
Ein neues System zur (öko-)effizienten Synthese von kleinen, chiralen
Alkoholen
Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der RWTH Aachen University zur Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften genehmigte Dissertation vorgelegt von
Master of Science Biotechnologie Dominik Benjamin Spickermann
aus Waiblingen
Berichter: Universitätsprofessor Dr. rer. nat. Ulrich Schwaneberg Universitätsprofessor Dr. rer. nat. Lothar Elling
Tag der mündlichen Prüfung: 13.04.2016
Diese Dissertation ist auf den Internetseiten der Universitätsbibliothek online
verfügbar.


Angefertigt bei der evocatal GmbH (Monheim) in Kooperation mit dem
Lehrstuhl für Biotechnologie an der RWTH Aachen University (Prof. Dr. rer.
nat. Ulrich Schwaneberg).
Zum Schutz der proprietären Gensequenzen (evocatal GmbH) werden einige
Enzyme nur durch ihre Katalognummern bezeichnet.


“If you take care of your microbial friends, they will take care of your future.”
David Perlman`s “Laws of Applied Microbiology”1


Mein Dank gilt - in besonderer Weise Herrn Prof. Dr. rer. nat. Ulrich Schwaneberg für die freundliche Übernahme des Referates sowie Herrn Prof. Dr. rer. nat. Lothar Elling für die freundliche Übernahme des Korreferates dieser Arbeit. - Herrn Dr. Thorsten Eggert für den Arbeitsplatz und die freundliche Überlassung dieses spannenden Themas. - Herrn Dr. Christian Leggewie für die wissenschaftliche Leitung dieser Arbeit, anregende Diskussionen und hilfreiche Ratschläge. - in besonderer Weise Frau Dr. Andrea Weckbecker für das freundschaftliche Arbeitsverhältnis, das während ihrer kompetenten wissenschaftlichen Betreuung dieser Arbeit entstand. - Herrn Dr. Frank Hollmann und Frau Dr. Selin Kara für die spannende und erfolgreiche Zusammenarbeit im Bereich der Cofaktor-Regenerierung. - Herrn Dr. Christian Degering für seine stete Hilfsbereitschaft bei der Lösung wissenschaftlicher Probleme. - Frau Daniela Christensen für ihre stete Hilfsbereitschaft bei der Lösung (nicht nur) wissenschaftlicher Probleme. Weiterhin danke ich dem gesamten evocatal-Team für die angenehme Arbeitsatmosphäre, die einen nicht unerheblichen Teil zum Erfolg dieses Projekts beigetragen hat, dabei insbesondere - Frau Dr. Stefanie Kind, Herrn Dr. Simon Eßer und Herrn Dr. Sascha Hausmann, die nie müde wurden, mir bei Problemen jeglicher Art weiterzuhelfen. - Herrn Jan Salmon und Herrn Bernd Beckers für den mentalen Beistand während dieser Arbeit. Abschließend danke ich Frau Brigitte Spickermann, Frau Dr. Andrea Weckbecker und Herrn Dr. Christian Degering für die Durchsicht des Manuskriptes dieser Arbeit.

Veröffentlichungen im Rahmen der Promotion Spickermann, D., Kara, S., Barackov, I., Hollmann, F., Schwaneberg, U., Duenkelmann, P., Leggewie, C. (2014). Alcohol dehydrogenase stabilization by additives under industrially relevant reaction conditions. J. Mol. Catal. B: Enzym., 103, 24-8, DOI: 10.1016/ j.molcatb.2013.11.015 Kara, S., Spickermann, D., Weckbecker, A., Leggewie, C., Arends, I. W. C. E., Hollmann, F. (2014). Bioreductions Catalysed by an Alcohol Dehydrogenase in Non-aqueous Media. ChemCatChem, 6(4), 973-6, DOI: 10.1002/cctc.201300841 Kara, S., Spickermann, D., Schrittwieser, J. H., Leggewie, C., van Berkel, W. J. H., Arends, I. W. C. E., Hollmann, F. (2013). More efficient redox biocatalysis by utilizing 1,4-butanediol as a ‘smart cosubstrate’. Green Chem., 15, 330, DOI: 10.1039/C2GC36797A Kara, S., Spickermann, D., Schrittwieser, J. H., Weckbecker, A., Leggewie, C., Arends, I. W. C. E., Hollmann, F. (2013). Access to Lactone Building Blocks via Horse Liver Alcohol Dehydrogenase - Catalysed Oxidative Lactonization. ACS Catal., 3(11), 2436-9, DOI: 10.1021/cs400535c Weitere Veröffentlichungen Spickermann, D., Hausmann, S., Eßer, S., Weckbecker, A., Leggewie, C., Eggert, T., Christensen, D. (2014). Variants of the Parvibaculum lavamentivorans short-chain alcohol dehydrogenase. Application No./Patent No. WO/2014/053558 Spickermann, D., Hausmann, S., Degering, C., Schwaneberg, U., Leggewie, C. (2014). Engineering of highly selective variants of Parvibaculum lavamentivorans alcohol dehydrogenase. ChemBioChem., 15(14), 2050-2, DOI: 10.1002/cbic.201402216 Tagungsbeiträge im Rahmen der Promotion Spickermann, D., Kara, S., Hollmann, F., Hausmann, S., Eggert, T., Puls, M., Leggewie, C.: Innovative approaches for the enzymatic synthesis of short-chain and cyclic chiral alcohols. DECHEMA-Jahrestagung, September 10th – 13th, 2012, Karlsruhe, Germany Spickermann, D., Kara, S., Hollmann, F., Hausmann, S., Eggert, T., Puls, M., Leggewie, C.: Innovative approaches for the enzymatic synthesis of short-chain and cyclic chiral alcohols. Gordon Conference, July 8th – 13th, 2012, Smithfield, USA Kara, S., Spickermann, D., Schrittwieser, J. H., Leggewie, C., van Berkel, W. J. H., Arends, I. W. C. E., Hollmann, F.: Overcoming the thermodynamic challenge in redox biocatalysis with ‘smart cosubstrates’. Biotrans Conference, July 21st – 25th, 2013, Manchester, United Kingdom Kara, S., Spickermann, D., Schrittwieser, J. H., Leggewie, C., van Berkel, W. J. H., Arends, I. W. C. E., Hollmann, F.: More efficient redox biocatalysis by utilising 1,4-butanediol as a smart cosubstrate. NCCC Conference, March 11th – 13th, 2013, Noordwijkerhout, Netherlands

Kara, S., Spickermann, D., Leggewie, C., Zuhse, R., Arends, I. W. C. E., Hollmann, F.: A Novel Eco-Efficient Approach for Biocatalytic Reductions: “smart cosubstrates”. BioCat Conference, September 2nd – 6th, 2012, Hamburg, Germany Kara, S., Spickermann, D., Leggewie, C., Arends, I. W. C. E., Hollmann, F.: Eco-efficient synthesis of chiral alcohols by combining biocatalysis with “smart cosubstrates“. DECHEMA-VAAM-Section Biotransformations, April 24th – 25th, 2012, Frankfurt, Germany Kara, S., Spickermann, D., Leggewie, C., Arends, I. W. C. E., Hollmann, F.: Eco-efficient synthesis of chiral alcohols by combining biocatalysis with “smart cosubstrates“. NCCC Conference, March 5th – 7th, 2012, Noordwijkerhout, Netherlands Puls, M., Duenkelmann, P., Spickermann, D., Leggewie, C.: Pushing the Limits - Innovative Biocatalysis by Engineered Enzymes and Use of “smart cosubstrates”. Zing Conference Biocatalysis, December 4th – 7th, 2012, Xcaret, Mexico


Inhaltsverzeichnis
I
1. Einleitung .............................................................................................................................. 1
1.1 Biokatalyse in der chemischen Industrie ...................................................................... 1
1.2 Alkoholdehydrogenasen (ADHs) ................................................................................... 3
1.3 Alkohole in der Industrie ............................................................................................... 5
1.4 Biologische Verfahren zur Gewinnung enantiomerenreiner Alkohole ..................... 5 1.4.1 „Chirale Pools“ ...................................................................................................................................... 6
1.4.2 Fermentation .......................................................................................................................................... 6 1.4.3 Asymmetrische Reduktion prochiraler Ketone ..................................................................................... 6
1.4.4 Kinetische Racematspaltung ................................................................................................................. 6
1.4.5 Vorteile enzymatischer Verfahren ......................................................................................................... 7
1.5 Chemische Methoden zur Synthese von Alkoholen ..................................................... 7
1.5.1 Nukleophile Substitution aus Halogenalkanen ...................................................................................... 7
1.5.2 Reduktion von Carbonylverbindungen .................................................................................................. 8
1.5.3 Reaktion metallorganischer Verbindungen (Grignard-Reaktion) .......................................................... 8 1.5.4 Aldolreaktion ......................................................................................................................................... 8 1.5.5 Vorteile klassisch-chemischer Verfahren .............................................................................................. 9
1.6 Cofaktor-Regenerierung ................................................................................................ 9
1.7 Identifizierung und Entwicklung neuer Enzyme ....................................................... 11
1.7.1 Metagenombanken .............................................................................................................................. 11
1.7.2 Entdeckung neuer Mikroorganismen................................................................................................... 12
1.7.3 Gentechnische Veränderung bereits bekannter Enzyme (Enzyme Engineering) ................................. 12
1.8 Zielsetzung der Arbeit .................................................................................................. 15
2. Material und Methoden....................................................................................................... 18
2.1 Materialien .................................................................................................................... 18
2.1.1 Geräte .................................................................................................................................................. 18 2.1.2 Chemikalien ........................................................................................................................................ 18
2.2 Mikrobiologische Methoden ........................................................................................ 19 2.2.1 Mikroorganismen und Vektoren .......................................................................................................... 19
2.2.2 Oligonukleotide ................................................................................................................................... 20
2.2.3 Medien ................................................................................................................................................. 21 2.2.4 Isolierung von Nukleinsäuren .............................................................................................................. 22
2.2.5 Gelelektrophorese von Nukleinsäuren ................................................................................................. 22
2.2.6 in vitro-Rekombination von Nukleinsäuren ........................................................................................ 22 2.2.7 Amplifikation von Nukleinsäuren durch Polymerase-Kettenreaktion (PCR) ...................................... 22 2.2.8 QuikChange-Mutagenese .................................................................................................................... 23
2.2.9 Sättigungsmutagenese ......................................................................................................................... 23
2.2.10 Ungerichtete Mutagenese mittels epPCR .......................................................................................... 23
2.2.11 DNA-Sequenzierungen...................................................................................................................... 24
2.2.12 Transformation von Bakterienzellen mit Plasmid-DNA durch Hitzeschock ..................................... 24 2.2.13 Transformation von Bakterienzellen mit Plasmid-DNA durch Elektroporation ............................... 24 2.2.14 Kultivierung von Bakterien und Proteinexpression ........................................................................... 25
2.2.15 Zellaufschluss/ Rohextraktgewinnung .............................................................................................. 25
2.2.16 Lagerung von Proteinlösungen .......................................................................................................... 26
2.2.17 Anzucht und Expression im Hochdurchsatzverfahren ....................................................................... 26
2.3 Biochemische Methoden............................................................................................... 27 2.3.1 ADH evo-040 Aktivitätsassay ............................................................................................................. 27
2.3.2 Hochdurchsatz-Screening von Variantenbibliotheken ........................................................................ 28 2.3.3 Quantitative Proteinmessungen ........................................................................................................... 28
2.3.4 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) ........................................................................ 29 2.3.5 Gaschromatographische Umsatz- und ee-Bestimmung ....................................................................... 29
2.3.6 Computerprogramme ........................................................................................................................... 30

Inhaltsverzeichnis
II
3. Ergebnisse Teil 1: Entwicklung eines Prozesses zur biokatalytischen Darstellung von (S)-2-Butanol ........................................................................................................................... 31
3.1 Identifizierung einer geeigneten Alkoholdehydrogenase (ADH) ............................. 31
3.1.1 Etablierung einer analytischen Methode zum Nachweis von 2-Butanon und der quantitativen Analyse der Stereoisomeren des 2-Butanols .............................................................................................................. 31
3.1.2 Identifizierung von Alkoholdehydrogenasen, die 2-Butanon als Substrat akzeptieren ....................... 32 3.1.3 Ermittlung der Enantioselektivitäten der aktiven Alkoholdehydrogenasen ........................................ 34 3.1.4 Screening nach lösemittelstabilen Alkoholdehydrogenasen ................................................................ 35
3.2 Produktracemisierung.................................................................................................. 37 3.2.1 Untersuchung der Produktstabilität ..................................................................................................... 37
3.2.2 Einfluss der Reaktionstemperatur auf die Produktracemisierung ........................................................ 37 3.2.3 Einfluss des pH-Wertes auf die Produktracemisierung ....................................................................... 38 3.2.4 Führt eine Schädigung der Strukturintegrität des Enzyms zur Erniedrigung der Selektivität? ........... 39 3.2.5 Einfluss des Cosubstrates auf die Produktracemisierung .................................................................... 40 3.2.6 Läuft die Rückoxidation des 2-Butanols enantioselektiv ab? .............................................................. 41
3.3 Festlegung eines geeigneten Ausgangsenzyms für die Prozessentwicklung ............ 42
3.4 Nähere Untersuchung der Lösemittelstabilität der ADH evo-040 ........................... 42
3.5 Enzymstabilisierung in organischem Lösemittel mit Hilfe von Additiven .............. 43 3.5.1 Stabilisierung der ADH evo-030 ......................................................................................................... 45
3.5.2 Stabilisierung der ADH evo-040 ......................................................................................................... 46
3.5.3 Stabilisierung der LB-ADH ................................................................................................................. 46
3.5.4 Stabilisierung der ADH evo-270 ......................................................................................................... 46
3.5.5 Stabilisierung der ADH evo-380 ......................................................................................................... 46
3.5.6 Up-scaling und Validierung der Ergebnisse ........................................................................................ 47
3.6 Bestimmung der optimalen Substratbeladung (2-Butanon) für die ADH evo-040 48
3.7 Bestimmung des pH-Optimums für die ADH evo-040 .............................................. 50
3.8 Zugabereihenfolge der organischen Komponenten................................................... 50
3.9 Einsatz von “smart cosubstrates” zur Erhöhung der Prozesseffizienz ................... 52
3.10 Prozess Up-scaling ...................................................................................................... 55
3.10.1 Produktion des Biokatalysators ......................................................................................................... 55
3.10.2 Durchführung der Biotransformation bis 4 Liter ............................................................................... 57
3.11 Produktaufreinigung .................................................................................................. 58 3.11.1 Produkttrocknung über ein Molekularsieb ........................................................................................ 58
3.11.2 Produkttrocknung mittels Azeotroprektifikation ............................................................................... 58
3.11.3 Produktaufreinigung mittels Destillation ........................................................................................... 58
Ergebnisse Teil 2: Enzymoptimierung durch Verfahren des Protein Engineerings ............ 60
3.12 Rationales Proteindesign durch den Einbau von Disulfidbrücken ........................ 60 3.12.1 Produktion der Enzymvarianten ........................................................................................................ 60
3.12.2 Untersuchung der Hitzestabilität der Disulfidvarianten .................................................................... 62 3.12.3 Kinetische Parameter der Disulfidvarianten ...................................................................................... 63
3.13 Semi-rationales Proteindesign mittels „Bio-Prodict 3DM“ ..................................... 64 3.13.1 Produktion der Enzymvarianten ........................................................................................................ 64
3.13.2 Untersuchung der Hitzestabilität der 3DM-Varianten ....................................................................... 67 3.13.3 Kinetische Parameter der 3DM-Varianten ........................................................................................ 68
3.13.4 Einsatz der 3DM-Varianten im (S)-2-Butanol Prozess ...................................................................... 69
3.14 Zufallsmutagenese mittels epPCR ............................................................................ 71 3.14.1 Entwicklung der Enzymvarianten ..................................................................................................... 71
3.14.2 Untersuchung der Hitzestabilität der Varianten aus der Zufallsmutagenese ..................................... 73 3.14.3 Kinetische Parameter der Varianten aus der Zufallsmutagenese ....................................................... 75 3.14.4 Einsatz der Varianten aus der Zufallsmutagenese im (S)-2-Butanol Prozess .................................... 76

Inhaltsverzeichnis
III
4. Diskussion ............................................................................................................................ 79
4.1 Identifizierung eines geeigneten Biokatalysators....................................................... 80
4.2 Produktracemisierung.................................................................................................. 82
4.3 Enzymstabilisierung in organischem Lösemittel ....................................................... 87
4.4 Prozessentwicklung ...................................................................................................... 90 4.4.1 Lösemittelstabilität der evo-040 .......................................................................................................... 92
4.4.2 Substratbeladung ................................................................................................................................. 93
4.4.3 Zugabereihenfolge der organischen Komponenten ............................................................................. 93 4.4.4 Bestimmung des pH-Optimums .......................................................................................................... 94
4.4.5 „smart cosubstrates“ ............................................................................................................................ 94
4.4.6 Prozess Up-scaling .............................................................................................................................. 95 4.4.7 Produktaufreinigung ............................................................................................................................ 96
4.5 Enzymoptimierung ....................................................................................................... 97 4.5.1 Rationales Proteindesign durch den Einbau von Disulfidbrücken ...................................................... 99
4.5.2 Semi-Rationales Proteindesign mittels „Bio-Prodict 3DM“ ............................................................. 101
4.5.3 Zufallsmutagenese mittels epPCR ..................................................................................................... 102
4.5.4 Interpretation der Mutationen ............................................................................................................ 106
Zusammenfassung................................................................................................................. 111
Summary ................................................................................................................................ 113
Literaturverzeichnis ............................................................................................................... 115
Anhang................................................................................................................................... 135

Inhaltsverzeichnis
IV

Abbildungen und Tabellen
V
Verzeichnis der Abbildungen und Tabellen
Abb. 1: Verteilung von Umsatz und F&E-Ausgaben dedizierter Biotechnologie-Unternehmen 2014 ............................................................................................................................................ 2
Abb. 2: Schematische Darstellung einer ADH-katalysierten Reduktionsreaktion. ................... 3
Abb. 3: Möglicher Übergangszustand im Reaktionsmechanismus der SDR-ADH aus Drosophila lebanonensis ............................................................................................................ 4 Abb. 4: Reaktionen der alkoholischen Funktion ........................................................................ 5 Abb. 5: Asymmetrische Reduktion mittels einer enantioselektiven ADH ................................. 6
Abb. 6: Kinetische Racematspaltung mittels einer enantioselektiven ADH. ............................. 7
Abb. 7: Beispiele zur Gewinnung von alkoholischen Verbindungen mittels nukleophiler Substitution ................................................................................................................................. 7
Abb. 8: A) 1,2-Diamin-Ruthenium(II)-Cl2-BINAP-Katalysator B) ein Oxazaborolidin. ......... 8
Abb. 9: Prinzip der Cofaktor-Regenerierung. .......................................................................... 10 Abb. 10: Schematische Darstellung der Cofaktor-Regenerierung mittels FDH ...................... 10
Abb. 11: Schematische Darstellung der Cofaktor-Regenerierung mittels G(-6-P-)DH-Reaktion .................................................................................................................................................. 11
Abb. 12: Gaschromato-graphische Analyse der Reaktionskomponenten. ............................... 32
Abb. 13: Analyse der Produktstabilität. ................................................................................... 37 Abb. 14: Einfluss der Cosubstrat-konzentration auf die Produkt-racemisierung. .................... 40
Abb. 15: Selektive Umsetzung von 2-Butanol in Abhängigkeit von seiner Stereo-konfiguration. ........................................................................................................................... 42
Abb. 16: Verhalten des Enzyms evo-040 bei unterschiedlichen Lösemittelbeladungen. ........ 43
Abb. 17: Umsatz von 2-Butanon im 50 ml Up-scaling Ansatz, erzielt durch die Sorbitol-stabilisierte evo-040 und das unstabilisierte Enzym (Referenz) .............................................. 48 Abb. 18: Bestimmung der optimalen Substrat-beladung (2-Butanon) für die ADH evo-040. 49
Abb. 19: Bestimmung des pH-Optimums für die ADH evo-040. ............................................ 50
Abb. 20: Auswirkung der Zugabereihenfolge der Reaktionskomponenten auf den Verlauf der Biokonversion. ......................................................................................................................... 52
Abb. 21: Prognostizierter Oxidationsmechanismus vom α,ω-Diol zum entsprechenden Lactonprodukt .......................................................................................................................... 53
Abb. 22: Screening von ADHs zur Oxidation von 1,4-Butandiol. .......................................... 53
Abb. 23: Substratumsatz nach 72 Stunden in der HL-ADH katalysierten Reduktion von Zimtaldehyd in Gegenwart von 0,5 Äquivalenten 1,4-BD (1,4-Butandiol) oder 0,5 – 5 Äquivalenten Isopropanol. ....................................................................................................... 54
Abb. 24: Expression der ADH evo-040 für die Biotransformation. ........................................ 56
Abb. 25: Verlauf der Bioreduktion von 2-Butanon im 1000 ml Maßstab ............................... 57
Abb. 26: Evaluierung der idealen QuikChange-Bedingungen zur Generierung der Disulfidvarianten. ..................................................................................................................... 61
Abb. 27: SDS-Gelelektrophorese der löslichen und unlöslichen Proteinfraktionen der Disulfidmutanten. ..................................................................................................................... 62
Abb. 28: Untersuchung der Temperaturstabilität der Disulfidvarianten. ................................. 63
Abb. 29: Bestimmung von Km und Vmax der Disulfidvarianten SSB5 und SSB6. ............... 64
Abb. 30: SDS-Gelelektrophorese der löslichen Proteinfraktionen einiger 3DM-Enzymvarianten. ....................................................................................................................... 67
Abb. 31: Untersuchung der Temperaturstabilität der 3DM-Varianten. ................................... 68
Abb. 32: Bestimmung von Km und Vmax der 3DM-Varianten. ............................................. 69
Abb. 33: Umsatzverläufe der 3DM-Varianten in einem wasserreichen Prozess zur Dar-stellung von 2-Butanol. ............................................................................................................ 70
Abb. 34: Einsatz der 3DM-Varianten im wasserarmen Reaktionsprozess. ............................. 71

Abbildungen und Tabellen
VI
Abb. 35: Screening-Ergebnisse der besten Varianten aus der Zufallsmutagenese. ................. 73
Abb. 36: SDS-Gelelektrophorese der löslichen Proteinfraktionen von Enzymvarianten aus der Zufallsmutagenese. ................................................................................................................... 74
Abb. 37: Untersuchung der Temperaturstabilität der Varianten aus der Zufallsmutagenese. . 75
Abb. 38: Bestimmung von Km und Vmax der Varianten aus der Zufallsmutagenese. ........... 76
Abb. 39: Umsatzverläufe der Varianten aus der Zufallsmutagenese im wasserreichen Prozess zur Darstellung von 2-Butanol. ................................................................................................ 77 Abb. 40: Einsatz der Varianten aus der Zufallsmutagenese im wasserarmen Reaktions-prozess. ..................................................................................................................................... 78
Abb. 41: Schematische Darstellung des Reaktionsweges einer ADH-katalysierten Substratreduktion sowie des beschriebenen Racemisierungseffekts nach Yang et al ............. 83 Abb. 42: Hypothetisches Verfahren zur dynamisch-kinetischen Racematspaltung nach Martín-Matute et al. ................................................................................................................. 91 Abb. 43: Schematische Übereinanderlegung des Proteinrückgrats der beiden erstellten Homologiemodelle von evo-040. ........................................................................................... 100 Abb. 44: Charakterisierung von 50 Sequenzen der ADH-Mutanten. .................................... 104
Abb. 45: Schematische Darstellung der 14 Mutationspositionen, die zu veränderten Eigenschaften des Enzyms führten. ....................................................................................... 108 Tab. 1: Beispiele industriell genutzter ADHs zur Produktion von chiralen Alkoholen ............. 4
Tab. 2: Verwendete Mikroorganismen ..................................................................................... 19 Tab. 3: Verwendete Vektoren .................................................................................................. 19 Tab. 4: Verwendete Oligonukleotide. ...................................................................................... 20 Tab. 5: Protokoll zur Durchführung der epPCR. ..................................................................... 24 Tab. 6: Verwendete Computerprogramme. .............................................................................. 30 Tab. 7: Übersicht über die im Initial-Screening aktiven Enzyme nach 24 Stunden Reaktionsdauer. ........................................................................................................................ 33
Tab. 8: Übersicht über die Aktivitäten der eingesetzten Enzympräparate. .............................. 34
Tab. 9: Übersicht über die Enantiomerenreinheiten der ADH-Reaktionen nach 24 Stunden. . 35
Tab. 10: Bewertung der Lösemitteltoleranz der ausgewählten ADHs. .................................... 36
Tab. 11: Enantiomerenreinheit am thermodynamischen Gleichgewicht bei unterschiedlichen Reaktionstemperaturen. ............................................................................................................ 38
Tab. 12: Enantiomerenreinheit in Abhängigkeit vom pH-Wert der Reaktionslösung. ............ 39
Tab. 13: Einfluss einer strukturschädigenden Vorinkubation auf die Selektivität der ADH evo-270. ........................................................................................................................................... 39
Tab. 14: Additive, die auf ihren stabilisierenden Effekt auf ADHs untersucht wurden .......... 44
Tab. 15: Konzentrationen, in denen die Additive im ersten Screening-Ansatz eingesetzt wurden. ..................................................................................................................................... 45
Tab. 16: Einfluss der effektivsten Additive auf den relativen Substratumsatz der ADHs. ...... 47
Tab. 17: Substratumsatz der ADH evo-040 bei der Reduktion von 2-Butanon (2,3 M) durch Einsatz verschiedener Cosubstrate. .......................................................................................... 55 Tab. 18: Destillationsprotokoll der Aufreinigung von 320 ml Reaktionslösung (mit 500 ml Cyclohexan vesetzt) ................................................................................................................. 59
Tab. 19: Disulfidvarianten, die zur Konstruktion ausgewählt wurden .................................... 61
Tab. 20: Die von „Bio-Prodict 3DM“ zur Sättigung vorgeschlagenen Positionen. ................. 65
Tab. 21: Ergebnisse der beiden Screenings der 3DM-Varianten. ............................................ 66
Tab. 22: Anteil der inaktiven Enzymvarianten je 3DM-Position ............................................. 66 Tab. 23: Bestimmung von Km und Vmax der 3DM-Varianten. .............................................. 68

Abbildungen und Tabellen
VII
Tab. 24: Bestimmung von Km und Vmax der Varianten aus der Zufallsmutagenese. ............ 76
Tab. 25: Mischverhältnisse und daraus resultierende Siedepunkte der Reaktionskomponenten .................................................................................................................................................. 92
Tab. 26: Übersicht über die Mutationen in den ADH evo-040 Varianten. ............................ 107

Abbildungen und Tabellen
VIII

Abkürzungen
IX
Abkürzungen
Im Folgenden sind die in dieser Arbeit verwendeten Abkürzungen aufgeführt. Aminosäuren
wurden nach dem gebräuchlichen Ein- oder Drei- Buchstabencode abgekürzt.
λ Wellenlänge
A. dest. Aqua destillata
ADH Alkoholdehydrogenase
AS Aminosäure
Abb. Abbildung
Amp Ampicillin
ATP Adenosintriphosphat
bp Basenpaar(e)
°C Grad Celsius
CBBG Coomassie-Brilliant-Blue G-250
C-Terminus Carboxyterminus
Da Dalton
DNA Desoxyribonukleinsäure
dNTP Desoxynucleosid-triphosphat
DWP Deepwell-Mikrotiterplatte, 96 x 2 ml
DTT Dithiothreitol
EC enzyme code (internat. Konvention)
EDTA Ethylendinitrilotetraacetat
ee enantiomeric excess (Enantiomerenüberschuss)
epPCR error prone polymerase chain reaction (fehlerhafte PCR)
EtOH Ethanol
FID Flammenionisationsdetektor
F&E Forschung und Entwicklung
g normale Erdbeschleunigung
g Gramm
GZE Ganzzellextrakt
h Stunde(n)
HTS Hochdurchsatz-Screening (high-throughput screening)
IPC in-process control (In-Prozess Kontrolle)

Abkürzungen
X
iPrOH 2-Propanol
IPTG Isopropyl-β-thiogalactosid
kb Kilobasen
kDa Kilodalton
l Liter
log P log (Octanol/Wasser-Verteilungskoeffizient)
Lsg. Lösung
LV Leervektor
Km Michaeliskonstante
M molar
MB Molenbruch
NAD Nicotinsäureamid-Adenin-Dinukleotid
NADP Nicotinsäureamid-Adenin-Dinukleotid-Phosphat
MCS multiple cloning site
min Minute(n)
ml Milliliter
MTP Mikrotiterplatte, 96 x 0,5 ml
µ mikro
n nano
nm Nanometer
N-Terminus Aminoterminus
org. organisch
O.D. optische Dichte
p.A. per analysis
PAGE Polyacrylamid-Gelelektrophorese
PCR Polymerase-Kettenreaktion
φ Volumenanteil
rpm Umdrehungen pro Minute (rounds per minute)
RT Raumtemperatur
s Sekunde(n)
SDS Natriumdodecylsulfat
SOP standard operating procedure (Standardvorgehensweise)
Tab. Tabelle
Tris Trishydroxymethylaminoethan

Abkürzungen
XI
ÜK Übernachtkultur
ü.N. über Nacht
U Unit (Enzymeinheit)
UpM Umdrehungen pro Minute
Vmax Maximalumsatzgeschwindigkeit
v/v Volumen pro Volumen
w/v Gewicht pro Volumen
WT Wildtyp

Abkürzungen
XII

Einleitung
1
1. Einleitung
1.1 Biokatalyse in der chemischen Industrie
Enzyme als biologische Katalysatoren sind in der chemischen Synthese längst keine
Besonderheit mehr. So bezieht eine stetig steigende Zahl chemischer Unternehmen
enzymkatalysierte Reaktionsschritte in die Planung ihrer Synthesen ein2. Dabei sind die
Einsatzgebiete vielfältig: von der Produktion pharmazeutischer und landwirtschaftlicher
Molekülzwischenstufen (building blocks)3 über Feinchemikalien bis hin zur Produktion von
Molekülblockbustern (bulk chemicals) und Biokraftstoffen4.
Besonders im Bereich der Feinchemiesynthese halten Enzyme Einzug. Feinchemikalien sind
nach Ciriminna et al. solche Verbindungen, die in reiner Form in batch-Reaktoren mit
limitiertem Volumen (< 5000 Tonnen/Jahr) und hohem Wert (> 10 $/kg) gewonnen werden5.
In diesem Fall ist der Einsatz von Enzymen besonders interessant, da sie eine sehr hohe
Stereoselektivität aufweisen können. Die dadurch generierte Enantiomerenreinheit des
Produkts ist vor allem im Bereich von Pharmaintermediaten, Aromen und Duftstoffen
unerlässlich6,7. Dies liegt in der Tatsache begründet, dass die Enantiomere eines Produktes oft
voneinander abweichende Eigenschaften aufweisen. Beispielsweise besitzt (R)-Limonen ein
orangenartiges Aroma, während die (S)-Form nach Terpentin riecht. Ähnliches gilt in der
Pharmaindustrie, wobei hier die Wirksamkeit aktiver Moleküle stark von ihrer
Stereokonformation abhängen kann. Der β-Blocker Propranolol ist beispielsweise in der
(-)-Form etwa 100fach stärker wirksam als in der (+)-Form8, was an der chiralen Natur seines
Rezeptors liegt.
Enzyme werden schätzungsweise bei der Synthese von 2/3 der chiralen Produkte im
industriellen Großmaßstab eingesetzt, da sie es ermöglichen, die Ausbeute von 50 % auf
100 % zu erhöhen9. Beispiele für chirale Medikamente, die mit Hilfe von Enzymen produziert
werden, sind Sitagliptin (Januvia), Rosuvastatin (Crestor), Atorvastatin (Lipitor) und
Montelukast (Singulair)5,10. Besonders bekannte Enzyme, die in der Industrie eingesetzt
werden, sind Penicillin-Amidase11, Xylose-Isomerase12 und Nitril-Hydratase9,13.
Allgemeine Vorteile von Biokatalysatoren gegenüber der klassisch-chemischen Synthese
liegen vor allem in milderen Reaktionsbedingungen. So sind Enzyme überwiegend bei
physiologischen pH-Werten, niedrigen Temperaturen und Drücken aktiv. Dadurch können
Synthesen energieeffizienter durchgeführt werden. Weiterhin können Enzyme auch
hochregioselektiv katalysieren, wodurch auf das Aktivieren beziehungsweise Schützen von
funktionellen Gruppen im umzusetzenden Molekül verzichtet werden kann.

Einleitung
2
Probleme der Biokatalyse liegen dagegen noch immer in ihrer begrenzten Einsetzbarkeit.
Diese kann je nach Enzym auf wenige Substrate beschränkt sein. Andere Enzyme hingegen,
die ein sehr großes Substratspektrum aufweisen, sind oft nur wenig spezifisch bezüglich ihrer
Stereo- oder Regioselektivität. Es gibt jedoch auch Ausnahmen, wie die ADH von
Parvibaculum lavamentivorans, die eine hohe Selektivität beim Umsatz einer Vielzahl von
Substraten aufweist14.
Zusammenfassend lässt sich festhalten, dass Enzyme bereits heute zur Synthese einer großen
Zahl von Molekülen eingesetzt werden7. Dadurch hat die Biokatalyse sowohl in der
akademischen als auch der industriellen Forschung den Stellenwert einer aussichtsreichen
Technologie erlangt, insbesondere bei der Entwicklung nachhaltiger Produktionssysteme3,15–
18. Dieser Trend spiegelt sich auch im wachsenden Wirtschaftszweig der Biotechnologie in
Deutschland wider. Sowohl Umsatz-, Mitarbeiter- als auch Finanzierungszahlen des Bereichs
stiegen im Jahr 2014 an. Die Zahl der Mitarbeiter in den dedizierten Unternehmen ist dabei
auf 17.930 (+ 5,8 %), die Zahl dieser Firmen auf 579 (+ 1,6 %) gestiegen19.
Dabei ist jedoch auch zu beachten, dass der Bereich der industriellen Biotechnologie im
Wirtschaftszweig Biotechnologie „lediglich“ 9,8 % ausmacht. Allerdings befindet sich dieser
Bereich auch in einem besonders starken und stabilen Wachstum. Der Umsatz stieg 2014 um
fast 4 % auf 214 Mio. Euro (Abb. 1)19.
Abb. 1: Verteilung von Umsatz und F&E-Ausgaben dedizierter Biotechnologie-Unternehmen 2014 (nach
biotechnologie.de)19

Einleitung
3
1.2 Alkoholdehydrogenasen (ADHs)
Alkoholdehydrogenasen bilden nach Einteilung der „International Union of Biochemistry“
die Enzymklasse EC 1.1.1.1.. Sie katalysieren Redoxreaktionen, bei denen
Reduktionsäquivalente zwischen Akzeptoren und Donoren ausgetauscht werden. Dabei
nutzen ADHs alkoholische Gruppen als Donoren und ketonische Gruppen als Akzeptoren. Sie
sind NAD(H)- oder NADP(H)-abhängig.
ADHs sind in Pflanzen, Tieren und Mikroorganismen sehr weit verbreitet20. Ihre
physiologischen Aufgaben bestehen in der Energieversorgung der Zelle mittels NAD(P)H-
Regenerierung. Weiterhin katalysieren sie oxidative Abbauprozesse der Zellabwehrfunktion,
Gärungsprozesse sowie assimilatorische und dissimilatorische Stoffwechselwege21. Industriell
interessant sind ADHs, da die Reduktion von Ketonen (Abb. 2) hoch enantiomerenselektiv
ablaufen kann.
Alkoholdehydrogenasen lassen sich in verschiedene Enzymsuperfamilien einteilen. Die
wichtigsten Vertreter sind die langkettigen Dehydrogenasen/ Reduktasen (LDR)22 mit einer
Aminosäure-Kettenlänge von über 450 AS, die mittelkettigen Dehydrogenasen/ Reduktasen
(MDR)23 mit 350 bis 400 AS und die kurzkettigen Dehydrogenasen/ Reduktasen (SDR)24 mit
unter 350 AS. Weitere wichtige Vertreter sind die Eisen-abhängigen Dehydrogenasen25 und
die Aldo-Keto-Reduktasen (AKR)26.
Abb. 2: Schematische Darstellung einer ADH-
katalysierten Reduktionsreaktion. Die
Rückreaktion, die Oxidation vom Alkohol zum
Keton, ist ebenso möglich (nicht dargestellt).
Die zwei am besten untersuchten Enzymsuperfamilien der ADHs sind die der MDR- und
SDR-Proteine:
Mittelkettige Dehydrogenasen/ Reduktasen sind Zink-abhängig und als Homodimere
oder -tetramere enzymatisch aktiv. Ein Monomer besteht aus 350 - 400 Aminosäuren und
setzt sich aus einer katalytischen und einer NAD(P)H-Bindedomäne zusammen27. Die
Struktur der katalytischen Domäne variiert stark zwischen den einzelnen Mitgliedern der
MDRs, wohingegen die der Cofaktor-Bindedomäne konserviert geblieben ist. Sie besteht aus
einer βαβ-Supersekundärstruktur27. Wichtige Vertreter dieser Gruppe sind beispielsweise die
Hefe-ADH, ein tetrameres Eukaryotenprotein, und Pferdeleber-ADH (HL-ADH), ein dimeres
Säugetierprotein28.

Einleitung
4
Kurzkettige Dehydrogenasen/ Reduktasen (SDR) liegen wie die MDRs in ihrer biologisch
aktiven Form als Homodimere oder Homotetramere vor24. Es gibt aber auch Ausnahmen, wie
die Carbonylreduktase aus dem Hausschwein, die als Monomer aktiv ist29. Eine Untereinheit
der SDRs ist aus etwa 250 AS aufgebaut. Ihre katalytische Aktivität ist Tyrosin-vermittelt und
sie enthält nur wenige Cysteine30. SDRs sind zwar wie die MDRs NAD(P)H-abhängig,
benötigen allerdings kein Metallion zur enzymatischen Aktivität. Die bisher einzigen
beschriebenen Metallionen-abhängigen SDR-ADHs stammt aus Lactobacillus brevis und
Lactobacillus kefir31.
Ubiquitär tritt in den SDRs ein konserviertes Substratbindungsmotiv auf: Tyr-X-X-X-Lys. Es
ist mit einem konservierten Serin-Rest im aktiven Zentrum lokalisiert und bildet eine
katalytische Triade, welche die enzymatische Reaktion katalysiert32,33. Daraus ist zu
schließen, dass allen SDRs der gleiche Reaktionsmechanismus zu Grunde liegt. Initiiert wird
die Reaktion durch das Binden des Cofaktors. Durch den Tyrosin-Rest der katalytischen
Triade kommt es parallel zum Hydridtransfer zu einer Protonenübertragung. Der Serinrest
übernimmt die Aufgabe der Orientierung und damit der Stabilisierung der
Reaktionsintermediate. Das Lysin ist hauptsächlich für die Stabilisierung des Nicotinamid-
Ribose-Endes des Cofaktors zuständig34–36 (Abb. 3).
Abb. 3: Möglicher Übergangszustand im
Reaktionsmechanismus der SDR-ADH
aus Drosophila lebanonensis36
Alkoholdehydrogenasen werden bereits heute industriell eingesetzt. Beispiele für
literaturbeschriebene Herstellungsverfahren zur Synthese chiraler Alkohole sind Tab. 1 zu
entnehmen.
Tab. 1: Beispiele industriell genutzter ADHs zur Produktion von chiralen Alkoholen
Organismus Stereospezifität Quelle
Thermoanaerobium brockii (S)-Alkohol 37
Pseudomonas sp. (R)-Alkohol 38
Lactobacillus kefir (R)-Alkohol 39

Einleitung
5
1.3 Alkohole in der Industrie
Alkohole dienen als wertvolle Syntheseintermediate in der chemischen Industrie. Die
alkoholische Funktion kann mittels klassisch-chemischer Methoden durch andere funktionelle
Gruppen ersetzt oder in diese überführt werden. Abb. 4 gibt einen Überblick über die
möglichen Reaktionen. Darstellbar sind beispielsweise Alkoxide, Alkene, Acetale und
Esterverbindungen. Besonders enantiomerenreine Alkohole erfreuen sich steigender
Beliebtheit40,41, da auch die daraus darstellbaren Produkte eine hohe Enantiomerenreinheit
aufweisen können (solange das chirale Zentrum bestehen bleibt).
Abb. 4: Reaktionen der alkoholischen Funktion42
1.4 Biologische Verfahren zur Gewinnung enantiomerenreiner Alkohole
Da die Enantiomeren optisch aktiver Alkohole sehr ähnliche physikalische Eigenschaften
aufweisen, ist ihre Darstellung und/ oder Aufreinigung nicht leicht durchführbar.
Grundsätzlich sind vier biotechnische Methoden anwendbar, um diese Substanzen in reiner
Form verfügbar zu machen43,44. Dabei ist die Anzahl und Art der durch die ersten beiden
Methoden (1.4.1 & 1.4.2) zu produzierenden Produkte jedoch gering. Daher wird in
biotechnologischen Prozessen zur Darstellung von enantiomerenreinen Alkoholen
hauptsächlich die kinetische Racematspaltung oder asymmetrische Reduktion angewendet
(1.4.3 & 1.4.4).

Einleitung
6
1.4.1 „Chirale Pools“
Als „chirale Pools“ werden natürliche Quellen bezeichnet, aus denen Substanzen wie
Milchsäure, Aminosäuren, Terpene oder Lipide extrahiert werden können45. Diese Moleküle
liegen in der natürlichen Quelle in reiner Form vor.
1.4.2 Fermentation
Die fermentative Anzucht von Mikroorganismen erlaubt die Gewinnung von optisch aktiven
Substanzen in reiner Form. Die Gewinnung ist jedoch auf die Stoffwechselwege des
fermentierten Organismus beschränkt. Mögliche Produkte sind somit Metabolite und
Sekundärmetabolite46.
1.4.3 Asymmetrische Reduktion prochiraler Ketone
Bei der asymmetrischen Reduktion wird eine ADH benötigt, die die Reduktion eines
prochiralen Edukts hochselektiv katalysiert. Es können auch nicht-natürliche Edukte
eingesetzt werden. Theoretisch ist bei dieser Methode eine Ausbeute von bis zu 100 %
möglich, da das gesamte Edukt zum gewünschten Produkt reduziert werden kann (Abb. 5)47.
Allerdings ist es oft schwierig, ein solch hochselektives Enzym zu identifizieren. Alternativ
kann die Selektivität eines Enzyms mittels Protein Engineering erhöht werden. Dies erfordert
jedoch einen hohen Forschungsaufwand und erhebliche Geldmittel.
Abb. 5: Asymmetrische
Reduktion mittels einer
enantioselektiven ADH
1.4.4 Kinetische Racematspaltung
Bei der kinetischen Racematspaltung wird ein racemisches Gemisch eines Alkohols
eingesetzt. Es können ebenfalls nicht-natürliche Edukte verwendet werden. Die benötigte
Alkoholdehydrogenase oxidiert in der folgenden Enantiomeren-differenzierenden Reaktion
das unerwünschte Enantiomer schneller als das gewünschte Enantiomer. Dieses bleibt somit
erhalten und kann mit einer maximalen Ausbeute von 50 % des eingesetzten Edukts
rückgewonnen werden (Abb. 6).
Um die Produktausbeute zu erhöhen, kann das entstandene Keton parallel rückreduziert
werden. Dabei entsteht auch das gewünschte Enantiomer erneut. Somit kann die

Einleitung
7
Produktausbeute theoretisch auf 100 % erhöht werden. In einem solchen Fall spricht man von
der „dynamisch-kinetischen Racematspaltung“48.
Abb. 6: Kinetische
Racematspaltung mittels
einer enantioselektiven
ADH.
1.4.5 Vorteile enzymatischer Verfahren
Der größte Vorteil der enzymatischen Verfahren im Vergleich zu klassisch-chemischen liegt
in einer teils herausragenden Selektivität der eingesetzten Biokatalysatoren. Optisch aktive
Alkohole lassen sich mit sehr hoher Enantiomerenreinheit gewinnen (ee bis ≥ 99 %)31,49,50.
Ein weiterer Vorteil liegt in den milden Bedingungen, unter denen die Reaktion abläuft. Dies
hat einen direkten positiven Einfluss auf die Produktionskosten. So wird in der Regel unter
neutralen bis schwach basischen/ sauren pH-Werten und unter atmosphärischem Druck
gearbeitet. Die erforderliche Temperatur liegt meist im physiologischen Bereich bei etwa
40 °C. Abhängig vom jeweiligen System und Enzym können die Reaktionen mit hohen
Ausbeuten und ohne störende Nebenprodukte ablaufen. Ein großer ökologischer Vorteil liegt
weiterhin im Verzicht auf umweltbelastende Schwermetallkatalysatoren.
1.5 Chemische Methoden zur Synthese von Alkoholen
Neben den beschriebenen biotechnischen Methoden existieren die folgenden klassisch-
chemischen Methoden zur Darstellung alkoholischer Verbindungen:
1.5.1 Nukleophile Substitution aus Halogenalkanen
Die einfachste Art der Darstellung alkoholischer Funktionen ist die Hydrolyse von
Halogenvorstufen mittels nukleophiler Substitution (Abb. 7). Es reagiert ein Nukleophil in
Form einer Lewis-Base (Elektronenpaardonator) mit einer organischen Verbindung vom Typ
R–X51.
Abb. 7: Beispiele zur Gewinnung von alkoholischen Verbindungen mittels nukleophiler Substitution

Einleitung
8
1.5.2 Reduktion von Carbonylverbindungen
Die Hydrierung von Carbonylverbindungen stellt eine weitere Möglichkeit zur Synthese von
alkoholischen Verbindungen dar. Unter Hydrierung versteht man im Allgemeinen die
Addition von Wasserstoff an andere chemische Elemente oder Verbindungen mittels Hydrid-
übertragender Reagenzien.
Zu den wichtigsten Hydrierungsreagenzien gehören Natriumborhydrid (NaBH4) oder
Lithiumaluminiumhydrid (LiAlH4). Dabei eignet sich NaBH4 vor allem für die Reduktion von
Aldehyden und Ketonen. LiAlH4 wird zur Reduktion von Carbonsäuren eingesetzt.
Die Verwendung dieser Katalysatoren führt allerdings zu einer unselektiven Darstellung der
Alkoholverbindung. Zur Gewinnung reiner Produkte werden spezielle Katalysatoren wie
1,2-Diamin-Ruthenium(II)-Cl2-BINAP52 benötigt (Abb. 8-A), die selbst einen chiralen
Charakter aufweisen. Eine weitere Möglichkeit zur Darstellung enantiomerenangereicherter
Alkohole ist die Nutzung von Boran in Gegenwart von Oxazaborolidin (Abb. 8-B).
Abb. 8: A) 1,2-Diamin-Ruthenium(II)-Cl2-
BINAP-Katalysator B) ein Oxazaborolidin.
Weitere Informationen: siehe 1.5.2.
1.5.3 Reaktion metallorganischer Verbindungen (Grignard-Reaktion)
Mittels metallorganischer Verbindungen lassen sich Alkohole ebenfalls darstellen. Bei der
sogenannten Grignard-Reaktion53 reagiert dabei eine Grignard-Verbindung (metallorganische
Verbindung) als Nukleophil mit einer elektrophilen Verbindung (z. B. der Carbonylgruppe in
einem Aldehyd oder Keton) zu einem Alkohol. Es wird allerdings auch ein Alkyl- oder Aryl-
Rest eingebracht.
1.5.4 Aldolreaktion
Ketone und Aldehyde können eine Aldolreaktion eingehen, wenn mindestens in einem der
Edukte in α-Stellung zur Carbonylgruppe ein H-Atom steht. Als Produkt ensteht ein
β-Hydroxyketon. Alternativ können bei einer gekreuzten Aldolreaktion zwei
Carbonylverbindungen miteinander reagieren. Dabei entstehen bis zu vier verschiedene
Aldole.

Einleitung
9
1.5.5 Vorteile klassisch-chemischer Verfahren
Klassisch-chemische Prozesse, welche nicht auf sehr hohe Enantiomerenreinheit abzielen,
sind oft ökonomischer. So benötigen sie, im Gegensatz zu ADH-katalysierten Synthesen,
keine teuren Cofaktoren wie NADH oder NADPH.
Weiterhin können die benötigten Enzyme an sich auch schon sehr teuer sein. So liegt zum
Beispiel der derzeitige Preis für das Enzym ADH von Parvibaculum lavamentivorans bei
60 €/ml Zellrohextrakt (≥ 40.0 U/ml) (Sigma-Aldrich, 2015).
Klassisch-chemische Prozesse sind oftmals schon lange etabliert53. Sie sind intensiv erforscht
und daher schnell zu adaptieren. Erste intensive Studien über ADHs wurden dagegen erst
gegen Ende der 40er Jahre des 20sten Jahrhunderts publiziert54–56. Es muss daher meist erst
nach einem geeigneten Enzymkandidaten und den anzuwendenden Prozessbedingungen
gesucht werden.
Weiterhin leiden viele ADHs unter einer niedrigen Lösemitteltoleranz57,58. Dies kann zu einer
geringen Substratbeladung des biokatalytischen Prozesses führen7. Darüber hinaus können
Enzyme sensibel gegen pH- und Temperaturveränderungen sein und dadurch im Prozess
inaktiviert oder denaturiert werden59.
1.6 Cofaktor-Regenerierung
Alkoholdehydrogenasen benötigen Cofaktoren für ihre katalytische Aktivität. Dabei dient
NAD(P)H zur Reduktion von ketonischen Funktionen und NAD(P)+ zur Oxidation von
alkoholischen. Diese Cofaktoren werden auch Coenzyme genannt, da sie für die Reaktionen
unabdingbar sind.
Industriell angewandt wird hauptsächlich der reduktive Schritt der ADHs von einer
Ketoverbindung zum entsprechenden Alkohol. In solchen Prozessen wird daher das reduzierte
Coenzym benötigt. Es überträgt während der Reaktion ein Hydrid-Ion auf die Ketogruppe.
Eine alkoholische Funktion und das nun oxidierte Coenzym (NAD(P)+) entstehen. Dabei wird
bei Monoalkoholen pro umgesetztem Substratmolekül ein Molekül des Cofaktors verbraucht.
Stöchiometrisch betrachtet müsste somit im präparativen Ansatz genauso viel Cofaktor wie
Substrat in die Reaktion eingebracht werden.
Um Produktionskosten zu minimieren, wird der Cofaktor in einer parallel laufenden Reaktion
wiederholt reduziert. Man spricht von einer in situ Cofaktor-Regenerierung. Mittels dieses
Verfahrens muss der Cofaktor lediglich in katalytischen Mengen eingesetzt werden. Ein
Beispiel ist in Abb. 9 dargestellt.

Einleitung
10
Abb. 9: Prinzip der Cofaktor-Regenerierung. In der
Hinreaktion (oben) wird Methylacetoacetat zum Methyl-
3-hydroxybutyrat unter NADH-Verbrauch reduziert.
Unter Oxidation von 2-Propanol wird gleichzeitig der
Cofaktor durch dasselbe Enzym regeneriert.
Es existieren verschiedene Möglichkeiten der Cofaktor-Regenerierung. Es wird zwischen
enzymatischen, chemischen, elektro- und photochemischen Methoden unterschieden60.
Letztere drei haben lediglich akademische Bedeutung, da sich der Cofaktor auf
enzymatischem Weg am effektivsten und preisgünstigsten regenerieren lässt.
Auf enzymatischem Weg lässt sich NAD(P)H auf zwei Arten zurückgewinnen:
a) Enzym-gekoppelte Regenerierung
In diesem Fall wird ein weiteres Enzym dem Reaktionsansatz beigesetzt, das in einer parallel
laufenden Reaktion die Reduktion des Cofaktors übernimmt.
Ein Beispiel dafür ist die Formiat-Dehydrogenase (FDH)61,62. Sie katalysiert die Oxidation
von Formiat zu CO2 (Abb. 10). Zur NADH-Regenerierung findet die FDH aus Candida
boidinii Verwendung, für NADPH wird FDH aus Pseudomonas sp. eingesetzt63. Einer der
größten Vorteile der FDH liegt in der Entstehung von Kohlendioxid als Produkt der
Formiatoxidation. Es verursacht keine Probleme bei der Produktaufreinigung, da es sich leicht
aus dem Ansatz entfernen und sich damit auch das Gleichgewicht der Gesamtreaktion in
Richtung NADH-Regenerierung verschieben lässt.
Abb. 10: Schematische Darstellung der Cofaktor-Regenerierung mittels FDH
Ein weiteres oft eingesetztes Enzym ist die Glucosedehydrogenase bzw. die
Glucose-6-Phosphat-Dehydrogenase64. Sie oxidiert Glucose(-6-Phosphat) unter
Regenerierung von NAD(P)H (Abb. 11). Verwendung findet vor allem die GDH aus Bacillus
subtilis65,66.

