Embed Size (px)
Citation preview

INTRODUCCION
Las técnicas voltamétricas tienen su origen en el año 1922, cuando el
químico Jaroslav Heyrovsky desarrolló la polarografía, una técnica
voltamétrica. Por esto recibió el Premio Nobel de Química en 1959. Se
sigue empleando el término de polarografía para la voltametría que
emplea electrodos de mercurio.
Las primeras técnicas de voltamperométricas tuvieron muchos
problemas, que limitaban su viabilidad para el uso diario en la química
analítica. En 1942 Hickling construyó el primer potenciostato de tres
electrodos. Los años 1960 y 1970 fueron testigos de muchos avances en
la teoría, la instrumentación, y la introducción de sistemas añadidos y
controlados por computadoras. Estos avances mejoraron la sensibilidad
y crearon nuevos métodos analíticos. La industria respondió con la
producción más barata de potenciostatos, de electrodos, y de las celdas
que podrían ser utilizados eficazmente en el trabajo analítico de rutina.
La voltametría abarca un grupo de métodos electroanalíticos en los que
la información sobre el analito se deduce de la medición de la corriente
en función del potencial aplicado, en condiciones que favorezcan la
polarización de un electrodo indicador o de trabajo. Cuando la corriente
proporcional a la concentración del analito se controla a potencial fijo, la
técnica se denomina amperometría. En general, con el objetivo de
aumentar la polarización, los electrodos de trabajo en voltametría y
amperometría tienen áreas superficiales pequeñas.
La polarografía es un tipo particular de la voltametría que difiere de
otros tipos de voltametría en que el electrodo de trabajo es electrodo de
goteo de mercurio (DME).
La polarografía ha venido en decadencia debido a cuestiones
relacionadas con las grandes cantidades de mercurio que se utilizan en

los laboratorios. Aunque la importancia de la polarografía ha declinado,
la voltametría y la amperometría en electrodos de trabajo diferentes al
de goteo de mercurio ha crecido a una velocidad impresionante. La
voltametría junto con la amperometría y la cromatografía de líquidos se
han convertido en herramientas poderosas para los análisis de mezclas
complejas.
Principios de la Voltametría
La voltametría es una técnica electro analítica en las que se aplica un
determinado potencial eléctrico a un electrodo (denominado electrodo
de trabajo) sumergido en una disolución que contiene una especie
electro activa y se mide la intensidad eléctrica que circula por este
electrodo. La intensidad medida es función del potencial aplicado y de la
concentración de la especie electro activa presente.
En voltametría se aplica una señal de excitación de potencial variable a
un electrodo de trabajo en una celda electroquímica. Esta señal de
excitación causa una respuesta de corriente característica, que es la
cantidad mensurable en este método.
El análisis de datos requiere la consideración de la cinética, además de
la termodinámica, debido al componente temporal de la
voltamperometría. Idealizadas relaciones termodinámicas
electroquímicas teóricas, tales como la ecuación de Nernst están
elaboradas sin una componente de tiempo. Aunque estos modelos son
insuficientes por sí mismos para describir los aspectos dinámicos de la
voltamperometría, sientan las bases para las relaciones de
voltamperometría modificadas que relacionan la teoría con los
resultados observados.

La representación gráfica de la relación entre el potencial y la corriente
eléctrica se denomina voltamograma.
En el proceso de voltametría se da una reacción de oxidación reducción,
para la cual definiremos la semireacción reversible que se lleva a cabo
en el electrodo de trabajo:
aA+ne−¿↔ pP ¿
Reversible, en el sentido electroquímico, es ligeramente distinto del
sentido de una reacción reversible. En la mayoría de los procesos
químicos, reversible significa que una reacción redox puede suceder en
ambas direcciones hacia delante y hacia atrás. En electroquímica
significa que la reacción redox se produce fácilmente en un sentido o en
otro. Consecuentemente, podemos deducir que la reversibilidad
electroquímica requiere que tanto las especies oxidadas como las
reducidas sean químicamente estables. Otro requisito para la
reversibilidad electroquímica es que la velocidad de transferencia de
electrones, tanto para la oxidación como para la reducción, sea
extremadamente rápida. En este sentido, relativamente pocas
reacciones electroquímicas son realmente reversibles. En disoluciones
acuosas, en casi todas las reacciones electroquímicas reversibles están
presentes iones metálicos. Las reacciones electroquímicas con molé-
culas orgánicas aromáticas tales como aquellas que contienen anillos de
benceno o de naftaleno sólo son reversibles con algunos electrodos o
con ciertos disolventes orgánicos.
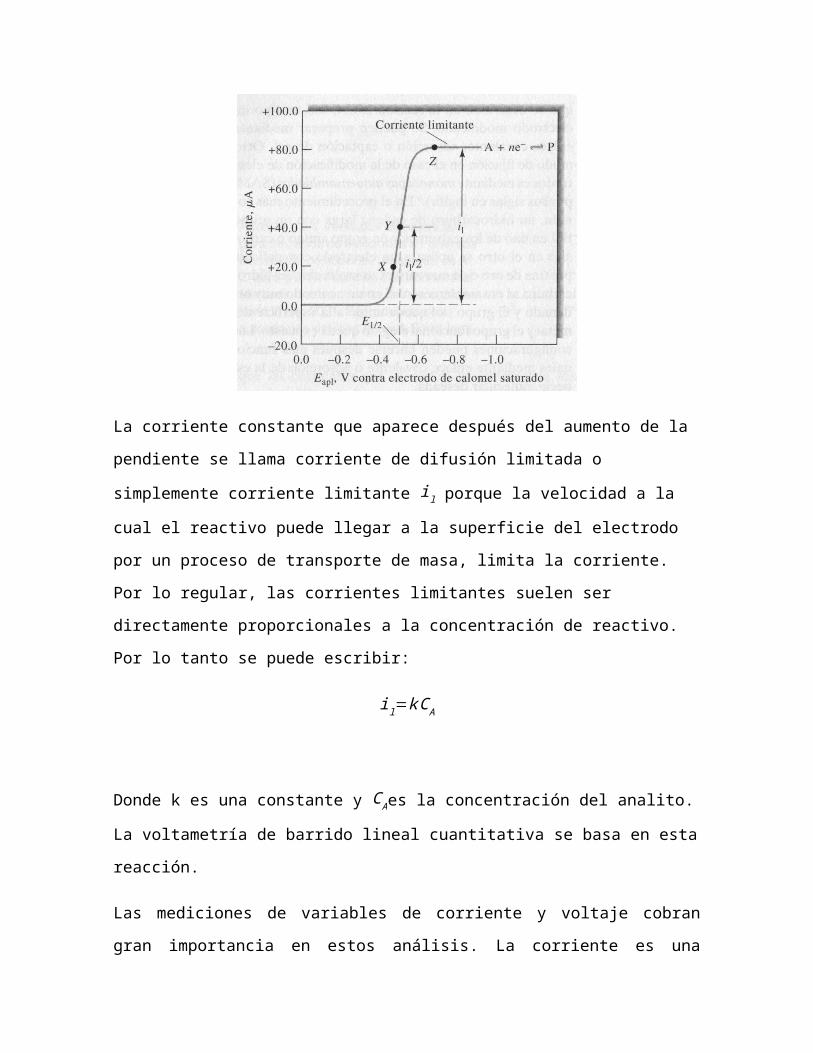
En la figura se ilustra la apariencia de un voltamograma de barrido lineal
típico para una electrólisis en la que hay reducción de una especie del
analito A para dar un producto P en un electrodo de película de
mercurio. En este caso se supone que el electrodo de trabajo está
conectado al polo negativo del generador de barrido lineal de manera
que los potenciales aplicados se dan con signo negativo tal como se

