
• 5 ans au diagnostic (0-53 ans)
• Infections bronchopulmonaires >90% des cas
• Hypertrophie ganglionnaire (75%)
• Autoimmunité (Uvéite, diabète) (25%)
• Arthrite aseptique, Méningite, Peau, adénite, ostéomyélite, Crohn
Déficits de production en Ig
CSH
T
B commutés, mémoires
Pro B Pre B
CLP
B
plasmocytes
IgGIgAIgE
IgM
Déficits en
CD40L et CD40
Déficits en
AID et UNG
Agammaglobulinémie
Déficit de l’immunité cellulaire = déficit immunitaire combiné
Plus grave mais moins fréquent
� Premiers signes< 3 mois (DICS)
� Germes : opportunistes et intra cellulaire
�Signes associés :
�infections pulmonaire et ORL,
�mycose,
�diarrhée,
�retard staturo-pondéral,
�auto-immunité (DIC)
7
Déficit Immunitaire Combiné Sévère (DISC)
Infections opportunistes (Pneumocystose…)
Infections fongiques (Candidose…)
Infections virales (Parainfluenzae, Adv, CMV,VRS…)
Infections bactériennes
BCGite
• Numération Formule Sanguine
lymphocytes +++
< 1500/mm3
• Absence de thymus
• Phénotypage lymphocytaire absence de lymphocyte T
• Hypogammaglobulinémie(hypoIgG, IgA et IgM)
Déficit immunitaire combiné sévère : Explorations
Age (mois) au diagnostic
Lymph
ocyte
s x 1
0-
3/m
m3
DICS (diagnostic)
Formule leucocytaire à interpréter en fonction de l’age
Déficit Immunitaire Combiné Sévère
CD8+ T cell
CD4+ T cell
NK cell
B cell
Common lymphoidprogenitor
Hematopoieticstem cell
Common myeloidprogenitor
macrophage
dendritic cell
PMN
Reticulardysgenesia
ADAdeficiency
SCID T-B+(γγγγc,Jak3)
SCID T-B+NK+IL7RA
SCID T-B-NK+Rag1, Rag2, Artemis
Cumulative probability of survival in SCID patients afterrelated HLA-mismatched HSCT according to year at
grafting
european registry
Déficits immunitaires combinés(immunités cellulaire et humorale touchées)
Nombreuses entités clinico-biologiques
4 exemples :- Déficit en CD40L (syndrome HyperIgM de type 1)
- Défaut d’expression des molécules HLA de classe II
- Syndrome de Wiskott-Aldrich
- Syndrome de Di George
Exploration :
Phénotypage lymphocytaire T, B
Proliférations lymphocytaires T
Exploration humorale (Ig et sérologies)

8
Défaut d’expression des molécules HLA Classe II
•Autosomique récessif•Signes cliniques:
–Infections pulmonaires (pneumocytoses)
–Diarrhée chronique
–Auto-immunité,
–Viroses
•Anomalie génétique d’un des facteurs de transcription (CIITA, RFX-ANK, RFXC ou RFXAP)
•Défaut de coopération T-B/monocytes
•Lymphopénie CD4
•Hypogammaglobulinémie, parfois IgM ↑↑↑↑
•Réponse spécifiques = absentes
– proliférations aux Ags, IDR, – fonctions anticorps
CD19
DR
CD19
DR
Syndrome de Wiskott-Aldrich (XR, 1/250 000)
• Anomalie génétique = WASP (polymérisation de l’actine)
• Signes cliniques: – Eczéma, – Auto-immunité, – Infections, – Hémorragie
• Signes biologiques : – Microplaquettes– Thrombopénie– Immunologique:
• Lymphopénie progressive (CD8), • Proliférations lymphocytaire N ou ↓↓↓↓ , • IgM ↓↓↓↓ IgA ↑↑↑↑ IgG N
Témoin
Patient
Wiskott-Aldrich
Syndrome de Di George (AD)
• Anomalie du développement 3 et 4ème arcs = µdélétion 22q11
• Signes cliniques (variabilité): Dysmorphie,Cardiopathie, Hypocalcémie
– Immunologique →→→→ Trois possibilités :• Absence de thymus complète =Di George complet = DICS• Présence d’un reliquat thymique= Di George partiel = DIC
transitoire• Thymus normal = absence de DI
