Embed Size (px)
Citation preview

RZECZPOSPOLITAPOLSKA
Urząd Patentowy Rzeczypospolitej Polskiej
(12) OPIS PATENTOWY (19)PL (11)165149(13)B1
(21) Numer zgłoszenia: 285552
(22) Data zgłoszenia: 08.06.1990
(51) IntCl5:C12N 15/55 C12N 9/86 C12P 21/02 A61K 39/00
(54) Sposób wytwarzania stabilnej termicznie dezaminazy cytozynowej
(30) Pierwszeństwo:01.06.1990, US,90 531646
(43)Zgłoszenie ogłoszono:25.02.1991 BUP 04/91
(45) O udzieleniu patentu ogłoszono: 30.11.1994 WUP 11/94
(73) Uprawniony z patentu:ONCOGEN LIMITED PARTNERSHIP, Seattle, US
(72) Twórcy wynalazku:Peter D. Senter, Seattle, US Peter Chong-Dug Su, Mercer Island, US Hans Marquardt, Mercer Island, US Martha S. Hayden, Seattle, US Peter Linsley, Seattle, US
(74) Pełnomocnik:PHZ POLSERVICE
PL 16
5149
B1
(57) 1. Sposób wytwarzania stabilnej termiczniedezaminazy cytozynowej drogą rekombinacji, zna-mienny tym, że eukariotyczne lub prokariotyczne komórki-gospodarcze transformuje się rekombinan- towym wektorem ekspresyjnym powodującym skute-czna ekspresję sekwencji nukleotydowej zasadniczo przedstawionej na fig. 10, po czym stransformowane komórki-gospodarze hoduje się w warunkach umożli-wiających ekspresję tej nukleotydowej sekwencji i ze stransformowanych wyhodowanych komórek-gospo- darzy wyodrębnia się stabilną termicznie dezaminazę cytozynową.
FIG. 10

SPOSÓB WYTWARZANIA STABILNEJ TERMICZNIE DEZAMINAZY CYTOZYNOWEJ
Z a s t r z e ż e n i a p a t e n t o w e
1. Sposób wytwarzania stabilnej termicznie dezaminazy cytozynowej drogę rekombinacji, z n a m i e n n y t y m , że eukariotyczne lub prokariotyczne komórki-gospodarza transfor-muje się rekombinantowym wektorem ekspresyjnym powodującym skuteczny ekspresję sekwencjl
nukleotydowej zasadniczo przedstawionej na fig. 10, po czym stransformowane komórki-gospo-darza hoduje się w warunkach umożliwiających ekspresję tej nukleotydowej sekwencji i ze
stransformowanych wyhodowanych komórek-gospodarzy wyodrębnia się stabilną termicznie deza-minazę cytozynową.
2. Sposób według zastrz. 1, z n a m i e n n y t y m , że jako komórki-gospodarza stosuje się komórki ssaków.
3. Sposób według zastrz. 1, z n a m i e n n y t y m , że jako komórki-gospodarza
stosuje się komórki drożdży.
* * *
Przedmiotem wynalazku jest sposób wytwarzania stabilnej termicznie dezaminazy cytozy-
nowej wyodrębnionej z drożdży. Bardziej szczegółowo, wynalazek niniejszy dotyczy wytwarzania techniką rekombinantową termicznie stabilnej dezaminazy cytozynowej pochodzącej z Saccha- romyces cerevisiae.
Dezaminaza cytozynowa /CDaza, EC, 3.5.4.1/ katalizuje hydrolizę cytozyny do uracylu na drodze reakcji przedstawionej schematem. Enzym ten, który gra ważną rolę w metabolizmie pi-rymidyny w drobnoustrojach /O'Donovan i Neuhard, 1970/, został wyodrębniony z kilku różnych
drobnoustrojów, lecz okazało się, że nie był obecny w komórkach ssaków /Nishiyama i in.,
1985/.Wykazano, że właściwości fizyczne CDazy z różnych organizmów różnię się znacznie jeśli
chodzi o masę cząsteczkową, stabilność i skład pod jednostek. I tak np., CDaze z Salmonella
typhimurlum oczyszczono do jednorodności /za pomocą elektroforezy w żelu poliakryloamidowym z siarczanem dodecylo-sodowym - SDS-PAGE/ i okazało się, że jest ona złożona z czterech pod-
jednostek o 54 kilodaltonach każda /West i in., 1982/, podczas gdy enzym z E. coli ma masę
cząsteczkową 200 kilodaltonów i jest złożony z pod jednostek o 35 do 46 kilodaltonów /Kateu-
ragi i in., 1986/. Obydwa te enzymy są w wysokim stopniu termicznie stabilne i utrzymują
wysoką aktywność w temperaturze 55°C.Jako źródła CDazy użyto także drożdży piekarniczych /Saccharomyces cerevisiae/. CDaza
otrzymana z nich uprzednio ma masę cząsteczkową 34 kilodaltony jak oznaczono za pomocą fil-tracji żelowej /Ipata i in., 1971, 1978/ i 32 - 33 kilodaltony jak oznaczono za pomocą
SDS-PAGE i analizy aminokwasów /Yergatian i in., 1977/. Enzym CDaza, który został uprzednio wyodrębniony z drożdży piekarniczych jest więc, jak się okazuje, białkiem monomerycznym.
Roztwory CDazy uprzednio wyodrębnionej z drożdży piekarniczych utrzymuję aktywność w cią-
gu co najmniej 48 godzin, gdy przechowywane są w temperaturze 4°C w pH 5 - 9./Ipata i in., 1971, 1978/. Jednakże, w temperaturze 37° surowy preparat CDazy z drożdży piekarniczych traci,
jak wykazano, połowę aktywności w ciągu godziny /Kream and Chargaff, 1952/, a w postaci oczyszczonej na pół okres trwania 30 minut. /Katsuragi, 1988/. Pół okres trwania w tempera-turze 37°C można zwiększyć do 28 dni za pomocą unieruchomienia enzymu na kuleczkach epoksy- akrylowych /Katauragi i in., 1987/. Tak więc, termiczna niestabilność CDazy z drożdży pie-karniczych, wraz z niską masą cząsteczkową, odróżnia ją od enzymów bakteryjnych wcześniej opisanych.

165 149 3
CDazy użyto terapeutycznie do konwersji proleku 5-fluorocytozyny /5-FC/ do przeciwrako- wego leku 5-fluorouracylu /5-FU/ /Katsuragi i in., 1987/; Nishyama i in., 1985 Sakai i in., 1985/: Senter i in., 1987/. Jednakże, bakteryjne źródła CDazy są niepraktyczne w tym zasto-sowaniu wymagając hodowli na wielką skalę w celu otrzymania odpowiedniej aktywności /Sakai i in., 1985/. Dodatkowo, ekstrakty bakteryjne mogą spowodować niepożądane działania ubocz-na u biorców.
W celu ominięcia tych problemów można użyć drożdży jako źródła CDazy. Jednak niestabil- ność termiczna uprzednio otrzymanego produktu z drożdży, wymaga, aby enzym ten został przed użyciem unieruchomiony /Katsuragi i in., 1987/. To też, wyodrębnienie i oczyszczenie termicz-
nie stabilnej drożdżowej CDazy dostarcza ważnego enzymu do użycia w terapii przeciwrakowej. Podobnie, klonowanie genu termicznie stabilnej CDazy z drożdży pozwala na wprowadzenia okreś-lonych zmian lub uzupełnień do samego genu, sekwencji kontrolujących jego ekspresję i sprzę-
żeń genowych utworzonych między genem z innymi cząsteczkami. Te nowe konstrukcje zwiększają
skuteczność lub użyteczność enzymu w terapii przeciwrakowej.Wynalazek niniejszy jest oparty na nieoczekiwanym odkryci u termicznie stabilnej CDazy
z drożdży piekarniczych. Analiza sekwencji aminokwasów tego enzymu nie ujawnia znacznej homo- logii ze znanymi sekwencjami innych białek.
Sposób wytwarzania stabilnej termicznie dezaminazy cytozynowej drogą rekombinacji polega
według wynalazku na tym, że eukariotyczne lub prokariotyczna komórki-gospodarza transformuje
się rekombinantowym wektorem ekspresyjnym powodującym skuteczną ekspresję sekwencji nukleo-
tydowej zasadniczo przedstawionej na fig, 10, po czym stransformowane komórki-gospodarze hodu-je się w warunkach umożliwiających ekspresję tej nukleotydowej sekwencji i ze stransformowa-
nych wyhodowanych komórek-gospodarzy wyodrębnia się stabilną termicznie dezaminazę cytozynową.
W sposobie według wynalazku jako komórki-gospodarze stosuje się komórki ssaków. W szcze-gólnie korzystnym sposobie realizacji wynalazku stabllną termicznie dezaminazę cytozynowę
wyodrębnia się z Saccharomyces cerevisies.Dodatkowe aspekty, korzyści i sposoby realizacji łatwo stanę się oczywiste dla zwykłych
fachowców w tej dziedzinie techniki w świetle ujawnienie w niniejszym opisie.W rysunkach stanowiących część niniejszego ujawnienia:
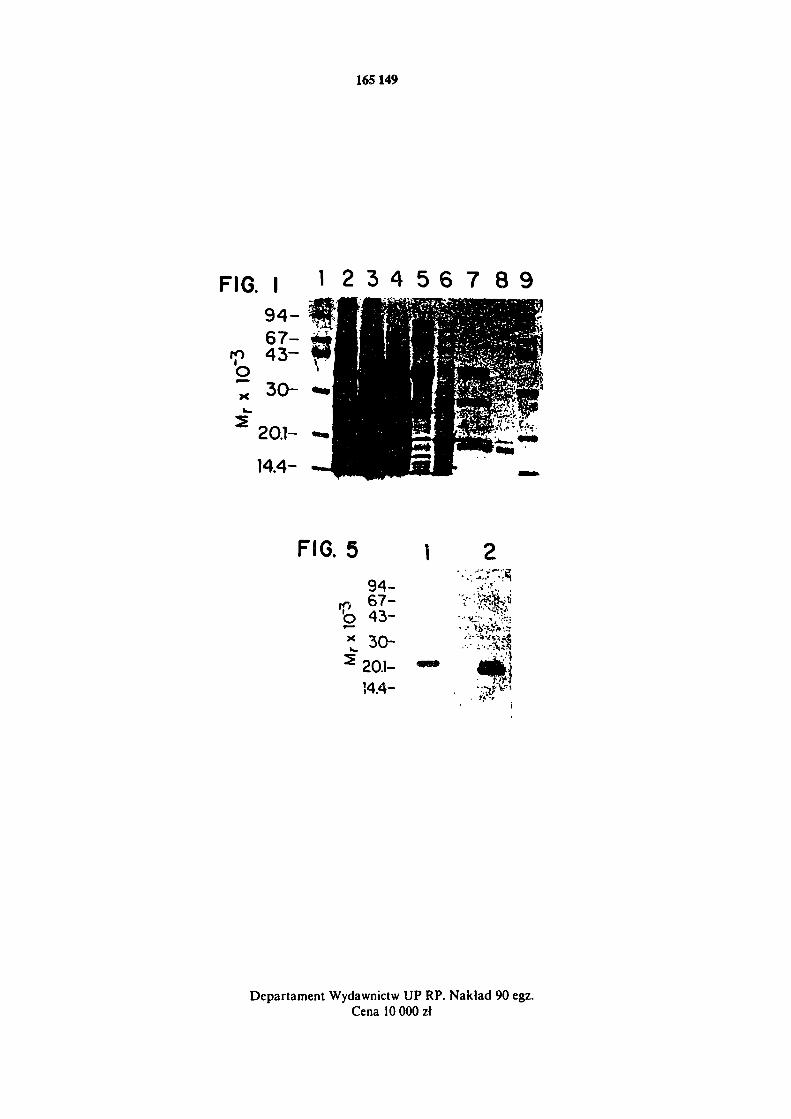
Figura 1 przedstawia analizę CDazy za pomocą SDS-PAGE /14% poliakryloamid w warunkach nieredukujących/. Substancje przedstawione na każdej ścieżce są jak następuje. Ścieżka 1 1 9 - białka markerowe ; ścieżka 2 - supernatant autolityczny z etapu 1 w przykładzie I;
ścieżka 3 - produkt po wytrąceniu /NH4/2SO4 / 7 0 % / w etapie 2 z przykładu I; ścieżka 4 - produkt po wytrąceniu /NH4/2SO4 /50 - 73%/ w etapie 2 z przykładu I; ścieżka 5 - produkt otrzymany po chromatografii na Q-Sepharose w etapie 3 z przykładu I, ścieżka 6 - produkt po
p r z e p r o w a d z e n i u przez kolumnę G-75 w etapie 4 z p r z y k ł a d u I ; ścieżka 7 - produkt po o c z y s z -
c z e n i u przy użyciu octyl-Sepharose w etapie 5 z przykładu I; ścieżka 8 - produkt finalny otrzymany po przeprowadzeniu przez drugą kolumnę G-75 w etapie 6 z przykładu I.
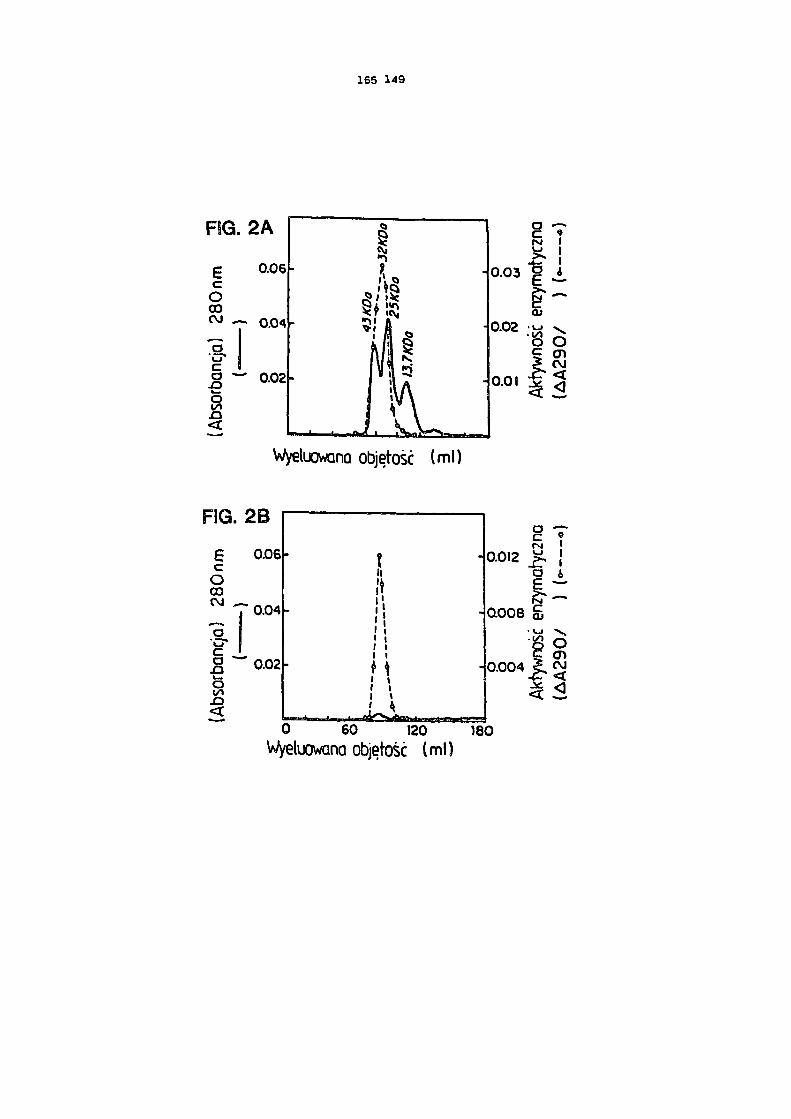
Figura 2 przedstawia profil elucjl CDazy z 1,5 x 100 cm kolumny Sephadex G-50.
/A/ 27 jednostek oczyszczonej CDazy zmieszano z 2 mg rybonukleazy A, 2 mg albuminy jaja ku-rzego, 1 mg chymotrypsynogenu A i eluowano PBS. Frakcje rejestrowano przy 280 na w celu
oznaczenia zawartości białka całkowitego i przy 290 nm, przy zastosowaniu 5FC jako substra- tu, w celu oznaczenia aktywności CDazy. /B/ Eluacja CDazy z powyższej kolumny bez standar-
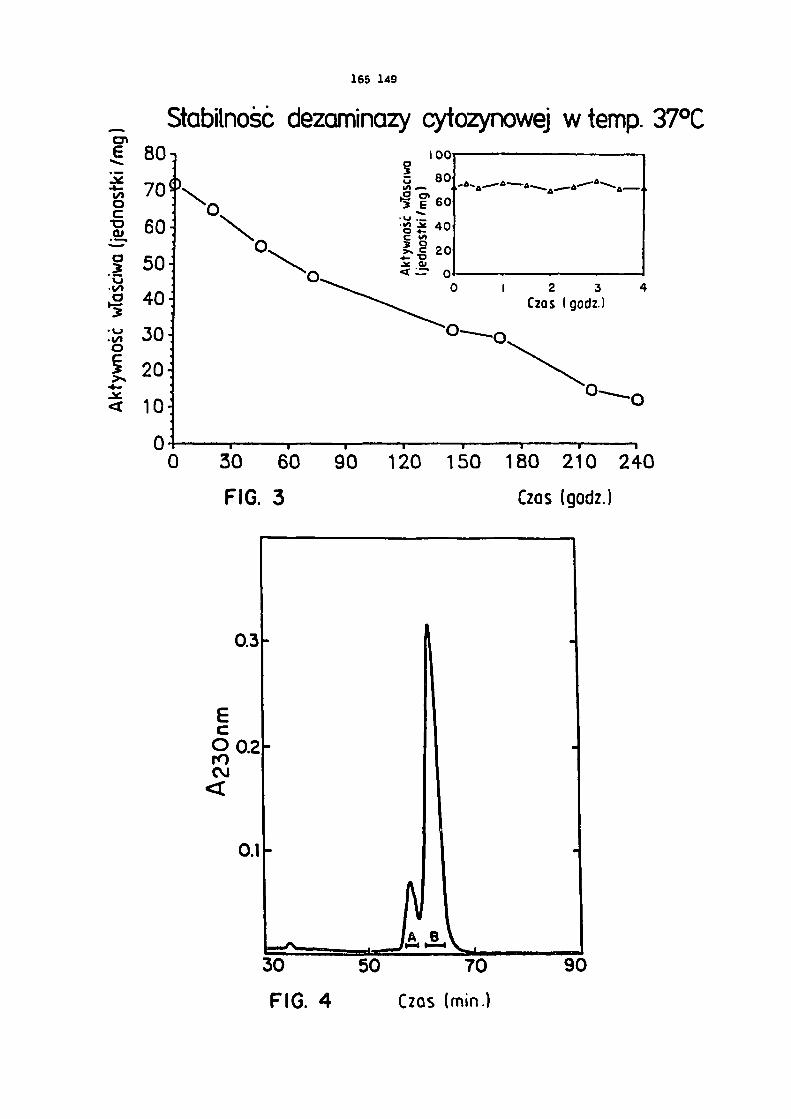
dów do kalibracji.Figura 3 przedstawia stabilność CDazy w temperaturze 37°C. CDazę /72 jednostki/mg/ w
PBS /który zawierał, w stężeniu 1 mg/ml, bydlącą albuminę surowiczą bez zanieczyszczeń pro- teazami/ inkubowano w temperaturze 37°C w probówce polipropylenowej. Aktywność CDazy ozna-czano w różnych odstępach czasu używając 10 /ul roztworu enzymu.
Figura 4 przedstawia profil CDazy oczyszczonej za pomocą chromatografii cieczowej wyso- kosprawnej /HPLC/. Odcinki poziome wskazują frakcje, które połączono do analizy sekwencji aminokwasów.
Figura 5 przedstawia analizę CDazy za pomocą SOS-PAGE po oczyszczeniu za pomocą HPLC / 15% poliakryloamid w warunkach nieredukujęcych/. Ścieżka 1 - pula A , ścieżka 2 - pula B.

