Embed Size (px)
DESCRIPTION
Graduation thesis - Padua University (Italy)
Citation preview

UNIVERSITA’ DEGLI STUDI DI PADOVA
FACOLTA’ DI FARMACIA
DIPARTIMENTO DI SCIENZE FARMACEUTICHE
CORSO DI LAUREA SPECIALISTICA IN CHIMICA E TECNOLOGIA FARMACEUTICHE
TESI DI LAUREA
SINTESI DI INIBITORI DI CK2
RELATORE : CHIAR.MO PROF. GIUSEPPE ZAGOTTO
LAUREANDA : ERICA TESCARI
ANNO ACCADEMICO 2010-2011

II

III
Nulla si crea,
nulla si distrugge,
tutto si trasforma.
(A. Lavoisier)

IV

1
SOMMARIO
1. INTRODUZIONE ............................................................................................ 3
1.1. CHINASI ............................................................................................................ 5
1.2. CASEIN CHINASI ............................................................................................. 8
1.3. CASEIN CHINASI 2 ........................................................................................ 10
1.3.1. CARATTERISTICHE STRUTTURALI DELLA CK2 ............................................. 10
1.3.1.1. STRUTTURA DELLE SUBUNITÀ CATALITICHE ( α/α′) ................................ 11
1.3.1.2. STRUTTURA DELLA SUBUNITÀ REGOLATORIA β ..................................... 12
1.4. RUOLO BIOLOGICO DELLA CK2 ................................................................ 13
1.4.1. SOPRAVVIVENZA DELLA CELLULA ................................................................... 14
1.4.2. CK2 E TUMORI ........................................................................................................... 15
1.4.3. CK2 E VIRUS ................................................................................................................ 16
1.5. SVILUPPO DI POTENZIALI INIBITORI DI CK2 ......................................... 17
1.5.1. ULTIMI SVILUPPI (2009-2010-2011) ........................................................................ 29
1.5.2. TRIALS CLINICI ......................................................................................................... 31
2. SCOPO DEL LAVORO E OBIETTIVI ........................................................... 33
2.1. SCOPO DEL LAVORO ................................................................................... 35
2.2. OBIETTIVI ...................................................................................................... 36
3. MATERIALI E METODI ............................................................................... 39
3.1. ABBREVIAZIONI ........................................................................................... 41
3.2. MATERIALI .................................................................................................... 43
4. DISCUSSIONE DEI RISULTATI .................................................................. 45
4.1. SCHEMA DI SINTESI 1 ................................................................................................... 47
4.1.1. RAZIONALE DELLO SCHEMA DI SINTESI 1 ...................................................... 49
4.2. SCHEMA DI SINTESI 2 ................................................................................................... 54
4.2.1. RAZIONALE DELLO SCHEMA DI SINTESI 2 ...................................................... 56
4.3. SCHEMA DI SINTESI 3 ................................................................................................... 58
4.3.1. RAZIONALE DELLO SCHEMA DI SINTESI 3 ...................................................... 60
4.4. SCHEMA DI SINTESI 4 ................................................................................................... 64
4.4.1. RAZIONALE DELLO SCHEMA DI SINTESI 4 ...................................................... 66
4.5. DISCUSSIONE DEI DATI BIOLOGICI .......................................................... 72
4.6. CONCLUSIONI ............................................................................................... 73

2
5. PROCEDURE SPERIMENTALI ................................................................... 75
5.1. SCHEMA 1 ....................................................................................................... 77
5.2. SCHEMA 2 ....................................................................................................... 85
5.3. SCHEMA 3 ....................................................................................................... 91
5.4. SCHEMA 4 ....................................................................................................... 99
BIBLIOGRAFIA.................................................................................................. 111

3
1. INTRODUZIONE

4

5
1.1. CHINASI
Le chinasi sono enzimi che catalizzano il trasferimento di un gruppo fosfato terminale
di una molecola di ATP, o più raramente di GTP, ad un substrato proteico (Schrenk &
Snaar Jagalska, 1999). La fosforilazione induce nella proteina delle modificazioni
strutturali, che si traducono in segnali di attivazione o di inibizione della stessa. Questo
processo è di natura reversibile e tale condizione è garantita dalla presenza di altri
enzimi, le fosfatasi, che catalizzano la reazione inversa (fig. 1).
Fig. 1. Rappresentazione schematica della reversibilità dell’azione chinasi-fosfatasi.
La fosforilazione proteica reversibile svolge un ruolo essenziale nella regolazione di
numerose funzioni cellulari comuni a tutti gli eucarioti come, ad esempio, il controllo
del ciclo cellulare (Pinna & Meggio, 1997), il trasporto intracellulare di proteine (Jans &
Hubner, 1996) e il metabolismo energetico (Ingebritsen & Cohen, 1983). Le protein
chinasi regolano tutti questi processi attraverso la fosforilazione di proteine substrato in
risposta a stimoli precisi, come quelli indotti da ormoni, fattori di crescita,
neurotrasmettitori ed antigeni (Beck, Weigel, & Edwards, 1992). Questi recettori
possono regolare l'attività di protein chinasi e fosfatasi attraverso la modulazione della
concentrazione citoplasmatica di secondi messaggeri, quali cAMP, che a sua volta
regola la protein chinasi A (PKA) (Walsh & Van Patten, 1994) e il sistema
Ca2+/diacilglicerolo, che attiva la protein chinasi C (PKC) (Nishizuka, 1995).

6
L’importanza dei processi di fosforilazione e defosforilazione nel controllo dei principali
processi cellulari si riflette nell’elevato grado di conservazione che è possibile
riscontrare nelle strutture delle protein chinasi e in quelle delle principali cascate
regolatorie.
Numerosi studi hanno posto in evidenza l'importanza dell'equilibrio dell'attività delle
chinasi e delle fosfatasi, per una corretta funzionalità cellulare; si è infatti verificato
come lo sbilanciamento di questi sistemi ricopra un ruolo nell’insorgenza di patologie.
Infatti, oltre 400 malattie umane, tra cui il diabete, alcune malattie neurodegenerative
come il morbo di Alzheimer (Selkoe, 1997) (Perez, Gil, & Martinez, 2010) e il morbo di
Parkinson (Guerra & Issinger, 2008), l’artrite reumatoide, molte neoplasie (leucemie e
linfomi) e malattie virali, sono imputabili a livelli anomali di fosforilazione (Cohen,
2001).
Nel genoma del lievito Saccharomyces cerevisiae (fig. 2), le chinasi rappresentano una
grande famiglia: 121 su 6144 geni di questo microorganismo (circa il 2%) codificano
per delle protein chinasi. Nel moscerino Drosophila melanogaster (fig. 3), 319 su 13338
geni codificano per protein chinasi; nel nematode Caenorhabditis elegans (fig. 4), sono
437 su 18366 i geni che codificano per queste proteine. Secondo una stima
approssimativa, circa un terzo delle proteine di mammifero contengono un gruppo
fosfato legato covalentemente.
Fig. 2-4. 2 - Saccharomyces cerevisiae; 3 - Drosophila melanogaster; 4 - Caenorhabditis elegans
(2) (3) (4)
7
Dagli studi riguardanti i genomi di lievito e di nematode, si ipotizzava che vi fossero non
meno di 1000 protein chinasi e 500 protein fosfatasi impegnate nella fosforilazione delle
proteine umane. Invece, state codificate in tutto circa 550 chinasi nel genoma umano,
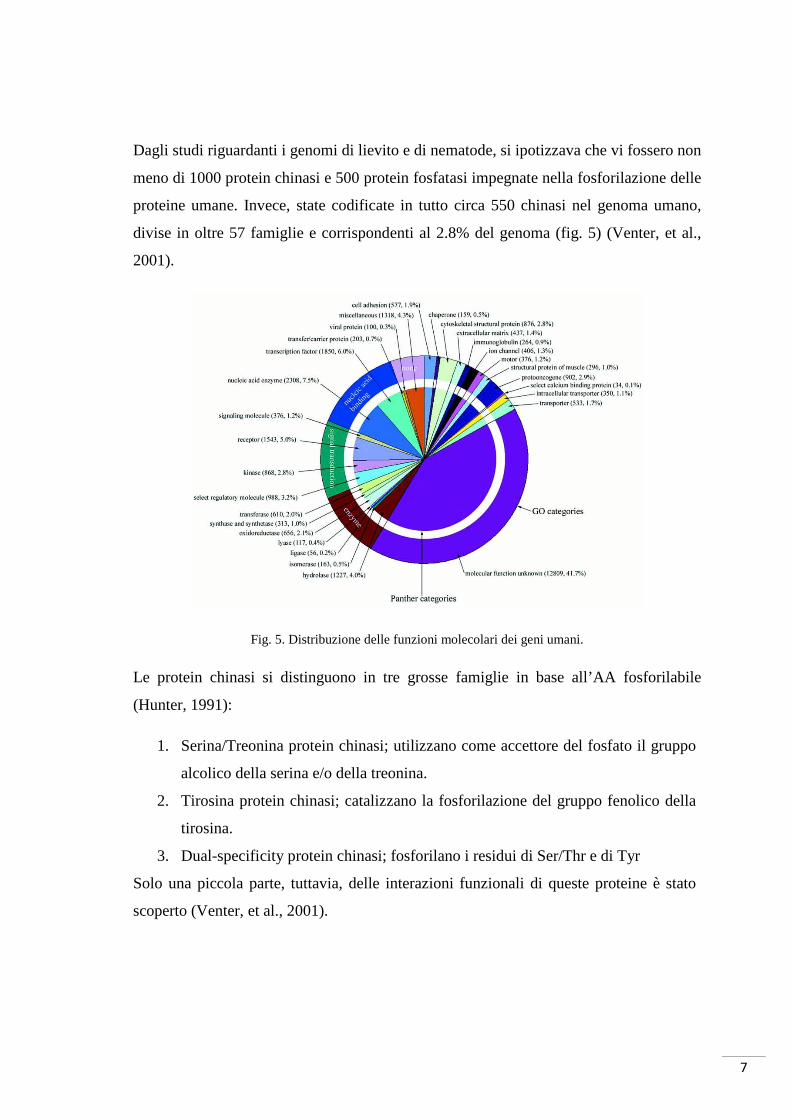
divise in oltre 57 famiglie e corrispondenti al 2.8% del genoma (fig. 5) (Venter, et al.,
2001).
Fig. 5. Distribuzione delle funzioni molecolari dei geni umani.
Le protein chinasi si distinguono in tre grosse famiglie in base all’AA fosforilabile
(Hunter, 1991):
1. Serina/Treonina protein chinasi; utilizzano come accettore del fosfato il gruppo
alcolico della serina e/o della treonina.
2. Tirosina protein chinasi; catalizzano la fosforilazione del gruppo fenolico della
tirosina.
3. Dual-specificity protein chinasi; fosforilano i residui di Ser/Thr e di Tyr
Solo una piccola parte, tuttavia, delle interazioni funzionali di queste proteine è stato
scoperto (Venter, et al., 2001).

8
1.2. CASEIN CHINASI
In letteratura, il primo esempio di enzima riportante l’attività chinasica è stato riportato
nel 1954 da Burnett e Kennedy mediante esperimenti condotti su fegato di ratto
utilizzando la caseina come substrato fosforilabile (Burnett & Kennedy, 1954); in
seguito si dimostrò che quella stessa attività era presente in molti altri tessuti. Tale
attività fu attribuita a un enzima che venne indicato come “casein-chinasi” o “ fosvitin-
chinasi”, il quale si distinse per la sua preferenza in vitro verso substrati acidi (tra cui
appunto la caseina del latte e la fosvitina del tuorlo d’uovo), rispetto a quelli basici
quali istoni e protammine (Hutti, Jarrell, Chang, Abbott, Storz, & Toker, 2004).
Solo nel 1969 si scoprì che l’attività era attribuibile a due enzimi distinti (Pinna,
Baggio, Moret, & Siliprandi, 1969), che furono denominati Casein Chinasi 1 e Casein
Chinasi 2 (CK1 – CK 2). Nonostante la caseina, come la fosvitina, sia un substrato
ideale per saggiarne l’attività, essa non è tuttavia fisiologica. La caseina costituisce il
vero substrato fisiologico solamente nella protein chinasi denominata G-CK (Golgi
Casein Kinase) (Lasa, Marin, Meggio, & Pinna, 1996)
Il termine casein chinasi, quindi, indica tre diverse classi di protein chinasi, distinte per
struttura, localizzazione e funzione:
1. Casein Chinasi 1 (CK1).
2. Casein Chinasi 2 (CK2).
3. Casein Chinasi della ghiandola mammaria (G-CK: Golgi Casein Kinase).
La numerazione di CK1 e CK2 è una convenzione che deriva dai primi lavori su tali
enzimi, e fa riferimento all’ordine di eluizione da una colonna di dietilaminoetil-
cellulosa (DEAE-cellulosa) usata durante il loro processo di purificazione. (Baggio,
Pinna, Moret, & Siliprandi, 1970). Queste due chinasi sono state trovate essere
ubiquitarie e agiscono su un numero elevato di enzimi e proteine non catalitiche
coinvolte in moltissime funzioni cellulari (Lasa, Marin, & Pinna, 1997).
La casein chinasi (G-CK) è una chinasi tessuto specifica, localizzata a livello delle
membrane dell’apparato del Golgi nella ghiandola mammaria. La sua funzione è quella
di fosforilare le proteine del latte appena sintetizzate, delle quali la caseina è la
principale componente (Duncan, Wilkinson, & Burgoyne, 2000). Recentemente è stato
dimostrato che anche nell’apparato di Golgi del fegato, della milza, e, in minor grado,

9
del rene e del cervello di ratto, è presente una protein chinasi, biochimicamente
indistinguibile dalla G-CK di ghiandola mammaria, il cui ruolo fisiologico, tuttavia, è
ancora oggetto di importanti studi (Lasa, Marin, & Pinna, 1997).
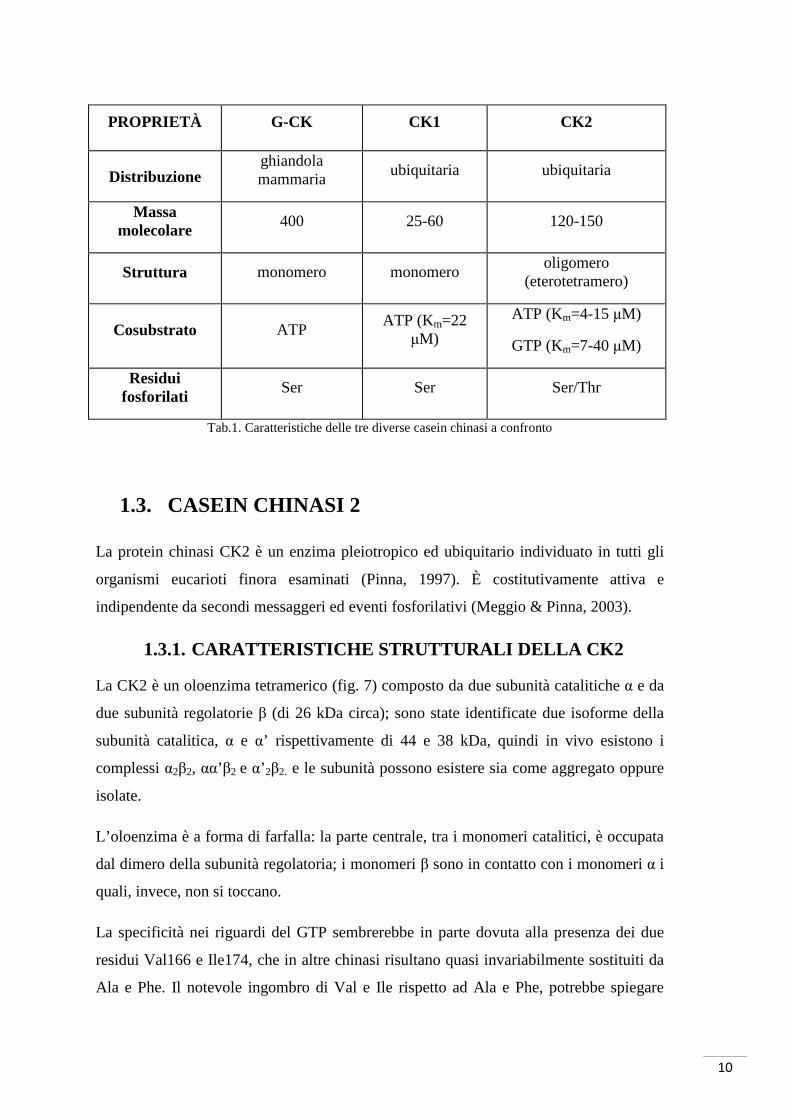
Le principali caratteristiche dei tre enzimi sono riportate nella tabella 1. In particolare è
possibile osservare che:
1. CK1 e CK2 sono profondamente diverse: CK1 è un monomero di PM compreso
tra 34 e 55 KDa; CK2 è un tetramero costituito da due subunità catalitiche α/α’
e due subunità regolatorie β (questa struttura eterotetramerica è rara tra le
protein chinasi) (Litchfield, 2003). I due enzimi fosforilano le stesse proteine in
siti diversi. Tra i substrati fisiologici riconosciuti da entrambe ricordiamo la
RNA-polimerasi, l’acetil-CoA-carbossilasi, la glicogeno sintetasi e il
fibrinogeno.
2. CK1 e G-CK sono in grado di utilizzare come donatore di fosfato solo ATP,
mentre la CK2 può utilizzare quasi con la stessa efficienza sia ATP che GTP. La
CK2 richiede la presenza di cationi bivalenti (soprattutto magnesio, ma anche
manganese e cobalto). È stato osservato che, in presenza di ioni magnesio,
l’affinità è maggiore per l’ATP rispetto al GTP, mentre è vero il contrario in
presenza di ioni manganese. È possibile che i complessi di coordinazione ATP-
Mg2+ e GTP-Mn2+, rispetto alle altre combinazioni, assumano la conformazione
ottimale tale da occupare in maniera idonea il sito di legame del donatore di
fosfato.
3. CK2 è insensibile alla staurosporina (fig. 6), uno dei più potenti inibitori
competitivi per l’ATP delle protein chinasi (Meggio, et al., 1995).
Fig. 6. Struttura della staurosporina (Streptomyces staurosporeus)
10
PROPRIETÀ G-CK CK1 CK2
Distribuzione ghiandola mammaria
ubiquitaria ubiquitaria
Massa molecolare
400 25-60 120-150
Struttura monomero monomero oligomero
(eterotetramero)
Cosubstrato ATP ATP (Km=22
µM)
ATP (Km=4-15 µM)
GTP (Km=7-40 µM)
Residui fosforilati
Ser Ser Ser/Thr
Tab.1. Caratteristiche delle tre diverse casein chinasi a confronto
1.3. CASEIN CHINASI 2
La protein chinasi CK2 è un enzima pleiotropico ed ubiquitario individuato in tutti gli
organismi eucarioti finora esaminati (Pinna, 1997). È costitutivamente attiva e
indipendente da secondi messaggeri ed eventi fosforilativi (Meggio & Pinna, 2003).
1.3.1. CARATTERISTICHE STRUTTURALI DELLA CK2
La CK2 è un oloenzima tetramerico (fig. 7) composto da due subunità catalitiche α e da
due subunità regolatorie β (di 26 kDa circa); sono state identificate due isoforme della
subunità catalitica, α e α’ rispettivamente di 44 e 38 kDa, quindi in vivo esistono i
complessi α2β2, αα’β2 e α’2β2. e le subunità possono esistere sia come aggregato oppure
isolate.
L’oloenzima è a forma di farfalla: la parte centrale, tra i monomeri catalitici, è occupata
dal dimero della subunità regolatoria; i monomeri β sono in contatto con i monomeri α i
quali, invece, non si toccano.
La specificità nei riguardi del GTP sembrerebbe in parte dovuta alla presenza dei due
residui Val166 e Ile174, che in altre chinasi risultano quasi invariabilmente sostituiti da
Ala e Phe. Il notevole ingombro di Val e Ile rispetto ad Ala e Phe, potrebbe spiegare

11
anche l’insensibilità della CK2 alla staurosporina, e la maggiore suscettibilità ai derivati
alogenati del benzimidazolo e del benzotriazolo (Meggio, Shugar, & Pinna, 1990)
(Ssyszka, Grakowski, Felczak, & Shugar, 1995).
Fig. 7. Struttura tetramerica dell’oloenzima della CK2, sono evidenziate le strutture α e β
1.3.1.1. STRUTTURA DELLE SUBUNITÀ CATALITICHE ( α/α′)
La descrizione dettagliata delle diverse regioni della subunità catalitica, è stata eseguita
per la prima volta con la definizione della struttura della subunità α della CK2 del
cereale Zea mays (il comune granturco) (fig. 8) (Niefind, Guerra, Pinna, Issinger, &
Schomburg, 1998).
Una caratteristica unica della CK2 è rappresentata dalla regione N-terminale (fig. 6);
questa porzione sembra avere un ruolo chiave nella stabilizzazione della conformazione
attraverso l’unione dei due lobi, formando così dei contatti ben definiti, principalmente
con il segmento di attivazione.

12
Fig 8. Struttura della subunità α di Zea mays
1.3.1.2. STRUTTURA DELLA SUBUNITÀ REGOLATORIA β
Diverse considerazioni devono essere prese per quanto riguarda la subunità β di CK2.
Questa regione è di per sé inattiva, ma riveste un ruolo assolutamente unico e
importante per la funzionalità del tetrametro enzimatico, il cui assemblaggio avviene a
partire dalla formazione di un suo dimero (Graham & Litchfield, 2000).
Il tetramero fosforila con più efficienza, rispetto alla singola subunità catalitica, la
maggior parte dei substrati e per alcuni di essi la presenza di CK2β è indispensabile.
Per altri substrati come la calmodulina ad esempio, invece, la presenza di CK2β ha un
effetto inibitorio (Marin, Meggio, & Pinna, 1999) (Ruzzene, Brunati, Sarno, Marin,
Donella, & Pinna, 2000)
. Il monomero della subunità β è costituito da 215 aminoacidi ed è organizzato in due
domini: dominio I e dominio II (Chantalat, et al., 1999). Il primo è interamente ad α-
elica e comprende la regione N-terminale; il secondo è costituito da foglietti β
antiparalleli nei quali uno ione Zn2+ viene coordinato tetraedricamente e comprende la
regione C-terminale, responsabile della dimerizzazione (fig. 9).

13
Fig. 9. Struttura del dimero CK2β. Si evidenziano gli atomi di Zn2+ coordinati dai residui glicinici.
1.4. RUOLO BIOLOGICO DELLA CK2
Abbiamo più volte ripetuto che la CK2 è una proteina pleiotropica, coinvolta in parecchi
e spesso fondamentali percorsi metabolici, nei quali ha, molte volte, un ruolo chiave (ciò
è evidenziato dal lungo elenco in espansione di substrati fosforilabili in vivo ed in vitro
(Meggio & Pinna, 2003)); spesso tali substrati sono degli enzimi, la cui regolazione
risulta cruciale in numerose vie metaboliche.
Molti studi hanno suggerito come questo enzima giochi un ruolo chiave in parecchie fasi
della regolazione cellulare; alcune ipotesi sostengono che CK2, sia fondamentale per la
sopravvivenza della cellula, nonché essere implicata fermamente in processi quali la
crescita di cellule sane e di cellule cancerose (Ahmed, Gerber, & Cochet, 2002). Tale
enzima inoltre sembra implicato in processi quali la sintesi di tRNA e rRNA (Ghavidel
& Schultz, 2001), l’apoptosi (Ahmad K. , Wang, Unger, Slaton, & Ahmedc, 2008), la
trasformazione cellulare (Ahmed, Gerber, & Cochet, 2002) e nella regolazione della
NADPH ossidasi (Kim, Jung, Niizuma, & Chan, 2009).

14
1.4.1. SOPRAVVIVENZA DELLA CELLULA
La produzione di entrambe le subunità (catalitica e regolatoria) della CK2 è essenziale
per la sopravvivenza cellulare: infatti, in Saccharomyces cerevisiae, l’interruzione dei
geni che codificano per la subunità catalitica della CK2, risulta letale (Padmnabha,
1990). CK2 α’ nel topo maschio è prodotta soprattutto nella fase finale della
spermatogenesi, e l’interruzione del gene che la codifica aumenta il numero di cellule
apoptotiche nei testicoli, causando l’infertilità dell’animale (Xu, Rich, & Seldin, 1998).
È evidente a questo punto la potenziale relazione tra CK2 e attività antiapoptotica e, di
conseguenza, tra elevata attività di CK2 e “anormale” proliferazione cellulare.
Tra tutti i numerosi substrati fosforilati da questa chinasi (tab. 2), tre meritano un cenno
particolare e sono: calmodulina, Rev ed il fattore eIF2β (Meggio & Pinna, 2003).
La calmodulina è una proteina chiave nella regolazione Ca2+-dipendente di molti enzimi
(tra cui varie protein chinasi). Essa rappresenta un substrato unico per la CK2, in quanto
la sua fosforilazione è strettamente legata alla struttura della chinasi: infatti, mentre la
subunità catalitica isolata è in grado di fosforilare la calmodulina, la CK2
eterotetramerica risulta del tutto incapace di farlo se non in presenza di composti di
natura policationica come la polilisina e gli istoni che inducono un’ottima attività
(Marin, Meggio, & Pinna, 1999) (Arrigoni, et al., 2004).
Rev è una delle proteine virali dell’HIV; al contrario della calmodulina, si è rivelato un
ottimo substrato solo per la forma oligomerica di CK2 suggerendo, per la prima volta,
che anche la subunità β di CK2 potesse essere coinvolta nelle fasi di riconoscimento del
substrato.
La terza proteina, il fattore eIF2β, è uno dei componenti dell’eIF2 che interviene nelle
fasi iniziali della sintesi proteica durante la formazione di complessi con il GTP e con il
Met-tRNA.

15
Enzimi coinvolti nella sintesi di acidi nucleici
RNA-polimerasi 1 e 2
DNA-polimerasi 1 e 2
Dna-ligasi
Enzimi chiave nelle vie metaboliche
Glicogeno sintetasi
Acetil CoA-carbossilasi
Ornitina decarbossilasi
Enzimi coinvolti nella traduzione del segnale
Calmodulina
Protein chinasi C
Recettore dell’insulina
Recettore di IGF-2
Recettore delle LDL
Sinaptogamina
Sintassina
Fattori della sintesi proteica
eIF-2
eIF-3
eIF-5
Fattori di trascrizione, oncogeni
e soppressori tumorali
c-Jun
c-Fos
c-Myc
p-53 Proteine del citoscheletro
Tubulina
Miosina
Spectrina
Troponina C
Proteine nucleari e nucleolari
Proteine nucleari di matrice
nucleolina
Tab.2. Alcuni substrati della CK2
1.4.2. CK2 E TUMORI
Da tempo è noto che la CK2 è coinvolta nel processo di divisione cellulare: la sua
concentrazione risulta essere particolarmente elevata nei tessuti in rapida proliferazione
come le cellule embrionali e la sua presenza è necessaria affinché il ciclo cellulare passi
dalla fase G1 alla fase S e dalla fase G2 alla fase M (Blanquet, 2000). Inoltre in molti
tumori umani esaminati la concentrazione e la attività di CK2 risultano essere aumentate
(Guerra & Issinger, 1999) (Pinna, 2002).
Il ruolo centrale della CK2 nella genesi dei tumori è confermato dall’osservazione che
l’introduzione del gene per la CK2α in topi transgenici, che porta alla sovraespressione
della subunità catalitica α induce l’insorgenza di leucemie (Pepperkok, Lorenz,
Ansonge, & Pyperin, 1994). Risulta evidente che l’evoluzione della neoplasia sia
direttamente proporzionale all’attività di CK2; per esempio l’attività di CK2 è elevata
nelle cellule leucemiche umane (Peña, Itarte, Domingo, & Cusso, 1983). Si ritiene,
quindi, che CK2 possa rappresentare un potenziale bersaglio per la ricerca di farmaci
anti-neoplastici (Ahmad K. , Wang, Slaton, Unger, & Ahmed, 2005) .

16
1.4.3. CK2 E VIRUS
La CK2 risulta anche essere coinvolta nella fosforilazione di proteine virali (tra le
proteine fosforilate fisiologicamente dalla CK2, almeno 40 sono di origine virale)
(Guerra & Issinger, 1999) (Ivanov, et al., 2003) (Meggio & Pinna, 2003). Il virus,
essendo privo di chinasi proprie, sfrutta la CK2 della cellula ospite per fosforilare le
proteine necessarie alla replicazione virale e alla progressione dell’infezione (Gurel, et
al., 2008).
Tra le proteine virali substrato della CK2 si possono annoverare la proteina Rev
espresse da HIV (Meggio, Marin, Boschett, Sarno, & Pinna, 2001), le proteine del
citomegalovirus (Alvisi, Jans, Guo, Pinna, & Ripalti, 2005), le oncoproteine E7 del
papilloma virus e Large T del virus SV40 (Allende & Allende, 1995), che possono
indurre la trasformazione neoplastica nell’ospite.
Tenendo conto di queste evidenze, si ritiene che la CK2 possa rappresentare un
importante bersaglio farmacologico, e che lo sviluppo di nuovi inibitori sempre più
efficaci e selettivi possa portare a nuovi farmaci utili nella terapia antitumorale e
antivirale (Sarno, et al., 2002).