muestra. Por convención, las corrientes catódicas se tratan siempre
como positivas y las corrientes anódicas se dan con signo negativo.
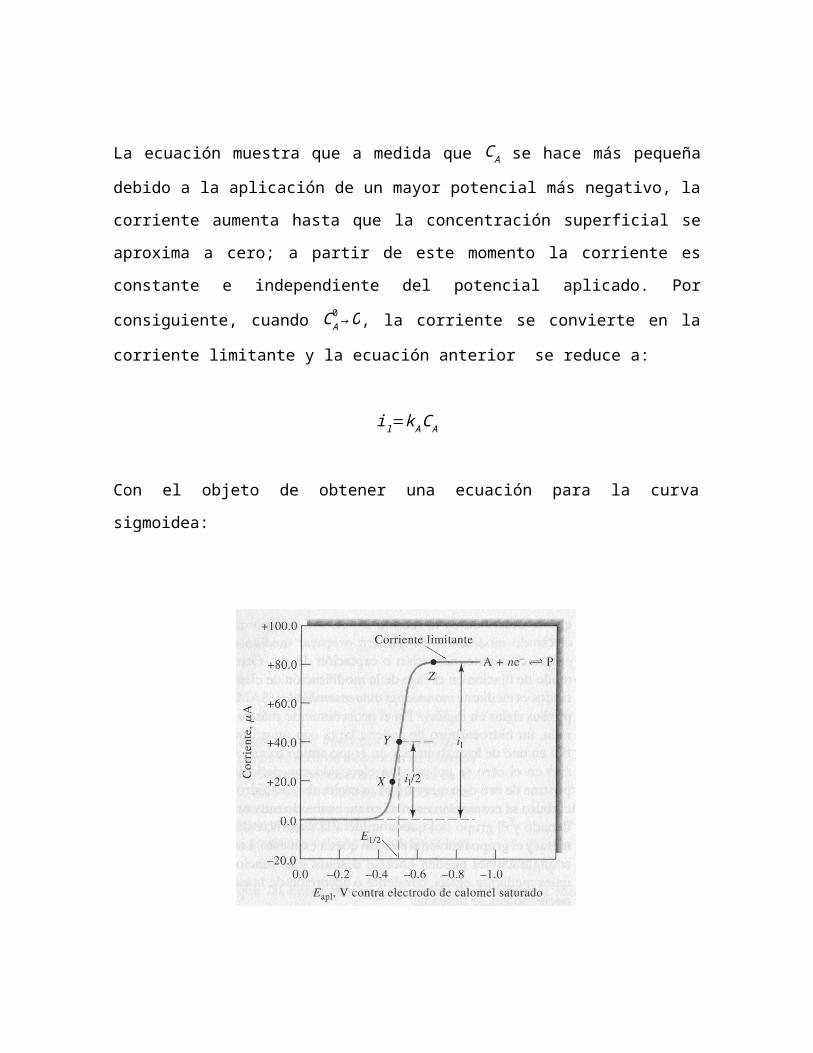
La corriente constante que aparece después del aumento de la
pendiente se llama corriente de difusión limitada o simplemente
corriente limitante il porque la velocidad a la cual el reactivo puede
llegar a la superficie del electrodo por un proceso de transporte de
masa, limita la corriente. Por lo regular, las corrientes limitantes suelen
ser directamente proporcionales a la concentración de reactivo. Por lo
tanto se puede escribir:
il=k C A
Donde k es una constante y C Aes la concentración del analito. La
voltametría de barrido lineal cuantitativa se basa en esta reacción.
Las mediciones de variables de corriente y voltaje cobran gran
importancia en estos análisis. La corriente es una medida que se
relaciona con la velocidad de la reacción ya que esta se define como:
i=dqdt
Y la cantidad de electrones (cargas) transferidos es proporcional al
avance de la reacción antes mencionada.
Se designan a las corrientes originadas por la electrólisis como
corrientes farádicas. Sin embargo, siempre que un electrodo tiene un
potencial aplicado que cambia, hay otra contribución de corriente,
aparte de la debida a la electrólisis, que se llama corriente no farádica;
es el resultado de tener que cargar el electrodo a medida que cambia el
potencial a un nuevo valor. La corriente total medida es la suma de los
dos tipos de corriente.
Durante una electrolisis el reactivo es transportado a la superficie del
electrodo mediante tres mecanismos:
1. Migración: A causa de la influencia de un campo eléctrico
2. Convección: Resultante de la agitación o la vibración
3. Difusión: por las diferencias de concentración entre la capa de
líquido en contacto con la superficie del electrodo y el seno de la
solución
En voltametría se pretende reducir al mínimo el efecto de la migración al
introducir un exceso de electrolito de soporte inactivo. Cuando la
concentración de electrolito soporte excede la del analito en 50 o 100
veces, la fracción de corriente total trasportada por el analito se
aproxima a cero. Como resultado la velocidad de migración del analito
hacia el electrodo de carga opuesta es prácticamente independiente del
potencial aplicado.
Cuando comienza el proceso las concentraciones son:
[A ]0=C A0
[P ]0=CP0=0