4 165 149
Figura 6 przedstawia częściową sekwencję aminokwasów CDazy. Wskazano peptydy otrzymane
przez rozszczepienie CNBr /seria M/, endoproteinazę Glu-C /seria E/, endoproteinazę Lys-C
/seria K/ i endoproteinazę Asp-N /seria D/.Figura 7 przedstawia sekwencję nukleotydów starterów CDA4R1 i CDA5AS, odpowiednie sek-
wencje aminokwasów i względne pozycje tych starterów w sekwencji aminokwasów CDazy. CDA4R1 jest zorientowany w kierunku od 5' do 3' w nici sensownej, podczas gdy CDA5AS jest zorien-
towany w kierunku od 5' do 3' w nici antysensownej.Figura 8 przedstawia sekwencję nukleotydów DNA z klonów genowych kodujących CDazę.
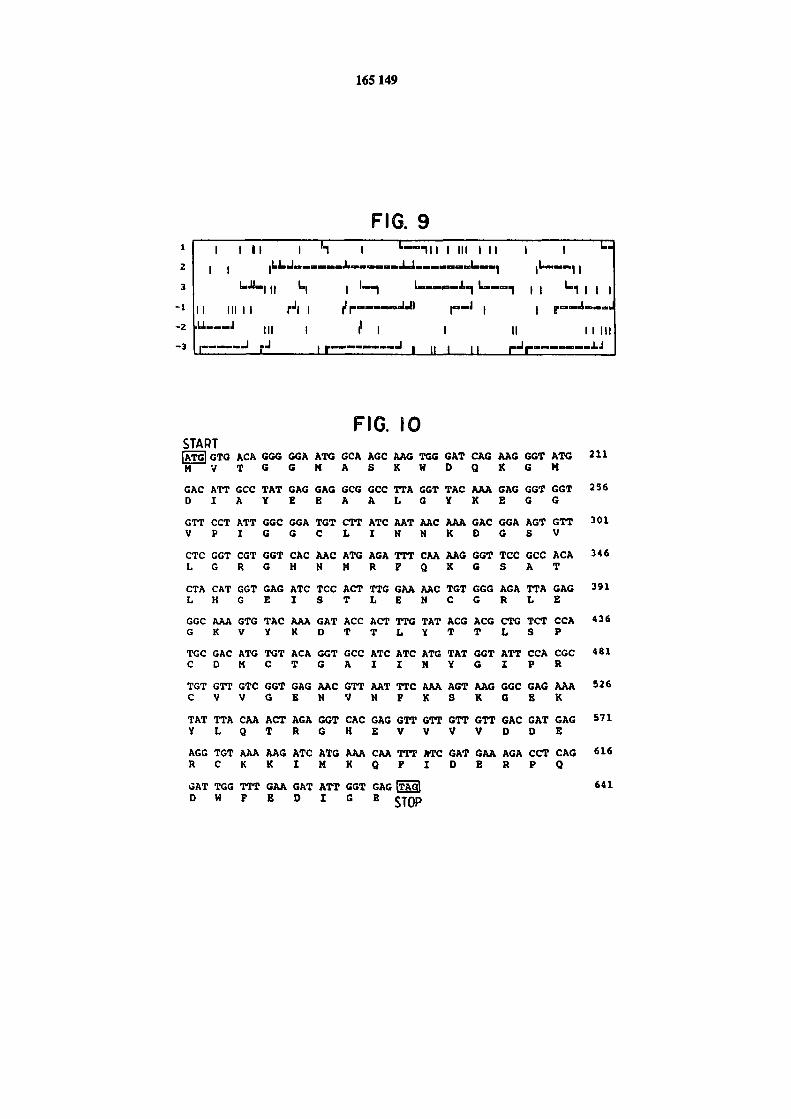
Figura 9 przedstawia otwarte ramki odczytu /ORF/ stwierdzone w genomowej sekwencji
CDazy.Figura 10 przedstawia sekwencję nukleotydów i odpowiednią sekwencję aminokwasów ORF 2.
Dodatkowe aminokwasy, przewidywane na podstawie sekwencji nukleotydów , ale nie wykryte w
sekwencjonowaniu peptydu są uwydatnione tłustą czcionką, a kodony START i STOP są ujęte
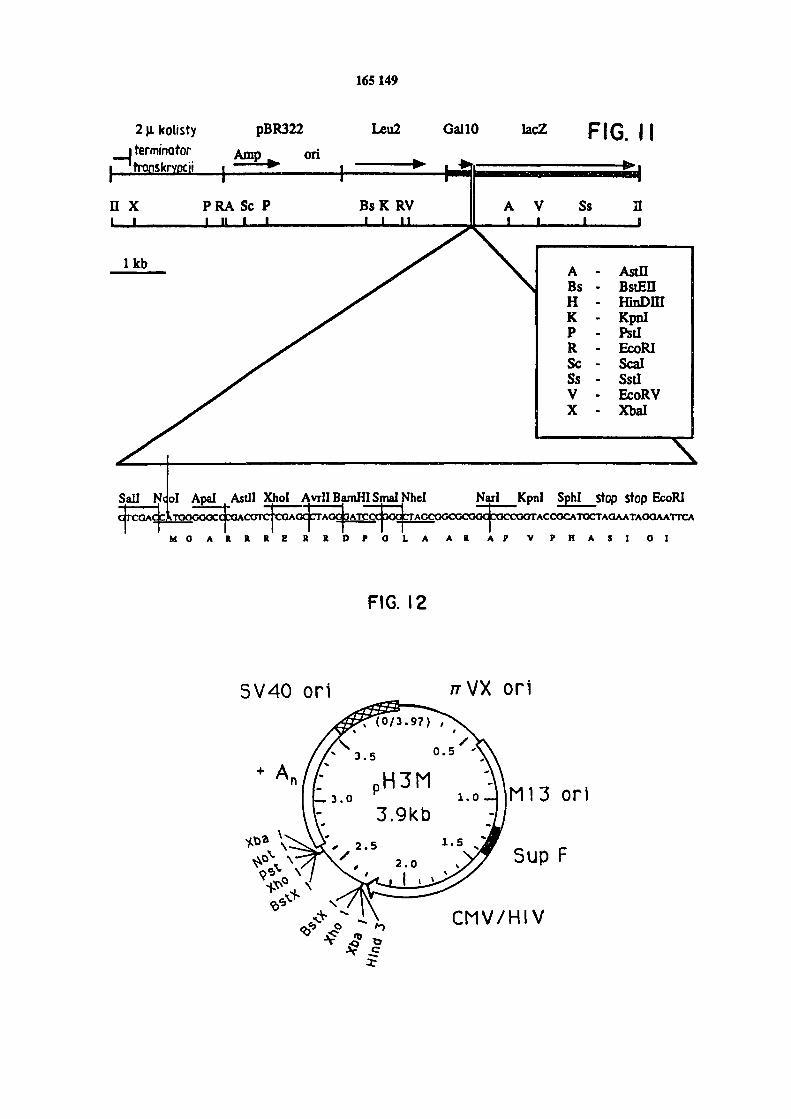
w ramki.Figura 11 przedstawia plazmidowy wektor ekspresyjny "sprzęgacz” zawierający gen CDazy.
Figura 12 przedstawia mapę wektora pH3M.W praktycznym zastosowaniu wynalazku stosuje się jeśli tego inaczej nie wskazano,
zwyczajne techniki chemii białek, biologii molekularnej, mikrobiologii i technologii rekom- binantowego DNA w zakresie znanym fachowcom. Techniki te są w pełni wyjaśnione w piśmien-
nictwie. Patrz np. R.K. Scopes, Protein Purlfication Principles and Practice, wyd. II
/Springer-Verlag 1987/) Mathods in Enzymology /wyd. S. Colowick i N. Kaplan, Academic Press, Inc.,/i Maniatis, Fritsch i Sambrock, Molecular Cloning, A. Laboratory Manual,
wyd. II /1989/; Oligonuclectide Synthesis /wyd. M.J.Gait 1984/.
Przy opisywaniu wynalazku używa się następujących terminów. Dla stosowania ich tu de-
finiuje się je jak wskazano poniżej."Termicznie stabilna dezaminaza cytozynowa'' odnosi się do CDazy, która zachowuje co
najmniej 50% aktywności w stanie wolnym, bez unieruchomienia, w ciągu ponad 12 godzin w
temperaturze 37°C, korzystnie w okresie dłuższym niż 1 dzień w temperaturze 37°C i naj-
korzystniej przed ponad 3 dni w temperaturze 37°C, co ustala się za pomocą oznaczenia konwersji 5FC do 5FU w obecności wyodrębnionego enzymu w badaniu objaśnionym pełniej poniżej.
Sekwencja aminokwasów lub białko jest "zasadniczo jednorodne" wobec innej sekwencji
aminokwasów lub białka, gdy co najmniej około 50%, korzystnie około co najmniej 85% i naj-
korzystniej co najmniej około 90 - 95% aminokwasów odpowiada określonej długości częstecz- ki. Dodatkowo, zmiany dotyczące aminokwasów mogą obejmować podstawienia, delecje lub dodania.
Termin "funkcjonalny odpowiednik" znaczy, że sekwencja aminokwasów lub białko określa łańcuch, który utworzy termicznie stabilny enzym, jak opisano powyżej, zdolny do konwersji
5-FC do 5-FU jak opisano w przykładach, białko, które jest funkcjonalnym odpowiednikiem
termicznie stabilnej CDazy nie musi mieć dokładnie takiej /lub całkowitej/ sekwencji jak przedstawiona w figurach w niniejszym opisie. Białko, raczej może istnieć jako biologicz-
nie aktywny fragment tej sekwencji, o aktywności zdefiniowanej jak powyżej. Dodatkowo, biał-
ko może obejmować dodania, delecje lub podstawienia w powyższej sekwencji, w takim zakresie, w jakim białko to pozostaje biologicznie aktywne.
"Oczyszczone białko" zasadniczo nie zawiera innych materiałów. I tak np., białko A zasad-
niczo nie zawiera B, gdzie 9 oznacza mieszaninę innych składników komórkowych 1 białek, gdy co najmniej 50% wag całości A + B, korzystniej co najmniej 75%, a najkorzystniej 90 - 95%i nawet 99% wag stanowi A. "Oczyszczone” nie odnosi się jednak do metody, jaką białko otrzy-
mano. Tak więc, oczyszczonym białkiem może być białko wytworzone technikami rekombinantowy- mi, wytworzone syntetycznie, albo wyodrębnione bezpośrednio z organizmu, w którym jest
znajdowane w naturze.Terminy "polipeptyd" i "białko" używane są w swoim rozszerzonym sensie, to jest ozna-
czają jakikolwiek polimer utworzony z aminokwasów /dipeptyd lub wyższy/ połączonych wiąza-
niami peptydowymi. Tak więc, terminy te obejmują oligopeptydy, fragmenty białka, analogi.

