Embed Size (px)
Citation preview

:Ministerio efeSa[uefSecretaria áe Po[iticas,'R!gufación e Institutos
)l.:N.:M.)l. 'T.
"2015- AÑO DEL BICENTENARIO DEL CONGRESO DE LOS PUEBLOS LIBRES",
DISPOSICiÓN N! '1 2 O if
BUENOSAIRES, o B SEP 2015
esta
VISTO el Expediente NO 1-47-0000-010339-14-4
ADMINISTRACIÓN NACIONAL DE MEDICAMENTOS,
del íegistro de
ALI~ENTOS y
TECNOLOGÍA MÉDICA Y
CONSIDERANDO:
Que por las presentes actuaciones la firma CILAG
FARMACÉUTICAS.A. solicita la autorización de nuevos proyectos de prospectos
para la Especialidad Medicinal denominada SIMPONI/ GOLIMUMAB, Forma
farmacéutica y concentración: SOLUCIÓN INYECTABLE 50 Y 100 mgJ autorizado
por el Certificado N° 55.910. ' : '
Que lo presentado se encuentra dentro de los alcances de las
normativas vigentes, Ley de Medicamentos 16463, Decreto 150/92.
Que los procedimientos para las modificaciones y/o rectificaciones
de los datos característicos correspondientes a un certificado de specialidad
Medicinal otorgado en los términos de la Disposición (ANMAT) N° 5755/96 se
encuentran establecidos en la Disposición (ANMAT) N° 6077/97.
-1-

:Ministerio áe Sa[lUfSecretaria áe '1'ofiticas,'1?fgutación e Institutos
)l.Jf.:M.)1 'T.
I- I
-2015- ANO DEL BICENTENARIO DEL CONGRESO DE LOS PUErOS LIBRES"".
DISPOSICaON N. 7 2 O1I
Que actúa en ejercicio de las facultades conferidas por los Decretos
Nros. 1490/92 Y 1886/14.
Por ello,
EL ADMINISTRADOR NACIONAL DE LA ADMINISTRACIÓN NACIO~AL DE
MEDICAMENTOS,ALIMENTOSY TECNOLOGÍA MÉDICA IDISPONE: l
ARTÍCULO 10.- Autorízase a la firma JANSSEN CILAG FARMACÉUTI,A S.A. los:
nuevos proyectos de prospectos para la Especialidad Medicinal Jenominada
SIMPONI/ GOLIMUMAB, Forma farmacéutica y concentración: SOLUCIÓN
INYECTABLE50 Y 100 mg, autorizada por el Certificado N° 55.910, duyos textos
obran a fojas: 212 a 245, 246 a 279 y 280 a 313; desglosándose las fojas 212 a
245.
ARTÍCULO 2°- Sustitúyase en el Anexo II de la Disposición autrrizant€ N°
7449/10 los prospectos autorizados por las fojas aprobadas en el artículo 1°.
ARTÍCULO 3°.- Acéptese el texto del Anexo de la Autorización de MLificaciones
el cual pasa a formar parte integrante de la presente Disposición y el! que deberá
agregarse al Certificado N° 55.910 en los términos de la Disposición 6077/97.
ARTICULO 4°. Regístrese; por la Mesa de Entradas notifíquese all interesado,
haciéndole entrega de la copia autenticada de la presente Disposición
-2-

:Ministerio áe SaÜufSecretaria efePofíticas,'í(fgufación e Institutos
;4.1{. :M.;4.'I.
I
"2015- AÑO DEL BICENTENARIO DEL CONGRESO DE LOS PUELos LIBRES".
DISPOSICION N~ 7 2 O .rz¡.
conjuntamente con los prospectos y Anexo, gírese a la Dirección de Gestión de
Información Técnica. Cumplido, archívese.
EXPEDIENTE NO1-47-0000-010339-14-4
log ROGELlO LOPEZAdmInistrador Nacional
A.N.M.A.T.
-3-

6'Ministerio áe Sa{wf
Secretaria áe PoCiticas,<R.fguCacióne Institutos
}!.JV. 'M.}l.'T.
I
"2015- AÑO DEL BICENTENARIO DEL CONGRESO DE LOS PUELos LIBRES".I
ANEXO DE AUTORIZACION DE MODIFICACIONES
El Administrador Nacional de la Administración Nacional de MedicamentosI '
Alimentos y Tecnología Médica (ANMAT), autorizó mediante ¡DiSPOSición
~"'2.0...710s efectos de su anexado en el Certificado de Autoriz~ción de la
Especialidad Medicinal N° 55.910 Y de acuerdo a lo solicitado por lb JANSSEN• I
CILAG FARMACEUTICA S.A., la modificación de los datos caracterí1sticoS, que
figuran al pie del producto inscripto en el Registro de Especialidades Medicinales
(REM) bajo:
Nombre comercial/ Genérico/s: SIMPONI/ GOLIMUMAB.
Forma farmacéutica: JERINGA PRELLENADA.
Disposición Autorizante de la Especialidad Medicinal N° 7449/10.
Tramitado por expediente N° 1-47-0000-022455-09-5
DATO A MODIFICAR DATO AUTORIZADO MODIFICACION AiTORIZADAHASTA LA FECHA
PROSPECTOS Anexo de Disposición De fojas 212 a 245, 246 aN° 6721/13. 279 Y 280 I a 313;
desglosándose las fojas 212a 245.
I
-4-
FARMACÉUTICA S.A., Titular delCILAG
IEl presente sólo tiene valor probatorio anexado al Certificado de ~utorización
antes mencionado. ISe extiende el presente Anexo de Autorización de Modificaciones del REM a la
ICeH:ificado de

"2015- AÑO DEL BICENTENARIO DEL CONGRESO DE LOS PUETOS LIBRES".
'Ministerio de Sa[uáSecretaria áe Poúticas,
'1?!gufacióne Institutos;4.:N.'M.;4. 'L
Autorización N° 55.910 en la Ciudad de Buenos Aires, a08 SEP 2015
de .
EXPEDIENTEN° 1-47-0000-010339-14-4
DISPOSICION N°
Ing. ROGELlO LOPEZAdminIstrador Nacional
A.N.M.A.T.
-5-
105 1"' .del
,
mes

Proyecto de Prospecto
SIMPONI@
7207a 8 SfP 2015
GOLIMUMAB
Solución inyectable(Para administración por vía subcutánea)
Industria Estadounidense
Jeringa PrellenadajAutoinyector SmartJect
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Venta Bajo Receta
iCada Autoinyector SmartJectjJeringa prellenada con una dosis de 50 mg contiene!:GOLIMUMABSorbitolL-HistidinaPolisorbato SOAgua para inyección csp
50,000 mg20,50 mg0,44 mg0,075 mg0,50 mL
Cada Autoinyector SmartJectjJeringa prellenada con una dosis de 100 mg contiene:GOLIMUMAB 100,00 mgSorbitol 41,00 mgL-Histidina 0,S7 mgPolisorbato SO 0,15 mgAgua para inyección csp 1,00 mL
DESCRIPCIÓN
Golimumab es un anticuerpo monoclonal IgG1k humano producido por una línea celularI
de hibridoma murino mediante tecnología de ADN recombinante. Cada jeringaprellenadajautoinyector SmartJect descartable de 0,5 mi contiene 50 mg de golim~mab.La solución es transparente y ligeramente opalescente, de incolora a amarillo claro.
ACCION TERAPEUTlCA
Golimumab es un anticuerpo monoclonal humano que forma complejos estables de altaafinidad con las formas solubles y bioactiva transmembrana del factor de nJcrosistumoral humano (TNF), lo que evita la unión del TNF a sus receptores. La exp~esiónelevada del TNF se ha vinculado con enfermedades inflamatorias crónicas colmo laartritis reumatoide (AR), y con espondiloartropatías, como la artritis psoriásica (+PS) yla espondilitis anquilosante (EA), y es un importante mediador de . maciónarticular y del daño estructural que caracterizan a estas enfermedad .
gina 1de 34

Código ATC: L04AB06.tumoral alfa (TNF-a)
Grupo farmacoterapéutico: Inhibid2s.2 Oto'!e
INDICACIONES
Artritis reumatoide (AR):SIMPONI@,en combinación con metotrexato (MTX), está indicado en:• el tratamiento de la artritis reumatoide activa, de moderada a grave, en pacientes
adultos cuando la respuesta a los fármacos antirreumáticos modificadores dé laenfermedad (FAMEs), incluido MTX, no ha sido adecuada. I
• el tratamiento de la artritis reumatoide activa, grave y progresiva, en pacientesadultos no tratados con anterioridad con MTX. i
SIMPONI@, en combinación con MTX, ha demostrado reducir la tasa de progresión deldaño articular medido por Rayos-X y mejorar la función física .
. Artritis Psoriásica (APs):SIMPONI@, solo o en combinación con MTX, está indicado en el tratamiento de artritispsoriásica activa y progresiva en adultos cuando la respuesta al tratamiento prev;~lioconFAMEs no ha sido adecuada.SIMPONI@ha demostrado reducir la tasa de progresión del daño articular peri érico,,medida por Rayos X en pacientes con subtipos de enfermedad poliarticular simétrica(ver propiedades farmacodinamicas) y mejorar la función física.
Espondilitis anquilosante (EA):SIMPONI@está indicado en el tratamiento de la espondilitis anquilosante activa, grave,en adultos que han respondido de forma inadecuada al tratamiento convencional.
Colitis Ulcerosa (CU)SIMPONI@está indicado para el tratamiento de la colitis ulcerosa activa de moderada agrave en pacientes adultos que han tenido una respuesta inadecuada al tratamientoconvencional, incluidos corticoides y 6-mercaptopurina (6-MP) o azatioprina (AZA),'o quepresentan intolerancia o contraindicaciones a dichas terapias.
PROPIEDADES FARMACOLÓGICAS
Propiedades FarmacodinámicasGrupo farmacoterapéutico: Inmunosupresores, inhibidores del factor de necrosistumoral alfa (TNF-a); código ATC: L04AB06
Mecanismo de acciónGolimumab es un anticuerpo monoclonal humano que forma complejos establ'es degran afinidad con las dos formas bioactivas del TNF-a humano, la solublel y latransmembranosa, impidiendo así la unión del TNF-a a sus receptores.
Efectos farmacodinámicos
íágína 2 de 34
,

7207 .~~Se ha demostrado que la unión de golimumab al TNF humano neutraliza la exp ~,¡,L0..-,¡inducida por el TNF-a de la molécula de adhesión selectina E, la molécula de ad~e' Ill: t."*,~endotelial 1 (VCAM-l) y la molécula de adhesión intercelular 1 (ICAM)-l de las células
Iendoteliales humanas. In vitro, golimumab también inhibe la secreción inducida por
ITNF de las interleucinas 6 y 8 (IL-6, IL-8) Y del factor estimulante de las colonias degranulocitos-macrófagos (GM-CSF) por parte de las células endoteliales humanas.1
Se ha observado una mejoría de la concentración de la proteína C reactiva (PdR) encomparación con los grupos placebo, y el tratamiento con SIMPONI@dio lugar la unareducción importante de la concentración sérica de IL-6, ICAM-l, matrizmetaloproteinasa 3 (MMP) -3 Y factor de crecimiento endotelial (VEGF) con resp~cto alos valores basales en comparación con el tratamiento de control. También se P~OdUjOuna disminución del TNF-a en los pacientes con AR y EA Y de la IL-8 en los pacientescon APs. Estos cambios se observaron en la primera evaluación (semana 4), ~ras ladosis inicial de SIMPONI@y por lo general se mantuvieron hasta la semana 24.
Eficacia clínicaArtritis reumatoideSe demostró la eficacia de SIMPONI@en tres ensayos multicéntricos, aleatorizados,doble ciego y controlados con placebo, con más de 1500 pacientes de edad i~ual osuperior a 18 años que presentaban AR activa de moderada a grave, diagnos\:¡cadasegún los criterios del American Col/ege of Rheumatology (ACR) como mínimb tresmeses antes de la selección. Los pacientes presentaban dolor en al menos tuatro
Iarticulaciones y tumefacción en al me'nos cuatro articulaciones. Se administróSIMPONI@o placebo, por vía subcutánea cada cuatro semanas.
En el ensayo GO-FORWARD se evaluaron 444 pacientes con AR activa a pesilr deltratamiento con MTX en dosis estables de al menos 15 mg por semana, que no habíanrecibido con anterioridad antagonistas del TNF. Los pacientes se distribuyeron a,1azarpara recibir placebo mas MTX, SIMPONI@50 mg mas MTX, SIMPONI@100 mg más MTXo SIMPONI@100 mg más placebo. Los pacientes tratados con placebo más MTX fueroncambiados a SIMPONI@50 mg más MTX después de la semana 24. En la semar\a 52,todos los pacientes entraron a formar parte de un estudio abierto de extensión a largoplazo.
En el ensayo GO-AFTER se evaluaron 445 pacientes previamente tratados con ~no omás de los antagonistas del TNF adalimumab, etanercept o infliximab. Los pacientes sedistribuyeron al azar para recibir placebo, SIMPONI@ 50 mg o SIMPONI@ lOb mg.Durante el ensayo se permitió a los pacientes mantener el tratamiento concomlitantecon FAMEs como MTX, sulfasalazina (SSZ) o hidroxicloroquina (HCQ). Las razonesaducidas para la suspensión del tratamiento previo con antagonistas del TNF fue'ron lafalta de eficacia (58%), la intolerancia (13%) y otras causas distintas de seguri1dad yeficacia (29%; en su mayoría motivos económicos).
Página 3 de 34