Antes del comienzo de la reacción el potencial en el electrodo es:
E0
Se supone que la reacción de reducción es rápida y reversible de modo
que las concentraciones de A y P en la capa de la solución
inmediatamente adyacente del electrodo se expresan en cada momento
mediante la ecuación de Nernst:
Eapl=E A0−
RTnFln(CP
0
CA0 )−E ref
Donde Eapl es el potencial entre el electrodo de trabajo y el electrodo de
referencia, F es la constante de Faraday, EA0 es el potencial de
semireacción en el electrodo, T es la temperatura absoluta, n es el
coeficiente estequiométrico de los electrones transferidos en la
semireacción, CP0 y C A
0 son las concentraciones molares de las especies P
y A en una capa delgada de solución solo en la superficie del electrodo.
Se supone que también que debido a que el electrodo es tan pequeño,
la electrólisis, durante cortos lapsos no altera de manera apreciable la
concentración en el seno de la solución.
Comportamiento de la solución el proceso analítico
Soluciones no agitadas
Cuando se aplica un potencial a un electrodo en ausencia de convección
(solución no agitada) el transporte de masa del analito hasta la
superficie del electrodo es solo por difusión.
Conforme avanza la reacción (después de aplicar un potencial) en la
solución se dan zonas donde las concentraciones de las especies A y P
varían según el tiempo, potencial aplicado y distancia del electrodo.

