Embed Size (px)
Citation preview

Theoretical study of the adsorption of urea related species onPt(1 0 0) electrodes
Maite Garc�õa-Hern�andez a,b, Uwe Birkenheuer a,1, Anguang Hu a,2,Francesc Illas b,*, Notker R�osch a
a Institut f �ur Physikalische und Theoretische Chemie, Technische Universit�at M�unchen, 85747 Garching, Germanyb Departament de Qu�õmica F�õsica & Centre Especial de Recerca en Qu�õmica Te�orica, Universitat de Barcelona, C/Mart�õ i Franqu�es 1,
08028 Barcelona, Spain
Received 14 August 2000; accepted for publication 10 October 2000
Abstract
We present a cluster model study on the two most likely urea adsorption complexes at Pt(1 0 0) electrodes. A goal of
the investigation is to determine whether urea or ureylene, a HNCONH biradical species, forms the most stable ad-
sorption complex. Geometry optimisations of both urea species adsorbed at various surface sites have been carried out
with the parallel PARAGAUSSPARAGAUSS program package using gradient-corrected density functionals. Scalar-relativistic all-
electron calculations as well as pseudopotential calculations have been performed. For each optimised structure, vib-
rational frequencies have been calculated. From energy considerations, ureylene adsorption is found in both types of
calculations to be preferred over urea adsorption, consistent with the interpretation of electrochemical measurements,
even though e�ective core potential results tend to underestimate the binding energies. This assignment is supported by
the calculated CO vibrational stretching frequency of chemisorbed ureylene which is closer to the value obtained from
``in situ'' Fourier transform infrared spectroscopy in an electrochemical environment than the CO frequency calculated
for adsorbed urea. Ó 2001 Elsevier Science B.V. All rights reserved.
Keywords: Platinum; Density functional calculations; Chemisorption
1. Introduction
There is little doubt that understanding ad-sorption processes at a molecular level requires fullcomprehension of the interactions between mole-cules and solid surfaces [1±3]. This claim for un-derstanding at a microscopic level is even morestringent for interactions occurring at electro-chemical interfaces since it is impossible to controlall parameters at a level similar to that whichcan be achieved in experiments carried out under
Surface Science 471 (2001) 151±162
www.elsevier.nl/locate/susc
* Corresponding author. Tel.: +34-93-402-1229; fax: +34-93-
402-1231.
E-mail address: [email protected] (F. Illas).1 Present address: Max-Planck-Institut f�ur Physik komplexer
Systeme, N�othnitzer Strasse 38, 01187 Dresden, Germany.2 Present address: Department of Chemistry, University of
Minnesota, Minneapolis, MN 55455 0431, USA.
0039-6028/01/$ - see front matter Ó 2001 Elsevier Science B.V. All rights reserved.
PII: S00 3 9-6 0 28 (0 0 )0 09 0 1- 8

ultra-high vacuum (UHV), conditions. A case inpoint is ``in situ'' Fourier transform infraredspectroscopy (FTIR), applied in an electrochemi-cal environment. While this technique enables toobtain precise experimental data about the speciesthat are adsorbed at the electrode, their interpre-tation is not simple at all and thus in situ FTIRdoes not permit an univocal assignment because ofthe di�culty, if not the impossibility, to compareto spectra of known species. Therefore, it is notsurprising that the interpretation of FTIR in situspectra is most often based on comparison to ex-isting inorganic complexes. However, this com-plex-to-surface analogy does not guarantee acorrect assignment [4]. As an alternative, quantumchemistry investigations applied to realistic surfacemodels can provide an adequate, yet ¯exible,computational approach to the chemistry of suchcomplex systems. Computational experiments per-mit to verify or discard a given hypothesis andsuggest new interpretations of experimental data.The present work is devoted to such a theoreticalstudy of the adsorption of urea and related specieson a single crystal Pt(1 0 0) electrode.
Several experimental studies [5±10] addressedstructural aspects of urea adsorption at plati-num electrodes and reactions of such adsorptionsystems. These investigations were carried out us-ing di�erent well-de®ned single-crystal surfacesand used a variety of experimental techniquesincluding voltammetry [5,6,8], radiochemistry [8],``ex situ'' low electron energy di�raction [6,8](LEED), and Auger electron spectroscopy [6,8].These data permitted to derive the dependenceof the potential on urea coverage, the potentialsaturation value, and the saturated urea adlayersurface structure. One particular feature of theadsorption of urea in the Pt(1 0 0) surface is thevery large excess charge compared to that mea-sured in other crystallographic surfaces. Thischarge excess due to the presence of urea in themedia indicates that the adsorbate undergoes acharge-transfer process at the electrode. The ad-sorption and desorption processes take place witha net charge transfer of two electrons per adsorbedmolecule [5,6,8]. This indicates that urea adsorp-tion may produce adsorbed ureylene according tothe following electrochemical process:
Hydrogen adsorption at the Pt(1 0 0) surfacemay be an important intermediate stage of thisreaction. The adsorption and desorption voltam-mogram peaks recorded on Pt(1 0 0) are very sharpand indicate that urea adsorption is reversible[5,6,8]. On the other hand, the stable c�5� 20�LEED pattern observed on a clean Pt(1 0 0) sur-face changes with urea adsorption to a c�2� 4�LEED pattern [6,8]. Radiochemistry measure-ments [8] gave a coverage of 0:26� 0:04 ML(number of molecules per Pt(1 0 0) unit cell) whichis fully consistent with the value of 0:24� 0:03 MLobtained from the Auger electron spectroscopycarbon signal [8]. Climent et al. reported comple-mentary studies of the adsorption of urea onPt(1 0 0) [11] and Pt(1 1 1) [12] electrodes. Thevoltammetric and charge displacement analysis onPt(1 0 0) are similar to those reported by Wiec-kowski et al. [5±10] under analogous conditions.Moreover, the integrated charge was found to belarger than the one corresponding to adsorptionand desorption of a single hydrogen atom by�50%, thus indicating the existence of additionalurea related surface processes. In situ FTIR spec-tra reported by Feliu et al. [11] provide additionalinformation on the nature and structure of theurea species adsorbed on Pt(1 0 0). A bipolar band,typical of potential dependent frequencies of thevibrational modes of adsorbed species, appearscentred at 1705 cm±1. Based on the similarity to theurea carbonyl group in coordination compounds[13,14] and on the fact that this peak does notappear at potentials at which there is no adsorbedurea, the peak at 1705 cm±1 was assigned to ureabonded at the Pt(1 0 0) surface through the nitro-gen atoms, in a bridge con®guration and with thecarbonyl group normal to the electrode surface.Likewise, once the electrode has been in contactwith the urea solution, the appearance of this peak
152 M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162

neither depends on the electrode potential nor onthe presence or absence of urea in the solutionwhere the Pt electrode is re-immersed.