".!l.A.c. ;;:,.,..~
fO\"\O'
¡2 O (l~,'?\< ~¡En el ensayo GO-BEFORE se evaluaron 637 pacientes con AR activa que no '1 abía J'4 DE ~~
recibido con anteriormente MTX ni antagonistas del TNF. Los pacientes se distribuyeronal azar para recibir placebo mas MTX, SIMPONI@50 mg mas MTX, SIMPONI@190 mgmás MTX o SIMPONI@100 mg más placebo. En la semana 52, los pacientes entrf.ron aformar parte de un estudio abierto de extensión a largo plazo en el que los pacientesque recibieron placebo más MTX que tenían al menos una articulación infla miada odolorida fueron cambiados a SIMPONI@50 mg más MTX.
En el GO-FORWARD, las variables (co-)principales de eficacia fueron el porcentaje depacientes que alcanzaban la respuesta ACR 20 en la semana 14 y la mejoría! en elcuestionario de evaluación de la salud (HAQ) en la semana 24 respecto al estado :baSal.En el ensayo GO-AFTER, la variable principal de eficacia fue el porcentaje de pacientes
I
que alcanzaban la respuesta ACR 20 en la semana 14. En el ensayo GO-BEFORE, lasI
variables ca-principales de eficacia fueron el porcentaje de pacientes que alcanzaban larespuesta ACR 50 en la semana 24 y el cambio respecto al estado basal en la ~scalavan der Heijde Sharp (VDH-S) modificada en la semana 52. Además de las va1iablesprincipales de eficacia, se llevaron a cabo otras evaluaciones del efecto del tratamiento
Icon SIMPONI@sobre los signos y síntomas de la artritis, la función física y la calidad devida relacionada con la salud.
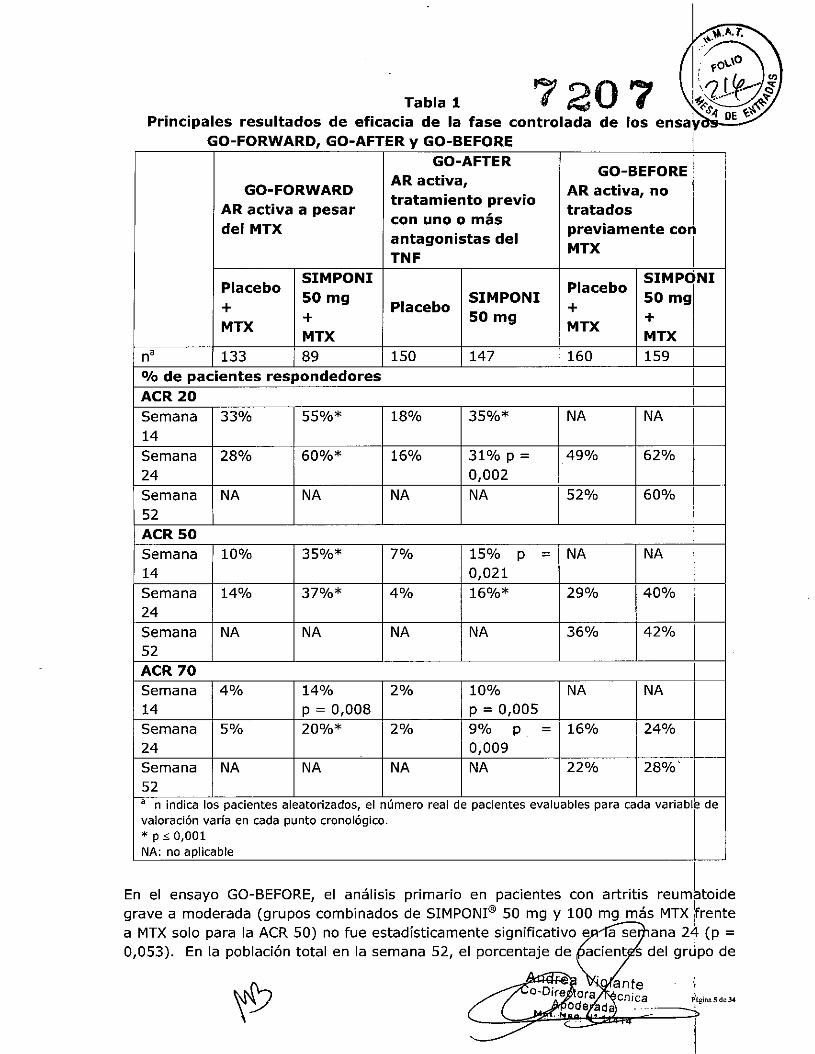
En general, no se observaron diferencias clínicamente significativas en términos deeficacia entre las pautas posológicas de SIMPONI@ 50 mg y SIMPONI@ 10:0 mgadministrados de forma concomitante con MTX, hasta la semana 104 en el ensaye GO-FORWARDy en el GO-BEFOREy hasta la semana 24 en el ensayo GO-AFTER. E~ cadauno de los estudios de AR, por díselo del estudio, los pacientes en la extensión a largoplazo pudieron cambiar entre las dosis de SIMPONI@50 mg y SIMPONI@100 mg Isegúnel criterio del médico del estudio. jSignos v síntomasEn la tabla 1 se recogen los principales resultados ACR con la dosis de SIMPO I@ 50
Img a las semanas 14, 24 Y 52 en los ensayos GO-BEFORE, GO-AFTER y GO-FORr'ARDque se describen a continuación. La respuesta se observó en la primera evaluación(semana 4) tras la dosis inicial de SIMPONI@. IEn el ensayo GO-FORWARD, entre los 89 pacientes tratados al azar con la dosis deSIMPONI@ 50 mg mas MTX, 48 siguieron con el tratamiento hasta la seman¡l 104.Entre ellos, 40, 33 Y 24 pacientes tuvieron una respuesta ACR de 20/50/70,respectivamente, en la semana 104. Entre los pacientes que seguían en el estLdio y
Ien tratamiento con SIMPONI@, se observaron tasas similares de respuesta ACR de20/50/70 desde la semana 104 hasta la semana 256.
Página 4 de 34
En el ensayo GO-AFTER, el porcentaje de pacientes que alcanzaron la respuesta ACR20 fue mayor en el grupo tratado con SIMPONI@que en el que recibió placebb, con
,
independencia del motivo aducido para la suspensión de uno o más tratamientosprevios con antagonistas del TNF.
NA
24%
NA
28%'
42%
40%
NA
22%
29%
NA
36%
160
GO-BEFORE!AR activa, no 1
Itratadospreviamente conMTX I
SIMPONI50 mg+MTX159
Placebo+MTX
= 16%
NA
10%P = 0,0059% P0,009NA
SIMPONI50 mg
15% P =0,02116%*
147
NA
NA
2%
II
18% 35%* NA NA
I16% 31% p- 49% 62%
I0,002NA NA 52% 60%
I
2%
150
7%
Placebo
4%
NA
NA
NA
35%*
55%*
14%P = 0,00820%*
60%*
37%*
GO-FORWARDAR activa a pesardel MTX
Placebo+MTX
10%
~,~
/', fOI.\O
7-2 O 7 \1 r [1....-; ~Tabla 1 \ ~ ",.,..'f' ~~~4 ~~Principales resultados de eficacia de la fase controlada de los ensa DE
GO-FORWARD, GO-AFTER y GO-BEFOREGO-AFTER
AR activa,tratamiento previocon uno o másantagonistas delTNF
Semana14
Semana 33%14Semana 28%24Semana NA52ACR 50
SIMPONI50 mg+MTX
na 133 89010 de pacientes respondedoresACR 20
Semana 14%24Semana NA52ACR 70Semana 4%14Semana 5%24Semana NA52a n indica los pacientes aleatorizados, el número real de pacientes evaluables para cada variable devaloración varía en cada punto cronológico.* p ,; 0,001NA: no aplicable
En el ensayo GO-BEFORE, el análisis primario en pacientes con artritis reumltoidegrave a moderada (grupos combinados de SIMPONI@50 mg y 100 mg más MTX (rentea MTX solo para la ACR 50) no fue estadísticamente significativo e a se ana 24 (p =0,053). En la población total en la semana 52, el porcentaje de acient s del grJpo de
!
, 'ante ¡o~DI(e ora cnica ~ágina5de,3,4
ode ada - ---,,"

I'.' fO\.\O
r, l-IJ:~;SIMPONI@50 mg más MTX que alcanzaron una respuesta ACR fue generalmen ~. ---- ~.•••alto, pero no significativamente diferente en comparación con MTX solo (ver tablJ 1 .11 DE ~~
Se realizaron análisis adicionales en subgrupos representativos de la población in~icadade pacientes con AR severa, activa y progresiva. Se demostró en general un rayorefecto de SIMPONI@ 50 mg más MTX frente a MTX solo en la población indicada encomparación con la población total.
En los ensayos GO-FORWARD y GO-AFTER se observaron respuestas clínica yestadísticamente significativas en la Escala de Actividad de la Enfermedad (DAS £8) entodas las evaluaciones programadas especificados en semanas 14 y 24 ( P ,; 0:,001).Entre los pacientes que permanecieron en el tratamiento de SIMPONI@ al que fueronasignados al azar el inicio del estudio, las respuestas DAS 28 se mantuvieron h~sta lasemana 104. Entre los pacientes que seguían en el estudio y en el tratamien~o conSIMPONI@, las respuestas DAS 28 fueron similares desde la semana 104 hJsta lasemana 256. .
iEn el ensayo GO-BEFORE, se midió la respuesta clínica significativa, definida cdmo elmantenimiento de una respuesta ACR 70 durante un periodo de 6 meses. I En lasemana 52, el 15% de los pacientes tratados con SIMPONI@ 50 mg más MTXconsiguieron una respuesta clínica significativa en comparación con el 7% be lospacientes del grupo de placebo más MTX (p = 0,018). Entre los 159 pacientes trÁtadosal azar con SIMPONI@50 mg mas MTX, 96 de ellos continuaron el tratamiento h~sta lasemana 104. Entre ellos, 85, 66 y 53 pacientes presentaron una respuestá ACR20/50/70, respectivamente, en la semana 104. Entre los pacientes que seguíarl en elestudio y en tratamiento con SIMPONI@, se observaron tasas similares de res~uestaACR 20/50/70 desde la semana 104 hasta la semana 256.
Respuesta radiográficaEn el ensayo GO-BEFOREse utilizó el cambio respecto al estado basal en la escala vdH-S, una escala compuesta de los daños estructurales, que radiográfica mente rrlide elnúmero y tamaño de las erosiones articulares y el grado de estrechamiento del ekpacioarticular en manos, muñecas y pies, para evaluar el grado de daño estructural.! En latabla 2 se presentan los resultados para la dosis de SIMPONI@50 mg en la semana 52.
El número de pacientes sin nuevas erosiones o con un cambio en la puntuacióri! totalvdH-S ::> O respecto al estado basal fue significativamente mayor en el grulpo detratamiento con SIMPONI@ que en el grupo control (p = 0,003). Los efectosradiográficos observados en la semana 52 se mantuvieron hasta la semana 104.1 Entrelos pacientes que seguían en el estudio y en tratamiento con SIMPONI@, los efec;:tosradiográficos fueron similares desde la semana 104 hasta la semana 256.
Página 6 de 34
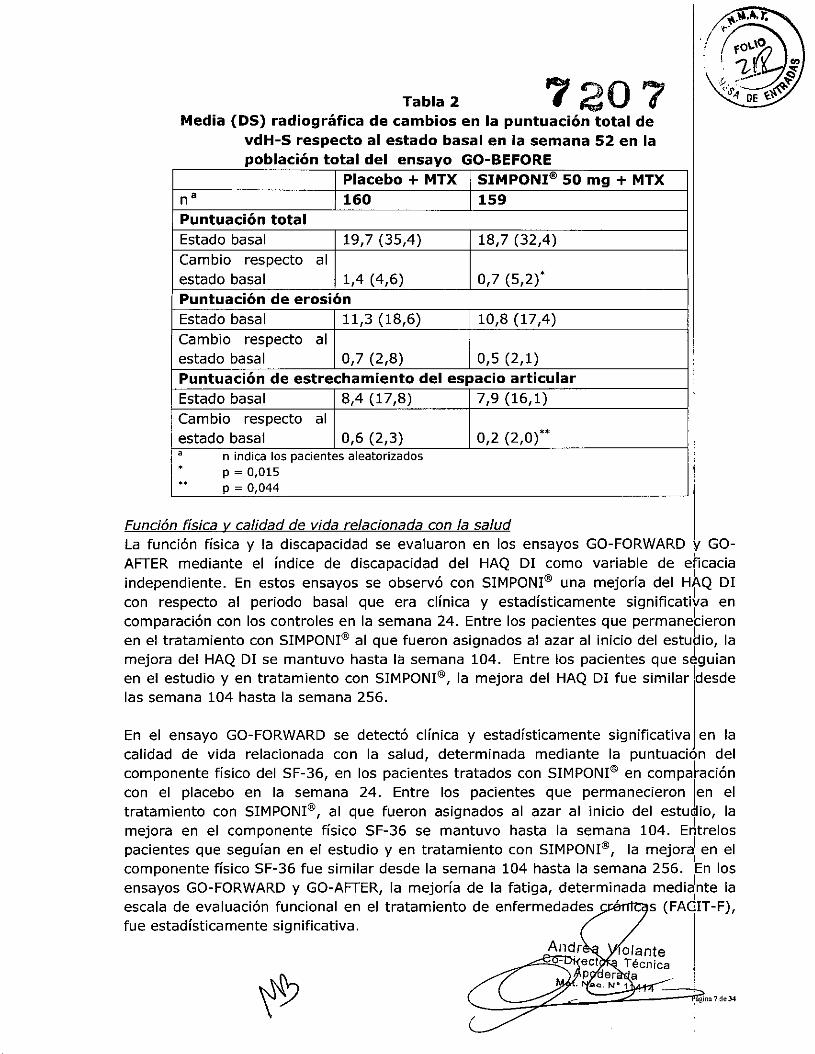
Tabla 2 720 7Media (OS) radiográfica de cambios en la puntuación total de
vdH-S respecto al estado basal en la semana 52 en lapoblación total del ensayo GO-BEFORE
Placebo + MTX SIMPONI@ 50 mg + MTXna 160 159Puntuación totalEstado basal 19,7 (35,4) 18,7 (32,4)Cambio respecto alestado basal 1,4 (4,6) 0,7 (5,2)'Puntuación de erosiónEstado basal 11,3 (18,6) 10,8 (17,4)Cambio respecto alestado basal 0,7 (2,8) 0,5 (2,1)Puntuación de estrechamiento del espacio articularEstado basal 8,4 (17,8) 7,9 (16,1)Cambio respecto alestado basal 0,6 (2,3) 0,2 (2,0)", n indica los pacientes aleatorizados. p = 0,015.. P = 0,044
Función física v calidad de vida relacionada con la saludLa función física y la discapacidad se evaluaron en los ensayos GO-FORWARD y GO-AFTER mediante el índice de discapacidad del HAQ DI como variable de eficaciaindependiente. En estos ensayos se observó con SIMPONI@una mejoría del HfQ DIcon respecto al periodo basal que era clínica y estadísticamente significativa encomparación con los controles en la semana 24. Entre los pacientes que permaneberonen el tratamiento con SIMPONI@al que fueron asignados al azar al inicio del estu~io, lamejora del HAQ DI se mantuvo hasta la semana 104. Entre los pacientes que sJguíanen el estudio y en tratamiento con SIMPONI@, la mejora del HAQ DI fue similar Idesdelas semana 104 hasta la semana 256.
En el ensayo GO-FORWARD se detectó clínica y estadísticamente significativa en lacalidad de vida relacionada con la salud, determinada mediante la puntuacion delcomponente físico del SF-36, en los pacientes tratados con SIMPONI@en compa~acióncon el placebo en la semana 24. Entre los pacientes que permanecieron len eltratamiento con SIMPONI@, al que fueron asignados al azar al inicio del estucilio, lamejora en el componente físico SF-36 se mantuvo hasta la semana 104. Eritrelospacientes que seguían en el estudio y en tratamiento con SIMPONI@, la mejoral en el
Icomponente físico SF-36 fue similar desde la semana 104 hasta la semana 256. En losensayos GO-FORWARD y GO-AFTER, la mejoría de la fatiga, determinada media1nte la
Iescala de evaluación funcional en el tratamiento de enfermedades ' s (FAGIT-F),fue estadísticamente significativa.
rgínU 7 de34