Einleitung
11
Abb. 11: Schematische Darstellung der Cofaktor-Regenerierung mittels G(-6-P-)DH-Reaktion
b) Substrat-gekoppelte Regenerierung65
In diesem Verfahren wird kein zusätzliches Enzym, jedoch ein zusätzliches Substrat
eingesetzt. Es handelt sich um eine alkoholische Verbindung, die von der im Ansatz
enthaltenen ADH unter NAD(P)H-Regenerierung oxidiert wird (Abb. 9).
Ein Problem besteht darin, dass das zweite Substrat (im Falle der Abb. 9: 2-Propanol) oft in
hohem Überschuss eingesetzt werden muss, um das Reaktionsgleichgewicht auf die Seite der
NADH-Regenerierung zu verschieben. Dadurch wird der organische Anteil im
Reaktionsansatz stark erhöht, wodurch es zu Stabilitätsproblemen des Enzyms kommen kann.
Dennoch wird dieses Verfahren aufgrund seiner Einfachheit und Effizienz oft angewandt,
besonders im Bereich der Reduktion wasserunlöslicher Ketone67.
1.7 Identifizierung und Entwicklung neuer Enzyme
Ein Nachteil der ADH-katalysierten, selektiven Biotransformation liegt immer noch in ihrer
begrenzten Einsetzbarkeit. Dies liegt daran, dass die Enzyme zwar oft ein breites
Substratspektrum aufweisen, aber meist nur wenige dieser Substrate hochselektiv umgesetzt
werden. Daher wird ständig an der Identifizierung und Entwicklung neuer Enzyme
geforscht68. Es existieren unterschiedliche Herangehensweisen, um die zur Verfügung
stehende Enzympalette zu erweitern69:
1.7.1 Metagenombanken
Bei der Erstellung von Metagenombanken werden enzymkodierende Gensequenzen aus der
Umwelt isoliert. Diese können daraufhin in Expressionsvektoren kloniert und die Proteine in
Expressionsstämmen wie E. coli BL21 heterolog exprimiert werden. Die auf diese Weise neu
gewonnenen Enzyme werden im Anschluss experimentell charakterisiert und damit
Eigenschaften wie Substratspektrum und Selektivität aufgedeckt68–71.

Einleitung
12
1.7.2 Entdeckung neuer Mikroorganismen
Aus allen erdenklichen Umweltisolaten wie Bodenproben, Wasserproben etc. kann versucht
werden, bisher unbekannte Mikroorganismen zu isolieren72,73. Besonders interessant sind
dabei Organismen aus extremen Ökosystemen wie der Tiefsee, der Arktis oder vulkanischen
Gebieten74–77, da die Biokatalysatoren dieser Organismen oft speziell an die extremen
Bedingungen angepasst sind72. So können sie beispielsweise besonders temperatur- und
lösemittelstabil sein oder auch ungewöhnliche Moleküle als Substrate akzeptieren69.
1.7.3 Gentechnische Veränderung bereits bekannter Enzyme (Enzyme Engineering)
Es steht heute bereits eine breite Palette an Enzymen zur Verfügung, die im industriellen
Maßstab hergestellt und zur Synthese von Chemikalien genutzt werden78–80. Diese Enzyme
können mittels verschiedener Methoden des Enzyme Engineerings verändert werden, um
optimierte Varianten zu generieren. Dabei werden meist Eigenschaften wie Selektivität,
Aktivität oder Stabilität der Katalysatoren als Ziel adressiert81–85. Mittels solcher
Optimierungen lassen sich die Enzyme an bestimmte Prozesse adaptieren, um sie
ökonomischer und/ oder ökologischer ablaufen zu lassen. Grundsätzlich werden die
Methoden des Enzyme Engineerings in a) gerichtete Evolution und b) rationales
Proteindesign unterteilt82,86–91.
a) Rationales Design: Um die Methoden des rationalen Proteindesigns anwenden zu können,
werden Kenntnisse über den atomaren Aufbau des Enzyms benötigt. Idealerweise steht eine
experimentell bestimmte Struktur zur Verfügung, beispielweise gewonnen mittels
Röntgenstrukturanalyse. Alternativ können jedoch auch Homologiemodelle berechnet
werden, welche, abhängig von den zur Verfügung stehenden templates, der Realität sehr nahe
kommen können92,93.
Anhand dieser Strukturinformationen kann rational in silico versucht werden,
Aminosäurepositionen zu identifizieren, die einen entscheidenden Einfluss auf die
Enzymeigenschaften nehmen könnten. Die daraus abgeleiteten Substitutionen können
definiert als Einzelaustausch oder semi-rational in Form einer Sättigungsmutagenese94 in das
Enzym eingefügt werden. Bei Kenntnis des Reaktionsmechanismusses können weiterhin
molekulardynamische Simulationen durchgeführt werden, um den Einfluss bestimmter
Substitutionen zu simulieren95. Nichtsdestotrotz sind die tatsächlichen Auswirkungen der
Mutationen auf die Eigenschaften des Enzyms nicht mit Sicherheit vorherzusehen, da die
atomaren und molekularen Mechanismen und Zusammenhänge sehr komplex sind. Daher
müssen die generierten Varianten im Anschluss in vitro oder in vivo genau charakterisiert

Einleitung
13
werden. Mit zunehmendem Verständnis der molekulardynamischen Zusammenhänge und
zunehmendem technischem Potential zur Berechnung der Simulationen ist jedoch
anzunehmen, dass die Vorhersagen präziser werden95,96. Erfolgreich eingesetzt wurde diese
Methode bisher meist bei lokalisierbaren Eigenschaften wie Substratselektivität
oder -spektrum97, die hauptsächlich durch den Aufbau der active site definiert werden98,99.
b) Gerichtete Evolution: Bei der gerichteten Evolution werden über die gesamte Gensequenz
oder in bestimmten, vorher definierten Bereichen auf zufällige Art Mutationen eingebracht88.
Dies kann mittels rekombinativer oder nicht-rekombinativer Methoden geschehen. Es werden
dabei meist große Variantenbanken des nativen Enzyms generiert100. Die Banken werden
nach gewünschten Eigenschaften durchmustert und positive Klone selektiert. Diese können in
einer nächsten Mutageneserunde als neue templates verwendet werden101. Mittels dieses
Verfahrens werden die Eigenschaften eines Enzyms Schritt für Schritt in eine gewünschte
Richtung verändert102,103. Im Prinzip ist das Verfahren vergleichbar mit dem Prozess der
natürlichen Evolution, welcher von Charles R. Darwin als „survival of the fittest“ bezeichnet
wurde104. Der Unterschied der gerichteten Evolution im Sinne des Protein Engineerings liegt
darin, dass die Selektionsmechanismen vom Experimentator und nicht durch
Überlebensvorteile in der Natur festgelegt werden.
Durch die Wahl der geeigneten Selektionsparameter kann rein theoretisch jede Eigenschaft
des Enzyms evolviert werden. Die Methode ist somit sehr weit anwendbar. Große Erfolge
wurden beispielsweise im Bereich der Lösemittelstabilität102,105,106 und
Thermostabilität85,107,108 erzielt, aber auch bei der Optimierung von Substratspektrum109,110
und Enantioselektivität14,91,102 eines Enzyms.
- Nicht-rekombinative, gerichtete Evolution
Die häufigste Technik, die bei der nicht-rekombinativen, gerichteten Evolution angewendet
wird, ist die fehlerhafte PCR (error-prone polymerase chain reaction). Es wird eine
Polymerase genutzt, die im Laufe der Genamplifikation zufällig falsche Basen in die Sequenz
einbaut111,112. Meist findet die Taq-Polymerase Anwendung. Durch Veränderung der
Reaktionsbedingungen (MgCl2- oder MnCl2-Konzentration) kann die Fehlerrate beeinflusst
werden. In der Regel liegt die gewünschte Mutationsrate bei 1-10 Basenaustauschen je
Kilobase113,114. Weiterhin kann durch den Einsatz von Nukleosidtriphosphat-Analoga oder
ungleichen Nukleosid-Konzentrationen die Fehlerrate beeinflusst werden115.

Einleitung
14
Der Erfolg der gerichteten Evolution mittels nicht-rekombinativer Methoden hängt dabei
entscheidend von der Qualität der generierten Variantenbanken ab. So muss eine vom zu
optimierenden Enzym abhängige, adäquate Mutationsrate ermittelt werden. Ist diese zu hoch
angesetzt, so droht die Inaktivierung eines Großteils der Mutanten. Ist sie dagegen zu niedrig
gewählt, so ist mit einer hohen Anzahl an Wildtyp-Enzymen und einer nur niedrigen Quote an
positiven Klonen zu rechnen14,116. Weiterhin muss beachtet werden, dass die Redundanz des
genetischen Codes bestimmte Aminosäuresubstitutionen aus statistischen Gründen besonders
bevorzugt. So ist Serin beispielsweise mit sechs verschiedenen Basentripletts im Vergleich zu
Histidin mit zwei Tripletts überrepräsentiert. Weiterhin treten bestimmte Basenaustausche mit
erhöhter Häufigkeit auf. So handelt es sich beispielsweise bei 40,9 % der Mutationen in der
Standard-epPCR um A↔T Transversionen117,118. Darüber hinaus können gewisse Bereiche
des Gens bevorzugt mutagenisiert werden. Dies alles führt zu Tendenzen in der Mutagenese,
durch die der abgedeckte Sequenzraum der Banken eingeschränkt wird118. Neue
Mutagenesetechniken wie SeSaM (Sequence Saturation Mutagenesis) überwinden diese
Nachteile, indem hot spots vermieden und mehrere nebeneinanderliegende DNA-Basen
gleichzeitig ausgetauscht werden118. Dadurch kann die Redundanz des genetischen Codes und
die Mutationstendenz überwunden und eine qualitativ hochwertige Mutantenbank generiert
werden.
- Rekombinative, gerichtete Evolution
Rekombinative Evolutionsmethoden basieren auf dem Prinzip der Fragmentrekombination.
Es werden also keine Punktmutationen in das Gen eingebracht, sondern ganze Genregionen
ausgetauscht. Dazu werden Gene unterschiedlicher Enzyme fragmentiert und die Fragmente
im Anschluss auf unterschiedliche Art neu reassembliert, wodurch Hybridgene entstehen. Die
ursprünglichste Technik ist das DNA-Shuffling109, welches auf DNase I Verdau beruht109.
Voraussetzung für diese Technik ist jedoch eine gewisse Sequenzhomologie.
Neben den sequenzhomologen Techniken wie DNA-Shuffling109, Staggered Extension
Process (StEP)119, Random Priming and Recombination (RPR)120, Nucleotide Excision and
Exchange (NExT)121 und weiteren existieren aber auch nicht-sequenzhomologe Verfahren.
Bekannte Vertreter solcher Techniken sind etwa Incremental Truncation for the creation of
Hybrid Enzymes (ITCHY)122, SCRATCHY123 oder Structure-based Combinatorial Protein
Engineering (SCOPE)124.
Es kann bei dieser evolutiven Methode nicht vorhergesagt werden, welche Neukombinationen
entstehen und ob diese Fusionsgene noch aktive Enzymvarianten codieren. Somit muss auch

Einleitung
15
in diesem Fall mittels Screening-Verfahrens die Variantenbank nach aktiven Enzymen
durchmustert werden. Die rekombinative, gerichtete Evolution wurde bereits vielfach
erfolgreich zur Veränderung nativer Enzyme eingesetzt, etwa zur Veränderung der
Selektivität125.
1.8 Zielsetzung der Arbeit
Chirale Alkohole dienen als wichtige Synthesebausteine für Feinchemikalien. Besonders im
Bereich der Pharma-, Agrar- sowie in der Aroma- und Duftstoffindustrie6,7 nehmen sie einen
bedeutenden Stellenwert ein. Dies führt zu einem wachsenden Bedarf an chiralen Alkoholen
in der Industrie126. Grundsätzlich sind diese Bausteine auch durch chemische Synthese
zugänglich. Die Produktionsverfahren leiden jedoch sowohl unter niedriger Selektivität als
auch unter dem Einsatz von giftigen und teuren Katalysatoren. Insbesondere kleine, chirale
Alkohole wie 2-Butanole sind nur schwer über chemische Prozesse in reiner Form
produzierbar. Daher stellen diese Moleküle interessante Ziele für eine biotechnologische
Synthese dar. Speziell Oxidotransferasen können zur Reduktion der Ketovorstufen eingesetzt
werden, bieten teilweise eine herausragende Selektivität und erfreuen sich daher steigender
Beliebtheit40,41. Dennoch leiden auch diese enzymatischen Redoxreaktionen unter einigen
Nachteilen, die in dieser Arbeit gelöst werden sollten. Das Ziel der Arbeit war, ein
ökonomisches sowie ökologisches Verfahren zur Synthese von enantiomerenreinem
(S)-2-Butanol zu entwickeln.
1) Identifizierung eines selektiven Enzyms
Für die Entwicklung des Prozesses zur Synthese von (S)-2-Butanol bot sich die Nutzung eines
Enzyms aus der Klasse der Alkoholdehydrogenasen (ADH) an. Diese katalysieren
Redoxreaktionen, bei denen Reduktionsäquivalente zwischen Akzeptoren und Donoren
ausgetauscht werden. Dadurch könnte die Darstellung des Butanols durch die Reduktion des
preisgünstigen Ketonsubstrats 2-Butanon realisiert werden.
Allerdings ist die Selektivität von ADHs meist nur auf wenige Substrate beschränkt. Dies
stellte die Suche nach einem passenden Biokatalysator an den Anfang der
Bioprozessentwicklung. Dabei war die geringe Größe des Substrates (2-Butanon) eine
besondere Herausforderung. So handelt es sich bei 2-Butanon um die kleinste vorkommende
achirale Kohlenwasserstoffverbindung ohne zusätzliche Heteroatome. Um dem Problem
angemessen zu begegnen, sollte eine breite Palette an Enzymen auf deren Fähigkeit zur
selektiven Umsetzung des Substrats untersucht werden.

Einleitung
16
2) Lösung des Problems der Produktaufreinigung
Ein besonderes Problem bei der Synthese von kurzkettigen Alkoholen und insbesondere von
2-Butanol ist die Bildung von azeotropen Mischungen der organischen Reaktions-
komponenten mit Wasser. Dies führt zu einer stark erschwerten destillativen
Produktaufreinigung, da diese Mischungen (und andere Reaktionskompenenten) sehr ähnliche
Siedepunkte aufweisen. Sie können daher nur noch mit großem Aufwand voneinander
getrennt werden.
Dieses Problem sollte durch den Einsatz hoher Volumenanteile der organischen Phase im
Reaktionsansatz gelöst werden. Durch den Verzicht auf Wasser könnte die
Azeotropenbildung unterbunden werden. Dadurch wäre das Problem der Produktaufreinigung
gelöst.
Allerdings stellt dieses organische Milieu eine große Herausforderung an das Enzym dar. So
werden die meisten Enzyme in Gegenwart großer Mengen an Lösemittel inaktiviert und/ oder
denaturiert. Es musste also ein Biokatalysator gefunden werden, der neben einer exzellenten
Selektivität auch eine enorme Lösemitteltoleranz aufweist.
3) Überwindung der ungünstigen Gleichgewichtslage
Als Reaktionstyp wurde ein System der asymmetrischen Reduktion127,128 gewählt. Dabei
werden prochirale Substrate eingesetzt, die entweder zum gewünschten Produkt oder seinem
(unerwünschten) Enantiomer umgesetzt werden. Theoretisch ist bei dieser Methode eine
Ausbeute von bis zu 100 % möglich, da bei genügend hoher Selektivität das gesamte Edukt
zum gewünschten Produkt reduziert werden kann.
Allerdings ist dieses System als Form der Meerwein-Ponndorf-Verley (MPV) Reaktion
anzusehen129–133. Diese erfordert aufgrund ihrer ungünstigen stöchiometrischen
Gleichgewichtslage den Einsatz hoher Überschüsse an Cosubstrat.
Dieses Problem könnte ebenfalls durch den geplanten hohen Anteil an organischer Phase im
Reaktionsmedium gelöst werden. Würde 2-Propanol als organisches Lösemittel eingesetzt
werden, so könnte es gleichzeitig als Cosubstrat dienen und das thermodynamische
Gleichgewicht stark auf die Produktseite verlagern Damit wäre die MPV Problematik
überwunden. Es würde auch den Einsatz von sehr hohen Mengen an Substrat ermöglichen.
4) Prozess Up-scaling
Das zu entwickelnde Verfahren musste auf die industrielle Tauglichkeit überprüft werden.
Dazu sollten Biotransformationen bis zu einem Volumen von vier Litern durchgeführt

Einleitung
17
werden. Gleichzeitig sollte damit auch genug Ausgangsmaterial produziert werden, um
signifikante Mengen des Produkts (S)-2-Butanol isolieren zu können.
5) Optimierung des Enzyms
Unter weitgehend wasserfreien Bedingungen war es wahrscheinlich, dass Aktivität und
Stabilität des Enzyms beeinträchtigt werden. Daher sollte der Biokatalysator (nach
eingehender Charakterisierung) mittels verschiedener Techniken des Protein Engineerings
hinsichtlich seiner Aktivität und/ oder Lösemittelstabilität optimiert werden.
6) Ökologische Aspekte
Die Verfahren nach Stand der Technik basieren hauptsächlich auf kinetischen
Racematspaltungen40,134,135. Diese produzieren signifikante Mengen an Nebenprodukten. Das
hypothetische Verfahren (basierend auf publizierten Modellreaktionen) zur dynamisch-
kinetischen Resolution (DKR) führt zu einer Produktion an Abfallstoffen von über 25 kg je
Kilogramm Produkt48,135 (23,5 kg Toluol/kg Produkt zur Verdünnung des racemischen
Substrats, dazu noch 1,8 kg Katalysator (Ru-Katalysator und Kalium-tert-Butylat) und 2 kg
Acylierungsreagenz). Darüber hinaus sind die Enantioselektivitäten oft nicht befriedigend. Es
ist davon auszugehen, dass weitere Reinigungsschritte mit dem entsprechenden Aufwand an
Energie und Zusatzstoffen notwendig sind.
ADH-katalysierte Verfahren wären prinzipiell geeignet, diesen Nachteil zu überwinden.
Allerdings beruhen die etablierten Verfahren auf wässrigen Reaktionssystemen, deren
Produktaufreinigung aufgrund der Azeotropenbildung nicht trivial ist. Dieses Problem wäre in
dem geplanten wasserarmen Verfahren nicht zu erwarten. Darüber hinaus würde der neue
Ansatz eine deutlich bessere Auslastung der Reaktoren ermöglichen (lösemittelfreie Systeme
statt einer 80 - 90 %igen Beladung mit Wasser), was sowohl aus ökonomischer als auch aus
ökologischer Sicht eine Verbesserung darstellt. Die generierte Menge an Abfallprodukten
könnte damit deutlich reduziert werden.

Material und Methoden
18
2. Material und Methoden
2.1 Materialien
2.1.1 Geräte
Analytik
Spektrometer UV-1800 Shimadzu
MTP Spektramax 250 (BC-MDSMX250) GMI
Gaschromatograph Focus GC mit FID Thermo Scientific
Gaschromatograph Autosampler TriPlus Thermo Scientific
Nanodrop 2000 Thermo Scientific
Zentrifuge
Ultrazentrifuge J2-21 M/E Beckmann Coulter
Tischzentrifuge 5810 R Eppendorf
Sonstige
Thermomixer compact Eppendorf
Thermostat CF31 Kryo-Kompakt Julabo
Multimagnetrührer Multipoint Komet Variomag
Rotationsverdampfer Rotavapor R-215 BÜCHI
Ultraschall Sonoplus Bandelin
Pipettierroboter Genesis Workstation 200 Tecan
Ultraschall-Homogenisator Sonopuls Bandelin Electronic
French-Press Homogenisator LM10 Microfluidics
PCR Mastercycler Gradient Eppendorf
Elektroporator MicroPulser BTX
Mikrotiterplattenschüttler Edmund-Bühler GmbH
Pickroboter Genetix Qpix Molecular Devices
SDS-PAGE Mini-Protean Bio-Rad
Lyophille Alpha 1-4 LSC Christ 2.1.2 Chemikalien
Alle im Rahmen dieser Doktorarbeit verwendeten Chemikalien und Medienkomponenten
wurden in p.A.- Qualität von folgenden Firmen erworben:

Material und Methoden
19
Antibiotika: Gerbu (Geilberg), Serva (Heidelberg), Sigma (Deisenhofen), Carl Roth
(Karlsruhe)
Chemikalien: Biomol (Hamburg), Fluka (Sternheim), Gibco BRL (Eggenstein),
Merck (Darmstadt), Pharmacia (Freiburg), Riedel-de-Haën (Seelze),
Carl Roth (Karlsruhe), Sigma (Deisenhofen), Serva (Heidelberg)
Enzyme: Restriktionsenzyme wurden von den Firmen MBI Fermentas
(St. Leon-Rot) und New England BioLabs (Schwalbach) bezogen.
Weitere Enzyme wurden von folgenden Firmen bezogen: Lysozym von
Sigma (Deisenhofen), T4-DNA-Ligase, Taq-Polymerase, Phusion-
Polymerase von New England BioLabs (Schwalbach), DNase I von
Promega (Mannheim).
Medien: Difco (Detroit, USA), Gibco BRL (Eggenstein)
2.2 Mikrobiologische Methoden
2.2.1 Mikroorganismen und Vektoren
Tab. 2: Verwendete Mikroorganismen
Stamm Genotyp Referenz
Escherichia coli DH5α
supE44 ∆(lacZYA-argF)U196
(Φ80∆lacZM15) hsdR17 recA1 endA1
gyrA96 thi-1 relA1
136
E. coli BL21 (DE3) F- dcm ompT hsdS(rB- mB-) gal (DE3) 137
E. coli T7 Shuffle
F ́lac, pro, lacIQ / ∆(ara-leu)7697
araD139 fhuA2 lacZ::T7 gene1
∆(phoA)PvuII phoR ahpC* galE (or U)
galK λatt::pNEB3-r1-
cDsbC (SpecR, lacIq) ∆trxB
rpsL150(StrR) ∆gor ∆(malF)3
New England BioLabs
Tab. 3: Verwendete Vektoren
Vektoren Genotyp Referenz
pET-21a(+) bla lacI PT7 137

Material und Methoden
20
2.2.2 Oligonukleotide
Sämtliche in dieser Arbeit verwendeten Oligonukleotide und Primer wurden lyophilisiert von
der Firma Microsynth (Lindau) bezogen. Zur Verwendung wurden sie in einer Konzentration
von 100 pmol/µl in A. dest. gelöst, die Lagerung erfolgte bei -20 °C.
Tab. 4: Verwendete Oligonukleotide.
Name Nukleotidsequenz
(unkenntlich gemacht zum Schutz der propriäteren Gensequenz (evocatal GmbH))
evo-040-Fwd GTGAGCGGATAACAATTCCC
evo-040-Rev TAGCAGCCGGATCTCAGTGG
SSB-E34C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-E34C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-G64C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-G64C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-V63C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-V63C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-A142C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-A142C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-V125C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-V125C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-A130C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-A130C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-D138C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-D138C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-Q141C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-Q141C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-G174C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-G174C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-G331C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-G331C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-T238C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-T238C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX
SSB-V261C-Fwd XXXXXXXXXXXXXXXXXXXXtgcXXXXXXXXXXXXXX
SSB-V261C-Rev XXXXXXXXXXXXXXXXXXXXgcaXXXXXXXXXXXXXX

Material und Methoden
21
3DM-1-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-1-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-2-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-2-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-3-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-3-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-4-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-4-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-5-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-5-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-6-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-6-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-7-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-7-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-8-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-8-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-9-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-9-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-10-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-10-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
3DM-11-Fwd XXXXXXXXXXXXXXXXXXXXnnkXXXXXXXXXXXXXX
3DM-11-Rev XXXXXXXXXXXXXXXXXXXXmnnXXXXXXXXXXXXXX
2.2.3 Medien
Alle Nähr- und Testmedien wurden für mindestens 20 Minuten bei 121°C und 200 kPa
autoklaviert. Hitzelabile Komponenten wurden sterilfiltriert (Millipore - Membranfilter:
0,22 µm Porendurchmesser) und den autoklavierten Medien bei < 60 °C zugefügt.
LB-Flüssigmedium138
10 g/l Trypton; 10 g/l NaCl; 5 g/l Hefeextrakt; pH 7,5

Material und Methoden
22
TB-Medium139
12 g/l Trypton; 24 g/l Hefeextrakt; 100 ml/l 10-fach konzentrierter KPi-Puffer
10-fach konz. KPi-Puffer (0,17 M KH2PO4; 0,72 M K2HPO4):
23 g/l KH2PO4; 164 g/l K2HPO4
2.2.4 Isolierung von Nukleinsäuren
Die Isolierung von Plasmid-DNA wurde nach der von Birnboim & Doly beschriebenen
Methode der alkalischen Lyse unter Verwendung eines Plasmid-Isolationskits der Firma
Fermentas (St. Leon-Rot) nach Herstellerangaben durchgeführt140. Die Konzentration von
DNA-Präparaten wurde durch analytische Gelelektrophorese oder durch einen Nanodrop
(Thermo Scientific) bestimmt.
2.2.5 Gelelektrophorese von Nukleinsäuren
Die Agarose-Gelelektrophorese wurde zur Analyse von DNA und der gezielten Isolierung
von DNA-Fragmenten durchgeführt. Dabei wurde nach der von Sambrock et al.
beschriebenen Methode vorgegangen138. Als Elektrophoresepuffer wurde 0,5 x TBE (45 mM
Tris-Base; 45 mM Borat; 1,25 mM EDTA; pH 8,3) benutzt. Als Größenstandard für die
DNA-Banden wurde, soweit nicht anders bezeichnet, die „1 kb-Ladder“ der Firma NEB
verwendet. Zur gezielten Isolierung von DNA aus Agarosegelen wurde das „Gel-Extraction-
Kit“ der Firma Eppendorf (Hamburg) benutzt.
2.2.6 in vitro-Rekombination von Nukleinsäuren
Die Restriktion und die Ligation von DNA-Fragmenten wurde nach Sambrock et al. sowie
nach den Angaben der Hersteller der jeweiligen Enzyme durchgeführt138. Die Reaktionen
fanden in den mitgelieferten Puffern statt.
2.2.7 Amplifikation von Nukleinsäuren durch Polymerase-Kettenreaktion (PCR)
Zur Amplifikation von DNA wurde die Polymerase-Kettenreaktion (PCR) nach Saiki et al.
durchgeführt141. Standardmäßig wurden PCR-Ansätze mit einem Volumen von 50 µl
angesetzt, die sich wie folgt zusammensetzten: 1-5 ng Plasmid, 25 pmol von jedem
Oligonukleotid, 0,2 mM dNTPs und 1 U „Phusion High Fidelity“-DNA-Polymerase im
Reaktionspuffer des Herstellers. Die PCR wurde in dem PCR-Automaten „Mastercycler ep
gradient“ der Firma Eppendorf (Hamburg) und, wenn nicht anders angegeben mit dem

Material und Methoden
23
folgenden Programm durchgeführt: 1 x (3 min, 98 °C), 35 x (1 min, 98 °C; 0,5 min, 58 °C;
1 min, 72 °C, abhängig von der Länge des zu amplifizierenden DNA-Fragments), 1 x (7 min,
72 °C).
2.2.8 QuikChange-Mutagenese
Zur Insertion einer oder mehrerer definierter Mutationen diente die QuikChange-Methode
nach Kunkel142. Bei der QuikChange-Mutagenese können Mutationen an beliebigen Stellen
der Nukleotidsequenz eingefügt werden, wobei als template ein Dam-methyliertes, zirkuläres
Doppelstrang-DNA-Plasmid dient. Durch zwei zueinander revers-komplementäre Primer wird
die Mutation in das Zielplasmid eingefügt. Die Mutagenese-Primer binden an die zu
mutierenden Bereiche des Plasmids und werden anschließend durch eine Polymerase zu
einem vollständigen, mutierten DNA-Strang aufgefüllt. Das template wird danach durch
Behandlung mit der Restriktionsendonuklease DpnI entfernt. Diese Nuklease hydrolysiert
spezifisch methylierte und hemimethylierte DNA an der häufig vorkommenden Zielsequenz
5’-Gm6ATC-3’143.
Das Produkt der QuikChange-Mutagenese kann durch Transformation in kompetente E. coli
Zellen eingebracht werden. Durch die zelleigenen Reparatursysteme werden die
Nukleotidstränge geschlossen und das Plasmid methyliert und repliziert. Aufgrund der
erneuten Methylierung kann das durch eine Mini-Präparation gewonnene Mutagenese-
Produkt als template für weitere Mutagenese-Runden eingesetzt werden.
2.2.9 Sättigungsmutagenese
Sättigungsmutagenesen wurden durchgeführt, um ein bestimmtes Basen-Triplett vollständig
zu mutagenisieren und so sämtliche natürliche Aminosäuren an dieser Position in das
Zielprotein einzubauen. Hierzu wurde die beschriebene Methode der QuikChange-
Mutagenese angewandt. Es wurde ein Gemisch aus Oligonukleotiden als Primer genutzt,
welche bis auf das Basentriplett, das sich an der zu sättigenden Position befand, identisch
waren. Dieses Triplett war randomisiert, das heißt jeder Primer des Gemisches codierte an
dieser Stelle für eine der 20 proteinogenen Aminosäuren in äquimolarem Verhältnis.
Der weitere Ablauf war identisch zu der Standard QuikChange-Mutagenese.
2.2.10 Ungerichtete Mutagenese mittels epPCR
Um ungerichtet über das gesamte evo-040-Strukturgen verteilt Basen zu substituieren, wurde
der Ansatz der fehlerhaften Polymerase-Kettenreaktion (epPCR = error-prone polymerase
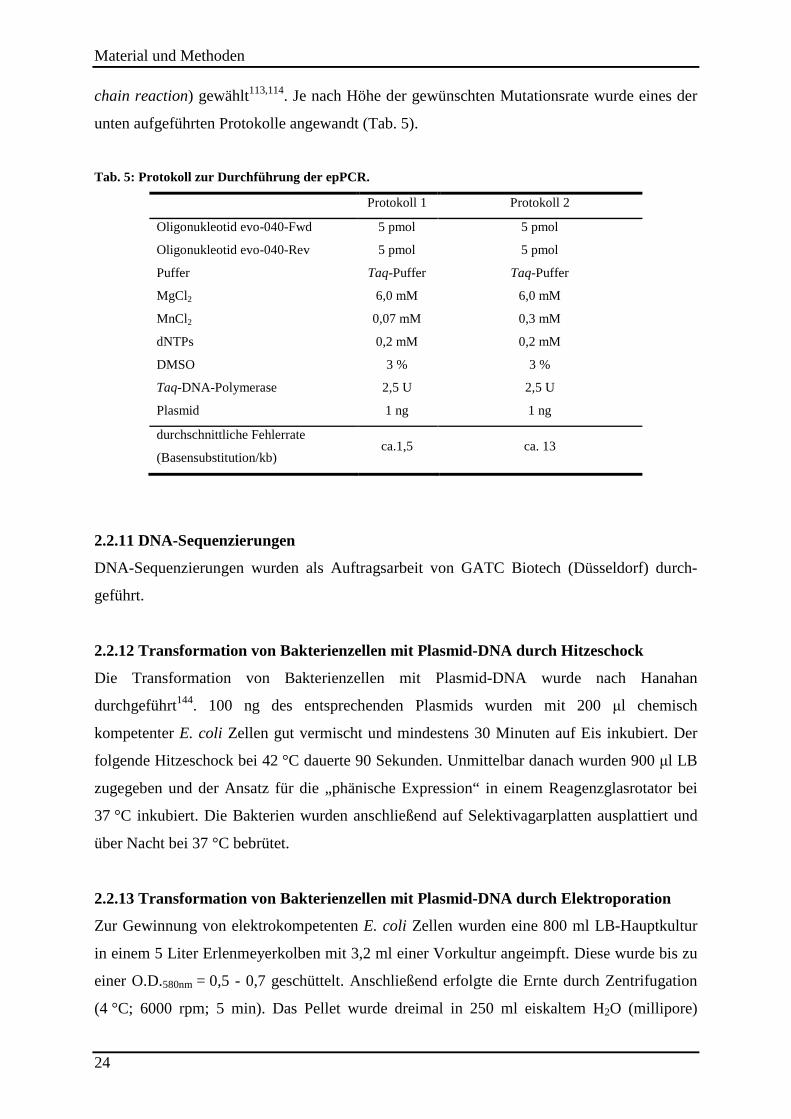
Material und Methoden
24
chain reaction) gewählt113,114. Je nach Höhe der gewünschten Mutationsrate wurde eines der
unten aufgeführten Protokolle angewandt (Tab. 5).
Tab. 5: Protokoll zur Durchführung der epPCR.
Protokoll 1 Protokoll 2
Oligonukleotid evo-040-Fwd 5 pmol 5 pmol
Oligonukleotid evo-040-Rev 5 pmol 5 pmol
Puffer Taq-Puffer Taq-Puffer
MgCl2 6,0 mM 6,0 mM
MnCl2 0,07 mM 0,3 mM
dNTPs 0,2 mM 0,2 mM
DMSO 3 % 3 %
Taq-DNA-Polymerase 2,5 U 2,5 U
Plasmid 1 ng 1 ng
durchschnittliche Fehlerrate
(Basensubstitution/kb) ca.1,5 ca. 13
2.2.11 DNA-Sequenzierungen
DNA-Sequenzierungen wurden als Auftragsarbeit von GATC Biotech (Düsseldorf) durch-
geführt.
2.2.12 Transformation von Bakterienzellen mit Plasmid-DNA durch Hitzeschock
Die Transformation von Bakterienzellen mit Plasmid-DNA wurde nach Hanahan
durchgeführt144. 100 ng des entsprechenden Plasmids wurden mit 200 µl chemisch
kompetenter E. coli Zellen gut vermischt und mindestens 30 Minuten auf Eis inkubiert. Der
folgende Hitzeschock bei 42 °C dauerte 90 Sekunden. Unmittelbar danach wurden 900 µl LB
zugegeben und der Ansatz für die „phänische Expression“ in einem Reagenzglasrotator bei
37 °C inkubiert. Die Bakterien wurden anschließend auf Selektivagarplatten ausplattiert und
über Nacht bei 37 °C bebrütet.
2.2.13 Transformation von Bakterienzellen mit Plasmid-DNA durch Elektroporation
Zur Gewinnung von elektrokompetenten E. coli Zellen wurden eine 800 ml LB-Hauptkultur
in einem 5 Liter Erlenmeyerkolben mit 3,2 ml einer Vorkultur angeimpft. Diese wurde bis zu
einer O.D.580nm = 0,5 - 0,7 geschüttelt. Anschließend erfolgte die Ernte durch Zentrifugation
(4 °C; 6000 rpm; 5 min). Das Pellet wurde dreimal in 250 ml eiskaltem H2O (millipore)

Material und Methoden
25
resuspendiert, gewaschen und erneut abzentrifugiert. Um die elektrokompetenten Zellen
lagern zu können, mussten sie daraufhin einmal in 40 ml 10 %igem Glycerin resuspendiert,
gewaschen und erneut geerntet werden (15 min; 7000 rpm; 4 °C). Das Zellpellet konnte dann
in 2 ml A. dest. mit 10 % Glycerin aufgenommen und in 50 µl Portionen aliquotiert werden.
Die Aliquots wurden zur längeren Lagerung bei – 80 °C eingefroren.
Für die Transformation wurden 5 µl dialysierter Plasmidlösung (100 ng/µl) zu einem Aliquot
kompetenter Zellen gegeben und das Gemisch wurde in eine vorgekühlte
Elektroporationsküvette (2 mm Spaltenbreite) überführt.
Unmittelbar danach fand die Transformation durch Anwenden eines elektrischen Pulses statt
(2,5 kV, 25 µF, 200 Ohm). Nach Zugabe von 800 µl SOC-Medium wurde die „phänische
Expression“ bei 37 °C durchgeführt, um den Resistenzmarker zu exprimieren (1 h).
Anschließend konnten die transformierten Zellen auf Selektionsagarplatten ausplattiert
werden.
2.2.14 Kultivierung von Bakterien und Proteinexpression
Kulturen von Escherichia coli wurden, wenn nicht anders angegeben, in LB-Medium bei
37 °C bebrütet. Unter Selektionsdruck wachsende Stämme wurden mit Antibiotika
(100 µg/ml Amp) angezogen. Die Anzucht von Flüssigkulturen erfolgte in zu maximal 10 %
befüllten Erlenmeyerkolben auf einem Rundschüttler mit 180 UpM.
Als Übernachtkulturen wurden solche Kulturen bezeichnet, die mindestens 16 h bebrütet
wurden. Hauptkulturen wurden aus einer solchen Übernachtkultur auf eine O.D.600nm = 0,05
beimpft. Die Bestimmung der Zelldichte von Kulturen erfolgte photometrisch bei einer
Wellenlänge von 600 nm gegen das entsprechende Medium als Referenz.
Als Expressionsinduktor wurde IPTG bei einer Zelldichte der Hauptkultur von
O.D.600nm = 0,5 - 0,7 mit einer Endkonzentration von 100 µM zugegeben. Die anschließende
Proteinexpression lief über insgesamt 16 h bei 30 °C im Rundschüttler (180 UpM).
Kulturen auf Festmedien wurden auf LB-Agarplatten ausgestrichen und bei 37 °C für ca. 16 h
inkubiert bzw. so lange, bis sich deutlich erkennbare Kolonien gebildet hatten.
2.2.15 Zellaufschluss/ Rohextraktgewinnung
Nach der Proteinexpression erfolgte der Aufschluss der E. coli Zellen per Ultraschall oder
mittels French-Press Homogenisator.

Material und Methoden
26
Die Bakterien wurden in der Ultrazentrifuge bei 4 °C über 20 min bei 21.000 UpM geerntet.
Die Zellmasse wurde bestimmt und in dreifacher Menge Puffer (100 mM Tris/ HCl; pH 7,2;
1 mM MgCl2) resuspendiert.
Ultraschall: Zum Zellaufschluss wurde die Lösung für 3 Zyklen von je 30 Sekunden
(Intensität 70 %) einer Ultraschallbehandlung unterzogen.
French-Press Homogenisator: Die Homogenisierung fand in zwei Durchläufen (900 bar) statt.
Der entstandene Zellrohextrakt wurde 20 Minuten bei 10.000 g zentrifugiert, um Zelltrümmer
zu entfernen.
2.2.16 Lagerung von Proteinlösungen
Der durch Ultraschallaufschluss gewonnene Rohextrakt wurde 20 min bei 14.000 g und 4 °C
zentrifugiert, der Überstand in 2 ml Eppendorf-Reaktionsgefäßen aliquotiert und bei -20 °C
aufbewahrt.
2.2.17 Anzucht und Expression im Hochdurchsatzverfahren
Die hergestellten Bibliotheken mutierter Gene der ADH evo-040 wurden mittels BamHi und
NdeI in den Expressionsvektor pET21(a)+ kloniert und der T7-Expressionsstamm E. coli
BL21(DE3) damit transformiert. Die Zellen wurden auf Selektiv-Agarplatten (100 µg/ml
Ampicillin) ausplattiert und nach ausreichender Entwicklung der Kolonien (37 °C)
automatisiert und in großer Zahl (500 – 5000 Kolonien pro Durchgang) per Pickroboter
(Qpix, Genetix) in 96-Deepwell-Mikrotiterplatten (Einzelgefäßvolumen je 2 ml (Greiner Bio-
One, Frickenhausen)) mit TB-Medium inklusive Ampicillin überführt.
Kulturen für Aktivitäts-Screening im Hochdurchsatz (HTS) wurden zu je 1 ml Volumen bei
37 °C im Mikrotiterplattenschüttler (TiMix 5, Edmund Bühler GmbH, Hechingen) bei
500 rpm angezogen. Die Proteinexpression erfolgte durch Induktion mit 0,1 mM IPTG
(Endkonzentration) für 5 h bei 30 °C.

Material und Methoden
27
2.3 Biochemische Methoden
2.3.1 ADH evo-040 Aktivitätsassay
Die Aktivität der Alkohol-Dehydrogenase evo-040 wurde photometrisch über die Abnahme
der NADH-Konzentration bei einer Wellenlänge von 340 nm gemessen.
Ablauf:
970 µl 10 mM trans-2-Hexen-1-al in 50 mM TEA-Puffer pH 7 mit 1 mM DTT
20 µl 12,5 mM NADH
10 µl Enzymlösung
Das Lyophilisat wurde eingewogen und in einer entsprechenden Menge A. dest. auf eine
Endkonzentration von 50 mg/ml gelöst. Puffer, Substrat- und NADH-Lösung wurden in eine
Küvette gegeben und 5 min bei 30 °C inkubiert. Nach Zugabe der Enzymlösung und
Durchmischen der Lösung wurde der Extinktionsverlauf über eine Minute bei 30 °C und
340 nm gemessen.
Für die Bestimmung der Aktivität der Dehydrogenase konnte die Umsetzung des Cofaktors
NAD+ bzw. NADH genutzt werden. Im Gegensatz zu NADH absorbiert NAD+ bei 340 nm
nicht, so dass bei dieser Wellenlänge im Photometer die Oxidation von NADH zu NAD+
beobachtet werden konnte. Die Aktivität des Enzyms wurde nach folgender Formel
berechnet:
�����������ä� � ���� =∆� ∙ �� ∙ � ∙ � ∙ �
∆E/∆t = Extinktionsänderung bei 340 nm (min-1)
V = Testvolumen (µl)
ε = molarer Extinktionskoeffizient für NAD(P)H (6,22 ml · mmol-1 · cm-1)
d = Schichtdicke der Küvette (cm)
v = Probenvolumen (µl)
f = Verdünnungsfaktor der Enzymlösung

Material und Methoden
28
2.3.2 Hochdurchsatz-Screening von Variantenbibliotheken
Die im Folgenden beschriebenen Pipettierschritte wurden in einem automatisierten Verfahren
in einer TECAN Workstation Genesis 200 der Firma TECAN (Crailsheim) durchgeführt:
ADH-Varianten wurden nach Anzucht in einer DWP mittels Zelllyse aufgeschlossen. Die
Lysislösung bestand aus 2 ml Aufschlusspuffer (30 mM Imidazol; 300 mM NaCl; 50 mM
NaH2PO4), 20 µl Lysozym (Stock 100 mg/ml), 2 µl MgCl2 und 1 ml A. dest..
40 µl Zellkultur wurden mit 120 µl Lysislösung vereint, durchmischt und der Ansatz zwei
Stunden bei RT inkubiert.
Assay 1: Thermostabilität
50 µl des Zelllysates wurden mit 50 µl KPi-Puffer (50 mM; pH 7,0) vermengt, in eine PCR-
Platte überführt und diese einem Hitzeschock unterzogen. Dieser fand separat in einem
„Mastercycler ep gradient“ der Firma Eppendorf (Hamburg) statt. Der Hitzeschock bei 50 °C
dauerte 5 Minuten mit einer anschließenden Rückkühlphase für 30 Sekunden bei 4 °C.
20 µl der behandelten Lösung wurden mit 180 µL Reaktionslösung (50 mM KPi pH 7,0;
10 mM 2-Butanon; 0,3 mM NADH) in eine flache, durchsichtige ELISA-Mikrotiterplatte
(96-Well MTP 0,3 ml (Greiner Bio-One, Frickenhausen)) überführt. Die MTP wurde zur
Messung ins Photometer (Sunrise, Tecan) transportiert, wo die Absorptionsänderung bei einer
Wellenlänge von 340 nm über 2 min aufgezeichnet wurde und mit Hilfe einer Software
(Magellan, Fa. TECAN, Crailsheim) ausgewertet werden konnte, um die Enzymaktivität zu
bestimmen.
Assay 2: Lösemittelstabilität
20 µl des Zelllysates wurden mit 180 µl Isopropanol in flache, durchsichtige ELISA-
Mikrotiterplatten (96-MTP 0,3 ml (Greiner Bio-One, Frickenhausen)) überführt und
5 Minuten bei RT inkubiert. 20 µl der so behandelten Lösung wurden mit 180 µL
Reaktionslösung (50 mM KPi pH 7,0; 10 mM 2-Butanon; 0,3 mM NADH) gemischt und die
MTP zur Messung ins Photometer (Sunrise, Tecan) transportiert, wo die Absorptionsänderung
bei 340 nm Wellenlänge über 2 min aufgezeichnet wurde und mittels Software (Magellan, Fa.
TECAN, Crailsheim) ausgewertet werden konnte, um die Enzymaktivität zu bestimmen.
2.3.3 Quantitative Proteinmessungen
Die Proteinbestimmung wurde photometrisch nach Bradford mit Roti Quant Fertiglösung
(Roth) in der Mikrotiterplatte durchgeführt. Gemessen wurde die Zunahme der Absorption bei

Material und Methoden
29
590 nm durch den CBBG-Protein-Komplex nach fünfminütiger Inkubationszeit.
Durch gemessene Absorptionswerte konnten die Proteinkonzentrationen der Enzymlösungen
berechnet werden. Dazu musste eine Kalibriergerade erstellt werden, anhand derer die
Absorptionswerte den Konzentrationen zugeordnet werden konnten. Die Kalibriergerade
wurde mittels Rinderalbumin (BSA) unter folgenden Standardkonzentrationen erstellt:
0,0 mg/ml; 0,02 mg/ml; 0,04 mg/ml; 0,05 mg/ml; 0,08 mg/ml; 0,10 mg/ml.
Verlässliche Proteinwerte konnten nur innerhalb dieses Kalibrationsspektrums bestimmt
werden, weshalb die Rohextrakte entsprechend ihrer Konzentration verdünnt werden mussten.
Die Reaktion selbst lief unter Raumtemperatur nach folgendem Schema ab:
50 µl Probe + 200 µl Reagenz (2 Teile Roti Quant + 5,5 Teile A. dest.).
2.3.4 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE)
Die Trennung von Proteinen erfolgte unter denaturierenden Bedingungen in Gegenwart von
SDS145. Das diskontinuierliche Gelsystem setzte sich aus einem 5 %igen Sammelgel und aus
einem 15 %igen Trenngel zusammen. Vor der elektrophoretischen Trennung wurden die
Proteinproben in SDS-Probenpuffer (50 mM Tris/ HCl; pH 6,8; 4 % (w/v) SDS; 10 % (v/v)
Glycerol; 2 % (v/v) β-Mercaptoethanol; 0,03 % (w/v) Bromphenol-Blau R-250)
aufgenommen und 10 min bei 98 °C denaturiert.
Zum Färben des Gels diente Coomassie-Färbelösung (Coomassie Brilliant Blue R250 1 g/l;
Methanol 40 % v/v; Eisessig 10 % v/v), zum Entfärben Coomassie-Entfärbelösung (Ethanol
40 % v/v; Eisessig 10 % v/v).
2.3.5 Gaschromatographische Umsatz- und ee-Bestimmung
Gaschromatographische Analysen wurden mit einem Gaschromatographen Focus GC
(Thermo Scientific) mit Flammenionisationsdetektor durchgeführt.
Geräteparameter: Trägergas: Helium; T(Injektor) = 200 °C; T(FID) = 250 °C
Temperaturprofile und Retentionszeiten sind unten aufgeführt.
1. IPC- (Umsatz) Analytik
Probenahme: 400 µl MtBE wurden mit 50 µl Reaktionslösung und 1 Spatelspitze Na2SO4
vermengt. Die Mischung wurde 3 Minuten bei 14.000 rpm zentrifugiert und 200 µl der
organischen Phase wurden in ein GC-Probengefäß überführt.

Material und Methoden
30
- Säule: FS-Innopeg 2000 (30 m x 0,25 mm x 0,25 µm)
- Flow: Helium const. 0,3 ml/min
- Split flow: 20 ml/min
- Ofen: Initial: 0 °C/min - 38 °C - 15,00 min
Rampe 1: 10 °C/min - 70 °C - 5,00 min
Rampe 2: 25 °C/min - 180 °C - 5,00 min
- Retentionszeiten: 2-Butanon: 8,49 min
2-Butanol: 15,89 min
2. Isomeren- (ee) Analytik
Probenahme: 400 µl MtBE wurden mit 50 µl Reaktionslösung und 1 Spatelspitze Na2SO4
vermengt. Die Mischung wurde 3 Minuten bei 14.000 rpm zentrifugiert und 200 µl der
organischen Phase in ein GC-Probengefäß überführt.
- Säule: CP-Chirasil-DEX CB (25 m x 0,25 mm x 0,25 µm)
- Flow: Helium const. 0,5 ml/min
- Split flow: 50 ml/min
- Ofen: Initial: 0 °C/min - 37 °C - 26,00 min
- Retentionszeiten: (R)-2-Butanol: 20,12 min
(S)-2-Butanol: 20,55 min
2.3.6 Computerprogramme
Tab. 6: Verwendete Computerprogramme.
Software Anwendung
MS-Office 2010 Textverarbeitung, Tabellenkalkulation
Softmax Pro 3.2.1 Auswertung Enzymaktivität
Chrom-card data system Ver. 2.7 GC Chromatogramme
Swiss pdb-Viewer 4.0.3 Kristallstrukturen
ChemSketch Molekülstrukturen
Pubmed, SciFinder Literaturrecherche
FACTS, Gemini, Magellan Bedienung Tecan-Pipettierautomat
In dieser Arbeit wurden Ergebnisse mit Hilfe eines Scanners, einer Digitalkamera oder durch
Videodokumentationsanlagen digitalisiert und eingefügt. Es wurden keine inhaltlichen
Änderungen der Abbildungen vorgenommen.

Ergebnisse
31
3. Ergebnisse Teil 1: Entwicklung eines Prozesses zur biokatalytischen Darstellung von
(S)-2-Butanol
3.1 Identifizierung einer geeigneten Alkoholdehydrogenase (ADH)
Das Ziel der Arbeit war die Entwicklung eines ökonomischen sowie ökologischen Verfahrens
zur Synthese von enantiomerenreinem (S)-2-Butanol. Um dieses Ziel zu erreichen, wurde
angestrebt, die enzymatische Reduktion des zugehörigen Ketons 2-Butanon in einem
wasserarmen Reaktionsmilieu stattfinden zu lassen. Dadurch sollten Verbesserungen
besonders in Bezug auf Prozessausbeute, Produktaufreinigung und Abfallvermeidung erreicht
werden146. Zu Beginn der Prozessentwicklung ergaben sich also zwei grundsätzliche
Herausforderungen: Der angemessene Biokatalysator musste zum einen eine sehr hohe
Stereoselektivität bei der Umsetzung des Ketons aufweisen (ee(Produkt) ≥ 99,5 %) und zum
anderen eine hohe Toleranz gegenüber organischen Lösemitteln besitzen
(φ(organische Phase) ≥ 80 %).
3.1.1 Etablierung einer analytischen Methode zum Nachweis von 2-Butanon und der
quantitativen Analyse der Stereoisomeren des 2-Butanols
Es wurde zunächst eine Analytik zur Detektion des Edukts und Produkts sowie der getrennten
Enantiomere entwickelt. Die Methode der Wahl war ein Nachweis mittels
Gaschromatographie, da diese eine schnelle und kostengünstige Detektion der Substanzen
ermöglicht. Die Trennung von Keton und Alkohol konnte mittels einer achiralen
FS-Innopeg-2000-Säule realisiert werden. Dabei musste der Ofen möglichst kühl temperiert
werden (40 °C), da die Analyten sowie die eingesetzten Lösemittel und
Cofaktorregeneratoren nahe beieinander liegende Siedepunkte besitzen (MtBE: 55 °C;
2-Butanon: 80 °C; 2-Propanol: 82 °C; 2-Butanol: 99 °C). Dadurch ergaben sich relativ lange
Retentionszeiten von 8,49 min für 2-Butanon und 15,89 min für 2-Butanol (Abb. 12-A).
Die Trennung der Produktenantiomere wurde über eine chirale CP-Chirasil-DEX CB Säule
realisiert. Auch hier ergaben sich durch ein möglichst niedriges Temperaturprofil lange
Retentionszeiten. Diese lagen bei 20,12 min für das (R)-Enantiomer sowie 20,55 min für das
(S)-Enantiomer, wobei sich die Retentionszeiten aufgrund von Peaktailing teils leicht
verschoben (± 0.5 min) (Abb. 12-B). Das Ausmaß des Tailings hing dabei von der injizierten
Probenmenge ab und konnte durch veränderte Elutionsprofile nicht behoben werden. Da es
die quantitative Analyse jedoch nicht beeinträchtigte, stellte es kein Problem bei der
Peakflächenbestimmung dar.