• Phénotypage lymphocytaire T – CD3+, CD4+, CD8+– Si lymphopénie études des populations T naïves et mémoires – +/- proliférations lymphocytaires T et dosage Ig
Ataxie télangiectasie (AR, 1/40000)
• télangiectasies cutanéomuqueuses
• ataxie cérébelleuse progressive
• Infections pulmonaires,
Pneumothorax
• hypo (IgA, IgG), puis immunité
cellulaire
• Cancer 10-38% :– lymphomes B & T, LA,Tumeurs solides
• diagnostic :↑↑↑↑αααα-fp et cytogénétique
• gène ATM (réparation des cassures double-brins, de recombinaisons au cours de la méïose ou de la maturation
des gènes d‘Ig )
TRAITEMENTS
DEFICITS HUMORAUX :
SUBSTITUTION EN IMMUNOGLOBULINES
DEFICIT CELLULAIRES :
GREFFE DE MO
DEFICIT COMPLEXES :
FONCTION DE LA GRAVITE ET DES POSSIBILITES
DI phagocytose et opsonisation
• Complément :� Méningite et septicémie à germes encapsulés (Pneumo, HI, Mén) :
� Voie classique (C3, C2, C4) : CH50
� Voie alterne du complément (I, H) : AP50
� Méningite à N.meningitidis:
� Complexe d’attaque membranaire (C5b, C6,C7,C8 ou C9) AR
� déficit en properdine (liée à l’X)
• Asplénie :� Méningite et septicémie à germes encapsulés
> Frottis sanguin (corps de Jolly+++) et échographie abdo.

9
Déficits héréditaires de la phagocytose
• Infections bactériennes et/ou fongiques
o Défaut quantitatif
�Neutropénie auto-immunes
�Neutropénies congénitales
o Défaut qualitatif
�Déficit d’adhésion leucocytaire (déficit CD18)
�Granulomatose septique chronique (CGD)
Neutropénies congénitales
� maladies métaboliques : glycogénose 1b
� maladie de Shwachman :
•AR, SDBS,
•Ins. pancréas exocrine, anomalies osseuses
� syndrome de Kostmann :
•AR, HAX1
� neutropénie cyclique :
•AD, ELA2
Déficit d’adhésion leucocytaire
� LFA1/ CD18
� Retard chute du cordon,
� Infections à bactériennes et
fongiques
� Hyperleucocytose (PN+++)
� Absence d’exp CD18/CD11
� Chimiotactisme PN anormal
CD18CD18
LymphosMonopn
LymphosMonopn
Patient Contrôle Migration orientée
Lecture de la distance parcourue
par le front de PN (mm)
PN fMLPPBS
Migration spontanée
Granulomatose septique chronique (CGD)
• Déficit phagocytaire
• Fréquence : 1/200 000
• XR (gp91phox),
• AR (p47phox, p22phox, p67phox)
• Porteur mutation récurrente
p47phox: 1/2000
• Anomalie de l’explosion oxydative
(NADPH)
Granulomatose septique chronique (CGD)
• Pneumopathies 79%
• Abcès 68%
• Adénites suppuratives 53%
• Ostéomyélites 25%
• Septicémies 18%
• Cellulites 5%
• Méningites 4%
• Otites 15%
• Germes : Aspergillus, S.aureus, entérobactéries
(Winkelstein et al. Medicine 2000)
Granulomatose septique chronique (CGD)
• Pneumopathie : Aspergillus (41%)
• Abcès :
� sous-cutané : Staphylococcus spp (27%),
� hépatique : Staphylococcus spp (50%)
� poumon : Aspergillus (23%)
� cérébraux : Aspergillus (58%)
• Adénite suppurative : Staphylococcus spp (26%)
• Ostéomyélite : Serratia (29%), Aspergillus (22%)
• Septicémie : Salmonelle (18%), Burkholderia (12%), Candida (11%)
• Méningite : Candida (20%)
• Aspergillus est responsable 1/3 des DC

10
Biopsie pulmonaire
Histologie : granulome Aspergillus fumigatus
Granulomatose septique chronique (CGD) Explorations : Fonctions
– Explosion oxydative Nitro Blue Tetrazolium (NBT)ou Di-Hydro-Rhodamine (DHR)
n = 74 patients rapportés dans la littérature
39 patients avec infections M. bovis BCG
4 patients avec infections à ME
7 patients avec infections à Mycobacterium spp.