7207 i~8Artritis psoriásica ,~~,¿'O~<r,~
La seguridad y eficacia de SIMPONI@ se evaluaron mediante un ensayo multicéntrico,aleatorizado, doble ciego y controlado por placebo (GO-REVEAL), en 405 adult~s conAPs activa (dolor en tres o más articulaciones y tumefacción en tres d másarticulaciones) a pesar del tratamiento con antiinflamatorios no esteroideos (AI~ES) ocon FAMEs. Los pacientes de este ensayo tenían una APs diagnosticada como n;'inimoseis meses antes y una psoriasis como mínimo leve. Se incluyeron pacientes con; todoslos subtipos de artritis psoriásica, como artritis poliarticular sin nódulos reumatoides(43%), artritis periférica asimétrica (30%), artritis de las articulaciones interfaláhgicasdistales (IFD) (15%), espondilitis con artritis periférica (11 %) Y artritis mutilante 1(1%).No estaba permitido el tratamiento previo con un antagonista del TNF. Se administróSIMPONI@ o placebo por vía subcutánea cada cuatro semanas. Los paciendes seasignaron al azar con placebo, SIMPONI@ 50 mg o SIMPONI@ 100 mg. Los padientesque recibieron placebo pasaron al grupo de SIMPONI@ 50 mg después de la sémana24. Los pacientes entraron en una ampliación del estudio de diseño abierto al largoplazo en la semana 52. Alrededor del 48% de los pacientes continuaron con una dosis
Iestable de metotrexato (::; 25 mg/semana). Las co-variables principales de eficaciafueron el porcentaje de pacientes que alcanzaban la respuesta ACR 20 en la semJna 14y el cambio desde el valor basal en el índice modificado total vdH-S para APs en lasemana 24.En general, las determinaciones de la eficacia no mostraron diferencias clínicamentesignificativas entre las pautas posológicas de SIMPONI@ de 50 mg y de 100 mg h~sta lasemana 104. Por diseño del estudio, los pacientes en la extensión a largo' plazopudieron cambiar entre las dosis de SIMPONI@ de 50 mg y de 100 mg según el criteriodel médico del estudio. ji
Signos y síntomasEn la tabla 3 se recogen los principales resultados con la dosis de 50 mg n lassemanas 14 y 24, que se describen a continuación.
Tabla 3.Principales resultados de eficacia en el ensayo GO-REVEAL
SIMPONI
1%1%
2%4%
Página 8 de34
12 %19 %
30 %32 %
51 %52 %
50 mg*146
9%12 %
Placebona 113% de pacientes respondedoresACR 20Semana 14Semana 24ACR 50Semana 14Semana 24ACR 70Semana 14Semana 24

7207PASlb 75c
Semana 14 3% 40 %Semana 24 1% 56 %* p < 0,05 en todas las comparacionesa n indicalospacientesaleatorizados;Elnúmerorealde pacientesevaluablesparacada variable de valoración varía en cada punto cronológico.b Índice de superficie afectada y gravedad de la psoriasis.o En ei subgrupo de pacientescon afectación2 3% del BSA(SuperficieCorporalAfectada)en el periodobasal: 79 pacientes(69,9%) del grupo placeboy 109pacientes(74,3%) delgrupodetratamientoconSIMPONI@50 mg.
Se observaron respuestas en la primera evaluación (semana 4) tras la administraciónde SIMPONI@. La respuesta ACR 20 en la semana 14 fue similar en los subti~oS deartritis poliarticular sin nódulos reumatoides y artritis periférica asimétrica. El nbmerode pacientes con otros subtipos de APs era demasiado bajo como para hacclr unaevaluación adecuada. No hubo diferencia en la respuesta observada en los gru~os detratamiento con SIMPONI@entre los pacientes que recibieron MTX concomitanteh,entey los que no lo recibieron. Entre los 146 pacientes distribuidos al azar para ~ecibir
ISIMPONI@ 50 mg, 70 mantuvieron el tratamiento hasta la semana 104. De estos 70
Ipacientes, 64, 46 Y 31 pacientes tuvieron una respuesta ACR 20/50/70,
Irespectivamente. Entre los pacientes que seguían en el estudio y en tratamiento conSIMPONI@,se observaron tasas similares de respuesta ACR 20/50/70 desde la sJmana104 hasta la semana 256. I
También se observaron respuestas estadísticamente significativas DAS28 t!n lassemanas 14 y 24 (p < 0,05).
En la semana 24, en los pacientes tratados con SIMPONI@se apreció una mejoría delos parámetros de actividad periférica de la artritis psoriásica (por ejemplo, número dearticulaciones con tumefacción, número de articulaciones con dolor, dactilitis yentesitis). El tratamiento con SIMPONI@produjo una mejoría importante de la filinciónfísica, evaluada mediante el HAQ DI, así como de la calidad de vida relacionada ton lasalud, determinada mediante la puntuación de los componentes físico y men~al delSF-36. Entre los pacientes que permanecieron en tratamiento con SIMPONI@, ~Iquefueron distribuidos al azar en el comienzo del estudio, las respuestas DAS28 y H!A.QDIse mantuvieron hasta la semana 104. Entre los pacientes que seguían en el estLdio yen tratamiento con SIMPONI@, las respuestas DAS 28 Y HAQ DI fueron similares desdela semana 104 hasta la semana 256.
Respuesta radiográficaEl daño estructural en ambas manos y pies se evaluó radiográfica mente por el cambio
I
en el índice vdH-S desde el valor basal, modificado para APs añadiendo la articuilacióninterfalángica distal (DIP).
Página 9 de 34
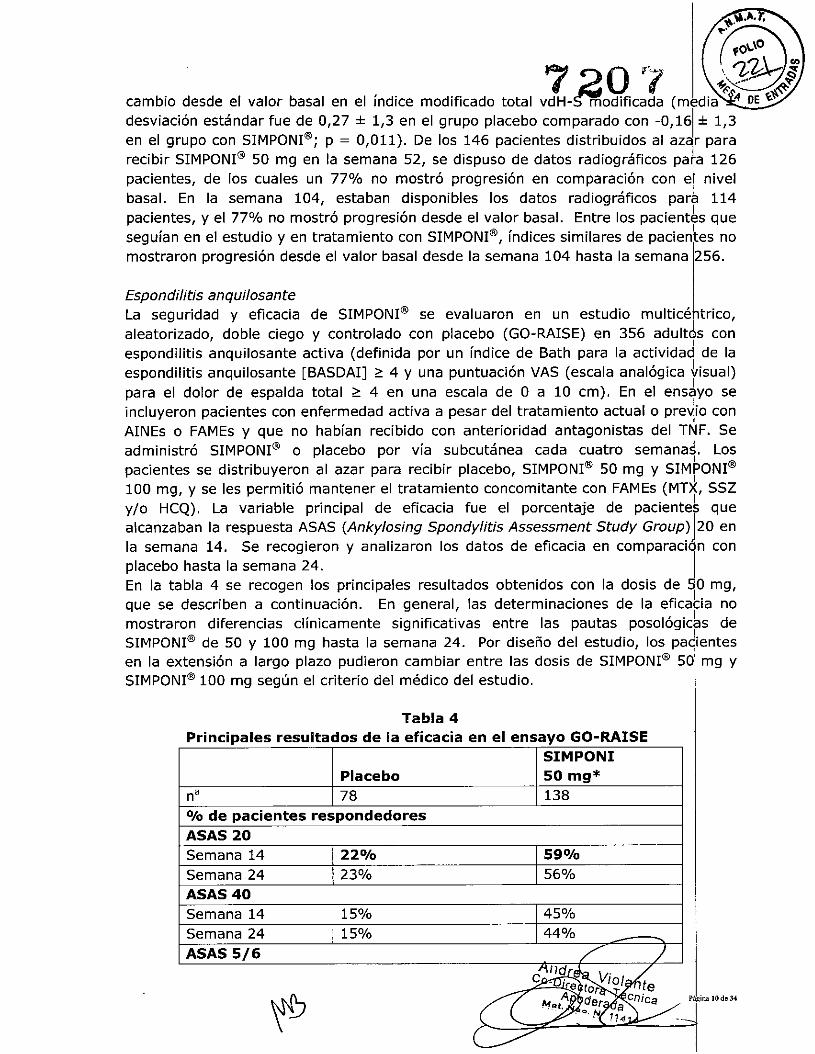
El tratamiento con SIMPONI@ 50 mg redujo la tasa de progreslon del daño articularperifériCO en comparación con el tratamiento placebo en la sema medido bar el
Andrea iollorader a
<-l!!~,N 1141._
•. .1\. r.•..fOI.\O. 7~O~ \~,(f ~~./ ~~
cambio desde el valor basal en el índice modificado total vdH- modificada (media !! DE ~~
desviación estándar fue de 0,27 :!:1,3 en el grupo placebo comparado con -0,161:!:1,3en el grupo con SIMPONI@; p = 0,011). De los 146 pacientes distribuidos al azar pararecibir SIMPONI@ 50 mg en la semana 52, se dispuso de datos radiográficos pa~a 126pacientes, de los cuales un 77% no mostró progresión en comparación con e! nivelbasal. En la semana 104, estaban disponibles los datos radiográficos para 114pacientes, y el 77% no mostró progresión desde el valor basal. Entre los pacientbs queseguían en el estudio y en tratamiento con SIMPONI@, índices similares de pacientes nomostraron progresión desde el valor basal desde la semana 104 hasta la semana t56.
Espondilítís anquílosanteLa seguridad y eficacia de SIMPONI@ se evaluaron en un estudio multicé trico,aleatorizado, doble ciego y controlado con placebo (GO-RAISE) en 356 adultJs con
Iespondilitis anquilosante activa (definida por un índice de Bath para la actividad de laespondilitis anquilosante [BASDAI] 2: 4 Y una puntuación VAS (escala analógica tisual)
Ipara el dolor de espalda total 2: 4 en una escala de ° a 10 cm). En el ensayo seincluyeron pacientes con enfermedad activa a pesar del tratamiento actual o pre~io conAINEs o FAMEs y que no habían recibido con anterioridad antagonistas del TNF. Seadministró SIMPONI@ o placebo por vía subcutánea cada cuatro semanaS. Los
I
pacientes se distribuyeron al azar para recibir placebo, SIMPONI@ 50 mg y SIMPONI@100 mg, y se les permitió mantener el tratamiento concomitante con FAMEs (MTX, SSZy/o HCQ). La variable principal de eficacia fue el porcentaje de pacientek quealcanzaban la respuesta ASAS (Ankylosíng Spondylítís Assessment Study Group) J20 enla semana 14. Se recogieron y analizaron los datos de eficacia en comparacion conplacebo hasta la semana 24. IEn la tabla 4 se recogen los principales resultados obtenidos con la dosis de 50 mg,que se describen a continuación. En general, las determinaciones de la eficaba nomostraron diferencias clínicamente significativas entre las pautas posOIÓ9iC~S deSIMPONI@ de 50 y 100 mg hasta la semana 24. Por diseño del estudio, los padientesen la extensión a largo plazo pudieron cambiar entre las dosis de SIMPONI@ 50! mg ySIMPONI@100 mg según el criterio del médico del estudio.
Página 10 de 34
45%44%
59%56%
22%23%
15%15%
Placebona 78010 de pacientes respondedoresASAS 20Semana 14Semana 24ASAS 40Semana 14Semana 24ASAS 5/6
Tabla 4Principales resultados de la eficacia en el ensayo GO-RAISE
SIMPONI50 rng*138