The ®rst theoretical study of urea adsorbed onPt(1 0 0) was reported by Rikvold and Wieckowski[7]. In this work urea adsorption on Pt(1 0 0) wasinvestigated within the gas-lattice model employ-ing a Monte Carlo simulation. This simple modelsuggests that urea is adsorbed along Pt±Pt bondsoccupying two adsorption sites and co-exists withhydrogen atoms adsorbed on top of the platinumatoms. Based on the fact that most of the surfacehydrogen atoms desorb at a potential close to thatcorresponding to urea adsorption, these authorssuggest that urea adsorbs on sites formerly occu-pied by hydrogen atoms. Further Monte Carlosimulations within the lattice gas-model [8±10]considered urea coordination at the platinumsurface through the nitrogen atoms. Clearly, anon-empirical approach appears desirable.
The present work is devoted to the analysis ofthe adsorption properties of urea and relatedspecies on Pt(100) electrodes. Binding energies,geometrical parameters, and vibrational frequen-cies of urea ± (H2N)CO(NH2) ± and ureylene ±(HN)CO(NH) ± the two most likely adsorbedspecies, are studied using a cluster model approachto represent the Pt(1 0 0) surface and gradient-corrected density functionals (DFs) to account forcorrelation. Relativistic e�ects due to the presenceof platinum atoms are also taken into account (fordetails see below).
2. Surface cluster models
The cluster model approach to chemisorptionassumes that the properties of interest are of localnature and, hence, restriction to a ®nite fraction ofthe extended surface does not introduce seriousartefacts. This approach has long been used tomodel surfaces and chemisorption phenomena andits success and limitations are well documented [1±3]. Reasonably large cluster models have beenemployed in the present study to simulate thevarious adsorption sites of urea and ureylene onthe Pt(1 0 0) surface. Adopting the Ptn�m1;m2;
m3; . . .� notation where n denotes the total numberof platinum atoms in the cluster and mi the num-ber of atoms in the ith crystal layer (starting withthe top-most one at the surface), the cluster modelschosen for the various adsorption sites of Pt(1 0 0)are Pt14(8,6), Pt9(5,4), and Pt9(4,5), respectively(see Fig. 1). These cluster models were consideredas rigid fractions of a Pt bulk crystal with thenearest-neighbour Pt distance set to the experi-mental value of 2.77 �A [15]. The geometry opti-misation for urea and ureylene on Pt(1 0 0) wascarried out using analytical energy gradients. C2
point group symmetry was imposed during ge-ometry optimisation with the only additionalconstraint ± beside the ®xed Pt±Pt distances ± thatthe orientation of the molecular N±CO±N plane ispreserved. In this way one ensures that the ad-sorbate will not change site during the geome-try optimisation. In all cases the calculated gasphase geometry of urea was taken as starting
Fig. 1. Schematic view of the cluster models used to represent
di�erent adsorption sites of the Pt(1 0 0) surface: (a) Pt14(8,6),
(b) Pt9(4,5), and (c) Pt9(5,4).
M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162 153

con®guration for both urea and, after abstractingtwo hydrogen atoms, for ureylene.
Four di�erent adsorption sites have been con-sidered, the corresponding positions of the ureaskeleton are indicated schematically in Fig. 2.These sites can be characterised as follows: (i) ontop site ± each nitrogen atom on top of a Pt atom(Fig. 2a), (ii) hollow site ± each nitrogen atomabove a four-fold hollow site (Fig. 2b), (iii) bridge-ontop site ± each nitrogen atom in a bridging po-sition above two surface Pt atoms such that thereis one Pt atom just below the C atom (Fig. 2c), and(iv) bridge-hollow site ± each nitrogen atom againin a bridging position, but with a four-fold hollowbelow the C atom (Fig. 2d).