Para este voltaje aplicado se dan estas condiciones:
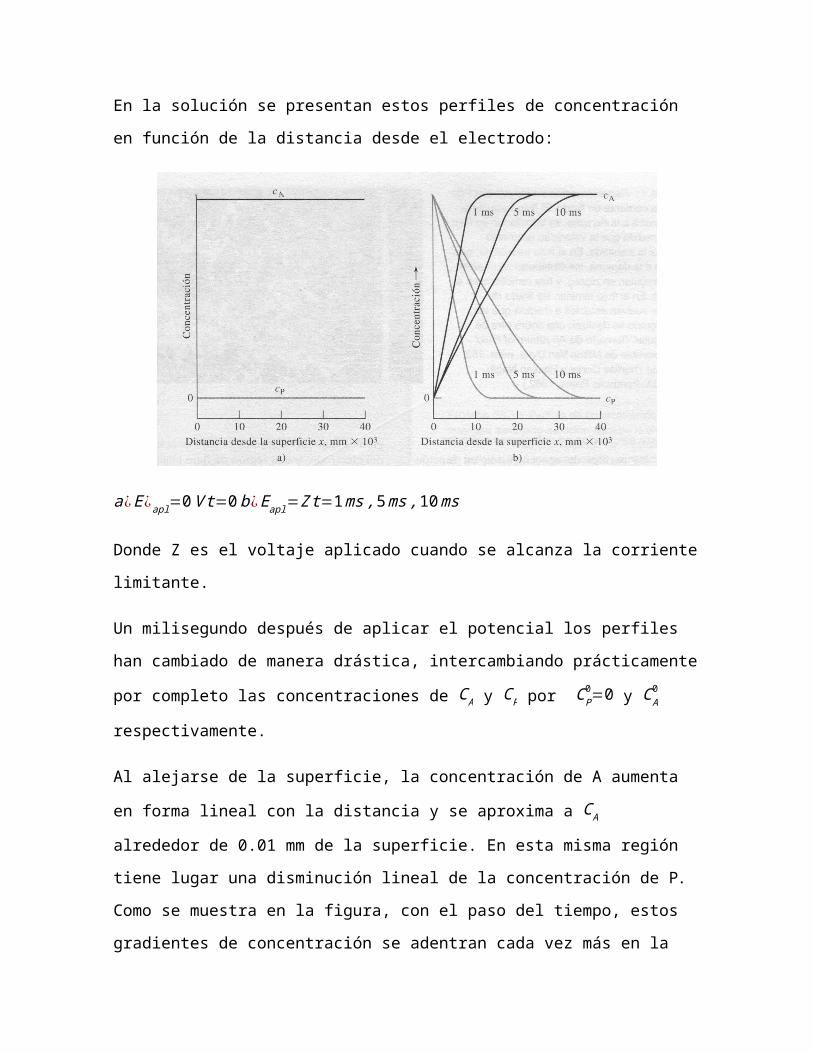
En la solución se presentan estos perfiles de concentración en función
de la distancia desde el electrodo:
a¿E ¿apl=0V t=0b¿Eapl=Z t=1ms,5ms ,10ms
Donde Z es el voltaje aplicado cuando se alcanza la corriente limitante.
Un milisegundo después de aplicar el potencial los perfiles han
cambiado de manera drástica, intercambiando prácticamente por
completo las concentraciones de C A y CP por CP0=0 y C A
0
respectivamente.
Potencial
Eapl
0 t
Corriente µA
0 t
Al alejarse de la superficie, la concentración de A aumenta en forma
lineal con la distancia y se aproxima a C A alrededor de 0.01 mm de la
superficie. En esta misma región tiene lugar una disminución lineal de la
concentración de P. Como se muestra en la figura, con el paso del
tiempo, estos gradientes de concentración se adentran cada vez más en
la solución. La corriente i necesaria para producir estos gradientes es
proporcional a las pendientes de los tramos rectos de las líneas
continuas de la figura. Y se puede modelar así:
i=nFA DA ( dC A
dx )Donde i es la corriente en amperes, n es el número de moles de
electrones por mol de analito, F es la constante de Faraday, A es el área
superficial del electrodo (cm2), DA es el coeficiente de difusión para A
(cm2/s) y C A es la concentración de A (mol/cm3). Como se puede ver en
la figura, estas pendientes dC A
dx se vuelven más pequeñas con el tiempo,
como lo hace la corriente. Al producto DA ( dC A
dx ) se le denomina flujo, el
cual es la cantidad de moles de A por unidad de tiempo por unidad de
área que se difunde hacia el electrodo.
Es impráctico obtener corrientes limitantes con electrodos planos en
soluciones sin agitación porque las corrientes disminuyen de manera
continua respecto al tiempo a medida que se reducen las pendientes de
los perfiles de concentración. Por lo que se vuelve casi necesario utilizar
soluciones agitadas.
Soluciones agitadas
Para entender el efecto de la agitación, es necesario tener una imagen
de los modelos de flujo de líquido en una solución agitada que contiene
un pequeño electrodo plano. Como se puede ver en la figura, es posible
identificar dos tipos de flujo que dependen de la velocidad de flujo
promedio. El flujo laminar se presenta a velocidades de flujo bajas y el
movimiento es uniforme y regular, como se puede ver a la izquierda de
la figura. En cambio, en el flujo turbulento hay altas velocidades, y el
movimiento es irregular y fluctuante, como se aprecia a la derecha.
En la celda electroquímica con agitación se tiene una región de flujo
turbulento en el seno de la solución, lejos del electrodo, y una región de
flujo laminar cerca del electrodo.
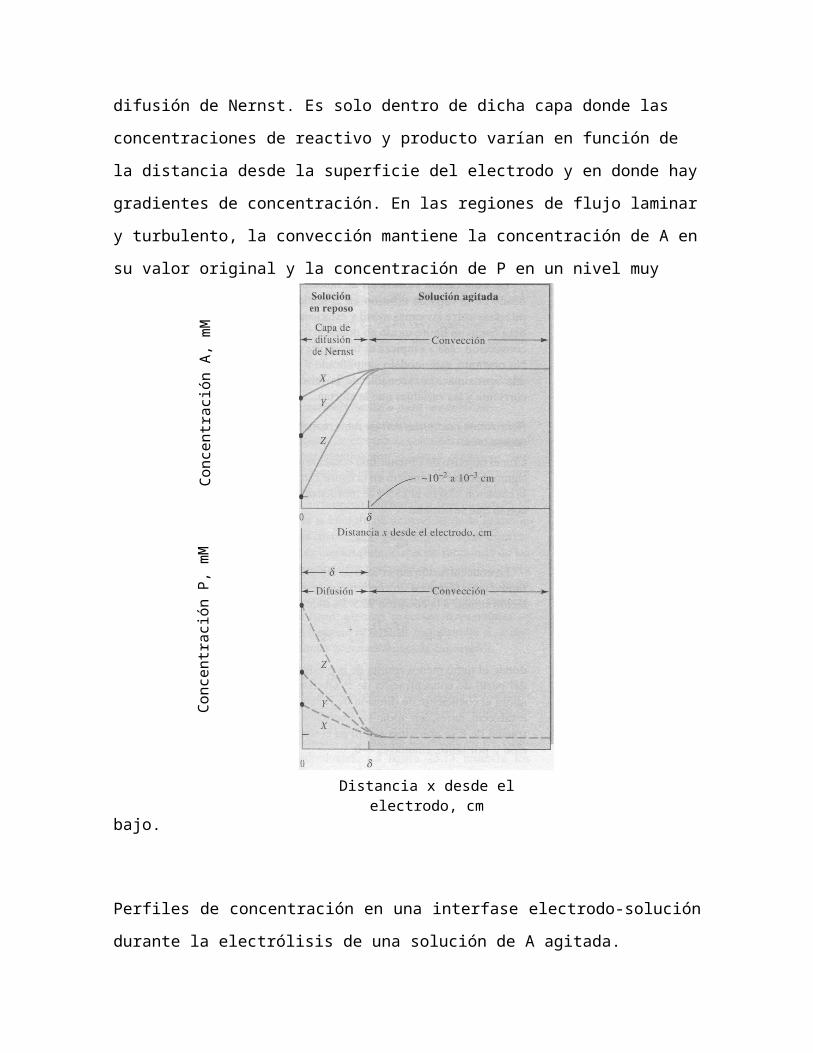
Muy cerca del electrodo, a una distancia δ de la superficie, las fuerzas de
fricción dan origen a una región donde la velocidad de flujo es en
esencia cero. La capa delgada de solución en esta región es una capa
estancada llamada capa de difusión de Nernst. Es solo dentro de dicha
capa donde las concentraciones de reactivo y producto varían en función
de la distancia desde la superficie del electrodo y en donde hay
gradientes de concentración. En las regiones de flujo laminar y
turbulento, la convección mantiene la concentración de A en su valor
original y la concentración de P en un nivel muy bajo.