165 149 5
muteiny, białka sprzężone itp. Terminy obejmują białka natywne i rekomibinantowe.
"Rekombinantowe" białka lub polipeptydy odnoszą się do białek powstałych w wyniku ekspre-sji rekombinantowej sekwencji nukleotydów, to jest wytworzonych przez komórki transformowane egzogenną konstrukcję ONA kodującą pożądany polipeptyd.
Termin "rekombinantowy" używany w niniejszym opisie charakteryzuje sekwencję nukleoty-dów kodującą CDazę i opisuje kwas nukleinowy pochodzenia genomowego, cDNA, półsyntetycznego lub syntetycznego, który z uwagi na swoje pochodzenie lub manipulację jest albo sekwencję nukleotydów nie występującą w naturze, albo sekwencję nukleotydów związaną z kwasami nuklei-nowymi innymi, niż kwasy z którymi jest związana w naturze.
"Replikon" jest to jakikolwiek element genetyczny, np. plazmid, chromosom, wirus, który
zachowuje się jako autonomiczna jednostka w replikacji polinukleotydów w komórce, tak więc jest on zdolny do replikacji pod swoją własną kontrolą.
"Wektor" jest replikonem, w którym przyłączony jest inny segment polinukleotydowy tak,- aby doprowadzić do replikacji i/lub ekspresji przyłączonego segmentu. "Wektor ekspresyjny"
odnosi się do wektora zdolnego do autonomicznej replikacji lub integracji, zawiera sekwen-cje kontrolne kierujące transkrypcją i translacją pożądanej sekwencji nukleotydów w orga-nizmie odpowiedniego gospodarza.
"Sekwencja kodująca" jest sekwencję nukleotydów, która ulega transkrypcji i/lub trans-lacji do polipeptydu.
"Sekwencja promotorowa" jest regionem regulatorowym zdolnym do przyłączania polimerazy
RNA i inicjowania transkrypcji "w dół", to jest w kierunku 3', sekwencji kodującej.Sekwencja kodująca znajduje się "pod kontrolą" sekwencji promotorowej w komórce, gdy
z przyłączenia polimerazy RNA do sekwencji promotorowej wynika translacja sekwencji kodu-jącej. Następnie translacja powstałego mRNA daje w wyniku polipeptyd zakodowany w sekwen-cji kodującej.
"Operatywnie połączony" odnosi się do zestawienia, w którym komponenty są tak ułożone względem siebie, że pozwala to na ich zwykłe działanie. Tak więc, sekwencje kontrolne ope-
ratywnie połączone z sekwencję kodującą są zdolne do wywoływania ekspresji sekwencji kodującej.
"Sekwencja kontrolne" odnoszę się do tych sekwencji, które kontroluję transkrypcję
i/lub translację sekwencji kodującej /sekwencji kodujących/. Mogę one obejmować, nie będąc do nich ograniczone, sekwencje promotorowe, transkrypcyjne sekwencje inicjacyjne i termi-
nacyjne, oraz translacyjne sekwencje inicjacyjne i terminacyjne. Oprócz tego, "sekwencje
kontrolne" odnoszą się do sekwencji kontrolujących przetwarzanie polipeptydu zakodowanego
w sekwencji kodującej. Mogą one obejmować, nie będąc do nich ograniczone, sekwencje kontro- lujące wydzielanie, rozszczepienie proteazaml i glikozylację polipeptydu.
"Sekwencja sygnałowa” może być włączona przed sekwencję kodującą. Ta sekwencja koduje peptyd sygnałowy, N-końcowy względem polipeptydu, który nadaje komórce-gospodarzowl tenden-
cję do skierowania polipeptydu na powierzchnię komórek lub wydzielenia polipeptydu do oto-czenia. Peptyd sygnałowy zostaje przez komórkę-gospodarza odszczepiony przez opuszczeniem
przez białko komórki. Sekwencje sygnałowe można znaleźć w połączeniu z różnymi białkami natywnymi wobec prokariontów i eukariontów. I tak np. czynnik α , natywne białko drożdżo-
we, jest wydzielane z drożdży, a jego sekwencję sygnałową można przyłączyć do białek hetero- logicznych, które maję być wydzielone do podłoża/patrz opis patentowy Stanów Zjednoczonych Ameryki nr 4546082/. Dalej, stwierdzono, że c z y n n i k αi jego analogi biorą udział w wydzie-laniu białek heterologicznych z rozmaitych drożdży, takich jak Saccharomycea i Kluyveromy-
ces /patrz np. EPO Pub. nr 0 301 669, data opublikowania 1 lutego 1989/."Transformacja" jest to insercja egzogennego polinukleotydu do komórki-gospodarza.
Egzogenny polinukleotyd może być utrzymywany jako plazmid, albo alternatywnie, może zostać zintegrowany z genomem gospodarza.
"Rekomblnentowa komórka gospodarza”, "komórki-gospodarze", "komórki" i inne tego rodza- ju terminy wskazujące drobnoustroje używane są wymiennie i odnoszą się do komórek, które mogę być, lub powinny być użyte jako biorcy wektorów rekombinantowych i innego przenoszonego

6 165 149
ONA i obejmuję potomstwo wyjściowej transfekowanej komórki. Rozumie się, że potomstwo poje-dynczej komórki macierzystej niekoniecznie musi być całkowicie identyczne pod względem ge- nomowego, lub całkowitego zespołu ONA z wyjściową komórką macierzystą z powodu przypadkowej lub zamierzonej mutacji. Tak więc, terminy te oznaczają potomstwo komórki macierzystej, któ-re jest do niej wystarczająco podobne, aby zostać scharakteryzowane przez istotną właściwość, na przykład zastąpienie natywnego genu, kodującego zasadniczy enzym, genem klonowanym, połą- czonym z genem struktury kodującym pożądany produkt genu.
"Terapeutycznie skuteczna ilość" CDazy jest to taka ilość, która, gdy zostanie podana wraz z substratem, na który CDaza działa, jest wystarczająca do konwersji substratu w aktyw-ny czynnik cytotoksyczny, który może z kolei hamować wzrost komórek nowotworowych.
Wynalazek jest ukierunkowany na sposób wytwarzania termicznie stabilnej dezaminazy cyto-
zynowej wyodrębnianej z drożdży. Ta CDaza wykazuje właściwości, które odróżniają ją od pier-wotnie wyodrębnionych z drożdży enzymów o charakterze CDazy. Masa cząstaczkowa ostatnio wy-odrębnionej drożdżowej CDazy wynosi około 32 kilodaltonów, jak to oznaczono za pomocą chroma-
tografii żelowej. SDS-PAGE wykazuje główne pasmo przy 17 kilodaltonach wskazując, że obecna
CDaza stanowi dimer z każdą z podjednostek o masie cząsteczkowej około 17 kilodaltonów.Oprócz tego, podczas gdy poprzednio wyodrębnione enzymy drożdżowe o charakterze CDazy były, jak wykazano, w wysokim stopniu termolabilne w temperaturze 37°C, oczyszczony enzym wytwo-
rzony sposobem według wynalazku jest stabilny w tej temperaturze. Dalej, sekwencja aminokwa-sów nowej CDazy nie wykazuje znacznej sekwencyjnej homologii z innymi znanymi, sekwencjono-
wanymi białkami. Klonowania i ekspresja genu kodującego tą unikalną CDazę dostarcza sekwen-
cji kodującej o długości około 474 par zasad, wyznaczającej białko o 158 aminokwasach o prze-widywanej masła cząsteczkowej wynoszącej około 17506 deltonów. Termicznie stabilną CDazę moż-na wyodrębnić z drożdży, w tym należących do askosporotwórczych, bazydiosporotwórczych i
drożdży niedoskonałych. Korzystnie, CDazę wyodrębnia się z organizmów należących do rodziny
Saccharomycetoideas, przy czym korzystne są gatunki należące do Saccharomyces, a szczególnie
korzystne są Saccharomyces cerevisiae /drożdże piekarnicze/. Stwierdzono, że drożdże praso-wane Fleishmanna są doskonałym źródłem termicznie stabilnej CDazy i są łatwo handlowo dostęp-ne w piekarniach i sklepach spożywczych.
Obecny sposób oczyszczania CDazy wytwarzanej sposobem według wynalazku różni się od spo-
sobów, o których donoszono /patrz np. Katsuragi i in., 1987/t Katsuragi i in., 1986; Ipatai ln., 1978; Ipata i ln., 1971/, jak również od sposobu Katsuragi /1988/. Specyficznie,
w sposobie tym występuję różne etapy oczyszczania, w tym dodatkowo żelowo-permeacyjne oczysz-
czanie na kolumnie, między etapami anlonitowej i hydrofobowej chromatografii /opisanymi po-
niżej/ jak również odmienne warunki oczyszczania, w tym różne bufory, wartości pH i składni-
ki buforów, takie jak EDTA i DTT. Oprócz tego, poprzednie sposoby nie okazuję się pomocne
w odniesieniu do termicznie stabilnego enzymu.
Pierwszy etap oczyszczania termicznie stabilnej CDazy z drożdży związany jest z rozbi-ciem komórek drożdży w celu otrzymania autolizatu zawierającego CDazę. Autolizę można uzys-kać stosując jakąkolwiek z kilku metod znanych w tej dziedzinie techniki. I tak np. komórki
drożdży można poddać plazmolizie z użyciem toluenu, metodę Kunitza /1947/. Szczególnie uży-
teczna jest ekspozycja drożdży na rozpuszczalnik organiczny, taki jak octan etylu, a następ-
nie dodanie buforu dla utrzymania roztworu w pH około 7. Do odpowiednich buforów należy bu-for z fosforanem potasu, roztwór soli buforowany fosforanami, bufor Tris, jak również kil-ka innych, dobrze znanych w tej dziedzinie techniki. Bufor korzystnie zawiera siarczan amo-
nowy w stężeniu 1 - 25%, korzystnie 15%, jak również EDTA i ditiotreitol /DTT/ w celu stabi-lizacji CDazy. Alternatywnie, EDTA i DTT mogą zostać wyeliminowane z tego i następnych bufo-rów. Stężenie EDTA, w razie jej obecności, mieści się w zakresie 0,1 - 10 mM, przy korzystnym stężeniu 5 mM, podczas gdy DTT może być obecny w stężeniu 0,01 - 10 mM, przy korzystnym stę-żeniu 0,1 mM. Tą mieszaninę miesza się w ciągu kilku dni, utrzymując pH około 7. Komórkowy
debris można usunąć za pomocą odwirowania.Po autolizie białko całkowite wytrąca się z autolizatu z użyciem siarczanu amonowego,
który dodaje się do stężenia wystarczającego do uzyskania wyższego stosunku CDazy do białka.