3. Computational details
Recall that relativistic e�ects are important forsystems containing heavy atoms; hence they mustbe included to describe the electronic structure ofthe Pt surface clusters used in the present study.Two di�erent approaches have been used forthat purpose. In a ®rst series of calculations allelectrons (AE), are treated explicitly within a sca-lar-relativistic variant [16] of the linear combina-tion of Gaussian-type orbitals DF method [17](LCGTO-DF) as implemented in the programPARAGAUSSPARAGAUSS for parallel computers [18,19]. Thesame computer code was used for a second seriesof calculations (the ECP approach) where scalar-relativistic e�ects are introduced indirectly throughthe usage of pseudopotentials [20]; in the presentstudy these are relativistic pseudopotentials ofthe Stuttgart type [21]. In both cases, a large and¯exible basis was employed to expand the Kohn±Sham orbitals. For the AE Pt atoms the basis set is(21s,17p, 12d,7f) contracted to [9s,8p,5d,2f]. Theprimitive basis set is derived from a (19s,14p,10d,5f) GTO basis [22] by adding two s, three p, two d,and two f exponents as proposed by Ferrari et al.[23] in their study of Pt clusters supported on ze-olites. For the other atoms ¯exible GTO basis setswere employed [24,25]. For C, N, and O, these are(9s,5p,1d) basis sets contracted to [5s,4p,1d], andfor H the basis is (6s,1p) contracted to [4s,1p].In the pseudopotential calculations a small core
containing 60 electrons was used for Pt; the 5s, 5p,5d, and 6s electrons are included in the valenceshell together with a (7s,6p,5d) primitive basisset contracted to [6s,3p,2d] [26]. The gradient-corrected BP exchange-correlation functional, acombination of BeckeÕs exchange functional [27]and PerdewÕs correlation functional, [28] was em-ployed throughout. All calculations were carriedout in spin-restricted fashion, since no open-shellcon®gurations were expected among the potentialadsorption species.
4. Structure and stability of urea species on Pt(1 0 0)
First, we brie¯y describe the results obtained forgas phase urea and compare them with availabletheoretical and experimental data. Both micro-wave spectroscopy [29] and matrix isolation stud-ies [30] have shown that, contrarily to organicchemistry textbook arguments, urea is not a planarmolecule. More recently Godfrey et al. [31] com-bined MP2/6-311++G(d,p) calculations with allprevious experimental data to fully characterisethe structure of the urea molecule. Later, Rous-seau et al. [32,33] reported a detailed assignment ofthe vibrational frequencies of urea and deuteratedurea. The geometric parameters of free gas phaseurea predicted by the present DF, calculations (seeTable 1) are in excellent agreement with the MP2results just mentioned as well as with experiment[31]. The accuracy of the DF calculations is about0.01 �A for distances, 1±2° for bond angles, and 5°for the pyramidalisation angle as de®ned in Ref.[29].
We now turn to the adsorption process. Withinthe ECP approach, the equilibrium geometry ofboth adsorption species, urea and ureylene, havebeen determined all for adsorption sites of Pt(1 0 0)described above. For ureylene, which is the mostinteresting species from the electrochemical pointof view, the adsorption geometries were also de-termined by scalar-relativistic AE calculations. Acomparison of these two approaches will be pre-sented later. The structural parameters of the ad-sorbed species are reported in Table 1 (for urea)and Table 2 (for ureylene). The ®rst surprisingresult, even without any reference to gas-phase
154 M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162
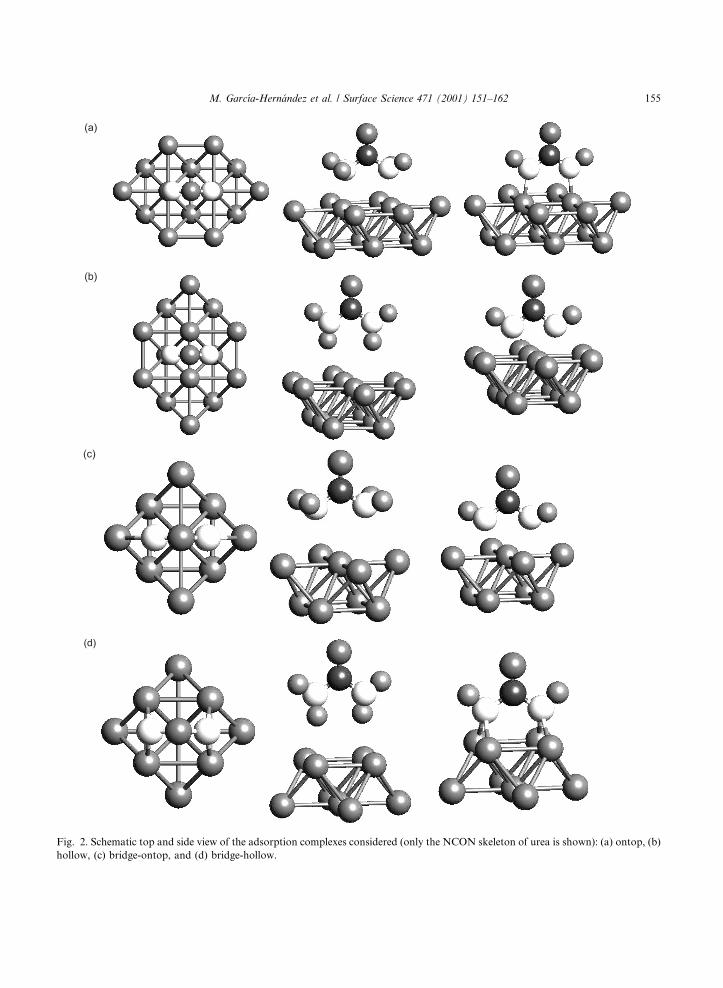
Fig. 2. Schematic top and side view of the adsorption complexes considered (only the NCON skeleton of urea is shown): (a) ontop, (b)
hollow, (c) bridge-ontop, and (d) bridge-hollow.