17
1.5. SVILUPPO DI POTENZIALI INIBITORI DI CK2
La CK2 rappresenta una delle protein chinasi maggiormente studiate come bersaglio
molecolare; il sito di legame del nucleotide (ATP o GTP) presenta alcuni elementi di
peculiarità che in alcuni casi sono stati proposti come spiegazione razionale di marcata
selettività per alcune molecole attive (Cozza, Bortolato, & Moro, 2009).
Gli inibitori di CK2 possono essere schematicamente divisi in cinque diversi gruppi :
1. Inibitori ATP-competitivi
2. Inibitori che si legano in parte ad una tasca allosterica
3. Inibitori che si legano in modo specifico ad una tasca allosterica vicina al sito di
legame dell’ATP
4. Inibitori substrato-competitivi
5. Inibitori allosterici che si legano ad una tasca non correlata con i siti di legame
dell’ATP o del substrato
La maggiore parte degli inibitori appartiene al gruppo di tipo 1, mentre nessun inibitore
di tipo 2 e 3 è noto finora (Cozza, Meggio, & Moro, 2011).
Le più rappresentative classi del gruppo 1 sono (Sarno, et al., 2011):
o Derivati polifenolici (Antrachinoni, Flavonoidi, Cumarine)
o Derivati polialogenati dell’acido cinnamico e dell’acido benzoico
o Derivati polialogenati del benzotriazolo e del benzimidazolo
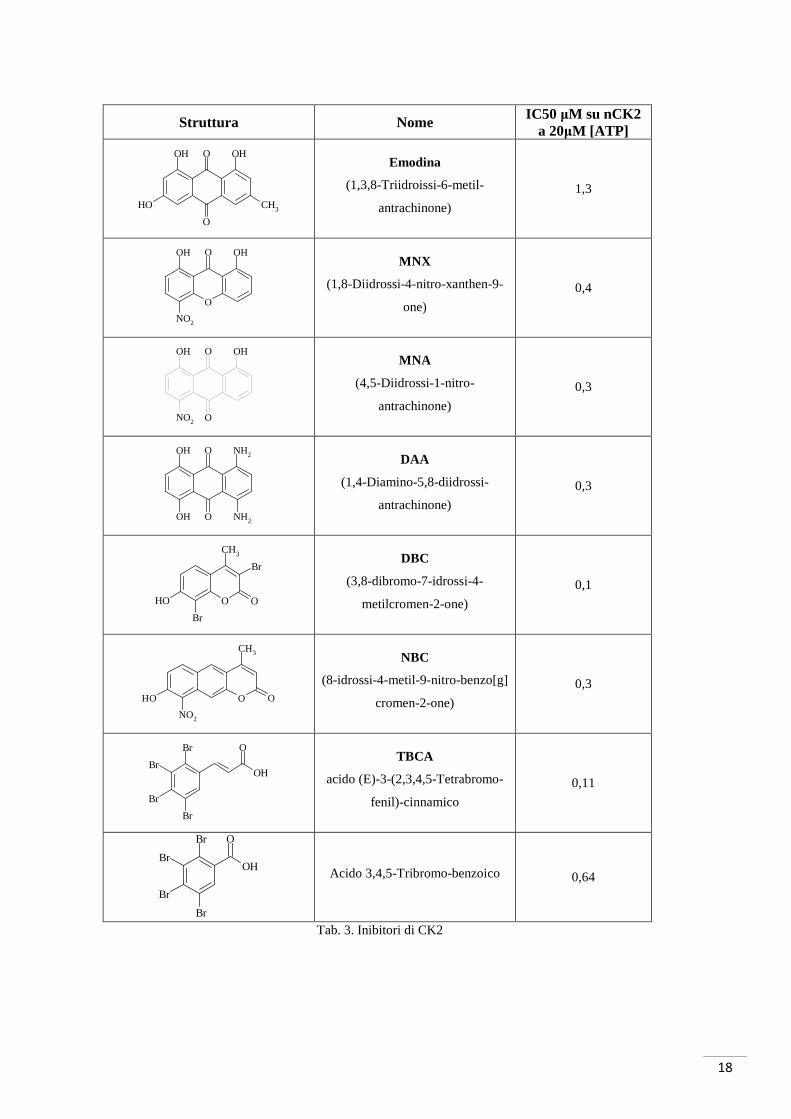
Punto di partenza per i derivati polifenolici condensati è stato l’emodina (principio attivo
estratto dal Rheum palmatum). Da uno studio computazionale su un modello umano
ottenuto per omologia dalla struttura cristallografica di CK2α di Zea mays (De Moliner,
et al., 2003), sono stati sviluppati nuovi inibitori quali l’MNA e l’MNX. Tali derivati
hanno dimostrato un interessante incremento dell’attività inibitoria; il composto
denominato DAA, infine, ha esibito l’attività inibitoria più alta fra questi (Meggio, et al.,
2004) (Chilin, Battistutta, Bortolato, Cozza, Zanatta, & Poletto, 2008) (tab. 3).
18
Struttura Nome IC50 µM su nCK2 a 20µM [ATP]
OH O OH
CH3OH
O
Emodina
(1,3,8-Triidroissi-6-metil-
antrachinone)
1,3
O
OH O OH
NO2
MNX
(1,8-Diidrossi-4-nitro-xanthen-9-
one)
0,4
O
O
OHOH
NO2
MNA
(4,5-Diidrossi-1-nitro-
antrachinone)
0,3
O
O
NH2OH
OH NH2
DAA
(1,4-Diamino-5,8-diidrossi-
antrachinone)
0,3
OOH
Br
CH3
Br
O
DBC
(3,8-dibromo-7-idrossi-4-
metilcromen-2-one)
0,1
O O
CH3
OH
NO2
NBC
(8-idrossi-4-metil-9-nitro-benzo[g]
cromen-2-one)
0,3
OH
O
Br
Br
Br
Br
TBCA
acido (E)-3-(2,3,4,5-Tetrabromo-
fenil)-cinnamico
0,11
Br
Br
Br
Br
OH
O
Acido 3,4,5-Tribromo-benzoico 0,64
Tab. 3. Inibitori di CK2
19
Struttura Nome IC 50 (µM) su nCK2
a 20 µM [ATP]
N
NHCl
Cl
OCH
2OH
OH OH
DRB
(dicloro-ribofuranosil-benzimidazolo)
23
N
N
Br
Br
Br
Br H
N
TBB
(4,5,6,7-Tetrabromo-1H-benzotriazolo)
0,60
NH
N
Br
Br
Br
Br
N
K25
Dimetil-(4,5,6,7-tetrabromo-1H-ben
zoimidazol-2-il)-ammina
0,14
NH
NS
Br
Br
Br
Br
K37
4,5,6,7-Tetrabromo-2-metilsulfanil
-1H-benzoimidazolo
0,25
N
Br
Br
Br
Br
NN
K44
5,6,7,8-Tetrabromo-1-metil-2,3-diidro-1H-
benzo[d]imidazo[1,2-a]imidazolo
0,74
NH
N
O
OH
O
IQA
acido (5-oxo-5,6-diidro-indolo
[1,2-a]chinazolin- 7-il) acetico
0,30
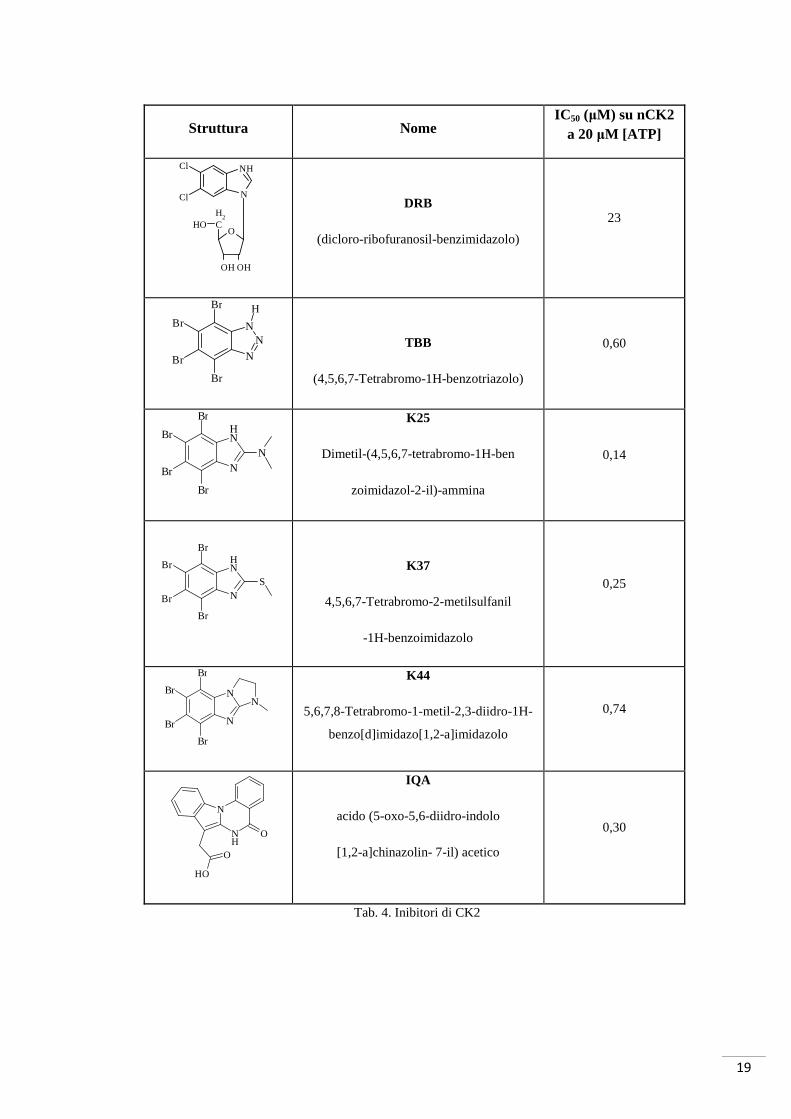
Tab. 4. Inibitori di CK2

20
Più recentemente sono stati individuati il DBC e alcuni suoi derivati (tab. 3) con
struttura cumarinica (Meggio, et al., 2004). Dall’apertura dell’estere ciclico della
cumarina è stata successivamente sviluppata una serie di acidi cinnamici E/Z
polibromurati, e fra questi il TBCA si è dimostrato essere il migliore (Pagano, et al.,
2007). Successivamente si è passati ad una semplificazione della struttura molecolare,
progettando una serie di acidi benzoici e derivati dell’acido salicilico polibromurati
(Pagano, et al., 2007).
Il gruppo di sintesi del professor Kazimierczuk (Università di Varsavia, Polonia),
apportando delle modifiche strutturali al DRB, ha ottenuto il TBB (Pagano, et al., 2004)
(tab. 4). Recentemente, le proprietà inibitorie del TBB sono state migliorate attraverso la
sintesi di nuovi derivati, alcuni dei quali mostrano selettività uguale al loro progenitore,
ma una maggiore efficacia. È il caso di K25, K37 e K44 (Battistutta, Mazzorana, Sarno,
Kazimierczuk, Zanotti, & Pinna, 2005) (Zawada, Wolniak, Kazimierczuk, & Wawer,
2009).
Utilizzando lo scaffold benzimidazolico come punto di partenza per tentare di
raggiungere anche l’esterno della tasca di legame per l’ATP, sono stati sintetizzati una
serie di nuovi inibitori. Lo scopo ultimo di questa strategia è stato quello di riuscire a
disegnare inibitori capaci di interagire contemporaneamente con la tasca dell’ATP e
con la zona di legame del substrato proteico. La TBB-propil-diammina e la TBB-etil-
diammina (fig. 10) possiedono un’ammina terziaria terminale facilmente sfruttabile per
legare catene carbossilate attraverso un legame ammidico (fig. 11). I risultati biochimici
ottenuti dai derivati GZ (tab. 5-6) dimostrano che questi composti mantengono una
discreta attività sulla CK2 che si attesta intorno a 0.2-0.3 µM (lavoro non pubblicato,
risultati preliminari da internato di tesi A. Bertamini, 2010)
. Fig. 10. (a) TBB-propil-diammina (IC50 = 0.12 µM, lavoro non pubblicato, risultati preliminari da
internato di tesi M. Fortuna, 2010). (b) TBB-etil-diammina.
NH
N
Br
Br
Br
Br
NH NH2
NH
N
Br
Br
Br
Br
NH NH2
(a) (b)
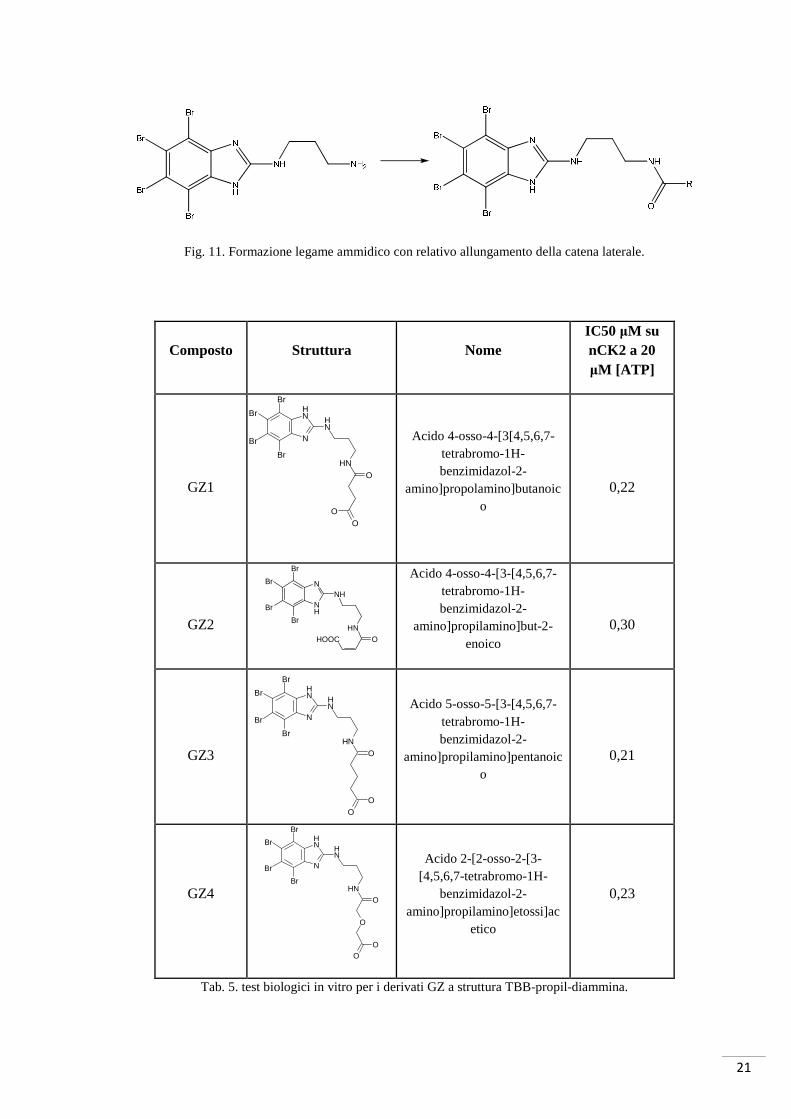
21
Fig. 11. Formazione legame ammidico con relativo allungamento della catena laterale.
Composto Struttura Nome IC50 µM su nCK2 a 20 µM [ATP]
GZ1
Acido 4-osso-4-[3[4,5,6,7-tetrabromo-1H-benzimidazol-2-
amino]propolamino]butanoico
0,22
GZ2
Br
Br
Br
Br
NH
NNH
HNOHOOC
Acido 4-osso-4-[3-[4,5,6,7-tetrabromo-1H-benzimidazol-2-
amino]propilamino]but-2-enoico
0,30
GZ3
Acido 5-osso-5-[3-[4,5,6,7-
tetrabromo-1H-benzimidazol-2-
amino]propilamino]pentanoico
0,21
GZ4
Br
Br
N
NH
NH
NH
O
OO
O
Br
Br
Acido 2-[2-osso-2-[3-[4,5,6,7-tetrabromo-1H-
benzimidazol-2-amino]propilamino]etossi]ac
etico
0,23
Tab. 5. test biologici in vitro per i derivati GZ a struttura TBB-propil-diammina.
Br
Br
N
NH
NH
NH
OO
O
Br
Br
Br
Br
N
NH
NH
NH
OO
O
Br
Br
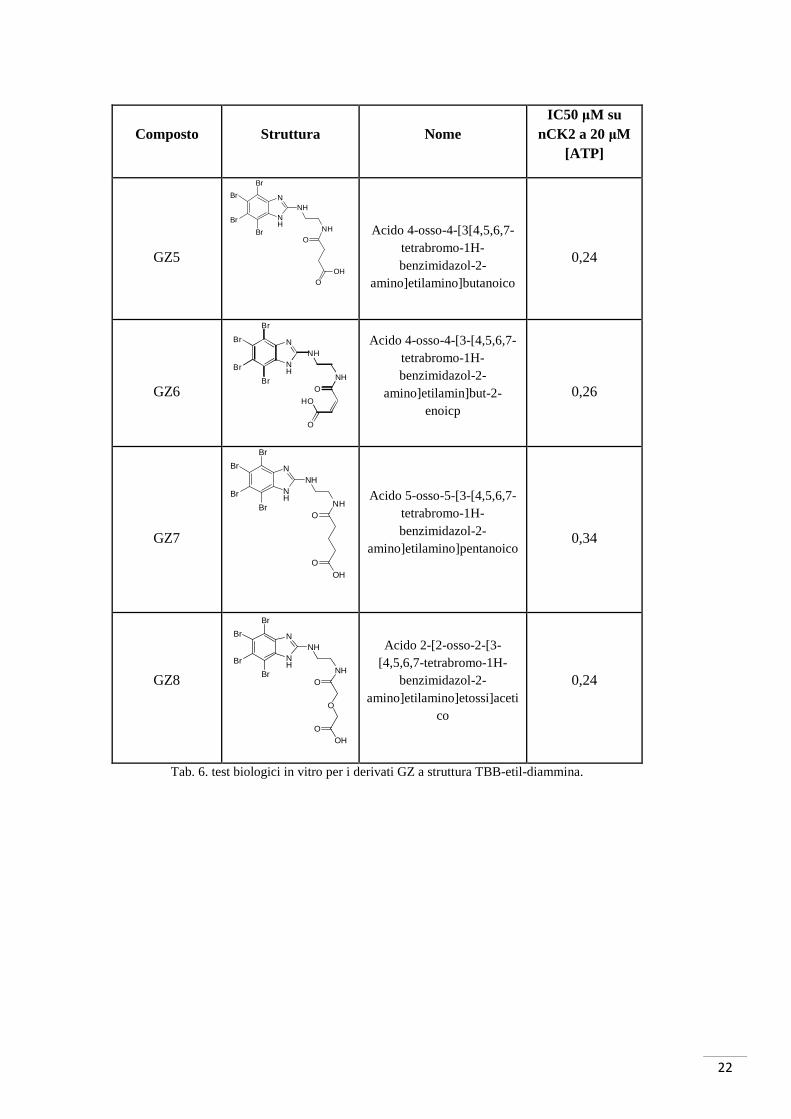
22
Composto Struttura Nome IC50 µM su
nCK2 a 20 µM [ATP]
GZ5
Acido 4-osso-4-[3[4,5,6,7-tetrabromo-1H-benzimidazol-2-
amino]etilamino]butanoico
0,24
GZ6
Br
Br
Br
Br
NH
NNH
NH
O
O
HO
Acido 4-osso-4-[3-[4,5,6,7-tetrabromo-1H-benzimidazol-2-
amino]etilamin]but-2-enoicp
0,26
GZ7
Acido 5-osso-5-[3-[4,5,6,7-tetrabromo-1H-benzimidazol-2-
amino]etilamino]pentanoico
0,34
GZ8
Br
Br
Br
Br
NH
NNH
NHO
O
OHO
Acido 2-[2-osso-2-[3-[4,5,6,7-tetrabromo-1H-
benzimidazol-2-amino]etilamino]etossi]aceti
co
0,24
Tab. 6. test biologici in vitro per i derivati GZ a struttura TBB-etil-diammina.
Br
Br
Br
Br
NH
NNH
NHO
OOH
Br
Br
Br
Br
NH
NNH
NHO
OHO

Altri potenti inibitori di CK2 di origine naturale
(Cozza, et al., 2006)
competitivo dell’ATP che va
e C-teminale dell’enzima
polifenolici (Cozza, et al., 2006)
Fig. 12. Rappresentazione della CK2 eterotetramero con l’acido ellagico legato al sito attivo della ATP.
Fig. 13. Docking dell’acido ellagico legato al sito attivo della subunità
di CK2 di origine naturale sono l’acido ellagico e
(Ghosal, 1990) (tab. 7). L’ acido ellagico è
che va ad occupare una regione di “binding” tra lobo N
teminale dell’enzima (fig. 12-13), in modo simile a TBB e ad altri composti
(Cozza, et al., 2006).
. Rappresentazione della CK2 eterotetramero con l’acido ellagico legato al sito attivo della ATP.
dell’acido ellagico legato al sito attivo della subunità α di CK2
23
sono l’acido ellagico e i suoi derivati
L’ acido ellagico è un inibitore
” tra lobo N-terminale
, in modo simile a TBB e ad altri composti
. Rappresentazione della CK2 eterotetramero con l’acido ellagico legato al sito attivo della ATP.
di CK2 (Cozza, et al., 2006).

24
Struttura Nome IC50 µM su nCK2 a 20 µM [ATP]
OO
OH
OH
OH
O O
OH
Acido Ellagico 0,04
O
OO
O
OH
OH
OH
OH
OH
Acido Flavellagico 2
OO
OH
OH
OH
OH OH
Monolattone dell'acido ellagico
0,2
OO
OH
OH
OH
OH
OH
OH
Monolattone dell'acido
flavellagico 2,2
Tab. 7. Inibizione di protein chinasi da parte di acido ellagico e derivati
La peculiarità dell’acido ellagico risiede nella sua capacità di legare
contemporaneamente sia la regione “hinge” che la porzione legante il fosfato nel sito per
l’ATP, caratteristica per il momento unica (Cozza, et al., 2006). Secondo questa ipotesi,
infatti,l’ossidrile in posizione 4 (fig. 14) dell’acido ellagico interagisce con il gruppo
carbonilico del Glu114 nella “hinge region”; questa è la stessa regione che interagisce
con l’adenina quando l’ATP si lega al sito attivo di CK2. I gruppi ossidrilici nelle
posizioni 3’ e 4’ interagiscono con il gruppo carbossilico di Asp175 attraverso un
doppio legame ad idrogeno. Inoltre, le interazioni idrofobiche con Val53, Val66,
Phe113, Met163 e Ile174 contribuiscono significativamente a stabilizzare il complesso
acido ellagico-CK2 (Cozza, et al., 2006).

25
Fig.14. Acido Ellagico
Al contrario, il carbonile del gruppo lattonico dell’acido flavellagico si distribuisce nelle
vicinanze dell’ossigeno carbonilico di Glu 144. Questa posizione sfavorevole e la
perdita di un legame ad idrogeno tra Glu144 e gruppo ossidrilico spiegano la
diminuzione di attività (IC50=2 µM) rispetto all’ acido ellagico, che presenta in questa
regione 2 legami ad idrogeno di due gruppi OH con gruppi carbonilici di Glu144 e
Val66. Dal lato opposto i gruppi ossidrilici interagiscono con Asp175 e Lys68 (legame
intermolecolare ione-dipolo); in più una molecola d’acqua forma legami ad idrogeno
con due ossidrili.
Il monolattone dell’acido flavellagico presenta un ossidrile in più rispetto quello
dell’acido ellagico. La IC50 misurata per questo composto è inferiore rispetto quella della
del monolattone dell’acido flavellagico (in press, G.Cozza, Università di Padova).
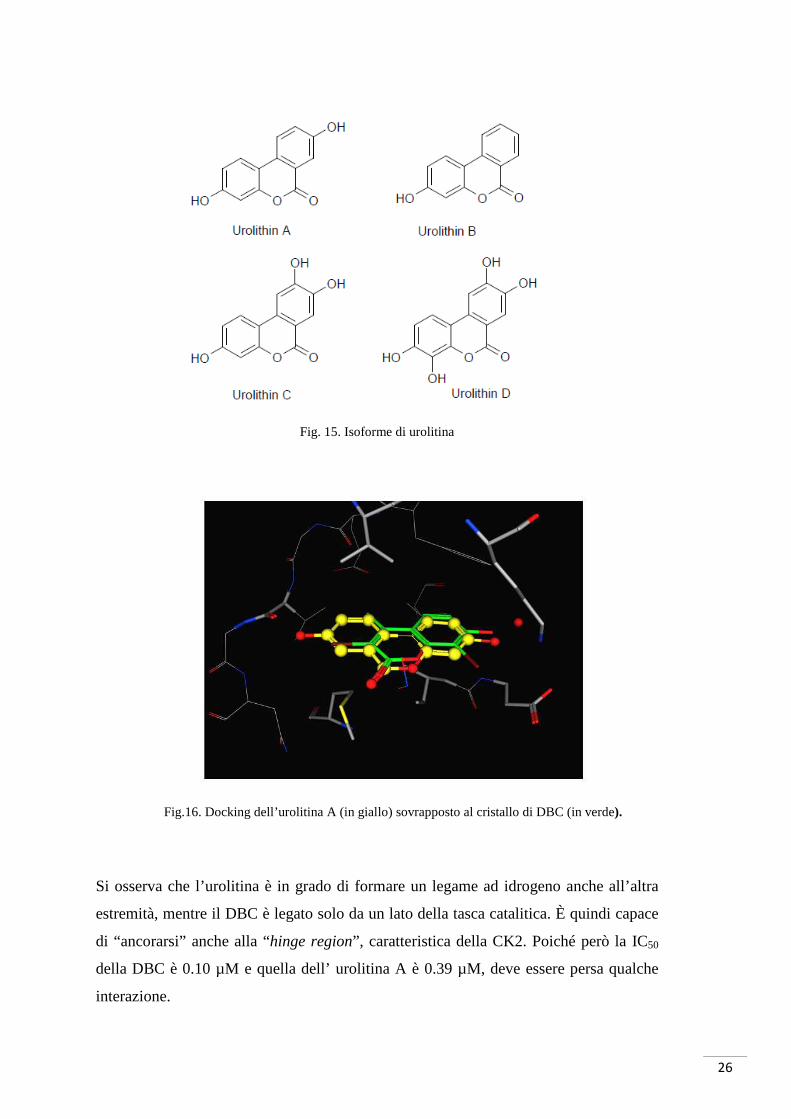
L’urolitina è un dibenzopiranone e quindi un monolattone rispetto all’acido ellagico che
presenta però solo due gruppi ossidrilici. In letteratura sono riportate quattro isoforme
di urolitina (Bialonska, Kasimsetty, Khan, & Ferreira, 2009) (fig. 15).
Il docking (effettuato presso il laboratorio di modellistica molecolare del Prof. Stefano
Moro) mostra la posizione dell’urolitina A nella tasca catalitica di CK2 di Zea mays in
relazione al composto DBC (fig. 16). Tale composto aveva dimostrato un’ottima
attività e quindi può essere un utile confronto.
26
Fig. 15. Isoforme di urolitina
Fig.16. Docking dell’urolitina A (in giallo) sovrapposto al cristallo di DBC (in verde).
Si osserva che l’urolitina è in grado di formare un legame ad idrogeno anche all’altra
estremità, mentre il DBC è legato solo da un lato della tasca catalitica. È quindi capace
di “ancorarsi” anche alla “hinge region”, caratteristica della CK2. Poiché però la IC50
della DBC è 0.10 µM e quella dell’ urolitina A è 0.39 µM, deve essere persa qualche
interazione.