Ergebnisse
32
Abb. 12: Gaschromato-
graphische Analyse der
Reaktionskomponenten. (A)
GC-Chromatogramm der
Trennung von 2-Butanon und
2-Butanol über eine achirale
FS-Innopeg-2000 Säule.
(B) Quantitative Enantiomeren-
analyse von (S)- und
(R)-2-Butanol über eine chirale
CP-Chirasil-DEX CB Säule.
3.1.2 Identifizierung von Alkoholdehydrogenasen, die 2-Butanon als Substrat
akzeptieren
Neben den in der Literatur beschriebenen Alkoholdehydrogenasen HL-ADH (Horseliver-
ADH) 28,147,148, LB-ADH (Lactobacillus brevis-ADH)149–152 und PL-ADH (Parvibaculum
lavamentivorans-ADH)14,153,154 wurden 21 proprietäre Enzyme der Firma evocatal GmbH
untersucht.
Aus diesem Portfolio sollte ein Enzym identifiziert werden, welches in der Lage war,
2-Butanon nicht nur selektiv, sondern auch mit hoher Aktivität zu reduzieren. Dass die
beschriebene katalytische Reaktion grundsätzlich möglich ist, wurde bereits in der
Vergangenheit dokumentiert40,134. Allerdings waren die beschriebenen (und ähnliche)
Prozesse stets durch niedrige Produktausbeute und -reinheit oder durch schwer zu
produzierende Biokatalysatoren gekennzeichnet.
Die insgesamt 24 in dieser Arbeit untersuchten Alkoholdehydrogenasen wurden in einem
initialen Screening auf ihre Eignung zur Reduktion von 2-Butanon untersucht. Dazu wurden
je 10 mg/ml lyophilisierter Enzymrohextrakt (heterolog exprimiert in E. coli BL21(DE3))
eingesetzt. Tab. 8 gibt einen Überblick über die Enzymaktivität der Lyophilisatpräparate.
Die Reaktionen liefen im 5 ml Maßstab (50 mM KPi-Puffer; pH 7,5) mit einer
Substratbeladung von 100 mM 2-Butanon ab (0,1 mM NAD+ bzw. 0,4 mM NADP+). Es
wurden Reaktionen zum einem mit einem Isopropanol (iPrOH)- und zum anderen mit einem
Glucosedehydrogenase (GDH)-Cofaktorregenerationssystem analysiert. Das iPrOH-
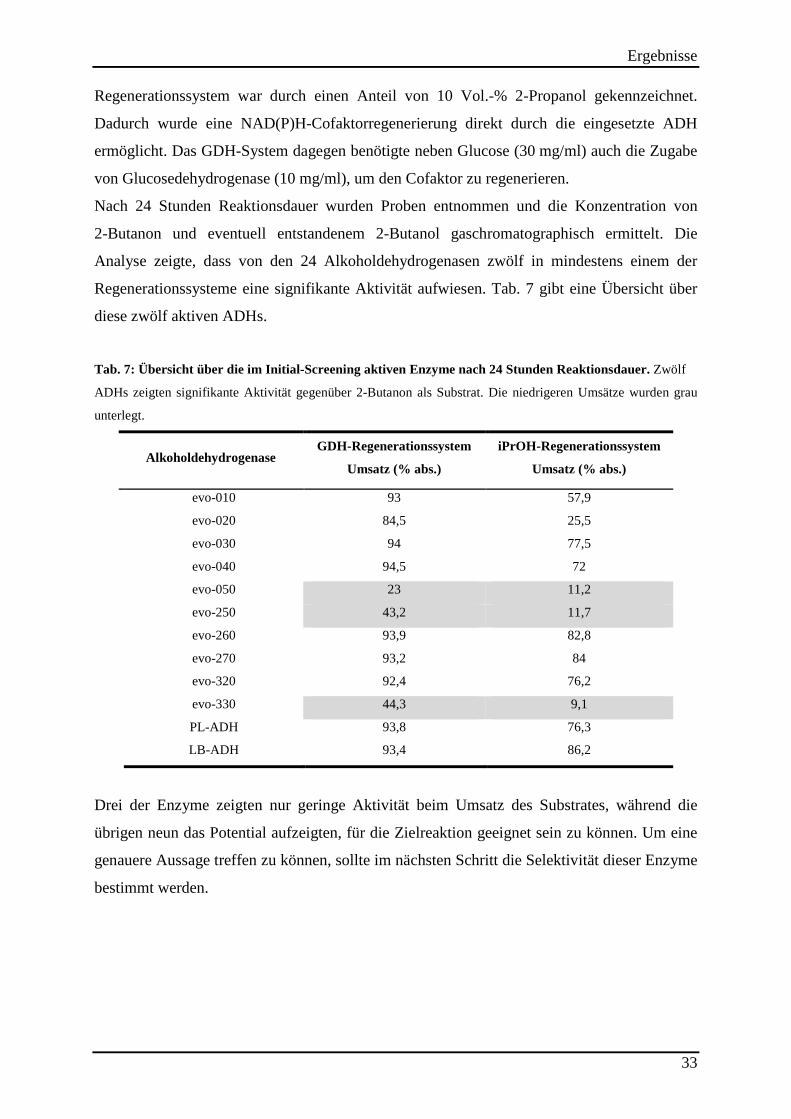
Ergebnisse
33
Regenerationssystem war durch einen Anteil von 10 Vol.-% 2-Propanol gekennzeichnet.
Dadurch wurde eine NAD(P)H-Cofaktorregenerierung direkt durch die eingesetzte ADH
ermöglicht. Das GDH-System dagegen benötigte neben Glucose (30 mg/ml) auch die Zugabe
von Glucosedehydrogenase (10 mg/ml), um den Cofaktor zu regenerieren.
Nach 24 Stunden Reaktionsdauer wurden Proben entnommen und die Konzentration von
2-Butanon und eventuell entstandenem 2-Butanol gaschromatographisch ermittelt. Die
Analyse zeigte, dass von den 24 Alkoholdehydrogenasen zwölf in mindestens einem der
Regenerationssysteme eine signifikante Aktivität aufwiesen. Tab. 7 gibt eine Übersicht über
diese zwölf aktiven ADHs.
Tab. 7: Übersicht über die im Initial-Screening aktiven Enzyme nach 24 Stunden Reaktionsdauer. Zwölf
ADHs zeigten signifikante Aktivität gegenüber 2-Butanon als Substrat. Die niedrigeren Umsätze wurden grau
unterlegt.
Alkoholdehydrogenase GDH-Regenerationssystem
Umsatz (% abs.)
iPrOH-Regenerationssystem
Umsatz (% abs.)
evo-010 93 57,9
evo-020 84,5 25,5
evo-030 94 77,5
evo-040 94,5 72
evo-050 23 11,2
evo-250 43,2 11,7
evo-260 93,9 82,8
evo-270 93,2 84
evo-320 92,4 76,2
evo-330 44,3 9,1
PL-ADH 93,8 76,3
LB-ADH 93,4 86,2
Drei der Enzyme zeigten nur geringe Aktivität beim Umsatz des Substrates, während die
übrigen neun das Potential aufzeigten, für die Zielreaktion geeignet sein zu können. Um eine
genauere Aussage treffen zu können, sollte im nächsten Schritt die Selektivität dieser Enzyme
bestimmt werden.
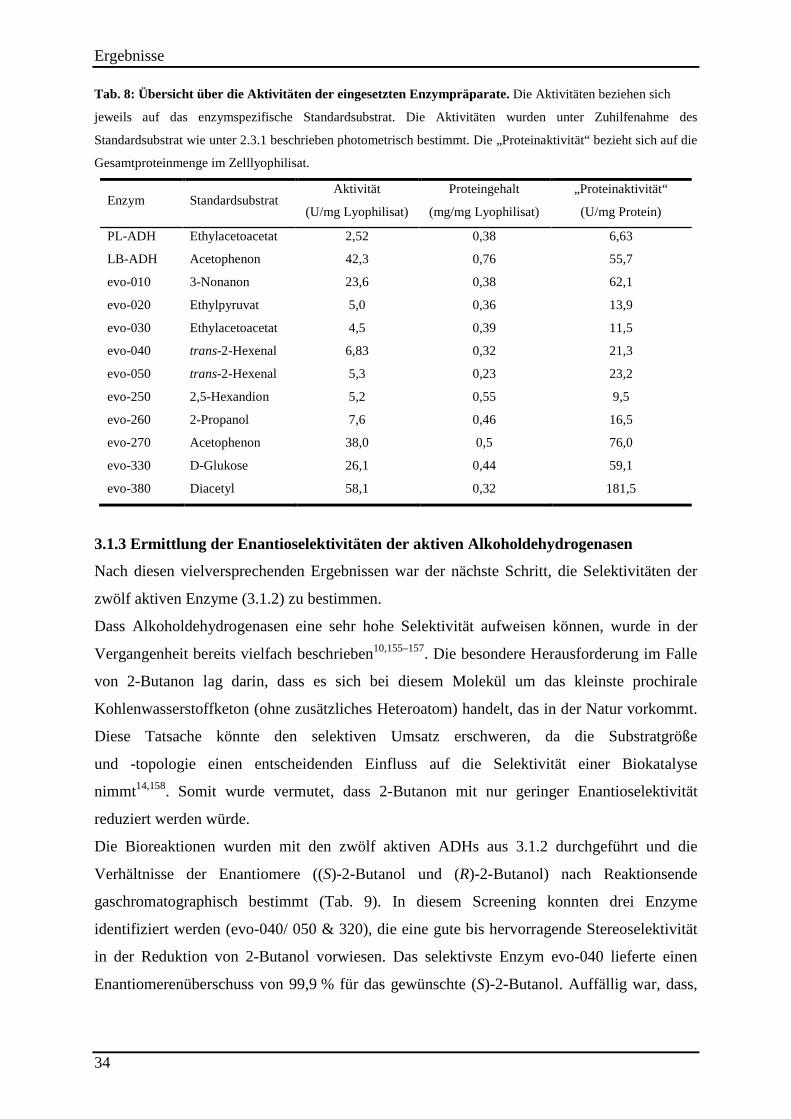
Ergebnisse
34
Tab. 8: Übersicht über die Aktivitäten der eingesetzten Enzympräparate. Die Aktivitäten beziehen sich
jeweils auf das enzymspezifische Standardsubstrat. Die Aktivitäten wurden unter Zuhilfenahme des
Standardsubstrat wie unter 2.3.1 beschrieben photometrisch bestimmt. Die „Proteinaktivität“ bezieht sich auf die
Gesamtproteinmenge im Zelllyophilisat.
Enzym Standardsubstrat Aktivität
(U/mg Lyophilisat)
Proteingehalt
(mg/mg Lyophilisat)
„Proteinaktivität“
(U/mg Protein)
PL-ADH Ethylacetoacetat 2,52 0,38 6,63
LB-ADH Acetophenon 42,3 0,76 55,7
evo-010 3-Nonanon 23,6 0,38 62,1
evo-020 Ethylpyruvat 5,0 0,36 13,9
evo-030 Ethylacetoacetat 4,5 0,39 11,5
evo-040 trans-2-Hexenal 6,83 0,32 21,3
evo-050 trans-2-Hexenal 5,3 0,23 23,2
evo-250 2,5-Hexandion 5,2 0,55 9,5
evo-260 2-Propanol 7,6 0,46 16,5
evo-270 Acetophenon 38,0 0,5 76,0
evo-330 D-Glukose 26,1 0,44 59,1
evo-380 Diacetyl 58,1 0,32 181,5
3.1.3 Ermittlung der Enantioselektivitäten der aktiven Alkoholdehydrogenasen
Nach diesen vielversprechenden Ergebnissen war der nächste Schritt, die Selektivitäten der
zwölf aktiven Enzyme (3.1.2) zu bestimmen.
Dass Alkoholdehydrogenasen eine sehr hohe Selektivität aufweisen können, wurde in der
Vergangenheit bereits vielfach beschrieben10,155–157. Die besondere Herausforderung im Falle
von 2-Butanon lag darin, dass es sich bei diesem Molekül um das kleinste prochirale
Kohlenwasserstoffketon (ohne zusätzliches Heteroatom) handelt, das in der Natur vorkommt.
Diese Tatsache könnte den selektiven Umsatz erschweren, da die Substratgröße
und -topologie einen entscheidenden Einfluss auf die Selektivität einer Biokatalyse
nimmt14,158. Somit wurde vermutet, dass 2-Butanon mit nur geringer Enantioselektivität
reduziert werden würde.
Die Bioreaktionen wurden mit den zwölf aktiven ADHs aus 3.1.2 durchgeführt und die
Verhältnisse der Enantiomere ((S)-2-Butanol und (R)-2-Butanol) nach Reaktionsende
gaschromatographisch bestimmt (Tab. 9). In diesem Screening konnten drei Enzyme
identifiziert werden (evo-040/ 050 & 320), die eine gute bis hervorragende Stereoselektivität
in der Reduktion von 2-Butanol vorwiesen. Das selektivste Enzym evo-040 lieferte einen
Enantiomerenüberschuss von 99,9 % für das gewünschte (S)-2-Butanol. Auffällig war, dass,

Ergebnisse
35
abhängig von den Cofaktor-Regenerationssystemen, teils starke Schwankungen der
Selektivitäten der Enzyme auftraten.
Tab. 9: Übersicht über die Enantiomerenreinheiten der ADH-Reaktionen nach 24 Stunden. Die höchsten
Enantiomerenüberschüsse sind grau unterlegt.
Alkoholdehydrogenase GDH-Regenerationssystem
ee (S)-2-Butanol (% rel.)
iPrOH-Regenerationssystem
ee (S)-2-Butanol (% rel.)
evo-010 44,1 20,5
evo-020 26,8 42
evo-030 -0,3 -0,6
evo-040 99,8 99,9
evo-050 92,1 99,8
evo-250 67 81,4
evo-260 1,6 0,1
evo-270 2,1 -0,4
evo-320 82,4 98,6
evo-330 32,2 29,3
PL-ADH 1,9 0
LB-ADH 1,2 -0,4
3.1.4 Screening nach lösemittelstabilen Alkoholdehydrogenasen
Die unter 3.1.2 identifizierten Enzyme wurden weiterhin auf ihre Stabilität gegenüber
2-Propanol untersucht. Angestrebt wurde ein sehr hoher Volumenanteil an organischer Phase,
um bestehende Probleme in der Meerwein-Ponndorf-Verley (MPV) Reduktion zu überwinden
und gleichzeitig die Produktaufreinigung zu erleichtern (siehe 4.1). Alle Reaktionen wurden
mit einer Enzymbeladung von 10 mg/ml Lyophilisat und 100 mM 2-Butanon in 50 mM
KPi-Puffer pH 7,5 durchgeführt (0,1 mM NAD+ bzw. 0,4 mM NADP+) (Tab. 8 gibt einen
Überblick über die Enzymaktivität der Lyophilisate).
Der Volumenanteil des Cosubstrats (iPrOH) im Reaktionsansatz wurde schrittweise erhöht.
Es wurden nach 24 Stunden Proben entnommen und sowohl Umsatz als auch
Enantiomerenreinheit bestimmt. Sechs Enzyme zeigten auch bei einem hohen organischen
Volumenanteil von 80 % noch Aktivität (Tab. 10). Daraufhin wurden diese ADHs in
Reaktionsmedien aus rein organischer Phase eingesetzt. Dadurch wurden allerdings alle
Enzyme nahezu komplett inaktiviert. Lediglich die PL-ADH generierte nach einer
verlängerten Reaktionsdauer (90 Stunden) noch relevante Produktmengen. Zur Steigerung des
Umsatzes wurde daraufhin versucht, die Enzyme zu stabilisieren. Dazu wurde
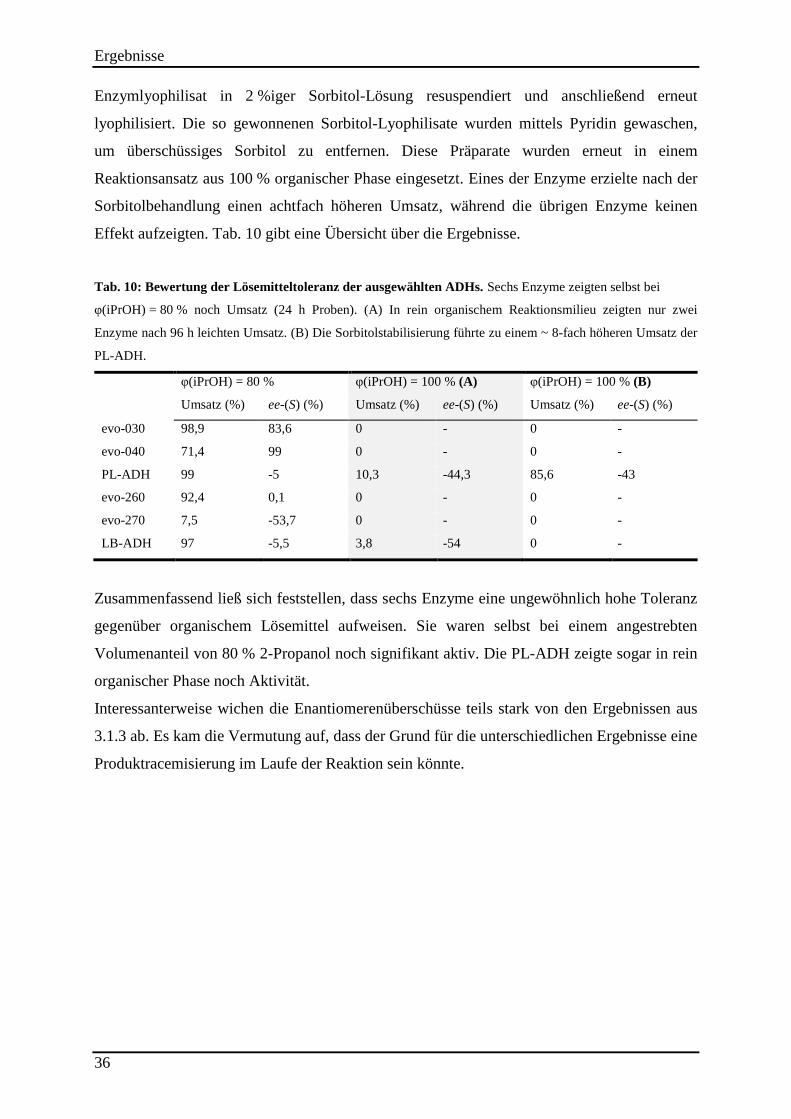
Ergebnisse
36
Enzymlyophilisat in 2 %iger Sorbitol-Lösung resuspendiert und anschließend erneut
lyophilisiert. Die so gewonnenen Sorbitol-Lyophilisate wurden mittels Pyridin gewaschen,
um überschüssiges Sorbitol zu entfernen. Diese Präparate wurden erneut in einem
Reaktionsansatz aus 100 % organischer Phase eingesetzt. Eines der Enzyme erzielte nach der
Sorbitolbehandlung einen achtfach höheren Umsatz, während die übrigen Enzyme keinen
Effekt aufzeigten. Tab. 10 gibt eine Übersicht über die Ergebnisse.
Tab. 10: Bewertung der Lösemitteltoleranz der ausgewählten ADHs. Sechs Enzyme zeigten selbst bei
φ(iPrOH) = 80 % noch Umsatz (24 h Proben). (A) In rein organischem Reaktionsmilieu zeigten nur zwei
Enzyme nach 96 h leichten Umsatz. (B) Die Sorbitolstabilisierung führte zu einem ~ 8-fach höheren Umsatz der
PL-ADH.
φ(iPrOH) = 80 % φ(iPrOH) = 100 % (A) φ(iPrOH) = 100 % (B)
Umsatz (%) ee-(S) (%) Umsatz (%) ee-(S) (%) Umsatz (%) ee-(S) (%)
evo-030 98,9 83,6 0 - 0 -
evo-040 71,4 99 0 - 0 -
PL-ADH 99 -5 10,3 -44,3 85,6 -43
evo-260 92,4 0,1 0 - 0 -
evo-270 7,5 -53,7 0 - 0 -
LB-ADH 97 -5,5 3,8 -54 0 -
Zusammenfassend ließ sich feststellen, dass sechs Enzyme eine ungewöhnlich hohe Toleranz
gegenüber organischem Lösemittel aufweisen. Sie waren selbst bei einem angestrebten
Volumenanteil von 80 % 2-Propanol noch signifikant aktiv. Die PL-ADH zeigte sogar in rein
organischer Phase noch Aktivität.
Interessanterweise wichen die Enantiomerenüberschüsse teils stark von den Ergebnissen aus
3.1.3 ab. Es kam die Vermutung auf, dass der Grund für die unterschiedlichen Ergebnisse eine
Produktracemisierung im Laufe der Reaktion sein könnte.
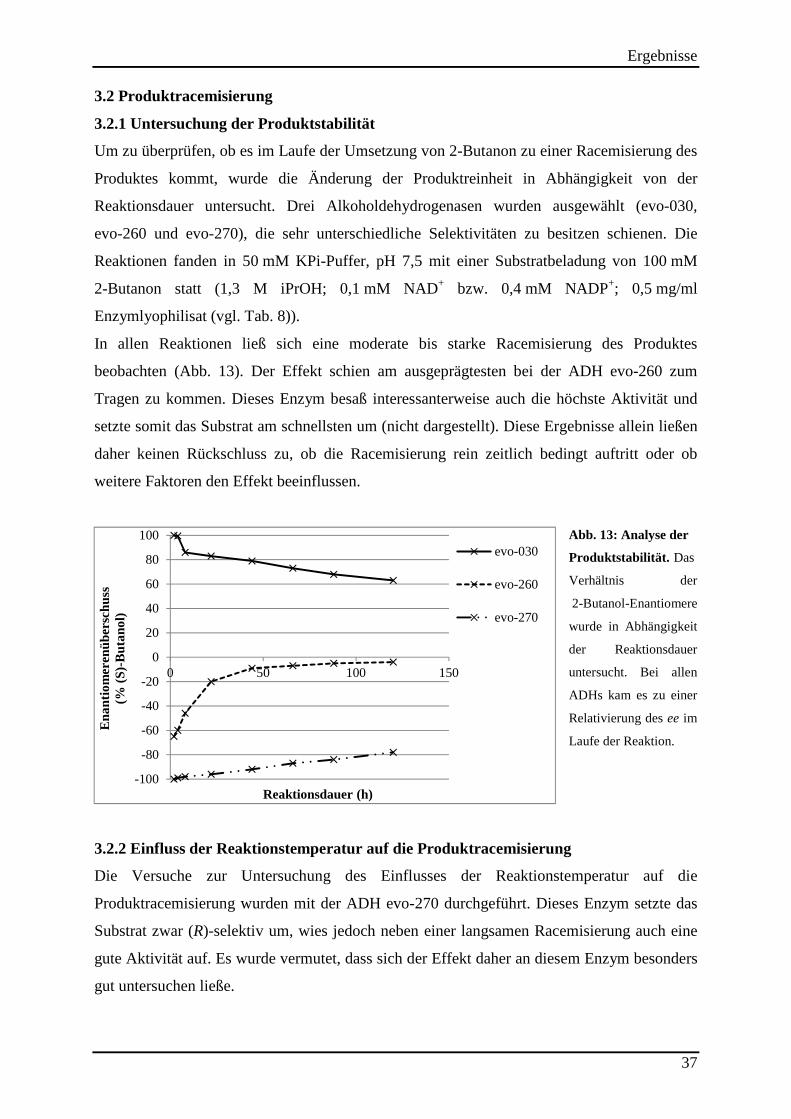
Ergebnisse
37
3.2 Produktracemisierung
3.2.1 Untersuchung der Produktstabilität
Um zu überprüfen, ob es im Laufe der Umsetzung von 2-Butanon zu einer Racemisierung des
Produktes kommt, wurde die Änderung der Produktreinheit in Abhängigkeit von der
Reaktionsdauer untersucht. Drei Alkoholdehydrogenasen wurden ausgewählt (evo-030,
evo-260 und evo-270), die sehr unterschiedliche Selektivitäten zu besitzen schienen. Die
Reaktionen fanden in 50 mM KPi-Puffer, pH 7,5 mit einer Substratbeladung von 100 mM
2-Butanon statt (1,3 M iPrOH; 0,1 mM NAD+ bzw. 0,4 mM NADP+; 0,5 mg/ml
Enzymlyophilisat (vgl. Tab. 8)).
In allen Reaktionen ließ sich eine moderate bis starke Racemisierung des Produktes
beobachten (Abb. 13). Der Effekt schien am ausgeprägtesten bei der ADH evo-260 zum
Tragen zu kommen. Dieses Enzym besaß interessanterweise auch die höchste Aktivität und
setzte somit das Substrat am schnellsten um (nicht dargestellt). Diese Ergebnisse allein ließen
daher keinen Rückschluss zu, ob die Racemisierung rein zeitlich bedingt auftritt oder ob
weitere Faktoren den Effekt beeinflussen.
Abb. 13: Analyse der
Produktstabilität. Das
Verhältnis der
2-Butanol-Enantiomere
wurde in Abhängigkeit
der Reaktionsdauer
untersucht. Bei allen
ADHs kam es zu einer
Relativierung des ee im
Laufe der Reaktion.
3.2.2 Einfluss der Reaktionstemperatur auf die Produktracemisierung
Die Versuche zur Untersuchung des Einflusses der Reaktionstemperatur auf die
Produktracemisierung wurden mit der ADH evo-270 durchgeführt. Dieses Enzym setzte das
Substrat zwar (R)-selektiv um, wies jedoch neben einer langsamen Racemisierung auch eine
gute Aktivität auf. Es wurde vermutet, dass sich der Effekt daher an diesem Enzym besonders
gut untersuchen ließe.
-100
-80
-60
-40
-20
0
20
40
60
80
100
0 50 100 150
Ena
ntio
mer
enüb
ersc
huss
(%
(S
)-B
utan
ol)
Reaktionsdauer (h)
evo-030
evo-260
evo-270

Ergebnisse
38
Es wurden vier identische Reaktionen durchgeführt, die sich lediglich in ihrer Temperatur
unterschieden. Die 20 ml Reaktionen bestanden zu 20 % aus wässrigem Anteil (50 mM
Kpi-Puffer; pH 7,5), 50 % iPrOH und 30 % 2-Butanon. Eingesetzt wurden je 40 mg evo-270
Lyophilisat aus Zellrohextrakt (vgl. Tab. 8). Da die Umsatzgeschwindigkeiten von der
Temperatur beeinflusst werden, wurden regelmäßig Proben genommen und die
Enantiomerenreinheit bei Erreichen des thermodynamischen Gleichgewichts miteinander
verglichen (Tab. 11).
Tab. 11: Enantiomerenreinheit am thermodynamischen Gleichgewicht bei unterschiedlichen
Reaktionstemperaturen. Beim -10 °C Ansatz wurde das Gleichgewicht nicht erreicht.
Temperatur ( °C) Umsatz (%) Reaktionsdauer (h) ee (R)-2-Butanol (%)
50 58 5 76
25 57 21 84
10 57 36 86
- 10 25 120 94
Erwartungsgemäß wurde das thermodynamische Gleichgewicht bei 50 °C am schnellsten
erreicht. In diesem Ansatz war der Enantiomerenüberschuss am Reaktionsgleichgewicht
jedoch auch am schlechtesten. Die Umsätze bei 25 °C und 10 °C wiesen am
Gleichgewichtspunkt sehr ähnliche ee-Werte auf und waren signifikant höher als in der 50 °C
Reaktion. Die Reaktion bei -10 °C erreichte den Gleichgewichtspunkt auch nach 120 Stunden
nicht, hatte zu diesem Zeitpunkt aber immer noch den mit Abstand höchsten Reinheitsgrad.
Zusammenfassend ließ sich eine Temperaturabhängigkeit der Racemisierung, zumindest im
Bereich hoher Temperaturen, feststellen. Inwiefern sich Temperaturen < 10 °C positiv auf die
Produktreinheit auswirken, konnte nicht abschließend geklärt werden.
3.2.3 Einfluss des pH-Wertes auf die Produktracemisierung
Um die pH-Abhängigkeit der Produktracemisierung zu untersuchen, wurden identische
Reaktionen mit der ADH evo-270 analysiert, die sich nur im pH-Wert des Mediums
unterschieden. Die 5 ml Reaktionen bestanden zu 20 % aus wässrigem Anteil (Kpi-Puffer), zu
50 % aus iPrOH und zu 30 % aus 2-Butanon. Eingesetzt wurden 10 mg evo-270 Lyophilisat
aus Zellrohextrakt. Nach 20 Stunden Reaktionsdauer wurden Proben entnommen und die
Enantiomerenüberschüsse verglichen (Tab. 12).
Es zeigte sich, dass der pH-Wert nur einen geringen Einfluss auf die Selektivität des Enzyms
zu nehmen scheint. Die Unterschiede in den ee-Werten waren gering. Die größte Differenz
Ergebnisse
39
zwischen pH 5 und dem Optimum bei pH 8 betrug 3 %. Interessant war jedoch, dass die
höchste Stereoselektivität bei dem pH-Wert erreicht wurde, unter dem das Enzym auch am
stabilsten ist (intern, unveröffentlicht), und dass evo-270 eine erstaunliche pH-Toleranz
aufweist.
Tab. 12: Enantiomerenreinheit in Abhängigkeit vom pH-Wert der Reaktionslösung. Nach 20 Stunden
wurden Proben entnommen und die Enantiomerenüberschüsse verglichen.
pH-Wert Umsatz (%) ee (R)-2-Butanol (%)
5 55 87
5,5 54 87
6 54 89
6,5 54 89
7 53 88
7,5 54 90
8 55 89
8,5 54 89
3.2.4 Führt eine Schädigung der Strukturintegrität des Enzyms zur Erniedrigung der
Selektivität?
In 3.2.3 konnte gezeigt werden, dass eine Abweichung vom optimalen pH-Wert zu leichter
Veränderung der Produktreinheit führen kann. Es kam die Vermutung auf, dass ein nicht
optimales Medium die Enzymstruktur negativ beeinflussen könnte und dadurch die
Selektivität des Katalysators verändert.
Um dies zu überprüfen, wurde das Enzym evo-270 (Endkonzentration 2 mg/ml, vgl. Tab. 8)
zwei Tage in einem harschen Reaktionsmilieu inkubiert, bevor die katalytische Reaktion
durch Zugabe des Cofaktors (0,4 mM NADP+) initiiert wurde. Die Reaktionslösung bestand
aus 80 % organischer Phase (φ(iPrOH) = 50 %, φ(2-Butanon) = 30 %), welche die Struktur
des Enzyms stark beanspruchen sollte159,160. Als Vergleich dazu wurde eine Parallelreaktion
durchgeführt, in welcher das Enzym nicht vorinkubiert wurde. Der Umsatz sowie der
Enantiomerenüberschuss wurden nach 24 Stunden Reaktionsdauer analysiert (Tab. 13).
Tab. 13: Einfluss einer strukturschädigenden Vorinkubation auf die Selektivität der ADH evo-270. Nach
24 Stunden wurden Umsatz und Enantiomerenüberschuss bestimmt.
Präparation Umsatz (%) ee (R)-2-Butanol (%)
Vorinkubierte Reaktion 51 93
Vergleichsreaktion 68 80,5
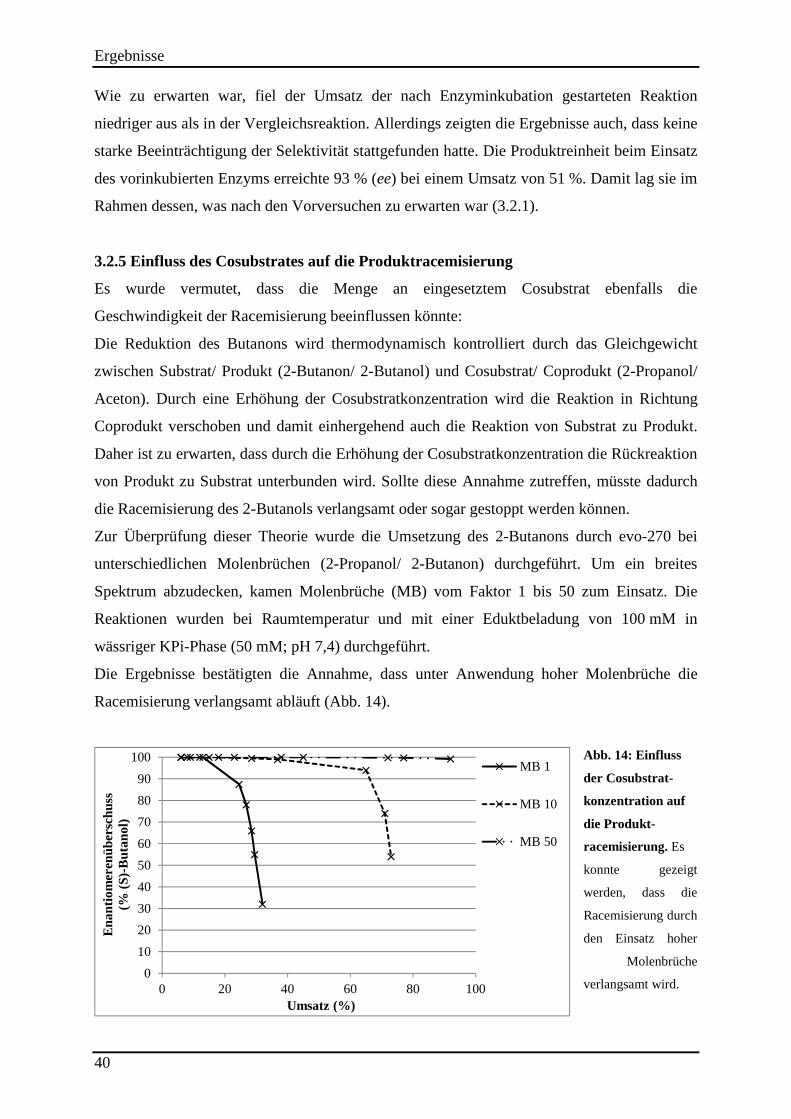
Ergebnisse
40
Wie zu erwarten war, fiel der Umsatz der nach Enzyminkubation gestarteten Reaktion
niedriger aus als in der Vergleichsreaktion. Allerdings zeigten die Ergebnisse auch, dass keine
starke Beeinträchtigung der Selektivität stattgefunden hatte. Die Produktreinheit beim Einsatz
des vorinkubierten Enzyms erreichte 93 % (ee) bei einem Umsatz von 51 %. Damit lag sie im
Rahmen dessen, was nach den Vorversuchen zu erwarten war (3.2.1).
3.2.5 Einfluss des Cosubstrates auf die Produktracemisierung
Es wurde vermutet, dass die Menge an eingesetztem Cosubstrat ebenfalls die
Geschwindigkeit der Racemisierung beeinflussen könnte:
Die Reduktion des Butanons wird thermodynamisch kontrolliert durch das Gleichgewicht
zwischen Substrat/ Produkt (2-Butanon/ 2-Butanol) und Cosubstrat/ Coprodukt (2-Propanol/
Aceton). Durch eine Erhöhung der Cosubstratkonzentration wird die Reaktion in Richtung
Coprodukt verschoben und damit einhergehend auch die Reaktion von Substrat zu Produkt.
Daher ist zu erwarten, dass durch die Erhöhung der Cosubstratkonzentration die Rückreaktion
von Produkt zu Substrat unterbunden wird. Sollte diese Annahme zutreffen, müsste dadurch
die Racemisierung des 2-Butanols verlangsamt oder sogar gestoppt werden können.
Zur Überprüfung dieser Theorie wurde die Umsetzung des 2-Butanons durch evo-270 bei
unterschiedlichen Molenbrüchen (2-Propanol/ 2-Butanon) durchgeführt. Um ein breites
Spektrum abzudecken, kamen Molenbrüche (MB) vom Faktor 1 bis 50 zum Einsatz. Die
Reaktionen wurden bei Raumtemperatur und mit einer Eduktbeladung von 100 mM in
wässriger KPi-Phase (50 mM; pH 7,4) durchgeführt.
Die Ergebnisse bestätigten die Annahme, dass unter Anwendung hoher Molenbrüche die
Racemisierung verlangsamt abläuft (Abb. 14).
Abb. 14: Einfluss
der Cosubstrat-
konzentration auf
die Produkt-
racemisierung. Es
konnte gezeigt
werden, dass die
Racemisierung durch
den Einsatz hoher
Molenbrüche
verlangsamt wird.
0
10
20
30
40
50
60
70
80
90
100
0 20 40 60 80 100
Ena
ntio
mer
enüb
ersc
huss
(%
(S
)-B
utan
ol)
Umsatz (%)
MB 1
MB 10
MB 50

Ergebnisse
41
Bei einem Molenbruch (MB) von 50 blieb bis zu einer Produktausbeute von ca. 80 % die
Enantiomerenreinheit über 99 %. Es ist jedoch zu beachten, dass solch hohe Molenbrüche nur
zu erreichen sind, wenn die Substratbeladung sehr niedrig gewählt wird. In industriell
interessanten Prozessen, die den Einsatz hoher Eduktmengen erfordern, können solch hohe
Molenbrüche nicht realisiert werden.
3.2.6 Läuft die Rückoxidation des 2-Butanols enantioselektiv ab?
Es ist zu vermuten, dass der Racemisierung eine Art „Ping-Pong“-Effekt zu Grunde liegt, bei
der repetitiv die Umsetzungen 1) Butanon ↔ Butanol und 2) 2-Propanol ↔ Aceton
ablaufen161. Es stand zur Diskussion, ob die Stereokonfiguration des Butanols Einfluss auf die
Geschwindigkeit der Rückoxidation des Produktes nimmt. Das würde bedeuten, dass eines
der Enantiomere schneller rückoxidiert werden könnte.
Um dies zu überprüfen, wurde ein Versuchsaufbau gewählt, der als organische Phasen
2-Butanol und Aceton (anstatt Butanon und iPrOH) enthielt. Somit wurde das 2-Butanol in
diesem Versuch zum Substrat (φ = 30 %), das aus thermodynamischen Gründen oxidiert wird.
Die Besonderheit bestand darin, dass zwei identische Versuche durchgeführt wurden, die
jeweils ein Enantiomer des 2-Butanols in angereicherter Form enthielten (je ~ 90 % ee). Sollte
das Butanol in einem der Fälle schneller umgesetzt werden, wäre dies ein Indiz dafür, dass die
Oxidation des 2-Butanols ebenfalls enantioselektiv stattfindet.
Weiterhin bestand der Versuchsaufbau aus 25 % Aceton, 50 mM Kpi-Puffer (pH 7,5) sowie
0,4 mM NADP+ und 2 mg/ml Enzymlyophilisat aus Zellrohextrakt.
Die Ergebnisse zeigten sehr deutlich, dass die Oxidation des (R)-Enantiomers schneller
abläuft als die seines Stereoanalogons (Abb. 15). Bereits bei der Entnahme der 16 Stunden
Probe wurde in diesem Fall das thermodynamische Gleichgewicht erreicht, während dies im
Falle des (S)-Enantiomers auch nach 170 Stunden noch nicht der Fall war. Es ist somit davon
auszugehen, dass die Oxidation von 2-Butanol enantiomerenselektiv abläuft.
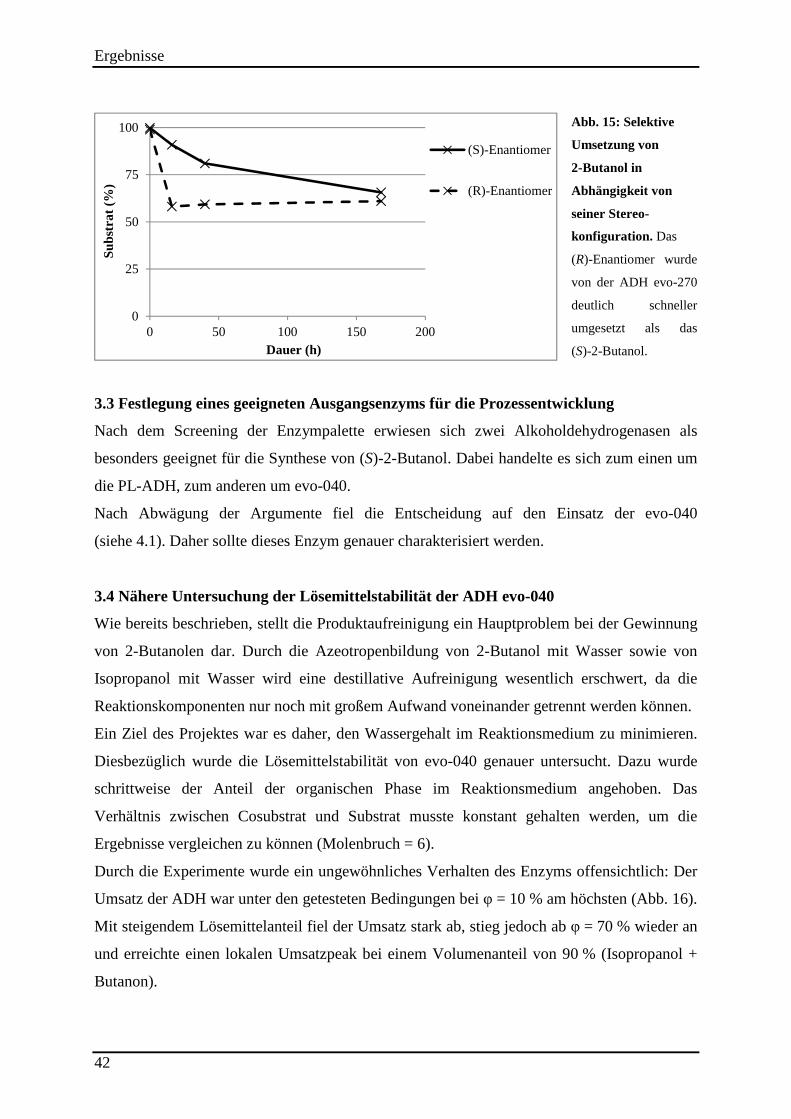
Ergebnisse
42
Abb. 15: Selektive
Umsetzung von
2-Butanol in
Abhängigkeit von
seiner Stereo-
konfiguration. Das
(R)-Enantiomer wurde
von der ADH evo-270
deutlich schneller
umgesetzt als das
(S)-2-Butanol.
3.3 Festlegung eines geeigneten Ausgangsenzyms für die Prozessentwicklung
Nach dem Screening der Enzympalette erwiesen sich zwei Alkoholdehydrogenasen als
besonders geeignet für die Synthese von (S)-2-Butanol. Dabei handelte es sich zum einen um
die PL-ADH, zum anderen um evo-040.
Nach Abwägung der Argumente fiel die Entscheidung auf den Einsatz der evo-040
(siehe 4.1). Daher sollte dieses Enzym genauer charakterisiert werden.
3.4 Nähere Untersuchung der Lösemittelstabilität der ADH evo-040
Wie bereits beschrieben, stellt die Produktaufreinigung ein Hauptproblem bei der Gewinnung
von 2-Butanolen dar. Durch die Azeotropenbildung von 2-Butanol mit Wasser sowie von
Isopropanol mit Wasser wird eine destillative Aufreinigung wesentlich erschwert, da die
Reaktionskomponenten nur noch mit großem Aufwand voneinander getrennt werden können.
Ein Ziel des Projektes war es daher, den Wassergehalt im Reaktionsmedium zu minimieren.
Diesbezüglich wurde die Lösemittelstabilität von evo-040 genauer untersucht. Dazu wurde
schrittweise der Anteil der organischen Phase im Reaktionsmedium angehoben. Das
Verhältnis zwischen Cosubstrat und Substrat musste konstant gehalten werden, um die
Ergebnisse vergleichen zu können (Molenbruch = 6).
Durch die Experimente wurde ein ungewöhnliches Verhalten des Enzyms offensichtlich: Der
Umsatz der ADH war unter den getesteten Bedingungen bei φ = 10 % am höchsten (Abb. 16).
Mit steigendem Lösemittelanteil fiel der Umsatz stark ab, stieg jedoch ab φ = 70 % wieder an
und erreichte einen lokalen Umsatzpeak bei einem Volumenanteil von 90 % (Isopropanol +
Butanon).
0
25
50
75
100
0 50 100 150 200
Sub
stra
t (%
)
Dauer (h)
(S)-Enantiomer
(R)-Enantiomer
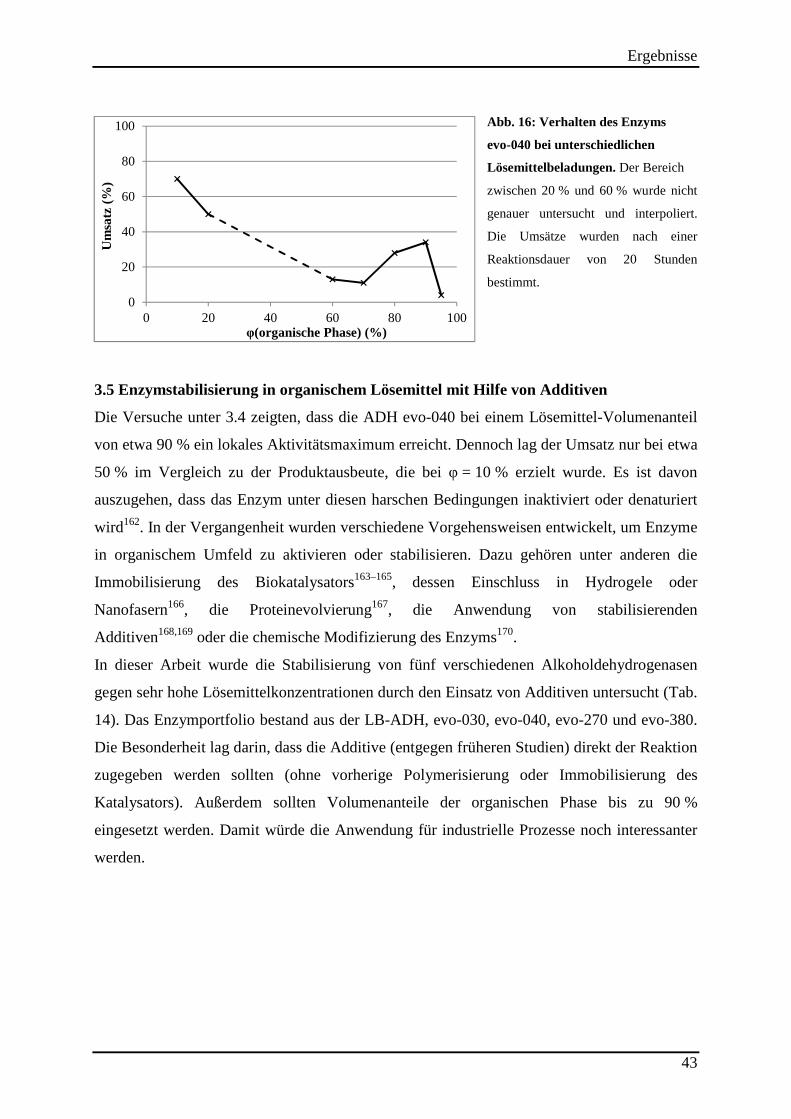
Ergebnisse
43
0
20
40
60
80
100
0 20 40 60 80 100
Um
satz
(%
)
φ(organische Phase) (%)
Abb. 16: Verhalten des Enzyms
evo-040 bei unterschiedlichen
Lösemittelbeladungen. Der Bereich
zwischen 20 % und 60 % wurde nicht
genauer untersucht und interpoliert.
Die Umsätze wurden nach einer
Reaktionsdauer von 20 Stunden
bestimmt.
3.5 Enzymstabilisierung in organischem Lösemittel mit Hilfe von Additiven
Die Versuche unter 3.4 zeigten, dass die ADH evo-040 bei einem Lösemittel-Volumenanteil
von etwa 90 % ein lokales Aktivitätsmaximum erreicht. Dennoch lag der Umsatz nur bei etwa
50 % im Vergleich zu der Produktausbeute, die bei φ = 10 % erzielt wurde. Es ist davon
auszugehen, dass das Enzym unter diesen harschen Bedingungen inaktiviert oder denaturiert
wird162. In der Vergangenheit wurden verschiedene Vorgehensweisen entwickelt, um Enzyme
in organischem Umfeld zu aktivieren oder stabilisieren. Dazu gehören unter anderen die
Immobilisierung des Biokatalysators163–165, dessen Einschluss in Hydrogele oder
Nanofasern166, die Proteinevolvierung167, die Anwendung von stabilisierenden
Additiven168,169 oder die chemische Modifizierung des Enzyms170.
In dieser Arbeit wurde die Stabilisierung von fünf verschiedenen Alkoholdehydrogenasen
gegen sehr hohe Lösemittelkonzentrationen durch den Einsatz von Additiven untersucht (Tab.
14). Das Enzymportfolio bestand aus der LB-ADH, evo-030, evo-040, evo-270 und evo-380.
Die Besonderheit lag darin, dass die Additive (entgegen früheren Studien) direkt der Reaktion
zugegeben werden sollten (ohne vorherige Polymerisierung oder Immobilisierung des
Katalysators). Außerdem sollten Volumenanteile der organischen Phase bis zu 90 %
eingesetzt werden. Damit würde die Anwendung für industrielle Prozesse noch interessanter
werden.
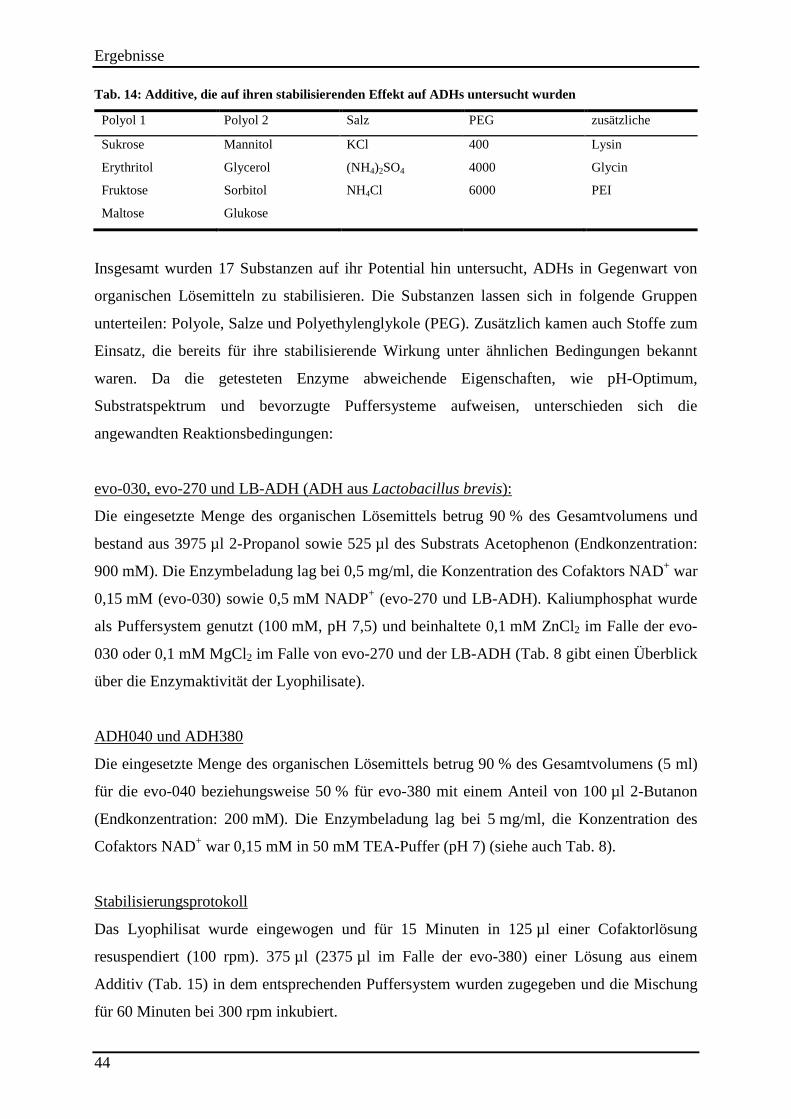
Ergebnisse
44
Tab. 14: Additive, die auf ihren stabilisierenden Effekt auf ADHs untersucht wurden
Polyol 1 Polyol 2 Salz PEG zusätzliche
Sukrose Mannitol KCl 400 Lysin
Erythritol Glycerol (NH4)2SO4 4000 Glycin
Fruktose Sorbitol NH4Cl 6000 PEI
Maltose Glukose
Insgesamt wurden 17 Substanzen auf ihr Potential hin untersucht, ADHs in Gegenwart von
organischen Lösemitteln zu stabilisieren. Die Substanzen lassen sich in folgende Gruppen
unterteilen: Polyole, Salze und Polyethylenglykole (PEG). Zusätzlich kamen auch Stoffe zum
Einsatz, die bereits für ihre stabilisierende Wirkung unter ähnlichen Bedingungen bekannt
waren. Da die getesteten Enzyme abweichende Eigenschaften, wie pH-Optimum,
Substratspektrum und bevorzugte Puffersysteme aufweisen, unterschieden sich die
angewandten Reaktionsbedingungen:
evo-030, evo-270 und LB-ADH (ADH aus Lactobacillus brevis):
Die eingesetzte Menge des organischen Lösemittels betrug 90 % des Gesamtvolumens und
bestand aus 3975 µl 2-Propanol sowie 525 µl des Substrats Acetophenon (Endkonzentration:
900 mM). Die Enzymbeladung lag bei 0,5 mg/ml, die Konzentration des Cofaktors NAD+ war
0,15 mM (evo-030) sowie 0,5 mM NADP+ (evo-270 und LB-ADH). Kaliumphosphat wurde
als Puffersystem genutzt (100 mM, pH 7,5) und beinhaltete 0,1 mM ZnCl2 im Falle der evo-
030 oder 0,1 mM MgCl2 im Falle von evo-270 und der LB-ADH (Tab. 8 gibt einen Überblick
über die Enzymaktivität der Lyophilisate).
ADH040 und ADH380
Die eingesetzte Menge des organischen Lösemittels betrug 90 % des Gesamtvolumens (5 ml)
für die evo-040 beziehungsweise 50 % für evo-380 mit einem Anteil von 100 µl 2-Butanon
(Endkonzentration: 200 mM). Die Enzymbeladung lag bei 5 mg/ml, die Konzentration des
Cofaktors NAD+ war 0,15 mM in 50 mM TEA-Puffer (pH 7) (siehe auch Tab. 8).
Stabilisierungsprotokoll
Das Lyophilisat wurde eingewogen und für 15 Minuten in 125 µl einer Cofaktorlösung
resuspendiert (100 rpm). 375 µl (2375 µl im Falle der evo-380) einer Lösung aus einem
Additiv (Tab. 15) in dem entsprechenden Puffersystem wurden zugegeben und die Mischung
für 60 Minuten bei 300 rpm inkubiert.
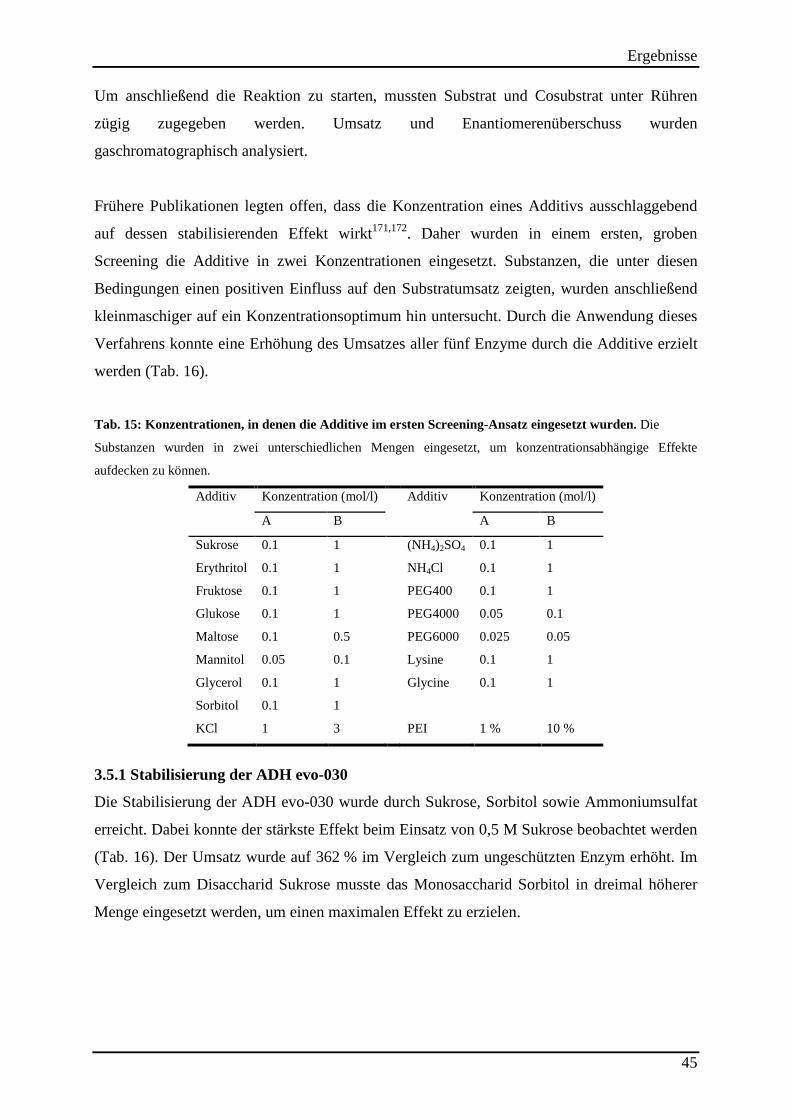
Ergebnisse
45
Um anschließend die Reaktion zu starten, mussten Substrat und Cosubstrat unter Rühren
zügig zugegeben werden. Umsatz und Enantiomerenüberschuss wurden
gaschromatographisch analysiert.
Frühere Publikationen legten offen, dass die Konzentration eines Additivs ausschlaggebend
auf dessen stabilisierenden Effekt wirkt171,172. Daher wurden in einem ersten, groben
Screening die Additive in zwei Konzentrationen eingesetzt. Substanzen, die unter diesen
Bedingungen einen positiven Einfluss auf den Substratumsatz zeigten, wurden anschließend
kleinmaschiger auf ein Konzentrationsoptimum hin untersucht. Durch die Anwendung dieses
Verfahrens konnte eine Erhöhung des Umsatzes aller fünf Enzyme durch die Additive erzielt
werden (Tab. 16).
Tab. 15: Konzentrationen, in denen die Additive im ersten Screening-Ansatz eingesetzt wurden. Die
Substanzen wurden in zwei unterschiedlichen Mengen eingesetzt, um konzentrationsabhängige Effekte
aufdecken zu können.
Additiv Konzentration (mol/l) Additiv Konzentration (mol/l)
A B A B
Sukrose 0.1 1 (NH4)2SO4 0.1 1
Erythritol 0.1 1 NH4Cl 0.1 1
Fruktose 0.1 1 PEG400 0.1 1
Glukose 0.1 1 PEG4000 0.05 0.1
Maltose 0.1 0.5 PEG6000 0.025 0.05
Mannitol 0.05 0.1 Lysine 0.1 1
Glycerol 0.1 1 Glycine 0.1 1
Sorbitol 0.1 1
KCl 1 3 PEI 1 % 10 %
3.5.1 Stabilisierung der ADH evo-030
Die Stabilisierung der ADH evo-030 wurde durch Sukrose, Sorbitol sowie Ammoniumsulfat
erreicht. Dabei konnte der stärkste Effekt beim Einsatz von 0,5 M Sukrose beobachtet werden
(Tab. 16). Der Umsatz wurde auf 362 % im Vergleich zum ungeschützten Enzym erhöht. Im
Vergleich zum Disaccharid Sukrose musste das Monosaccharid Sorbitol in dreimal höherer
Menge eingesetzt werden, um einen maximalen Effekt zu erzielen.