M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162 155
urea, is that the relative orientation of the hydro-gen atoms of the amide groups with respect to thesurface di�ers substantially among the four dif-ferent sites, see Fig. 2. This is at variance with thecase of ureylene precisely because of the lack ofone hydrogen atom in each amide group (see be-low). For urea in the ontop and bridge-ontop sites,the lone pairs of the nitrogen atoms point towards
the surface, and the hydrogen atoms of each amidegroup point away from the surface (see middlecolumns of Fig. 2a and c), i.e. the surface bond ismediated through the lone-pairs on the amidenitrogen atoms. Compared to the gas phase, theC±O distance decreases concomitantly with anincrease of the C±N and the two N±H distances.Note also that for the bridge-ontop adsorption site
Table 1
Equilibrium structure of urea on Pt(1 0 0) from a relativistic pseudopotential description of the Pt core electrons
Site cluster Ontop Hollow Bridge-ontop Bridge-hollow Free urea
Pt14(8,6) Pt14(8,6) Pt9(5,4) Pt9(5,4)
DistancesC±O 1.207 1.235 1.212 1.232 1.228
C±N 1.459 1.386 1.457 1.390 1.396
N±Hup 1.040 1.017 1.029 1.030 1.021
N±Hdown 1.030 1.024 1.029 1.018 0.998
N±N 2.463 2.343 2.423 2.337 2.338
N±Pt 2.205 3.853 2.946 3.525 ±
N±surf a 2.200 3.399 2.685 3.234 ±
H±Pt ± 2.899 ± 2.504 ±
H±surf a ± 2.420 ± 2.265 ±
AnglesO±C±N 122.4 122.3 123.7 122.8 123.1
C±N±Hup 106.8 114.1 107.5 118.9 112.8
C±N±Hdown 108.6 120.6 107.5 114.2 119.2
N±C±N 115.2 102.4 112.5 114.4 113.7
O±C±N±Hup ÿ73.1 14.2 56.6 ÿ157.3 12.8
O±C±N±Hdown 41.7 160.2 ÿ56.5 ÿ14.2 148.9
Distances in �A, angles in degrees. A C2 symmetry constraint was imposed during the geometry optimisation.a Distance to the top ``crystal'' plane of the cluster model.
Table 2
Equilibrium structure of the ureylene radical on Pt(1 0 0) from a relativistic pseudopotential description of the Pt core electrons
Site cluster Ontop Hollow Bridge-ontop Bridge-hollow Free urea
Pt14(8,6) Pt14(8,6) Pt9(5,4) Pt9(5,4)
DistancesC±O 1.244 1.227 1.224 1.221 1.228
C±N 1.375 1.399 1.404 1.450 1.396
N±H 1.021 1.028 1.031 1.030 1.018
N±N 2.338 2.235 2.311 2.491 2.338
N±Pt 1.993 2.459 2.208 2.120 ±
N±surf a 1.981 1.696 1.882 1.598 ±
AnglesO±C±N 121.8 127.0 124.6 120.7 123.1
C±N±H 113.6 111.6 109.2 106.5 112.5
N±C±N 116.4 106.0 110.8 118.5 113.7
O±C±N±H 0.0 0.1 50.5 0.0 ÿ12.8
Distances in �A, angles in degrees. A C2 symmetry constraint was imposed during the geometry optimisation.a Distance to the top ``crystal'' plane of the cluster model.
156 M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162

the urea molecule becomes C2v symmetric, eventhough this is not enforced by symmetry con-straints. At the hollow and bridge-hollow sites,apart from a slight elongation of the N±H andC±O bonds and a shortening of the C±N bonds,the most interesting feature is that the atomsclosest to the surface are two hydrogen atoms (seemiddle columns of Fig. 2b and d). Hence, at thesetwo sites urea interacts with the surface directlythrough one hydrogen atom per amide grouprather than through the nitrogen lone pairs. Ap-parently a bond con®guration is preferred whichmay be regarded as a precursor for dehydrogena-tion of urea via hydrogen adsorption on Pt(1 0 0).
In spite of having lost two hydrogen atoms, thegeometry of adsorbed ureylene is rather reminis-cent of that of gas-phase urea. In fact, in adsorbedureylene the C±O distance remains almost un-changed with respect to free urea as do most of thebond angles (see Table 2). Much more interestingis the position of the remaining hydrogen centresrelative to the urea skeleton, right column of Fig.2. Except for the bridge-ontop site, the hydrogenatoms lay almost perfectly in the N±(CO)±Nplane, indicating a symmetric two-fold coordina-tion (including the lone-pair) of the nitrogen atomstoward the Pt surface. The resulting coordinationof the ureylene nitrogen atoms can be rationalisedin terms of an almost sp3 hybridisation. Two of thesp3 hybrid orbitals form the N±C and the N±Hbonds while the two remaining hybrids establishthe surface bonding. Hereby, the platinum surfaceis meant to provide the one extra electron neces-sary to form the two equivalent single bonds.Notice that the participation of the extra electronto the surface bond does not necessarily imply acharge donation to the adsorbate. The formationof such a two-fold coordination is obvious for themost stable adsorption site, the bridge-hollow site,which in turn rationalises the energetic preferenceof that site. For the hollow and ontop sites, thistwo-fold coordination is not apparent, indicating amore delocalised bonding mechanism. For thebridge-ontop site, on the other hand, the dihedralOCNH angle is about 50°, indicating a deviationform planarity, at variance from the value zero forthe other cases discussed above. The correspond-ing structure is again compatible with a typical
sp3-type hybridisation of the amide nitrogen at-oms, with one lone pair of each nitrogen atomoriented almost perpendicularly to the molecularplane and the other one pointing directly towardsthe surface. Finally, it is worth mentioning thatthe surface bonding mechanism for ureylene to thebridge-hollow site is favoured with respect to thedirect hydrogen-surface interaction encountered inurea adsorption at that site simply because of theneed to saturate the free valences of the ureyleneradical.