165 149 7
I tak np., siarczan amonowy można najpierw dodać do osiągnięcia 60 - 80% nasycania, korzyst-nie 70% nasycenia. EDTA korzystnie włącza się do mieszaniny reakcyjnej w stężeniu 1 - 3 g/ litr, korzystnie 1,5 - 2,0 g/lltr, dopuszcza się, aby reakcja zachodziła w ciągu kilku go-dzin i zbiera się osad po odwirowaniu. Osad rozpuszcza się w odpowiednim buforze, takim jak PBS przy pH około 7, przy czym bufor korzystnie zawiera EDTA i DTT, jak powyżej. Roztwór dia-lizuje się wobec tego samego buforu. Na dializat można podziałać drugi raz siarczanem amono-wym, w celu osiągnięcia stężenia około 50%, w obecności EDTA jak powyżej. Osad ponownie zbie-ra się za pomocą odwirowania i do supernatantu dodaje siarczan amonowy do osiągnięcia około 73% nasycenia. Osad zbiera się i rozpuszcza w odpowiednim buforze o pH około 6,6 - 8,5, ko-rzystnie w buforze Tris o pH 8,0, zawierającym DTT w stężeniu wyżej opisanym. Rekonstytuo-wany osad dializuje się następnie wobec tegoż buforu w ciągu kilku godzin. CDazę z dializatu można dalej oczyszczać za pomocą chromatografii na anioncie stosując np. sieciowaną agaro- zę lub celulozowy materiał do upakowania. Szczególnie użyteczne są anionity, takie jak Q-Sepharose i DEAE-Sepharose, dostępne z firmy Pharmacia. Do odpowiednich buforów równowa-żących należy np. bufor Tris lub bufor fosforanowy, z przeprowadzeniem elucji liniowym gra-
dientem. Szczególnie stosowne jest użycie 20 mM Tris zawierającego 0,1 mM DTT przy pH 8,0z liniowym gradientem O - 03 M KCl w tym buforze.
Po chromatografii na anionicie, frakcje wykazujące aktywność CDazy /oszacowaną jak opi-
sano poniżej/ zbiera się i zatęża za pomocą ultrafiltracji, stosując np. filtr z odcięciem
przy masie cząsteczkowej około 30000 daltonów, lub niższej.Następnie, roztwór zawierający CDazę przenosi się na kolumnę żelowo-permeacyjnę. Szcze-
gólnie użyteczny jest sieciowany dekstran, agaroza lub żele dekstran/bisakryloemid, przy czym
korzystne są Sephadex G-75, G-100 lub Sephacryl S-300. Eluantem może być każdy zwykły bu-for, dobrze znany w tej dziedzinie techniki, o pH około 7, przy czym korzystny jest PBS za-
wierający DTT jak uprzednio opisano. Frakcje wykazujące aktywność CDazy łączy się i dializu-je wobec buforu takiego jak bufor z fosforanem potasowym. Korzystnie, bufor zawiera 1 - 2 M siarczan amonowy oraz EDTA i DTT jak opisano powyżej w odniesieniu do buforu do autolizy.
Można przeprowadzić drugi rozdział na kolumnie żelowo-permeacyjnej, jeśli jest to pożądane, i zatężyć aktywne frakcje za pomocą ultrafiltracji, stosując filtr z odcięciem przy masie
cząsteczkowej np, 5000 daltonów.Następnie materiał zawierający CDazę można nanieść na kolumnę, która rozdziela substancje
w oparciu o hydeofobowość i eluować np. odwróconym gradientem siarczanu amonowego w buforze z fosforanem potasowym. Frakcje wykazujące aktywność CDazy łączy się i zatęża za pomocą ul-
trafiltracji stosując filtr z odcięciem przy masie cząsteczkowej 5000 dalton. Koncentrat
można przechowywać- zamrożony do dalszego użycia. Jednak jeżeli nie przeprowadzono rozdziału
na drugiej kolumnie żelowo-permeacyjnej i aktywność właściwa CDazy jest niska, można przepro-
wadzić rozdział na innej kolumnie żelowo-permeacyjnej, jak opisano powyżej. Tak oczyszczona
CDaza jest termicznie stabilna i może wobec tego być przechowywana, w etanie zamrożonym lub
zliofilizowana, w wolnej postaci.Aktywność CDazy można kontrolować podczas oczyszczania kilkoma metodami znanymi w tej
dziedzinie techniki. I tak np,, konwersję cytozyny do uracylu, albo jego pochodnych w obec-ności CDazy można kontrolować w bezpośrednim badaniu spektrofotometrycznym ze spadku absor-
bancji przy 286 nm po konwersji. /Patrz np. Ipata i Carcignani, 1978/. Alternatywnie, aktyw-
ność można określić za pomocą kontrolowania konwersji 5FC do SFU spektrofotometrycznie jak
opisali Nishyama i in./1985/, co włączono do niniejszego opisu Jako odnośnik.Termicznie etabilną CDazę oczyszczoną w ten sposób sekwencjonowano jak opisano w części
eksperymentalnej i częściowa sekwencja aminokwasów przedstawiona jest na fig. 6. Sekwencja nie wykazuje zasadniczej homologil z innymi sekwencjonowanymi białkami. Na podstawie tych informacji termicznie stabilną CDazę można wytwarzać technikami rekombinantowymi. I tak np., sekwencje DNA kodujące CDazę można otrzymywać syntetycznie w oparciu o otrzyma n ą sekwencję
aminokwasów, stosując odpowiednie kodony. Na ogół selekcjonuje się korzystne kodony ze wzglę-du na przewidywanego gospodarza używanego do ekspresji CDezy. Całkowitą sekwencję montuje się z zachodzących na siebie nukleotydów otrzymanych metodami standardowymi. Patrz np, Edge, Nature,

8 165 149
292, 756 /1981/i Nambair i in., Science, 223, 1299 /1984/t Jay i in., J. Biol. Cham.,
259, 6311 /1984/.
Alternatywnie, rekombinantową, termicznie stabilną CDazę można otrzymać jak następuje. Sondy oligonukleotydowe zawierające kodony dla części oznaczonej sekwencji aminokwasów moż-na otrzymać i użyć do screeningu zbiorów genomowych lub cDNA pod względem genu kodującego CDazę, Zasadnicze strategie otrzymywania sond oligonukleotydowych i zbiorów DNA, jak rów-nież przeprowadzanie ich screeningu przez hybrydyzację kwasów nukelinowych, są dobrze znane fachowcom w tej dziedzinie techniki. Patrz np. Oligonucleotide Synthesis, wyżej; T. Maniatis i in., wyżej i gdy klon z poddanego screningowi zbioru zidentyfikowano na podstawie pozytyw-nej hybrydyzacji, można przeprowadzić analizę enzymami restrykcyjnymi i screening DNA w celu
potwierdzenia, że określony insert ze zbioru zawiera gen kodujący CDazę. Dodatkowo polimery-
zowej reakcji łańcuchowej /PCR/ można użyć do amplifikacji i następnie wykrycia sekwencji nukleotydów kodującej CDazę. Metoda jest opisana przez Salki i in., /1986/ oraz w opisach
patentowych Stanów Zjednoczonych Ameryki nr nr 4683195 i 4683202, które to ujawnienia włą- czono do niniejszego opisu jako odnośnik. Analizę sekwencji nukleotydów PCR-amplifikowanych
produktów można przeprowadzić za pomocą bezpośredniej analizy sekwencji jak opisali Saiki
1 in. /1988/. Alternatywnie, amplifikowaną docelową sekwencję /sekwencje/ można klonować
przed analizę sekwencji. Metoda bezpośredniego klonowania i analizy sekwencji enzymatycz-nie amplifikowanych segmentów genonowych opisana została przez Scharfa i in. /1986/. W me-
todzie tej, startery użyte w technice PCR są zmodyfikowane blisko ich końca 5' w celu wy-tworzenia dogodnych miejsc restrykcyjnych do klonowania bezpośrednio do np. wektora sekwen-
cjonującego M13. Po amplifikacji produkty PCR rozszczepia się odpowiednimi enzynami restryk-cyjnymi. Fragmenty restrykcyjne liguje się do wektora M13 i transformuje nim np. gospoda-
rza IM103, nanosi na płytki i powstałe łysinki poddaje screeningowi na drodze hybrydyzacji
ze znakowaną sondę oligonukleotydową. Inne metody klonowania i analizy sekwencji są też znane w tej dziedzinie techniki.
W szczególnie korzystnym sposobie realizacji wynalazku zsyntetyzowano dwa startery oli-gonukleotydowe przy zastosowaniu częściowej sekwencji aminokwasów i modelu użycia kodonów
przez S. cerevisiae /Guthrie i Abelson, 1982/ jako klucza do wyznaczania sekwencji "guess- ner" sekwencji DNA. Tych starterów użyto do PCR, w której klonowany drożdżowy zbiór genomo- wego DNA był obecny jako matryca. Oligonukleotydy syntetyzowano z regionów CDazy, w których
aminokwasy wykazują znaczniejszą tendencję w zużytkowaniu kodonów i/lub siniej degeneracji.
Pierwszy starter, CDA4R1, był to 42-mer zawierający 33 nukleotydy odpowiadające eminowemu
końcowi sekwencji aminokwasów, podczas gdy drugi, CDA5AS, zawierał nukleotydy komplementar-
ne z sekwencją znajdującą się blisko karboksy-końca białka. Sekwencja tych oligonukleotydów
przedstawiona jest na fig. 7.Jako matryca DNA w reakcjach PCR wykorzystano dwie genomowe biblioteki i stwierdzono,
że obie dały pojedyńczy określony fragment, w przybliżeniu o długości 350 par zasad, wymiar przewidziany na podstawie dostępnej odpowiedniej sekwencji aminokwasów. Primery PCR obro-
biono za pomocą EcoRI miejąc restrykcyjnych na ich 5' końcach aby ułatwić klonowanie fragmen-tów generowanych przez PCR.
Fragment pochodzący z PCR o 350 parach zasad oczyszczono za pomocą elektroforezy żelowej, po czym subklonowano go do właściwego wektora do ustalania sekwencji DNA. Fragment pochodzą-cy z PCR wykorzystano również jako swoistą sondę CDazy dla skriningu bibliotek drożdży za
pomocą technik hybrydyzacji kolonii na filtrze. Początkowa usiłowania wykorzystania oligonu- kleotydów o przypadkowej długości jako sond nie powiodły się z powodu niskiego stosunku syg-
nał ;zakłócenia po hybrydyzacji. Zebrano wszystkie potencjalne klony i dwukrotnie je reskri- ningowano w celu sprawdzenia sygnału pozytywnej hybrydyzacji.
Plazmidy oczyszczono z pojedynczych klonów i restrykcje odwzorowano przez trawienie za
pomocą serii enzymów restrykcyjnych zarówno w pojedynczych jak i w różnych reakcjach wielo-
krotnego trawienia. Oba, klonowane fragmenty z PCR i genomowe klony kodujące CDazę poddano analizie sekwencji DNA. Zmiany warunków reakcji pozwoliły na analizę sekwencji bezpośrednio przylegających do primeru jak i tych oddalonych o kilka setek par zasad. Uzyskaną sekwencję