Ergebnisse
46
3.5.2 Stabilisierung der ADH evo-040
Da die Umsatzrate der ADH evo-040 sehr niedrig war, wurde die Reaktionszeit auf
140 Stunden erhöht. Eine Sorbitolzugabe (1 M) erhöhte den Umsatz auf 157 % im Vergleich
zum ungeschützten Enzym (Tab. 16). Der Enantiomerenüberschuss blieb bei allen Ansätzen
hervorragend und belief sich auf über 99,9 % für das (S)-2-Butanol.
3.5.3 Stabilisierung der LB-ADH
Der stabilisierende Effekt der Additive auf die LB-ADH war signifikant, jedoch der niedrigste
im Vergleich zu den anderen Enzymen. Der Umsatz in Gegenwart von Ammoniumsulfat
belief sich im Maximum auf 152 % (Tab. 16) und war somit 52 % höher als im ungeschützten
Zustand.
3.5.4 Stabilisierung der ADH evo-270
Die ADH evo-270 konnte durch fünf verschiedene Additive erfolgreich stabilisiert werden:
Ammoniumsulfat, PEG4000, PEG6000, PEI und Glycin (Tab. 16). Der stärkste Effekt konnte
mit PEG6000 bei einer Konzentration von 0,01 M erreicht werden (159 %).
3.5.5 Stabilisierung der ADH evo-380
Im Falle der ADH evo-380 konnte der signifikanteste Effekt erzielt werden. In Gegenwart
von 0,1 M PEG4000 wurde eine fast siebenfache Steigerung des Umsatzes erzielt (690 %,
Tab. 16). Das kurzkettigere PEG400 musste im Vergleich dazu in vielfach höherer Menge
eingesetzt werden, um einen maximalen Einfluss zu nehmen (2,5 M).
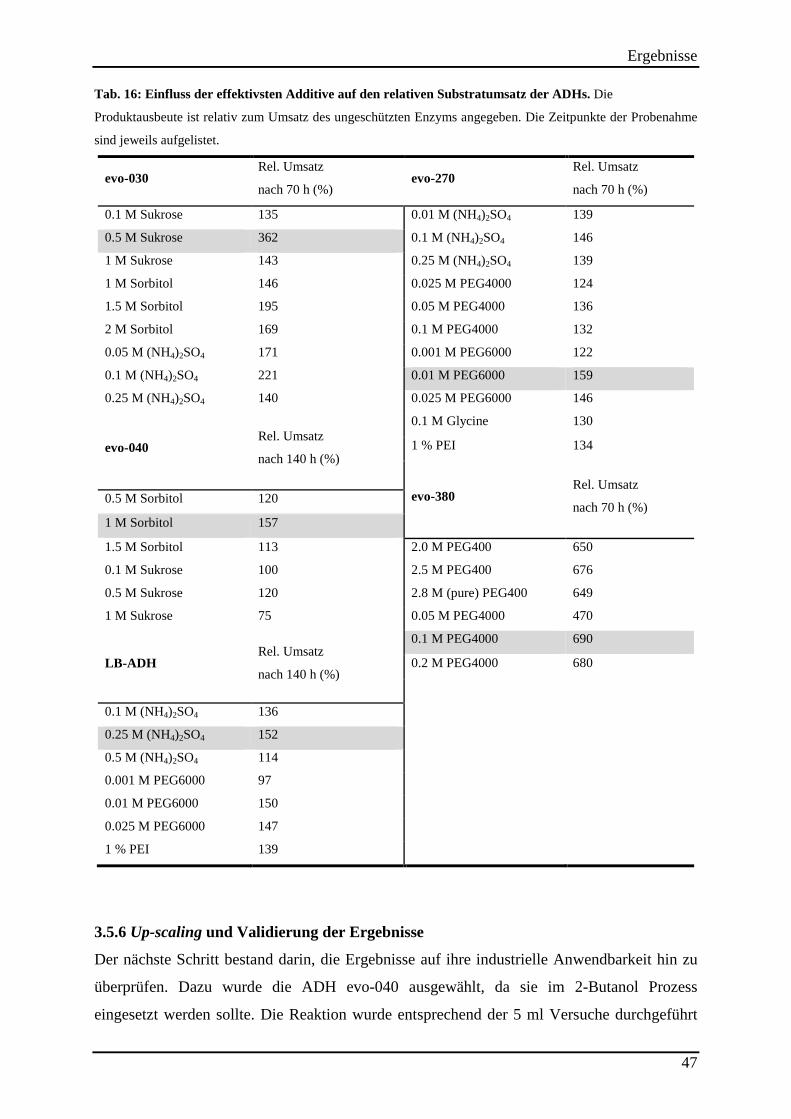
Ergebnisse
47
Tab. 16: Einfluss der effektivsten Additive auf den relativen Substratumsatz der ADHs. Die
Produktausbeute ist relativ zum Umsatz des ungeschützten Enzyms angegeben. Die Zeitpunkte der Probenahme
sind jeweils aufgelistet.
evo-030 Rel. Umsatz
nach 70 h (%) evo-270
Rel. Umsatz
nach 70 h (%)
0.1 M Sukrose 135 0.01 M (NH4)2SO4 139
0.5 M Sukrose 362 0.1 M (NH4)2SO4 146
1 M Sukrose 143 0.25 M (NH4)2SO4 139
1 M Sorbitol 146 0.025 M PEG4000 124
1.5 M Sorbitol 195 0.05 M PEG4000 136
2 M Sorbitol 169 0.1 M PEG4000 132
0.05 M (NH4)2SO4 171 0.001 M PEG6000 122
0.1 M (NH4)2SO4 221 0.01 M PEG6000 159
0.25 M (NH4)2SO4 140 0.025 M PEG6000 146
evo-040 Rel. Umsatz
nach 140 h (%)
0.1 M Glycine 130
1 % PEI 134
evo-380 Rel. Umsatz
nach 70 h (%) 0.5 M Sorbitol 120
1 M Sorbitol 157
1.5 M Sorbitol 113 2.0 M PEG400 650
0.1 M Sukrose 100 2.5 M PEG400 676
0.5 M Sukrose 120 2.8 M (pure) PEG400 649
1 M Sukrose 75 0.05 M PEG4000 470
LB-ADH Rel. Umsatz
nach 140 h (%)
0.1 M PEG4000 690
0.2 M PEG4000 680
0.1 M (NH4)2SO4 136
0.25 M (NH4)2SO4 152
0.5 M (NH4)2SO4 114
0.001 M PEG6000 97
0.01 M PEG6000 150
0.025 M PEG6000 147
1 % PEI 139
3.5.6 Up-scaling und Validierung der Ergebnisse
Der nächste Schritt bestand darin, die Ergebnisse auf ihre industrielle Anwendbarkeit hin zu
überprüfen. Dazu wurde die ADH evo-040 ausgewählt, da sie im 2-Butanol Prozess
eingesetzt werden sollte. Die Reaktion wurde entsprechend der 5 ml Versuche durchgeführt
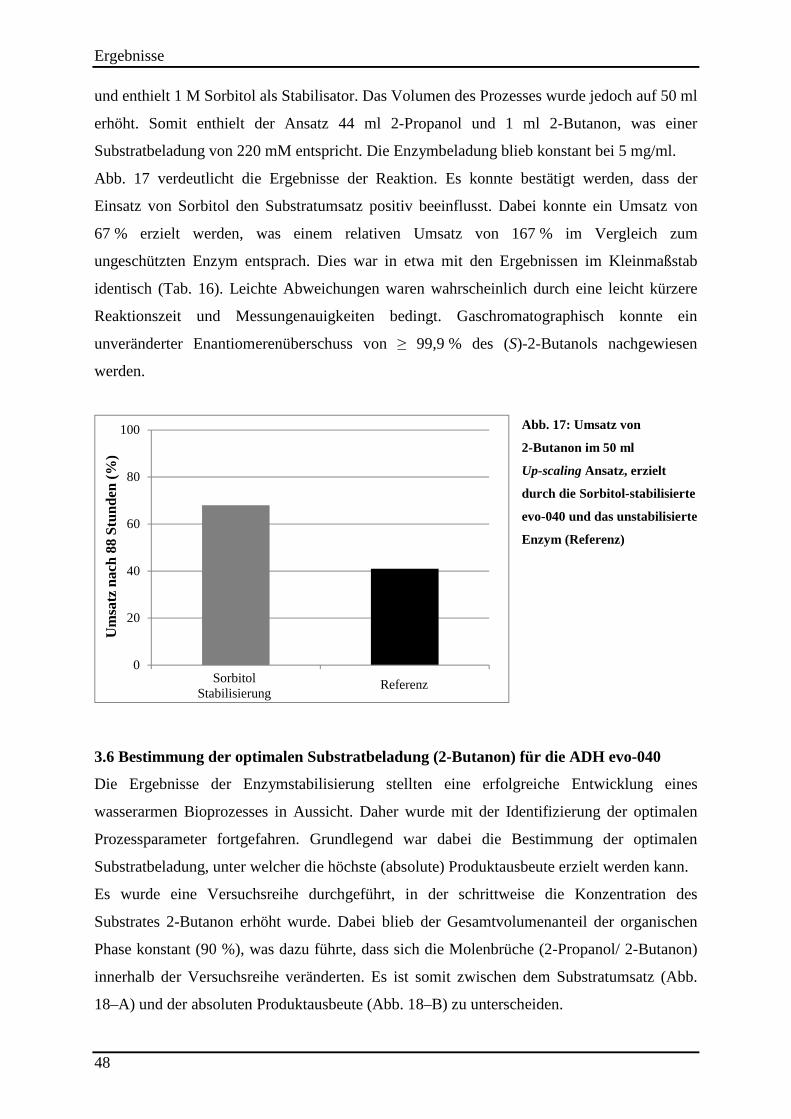
Ergebnisse
48
und enthielt 1 M Sorbitol als Stabilisator. Das Volumen des Prozesses wurde jedoch auf 50 ml
erhöht. Somit enthielt der Ansatz 44 ml 2-Propanol und 1 ml 2-Butanon, was einer
Substratbeladung von 220 mM entspricht. Die Enzymbeladung blieb konstant bei 5 mg/ml.
Abb. 17 verdeutlicht die Ergebnisse der Reaktion. Es konnte bestätigt werden, dass der
Einsatz von Sorbitol den Substratumsatz positiv beeinflusst. Dabei konnte ein Umsatz von
67 % erzielt werden, was einem relativen Umsatz von 167 % im Vergleich zum
ungeschützten Enzym entsprach. Dies war in etwa mit den Ergebnissen im Kleinmaßstab
identisch (Tab. 16). Leichte Abweichungen waren wahrscheinlich durch eine leicht kürzere
Reaktionszeit und Messungenauigkeiten bedingt. Gaschromatographisch konnte ein
unveränderter Enantiomerenüberschuss von ≥ 99,9 % des (S)-2-Butanols nachgewiesen
werden.
Abb. 17: Umsatz von
2-Butanon im 50 ml
Up-scaling Ansatz, erzielt
durch die Sorbitol-stabilisierte
evo-040 und das unstabilisierte
Enzym (Referenz)
3.6 Bestimmung der optimalen Substratbeladung (2-Butanon) für die ADH evo-040
Die Ergebnisse der Enzymstabilisierung stellten eine erfolgreiche Entwicklung eines
wasserarmen Bioprozesses in Aussicht. Daher wurde mit der Identifizierung der optimalen
Prozessparameter fortgefahren. Grundlegend war dabei die Bestimmung der optimalen
Substratbeladung, unter welcher die höchste (absolute) Produktausbeute erzielt werden kann.
Es wurde eine Versuchsreihe durchgeführt, in der schrittweise die Konzentration des
Substrates 2-Butanon erhöht wurde. Dabei blieb der Gesamtvolumenanteil der organischen
Phase konstant (90 %), was dazu führte, dass sich die Molenbrüche (2-Propanol/ 2-Butanon)
innerhalb der Versuchsreihe veränderten. Es ist somit zwischen dem Substratumsatz (Abb.
18–A) und der absoluten Produktausbeute (Abb. 18–B) zu unterscheiden.
Sorbitol Stabilisierung
Referenz
0
20
40
60
80
100
Um
satz
nac
h 88
Stu
nden
(%
)
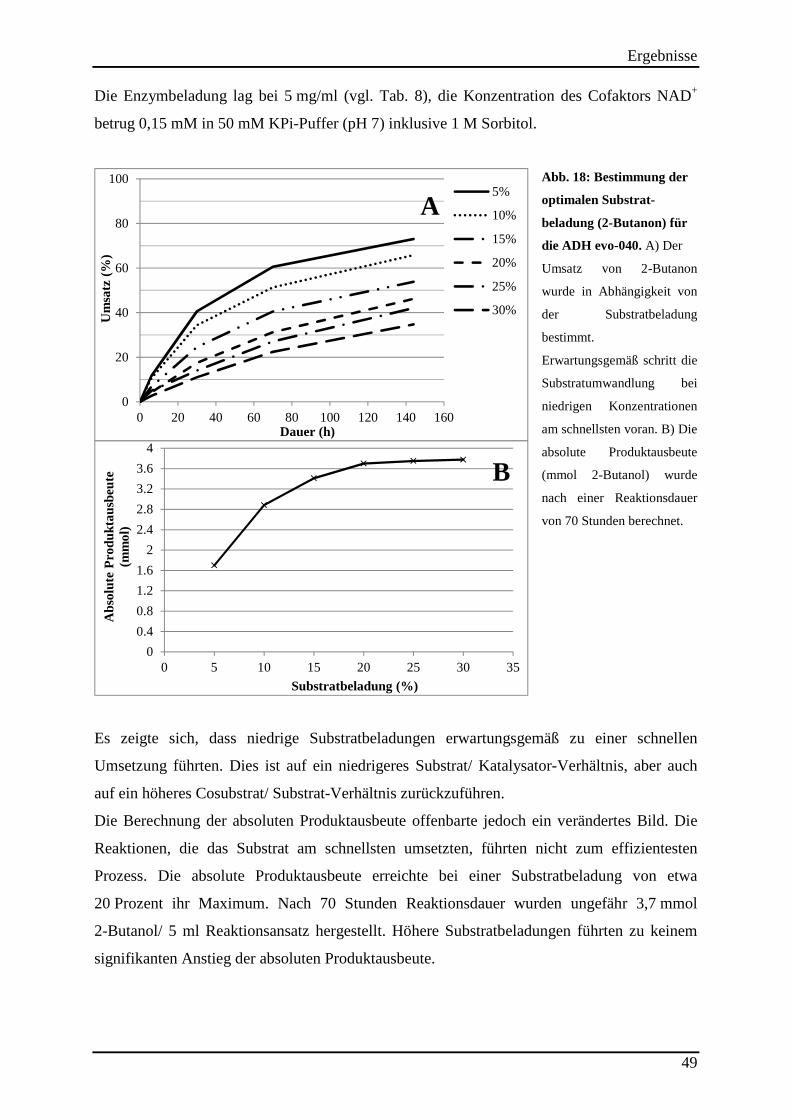
Ergebnisse
49
Die Enzymbeladung lag bei 5 mg/ml (vgl. Tab. 8), die Konzentration des Cofaktors NAD+
betrug 0,15 mM in 50 mM KPi-Puffer (pH 7) inklusive 1 M Sorbitol.
Abb. 18: Bestimmung der
optimalen Substrat-
beladung (2-Butanon) für
die ADH evo-040. A) Der
Umsatz von 2-Butanon
wurde in Abhängigkeit von
der Substratbeladung
bestimmt.
Erwartungsgemäß schritt die
Substratumwandlung bei
niedrigen Konzentrationen
am schnellsten voran. B) Die
absolute Produktausbeute
(mmol 2-Butanol) wurde
nach einer Reaktionsdauer
von 70 Stunden berechnet.
Es zeigte sich, dass niedrige Substratbeladungen erwartungsgemäß zu einer schnellen
Umsetzung führten. Dies ist auf ein niedrigeres Substrat/ Katalysator-Verhältnis, aber auch
auf ein höheres Cosubstrat/ Substrat-Verhältnis zurückzuführen.
Die Berechnung der absoluten Produktausbeute offenbarte jedoch ein verändertes Bild. Die
Reaktionen, die das Substrat am schnellsten umsetzten, führten nicht zum effizientesten
Prozess. Die absolute Produktausbeute erreichte bei einer Substratbeladung von etwa
20 Prozent ihr Maximum. Nach 70 Stunden Reaktionsdauer wurden ungefähr 3,7 mmol
2-Butanol/ 5 ml Reaktionsansatz hergestellt. Höhere Substratbeladungen führten zu keinem
signifikanten Anstieg der absoluten Produktausbeute.
0
20
40
60
80
100
0 20 40 60 80 100 120 140 160
Um
satz
(%
)
Dauer (h)
A5%
10%
15%
20%
25%
30%
0
0.4
0.8
1.2
1.6
2
2.4
2.8
3.2
3.6
4
0 5 10 15 20 25 30 35
Abs
olut
e P
rodu
ktau
sbeu
te
(mm
ol)
Substratbeladung (%)
B
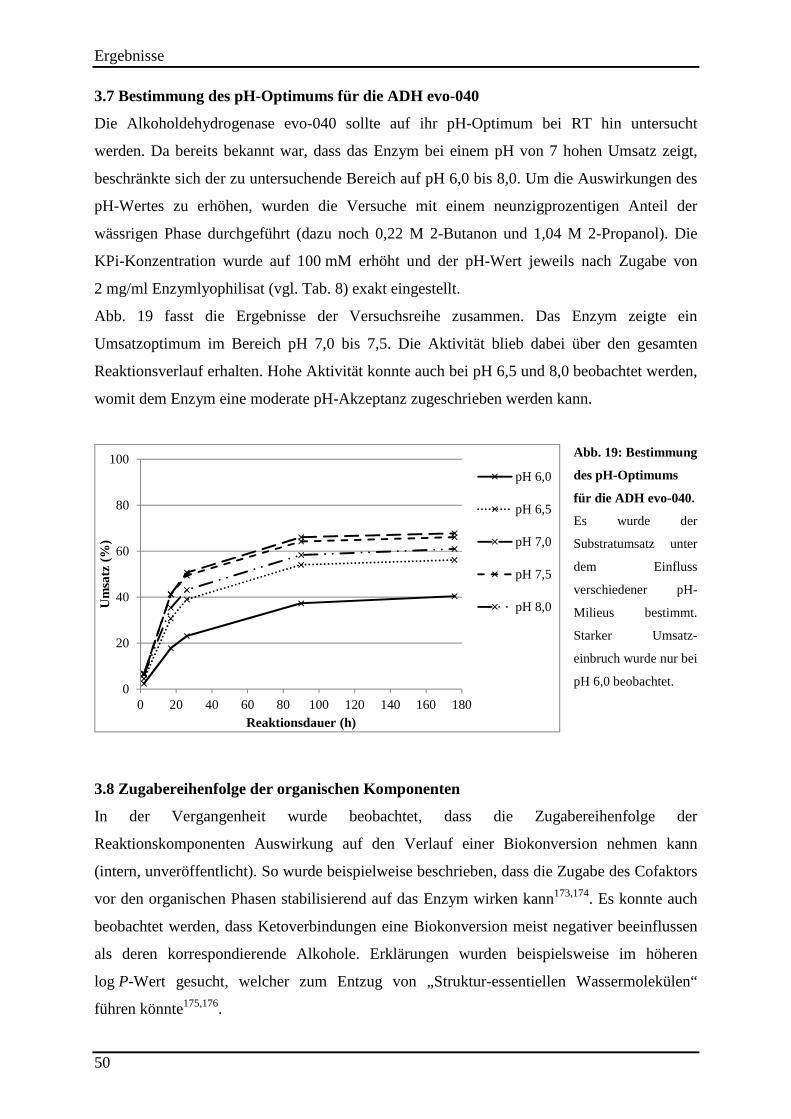
Ergebnisse
50
3.7 Bestimmung des pH-Optimums für die ADH evo-040
Die Alkoholdehydrogenase evo-040 sollte auf ihr pH-Optimum bei RT hin untersucht
werden. Da bereits bekannt war, dass das Enzym bei einem pH von 7 hohen Umsatz zeigt,
beschränkte sich der zu untersuchende Bereich auf pH 6,0 bis 8,0. Um die Auswirkungen des
pH-Wertes zu erhöhen, wurden die Versuche mit einem neunzigprozentigen Anteil der
wässrigen Phase durchgeführt (dazu noch 0,22 M 2-Butanon und 1,04 M 2-Propanol). Die
KPi-Konzentration wurde auf 100 mM erhöht und der pH-Wert jeweils nach Zugabe von
2 mg/ml Enzymlyophilisat (vgl. Tab. 8) exakt eingestellt.
Abb. 19 fasst die Ergebnisse der Versuchsreihe zusammen. Das Enzym zeigte ein
Umsatzoptimum im Bereich pH 7,0 bis 7,5. Die Aktivität blieb dabei über den gesamten
Reaktionsverlauf erhalten. Hohe Aktivität konnte auch bei pH 6,5 und 8,0 beobachtet werden,
womit dem Enzym eine moderate pH-Akzeptanz zugeschrieben werden kann.
Abb. 19: Bestimmung
des pH-Optimums
für die ADH evo-040.
Es wurde der
Substratumsatz unter
dem Einfluss
verschiedener pH-
Milieus bestimmt.
Starker Umsatz-
einbruch wurde nur bei
pH 6,0 beobachtet.
3.8 Zugabereihenfolge der organischen Komponenten
In der Vergangenheit wurde beobachtet, dass die Zugabereihenfolge der
Reaktionskomponenten Auswirkung auf den Verlauf einer Biokonversion nehmen kann
(intern, unveröffentlicht). So wurde beispielweise beschrieben, dass die Zugabe des Cofaktors
vor den organischen Phasen stabilisierend auf das Enzym wirken kann173,174. Es konnte auch
beobachtet werden, dass Ketoverbindungen eine Biokonversion meist negativer beeinflussen
als deren korrespondierende Alkohole. Erklärungen wurden beispielsweise im höheren
log P-Wert gesucht, welcher zum Entzug von „Struktur-essentiellen Wassermolekülen“
führen könnte175,176.
0
20
40
60
80
100
0 20 40 60 80 100 120 140 160 180
Um
satz
(%
)
Reaktionsdauer (h)
pH 6,0
pH 6,5
pH 7,0
pH 7,5
pH 8,0

Ergebnisse
51
Daher galt es zu untersuchen, ob eine Zugabevariation der organischen Komponenten
Einfluss auf die Effizienz des 2-Butanol Prozesses nimmt. Um den potentiellen Effekt besser
beobachten zu können, wurde die Konzentration des Enzymlyophilisats auf 2 mg/ml
erniedrigt (vgl. Tab. 8). Die Substratbeladung betrug 20 % des Gesamtvolumens, die des
Cosubstrates 70 % (wässrige Phase: 50 mM KPi; pH 7,0 inklusive 1 M Sorbitol; 0,15 mM
NAD+).
Nach der Durchführung des unter 3.5 beschriebenen Stabilisierungsprotokolls wurde die
Enzymlösung a) direkt mit der gesamten organischen Phase vereint, b) 2 Minuten mit der
2-Propanol Fraktion inkubiert, c) 5 Minuten mit der 2-Propanol Fraktion inkubiert,
d) 2 Minuten mit der 2-Butanon Fraktion inkubiert oder, e) 5 Minuten mit der 2-Butanon
Fraktion inkubiert, bevor die noch fehlende Komponente zugegeben wurde.
Die Ergebnisse sind in Abb. 20 dargestellt. Es zeigte sich, dass die separate Zugabe von
2-Propanol vor 2-Butanon Zugabe zu signifikant höherem Umsatz führte. Erstaunlicherweise
lag dieser sogar über dem der Inkubations-freien Reaktion. Die Inkubation mit 2-Butanon
dagegen schien den Prozess negativ zu beeinflussen. Der Effekt verstärkte sich bei
verlängerter Inkubation (5 Minuten). Dieses Verhalten zeigte Isopropanol dagegen nicht, es
spielte keine signifikante Rolle, ob das Enzym für kurze oder längere Zeit inkubiert wurde.
Weiterhin muss jedoch auch beachtet werden, dass während der unterschiedlichen
Inkubationsphasen (a - e) auch unterschiedliche Verhältnisse der wässrigen zur organischen
Phase herrschten, da (prozessbedingt) 2-Butanon und Isopropanol in unterschiedlichen
Volumina eingesetzt werden. Dies könnte ebenfalls Einfluss auf die Enzymintegrität
genommen haben.
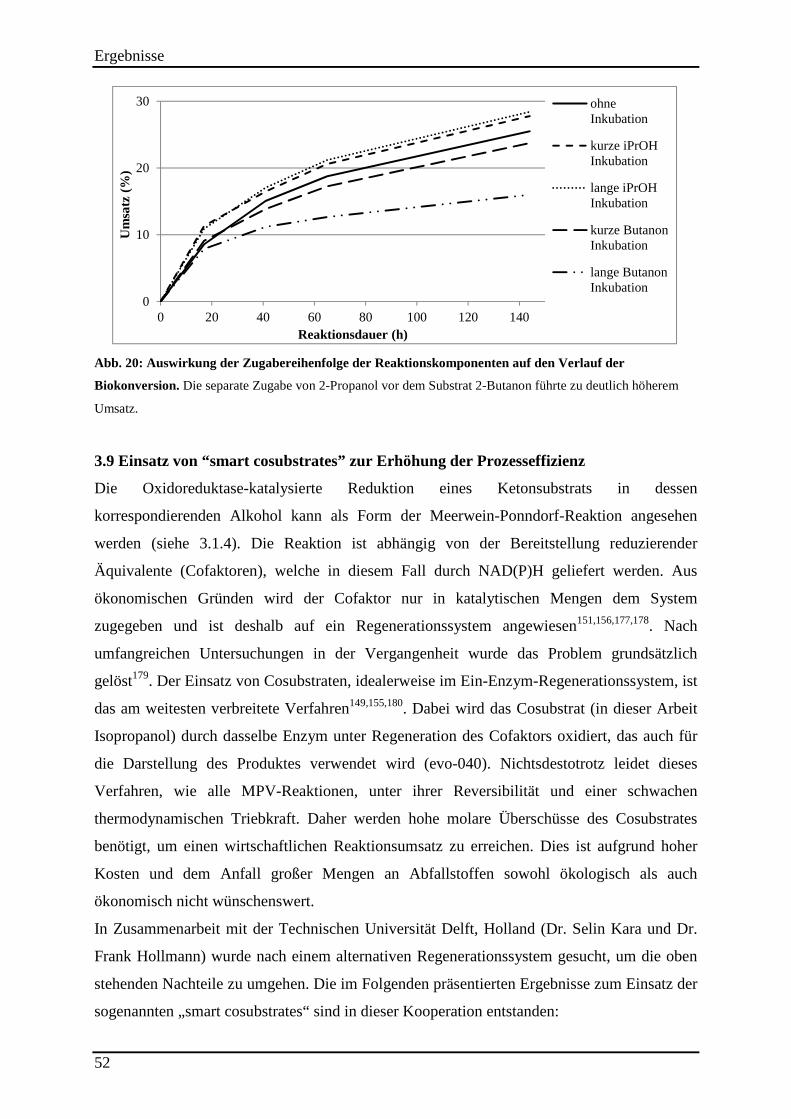
Ergebnisse
52
Abb. 20: Auswirkung der Zugabereihenfolge der Reaktionskomponenten auf den Verlauf der
Biokonversion. Die separate Zugabe von 2-Propanol vor dem Substrat 2-Butanon führte zu deutlich höherem
Umsatz.
3.9 Einsatz von “smart cosubstrates” zur Erhöhung der Prozesseffizienz
Die Oxidoreduktase-katalysierte Reduktion eines Ketonsubstrats in dessen
korrespondierenden Alkohol kann als Form der Meerwein-Ponndorf-Reaktion angesehen
werden (siehe 3.1.4). Die Reaktion ist abhängig von der Bereitstellung reduzierender
Äquivalente (Cofaktoren), welche in diesem Fall durch NAD(P)H geliefert werden. Aus
ökonomischen Gründen wird der Cofaktor nur in katalytischen Mengen dem System
zugegeben und ist deshalb auf ein Regenerationssystem angewiesen151,156,177,178. Nach
umfangreichen Untersuchungen in der Vergangenheit wurde das Problem grundsätzlich
gelöst179. Der Einsatz von Cosubstraten, idealerweise im Ein-Enzym-Regenerationssystem, ist
das am weitesten verbreitete Verfahren149,155,180. Dabei wird das Cosubstrat (in dieser Arbeit
Isopropanol) durch dasselbe Enzym unter Regeneration des Cofaktors oxidiert, das auch für
die Darstellung des Produktes verwendet wird (evo-040). Nichtsdestotrotz leidet dieses
Verfahren, wie alle MPV-Reaktionen, unter ihrer Reversibilität und einer schwachen
thermodynamischen Triebkraft. Daher werden hohe molare Überschüsse des Cosubstrates
benötigt, um einen wirtschaftlichen Reaktionsumsatz zu erreichen. Dies ist aufgrund hoher
Kosten und dem Anfall großer Mengen an Abfallstoffen sowohl ökologisch als auch
ökonomisch nicht wünschenswert.
In Zusammenarbeit mit der Technischen Universität Delft, Holland (Dr. Selin Kara und Dr.
Frank Hollmann) wurde nach einem alternativen Regenerationssystem gesucht, um die oben
stehenden Nachteile zu umgehen. Die im Folgenden präsentierten Ergebnisse zum Einsatz der
sogenannten „smart cosubstrates“ sind in dieser Kooperation entstanden:
0
10
20
30
0 20 40 60 80 100 120 140
Um
satz
(%
)
Reaktionsdauer (h)
ohneInkubation
kurze iPrOHInkubation
lange iPrOHInkubation
kurze ButanonInkubation
lange ButanonInkubation
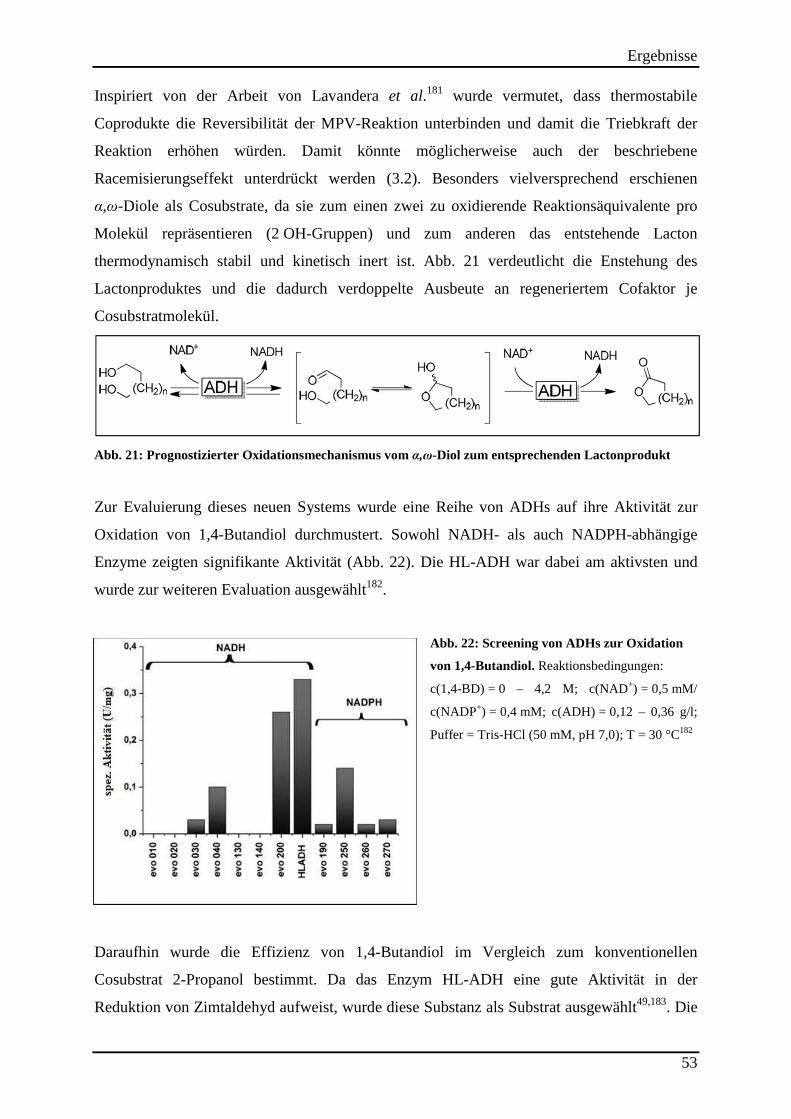
Ergebnisse
53
Inspiriert von der Arbeit von Lavandera et al.181 wurde vermutet, dass thermostabile
Coprodukte die Reversibilität der MPV-Reaktion unterbinden und damit die Triebkraft der
Reaktion erhöhen würden. Damit könnte möglicherweise auch der beschriebene
Racemisierungseffekt unterdrückt werden (3.2). Besonders vielversprechend erschienen
α,ω-Diole als Cosubstrate, da sie zum einen zwei zu oxidierende Reaktionsäquivalente pro
Molekül repräsentieren (2 OH-Gruppen) und zum anderen das entstehende Lacton
thermodynamisch stabil und kinetisch inert ist. Abb. 21 verdeutlicht die Enstehung des
Lactonproduktes und die dadurch verdoppelte Ausbeute an regeneriertem Cofaktor je
Cosubstratmolekül.
Abb. 21: Prognostizierter Oxidationsmechanismus vom α,ω-Diol zum entsprechenden Lactonprodukt
Zur Evaluierung dieses neuen Systems wurde eine Reihe von ADHs auf ihre Aktivität zur
Oxidation von 1,4-Butandiol durchmustert. Sowohl NADH- als auch NADPH-abhängige
Enzyme zeigten signifikante Aktivität (Abb. 22). Die HL-ADH war dabei am aktivsten und
wurde zur weiteren Evaluation ausgewählt182.
Abb. 22: Screening von ADHs zur Oxidation
von 1,4-Butandiol. Reaktionsbedingungen:
c(1,4-BD) = 0 – 4,2 M; c(NAD+) = 0,5 mM/
c(NADP+) = 0,4 mM; c(ADH) = 0,12 – 0,36 g/l;
Puffer = Tris-HCl (50 mM, pH 7,0); T = 30 °C182
Daraufhin wurde die Effizienz von 1,4-Butandiol im Vergleich zum konventionellen
Cosubstrat 2-Propanol bestimmt. Da das Enzym HL-ADH eine gute Aktivität in der
Reduktion von Zimtaldehyd aufweist, wurde diese Substanz als Substrat ausgewählt49,183. Die
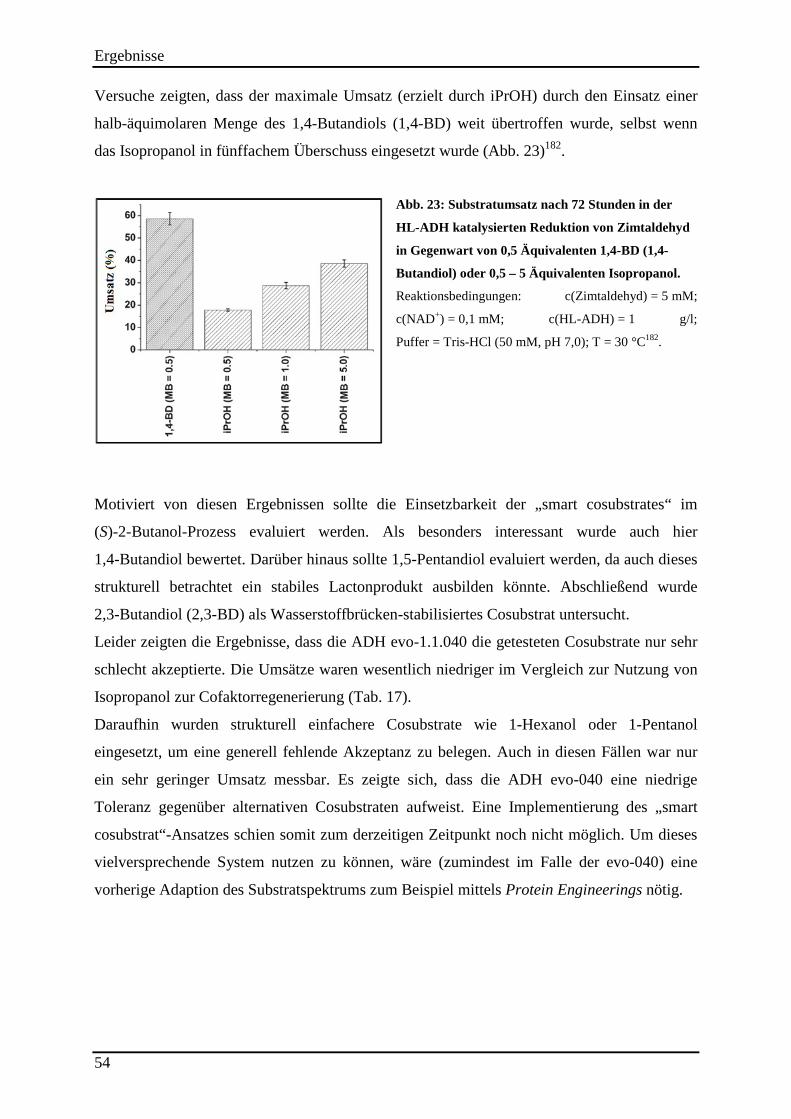
Ergebnisse
54
Versuche zeigten, dass der maximale Umsatz (erzielt durch iPrOH) durch den Einsatz einer
halb-äquimolaren Menge des 1,4-Butandiols (1,4-BD) weit übertroffen wurde, selbst wenn
das Isopropanol in fünffachem Überschuss eingesetzt wurde (Abb. 23)182.
Abb. 23: Substratumsatz nach 72 Stunden in der
HL-ADH katalysierten Reduktion von Zimtaldehyd
in Gegenwart von 0,5 Äquivalenten 1,4-BD (1,4-
Butandiol) oder 0,5 – 5 Äquivalenten Isopropanol.
Reaktionsbedingungen: c(Zimtaldehyd) = 5 mM;
c(NAD+) = 0,1 mM; c(HL-ADH) = 1 g/l;
Puffer = Tris-HCl (50 mM, pH 7,0); T = 30 °C182.
Motiviert von diesen Ergebnissen sollte die Einsetzbarkeit der „smart cosubstrates“ im
(S)-2-Butanol-Prozess evaluiert werden. Als besonders interessant wurde auch hier
1,4-Butandiol bewertet. Darüber hinaus sollte 1,5-Pentandiol evaluiert werden, da auch dieses
strukturell betrachtet ein stabiles Lactonprodukt ausbilden könnte. Abschließend wurde
2,3-Butandiol (2,3-BD) als Wasserstoffbrücken-stabilisiertes Cosubstrat untersucht.
Leider zeigten die Ergebnisse, dass die ADH evo-1.1.040 die getesteten Cosubstrate nur sehr
schlecht akzeptierte. Die Umsätze waren wesentlich niedriger im Vergleich zur Nutzung von
Isopropanol zur Cofaktorregenerierung (Tab. 17).
Daraufhin wurden strukturell einfachere Cosubstrate wie 1-Hexanol oder 1-Pentanol
eingesetzt, um eine generell fehlende Akzeptanz zu belegen. Auch in diesen Fällen war nur
ein sehr geringer Umsatz messbar. Es zeigte sich, dass die ADH evo-040 eine niedrige
Toleranz gegenüber alternativen Cosubstraten aufweist. Eine Implementierung des „smart
cosubstrat“-Ansatzes schien somit zum derzeitigen Zeitpunkt noch nicht möglich. Um dieses
vielversprechende System nutzen zu können, wäre (zumindest im Falle der evo-040) eine
vorherige Adaption des Substratspektrums zum Beispiel mittels Protein Engineerings nötig.
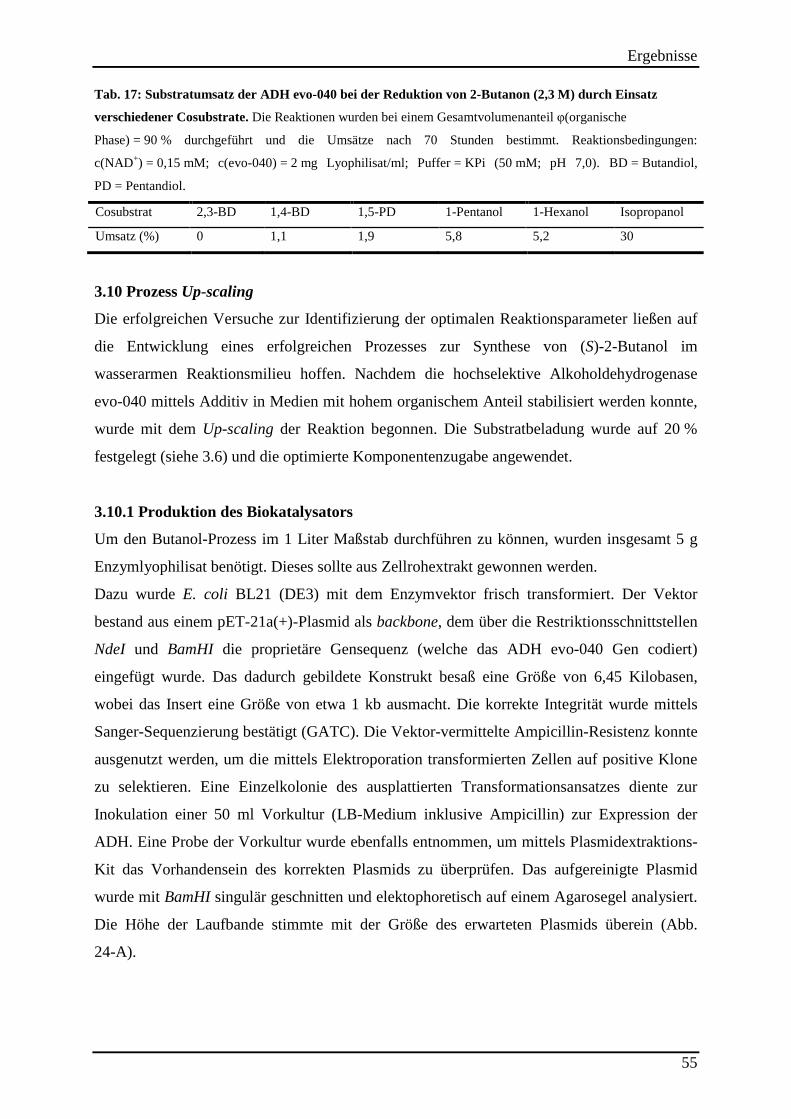
Ergebnisse
55
Tab. 17: Substratumsatz der ADH evo-040 bei der Reduktion von 2-Butanon (2,3 M) durch Einsatz
verschiedener Cosubstrate. Die Reaktionen wurden bei einem Gesamtvolumenanteil φ(organische
Phase) = 90 % durchgeführt und die Umsätze nach 70 Stunden bestimmt. Reaktionsbedingungen:
c(NAD+) = 0,15 mM; c(evo-040) = 2 mg Lyophilisat/ml; Puffer = KPi (50 mM; pH 7,0). BD = Butandiol,
PD = Pentandiol.
Cosubstrat 2,3-BD 1,4-BD 1,5-PD 1-Pentanol 1-Hexanol Isopropanol
Umsatz (%) 0 1,1 1,9 5,8 5,2 30
3.10 Prozess Up-scaling
Die erfolgreichen Versuche zur Identifizierung der optimalen Reaktionsparameter ließen auf
die Entwicklung eines erfolgreichen Prozesses zur Synthese von (S)-2-Butanol im
wasserarmen Reaktionsmilieu hoffen. Nachdem die hochselektive Alkoholdehydrogenase
evo-040 mittels Additiv in Medien mit hohem organischem Anteil stabilisiert werden konnte,
wurde mit dem Up-scaling der Reaktion begonnen. Die Substratbeladung wurde auf 20 %
festgelegt (siehe 3.6) und die optimierte Komponentenzugabe angewendet.
3.10.1 Produktion des Biokatalysators
Um den Butanol-Prozess im 1 Liter Maßstab durchführen zu können, wurden insgesamt 5 g
Enzymlyophilisat benötigt. Dieses sollte aus Zellrohextrakt gewonnen werden.
Dazu wurde E. coli BL21 (DE3) mit dem Enzymvektor frisch transformiert. Der Vektor
bestand aus einem pET-21a(+)-Plasmid als backbone, dem über die Restriktionsschnittstellen
NdeI und BamHI die proprietäre Gensequenz (welche das ADH evo-040 Gen codiert)
eingefügt wurde. Das dadurch gebildete Konstrukt besaß eine Größe von 6,45 Kilobasen,
wobei das Insert eine Größe von etwa 1 kb ausmacht. Die korrekte Integrität wurde mittels
Sanger-Sequenzierung bestätigt (GATC). Die Vektor-vermittelte Ampicillin-Resistenz konnte
ausgenutzt werden, um die mittels Elektroporation transformierten Zellen auf positive Klone
zu selektieren. Eine Einzelkolonie des ausplattierten Transformationsansatzes diente zur
Inokulation einer 50 ml Vorkultur (LB-Medium inklusive Ampicillin) zur Expression der
ADH. Eine Probe der Vorkultur wurde ebenfalls entnommen, um mittels Plasmidextraktions-
Kit das Vorhandensein des korrekten Plasmids zu überprüfen. Das aufgereinigte Plasmid
wurde mit BamHI singulär geschnitten und elektophoretisch auf einem Agarosegel analysiert.
Die Höhe der Laufbande stimmte mit der Größe des erwarteten Plasmids überein (Abb.
24-A).