27
Il sito catalitico è una zona basica e polare e quindi favorevole alla presenza di gruppi
acidi o in grado di stabilire legami ad idrogeno; ad esempio l’IQA (tab. 4) posiziona
proprio in questa zona il proprio gruppo carbossilico. Nel caso dell’urolitina, invece,
l’ossigeno del gruppo fenolico si comporta da donatore di legami ad idrogeno come
accade anche nel caso della DBC. A questo punto si può dire che l’urolitina rappresenta
il derivato dell’acido ellagico con il minor numero di ossidrili che mantiene comunque
una buona attività.
È stata quindi valutata presso il laboratorio del prof. Meggio del Dipartimento di
Biochimica dell’ Università di Padova l’effettiva attività inibitoria nei confronti di CK2
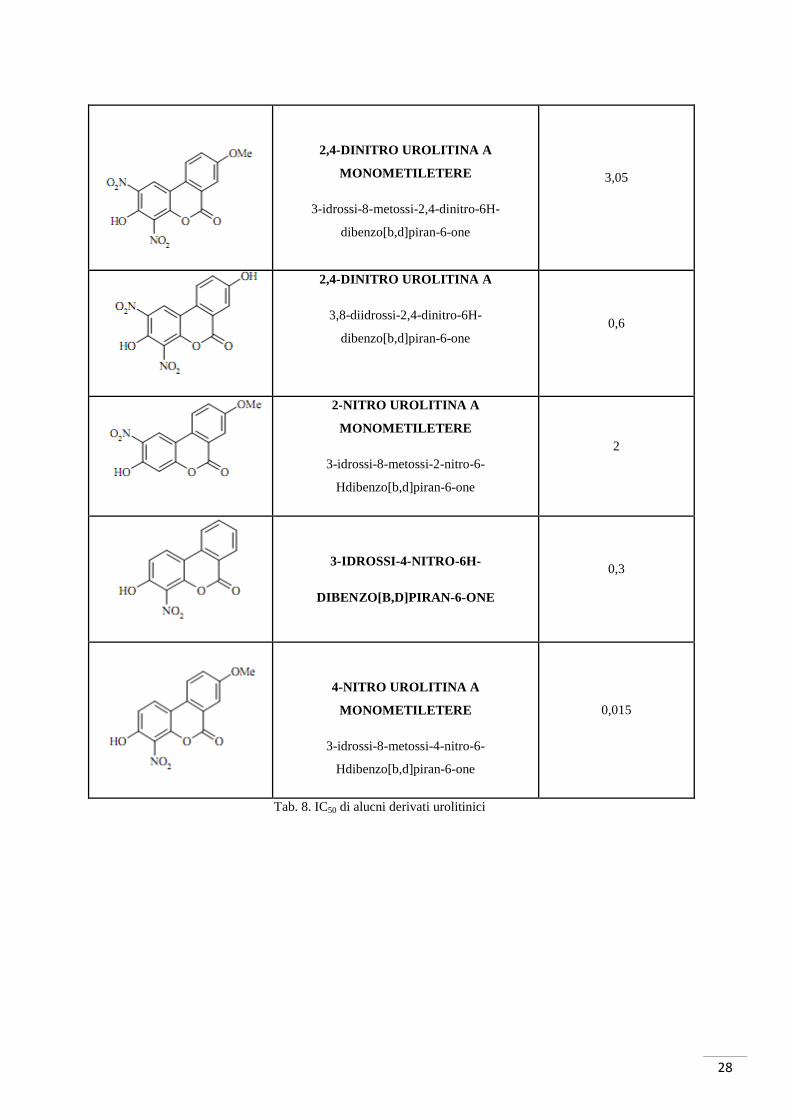
dei derivati urolitinici sintetizzati precedentemente (tab. 8).
Molecola Nome IC50 µM su nCK2 a
20 µM [ATP]
0,39
UROLITINA A
3,8-diidrossi-6H-dibenzo[b,d]piran-6-one
3,47
UROLITINA A MONOMETILETERE
3-idrossi-8-metossi-6H-dibenzo[b,d]piran-6-
one
UROLITINA A DIMETILETERE
3,8-dimetossi-6H-dibenzo[b,d]piran-6-one >40
>40
2,4,7,9-TETRANITRO UROLITINA A
3-idrossi-8-metossi-2,4,7,9-tetranitro-6H-
dibenzo[b,d]piran-6-one
28
3,05
2,4-DINITRO UROLITINA A
MONOMETILETERE
3-idrossi-8-metossi-2,4-dinitro-6H-
dibenzo[b,d]piran-6-one
2,4-DINITRO UROLITINA A
3,8-diidrossi-2,4-dinitro-6H-
dibenzo[b,d]piran-6-one 0,6
2-NITRO UROLITINA A
MONOMETILETERE
3-idrossi-8-metossi-2-nitro-6-
Hdibenzo[b,d]piran-6-one
2
3-IDROSSI-4-NITRO-6H-
DIBENZO[B,D]PIRAN-6-ONE
0,3
4-NITRO UROLITINA A
MONOMETILETERE
3-idrossi-8-metossi-4-nitro-6-
Hdibenzo[b,d]piran-6-one
0,015
Tab. 8. IC50 di alucni derivati urolitinici

29
1.5.1. ULTIMI SVILUPPI (2009-2010-2011)
Sono di seguito elencati una serie di inibitori CK2 risultanti dai più recenti studi riportati
in letteratura; i seguenti composti sono stati sintetizzati da gruppi di ricerca diversi da
quelli dell’Università di Padova.
• Emateina (Hung, et al., 2009)
L’emateina (fig. 17) è un composto naturale presente in Caesalpinia sappan, un’erba
medicinale orientale. L’IC50 di questo composto risulta essere di 0.55 µM. Si tratta di
un inibitore ATP non competitivo il cui meccanismo d’azione non è ancora chiaro
(Cozza, Meggio, & Moro, 2011).
Fig. 17. Emateina.
• AMR (Ramos, et al., 2010)
Si tratta di un derivato del naftalene (fig. 18a) a cui sono stati aggiunti due anelli furanici
dotati entrambi di gruppi carbossilici (fig. 17c). Il derivato del naftalene in fig. 17b è,
invece, risultato inattivo. L’IC50 dell’AMR è risultata essere di 200 nM. Docking
molecolare in fig. 19.
Fig. 18. (a) 2,7-diidrossinaftalene; (b) 2,6-diidrossinaftalene; (c) AMR.
30
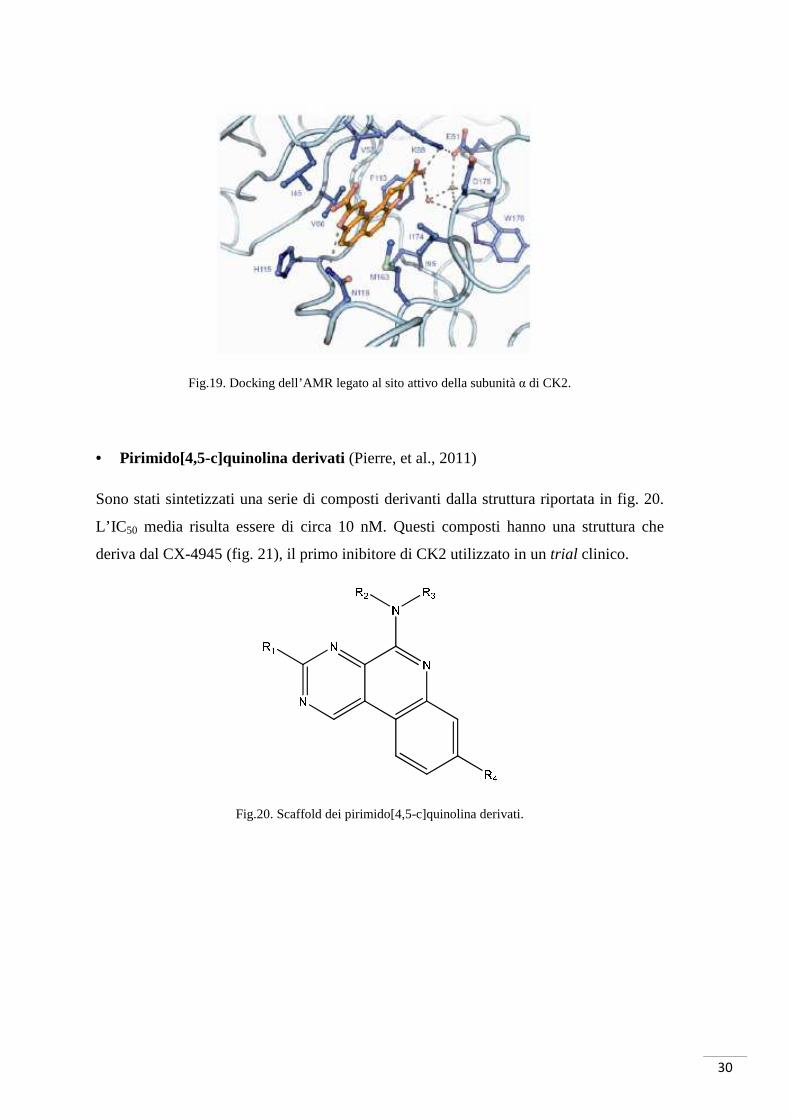
Fig.19. Docking dell’AMR legato al sito attivo della subunità α di CK2.
• Pirimido[4,5-c]quinolina derivati (Pierre, et al., 2011)
Sono stati sintetizzati una serie di composti derivanti dalla struttura riportata in fig. 20.
L’IC 50 media risulta essere di circa 10 nM. Questi composti hanno una struttura che
deriva dal CX-4945 (fig. 21), il primo inibitore di CK2 utilizzato in un trial clinico.
Fig.20. Scaffold dei pirimido[4,5-c]quinolina derivati.

31
1.5.2. TRIALS CLINICI
Recentemente un inibitore di CK2 ATP competitivo è stato sperimentato in fase clinica
I come antitumorale (Pierre, et al., 2011): Si tratta del CX-4945 (fig. 21), progettato da
Cylene Pharmaceuticals per il trattamento del mieloma multiplo, del tumore al seno,
della malattia di Castlaman e dei tumori solidi avanzati (clinicaltrials.gov).
Questo composto risulta avere una IC50 = 1 nM (Pierre, et al., 2011). Durante il trial in
fase I il 20% dei pazienti valutati hanno mostrato una stabilizzazione della malattia per
almeno 16 settimane, il 67% di questi pazienti sono rimasti in studio per almeno sei
mesi. Questi risultati stabiliscono che CX-4945, è un promettente agente terapeutico
(Cylene Pharmaceuticals).
La fase I del trial è iniziata nel Febbraio 2009 negli Stati Uniti (Arizona, Colorado e
Texas), la formulazione è di tipo orale e attualmente partecipano 55 pazienti; si stima
che questa fase dello studio terminerà del Dicembre 2011. (clinicaltrials.gov).
Fig. 21. (a) CX-4945 legato alla subunità α di CK2 (Ferguson, et al., 2011). (b) Struttura del CX-4945.

32

33
2. SCOPO DEL LAVORO E OBIETTIVI

34

35
2.1. SCOPO DEL LAVORO
Nell’introduzione è stato evidenziato il ruolo fondamentale che ricopre la CK2. È
importante ricordare come questo enzima sia coinvolto in reazioni chiave della
sopravvivenza cellulare (punto 1.4 del presente testo), per questo è protagonista
nell’insorgenza di patologie quali, ad esempio, i tumori.
È necessario inibire la CK2 in quanto esiste una stretta relazione tra un’elevata attività di
questo enzima e un’anormale proliferazione cellulare (Unger, Davis, Slaton, & Ahmed,
2004).
Questo lavoro è inserito in un contesto di sintesi di derivati il cui scopo è approfondire le
conoscenze riguardanti lo spazio sterico ed elettronico della cavità catalitica
dell’enzima. La ricerca è stata indirizzata verso la sintesi di potenziali inibitori
bifunzionali (questo termine sta ad indicare la capacità del presunto inibitore di
interagire contemporaneamente con la tasca dell’ATP e con la zona di legame del
substrato proteico) schematizzati in fig. 22:
.
Fig. 22. Bifunzionalità di un inibitore CK2.

36
2.2. OBIETTIVI
L’obiettivo ultimo di questo lavoro di tesi è stato la sintesi di nuovi inibitori di CK2.
Sono state progettate delle strategie di sintesi per i seguenti composti (tab. 9):
COMPOSTO STRUTTURA
a
b
c
Tab. 9. Composti obiettivo di questo lavoro di tesi.

37
Il composto a è un derivato dell’urolitina A; le strutture ad esso correlate hanno una IC50
di 4,5 µM (fig. 3a) e 0,026 µM (fig. 3b) sulla CK2 (lavoro non pubblicato, risultati
preliminari da internato di tesi A. Bertamini, 2010). La presenza di un alogeno permette
di esplorare lo spazio sterico ed elettronico della cavità catalitica in modo più
approfondito. Gli aspetti da considerare relativamente al bromo sono:
1. Aspetto elettronico; Br è un atomo tipicamente elettronegativo.
2. Aspetto sterico; la sua presenza comporta un notevole ingombro sterico.
3. Lipofilia; modifica la logP aumentando la lipofilia del composto a cui è legato,
può dare legami idrofobici.
Il composto in fig. 23a è risultato avere un’attività inferiore al suo derivato in fig. 23b;
questo è dovuto alla presenza di un metile in posizione 4 che difficilmente permette la
formazione di interazioni idrofobiche con il sito di legame per l’ATP. Al contrario, il
composto in fig. 23b, è risultato essere attivo grazie alla presenza del Br. Il composto a
(tab. 9) presenta in posizione 4 un bromometile. È stato ipotizzato che tale gruppo
impedisca il legame inibitore-enzima in quanto caratterizzato da un notevole ingombro
sterico. Il composto, quindi, dovrebbe risultare inattivo.
Fig.23.(a) 4-metil urolitina A; (b) 4-bromo urolitina A.
Il composto b è un derivato della TBB-propil-diammina consecutivo alla serie GZ1-4. Si
è voluto allungare ulteriormente la catena laterale inserendo due gruppi carbossilici
terminali in grado di favorire l’interazione inibitore-enzima: si è supposto che avvenga
un’interazione con l’Asp presente nel sito di legame per il substrato.
Il composto c è un N-derivato dell’urolitina A: all’azoto presente nella struttura triciclica
è legato un benzile. E’ stato ipotizzato che la presenza di tale sostituente impedisca
l’accesso al sito per il legame dell’ATP a causa di un elevato ingombro sterico. Il
composto, quindi, dovrebbe risultare inattivo.
(a) (b)

38

39
3. MATERIALI E METODI

40
41
3.1. ABBREVIAZIONI
°C gradi centigradi
Ac2O anidride acetica
AIBN azobisisobutirnitrile
AlCl 3 cloruro di alluminio
Ar aromatico
BBr3 tribromuro di boro
BPO perossido di benzoile Br bromo C carbonio
CCl4 tetracloruro di carbonio
CDCl3 cloroformio deuterato
CH3COOH acido acetico
CHCl3 cloroformio
Cu rame CuCl cloruro di rame CuI ioduro di rame
CuSO4 solfato di rame
Da dalton DCC N,N′-Dicicloesilcarbodiimmide DCM diclorometano DCU N,N’-Dicicloesilurea dd doppietto di doppietti δ chemical shift DIEA N,N-diisopropiletilamina DMAP 4-(dimetilamino) piridina DMF dimetilformammide Eq equivalenti
Et2O etere etilico
Et3N trietilammina
EtOAc etilacetato g grammi H idrogeno
H2O acqua
HCl acido cloridrico Hz Hertz J costante di accoppiamento
K2CO3 carbonato di potassio
42
KOH idrossido di potassio m multipletto Me metile MeOH metanolo mg milligrammi ml millilitri mmol millimoli
Na2SO4 solfato di sodio
NaCl cloruro di sodio
NaHCO3 bicarbonato di sodio
NaOAc acetato di sodio NaOH idrossido di sodio NBS N-bromosuccinimmide
NH3 ammoniaca
NH4OAc acetato di ammonio
NMR risonanza magnetica nucleare Nu nucleofilo Pd palladio
Pd(OAc)2 palladio acetato
PM peso molecolare
PPh3 trifenilfosfina
ppm parti per milione RF fattore di ritenzione s singoletto Sn stagno
SOCl2 cloruro di tionile
t tripletto T temperatura TBT tributilstagno THF tetraidrofurano TLC cromatografia su strato sottile

43
3.2. MATERIALI
Reagenti: Sono stati usati reagenti: Sigma Aldrich, Fluka senza ulteriori purificazioni.
Solventi: Sono stati usati solventi delle ditte Sigma Aldrich, Fluka.
Solventi deuterati: Sono stati utilizzati solventi deuterati della ditta Aldrich.
Fasi stazionarie per cromatografia su colonna: E’ stato utilizzato gel di silice Silica
Gel 60 (230-400 Mesh) della Fluka.
Lastre per TLC: Sono state usate lastre con supporto in vetro Silica Gel 60 F254
della Merck KGaA e lastre con supporto in alluminio Silica Gel.
METODI E STRUMENTAZIONE
Spettrometria di massa
Gli Spettri di Massa sono stati eseguiti utilizzando lo strumento Applied Biosystems
5220 basato sulla tecnica ESI-TOF.
Misure di Spettroscopia di Risonanza Magnetica Nucleare (NMR)
Gli spettri NMR 1H e 13C sono stati ottenuti alla temperatura T 298 K sciogliendo i
campioni in solventi deuterati anidri lavorando in atmosfera inerte di azoto al fine di
evitare contaminazioni da H2O.
Lo strumento utilizzato è lo spettrometro NMR Bruker Avance III, operante alle
frequenze-base di 400 MHz sul nucleo 1H e 100 MHz sul nucleo 13C. I valori di
chemical shift sono dati in unità δ riferiti alla posizione del segnale del tetrametilsilano
quale standard interno (δ=0).

44

45
4. DISCUSSIONE DEI RISULTATI

46
47
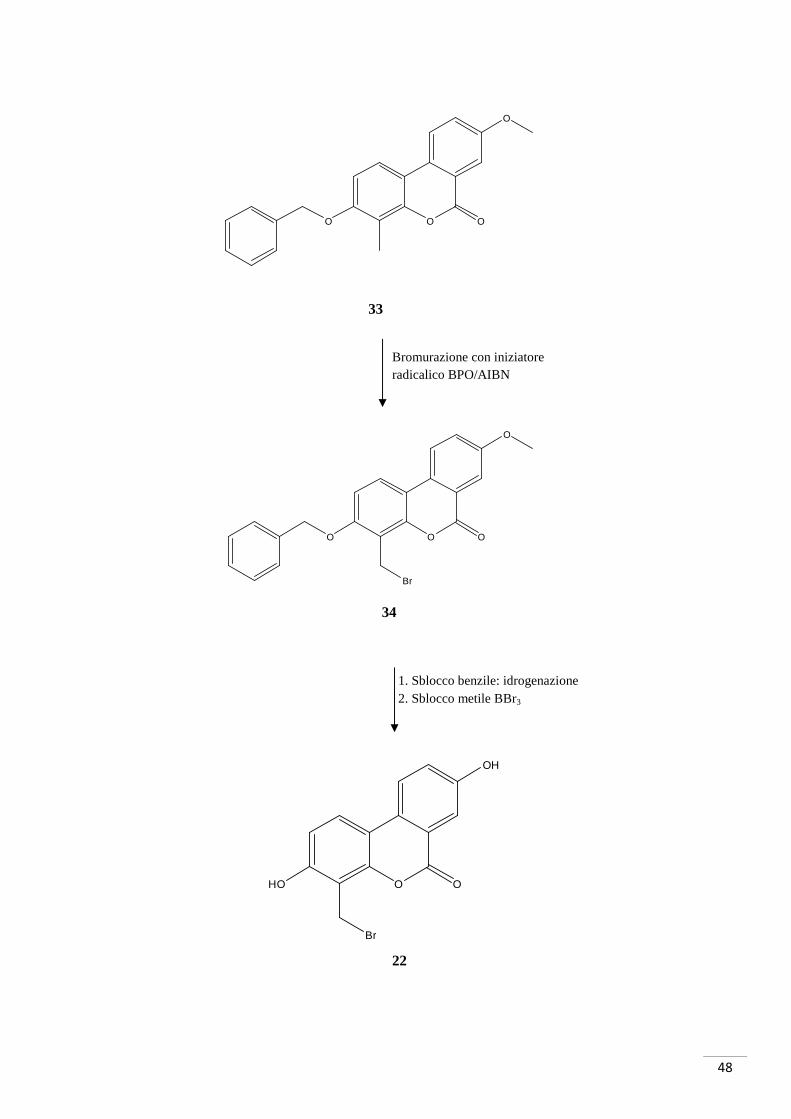
4.1. SCHEMA DI SINTESI 1
Sintesi di 4-(bromometil)-3,8-diidrossi-6H-benzo[c]cromen-6-one
Metodo A
(4-BROMOMETIL UROLITINA A)
NaOAc, Pd(OAc)2, PPh3, reflusso 130°C , atm N2
comp. 29, comp. 31, DCC,DMAP T ambiente
1. K2CO3, acetone, comp. 30 (benzilbromuro), reflusso 50°C Br2, CH3COOH
Reflusso 100°C
O
O OH
HO OH
HO O
O
O OH
Br
OO O
Br
O
27 28
29 31
32
48
1. Sblocco benzile: idrogenazione 2. Sblocco metile BBr3
Bromurazione con iniziatore radicalico BPO/AIBN
OO O
O
OO O
O
Br
OHO O
OH
Br
33
34
22

49
4.1.1. RAZIONALE DELLO SCHEMA DI SINTESI 1
La procedura di sintesi è iniziata con la preparazione degli intermedi base per la
formazione dello scheletro dell’urolitina. L’acido 2-bromo-5-metossibenzoico
(composto 29) è stato sintetizzato tramite bromurazione in posizione orto al carbossile
dell’acido 3-metossibenzoico (composto 27). Per facilitare la successiva bromurazione
del composto 33 e, alla stesso tempo permettere l’esterificazione per la formazione del
composto 32, è stato benzilato un unico idrossile del 2-metilresorcinolo (composto 28)
tramite l’utilizzo di benzilbromuro.
Facendo reagire gli intermedi così ottenuti (composti 29 e 31) è stato sintetizzato il
composto 32: è stata effettuata un’attivazione dell’acido 2-bromo-5-metossibenzoico
con DCC (esterificazione di Steglich). Lo step successivo ha previsto la chiusura
dell’anello centrale con una reazione Pd-catalizzata avvenuta a 130°C e sotto atmosfera
di azoto (reazione di Heck).
La bromurazione del composto 33 è stata tentata con due diversi iniziatori radicalici:
BPO e AIBN in CCl4 e utilizzando un equivalente di NBS come “donatore di Br”.
Entrambe le reazioni non hanno dato esito positivo.
Pertanto non è stato possibile sintetizzare il composto 34 e il suo derivato finale
sbloccato (4-bromometil-urolitina A).
50
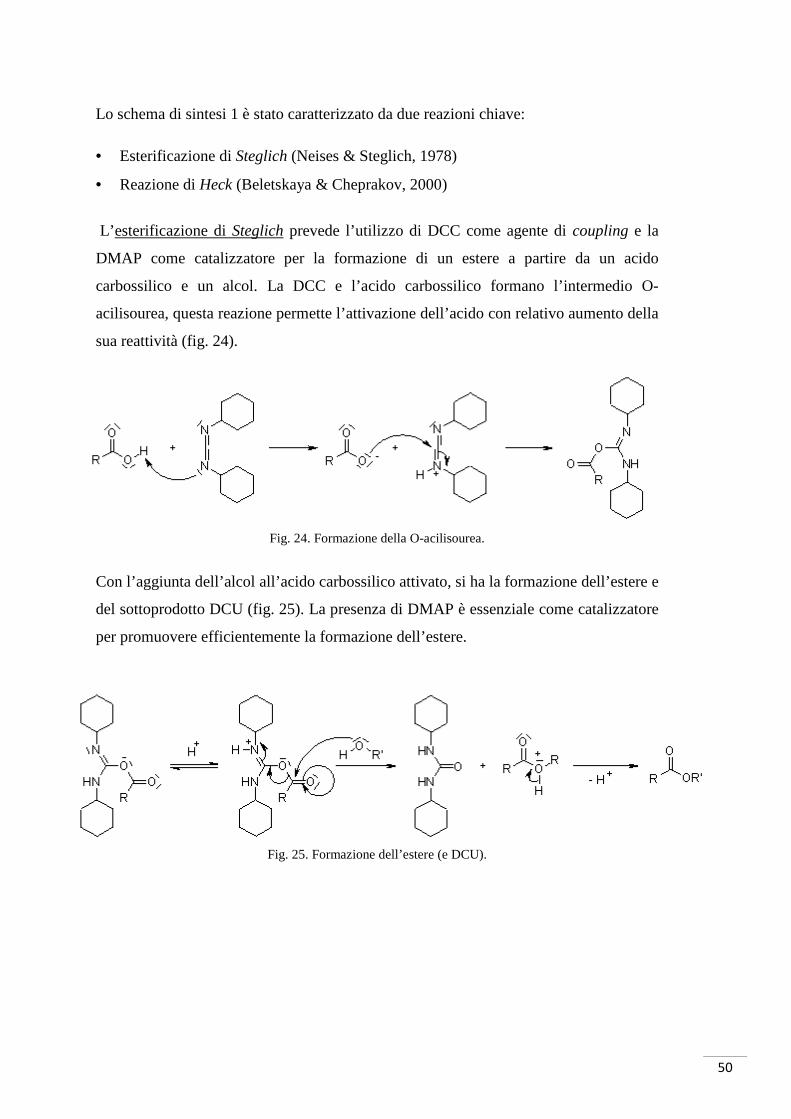
Lo schema di sintesi 1 è stato caratterizzato da due reazioni chiave:
• Esterificazione di Steglich (Neises & Steglich, 1978)
• Reazione di Heck (Beletskaya & Cheprakov, 2000)
L’esterificazione di Steglich prevede l’utilizzo di DCC come agente di coupling e la
DMAP come catalizzatore per la formazione di un estere a partire da un acido
carbossilico e un alcol. La DCC e l’acido carbossilico formano l’intermedio O-
acilisourea, questa reazione permette l’attivazione dell’acido con relativo aumento della
sua reattività (fig. 24).
Fig. 24. Formazione della O-acilisourea.
Con l’aggiunta dell’alcol all’acido carbossilico attivato, si ha la formazione dell’estere e
del sottoprodotto DCU (fig. 25). La presenza di DMAP è essenziale come catalizzatore
per promuovere efficientemente la formazione dell’estere.
Fig. 25. Formazione dell’estere (e DCU).

51
La reazione di Heck si colloca subito dopo la formazione dell’estere ed è responsabile
della chiusura dell’anello centrale dell’urolitina. In questa reazione si ha la sostituzione
del gruppo R di un alo-alchene (o di un alo-arene) con un H di un alchene (H vinilico)
(Brown, Foote, Iverson, & Anslyn, 2011). Si tratta di una reazione palladio-catalizzata:
la forma più comune di Pd utilizzata è il Pd(II)- acetato che viene complessato in situ da
PPh3. Il meccanismo di reazione è di tipo ossido-riduttivo, dove il Pd(II) viene ridotto a
Pd(0) formando un complesso con due molecole di ligando (PPh3) in questo modo
(fig.26):
Fig. 26. In alto: formazione del complesso del Pd(0) per riduzione del Pd(II). In basso: il complesso
Pd(0)PPh3 reagisce con un alo-arene.
Il fine di questa reazione consiste nella formazione di un legame C-C che, nella
reazione svolta in questo lavoro di tesi, ha permesso il coupling del composto con
l’ottenimento della classica struttura triciclica dell’urolitina.

52
Altre reazioni di formazione di legami C-C catalizzate da metalli di transizione sono la
reazione di Hurtley (Aalten, Koten, Riethorst, & Stamlc, 1989) e la reazione di Stille
(Stille, 1986).
In questo lavoro di tesi è stata utilizzata la reazione di Heck per i seguenti motivi:
o La reazione di Hurtley prevede l’utilizzo di Cu(II) e NaOH. Tale metodo ha
dimostrato avere una resa minore rispetto a quella di Heck: infatti il Pd(OAc)2 è
un catalizzatore più stabile termodinamicamente e il Pd2+ coordina in modo più
efficace la nuvola π dell’anello aromatico.
o La reazione di Stille prevede l’utilizzo di organo-stannici. Tali composti hanno
una preparazione complessa e sono tossici per l’ambiente; sono dannosi in
particolar modo per i molluschi (Haggera, Depledgeb, & Gallowaya, 2005) e si
depositano nei tessuti biologici di pesci e mammiferi (Harino, Fukushima, &
Kawai, 2000).
Per la strategia di sintesi della 4-bromometilurolitina A è stato necessario proteggere gli
ossidrili degli starting materials 27 e 28 in previsione della successiva reazione di
bromurazione del composto 33. La protezione è stata effettuata a monte delle reazioni
chiave: come prodotti sono stati ottenuti gli intermedi acido 2-bromo-5-
metossibenzoico (composto 29) e 3-metossibenzil-2-metilfenolo (composto 31). La
necessità di effettuare la protezione prima delle reazioni di esterificazione e di coupling
si spiega anche con l’esempio della mono-benzilazione del 2-metilresorcinolo: è stata
eseguita prima dell’esterificazione in quanto, a causa delle condizioni di reazione (la
presenza di K2CO3 porta ad un ambiente basico), l’estere si sarebbe potuto idrolizzare
(Bronsted, 1928).

53
Facendo un quadro generale dello schema di sintesi 1 si può dire che i suoi punti critici
sono stati:
o Benzilazione del 2-metilresorcinolo (resa 18%)
o Reazione di Heck (sensibile calo di resa che avrebbe potuto incidere sull’intero
processo di sintesi)
o Bromurazione del metile in posizione 4 (non riuscita)
Le motivazioni che possono spiegare la non riuscita bromurazione del metile in
posizione 4 sono probabilmente l’ingombro sterico e la densità elettronica dell’anello: a
causa della presenza del sostituente aromatico (benzile) diminuisce l’efficienza
dell’addizione radicalica di bromo (fig. 27).
Fig. 37. Raffigurazione schematica della zona ad alta densità elettronica.
La mancata bromurazione del composto 33 è avvalorata dal fatto che in entrambi i
tentativi con i due diversi iniziatori radicalici (BPO/AIBN), lo spettro NMR
evidenziava la presenza del picco a δ 2,45 ppm (s, 3H) caratteristico del metile in
posizione 4: l’eventuale bromurazione avrebbe comportato un segnale con chemical
shift più elevato.
54
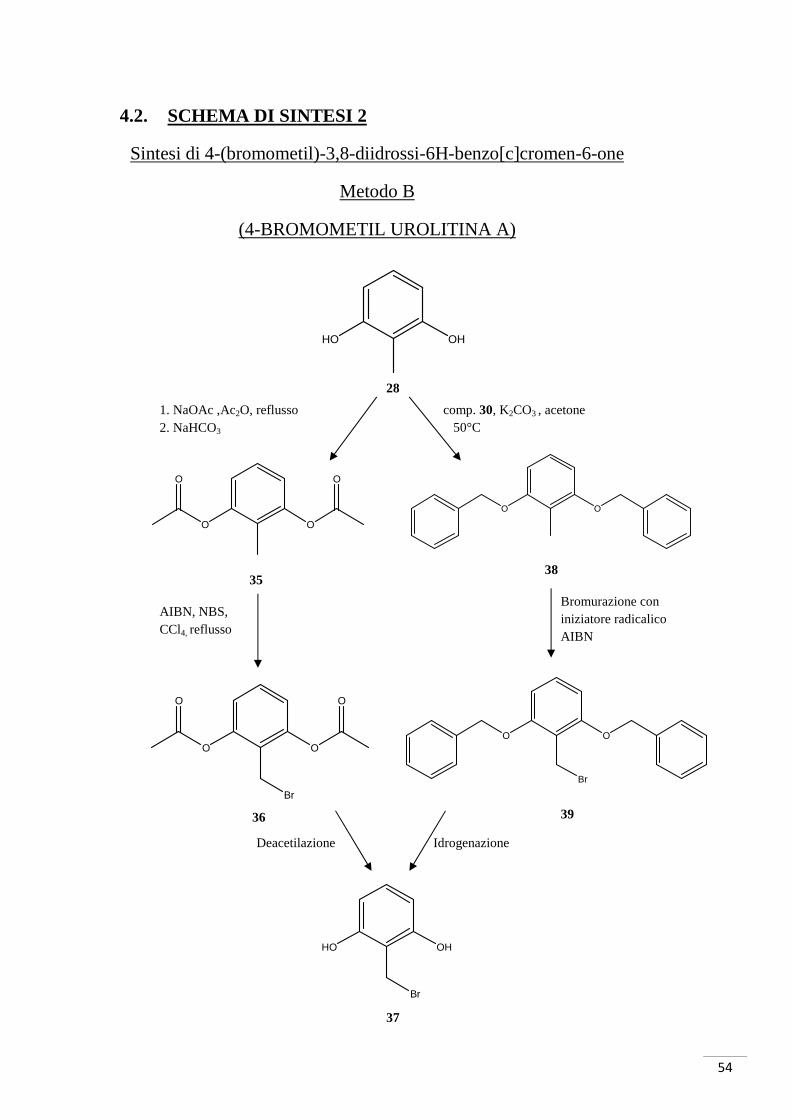
4.2. SCHEMA DI SINTESI 2
Sintesi di 4-(bromometil)-3,8-diidrossi-6H-benzo[c]cromen-6-one
Metodo B
(4-BROMOMETIL UROLITINA A)
Deacetilazione Idrogenazione
Bromurazione con iniziatore radicalico AIBN
AIBN, NBS, CCl4, reflusso
1. NaOAc ,Ac2O, reflusso 2. NaHCO3
comp. 30, K2CO3 , acetone 50°C
28
O O
O O
O O
O O
O O
Br
O O
Br
HO OH
Br
HO OH
35 38
36 39
37
55
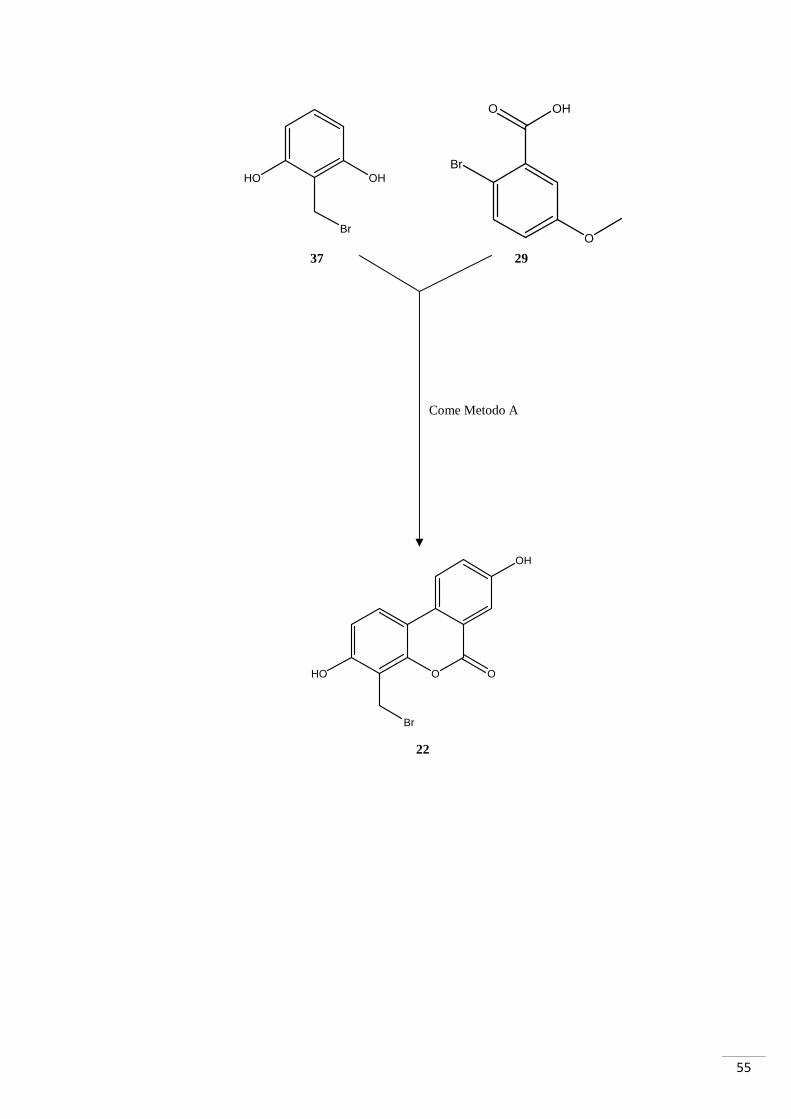
Come Metodo A
37
HO OH
Br
O OH
Br
O
HO
Br
O O
OH
29
22