Now we turn to a discussion of the relativestability of urea and ureylene adsorbed at thedi�erent surface sites. In order to compare theadsorption energies of the various species, a ther-modynamic cycle has been used. In this way, oneavoids any explicit reference to the energy of thegas-phase biradical and at the same time one doesnot introduce any bias on the relative stability. Foradsorbed urea, the interaction energy is given bythat of the simple reaction:
CO�NH2�2 � Ptn�m1;m2� ! CO�NH2�2�ads� �1�
As usual, a negative value indicates that productsare more stable than reactants. However, for ad-sorbed ureylene it is convenient to refer to theadsorbed species plus adsorbed hydrogen. There-fore, the interaction energy is calculated as theenergy of the following reaction:
CO�NH2�2 � Ptn�m1;m2�! CO�NH2�2�ads� � 2H�ads� �2�
Here, H�ads� designates adsorbed atomic hydrogenalthough one must realise that in the electro-chemical environment this adsorbed speciesbecomes oxidised. In order to obtain a uniquereference energy for H�ads�, the interaction energyof atomic hydrogen was computed for di�erenthigh symmetry sites of Pt(1 0 0) using the variouscluster models shown in Fig. 1. The largest inter-action energy, 69.0 kcal molÿ1 for the ECP ap-proach (72.6 kcal molÿ1 with the AE treatment),was found for adsorption of a single H at thebridge site of the Pt14(8,6) cluster which falls wellinto the range of about 60±80 kcal molÿ1 for hy-drogen-metal binding energies as typically ob-served in experiment [34,35]. This value was taken
M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162 157

to compute the interaction energies according toreaction (2).
It is worth comparing the present cluster cal-culations to recent periodic DF calculations usingthe ADF-Band code [36]. These authors reportadsorption energies for H on Pt(1 1 1) relative togas phase H2. For a 0.25 coverage the adsorptionenergy calculated obtained with the BP functionalis about 11 kcal molÿ1. Using the experimentalbinding energy of ÿ103.3 kcal molÿ1 for the hy-drogen molecule and the results reported by Olsenet al. [36], it is straightforward to obtain the cor-responding atomic hydrogen chemisorption en-ergy. This leads to a value of 63 kcal molÿ1 thatcan be directly compared to the present clustermodel results reported above. This result corrob-orates the cluster estimates discussed above. Note,however, that the slab model calculations refer toPt(1 1 1), use a frozen core approximation, and adi�erent basis set whereas the present calculationsare for H adsorption on Pt(1 0 0). Therefore, onlyan approximate comparison is meaningful. Giventhe di�erences between both theoretical studies,the agreement between cluster and periodic cal-culations is rather satisfactory.
For adsorbed urea, both hollow and bridge-hollow sites are most stable (�ÿ5 kcal molÿ1) andenergetically nearly equivalent, the former beinglower in energy by 0.7 kcal molÿ1 only (Table 3).Adsorption in the ontop site is endothermic by�0.6 kcal molÿ1 and, ®nally, the interaction abovethe bridge-ontop site is highly disfavoured by morethan 25 kcal molÿ1. Moreover, given the well-known limitations of cluster models when ad-sorption energies are to be determined [1±3], weconsider these ®rst three cases as comparable intheir binding energy (see below).
A rather di�erent situation occurs when weconsider the relative stability of adsorbed ureylene.
With an interaction energy of �27 kcal molÿ1, themost stable adsorption complex is at the bridge-hollow site (Table 3). The adsorption processdescribed by reaction (2) is preferred over non-dissociative adsorption of urea ± reaction (1) ± atthe bridge hollow site by about 22 kcal molÿ1. Thisis consistent with the hypothesis that upon adsorp-tion urea looses two hydrogen atoms [11]. Inter-action at the three other sites is far less favourablethan in the case of urea. This result is of specialrelevance since it is in agreement with the experi-mental observation of a urea adlayer which isstrongly bound to the Pt electrode [11]. In fact, thisadlayer is so tightly bound to the electrode surfacethat it is detected even after the electrode is re-moved from the urea solution and an in situ FTIRmeasurement is performed after inserting theelectrode into a urea-free solution [11]. Therefore,the bridge hollow adsorption of the urea moleculecan be regarded as a ®rst step of a more complexadsorption mechanism in an electrochemical en-vironment.
Next, we will address the accuracy of thepseudopotential approach compared to scalar-relativistic AE calculations. This comparison hasbeen carried out for ureylene on the four activesites described above. Overall, the adsorbatestructures arising from the more accurate AE cal-culations are very similar to those obtained withthe ECP approach. The changes in the geometry ofadsorbed ureylene are essentially negligible. Theonly noticeable geometrical variation is found forthe Pt±N distance; it is by 0.03±0.06 �A shorter inthe AE calculations. Quite noticeable di�erencesare obtained for the adsorption energies. The AEcalculations result in an additional stabilisation ofthe adsorbed species by 18±29 kcal molÿ1 com-pared to pseudopotential results (Table 3). Thisenergy di�erence is larger than usually encoun-
Table 3
Interaction energies (in kcal molÿ1) of urea and ureylene on a Pt(1 0 0) surface according to the reactions (1) and (2) (see text) as
obtained from relativistic pseudo potential (ECP) and AE calculationsa
Ontop Hollow Bridge-ontop Bridge-hollow
Urea ECP 0.6 ÿ5.2 26.6 ÿ4.6
Ureylene ECP ÿ10.1 54.3 38.0 ÿ26.5
AE ÿ30.1 41.6 20.1 ÿ55.4
a Negative energies imply products being more stable than reactants.
158 M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162

tered in transition metal compounds and indicatessome limitation of the Pt pseudopotential repre-sentation (likely due to the ECP basis set). Note,however, that from Table 3 one can see that theseenergy di�erences are fairly uniform over thewhole series of adsorption sites investigated andhence, the relative stability of the di�erent ad-sorption complexes is not a�ected.