Ergebnisse
56
Abb. 24: Expression der ADH evo-040 für die Biotransformation. A) Testrestriktion des Konstrukts
pET21a(+)-evo-040. Die Laufhöhe der mit BamHI geschnittenen DNA stimmt mit dem erwarteten Plasmid
überein (6,45 kb). B) SDS-Elektrophoresegel des Zellrohextrakts. Es ist eine Überexpressionsbande auf Höhe
des erwarteten Enzyms zu erkennen.
Je 10 ml der Vorkultur wurden genutzt, um vier Hauptkulturen aus 1 Liter TB-Medium
(inklusive Ampicillin) zu inokulieren (37 °C; 180 rpm). Die Hauptkulturen wuchsen bis zu
einer OD600 = 0,9, bevor die Expression durch Zugabe von IPTG
(Endkonzentration = 0,1 mM) induziert wurde. Nach 20 Stunden der Expression bei 30 °C
wurde die gesamte Zellmasse durch Zentrifugation (15 min, 8000 rpm) geerntet. Sie ergab
eine Zellfeuchtmasse von 72 g. Für den Zellaufschluss wurde das Pellet in 216 ml TEA-
Puffer (50 mM, pH 7,0) resuspendiert, 280 µl einer 1 M MgCl2 sowie 3 U/ml DNase I
zugegeben.
Durch French-Press Homogenisierung in zwei Durchläufen (900 bar) sollten die Zellen
aufgeschlossen werden. Der entstandene Zellrohextrakt wurde 20 Minuten bei 10.000 g
zentrifugiert, um Zelltrümmer zu entfernen. Die Durchführung einer SDS-Gelelektrophorese
bestätigte die Überexpression des Zielproteins (Abb. 24-B).
Der dadurch gewonnene Rohextrakt wurde photometrisch auf Oxidoreduktaseaktivität unter
Ausnutzung des evo-040 Aktivitätsassays untersucht. Die Lösung besaß eine Aktivität von
1110 U/ml (Substrat: trans-2-Hexenal) bei einem Proteingehalt von 22 mg/ml, was einer
Proteinaktivität von 50 U/mg Protein entspricht.
Zur Lyophilisierung wurde der Rohextrakt bei -20 °C eingefroren und anschließend für 48
Stunden gefriergetrocknet. Das Lyophilisat besaß eine Gewichtsaktivität von 8,2 U/mg
Lyophilisat beziehungsweise eine Proteinaktivität von 28,2 U/mg Protein.
Die Gesamtausbeute an Lyophilisat betrug 13,4 g, womit genug Produkt gewonnen werden
konnte, um den Bioprozess im 1 Liter Maßstab durchführen zu können.
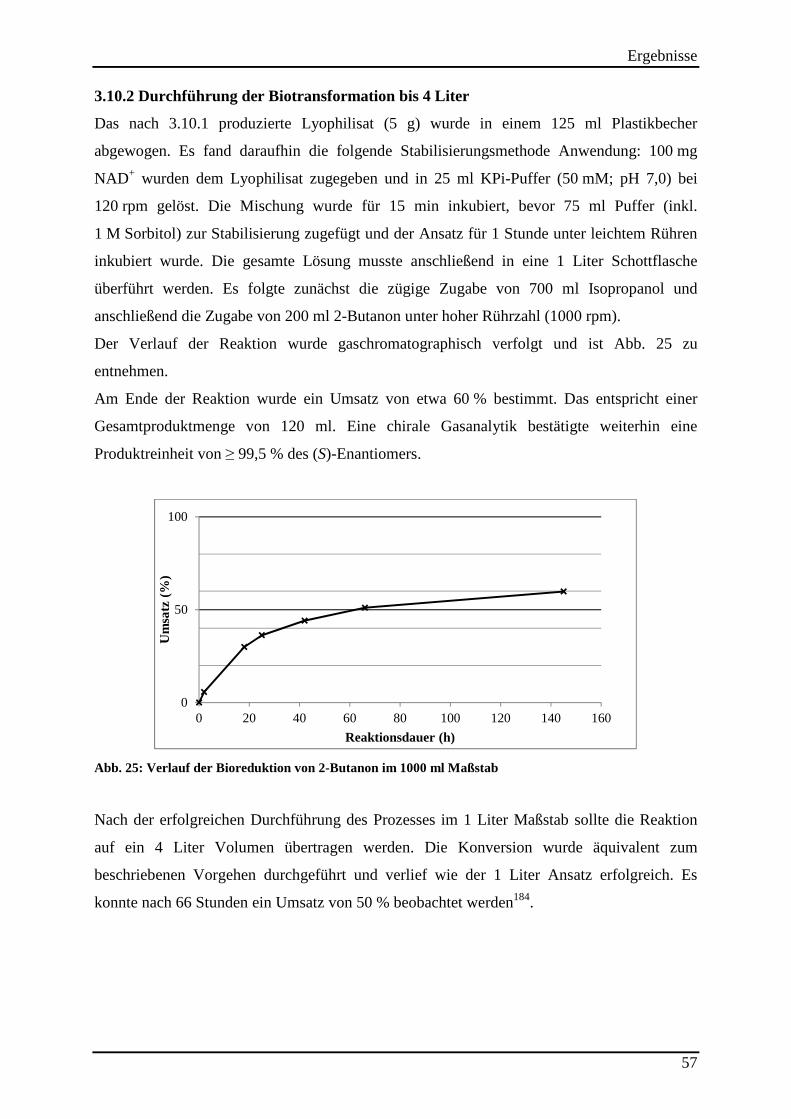
Ergebnisse
57
3.10.2 Durchführung der Biotransformation bis 4 Liter
Das nach 3.10.1 produzierte Lyophilisat (5 g) wurde in einem 125 ml Plastikbecher
abgewogen. Es fand daraufhin die folgende Stabilisierungsmethode Anwendung: 100 mg
NAD+ wurden dem Lyophilisat zugegeben und in 25 ml KPi-Puffer (50 mM; pH 7,0) bei
120 rpm gelöst. Die Mischung wurde für 15 min inkubiert, bevor 75 ml Puffer (inkl.
1 M Sorbitol) zur Stabilisierung zugefügt und der Ansatz für 1 Stunde unter leichtem Rühren
inkubiert wurde. Die gesamte Lösung musste anschließend in eine 1 Liter Schottflasche
überführt werden. Es folgte zunächst die zügige Zugabe von 700 ml Isopropanol und
anschließend die Zugabe von 200 ml 2-Butanon unter hoher Rührzahl (1000 rpm).
Der Verlauf der Reaktion wurde gaschromatographisch verfolgt und ist Abb. 25 zu
entnehmen.
Am Ende der Reaktion wurde ein Umsatz von etwa 60 % bestimmt. Das entspricht einer
Gesamtproduktmenge von 120 ml. Eine chirale Gasanalytik bestätigte weiterhin eine
Produktreinheit von ≥ 99,5 % des (S)-Enantiomers.
Abb. 25: Verlauf der Bioreduktion von 2-Butanon im 1000 ml Maßstab
Nach der erfolgreichen Durchführung des Prozesses im 1 Liter Maßstab sollte die Reaktion
auf ein 4 Liter Volumen übertragen werden. Die Konversion wurde äquivalent zum
beschriebenen Vorgehen durchgeführt und verlief wie der 1 Liter Ansatz erfolgreich. Es
konnte nach 66 Stunden ein Umsatz von 50 % beobachtet werden184.
0
50
100
0 20 40 60 80 100 120 140 160
Um
satz
(%
)
Reaktionsdauer (h)

Ergebnisse
58
3.11 Produktaufreinigung
Auch wenn bereits ein Prozess etabliert wurde, in dem der Wasseranteil auf nur 10 %
erniedrigt war, stellte dieser immer noch ein Problem bei der Aufreinigung des Butanols dar
(siehe 3.4). Zum Entfernen des restlichen Wassers wurden zwei Strategien entwickelt:
3.11.1 Produkttrocknung über ein Molekularsieb
Die erste Strategie beruhte auf dem Entzug des Wassers nach Reaktionsende durch Zugabe
von Salz und eines Molekularsiebs. Dazu wurden zuerst 10 % w/w Kaliumcarbonat dem
Reaktionsmedium beigemischt, gut durchmengt und anschließend über eine Nutsche
abfiltriert. Die Feintrocknung des Filtrats erfolgte anschließend über Nacht über einem 3 Å
Molekularsieb. Der Wassergehalt konnte durch diese Methode auf unter 0,1 % reduziert
werden. Allerdings ergaben sich einige Nachteile: So flockte das Salz sehr stark aus und es
blieb ein Teil der Reaktionslösung an diesem anhaften. Deshalb musste das Kaliumcarbonat
mittels Dichlormethan nachgespült werden. Weiterhin erwies sich auch das Molekularsieb,
insbesondere in Hinblick auf ein Up-scaling, als unpraktisch, da es ebenfalls nachgespült und
nach Gebrauch aufwändig regeneriert werden musste.
3.11.2 Produkttrocknung mittels Azeotroprektifikati on
Die zweite Methode zur Trocknung beruhte auf dem Prinzip der Azeotroprektifikation. Dazu
wurde dem azeotropen Gemisch ein Lösemittel zugesetzt, welches das Wasser aus dem
Reaktionsmedium bei einer niedrigen Siedetemperatur ausschleppt. Es wurde Cyclohexan im
Verhältnis 1,5:1 mit dem Reaktionsmedium vereint und anschließend die gesamte
Reaktionslösung über eine Füllkörperkolonne abdestilliert. Der Wassergehalt des so
erhaltenen Produktes belief sich auf unter 0,1 %.
3.11.3 Produktaufreinigung mittels Destillation
320 ml der filtrierten Reaktionslösung wurden mit 500 ml Cyclohexan versetzt und über drei
50 cm Füllkörperkolonnen mit 5 mm Raschig-Ringen destilliert. Das Destillationsprotokoll ist
Tab. 18 zu entnehmen.
Der Sumpf (S2) wurde separat erneut destilliert. Es ergab sich eine Fraktion ohne Vorlauf mit
einem Siedepunkt von 99 °C. Auswaage: 32 g farblose, klare Flüssigkeit (40 ml). Die GC-
Analytik bezifferte das Produkt auf eine Reinheit von ≥ 99 % 2-Butanol sowie eine
Enantiomerenreinheit von ≥ 99,5 % des (S)-Enantiomers. Durch die Karl-Fischer-Titration
konnte ein Wassergehalt von 0,05 % bestimmt werden.
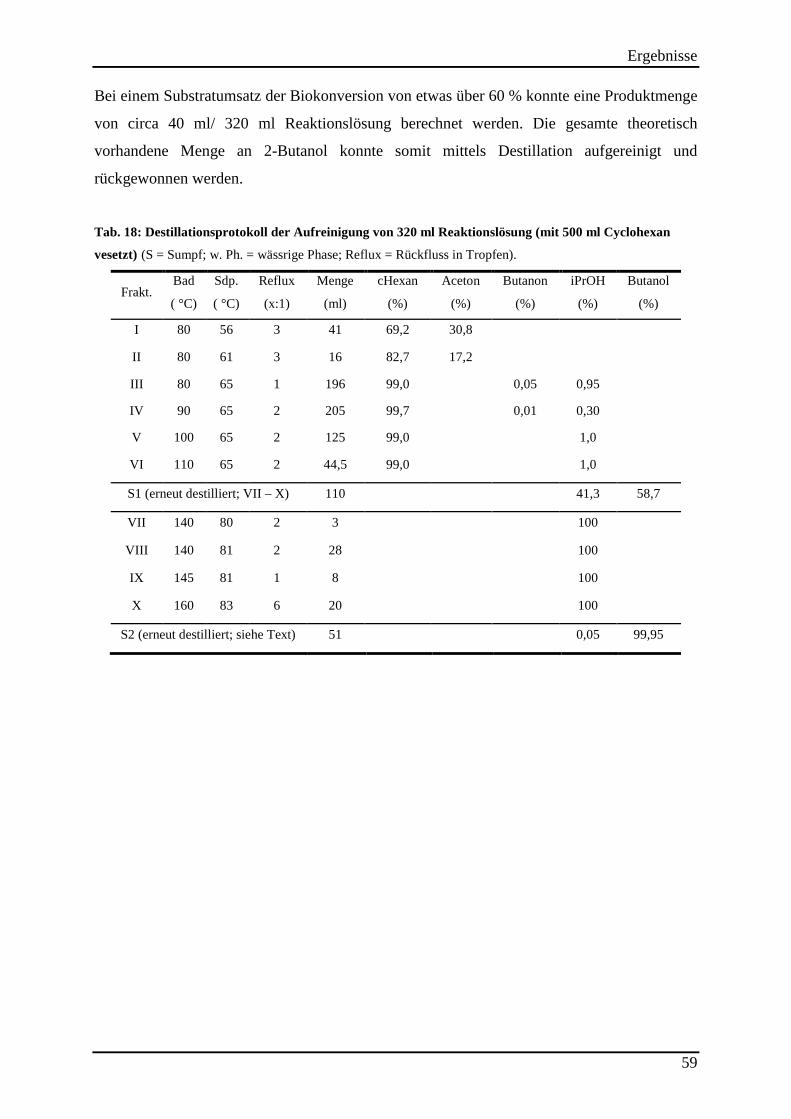
Ergebnisse
59
Bei einem Substratumsatz der Biokonversion von etwas über 60 % konnte eine Produktmenge
von circa 40 ml/ 320 ml Reaktionslösung berechnet werden. Die gesamte theoretisch
vorhandene Menge an 2-Butanol konnte somit mittels Destillation aufgereinigt und
rückgewonnen werden.
Tab. 18: Destillationsprotokoll der Aufreinigung von 320 ml Reaktionslösung (mit 500 ml Cyclohexan
vesetzt) (S = Sumpf; w. Ph. = wässrige Phase; Reflux = Rückfluss in Tropfen).
Frakt. Bad
( °C)
Sdp.
( °C)
Reflux
(x:1)
Menge
(ml)
cHexan
(%)
Aceton
(%)
Butanon
(%)
iPrOH
(%)
Butanol
(%)
I 80 56 3 41 69,2 30,8
II 80 61 3 16 82,7 17,2
III 80 65 1 196 99,0 0,05 0,95
IV 90 65 2 205 99,7 0,01 0,30
V 100 65 2 125 99,0 1,0
VI 110 65 2 44,5 99,0 1,0
S1 (erneut destilliert; VII – X) 110 41,3 58,7
VII 140 80 2 3 100
VIII 140 81 2 28 100
IX 145 81 1 8 100
X 160 83 6 20 100
S2 (erneut destilliert; siehe Text) 51 0,05 99,95

Ergebnisse
60
Ergebnisse Teil 2: Enzymoptimierung durch Verfahren des Protein Engineerings
In diesem Arbeitsabschnitt sollte die Alkoholdehydrogenase evo-040 mittels Protein
Engineering optimiert werden. Ziel war es, den entwickelten Prozess zur Darstellung von
enantiomerenreinem (S)-2-Butanol durch den Einsatz einer optimierten Enzymvariante noch
effizienter zu machen. Idealerweise sollte das Enzym gegen die organischen Lösemittel
stabilisiert werden. Allerdings würde auch eine höhere Aktivität der evo-040 die
Prozessausbeute verbessern.
Zur Entwicklung einer optimierten Variante wurden drei Ansätze verfolgt (siehe auch 4.5.1):
A) Rationale Proteinentwicklung: Durch die Integration von Disulfidbrücken sollte die
Proteinstruktur stabilisiert werden. Um die Etablierung solcher Brücken zu bewerkstelligen,
mussten je Variante zwei Aminosäureaustausche mittels QuikChange-Mutagenese
durchgeführt werden.
B) Semi-rationale Proteinentwicklung: Es sollte das Bioinformatikprogramm Bio-Prodict
3DM verwendet werden, um Aminosäureaustausche vorherzubestimmen, die vorteilhaft sein
könnten, da sie in homologen Proteinen zu verbesserten Eigenschaften geführt haben. Die
dadurch bestimmten Aminosäuren sollten durch Sättigungsmutagenese mittels QuikChange-
Technik ausgetauscht und die Enzymvarianten im Anschluss charakterisiert werden.
C) Zufallsmutagenese: In diesem Ansatz sollten durch das Anwenden der error-prone PCR
Technik111 zufällig Mutationen in die DNA-Sequenz der Alkoholdehydrogenase evo-040
eingeführt werden. Die generierten Mutanten sollten anschließend in einem high-throughput
Verfahren auf verbesserte Eigenschaften durchmustert und die Gensequenzen dieser
Varianten aufgeklärt werden.
3.12 Rationales Proteindesign durch den Einbau von Disulfidbrücken
3.12.1 Produktion der Enzymvarianten
Zur Identifizierung von Aminosäurepositionen, die theoretisch im Stande sind,
Disulfidbrücken auszubilden, wurde das Programm „Disulfide by Design Version 1.2“
(http://cptweb.cpt.wayne.edu/DbD/) genutzt185 (siehe auch 4.5.1). Um die Berechnungen
durchzuführen, wurden im Vorfeld zwei Homologiemodelle der ADH evo-040 erstellt. Dazu
wurde zum einen das Programm „Swiss-Model“ (http://swissmodel.expasy.org/)92,186, zum
anderen das Programm „CPHmodels 3.2“ (http://www.cbs.dtu.dk/services/CPHmodels/“)93
genutzt. Die beiden erstellten Homologiemodelle zeigten untereinander eine sehr hohe
Strukturübereinstimmung. Die erstellten Modelle wurden in das Programm „Disulfide by

Ergebnisse
61
Design Version 1.2“ eingepflegt. Dieses berechnete eine Vielzahl möglicher Positionspaare,
die theoretisch zur Ausbildung einer Schwefelbrücke imstande sein könnten. Unter
Berücksichtigung des idealen Torsionswinkels fand eine Selektion von insgesamt sechs
Positionspaaren statt (Tab. 19).
Tab. 19: Disulfidvarianten, die zur Konstruktion ausgewählt wurden
Disulfid-Variante Mutation A Mutation B
SSB1 E34C G64C
SSB2 V63C A142C
SSB3 V125C A130C
SSB4 D138C Q141C
SSB5 G174C G331C
SSB6 T238C V261C
Die Disulfidvarianten wurden mittels QuikChange-Technik generiert. Dazu mussten zunächst
die optimalen Mutagenesebedingungen evaluiert werden. Je ein Primerpaar (SSB-xxC-Fwd
und SSB-xxC-Rev) diente zur Insertion einer Mutation. Bereits unter Standardbedingungen
waren zehn der zwölf PCR-Reaktionen erfolgreich (Abb. 26). Die QuikChange-PCR der
beiden Mutanten A142C und T238C konnte durch eine zehnfach erniedrigte
Primerkonzentration realisiert werden.
Abb. 26: Evaluierung der idealen QuikChange-Bedingungen zur Generierung der Disulfidvarianten.
Lediglich die Mutationen A142C und T238C konnten nicht unter Standardbedingungen etabliert werden.
Im Folgenden wurden die Doppelmutanten der evo-040 durch je zwei aufeinander folgende
QuikChange-Mutageneseschritte konstruiert. Die Korrektheit der Varianten konnte durch
Sanger-Sequenzierung bestätigt werden. Als Expressionswirt diente E. coli SHuffle T7
(NEB), da dieser Stamm im Stande ist, Disulfidbrücken auszubilden. Die Transformation
konnte durch Elektroporation erfolgreich durchgeführt werden.
Nach erfolgreicher Entwicklung der neuen Stämme wurden die Enzyme im 500 ml Maßstab
exprimiert und Lyophilisate hergestellt. Eine SDS-Gelelektrophorese der löslichen und
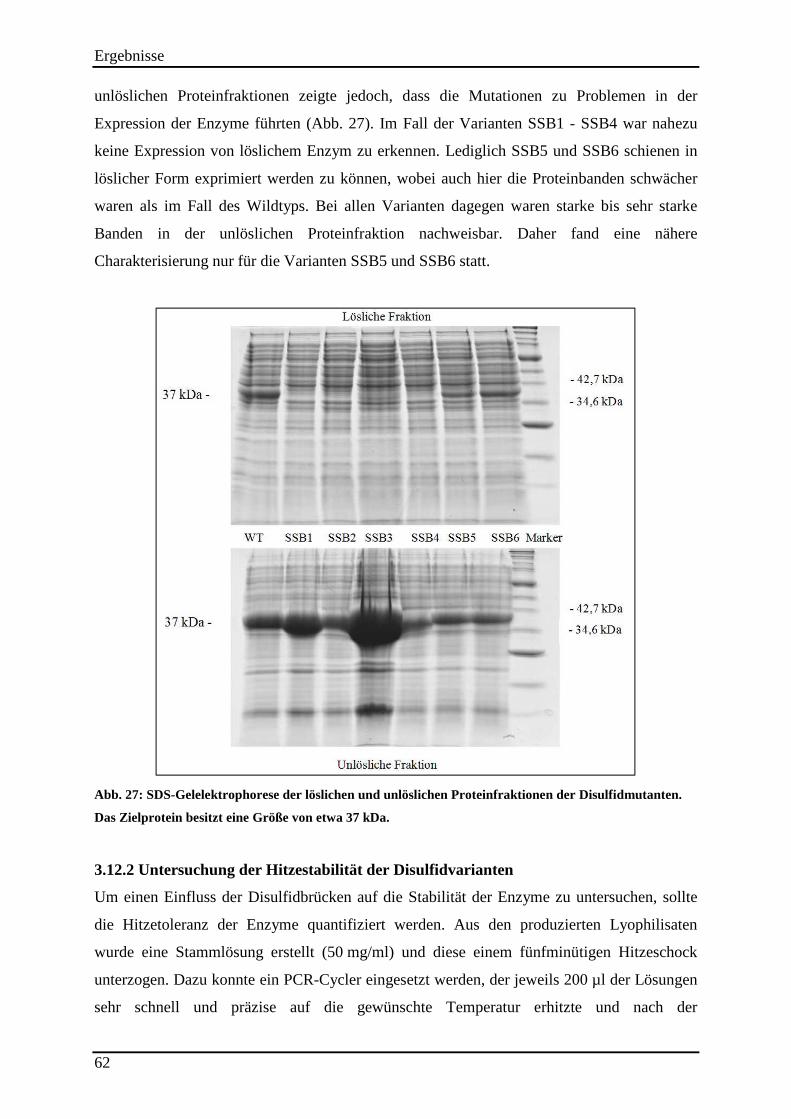
Ergebnisse
62
unlöslichen Proteinfraktionen zeigte jedoch, dass die Mutationen zu Problemen in der
Expression der Enzyme führten (Abb. 27). Im Fall der Varianten SSB1 - SSB4 war nahezu
keine Expression von löslichem Enzym zu erkennen. Lediglich SSB5 und SSB6 schienen in
löslicher Form exprimiert werden zu können, wobei auch hier die Proteinbanden schwächer
waren als im Fall des Wildtyps. Bei allen Varianten dagegen waren starke bis sehr starke
Banden in der unlöslichen Proteinfraktion nachweisbar. Daher fand eine nähere
Charakterisierung nur für die Varianten SSB5 und SSB6 statt.
Abb. 27: SDS-Gelelektrophorese der löslichen und unlöslichen Proteinfraktionen der Disulfidmutanten.
Das Zielprotein besitzt eine Größe von etwa 37 kDa.
3.12.2 Untersuchung der Hitzestabilität der Disulfidvarianten
Um einen Einfluss der Disulfidbrücken auf die Stabilität der Enzyme zu untersuchen, sollte
die Hitzetoleranz der Enzyme quantifiziert werden. Aus den produzierten Lyophilisaten
wurde eine Stammlösung erstellt (50 mg/ml) und diese einem fünfminütigen Hitzeschock
unterzogen. Dazu konnte ein PCR-Cycler eingesetzt werden, der jeweils 200 µl der Lösungen
sehr schnell und präzise auf die gewünschte Temperatur erhitzte und nach der
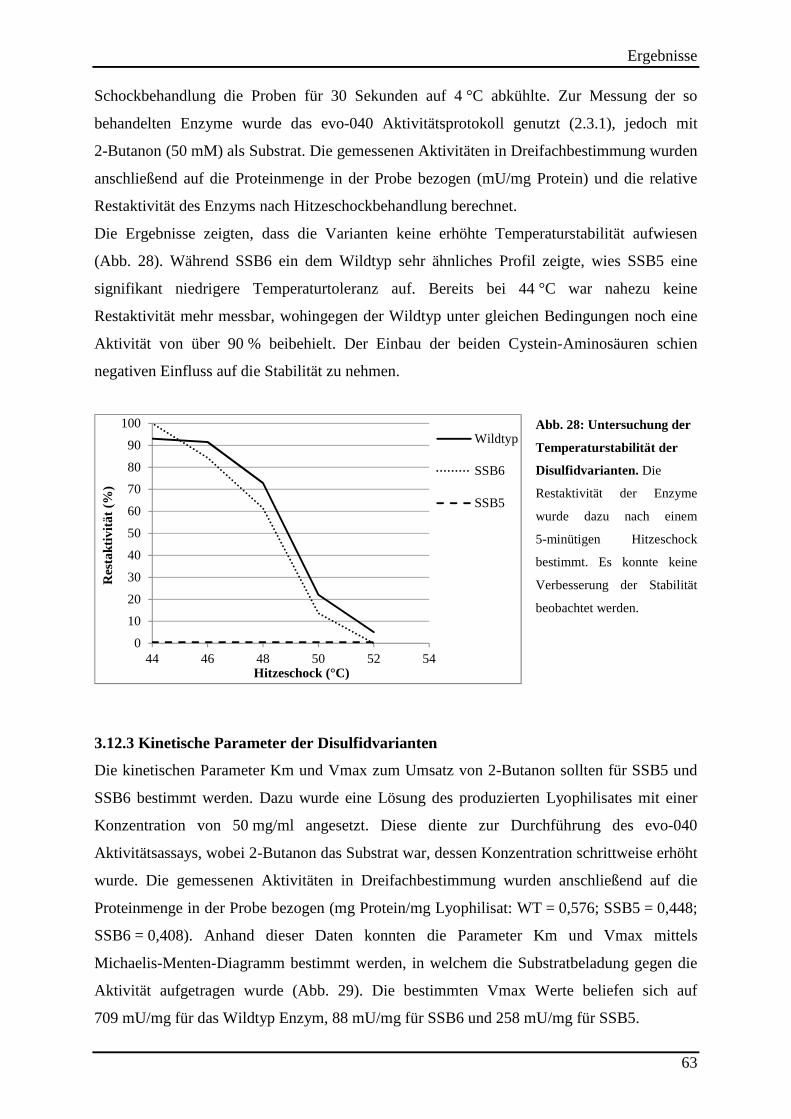
Ergebnisse
63
Schockbehandlung die Proben für 30 Sekunden auf 4 °C abkühlte. Zur Messung der so
behandelten Enzyme wurde das evo-040 Aktivitätsprotokoll genutzt (2.3.1), jedoch mit
2-Butanon (50 mM) als Substrat. Die gemessenen Aktivitäten in Dreifachbestimmung wurden
anschließend auf die Proteinmenge in der Probe bezogen (mU/mg Protein) und die relative
Restaktivität des Enzyms nach Hitzeschockbehandlung berechnet.
Die Ergebnisse zeigten, dass die Varianten keine erhöhte Temperaturstabilität aufwiesen
(Abb. 28). Während SSB6 ein dem Wildtyp sehr ähnliches Profil zeigte, wies SSB5 eine
signifikant niedrigere Temperaturtoleranz auf. Bereits bei 44 °C war nahezu keine
Restaktivität mehr messbar, wohingegen der Wildtyp unter gleichen Bedingungen noch eine
Aktivität von über 90 % beibehielt. Der Einbau der beiden Cystein-Aminosäuren schien
negativen Einfluss auf die Stabilität zu nehmen.
Abb. 28: Untersuchung der
Temperaturstabilität der
Disulfidvarianten. Die
Restaktivität der Enzyme
wurde dazu nach einem
5-minütigen Hitzeschock
bestimmt. Es konnte keine
Verbesserung der Stabilität
beobachtet werden.
3.12.3 Kinetische Parameter der Disulfidvarianten
Die kinetischen Parameter Km und Vmax zum Umsatz von 2-Butanon sollten für SSB5 und
SSB6 bestimmt werden. Dazu wurde eine Lösung des produzierten Lyophilisates mit einer
Konzentration von 50 mg/ml angesetzt. Diese diente zur Durchführung des evo-040
Aktivitätsassays, wobei 2-Butanon das Substrat war, dessen Konzentration schrittweise erhöht
wurde. Die gemessenen Aktivitäten in Dreifachbestimmung wurden anschließend auf die
Proteinmenge in der Probe bezogen (mg Protein/mg Lyophilisat: WT = 0,576; SSB5 = 0,448;
SSB6 = 0,408). Anhand dieser Daten konnten die Parameter Km und Vmax mittels
Michaelis-Menten-Diagramm bestimmt werden, in welchem die Substratbeladung gegen die
Aktivität aufgetragen wurde (Abb. 29). Die bestimmten Vmax Werte beliefen sich auf
709 mU/mg für das Wildtyp Enzym, 88 mU/mg für SSB6 und 258 mU/mg für SSB5.
0
10
20
30
40
50
60
70
80
90
100
44 46 48 50 52 54
Res
takt
ivitä
t (%
)
Hitzeschock (°C)
Wildtyp
SSB6
SSB5
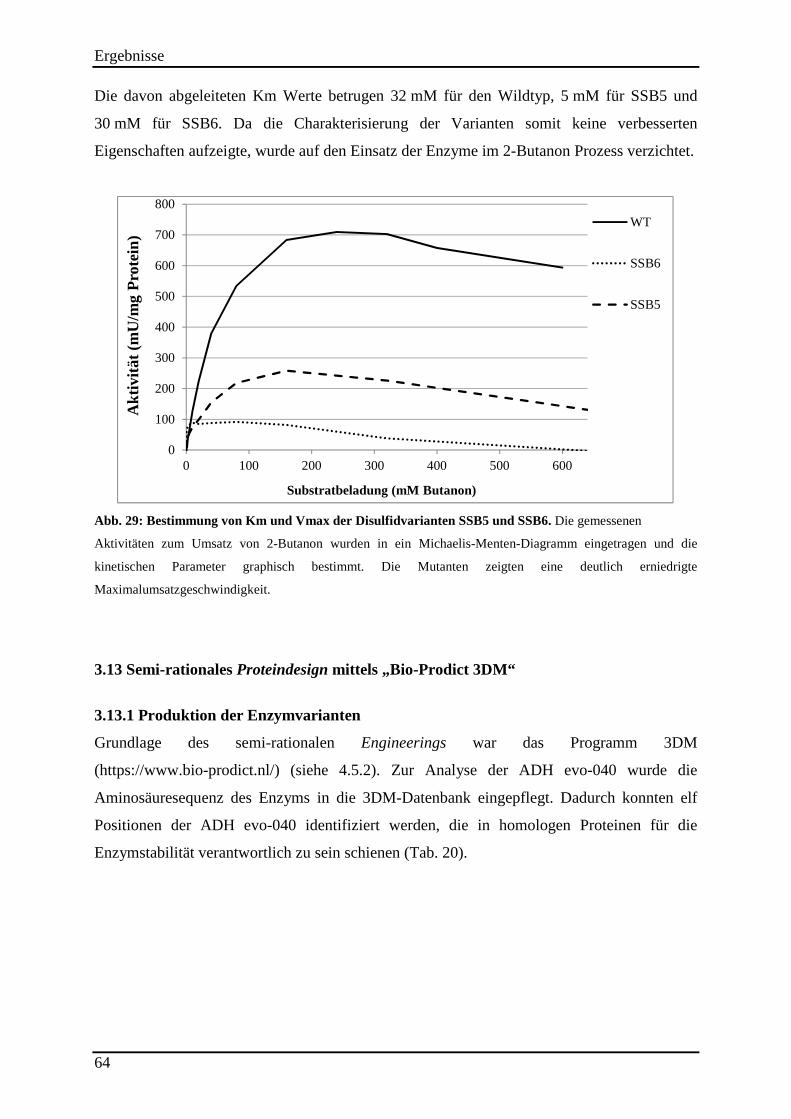
Ergebnisse
64
Die davon abgeleiteten Km Werte betrugen 32 mM für den Wildtyp, 5 mM für SSB5 und
30 mM für SSB6. Da die Charakterisierung der Varianten somit keine verbesserten
Eigenschaften aufzeigte, wurde auf den Einsatz der Enzyme im 2-Butanon Prozess verzichtet.
Abb. 29: Bestimmung von Km und Vmax der Disulfidvarianten SSB5 und SSB6. Die gemessenen
Aktivitäten zum Umsatz von 2-Butanon wurden in ein Michaelis-Menten-Diagramm eingetragen und die
kinetischen Parameter graphisch bestimmt. Die Mutanten zeigten eine deutlich erniedrigte
Maximalumsatzgeschwindigkeit.
3.13 Semi-rationales Proteindesign mittels „Bio-Prodict 3DM“
3.13.1 Produktion der Enzymvarianten
Grundlage des semi-rationalen Engineerings war das Programm 3DM
(https://www.bio-prodict.nl/) (siehe 4.5.2). Zur Analyse der ADH evo-040 wurde die
Aminosäuresequenz des Enzyms in die 3DM-Datenbank eingepflegt. Dadurch konnten elf
Positionen der ADH evo-040 identifiziert werden, die in homologen Proteinen für die
Enzymstabilität verantwortlich zu sein schienen (Tab. 20).
0
100
200
300
400
500
600
700
800
0 100 200 300 400 500 600
Akt
ivitä
t (m
U/m
g P
rote
in)
Substratbeladung (mM Butanon)
WT
SSB6
SSB5
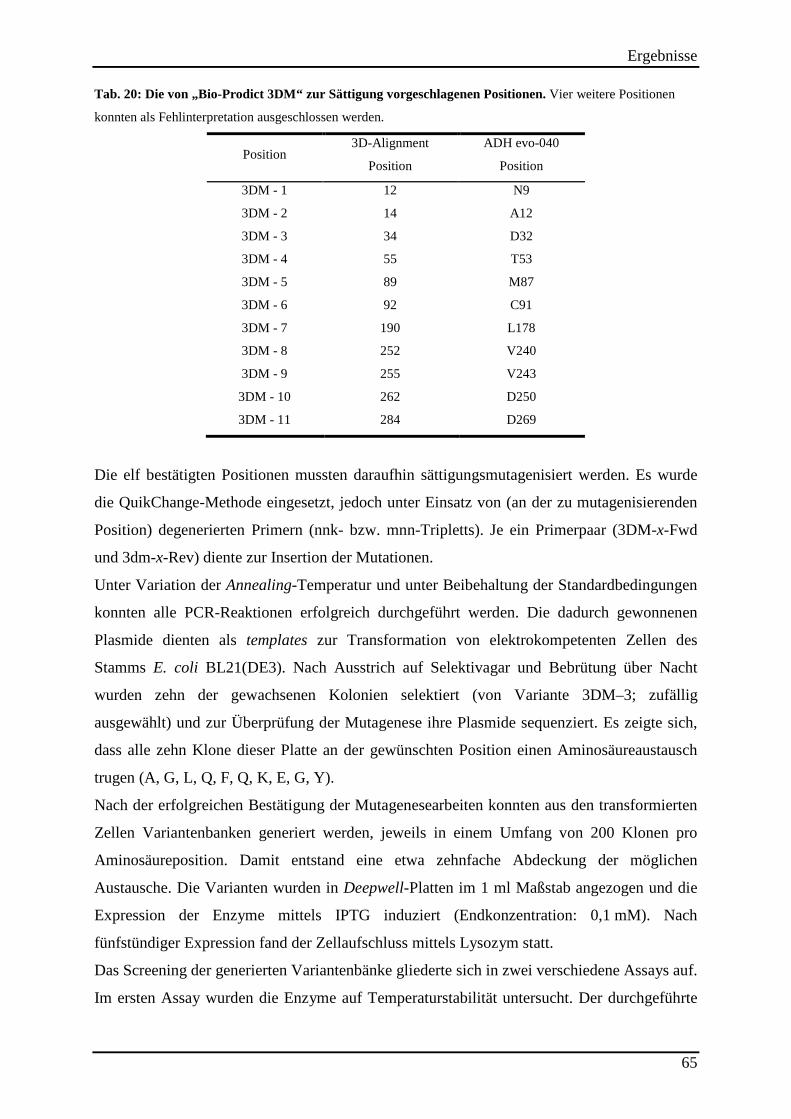
Ergebnisse
65
Tab. 20: Die von „Bio-Prodict 3DM“ zur Sättigung vorgeschlagenen Positionen. Vier weitere Positionen
konnten als Fehlinterpretation ausgeschlossen werden.
Position 3D-Alignment
Position
ADH evo-040
Position
3DM - 1 12 N9
3DM - 2 14 A12
3DM - 3 34 D32
3DM - 4 55 T53
3DM - 5 89 M87
3DM - 6 92 C91
3DM - 7 190 L178
3DM - 8 252 V240
3DM - 9 255 V243
3DM - 10 262 D250
3DM - 11 284 D269
Die elf bestätigten Positionen mussten daraufhin sättigungsmutagenisiert werden. Es wurde
die QuikChange-Methode eingesetzt, jedoch unter Einsatz von (an der zu mutagenisierenden
Position) degenerierten Primern (nnk- bzw. mnn-Tripletts). Je ein Primerpaar (3DM-x-Fwd
und 3dm-x-Rev) diente zur Insertion der Mutationen.
Unter Variation der Annealing-Temperatur und unter Beibehaltung der Standardbedingungen
konnten alle PCR-Reaktionen erfolgreich durchgeführt werden. Die dadurch gewonnenen
Plasmide dienten als templates zur Transformation von elektrokompetenten Zellen des
Stamms E. coli BL21(DE3). Nach Ausstrich auf Selektivagar und Bebrütung über Nacht
wurden zehn der gewachsenen Kolonien selektiert (von Variante 3DM–3; zufällig
ausgewählt) und zur Überprüfung der Mutagenese ihre Plasmide sequenziert. Es zeigte sich,
dass alle zehn Klone dieser Platte an der gewünschten Position einen Aminosäureaustausch
trugen (A, G, L, Q, F, Q, K, E, G, Y).
Nach der erfolgreichen Bestätigung der Mutagenesearbeiten konnten aus den transformierten
Zellen Variantenbanken generiert werden, jeweils in einem Umfang von 200 Klonen pro
Aminosäureposition. Damit entstand eine etwa zehnfache Abdeckung der möglichen
Austausche. Die Varianten wurden in Deepwell-Platten im 1 ml Maßstab angezogen und die
Expression der Enzyme mittels IPTG induziert (Endkonzentration: 0,1 mM). Nach
fünfstündiger Expression fand der Zellaufschluss mittels Lysozym statt.
Das Screening der generierten Variantenbänke gliederte sich in zwei verschiedene Assays auf.
Im ersten Assay wurden die Enzyme auf Temperaturstabilität untersucht. Der durchgeführte

Ergebnisse
66
Hitzeschock der Zellrohextrakte bei 50 °C dauerte fünf Minuten, gefolgt von einer
Rückkühlphase von 30 Sekunden bei 4 °C. Daraufhin wurde die Restaktivität der Rohextrakte
im Mikroplatten-Photometer mittels NADH-Abnahme bestimmt und mit der Restaktivität des
auf gleiche Weise behandelten Wildtyps verglichen.
Der zweite Assay zielte auf die Lösemittelstabilität der Varianten ab. Dazu wurden die Klone
5 Minuten in einer Lösung aus 90 % Isopropanol inkubiert. Auch hier wurde die Restaktivität
der Enzyme photometrisch bestimmt und mit der des Wildtyps verglichen.
Die beiden beschriebenen Assays wurden mit den Varianten der Enzymbank dreimal
durchgeführt. Klone, die in einer Bank bei allen drei Wiederholungen des Screenings die
höchste Aktivität zeigten, wurden selektiert und die Gensequenz mittels Sequenzierung
offengelegt (Tab. 21).
Tab. 21: Ergebnisse der beiden Screenings der 3DM-Varianten. Die aktivsten Klone jeder Bank wurden
sequenziert und sind unten aufgeführt. Die angegebenen relativen Restaktivitäten sind auf die Restaktivität des
Wildtyps nach identischer Behandlung bezogen.
Variante Mutation Hitzeschock-Behandlung
Restaktiv. rel. zum Wildtyp (%)
Lösemittelinkubation
Restaktiv. rel. zum Wildtyp (%)
3DM - 1 N9G 125 263
3DM - 2 A12Q 141 246
3DM - 8 V240N 120 306
3DM - 9 V243G < 100 148
3DM - 10 D250W 141 255
Insgesamt wurden aus den elf Banken auf diese Weise fünf Enzymvarianten isoliert. Die
restlichen sechs Enzymbanken zeigten keine Mutanten, die in einem der beiden Assays
höhere Restaktivität als der Wildtyp aufwiesen. Der Anteil der durch die Mutagenese
inaktivierten Enzyme war in allen Bänken sehr niedrig (Tab. 22).
Tab. 22: Anteil der inaktiven Enzymvarianten je 3DM-Position
Mutantenbank 1 2 3 4 5 6 7 8 9 10 11
Inaktive
Mutanten (%) 4,3 1 0 0 2 0,3 0,6 1,6 1,6 7,6 0
Da nicht ausgeschlossen werden konnte, dass die Aktivitätsunterschiede auf veränderte
Expressionsraten zurückzuführen waren, musste von den Rohextrakten auch ein SDS-Gel
erstellt werden. Dieses zeigte nur leichte Variationen der Enzymeexpressionen (Abb. 30).

Ergebnisse
67
Abb. 30: SDS-Gelelektrophorese der löslichen Proteinfraktionen einiger 3DM-Enzymvarianten. Die
Überexpressionsbanden der ADHs (37 kDa) sind nicht sehr ausgeprägt, da die Expression nur unter
suboptimalen Bedingungen im Deepwell-Format durchgeführt wurde.
3.13.2 Untersuchung der Hitzestabilität der 3DM-Varianten
Die fünf unter 3.13.1 identifizierten Enzymvarianten wurden zur genaueren Charakterisierung
im 500 ml Maßstab exprimiert und der Rohextrakt nach Zellaufschluss mittels Homogenisator
lyophilisiert.
Die produzierten Lyophilisate dienten zur Untersuchung der Hitzestabilität der Enzyme. Die
Versuche verliefen parallel zu dem unter 3.12.2 vorgestellten Verfahren. Auch in diesem Fall
handelte es sich um die Analyse der Restaktivitäten in Dreifachbestimmung nach Behandlung
der Lyophilisatlösung durch Hitzeschock. Die Ergebnisse zeigten, dass zwei der Varianten
(N9G und V240N) eine signifikant höhere Temperaturtoleranz besaßen (etwa 3 - 4 °C) als der
Wildtyp (Abb. 31). Die übrigen Varianten dagegen zeigten eine leichte bis signifikante
Erniedrigung ihrer Temperaturstabilität.
Diese Ergebnisse standen teilweise im Widerspruch mit den Ergebnissen des Screenings der
Variantenbanken (Tab. 21). Allerdings wurde das vorhergehende Screening in sehr viel
kleinerem Volumen durchgeführt, als die hier beschriebene genauere Charakterisierung der
Enzyme. Daher sind die hier generierten Ergebnisse als verlässlicher anzusehen.
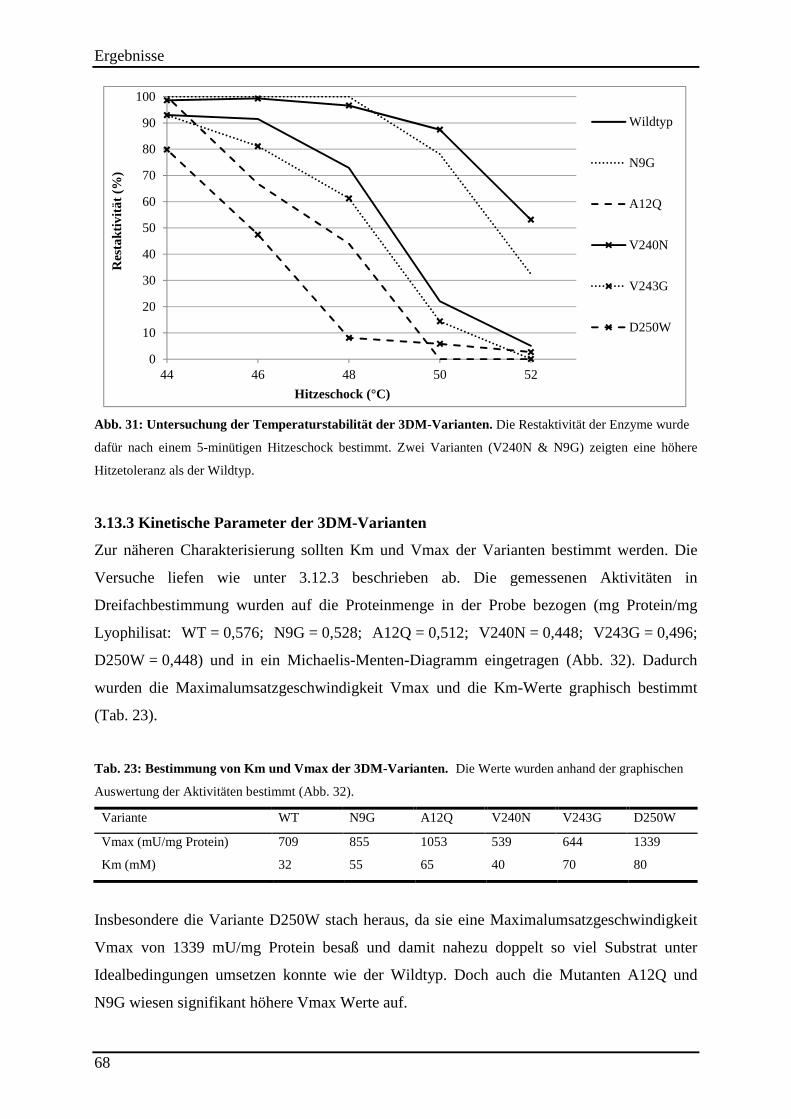
Ergebnisse
68
Abb. 31: Untersuchung der Temperaturstabilität der 3DM-Varianten. Die Restaktivität der Enzyme wurde
dafür nach einem 5-minütigen Hitzeschock bestimmt. Zwei Varianten (V240N & N9G) zeigten eine höhere
Hitzetoleranz als der Wildtyp.
3.13.3 Kinetische Parameter der 3DM-Varianten
Zur näheren Charakterisierung sollten Km und Vmax der Varianten bestimmt werden. Die
Versuche liefen wie unter 3.12.3 beschrieben ab. Die gemessenen Aktivitäten in
Dreifachbestimmung wurden auf die Proteinmenge in der Probe bezogen (mg Protein/mg
Lyophilisat: WT = 0,576; N9G = 0,528; A12Q = 0,512; V240N = 0,448; V243G = 0,496;
D250W = 0,448) und in ein Michaelis-Menten-Diagramm eingetragen (Abb. 32). Dadurch
wurden die Maximalumsatzgeschwindigkeit Vmax und die Km-Werte graphisch bestimmt
(Tab. 23).
Tab. 23: Bestimmung von Km und Vmax der 3DM-Varianten. Die Werte wurden anhand der graphischen
Auswertung der Aktivitäten bestimmt (Abb. 32).
Variante WT N9G A12Q V240N V243G D250W
Vmax (mU/mg Protein) 709 855 1053 539 644 1339
Km (mM) 32 55 65 40 70 80
Insbesondere die Variante D250W stach heraus, da sie eine Maximalumsatzgeschwindigkeit
Vmax von 1339 mU/mg Protein besaß und damit nahezu doppelt so viel Substrat unter
Idealbedingungen umsetzen konnte wie der Wildtyp. Doch auch die Mutanten A12Q und
N9G wiesen signifikant höhere Vmax Werte auf.
0
10
20
30
40
50
60
70
80
90
100
44 46 48 50 52
Res
takt
ivitä
t (%
)
Hitzeschock (°C)
Wildtyp
N9G
A12Q
V240N
V243G
D250W