56
4.2.1. RAZIONALE DELLO SCHEMA DI SINTESI 2
Lo schema di sintesi 2 consiste in una strategia alternativa al precedente metodo di
sintesi della 4-bromometil urolitina A. Date le difficoltà incontrate nella bromurazione
del metile in posizione 4 del composto 33, è stata progettata una via di alogenazione per
modificare direttamente lo starting material 2-metiresorcinolo. L’obiettivo è stato
quello di anteporre la bromurazione alla reazione di esterificazione.
Il metodo A e il metodo B differiscono solamente nella parte iniziale del processo;
infatti, l’unica reazione della precedente strategia in cui sono state incontrate difficoltà
non aggirabili è stata la formazione del composto 34 (bromurazione).
Per facilitare tale bromurazione sono stati protetti entrambi gli ossidrili (fig. 28) del 2-
metilresorcinolo:
Fig. 28. Protezione OH.
Sono state utilizzate due tipologie di protezione:
o Acetilazione (fig. 29)
Fig.29. Protezione e bromurazione del 2-metilresorciolo
HO
CH3
OH O
CH3
O
O O
O O
O O
Br
28 35 36

57
L’acetilazione del 2-metilresorcinolo è stata effettuata con NaOAc e Ac2O a reflusso
per tutta la notte. Una volta purificato il composto di-acetilato è stata eseguita la
bromurazione del metile in posizione 2: la reazione ha previsto l’utilizzo dell’iniziatore
radicalico AIBN e del “donatore” di bromo NBS; è stato così ottenuto il composto 36.
Il successivo sblocco degli acetili è stato tentato con sette diverse procedure, ma in
nessun caso è stato ottenuto il composto 37. In generale, la deprotezione del composto
avrebbe previsto l’idrolisi dell’estere e la successiva acidificazione per la formazione
degli ossidrili.
o Benzilazione (fig. 30)
Fig. 30. Benzilazione del 2-metilresorcinolo.
La benzilazione del 2-metilresorcinolo è stata effettuata con benzilbromuro come per il
composto 31 ma ottenendo la doppia sostituzione degli ossidrili. La successiva
bromurazione del composto 38 ha avuto esito negativo: la causa di ciò è data dal
consistente ingombro sterico; infatti la presenza dei due benzili ha reso difficile
l’attacco del bromo (altro atomo con notevole ingombro sterico).
58
4.3. SCHEMA DI SINTESI 3
Sintesi di acido (S)-2-(4-osso-4-(3-(4,5,6,7-tetrabromo-1H-
benzo[d]imidazol-2-amino)propilamino)butanammido)pentandioico
O
O
NH2
O
O
O
O
O
O
O
NH
O
O
CH2
O
CH2
O OH
NH
N
Br
Br
Br
Br
NH NH2
40 41
1.DIEA, T ambiente 2. HCl
42
12
DCC, DIEA, comp. 12 T ambiente
59
1. NaOH 1M, T ambiente 2. HCl 10% v/v
NH
N
Br
Br
Br
Br
NHHN
O
O
HN
OO
CH2
CH2
O O
NH
N
Br
Br
Br
Br
NHHN
O
O
HN
OHO
CH2
CH2
O OH
43
23

60
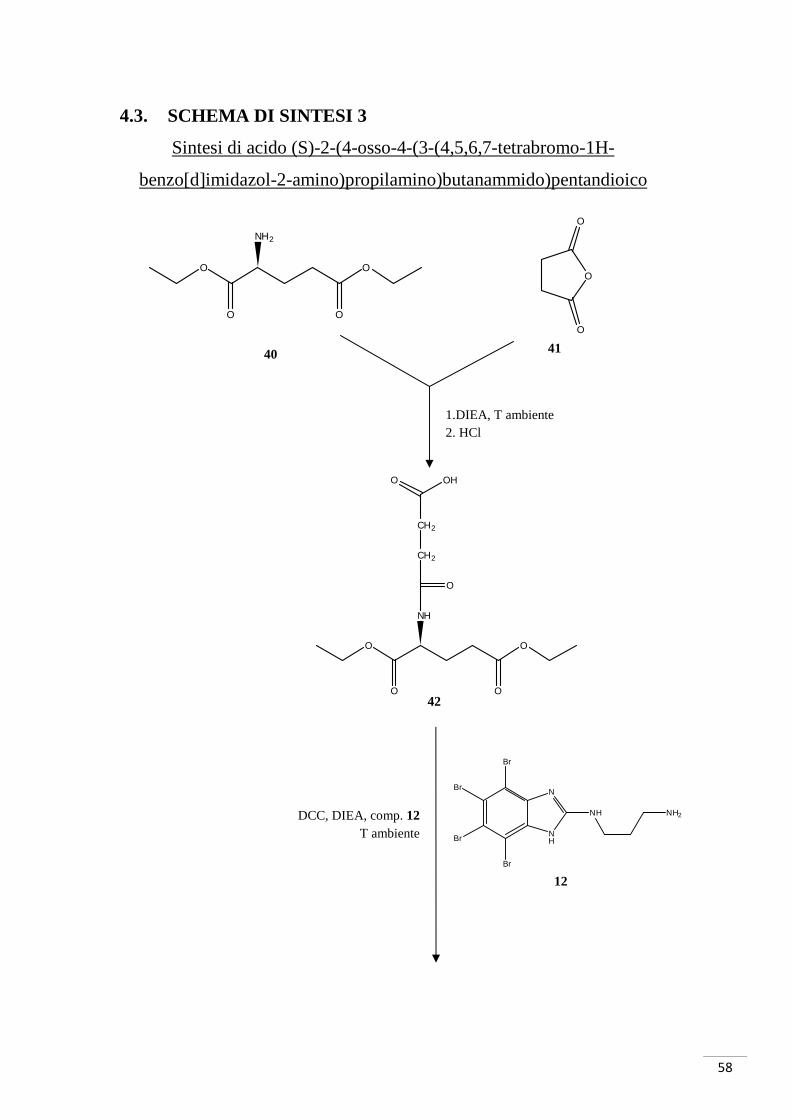
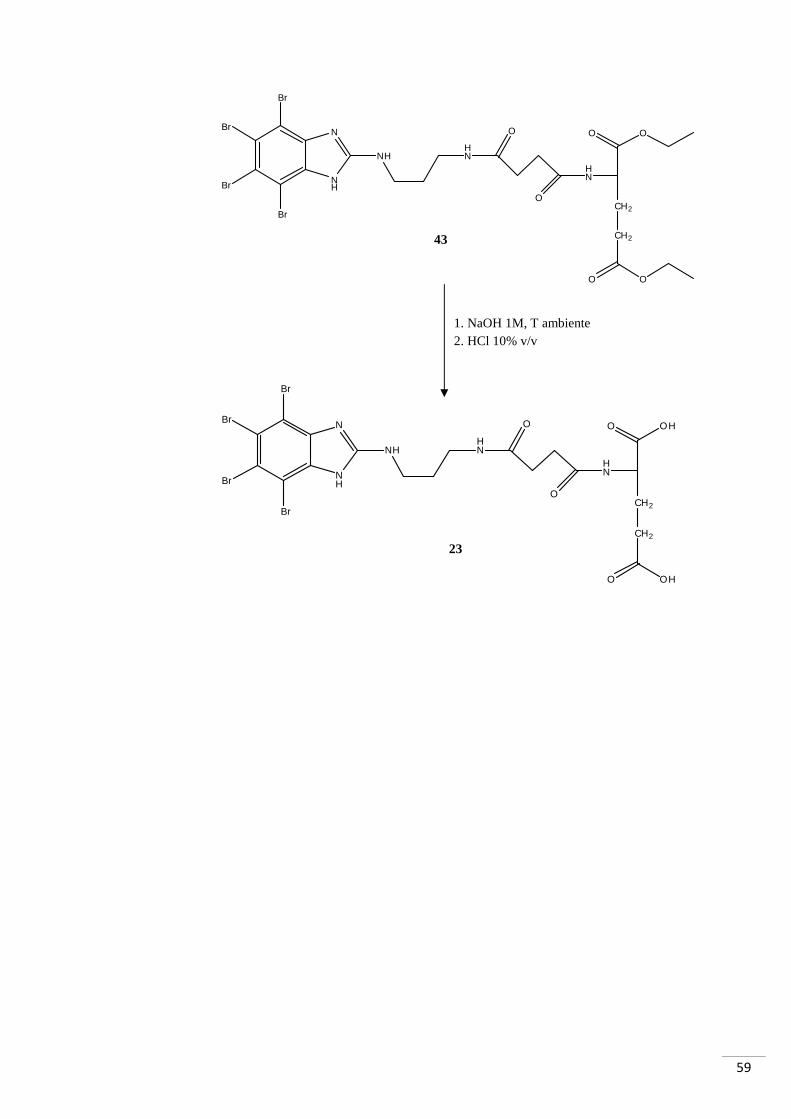
4.3.1. RAZIONALE DELLO SCHEMA DI SINTESI 3
La sintesi è iniziata con la reazione tra anidride succinica (composto 41) e acido
glutammico cloridrato (composto 40, i gruppi carbossilici in posizione 1 e 5 sono
protetti da etili). Il composto così ottenuto è stato fatto reagire con il composto 12 per la
formazione di un nuovo legame ammidico: in questo caso è stato necessario attivare
l’acido carbossilico del composto 42 con DCC e la reazione è avvenuta a temperatura
ambiente. Il successivo sblocco dei gruppi etilici avrebbe dovuto portare alla
formazione del composto 23, ma è stato possibile isolare solamente il mono-derivato
(composto 44 - GZ9) (fig. 31a-b).
Fig. 31. In alto: mono-sblocco tipo 1. Al centro: mono-sblocco tipo 2. In basso: sblocco totale.
61
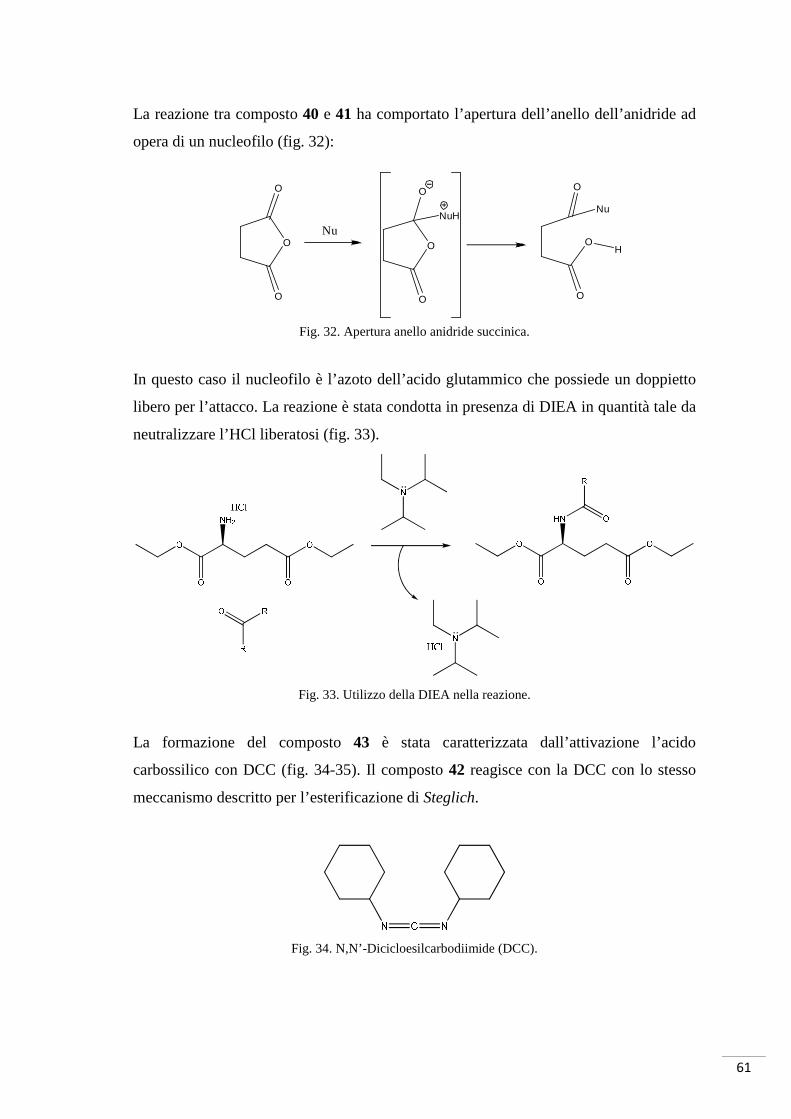
La reazione tra composto 40 e 41 ha comportato l’apertura dell’anello dell’anidride ad
opera di un nucleofilo (fig. 32):
Fig. 32. Apertura anello anidride succinica.
In questo caso il nucleofilo è l’azoto dell’acido glutammico che possiede un doppietto
libero per l’attacco. La reazione è stata condotta in presenza di DIEA in quantità tale da
neutralizzare l’HCl liberatosi (fig. 33).
Fig. 33. Utilizzo della DIEA nella reazione.
La formazione del composto 43 è stata caratterizzata dall’attivazione l’acido
carbossilico con DCC (fig. 34-35). Il composto 42 reagisce con la DCC con lo stesso
meccanismo descritto per l’esterificazione di Steglich.
Fig. 34. N,N’-Dicicloesilcarbodiimide (DCC).
O
O
O
NuO
O
O
NuH
O
O
Nu
H
O

62
Fig. 35. Reazione tra R-NH2 e acido attivato da DCC.
La formazione del sottoprodotto DCU (fig. 36) non ha permesso l’isolamento del
prodotto puro e pertanto ha reso complicato il calcolo della resa; non è stato possibile,
anche per le esigue quantità di prodotto, purificare il residuo. La DCU viene in genere
rimossa tramite filtrazione, ma le sue tracce sono state difficili da eliminare. In questo
caso, il crudo di reazione è stato purificato mediante cromatografia flash, ma le tracce
non sono state rimosse.
Fig. 36. N,N’-Dicicloesilurea (DCU).
Per questo tipo di reazione non potevano essere utilizzate altre strategie di attivazione
dell’acido; l’uso di SOCl2 avrebbe potuto rendere troppo “violento” l’ambiente di
reazione portando all’idrolisi dell’estere formatosi nelle precedente fase di sintesi.
Lo sblocco del composto 43 ha previsto l’idrolisi dell’estere in ambiente alcalino; con
la successiva acidificazione è stato ottenuto il gruppo carbossilico.

63
Tramite successiva cristallizzazione con CHCl3/n-esano è stato possibile purificare il
composto 44. Dall’analisi di massa di tale composto è stata evidenziata la presenza del
solo composto monosbloccato, come già precedentemente menzionato. La motivazione
potrebbe risiedere nella quantità di NaOH utilizzata: le moli di NaOH (0,05 mmol)
sono minori di quelle necessarie (0,06 mmol); nonostante la presenza dell’83% delle
moli necessarie alla reazione, però, non è stata comunque evidenziata la formazione del
composto di-sbloccato.
Considerata la minima quantità di prodotto ottenuta (2,5 mg) si è optato per
l’immediato test biochimico senza ulteriori analisi di caratterizzazione. Per questo
motivo non è stato possibile determinare quale dei due esteri si sia sbloccato. È lecito
ipotizzare, quindi, come il prodotto sia una miscela di composti aventi il medesimo
peso molecolare, ma con la porzione carbossilica libera in diverse posizioni.
64
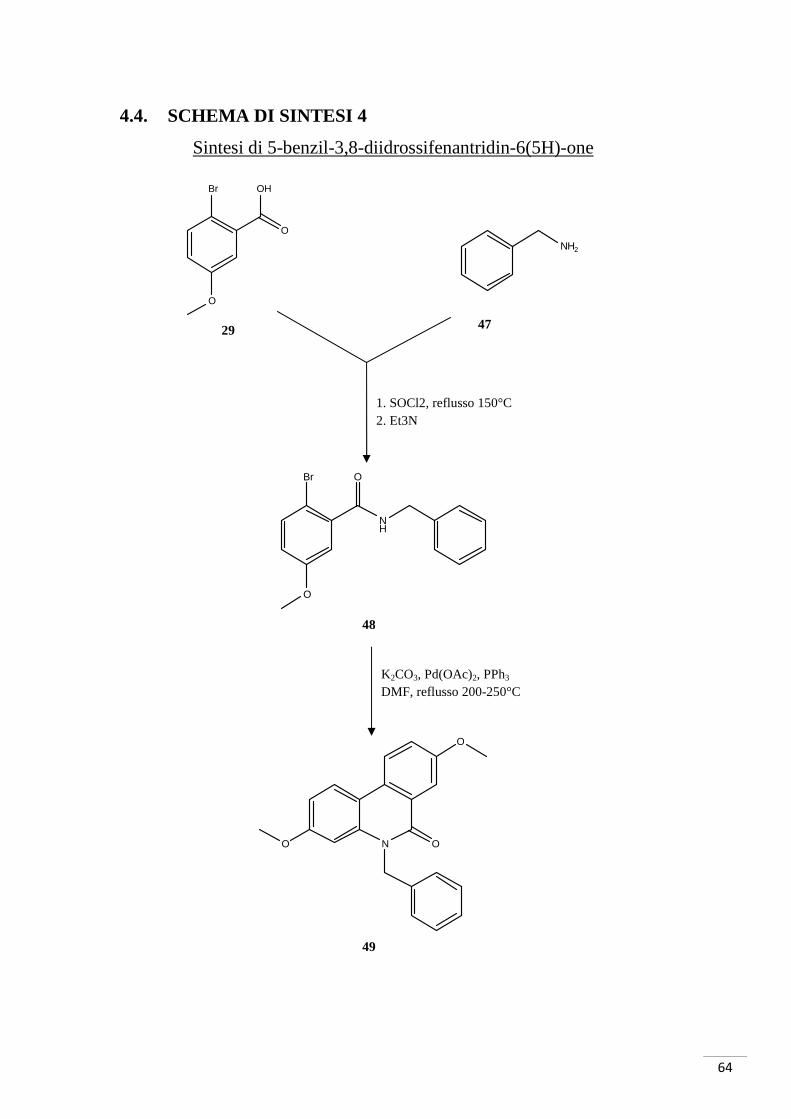
4.4. SCHEMA DI SINTESI 4
Sintesi di 5-benzil-3,8-diidrossifenantridin-6(5H)-one
K2CO3, Pd(OAc)2, PPh3 DMF, reflusso 200-250°C
1. SOCl2, reflusso 150°C 2. Et3N
Br
O
OH
O
NH2
Br
NH
O
O
N O
O
O
29 47
48
49
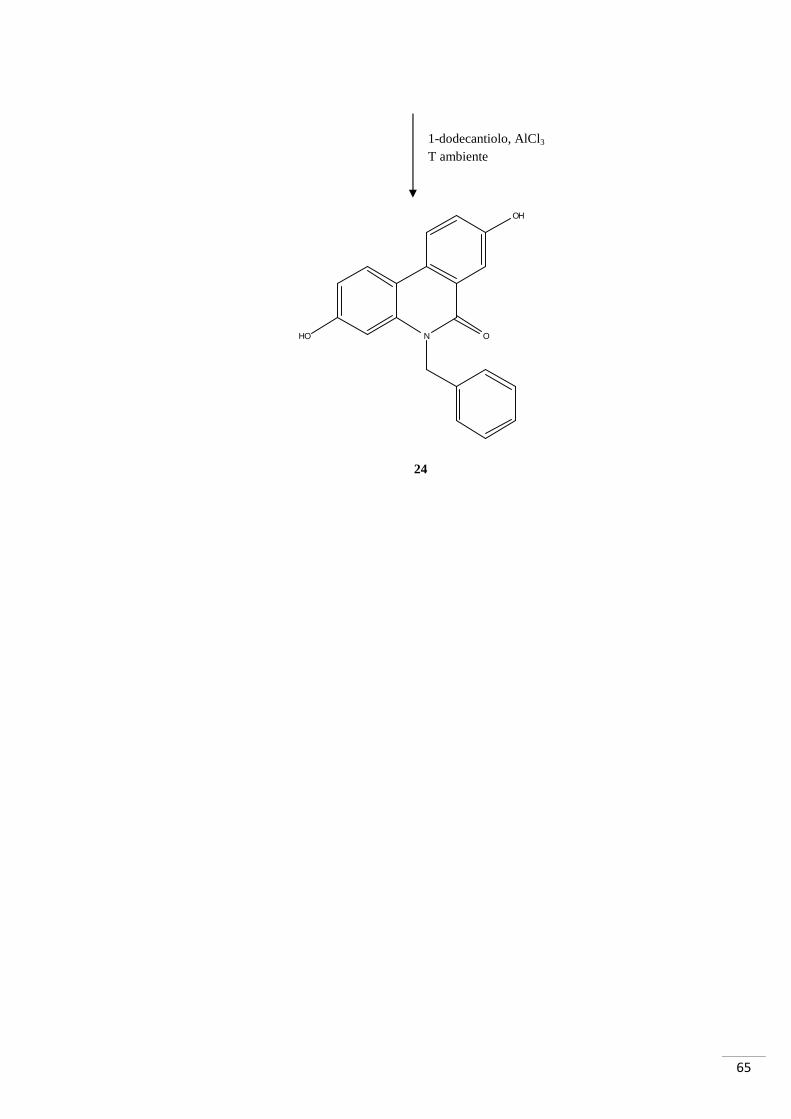
65
N O
OH
HO
24
1-dodecantiolo, AlCl3 T ambiente

66
4.4.1. RAZIONALE DELLO SCHEMA DI SINTESI 4
La procedura per la sintesi dell’N-derivato dell’urolitina A si è articolata in tre passaggi
per giungere al composto 24. Inizialmente si sono fatti reagire gli starting materials
(composto 29 e composto 47) per la formazione del legame ammidico: l’attivazione
dell’acido è stata effettuata con SOCl2 in ambiente anidro a 150°C. Tramite una
reazione domino, il composto 48 così ottenuto, è stato fatto reagire con Pd(OAc)2, PPh3
e K2CO3 in DMF a 200-250°C. Il prodotto di tale reazione è stato sbloccato a livello dei
metossili con AlCl3 in 1-dodecantiolo: è stata isolata e purificata l’urolitina N-benzilata.
L’acido 2-bromo-5-metossibenzoico e la benzilammina hanno partecipato alla
formazione dell’ammide: a differenza dei casi riportati nelle altre sintesi di questo
lavoro, è stato utilizzato SOCl2 (cloruro di tionile) per attivare l’acido. Questo
composto inorganico converte gli acidi carbossilici in cloruri acilici (fig. 37):
Fig. 37. Azione del cloruro di tionile.
La fig. 37 evidenzia la formazione di HCl come sottoprodotto a partire da SOCl2;
questa acidificazione è stata neutralizzata con Et3N. Il cloruro acilico ha un gruppo
uscente migliore dell’acido carbossilico: avviene quindi un attacco nucleofilo da parte
dell’azoto della benzilammina sul carbonio carbonilico. Si ottiene così il composto 48.
La fase critica di questa strategia di sintesi è stata la reazione domino necessaria per la
formazione del composto 49; è stato possibile utilizzare questo metodo in quanto il
composto che si voleva ottenere era un composto simmetrico. Per prodotti non
simmetrici si sarebbe dovuta utilizzare la strategia di sintesi riportata nello schema 1.

67
Una reazione domino è un processo che coinvolge due o più trasformazioni di legame
(solitamente C-C) che avvengono nelle stesse condizioni di reazione senza l’aggiunta di
ulteriori reagenti o catalizzatori, e in cui il risultato di reazioni concatenate è la
conseguenza del passaggio precedente (Tietze, 1996).
La classificazione delle reazioni domino si basa sull’intermedio generato nel primo step
di reazione. Sono stati riconosciuti vari tipi di reazioni domino (Atta-ur-Rahman, 2008)
(Tietze, 1996), i più significativi sono:
o Reazioni domino cationiche
o Reazioni domino anioniche
o Reazioni domino radicaliche
o Reazioni domino pericicliche
o Reazioni domino enzimatiche
o Reazioni domino catalizzate da metalli di transizione
La prima reazione domino di un prodotto presente in natura è stata eseguita da Schöpf e
Robinson (Robinson, 1917) (Schopf, Lehmann, & Arnold, 1937): da una miscela di
succindialdeide, metilamina e acido acetondicarbossilico è stato ottenuto il tripinone
biciclico (alcaloide precursore dell’atropina) (fig.38).
Fig. 38. Sintesi del tropi none.
La sintesi del composto 49 ha previsto una reazione domino di coupling Pd-catalizzata
in cui si sono formati in concomitanza legami C-C e legami C-N (si tratta quindi di una
reazione domino catalizzata da metalli di transizione).
O
O
H
HH2N-Me
OO
HO
O
OH
N
O
H3C

68
Il Pd(OAc)2 in presenza di PPh3 forma il complesso Pd(0)(PPh3)2 visto in fig. 27.
Attraverso la transmetallazione, il Pd si inserisce fra l’atomo di bromo e l’anello
aromatico dell’acido 2-bromo-5-metossibenzoico; ottenendo, quindi, l’unione di due
molecole di acido. Grazie all’ambiente basico (K2CO3) e all’alta temperatura si ottiene
il coupling: l’atomo di C aromatico diviene meno elettrondenso a causa della
dislocazione della carica, ciò permette la sostituzione nucleofila aromatica. In più, in
base alla temperatura, si ha la decomposizione dell’intermedio con liberazione di N-
benzilformammide. Questo meccanismo non è riportato in letteratura ed è stato
ipotizzato secondo la fig. 39.
Il vantaggio dell’utilizzo di una reazione domino sta nel fatto che questa è una reazione
one pot in cui il primo prodotto che si forma ha una struttura tale da subire, nelle stesse
condizioni di reazione, un’ulteriore reazione e così via. Il meccanismo che sta alla base
della reazione effettuata è lo stesso della reazione di Heck.
Dai vantaggi della reazione domino però nascono anche alcuni dei suoi problemi: la
consecutività delle reazioni non può essere controllata come in una normale reazione di
Heck e l’ambiente di reazione influenza l’andamento dell’intero processo e non di un
singolo step.
Proprio per quanto riguarda l’ambiente di reazione, sono stati incontrati dei problemi
nella definizione delle condizioni ideali in cui svolgere la domino.
La reazione in questione è stata tentata in due differenti condizioni: in THF a 150°C
(metodo A) e in DMF a 200-250°C (metodo B). Con il metodo A si sono potuti isolare
sia il composto 49 che il composto 50, quest’ultimo altro non è che l’intermedio non
decomposto della reazione domino. Con il metodo B, al contrario,si riporta la sola
presenza del composto 49 come prodotto. La spiegazione a questa differenza è data
dalla temperatura di reazione: il THF e la DMF sono solventi con temperatura di
ebollizione rispettivamente di 65-67°C e 153°C; la DMF, quindi, permette di
raggiungere temperature di reazione più alte del THF. La decomposizione spontanea
necessaria per la trasformazione del composto 50 nel composto 49 è, quindi,
temperatura-dipendente: con il metodo B tale reazione è avvenuta in modo completo.
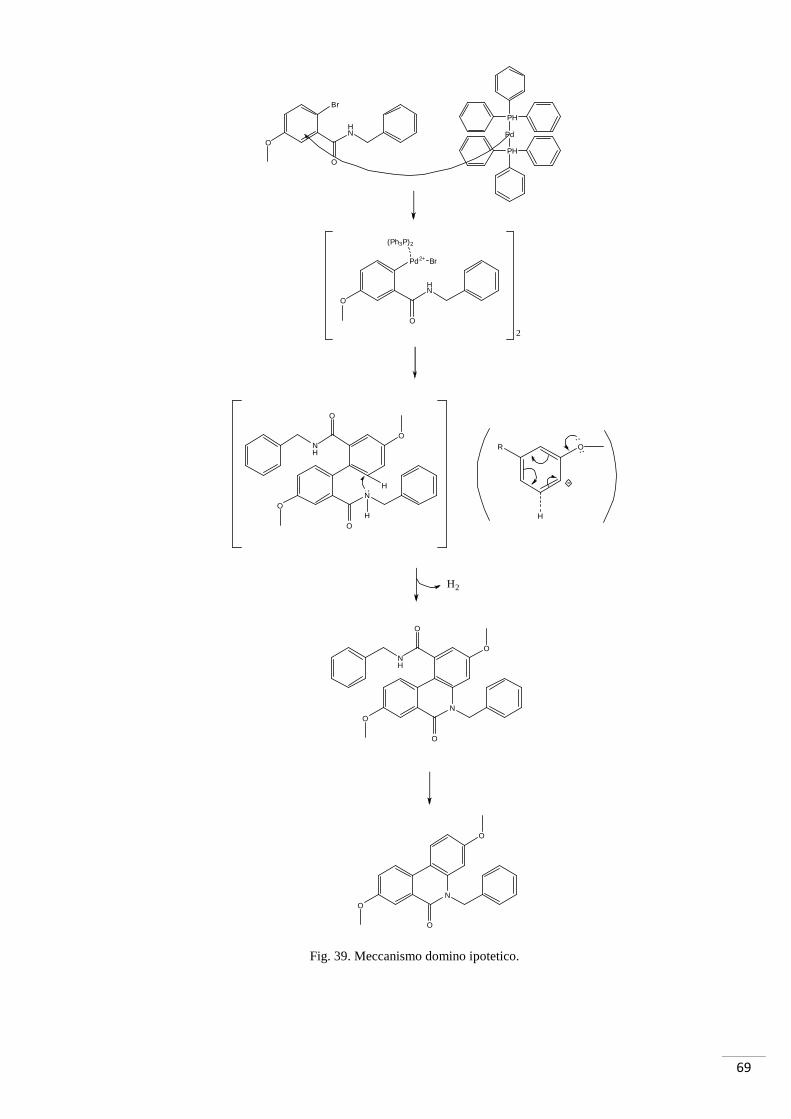
69
Fig. 39. Meccanismo domino ipotetico.
O
Br
HN
O
Pd
PH
PH
O
HN
O
Pd2+ Br
(Ph3P)2
2
ON
O
H
H
ONH
O
O
H
R
ON
O
ONH
O
H2
ON
O
O