165 149 9
ONA przedstawiono na rysunku 8. Jak można zauważyć sekwencje są homologiczno w 93% posiada-jąc 23 nukleotydy niezgodne i 330 nukleotydy zgodne. ORF-y znalezione w tej sekwencji poka-zano na rysunku 9. Rysunek 10 przedstawia sekwencję nukleotydu i przewidzianą sekwencję ami-
nokwasu z ORF 2, potwierdzającą uprzednio określoną sekwencję aminokwasu i przewidującą do-danie tylko kilku aminokwasów do któregokolwiek końca cząstkowej sekwencji uzyskanej za po-mocą analizy oczyszczonego białka.
Dane sekwencji DNA wykorzystano następnie do projektowania nowych primerów PCR dla gene-rowania wzmocnionych kasetek DNA kodujących CDazę. Takie kasetki można stosować w konstruk- tach genetycznych zaprojektowanych do produkcji wysokich poziomów ekspresji genów w których- kolwiek prokaryotycznych lub eukaryotycznych komórkach oraz do generowania fuzji genowych
między CDazę a innymi cząsteczkami interesującymi z punktu widzenia biologii, lecz nieogra-niczonymi do cząsteczek immunoglobulin wycelowanych w antygeny raka. Sekwencją kodującą CDazę można klonować w jakikolwiek wektor lub replikon dobrze znany w tej dziedzinie wiedzy.
Przykłady wektorów będących produktami rekombinacji DNA służących do klonowania oraz
komórek gospodarzy, które mogą one przekształcać, w nawiasach, obejmują bakteriofagi lamb-da /E. coli/, pBR322 /E. coli/, pACYCl77 /E. coli/, pKT230 /bakterie gram-ujemne/, pCVll06
/bakterie gram-ujemne/, /pLAFR1 /bakterie gram-ujemne/, pME290 /nie- E.coli bakterie gram-
ujemne/, pHV14 /E.coli i Bacillus subtilis/, pBD9 /Bacillus/, pIJ61 /Streptomyces/, pUC6 /Streptomyces/, YIp5 /Saccharomyces/, YCp19 /Saccharomyces/, oraz wirus brodawczaka woło-
wego /komórki ssaka/.Sekwencję kodującą białko CDazy można umieścić pod kontrolą promotora, miejsca więżą-
cego rybosom /dla ekspresji bakteryjnej/ i alternatywnie operatora /zbiorczo określonych
tutaj Jako elementy "kontrolne", tak aby sekwencja ONA kodująca białko była skopiowana w RNA w komórce gospodarza przekształconej przez wektor zawierający tę konstrukcję ekspresji. Sekwencja kodująca może lub nie może zawierać peptyd sygnalny lub sekwencję wiodącą. Sek-
wencje wiodące można usuwać za pomocą bakteryjnego gospodarza w obróbce potrenslacyjnej.Patrz np, patent USA nr 4431739, 4425437, 4338397.
Dodatkowo, oprócz sekwencji kontrolnych, pożądane może być dodanie sekwencji regula-
cyjnych, które pozwalają na regulację sekwencji wyrażających CDazę zależnie od wzrostu komórki gospodarza. Sekwencje regulacyjne są dobrze znane specjalistom w tej dziedzinie wiedzy i przykłady zawierają te, które powoduję włączanie lub wyłączanie ekspresji genu
w odpowiedzi na chemiczny lub fizyczny bodziec, w tym obecność związku regulacyjnego. Inne
typy elementów regulacyjnych, na przykład sekwencje wzmacniające również mogę być obec-
ne w wektorze. Wektor ekspresyjny jest tak skonstruowany aby sekwencja kodująca CDazę była umieszczona w wektorze z właściwymi sekwencjami regulacyjnymi, usytuowanie i orientacja
sekwencji kodującej w odniesieniu do sekwencji kontrolnej są takie aby sekwencja kodująca była kopiowana pod "kontrolą" sekwencji kontrolnych /tzn. polimeraza RNA, które przyłącza
się do cząsteczki DNA w sekwencjach kontrolnych kopiuje sekwencję kodującą/. Aby uzyskać ten koniec pożądana może być modyfikacja sekwencji kodujących CDazę. Na przykład, w nie-
których przypadkach niezbędne może być modyfikowanie sekwencji tak aby mogła być ona przy-czepiona z właściwą orientację do sekwencji kontrolnych to znaczy, aby zachować ramkę od-
czytu. Sekwencje kontrolne i inne sekwencje regulacyjne można podwiązać do sekwencji ko-dującej przed wprowadzeniem do wektora, tak jak wektory klonujące opisane wcześniej.
Alternatywnie sekwencję kodującą można klonować bezpośrednio w wektor ekspresji, który zawiera już sekwencje kontrolne i odpowiednie miejsca restrykcyjne.
W tej dziedzinie znana jest pewna liczba prokaryotycznych wektorów ekspresji. Patrz
np. patenty USA nr 4440859, 4436815, 4431740, 4431739, 4428941, 4425437, 4418149, 4411994,
4366246, 4342832, zobacz również zgłoszenia patentowe Zjednoczonego Królestwa G3 2121054,GB 2008123, GB 2007675, i Europejskie zgłoszenie patentowe 103395. Wektory ekspresyjne drożdży na również znane. Patrz np. patent USA nr 4446235, 4443539, 4430428, zobacz też
Europejskie zgłoszenie patentowe 103409, 100561, 96491. Wektory ekspresyjne odnoszące się do ssaków są również znane.

10 165 149
W zależności od systemu ekspresji i wybranego gospodarza CDazę produkuje się hodując w warunkach ekspresji CDazy komórki gospodarzy przekształcone przez wektor ekspresyjny opisa-ny powyżej. Później białko wyodrębnia się z komórek gospodarzy i oczyszcza. Jeśli system ekspresji wydziela białko do środowiska hodowli, to może być one oczyszczane bezpośred-nio z niego. Jeśli białko nie jest wydzielane, wyodrębnia się je z lizatów komórek. Dobór odpowiednich warunków hodowli oraz sposobów odzyskiwania są dobrze znane specjalistom w tej dziedzinie.
Przykład jednego konstruktu do kontrolowanej indukcji sekwencji CDazy w drożdżach pole-ga na wprowadzeniu kasetki CDazy do wektora drożdży zwanego "fuzjonator". Wektor ten dostęp-ny jest w Department of Cenetics, University of Washington, Seattle, Wachington, oraz w NIH.
Taki konstrukt przedstawiony jest na rysunku 11. W konstrukcie tym CDazę wprowadza się do polilinkera, gdzie CDaza jest pod kontrolę silnego promotora z drożdży /Johnston, M. 1987/, który jest ściśle regulowany białkiem CALlO. Promotor CAL10 jest czuły na galaktozę, indu-
kując pod jej kontrolę ekspresję genu o wysokim poziomie. Wektor ten można również wykorzy-
stać do stworzenia fuzji CDeza-gen lac2 poprzez właściwe zmiany w sekwencji ONA. Białko ge-
nerowane fuzję może ułatwić ilościowe określenie indukcji, oczyszczanie i biochemiczną ana-lizę. Te konstrukty można transformować w dogodny szczep drożdży. Ten szczep drożdży zawie-
ra pewną liczbę mutacji, które sprawiają, że jest on wskazany do stosowania z takim kon- struktem jak opisano wyżej. Dokładniej, obecna jest mutacja gali powodująca deficyt dla
enzymu, który katalizuje pierwszy etap utylizacji galaktozy zapobiegając w ten sposób wyczer-
paniu w środowisku substancji indukującej /galaktozy/. Obecna jest również mutacja regi-501, która eliminuje represję glukozy ekspresji genowej galaktozy i pozwala na zakładanie kultur w warunkach optymalnych dla rozwoju i żywotności.
Indukcja może być łatwo osiągnięta przez dodanie galaktozy do środowiska opartego na
glukozie. Wreszcie w tym szczepie obecnych jest kilka mutacji z niedoborem proteazy, zwięk-szających stabilność jakichkolwiek białek heterologicznych lub ich fragmentów produkowanych
w czasie wzrostu. Taki konstrukt ekspresji powinien ułatwiać wyodrębnianie i oczyszczanie CDazy z drożdży w ilościach wcześniej niemożliwych do osiągnięcia, stosując poziomy ekspresji
uzyskane przy genie endogennym. Do stosowania w systemach kultur komórek ssaka można stwo-rzyć inne konstrukty. W jednym korzystnym wcieleniu wynalazku kasetka kodująca CDozę przy-
czepiona jest przy użyciu technologii PCR do sygnalnego peptydu wydzielania onkostatyny
M /Malik, 1989/ i może być później wprowadzona do wektora ekspresyjnego odnoszącego się do ssaka pH3MPy /Stamenkovic, /1990/, który zawiera właściwe wzmacniacze, promotory, zakoń-
czenia i sygnały obrabiające dla ekspresji ssaka. Konstrukt można transfektować w komórki
COS /Aruffo i Seed, 1987/, zebrane środowisko kultury pozbawione surowicy i zanalizować pod
względem aktywności CDazy w systemie pozbawionym czynności enzymu endogennego. Podobnie, konstrukty cDNA do fuzji genów między CDazą i innymi cząsteczkami będącymi przedmiotem
zainteresowania można konstruować za pomocą standardowych technik, transfektować do komó-rek COS a ekstrakty komórek lub ciecze znad osadów można analizować pod względem białek i ich czynności.
Inne fuzje genów mogę również znaleźć zastosowania w rekombinacyjnej produkcji CDazy.Na przykład sekwencja nukleotydowa kodująca CDazę lub funkcjonalny butant albo jej fragment może być przyłączony do cząsteczek immunoglobuliny kodujących cDNA wymierzonych w antygeny
raka, aby stworzyć fuzyjne białko przeciwciało-enzym. Uzyskano monoklonalne przeciwciała, które rozpoznaję determinanty korzystnie ulegające ekspresji w komórkach raka. /Hellatrom, 1984/. Zatem, można stworzyć białka fuzyjne, któro specyficznie celuję w wybrane komórki
raka. Patrz np. patent USA nr 4906562 i Hallstrom 1985. Enzym może działać bezpośrednio na siedlisko komórki raka aby zmienić prolek 5FC w przeciwrakowy czynnik 5FU. Genomowe fuzje między genami kodującymi enzym, będący obiektem zainteresowania a przeciwciałem wymierzonym
w komórki raka eliminuję konieczność chemicznego sprzężenia dwóch białek. Podobnie, kon-strukty cDNA między dwiema cząsteczkami będącymi przedmiotem zainteresowania mogą zwięk-szyć giętkość takich konstruktów w systemach ekspresji hoterologicznej.