50%49%
7Semana 14 8%Semana 24 13%* p ~ 0,001 en todas las comparaciones, n indica los pacientes aieatorizados; El número real de pacientes evaluables paracada variable de valoración varía en cada punto .cronológico.
Entre los pacientes que seguían en el estudio y en tratamiento con SIMPO I@, elporcentaje de pacientes que alcanzaban la respuesta ASAS 20 Y ASAS 40 fue ~imilardesde la semana 24 hasta la semana 256. 1También se observaron diferencias significativas en BASDAI 50, 70 Y 90 (p ,; 0,0 7) enlas semanas 14 y 24. En la primera evaluación (semana 4) tras la adminis~racióninicial de SIMPONI@se detectó una mejoría en los principales indicadores de adividad
Ide la enfermedad que se mantuvo hasta la semana 24. Entre los pacientes queseguían en el estudio y en tratamiento con SIMPONI@,se observaron índices si1i1aresde cambios desde las visita basal en BASDAI desde la semana 24 hasta la semana
I256. La eficacia en los pacientes según la respuesta ASAS 20 en la semana +4 fueuniforme con independencia del tratamiento con FAMEs (MTX, SSZ y/o HCQ), lapresencia o ausencia del antígeno HLA-B27 y la concentración de PCR en el p~riodobasal.
El tratamiento con SIMPONI@indico mejoras significativas en la función física evaluadapor los cambios desde la visita basal en BASFI (Índice funcional de Bath p~ra laespondilitis anquilosante) a las semanas 14 y 24. En las semanas 14 y 24 se o~servóuna mejoría significativa de la calidad de vida relacionada con la salud deternl
linada
mediante del componente físico del SF-36. Entre los pacientes que seguían, en elestudio y en tratamiento con SIMPONI@, las mejoras en la función física y la relaciónentre la calidad de vida y la salud fueron similares desde la semana 24 hakta lasemana 256.
Colitis ulcerosaLa eficacia de SIMPONI@se evaluó en dos ensayos clínicos aleatorizados, doble ciego,controlados con placebo en pacientes adultos.
El estudio de inducción (PURSUIT-Inducción) evaluó a los pacientes con colitis ul(¡erosaactiva moderada a grave (puntuación Mayo 6 a 12; subpuntuación endoscóPid ~ 2)
Ique habían tenido una respuesta inadecuada o que no habían tolerado los tratamientosconvencionales o que eran dependientes de corticosteroides. En la parte del estudio deconfirmación de la dosis, 761 pacientes se aleatorizaron para recibir 400 rl,g deSIMPONI@SC en la semana O y 200 mg en la semana 2, 200 mg de SIMPONI@SC en lasemana O y 100 mg en la semana 2, o placel:>0 SC en las semanas O y 12. Sepermitieron dosis estables concomitantes de aminosalicilatos orales, corticoste~oidesy/o agentes inmunomoduladores. En este estudio se evaluó la eficacia de Si~ponihasta la semana 6.
Pagina JI de 34
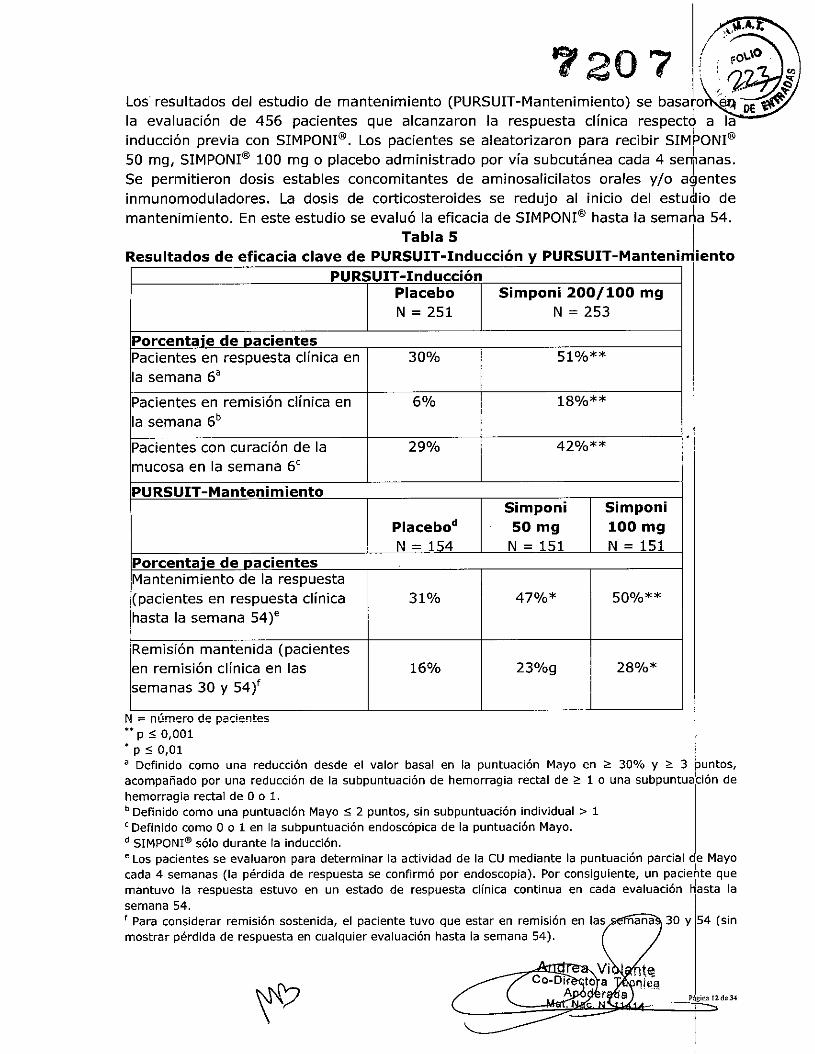
7207 lé~~Los resultados del estudio de mantenimiento (PURSUIT-Mantenimiento) se basa~o .~ DE ~la evaluación de 456 pacientes que alcanzaron la respuesta clínica respectó a lainducción previa con SIMPONI@. Los pacientes se aleatorizaron para recibir SIM~ONI@50 mg, SIMPONI@100 mg o placebo administrado por vía subcutánea cada 4 sefanas.Se permitieron dosis estables concomitantes de aminosalicilatos orales y/o agentesinmunomoduladores. La dosis de corticosteroides se redujo al inicio del estudio demantenimiento. En este estudio se evaluó la eficacia de SIMPONI@hasta la sema~la 54.
Tabla 5Resultados de eficacia clave de PURSUIT-Inducción y PURSUIT-Mantenimiento
PURSUIT -InducciónPlacebo Simponi 200/100 mgN = 251 N = 253
Porcentaie de DacientesPacientes en respuesta clínica en 30% 51%**la semana 6a
Pacientes en remisión clínica en 6% 18%**la semana 6b
Pacientes con curación de la 29% 42%**mucosa en la semana 6c
PURSUIT -MantenimientoSimponi Simponi
Placebod 50 mg 100 mgN - 154 N - 151 N - 151
Porcentaie de DacientesMantenimiento de la respuesta(pacientes en respuesta clínica 31% 47%* 50%**hasta la semana 54)"
Remisión mantenida (pacientesen remisión clínica en las 16% 23%g 28%*semanas 30 y 54)f
. !,
P ¡na 12de34
N = número de pacientes** p :50,001'p:SO,Ola Definido como una reducción desde el valor basal en la puntuación Mayo en ;;:::30% y ~ 3 puntos,acompañado por una reducción de la subpuntuación de hemorragia rectal de ~ 1o una sUbpuntuabón dehemorragia rectai de ° o 1.b Definido como una puntuación Mayo s: 2 puntos, sin subpuntuación individual> 1e Definido como ° o 1 en la subpuntuación endoscópica de la puntuación Mayo.d SIMPONI@sólo durante la inducción.e Los pacientes se evaluaron para determinar la actividad de la CU mediante la puntuación parcial de Mayocada 4 semanas (la pérdida de respuesta se confirmó por endoscopia). Por consiguiente, un paciehte quemantuvo la respuesta estuvo en un estado de respuesta clínica continua en cada evaluación Hasta lasemana 54.f Para considerar remisión sostenida, el paciente tuvo que estar en remisión en lasmostrar pérdida de respuesta en cualquier evaluación hasta la semana 54).

720'1 (~j-.y" ~
g En pacientes con un peso inferior a 80 kg, una mayor proporción de pacientes que recibieron 50 mg éi'" DE ~:~tratamiento de mantenimiento mostraron remisión clínica mantenida en comparación con 1105querecibieron placebo.
Más pacientes tratados con SIMPONI@ demostraron curación de la mucosa mantenidaI
(pacientes con curación de la mucosa en las semanas 30 y 54) en el grupo de 50 mgI
(42%, nominal p < 0,05) Y en el grupo de 100 mg (42%, p < 0,005) comparado conlos pacientes del grupo placebo (27%).
Entre el 54% de los pacientes (247/456) que recibieron corticosteroides concomitanteal inicio de PURSUIT-Mantenimiento, el porcentaje de pacientes que mantenlían larespuesta clínica hasta la semana 54 y que no recibían corticosteroides concomitante
I
en la semana 54 era mayor en el grupo de 50 mg (38%, 30/78) Y en el grupo de 100mg (30%, 25/82) comparado con el grupo del placebo (21%,18/87). El porcentaje de,pacientes a los que se eliminó los corticosteroides en la semana 54 era mayorl en elgrupo de 50 mg (41%, 32/78) Y en el grupo de 100 mg (33%, 27/82) comparado conel grupo placebo (22%, 19/87).
En la semana 6, SIMPONI@mejoró significativamente la calidad de vida medida por elcambio desde el momento basal en una medida específica de la enfermedad, IBDQ(cuestionario de la enfermedad intestinal inflamatoria). Entre los pacientes querecibieron tratamiento de mantenimiento con SIMPONI@'lamejoríaenlacalid¡laddevida medida por el IBDQ se mantuvo hasta la semana 54.
InmunogenicidadEn los ensayos de fase In en AR, APs y EA hasta la semana 52, se dete taronanticuerpos frente a golimumab en el 5% (105 de 2.115) de los pacientes tratadbs congolimumab y cuando se analizaron, casi todos los anticuerpos fueron neutraliza~tes invitro. La tasa fue similar en todas las indicaciones reumatológicas. El tratarhiento,concomitante con MTX dio lugar a una menor proporción de pacientes con anticuerposfrente a golimumab en comparación con los que recibieron golimumab si~ MTX(alrededor del 3% [41 de 1.262] frente al 8% [64 de 853], respectivamente). IEn los ensayos de fase n y In en CU hasta la semana 54, se detectaron anticuerposfrente a golimumab en el 3% (26 de 946) de los pacientes tratados con gOlimurrJab. Elsesenta y ocho por ciento (21 de 31) de los pacientes que dieron positivo I a losanticuerpos tenían anticuerpos neutralizantes in vitro. El tratamiento concomitante coninmunomoduladores (azatioprina, 6-mercaptopurina y MTX) dio lugar a una fenorproporción de pacientes con anticuerpos frente a golimumab en comparación cbn losque recibieron golimumab sin inmunomoduladores (el 1% (4 de 308) frente al 36/0 (22de 638), respectivamente).
La presencia de anticuerpos frente a golimumab puede incrementar el riesgo deaparición de reacciones en la zona de inyección (Ver Precauciones y advertencias). Elbajo número de pacientes que dio positivo para anticuerpos frente a golimumab limita

7207la capacidad de extraer conclusiones definitivas sobre la relación entreanticuerpos y la eficacia clínica o los datos de seguridad.Las pruebas de inmunogenicidad son específicas para cada producto y tipo de ensayoanalítico, por lo que no es adecuado comparar la tasa de anticuerpos con las dE! otrosproductos.
Población pediátricaLa Agencia Europea de Medicamentos ha eximido al titular de la obligación depresentar los resultados de los ensayos realizados con SIMPONI@ en los dife~entesgrupos de la población pediátrica en espondilitis anquilosante y en artritis reumbtoide(ver Posología y Forma de administración para consultar la información sobre el ¿so enpoblación pediátrica).
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento parapresentar los resultados de los ensayos realizados con SIMPONI@en uno o más druposde la población pediátrica en artritis idiopática juvenil, en artritis psoriásica y colitisulcerosa (ver Posología y Forma de administración para consultar la información sobreel uso en población pediátrica).
Propiedades FarmacocinéticasAbsorciónLa mediana del tiempo hasta alcanzar la concentración senca maxlma (Tmáx)tras laadministración subcutánea de una dosis única de golimumab a sujetos sahos opacientes con AR varió entre dos y seis días. En sujetos sanos, una iny~cciónsubcutánea de 50 mg de golimumab produjo una concentración sérica máXimaj(cmáX)media :lo desviación estándar de 3,1 :lo 1,4 IJg/ml.
La absorción de golimumab tras una sola inyección subcutánea de 100 mg fue imilarcuando se inyectó en el brazo, el abdomen o el muslo, con una biodisponifuilidadabsoluta media del 51%. Dado que la farmacocinética de golimumab t{as laadministración subcutánea de una dosis era casi proporcional a la dosis, es previsibleque la biodisponibilidad absoluta de una dosis de 50 mg o 200 mg sea similar.
DistribuciónEl volumen de distribución medio tras una única administración IV fue 115 :lo 19 mi/kg.
Eliminación
El aclaramiento sistémico de golimumab se calculó en 6,9 :lo 2,0 mI/día/kg. La vidamedia de eliminación estimada fue de aproximadamente 12 :lo 3 días en los s~jetossanos, y en los pacientes con AR, APs, EA o CU presentó valores parecidos. ICuando se administraron 50 mg de golimumab por vía subcutánea cada cuatrosemanas a pacientes con AR, APs o EA, la concentración s" el éstadoestacionario se alcanzó en la semana 12. Con la administración c ncomit te dJ MTX,
Andr o/ante
~
DAlfec a TécnicaP e da
t.N c.