70
Con la determinazione della resa è stato possibile fare un ulteriore confronto tra i due
metodi: con il metodo A la resa è stata del 12%, mentre con il metodo B è stata ottenuta
una resa del 56% nettamente superiore alla precedente.
Ottenuto il composto 49 è stato necessario sbloccare gli ossidrili precedentemente
bloccati tramite etossilazione a livello dell’acido 2-bromo-5-metossibenzoico.
L’utilizzo di eteri per proteggere gli idrossili è una pratica molto utilizzata nelle sintesi
organiche. Il metodo più comune per sbloccare dei metossili consiste nell’utilizzo di
acidi di Lewis come BBr3 o AlCl3.
Sono state incontrate delle difficoltà in questa fase della strategia di sintesi; infatti sono
stati effettuati vari tentativi di sblocco per arrivare al composto 24. Un elenco generale
è di seguito riportato:
- BBr3 in benzene anidro (McOmie, Watts, & West, 1968)
- BBr3 in DCM anidro (Punna, Meunier, & Finn, 2004)
- AlCl 3 in etanditiolo (Inaba, Umezawa, Yusa, Mihashi, & H.Itokawa, 1987)
- AlCl 3 in 1-dodecantiolo (Kale, Shinde, Sonar, Shingate, Kumar, & Ghosh,
2010)
Tutti i tentativi di sblocco effettuati con BBr3 hanno dato esito negativo: infatti è stato
visto come il tribromuro di boro causi la debenzilazione di N-benzil e O-benzil gruppi
(Paliakov & Strekowski, 2004).
Nelle varie prove effettuate, ne è stata fatta una direttamente sull’ammide (composto
48) con AlCl3 in etanditiolo; quest’ultimo si prestava come solvente e reattivo. Questa
strategia ha dato esito positivo ed è quindi stata adattata allo sblocco del composto 49: è
stato utilizzato 1-dodecantiolo al posto dell’etanditiolo perché quest’ultimo era difficile
da manipolare dato il suo forte cattivo odore.
Relativamente al meccanismo di sblocco, il metile può diventare un buon gruppo
uscente tramite attivazione con un acido di Lewis (AlCl3); il gruppo uscente (fig.40) si
lega al nucleofilo (tiolo) e, successivamente con agitazione in H2O, si ha la
trasformazione del metossile in ossidrile.
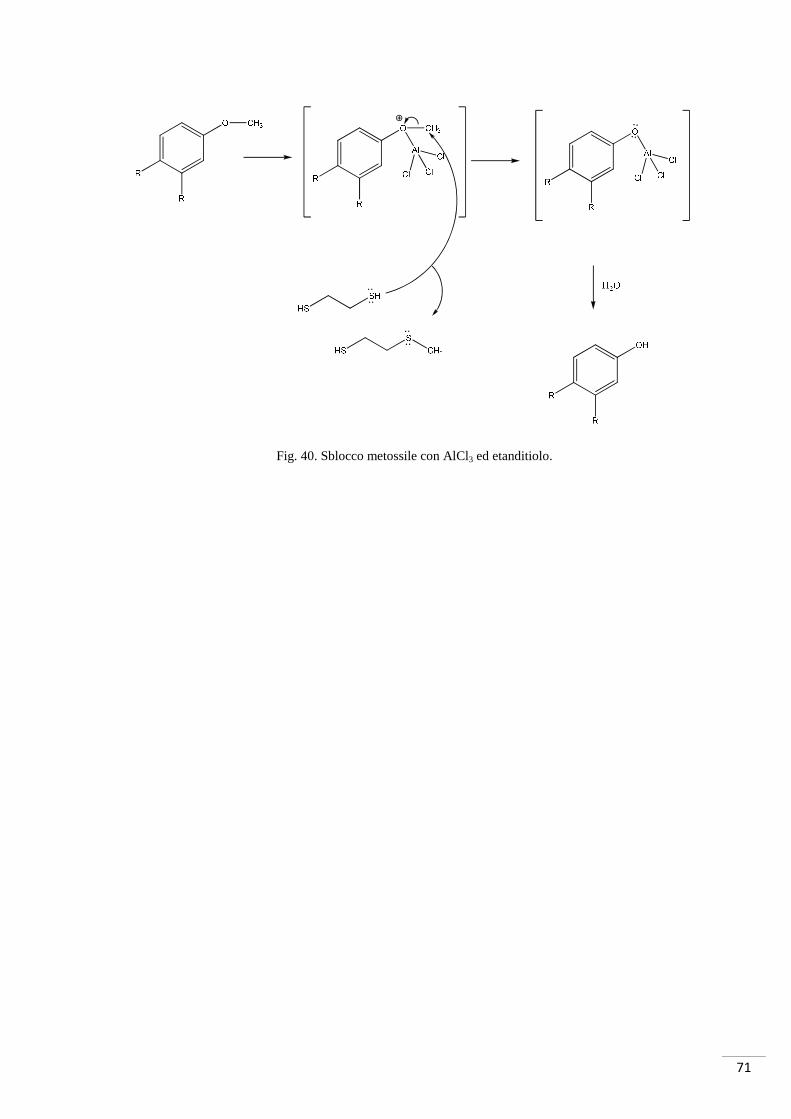
71
Fig. 40. Sblocco metossile con AlCl3 ed etanditiolo.

72
4.5. DISCUSSIONE DEI DATI BIOLOGICI
Il composto 44 (GZ9) è stato testato presso il laboratorio di G. Cozza del Dipartimento
di Biochimica dell’Università di Padova; è stata valutata l’attività inibitoria dei
composti sintetizzati rispetto alla CK2. Il test ha mostrato la seguente attività espressa
come IC50 ([ATP]= 20 µM che corrisponde alla Km della CK2 nei confronti dell’ATP):
IC50 = 31 µM
Si ipotizza che l’attività molto più alta rispetto ai derivati GZ 1-8 (0.2-0.3 µM) sia
dovuta al fatto che questo composto è solo in parte sbloccato. Dagli esperimenti di
docking effettuati da G. Cozza (Università di Padova) su entrambe le varianti (fig. 31a-
b) si è visto che l’estere rimasto è troppo ingombrante e non agevola l’aggancio del
composto all’enzima. Tale osservazione è confermata dal comportamento analogo del
TBCA: questo è un composto molto attivo (0.11 µM), ma diventa inefficace se al posto
dell’acido è presente un estere (>40 µM) (Pagano, et al., 2007).
Il composto 24 è in fase di testing presso il laboratorio sopra citato. Non è pertanto
possibile fornire i dati della sua attività. Dai precedenti risultati ottenuti da composti
simili (caratterizzati da un elevato ingombro sterico) si ipotizza, come già esposto al
punto 2.2 del presente lavoro di tesi, che il composto risulterà inattivo: il sostituente
benzilico dovrebbe impedire l’accesso al sito per il legame dell’ATP.

73
4.6. CONCLUSIONI
Riassumendo i risultati ottenuti:
o Non è stata sintetizzata la 4-bromometilurolitina A. Sono state ideate due
diverse strategie di sintesi ma entrambe sono risultate essere inconcludenti; nel
metodo A non è stata portata a termine la bromurazione del metile in posizione
4 del composto 33, mentre nel metodo B alternativo non è stato ottenuto il 2-
bromometilrsorcinolo.
o Per quanto riguarda il derivato della TBB-propil-diammina, nella reazione di
sblocco per ottenere il composto finale è stato possibile isolare solamente il
mono-derivato GZ9 (composto 44). Tale composto è risultato avere una IC50 di
31 µM sulla CK2; la sua attività bassa è probabilmente dovuta alla presenza
dell’estere che crea ingombro sterico e non facilita l’interazione inibitore-
enzima.
o L’N-derivato benzilato dell’urolitina A (composto 24) è stato sintetizzato
tramite una reazione domino che ha permesso di aumentare la resa totale del
processo. Attualmente i test biochimici su tale composto sono ancora in corso.
Concludendo, le procedure di sintesi utilizzate in questo lavoro hanno portato
all’ottenimento di un solo composto dei tre prefissati, ma sono state utili per
l’ideazione di nuove strategie da utilizzare in lavori futuri. Infatti, visto l’interesse
generale sull’argomento trattato, si può ipotizzare che nel futuro prossimo saranno
continuate le attività di ricerca di nuovi inibitori di CK2. Con il metodo domino
testato in questo lavoro sarà possibile sintetizzare più facilmente nuovi derivati
urolitinici simmetrici; sarà inoltre possibile risolvere il problema di resa bassa che si
aveva con i metodi precedenti (reazione di Hurtley e reazione di Heck). È stato
anche possibile formulare una nuova strategia di sblocco di eteri (in presenza di N-
benzili).
Questo “bagaglio” di reazioni testate potrà essere uno strumento di sostegno e
confronto per chi dovrà sintetizzare composti simili.

74

75
5. PROCEDURE SPERIMENTALI

76
77
HO O
O
Br
5.1. SCHEMA 1
SINTESI DI ACIDO 2-BROMO-5-METOSSIBENZOICO
reagente PM MASSA
(mg) VOLUME
(ml) MOLI (mmol)
densità Eq.
27 152,15 687,00 4,50 1,00
Br2 159,81 72,00 0,23 4,50 3,11 1,00
CH3COOH 6,90
H2O 4,60
prodotto 29 231,05 625,00 2,70
PROCEDIMENTO:
Il composto 27 (687 mg) è stato posto in un pallone in presenza di 4,5 ml di CH3COOH
sotto agitazione. A parte è stata preparata una soluzione di Br2 (0,23 ml), H2O (4,6 ml)e
i rimanenti 2,4 ml di CH3COOH, la quale è stata aggiunta goccia a goccia alla prima
soluzione e portata a riflusso. A reazione terminata la soluzione è diventata limpida, è
stata posta in ghiaccio per ottenere un precipitato. Quest’ultimo è stato filtrato sotto
vuoto e lavato con H2O e metanolo, è stato poi posto in stufa.
Sono stati ottenuti 625 mg del composto 29 puro; resa 60%.
29
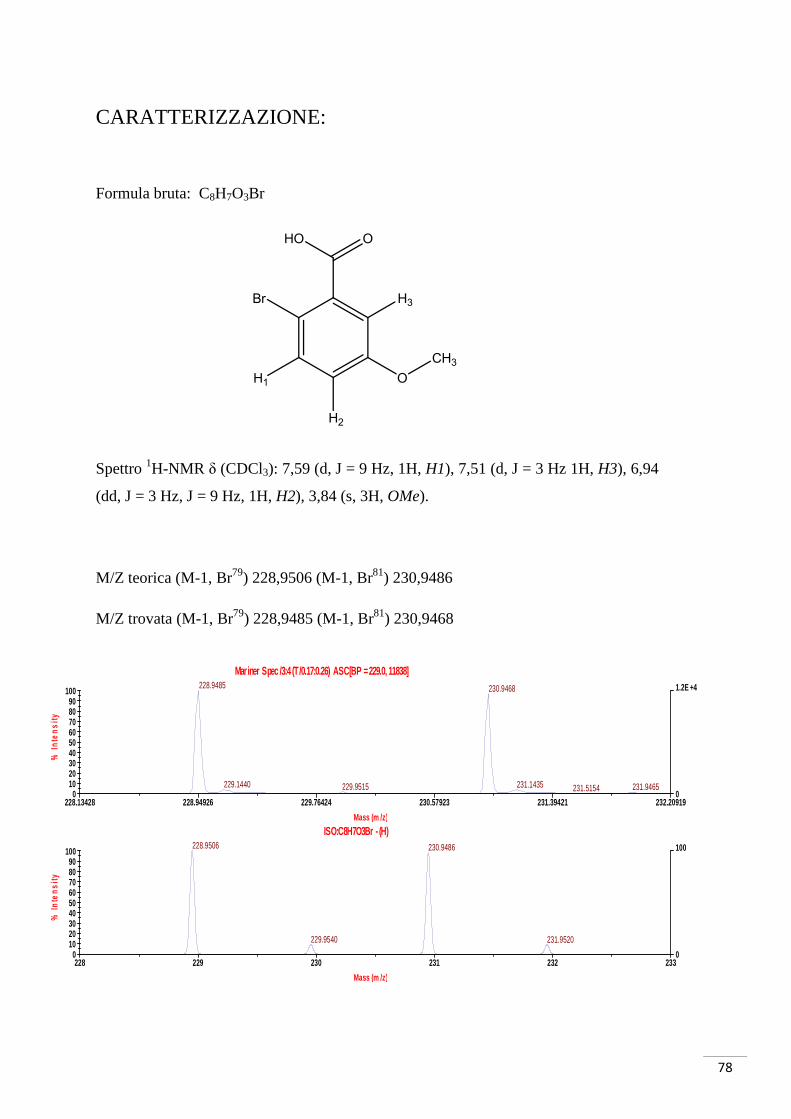
78
228 229 230 231 232 233
Mass (m /z)
0
100
0102030405060708090
100
% In
tens
ity
ISO:C8H7O3Br - (H)228.9506 230.9486
229.9540 231.9520
228.13428 228.94926 229.76424 230.57923 231.39421 232.20919
Mass (m /z)
0
1.2E +4
0102030405060708090
100
% In
tens
ity
Mariner Spec /3:4 (T /0.17:0.26) ASC[BP = 229.0, 11838]228.9485 230.9468
229.1440 231.1435229.9515 231.9465231.5154
CARATTERIZZAZIONE:
Formula bruta: C8H7O3Br
Spettro 1H-NMR δ (CDCl3): 7,59 (d, J = 9 Hz, 1H, H1), 7,51 (d, J = 3 Hz 1H, H3), 6,94
(dd, J = 3 Hz, J = 9 Hz, 1H, H2), 3,84 (s, 3H, OMe).
M/Z teorica (M-1, Br79) 228,9506 (M-1, Br81) 230,9486
M/Z trovata (M-1, Br79) 228,9485 (M-1, Br81) 230,9468
HO O
O
CH3
Br H3
H1
H2
79
SINTESI DI 3-METOSSIBENZIL-2-METILFENOLO
reagente PM MASSA
(g) VOLUME
(ml) MOLI (mmol)
densità Eq.
28 124,14 3,72 30,00 1,00
30 171,03 1,19 10,00 0,33
K2CO3 138,21 2,76 20,00 0,50
acetone 30,00
DMF 95,00
prodotto 31 214,26 1,41 5,33
PROCEDIMENTO:
(Suzuki, Okada, Arai, & Awaji, 2001)
Al composto 28 (3,72 g) posto in un pallone, è stato aggiunto K2CO3 (2,76 mg) e
acetone (30 ml) sotto agitazione; successivamente è stato aggiunto il composto 30 (1,19
ml) lasciando tutta la notte a riflusso a 50°C.
La reazione è stata monitorata tramite TLC n-esano/EtOAc 4:1, una volta terminata
sono stati aggiunti 30 ml di una soluzione 2N di NaOH ed è stata effettuata una
31
HO O

80
estrazione con EtOAc; la fase organica è stata lavata con H2O e con una soluzione
satura di NaCl, poi è stata anidrificata con Na2SO4 per poi filtrarla e portarla a secco.
La TLC sulla soluzione ottenuta dopo la filtrazione ha rivelato la presenza di due
macchie:
RFx1= 0,68 (monosostituito)
RFx2= 0,4 (disostituito)
E’ stata isolata la macchia X1 tramite colonna cromotografica n-esano/EtOAc 4:1; resa
18%.
CARATTERIZZAZIONE:
Formula bruta: C14H14O2
Spettro 1H-NMR δ (CDCl3): 7,51-7,33 (m, 6H, HAr), 7,05 (t, J= 8,2 Hz, 1H, H1), 6,54
(dd, J= 29,9 Hz, J= 8,0 Hz, 2H, HAr), 5,10 (s, 2H, CH2), 2,22 (s, 3H, CH3).
81
32
SINTESI DI 3-(BENZILOSSI)-2-METILFENIL 2-BROMO-5-
METOSSIBENZOATO
reagente PM
MASSA (mg)
VOLUME (ml)
MOLI (mmol)
densità Eq.
31 214,26 468,30
2,18
1,00
29 231,05 500,00
2,16
0,99
DCC
490,20
2,38
1,09
DMAP
52,80
0,43
0,20
DCM
22,00
prodotto 32 427,28 741,00
1,74
PROCEDIMENTO:
(Enkvist, et al., 2006)
A una sospensione del composto 31 (468,3 mg), DCC (490,2 mg) e DMAP (52,8 mg)
in DCM (11 ml) anidro è stata aggiunta una soluzione del composto 29 (500 mg) in
DCM (11 ml) e la reazione è stata posta sotto N2 e agitata a temperatura ambiente per
tutta la notte. L’eventuale precipitato (DCU) che si è formato viene filtrato e la
soluzione rimasta è stata purificata tramite colonna cromatografica toluene/EtOAc 9:1.
Sono stati ottenuti 741 mg del composto 32 puro; resa 81%.
O O O
Br
O

82
CARATTERIZZAZIONE:
Formula bruta: C22H19O4Br
Spettro 1H-NMR δ (CDCl3): 7,65 (d, J= 8,8 Hz, 1H, HAr), 7,62 (d, J= 3,1 Hz, 1H,
HAr), 7,52-7,35 (m, 5H, HAr), 7,25 (t, J= 8,1 Hz, 1H, HAr), 7,01 (dd, J= 8,8 Hz, J= 3,1
Hz, 1H, HAr), 6,90 (d, J= 8,2 Hz, 2H, HAr), 5,16 (s, 2H, CH2), 3,89 (s, 3H, OCH3),
2,26 (s, 3H, CH3-Ar).
O
CH3
O O
Br
O
CH3
H
H
H
H
H
H
H
H
H
H
H
83
33
SINTESI DI 3-(BENZILOSSI)-8-METOSSI-4-METIL-6H-
BENZO[C]CROMAN-6-ONE
reagente PM
MASSA (mg)
VOLUME (ml)
MOLI (mmol)
densità Eq.
32 427,28 741,00
1,74
1,00
Pd(OAc)2 224,51 39,00
0,17
0,10
PPh3 262,30 91,30
0,35
0,20
NaOAc 285,50 285,50
3,48
2,00
DMF
25,00
prodotto 33 346,12 189,00
0,55
PROCEDIMENTO:
(Enkvist, et al., 2006)
Ad una soluzione di Pd(OAc)2 (39 mg) e PPh3 (91,3 mg) in DMF anidra (10 ml) è stata
aggiunta una sospensione del composto 32 (741 mg) e NaOAc in DMF anidra (15 ml)
in un pallone a 3 colli. La reazione è stata posta sotto agitazione a 130°C per tutta la
notte sotto atmosfera di azoto. Successivamente è stata fatta un’estrazione con
EtOAc/H2O, si è anidrificato con Na2SO4 e il solvente è stato rimosso sotto vuoto. Il
composto è stato purificato mediante cristallizzazione con DCM/n-esano.
Sono stati ottenuti 189 mg del composto 33 puro; resa 31,6%.
O O O
O

84
CARATTERIZZAZIONE:
Formula bruta: C22H18O4
Spettro 1H-NMR δ (CDCl3): 7,96 (d, J= 8,9 Hz, 1H, HAr), 7,81-7,76 (m, 2H, HAr),
7,52-7,33 (m, 6H, HAr), 6,95 (d, J= 8,8 Hz, 1H, HAr), 5,21 (s, 2H, CH2), 3,96 (s, 3H,
OCH3), 2,45 (s, 3H, CH3-Ar).
O
CH3
O O
O
CH3
H
H
H
H
H
H
H
H
H
H
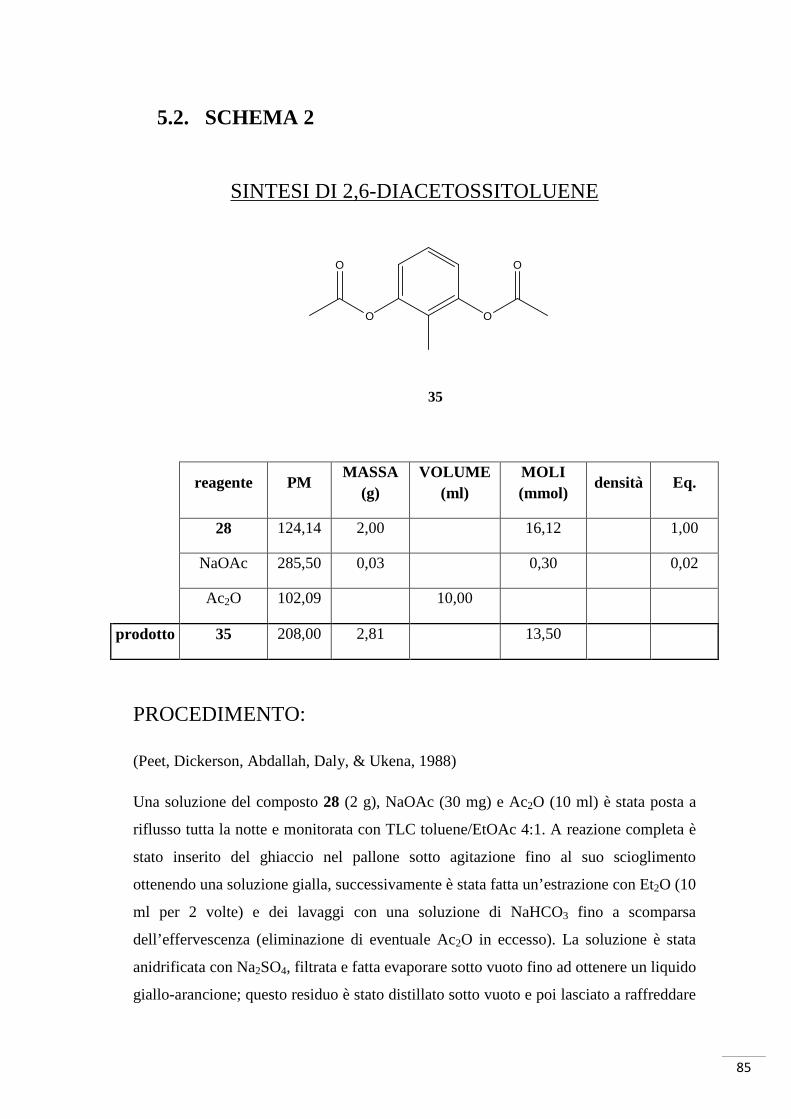
85
5.2. SCHEMA 2
SINTESI DI 2,6-DIACETOSSITOLUENE
reagente PM
MASSA (g)
VOLUME (ml)
MOLI (mmol)
densità Eq.
28 124,14 2,00
16,12
1,00
NaOAc 285,50 0,03
0,30
0,02
Ac2O 102,09
10,00
prodotto 35 208,00 2,81
13,50
PROCEDIMENTO:
(Peet, Dickerson, Abdallah, Daly, & Ukena, 1988)
Una soluzione del composto 28 (2 g), NaOAc (30 mg) e Ac2O (10 ml) è stata posta a
riflusso tutta la notte e monitorata con TLC toluene/EtOAc 4:1. A reazione completa è
stato inserito del ghiaccio nel pallone sotto agitazione fino al suo scioglimento
ottenendo una soluzione gialla, successivamente è stata fatta un’estrazione con Et2O (10
ml per 2 volte) e dei lavaggi con una soluzione di NaHCO3 fino a scomparsa
dell’effervescenza (eliminazione di eventuale Ac2O in eccesso). La soluzione è stata
anidrificata con Na2SO4, filtrata e fatta evaporare sotto vuoto fino ad ottenere un liquido
giallo-arancione; questo residuo è stato distillato sotto vuoto e poi lasciato a raffreddare
OO
OO
35