Ergebnisse
69
Abb. 32: Bestimmung von Km und Vmax der 3DM-Varianten. Die gemessenen Aktivitäten zum Umsatz von
2-Butanon wurden in ein Michaelis-Menten-Diagramm eingetragen und die kinetischen Parameter graphisch
bestimmt.
3.13.4 Einsatz der 3DM-Varianten im (S)-2-Butanol Prozess
Die Enzymvarianten wurden zunächst in einem Prozess mit niedrigem Anteil an organischer
Phase eingesetzt. Der Volumenanteil des Isopropanols lag bei 8,5 %, die Substratbeladung bei
φ(2-Butanon) = 1,5 %. Die Menge des eingesetzten Enzyms betrug 0,625 g Protein/ml
Reaktionsansatz. Weitere Parameter waren: c(NAD+) = 0,1 mM; Puffer = KPi (50 mM,
pH 7,0); T = RT.
Es zeigte sich, dass insbesondere die Variante D250W wesentlich aktiver war als der Wildtyp
(Abb. 33). Nach sechs Stunden lag der Umsatz bereits bei 64 % (vgl. Wildtyp: 28 %). Die
Varianten N9G, A12Q und V240N waren leicht aktiver als der Wildtyp, während die Variante
V243G leicht stabiler zu sein schien als die übrigen Enzyme. Alle Enzyme lieferten einen
unveränderten Enantiomerenüberschuss von ≥ 99,5 % für das (S)-Enantiomer.
0
200
400
600
800
1000
1200
1400
1600
0 100 200 300 400 500 600
Akt
ivitä
t (m
U/m
g P
rote
in)
Substratbeladung (mM Butanon)
WT
N9G
A12Q
V240N
V243G
D250W
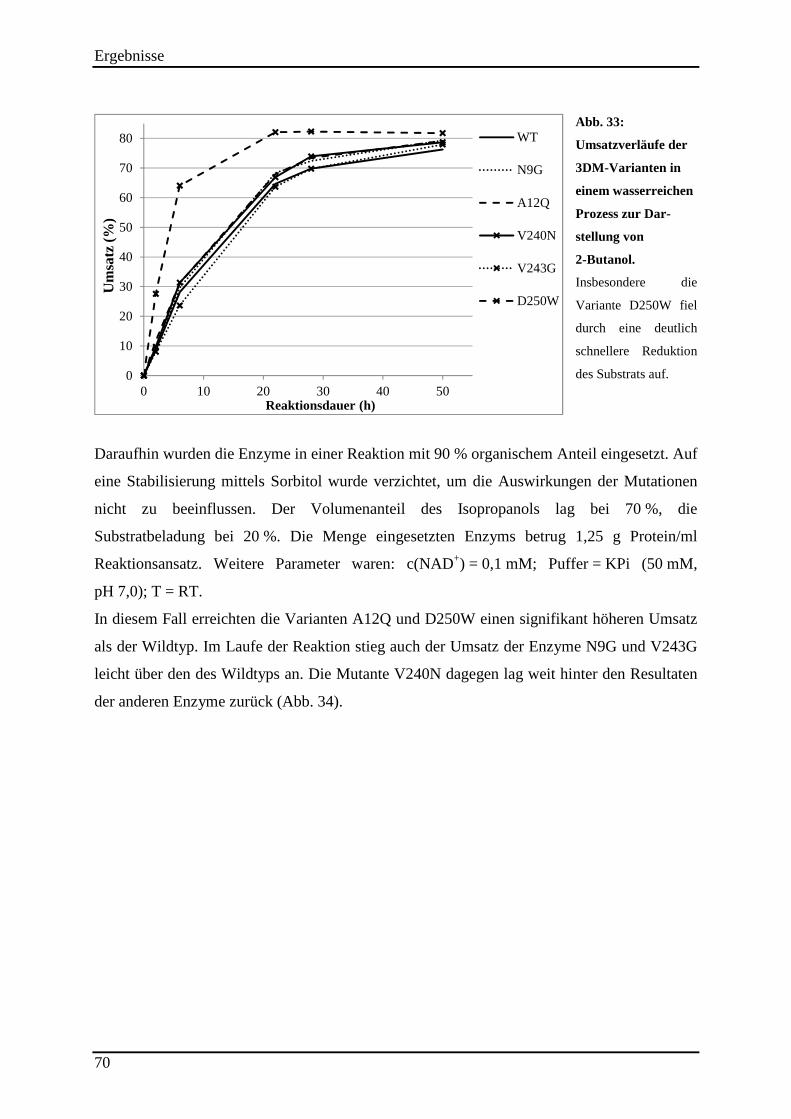
Ergebnisse
70
Abb. 33:
Umsatzverläufe der
3DM-Varianten in
einem wasserreichen
Prozess zur Dar-
stellung von
2-Butanol.
Insbesondere die
Variante D250W fiel
durch eine deutlich
schnellere Reduktion
des Substrats auf.
Daraufhin wurden die Enzyme in einer Reaktion mit 90 % organischem Anteil eingesetzt. Auf
eine Stabilisierung mittels Sorbitol wurde verzichtet, um die Auswirkungen der Mutationen
nicht zu beeinflussen. Der Volumenanteil des Isopropanols lag bei 70 %, die
Substratbeladung bei 20 %. Die Menge eingesetzten Enzyms betrug 1,25 g Protein/ml
Reaktionsansatz. Weitere Parameter waren: c(NAD+) = 0,1 mM; Puffer = KPi (50 mM,
pH 7,0); T = RT.
In diesem Fall erreichten die Varianten A12Q und D250W einen signifikant höheren Umsatz
als der Wildtyp. Im Laufe der Reaktion stieg auch der Umsatz der Enzyme N9G und V243G
leicht über den des Wildtyps an. Die Mutante V240N dagegen lag weit hinter den Resultaten
der anderen Enzyme zurück (Abb. 34).
0
10
20
30
40
50
60
70
80
0 10 20 30 40 50
Um
satz
(%
)
Reaktionsdauer (h)
WT
N9G
A12Q
V240N
V243G
D250W
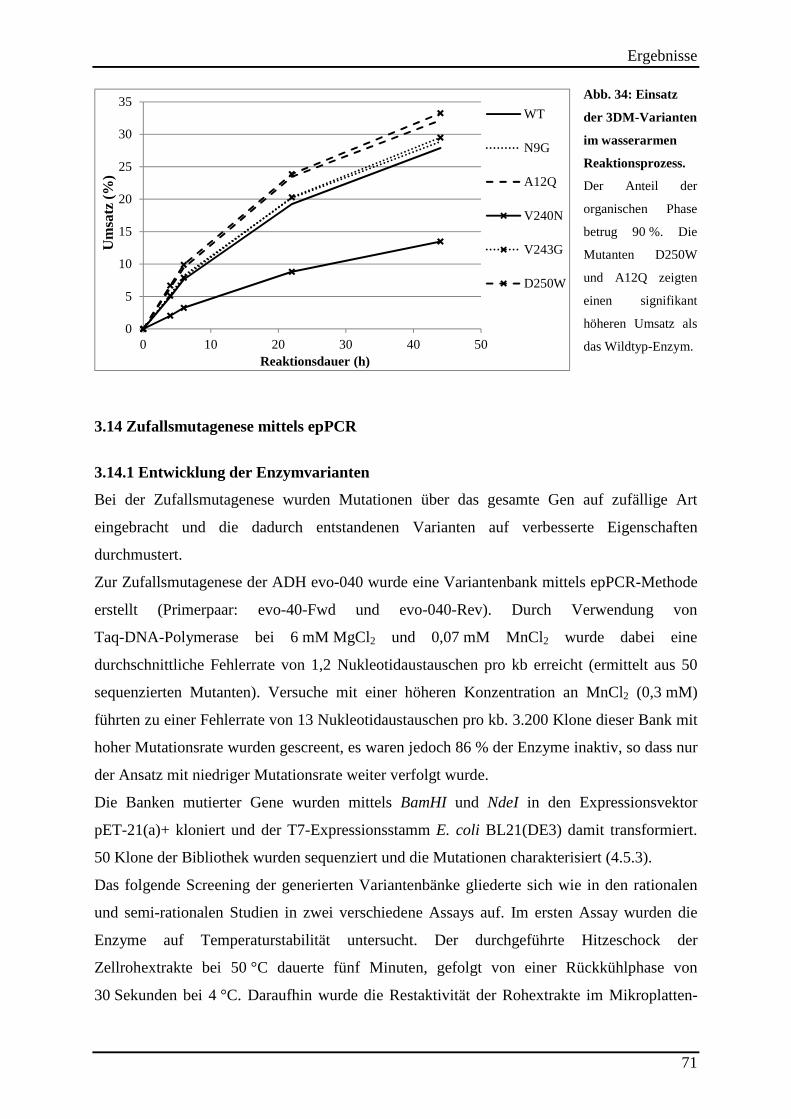
Ergebnisse
71
Abb. 34: Einsatz
der 3DM-Varianten
im wasserarmen
Reaktionsprozess.
Der Anteil der
organischen Phase
betrug 90 %. Die
Mutanten D250W
und A12Q zeigten
einen signifikant
höheren Umsatz als
das Wildtyp-Enzym.
3.14 Zufallsmutagenese mittels epPCR
3.14.1 Entwicklung der Enzymvarianten
Bei der Zufallsmutagenese wurden Mutationen über das gesamte Gen auf zufällige Art
eingebracht und die dadurch entstandenen Varianten auf verbesserte Eigenschaften
durchmustert.
Zur Zufallsmutagenese der ADH evo-040 wurde eine Variantenbank mittels epPCR-Methode
erstellt (Primerpaar: evo-40-Fwd und evo-040-Rev). Durch Verwendung von
Taq-DNA-Polymerase bei 6 mM MgCl2 und 0,07 mM MnCl2 wurde dabei eine
durchschnittliche Fehlerrate von 1,2 Nukleotidaustauschen pro kb erreicht (ermittelt aus 50
sequenzierten Mutanten). Versuche mit einer höheren Konzentration an MnCl2 (0,3 mM)
führten zu einer Fehlerrate von 13 Nukleotidaustauschen pro kb. 3.200 Klone dieser Bank mit
hoher Mutationsrate wurden gescreent, es waren jedoch 86 % der Enzyme inaktiv, so dass nur
der Ansatz mit niedriger Mutationsrate weiter verfolgt wurde.
Die Banken mutierter Gene wurden mittels BamHI und NdeI in den Expressionsvektor
pET-21(a)+ kloniert und der T7-Expressionsstamm E. coli BL21(DE3) damit transformiert.
50 Klone der Bibliothek wurden sequenziert und die Mutationen charakterisiert (4.5.3).
Das folgende Screening der generierten Variantenbänke gliederte sich wie in den rationalen
und semi-rationalen Studien in zwei verschiedene Assays auf. Im ersten Assay wurden die
Enzyme auf Temperaturstabilität untersucht. Der durchgeführte Hitzeschock der
Zellrohextrakte bei 50 °C dauerte fünf Minuten, gefolgt von einer Rückkühlphase von
30 Sekunden bei 4 °C. Daraufhin wurde die Restaktivität der Rohextrakte im Mikroplatten-
0
5
10
15
20
25
30
35
0 10 20 30 40 50
Um
satz
(%
)
Reaktionsdauer (h)
WT
N9G
A12Q
V240N
V243G
D250W

Ergebnisse
72
Photometer mittels NADH-Abnahme bestimmt und mit der Restaktivität des auf gleiche
Weise behandelten Wildtyps verglichen.
Der zweite Assay zielte auf die Lösemittelstabilität der Varianten ab. Dazu wurden die Klone
5 Minuten in einer Lösung aus 90 % Isopropanol inkubiert. Auch hier wurde die Restaktivität
der Enzyme photometrisch bestimmt und mit der des Wildtyps verglichen. Die Gesamtanzahl
der auf diese Weise untersuchten Varianten betrug circa 13.000 Mutanten.
Nach jedem Screening wurden etwa 10 % der aktivsten Klone isoliert und nach Neuanzucht
in Deepwell-Platten erneut dem Screening unterzogen. Auf diese Weise kam es zu drei
Screening-Runden, aus denen am Schluss acht Klone ausgewählt und deren
Nukleotidsequenzen bestimmt wurden. Diese Enzyme wiesen in mindestens je einem der
beiden Assays eine höhere Restaktivität als der Wildtyp auf (Abb. 35).
Das beste Enzym, welches aus der Hitzeschockbehandlung hervorging, war die Mutante
D250R. Sie lieferte eine Restaktivität von 155 % verglichen mit dem Wildtyp. Auch die
Variante M87V war deutlich aktiver, ihre Restaktivität belief sich auf 151 %.
Aus dem Screening der Lösemittelstabilität gingen vier Enzyme mit deutlich erhöhter
Restaktivität hervor. Diese waren Y182F (172 %), M87V (171 %), P273L (154 %) und N7S
(143 %).
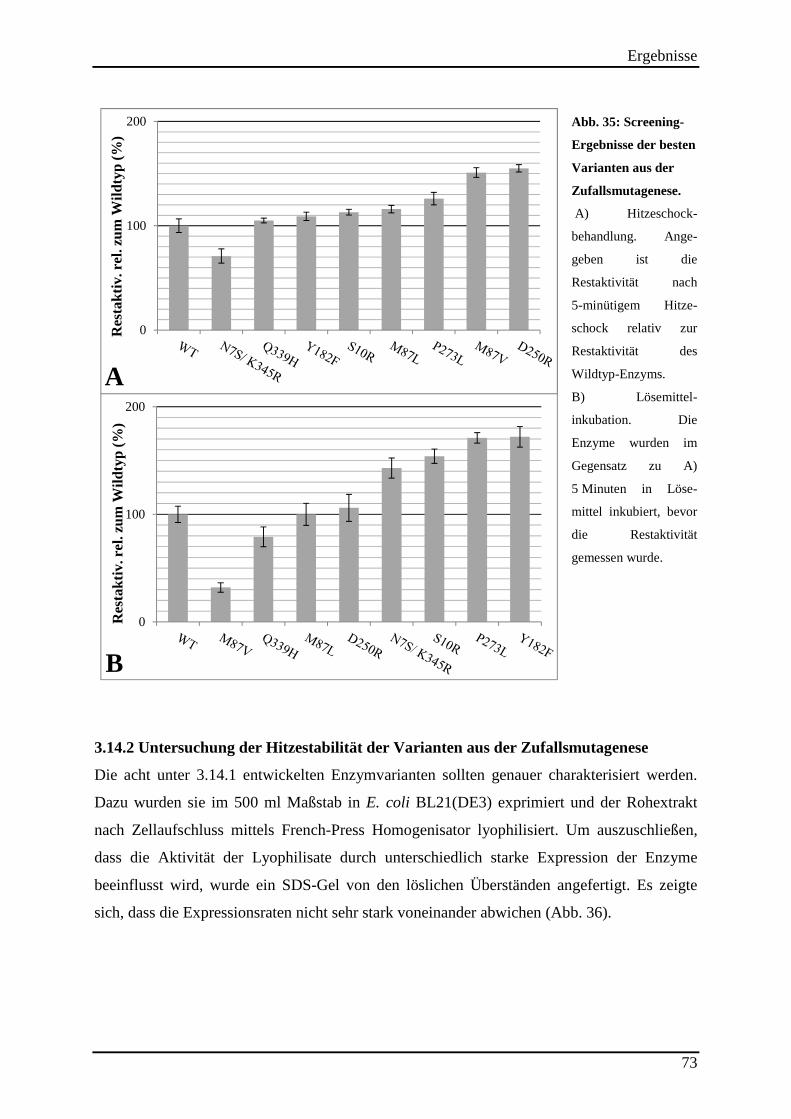
Ergebnisse
73
0
100
200R
esta
ktiv
. rel
. zum
Wild
typ
(%)
A
Abb. 35: Screening-
Ergebnisse der besten
Varianten aus der
Zufallsmutagenese.
A) Hitzeschock-
behandlung. Ange-
geben ist die
Restaktivität nach
5-minütigem Hitze-
schock relativ zur
Restaktivität des
Wildtyp-Enzyms.
B) Lösemittel-
inkubation. Die
Enzyme wurden im
Gegensatz zu A)
5 Minuten in Löse-
mittel inkubiert, bevor
die Restaktivität
gemessen wurde.
3.14.2 Untersuchung der Hitzestabilität der Varianten aus der Zufallsmutagenese
Die acht unter 3.14.1 entwickelten Enzymvarianten sollten genauer charakterisiert werden.
Dazu wurden sie im 500 ml Maßstab in E. coli BL21(DE3) exprimiert und der Rohextrakt
nach Zellaufschluss mittels French-Press Homogenisator lyophilisiert. Um auszuschließen,
dass die Aktivität der Lyophilisate durch unterschiedlich starke Expression der Enzyme
beeinflusst wird, wurde ein SDS-Gel von den löslichen Überständen angefertigt. Es zeigte
sich, dass die Expressionsraten nicht sehr stark voneinander abwichen (Abb. 36).
0
100
200
Res
takt
iv. r
el. z
um W
ildty
p (%
)
B
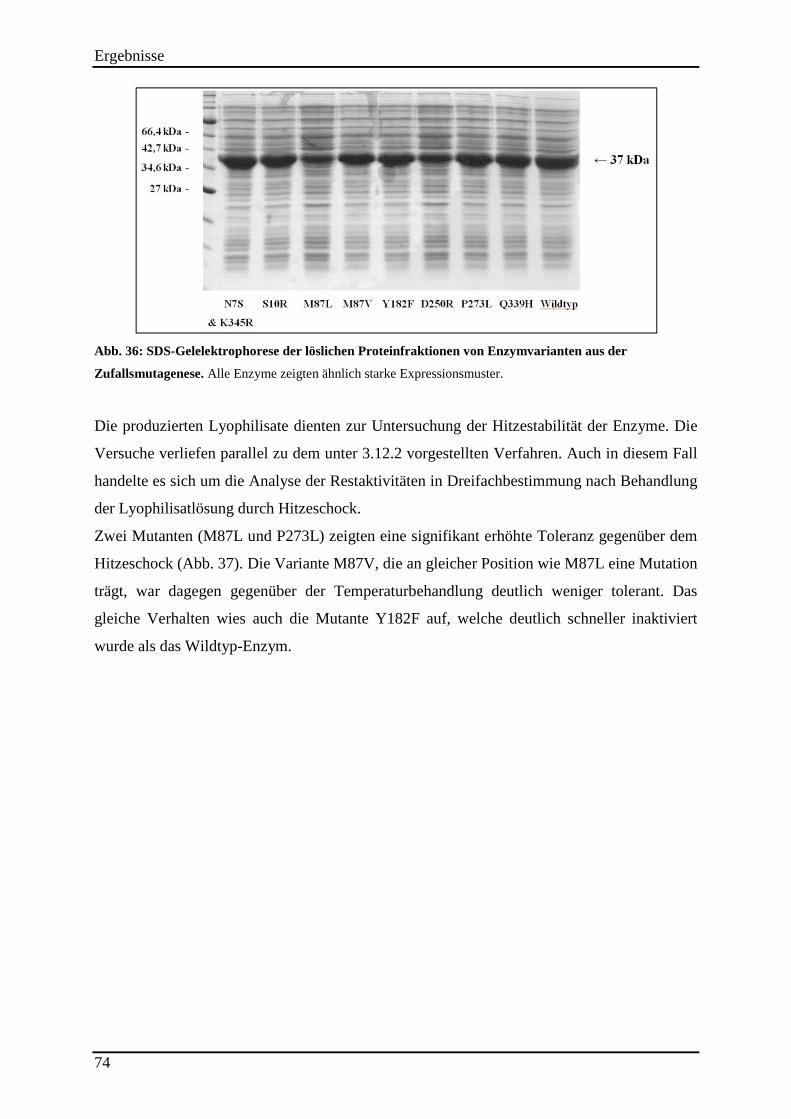
Ergebnisse
74
Abb. 36: SDS-Gelelektrophorese der löslichen Proteinfraktionen von Enzymvarianten aus der
Zufallsmutagenese. Alle Enzyme zeigten ähnlich starke Expressionsmuster.
Die produzierten Lyophilisate dienten zur Untersuchung der Hitzestabilität der Enzyme. Die
Versuche verliefen parallel zu dem unter 3.12.2 vorgestellten Verfahren. Auch in diesem Fall
handelte es sich um die Analyse der Restaktivitäten in Dreifachbestimmung nach Behandlung
der Lyophilisatlösung durch Hitzeschock.
Zwei Mutanten (M87L und P273L) zeigten eine signifikant erhöhte Toleranz gegenüber dem
Hitzeschock (Abb. 37). Die Variante M87V, die an gleicher Position wie M87L eine Mutation
trägt, war dagegen gegenüber der Temperaturbehandlung deutlich weniger tolerant. Das
gleiche Verhalten wies auch die Mutante Y182F auf, welche deutlich schneller inaktiviert
wurde als das Wildtyp-Enzym.
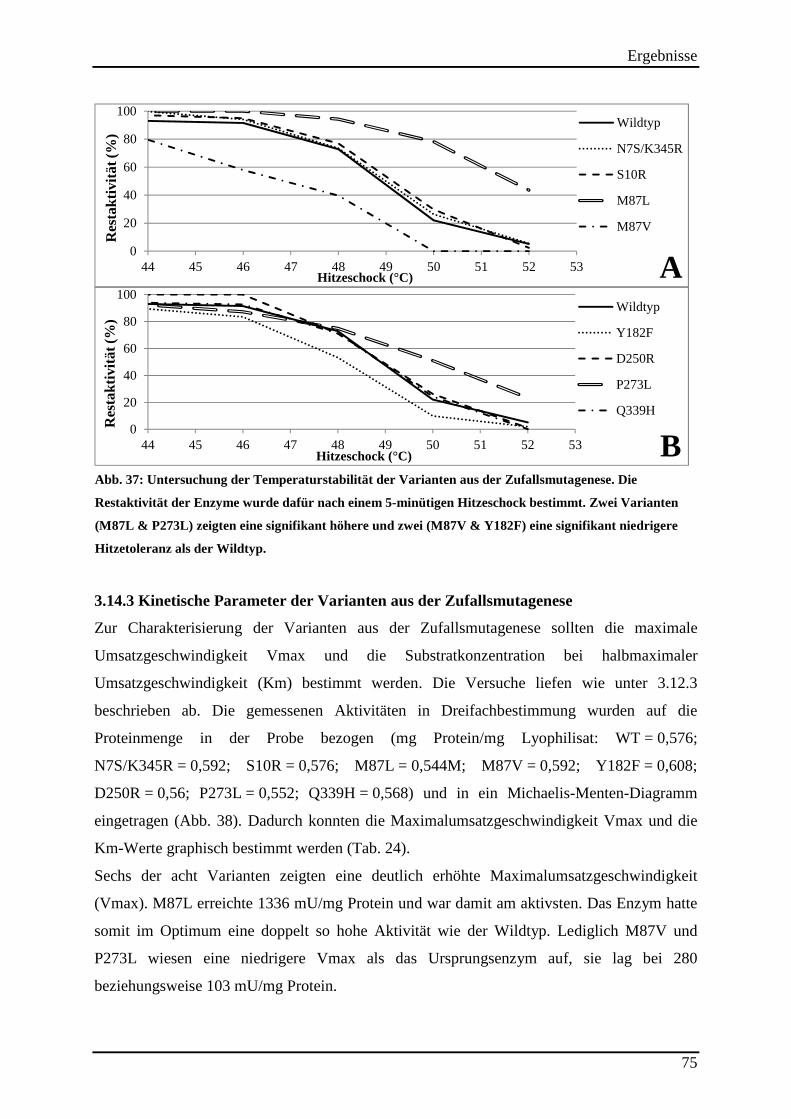
Ergebnisse
75
3.14.3 Kinetische Parameter der Varianten aus der Zufallsmutagenese
Zur Charakterisierung der Varianten aus der Zufallsmutagenese sollten die maximale
Umsatzgeschwindigkeit Vmax und die Substratkonzentration bei halbmaximaler
Umsatzgeschwindigkeit (Km) bestimmt werden. Die Versuche liefen wie unter 3.12.3
beschrieben ab. Die gemessenen Aktivitäten in Dreifachbestimmung wurden auf die
Proteinmenge in der Probe bezogen (mg Protein/mg Lyophilisat: WT = 0,576;
N7S/K345R = 0,592; S10R = 0,576; M87L = 0,544M; M87V = 0,592; Y182F = 0,608;
D250R = 0,56; P273L = 0,552; Q339H = 0,568) und in ein Michaelis-Menten-Diagramm
eingetragen (Abb. 38). Dadurch konnten die Maximalumsatzgeschwindigkeit Vmax und die
Km-Werte graphisch bestimmt werden (Tab. 24).
Sechs der acht Varianten zeigten eine deutlich erhöhte Maximalumsatzgeschwindigkeit
(Vmax). M87L erreichte 1336 mU/mg Protein und war damit am aktivsten. Das Enzym hatte
somit im Optimum eine doppelt so hohe Aktivität wie der Wildtyp. Lediglich M87V und
P273L wiesen eine niedrigere Vmax als das Ursprungsenzym auf, sie lag bei 280
beziehungsweise 103 mU/mg Protein.
0
20
40
60
80
100
44 45 46 47 48 49 50 51 52 53
Res
takt
ivitä
t (%
)
Hitzeschock (°C) B
Wildtyp
Y182F
D250R
P273L
Q339H
0
20
40
60
80
100
44 45 46 47 48 49 50 51 52 53
Res
takt
ivitä
t (%
)
Hitzeschock (°C) A
Wildtyp
N7S/K345R
S10R
M87L
M87V
Abb. 37: Untersuchung der Temperaturstabilität der Varianten aus der Zufallsmutagenese. Die
Restaktivität der Enzyme wurde dafür nach einem 5-minütigen Hitzeschock bestimmt. Zwei Varianten
(M87L & P273L) zeigten eine signifikant höhere und zwei (M87V & Y182F) eine signifikant niedrigere
Hitzetoleranz als der Wildtyp.
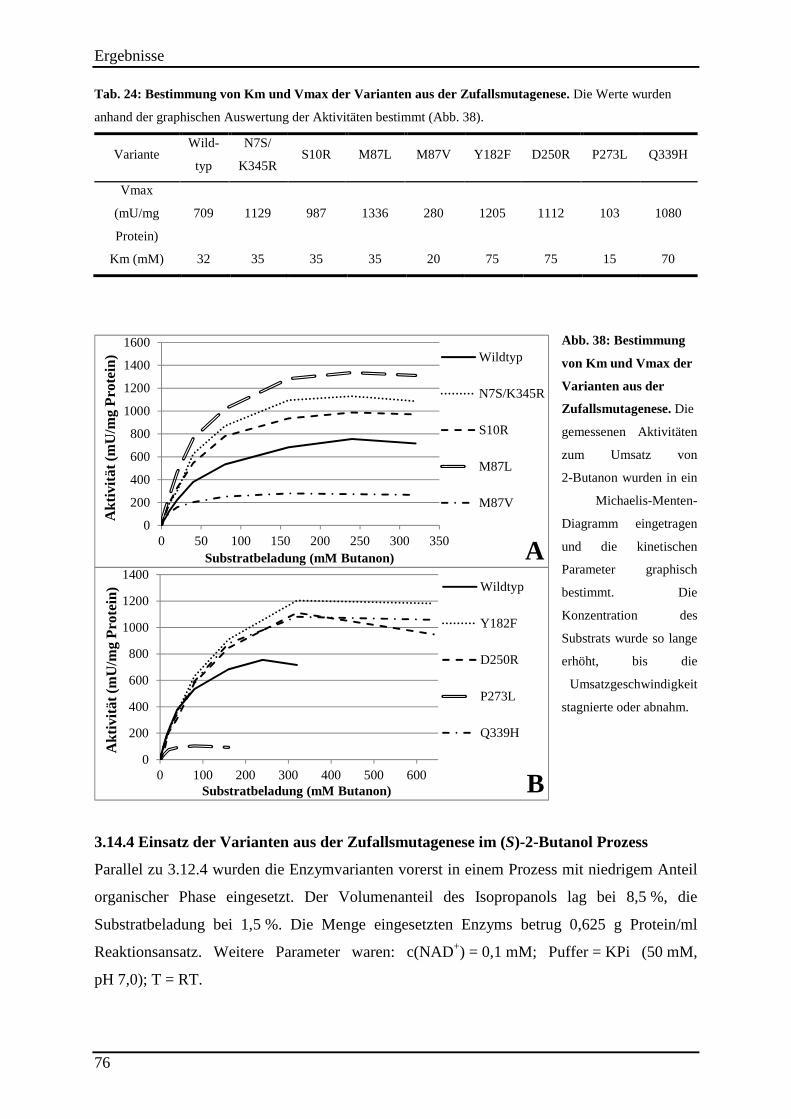
Ergebnisse
76
Tab. 24: Bestimmung von Km und Vmax der Varianten aus der Zufallsmutagenese. Die Werte wurden
anhand der graphischen Auswertung der Aktivitäten bestimmt (Abb. 38).
Variante Wild-
typ
N7S/
K345R S10R M87L M87V Y182F D250R P273L Q339H
Vmax
(mU/mg
Protein)
709 1129 987 1336 280 1205 1112 103 1080
Km (mM) 32 35 35 35 20 75 75 15 70
Abb. 38: Bestimmung
von Km und Vmax der
Varianten aus der
Zufallsmutagenese. Die
gemessenen Aktivitäten
zum Umsatz von
2-Butanon wurden in ein
Michaelis-Menten-
Diagramm eingetragen
und die kinetischen
Parameter graphisch
bestimmt. Die
Konzentration des
Substrats wurde so lange
erhöht, bis die
Umsatzgeschwindigkeit
stagnierte oder abnahm.
3.14.4 Einsatz der Varianten aus der Zufallsmutagenese im (S)-2-Butanol Prozess
Parallel zu 3.12.4 wurden die Enzymvarianten vorerst in einem Prozess mit niedrigem Anteil
organischer Phase eingesetzt. Der Volumenanteil des Isopropanols lag bei 8,5 %, die
Substratbeladung bei 1,5 %. Die Menge eingesetzten Enzyms betrug 0,625 g Protein/ml
Reaktionsansatz. Weitere Parameter waren: c(NAD+) = 0,1 mM; Puffer = KPi (50 mM,
pH 7,0); T = RT.
0
200
400
600
800
1000
1200
1400
1600
0 50 100 150 200 250 300 350
Akt
ivitä
t (m
U/m
g P
rote
in)
Substratbeladung (mM Butanon) A
Wildtyp
N7S/K345R
S10R
M87L
M87V
0
200
400
600
800
1000
1200
1400
0 100 200 300 400 500 600
Akt
ivitä
t (m
U/m
g P
rote
in)
Substratbeladung (mM Butanon) B
Wildtyp
Y182F
D250R
P273L
Q339H
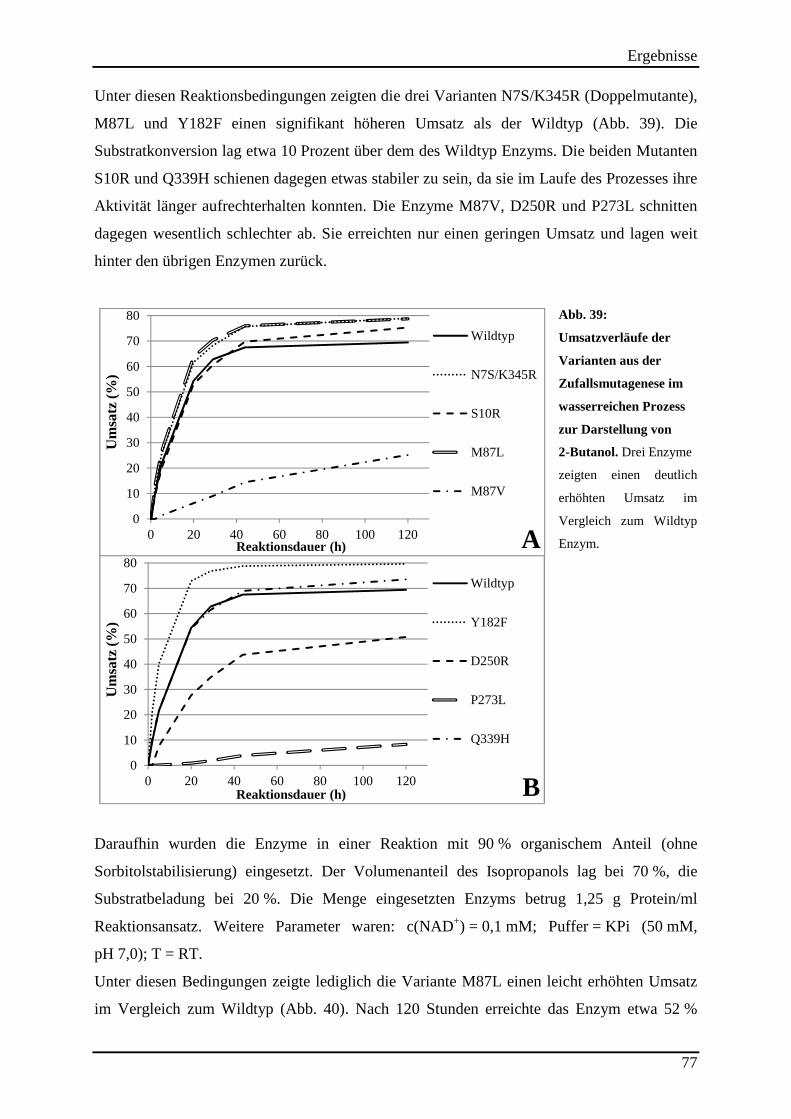
Ergebnisse
77
Unter diesen Reaktionsbedingungen zeigten die drei Varianten N7S/K345R (Doppelmutante),
M87L und Y182F einen signifikant höheren Umsatz als der Wildtyp (Abb. 39). Die
Substratkonversion lag etwa 10 Prozent über dem des Wildtyp Enzyms. Die beiden Mutanten
S10R und Q339H schienen dagegen etwas stabiler zu sein, da sie im Laufe des Prozesses ihre
Aktivität länger aufrechterhalten konnten. Die Enzyme M87V, D250R und P273L schnitten
dagegen wesentlich schlechter ab. Sie erreichten nur einen geringen Umsatz und lagen weit
hinter den übrigen Enzymen zurück.
Abb. 39:
Umsatzverläufe der
Varianten aus der
Zufallsmutagenese im
wasserreichen Prozess
zur Darstellung von
2-Butanol. Drei Enzyme
zeigten einen deutlich
erhöhten Umsatz im
Vergleich zum Wildtyp
Enzym.
Daraufhin wurden die Enzyme in einer Reaktion mit 90 % organischem Anteil (ohne
Sorbitolstabilisierung) eingesetzt. Der Volumenanteil des Isopropanols lag bei 70 %, die
Substratbeladung bei 20 %. Die Menge eingesetzten Enzyms betrug 1,25 g Protein/ml
Reaktionsansatz. Weitere Parameter waren: c(NAD+) = 0,1 mM; Puffer = KPi (50 mM,
pH 7,0); T = RT.
Unter diesen Bedingungen zeigte lediglich die Variante M87L einen leicht erhöhten Umsatz
im Vergleich zum Wildtyp (Abb. 40). Nach 120 Stunden erreichte das Enzym etwa 52 %
0
10
20
30
40
50
60
70
80
0 20 40 60 80 100 120
Um
satz
(%
)
Reaktionsdauer (h) A
Wildtyp
N7S/K345R
S10R
M87L
M87V
0
10
20
30
40
50
60
70
80
0 20 40 60 80 100 120
Um
satz
(%
)
Reaktionsdauer (h) B
Wildtyp
Y182F
D250R
P273L
Q339H

Ergebnisse
78
Substratkonversion, während der Wildtyp zu diesem Zeitpunkt 47 % umgesetzt hatte. Die drei
Mutanten M87V, D250R und Q339H waren in diesem Reaktionsmilieu kaum aktiv und
erreichten nach 120 Stunden Umsätze zwischen 4 und 12 %. Die übrigen Enzyme lagen etwa
im Bereich des Umsatzes des Wildtyp Enzyms.
Abb. 40: Einsatz der
Varianten aus der
Zufallsmutagenese im
wasserarmen Reaktions-
prozess. Der Anteil der
organischen Phase betrug
90 %. Nur die Variante
M87L zeigte unter diesen
Bedingungen einen
höheren Umsatz als das
Wildtyp Enzym.
0
10
20
30
40
50
60
0 20 40 60 80 100 120
Um
satz
(%
)
Reaktionsdauer (h) A
Wildtyp
N7S/K345R
S10R
M87L
M87V
0
10
20
30
40
50
60
0 20 40 60 80 100 120
Um
satz
(%
)
Reaktionsdauer (h) B
Wildtyp
Y182F
D250R
P273L
Q339H

Diskussion
79
4. Diskussion
Chirale Alkohole dienen als wichtige Synthesebausteine für Feinchemikalien. Besonders im
Bereich der Pharma-, Agrar- sowie in der Aroma- und Duftstoffindustrie6,7 nehmen sie einen
bedeutenden Stellenwert ein. Dies liegt an der Tatsache, dass unerwünschte Enantiomere
eines Moleküls oft abweichende Eigenschaften aufweisen. Beispielweise besitzt Limonen
zwei Enantiomere, wobei die (R)-Form ein orangenartiges Aroma aufweist, während das
(S)-Enantiomer nach Terpentin riecht. Einen besonders hohen Stellenwert nimmt die
Enantiomerenreinheit jedoch in der Pharmaindustrie ein. Der β-Blocker Propranolol ist in
seiner (-)-Form etwa 100fach stärker wirksam als die (+)-Form8, was an der chiralen Natur
seines Rezeptors liegt. Weitere wichtige Beispiele für chirale Wirkstoffe aus der
Pharmaindustrie sind die Progesteron-Rezeptor Antagonisten in der Krebstherapie
(modifizierte Indolsulfonamide)187,188 oder auch 4-Chromenonyl-1,4-dihydropyridin zur Herz-
Kreislauf-Therapie189.
Somit besteht ein wachsender Bedarf an chiralen Alkoholen in der Industrie126. Grundsätzlich
sind diese Bausteine auch durch eine chemische Synthese darstellbar. Die
Produktionsverfahren leiden jedoch sowohl unter niedriger Selektivität als auch unter dem
Einsatz von giftigen und teuren Katalysatoren. Insbesondere kleine, chirale Alkohole wie
2-Butanole sind nur schwer über chemische Prozesse in reiner Form zu synthetisieren. Daher
stellen diese Moleküle interessante Ziele für eine biotechnologische Synthese dar. Speziell
Oxidotransferasen können zur Reduktion der Ketovorstufen eingesetzt werden, bieten
teilweise eine herausragende Selektivität und erfreuen sich daher steigender Beliebtheit40,41.
Dennoch leiden auch biokatalytische Reduktionsreaktionen unter einigen Nachteilen, wie
ungünstigen Gleichgewichtslagen, erschwerter Produktaufreinigung oder instabilen und/ oder
unselektiven Enzymen.
Diese Probleme sollten in dieser Arbeit durch die Entwicklung eines neuen
Biotransformationsverfahrens gelöst werden. Es war geplant, die Produktion von
enantiomerenreinem 2-Butanol durch den Einsatz von hochselektiven Oxidoreduktasen in
einem wasserarmen System durchzuführen. Dadurch könnte die Produktausbeute stark erhöht,
die Aufreinigung erleichtert und die Abfallmenge reduziert werden. Idealerweise sollten auch
neue „smart cosubstrates“ zum Einsatz kommen, welche die Menge an Cosubstrat auf ein
Minimum reduzieren sollten. Der dazu benötigte Biokatalysator musste neben einer
exzellenten Selektivität auch eine enorme Lösemitteltoleranz aufweisen. Unzureichende
Eigenschaften sollten durch Techniken des Protein Engineerings optimiert werden.

Diskussion
80
4.1 Identifizierung eines geeigneten Biokatalysators
Am Anfang der Prozessentwicklung stand die Suche nach einem geeigneten Enzym zur
Reduktion von 2-Butanon. Dabei stellte die geringe Größe des Substrates eine besondere
Herausforderung dar. So handelt es sich um die kleinste vorkommende achirale
Kohlenwasserstoffverbindung ohne zusätzliche Heteroatome. In der Literatur ist lediglich
eine einzige Alkoholdehydrogenase beschrieben, die die Reduktion selektiv katalysiert. Dies
ist die Candida parapsilosis ADH41. Leider ist dieses Enzym nur schwer zu exprimieren und
besitzt keine hohe Lösemittelstabilität. Dennoch belegt es die grundsätzliche Verfügbarkeit
von Katalysatoren, die in dieser Reaktion eingesetzt werden könnten. Im Gegensatz dazu gibt
es eine Vielzahl von ADHs, welche laut Literatur nur eine unzureichende Enantioselektivität
bezüglich des Umsatzes von 2-Butanon aufweisen. Dazu gehören beispielsweise die Enzyme
von Rhodococcus ruber, Saccharomyces cerevisiae, Thermoanaerobium brockii, Thermus
ethanolicus und viele weitere134,161,190–192. Diese Tatsache verdeutlicht die Herausforderung,
die das kleine Substrat an das Enzym stellt.
Um dem Problem angemessen zu begegnen, wurde eine breite Palette an Enzymen auf deren
Fähigkeit zur selektiven Umsetzung des Substrats untersucht. Neben den in der Literatur
beschriebenen Alkoholdehydrogenasen HL-ADH (Horseliver-ADH)28,147,148, LB-ADH
(Lactobacillus brevis-ADH)149–152 und PL-ADH (Parvibaculum
lavamentivorans-ADH)14,153,154 gehörten 21 proprietäre Enzyme der Firma evocatal GmbH
dazu. Zu Beginn wurden diese 24 Enzyme in einem Initial-Screening auf ihre grundsätzliche
Eignung zum Umsatz von 2-Butanon untersucht. Es zeigte sich, dass zwölf der Katalysatoren
einen signifikanten Umsatz erzielten (Tab. 7). Diese dienten als Grundlage für die weiteren
Untersuchungen. Interessanterweise lag der Umsatz in allen Fällen bei Einsatz des GDH-
Regenerationssystems deutlich über dem des 2-Propanol-Regenerationssystems. Dies ist
dadurch zu erklären, dass es sich im ersten Fall nicht um ein Cosubstrat-gekoppeltes, sondern
um ein Enzym-gekoppeltes Regenerierungssystem handelt. Dabei wird ein zweiter
Biokatalysator, hier eine GDH, eingesetzt, um in einer unabhängigen Nebenreaktion unter
Oxidation von Glukose den Cofaktor NAD(P)H zu regenerieren. Somit besteht die Aufgabe
der ADH in diesem Fall nur darin, das Butanon zu reduzieren und nicht, wie im Falle des
Cosubstrat-Systems darin, auch das 2-Propanol zu oxidieren. Weiterhin leidet es unter einem
höheren Prozentanteil an organischer Phase, was die Struktur des Enzyms stören und damit
dessen Aktivität beeinflussen kann.
Die zwölf aktiven Dehydrogenasen wurden daraufhin auf ihre Selektivität untersucht.
Angestrebt wurde dabei, das (S)-Enantiomer zu synthetisieren. Hier spiegelte sich die

Diskussion
81
Problematik des sehr kleinen Substrates in den erzielten Ergebnissen wider (Tab. 9). Nur zwei
der untersuchten Enzyme erreichten die geforderte Selektivität von über 99,5 %. Eines davon
erzielte diese Vorgabe jedoch nur im 2-Propanol Regenerationssystem, womit die
Reproduzierbarkeit in Frage gestellt wurde. Auffällig war dabei, dass in fast allen Fällen die
Produktreinheit der beiden Regenerationssysteme teils stark variierte. Da in den
vorhergehenden Versuchen bereits beobachtet wurde, dass die Systeme auch unterschiedliche
Umsätze erzielten, stellte sich zu diesem Zeitpunkt bereits die Frage, ob die
Enantiomerenreinheit auch abhängig vom generierten Substratumsatz sein könnte.
Das Enzym-Screening beinhaltete auch die Untersuchung der Lösemittelstabilität der
Enzyme, da ein hoher Volumenanteil an organischer Phase im Prozess angestrebt wurde. Die
Auswahl eines geeigneten Katalysators beschränkte sich damit auf lösemitteltolerante ADHs.
Dass diese unter solch harschen Bedingungen zwar meist inaktiviert werden, es aber auch
tolerante Vertreter gibt, wurde in der Vergangenheit beschrieben57,162,169,193,194.
Die Ergebnisse der Experimente bescheinigten insgesamt sechs Enzymen eine ungewöhnliche
Toleranz gegenüber hohen Anteilen an 2-Propanol (3.1.4). Diese lag bei mindestens achtzig
Prozent des Gesamtvolumens. Dennoch wurde ein signifikanter Umsatz erzielt, welcher in
vier Fällen sogar über 90 % lag, auch wenn zu beachten ist, dass die Substratbeladung
lediglich 100 mM betrug (Tab. 10).
Im wasserfreien Medium konnte dagegen kaum noch Aktivität gemessen werden. Lediglich
die PL-ADH und die LB-ADH zeigten unter diesen Umständen einen leichten Umsatz. Grund
für diesen abrupten Aktivitätsverlust im Vergleich zu den vorhergehenden Versuchen lag
wahrscheinlich am Entzug von benötigten Wassermolekülen aus der Enzymstruktur. Diese
dienen als eine Art Schmiermittel und gewährleisten die Elastizität der Strukur. Daher sind sie
für den Ablauf der Katalyse unabdingbar195,196. Der Versuch, die Enzyme durch Zugabe von
Sorbitol während der Lyophilisierung unter diesen Bedingungen zu stabilisieren, führte im
Falle der PL-ADH zum Erfolg. Das Enzym erzielte damit trotz der erneuten Lyophilisierung
einen etwa achtfach höheren Umsatz. Es ist davon auszugehen, dass Sorbitol als sogenanntes
„Lyoprotectant“197 die Struktur vor Wasserentzug schützt. Die genaueren Hintergründe
werden unter 4.3 diskutiert.
Nach dem Screening der Enzympalette stellten sich somit zwei Alkoholdehydrogenasen als
besonders geeignet für die Synthese von (S)-2-Butanol dar. Dabei handelte es sich zum einen
um die PL-ADH, zum anderen um die evo-040.
Die Ergebnisse zeigten, dass die PL-ADH eine ungewöhnlich hohe Lösemitteltoleranz besitzt.
Dies wäre ein großer Vorteil bei der Entwicklung eines wasserarmen Prozesses.

Diskussion
82
Nichtsdestotrotz produzierte das Enzym unter den genannten Bedingungen keine
zufriedenstellende Enantiomerenreinheit (Tab. 9).
Die Alkoholdehydrogenase evo-040 wurde dagegen als einziges Enzym identifiziert, das
einen exzellenten und reproduzierbaren Enantiomerenüberschuss generiert (siehe 4.2). Der ee
belief sich auf > 99,5 % für das (S)-Enantiomer.
Da nach der Prozessentwicklung eine Enzymevolution geplant war, boten sich damit zwei
Herangehensweisen an: Die erste Möglichkeit bestand im Einsatz und der späteren Evolution
der PL-ADH in Richtung einer verbesserten Stereoselektivität, die zweite beruhte auf einer
Nutzung und Evolution der ADH evo-040 in Bezug auf ihre Lösemittelstabilität.
Die Entscheidung fiel letztlich auf die Nutzung der evo-040. Ausschlaggebend dafür war die
Tatsache, dass sich eine Optimierung der Enzymstabilität in der Vergangenheit oftmals
einfacher und erfolgversprechender als die der Selektivität gestaltete. Auch die Tatsache, dass
alle Enzyme außer der evo-040 unter schwankender Produktreinheit litten (4.2), die bisher
nicht erklärt werden konnte, bestärkte diese Entscheidung. Weiterhin ist der Wildtyp der
evo-040 bereits an sich auch schon relativ lösemitteltolerant (Tab. 10).
In Hinblick auf ein kommendes high-throughput Screening der Mutantenbanken war
außerdem von Vorteil, dass sich die Analyse von Enzymaktivitäten wesentlich einfacher
gestaltet als die Bestimmung einer veränderten Selektivität. Im letzteren Fall müsste eine
zeitintensive chromatographische Bestimmung der Enantiomerenreinheit für jede
Enzymvariante durchgeführt werden, während Aktivitäten schnell und parallel im
Photometerassay im 96-well Format durchgeführt werden können.
Somit lässt sich abschließend festhalten, dass eine Enzymevolution der ADH evo-040 sowohl
aus proteinchemischen als auch aus praktischen Gründen bevorzugt wurde.
4.2 Produktracemisierung
Während der Versuche zur Identifizierung eines geeigneten Enzymkandidaten zeigte sich,
dass die Enantiomerenreinheit des Produkts auch unter Nutzung desselben Enzyms teilweise
stark schwankte. Es kam die Vermutung auf, dass es im Laufe der Reaktion zu einer
Racemisierung des Produktes kommt. Diese Problematik könnte die vielversprechenden
Ergebnisse des Enzym-Screenings relativieren und musste daher untersucht werden.
Das Phänomen der Produktracemisierung in ADH-Reaktionen wurde auch von Yang et al.
beobachtet161. In der genannten Arbeit wurde ein Mechanismus vorgeschlagen, welcher
versucht, den Effekt zu erklären.

Diskussion
83
In der vorliegenden Arbeit sollte nun geklärt werden, ob sich der vorgeschlagene
Mechanismus auf die hier untersuchten Biokatalysatoren übertragen lässt, welche weiteren
Faktoren die Racemisierungsgeschwindigkeit beeinflussen könnten, und ob der
vorgeschlagene Mechanismus eventuell erweitert werden kann.
In der Arbeit von Yang et al. wurde die Reduktion von 2-Butanon durch die ADH von
Thermoanaerobium brockii (TB-ADH) untersucht. Erste Umsetzungen ergaben dabei einen
Enantiomerenüberschuss von 55 % für die (R)-Form. Dieser Wert erniedrigte sich jedoch im
Laufe der Reaktion bis hin zu einem racemischen Gemisch. Es wurde gefolgert, dass die
Reduktion des Substrates reversibel ist, und somit ein einmal entstandenes Produkt
rückoxidiert und anschließend erneut reduziert werden könnte. Der Effekt ist schematisch
dargestellt Abb. 41 zu entnehmen. Wie dargestellt, wird davon ausgegangen, dass der
Cofaktor entweder dauerhaft oder zumindest über einen bestimmten Zeitraum mit dem
Substrat verbunden bleibt, wie an anderer Stelle auch belegt wurde198,199. Dadurch entstehen
die Komplexe E●NADPH oder E●NADP+. An diese können nun die Redoxpartner binden,
wodurch die Komplexe E●NADPH●ket(S), E●NADPH●ket(R) und E●NADPH●Aceton
entstehen (bzw. die entsprechenden Gegenpaare). Da die nun folgende Redoxreaktion eine
MPV-Gleichgewichtsreaktion darstellt, findet sie auch im scheinbar ruhenden System ständig
als Hin- und Rückreaktion statt.
Abb. 41: Schematische Darstellung des Reaktionsweges einer ADH-katalysierten Substratreduktion sowie
des beschriebenen Racemisierungseffekts nach Yang et al 161
Um zu verstehen, weshalb die Rückoxidation und damit die Racemisierung erst im Laufe des
Prozesses stattfindet, müssen zwei Situationen im Reaktionsverlauf gesondert betrachtet
werden. Zu Beginn zeigt sich folgendes Bild: Das Substrat 2-Butanon sowie das Cosubstrat
2-Propanol stehen in hohem Überschuss zur Verfügung. Thermodynamisch betrachtet wird

Diskussion
84
ein Gleichgewicht angestrebt, woraus sich eine starke Triebkraft zur Reduktion des Butanols
und zur Oxidation des 2-Propanols ergibt. Das führt zur Bildung von Produkt (2-Butanol) und
Coprodukt (Aceton).
In einem weiter fortgeschrittenen Zustand der Reaktion ergibt sich ein anderes Bild: Als
alkoholische Komponenten stehen nun entstandenes (S)-2-Butanol und (R)-2-Butanol sowie
immer noch 2-Propanol zur Verfügung, welche oxidiert werden können. Reduziert werden
können dagegen 2-Butanon und das entstandene Aceton. Da die anfänglichen
Konzentrationsüberschüsse von Substrat und Cosubstrat nicht mehr vorhanden sind, fehlt die
Gleichgewichtstriebkraft, so dass eine thermodynamisch bedingte Selektion nicht mehr
stattfindet. Damit erfolgt die Oxidation des 2-Propanols nun kompetitiv mit der des
(R)-Butanols und (S)-Butanols.
Während eine Oxidation des Propanols die Stereomerenreinheit des Produkts in der Reaktion
nicht verändert, so ist dies bei der Oxidation eines Enantiomers durchaus der Fall. Da damit
gerechnet werden kann, dass bei einer selektiven Umsetzung ein Enantiomer im Überschuss
vorhanden ist, wird dieses mit höherer Wahrscheinlichkeit rückoxidiert, was nach Yang et al.
zum Racemisierungseffekt führt.
Um zu überprüfen, ob ein ähnlicher Effekt auch bei den Enzymen in der vorliegenden Arbeit
zum Tragen kam, wurde die Stereomerenreinheit des Produkts in Abhängigkeit von der
Reaktionsdauer analysiert. Es ergab sich ein eindeutiges Bild, das eine Racemisierung auch
bei den getesteten ADHs nachwies (Abb. 13). Der Effekt trat sowohl bei (R)- als auch bei
(S)-selektiven Enzymen auf und die Geschwindigkeit der Racemisierung schien
enzymabhängig zu sein. So verlief die Isomerisierung bei der evo-260 beispielsweise deutlich
schneller ab als bei der evo-030 oder evo-270.
Weiterhin wurde ein Einfluss der Cosubstratbeladung auf die Racemisierungsgeschwindigkeit
vermutet161. Dieser Effekt konnte in Versuchen mit der evo-270 nachvollzogen werden
(3.2.5). Wurde dem Reaktionsansatz mehr Cosubstrat zugesetzt, so lief die Racemisierung
stark verlangsamt ab. Unter Hinzunahme der oben erläuterten Hypothese des
Reaktionsablaufs ist dieses Ergebnis verständlich. So wird die Reduktion des Butanons
thermodynamisch kontrolliert durch das Gleichgewicht zwischen Substrat/ Produkt
(2-Butanon/ 2-Butanol) und Cosubstrat/ Coprodukt (2-Propanol/ Aceton). Durch eine
Erhöhung der Cosubstratkonzentration wird die Reaktion in Richtung Coprodukt verschoben
und damit einhergehend auch die Reaktion vom Substrat zum Produkt. Eine Rückreaktion des
Produktes zum Substrat und die damit einhergehende Racemisierung wird somit
unwahrscheinlicher.