! ".~.A.r.
7 ~o7 (~co~ ~~
el tratamiento con 50 mg de golimumab por vía subcutánea cada 4 semanas dio 1"'(0' DEfe.~~'"a una concentración sérica media (:l: desviación estándar) en el estado estacionJrio deaproximadamente 0,6 :l: 0,4 ~g/ml en los pacientes con AR activa a pes~r deltratamiento con MTX, de aproximadamente 0,5 :l: 0,4 ~g/ml en los pacientes cÓn APsactiva y de aproximadamente 0,8 :l: 0,4 ~g/ml en los pacientes con EA.
En los pacientes con AR, APs o EA que no recibieron MTX concomitante, laconcentración en el estado estacionario de golimumab fue aproximadamente u~ 30%menor que en los que recibieron golimumab más MTX. En un número limitabo depacientes con AR tratados con golimumab por vía subcutánea durante un periodb de 6meses, el tratamiento concomitante con MTX redujo el aclaramiento aparerlte de
1
golimumab aproximadamente en un 36%. Sin embargo, el análisis farmacocinéticoI
poblacional indicó que la administración concomitante de AINEs, corticoesteroidesorales o sulfasalazina no afectó al aclaramiento aparente de golimumab. ITras las dosis de inducción de 200 mg y 100 mg de golimumab en la semana O y 2,respectivamente y las dosis de mantenimiento de 50 mg o 100 mg de golimUméb porvía subcutánea cada 4 semanas en adelante a pacientes con CU, las concentrabonesséricas de golimumab alcanzaron el estado estacionario aproximadamente 14 seranasdespués de iniciar la terapia. El tratamiento con 50 mg o 100 mg de golimumabsubcutáneo cada 4 semanas durante el mantenimiento tuvo como resultadb unaconcentración sérica media en el estado estacionario de aproximadamente 0,9 :l: 0,5~g/ml y 1,8 :l: 1,1 ~g/ml, respectivamente.
En pacientes con CU tratados con 50 mg o 100 mg de golimumab subcutáneo cada 4semanas, el uso concomitante de inmunomoduladores no tuvo ningún efecto sustancialsobre las concentraciones valle en el estado estacionario de golimumab. lEn general, los pacientes que desarrollaron anticuerpos antigolimumab tenía unaconcentración séricade golimumab en el estado estacionario más baja (ver propi~,dadesfarmacodinamicas).
LinealidadEn los pacientes con AR, la farmacocinética de golimumab tras la administración deuna dosis intravenosa única fue casi proporcional a la dosis en el intervalo posolÓ~icode 0,1 a 10,0 mg/kg. ITras la administración de una dosis se única en sujetos sanos, también se observó unafarmacocinética casi proporcional a la dosis en el intervalo posológico de 50 mg a 400mg.
Efecto del peso sobre la farmacocinéticaSe puso de manifiesto que el aclaramiento aparente de golimumab tiende a aumentarcon el peso corporal (ver Posología y forma de administración).
DATOS PRECLÍNICOS SOBRE SEGURIDAD
Los datos de los estudios no clínicos no muestran los seres
't"."

7207
No se han llevado a cabo con golimumab estudios de mutagenicidad, estudios defertilidad animal ni estudios carcinogénicos a largo plazo. jEn un estudio sobre la fertilidad y la función reproductora general en el ratón,utilizando un anticuerpo análogo que inhibe selectivamente la actividad funcio al delTNFa del ratón, el número de ratones preñados se redujo. Se desconoce si estk datofue debido a los efectos sobre los machos y/o las hembras. En un estudio de to~icidadsobre el desarrollo embrionario llevado a cabo en el ratón tras la administraci6n del,mismo anticuerpo análogo, y en el mono cynomolgus tras la administraci~'>n degolimumab, no hubo indicación de toxicidad materna, embriotoxicidad oteratogenicidad.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN
El tratamiento con SIMPONI@se debe iniciar y supervisar por médicos calificado1s, conexperiencia en el diagnóstico y tratamiento de la artritis reumatoide, artritis psoriásica,espondilitis anquilosante o colitis ulcerosa. A los pacientes tratados con SIMPONI@se lesdeberá entregar la Tarjeta de Alerta para el Paciente.
PosologíaArtritis reumatoideSe debe administrar SIMPONI@50 mg una vez al mes, el mismo día de cada mes.SIMPONI@debe administrarse de forma conjunta con MTX.
Artritis psoriásicaSe debe administrar SIMPONI@50 mg una vez al mes, el mismo día de cada mes.
Espondilitis anquilosan teSe debe administrar SIMPONI@50 mg una vez al mes, el mismo día de cada mes.
Página 16 de 34
iPara todas las indicaciones anteriores, los datos disponibles sugieren que la respuestaclínica se alcanza generalmente dentro de las 12 a 14 semanas de tratamiento (dJspuésde 3-4 dosis). Se debe reconsiderar continuar con el tratamiento en pacientes en lbs queno se observe beneficio terapéutico tras ese periodo de tiempo. 1Pacientes con peso corporal superior alOa kgPara todas las indicaciones anteriores, en pacientes con artritis reumatoide, rtritispsoriásica o espondilitis anquilosante con un peso corporal de más de 100 kg Y ~ue noalcancen una respuesta clínica adecuada después de 3 ó 4 dosis, se puede consid~rar elaumentar la dosis de golimumab alOa mg administrados una vez al mes, tenierido en
Icuenta el aumento del riesgo de ciertas reacciones adversas grav la dosis de 100mg en comparación con la dosis de 50 mg (ver Reaccio es Adv rsas). Se debe

reconsiderar continuar el tratamiento en pacientes en los que no se observeterapéutico después de recibir entre 3 y 4 dosis adicionales de 100 mg.
Colitis ulcerosaPacientes con peso corporal inferior a 80 kgSIMPONI@, se administra como una dosis inicial de 200 mg, seguido de 100 mg en lasemana 2, y posteriormente 50 mg cada 4 semanas (ver PropiJdadesFarmacodinamicas).Pacientes con peso corporal superior o igual a 80 kgSIMPONI@ se administra como una dosis inicial de 200 mg seguido de 100 mg en lasemana 2, y posteriormente 100 mg cada 4 semanas (ver PropiedadesFarmacodinamicas).
Durante el tratamiento de mantenimiento, los corticosteroides se pueden reducir deacuerdo con las directrices de la práctica clínica.
Los datos disponibles sugieren que la respuesta clínica se alcanza habitualmente dentrode las 12-14 semanas de tratamiento (después de 4 dosis). Se deberá considJrar lacontinuación del tratamie~to en pacientes que no muestran evidencia de beheficioterapéutico dentro de este período de tiempo.
Dosis olvidadasSi un paciente olvida inyectarse SIMPONI@en la fecha programada, se debe inyectar la
I
dosis olvidada tan pronto como el paciente lo recuerde. Se debe avisar a los pacientes,que no se inyecten una dosis doble para compensar la dosis olvidada.
La siguiente dosis se debe administrar conforme a las siguientes recomendaciones:• Si la dosis se ha retrasado menos de 2 semanas, el paciente debe inyectarse la
dosis olvidada y mantener su calendario original. I• Si la dosis se ha retrasado más de 2 semanas, el paciente debe inyectarse IJ dosis
olvidada y se debe establecer un nuevo calendario a partir de la fecha d~ estainyección.
Poblaciones especialesPacientes de edad avanzada (?: 65 años)No se requiere un ajuste de la dosis en pacientes de edad avanzada.
Insuficiencia renal y hepáticaNo se ha estudiado SIMPONI@ en estas poblaciones de pacientes. No se pueden i hacerrecomendaciones de dosis. :,
Población pediátrica 1No se ha establecido todavía la seguridad y eficacia de SIMPONI@ en pacientes m noresde 18 años. No se dispone de datos.
P ina17de34

720Forma de administraciónSIMPONI@,sedebe administrar por vía subcutánea. Tras un entrenamiento adecu Ido enla técnica de la inyección subcutánea, los pacientes se pueden autoinyectar SIMPONI@sisu médico lo considera apropiado, con seguimiento médico según sea necesario. S~ debeenseñar a los pacientes a inyectarse el volumen total de SIMPONI@de acuerdo don lasinstrucciones de administración que figuran en el prospecto. Si son necesarias Ivariasinyecciones a la vez, las inyecciones deben administrarse en diferentes zonas del cuerpo.
Precauciones especiales de eliminación y otras manipulaciones lDespués de sacar la Jeringa Prellenada/Autoinyector SmartJectde la heladera se I ebenesperar 30 minutos para que ésta alcance temperatura ambiente antes de inwectarSIMPONI@,No debe agitarse la Jeringa Prellenada/Autoinyector SmartJect. _La solución es entre transparente y ligeramente opalescente, de incolora a ararilloclaro y puede contener algunas partículas de proteína pequeñas -translúcidas o blancas.Este aspecto es habitual en soluciones que contienen proteína. SIMPONI@ no! debeutilizarse si la solución cambia de color, está turbia o contiene partículas ex~rañasvisibles.
CONTRAINDICACIONES
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en laComposición Cualitativa y Cuantitativa
Tuberculosis (TB) activa u otras infecciones graves como sepsis, e infecciones
oportunistas (ver Precauciones y Advertencias). NIYHA)
Insuficiencia cardiaca de moderada o grave (clase IU/IV segúnCClaclasificación(ver Precauciones y Advertencias).
PRECAUCIONES Y ADVERTENCIAS
Infecciones
Antes, durante y tras el tratamiento con SIrJlPONI@,debe vigilarse cuidadosam~nte alos pacientes en relación a la aparición de infecciones incluida la tuberculosis. Dado quela eliminación de golimumabpuede llevar hasta 5 meses, se deberá continuar el dontrola lo largo de este periodo. Si un paciente desarrolla una infección grave o sepsis no sedebe continuar el tratamiento con SIMPONI@(ver Contraindicaciones).
No se debe administrar SIMPONI@a pacientes con una infección activa, clínicamenteimportante. Se debe tener precaución al considerar la utilización de SIMPONI@ enpacientes con infección crónica o historia de infección recurrente. Según sea necJsario,se deberá recomendar a los pacientes, que eviten la exposición -a factores de r1!iesgo,potenciales para la infección.
Los pacientes que estén utilizando antagonistas del TNF son - les a p decerinfecciones graves.
P inal8de34

••.\I.M ..-. J;
Eo P"'oo'" t~"d" 000SIMPON!"~ h" ootifi"do ¡Of''':'~'?'¡~' "q""••~.sepsis y neumonía), por micobacterias (incluida la tuberculosis), fúngicas invaSivas yoportunistas, algunas de las cuales condujeron a la muerte. Algunas de I estasinfecciones graves se han producido en pacientes con terapia inmunosupresoraconcomitante que, junto con la enfermedad subyacente, puede predispone~ a laaparición de infecciones. Aquellos pacientes que desarrollen una nueva infeccióncuando están en tratamiento con SIMPONI@debenser cuidadosamente monitoriz~dos ysometerse a una completa evaluación diagnóstica. Si el paciente desarrolla una ¡nuevainfección grave o sepsis, se debe suspender la administración de SIMPONI@e iniciarseel tratamiento antimicrobiano o antifúngico adecuada hasta que la infección estécontrolada. En el caso de pacientes que hayan residido o viajado a regiones donbe lasinfecciones fúngicas invasivas como histoplasmosis, coccidiodomicosis o blasto~icOSiSson endémicas, deben evaluarse cuidadosamente los riesgos y los beneficicl>s deltratamiento con SIMPONI@antes de iniciar dicho tratamiento.
Tuberculosis
Se han notificado casos de tuberculosis en pacientes que recibieron [email protected] haobservado que en la mayoría de estos casos la tuberculosis fue extra pulmonar" y sepresentó como enfermedad local o diseminada. IAntes de iniciar el tratamiento con SIMPONI@,se debe evaluar en todos los pacie~tes laexistencia de infección tuberculosa activa e inactiva ('latente'). Esta evaluación deberáincluir una detallada historia clínica con antecedentes personales de tUberculbsis o
Iposible contacto previo con la enfermedad y terapia inmunosupresora previa y/oactual. Se deberán realizar en todos los pacientes pruebas de detección adeciliadas,esto es prueba cutánea de la tuberculina o análisis de sangre y radiografía de tórax(conforme a las recomendaciones locales). Se recomienda anotar en la tarjeta de alertapara el paciente la realización de estas pruebas. Se recuerda a los médicos el riesgo defalsos negativos en la prueba cutánea de la tuberculina, especialmente en pacientesque están gravemente enfermos o inmunodeprimidos. iSi se diagnostica una tuberculosis activa, no debe iniciarse el tratamientb conSIMPONI@(ver Contraindicaciones). ISi se sospecha de tuberculosis latente, se debe consultar a un médico con expe~ienciaen el tratamiento de tuberculosis. En todas las situaciones que se describen acontinuación, se debe considerar muy cuidadosamente la relación beneficio/ries6o deltratamiento con [email protected] se diagnostica tuberculosis inactiva ("latente"), se debeiniciar el tratamiento para la misma con medicamentos para la tuberculosis an~es deiniciar el tratamiento con SIMPONI@,y de acuerdo con las recomendaciones local~s. Sedebe considerar la terapia frente a la tuberculosis antes de iniciar el tratamien~o conSIMPONI@ en pacientes que presenten varios o importantes factores de riesbo de,tuberculosis y una prueba negativa para tuberculosis latente. Se debe considerartambién la utilización de terapia frente a la tuberculosis antes del inicio del tratarhientocon SIMPONI@en pacientes con historia de tuberculosis latente o activa en los due nose puede confirmar un curso adecuado de tratamiento.
Pá ína19de34