86
fino alla formazione dei cristalli, i quali sono stati lavati con H2O e messi in stufa ad
asciugare (bisogna fare attenzione a non eccedere con la temperatura in quanto i
cristalli rifondono a 42-43,°C, è stata mantenuta una temperatura minore di 30°C).
Sono stati ottenuti 2,81 g del composto 35 puro; resa 83,8%.
CARATTERIZZAZIONE:
Formula bruta: C11H12O4
Spettro 1H-NMR δ (CDCl3): 7,24 (t, J= 8,1 Hz, 1H, H2), 6,97 (d, J= 8,2 Hz, 2H, H1-2),
2,35 (s, 6H, COOCH3), 2,03 (s, 3H, ArCH3).
OO
OO
H1
H2
H3
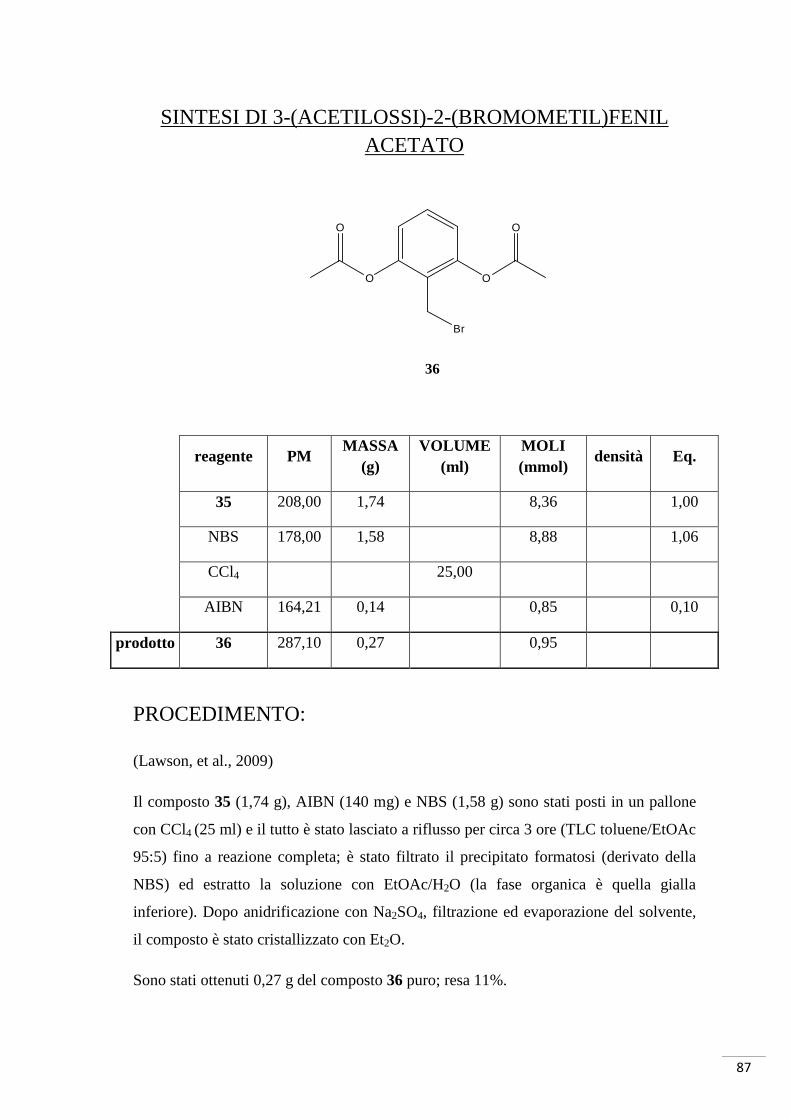
87
SINTESI DI 3-(ACETILOSSI)-2-(BROMOMETIL)FENIL ACETATO
reagente PM
MASSA (g)
VOLUME (ml)
MOLI (mmol)
densità Eq.
35 208,00 1,74
8,36
1,00
NBS 178,00 1,58
8,88
1,06
CCl4
25,00
AIBN 164,21 0,14
0,85
0,10
prodotto 36 287,10 0,27
0,95
PROCEDIMENTO:
(Lawson, et al., 2009)
Il composto 35 (1,74 g), AIBN (140 mg) e NBS (1,58 g) sono stati posti in un pallone
con CCl4 (25 ml) e il tutto è stato lasciato a riflusso per circa 3 ore (TLC toluene/EtOAc
95:5) fino a reazione completa; è stato filtrato il precipitato formatosi (derivato della
NBS) ed estratto la soluzione con EtOAc/H2O (la fase organica è quella gialla
inferiore). Dopo anidrificazione con Na2SO4, filtrazione ed evaporazione del solvente,
il composto è stato cristallizzato con Et2O.
Sono stati ottenuti 0,27 g del composto 36 puro; resa 11%.
OO
OO
Br
36
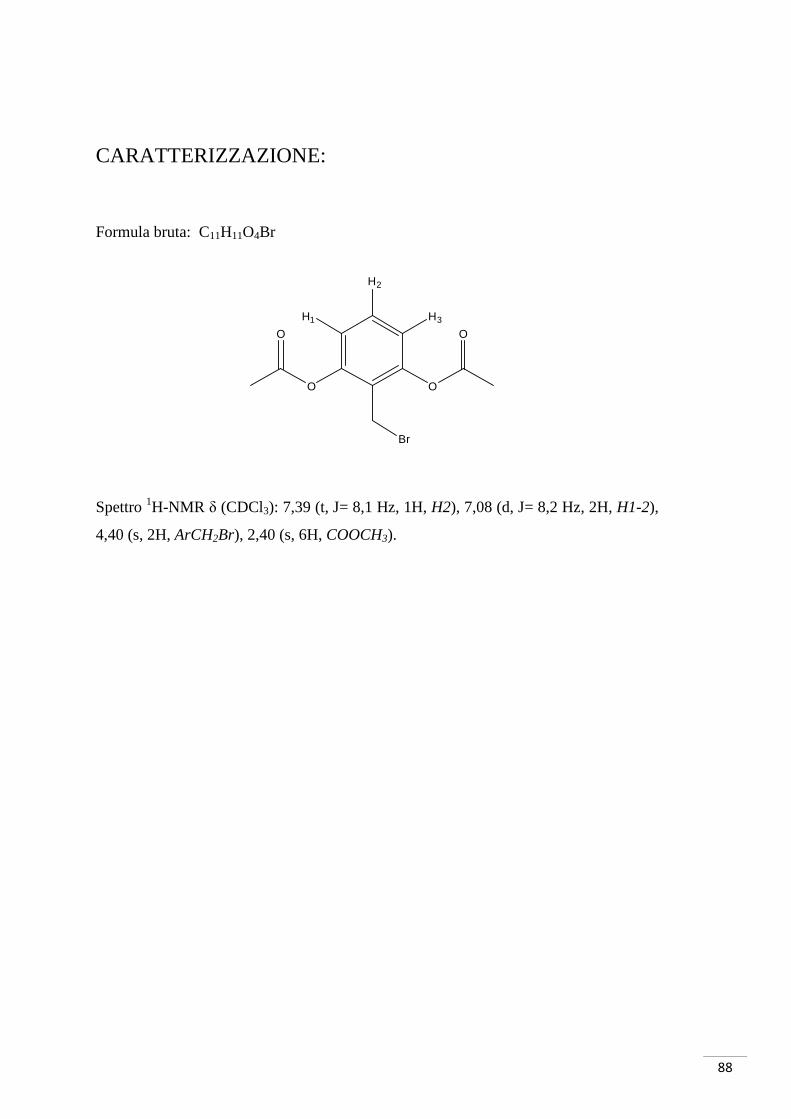
88
CARATTERIZZAZIONE:
Formula bruta: C11H11O4Br
Spettro 1H-NMR δ (CDCl3): 7,39 (t, J= 8,1 Hz, 1H, H2), 7,08 (d, J= 8,2 Hz, 2H, H1-2),
4,40 (s, 2H, ArCH2Br), 2,40 (s, 6H, COOCH3).
OO
OO
Br
H1
H2
H3
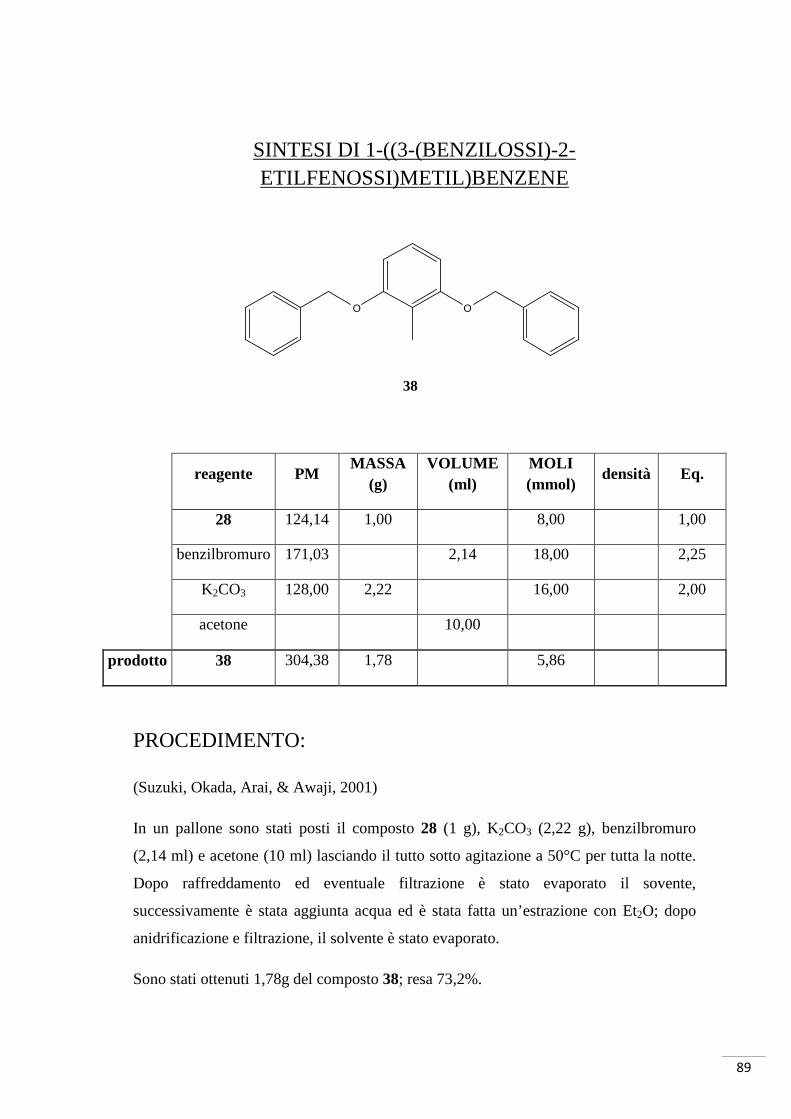
89
SINTESI DI 1-((3-(BENZILOSSI)-2-ETILFENOSSI)METIL)BENZENE
reagente PM
MASSA (g)
VOLUME (ml)
MOLI (mmol)
densità Eq.
28 124,14 1,00
8,00
1,00
benzilbromuro 171,03
2,14 18,00
2,25
K2CO3 128,00 2,22
16,00
2,00
acetone
10,00
prodotto 38 304,38 1,78
5,86
PROCEDIMENTO:
(Suzuki, Okada, Arai, & Awaji, 2001)
In un pallone sono stati posti il composto 28 (1 g), K2CO3 (2,22 g), benzilbromuro
(2,14 ml) e acetone (10 ml) lasciando il tutto sotto agitazione a 50°C per tutta la notte.
Dopo raffreddamento ed eventuale filtrazione è stato evaporato il sovente,
successivamente è stata aggiunta acqua ed è stata fatta un’estrazione con Et2O; dopo
anidrificazione e filtrazione, il solvente è stato evaporato.
Sono stati ottenuti 1,78g del composto 38; resa 73,2%.
OO
38
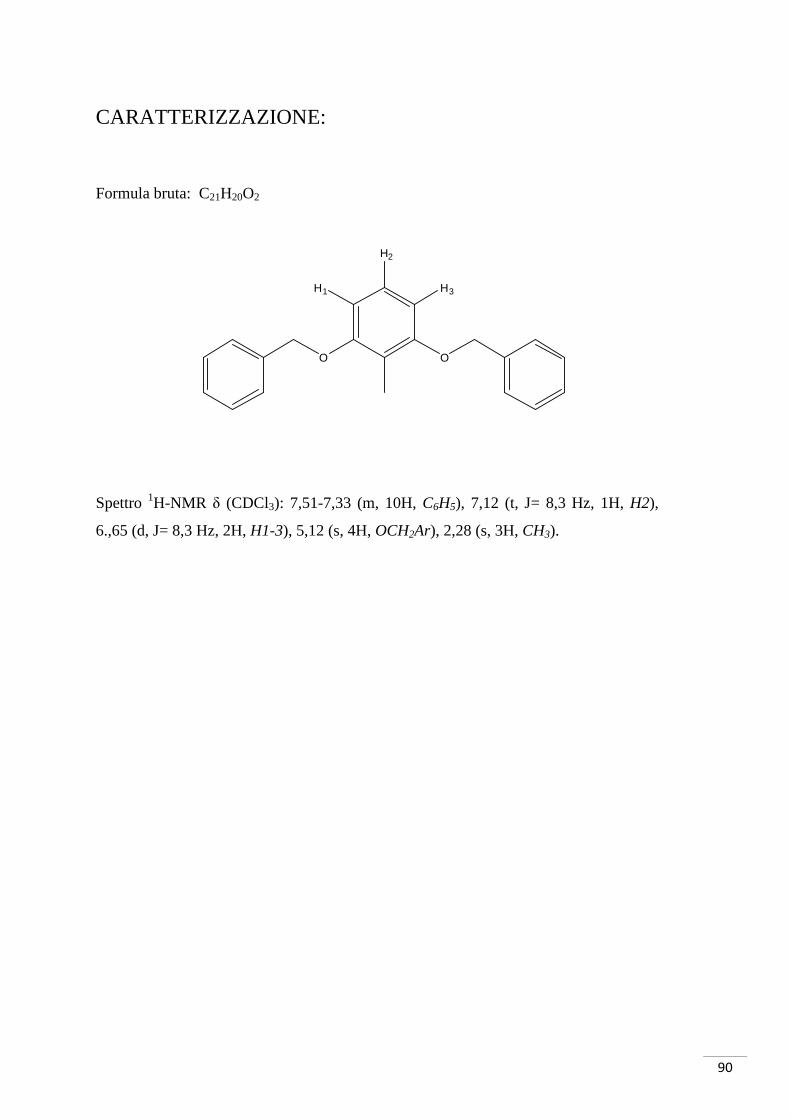
90
CARATTERIZZAZIONE:
Formula bruta: C21H20O2
Spettro 1H-NMR δ (CDCl3): 7,51-7,33 (m, 10H, C6H5), 7,12 (t, J= 8,3 Hz, 1H, H2),
6.,65 (d, J= 8,3 Hz, 2H, H1-3), 5,12 (s, 4H, OCH2Ar), 2,28 (s, 3H, CH3).
OO
H1
H2
H3
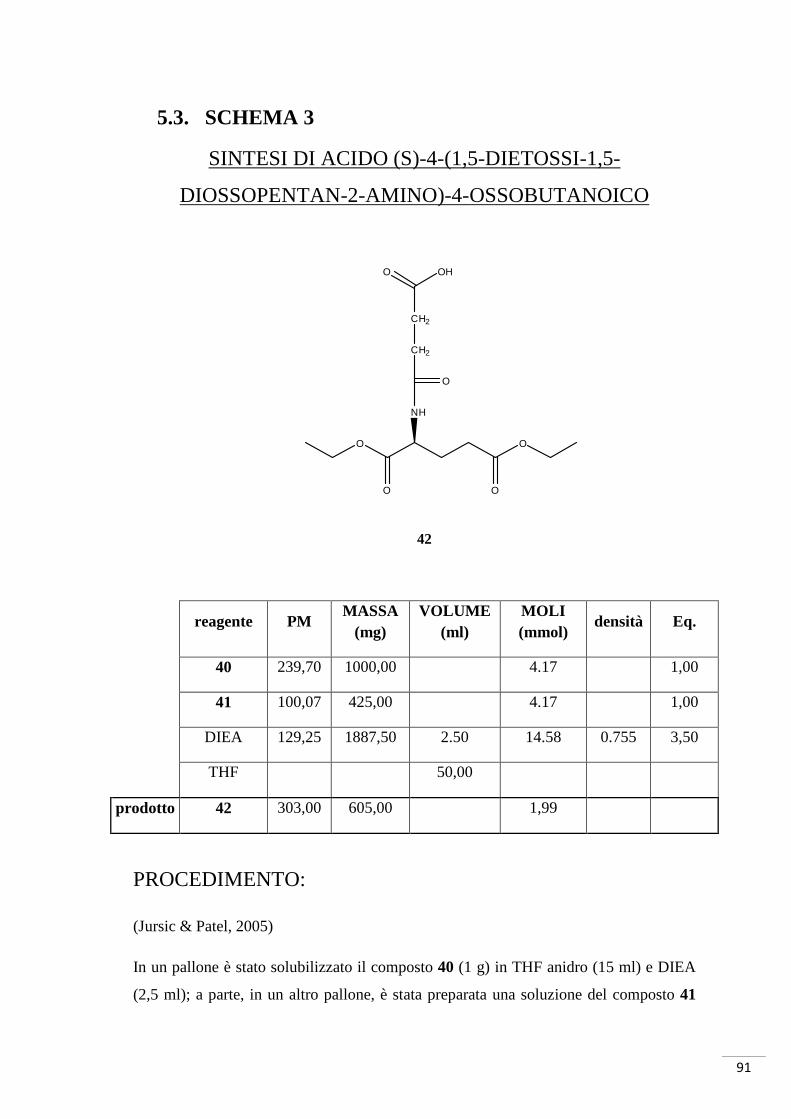
91
5.3. SCHEMA 3
SINTESI DI ACIDO (S)-4-(1,5-DIETOSSI-1,5-
DIOSSOPENTAN-2-AMINO)-4-OSSOBUTANOICO
reagente PM
MASSA (mg)
VOLUME (ml)
MOLI (mmol)
densità Eq.
40 239,70 1000,00
4.17
1,00
41 100,07 425,00
4.17
1,00
DIEA 129,25 1887,50 2.50 14.58 0.755 3,50
THF
50,00
prodotto 42 303,00 605,00
1,99
PROCEDIMENTO:
(Jursic & Patel, 2005)
In un pallone è stato solubilizzato il composto 40 (1 g) in THF anidro (15 ml) e DIEA
(2,5 ml); a parte, in un altro pallone, è stata preparata una soluzione del composto 41
O
O
NH
O
O
CH2
O
CH2
O OH
42

92
(425 mg) in THF anidro (35 ml). Sotto agitazione e a temperatura ambiente la seconda
soluzione è stata aggiunta alla prima con un imbuto gocciolatore in 10-15 minuti; la
reazione è stata lasciata in agitazione per 18 ore e monitorata con TLC CHCl3/MeOH
8:2 in fosfomolibdato. Successivamente, il precipitato è stato eliminato per filtrazione e
il solvente è stato eliminato per evaporazione a pressione ridotta. Quindi, è stata
aggiunta H2O e acidificato con HCl 37% fino a raggiungere pH 1-2. Con un’estrazione
con EtOAc è stato eliminato l’eventuale glutammico non reagito, la fase organica è
stata anidrificata e portata a secco (si ottiene un liquido).
Sono stati ottenuti 0,605g del composto 42 puro; resa 46,28%.
CARATTERIZZAZIONE:
Formula bruta: C13H21NO7
Spettro 1H-NMR δ (CDCl3): 4,51 (t, J= 8,1 Hz, 1H, OCOCHNH), 4,27-4,09 (m, 4H,
HCHR), 2,72 (t, J= 7,1 Hz, 2H, HCHR), 2,41-2,28 (m, 6H, HCHR), 1,17 (t, J= 7,8 Hz,
6H, HCHR).
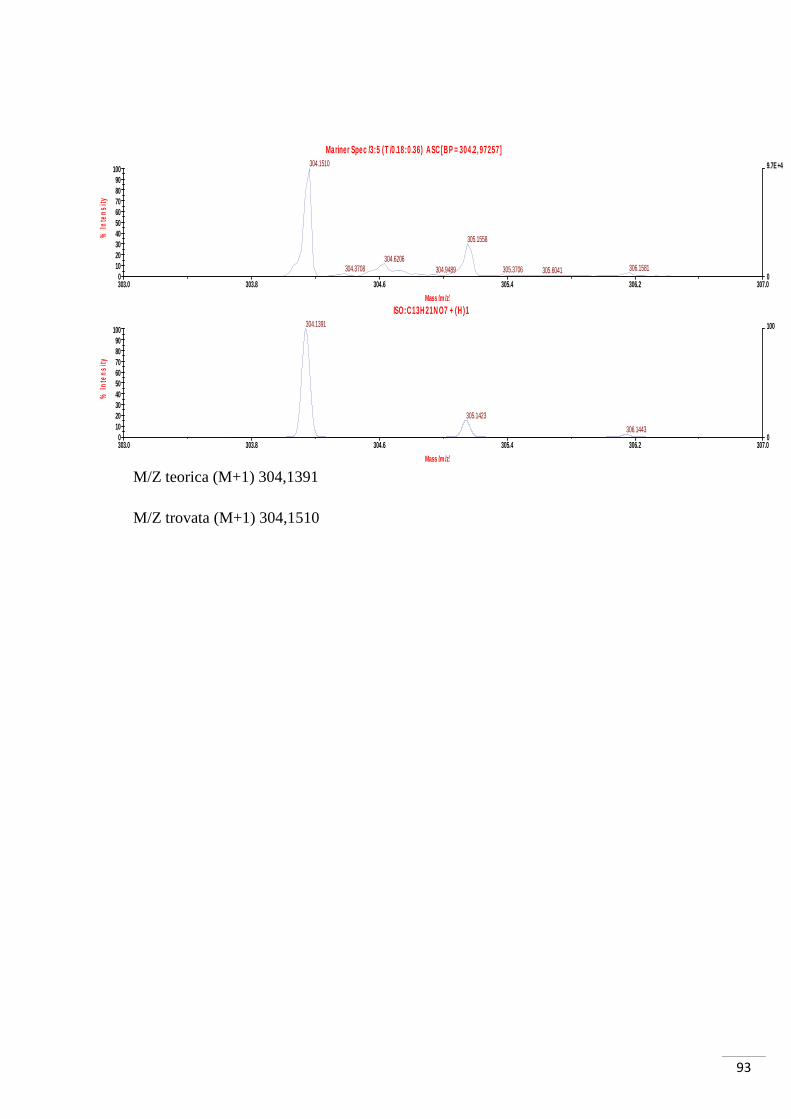
93
303.0 303.8 304.6 305.4 306.2 307.0
Mass (m /z)
0
100
0102030405060708090
100
% In
tens
ity
ISO:C13H 21N O7 + (H )1304.1391
305.1423
306.1443
303.0 303.8 304.6 305.4 306.2 307.0
Mass (m /z)
0
9.7E+4
0102030405060708090
100
% In
tens
ity
Mariner Spec /3:5 (T /0.18:0.36) ASC [BP = 304.2, 97257]304.1510
305.1558
304.6206306.1581304.3708 305.3706304.9489 305.6041
M/Z teorica (M+1) 304,1391
M/Z trovata (M+1) 304,1510
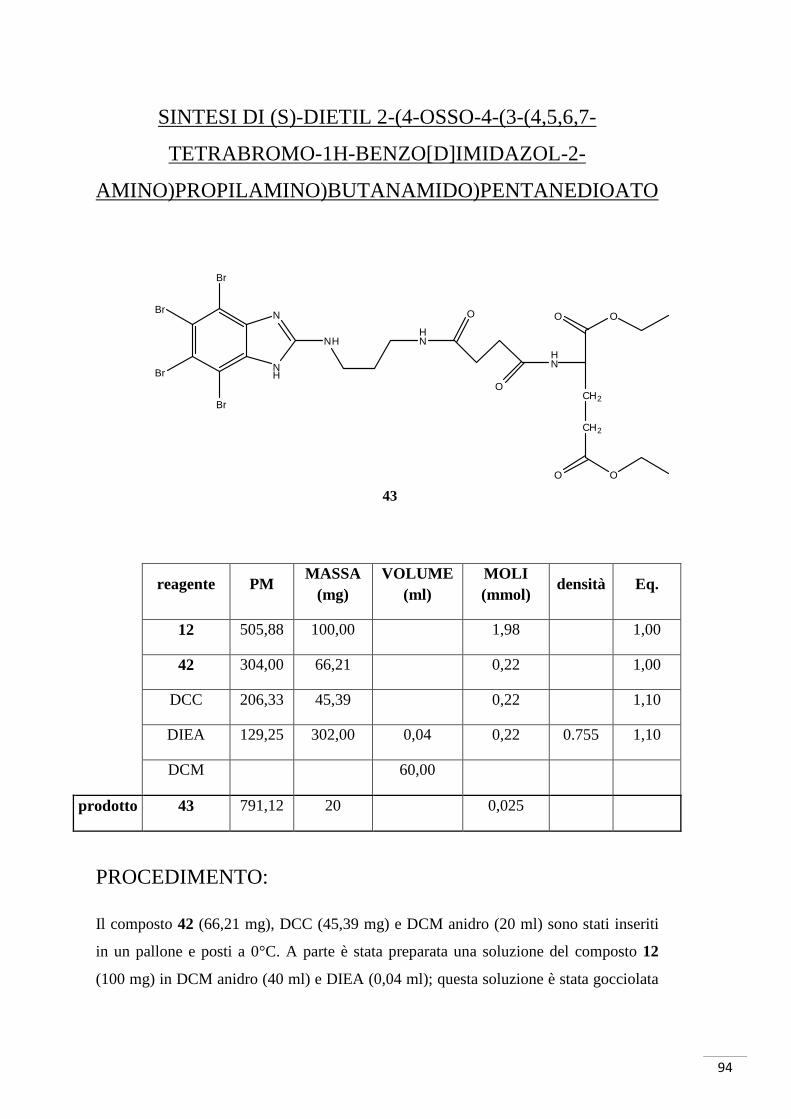
94
SINTESI DI (S)-DIETIL 2-(4-OSSO-4-(3-(4,5,6,7-
TETRABROMO-1H-BENZO[D]IMIDAZOL-2-
AMINO)PROPILAMINO)BUTANAMIDO)PENTANEDIOATO
reagente PM
MASSA (mg)
VOLUME (ml)
MOLI (mmol)
densità Eq.
12 505,88 100,00
1,98
1,00
42 304,00 66,21
0,22
1,00
DCC 206,33 45,39
0,22
1,10
DIEA 129,25 302,00 0,04 0,22 0.755 1,10
DCM
60,00
prodotto 43 791,12 20
0,025
PROCEDIMENTO:
Il composto 42 (66,21 mg), DCC (45,39 mg) e DCM anidro (20 ml) sono stati inseriti
in un pallone e posti a 0°C. A parte è stata preparata una soluzione del composto 12
(100 mg) in DCM anidro (40 ml) e DIEA (0,04 ml); questa soluzione è stata gocciolata
NH
N
Br
Br
Br
Br
NHHN
O
O
HN
OO
CH2
CH2
O O
43
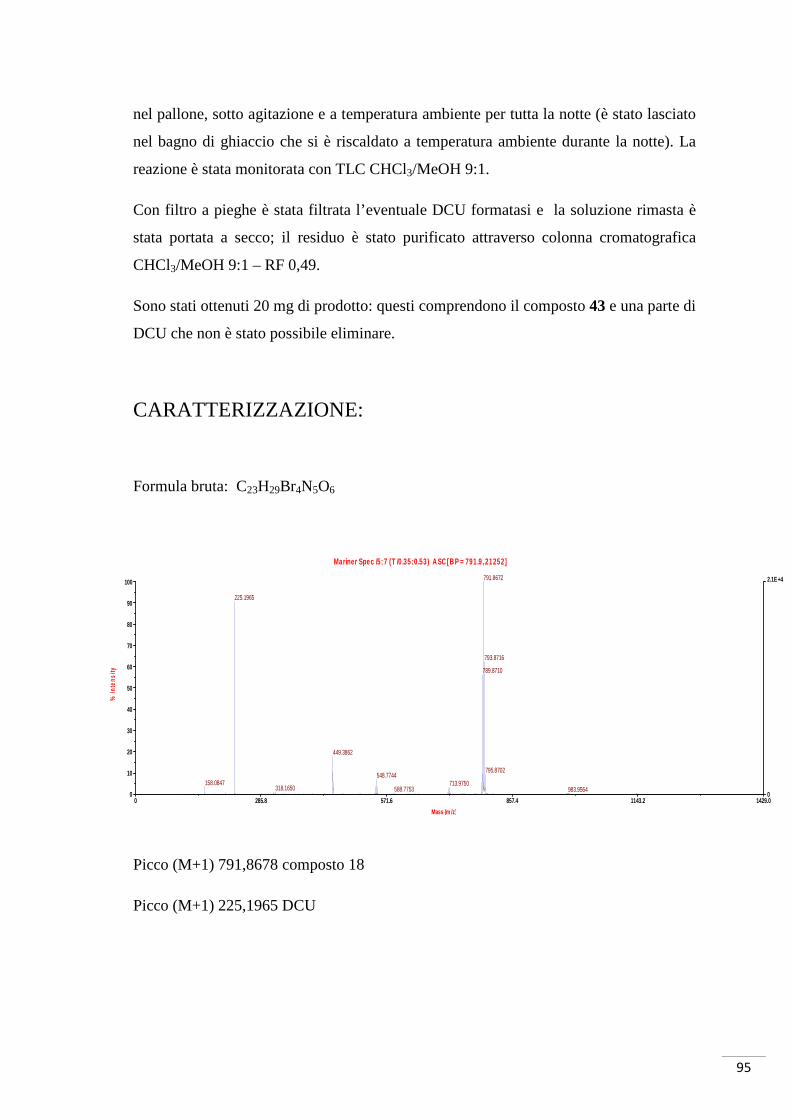
95
nel pallone, sotto agitazione e a temperatura ambiente per tutta la notte (è stato lasciato
nel bagno di ghiaccio che si è riscaldato a temperatura ambiente durante la notte). La
reazione è stata monitorata con TLC CHCl3/MeOH 9:1.
Con filtro a pieghe è stata filtrata l’eventuale DCU formatasi e la soluzione rimasta è
stata portata a secco; il residuo è stato purificato attraverso colonna cromatografica
CHCl3/MeOH 9:1 – RF 0,49.
Sono stati ottenuti 20 mg di prodotto: questi comprendono il composto 43 e una parte di
DCU che non è stato possibile eliminare.
CARATTERIZZAZIONE:
Formula bruta: C23H29Br4N5O6
Picco (M+1) 791,8678 composto 18
Picco (M+1) 225,1965 DCU
0 285.8 571.6 857.4 1143.2 1429.0
Mass (m /z)
0
2.1E+4
0
10
20
30
40
50
60
70
80
90
100
% In
tens
ity
Mariner Spec /5:7 (T /0 .35:0.53) A SC [B P = 791.9, 21252]
791.8672
225.1965
793.8716
789.8710
449.3862
795.8702548.7744
158.0847 713.9750318.1650 588.7753 983.9564
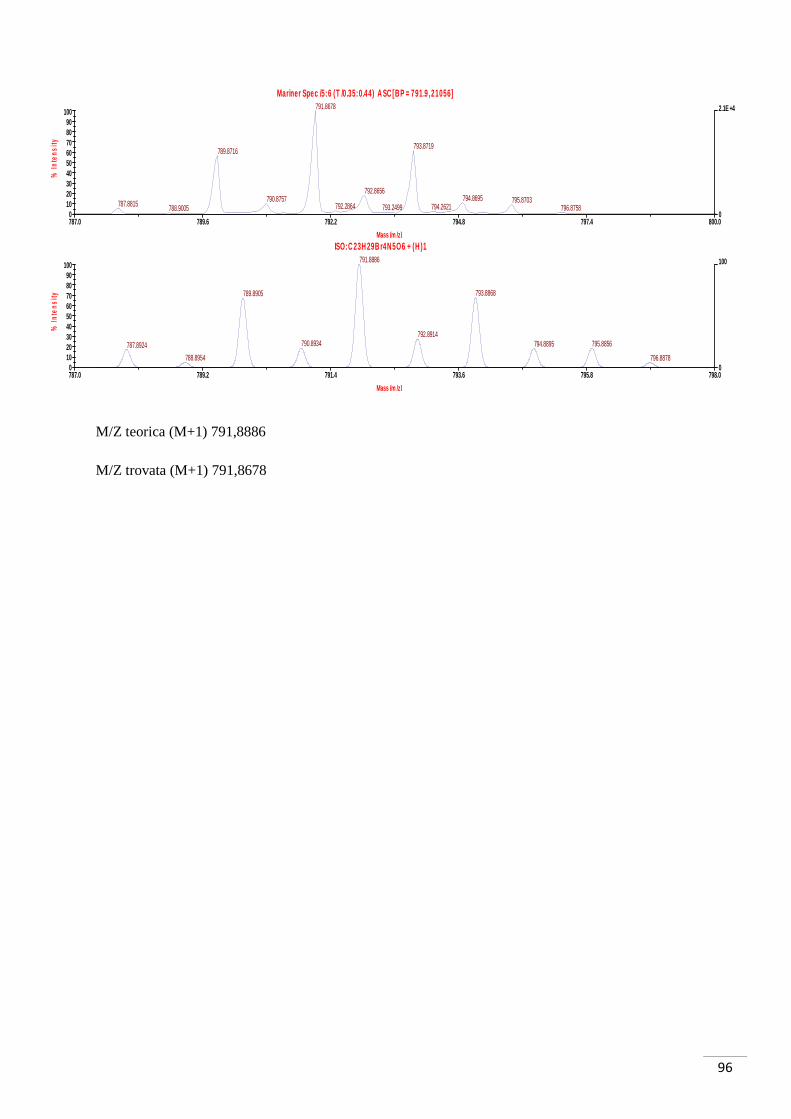
96
787.0 789.2 791.4 793.6 795.8 798.0
Mass (m /z)
0
100
0102030405060708090
100
% In
tens
ity
ISO:C 23H 29B r4N5O6 + (H )1791.8886
793.8868789.8905
792.8914790.8934 795.8856794.8895787.8924
788.8954 796.8878
787.0 789.6 792.2 794.8 797.4 800.0
Mass (m /z)
0
2.1E +4
0102030405060708090
100
% In
tens
ity
Mariner Spec /5:6 (T /0.35:0.44) A SC [BP = 791.9 , 21056]791.8678
793.8719789.8716
792.8656794.8695790.8757 795.8703787.8815 792.2864 794.2621793.2499 796.8758788.9005
M/Z teorica (M+1) 791,8886
M/Z trovata (M+1) 791,8678
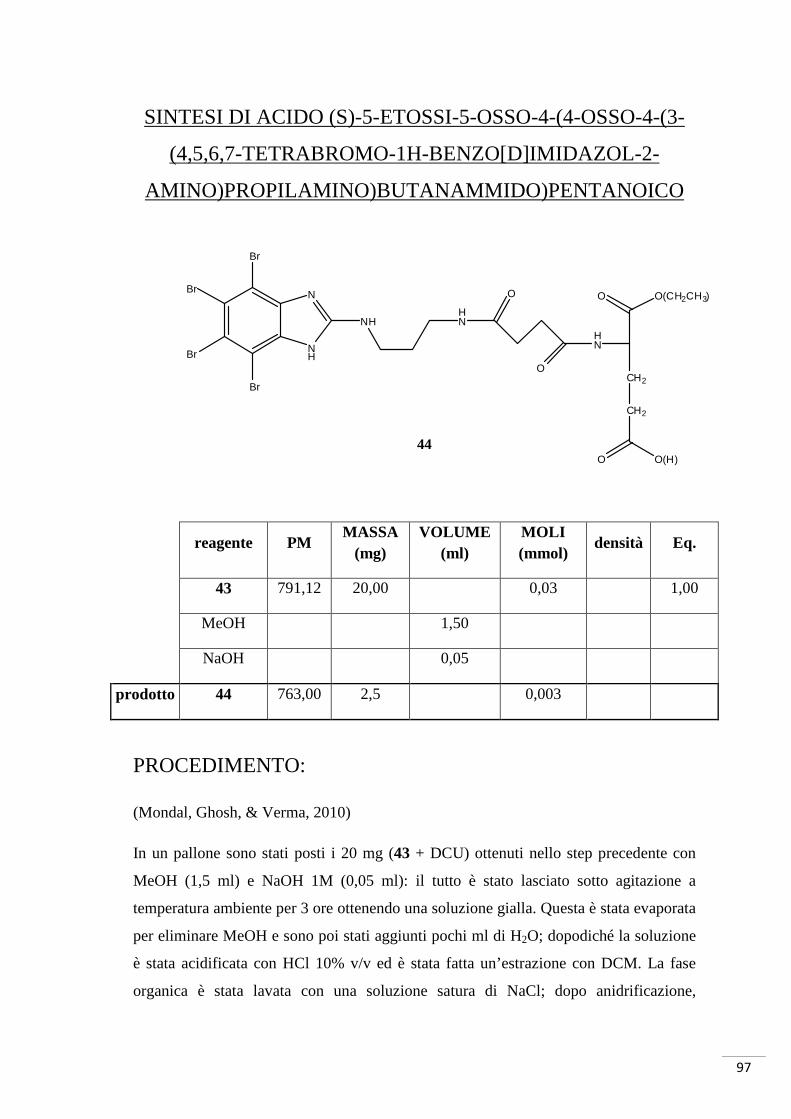
97
SINTESI DI ACIDO (S)-5-ETOSSI-5-OSSO-4-(4-OSSO-4-(3-
(4,5,6,7-TETRABROMO-1H-BENZO[D]IMIDAZOL-2-
AMINO)PROPILAMINO)BUTANAMMIDO)PENTANOICO
reagente PM
MASSA (mg)
VOLUME (ml)
MOLI (mmol)
densità Eq.
43 791,12 20,00
0,03
1,00
MeOH
1,50
NaOH
0,05
prodotto 44 763,00 2,5
0,003
PROCEDIMENTO:
(Mondal, Ghosh, & Verma, 2010)
In un pallone sono stati posti i 20 mg (43 + DCU) ottenuti nello step precedente con
MeOH (1,5 ml) e NaOH 1M (0,05 ml): il tutto è stato lasciato sotto agitazione a
temperatura ambiente per 3 ore ottenendo una soluzione gialla. Questa è stata evaporata
per eliminare MeOH e sono poi stati aggiunti pochi ml di H2O; dopodiché la soluzione
è stata acidificata con HCl 10% v/v ed è stata fatta un’estrazione con DCM. La fase
organica è stata lavata con una soluzione satura di NaCl; dopo anidrificazione,
NH
N
Br
Br
Br
Br
NHHN
O
O
HN
O(CH2CH3)O
CH2
CH2
O O(H)44
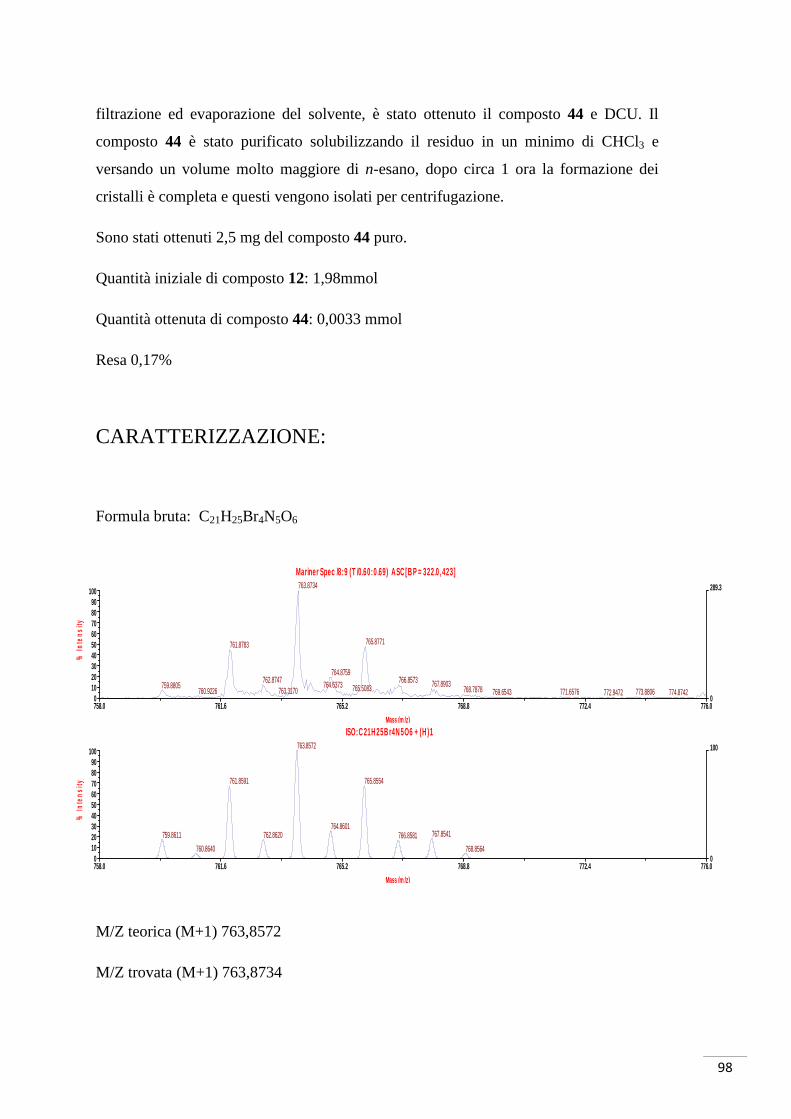
98
filtrazione ed evaporazione del solvente, è stato ottenuto il composto 44 e DCU. Il
composto 44 è stato purificato solubilizzando il residuo in un minimo di CHCl3 e
versando un volume molto maggiore di n-esano, dopo circa 1 ora la formazione dei
cristalli è completa e questi vengono isolati per centrifugazione.
Sono stati ottenuti 2,5 mg del composto 44 puro.
Quantità iniziale di composto 12: 1,98mmol
Quantità ottenuta di composto 44: 0,0033 mmol
Resa 0,17%
CARATTERIZZAZIONE:
Formula bruta: C21H25Br4N5O6
M/Z teorica (M+1) 763,8572
M/Z trovata (M+1) 763,8734
758.0 761.6 765.2 768.8 772.4 776.0
Mass (m /z)
0
100
0102030405060708090
100
% In
tens
ity
ISO:C 21H 25B r4N 5O6 + (H )1763.8572
761.8591 765.8554
764.8601767.8541762.8620759.8611 766.8581
760.8640 768.8564
758.0 761.6 765.2 768.8 772.4 776.0
Mass (m /z)
0
289.3
0102030405060708090
100
% In
tens
ity
Mariner Spec /8:9 (T /0.60:0.69) ASC [B P = 322.0, 423]763.8734
765.8771761.8783
764.8759766.8573762.8747 767.8903764.6373759.8805 765.5083 768.7878763.3170760.9226 773.8806771.6576769.6543 774.8742772.9472
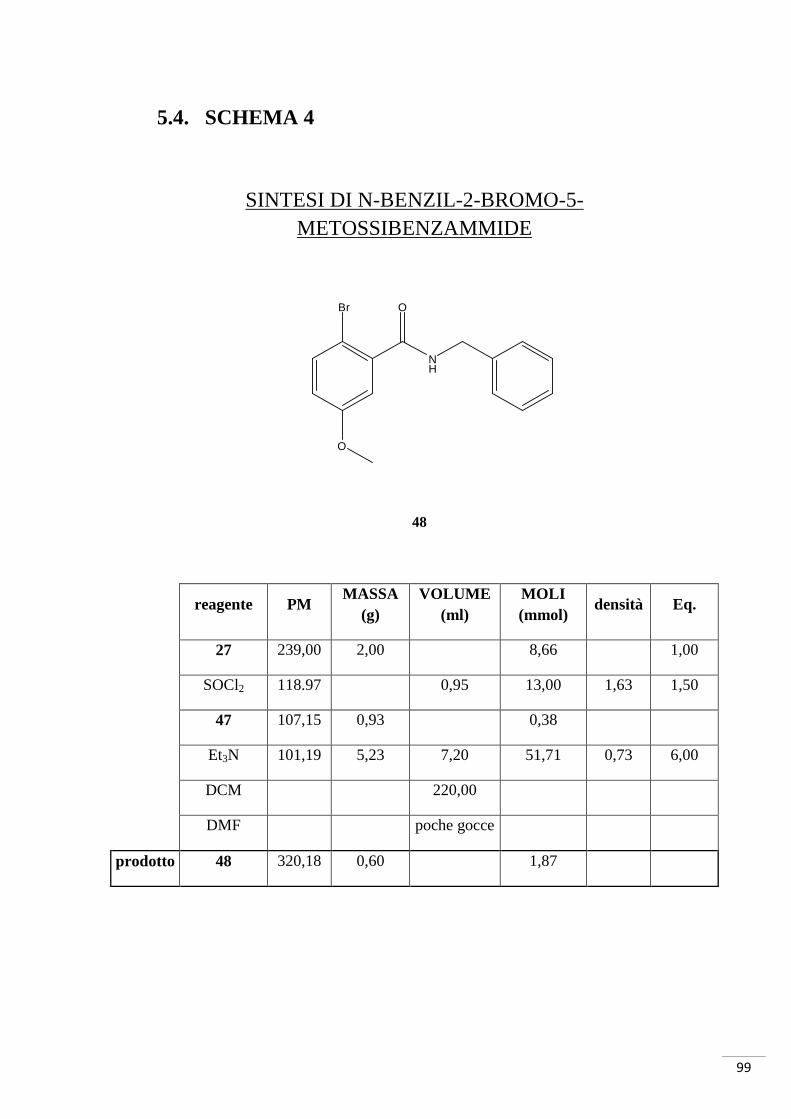
99
5.4. SCHEMA 4
SINTESI DI N-BENZIL-2-BROMO-5-METOSSIBENZAMMIDE
reagente PM
MASSA (g)
VOLUME (ml)
MOLI (mmol)
densità Eq.
27 239,00 2,00
8,66
1,00
SOCl2 118.97
0,95 13,00 1,63 1,50
47 107,15 0,93
0,38
Et3N 101,19 5,23 7,20 51,71 0,73 6,00
DCM
220,00
DMF
poche gocce
prodotto 48 320,18 0,60
1,87
Br
O
NH
O
48