Diskussion
85
Weitere Versuche zeigten auch einen Einfluss der Reaktionstemperatur. Umsetzungen bei
50 °C erzielten niedrigere Enantiomerenüberschüsse am Reaktionsgleichgewicht als bei 25 °C
oder 10 °C (Tab. 11). Dieses Ergebnis ist thermodynamisch erklärbar191,192,199–201. So kann die
stereochemische Reinheit einer Substanz als E beschrieben werden, was dem Verhältnis der
Enantiomere enstpricht.
Dabei gilt: E = (R/S) = (kcat/Km)R / (kcat/Km)S (1)
Weiter kann E kinetisch beschrieben werden als Differenz der freien Aktivierungsenergie der
Enantiomere: ∆∆G = -RTlnE (2)
Ist ein Enyzm unselektiv, so ergibt sich aus (2), dass der Unterschied ∆∆G = 0, da E = 1.
Wird ∆G durch die Eyring Aktivierungsparameter dargestell, ergibt sich:
∆∆G = ∆∆H - T∆∆S (3)
Wird nun wieder angenommen, dass ein Enzym unselektiv ist, so ergibt sich aus (2), dass
∆∆G = 0 und damit aus (3):
∆∆H = T∆∆S (4)
Das bedeutet, dass die Beiträge aus Enthalpie und Entropie gleichwertig sind.
Da die Reaktionstemperatur T variabel ist und der Entropiebeitrag ∆∆S von T abhängt, lässt
sich daraus schließen, dass für jede Reaktion im Sinne von (1) gilt, dass eine Temperatur Tr
existiert, unter welcher Gleichung (4) erfüllt ist. Es ergibt sich die „racemische Temperatur“
Tr als:
Tr = ∆∆H / ∆∆S (201) (5)
Dies verdeutlicht die Temperaturabhängigkeit der Reaktion. Bei Temperaturen unter Tr wird
∆∆G (und damit E) durch ∆∆H bestimmt. Bei Temperaturen über Tr wird ∆∆G durch ∆∆H
bestimmt201.
Weitere Experimente zeigten, dass der pH-Wert des Reaktionsmediums ebenfalls einen
Einfluss auf die Stereoisomerenreinheit des Produktes nehmen kann (3.2.3). Auch wenn die
Unterschiede gering waren, erreichte die untersuchte ADH evo-270 bei dem pH-Wert ihrer
größten Stabilität (pH = 7,5) den höchsten ee mit einem Wert von 90 % nach 20 Stunden
Reaktionsdauer. Bei Reaktionen in basischerem oder saurerem Milieu sank die
Produktreinheit leicht ab, auch wenn der gleiche Umsatz erzielt wurde (Tab. 12). Durch diese
Ergebnisse kam die Vermutung auf, dass ein nicht optimales Medium die Enzymstruktur
negativ beeinflussen könnte und dadurch die katalytischen Eigenschaften des Katalysators
verändert.

Diskussion
86
Versuche zur Überprüfung dieser Hypothese widerlegten jedoch diese Annahme (3.2.4). Ein
durch Inkubation (48 Stunden) in Lösemittel „geschädigtes“ Enzym zeigte eine ähnliche
Selektivität wie dasselbe Enzym ohne Vorinkubation.
Diese Ergebnisse zeigten, dass das Enzym auch nach längerer Reaktionsdauer noch dieselben
selektiven Eigenschaften aufweist, wie zu Beginn der Reaktion. Daher erschien es fraglich,
ob, wie von Yang et al.161 vermutet, in erster Linie die Konkurrenz der vorhandenen
Enantiomere um einen freien Enzymkomplex die Racemisierung vorantreibt. Nach dieser
Theorie wäre die Rückoxidation eines bestimmten Enantiomers umso wahrscheinlicher, je
höher dessen Konzentration in der Reaktion ist. Dies erscheint plausibel, da es sich um ein
zufälliges und nicht gelenktes Aufeinandertreffen handelt. Somit würde bei einem Enzym mit
E = 95 % die Rückoxidation des überwiegenden Enantiomers auch mit einer
Wahrscheinlichkeit von 95 % stattfinden (da es sich in 95 %igem Überschuss befindet). Das
zweite Enantiomer würde somit mit einer Wahrscheinlichkeit von 5 % rückoxidiert werden
(in einem rein theoretischen Reaktionszustand, in welchem die Racemisierung beginnt). Das
kann jedoch die Isomerisierung nicht vollends erklären, da gezeigt werden konnte, dass das
Enzym seine Selektivität beständig beibehält. Somit würde das rückoxidierte Produkt in
einem nächsten Reduktionsschritt auch mit 95 % Wahrscheinlichkeit wieder zum präferierten
Enantiomer reduziert werden. Es würde sich somit ein Gleichgewichtszustand ergeben, bei
dem der ee beständig bleibt.
Eine Erklärung, wie es dennoch zur Racemisierung kommen kann, könnte möglicherweise in
der unterschiedlichen Kinetik der zwei Rückoxidationsreaktionen liegen. Die zugehörigen
Versuche zeigten, dass im Falle der evo-270 das (R)-Enantiomer mindestens zehnmal
schneller rückoxidiert wird als die (S)-Form (3.2.6). Dieses Ergebnis ist durchaus
nachvollziehbar, da die Reaktionsgeschwindigkeit nach Arrhenius202,203
= � �� ���� (6)
von der Aktivierungsenergie EA abhängig ist, welche für die beiden Enantiomere
unterschiedlich ist191,192,200.
Es lässt sich daraus schließen, dass zwei Tatsachen zur Racemisierung führen: 1) Das
überwiegende Enantiomer wird mit höherer Wahrscheinlichkeit auf einen freien
Enzymkomplex treffen. 2) Unter gleichen Bedingungen wird das überwiegende Enantiomer
mit vielfach höherer Geschwindigkeit als das mengenmäßig unterlegene Enantiomer
rückoxidiert.
Weiterhin konnte gezeigt werden, dass Reaktionsparameter wie Temperatur, pH-Wert und
Cosubstratkonzentration die Racemisierungsgeschwindigkeit beeinflussen. Die Auswirkungen

Diskussion
87
waren aber eher gering. Die Racemisierung konnte nur durch das Anwenden hoher
Molenbrüche (Cosubstrat/ Substrat) deutlich verlangsamt werden. Allerdings ist dies nur bei
Prozessen mit niedriger Substratbeladung möglich, welche dann allerdings unter
ökonomischer Ineffizienz leiden.
4.3 Enzymstabilisierung in organischem Lösemittel
Die Optimierung der Enzymstabilität ist eine Grundvoraussetzung zur Entwicklung
ökonomischer biokatalytischer Prozesse. Die dabei am häufigsten adressierten Ziele sind eine
verlängerte Lagerstabilität oder eine erhöhte Resistenz gegenüber hohen Temperaturen,
extremen pH-Werten und Lösemitteln.
Die Stabilisierung gegen hohe Konzentrationen an organischem Lösemittel ist dabei
besonders interessant, da der Einsatz solcher Reaktionsmedien einige Vorteile gegenüber der
Arbeit in rein wässrigem Milieu besitzt:
Ein mengenmäßig höherer Einsatz von organischen Lösemitteln erlaubt beispielsweise die
Anwendung höherer Substratbeladungen, was zu besseren Reaktionsausbeuten führen kann.
Im Hinblick auf die Thermodynamik einer Reaktion führen hohe Mengen an Cosubstrat durch
die Verschiebung des thermodynamischen Gleichgewichts in der Regel ebenso zu höheren
Produktausbeuten146. Ein weiterer interessanter Aspekt ist eine erleichterte
Produktaufreinigung, wenn Lösemittel mit niedrigem Siedepunkt eingesetzt werden. Auf den
Reaktionsmechanismus bezogen, unterdrücken niedrige Wasserkonzentrationen unerwünschte
Nebenreaktionen204. Diese Faktoren können die Ökonomie eines Prozesses signifikant
verbessern und ihn damit wettbewerbsfähiger gegenüber klassisch-chemischen Synthesen
machen.
Darüber hinaus existieren auch bestimmte Reaktionen wie Veresterung und Transveresterung,
die eine niedrige Wasseraktivität benötigen, um stattfinden zu können146,205.
Grundsätzlich gibt es verschiedene Techniken, um Proteine zu stabilisieren. Ein Ansatz ist,
die Enzyme mittels Protein Engineering zu optimieren, etwa über gerichtete Evolution oder
durch rationale Methoden83,105. Beispiele dafür sind das Einfügen von Prolin Aminosäuren
oder Disulfidbrücken durch den Austausch flexibler Aminosäuren (hoher B-Faktor) oder
durch Polypeptid Chain Extension 172,206,207.
Eine zweite Stabilisierungsmethode ist die Nutzung chemischer Modifikationen von
Enzymen. Beispiele sind die Monofunktionalisierung durch Aminosäurederivate, Acylierung

Diskussion
88
oder Alkylierung171 oder die Bifunktionalisierung durch verknüpfende Reagenzien wie
Glutaraldehyd oder Diimidate.
Eine dritte und vielgenutzte Technik ist die Immobilisierung von Enzymen. Es handelt sich
um ein sehr beliebtes Verfahren, da weitere Vorteile enstehen208: Es ermöglicht eine leichte
Rückgewinnung des Katalysators, vereinfachte Produktaufreinigung und ein flexibleres
Reaktor-Design.
In der vorliegenden Arbeit wurde die Stabilisierung von Biokatalysatoren gegen organische
Lösemittel durch den Einsatz von stabilisierenden Additiven untersucht. Diese Methode
wurde früher überwiegend angewendet, um die Lagerstabilität von Enzymen in wässrigen
Medien zu verlängern209,210, oder um Peptide während der Lyophilisierung zu schützen197,211.
Nun sollte jedoch das gelöste Enzymlyophilisat direkt im Reaktionsansatz gegenüber
extremen Reaktionsmilieus stabilisiert werden. Diese Milieus bestanden aus einem
Volumenanteil der organischen Phase von bis zu 90 %. Für die Arbeit wurden fünf Enzyme
(evo-030, evo-040, evo-270, evo-380 und LB-ADH) ausgewählt, die mit verschiedenen
Selektivitäten und Substratspektren eine Bandbreite unterschiedlicher
Alkoholdehydrogenasen abdecken sollten. Die Erhöhung des Reaktionsmaßstabs auf bis zu
vier Liter sollte weiterhin die Anwendbarkeit des Verfahrens für industrielle Zwecke
überprüfen.
Insgesamt wurden 17 Substanzen auf ihr Potential hin untersucht, ADHs in Gegenwart von
organischen Lösemitteln zu stabilisieren. Die Substanzen ließen sich in folgende Gruppen
unterteilen: Polyole, Salze und Polyethylenglykole (PEG) (Tab. 14). Frühere Publikationen
legten offen, dass die Konzentration eines Additivs ausschlaggebend auf dessen
stabilisierenden Effekt wirkt171,172. Daher wurden in einem ersten, groben Screening die
Additive in zwei Konzentrationen eingesetzt. Substanzen, die unter diesen Bedingungen einen
positiven Einfluss auf den Substratumsatz zeigten, wurden anschließend kleinmaschiger auf
ein Konzentrationsoptimum durchmustert. Durch die Anwendung dieses Verfahrens konnte
eine positive Beeinflussung aller fünf Enzyme durch spezifische Additive erzielt werden
(Tab. 16).
Der größte Erfolg konnte dabei mit der evo-380 erzielt werden. Der Umsatz war fast
siebenfach erhöht, wenn der Katalysator mit 0,1 mM PEG4000 stabilisiert worden war. Auch
die ADH evo-030 erreichte eine sehr viel höhere Substratumsetzung. Sie lag bei 360 % im
Vergleich zum ungeschützten Enzym. Die übrigen Enzyme evo-040 und evo-270 und
LB-ADH erreichten Werte von etwa 150 % Umsatz im Vergleich zum ungeschützten Enzym,
womit auch hier von einer erfolgreichen Stabilisierung gesprochen werden kann.

Diskussion
89
Bis heute ist der Einfluss von stabilisierenden Substanzen nicht sicher geklärt. Dabei spielt es
auch eine Rolle, welcher Typ der Stabilisierung adressiert wird. So ist zu unterscheiden, ob
die Additive (i) in der Produktion des Katalysators bei der Lyophilisierung197 (ii), während
der Lagerung zur Erhöhung der Haltbarkeit212 oder (iii) direkt in der Reaktion zur Reduktion
der Enzyminaktivierung168 eingesetzt werden. Die Effekte in der vorliegenden Arbeit sind
dabei der Gruppe (iii) zuzuordnen, da die Substanzen dem Reaktions-Batch direkt zugesetzt
wurden.
Es existieren unterschiedliche Hypothesen, wie die Additive im Reaktionsbatch wirken. Die
am weitesten verbreitete beruht darauf, dass die Substanzen an die Enzymstruktur binden und
dadurch eine Aufrechterhaltung der Hydrathülle bewirken169,213,214. Andere Studien dagegen
ordnen den verschiedenen Klassen der Additive auch unterschiedliche Wirkweisen zu205:
(a) Unspezifische ionische Effekte von Salzen, die an geladene Aminosäuren binden. Dadurch
könnte es zu einem Aussalzeffekt der Proteine kommen, welcher die Steifheit der Struktur
erhöht215. (b) Polyole und Zucker zeigten die Fähigkeiten, hydrophobe Interaktionen von
Aminosäuren zu stärken204. (c) Niedermolekulare Additive führten zu einer bevorzugten
Hydratation von Enymen im Reaktionsmilieu207. Vermutlich entsteht der Effekt durch
Wechselwirkungen der Additive mit dem Proteinrückgrat. Da diese thermodynamisch
ungünstig sind, werden die Additive abgesondert. Dies erhöht die Wasseraktivität des
Mediums und führt zur gewünschten Hydratation des Enzyms216,217. (d) Polymere wie PEG
könnten das Enzym durch unspezifische Wechselwirkungen teilweise aus dem Lösemittel
absondern204,218.
In der vorliegenden Arbeit zeigten alle Enzyme je eine Präferenz entweder für PEG oder für
Zuckeradditive. Keines erzielte mit beiden Additivklassen erhöhten Umsatz. Es ist zu
vermuten, dass dieses Verhalten auch auf andere Alkoholdehydrogenasen übertragbar ist und
die Stabilisatoren somit sehr spezifisch mit den Enzymen interagieren. Nichtsdestotrotz
beantwortet das nicht die Frage, ob die Additive direkt an das Enzym binden und damit die
Hydrathülle aufrechterhalten, welche für die Aktivität unabdingbar ist219, oder ob sie Einfluss
auf die Eigenschaften des Reaktionsmediums nehmen und dadurch nur indirekt auf die
Enzyme wirken204.
Im Gegensatz zu den vorherigen Substanzen wirkte Ammoniumsulfat auf beide Enzymtypen,
auf solche, die von PEG stabilisiert werden und auf solche, die durch Zucker stabilisiert
werden. Dies könnte die Hypothese untermauern, dass Salze wiederum einen anderen
Wirkmechanismus besitzen. Nichtsdestotrotz kann allen Substanzen, die positiven Einfluss
zeigten, eine hygroskopische Eigenschaft zugeschrieben werden. Das würde wiederum dafür

Diskussion
90
sprechen, dass die Additive direkt an die Enzymstruktur binden und dort die Wasserhülle
aufrechterhalten. Dabei kann angenommen werden, dass die enzymspezifische
Oberflächentopologie und –ladungsverteilung die Interaktion mit bestimmten Additivklassen
begünstigt und mit anderen wiederum behindert.
Weiterhin wurden in dieser Arbeit auch Hinweise gefunden, dass ein Zusammenhang
zwischen der Molekülgröße des Additivs und dessen idealer Konzentration besteht. Im Falle
der evo-040 konnte beispielsweise beobachtet werden, dass das Monosaccharid in doppelt so
hoher Konzentration wie das Disaccharid eingesetzt werden musste, um einen optimalen
Effekt zu erzeugen (Tab. 16). Im Falle der evo-030 musste das Monosaccharid sogar in
dreifacher Menge zugefügt werden. Im Vergleich dazu fiel bei der evo-380 auf, dass
kurzkettige PEGs in vielfach höherer Konzentration eingesetzt werden mussten als
langkettige.
Im Falle der evo-040, die zur Prozessentwicklung genutzt werden sollte, konnte durch die
Zugabe von 1 M Sorbitol der Umsatz um 57 % (relativ) erhöht werden. Der
Enantiomerenüberschuss blieb erfreulicherweise exzellent (ee ≥ 99,5 % für (S)-2-Butanol).
Daher wurde diese Stabilisierungstechnik im 1 Liter Reaktionsmaßstab angewendet (3.5.6).
Die Ergebnisse bestätigten die Versuche im Kleinmaßstab, woraufhin eine 4 Liter Reaktion
durchgeführt wurde. Auch diese erzielte den gewünschten Umsatzanstieg. Das Ergebnis
bestätigte die Reproduzierbarkeit der entwickelten Technik.
Es ist davon auszugehen, dass durch den Einsatz dieser Technik die Effizienz des
(S)-2-Butanol Prozesses steigt, wodurch die absolute Abfallproduktion pro generierter Menge
Produkt sinkt. Dies würde neben einer erhöhten ökonomischen Relevanz auch die
Umweltverträglichkeit verbessern.
4.4 Prozessentwicklung
Es stehen bereits chemische Verfahren zur Herstellung von 2-Butanol zur Verfügung. Diese
lassen sich in zwei Gruppen unterteilen. Die erste nutzt die enantioselektive Reduktion von
2-Butanon, die zweite beruht auf einer kinetischen Resolution von racemischem 2-Butanol.
Letztere stellt dabei die derzeit etablierte und industriell bevorzugte Technik zur
enantiomerenreinen Synthese dar43. Atomökonomisch betrachtet leidet dieses Verfahren
jedoch unter einer maximalen Produktausbeute von nur 50 % des eingesetzten Substrats. Um
dieses Problem zu umgehen kann zusätzlich die Technik der in situ Racemisierung eingesetzt
werden, womit die dynamisch-kinetische Racematspaltung erreicht wird. Das erfordert jedoch
den Einsatz von Übergangsmetallkatalysatoren wie Ruthenium in relativ großen Mengen und

Diskussion
91
kann zur Bildung von Esternebenprodukten führen48. Bei der beschriebenen Technik werden
in der Regel Lipasen eingesetzt, wie in Abbildung Abb. 42 dargestellt135.
Abb. 42: Hypothetisches
Verfahren zur dynamisch-
kinetischen Racematspaltung
nach Martín-Matute et al.135
Dies führt zu großen Mengen an Abfallprodukten, da das racemische Substrat verdünnt
werden muss, der beschriebene Rutheniumkatalysator und außerdem ein Acylierungsreagenz
benötigt werden. Das wiederum führt zu einer hypothetischen Produktion an Abfallstoffen
von über 25 kg je Kilogramm Produkt. Die genauen Reaktionsparameter sind jedoch nicht
öffentlich zugänglich (z.B. ChiprosTM, BASF). Ein weiterer Nachteil liegt in der niedrigen
Enantioselektivität vieler Lipasen220–222. Beispiel für einen solchen Prozess beschreibt Patent
US2007/02207529134, welches ein Verfahren zur Synthese von 2-Butanol mittels dynamisch-
kinetischer Racematspaltung beschreibt. Es wird eine Produktreinheit von ≥ 98 % bei einer
Ausbeute von maximal 50 % beschrieben.
Eine Alternative zur kinetischen Racematspaltung wird durch die enantioselektive Reduktion
von 2-Butanon geboten. Diese ist aus atomökonomischer Sicht sinnvoller, da theoretisch eine
Produktausbeute von bis zu 100 % erreicht werden kann. Die chemische Meerwein-Ponndorf-
Verley Reduktion (MPV) liefert allerdings nur eine ungenügende Enantiomerenreinheit und
benötigt ebenfalls Al-, Rh oder Ru-Katalysatoren133. Daher ist an dieser Stelle der Einsatz von
biologischen Katalysatoren interessant. Diese können (abhängig vom Enzym) hochselektiv
ablaufen223–225. Beispiel für ein Verfahren, welches die enantioselektive Reduktion nutzt, wird
in Patent WO 2006/053713 beschrieben40. Das eingesetzte Enzym erreicht dabei einen
Enantiomerenüberschuss von ≥ 98 %. Auch wenn diese Reinheit bereits sehr hoch ist, so
reicht sie für viele Anwendungen, wie etwa im Pharmabereich, nicht aus. Es wird deutlich,
wie schwierig es ist, einen Biokatalysator zu finden, der das sehr kleine Substrat 2-Butanon
enantiomerenrein reduziert. Ein weiterer Nachteil in der Nutzung von Enzymen liegt darin,
dass sie meist nur in wässrigen Reaktionsmilieus aktiv sind. Dadurch können sich azeotrope
Mischungen aus organischen Komponenten mit Wasser bilden, wodurch die
Produktaufreinigung stark erschwert wird, da eine einfache destillative Aufreinigung nicht

Diskussion
92
anwendbar ist. Tab. 25 veranschaulicht die sehr nahe beieinanderliegenden Siedepunkte der
Reaktionskomponenten im 2-Butanol Prozess.
Tab. 25: Mischverhältnisse und daraus resultierende Siedepunkte der Reaktionskomponenten
Komponenten
Mischverhältnis Siedepunkt
Aceton
56,0 °C
2-Butanol/ Wasser
79 %/ 21 % 73,0 °C
2-Propanol/ 2-Butanon 32 %/ 68 % 78,0 °C
2-Butanon
80,0 °C
2-Propanol
82,0 °C
2-Propanol/ Wasser
72 %/ 28 % 88,1 °C
2-Butanol/ Wasser
68 %/ 32 % 88,5 °C
2-Butanol
99,0 °C
Weiterhin leidet dieses Verfahren, wie alle MPV-Reaktionen, unter ihrer Reversibilität und
einer schwachen thermodynamischen Triebkraft.
Die Probleme sollten nun durch den Einsatz von sehr hohen Mengen an Cosubstrat
(2-Propanol) in der Reaktion umgangen werden. Da durch das fehlende Wasser die
Azeotropenbildung unterbunden wird, könnte dies zu einer stark erleichterten
Produktaufreinigung führen. Weiterhin würde das thermodynamische Gleichgewicht auf die
Produktseite verschoben, was zu einer erhöhten Raum-/ Zeitausbeute führt.
4.4.1 Lösemittelstabilität der evo-040
Wie in 4.1 beschrieben, wurde die Alkoholdehydrogenase evo-040 ausgewählt, um einen
biokatalytischen Prozess zur Synthese von enantiomerenreinem (S)-2-Butanol zu entwickeln.
Das Enzym zeigte in den Vorversuchen bereits eine ungewöhnlich hohe Lösemitteltoleranz.
Aktivität konnte bis zum einem Anteil der organischen Phase von bis 80 % nachgewiesen
werden (3.1.4). Eine genauere Charakterisierung der Stabilität des Enzyms offenbarte einen
ungewöhnlichen Effekt: Der erzielte Umsatz des Enzyms sank, wie zu erwarten war, mit
steigender organischer Beladung. Dies ist auf Deaktivierung oder Denaturierung durch das
Eindringen von Lösemittelmolekülen in die Enzymstruktur zu erklären226. Die organischen,
apolaren Moleküle stören dabei intramolekulare Bindungen in der Enzymstruktur und
zerstören damit den Tertiäraufbau. Ab einer bestimmten Lösemittelkonzentration stieg der
Umsatz der evo-040 jedoch wieder an. Ein lokales Umsatzmaximum lag bei 90 %
Gesamtanteil der organischen Phase (Abb. 16). Zwei unterschiedliche Mechanismen könnten
der Grund für diesen Effekts sein: Zum einen führt die sinkende Wasserkonzentration im

Diskussion
93
Medium zu einem Entzug von Wassermolekülen aus der Enzymstruktur. Dadurch wird deren
Starrheit erhöht227–229. Ein Eindringen von Lösemittelmolekülen in die Struktur wird
erschwert und damit die Denaturierung verlangsamt230. Eine andere Theorie ist, dass sich ab
einer kritischen Konzentration die Wassermoleküle im Reaktionsmedium zu Mizellen
zusammenfinden und damit wässrige „Inseln“ bilden, in denen die Proteine akkumulieren und
somit vor dem Lösemittel geschützt sind146.
Das beschriebene Umsatzmaximum bei 90 % organischer Phase wurde ausgenutzt, um einen
Prozess unter diesen Bedingungen zu entwickeln. Weiterhin konnte eine zusätzliche
Stabilisierung der evo-040 unter diesen harschen Bedingungen erreicht werden, wie unter 4.3
beschrieben.
4.4.2 Substratbeladung
Der nächste Schritt der Prozessentwicklung war die Bestimmung der optimalen
Substratbeladung. Es handelt sich um einen kritischen Parameter, da hohe Substratbeladungen
auch eine hohe Produktausbeute ermöglichen. Nachdem die absolute Menge an organischer
Phase jedoch festgelegt war (4.4.1), nahm die Menge an Cosubstrat mit der Zunahme der
Substratkonzentration ab, wodurch die thermodynamische Triebkraft nachließ. Somit musste
ein idealer Mittelwert ermittelt werden, der den höchsten Umsatz gewährleistete.
Es ließ sich anhand der Umsätze erkennen, dass die ADH evo-040 bei allen untersuchten
Beladungen aktiv war (3.6). Da der Gesamtanteil der organischen Phase konstant gehalten
wurde, änderte sich durch steigende Substratkonzentrationen der Molenbruch (2-Propanol/
2-Butanon). Der Umsatz war bei hohen Molenbrüchen und damit niedrigen
Substratbeladungen erwartungsgemäß höher, auch wenn die eingesetzte Enzymmenge die
gleiche blieb. Um aussagekräftige Werte zu erhalten, mussten daher die relativen Umsätze in
absolute Werte umgerechnet werden (Abb. 18-B). Hier zeigte sich, dass niedrigere
Molenbrüche höhere Ausbeuten bewirkten (auch wenn der prozentuale Umsatz geringer war).
Das Optimum wurde ab einer Butanonbeladung von 20 % erreicht.
4.4.3 Zugabereihenfolge der organischen Komponenten
Es konnte festgestellt werden, dass 2-Butanon einen schädlicheren Einfluss auf evo-040
ausübt als 2-Propanol (3.8). Die Inkubation in 2-Propanol vor Reaktionsstart führte sogar zu
einem relativen Umsatzanstieg. Die Inkubation mit 2-Butanon dagegen schien den Prozess
negativ zu beeinflussen. Der Effekt verstärkte sich bei verlängerter Inkubation (5 Minuten).

Diskussion
94
Ein möglicher Grund dafür könnte die höhere Hydrophobizität von 2-Butanon sein176. Es
besitzt einen log P von 0,25, während der von Isopropanol bei 0,17 liegt. Dabei bedeutet ein
höherer Wert für log P auch eine erhöhte Hydrophobizität231. Dadurch besitzt 2-Propanol
möglicherweise eine niedrigere wasserentziehende Wirkung auf das Enzym und wirkt
weniger strukturschädigend. Prozesstechnisch empfiehlt es sich daher, nach der Vereinigung
von Puffer, Cofaktor und Enzym zuerst das 2-Propanol und erst nach kurzer Inkubationszeit
2-Butanon zuzugeben.
4.4.4 Bestimmung des pH-Optimums
Enzyme sind in der Regel nur in einem engen pH-Bereich maximal aktiv und stabil. Dabei
handelt es sich meistens um die H+-Konzentration, die in dem Mikroorganismus vorliegt, aus
dem sie stammen. Liegt der pH-Wert unter oder über diesem Optimum, so leidet die Aktivität
und/ oder Stabilität des Enzyms und damit der zu erwartende Umsatz eines Prozesses. Gründe
dafür liegen in einer pH-bedingten Änderung der Raumstruktur des Enzyms, verursacht durch
Ladungsveränderungen der Aminosäuren sowie deren Seitenketten, und in veränderten
Wechselwirkungen des Enzyms mit dem Reaktionsmedium. Das kann bis zur Denaturierung
führen, wodurch das Enzym irreversibel inaktiviert wird. Daher ist es für Reaktionen
unabdingbar, das pH-Optimum des eingesetzten Biokatalysators zu kennen.
Die ADH evo-040 zeigte den höchsten Umsatz bei einem pH-Wert von 7,0 (Abb. 19). Eine
minimal erniedrigte Produktausbeute war laut GC-Messung im pH 7,5-Ansatz zu erkennen.
Dieser Unterschied könnte jedoch auf Reaktionsungenauigkeiten zurückzuführen sein. Solche
entstehen durch eine ungenaue Einwaage des Enzymlyophilisats oder durch
Pipettierabweichungen bei der Zugabe der Reaktionskomponenten. Auch die
gaschromatographische Bestimmung der Edukt- und Produktmenge zeigte leichte
Schwankungen (≤ 1 %), die zu Ungenauigkeiten führen können. Relevante Umsatzverluste
konnten erst bei pH 6,5 und 8,0 beobachtet werden, so dass hier die Grenze der pH-Toleranz
der evo-040 anzusetzen war.
4.4.5 „smart cosubstrates“
Auf den Einsatz der neuen „smart cosubstrates“182,232 musste in diesem Prozess leider
verzichtet werden. Grund dafür war die mangelnde Akzeptanz der ADH evo-040 für die
untersuchten Cosubstrate (3.9). Nichtsdestotrotz konnte für andere Enzyme die grundsätzliche
Anwendbarkeit und der Nutzen des Regenerierungssystems verdeutlicht werden. In
Reaktionen zur Reduktion von Carbonylverbindungen und Kohlenstoffdoppelbindungen oder

Diskussion
95
der aromatischen Hydroxylierung konnte gezeigt werden, dass die diolischen Cosubstrate wie
1,4-Butandiol in halbäquimolaren Mengen bereits zum Vollumsatz führten182. Weiterhin
stellen die generierten Coprodukte an sich auch schon wertvolle Synthesebausteine dar und
könnten die Ökonomie von Bioreduktionen stark erhöhen232.
4.4.6 Prozess Up-scaling
Die erfolgreichen Versuche zur Identifizierung der optimalen Reaktionsparameter ließen auf
die Entwicklung eines erfolgreichen Prozesses zur Synthese von (S)-2-Butanol im
wasserarmen Reaktionsmilieu hoffen.
Die Alkoholdehydrogenase evo-040 wurde als Biokatalysator ausgewählt, da sie als einziges
Enzym einen stabilen Enantiomerenüberschuss ≥ 99,5 % lieferte. Weiterhin konnte gezeigt
werden, dass die Katalyse bei einem Gesamtvolumenanteil φ(organische Phase) = 90 %
durchgeführt werden kann, da das Enzym in diesem Bereich ein lokales Aktivitätsmaximum
besitzt. Die optimale Substratbeladung wurde auf 20 % festgelegt und darüber hinaus ein
Protokoll entwickelt, welches den Katalysator im wasserarmen Milieu stabilisiert. Weitere
Parameter wie der optimale pH-Wert und die beste Komponentenzugabereihenfolge wurden
ebenfalls festgelegt.
Als nächstes musste die Durchführbarkeit des Prozesses im größeren Maßstab bewertet
werden. Für die 1 Liter Reaktion wurden 5 g Lyophilisat benötigt (3.10.1). Erfreulicherweise
produzierte E. coli kein Enzym, das durch eine Hintergrundaktivität den Prozess negativ
beeinflusste (z.B. bezüglich der Enantiomerenreinheit des Produkts), wie es in ähnlichen
Prozessen vorkommen kann233. Daher konnte das Lyophilisat aus Zellrohextrakt gewonnen
werden. Mit einer Lyophilisatbeladung von 5 g/l, was einer Aktivität von 2,5 kU je Liter
entspricht, scheint die eingesetzte Menge an Enzym hoch zu sein, liegt jedoch im Rahmen
vergleichbarer Biotransformationen38,40,41,134. Die Reaktion lief erfolgreich und in einer
Quantität ab, welche der der Vorversuche entsprach (3.10.2). Die Hälfte des eingesetzten
Substrats war nach 62 Stunden umgesetzt (Abb. 25). Der Prozess wurde nach 150 Stunden
beendet, auch wenn die Reaktion noch nicht am thermodynamischen Gleichgewicht
angekommen war. Jedoch sank die Umsatzgeschwindigkeit stark ab, wahrscheinlich aufgrund
einer langsamen Inaktivierung des Enzyms. Zum Ende der Reaktion wurde ein Umsatz von
etwa 60 % bestimmt. Das entspricht einer Gesamtproduktmenge von 120 ml. Eine chirale
GC-Analytik bestätigte weiterhin eine Reinheit von ≥ 99,5 % des (S)-Enantiomers. Mit einer
Gesamtreaktionsdauer von 62 Stunden liegt der Prozess etwas über dem Rahmen
vergleichbarer Biokonversionen. So wird in EP174513441 von einem 70 %igen Umsatz nach

Diskussion
96
maximal 48 Stunden berichtet. Aus Patent WO2006/05371340 sind keine genauen Angaben
über die Reaktionsdauer zu entnehmen, wobei schematisch dargestellt wird, dass 400 mM
2-Butanon nach 30 Stunden voll umgesetzt werden. Der in der vorliegenden Arbeit
entwickelte Prozess erreicht jedoch mit einer Substratkonzentration von etwa 2200 mM eine
weit höhere Eduktbeladung, was die Vergleichbarkeit mit Patent WO2006/053713
erschwert40.
Nach der Durchführung der Reaktion konnte der Prozess weiterhin erfolgreich auf den 4 Liter
Maßstab übertragen werden. Es traten keine Probleme auf und die Produktausbeute war im
Rahmen der vorherigen Experimente. Dadurch konnte gezeigt werden, dass der Prozess
reproduzierbar in einen größeren Maßstab übertragen werden kann. Es ist anzunehmen, dass
auch die Übertragung in noch höhere Volumina unproblematisch verlaufen sollte.
Es lässt sich zusammenfassend sagen, dass je Liter Reaktionsansatz insgesamt 120 ml
Produkt gewonnen werden konnten. Das entspricht hochgerechnet einer Generierung von
etwa 7 kg Abfall pro Kilogramm Produkt. Damit stellt das hier entwickelte System (neben
ökonomischen Vorteilen) ein wesentlich umweltfreundlicheres Verfahren zur Synthese von
(S)-2-Butanol im Vergleich zu dem oben beschriebenen (25 kg Abfall pro Kilogramm
Produkt) dar.
4.4.7 Produktaufreinigung
Auch wenn bereits ein Prozess etabliert wurde, in dem der Wasseranteil auf nur 10 %
erniedrigt war, stellte dieser Wert immer noch ein Problem bei der Aufreinigung des Butanols
dar. Durch die Anwendung der Azeotroprektifikation konnte das Problem gelöst werden.
Allerdings verlangte dies den Einsatz von großen Mengen an Cyclohexan im destillativen
Reinigungsschritt, was die Umweltbilanz der Reaktion zu verschlechtern drohte. Es zeigte
sich jedoch, dass während der Aufreinigung (3.11.3) 75 % des eingesetzten Cyclohexans
hochrein rückgewonnen werden konnten (375 ml von 500 ml, Tab. 18). Dennoch steigt die
Abfallmenge um etwa 2,5 kg je Kilogramm Produkt auf nun insgesamt 9,5 kg Abfall/
Kilogramm Produkt an.
Doch auch damit liegt das Verfahren noch deutlich unter den errechneten 25 kg an Abfall in
ähnlichen Prozessen. Es muss jedoch betont werden, dass die genauen Kennzahlen der
konkurrierenden Synthesewege der Geheimhaltung unterliegen und hier deshalb mit
hypothetischen Werten gerechnet werden muss.
Während der Produktaufreinigung zeigte sich auch, dass ein großer Teil des 2-Propanols
rückgewonnen werden konnte. Von den enthaltenen 210 ml 2-Propanol (gaschromatograpisch

Diskussion
97
bestimmt) konnten 59 ml hochrein isoliert werden und würden somit theoretisch aus der
Abfallberechnung entfallen. Es ist jedoch davon auszugehen, dass auch Vergleichsprozesse
durch eine solche Rückgewinnung ihre absolute Abfallgenerierung senken. Da die genauen
Kennzahlen jedoch leider nicht bekannt sind, wurde die Rückgewinnung von
Reaktionskomponenten aus der Berechnung ausgenommen.
Abschließend lässt sich festhalten, dass die komplette Produktmenge mittels Destillation über
eine Füllkörperkolonne rückgewonnen werden konnte. Die chemische Reinheit betrug
99,95 %, der Enantiomerenüberschuss ≥ 99,5 % und der Wassergehalt nach Karl-Fischer-
Titration 0,3 %. Es kann somit zusammengefasst werden, dass mit einer Produktausbeute von
120 ml (S)-2-Butanol je Liter Reaktionsansatz ein überaus ökonomisches Verfahren
entwickelt wurde. Auch aus ökologischer Sicht scheint es vergleichbare Synthesen zu
übertreffen, wobei hier nur mit hypothetischen Werten gerechnet werden konnte.
4.5 Enzymoptimierung
Die Natur stellt eine riesige Anzahl an Enzymen zur Verfügung, die zur Entwicklung von
Bioprozessen genutzt werden können. Dadurch ergibt sich auch ein sehr großes Spektrum an
Reaktionen, die katalysiert werden, und an Substraten, die umgesetzt werden. Allerdings sind
diese nativen Enzyme durch natürliche Evolution meist hochspezifisch an physiologisch
relevante Reaktionen adaptiert104. Anforderungen, wie sie in chemischen Synthesen gestellt
werden, können daher meist von den natürlichen Enzymen nicht erbracht werden. So sind sie
entweder nicht stabil genug, um den harschen Reaktionsbedingungen standzuhalten, oder die
gewünschte Umsetzung liegt außerhalb ihres Substrat- oder Selektionsspektrums. Daher
nimmt die Optimierung eines Enzyms einen immer höheren Stellenwert in der
Prozessentwicklung ein. Dies spiegelt sich auch in der wachsenden Zahl an Unternehmen
wider, die sich auf Enzymevolution spezialisiert haben78–80,234.
Das Verändern und Optimieren eines nativen Enzyms wird unter dem Oberbegriff Protein
Engineering zusammengefasst. Dabei werden durch Mutagenesearbeiten neue Varianten
bekannter Enzyme entwickelt, um deren Eigenschaften zu verändern. Das Vorgehen dabei
lässt sich in a) rationales Engineering (bzw. semi-rationales Engineering) oder b) gerichtete
Evolution unterteilen96–102. Angriffspunkte sind dabei, wie schon angesprochen, die
Enzymselektivität57,97,103, -aktivität239–242 und –stabilität83,84,243–245, wobei die Wahl der zu
verändernden Eigenschaft auch die Wahl der Mutagenesetechnik beeinflusst. So hat sich
beispielsweise zur Erhöhung der Selektivität die Methode des CASTings14,206,246 (neben
anderen) bewährt.

Diskussion
98
Um den in dieser Arbeit entwickelten Prozess zur Synthese von (S)-2-Butanol weiter zu
optimieren, sollte auch die Alkoholdehydrogenase evo-040 durch Techniken des Protein
Engineerings effizienter gemacht werden. Da sie bereits über eine hervorragende Selektivität
verfügt, waren die Angriffspunkte für eine Optimierung eine Erhöhung der Enzymstabilität
oder der –aktivität. Dazu wurden drei Ansätze verfolgt:
A) Rationale Proteinentwicklung: Die Idee bestand darin, dass die Integration von
Disulfidbrücken stabilisierenden Einfluss auf die Proteinstruktur nehmen könnte. Die
Anwendung eines solchen Verfahrens führte bereits in der Vergangenheit zur erfolgreichen
Stabilisierung verschiedener Enzyme107,185,247,248. Um die Etablierung solcher Brücken zu
bewerkstelligen, mussten je Variante zwei Aminosäureaustausche mittels QuikChange-
Mutagenese durchgeführt werden. Diese Aminosäuren wurden durch Cysteine ersetzt, die
dank ihrer räumlichen Lage dazu fähig waren, zusammen eine Disulfidbrücke auszubilden.
Auf diese Weise sollten mehrere Varianten entwickelt und anschließend charakterisiert
werden.
B) Semi-rationale Proteinentwicklung: In der Vergangenheit wurde eine hohe Anzahl von
Studien zur Optimierung von Biokatalysatoren veröffentlicht14,83,85,236,249,250. Das dadurch
generierte Wissen und Verständnis der Einflussnahme von Mutationen auf die
Enzymcharakteristika kann genutzt werden, um abzuschätzen, ob Mutationen in
strukturverwandten Enzymen auch zu verbesserten Eigenschaften führen könnten. Ein solcher
Algorithmus wird beispielsweise von dem Bioinformatikprogramm „Bio-Prodict 3DM“
eingesetzt (https://www.bio-prodict.nl/). Es wurde in dieser Arbeit verwendet, um
Aminosäureaustausche vorherzubestimmen, die vorteilhaft sein könnten, da sie in homologen
Proteinen zu verbesserten Eigenschaften geführt haben. Die so bestimmten Aminosäuren
sollten durch Sättigungsmutagenese mittels QuikChange-Technik ausgetauscht und die
Enzymvarianten im Anschluss charakterisiert werden.
C) Zufallsmutagenese: In diesem Ansatz sollten durch das Anwenden der error-prone PCR
Technik111 zufällig Mutationen in die DNA-Sequenz der Alkoholdehydrogenase evo-040
eingeführt werden. Im Gegensatz zum rationalen Design sind hierbei keine Kenntnisse über
die Struktur des Enzyms erforderlich. Die generierten Mutanten werden in einem high-
throughput-Verfahren auf verbesserte Eigenschaften durchmustert und die Gensequenz dieser

Diskussion
99
Varianten im Anschluss aufgeklärt. Diese Methode wurde bereits erfolgreich zur Optimierung
verschiedener Eigenschaften von Enzymen eingesetzt95,236,251,252.
4.5.1 Rationales Proteindesign durch den Einbau von Disulfidbrücken
Zur Identifizierung von Aminosäurepositionen, die theoretisch im Stande sind,
Disulfidbrücken auszubilden, wurde das Programm „Disulfide by Design Version 1.2“
(http://cptweb.cpt.wayne.edu/DbD/) genutzt185. Anhand eines Proteinstrukturmodells
durchmustert das Programm alle Aminosäure-Paare bezüglich ihrer räumlichen Lage und
Ausrichtung und bewertet daran (angenommen die Aminosäuren würden durch Cysteine
substituiert werden), ob stabile Disulfidbrücken ausgebildet werden könnten.
Um die Berechnungen durchzuführen, wurden im Vorfeld zwei Homologiemodelle der ADH
evo-040 erstellt. Dazu wurde zum einen das Programm „Swiss-Model“
(http://swissmodel.expasy.org/)92,186, zum anderen das Programm „CPHmodels-3.2“
(http://www.cbs.dtu.dk/services/CPHmodels/“)93 genutzt.
Das Programm „Swiss-Model“ wählte das PDB template 1rjw Chain A zum Modellieren aus
(Auflösung 2,34 Å). Es besitzt mit einer Sequenzidentität von 50 % ausreichende Homologie,
um eine zuverlässige Struktur berechnen zu können. Der QMEAN Z-score des
Homologiemodells wurde mit -1,86 angegeben und kann somit als „good model“ klassifiziert
werden253.
„CPHmodels-3.2“ nutzte zur Modellierung das template 4eez Chain B. Es besitzt eine höhere
Sequenzidentität von 64,5 % und eine Auflösung von 1,9 Å. Damit weist es sehr gute
Voraussetzungen auf, um ein verlässliches Homologiemodell zu entwickeln. Daten zur
Qualität der berechneten Struktur wurden nicht angegeben.
Die beiden erstellten Homologiemodelle zeigten untereinander eine sehr hohe
Strukturübereinstimmung. Die Proteinrückgrate ließen sich übereinanderlegen (Abb. 43) und
zeigten auch in Bereichen des random coils nur geringe Abweichungen.

Diskussion
100
Abb. 43: Schematische
Übereinanderlegung des
Proteinrückgrats der beiden
erstellten Homologiemodelle von
evo-040. Schwarz dargestellt ist die
Struktur, die durch das Programm
„Swiss-Model“ berechnet wurde.
Grau dargestellt ist jene, die durch
„CPHmodels-3.2“ entstand. Die
Abbildung wurde mit Hilfe der
Programme „DeepView - Swiss-
PdbViewer“ (http://spdbv.vital-
it.ch/)186 und „POV-Ray 3.7“
(http://www.povray.org/) erstellt.
Die erstellten Modelle wurden in das Programm „Disulfide by Design Version 1.2“
eingepflegt. Dieses berechnete eine Vielzahl möglicher Positionspaare, die theoretisch zur
Ausbildung einer Schwefelbrücke imstande wären. Unter Berücksichtigung des idealen
Torsionswinkels χ3, welcher bei +100° oder -80° liegt254, und der berechneten Energiewerte
der Disulfidbrücken, wobei hier niedrige Werte bevorzugt wurden, fand eine Selektion von
insgesamt sechs Positionspaaren statt (Tab. 19).
Die Doppelmutanten wurden mittels QuikChange-Mutagenese erstellt. Allerdings zeigte sich,
dass die Aminosäureautausche dazu führten, dass sich vier der Varianten überhaupt nicht
mehr und die übrigen zwei schlechter als der Wildtyp in löslicher Form exprimieren ließen.
Es ist zu vermuten, dass die Austausche die Ausbildung der intakten Tertiärstruktur so weit
störten, dass sich die Proteine nicht mehr korrekt falten konnten. Ein Hinweis darauf war auch
in der Analyse der unlöslichen Zellbestandteile zu finden. Das SDS-Gel (Abb. 27) zeigte
starke Banden auf Höhe des Zielproteins - auch in den Stämmen, die kein lösliches Protein
mehr produzierten.
Die Varianten SSB5 und SSB6 (welche sich in löslicher Form exprimieren ließen) zeigten
jedoch bei näherer Charakterisierung schlechtere Eigenschaften als das Wildtypenzym. In
beiden Fällen war sowohl die Hitzestabilität als auch die maximale Umsatzgeschwindigkeit
Vmax teils stark erniedrigt (Abb. 28 & Abb. 29). Es lässt sich daher auch hier vermuten, dass
die eingebrachten Mutationen zu einer Fehlfaltung der Tertiärstruktur führten. Dies könnte die
sowohl niedrige Stabilität als auch Aktivität erklären.

Diskussion
101
4.5.2 Semi-Rationales Proteindesign mittels „Bio-Prodict 3DM“
Grundlage des semi-rationalen Engineerings war die 3DM-Datenbank
(https://www.bio-prodict.nl/). Das 3DM-Tool analysiert und kategorisiert einen Großteil der
online verfügbaren Wissenschaftsliteratur. Dabei durchmustert ein automatisierter
Algorithmus die publizierten Daten nach Enzym-Mutationsstudien. Solche Mutationen
werden in der Datenbank zusammen mit Informationen über die veränderten
Proteineigenschaften abgelegt. Wird das 3DM-Tool für die Planung eines neuen Engineerings
eingesetzt, so wird das zu optimierende Protein mit dieser Datenbank abgeglichen.
Zur Analyse der ADH evo-040 wurde die Aminosäuresequenz des Enzyms in die Software
eingepflegt. Mit dieser Information wurde ein dreidimensionales Strukturalignment
durchgeführt, bei dem die evo-040 mit allen hinterlegten strukturhomologen Enzymen
verglichen wurde. Die in der Datenbank hinterlegten Mutationsstudien wurden auf die
Struktur der ADH übertragen. Damit konnten Vorschläge generiert werden, welche
Aminosäureaustausche vorteilhaft sein könnten, da sie in homologen Proteinen zu
verbesserten Eigenschaften geführt haben.
Durch den Einsatz dieses Verfahrens identifizierte die Software 15 Positionen der ADH
evo-040, die in homologen Proteinen für die Enzymstabilität verantwortlich zu sein schienen.
Durch die Analyse der zugehörigen Publikationen konnten vier der Vorschläge als
Fehlinterpretationen identifiziert werden. Die übrigen elf Positionen wurden als Teil von
Mutationsstudien bestätigt, die das Engineering von homologen Enzymen beschreiben (Tab.
20)108,255–261.
Diese elf Positionen wurden mittels QuikChange-Technik und degenerierter Primer (nnk)
sättigungsmutagenisiert. Es entstanden Banken aus je 200 Klonen pro Position. Ein high-
troughput Screening identifizierte aus diesen Banken fünf Varianten, die in den angewandten
Assays besonders positiv abschnitten. Dabei muss beachtet werden, dass je Bank maximal 1
verbessertes Enzym ausgewählt wurde. Es kann vermutet werden, dass Banken, aus denen ein
verbessertes Enzym gewonnen werden konnte, auch über weitere Varianten verfügten, welche
zwar besser als der Wildtyp abschnitten, jedoch hinter den Ergebnissen der besten Variante
zurücklagen.
Die fünf Enzymvarianten trugen die Mutationen N9G, A12Q, V240N, V243G und D250W.
Zwei davon (N9G und V240N) stachen durch ihre erhöhte Temperaturstabilität von etwa
2 - 3 °C heraus (3.13.2). Dieser Anstieg bezieht sich dabei auf jene Temperatur, die zu einer
50 %igen Inaktivierung des Enzyms führte. Die beobachteten Stabilitätssteigerungen lagen in
dem zu erwartenden Bereich262. Die übrigen Enzyme dagegen waren instabiler als der

Diskussion
102
Wildtyp. Allerdings zeigten zwei dieser instabileren Varianten eine verbesserte
Maximalumsatzgeschwindigkeit. Sie lag bei 149 % (A12Q) beziehungsweise 189 %
(D250W) im Vergleich zum nativen Enzym (Tab. 23).
Dies spiegelte sich auch in den daraufhin durchgeführten Prozessversuchen wieder. Im
wasserreichen Medium stach die Variante D250W besonders heraus und erreichte das
thermodynamische Gleichgewicht bereits nach 20 Stunden, während dieses vom Wildtyp
auch nach 50 Stunden noch nicht erreicht war (Abb. 33). Die übrigen Varianten zeigten im
Prozess nur leichte Verbesserungen. So war der Umsatz nach 50 Stunden in allen Fällen um
etwa 4 % erhöht. Interessanterweise zeigte Variante V243G trotz ihrer erniedrigten
Maximalumsatzgeschwindigkeit am Ende der Reaktion einen höheren Umsatz als der
Wildtyp. Anscheinend besitzt dieses Enzym eine höhere Stabilität gegenüber diesem
Reaktionsmilieu. Dieses Verhalten war aus den vorhergehenden Untersuchungen zur
Temperaturstabilität nicht ersichtlich.
Unter realen, wasserarmen Prozessbedingungen stellten sich die zwei Varianten D250W und
A12Q als besonders geeignet heraus (Abb. 34). Dabei ist zu beachten, dass in diesen
Versuchen auf die entwickelte Sorbitolstabilisierung verzichtet wurde, um die Eigenschaften
der Enzyme nicht zu beeinflussen. Interessanterweise waren hier die besten Varianten auch
jene, die die höchste Maximalumsatzgeschwindigkeit besaßen. Leicht erhöhte Umsätze
konnten noch von den Varianten N9G und V243G erzielt werden, wobei es sich bei N9G um
eine temperaturstabile Variante handelt. Die stabile Variante V240N erreichte im
wasserarmen Prozess jedoch den mit Abstand schlechtesten Umsatz.
Zusammenfassend lässt sich aus den Ergebnissen schließen, dass die erhöhte
Temperaturstabilität in einem der beiden Fälle anscheinend auch Vorteile im
lösemittelreichen Milieu mit sich brachte (N9G), während dies für das zweite stabilere Enzym
nicht der Fall war (V240N). Weiterhin zeigte sich, dass die Enzyme mit höchster
Maximalumsatzgeschwindigkeit auch im wasserarmen Prozess den höchsten Umsatz
erzielten. Dies scheint naheliegend, da die Substratbeladung mit 20 Volumenprozent sehr
hoch war und damit die Maximalgeschwindigkeit auch zum Tragen kam.
4.5.3 Zufallsmutagenese mittels epPCR
Bei der Zufallsmutagenese werden Mutationen über das gesamte Gen auf zufällige Art
eingebracht und anschließend die generierten Varianten auf verbesserte Eigenschaften
durchmustert. Eine mögliche Technik zum Einfügen der zufälligen Mutationen ist die error-
prone PCR, welche hier Anwendung fand113,114.