Finally, we come to a discussion of the ad-sorption energetics as it emerges from the resultsobtained with ECP and AE models. Besides theincrease of the binding energies in AE calculations,we need to discuss two further limitations of thecomputational methodology employed. Since, sofar the basis set superposition error (BSSE) hasbeen neglected, the calculated interaction energiesmay be somewhat too large. On the other hand,common exchange-correlation approximations(such as the BP functional used in the presentwork) are known to underestimate weak interac-tions of about 5 kcal molÿ1 or less as found herefor some adsorption complexes (Table 3) [37].
To estimate the basis set superposition error, weapplied the counterpoise correction [38] to ureaadsorbed in the most stable hollow adsorptionsite. This correction decreases the ECP bindingenergy by about 3 kcal molÿ1, from ÿ5.2 to ÿ2.4kcal molÿ1. On the other hand, binding energiesobtained from the relativistic AE calculations areexpected to exhibit a larger BSSE e�ect. Indeed,with the counterpoise correction, the interactionenergy of ureylene at the bridge-hollow site is re-duced by about 7 kcal molÿ1, while the ECP resultchanges by about 2 kcal molÿ1 only. However, it isobvious that even with the BSSE correction takeninto account, ECP binding energies are notablyunderestimated compared to the correspondingAE results, by about 10±25 kcal molÿ1. With suchincreased interaction energies (as they are expectedto derive from AE calculations even in cases wherethe ECP approach yields weak interactions only),the above caveat concerning weak interactionsdoes not seem pertinent in the present context.
In any case, the stability trend of the varioussites is expected to be identical for relativistic AEand relativistic ECP results, but some caution isrequired when discussing and interpreting absolutevalues of the interaction energies.
5. Vibrational frequencies of urea and urea species
on Pt(1 0 0)
Despite its apparent simplicity and the consid-erable amount of theoretical and experimentalwork [31±33], the vibrational spectrum of gas-phase urea is still far from being completelyinterpreted. Interestingly, the present DF calcula-tions are in very good agreement with experimen-tal data, especially for the frequencies in the rangefrom 1000 to 3500 cmÿ1, corroborating someprevious assignments that had to be quali®ed astentative [30]. The main interest of the presentwork lies in the mCO stretching frequency of theadsorbed species because this is the only frequencythat is identi®ed in the in situ FTIR experiments.Nevertheless, the gas phase vibrational spectrum isof interest for comparison, too. The calculatedvibrational frequencies together with other avail-able data are reported in Table 4. For gas-phaseurea, the accuracy of the present BP results isnoteworthy, with a mean absolute deviation of 22cmÿ1 and maximum deviations of at most 50 cmÿ1
in the less favourable case. It is also worth pointingout that this accuracy is signi®cantly better thanthat of the MP2 method for the soft modes of urea[31] and comparable to that of empirically cor-rected HF results [32].
For the adsorption complexes, full vibrationalanalyses have been carried out for both adsorbedurea and ureylene. However, since the experi-mental information is rather limited there is littlevalue in reporting the full series of data here. In-stead, selected vibrational frequencies, namely forthe C±O, C±N and N±H symmetric stretchingmotions, are reported in Table 5. Unfortunately,several di�erent adsorption complexes exhibitcalculated C±O stretching frequencies near 1700cmÿ1. For urea, somewhat higher CO frequenciesare calculated, from about 1700±1800 cmÿ1, thanfor ureylene, where the CO frequencies of thevarious cluster models range from about 1600±1700 cmÿ1 (Table 5). Therefore, from these com-puted vibrational data it is impossible to infereither the nature of the adsorbed species or to theactive site. However, for the most stable speciesand adsorption site, ureylene at the bridge-hollowsite, the calculated C±O stretching frequency
M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162 159

amounts to 1693 cmÿ1, which is very close to 1705cmÿ1, the frequency experimentally observed byCliment et al. [11] in in situ FTIR experiments.Remarkably, by comparison to di�erent inorganiccomplexes, this experimental group deduced pre-cisely the same assignment.
Notice that, after urea adsorption as ureylene atthe bridge-hollow site, the C±N stretching modeundergoes a shift to a lower frequency, consistentwith the trend observed in coordination com-pounds having urea as an N-bonded ligand [14].However, the fact that the surface-complex anal-
Table 5
Calculated pseudopotential BP values of the symmetric vibrational frequencies of urea and ureylene (in cmÿ1) at various sites of the
Pt(1 0 0) surfacea
Assignment Ontop
Pt14(8,6)
Hollow
Pt14(8,6)
Bridge-ontop
Pt9(5,4)Bridge-hollow Gas phase urea
Pt9(5,4) Assignment
Ureams(CN) 707 930 737 933 919 ms(CN)
m(CO) 1772 1646 1736 1681 1748 m(CO)
ms(NH2)s 3139 3391 3313 3264 3473 ms(NH2)s
ma(NH2)s 3362 3584 3347 3568 3591 ma(NH2)s
Ureylenems(CN) 1046 772 860 764 (773) 919 ms(CN)
m(CO) 1583 1646 1658 1693 (1686) 1748 m(CO)
ms(NH)s 3491 3410 3280 3395 (3552) 3473 ms(NH2)s
a Frequencies calculated at the all-electron level are given in parentheses. Calculated gas phase frequencies of urea and their as-
signment are given for comparison.