100
PROCEDIMENTO:
(Furuta, Kitamura, Hashimoto, Fuji, Tanaka, & Kan, 2007)
• FASE 1: ATTIVAZIONE ACIDO
Il composto 27 (2 g) è stato posto in un pallone con DCM anidro (120 ml), SOCl2 (0,95
ml) e alcune gocce di DMF a 150°C per circa 3 ore fino a scomparsa del composto di
partenza (TLC DCM/MeOH 4:1, il campione è stato preparato con l’aggiunta di EtOH
che produceva un estere in situ). Il tutto è stato lasciato raffreddare a temperatura
ambiente e poi messo in bagno di ghiaccio, sono stati aggiunti 6 ml di Et3N sotto
agitazione ottenendo una soluzione marrone.
• FASE 2: FORMAZIONE AMMIDE
È stata preparata una soluzione del composto 47 in DCM anidro (100 ml) e Et3N (1,2
ml); questa è stata gocciolata con apposito imbuto nel pallone della fase 1 in bagno di
ghiaccio.
La reazione è stata lasciata sotto agitazione tutta la notte e monitorata con TLC
CHCl3/MeOH 8:2.
Successivamente, la soluzione è stata portata a secco e con colonna cromatografica è
stato isolato il composto con RF 0,82 (con altre 2 impurezze troppo vicine per
separarle); il residuo è stato cristallizzato con CHCl3/n-esano, filtrato con bukner e
lavato con n-esano e acqua.
Sono stati ottenuti 597 mg del composto 48 puro (cristalli giallo-marroni); resa 21,5%.
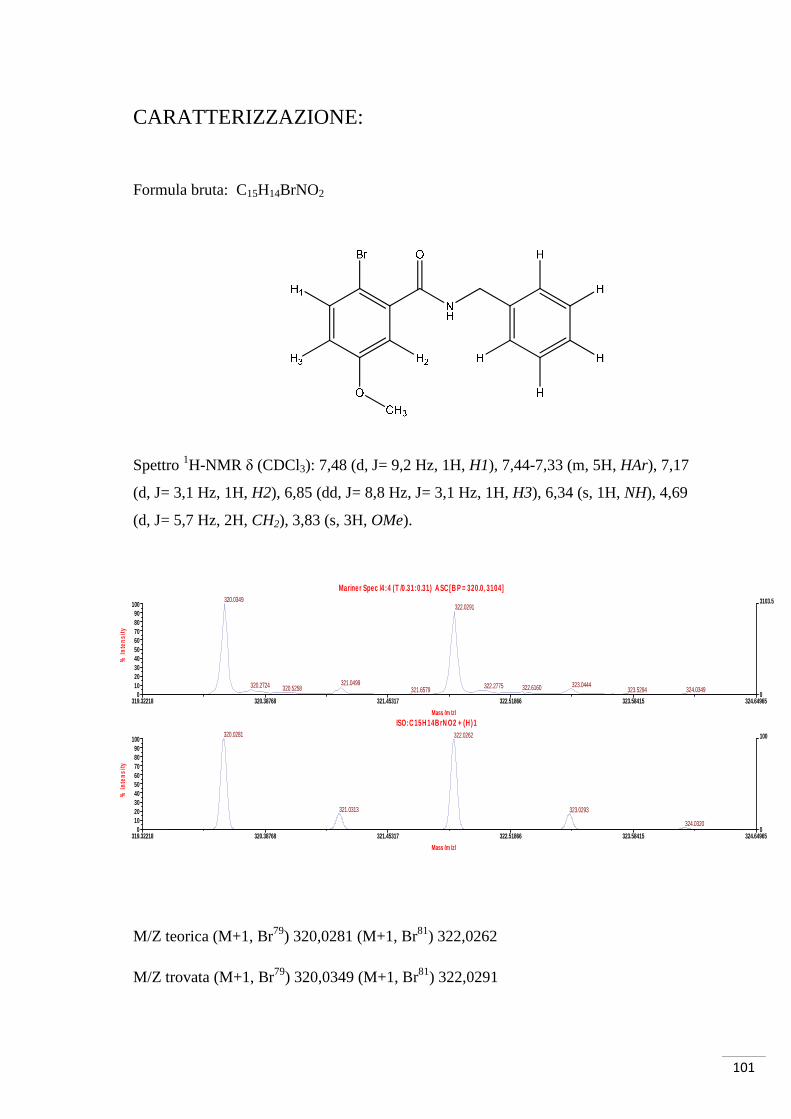
101
319.32218 320.38768 321.45317 322.51866 323.58415 324.64965
Mass (m /z)
0
100
0102030405060708090
100
% In
tens
ity
ISO:C 15H 14B rN O2 + (H )1320.0281 322.0262
321.0313 323.0293
324.0320
319.32218 320.38768 321.45317 322.51866 323.58415 324.64965
Mass (m /z)
0
3103.5
0102030405060708090
100
% In
tens
ity
Mariner Spec /4 :4 (T /0.31:0.31) A SC [B P = 320.0 , 3104]320.0349
322.0291
321.0499 323.0444320.2724 322.2775 322.6160320.5258 324.0349323.5294321.6579
CARATTERIZZAZIONE:
Formula bruta: C15H14BrNO2
Spettro 1H-NMR δ (CDCl3): 7,48 (d, J= 9,2 Hz, 1H, H1), 7,44-7,33 (m, 5H, HAr), 7,17
(d, J= 3,1 Hz, 1H, H2), 6,85 (dd, J= 8,8 Hz, J= 3,1 Hz, 1H, H3), 6,34 (s, 1H, NH), 4,69
(d, J= 5,7 Hz, 2H, CH2), 3,83 (s, 3H, OMe).
M/Z teorica (M+1, Br79) 320,0281 (M+1, Br81) 322,0262
M/Z trovata (M+1, Br79) 320,0349 (M+1, Br81) 322,0291
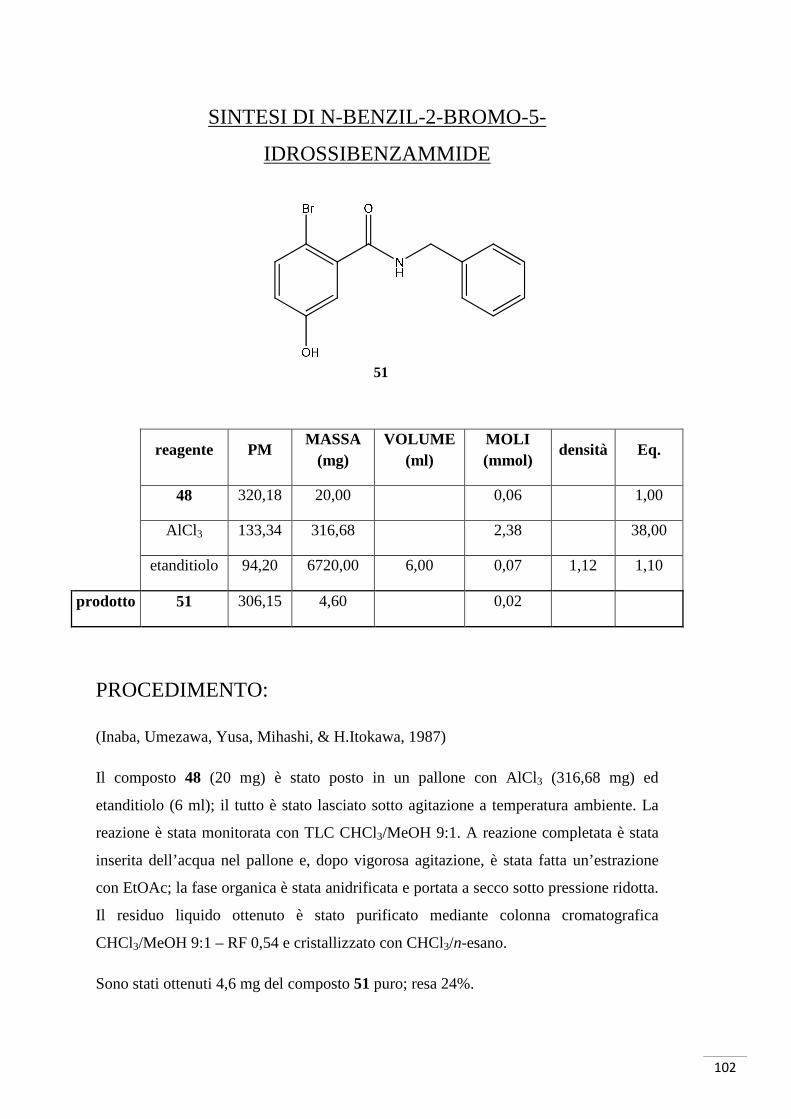
102
SINTESI DI N-BENZIL-2-BROMO-5-
IDROSSIBENZAMMIDE
reagente PM
MASSA (mg)
VOLUME (ml)
MOLI (mmol)
densità Eq.
48 320,18 20,00
0,06
1,00
AlCl 3 133,34 316,68
2,38
38,00
etanditiolo 94,20 6720,00 6,00 0,07 1,12 1,10
prodotto 51 306,15 4,60
0,02
PROCEDIMENTO:
(Inaba, Umezawa, Yusa, Mihashi, & H.Itokawa, 1987)
Il composto 48 (20 mg) è stato posto in un pallone con AlCl3 (316,68 mg) ed
etanditiolo (6 ml); il tutto è stato lasciato sotto agitazione a temperatura ambiente. La
reazione è stata monitorata con TLC CHCl3/MeOH 9:1. A reazione completata è stata
inserita dell’acqua nel pallone e, dopo vigorosa agitazione, è stata fatta un’estrazione
con EtOAc; la fase organica è stata anidrificata e portata a secco sotto pressione ridotta.
Il residuo liquido ottenuto è stato purificato mediante colonna cromatografica
CHCl3/MeOH 9:1 – RF 0,54 e cristallizzato con CHCl3/n-esano.
Sono stati ottenuti 4,6 mg del composto 51 puro; resa 24%.
51
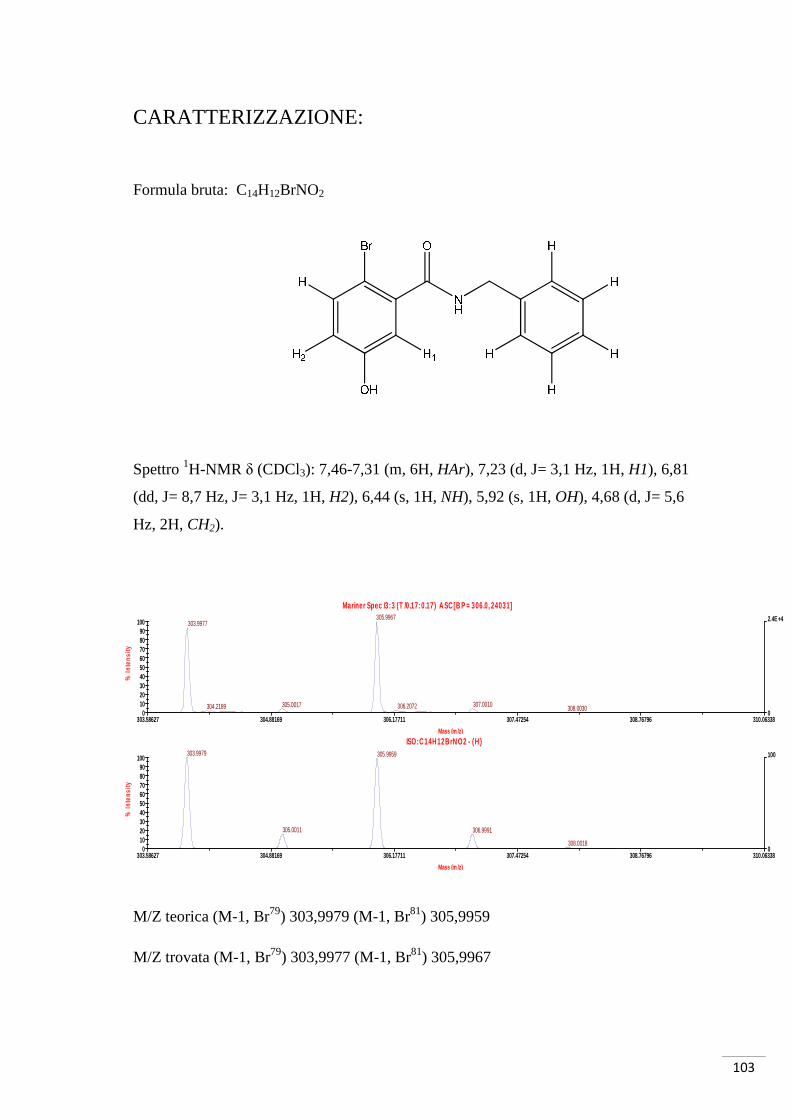
103
303.58627 304.88169 306.17711 307.47254 308.76796 310.06338
Mass (m /z)
0
100
0102030405060708090
100
% In
tens
ity
ISO:C 14H 12B rN O2 - (H )303.9979 305.9959
305.0011 306.9991
308.0018
303.58627 304.88169 306.17711 307.47254 308.76796 310.06338
Mass (m /z)
0
2.4E+4
0102030405060708090
100
% In
tens
ity
Mariner Spec /3 :3 (T /0.17:0.17) A SC [B P = 306.0, 24031]305.9967
303.9977
307.0010305.0017 306.2072304.2189 308.0030
CARATTERIZZAZIONE:
Formula bruta: C14H12BrNO2
Spettro 1H-NMR δ (CDCl3): 7,46-7,31 (m, 6H, HAr), 7,23 (d, J= 3,1 Hz, 1H, H1), 6,81
(dd, J= 8,7 Hz, J= 3,1 Hz, 1H, H2), 6,44 (s, 1H, NH), 5,92 (s, 1H, OH), 4,68 (d, J= 5,6
Hz, 2H, CH2).
M/Z teorica (M-1, Br79) 303,9979 (M-1, Br81) 305,9959
M/Z trovata (M-1, Br79) 303,9977 (M-1, Br81) 305,9967
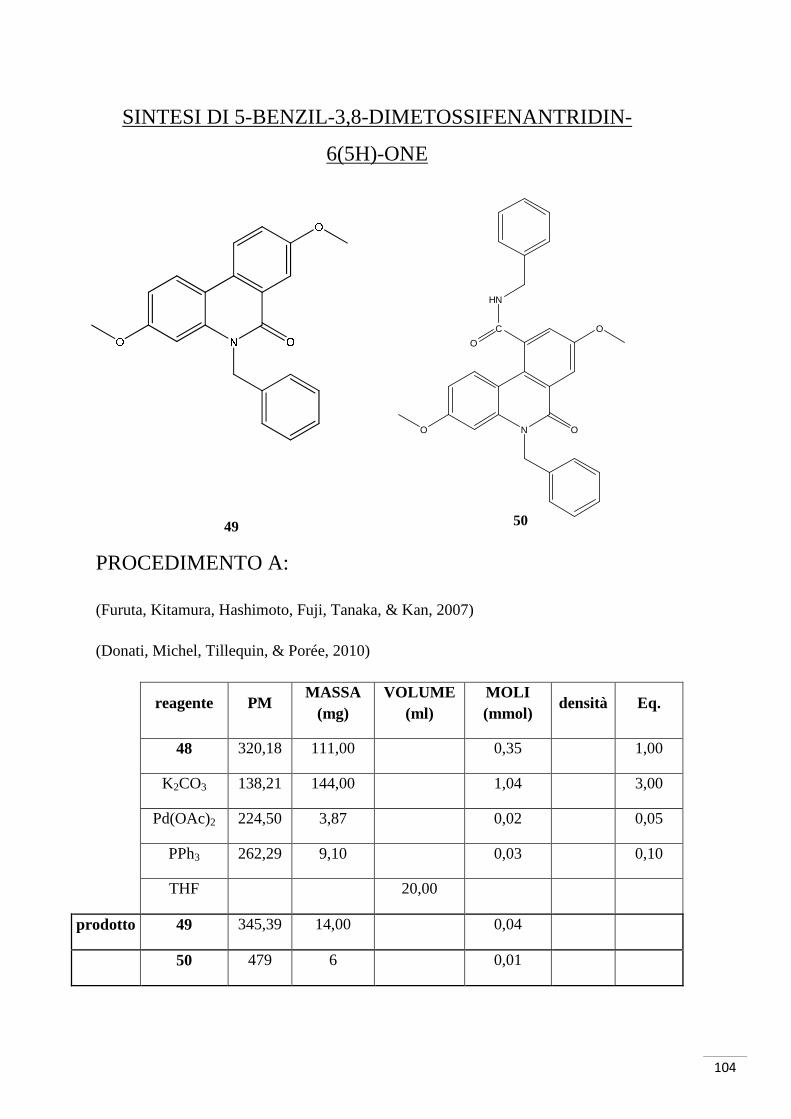
104
SINTESI DI 5-BENZIL-3,8-DIMETOSSIFENANTRIDIN-
6(5H)-ONE
PROCEDIMENTO A:
(Furuta, Kitamura, Hashimoto, Fuji, Tanaka, & Kan, 2007)
(Donati, Michel, Tillequin, & Porée, 2010)
reagente PM
MASSA (mg)
VOLUME (ml)
MOLI (mmol)
densità Eq.
48 320,18 111,00
0,35
1,00
K2CO3 138,21 144,00
1,04
3,00
Pd(OAc)2 224,50 3,87
0,02
0,05
PPh3 262,29 9,10
0,03
0,10
THF
20,00
prodotto 49 345,39 14,00
0,04
50 479 6
0,01
49 50
NO
O
O
C
HN
O
105
In un pallone sono stati inseriti il composto 48 (111 mg), K2CO3 (144 mg), Pd(OAc)2
(3,87 mg), PPh3 (9,10 mg) e THF anidro (20ml). E’ stato portato a riflusso a 150°C
sotto agitazione in atmosfera di azoto (TLC n-esano/EtOAc 7:3). Dopo circa 4 ore la
soluzione è stata portata a secco (nonostante la presenza del composto di partenza), è
stata fatta un’estrazione con DCM/H2O e lavaggi con una soluzione satura di NaCl. La
fase organica è stata anidrificata ed evaporata a pressione ridotta. Il residuo è stato
purificato tramite colonna cromatografica n-esano/EtOAc 7:3:
RF composto 21 – 0,34
RF composto 22 – 0,47
RF composto 23 – 0,16
Sono stati ottenuti 14 mg del composto 49 (resa 11,7%) e 6 mg del composto 50 (resa
3,61%).
Sono stati recuperati inoltre 66 mg del composto 48 non reagiti.
PROCEDIMENTO B:
(Donati, Michel, Tillequin, & Porée, 2010)
reagente PM
MASSA (mg)
VOLUME (ml)
MOLI (mmol)
densità Eq.
48 320,18 1000,00
3.13
1,00
K2CO3 138,21 1340,00
9.73
3,10
Pd(OAc)2 224,50 79,50
0.35
0,11
PPh3 262,29 185,50
0.71
0,22
DMF
40,00
prodotto 49 345,39 310,00
0,89