7 patients avec infections àM.tuberculosis +
M. bovis BCG
16 patients avec infections à M. tuberculosis
GSC et infections mycobactériennes Diagnostic DI : Phagocytes et opsonisation
Phagocytes Complément Asplénie
• Germes : Pyogènes Neisseria S.pneumoniaeFungiques S.pn, HI H.Influenzae
• Localisat.: Abcès Méningite Septicémie
Poumons Septicémie
• S.associés : Granulomes Auto-Immu. Cardiopathie
Chute tardive (S. d’Ivemark)
cordon
DIH inmunité innée
• Syndrome hyperIgE (Job ou Buckley)
• Susceptibilité aux bactéries pyogènes– Déficit en IRAK-4
• Susceptibilité Mendélienne aux mycobactéries
Syndrome hyperIgE
• Syndrome de Job ou Buckley
•AD, > 1/ 1 000 000
• gène STAT3
•Rash à la naissance : 75%
•Eczéma récurrent 82%
•Infections cutanées et pneumopathies récurrentes >70%
•Pneumatocèle 87% avec dans 42% des cas Aspergillus
•Infections fungiques récurrentes : 78%
•Hyperlaxité, Anomalies osseuses et dentaires
•Dysmorphie

11
Lung-complications
16/20 patients :
Bronchiectasie: 7 pts Pneumatocèle: 12 pts(Aspergillus (3), P.aeruginosa + Serratia + Stenotrophomonas (1))
Pneumotharax: 1 pts Lobectomy: 4 pts
Syndrome hyperIgE
B.Grimbacher, NEJM 1999
DIH > susceptibilité génétique
DICSM.Bruton
Déficit C’Susceptibilitéà 1 germe
Phagocytes - Reconnaissance :Toll-like récepteurs (TLR)