Perfiles de concentración en una interfase electrodo-solución durante la
electrólisis de una solución de A agitada.
La Figura muestra dos grupos de perfiles de concentración uno para A y
otro para P a los tres potenciales que se indican como X, Y y Z en la Fi-
gura. La disolución se divide en dos regiones. Una representa el seno de
la disolución y se compone de las dos regiones de flujo turbulento y flujo
laminar, donde el transporte de masa tiene lugar por convección
mecánica producida por el agitador. La capa de difusión de Nernst, que
está inmediatamente adyacente a la superficie del electrodo y tiene un
espesor de δ cm. Normalmente, δ oscila entre 10−2 y 10−3 cm,
C A0=
C A
2
Concentración A, mM
CP0=CA
CP0=
C A
2
Concentración P, mM
Distancia x desde el electrodo, cm
CP
C A

dependiendo de la eficacia de la agitación y de la viscosidad del líquido.
En la capa estática de difusión, el transporte de masa tiene lugar sólo
por difusión, igual que en el caso de una disolución sin agitación. Sin
embargo, si se agita la disolución, la difusión se limita a una capa
estrecha de líquido, que no puede ampliarse hacia la disolución ni con el
transcurso del tiempo. Como consecuencia, muy poco después de
aplicar un potencial, aparecen intensidades de corriente constantes
controladas por difusión.
La corriente en cualquier momento de la electrólisis está determinada
por la velocidad de transporte de A desde el límite exterior de la capa de
difusión hasta la superficie del electrodo. Debido a que el producto de la
electrólisis P se difunde desde la superficie y es barrido por convección,
se requiere una corriente continua para mantener las concentraciones
superficiales que demanda la ecuación de Nernst. Sin embargo, la
convección mantiene un suministro constante de A en el borde externo
de la capa de difusión. De este modo, se obtiene una corriente de
estado estable que está determinada por el potencial aplicado. Esta
corriente es una medida cuantitativa de la rapidez con que A está siendo
transportado a la superficie del electrodo y esta velocidad viene dada
por dC A
dx donde x es la distancia en centímetros desde la superficie del
electrodo. En el caso de un electrodo plano, la corriente se obtiene con
la ecuación:
i=nFA DA ( dC A
dx )
Se debe tener en cuenta que dC A
dx es la pendiente de la parte inicial de
los perfiles de concentración, y se puede obtener el valor aproximado de
estas pendientes con:
CA−C A0
δ
Cuando esta aproximación es válida, y la ecuación se reduce a:
i=nFA DA (CA−C A0
δ )=k A(CA−C A0 )
La ecuación muestra que a medida que C A se hace más pequeña debido
a la aplicación de un mayor potencial más negativo, la corriente
aumenta hasta que la concentración superficial se aproxima a cero; a
partir de este momento la corriente es constante e independiente del
potencial aplicado. Por consiguiente, cuando C A0 →O, la corriente se
convierte en la corriente limitante y la ecuación anterior se reduce a:
il=k ACA
Con el objeto de obtener una ecuación para la curva sigmoidea:

Se sustituye las dos últimas ecuaciones y reordenando, se obtiene:
C A0=
il−i
k A
La concentración superficial de P puede expresarse también en términos
de intensidad de corriente utilizando una relación similar a la de A,
expuesta anteriormente:
i=−nFA DP
δ(CP−CP
0 )
Donde el signo menos indica la pendiente negativa del perfil de
concentración de P. Obsérvese que DP es ahora el coeficiente de
difusión de P. Pero ya hemos indicado anteriormente que durante la
electrólisis la concentración de P se aproxima a cero en el seno de la
disolución, por tanto, cuando CP≈0:
i=−nFA DP
δ(CP−CP
0 )=k PC P0
Con las nuevas expresiones se sustituyen en esta ecuación:
Eapl=E A0−
RTnFln(CP
0
CA0 )−E ref
Y se genera esta expresión:
Eapl=E A0− RT
nFln( k A
kP )− RTnFln( i
il−i )−E ref
Cuando i=il2 el tercer término del lado derecho de esta ecuación se hace
igual a cero y, por definición Eapl , es el potencial de semionda:

Eapl=E1/2=EA0−RT
nFln ( k A
k P)−Eref
Reacomodando las dos últimas ecuaciones da una expresión para el
voltamograma:
Eapl=E1/2−RTnFln( i
il−i )A menudo la relación
k A
k P se aproxima a uno por lo que se puede escribir
para la especie A:
E1 /2≈ EA0−Eref
Reacciones irreversibles
Muchos proceros de electrodo voltamétricos, en particular los
relacionados con sistemas orgánicos, son irreversibles, lo que da lugar a
ondas alargadas y no tan bien definidas. La descripción cuantitativa de
tales ondas requiere un término adicional en la ecuación:
Eapl=E1/2=EA0−RT
nFln ( k A
k P)−Eref
que involucre la energía de activación de la reacción tomar en cuenta la
cinética del proceso de electrodo. A pesar de que los potenciales de
semionda de las reacciones irreversibles normalmente muestran cierta
dependencia de la concentración, las corrientes de difusión conservan
una relación lineal con la concentración. Por tanto, algunos procesos
irreversibles se adaptan con facilidad al análisis cuantitativo si se dis-
pone de patrones adecuados para la calibración.
Voltamogramas para mezclas de reactivos
Por lo regular, los reactivos de una mezcla se comportan independientes
unos de otros en un electrodo de trabajo. Por lo consiguiente, el
voltamograma de una mezcla
es simplemente la
suma de las ondas de los
componentes individuales.
Valoraciones
amperométricas
La voltametría hidrodinámica (voltametría de barrido lineal en la cual la
solución o el electrodo de trabajo se mantiene en movimiento) se puede
utilizar para estimar el punto de equivalencia de valoraciones, siempre
que al menos uno de los participantes o de los productos de la reacción
considerada se oxide o reduzca en el microelectrodo. En este caso, se
mide la intensidad de corriente a un potencial fijado de la región de
corriente límite en función del volumen de reactivo (o del tiempo si el
reactivo se genera por un proceso culombimétrico a intensidad
constante). Las representaciones de los datos a ambos lados del punto
de equivalencia son líneas rectas con diferentes pendientes; el punto
final se establece por extrapolación hasta su intersección.
Curvas típicas de valoración amperométrica:

(a) El analito se reduce y no se reduce el reactivo
(b) El reactivo se reduce y no lo hace el analito
(c) Ambos se reducen
Con una notable excepción, las valoraciones amperométricas con un
electrodo indicador se emplean tan sólo en los casos en los que el
producto es un precipitado o un complejo estable. Los reactivos
precipitantes utilizados incluyen el nitrato de plata para los iones haluro,
el nitrato de plomo para el ion sulfato y diversos reactivos orgánicos,
tales como la 8-hidroxiquinoleína, la dimetilglioxima y el cupferrón, para
varios iones metálicos que son reducibles en los microelectrodos.
La utilización de un par de microelectrodos metálicos idénticos para
establecer el punto de equivalencia en las valoraciones amperométricas
ofrece la ventaja de la sencillez del equipo y evita el tener que preparar
y mantener un electrodo de referencia. Este tipo de sistemas se ha
incorporado a los equipos diseñados para las determinaciones
automáticas de rutina de una sola especie, empleando normalmente un
reactivo generado culombimétricamente.
Electrodos giratorios
Para llevar a cabo estudios teóricos de reacciones de oxidación-
reducción, a menudo es de interés conocer de qué manera se ve
afectado el término k A de la ecuación:
il=k ACA
por las condiciones hidrodinámicas del sistema. Un método común para
obtener una descripción rigurosa del flujo hidrodinámico de una solución
agitada se basa en mediciones efectuadas con un electrodo de disco
rotatorio como el que se ilustra en la figura. Cuando el electrodo de

disco gira con rapidez, se establece el modelo de flujo que señalan las
flechas en la figura:
(a) Vista lateral de un electrodo de disco rotatorio que muestra el
modelo de flujo de la disolución
(b) Vista de la base de un electrodo de disco
(c) Vista de la base de un electrodo de anillo-disco
En este caso, el líquido que está en la superficie del disco se mueve
hacia afuera en dirección horizontal desde el centro del dispositivo, lo
que da lugar a un flujo axial hacia arriba para compensar el líquido
desplazado. En este caso es posible plantear un tratamiento riguroso de
la hidrodinámica, el cual origina la ecuación de Levich.
il=0.620nFADω1 /2 v−1 /6C A
Dónde iles la corriente limitante,n es el número de moles de electrones
por mol de analito, F es la constante de Faraday, A es el área superficial
del electrodo, DA es el coeficiente de difusión para la especie A, C A es la
concentración de la especie A, ω es la velocidad angular del disco en

radianes por segundo y v es la viscosidad cinemática (relación entre la
viscosidad de la solución y su densidad).
La ecuación Levich da un modelo de la difusión y el flujo de condiciones
de la solución en torno a un electrodo de disco rotatorio (RDE). La
ecuación Levich da la altura de la onda sigmoidal observada en rotación
voltamperometría disco.
El electrodo de disco giratorio es útil para el estudio de reacciones de
electrodo, pero tiene poca utilidad en análisis. Estudios de este tipo
proporcionan mucha información útil sobre mecanismos y productos
intermedios en reacciones electroquímicas.
Modelo de un circuito de un electrodo de trabajo
A menudo es útil e instructivo representar la celda electroquímica como
un circuito eléctrico que corresponde a la excitación de la misma
manera que la celda. En este análisis la atención se centra sólo en de
trabajo y se supone que el contraelectrodo de trabajo y se supone que
es inerte, grande, no polarizable y sólo sirve para hacer contacto con la
solución del analito. En la figura se muestran tres modelos de circuitos
posibles para la celda electroquímica.

El diagrama físico del electrodo que está arriba del circuito indica la
correspondencia entre los elementos del circuito y las características del
electrodo. Aunque RΩ y Cd representan el comportamiento de los
electrodos reales con mucha exactitud en un amplio intervalo de
frecuencias y sus valores son independientes de la frecuencia, la
impedancia Farádica no lo es.
La impedancia es una magnitud que establece la relación (cociente)
entre la tensión y la intensidad de corriente. Tiene especial importancia
si la corriente varía en el tiempo, en cuyo caso, ésta, la tensión y la
propia impedancia se describen con números complejos o funciones del
análisis armónico.