106
NO
H3C
O
CH3
H2C
O
H
H
H
H
H
H
H H
H
H
H
In un pallone a due colli sono stati sono stati introdotti il composto 48 (1,0 g), K2CO3
(1,34 g), Pd(OAc)2 (79,5·mg,), PPh3 (185,5·mg) e DMF anidro (40 ml) mantenendo il
tutto sotto agitazione e sotto atmosfera di N2. La miscela di reazione è stata quindi
riscaldata a 200-250°C per 4 ore e monitorata tramite TLC n-esano/EtOAc 7:3,
raffreddata a temperatura ambiente, estratta in DCM/H2O e lavata con una soluzione
satura di NaCl. La fase organica è stata anidrificata con Na2SO4, evaporata a pressione
ridotta ed il residuo è stato purificato via colonna cromatografia n-esano/EtOAc 7:3.
Sono stati ottenuti 310 mg del composto 49 puro (solido bianco); resa 56%.
CARATTERIZZAZIONE COMPOSTO 49:
Formula bruta: C22H19NO3
Spettro 1H-NMR δ (CDCl3): 8,13 (d, J= 8,8 Hz, 1H, HAr), 8,12 (d, J= 8,8 Hz, 1H,
HAr), 8,02 (d, J= 2,8 Hz, 1H, HAr), 7,4 (dd, J= 2,8, J= 8,8 Hz, 1H, HAr), 7,3-7,2 (m,
5H, HAr), 6,87 (dd, J= 2,4, J= 8,8 Hz, 1H, HAr), 6,83 (d, J= 2,4 Hz, 1H, HAr), 5,67 (s,
2H, CH2), 3,99 (s, 3H, OMe), 3,78 (s, 3H, OMe).
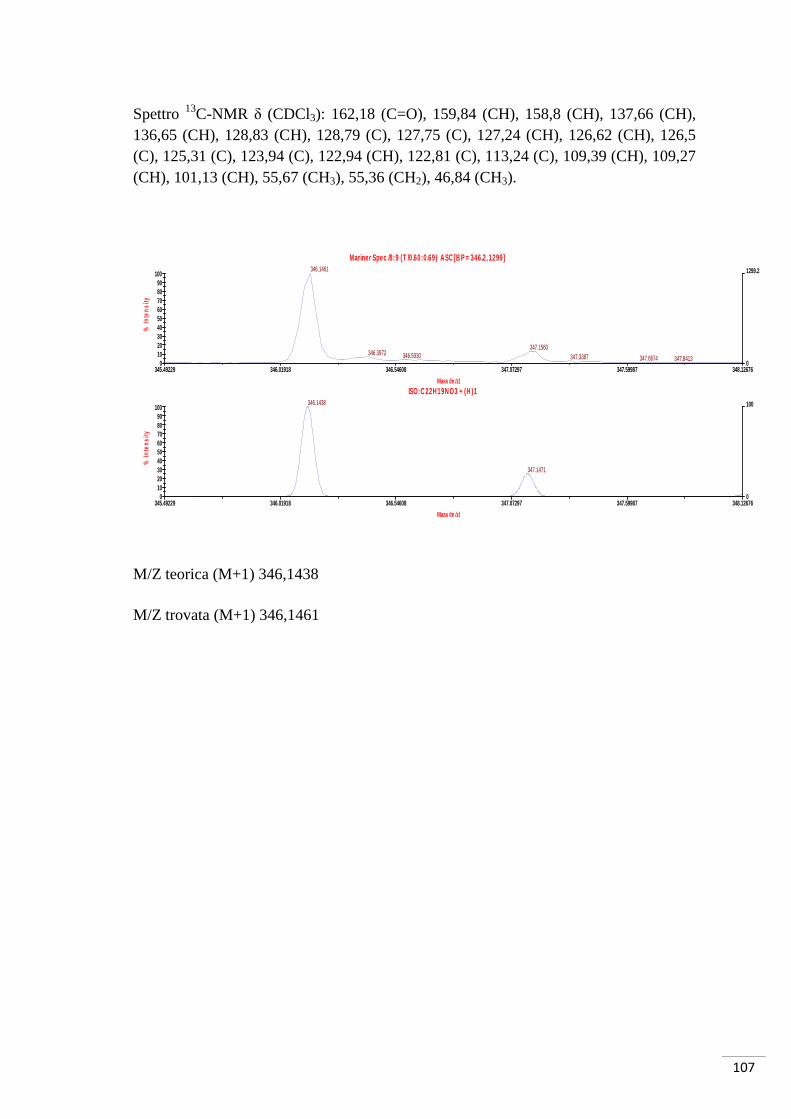
107
Spettro 13C-NMR δ (CDCl3): 162,18 (C=O), 159,84 (CH), 158,8 (CH), 137,66 (CH), 136,65 (CH), 128,83 (CH), 128,79 (C), 127,75 (C), 127,24 (CH), 126,62 (CH), 126,5 (C), 125,31 (C), 123,94 (C), 122,94 (CH), 122,81 (C), 113,24 (C), 109,39 (CH), 109,27 (CH), 101,13 (CH), 55,67 (CH3), 55,36 (CH2), 46,84 (CH3).
M/Z teorica (M+1) 346,1438
M/Z trovata (M+1) 346,1461
345.49229 346.01918 346.54608 347.07297 347.59987 348.12676
Mass (m /z)
0
100
0102030405060708090
100
% In
tens
ity
ISO:C 22H 19N O3 + (H )1346.1438
347.1471
345.49229 346.01918 346.54608 347.07297 347.59987 348.12676
Mass (m /z)
0
1299.2
0102030405060708090
100
% In
tens
ity
Mariner Spec /8:9 (T /0.60:0.69) A SC [B P = 346.2, 1299]346.1461
347.1560346.3972 346.5930 347.3387 347.6674 347.8413
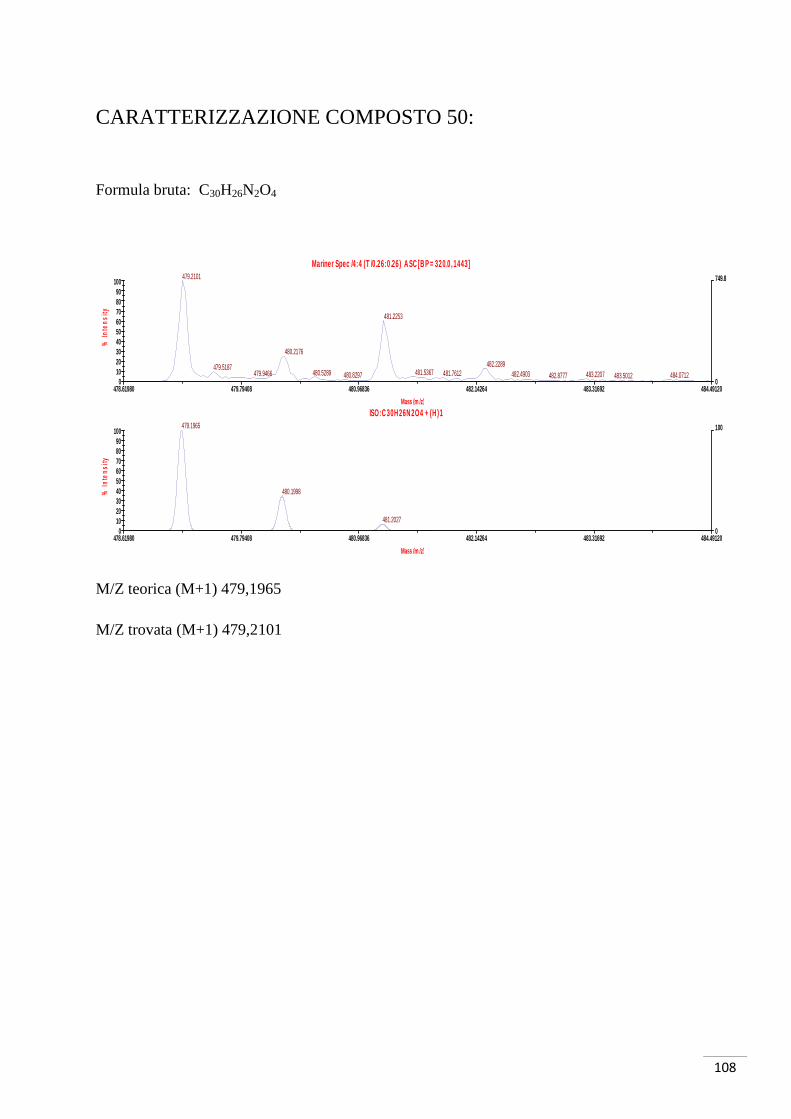
108
CARATTERIZZAZIONE COMPOSTO 50:
Formula bruta: C30H26N2O4
M/Z teorica (M+1) 479,1965
M/Z trovata (M+1) 479,2101
478.61980 479.79408 480.96836 482.14264 483.31692 484.49120
Mass (m /z)
0
100
0102030405060708090
100
% In
tens
ity
ISO:C 30H 26N 2O4 + (H )1479.1965
480.1998
481.2027
478.61980 479.79408 480.96836 482.14264 483.31692 484.49120
Mass (m /z)
0
749.8
0102030405060708090
100
% In
tens
ity
Mariner Spec /4:4 (T /0.26:0 .26) A SC [B P = 320.0 , 1443]479.2101
481.2253
480.2176
482.2289479.5187481.5367480.5289 481.7612479.9466 482.4903 483.2207480.8297 484.0712482.8777 483.5012
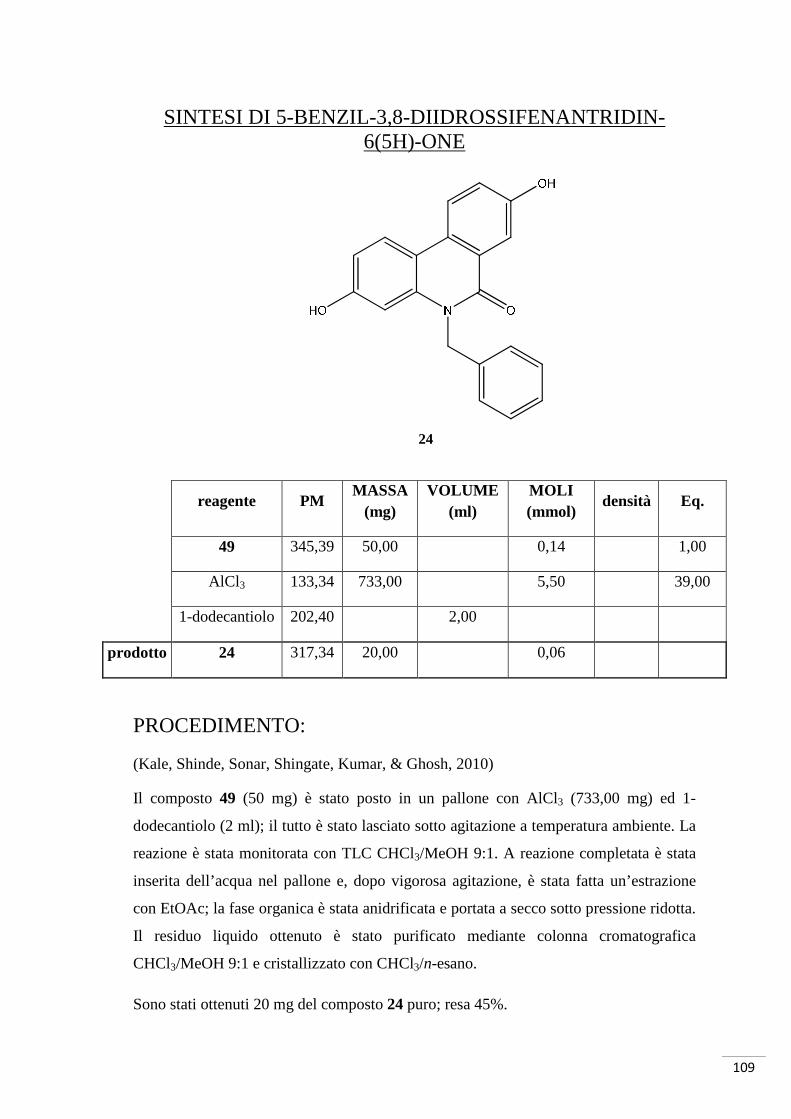
109
SINTESI DI 5-BENZIL-3,8-DIIDROSSIFENANTRIDIN- 6(5H)-ONE
reagente PM
MASSA (mg)
VOLUME (ml)
MOLI (mmol)
densità Eq.
49 345,39 50,00
0,14
1,00
AlCl 3 133,34 733,00
5,50
39,00
1-dodecantiolo 202,40
2,00
prodotto 24 317,34 20,00
0,06
PROCEDIMENTO:
(Kale, Shinde, Sonar, Shingate, Kumar, & Ghosh, 2010)
Il composto 49 (50 mg) è stato posto in un pallone con AlCl3 (733,00 mg) ed 1-
dodecantiolo (2 ml); il tutto è stato lasciato sotto agitazione a temperatura ambiente. La
reazione è stata monitorata con TLC CHCl3/MeOH 9:1. A reazione completata è stata
inserita dell’acqua nel pallone e, dopo vigorosa agitazione, è stata fatta un’estrazione
con EtOAc; la fase organica è stata anidrificata e portata a secco sotto pressione ridotta.
Il residuo liquido ottenuto è stato purificato mediante colonna cromatografica
CHCl3/MeOH 9:1 e cristallizzato con CHCl3/n-esano.
Sono stati ottenuti 20 mg del composto 24 puro; resa 45%.
24
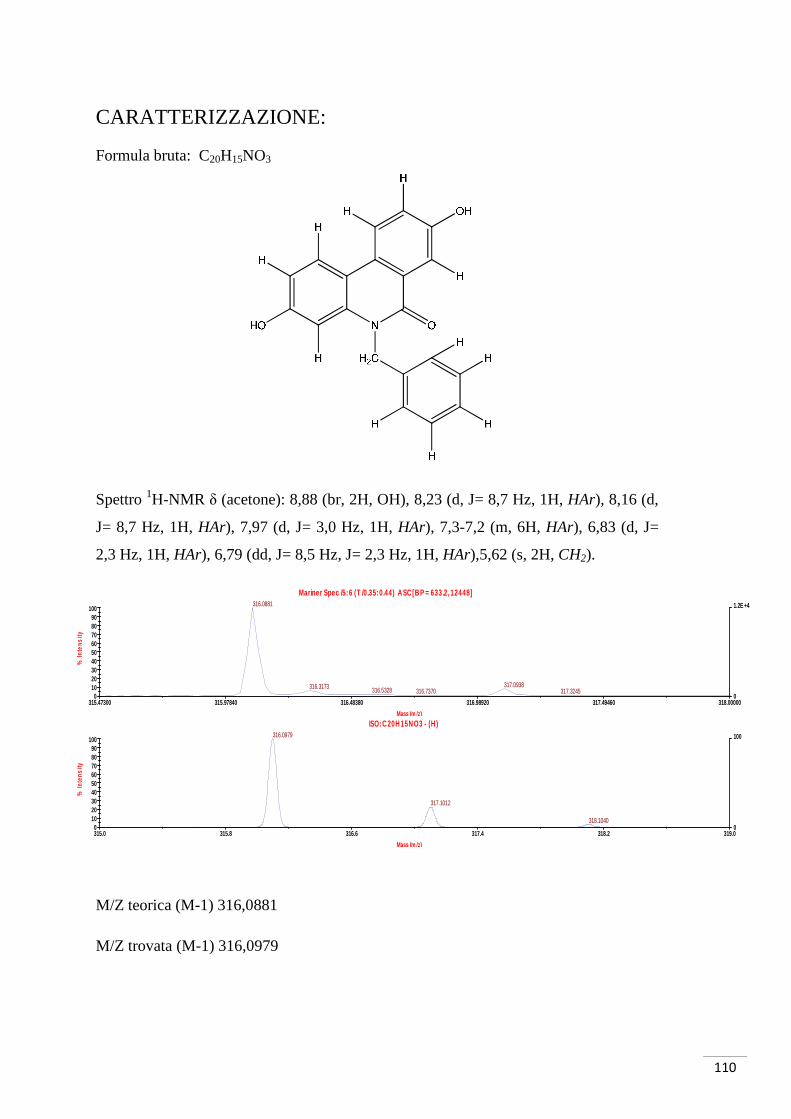
110
CARATTERIZZAZIONE:
Formula bruta: C20H15NO3
Spettro 1H-NMR δ (acetone): 8,88 (br, 2H, OH), 8,23 (d, J= 8,7 Hz, 1H, HAr), 8,16 (d,
J= 8,7 Hz, 1H, HAr), 7,97 (d, J= 3,0 Hz, 1H, HAr), 7,3-7,2 (m, 6H, HAr), 6,83 (d, J=
2,3 Hz, 1H, HAr), 6,79 (dd, J= 8,5 Hz, J= 2,3 Hz, 1H, HAr),5,62 (s, 2H, CH2).
M/Z teorica (M-1) 316,0881
M/Z trovata (M-1) 316,0979
315.0 315.8 316.6 317.4 318.2 319.0
Mass (m /z)
0
100
0102030405060708090
100
% In
tens
ity
ISO:C 20H 15N O3 - (H )316.0979
317.1012
318.1040
315.47300 315.97840 316.48380 316.98920 317.49460 318.00000
Mass (m /z)
0
1.2E +4
0102030405060708090
100
% In
tens
ity
Mariner Spec /5:6 (T /0.35:0.44) A SC [B P = 633.2, 12448]316.0881
317.0938316.3173 316.5328 317.3245316.7370

111
BIBLIOGRAFIA
Aalten, H. L., Koten, G. V., Riethorst, E., & Stamlc, C. (1989). Inorganic Chemistry , 28 (22), 4140-4146.
Ahmad, K., Wang, G., Slaton, J., Unger, G., & Ahmed, K. (2005). Anti-Cancer Drugs , 16, 1037-1043.
Ahmad, K., Wang, G., Unger, G., Slaton, J., & Ahmedc, K. (2008). Adv Enzyme Regul. , 48, 179–187.
Ahmed, K., Gerber, D. A., & Cochet, C. (2002). Trends Cell. Biol. , 12, 226-230.
Allende, J., & Allende, C. (1995). FASEB J. , 9, 313-323.
Alvisi, G., Jans, D., Guo, J., Pinna, L., & Ripalti, A. (2005). Traffic , 6, 1002–1013.
Arrigoni, G., Marin, O., Pagano, M., Settimo, L., Paolin, B., Meggio, F., et al. (2004). Biochemistry , 43, 12788–12798.
Atta-ur-Rahman. (2008). Studies in Natural Products Chemistry (Vol. 35).
Baggio, B., Pinna, L. A., Moret, V., & Siliprandi, N. (1970). Biochem. Biophys. Acta , 212, 515-517.
Battistutta, R., Mazzorana, M., Sarno, S., Kazimierczuk, Z., Zanotti, G., & Pinna, L. A. (2005). Chem. Biol. , 12, 1211-1219.
Beck, C. A., Weigel, L. N., & Edwards, D. P. (1992). Molecular Endocrinology , 6, 607-20.
Beletskaya, I., & Cheprakov, A. (2000). Chem. Rev. , 100, 3009-3066.
Bialonska, D., Kasimsetty, S. G., Khan, S. I., & Ferreira, D. (2009). J.Agric.Food Chem. , 57, 10181-10186.
Blanquet, P. R. (2000). Progress in Neurobiology , 60, 211-246.
Bronsted, J. N. (1928). Chem. Rev. , 5 (3), 231–338.
Brown, W., Foote, C., Iverson, B., & Anslyn, E. (2011). Organic Chemistry (6 ed.).
Buech, G., & Weinreb, S. (1971). JACS , 93, 746–752.
Burnett, G., & Kennedy, E. P. (1954). J. Biol. Chem. , 211, 969-980.
Casado, A., & Espinet, P. (1998). J. Am. Chem. Soc. , 120, 8978-8985.
Chantalat, L., Leroy, D., Filhol, O., Nueda, A., Benitez, M., Chambaz, E., et al. (1999). EMBO J. , 18, 2930-2940.
Chilin, A., Battistutta, R., Bortolato, A., Cozza, G., Zanatta, S., & Poletto, G. (2008). J. Med. Chem. , 51, 752–759.
clinicaltrials.gov. (s.d.). identifier: NCT00891280. Tratto da http://clinicaltrials.gov/ct2/show/NCT00891280?term=NCT00891280&rank=1
Cohen, P. (2001). Eur. J. Biochem. , 268, 5001-5010.
Cozza, G., Bonvini, P., Zorzi, E., Poletto, G., Pagano, M. A., Sarno, S., et al. (2006). J Med. Chem. , 49, 2363-2366.

112
Cozza, G., Bortolato, A., & Moro, S. (2009). Medicinal Research Reviews , 30.
Cozza, G., Meggio, F., & Moro, S. (2011). Current Medicinal Chemistry , 18, 2867-2884.
Cylene Pharmaceuticals. (s.d.). Tratto da http://www.cylenepharma.com/newsroom/cylene-presents-encouraging-clinical-data-oral-ck2-inhibitor-asco
De Moliner, E., Moro, S., Sarno, S., Zagotto, G., Zanotti, G., Pinna, L., et al. (2003). J. Biol.Chem. , 278, 1831–1836.
Dixon, D., Sethumadhavan, D., Benneche, T., Banaag, A., Tius, M., Thakur, G., et al. (2010). J. Med. Chem. , 53, 5656–5666.
Donati, L., Michel, S., Tillequin, F., & Porée, F. (2010). Org. Lett. , 12, 156-158.
Duncan, J., Wilkinson, M., & Burgoyne, R. (2000). Biochem J. , 350, 463–468.
Enkvist, E., Lavogina, D., Raidaru, G., A.Vassa, Viil, I., Lust, M., et al. (2006). J.Med.Chem. , 49, 7150-7159.
Fanta, P. (1974). Synthesis , 9-21.
Ferguson, A., Sheth, P., Basso, A., Paliwal, S., Gray, K., Fischmann, T., et al. (2011). FEBS Letters , 585, 104–110.
Furuta, T., Kitamura, Y., Hashimoto, A., Fuji, S., Tanaka, K., & Kan, T. (2007). Org.Lett. , 9, 183-186.
Ghavidel, A., & Schultz, M. C. (2001). Cell. , 106, 575-584.
Ghosal, S. (1990). Pure & Applied Chemistry , 62, 1285-1288.
Graham, K., & Litchfield, D. (2000). J. Biol. Chem. , 275, 5003–5010.
Guerra, B., & Issinger, O. (1999). Electrophoresis , 20, 391-408.
Guerra, B., & Issinger, O. (2008). Current Medicinal Chemistry , 15, 1870-1886.
Gurel, Z., Ronni, T., Ho, S., Kuchar, J., Payne, K., Turk, C., et al. (2008). J. Biol. Chem. , 283, 8291-8300.
Haggera, J., Depledgeb, M., & Gallowaya, T. (2005). Marine Pollution Bulletin , 51, 811-816.
Han, X., Stoltz, B., & Corey, E. (1999). J. Am. Chem. Soc. , 121, 7600-7605.
Harino, H., Fukushima, M., & Kawai, S. (2000). Arch. Environ. Contam. Toxicol. , 39, 13-19.
Haslam, E., Makinson, G., Naumann, M., & Cunningham, J. (1964). J. Chem. Soc. , 2137.
Hung, M., Xu, Z., Lin, Y., Mao, J., Yang, C., Chang, P., et al. (2009). BMC Cancer , 9.
Hunter, T. (1991). Methods in Enzymology , 200, 3-37.
Hutti, J., Jarrell, E., Chang, J., Abbott, D., Storz, P., & Toker, A. (2004). Nature Methods. 1, 27–29.
Inaba, T., Umezawa, I., Yusa, M., Mihashi, S., & H.Itokawa. (1987). J.Org.Chem. , 52, 2957.
Ingebritsen, T. S., & Cohen, P. (1983). Science , 221, 331-338.

113
Ivanov, K., Puustinen, P., Gabrenaitea, R., Vihinena, H., Rönnstrand, L., Valmua, L., et al. (2003). The Plant Cell , 15, 2124-2139.
Jans, D. A., & Hubner, S. (1996). Physiol. Rev. , 76 (3), 651-685.
Jursic, B., & Patel, P. (2005). Tetrahedron , 61, 919-926.
Kale, B., Shinde, A., Sonar, S., Shingate, B., Kumar, S., & Ghosh, S. (2010). Tetrahedron Letters , 51, 3075–3078.
Kim, G., Jung, J., Niizuma, K., & Chan, P. (2009). J Neurosci. , 29, 14779–14789.
Lasa, M., Marin, O., & Pinna, L. A. (1997). Eur. J. Biochem. , 243, 719-725.
Lasa, M., Marin, O., Meggio, F., & Pinna, L. A. (1996). FEBS Lett. , 382, 149-152.
Lawson, E., Luci, D., Ghosh, S., Kinney, W., Reynolds, C., Qi, J., et al. (2009). J. Med. Chem. , 52, 7432–7445.
Litchfield, D. W. (2003). Biochem. J. 369, 1-15.
Marin, O., Meggio, F., & Pinna, L. (1999). Biochem. Biophys. Res. Commun. , 256, 442-446.
McOmie, J., Watts, M., & West, D. (1968). Tetrahedron , 24, 2289.
Meggio, F., & Pinna, L. (2003). FASEB J. , 17, 349-368.
Meggio, F., Donella Deana, A., Ruzzene, M., Brunati, A. M., Cesaro, L., Guerra, B., et al. (1995). Eur. J. Biochem. , 234, 317-322.
Meggio, F., Marin, O., Boschett, M., Sarno, S., & Pinna, L. A. (2001). Molecular and Cellular Biochemistry , 227, 145–151.
Meggio, F., Pagano, M. A., Moro, S., G., Z., Ruzzene, M., Sarno, S., et al. (2004). Biochemistry , 43, 12931-12936.
Meggio, F., Shugar, D., & Pinna, L. (1990). Eur. J. Biochem. , 187, 89-94.
Mondal, S., Ghosh, S., & Verma, S. (2010). Tetrahedron Letters , 51 (5), 856-859.
Morgan, B., Mulrooney, C., O’Brien, E., & Kozlowski, M. (2010). J. Org. Chem. , 75, 30–43.
Neises, B., & Steglich, W. (1978). Angew. Chem. Int. , 17, 522–524.
Niefind, K., Guerra, B., Pinna, L. A., Issinger, O., & Schomburg, D. (1998). Embo J. , 17, 2451-2462.
Nishizuka, Y. (1995). 9, 484-496.
Padmnabha, R. (1990). Mol. Cell. Biol. , 10, 4089-4099.
Pagano, M. A., Andrzejewska, M., Ruzzene, M., Sarno, S., Cesaro, L., Bain, J., et al. (2004). J. Med Chem. , 47, 6239-6247.
Pagano, M. A., Poletto, G., Di Maria, G., Cozza, G., Ruzzene, M., Sarno, S., et al. (2007). Chembiochem , 8, 129-139.
Paliakov, E., & Strekowski, L. (2004). Tetrahedron Letters , 45, 4093–4095.

114
Peet, N., Dickerson, G., Abdallah, A., Daly, J., & Ukena, D. (1988). J . M e d . C h e m . , 31, 2034-2039.
Peña, J. M., Itarte, E., Domingo, A., & Cusso, R. (1983). Cancer Res. , 43, 1172-1175.
Pepperkok, R., Lorenz, P., Ansonge, W., & Pyperin, W. (1994). J. Biol. Chem , 269, 6986-6991.
Perez, D. I., Gil, C., & Martinez, A. (2010). Med. Research Reviews .
Pierre, F., Chua, P., O’Brien, S., Siddiqui-Jain, A., Bourbon, P., Haddach, M., et al. (2011). J. Med. Chem. , 54, 635–654.
Pierre, F., O’Brien, S., Haddach, M., Bourbon, P., Schwaebe, M., Stefan, E., et al. (2011). Bioorg. Med. Chem. Lett , 21, 1687–1691.
Pinna, L. A. (1997). Int. J. Biochem. Cell. Biol. , 29, 551-554.
Pinna, L. A. (2002). Journal of Cell Science , 115, 3873-3876.
Pinna, L. A., & Meggio, F. (1997). Prog Cell Cycle Res. , 3, 77-97.
Pinna, L. A., Baggio, B., Moret, V., & Siliprandi, N. (1969). Biochem. Biophys. Acta , 178, 199-201.
Punna, S., Meunier, S., & Finn, M. (2004). Org. Lett. , 6 (16), 2777–2779.
Ramos, M., Prudent, R., Moucadel, V., Sautel, C., C.Barette, Lafanechère, L., et al. (2010). FASEB J. , 24.
Robinson, R. (1917). Chem. Soc. , 111, 762.
Ruzzene, M., Brunati, A., Sarno, S., Marin, O., Donella, A., & Pinna, L. (2000). Eur J Biochem. , 267, 3065–3072.
Sarno, S., Moro, S., Meggio, F., Zagotto, G., Benb, D. D., Ghisellini, P., et al. (2002). Pharmacology & Therapeutics , 93, 159–168.
Sarno, S., Papinutto, E., Franchin, C., Bain, J., Elliot, M., Meggio, F., et al. (2011). Current Topics in Medicinal Chemistry , 11 (11), 1340-1351.
Schopf, C., Lehmann, G., & Arnold, W. (1937). Angew. Chem. , 50, 779.
Schrenk, W., & Snaar Jagalska, B. (1999). Biochimica et Biophysica Acta , 1449, 1-24.
Selkoe, D. (1997). Science , 275, 630-631.
Ssyszka, R., Grakowski, N., Felczak, K., & Shugar, D. (1995). Biochem. Biophys. Res. Commun. , 208, 418-424.
Stille, J. (1986). Angewandte Chemie Int. Ed. , 25, 508–524.
Suzuki, T., Okada, C., Arai, K., & Awaji, A. (2001). J. Heterocyclic Chem. , 38, 1409.
Tian, H., Vogel, R., Amic, L., Tamagnan, G., & Baldwin, R. (2006). J Label Compd Radiopharm , 49, 1247–1258.
Tietze, L. (1996). Chem.REv. , 96, 115-136.
Unger, G., Davis, A., Slaton, J., & Ahmed, K. (2004). Current Cancer Drug Targets , 4, 77-84.

115
Venter, J. C., Adams, M., Myers, E., Li, P., Mural, R., Sutton, G., et al. (2001). Science , 291, 1304-1351.
Walsh, D. N., & Van Patten, S. M. (1994). FASEB J. , 8, 1227-1236.
Xu, X., Rich, E. S., & Seldin, D. C. (1998). Genomics , 48, 79-86.
Ye, J., Abiman, P., Crossley, A., Jones, J., Wildgoose, G., & Compton, R. (2010). Langmuir , 26 (3), 1776–1785.
Zawada, K., Wolniak, M., Kazimierczuk, K., & Wawer, I. (2009). J. of Mol. Struct. , 918, 174–182.

116

117
RINGRAZIAMENTI:
Un ringraziamento particolare al Prof. Zagotto, a Riccardo e a Francesca per aver fatto
di questo periodo di tesi un’esperienza unica, grazie per i vostri preziosi aiuti.
Grazie anche ai miei colleghi di laboratorio Martina, Giovanni, Arianna ed Alessandro:
ho passato momenti bellissimi, etanditiolo a parte, anche nelle fasi “buie” delle mie
sintesi (so che potete capirmi).
Un altro ringraziamento speciale ai miei genitori, che mi hanno sostenuta in questi
cinque anni, senza di voi forse non ce l’avrei fatta. Grazie anche ai miei nonni…grazie
a tutti quelli che hanno creduto in me.
“Ultimo ma non ultimo” grazie alle mie amiche nonché compagne di corso Elisa, Elena
ed Anna, per aver rallegrato mattine, pomeriggi e serate di questi ultimi cinque anni. Ed
ora…che la festa abbia inizio.





