• 11 chez l’homme
• glycoprotéines membranaires de type 1
•90-115 kD
•Leucin rich repeat (LRR)
•Reconnaissance de motifs microbiens
•Rôle dans l’initiation de la réponse inflammatoire
Défenses anti-infectieuses:Toll-like récepteurs (TLR) Déficits en IRAK4
• Pas d’anomalie de développement
•Infections multiples à bactéries pyogènes précoces (S.pneumoniae et S. aureus) :
•Méningite•Sepsis•Arthrite•Abcès
• Réponse inflammatoire initiale peu importante (neutropénie, fièvre)
• Explorations immunologiques normales (T, B, C’)
12
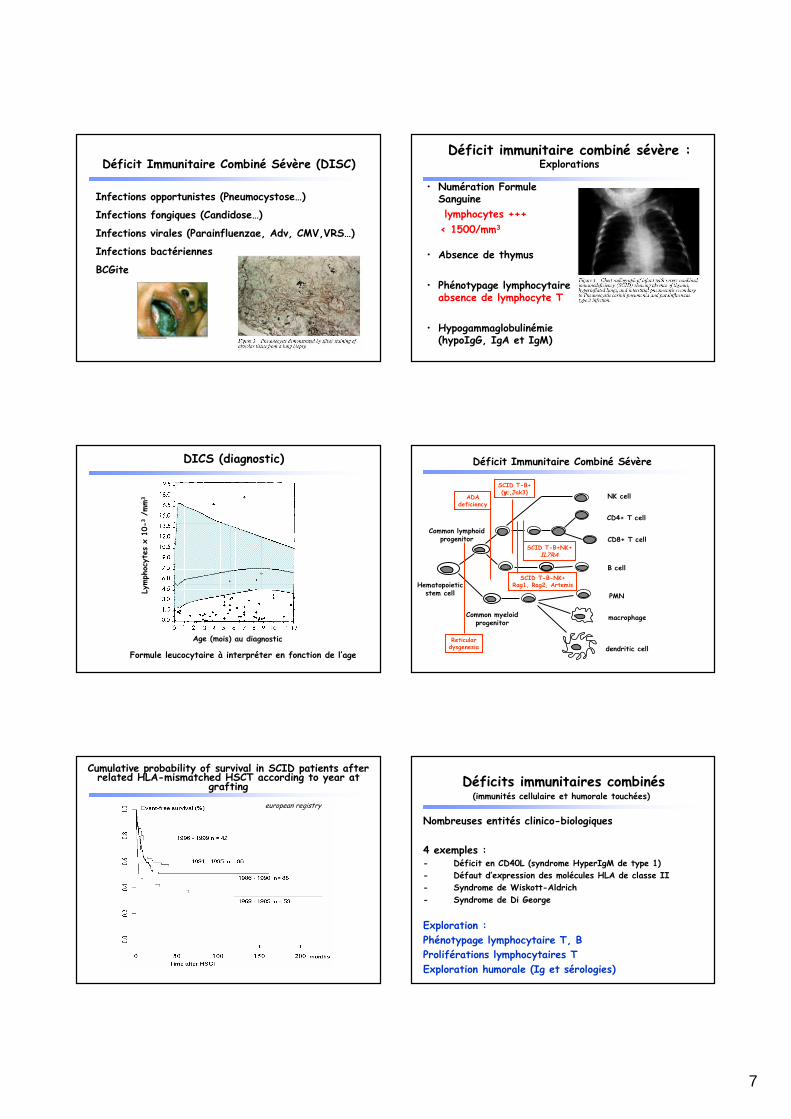
Défaut de réponse aux ligands des TLRs et aux bactéries (sang total)
Défaut de réponse aux ligands des TLRs et à l’IL-1ββββ (sang total)
TNFR-S
MAPK
p p
p50p65
IκκκκBαααα
αααα ββββ
p50p65
Iκκκκ Bαααα
IkBαααα
AP-1
p50p65 AP-1
TCR/BCR
Gene
transcription
NOD
IKKNEMO
RANK/VEGFR3/EDAR
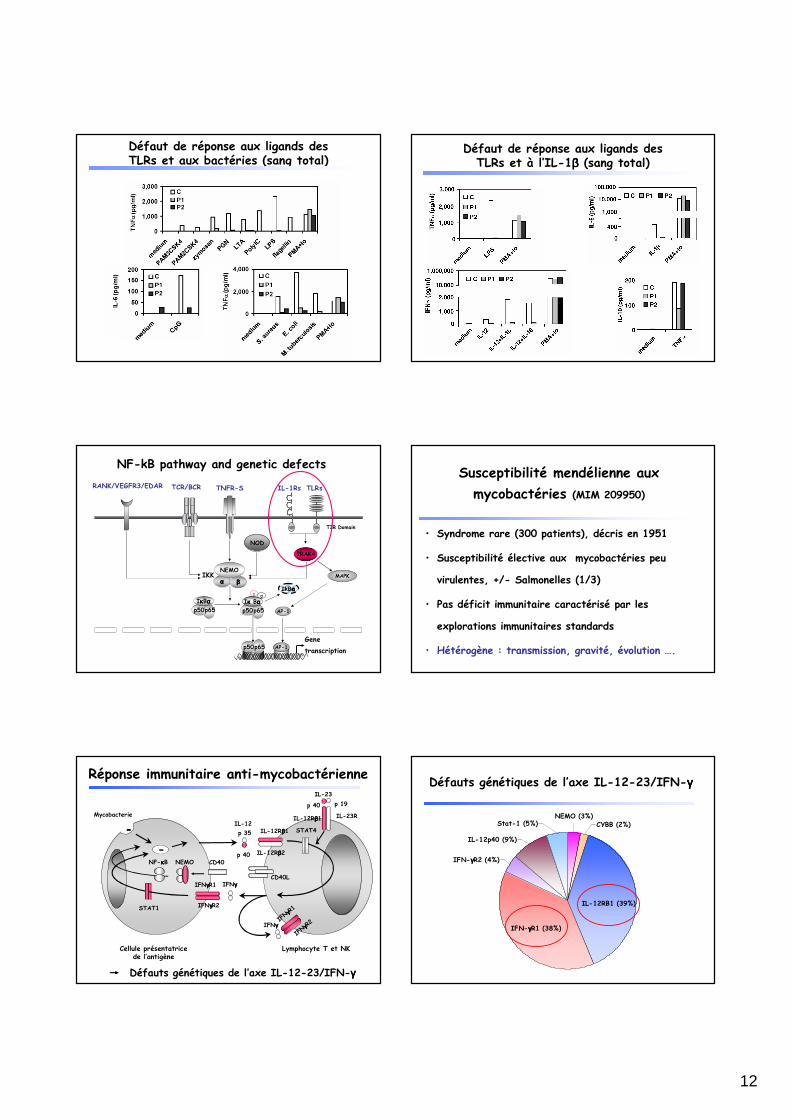
NF-kB pathway and genetic defects
TIR Domain
TLRsIL-1Rs
IRAK4
Susceptibilité mendélienne aux
mycobactéries (MIM 209950)
• Syndrome rare (300 patients), décris en 1951
• Susceptibilité élective aux mycobactéries peu
virulentes, +/- Salmonelles (1/3)
• Pas déficit immunitaire caractérisé par les
explorations immunitaires standards
• Hétérogène : transmission, gravité, évolution ….