•..". ..4.(imollO
.'"~,7. ~il'4 ~'i
Se han producido casos de tuberculosis activa en pacientes tratados con SIM ONI DE ~1Idurante y después del tratamiento de tuberculosis latente. Los pacientes que r~cibenSIMPONI@deben ser monitorizados cuidadosamente para detectar cualquier signo y/osíntoma de tuberculosis activa, incluyendo pacientes con prueba negatival paratuberculosis latente, pacientes que están en tratamiento de tuberculosis latente, opacientes que fueron tratados previamente de infección tuberculosa. 1Se deben dar instrucciones a todos 105pacientes para que consulten con su mé ico siapareciesen Signos/síntomas indicativos de tuberculosis (por ejemplo, tos persi~tente,debilidad/pérdida de peso, febrícula) durante o después del tratamiento con SIMPbNI@.
Reactivación del virus de la hepatitis B
Se ha producido reactivación de la hepatitis B en pacientes que recibieron unantagonista TNF incluyendo SIMPONI@, y que son portadores crónicos de estJ virus(esto es, positivos para el antígeno de superficie). Algunos casos tuvieron un des~nlacemortal. ILos pacientes deben hacerse la prueba de infección por VHB antes de inidiar eltratamiento con SIMPONI@. En aquellos pacientes que den positivo a la prueba deinfección por VHB, se recomienda la consulta con un médico con experiencia en eltratamiento de la hepatitis B.
Los portadores del VHB que precisen tratamiento con SIMPONI@ deben sercuidadosamente monitorizados para detectar cualquier signo y/o síntoma de inf~cciónactiva por VHB durante el tratamiento, y durante 105 meses siguientes a la finalikacióndel tratamiento. No se dispone de suficientes datos sobre el tratamiento de pa6ientesportadores de VHB que reciben de forma conjunta tratamiento con antivir~les yantagonistas TNF para evitar la reactivación del VHB. En pacientes que desarrollJn unareactivación del VHB, se debe interrumpir el tratamiento con SIMPONI@y se inici~rá untratamiento antiviral eficaz junto con un tratamiento de soporte adecuado.
Neoplasias v trastornos linfoproliferativos
Se desconoce el posible papel del tratamiento con antagonistas del TNF en el desarrollode neoplasias. IEn base a 105 conocimientos actuales, no se puede excluir el riesgo de desarrollo delinfomas, leucemia u otras neoplasias en pacientes tratados con un agente antagpnistaTNF. Se debe tener precaución al considerar el tratamiento con antagonistas dél TNFen pacientes con antecedentes de neoplasia o cuando se valore si continuar ton eltratamiento en pacientes que desarrollen neoplasia.
Neoplasias pediátricas
Durante la fase postcomercialización se han notificado neoplasias, algunas mortales, enniños, adolescentes y adultos jóvenes (hasta 22 años) tratados con antagonisths delTNF (inicio de la terapia ~ 18 años de edad). Aproximadamen 'tad de 105 casos

FOl.IO
7 20 (f ~~}L.-;oíl'", ~
fueron Iinfomas. Los otros casos se correspondían con distintas neoplasias inclui !JE £11"\'1'neoplasias raras normalmente asociadas con inmunosupresión. No puede excluirse elriesgo de desarrollo de neoplasias en niños y adolescentes tratados con antagdnistasdel TNF.
Linfoma y leucemia
En las fases controladas de los ensayos clínicos de todos los antagonistas de TNF,entre ellos SIMPONI@,se han observado más casos de linfomas entre los pacientes querecibieron tratamiento anti- TNF que en los pacientes control. Durante los er\sayosclínicos fase IIb y fase III en AR, APs y EA con SIMPONI@, la incidencia de Iinfolna enpacientes tratados con SIMPONI@fue superior a la esperada en la población gJneral.Durante la fase postcomercialización, se han notificado casos de leucemia en padientestratados con un antagonista del TNF. Existe un mayor riesgo basal de IinfOraS yleucemia en pacientes con artritis reumatoide (con enfermedad inflamatoria de largaduración y de alta actividad), lo que complica la estimación del riesgo. IDurante la fase postcomercialización se han notificado en raras ocasiones casos delinfoma de células T hepatoesplénico (HSTCL) en pacientes tratados con antag~nistasdel TNF (ver sección 4.8). Este tipo raro de Iinfoma de células T, tiene un curso~'de laenfermedad muy agresivo y habitualmente provoca la muerte. La mayoría de los casoshan ocurrido en varones adolescentes y adultos jóvenes, mientras casi todo ellosrecibían tratamiento concomitante con azatioprina (AZA) o 6-mercaptopurina (16-MP)para la enfermedad inflamatoria intestinal. El riesgo potencial de la combinación deAZA o 6-MP con SIMPONI@debe considerarse cuidadosamente. El riesgo de des~rrollode Iinfoma de células T hepatoesplénico en pacientes tratados con antagonistas d~1TNFno puede excluirse.
Neoplasias distintas allinfoma
En las fases controladas de los ensayos clínicos fase IIb y fase 111 con SIMPONI@en AR,APs, EA Y CU, la incidencia de neoplasias distintas al linfoma (excluyendo el cánter depiel no melanoma) fue similar entre los grupos de SIMPONI@y control.
r'"''''
Displasia/carcinoma de colon
Se desconoce si el tratamiénto con golimumab influye en el riesgo de desa~rollardisplasia o cáncer de colon. Todos los pacientes con colitis ulcerosa que presentan unriesgo elevado de displasia o carcinoma de colon (por ejemplo, pacientes conlcolitisulcerosa de larga evolución o colangitis esclerosante primaria), o que han presJntadohistoria previa de displasia o carcinoma de colon deberán someterse a una revikión aintervalos regulares para el diagnóstico de displasia, antes de recibir el tratamienito y alo largo del curso de su enfermedad. Esta evaluación debería incluir colonoscbpia ybiopsia según recomendaciones locales. En pacientes con displasia de huevodiagnóstico, tratados con SIMPONI@, se debe revisar cuidadosa I riesgo y losbeneficios para los pacientes y se debe considerar la interrupció del tr miento.

7207 '~$En un ensayo clínico preliminar que evaluaba el uso de SIMPONI@ en lIE fJ\~'l'asma persistente grave, se notificaron más neoplasias en pacientes tratados conSIMPONI@ en comparación con los pacientes control (ver Reacciones Adversak). Sedesconoce la relevancia de este dato. IEn un ensayo clínico preliminar en el cual se evaluaba el uso de otro agente anti- TNF,
. I
infliximab, en pacientes con enfermedad pulmonar obstructiva crónica (EPOC)I
moderada a grave, se notificaron más neoplasias, principalmente de pulmón o cabeza ycuello, en los pacientes tratados con infliximab en comparación con los paCientes
I
control. Todos los pacientes tenían un historial de tabaquismo importante. Por lo ¡tanto,se debe tener precaución al usar cualquier antagonista TNF en pacientes con EPOC, asícomo en pacientes con riesgo elevado de neoplasia por tabaquismo importante.
Cánceres de piel
Se ha notificado melanoma en pacientes tratados con antagonistas del TNF, incluidoSIMPONI@. Se ha notificado carcinoma de células de Merkel en pacientes tratad¿s conotros antagonistas del TNF (ver Reacciones Adversas). Se recomiendan exárenesperiódicos de la piel, especialmente en pacientes con factores de riesgo para cáncer depiel.
PlJgina 22 de 34
Cirugía
Insuficiencia cardiaca congestiva ([CC)
Con los antagonistas del TNF, incluyendo SIMPONI@, se han comunicado cases deempeoramiento de la insuficiencia cardíaca congestiva (ICC) y de nueva aparición deICe. En un ensayo clínico con otro antagonista TNFse ha observado empeoramiehto dela insuficiencia cardiaca congestiva y aumento de la mortalidad por ICe. No Ise haestudiado SIMPONI@en pacientes con ICe. SIMPONI@debe utilizarse con precaución enpacientes con insuficiencia cardiaca leve (clase I/I! según la clasificación NYHA~. Lospacientes deberán ser cuidadosamente vigilados y no se deberá continJar eltratamiento con SIMPONI@en pacientes que desarrollen síntomas nuevos o en Iqs quese observe un empeoramiento de la insuficiencia cardiaca (ver ContraindicacionesD.
Trastornos neurológicos lEl uso de antagonistas del TNF, incluyendo SIMPONI@, ha sido asociado con la uevaaparición de casos o exacerbación de los síntomas clínicos y/o evidencia radiOgrá~iCadeenfermedades desmielinizantes del sistema nervioso central, incluida la esclerosismúltiple y enfermedades desmielinizantes periféricas. En pacientes con enfermebadesdesmielinizantes preexistentes o de reciente aparición, se deberán con~iderarcuidadosamente los beneficios y riesgos del tratamiento con anti- TNF antes del; iniciodel tratamiento con [email protected] aparecen estas enfermedades, se debe considJrar lainterrupción de SIMPONI@(ver Reacciones Adversas). ¡

•..".11.••.1.'
71207 (~~~~
La experiencia sobre la seguridad del tratamiento con SIMPONI@ en pacien OE f.lI.~~sometidos a intervenciones quirúrgicas, incluida la arterioplastia, es limitada. I Si seplanea una intervención quirúrgica se deberá tener en cuenta la larga semivida df! estemedicamento. El paciente que requiera cirugía durante el tratamiento con SIMPONI@
Ideberá ser cuidadosamente controlado en relación con la aparición de infecciones, y sedeberán adoptar las medidas adecuadas.
Inmunosupresión
Existe la posibilidad de que los antagonistas del TNF, incluyendo SIMPONI@, afecten alas defensas del huésped frente a infecciones y neoplasias, ya que el TNF ~s unmediador de la inflamación y modula la respuesta inmunitaria celular.
Procesos autoinmunes
La deficiencia relativa de TNFO que provoca el tratamiento anti-TNF puededesencadenar un proceso autoinmune. Si un paciente desarrolla síntomas indic'ativosde un síndrome tipo lupus después del tratamiento con SIMPONI@y es positivJ paraanticuerpos anti-DNA de doble cadena, se debe interrumpir el tratamient6 conSIMPONI@(ver Reacciones Adversas).
Reacciones hematológicas
Se han notificado durante la fase postcomercialización casos de pancitopenia,leucopenia, neutropenia, anemia aplásica y trombocitopenia en pacientes tratad6s conantagonistas del TNF. Durante los ensayos clínicos con SIMPONI@se han notifiddo demanera poco frecuente citopenias como pancitopenia. Todos los pacientes debén ser
Iinformados de que deben acudir en busca de asistencia médica inmediatamente sidesarrollan signos y presentan síntomas indicativos de discrasias sanguíneaJ (porejemplo fiebre persistente, sangrado, cardenales, palidez). Se debe con~iderarinterrumpir la administración de SIMPONI@en pacientes en los cuales se conftirmenalteraciones hematológicas significativas.
Administración concomitante de antagonistas del TNF y anakinra
Se observaron infecciones graves y neutropenia en ensayos clínicos con lusoconcomitante de anakinra y otro antagonista del TNF, etanercept, sin beneficio tlínicoañadido. Debido a la naturaleza de los acontecimientos adversos observados ton laterapia de combinación, pueden aparecer toxicidades similares también cpn lacombinación de anakinra y otros antagonistas del TNF. No se recomientla laadministración conjunta de SIMPONI@y anakinra.
Administración concomitante del antagonista del TNF y abatacept
En los ensayos clínicos la administración conjunta de antagonistas dse ha asociado con un incremento en el riesgo de infecciones, nclui
aba acept, ;Ofto~P4glltD. 23 de 34

Administración concomitante con otras terapias biológicas
No hay información suficiente relativa al uso concomitante de SIMPONI@ con' otrasterapias biológicas utilizadas para tratar las mismas afecciones que SIMPONI@.,No serecomienda el uso concomitante de SIMPONI@ con estos medicamentos biológicos
I
debido a la posibilidad de un aumento del riesgo de infección y otras interaccionesfarmacológicas potenciales.
Cambio entre FAMEsbiológicos
Se debe tener cuidado y los pacientes deben ser monitorizados cuando se cam ia deun medicamento biológico a otro, ya que la superposición de la actividad bi91ÓgiCapuede aumentar todavía más el riesgo de acontecimientos adversos, incluida lainfección.
Vacunas/agentes infecciosos terapéuticos
Los pacientes tratados con SIMPONI@ pueden recibir simultáneamente vali:Unas,excepto vacunas de microorganismos vivos (ver secciones 4.5 y 4.6). En pacientés queestán recibiendo terapia anti-TNF, los datos disponibles sobre la respuestal a lavacunación con vacunas de microorganismos vivos o sobre la transmisión secuhdariade la infección por vacunas de microorganismos vivos son limitados. lEl uso de vacunas de microorganismos vivos puede producir infecciones, i c1usodiseminadas. IOtros usos de los agentes infecciosos terapéuticos como bacterias vivas atenuadas (porej., la instilación de BCG en la vejiga para el tratamiento del cáncer) pueden prbducirinfecciones, incluso diseminadas. No se recomienda la administración concomitahte deagentes infecciosos terapéuticos con SIMPONI@.
Reacciones alérgicas
En la experiencia postcomercialización, se han observado reacciones grav s dehipersensibilidad sistémica (incluyendo reacción anafiláctica) tras la administración [email protected] de estas reacciones se produjeron tras la primera administ~aciónde SIMPONI@. Si se produce una reacción anafiláctica u otras reacciones al~rgiCaSgraves, se debe interrumpir inmediatamente la administración de SIMPONI@e inifiar eltratamiento adecuado. I
Sensibilidad al látex