Table 4
Vibrational frequencies (in cmÿ1) of gas-phase urea as obtained from Becke±Perdew, BP, DF calculations compared to available
experimental dataa
Present work Assignment BP Experimental
Ref. [33] Ref. [32] Ref. [30]
ss(NH2) 426 (A)
xa(NH2) 458 (B) 410 227
d(CN) 475 (A) 410?
sa(NH2) 537 (B) 542?
xs(NH2) 557 (B) 578?
d(CO) 585 (A) 578 618?
x(CO) 747 (B) 790 775 790?
ms(CN) 919 (A) 960 1032 1014
qa(NH2) 1014 (B) 1014
qs(NH2) 1148 (A) 1157
ma(CN) 1370 (B) 1394 1394 1394
ds(NH2) 1584 (A) 1594 1604 1594
da(NH2) 1587 (B) 1594 1749 1594
m(CO) 1748 (A) 1734 1776 1734
ms(NH2)a 3466 (B) 3440 3434 3440
ms(NH2)s 3473 (A) 3440 3460 3440
ma(NH2)a 3590 (B) 3548 3533 3448
ma(NH2)s 3591 (A) 3548 3559 3548
a The question marks indicate tentative experimental assignments. The symmetry character of the calculated modes, A or B of the C2
point group, is also given.
160 M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162

ogy permits, in this case, a correct assignment ofthe coordination mode and of the observed vib-rational frequency does not validate this approachin general. It is important to re-iterate that anadsorbate frequency assignment by comparison tothe vibrational frequencies of these adsorbatesacting as ligands in complexes can be very mis-leading [4]. In the present case of urea adsorbed onPt(1 0 0) electrodes, both frequencies obtained ei-ther from the in situ FTIR measurements or in Pt-complexes are fortuitously close, but this is by nomeans a strict rule. Actually, inspection of Table 5reveals that variations of the order of 100 cmÿ1 aretypically encountered for the C±O stretching fre-quency of the two urea species as a function ofcoordination to the Pt atoms of the electrode. Thisclearly indicates the limitation of the surface-complex analogy. Rather, comparison to reason-able theoretical models of the type used in thepresent work provides a more adequate frame-work for understanding the adsorbate±substratechemical bonding that occurs in these adsorptioncomplexes.
Finally, it is worth pointing out that the presentanalysis remains largely unchanged if the compu-tationally more accurate AE approach is em-ployed. In fact, in Table 5 we also present acomparison of the frequencies for the most stablebridge-hollow site obtained either by the ECPapproach or by the scalar-relativistic AE ap-proach. The vibrational frequencies of interestdi�er by less than 4%. Therefore, we conclude thatas far as vibrational frequencies are concerned, theapproximate treatment of the platinum core via arelativistic pseudopotential results in a consider-able saving of computational time without anysigni®cant loss of accuracy.
6. Summary and conclusions
The adsorption of urea and ureylene on aPt(1 0 0) single crystal electrode has been studiedusing two di�erent DF based quantum chemistryapproaches (AE scalar-relativistic and pseudopo-tential) as well as reasonably large cluster modelrepresentations of di�erent active sites of Pt(1 0 0).
The choice for urea and ureylene is related to thefact that electrochemical experiments suggest ureato lose two H atoms in the adsorption processleading to adsorbed ureylene. Four di�erent ad-sorption sites have been modelled and the geom-etry, the interaction energy and the vibrationalfrequencies of both adsorbed species have beencalculated. In the present study, dissociative ureaadsorption ± with two removed hydrogen atoms toadsorbed as atomic hydrogen on Pt(1 0 0) ± isfound to be the most favourable adsorption pro-cess. Thus the present study corroborates previousinterpretations derived from electrochemical ex-periments [11]. The bridge-hollow site, schemati-cally shown in Fig. 2d, turns out to be the moststable adsorption complex of ureylene. Further-more, calculated stretching frequencies of the C±Ovibrations of adsorbed urea and ureylene, also atdi�erent sites, exhibit similar values, all of themrather close to the value obtained in the in situFTIR experiment. Hence, the calculated vibra-tional frequencies alone do not provide enoughinformation for deciding whether the adsorbedspecies is urea or ureylene and even less on thenature of the adsorption site. Yet, the clustermodel calculations permit an assignment of theadsorbed species and the active site based on cal-culated interaction energies. Interestingly, for themost stable species, adsorbed ureylene, at the mostfavourable site, the bridge-hollow site, the calcu-lated value for the C±O stretching frequency isclosest to the experimental value.
To summarise, the relative stability of thedi�erent adsorbed species and the vibrationalfrequency analysis support the hypotheses byCliment et al. [11] that urea adsorbs as ureylene,that this species is bonded to the surface throughthe two nitrogen atoms, and that adsorption oc-curs at the bridge-hollow site. Nevertheless, it isworth pointing out that the hypotheses suggestedin the work of Climent et al. [11] were mostlybased in chemical intuition and on invoking theso-called surface-complex analogy whose success isby far not guaranteed in general [4]. Here, it wasdemonstrated that ``®rst principles'' calculationslead to the same ®nal picture, advocating thattheoretical investigations on models of adsorbatesat surfaces provide an alternative and more secure
M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162 161

way to interpreting in situ electrochemical inter-faces at a molecular level.
Acknowledgements
The authors are indebted to Prof. Juan Feliuand Dr. Victor Climent for bringing the problemof urea adsorption to their attention. M. Garc�õa-Hern�andez is grateful to the ``Generalitat de Ca-talunya'' for a predoctoral grant. This work hasbeen supported by Deutsche Forschungsgemeins-chaft, Fonds der Chemischen Industrie, Spanish``Ministerio de Educaci�on y Ciencia'', project CI-CyT PB98-1216-C02-01, and ``Generalitat de Ca-talunya'', project 1999-SGR-00040.
References
[1] J.L. Whitten, H. Yang, Surf. Sci. Rep. 24 (1996) 59.
[2] P.S. Bagus, F. Illas, in: P.R. Schleyer, N.L. Allinger, T.
Clark, J. Gasteiger, P.A. Kollman, H.F. Schae�er III, P.R.
Schreiner (Eds.), Encyclopedia of Computational Chemis-
try, Encyclopedia of Computational Chemistry, vol. 4,
Wiley, Chichester, 1998, p. 2870.