Réponse immunitaire anti-mycobactérienne
Cellule présentatrice de l’antigène
Lymphocyte T et NK
Mycobacterie
IL-12
p 35
p 40
IL-12Rββββ1
IL-12Rββββ2
STAT4
IFNγγγγ
IFNγγγγ
STAT1 IFNγγγγR2
IFNγγγγR1
IFNγγγγR
2IFNγγγγR
1
CD40
CD40L
NF-κκκκB NEMO
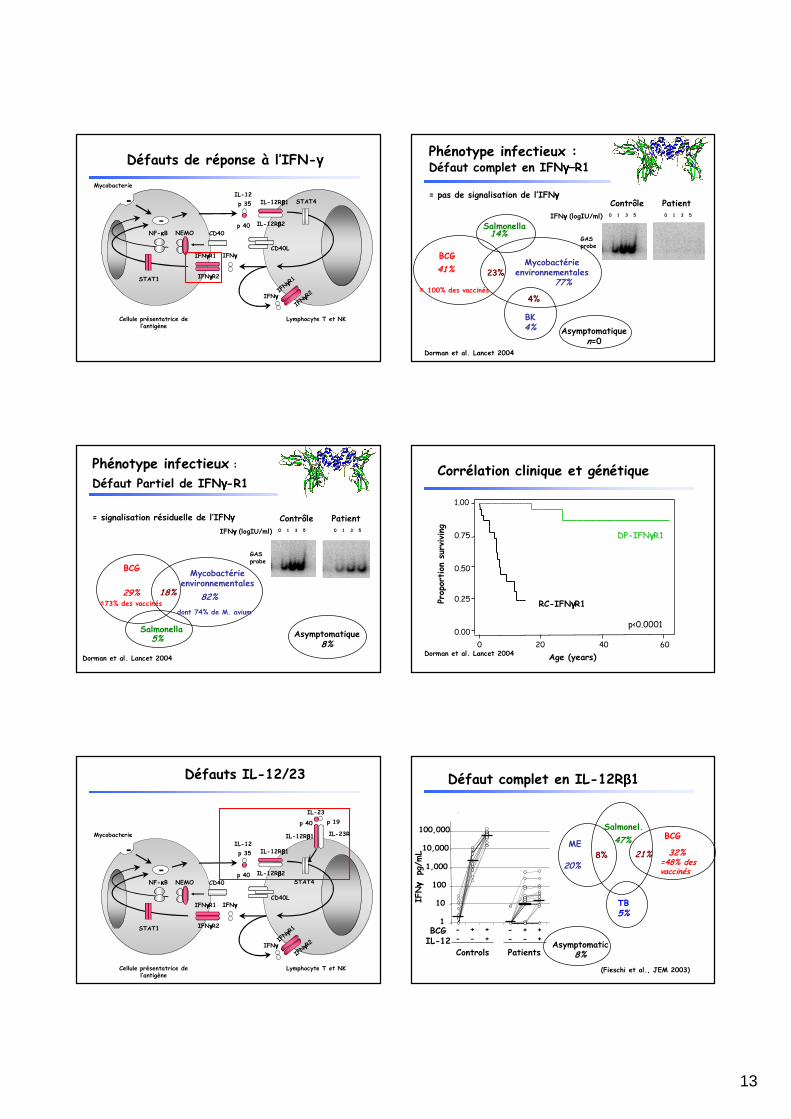
→→→→ Défauts génétiques de l’axe IL-12-23/IFN-γγγγ
IL-12Rββββ1 IL-23R
p 40 p 19
IL-23
CYBB (2%)NEMO (3%)
Stat-1 (5%)
IL-12p40 (9%)
IFN-γγγγR2 (4%)
IL-12RB1 (39%)
IFN-γγγγR1 (38%)
Défauts génétiques de l’axe IL-12-23/IFN-γγγγ
13
Cellule présentatrice de l’antigène
Lymphocyte T et NK
Mycobacterie
IL-12
p 35
p 40
IL-12Rββββ1
IL-12Rββββ2
STAT1
STAT4
IFNγγγγ
IFNγγγγ
IFNγγγγR2
IFNγγγγR1
IFNγγγγR
2IFNγγγγR
1
CD40
CD40L
NF-κκκκB NEMO
Défauts de réponse à l’IFN-γγγγ
Mycobactérie environnementales
BCG
Salmonella
BK4% Asymptomatique
n=0
41%
= 100% des vaccinés
23%77%
14%
Phénotype infectieux : Défaut complet en IFNγγγγ−−−−R1
= pas de signalisation de l’IFNγγγγ
4%
Dorman et al. Lancet 2004
Contrôle Patient
IFNγγγγ (logIU/ml) 0 1 3 5 0 1 3 5
GAS probe
BCG
SalmonellaAsymptomatique
8%
29%=73% des vaccinés
18% 82%
dont 74% de M. avium
5%
Phénotype infectieux :
Défaut Partiel de IFNγγγγ-R1
= signalisation résiduelle de l’IFNγγγγ
Dorman et al. Lancet 2004
Mycobactérie environnementales
Contrôle Patient
IFNγγγγ (logIU/ml) 0 1 3 5 0 1 3 5
GAS probe
Corrélation clinique et génétique
0 20 40 60
0.00
0.25
0.50
0.75
1.00
Prop
ortion
sur
viving
Age (years)
DP-IFNγγγγR1
RC-IFNγγγγR1
p<0.0001
Dorman et al. Lancet 2004