7 20 7 FO~IO~z_ ~La tapa de la aguja en la pluma precargada se fabrica a partir de goma seca ~ ~:~que contiene látex, y puede producir reacciones alérgicas en personas con sensibilid El!
al látex.
Poblaciones especiales
Pacientes de edad avanzada (2: 65 años)
En los ensayos de fase III en AR, APs, EA y CU, no se observaron diferencias globalesen reacciones adversas (RAs), reacciones adversas graves (RAGs) e infecciones ~ravesen pacientes de edad igual o superior a 65 años que recibieron SIMPON~@encomparación con pacientes más jóvene's. No obstante, se deben tomar precaufionescuando se trate pacientes de edad avanzada y prestar especial atención en relaci0n conla aparición de infecciones.
Insuficiencia renal y hepática '
No se han llevado a cabo ensayos específicos de SIMPONI@ en pacientek coninsuficiencia renal o hepática. SIMPONI@debe utilizarse con precaución en padentescon insuficiencia hepática (ver Posología y Forma de Administración).
Excipientes
SIMPONI@ contiene sorbitol (E420). Los pacientes con intolerancia hereditaria a lafructosa no deben usar SIMPONI@.
Potenciales errores de medicación
SIMPONI@ está registrado en dosis de 50 mg y 100 mg para administraciónsubcutánea, Es importante utilizar la dosis adecuada para administrar la dosis c+rectasegún se indica en la posología (ver Posología y Forma de Administración). Se debetener cuidado al proporcionar
INTERACCIONES MEDICAMENTOSAS
No se han efectuado estudios de interacciones.
Uso concomitante con otras terapias biológicasNo se recomienda la administración conjunta de SIMPONI@con otras terapiasbiológicas utilizadas para tratar las mismas afecciones que SIMPONI@, incluidasanakinra y abatacept (ver Precauciones y Advertencias).
Vacunas de microorganismos vivos/Agentes infecciosos TerapeúticosLas vacunas de microorganismos vivos no se deben administrar simultáneamente conSIMPONI@(ver Precauciones y Advertencias y Fertilidad, embarazo y ancia). 1
,
Página 25 de 34

No se recomienda la administración simultanea de agentesSIMPONI@(ver Precauciones y Advertencias)
MetotrexatoAunque el uso concomitante de MTX provoca un aumento de las concentraciores en. estado estacionario de SIMPONI@en pacientes con AR, APs o EA, los datos no sU~ierenque sea necesario realizar un ajuste la dosis de SIMPONI@ni de MTX (ver PropiedadesFarmacocinéticas)
Fertilidad, embarazo y lactanciaMujeres en edad fértilLas mujeres en edad fértil deben utilizar métodos anticonceptivos adecuados paraprevenir el embarazo y continuar su uso durante al menos 6 meses después del dltimotratamiento con golimumab, I
EmbarazoNo existen datos suficientes sobre la utilización de golimumab en mujeresembarazadas. Debido a su inhibición del TNF, la administración de golimumab durranteel embarazo podría afectar a la respuesta inmunitaria normal en el recién nacido. Losestudios en animales no muestran efectos dañinos directos o indirectos sobre elembarazo, desarrollo embrional/fetal, parto o desarrollo posnatal (ver Datos Precl,ínicossobre seguridad). No se recomienda el uso de golimumab en mujeres embarazadas;solamente se debe administrar golimumab a una mujer embarazada si fueseestrictamente necesario.
Golimumab atraviesa la placenta. Tras el tratamiento con anticuerpos monoclonal1esantagonistas del TNF durante el embarazo, se han detectado anticuerpos hasta 6meses en el suero de los bebés nacidos de mujeres tratadas. Por lo tanto, estos niñospueden tener un mayor riesgo de infección. No se recomienda la administración dbvacunas de virus vivos a los bebés expuestos a golimumab en el útero durante lo 6meses después de la última inyección de golimumab de la madre durante el emb raza(ver Precauciones y Advertencias e Interacciones Medicamentosas).
LactanciaSe desconoce si golimumab es excretado en la leche materna o si se absorbesistémicamente después de la ingestión. Se ha observado que golimumab pasa a laleche materna en el mono, y como las inmunoglobulinas humanas se excretan en laleche, las mujeres no deben dar el pecho durante y al menos 6 meses después deltratamiento con golimumab.
FertilidadNo se han realizado estudios de fertilidad en animales con golimumab. Un estudio, defertilidad en ratones, utilizando un anticuerpo análogo que inhibe selectivamente la
Iactividad funcional del TNFo del ratón, no mostró efectos rel obre la fertilidad(ver Datos Preclínicos sobre seguridad).
P~gina 26 de 34

Efectos sobre la capacidad para conducir y utilizar máquinas
72017
pequeña. Se puedeReacciones Adversas).
REACCIONES ADVERSAS
Resumen del perfil de seguridad
Tabla 6Lista de las RAM
Frecuentes:
Raras:
Raras:Frecuencia no conocida:
Poco frecuentes:
La reacción adversa (RA) más frecuente notificada en el período controlado de losensayos pivota les en AR, APs, EA Y CU fue la infección del tracto respiratorio superior,produciéndose en el 12,6% de los pacientes tratados con golimumab, en compalracióncon el 10,7% de los pacientes control. Las reacciones adversas más graves que se hannotificado con golimumab son infecciones graves (como sepsis, neumoní~, TB,infecciones fúngicas invasivas e infecciones oportunistas), trastornos desmielinizbntes;Iinfoma, la reactivación del VHB, ICC, procesos autoinmunes (síndrome de tipo lubus) yreacciones hematológicas (ver Precauciones y Advertencias). !
¡
IInfecciones e infestaciones IMuy frecuentes: Infección del tracto respiratorio superior
(nasofaringitis, faringitis, laringitis y rinitis) IInfecciones bacterianas (como celulitis), infecCióndel tracto respiratorio inferior (como neumofía),infecciones víricas (tales como influenza y herP1es),bronquitis, sinusitis, infecciones fúngicassuperficiales, abscesos ISepsis incluyendo shock séptico, infecciónesoportunistas (tales como infecciones fúndicasinvasivas [histoplasmosis, COCcidioidomicosij yneumocistiasis], bacterianas, infección pormicobacterianas atípicas y protozoos), a ,ritisbacteriana, pielonefritisReactivación de la hepatitis B, tuberculosis,infecciosa
Neoplasias benignas, malignas y no especificadasPoco frecuentes: Neoplasias (tales como cáncer de piel, carcinoma de
células escamosas, nevo melanocitico) ILinfoma, leucemia, melanoma.Carcinoma de células de Merkel*, linfoma de célulasT hepatoesplenico* I
Trastornos de la sangre y del sistema linfaticoFrecuentes: Anemia
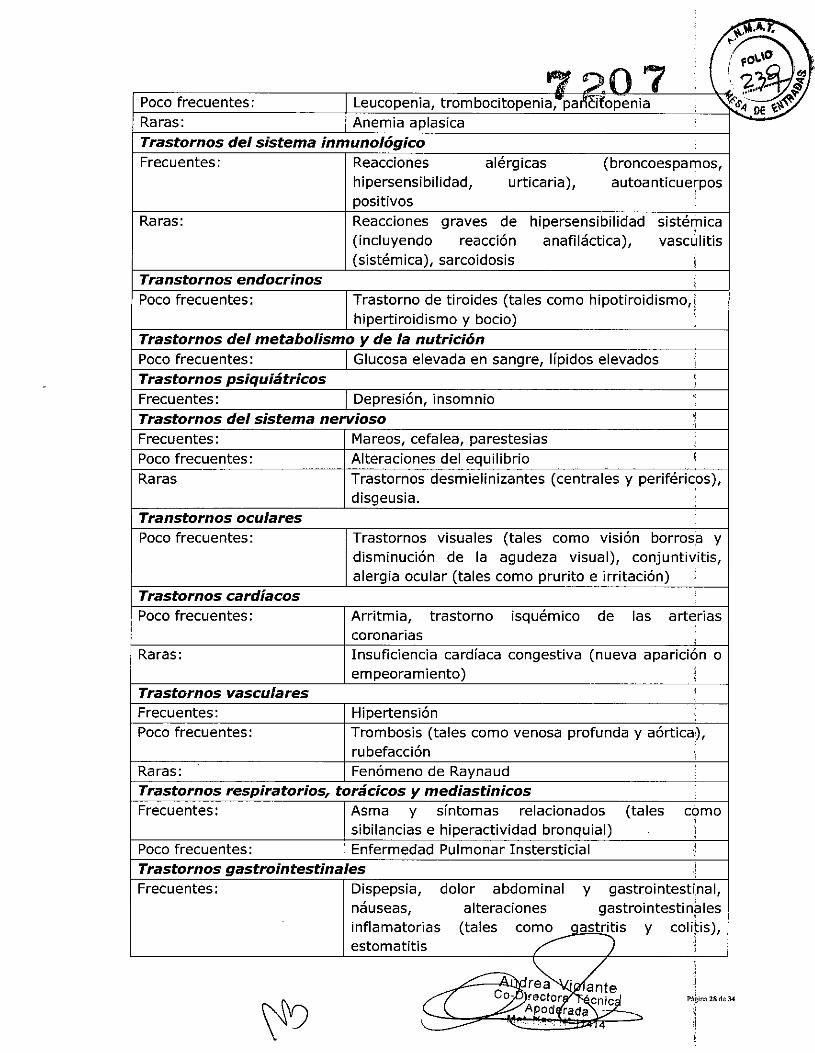
.,",A:r.;~\t#J 90 7 ! ",f, '1.) i
Poco frecuentes: Leucopen ia, trom bocitopenia,.pa /ícfropenia '\ &'~DE "t;~~Raras: Anemia aplasicaTrastornos del sistema inmunológicoFrecuentes: Reacciones alérgicas (broncoespamos,
hipersensibilidad, urticaria), autoanticue~pospositivos I
Raras: Reacciones graves de hipersensibilidad sistémica(incluyendo reacción anafiláctica),
Ivascl!llitis
(sistémica), sarcoidosis ITranstornos endocrinos I
IPoco frecuentes: Trastorno de tiroides (tales como hipotiroidismo, I
hipertiroidismo y bocio) I
Trastornos del metabolismo y de la nutriciónt
Poco frecuentes: Glucosa elevada en sangre, lípidos elevadosTrastornos psiquiátricosFrecuentes: Depresión, insomnioTrastornos del sistema nervioso jFrecuentes: Mareos, cefalea, parestesiasPoco frecuentes: Alteraciones del equilibrio IRaras Trastornos desmielinizantes (centrales y periférióos),
disgeusia. ,,Transtornos oculares I
Poco frecuentes: Trastornos visuales (tales.. ,
borros'acomo vlslon ydisminución de la agudeza visual), conjuntivitis,alergia ocular (tales como prurito e irritación)
Trastornos cardíacosPoco frecuentes: Arritmia, trastorno isquémico de las arterias
Icoronarias t
Raras: Insuficiencia cardíaca congestiva (nueva aparición oempeoramiento) I
Trastornos vasculares iFrecuentes: Hipertensión I
I
Poco frecuentes: Trombosis (tales como venosa profunda y aórtica'),rubefacción ¡"
Raras: Fenómeno de Raynaud ITrastornos respiratorios, torácicos y mediastinicos :Frecuentes: Asma y síntomas relacionados (tales como
;sibilancias e hiperactividad bronquial) I
Poco frecuentes: Enfermedad Pulmonar Instersticial ,1
Trastornos gastrointestinales IFrecuentes: Dispepsia, dolor abdominal y gastrointestinal,
náuseas, alteraciones gastrointestin~lesinflamatorias (tales como qastritis y cOlibs),estomatitis /' )
/ ~~~~~ antec( ~iector c~LiL Página 28 de 34
Apod rada .--- . "., '.

".IU.,
720 7@\~ 2.'tJ,Q...-¡Poco frecuentes Estreñimiento, enfermedad por refl uj~ DE <1l~
gastroesofágicoTranstornos hepatobiliaresFrecuentes: Alanina aminotransferasa elevada, aspafato
aminotransferase elevadaPoco frecuentes: Colelitiasis, trastornos hepáticos ITrastornos de la piel y del tejido subcutáneo IFrecuentes: Prurito, rash, alopecia, dermatitisPoco frecuentes: Reacción cutánea ampollosa, psoriasis (por nueva
aparición o empeoramiento de psoriasis bre-existente, palmar/plantar y pustular), urticaria
Raras: Exfoliación de la piel, vasculitis (cutánea)Trastornos musculoesque/éticos y del tejido conectivoRaras: I Síndrome tipo lupus ITrastornos renales y urinarios IRaras: Trastorno vesical, trastornos renales ITrastornos del aparato reproductor y de la mama IPoco frecuentes: Trastornos de la mama, trastornos menstrualesTrastornos generales y alteraciones en el lugar de administraciónFrecuentes: Pirexia, astenia, reacción en la zona de inyecCión
(tales como eritema, urticaria, induración, d~lor,cardenales, prurito, irritación y parestesias el, lazona de inyección), malestar torácico
Raras: Alteración de la cicatrizaciónLesiones traumáticas, intoxicaciones y complicaciones de procedimientosterapéuticos: IFrecuentes: I Fracturas óseas I,* Observado con otros antagonistas del TNF, pero no observados en los ensayos clínicos: congolimumab. i
I
Descripción de reacciones adversas seleccionadasInfeccionesLa reacción adversa más frecuente notificada en el período controlado de los ensayospivotales fue la infección del tracto respiratorio superior, presente en el 12,6% ~e lospacientes tratados con gotimumab (incidencia por cada 100 pacientes-año: 160,9;intervalo de confianza [IC] del 95%: 54,9, 67,3) en comparación con el 10,7% de lospacientes control (incidencia por cada 100 pacientes-año: 53,2; lC del 95%: 144,4,63,2). En las fases controladas y no controladas de estos ensayos con una mediana deseguimiento de aproximadamente 2 años, la incidencia por cada 100 pacientes-Jño deinfecciones del tracto respiratorio superior fue de 35,9 episodios, lC del 95%: 34,8,37,1 en los pacientes tratados con golimumab.
En el período controlado de los ensayos pivota les, se observaron infecciones en el22,8% de los pacientes tratados con golimumab (incidencia por cada 100 paci ntes-
I
año: 130,4; lC del 95%: 121,6, 139,7) en comparación con 9,9% e los pacientesAlldrea Violanl I
. acto Té Icapodara Piiginn 29 de 34
.-dQ. M.