165 149 11
W jednym systemie cDNA ciężkiego łańcucha, przeciwciała raka uzyskuje się z produkują-cych je linii komórek. Fragmenty PCR kodujące całość lub części tego cDNA można generować stosując dobrze znane w tej dziedzinie sposoby. Fuzję dwóch kasetek w ramce można uzyskać w kilku położeniach stosując standardowe techniki a powstałe fuzje zbadać pod względem produkcji białek o pożądanej czynności biologicznej. Kotransfekcja z drugim plazmidem nio- sącym cDNA kodujące lekki łańcuch będący częścią cząsteczki przeciwciała pozwala na wyo-drębnienie fragmentu przeciwciała czynnego biologicznie poddanego fuzji z enzymem czynnej CDazy. Zatem będzie można osiągnąć wycelowanie enzymu w pożądane miejsce. Produkcja mutan-tów lub analogów stabilnej termicznie CDazy jest również pożądana, z tym, że muszą być one funkcjonalnie z nią równoważne. Można je wytworzyć wymazując część sekwencji kodującej CDazę, wprowadzając jakąś sekwencję, i/lub przez podstawienie jednego lub więcej nukleoty-
dów w sekwencji. Specjaliści w tej dziedzinie dobrze znają techniki modyfikowania sekwencji
nukleotydu, takie jak skierowana w określone miejsce mutageneza albo mutageneza nukleotydu PCR.CDazę będącą przedmiotem niniejszego wynalazku syntetyzuje się chemicznie stosując ta-
kie syntezy jak synteza peptydów w fazie stałej i znane sekwencje aminokwasów, bądź też
sekwencja aminokwasów pochodzących z sekwencji DNA genu będącego obiektem zainteresowania.Takie sposoby znane są osobom wyszkolonym w tej dziedzinie. Termicznie stabilną CDazę można
stosować jako środek chemoterapeutyczny. A dokładniej, enzym ten można użyć in vivo do prze-
miany proleku 5FC w środek przeciwrakowy 5 FU. CDazę można więc podawać wspólnie z 5FC po-przez chirurgiczne wprowadzenie CDazy przy lub blisko siedliska raka i doustne podawanie 5FC, jak to opisał Katsuragi 1987, w pracy której tytuł można znaleźć w odnośnikach. Alter-
natywnie, enzym można dostarczać do siedliska raka przez wycelowanie dostarczania kompleksu
COaza/przeciwciało, na raka przy zastosowaniu przeciwciał specyficznych dla tych typów raka. Kompleks ten można wytworzyć sposobami chemicznymi, lub genetycznie konstruując fuz-je genów między właściwym genem immunoglobulinowym i genem CDazy, jak to opisano wcześniej.
Poniżej zamieszczono przykłady szczegółowych zastosowań niniejszego wynalazku. Mają one na
celu jedynie zilustrowanie go a nie ograniczenie jego zakresu w jakikolwiek sposób.
P r z y k ł a d I. Oczyszczanie enzymu, oznaczanie czynności i charakteryzacja CDazy.CDazę oczyszczono za sprasowanych drożdży piekarskich Flelshmann'a stosując następujący
sposób. Wszystkie etapy oczyszczania przeprowadzono w 4°C. Czynność enzymu oznaczono za pomocą 5' fluorocytozyny /5FC/ przy 3 mM w roztworze soli zbuforowanym fosforanem /PBS/ w
37°C /Nishiyama, 1985/. Po dodaniu roztworów enzymu przebieg reakcji śledzono spektrofoto- metrycznie na podwielokrotnych częściach próbek, w których przerwano reakcję stosując 0, 1 N
roztwór HCl. Do zmierzenia ilości utworzonego 5-fluorouracylu /5FU/ wykorzystano stosunek absorbancjl 255/290. Jednostkę czynności enzymu zdefiniowano jako 1 mol utworzonego 5FU
na minutę w 37°C. Stężenie białka zmierzono stosując analizę BCA dostępną w Pierce /Rock-
ford, II/. Do monitorowania składu białka po każdym etapie oczyszczania wykorzystano SOS-PAGE.
Etap 1. Wytwarzanie autolizatu drożdży.Drożdże piekarniane /2,25 kg/ dodano do octanu etylu /225 ml/, po czym mieszano 30 mi-
nut. Następnie przy pH 7,2 dodano 2,25 1 50 mM buforu fosforanu potasu zawierającego 15%
siarczanu amonu oraz 5 mM EDTA i 0,1 mM ditiotreitolu /DTT/. Po 3 dniach mieszania i do-prowadzania codziennie pH do wartości 7,2 za pomocą stałego trihydroksymetyloaminometanu / T r i s / , usunięto resztki komórek przez trwające 15 ninut odwirowanie przy 10000 rpm.
Etap 2. Frakcjonowanie siarczanem amonu
Z cieczy sklarowanej osadem z etapu 1 wytrącono całe białko dodając EDTA /1,8 g/l/ i siarczanu amonu /371 g/l/, tak, aby stężenie siarczanu amonu było równe pod koniec 70% roztworu nasyconego. Roztwór trzymano 16 godzin w 4°C, po czyn osad zebrano po odwirowa- niu. G r u d k ę rozpuszczono w 1,5 l 50 mM roztworu buforowego fosforanu potasu przy pH 7,2
zawierającego 5 mM EDTA i 0,1 mM DTT, a następnie przez około 12 do 16 godzin poddawano go dializie. Do dializatu dodano EDTA /1,8 g/l/ i siarczanu amonu w takiej ilości aby osiągnąć
50% nasycenia /314 g siarczanu amonu/litr dializatu/. Po 1 godzinie strącony osad odwirowa-
no i do sklarowanej cieczy dodano dod8tkowę porcję siarczanu amonu aby jego końcowa stęże-
nie wynosiło 73% nasycenia. Strącony osad zebrano, rozpuszczono w 1 litrze 20 mM roztworu
12 165 149
buforowego Tris przy pH 8,0 zawierającego 0,1 mM DTT i przez około 12 do 16 godzin poddawano
go dializie.Etap 3. ChromatografiaDializat z etapu 2 naniesiono na kolumnę 4,8 x 25 cm Q-Sepharosa /Pharmacia/, którą
zrównoważono w 20 mM Tris zawierającym 0,1 mM DTT, przy pH 8,0. Po przemyciu kolumny wymywa-no enzym stosując powyższy bufor o linearnym gradiencie 0-0,3 M KCl. Frakcje zawierające CDazę zlano i zatężono przez ultrafiltrację /Amicon, PM 30-filtr/ do, w przybliżaniu 15 ml.
Etap 4. Chromatografia żelowaRoztwór z etapu 3 zawierający CDazę naniesiono na kolumnę G-75 Sephadex /2,5x100 cm/ i
eluowano stosując PBS zawierający 0,1 mM DTT. Frakcje z CDazę zlano i poddano dializie w 4 litrach 100 mM roztworu buforowego fosforanu potasu zawierającego 1,8 M siarczanu amonu,
5 mM roztwór EDTA oraz 0,1 mM DTT, przy pH 7,0.Etap 5, Chromatografia hydrofobowa
Materiał z etapu 4 naniesiono na kolumnę /2,5x15 cm/ oktyl-Sepharose /Pharmacia/, którą zrównoważono 100 mM fosforanu potasu zawierającego 1,8 M siarczanu amonu, 5 mM EDTA i 0,1
mM DTT, przy pH 7,0. Kolumnę przemyto tym buforem, a enzym eluowano stosując linearny gra-dient 1,8-0 M siarczanu amonu w powyższym buforze fosforanowym. Frakcje zawierające CDazę
połączono i zatężono przez ultrafiltrację /Amicon, filtr YM5/.
Etap 6. Chromatografia żelowaKońcową filtrację żelową materiału z etapu 5 wykonano na kolumnie G-75 Sephadex stosu-
jąc jako eluent PBS w sposób opisany w etapie 4. Oczyszczony enzym zatężono przez ultrafil-
trację i przechowywano w -70°C.
Rezultaty oczyszczania w każdym etapie są zamieszczone w tabeli 1. Profile SDS-PAGE służące
do kontrolowania składu białka po każdym stenie oczyszczanie podano na rysunku 1. Końcowy preparat enzymu składał się z głównego pasma przy około 17 kDa oraz pasma mniejszego wystę- pującego przy około 19 kDa.
T a b e l a 1
Oczyszczanie CDezy otrzymanej z 2,25 kg drożdży piekarnianych
Etap Białko sumarycznie
/mg/1
Czynnośćsumaryczna
/U/2
Czynnośćwłaściwa/U/mg/
Stopieńoczyszcze-nia
ekstrat pozbawiony komórek 120000 1750 0,014 1
/NH4/2so4 36000 1274 0,035 2,5
Q-Sepharosa 4200 685 0,16 11,4
G-5 Sephadex 184 648 3,5 250
Oktyl-Sepharose 20 495 25 1800
G-75 Sephadex 6 394 67 4800
1 Stężenie białka oznaczono za pomocą BCA /Pierce/.
2 Jednostkę czynności enzymu zdefiniowano jako 1 μ mol 5 FU utworzonego w ciągu jednej
minuty w 37°C.
Masę cząsteczkową CDazy wyznaczono nanosząc oczyszczony enzym na kolumnę G-60 Sephadex razem z rybonukleazą A /13,7 kDa/, chymotrypsynogenem A /25 kDa/ i albuminę jaja kurzego /43 kDa/, Frakcje kontrolowano przy 280 nm aby zmierzyć białko sumaryczna i czynność CDazy przy zastosowaniu 5FC jako substratu. Czynność enzymu CDazy skoncentrowała się przy w przy-bliżeniu 32 kDa /Rysunek 2A/. Enzym eluowano dokładnie w takiej samej objętości w jakiej go naniesiono na kolumnę bez wzorców kalibracyjnych /rysunek 2B/.

165 149 13
Stabilność oczyszczonej CDazy w 37°C w roztworze soli zbuforowanej fosforanom przy pH
7,2 oznaczono za pomocą 5 FC jako substratu. Zaobserwowano niewielkie zmniejszenie czynnoś-ci enzymu po przedłużonej inkubacji /rysunek 3/. W tych warunkach czynność enzymu obniżyła się o połowę po upływie 5,2 dnia. W pierwszych 4 godzinach inkubacji nie zachodziło widocz-ne zmniejszenie czynności enzymu /rysunek 3/.
P r z y k ł a d II. Sekwencja aminokwasów w CDazieReagenty wykorzystane przy ustaleniu sekwencji CDezy uzyskano z Apolied Biosystems, Inc.
Rozpuszczalniki stosowane w HPLC o odwróconych fazach pochodziły z firmy Budrick and Jack-
son, 4-winyloplrydyna pochodziła z Aldrich Chemical Co, CNBr z Kodak i wszystkie inne sub-stancje chemiczne miały stopień czystości odczynników. Endoproteinazę Glu-C z Stephylococ-
cus sureus uzyskano w Miles Leboratories, Enkoproteinazę Asp-N z Pseudomonas fragi i endo-proteinazę Lys-C otrzymano w Boehringer Yannheim.
Automatyczną analizę sekwencji wykonano na aparacie /ABI/ model 475A stosując programy
RUN470-L lub PRC470-L, Przed naniesieniem próbki użyto 1,5 mg BioBrena /ABI/ i poddano go
dwum lub trzem precyklom degradacji Edman'a. Konwersja pochodnych tiazolinonu do fenylotio-
hydantoino aminokwasów została przeprowadzona za pomocą 25% TFA w 61°C. Fenylotriohydantoi- nowe pochodne aminokwasów rozdzielono za pomocą HPLC o odwróconych fazach na kolumnie PTH
C1B /2,1x220 mm, ABI/ stosując jako bufor rozpoczynający roztwór buforowy octanu sodu zawie-
rający 5% /objętość/ objętość/tetrahydrofuranu a jako bufor ograniczający acetonitryl zawie-rający 500 nM dimetylofenylotiomocznika /ABI/. Wykorzystano analizator 9/ABI/ model 120A PTH. CDazę i fragmenty białka oczyszczono za pomocą HPLC o odwróconych fazach na urządzeniu do
rozdzielania model 130A /Apolied Biosystems, Inc./. Rozdzielanie przeprowadzono w 40°C na kolumnie RP-300 /2,1x30 mm) ABI/. Do wymycia białek i fragmentów peptydowych stosowano linio-
wy gradient 0-60% acetonitrylu w 0,1% wodnym roztworze kwasu trifluorooctowego /TFA/ w cza-sie 2 godzin z prędkością 0,1 ml/minutę. Peptydy CNBr, endoproteinazę Lys-C, oraz peptydy
endoproteinezy Clu-C i endoproteinazy Asp-N użyto w analizie sekwencji bez dalszego oczysz-
czania.Reakcja CDazy z 4-winylopirydyną
CDaza z przykładu 1 etap 6 została zmodyfikowana przez dodanie 4-winylopirydyny w na-stępujący sposób. CDazę zredukowano za pomocą 20 mM ditiotreitolu w 0,1 ml 0,4 M buforu
Tris-HCl, pH 8,5, zawierającego 6 M roztwór guanidyna-HCl, 0,1% Na2EDTA. Reakcję prowadzo-no 2 godziny w 50°C. Następnie dodano 100 mM 4-winylopirydyny i pozostawiono na całą noc w
temperaturze pokojowej. Mieszaninę reakcyjną zakwaszono do pH 2,0 stosując 20% TFA, po czym
ją odsolono i oczyszczono za pomocą HPLC o odwróconych fazach jak to opisano wcześniej. Ana-
liza HPLC powstałych produktów wykazała obecność dwóch wyraźnych pików /rysunek 4/, które
rozdzielono i ponownie analizowano przy użyciu SDS-PAGE /rysunek 5/. Mniejszy pik odpowia-dał pasmu 19 kDa /pole A/ z rysunku 1, a pik większy odpowiadał pasmu 17 kDa.
Końcową grupę aminową sekwencji aminokwasów białek w polu A /rysunek 4/ uzyskano przezidentyfikację pochodnych aminokwasów fenylotiohydantoinowych aż do pozostałości 27. Stwier-
dzono, że sekwencja ta jest identyczna z sekwencją N-terminalną ponadtlenku diamutazy z drożdży piekarnianych /EC 1.15.1.1, Steinman, 1980/. To dowodzi identyczności pasma mniej-szego obserwowanego w żelach oczyszczonej CDazy /rysunek 1,5/.
Z degradacji Edman'a dużego piku z rysunku 4 /pole B/ nie uzyskano żadnej sekwencji ami-nokwasów. To sugerowało, że zakończenie aminowe CDazy było zablokowane. Dlatego też częściową sekwencję aminokwasów CDazy uzyskano ustalając sekwencję wybranych fragmentów z enzymatycz-nej i bromo cyjanowej degradacji, w sposób opisany poniżej.
Enzymatyczne i chemiczne rozszczepienia CDazy.Rozkładu CDazy zmodyfikowanej 4-winylopirydyną dokonano za pomocą endoproteinazy Lys-C
i endoproteinezy Glu-C w 40 l 0,1 M roztworze Tris-kwas octowy /pH 8,0/. Reakcję prowadzono w 37°C przez 12 do około 16 godzin. Stosunek enzymu do substratu wynosił odpowiednio 1,10 /masa/masę/. Rozkładu endoproteinazą Asp-N dokonano w podobny sposób przy stosunku enzym/ substrat 1:100 w 37°C w ciągu około 12 do 16 godzin. Mieszaniny po trawieniu enzymatycznym
zakwaszono 20% TFA do 2,0 pH i rozdzielono za pomocą HPLC o odwróconych fazach.