Cellule présentatrice de l’antigène
Défauts IL-12/23
Lymphocyte T et NK
Mycobacterie
IL-12
p 35
p 40
IL-12Rββββ1
IL-12Rββββ2
STAT1
STAT4
IFNγγγγ
IFNγγγγ
IFNγγγγR2
IFNγγγγR1
IFNγγγγR
2IFNγγγγR
1
CD40
CD40L
NF-κκκκB NEMO
IL-12Rββββ1 IL-23R
p 40 p 19
IL-23
Défaut complet en IL-12Rββββ1
IFN
γγ γγpg
/mL
1
10
100
1,000
10,000
100,000
BCGIL-12
+-
--
++
--
+-
++
Controls Patients
MEBCG
Salmonel.
TB5%
Asymptomatic8%
32%=48% des vaccinés
21%8%20%
47%
(Fieschi et al., JEM 2003)
14
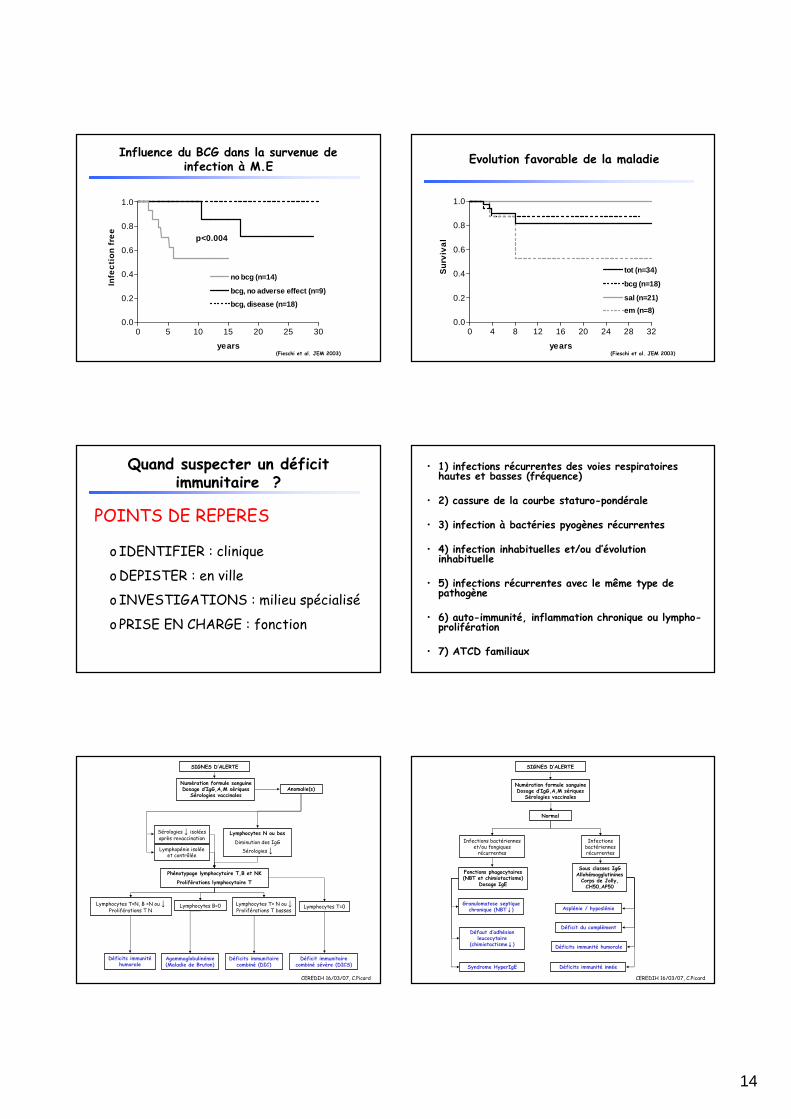
Influence du BCG dans la survenue de infection à M.E
0 5 10 15 20 25 300.0
0.2
0.4
0.6
0.8
1.0
no bcg (n=14)
bcg, disease (n=18)
bcg, no adverse effect (n=9)
p<0.004
years
Infe
cti
on
fre
e
(Fieschi et al. JEM 2003)
Evolution favorable de la maladie
0 4 8 12 16 20 24 28 320.0
0.2
0.4
0.6
0.8
1.0
tot (n=34)
em (n=8)
sal (n=21)
bcg (n=18)
years
Su
rviv
al
(Fieschi et al. JEM 2003)
Quand suspecter un déficit immunitaire ?
POINTS DE REPERES
o IDENTIFIER : clinique
o DEPISTER : en ville
o INVESTIGATIONS : milieu spécialisé
o PRISE EN CHARGE : fonction
• 1) infections récurrentes des voies respiratoires hautes et basses (fréquence)
• 2) cassure de la courbe staturo-pondérale
• 3) infection à bactéries pyogènes récurrentes
• 4) infection inhabituelles et/ou d’évolution inhabituelle
• 5) infections récurrentes avec le même type de pathogène
• 6) auto-immunité, inflammation chronique ou lympho-prolifération
• 7) ATCD familiaux
SIGNES D’ALERTE
Numération formule sanguineDosage d‘IgG,A,M sériques