De los componentes del diagrama físico del electrodo falta por definir la
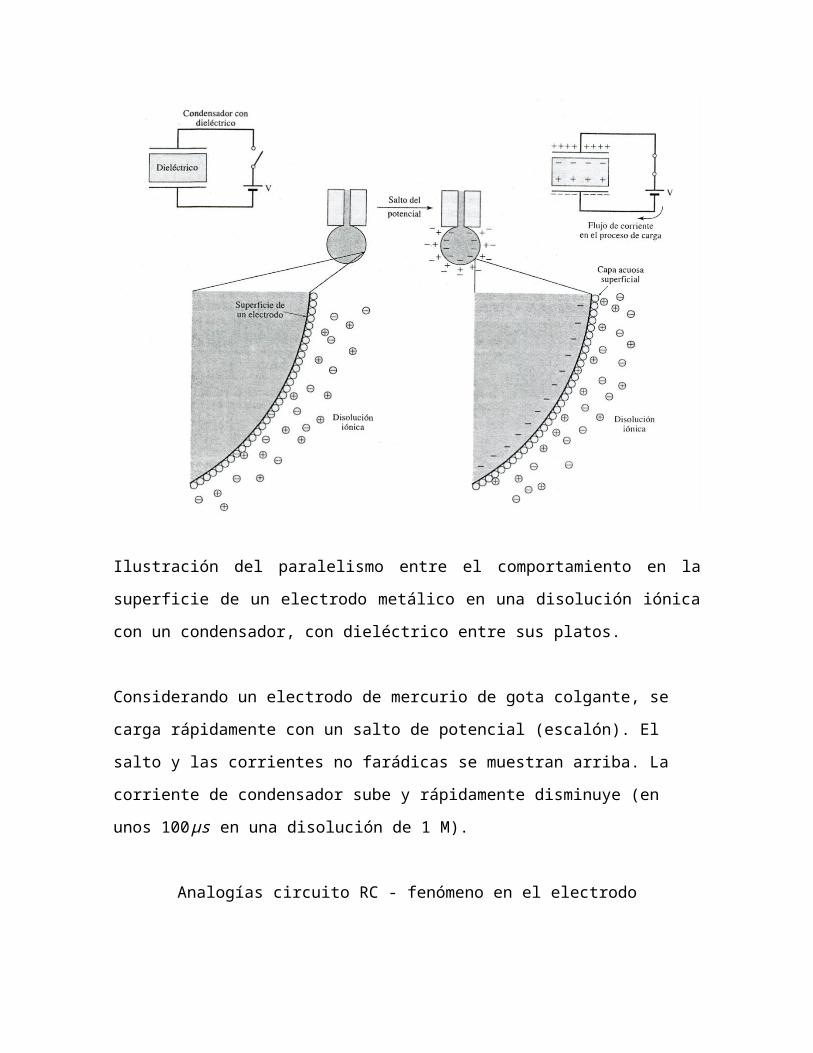
doble capar eléctrica:
La doble capa eléctrica describe la variación del potencial eléctrico
próximo a una superficie. La doble capa es un término que describe el
arreglo que presentan los iones y las moléculas de solvente en solución
al aproximarse a la superficie de un electrodo cargado eléctricamente,
de tal forma que se presentan dos capas con polaridad distinta
separadas por una distancia de orden molecular.
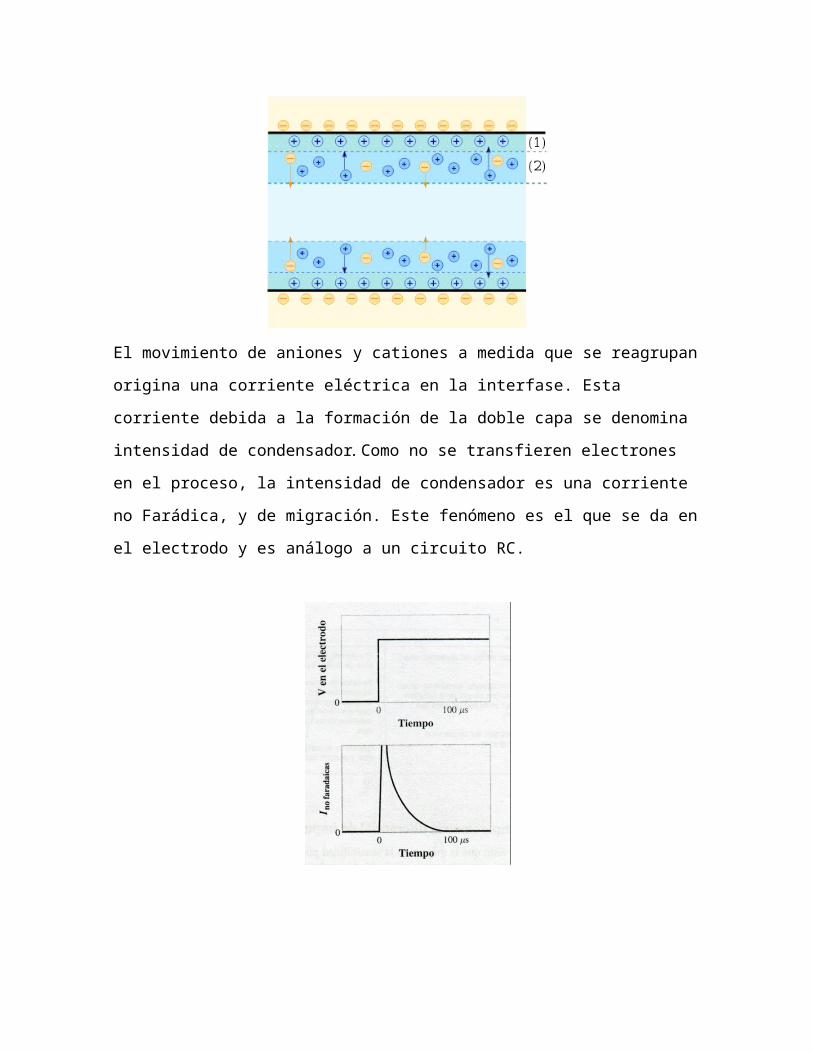
El movimiento de aniones y cationes a medida que se reagrupan origina
una corriente eléctrica en la interfase. Esta corriente debida a la
formación de la doble capa se denomina intensidad de condensador.
Como no se transfieren electrones en el proceso, la intensidad de
condensador es una corriente no Farádica, y de migración. Este
fenómeno es el que se da en el electrodo y es análogo a un circuito RC.