14 165 149
Do rozkładu CDazy w resztkach metionylowych użyto bromocyjanu/CNBr/. CDazę /2,25 moli/, która była zmodyfikowana 4-winylopirydynę rozpuszczono w 40 l 70% roztworu kwasu mrówkowego, po czym dodano 200-krotny molarny nadmiar CNBr w 70% kwasie mrówkowym. Przez pierwsze 2 godzi-ny mieszaninę reakcyjną utrzymywano w 35°C a przez następne około 12 do 16 godzin w tempera-turze pokojowej. Po rozcieńczeniu wodą i suszeniu pod próżnią w celu usunięcia nadmiaru CNBr, przygotowano roztwór do analizy HPLC o odwróconych fazach stosując 1% TFA.Rysunek 6 przedstawia częściową sekwencję aminokwasów CDazy uzyskaną w oparciu o fragmenty otrzymane z rozkładu za pomocą bromocyjanu i rozkładu proteolitycznego z zastosowaniem enzy- mów: andoproteinazy Glu-C, endoproteinazy Lys-C oraz endoproteinazy Asp-N. Zidentyfikowano
154 aminokwasy, które składają się na masę białka. Numeracja resztek aminokwasów nie obejmu-je tych aminokwasów blisko zakończeń aminowych, które nie były dotychczas zidentyfikowane.
Porównanie częściowej sekwencji CDazy z jakimikolwiek innymi znanymi sekwencjami, w tym adenozynodeaminazę myszy /Young, 1985/ i deaminazę drożdży piekarnianych dCMP /McIntosh and Haynes, 1986/ nie ujawniły istotnej homologii.
P r z y k ł a d III. Klonowanie i ekspresja genu CDazy
Gen kodujący CDazę wyodrębniono ze szczepów laboratoryjnych S.cerevisiae /drożdże pieka rniane/ i ekspresjonowano w komórki ssaka postępując w następujący sposób :
Etap 1: Generowanie specyficznej sondy CDazy za pomocą PCR
Dwa primery nukleotydowe zsyntetyzowano na zautomatyzowanym syntetyzerze DNA stosując
częściową sekwencję aminokwasów oraz wzory kodonów z S.cerevisiae /Guthris and Abelson, 1982/ jako przewodnik w projektowaniu przypadkowych segmentów sekwencji DNA. Te oligonukleotydy
wykorzystano później jako primery w PCR z genomową bibliotekę DNA klonowanych drożdży obec- ną jako matryca. Primery nazwane CDA4R1 i CDA5AS, oba były 42-merami zawierając 33 nukleo
tydy o sekwencji odpowiadającej niciom CDazy odpowiednio sensownej i antysensownej. Pozosta-łość każdego nukleotydu kodowała miejsce enzymu restrykcyjnego dla EcoRI. Sekwencja dwóch primerów przedstawiona jest na rysunku 7.
Oba primery obecne są w reakcji w ilości albo 50 albo 100 pmoli. Dwie genomowe biblio-teki użyte jako matryce DNA w reakcjach PCR skonstruowano z genomowych DNA drożdży stosując
Sau3A częściowe trawienie restrykcyjne, a następnie podłączanie do miejsc BamHI wektora bakteryjno-drożdżowego typu CV13 lub YCp50. Te dwa wektory oraz biblioteki genomowe są ogólnie dostępne i uzyskano je z Departament of Genetics, University of Washington. Obie biblioteki były już przetransformowane do gospodarzy bakteryjnych. Bibliotekę DNA wyodręb-
niono przez zasadową lizę preparatów plazmidowych, które oczyszczono następnie przez odwiro-
wanie stosując gradienty gęstości chlorek cezu-bromek etydyny. Nastawiono reakcje PCR uży-wając 1 g CV13 oczyszczonego za pomocą CaCl lub YCp50 genomowych bibliotek DNA z drożdży,
50 lub 100 pmoli każdego primeru, 1/10 objętości 10x bufor reakcji PCR /Stratagene/, 16/100
objętości 1,25 mM dNTP's /A, G, T, C/, destylowanej wody klasy PCR oraz 0,5 μ l Taq DNA polimerazy /5U/ /μl, Stratagene/ do końcowej objętości 100 μl. Reakcje przeprowadzono w
sterylnych mikroprobówkach do wirówki pod 75 l warstwą oczyszczonego oleju mineralnego aby
zapobiec odparowaniu. Reakcje PCR prowadzono w Perkin-Elmer Cetus Thermal Cycler stosując
program 30 cyklowy z następującymi zmianami temperatury: etap cyklu 94°C przez 30 sekund
aby zdenaturować, 55°C przez 45 sekund aby wystudzić, 72°C przez 1,5 minuty aby /90 se-
kund/ aby przedłużyć. Podwielokrotności próbek z reakcji PCR analizowano na żelach aga- rozowych aby uzyskać dowód na istnienie przewidzianego fragmentu o 350 parach zasad kodu- jącego większą część CDazy. Następnie fragmenty o 350 parach zasad oczyszczono za pomocą
preparatywnej elektroforezy na żelu agarozowym, po czym eluowano je wykorzystując sposób
postępowania i odczynniki dostarczone w zestawie GeneClean /BIOlOl/. W ten sposób usunięto
primery i fragmenty heterologiczne z preparatu.Etap 2: Subklonowanie fragmentów PCR do ustalania sekwencjiPrimery PCR z etapu 1 obrobiono w EcoRI miejscach restrykcji na ich 5' końcach aby
ułatwić klonowanie jakichkolwiek wygenerowanych fragmentów. Podwielokrotność próbki oczysz-czonego fragmentu o 350 parach zasad pochodzącego z PCR trawiono enzymem restrykcyjnym EcoRI i podłączono do cięcia EcoRI wektor kosmidowy pBSII-SK+ /Stratagene/. Enzymy restryk-

165 149 15
cyjne uzyskano z Boehringer Mainnheim Biochemicals /BMB/. Po dwugodzinnym inkubowaniu w 37°C
przeprowadzono ekstrakcję stosując równe objętości fenolu i chloroform j: alkohol izoamylowy, a następnie zastosowano wytrącanie etanolem. Fragmenty ponownie zawieszono w TE /10 mM Tris-HCl pH 8,0; 1 mM EDTA pH 8,0/, po czym złączono razem wykorzystując T4 DNA Ligase /BMB/. Kompetentne bakterie JM109 przetransformowano za pomocą reakcji podwiązania i w od-powiednich rozcieńczeniach położono na płytki z IB+ampicylina /100 μg / m l / uzupełnione
XGal /5O μ l 2% zapas w dimetyloformamidzie/ oraz IPTC /10 μ l100 mM zapasu/. Zabrano białe kolonie i wykonano preparaty plazmidów w małej skali według przepisu podanego przez Sambrook'a /1989/. Plazmidy skriningowano pod względem wstawek 350 par zasad poprzez tra-wienie EcoRI. Wybrano kilka różnych izolatów, wyodrębniono plazmidy ONA, oczyszczono je i poddano analizie sekwencji DNA.
Etap 3: Wykorzystanie fragmentu PCR jako specyficznej sondy CDazy do skriningowania genomowych bibliotek drożdży
Fragment PCR uzyskany w etapie 1 wykorzystano również jako sondę do detekcji sekwencji kodującej CDazę w skriningowaniu bibliotek genomowych pod względem klonów zawierających
ten gen. Oczyszczony fragment oznakowano za pomocą α - 3 2 -dCTP i zestawu do oznakowania
ONA przypadkowego primeru z firmy Boehringer Mannheim Biochemicals. Biblioteki drożdży ho-
dowano w ciągu nocy w LB+amp i w postaci rozcieńczonej umieszczono je na płytkach z LB+amp.,
tak aby na płytce znajdowało się 250-2000 kolonii. Kolonie przenoszono za pomocą krążków z nitrocelulozy 0,45 μ m lub kwadracików produkowanych przez Schliecher and Schuell.
Sączki przerobiono w ten sposób, że położono je koloniami na papier 3MM Whatman nasycony
przez 5 minut 10% SDS aby związać, 0,5M NaOH, 1,5M NaCl przez 5 minut aby zdenaturować,1,5 M NaCl, 0,5M Tris-HCl pH 7,4 przez 5 minut aby zneutralizować i 2 X SSC przez 5 minut przed poprzecznym usieciowaniem UV za pomocą Statalinker /Stratagene/, stosownie do instruk-
cji producenta. Sączki następnie zanużono w 2 X SSC przez 5 minut i przemyto 5 X SSC, 0,5%
SDS, 1 mM EDTA w temperaturze 50°C.Prehybrydyzację wykonano w zgrzanych torebkach plastykowych zawierających 20 ml roztwo-
ru na torebkę z 12 sączkami. Roztwór prehybrydyzacyjny zawierał 50% formamidu, 6 X SSC, 0,01M NaP pH 6,8, 1 mM EDTA /pH 8,0/, 0,5% SDS, 100 μg/ml DNA zdenaturowanej spermy łososia,oraz 5X Denhardt's Solution. Plastikowe torebki zanurzono w kąpieli wodnej o temperaturze
42°C i inkubowano przez około 12 do 16 godzin. Oznakowaną sondę oczyszczono z nie wprowa-dzonych nukleotydów poprzez elucję z Push-Columns /Stratagene/. Sondę dodano w przybliżo-
nym stężeniu 10*6 cpm/ml cieczy i inkubowano przez około 12 do 16 godzin w 42°C aby nastąpiła
hybrydyzacja. Sączki przemyto kilkakrotnie w 2 X SSC, 0,1% SDS w temperaturze pokojowej,
a następnie pozostawiono w kąpieli 1 X SSC, 0,1% SDS w 55°C przez 1 godzinę. Sączki wysu-szono na powietrzu, opakowano w folię saran. Błonę rentgenowską z ekranami wzmacniającymi
wystawiono na działanie sączków w - 7O°C. Autoradiografy ustawiono w szeregu z sączkami i macierzystymi płytkami w celu wybrania klonów dających sygnał pozytywnej hybrydyzacji.Przed wyodrębnieniem plazmidu pozytywne klony dwukrotnie reskriningowano w podany wcześniej
sposób. Plazmidy DNA wyodrębniono przez hodowlę kultur na 500 ml roztworu LB+ampicilyna, wzmocnionego chloramfenikolem a następnie przez ich oczyszczenie za pomocą zasadowej lizy, w sposób opisany przez Sambrook'a /1989/. Niektóre plazmidy oczyszczono stosując odwirowa-nia równowagowe z chlorkiem cezu/bromkiem etydyny, natomiast inne eluowano z kolumn PZ253 /5 ' — >3'/ a następnie wytrącono glikolem polietylenowym, ekstrahowano mieszeninę fenol:
:chloroform po czym wytrącono etanolem.Etap 4: Ustalenia sekwencji klonów pochodzących z PCR oraz klonów genomowych kodują-
cych CDazęW celu ustalenia sekwencji DNA z klonów PCR i genomowych klonów CDazy zsyntetyzowano
lub zamówiono kilka 18-merowych oligonukleotydów. Niektóre primery generowały sekwencję z nici kodującej, a inne dawały sekwencję z nici komplementarnej. Reakcje ustalające sek-wencje prowadzono według przepisów i na odczynnikach dostarczonych z wyposażeniem Sequenase
Version 2,0 Kit/ United States Biochemical/. Przed reakcjami ustalającymi sekwencję zdena- turowano DNA w roztworze zasadowym, stosując 0,2 N NaOH. Rozcieńczenie mieszanin reakcji

16 165 149
znakującej oraz czasy trwania reakcji zmieniano zależnie od obszaru, w którym miała być usta-lana sekwencja. W większości reakcji użyto po 1-1,5 pmoli matrycy DNA i primerów z 10-15 μ Ci
α-35S-dATP. Próbki naniesiono na żele do ustalania sekwencji, składająca się z 8% polia- kryloamid-mocznlk i rozwijano je przez 1-7 godzin zależnie od sekwencji, która miała być odczytana. Żele utrwalono w 10% metanolu, 10% kwasie octowym przez 30 minut i suszono 45 mi-nut w 60°C za pomocą suszarki Slab-Gel połączonej z pompą próżniową. Następnie błonę Kodak XAR-5 wystawiono na działanie wysuszonych żeli w temperaturze pokojowej przez 16-72 godzin. Sekwencje odczytano manualnie. Orientację i analizę danych sekwencji wykonano za pomocą kom-putera Gene Pro Software /Riverside Scientific, Seattle, Washington/. Rysunek 8 przedstawia sekwencję nukleotydów genomowych klonów kodujących CDazę. Rysunek 10 obrazuje przewidywaną
sekwencję aminokwasów. Jak można zauważyć na rysunkach sekwencja kodująca składa się w przy-
bliżeniu z 474 par zasad i określa białko o 158 aminokwasach. Przewidywana masa cząsteczkowa uzyskanej w ten sposób CDazy wynosi około 17506 daltonów.
Etap 5: Konstruowanie kasetek kodujących CDazę do ekspresji w drożdżach i systemach
ssaków.Sekwencję DNA uzyskaną w etapie 4 wykorzystano przy projektowaniu primerów oligonukle-
otydowych do generowanych za pomocą PCR kasetek kodujących CDazę. Niektóre z tych oligonukle-
otydów zawierały w sobie dodatkowe sekwencje takie jak peptydy sygnalne wydzielania, oraz miejsca restrykcyjne, które mogą być wykorzystane przy klonowaniu lub ekspresji. Oligonukle-
otydowe primary stosowano w parach do generowania fragmentów PCR z właściwymi sekwencjami dodanymi na końcach.
Dokładniej kasetę kodującą CDazę skonstruowano za pomocą PCR stosując dwa primery za-wierająca zarówno specyficzną sekwencję CDazy jak i dodatkową sekwencję kodującą miejsca restrykcyjne do klonowania. Primer 5' był 65-merowy i zawierał 45 zasad o sekwencji homo-logicznej, lecz niecałkowicie identycznej z końcem 5' genomowej kopii CDazy przylegającej
do ogonka 5' zawierającego miejsca restrykcyjne HindIII, Sali i NcoI. 39-merowy primer 3' był identyczny z antysensowną nicią C-zakończenia CDazy, z ogonkiem XbaI przyłączonym do jego końca 5; Jako matrycę stosowano genomowy klon CDazy w ilości, w przybliżeniu 1 ng na reakcję. Każdy primer obecny był w stężeniu 75 pmol. Reakcje PCR prowadzono w sposób
opisany w etapie 1. Produkty reakcji PCR oczyszczono ekstrahując chloroformem a następ-nie wytrącając etanolem. DNA trawiono enzymami restrykcyjnymi HindIII, i XbaI, poddano
elektroforezie żelowej i oczyszczono przez elucję ze pomocą GeneClean /B10101/ stosownie
do instrukcji producenta.
Etap 6: Ekspresja CDazy w systemie ssakaOczyszczony fragment z etapu 5 podwiązano do wektora plazmidowego CDM8 lub pH3M,
który był trawiony HindIII-XbaI i przetransformowano go do gospodarza bakteryjnego MC1061
/Aruffo and Seed, 1987/. Mapa wektora pH3M przedstawiona jest na rysunku 12. Zebrano 20 transformantów, przeprowadzono na małą skalę zasadową lizę preparatów plazmidowych a pod-
wielokrotności próbki trawiono HindIII i XBaI, aby sprawdzić ich budowę. Pozostały z 3
preparatów plazmid użyto do transfekcji komórek COS metodą DEAE-Dextran /Ausubel, Cur-
rent Protocols in Molecular Biology/. Opisując w skrócie komórki COS w ilości 3,5x10*5
włożono do 6 cm naczyń z DMEM/ 10% surowicy z płodu wołu /FBS/ i inkubowano przez noc w 6% CO2 w 37°C. DNA z mini-preparatów wymieszano z PBS i 50 mg/ml DEAE-Dextran do końco-wej objętości 100 l w ilościach wyszczególnionych poniżej.
Próbka DNA FBS DAEDEAE-Dextran /50 mg/ml/
1 2 3 4
Próbki kontrolne Rzekome 0 μ l
BB1 4 μ l80 μ l 20 μl
76 μl 20 μ l