[3] G. Pacchioni, Heterog. Chem. Rev. 2 (1996) 213.
[4] A. Markovits, M. Garc�õa-Hern�andez, J.M. Ricart, F. Illas,
J. Phys. Chem. B 103 (1999) 509.
[5] M. Rubel, C.K. Rhee, A. Wieckowski, P.A. Rikvold,
J. Electroanal. Chem. 315 (1991) 301.
[6] C.K. Rhee, J. Electrochem. Soc. 139 (1992) 13C.
[7] P.A. Rikvold, A. Wieckowski, Phys. Scripta T 44 (1992) 71.
[8] M. Gamboa-Aldeco, P. Mrozek, C.K. Rhee, A. Wieckow-
ski, P.A. Rikvold, Q. Wang, Surf. Sci. Lett. 297 (1993)
L135.
[9] P.A. Rikvold, M. Gamboa-Aldeco, J. Zhang, M. Han, Q.
Wang, H.L. Richards, A. Wieckowski, Surf. Sci. 335 (1995)
389.
[10] P.A. Rikvold, J. Zhang, Y.-E. Sung, A. Wieckowski,
Electrochim. Acta 41 (1996) 2175.
[11] V. Climent, A. Rodes, J.M. Orts, J.M. Feliu, J.M. P�erez,
A. Aldaz, Langmuir 13 (1997) 2380.
[12] V. Climent, A. Rodes, J.M. Orts, A. Aldaz, J.M. Feliu, J.
Electroanal. Chem. 461 (1999) 65.
[13] K. Nakamoto, Infrared and Raman Spectra of Inorganic
and Coordination Compounds, Wiley, New York, 1986.
[14] R.B. Penland, S. Mizushima, C. Curran, J.V. Quagliano,
J. Am. Chem. Soc. 79 (1957) 1575.
[15] R.W.G. Wycko�, Crystal Structures, second edition, vol.
1, Interscience Publishers, New York, 1965.
[16] N. R�osch, K. Kr�uger, M. Mayer, V.A. Nasluzov, in: J.M.
Seminario (Ed.), Recent Development and Applications of
Modern Density Functional Theory, vol. 4, Theoret.
Comput. Chem. Elsevier, Amsterdam, 1996, p. 497.
[17] B.I. Dunlap, N. R�osch, Adv. Quant. Chem. 21 (1990) 317.
[18] T. Belling, T. Grauschopf, S. Kr�uger, F. N�ortemann, M.
Staufer, M. Mayer, V.A. Nasluzov, U. Birkenheuer, A.
Hu, A.V. Matveev, N. R�osch, Program ParaGauss 2.1,
Technische Universit�at M�unchen, 1999.
[19] T. Belling, T. Grauschopf, S. Kr�uger, M. Mayer, F.,
N�ortemann, M. Staufer, C. Zenger, N. R�osch, in: High
Performance Scienti®c and Engineering Computing, Pro-
ceedings of the First International FORTWIHR Con-
ference, February 1998, in: H.-J. Bungartz, F. Durst,
C. Zenger (Eds.), Lecture Notes in Computational Sci-
ence and Engineering, vol. 8, Springer, Heidelberg, 1999,
p. 439.
[20] A. Hu, M. Staufer, U. Birkenheuer, V. Igoshine, N. R�osch,
Int. J. Quant. Chem. 79 (2000) 209.
[21] D. Andrae, U. Haeussermann, M. Dolg, H. Stoll, H.
Preuss, Theor. Chim. Acta 77 (1990) 123.
[22] O. Gropen, J. Comput. Chem. 8 (1987) 982.
[23] A.M. Ferrari, K.M. Neymann, T. Belling, M. Mayer, N.
R�osch, J. Phys. Chem. B 103 (1999) 216.
[24] F.B. van Duijneveldt, IBM Res. Rep. RJ945, 1971.
[25] S. Huzinaga (Ed.), Gaussian Basis Sets, Elsevier, Amster-
dam, 1984.
[26] M. Dolg, H. Stoll, H. Preuss, R.M. Pitzer, J. Phys. Chem.
97 (1993) 5852.
[27] A.D. Becke, Phys. Rev. A 38 (1988) 3098.
[28] J.P. Perdew, Phys. Rev. B 33 (1986) 8622; Erratum 34
(1986) 7406.
[29] R.D. Brown, P.D. Godfrey, J. Storey, J. Mol. Spectrosc. 58
(1975) 445.
[30] S.T. King, Spectrochim. Acta 28A (1972) 165.
[31] P.D. Godfrey, R.D. Brown, A.N. Hunter, J. Mol. Struct.
413±414 (1997) 405.
[32] B. Rousseau, C. Van Alsenoy, R. Keeulers, H.O. Desseyn,
J. Phys. Chem. A 102 (1998) 6540.
[33] R. Keeulers, H.O. Desseyn, B. Rousseau, C. Van Alsenoy,
J. Phys. Chem. A 103 (1999) 4621.
[34] K. Christmann, Prog. Surf. Sci. 48 (1995) 15.
[35] K. Christmann, Rep. Surf. Sci. 9 (1998) 1.
[36] R.A. Olsen, G.J. Kroes, E.J. Baerends, J. Chem. Phys. 111
(1999) 11155.
[37] A. G�orling, S.B. Trickey, P. Gisdakis, N. R�osch, in:
J. Brown, P. Hofmann (Eds.), Topics in Organometal-
lic Chemistry, vol. 4, Springer, Heidelberg, 1999,
p. 109.
[38] F. Jensen, Introduction to Computational Chemistry,
Wiley, Chichester, 1999.
162 M. Garc�õa-Hern�andez et al. / Surface Science 471 (2001) 151±162





