Ilustración del paralelismo entre el comportamiento en la superficie de
un electrodo metálico en una disolución iónica con un condensador, con
dieléctrico entre sus platos.

Considerando un electrodo de mercurio de gota colgante, se carga
rápidamente con un salto de potencial (escalón). El salto y las corrientes
no farádicas se muestran arriba. La corriente de condensador sube y
rápidamente disminuye (en unos 100 μs en una disolución de 1 M).
Analogías circuito RC - fenómeno en el electrodo
Condensador Electrodo
El condensador está descargado, y
las cargas dentro del dieléctrico
están distribuidas al azar.
El potencial que se aplica a las placas
del condensador produce un campo
eléctrico que aparece entre las
placas. Así surgen cargas en la
superficie del dieléctrico. Por tanto,
para que aparezcan estas cargas,
tiene que atravesar corriente
eléctrica por el dieléctrico.
Cuando las placas del condensador se
descargan, las cargas en el dieléctrico
revierten a la distribución original al
azar.
El electrodo está descargado, y los
iones están distribuidos al azar en la
zona de la interface. Si los iones son
adsorbidos, tienden a ser aniones.
Se aplica un potencial negativo al
electrodo, originando un movimiento
de los iones positivos cercanos, que
tienden a moverse hacia la superficie,
y los iones negativos acompañantes
que tienden a alejarse. Este
reagrupamiento de cargas produce
una corriente, a la vez que se
redistribuyen las cargas de forma
menos al azar en la interface: la doble
capa.
Cuando se descarga el electrodo, los
iones vuelven a la distribución más al
azar, y la doble capa se vuelve a
“colapsar”, produciéndose una
corriente.

Voltametría de impulsos
Siempre que un electrodo tiene un potencial aplicado que cambia, hay
otra contribución de corriente, aparte de la debida a la electrólisis, que
se llama corriente no Farádica; es el resultado de tener que cargar el
electrodo a medida que cambia el potencial a un nuevo valor. La
corriente total medida es la suma de los dos tipos de corriente.

Después de estar cargado el electrodo, cuando se descarga la superficie
del electrodo, la doble capa desaparece con la misma rapidez con la que
se cargó. Después de un cambio rápido (un escalón) en potencial, las
corrientes no farádicas aumentan y disminuyen en el tiempo con una
simple dependencia exponencial.
Para la determinación cuantitativa de muchos analitos por
voltamperometría y dentro de un intervalo comprendido entre 10−4 y 10−5
M, no tenemos que preocuparnos de la corriente no farádica; las
corrientes farádicas de la electrólisis del analito son mucho mayores y
duran más tiempo. Sin embargo, para determinar concentraciones de
disoluciones más diluidas, del orden de 10−6 M o inferiores, hay que
poner especial cuidado para separar las corrientes farádicas de las no
farádicas y de las corrientes residuales, que están presentes
simultáneamente. Para realizar esto se utilizan principalmente dos
métodos distintos de voltamperometría de impulsos (Con un electrodo
de gota de mercurio colgante se utiliza el término de polarografía de
impulsos) pero que se basan en los mismos principios básicos:
-Voltametría de onda diferencial de pulsos
-Voltametría de onda cuadrada
El objetivo de la utilización de métodos voltamétricos de impulsos es
disminuir la influencia de las corrientes no farádicas en las medidas.
Basándonos en los comentarios anteriores, podemos establecer dos
propiedades de las corrientes no farádicas:
a) Cuanto más rápido es el cambio de potencial, mayores serán las
corrientes no farádicas.
b) Después de un cambio de potencial, las corrientes no farádicas
disminuyen muy rápidamente en relación con las corrientes
farádicas.
Muchas de las limitaciones de la voltametría tradicional de barrido lineal
fueron eliminadas con el perfeccionamiento de los métodos de pulsos.
La idea en la que se apoyan todos los métodos voltamétricos de pulsos
es medir la corriente en el momento en el que sea grande la diferencia
entre la curva farádica deseada y la corriente de carga que interfiere.
Estos métodos se usan con muy diferentes tipos de electrodos sólidos, el
electrodo de gota colgante de mercurio y los electrodos giratorios.
Para minimizar la interferencia de las corrientes de condensador, la
corriente total se mide justo antes del impulso y de nuevo hacia el final
del mismo (después de cientos de microsegundos). La primera medida
proporciona la línea de base, y las siguientes ocurren cuando las
corrientes no farádicas disminuyen significativamente. Esta mani-
pulación del potencial en el electrodo de trabajo y la toma de datos se
realiza electrónicamente.
En la figura hay un polarógrama de corriente continua (cc) con las
condiciones expresadas en la gráfica. La cuantificación de la cc se
realiza utilizando la corriente límite de la parte superior de la oscilación.





