165 149 17
1 2 3 4
TransfektantyCDazy
1-1 13 μl 67 μl 20 μl1-4 13 f i l 67 μl 20 μl2-6 13 μl 67 μl 20 μl
Każda transfekcja została zdublowana. Środowiska kultur hodowanych 1 dzień wyssano z na-czyń a komórki myto trzy razy w 2 ml PBS. Medium do transfekcji DMEM - chlorokin naniesiono
w ilości 1,7 ml na płytkę, po czym wkroplono do niego koktajl DNA. Transfekcje inkubowano
3 godziny, środowisko usunięto, komórki na 2 minuty poddano działaniu mieszaniny FBS /10% DMSO, wypłukano trzykrotnie w medium pozbawionym surowicy i inkubowano 3 dni w mieszaninie
3,5 ml DMEM/ 10% PBS.
Etap 7: Charakterystyka produktów rekombinacyjnych ekspresji
1. Reakcja strącania przy użyciu radioaktywnie znakowanych przeciwciał
Płytki z etapu 6, które miały być użyte do wytrącania radioimmunologicznego /RIPS/ inkubowa-no około 12 do 16 godzin w świeżym DMEM /10% FBS zawierającym 200 μ Ci/ml 35S-metloniny, RIPS wykonano zasysając oczyszczone środowiska po okresie około 12 do 16 godzinnej inkubacji
w 37°C, dodając 1 ml 1% 0G-P04RIPAE / 50 mM Na2HPO4, 1% Nadeoxycholate, 1% trytonu X-100,0,1% SDS, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 2 mM PMSF i 1 μg/ml aprotinin=P04-RIPAE z okto- glukozydem dodanym do 1% /buforu do każdego naczynia z kulturę, i inkubując 10 minut na lo-dzie. Lizaty przeniesiono do 10 ml probówek do wirówek Dak Ridge i odwirowywano przy 40000 rpm w 4°C przez 40 minut. Ciecze sklarowane osadem przeniesiono do dwóch probówek do mikro- wirówek na lodzie /0,4 ml na probówkę/. Transfektanty BB1 inkubowano za pomocą 2 g przeciw-
ciała, natomiast do innych dodano 17,5 lub 31,5 g króliczej surowicy przeciwko poliklonalnym przeciwciałom CDazy. RIPS inkubowano na lodzie przez 1 godzinę. Do probówki BB1 jako drugo-rzędnego przeciwciała dodano koziej surowicy przeciwko myszy i inkubowano 20 minut na lodzie.
Staphylococcus aureus /Calbiochem/ wypłukano w 700 μ lbuforu, 0,5% NP-40 /TES, potem w bu-
forze 0,05% NP-40 /TES i zawieszono w buforze P04-RIPAE z 1 mg/ml białka jaja kurzego /TES = 50 mM Tis-HCl, pH7,4, 1 mM EDTA, 150 mM NaCl/. Próbki 50 mikrolitrowe bakterii przemytych
StephA dodano do materiału RIP i inkubowano 10 minut na lodzie. RIPS zgranulowano w mikro-
wirówce i trzykrotnie przemyto 0,5 ml TWEN /20 mM Tris-HCl pH 8,0, 100 mM NaCl, 1 mM EDTA, 0,5% NP-40/. Grudki z w y j ś c i o w y c h 400 l podwójnych próbek zawieszono albo w 35 l redukują
cego lub w 35 l nieredukującego buforu roboczego SDS-PAGE. Próbki naniesiono na żel z gra-dientem 10-20% SDS-PAGE wraz z markerami białka o niskiej masie cząsteczkowej z Bio-Rad i
poddawano elektroforezie przy 300 V przez, w przybliżeniu 2 godziny. Żele barwiono w Coomassie Blue przez 1 godzinę, odbarwiono w 10% metanolu, 5% kwasie octowym przez około 12 do 16
godzin, wystawiono do wzmocnienia przez 30 minut, później płukano 3 razy w wodzie i suszono 1 godzinę w 60°C. Wystawienie błony rentgenowskiej przez 4 godziny na działanie żelu wykazało
nieobecność w próbkach rzekomych pasma przy 17000 daltonów, brak pasma 17000 daltonów w prób-kach BB1, w których natomiast obecne było specyficzne pasmo około 45000 daltonów, podczas gdy wszystkie 3 transfektanty wykazywały specyficzne pasmo przy około 17000 daltonów.
2. Analiza Western:Identyczne płytki do transfekcji z etapu 6 pozostawiono nieoznakowane i zebrano je po trzy-dniowym okresie inkubacji. Środowiska wyssano, komórki przepłukano w PBS, i zawieszono w 0,5 ml 10 mM Tris-HCl /pH 8,0/ zawierającego 1 μg/ml aprotyniny i 30 μg/ml PMSF. Komórki prze-niesiono z naczyń do hodowli za pomocą sterylnych skrobaczek do sterylnych probówek do mikro- wirówek na łodzie. Każdą zawiesinę poddawano działaniu dźwięków w 2-15 sekundowych uderzeniach z całą mocą. Lizaty odwirowywano 20 minut w chłodzonej mikrowirówce, aby usunąć szczątki ko-mórek, a ciecze sklarowane osadem przeniesiono do świeżych probówek. Następnie przeprowadzono

18 165 149
oznaczanie enzymów w sposób opisany poniżej. Próbki tych lizatów również stosowano do wyko-nania Western blot-ów używając króliczej poliklonalnej przeciwsurowicy wyhodowanej przeciw-ko oczyszczonej CDazie. Dwudziestopięciomikrolitrowe próbki z nierozcieńczonych transfekto- wanych lizatów poddano elektroforezie SDS-PAGE, tak jak to opisano dla RIPS, z takim wyjątkiem, że jako standardy stężenia na kolumnę naniesiono serię roztworów oczyszczonej CDazy. Stężo- ną CDazę przy 500 ng/tor rozcieńczono w 5-krotnych przyrostach do 100 ng, 20 ng, 4 ng i 0,8 ng na tor. Następnie żel został przeniesiony na filtr z bibuły za pomocą prądu elektrycz-nego zgodnie z przepisami stosowanymi w ręcznym klonowaniu CSH /Sambrock, 1989/. Bloty za-
blokowano w Blotto przez 1 godzinę /Blotto-PBS+ 1% mleko bez tłuszczu + 0,5% NP-40/. Świeże Blotto zawierające 2,5 g/ml poliklonalnej króliczej surowicy z przeciwciałami CDazy dodano do filtra i inkubowano 1 godzinę w temperaturze pokojowej. Filtr wypłukano trzy razy po
5 minut w Blotto i inkubowano w roztworze przeciwciał sprzężonych z alkaliczną fosfatazą /Boehringer Mannheim Biochetnicals/ rozcieńczonym w stosunku 1:1000 w Blotto. Nadmiar sprzę-
żonych przeciwciał usunięto za pomocą trzech kąpieli w Blotto i końcowe przepłukanie w bu-
forze o podłożu z zasadowej fosfatazy /100 mM Tris-HCl pH 9,5, 100 mM NeCl, 5 mM MgCl2/.Sączek inkubowano 15 minut w 40 ml buforu do którego dodano 12 mg fosforanu bromochorin- dolilu i 7 mg błękitu nitrotetrazoliowego aby wywołać kolor. Reakcje zatrzymano przemywa-
jąc sączek destylowaną wodą. Po wywołaniu barwy wszystkie 3 transfektanty ukazały pasma migrujące przy 17000 daltonów o intensywności większej od 0,8 ng i mniejszej od 4 ng stan-
dardowego stężenia oczyszczonej CDazy.
3. Pomiary czynności rekombinacyjnie ekspresjonowanej CDazyW ekstraktach z transformowanych wcześniej komórek COS zmierzono konwersję 5-fluoro-
cytozyny /5-FC/ w 5-fluoro uracyl /5-FU/. Transfektanty /3x10*5 komórek każdy/ zawierające CDazę /1-1, 1-4, 2-6/, transfekcję rzekomą lub transfekcję plazmidów kontrolnych /BB1/ pod-dano działaniu dźwięków w buforze 0,5 ml 10 mM TRIS a następnie odwirowano, Do 100 μlkaż-dej cieczy i 1 μ l/ml roztworu CDazy dodano 170 μ l roztworu soli zbuforowanego fosforanem /PBS/ i 30 μ l 30 mM roztworu 5-FC. W wyniku tego otrzymano 300 μ l mieszaniny reakcyj-nej z 3 mM 5-FC. Wykonano też reakcję kontrolną bez enzymu. Probówki te inkubowano 24 go-
dziny w 37°C, po czym pobrano z nich 50 każdego roztworu i reakcję przerwano w 1 ml 0,1 N roztworu HCl. Próbki te mierzono na spektrofotometrze UV przy 255 i 290 nm. Wykorzy-
stując równania:
/0,1191 X OD290 - 0,02485 X 0D255 / X 20 = mM 5-FC
i
/0,1849 X 0D255 - 0,04907 X OD290/ X 20 = mM 5-FU
zmierzono ilość konwersji 5-FC w 5-FU. Następnie obliczono liczbę jednostek czynności w
ekstrakcie komórki pierwotnej /1 Jednostka = 1 μ mol/minutę przemiany 5-FC w 5-FU. /Trans- fekcje rzekome i BB1 oraz reakcja bez enzymów nie wykazały się czynnością, natomiast wszyst-
kie trzy transfektanty CDazy były czynne. Transfektant 1-1 miał 0,41 X 10 3̄ jednostek, 1-4miał 0,54 x 10ˉ3 jednostek a 2-6 miał 0,26 X 10̄ 3 jednostek. Kontrolne oznaczanie za pomo-cą 1 μ g/ml CDazy przemieniło 5-FC w 5-FU w całości. Zakładając, że czynną CDazę odznacza się czynnością 40 U/mg, czynną CDazę otrzymano w ilości 10 ng /1-1/, 13,5 ng /1-4/,
7,3 ng /2-6/.Te same próbki zbadano również, stosując oznaczanie niezależne. Każda badana probówka
zawierała 50 μl cieczy sklarowanej osadem lub 1 g/ml CDazy, 130 μ l PBS, 20 μ l 30mM 5-FC oraz 1 μ Cl / 3H / 5-FC. Mieszaniny reakcyjne inkubowano 24 godziny, odparowano,a pozostałość rozpuszczono w 20 μ lmetanolu, po czym naniesiono je na płytki do chromato- grafii cienkowarstwowej na żełu krzemionkowym /TCL/. Płytki rozwinięto stosując mieszaninę 96:4 aceton/woda, w której współczynniki Rf dla 5-FC i 5-FU wynosiły odpowiednio 0,2 i 0,8.Z płytek TLC wycięto odpowiednie porcje, które włożono do cieczy scyntylacyjnej. Próbki zliczono w liczniku scyntylacyjnym i dla każdej z nich wyznaczono relatywna ilości 5-FC i 5-FU. Oznaczenie próbek rzekomych i BB1 nie wykazało obecności 5-FU, natomiast wszystkie
165 149 19
transfektanty zmieniły 14% 5-FC w 5-FU. To odpowiada 0,6 X 10‾3 jednostkom w każdej próbce,
potwierdzając ilości wyliczone w doświadczaniu z UV. Reakcje kontrolne z 1 g/ml CDazy przeksz-
tałciły 5-FC w całości w 5-FU,Doświadczenia wykazują, że tranasfektanty CDazy rzeczywiście produkują czynny enzym. Ten
fakt w połączeniu z analizą Western blot wskazuje na to, że specyficzna czynność enzymów rekombinacyjnych podobna jest do wykazywanej przez enzymy wyodrębnione z drożdży
Ujawniono zatem termicznie stabilną CDazę oraz sposoby oczyszczania jej jak i rekombi- nacyjnego wytwarzania. Chociaż korzystne aspekty wynalazku zostały szczegółowo opisane nale-ży uważać je za przykładowe i nie ograniczają one zakresu wynalazku zdefiniowanego w dołą- czonych zastrzeżeniach.
S c h e m a t
165 149
FIG. 11
FIG. 12
165 149
FIG. 9
FIG. 10

165 149
FIG. 7
FIG. 8

165 149
FIG. 6
165 149
FIG. 3
FIG. 4
165 149
FIG. 2A
FIG. 2B
165 149
FIG. I
FIG. 5
Departament Wydawnictw UP RP. Nakład 90 egz. Cena 10 000 zł







![RZECZPOSPOLITA TŁUMACZENIE PATENTU …public.sds.tiktalik.com/patenty/pdf/252760.pdf · miot rosnących obaw związanych z promowaniem oporności bakterii. [0005] Niedawno podjęto](https://img.pdfslide.tips/doc/110x75/5d07c52c88c993ea1b8cb488/rzeczpospolita-tlumaczenie-patentu-miot-rosnacych-obaw-zwiazanych-z-promowaniem.jpg)








![RZECZPOSPOLITA TŁUMACZENIE PATENTU …public.sds.tiktalik.com/patenty/pdf/261096.pdf · [0002] Choroba Charcota-Mariego-Tootha („CMT”) jest genetyczn ą sieroc ą polineuropati](https://img.pdfslide.tips/doc/110x75/5c7ad47809d3f24e7d8cc038/rzeczpospolita-tlumaczenie-patentu-0002-choroba-charcota-mariego-tootha-cmt.jpg)