".tU.,(::;\720 ~~¡
control (incidencia por cada 100 pacientes-año: 123,0; re del 95%: 109,4, 137,8). <l' DE E1\~'"las fases controladas y no controladas de estos ensayos con una media ha deseguimiento de aproximadamente 2 años, la incidencia por cada 100 pacientes-clño deinfecciones fue de 83,5 episodios, rc del 95%: 81,8, 85,3 en los pacientes tratad6s congolimumab.
En el período controlado de los ensayos en AR, APs y EA, se observaron infeccionesgraves en el 1,4% de los pacientes tratados con golimumab y en el 1,3% be lospacientes control. Durante el seguimiento la incidencia de infecciones graves po~ cada100 pacientes-año en el período controlado de los ensayos en AR, APs y EA fue ~e 7,4eventos; rc del 95%: 4,6, 11,1 en el grupo de tratamiento con golimumab 10b mg,3,3; rc del 95%: 1,3, 6,9 en el grupo de tratamiento con golimumab 50 mg y 41,2; rcdel 95%: 1,8, 8,2 en el grupo placebo. En el período controlado de los ensayos be cude inducción con golimumab, se observaron infecciones graves en el 0,8% be lospacientes tratados con golimumab en comparación con el 1,5% de los padientestratados con placebo. Las infecciones graves detectadas en los pacientes tratadbs congolimumab fueron tuberculosis, infecciones bacterianas, incluida sepsis y neurhonía,infecciones fúngicas invasivas y otras infecciones oportunistas. Algunas de lestasinfecciones han tenido un desenlace mortal. En las fases controladas y no contr ladasde los ensayos pivotales con una mediana de seguimiento de aproximadam~nte 2años, se observó una mayor incidencia de infecciones graves, incluyendo infecbones
I
oportunistas y TB en pacientes tratados con golimumab 100 mg en comparación conlos pacientes tratados con golimumab 50 mg. La incidencia por cada 100 pacient~s-año
Ide todas las infecciones graves fue de 4,3 episodios, rc del 95%: 3,8, 4,8 t;ln lospacientes tratados con golimumab 100 mg y de 2,7 episodios, rc del 95%: 2), 3,3 enlos pacientes tratados con golimumab 50 mg. I
Neoplasias lLinfoma .En los ensayos pivota les, la incidencia de linfoma en los pacientes tratado congolimumab fue mayor que la esperada en la población general. En las fases contrbladasy no controladas de estos ensayos con una mediana de seguimientio deaproximadamente 2 años, se observó una mayor incidencia de Iinforna en pacientestratados con golimumab 100 mg comparada con los pacientes tratados con golimilimab50 mg. Se diagnosticó Iinfoma en 9 pacientes (uno en el grupo de tratadds congolimumab 50 mg y 8 en los grupos de tratados con golimumab 100 mg) coh unaincidencia (rC del 95%) por cada 100 pacientes-año durante el seguimiento d~ 0,03
I(0,00, 0,16) Y de 0,12 (0,05, 0,24) episodios con golimumab 50 mg y 10m mg,respectivamente, y de 0,00 (0,00, 0,63) episodios con placebo. La mayoría be loslinfomas se produjeron en el estudio GO-AFTER, que reclutó pacientes previarenteexpuestos a agentes anti- TNF, siendo su enfermedad de mayor duración ~ másrefractaria (ver Precauciones y Advertencias).
Neoplasias distintas allinfoma

~
,,'.lU.r.
( fOLIO
7 2 O 7 ~..?.~~.~~"'<5' ",v
En los períodos controlados de los ensayos pivotales y durante aproximadament DE ~)\~años de seguimiento, la incidencia de neoplasias distintas al linfoma (excluido el ~áncerde piel no melanoma) fue similar en el grupo tratado con golimumab y en los Jruposcontrol. Durante aproximadamente 2 años de seguimiento, la incidencia de neo~lasiasdistintas al Iinfoma (excluyendo cáncer de piel no melanoma) fue similar a la de lapoblación general.
En los períodos controlados y no controlados de los ensayos pivotales con una medianade seguimiento de aproximadamente 2 años, se diagnosticó cáncer de piel nomelanoma en cinco pacientes tratados con placebo, 10 con golimumab 50 mg y 19 congolimumab 100 mg, siendo la incidencia (IC del 95%) por 100 pacientes y a!ño deseguimiento de 0,39 (0,28, 0,53) con golimumab combinado y de 0,95 (0,31, 2,22)con placebo.
En los períodos controlados y no controlados de los ensayos pivotales con una medianaI
de seguimiento de aproximadamente 2 años, se diagnosticaron neoplasias, sin incluirlinfoma ni cáncer de piel no mela noma, en cinco pacientes tratados con place~o, 20con golimumab 50 mg y 32 con golimumab 100 mg, siendo la incidencia (IC deI195%)por 100 pacientes y año de seguimiento de 0,51 (0,38, 0,67) con golimumabcombinado y de 0,95 (0,31, 2,22) con placebo (ver Precauciones y Advertencias).
Casos notificados en los ensayos clínicos en asmaEn un ensayo clínico preliminar, los pacientes con asma grave persistente recibieronuna dosis de carga de golimumab (150% de la dosis asignada) por vía SUbcutá~ea enla semana O, seguida de golimumab 200 mg, golimumab 100 mg o golimumab 50 mgpor vía subcutánea cada cuatro semanas hasta la semana 52. Se diagnosticaro~ ochoneoplasias en el grupo combinado de tratamiento con golimumab (n = 230) Y nihguno
Ien el grupo placebo (n = 79). Se notificó linfoma en un paciente, cáncer de piel nomelanoma en dos pacientes y otras neoplasias en cinco pacientes. No se obser1vóunagrupamiento específico de ningún tipo de neoplasia. I
Durante la fase controlada con placebo del ensayo, la incidencia (IC del 95%) de todaslas neoplasias por 100 pacientes y año de seguimiento en el grupo de tratamiento congolimumab fue de 3,19 (1,38, 6,28). En este ensayo, la incidencia (IC del 95°)0) por100 pacientes y año de seguimiento en los tratados con golimumab fue de 0,40 (0,01,2,20) para el linfoma, de 0,79 (0,10, 2,86) para el cáncer de piel no melanomJ y de1,99 (0,64, 4,63) para otras neoplasias. En los pacientes que recibieron placebo, laincidencia (IC del 95%) por 100 pacientes y año de seguimiento de estas neoJlasiasfue de 0,00 (0,00, 2,94). Se desconoce la relevancia de este dato. jTrastornos neurológicosEn los períodos controlados y no controlados de los ensayos pivota les con una m dianade seguimiento de aproximadamente 2 años, se observó una mayor incidenJia deenfermedad desmielinizante en pacientes tratados con goli ab 00 rrig en
ina31 de 34

720comparación con pacientes tratados con golimumab 50Advertencias).
Elevación de las enzimas hepáticasEn el período controlado de los ensayos pivotales en AR y APs, la proporción depacientes tratados con golimumab y de controles que presentaron elevación levJ de laALT (> 1 y < 3 x límite superior de la normalicjad [L5NJ) fue similar (del 22,11% al27,4%); en el ensayo en EA, se observó una elevación leve de la ALT en un mayornúmero de pacientes tratados con golimumab (25,6%) que de controles (3,9%).iEn losperíodos controlados y no controlados de los ensayos pivota les en AR y APs, con unamediana de seguimiento de aproximadamente 5 años la incidencia de la elevació'n levede la ALT también fue similar en los pacientes tratados con golimumab y én lospacientes control. En la población con EA, la elevación leve de la ALT tuvb unaincidencia más alta en los tratados con golimumab que en el grupo control. I En elperíodo controlado de los ensayos pivótales en CU de inducción con golimumab, seobservaron elevaciones leves de la ALT (> 1 y < 3x LSN) en proporciones si1i1aresentre pacientes tratados con golimumab y pacientes control (de 8,0% a 16,9%,respectivamente). En los períodos controlados y no controlados de los ensayospivotales en CU, con una mediana de seguimiento de 1 año, la incidencia Ide laelevación leve de ALT fue 17,4% en pacientes que reciben golimumab durante elperíodo de mantenimiento del estudio en CU. IEn el período controlado de los ensayos pivota les en AR y EA, la elevación de la ALT ~5 x LSN fue poco frecuente y se detectó en más pacientes tratados con golimumJb (del0,4% al 0,9%) que pacientes control (0,0%). No se observó esta tendencia Ien lapoblación con APs. En los períodos controlados y no controlados de los ensayos
Ipivótales en AR, APs y EA, con una mediana de seguimiento de 5 años, la elevación de
Ila ALT ~5 x LSN tuvo una incidencia similar en los grupos tratados con golimumab y decontrol. Por lo general, estas elevaciones fueron asintomáticas y las alterabonesdisminuyeron o desaparecieron al continuar o suspender el tratamiento con gOlirrlumabo al modificar los medicamentos concomitantes. En los períodos controlados ~e losensayos pivota les en CU de inducción con golimumab, se observaron elevaciones de laALT ~ 5 x LSN en proporciones similares entre pacientes tratados con golimumab ytratados con placebo (de 0,3% a 1,0%, respectivamente). En los períodos contrQladosy no controlados de los ensayos pivotales en CU, con una mediana de seguimient6 de 1año, la incidencia de la elevación de ALT ~ 5 x LSN fue 0,7% en pacientes que récibengolimumab durante el período de mantenimiento del estudio en CU.
Dentro de los ensayos pivota les en AR, APs y EA, un paciente con alteracioneshepáticas previas y medicamentos como factor de confusión que había r~cibidogolimumab sufrió una hepatitis ictérica no infecciosa que resultó mortal. No buedeexcluirse el papel de golimumab como un factor contribuyente o agravante.
Reacción en la zona de inyecciónEn los períodos controlados de los ensayos pivota les, el 5,1% de I pacien es tratadoscon golimumab presentaron reacciones en la zona de inyección, frente a I

FOLIO~ t"')IO r,,; \"Z~ '", G.ít ( ~ ...-- ~~
pacientes control. La presencia de anticuerpos anti-golimumab pude incrementar il' DE f.~~'"riesgo de reacciones en la zona de inyección. La mayoría de las reacciones en I~ zonade inyección fueron leves o moderadas, siendo la manifestación más frecuehte eleritema en la zona de inyección. Las reacciones en la zona de inyección no ~uelenrequerir que se interrumpa el tratamiento.
En los ensayos controlados de fase IIb y III en AR, APs, EA, asma persistente grave yen los ensayos de fase II/I1I en CU, ningún paciente tratado con golimumab sufrióninguna reacción anafiláctica.
AutoanticuerposDurante el año de seguimiento de los períodos controlados y no controlados de losensayos pivota les, el 3,5% de los pacientes tratados con golimumab y el 2,3% ~e lospacientes control tuvieron un resultado positivo para ANA (título de 1:160 o mayJr). Lapresencia de anticuerpos anti-DNA de doble cadena (anti-dsDNA) al aRo deseguimiento fue poco frecuente en los pacientes que en al inicio del tratamiento ~abíansido negativos para dichos anticuerpos.
Notificación de sospechas de reacciones adversasEs importante notificar sospechas de reacciones adversas al medicamento t~as suautorización. Ello permite una supervisión continuada de la relación beneficio/riesgo delmedicamento. Se invita a los profesionales sanitarios a notificar las sospech~s dereacciones adversas a través del sistema nacional de notificación. .
SOBREDOSIFICACIÓN
Se han administrado dosis únicas de hasta 10 mg/kg por vía intravenosa en un ensayoclínico sin que se haya establecido toxicidad Iimitante de la dosis. En ca~o desobredosis, se recomienda la monitorización del paciente para detectar cualquierl signoo síntoma de reacciones adversas y se instaurará inmediatamente el tratamientosintomático adecuado. lAnte la eventualidad de una sobredosificación, concurrir al hospital más cerc no ocomunicarse con los Centros de Toxicología de:
Hospital de Pediatría Dr. Ricardo Gutiérrez - Tel: (011) 4962-6666/2247
Hospital A. Posadas - Tel: (011) 4654-6648 Y 4658-7777
PRESENTACIÓN
SIMPONI@ 50 mg de solución inyectable en jeringa prellenada, envases de 1- o 3jeringa(s) prellenada(s). Es posible que no se comercialicen todos los tamañbs deenvases.
SIMPONI@50 mg de solución inyectable en autoinyector SmartJect, envase de 1 o 3autoinyector(es). Es posible que no se comercialicen todos los tam - s envases.
SIMPONI@ 100 mg de solución inyectable en jeringa o 3
'Pi ina 33 de 34

jeringa(s) prellenada(s). Es posible que no se comercialicen todos losenvases.
SIMPONI@ 100 mg de solución inyectable en autoinyector SmartJect, envase de 1 o 3autoinyector(es). Es posible que no se comercialicen todos los tamaños de envas s.
CONDICIONES DE CONSERVACIÓN Y ALMACENAMIENTO
Conservar en heladera (entre 2°C y 8°C).
No congelar.
Conservar el autoinyector SmartJect/la jeringa prellenada en la caja original paraprotegerlos de la luz.
MANTENER FUERA DEL ALCANCE DE LOS NIÑOS
Especialidad Medicinal autorizada por el Ministerio de Salud.Certificado NO55.910Director Técnico: Georgina Rodriguez, Farmacéutica-Bioquímica
Elaborado en Baxter Pharmaceutical Solutions,927 S Curry Pike, Bloomington, IN 47403,EEUU
Importado por JANSSEN CILAG Farmacéutica S.A.,
Mendoza 1259, C1428DJG Ciudad Autónoma de Buenos Aires
ARGENTINA
Fecha de última revisión: ---.1----1 __
Página 34 de 34





