Sérologies vaccinales
Lymphocytes T=0Lymphocytes B=0
Agammaglobulinémie (Maladie de Bruton)
Déficit immunitaire combiné sévère (DICS)
Lymphocytes T=N, B =N ou ↓Proliférations T N
Déficits immunitéhumorale
Déficits immunitaire combiné (DIC)
Phénotypage lymphocytaire T,B et NK
Proliférations lymphocytaire T
Lymphocytes T= N ou ↓Proliférations T basses
Anomalie(s)
Lymphopénie isolée et contrôlée
Sérologies ↓ isolées après revaccination
Lymphocytes N ou bas
Diminution des IgG
Sérologies ↓
CEREDIH 16/03/07, C.Picard
Infections bactériennes récurrentes
SIGNES D’ALERTE
Numération formule sanguineDosage d‘IgG,A,M sériques
Sérologies vaccinales
Sous classes IgGAllohémagglutinines
Corps de Jolly,CH50,AP50
Déficits immunité humorale
Déficit du complément
Asplénie / hyposlénie
Déficits immunité innée
Normal
Infections bactériennes et/ou fongiques
récurrentes
Fonctions phagocytaires(NBT et chimiotactisme)
Dosage IgE
Syndrome HyperIgE
Défaut d’adhésion leucocytaire
(chimiotactisme↓↓↓↓)
Granulomatose septique chronique (NBT↓↓↓↓)
CEREDIH 16/03/07, C.Picard

15
Conclusions
• Pensez à éliminer un DI devant des infections sévères et récurrentes
• Explorations des infections idiopathiques de l’enfant conduit à l’identification de nouveaux DI
• Ne pas éliminer DI devant la normalité des
explorations immunologiques :
la définition d’un DI est avant tout clinique
(toute infection sévère)
Caractérisation du déficit immunitaire permet une meilleure prise en charge et un Conseil génétique