Diskussion
103
Durch die Wahl der angemessenen Konzentration an MnCl2 konnte die Mutationsrate so
eingestellt werden, dass pro Kilobase im Durchschnitt 1,2 Basenaustausche stattfanden, was
ähnlichen Studien entspricht117. Damit wurde je Enzymvariante im Durchschnitt etwa auch
ein Aminosäureautausch erzielt (Abb. 44). Versuche mit höherer Fehlerrate von 13
Basenfehlpaarungen pro Kilobase wurden ebenfalls durchgeführt. Allerdings waren 86 %
dieser generierten Mutanten inaktiv.
Die Sequenzierung von 50 zufällig ausgewählten Mutanten konnte genutzt werden, um die
Qualität der Banken zu beurteilen. Dabei ist zu beachten, dass die geringe Anzahl von 50
Sequenzen keinesfalls einen statistisch abgesicherten Nachweis, sondern lediglich einen
groben Einblick erbringen kann. Es konnte gezeigt werden, dass es eindeutig präferierte
Bereiche in der Gensequenz gibt, die bevorzugt der Mutagenese unterliegen (Abb. 44), wie
auch in ähnlichen Studien beschrieben118. So wurden etwa doppelt so viele Mutationen im
Bereich 201-300 und 401-500 gefunden wie im Bereich 701-800 oder 901-1000. Dennoch
wurden die Mutationen über den gesamten Sequenzbereich in einem solchen Maße
eingebracht, dass kein Bereich komplett herausfiel. Weiterhin wurde auch gezeigt, dass A↔T
Transversionen die am häufigsten auftretenden Basenaustausche waren. Das stimmt mit
ähnlichen Studien überein, die von einer Häufigkeit der A↔T Transversionen von 40,9 %
berichten117,118. Überraschend hoch dagegen war die Anzahl der A→G Transitionen, wobei
dies auch an der geringen Anzahl an sequenzierten Genen gelegen haben könnte.

Diskussion
104
Abb. 44: Charakterisierung
von 50 Sequenzen der ADH-
Mutanten. 8 % der Sequenzen
trugen einen defekten open
reading frame aufgrund von
frame shifts oder eingefügten
STOP Tripletts. A) A↔T
Transversionen traten am
häufigsten auf, G↔C
Transversionen dagegen
überhaupt nicht. Das Auftreten
von A→G Transitionen war
ungewöhnlich hoch. B) Es zeigte
sich, dass die
Nukleotidaustausche über den
gesamten Genbereich eingefügt
wurden, wenn auch einige
Bereiche bevorzugt wurden.
Das folgende high-troughput Screening der generierten Variantenbänke erbrachte insgesamt
acht Enzymvarianten, die entweder im Hitzeschock- oder im Lösemittelassay eine höhere
Restaktivität besaßen als der Wildtyp (Abb. 35). Auffällig dabei war, dass kein direkter
Zusammenhang der Ergebnisse der beiden Assays hergestellt werden konnte. So war Variante
M87V beispielsweise im Hitzeschock-Assay besonders stabil, während sie nach
Lösemittelinkubation die niedrigste Restaktivität aller Enzyme aufwies. Die einzige
Ausnahme bildete das Enzym P273L, welches sowohl hitze- als auch lösemittelstabiler zu
sein schien.
Daraufhin wurden die Enzyme parallel zu den Versuchen der 3DM-Varianten als Lyophilisat
produziert und genauer charakterisiert. Bei der Untersuchung der Hitzestabilität zeigte sich
1 - 100
101-200
201-300
301-400
401-500
501-600
601-700
701-800
801-900
901-1000
- 5 10 15
Gen
bere
ich
(Nuk
leot
ide)
Mutationswahrscheinlichkeit (%)B
AA T G C
A - 10 4 0
T 10 - 3 2
G 14 6 - 0
C 4 6 0 -
Wildtyp-Anteil: 21 %
Durchschnitt Nukleotidaustausche: 1,2 / 1 kB
Durchschnitt Aminosäureaustausche: 0,29 / 100 AS
Silent mutations: 34,4 % aller AS Mutationen
Frame shifts: 3,3 % aller AS Mutationen
Transversionen: 57 %
Transitionen: 43 %
UrsprungM
uta
tion

Diskussion
105
ein anderes Bild als im Deepwell-Assay (Abb. 37). Auch jetzt waren zwar die Varianten
P273L und M87L deutlich stabiler als der Wildtyp, allerdings zeigten die Mutanten D250R
und M87V eine deutlich herabgesetzte Stabilität im Vergleich zu den high-troughput
Ergebnissen (Abb. 35). Dies lässt sich eventuell durch den veränderten Maßstab erklären. So
fanden im high-troughput Screening alle Schritte - von der Zellanzucht über die
Enzymexpression bis zur Aktivitätsbestimmung - im 1 ml Maßstab statt. Bei einem solch
kleinen Volumen können Ungenauigkeiten wie Pipettierfehler, Temperaturabweichungen
oder ungenügendes Vermischen einen wesentlich größeren Einfluss nehmen als bei einem
größeren Volumen. Daher ist anzunehmen, dass die Ergebnisse der Charakterisierung in
größerem Maßstab verlässlicher sind.
Nach diesen Ergebnissen waren nur die Varianten P273L und M87L hitzestabiler als das
Wildtyp-Enzym. Die beobachtete Stabilitätssteigerung lag bei 1 – 2 °C und damit in dem zu
erwartenden Bereich262. Besonders interessant war, dass mit der Aminosäure M87
anscheinend eine besonders signifikante Position bezüglich der Stabilität aufgedeckt wurde.
So war die Mutante M87V wesentlich instabiler als der Wildtyp, M87L dagegen deutlich
stabiler (Abb. 37). An dieser Position wäre somit eine Sättigungsmutagenese besonders
interessant, um die optimale Aminosäurebesetzung zu identifizieren.
Bei der Bestimmung der Maximalumsatzgeschwindigkeit zeigte sich, dass fast alle Varianten
eine deutlich höhere Vmax aufwiesen als der Wildtyp (Tab. 24). Lediglich die zwei Enzyme
M87V und P273L waren diesbezüglich schlechter als der Wildtyp. Besonders positiv stach
die Mutante M87L mit einer Umsatzgeschwindigkeit von 1336 mU/mg Protein heraus (188 %
im Vergleich zum Wildtyp). Erneut zeigte sich die Bedeutung der Position M87. So war die
Mutante M87V schwächer aktiv als der Wildtyp, M87L dagegen stark aktiviert.
Die hohen Maximalumsatzgeschwindigkeiten spiegelten sich auch in den daraufhin
durchgeführten Prozessversuchen wider. Im wasserreichen Reaktionsmedium erreichten drei
der Mutanten nach knapp 50 Stunden das thermodynamische Gleichgewicht, während dies
vom Wildtyp auch nach 120 Stunden nicht erreicht wurde (Abb. 39). Die zwei Enzyme
P273L und M87V waren dagegen schwächer aktiv als der Wildtyp, was im Einklang steht mit
den gemessenen, sehr niedrigen Maximalumsatzgeschwindigkeiten. Auch die Variante
D250R erreichte nicht das Wildtypniveau. Dies ist durch die vorherigen Versuche nicht zu
erklären, da sie sowohl eine gute Stabilität als auch eine sehr hohe Aktivität aufwies.
Möglicherweise ist diese Variante besonders anfällig gegenüber einer Inaktivierung in dem
angelegten wässrig-organischen Reaktionsmilieu (Volumenanteil der organischen
Phase = 10 %).

Diskussion
106
Anschließend wurden die Enzyme unter wasserarmen Prozessbedingungen untersucht (Abb.
40). Dabei ist zu beachten, dass an dieser Stelle auf die entwickelte Sorbitolstabilisierung
verzichtet wurde, um die Eigenschaften der Varianten nicht zu beeinflussen. Unter diesen
Bedingungen zeigte lediglich die Variante M87L einen erhöhten Umsatz im Vergleich zum
Wildtyp. Der Substratumsatz nach 120 Stunden war in Relation zum Wildtyp um 10 %
erhöht. Die drei Mutanten M87V, D250R und Q339H waren in diesem Reaktionsmilieu kaum
aktiv und erreichten nur Umsätze zwischen 4 und 12 % nach 120 Stunden. Die übrigen
Enzyme lagen etwa im Bereich des Umsatzes des Wildtyp Enzyms.
Abschließend lässt sich festhalten, das durch die Zufallsmutagenese lediglich ein Enzym
generiert werden konnte, dass durch seine verbesserten Eigenschaften die Effizienz des
2-Butanol Prozesses erhöhte. Besonders interessant wäre eine Kombination dieser Mutante
mit den Mutationen, die in der 3DM-Studie zu Verbesserungen geführt haben. Diese waren
D250W und A12Q. Beide führten zu einer relativen Umsatzerhöhung von 20 % im Vergleich
zum Wildtyp. Möglicherweise summieren sich die positiven Eigeschaften dieser Mutationen
auf und würden damit ein wesentlich effizienteres Enzym ergeben. Diese Arbeiten konnten
aus zeitlichen Gründen in dieser Studie nicht mehr durchgeführt werden.
4.5.4 Interpretation der Mutationen
Durch das Protein Engineering mittels Zufallsmutagenese und semi-rationalem Design
wurden in dieser Arbeit 13 Varianten der ADH evo-040 generiert, die verbesserte
Eigenschaften aufwiesen. Eine Variante trug zwei Aminosäuresubstitutionen (N7S/ K345R),
so dass insgesamt 14 Mutationen zu analysieren waren. Einen Überblick über diese gibt Tab.
26.
Es war möglich, die 14 Mutationen in insgesamt sechs Gruppen zu unterteilen. Positionen
einer Gruppe ähnelten sich dabei in ihrer räumlichen Lage in der Enzymstruktur. Somit war
denkbar, dass sie eine ähnliche Wirkungsweise entfalteten. Die Gruppen bestehen aus
folgenden Positionen:
A) N7, N9, S10, A12 B) M87 C)Y182
D) V240, V243, D250 E) P273 F) Q339, K345
Schematisch dargestellt ist die Lage der Aminosäuren sowie die Gruppenaufteilung auch Abb.
45 zu entnehmen.
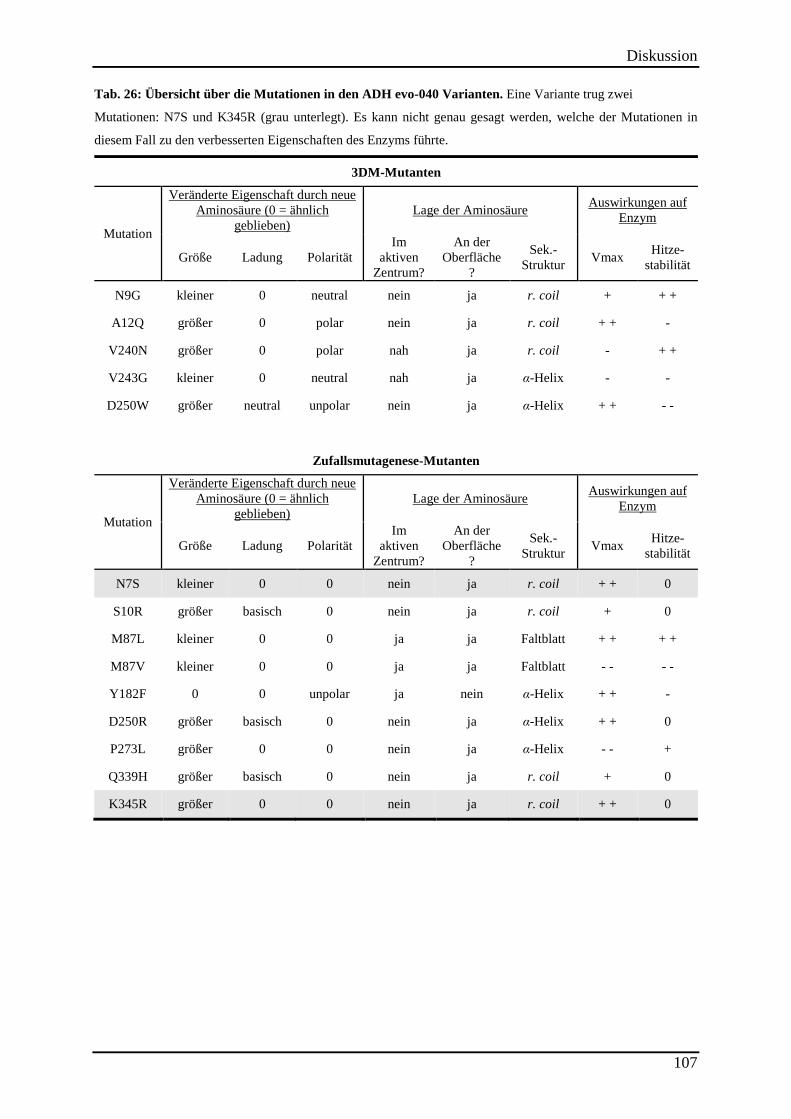
Diskussion
107
Tab. 26: Übersicht über die Mutationen in den ADH evo-040 Varianten. Eine Variante trug zwei
Mutationen: N7S und K345R (grau unterlegt). Es kann nicht genau gesagt werden, welche der Mutationen in
diesem Fall zu den verbesserten Eigenschaften des Enzyms führte.
3DM-Mutanten
Mutation
Veränderte Eigenschaft durch neue Aminosäure (0 = ähnlich
geblieben) Lage der Aminosäure
Auswirkungen auf Enzym
Größe Ladung Polarität Im
aktiven Zentrum?
An der Oberfläche
?
Sek.- Struktur
Vmax Hitze-
stabilität
N9G kleiner 0 neutral nein ja r. coil + + +
A12Q größer 0 polar nein ja r. coil + + -
V240N größer 0 polar nah ja r. coil - + +
V243G kleiner 0 neutral nah ja α-Helix - -
D250W größer neutral unpolar nein ja α-Helix + + - -
Zufallsmutagenese-Mutanten
Mutation
Veränderte Eigenschaft durch neue Aminosäure (0 = ähnlich
geblieben) Lage der Aminosäure
Auswirkungen auf Enzym
Größe Ladung Polarität Im
aktiven Zentrum?
An der Oberfläche
?
Sek.- Struktur
Vmax Hitze-
stabilität
N7S kleiner 0 0 nein ja r. coil + + 0
S10R größer basisch 0 nein ja r. coil + 0
M87L kleiner 0 0 ja ja Faltblatt + + + +
M87V kleiner 0 0 ja ja Faltblatt - - - -
Y182F 0 0 unpolar ja nein α-Helix + + -
D250R größer basisch 0 nein ja α-Helix + + 0
P273L größer 0 0 nein ja α-Helix - - +
Q339H größer basisch 0 nein ja r. coil + 0
K345R größer 0 0 nein ja r. coil + + 0

Diskussion
108
Abb. 45: Schematische Darstellung der 14 Mutationspositionen, die zu veränderten Eigenschaften des
Enzyms führten. Die Positionen wurden in sechs Gruppen (A-F) aufgeteilt, die jeweils Positionen ähnlicher
räumlicher Lage vereinten. Weiß: Ausschnitt des Grundgerüsts des Proteinrückgrat mit Sekundärstrukturen /
Grau: Moleküloberfläche des gebundenen NADH-Moleküls / Stern: ungefährer Ort des katalytischen Zentrums.
Nähere Erläuterungen sind dem Text zu entnehmen (Strukturen dargestellt in einem Homologiemodell nach
„CPHmodels 3.2“, http://www.cbs.dtu.dk/services/CPHmodels/).
Gruppe A: Die vier Positionen N7, N9, S10 und A12 führten alle nach Mutagenese zu
aktiveren Enzymvarianten (gegenüber 2-Butanon) (Tab. 26). Die Betrachtung der
Enzymstruktur zeigte, dass die Positionen in exponierter Lage an der Enzymoberfläche

Diskussion
109
lokalisiert sind. Abb. 45-A legt weiterhin offen, dass sie nicht unmittelbar an der Ausbildung
der katalytischen Tasche beteiligt sind. In der Abbildung Abb. 45-A ist diese im Vordergrund
links unten zu finden. Zwischen dieser Tasche und den vier Positionen liegt somit noch ein
random coil Bereich, welcher die Oberfläche der Tasche ausbildet. Es kann vermutet werden,
dass die vier Aminosäuren Einfluss auf die Form dieses random coil Bereichs nehmen
können. Dadurch würden sie die Ausbildung der katalytischen Tasche indirekt modifizieren.
Weiterhin liegen sie innerhalb eines Bereichs von 15 Å zum aktiven Zentrum. Die Distanz ist
somit so klein, dass Wechselwirkungen zugesprochen werden können206. Die erhöhte
Aktivität könnte beispielsweise durch eine räumliche Vergrößerung der active site zustande
kommen263, was den Eintritt des Substrats erleichtert.
Gruppe B: Sie beinhaltet lediglich eine Aminosäureposition: M87. Es konnte jedoch gezeigt
werden, welch hohen Einfluss die Position sowohl auf die Enzymaktivität als auch –stabilität
nimmt (vergl. M87L und M87V: Abb. 37 und Tab. 24). Sie ist auch die einzige identifizierte
Position, die direkt im katalytischen Zentrum lokalisiert ist. Mutationen an dieser Stelle
können somit großen Einfluss auf den katalytischen Mechanismus nehmen. Veränderungen
können dabei sowohl sterisch als auch ladungsbedingt sein263. Mutationen im aktiven
Zentrum beeinflussen jedoch auch oft die Selektivität des Enzyms81, was in diesem Fall
erfreulicherweise nicht beobachtet wurde.
Gruppe C: Ähnlich wie Gruppe B könnte die Position Y182 die katalytische Reaktion
beeinflussen. Sie liegt in der zweiten Sphäre um das aktive Zentrum und mit einem Abstand
von 12,5 Å in einem einflussnehmenden Bereich206. Der Bereich der sekundären Sphäre wird
oft in der Generierung von smart libraries adressiert, zum Beispiel beim sogenannten
CASTing14,246,264. Dieser Bereich wird ausgewählt, da die Mutagenese von Positionen in der
ersten Sphäre das Enzym oft inaktivieren.
Auch die Mutagenese von Y182 hätte zu veränderter Selektivität führen können81, was jedoch
nicht eintrat. Die beobachtete erhöhte Aktivität der Mutante Y182F könnte durch sterische
Veränderungen oder die erniedrigte Polarität stattgefunden haben.
Gruppe D: Diese Gruppe beinhaltet die Postitionen V240, V243 und D250. Sie sind räumlich
betrachtet nicht am katalytischen Zentrum, sondern am hinteren Teil der NADH-Bindetasche
lokalisiert. Dies macht es schwierig, ihren genauen Effekt zu deuten. Mutationen an diesen
Positionen führten außerdem zu sehr divergenten Auswirkungen auf das Enzym. So war

Diskussion
110
V240N wesentlich hitzestabiler, V243G dagegen hitzeinstabiler, dafür aber lösemittelstabiler
und D250W deutlicher aktiver als das Wildtyp Enzym.
Es kann nur vermutet werden, dass diese Positionen die Bindung des Cofaktors beeinflussen
könnten. Eventuell könnte die Mutation D250W die Bindung zwischen Enzym und Cofaktor
durch die neutrale Ladung und Unpolarität des Tryptophans verstärken, wodurch das Enzym
auch länger in aktivierter Form vorliegt. Insgesamt sind die Auswirkungen dieser Gruppe aber
nur schwer zu deuten.
Gruppe E: Zu dieser Gruppe gehört nur die Position P273. Die Mutante P273L führte zu einer
erhöhten Hitze- und Lösemittelstabilität des Enzyms (Tab. 26 & Abb. 35-B). Die Position
befindet sich in einer exponierten Lage an der Enzymoberfläche und in einer α-Helix.
Allerdings wird Prolin eine helixbrechende Funktion zugeschrieben265,266. Es kann somit
angenommen werden, dass die α-Helix und damit auch dieser Enzymbereich durch den
Austausch des Prolins stabilisiert wurde.
Gruppe F: Die Positionen Q339 und K345 dieser Gruppe führten nach Mutation zu aktiveren
Enzymvarianten. Diese Aminosäuren liegen am C-terminalen Ende des Enzyms. Leider war
dieser Bereich nicht modellierbar, da ihn die templates für das Homologie-Modelling
aufgrund ihrer geringeren Größe nicht abdeckten. Es ist daher nicht klar, wie ihre genaue
Lage im Raum ist. Allerdings befinden sie sich in Nachbarschaft zu den Positionen aus
Gruppe A, weshalb sie eventuell einen ähnlichen Einfluss nehmen wie N7, N9, S10 und A12.
Dafür würde auch sprechen, dass Q339H und K345R wie die Mutanten aus Gruppe A zu
einer erhöhten Aktivität des Enzyms führten.

Zusammenfassung
111
Zusammenfassung
Chirale Alkohole dienen als wichtige Synthesebausteine für Feinchemikalien. Besonders im
Bereich der Pharma-, Agrar- sowie in der Aroma- und Duftstoffindustrie nehmen sie einen
hohen Stellenwert ein. Dabei ist eine hohe Enantiomerenreinheit des eingesetzten Alkohols
von entscheidender Bedeutung. Dies liegt an der Tatsache, dass unerwünschte Enantiomere
eines Moleküls oft voneinander abweichende Eigenschaften aufweisen.
Grundsätzlich sind chirale Alkohole auch durch eine chemische Synthese darstellbar. Die
Produktionsverfahren leiden jedoch sowohl unter niedriger Selektivität als auch unter dem
Einsatz von giftigen und teuren Katalysatoren. Insbesondere kleine, chirale Alkohole wie
2-Butanole sind nur schwer über chemische Prozesse in reiner Form darstellbar. Daher stellen
diese Moleküle interessante Ziele für eine biotechnologische Synthese dar. Dabei bietet sich
insbesondere die Nutzung von Alkoholdehydrogenasen an, welche die Reduktion der
prochiralen Ketonvorstufe hochselektiv katalysieren können.
Dennoch leiden auch diese enzymatischen Redoxreaktionen unter einigen Nachteilen, die in
dieser Arbeit durch die Entwicklung eines neuen Verfahrens umgangen werden sollten.
Zur Identifizierung eines Biokatalysators, der zur Reduktion von 2-Butanon geeignet ist,
wurde eine breite Palette von insgesamt 24 Enzymen durchmustert. Durch die Bestimmung
der Selektivitäten und Umsatzraten konnte ein Enzym identifiziert werden, welches die
Reduktion von 2-Butanon hochselektiv und effizient katalysiert und dabei einen
Enantiomerenüberschuss von ≥ 99,5 % für das (S)-Enantiomer generiert. Alle übrigen
Enzyme zeigten einen starken Racemisierungseffekt, der dazu führte, dass sich die
Produktreinheit im Laufe der Reaktion stark verringerte. Die Aufklärung dieses
Racemisierungseffekts war Teil der vorliegenden Arbeit. Es gelang, die potentiell zu Grunde
liegenden Mechanismen aufzudecken und entscheidende Parameter für die
Racemisierungsgeschwindigkeit zu identifizieren.
Ein weiteres Problem bei der Synthese von enantiomerenreinem 2-Butanol stellte die
schwierige Produktrückgewinnung dar, da sich aus den organischen Reaktionskomponenten
azeotrope Mischungen mit Wasser bilden, die nahe beieinanderliegende Siedepunkte
aufweisen. Daher wurde ein Reaktionsmilieu gewählt, das zu einem Anteil von 90 % aus
organischer Phase bestand, um die Bildung der Azeotrope zu minimieren. Es konnte
außerdem eine neuartige Stabilisierungstechnik entwickelt werden, die es ermöglicht, den

Zusammenfassung
112
Umsatz von Alkoholdehydrogenasen unter diesen anspruchsvollen Reaktionsbedingungen
stark zu erhöhen.
Das organische Reaktionsmilieu erlaubte weiterhin den Einsatz von hohen Mengen an
Substrat und Cosubstrat. Dies war einer der Schlüsselfaktoren zur Entwicklung eines
Prozesses der asymmetrischen Reduktion, der zu einer Produktausbeute von 120 ml
(= 1,34 mol) hochreinem (S)-2-Butanol pro Liter Reaktionsansatz führte. Die
Enantiomerenreinheit des Produkts lag bei hervorragenden ≥ 99,5 % und die chemische
Reinheit bei ≥ 99 %. Gleichzeitig wurde die theoretische Menge an generiertem Abfall von
25 kg (hypothetische Vergleichsreaktion) auf etwa 9,5 kg Abfall pro Kilogramm Produkt
reduziert.
Der gewählte Biokatalysator wurde weiterhin mittels verschiedener Techniken des Protein
Engineerings für den entwickelten Prozess optimiert. Es gelang, drei Varianten des Enzyms
zu generieren, die zu deutlich höheren Umsatzraten führten. Durch eine Auswertung der
eingefügten Mutationen wurden Hypothesen entwickelt, wie bestimmte
Aminosäuresubstitutionen zu einem Anstieg der Aktivität oder Stabilität beitragen könnten.
Durch den Einsatz der entwickelten Enzymvarianten konnte die Produktausbeute des
Prozesses um bis zu 20 % erhöht werden.

Summary
113
Summary
Chiral alcohols serve as important intermediates for the synthesis of fine chemicals. These
molecules are crucial for industrial supplies in a large variety of applications including
pharmaceuticals, pesticides, flavors and fragrances. However, undesired enantiomers of a
molecule can exhibit divergent properties of a product. Thus, a great enantiomeric purity is
highly important.
In principal, chiral alcohols are accessible by classical chemical synthesis. However, these
processes are low in selectivity and require expensive and toxic catalysts. Additionally, small
chiral compounds like 2-butanols are difficult to produce by chemical processes with high
purity. Biotechnological synthesis has the potential to increase the production efficiency of
these molecules.
One reason is that enzymes like alcohol dehydrogenases have the ability to reduce ketone
substrates with high selectivity. Thus, this class of enzymes is a good choice for the
production of enantiomerically pure 2-butanol. Nevertheless, there are some drawbacks in
using alcohol dehydrogenases like low stability in organic solvents or the need for inefficient
cofactor regeneration systems. These drawbacks hindered the development of successful
processes in the past. Now, a novel process approach was established to circumvent these
drawbacks.
To identify an appropriate biocatalyst for the reduction of 2-butanone, a large array of 24
different alcohol dehydrogenases was screened. Their selectivity and conversion rates were
determined. One enzyme was identified that catalyzed actively and highly selective the
conversion. The generated enantiomeric excess exhibited a value of ≥ 99.5 % for the
(S)-enantiomer. In contrast, all other enzymes displayed a racemization effect during the
reaction, which led to a decrease of product purity. The elucidation of this effect was part of
the present study. Several potential causes that may account for the racemization were
identified. Also, some critical parameters for the velocity of racemization were disclosed.
One central issue in the synthesis of 2-butanol was the impaired product recovery. This is due
to the formation of azeotropes of the reaction compounds. Those mixtures possess very
similar boiling points. The problem was solved by choosing a reaction environment with a
ratio of 90 % organic phase. Following this, the formation of azeotropic mixtures was
suppressed in large part. For that purpose, a novel enzyme stabilization technique was
developed. This increased substrate conversion under the harsh conditions dramatically.

Summary
114
Furthermore, the organic environment facilitated the usage of high loadings of substrate and
cosubstrate. This was a key factor for the development of asymmetric reduction process. The
yield of recovered product was 120 ml of (S)-2-butanol per liter of reaction broth. The
enantiomeric excess of the product was ≥ 99.5 % and the chemical purity ≥ 99 %.
Concomitantly, the amount of generated waste was reduced to 9,5 kg per kilogram of product.
In contrast, the comparative reaction leads to 25 kg of waste production.
Furthermore, the chosen biocatalyst was subject to different techniques of protein engineering
to improve the overall efficiency. Successfully, three variants of the enzyme were generated
that yielded significantly increased substrate conversion. Two of them elevated the conversion
by up to 20 %. Evaluation of the introduced mutations showed how amino acid substitutions
may act on the protein activity and/ or stability.

Literaturverzeichnis
115
Literaturverzeichnis
1. Perlman, D. Some problems on the new horizons of applied microbiology. Dev. Ind. Microbiol. 21, 15–23 (1980).
2. Clouthier, C. M. & Pelletier, J. N. Expanding the organic toolbox: a guide to integrating biocatalysis in synthesis. Chem. Soc. Rev. 41, 1585–605 (2012).
3. Schmid, A., Dordick, J. S., Hauer, B., Kiener, A., Wubbolts, M. & Witholt, B. Industrial biocatalysis today and tomorrow. Nature 409, 258–68 (2001).
4. Turner, N. J. Directed evolution drives the next generation of biocatalysts. Nat. Chem. Biol. 5, 567–73 (2009).
5. Ciriminna, R. & Pagliaro, M. Green Chemistry in the Fine Chemicals and Pharmaceutical Industries. Org. Process Res. Dev. 17, 1479–84 (2013).
6. Solodenko, W., Mennecke, K., Vogt, C., Gruhl, S. & Kirschning, A. Polyvinylpyridine, a Versatile Solid Phase for Coordinative Immobilisation of Palladium Precatalysts - Applications in Suzuki-Miyaura Reactions. Synth. 11, 1873–81 (2006).
7. Nising, C. F., Schmid, U. K., Nieger, M. & Bräse, S. A new protocol for the one-pot synthesis of symmetrical biaryls. J. Org. Chem. 69, 6830–3 (2004).
8. Lüllmann, H., Mohr, K. & Hein, L. Pharmakologie und Toxikologie. (Thieme, 2010).
9. Downey, W. Trends in Biopharmaceutical Contract Manufacturing. Chim. Oggi-Chemistry Today 31, 19–24 (2013).
10. Panke, S., Held, M. & Wubbolts, M. Trends and innovations in industrial biocatalysis for the production of fine chemicals. Curr. Opin. Biotechnol. 15, 272–9 (2004).
11. Boesten, W. H. J. Process for the recovery of ampicillin. EP 0 869 960 B1 (2002).
12. Jensen, V. J. & Rugh, S. Industrial-scale production and application of immobilized glucose isomerase. Methods Enzymol. 136, 356–70 (1987).
13. Nagasawa, T. & Yamada, H. Application of nitrile converting enzymes for the production of useful compounds. Pure Appl. Chem. 62, 1441–4 (1990).
14. Spickermann, D., Hausmann, S., Degering, C., Schwaneberg, U. & Leggewie, C. Engineering of Highly Selective Variants of Parvibaculum lavamentivorans Alcohol Dehydrogenase. ChemBioChem 15, 2050–2 (2014).
15. Vasic-Racki, D. in Ind. Biotransformations (Liese, A., Seelbach, K. & Wandrey, C.) 1–29 (Wiley-VCH Verlag GmbH & Co. KGaA, 2006).
16. Schoemaker, H. E., Mink, D. & Wubbolts, M. G. Dispelling the Myths - Biocatalysis in Industrial Synthesis. Science. 299, 1694–7 (2003).

Literaturverzeichnis
116
17. Schmid, A., Hollmann, F., Park, J. B. & Bühler, B. The use of enzymes in the chemical industry in Europe. Curr. Opin. Biotechnol. 13, 359–66 (2002).
18. Shaw, N. M., Robins, K. T. & Kiener, A. Lonza: 20 Years of Biotransformations. Adv. Synth. Catal. 345, 425–35 (2003).
19. http://www.biotechnologie.de. Die deutsche Biotechnologie-Branche 2015. (2015).
20. Reid, M. F. & Fewson, C. A. Molecular characterization of microbial alcohol dehydrogenases. Crit. Rev. Microbiol. 20, 13–56 (1994).
21. Danielsson, O., Atrian, S., Luque, T., Hjelmqvist, L., Gonzàlez-Duarte, R. & Jörnvall, H. Fundamental molecular differences between alcohol dehydrogenase classes. Proc. Natl. Acad. Sci. USA 91, 4980–4 (1994).
22. Inoue, T., Sunagawa, M., Mori, A., Imai, C., Fukuda, M., Takagi, M. & Yano, K. Cloning and sequencing of the gene encoding the 72-kilodalton dehydrogenase subunit of alcohol dehydrogenase from Acetobacter aceti. J. Bacteriol. 171, 3115–22 (1989).
23. Nordling, E., Oppermann, U. C. T., Jörnvall, H. & Persson, B. Human type 10 17β-hydroxysteroid dehydrogenase: molecular modelling and substrate docking. J. Mol. Graph. Model. 19, 514–20 (2001).
24. Jörnvall, H., Persson, B., Krook, M., Atrian, S., Gonzàlez-Duarte, R., Jeffery, J. & Ghosh, D. Short-Chain Dehydrogenases/Reductases (SDR). Biochemistry 34, 6003–13 (1995).
25. Scopes, R. K. An iron-activated alcohol dehydrogenase. FEBS Lett. 156, 303–6 (1983).
26. Crosas, B., Cederlund, E., Torres, D., Jornvall, H., Farrés, J. & Parés, X. A vertebrate aldo-keto reductase active with retinoids and ethanol. J. Biol. Chem. 276, 19132–40 (2001).
27. Rossmann, M. G., Liljas, A., Bränden, C. I. & Banaszak, L. J. in Enzymes (Boyer, P. D.) 61–102 (Academic Press, 1975).
28. Eklund, H., Nordström, B., Zeppezauer, E., Söderlund, G., Ohlsson, I., Boiwe, T., Söderberg, B.-O., Tapia, O. & Brändén, C.-I. Three-dimensional structure of horse liver alcohol dehydrogenase at 2-4 A resolution. J. Mol. Biol. 102, 27–59 (1976).
29. Ghosh, D., Sawicki, M., Pletnev, V., Erman, M., Ohno, S., Nakajin, S. & Duax, W. L. Porcine carbonyl reductase structural basis for a functional monomer in short chain dehydrogenases/reductases. J. Biol. Chem. 276, 18457–63 (2001).
30. Schwartz, M. F. & Jörnvall, H. Structural analyses of mutant and wild-type alcohol dehydrogenases from Drosophila melanogaster. Eur. J. Org. Chem. 68, 159–68 (1976).
31. Weckbecker, A. & Hummel, W. Cloning, expression, and characterization of an (R)-specific alcohol dehydrogenase from Lactobacillus kefir. Biocatal. Biotransform. 24, 380–9 (2006).

Literaturverzeichnis
117
32. Ensor, C. M. & Tai, H. H. Site-Directed Mutagenesis of the conserved Tyrosine 151 of Human Placental NAD+-dependent 15-Hydroxyprostaglandin Dehydrogenase Yields a Catalytically Inactive Enzyme. Biochem. Biophys. Res. Commun. 176, 840–5 (1991).
33. Albalat, R., Gonzàlez-Duarte, R. & Atrian, S. Protein engineering of Drosophila alcohol dehydrogenase. The hydroxyl group of Tyr 152 is involved in the active site of the enzyme. FEBS Lett. 308, 235–39 (1992).
34. Ghosh, D., Wawrzakt, Z., Weeks, C. M., Duax, W. L. & Erman, M. The refined three-dimensional structure of 3α, 20β-hydroxysteroid dehydrogenase and possible roles of the residues conserved in short-chain dehydrogenases. Structure 2, 629–40 (1994).
35. Auerbach, G., Herrmann, A., Gütlich, M., Fischer, M., Jacob, U., Bacher, A. & Huber, R. The 1.25 Å crystal structure of sepiapterin reductase reveals its binding mode to pterins and brain neurotransmitters. EMBO J. 16, 7219–30 (1997).
36. Benach, J., Atrian, S., Gonzàlez-Duarte, R. & Ladenstein, R. The catalytic reaction and inhibition mechanism of Drosophila alcohol dehydrogenase: observation of an enzyme-bound NAD-ketone adduct at 1.4 Å resolution by X-ray crystallography. J. Mol. Biol. 289, 335–55 (1999).
37. Keinan, E., Sinha, S. C. & Sinha-Bagchi, A. Thermostable enzymes in organic synthesis. 7. Total synthesis of the western corn rootworm sex pheromone 8-methyldec-2-yl propanoate using a TBADH-generated C2-Bifunctional Chiron. J. Org. Chem. 57, 3631–6 (1992).
38. Bradshaw, C. W., Fu, H., Shen, G.-J. & Wong, C.-H. A Pseudomonas sp. alcohol dehydrogenase with broad substrate specificity and unusual stereospecificity for organic synthesis. J. Org. Chem. 57, 1526–32 (1992).
39. Bradshaw, C. W., Hummel, W. & Wong, C.-H. Lactobacillus kefir alcohol dehydrogenase: a useful catalyst for synthesis. J. Org. Chem. 57, 1532–6 (1992).
40. Stürmer, R., Hauer, B., Drew, D., Breuer, M., Schröder, H., Dejana, D., Breuer, M. & Schröder, H. Method for the production of optically-active alcohols. WO 2006/053713 A1 (2006).
41. Gupta, A., Tschenktscher, A. & Bobkova, M. Verfahren zur Herstellung von 2-Butanol durch enzymatische Reduktion von 2-Butanon in einem Zwei-Phasen-System. EP 1 745 134 B1 (2009).
42. Vollhardt, K. P. C. & Schore, N. E. Organische Chemie. (Wiley-VCH Verlag GmbH & Co. KGaA, 1995).
43. Breuer, M., Ditrich, K., Habicher, T., Hauer, B., Keßeler, M., Stürmer, R. & Zelinski, T. Industrial methods for the production of optically active intermediates. Angew. Chem. Int. Ed. 43, 788–824 (2004).
44. Patel, R. N. Enzymatic synthesis of chiral intermediates for drug development. Adv. Synth. Catal. 343, 527–46 (2001).

Literaturverzeichnis
118
45. Collins, A. L., Sheldrake, G. & Crosby, J. Chirality in Industry. (John Wiley & Sons, 1998).
46. Puls, M. Evolutive Optimierung von Lipasen aus Bacillus subtilis und Pseudomonas aeruginosa. PhD Thesis - Düsseld. Univ. (2007). at <http://docserv.uni-duesseldorf.de/servlets/DocumentServlet?id=6818>
47. Reetz, M. T. in Asymmetric Synth. - Essentials (Christmann, M. & Bräse, S.) (Wiley-VCH, 2006).
48. Martín-Matute, B. & Bäckvall, J. E. Dynamic kinetic resolution catalyzed by enzymes and metals. Curr. Opin. Chem. Biol. 11, 226–32 (2007).
49. Giacomini, D., Galletti, P., Quintavalla, A., Gucciardo, G. & Paradisi, F. Highly efficient asymmetric reduction of arylpropionic aldehydes by horse liver alcohol dehydrogenase through dynamic kinetic resolution. Chem. Commun. 4038–40 (2007).
50. Itoh, N. Use of the anti-Prelog stereospecific alcohol dehydrogenase from Leifsonia and Pseudomonas for producing chiral alcohols. Appl. Microbiol. Biotechnol. 98, 3889–904 (2014).
51. Ernest, I. Bindung, Struktur und Reaktionsmechanismen in der organischen Chemie. (Springer-Verlag, 1972).
52. Sandoval, C. A., Ohkuma, T., Muñiz, K. & Noyori, R. Mechanism of Asymmetric Hydrogenation of Ketones Catalyzed by BINAP/1,2-Diamine-Ruthenium (II) Complexes. J. Am. Chem. Soc. 125, 13490–503 (2003).
53. Grignard, V. in CR Hebd. Séances Acad. Sci. 1322–4 (1900).
54. Kaplan, N. O., Ciotti, M. M. & Stolzenbach, F. E. The action of hydroxylamine and cyanide on alcohol dehydrogenase of horse liver. J. Biol. Chem. 211, 419–29 (1954).
55. Kendal, L. P. & Ramanathan, A. N. Liver alcohol dehydrogenase and ester formation. Biochem. J. 52, 430–8 (1952).
56. Racker, E. Crystalline Alcohol Dehydrogenase from Bakers’ Yeast. J. Biol. Chem. 184, 313–312 (1950).
57. Grunwald, J., Wirz, B., Scollar, M. P. & Klibanov, A. M. Asymmetric oxidoreductions catalyzed by alcohol dehydrogenase in organic solvents. J. Am. Chem. Soc. 108, 6732–4 (1986).
58. Laane, C., Boeren, S., Vos, K. & Veeger, C. Rules for optimization of biocatalysis in organic solvents. Biotechnol. Bioeng. 30, 81–7 (1987).
59. May, S. W. & Padgette, S. R. Oxidoreductase enzymes in biotechnology: Current status and future potential. Nature 1, 677–86 (1983).
60. Wichmann, R. & Vasci-Racki, D. Cofactor Regeneration at the Lab Scale. Adv. Biochem. Engin./Biotechnol. 92, 225–60 (2005).

Literaturverzeichnis
119
61. Schüte, H., Flossdorf, J., Sahm, H. & Kula, M.-R. Purification and properties of formaldehyde dehydrogenase and formate dehydrogenase from Candida boidinii. Eur. J. Org. Chem. 62, 151–60 (1976).
62. Galkin, A., Kulakova, L., Yoshimura, T., Soda, K. & Esaki, N. Synthesis of optically active amino acids from alpha-keto acids with Escherichia coli cells expressing heterologous genes. Appl. Environ. Microbiol. 63, 4651–6 (1997).
63. Kruse, W., Hummel, W. & Kragl, U. Alcohol-dehydrogenase-catalyzed production of chiral hydrophobic alcohols. A new approach leading to a nearly waste-free process. Recl. Trav. Chim. Pays-Bas 115, 239–43 (1996).
64. Kataoka, M., Rohani, L. P. S., Wada, M., Kita, K., Yanase, H., Urabe, I. & Shimizu, S. Escherichia coli transformant expressing the glucose dehydrogenase gene from Bacillus megaterium as a cofactor regenerator in a chiral alcohol production system. Biosci. Biotechnol. Biochem. 62, 167–9 (1998).
65. Ikemi, M., Ishimatsu, Y. & Kise, S. Sorbitol production in charged membrane bioreactor with coenzyme regeneration system: II. Theoretical analysis of a continuous reaction with retained and regenerated NADPH. Biotechnol. Bioeng. 36, 155–65 (1990).
66. Hainke, M., Ammon, K. & Onken, U. Simultane Messung von Gelöstsauerstoffkonzentration und Hydrodynamik in aeroben Submersfermentationen. Chem. Ing. Tech. 70, 299–301 (1998).
67. Hummel, W. & Riebel, B. Chiral Alcohols by Enantioselective Enzymatic Oxidation. Ann. N.Y. Acad. Sci. 799, 713–6 (1996).
68. Demain, A. L. Small bugs, big business: The economic power of the microbe. Biotechnol. Adv. 18, 499–514 (2000).
69. Eggert, T., Leggewie, C., Puls, M., Streit, W., van Pouderoyen, G., Dijkstra, B. W. & Jaeger, K.-E. Novel biocatalysts by identification and design. Biocatal. Biotransform. 22, 139–44 (2004).
70. Leggewie, C., Puls, M. & Eggert, T. Identifizierung und Expression neuer Biokatalysatoren. Chem. Ing. Tech. 82, 27–34 (2010).
71. Rondon, M. R., August, P. R., Bettermann, A. D., Brady, S. F., Grossman, T. H., Liles, M. R., Loiacono, K. A., Lynch, B. A., Macneil, I. A., Minor, C., Tiong, C. L., Gilman, M., Osburne, M. S., Clardy, J., Handelsman, J. & Goodman, R. M. Cloning the Soil Metagenome: a Strategy for Accessing the Genetic and Functional Diversity of Uncultured Microorganisms. Appl. Environ. Microbiol. 66, 2541–7 (2000).
72. Dalbøge, H. & Lange, L. Using molecular techniques to identify new microbial biocatalysts. Trends Biotechnol. 16, 265–72 (1998).
73. Demain, A. L. Microbial biotechnology. Trends Biotechnol. 18, 26–31 (2000).

Literaturverzeichnis
120
74. Ohmae, E., Miyashita, Y. & Kato, C. Thermodynamic and functional characteristics of deep-sea enzymes revealed by pressure effects. Extremophiles 17, 701–9 (2013).
75. Davydov, D. R., Sineva, E. V, Davydova, N. Y., Bartlett, D. H. & James, J. R. CYP261 enzymes from deep sea bacteria: a clue to conformational heterogeneity in cytochromes P450. Biotechnol. Appl. Biochem. 60, 30–40 (2013).
76. Arnosti, C. & Jørgensen, B. B. High activity and low temperature optima of extracellular enzymes in Arctic sediments: implications for carbon cycling by heterotrophic microbial communities. Mar Ecol Prog Ser 249, 15–24 (2003).
77. Wallenstein, M. D., McMahon, S. K. & Schimel, J. P. Seasonal variation in enzyme activities and temperature sensitivities in Arctic tundra soils. Glob. Chang. Biol. 15, 1631–9 (2009).
78. Webpage. evocatal GmbH. at <www.evocatal.de>
79. Webpage. BRAIN AG. at <www.brain-biotech.de>
80. Webpage. c-LEcta GmbH. at <www.c-lecta.com>
81. Morley, K. L. & Kazlauskas, R. J. Improving enzyme properties: when are closer mutations better? Trends Biotechnol. 23, 231–7 (2005).
82. Jaeger, K.-E., Eggert, T., Eipper, A. & Reetz, M. T. Directed evolution and the creation of enantioselective biocatalysts. Appl. Microbiol. Biotechnol. 55, 519–30 (2001).
83. Jakoblinnert, A., van den Wittenboer, A., Shivange, A. V, Bocola, M., Heffele, L., Ansorge-Schumacher, M. & Schwaneberg, U. Design of an activity and stability improved carbonyl reductase from Candida parapsilosis. J. Biotechnol. 165, 52–62 (2013).
84. Eijsink, V. G. H., Bjørk, A., Gåseidnes, S., Sirevåg, R., Synsta, B., van den Burg, B. & Vriend, G. Rational engineering of enzyme stability. J. Biotechnol. 113, 105–20 (2004).
85. Martinez, R., Jakob, F., Tu, R., Siegert, P., Maurer, K.-H. & Schwaneberg, U. Increasing activity and thermal resistance of Bacillus gibsonii alkaline protease (BgAP) by directed evolution. Biotechnol. Bioeng. 110, 711–20 (2013).
86. Jaeger, K.-E. & Reetz, M. T. Directed evolution of enantioselective enzymes for organic chemistry. Curr. Opin. Chem. Biol. 4, 68–73 (2000).
87. Reetz, M. T. & Jaeger, K. E. Enantioselective enzymes for organic synthesis created by directed evolution. Chem. Eur. J. 6, 407–12 (2000).
88. Arnold, F. H. & Volkov, A. A. Directed evolution of biocatalysts. Curr. Opin. Chem. Biol. 3, 54–9 (1999).
89. Ruff, A. J., Dennig, A. & Schwaneberg, U. To get what we aim for - progress in diversity generation methods. FEBS J. 280, 2961–78 (2013).

Literaturverzeichnis
121
90. Wong, T. S., Roccatano, D. & Schwaneberg, U. Steering directed protein evolution: strategies to manage combinatorial complexity of mutant libraries. Environ. Microbiol. 9, 2645–59 (2007).
91. Bornscheuer, U. T. & Pohl, M. Improved biocatalysts by directed evolution and rational protein design. Curr. Opin. Chem. Biol. 5, 137–43 (2001).
92. Arnold, K., Bordoli, L., Kopp, J. & Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 (2006).
93. Nielsen, M., Lundegaard, C., Lund, O. & Petersen, T. N. CPHmodels-3.0-remote homology modeling using structure-guided sequence profiles. Nucleic Acids Res. 38, 576–81 (2010).
94. Dennig, A., Marienhagen, J., Ruff, A. J. & Schwaneberg, U. in Dir. Evol. Libr. Creat. (Gillam, E. M. J., Copp, J. N. & Ackerley, D.) 1179, 139–49 (Springer New York, 2014).
95. Dalby, P. A. Engineering enzymes for biocatalysis. Recent Pat. Biotechnol. 1, 1–9 (2007).
96. Hibbert, E. G. & Dalby, P. A. Directed evolution strategies for improved enzymatic performance. Microb. Cell Fact. 4, http://www.microbialcellfactories.com/content/4/1/ (2005).
97. Henze, M., You, D.-J., Kamerke, C., Hoffmann, N., Angkawidjaja, C., Ernst, S., Pietruszka, J., Kanaya, S. & Elling, L. Rational design of a glycosynthase by the crystal structure of β-galactosidase from Bacillus circulans (BgaC) and its use for the synthesis of N-acetyllactosamine type 1 glycan structures. J. Biotechnol. 191, 78–85 (2014).
98. Scheib, H., Pleiss, J., Stadler, P., Kovac, A., Potthoff, A. P., Haalck, L., Spener, F., Paltauf, F. & Schmid, R. D. Rational design of Rhizopus oryzae lipase with modified stereoselectivity toward triradylglycerols. Protein Eng. 11, 675–82 (1998).
99. Schulz, T., Pleiss, J. & Schmid, R. D. Stereoselectivity of Pseudomonas cepacia lipase toward secondary alcohols: a quantitative model. Protein Sci. 9, 1053–62 (2000).
100. Miyazaki, K. in Methods Mol. Biol. vol. 231 Dir. Evol. Libr. Creat. Methods Protoc. (Arnold, F. H. & Georgiou, G.) (Humana Press Inc., 2003).
101. Cheng, F., Zhu, L. & Schwaneberg, U. Directed evolution 2.0: improving and deciphering enzyme properties. Chem. Commun. 51, 9760–72 (2015).
102. Tracewell, C. A. & Arnold, F. H. Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr. Opin. Chem. Biol. 13, 3–9 (2009).
103. Reetz, M. T. Controlling the enantioselectivity of enzymes by directed evolution: practical and theoretical ramifications. Proc. Natl. Acad. Sci. USA 101, 5716–22 (2004).

Literaturverzeichnis
122
104. Darwin, C. R. The origin of species by means of natural selection. (John Murray, 1859).
105. Frauenkron-Machedjou, V. J., Fulton, A., Zhu, L., Anker, C., Bocola, M., Jaeger, K.-E. & Schwaneberg, U. Towards Understanding Directed Evolution: More than Half of All Amino Acid Positions Contribute to Ionic Liquid Resistance of Bacillus subtilis Lipase A. ChemBioChem 16, 937–45 (2015).
106. Eijsink, V. G. H., Gåseidnes, S., Borchert, T. V & van den Burg, B. Directed evolution of enzyme stability. Biomol. Eng. 22, 21–30 (2005).
107. Han, Z. L., Han, S. Y., Zheng, S. P. & Lin, Y. Enhancing thermostability of a Rhizomucor miehei lipase by engineering a disulfide bond and displaying on the yeast cell surface. Appl. Microbiol. Biotechnol. 85, 117–126 (2009).
108. Rellos, P., Pinheiro, L. & Scopes, R. K. Thermostable variants of Zymomonas mobilis alcohol dehydrogenase obtained using PCR-mediated random mutagenesis. Protein Express. Purif. 12, 61–6 (1998).
109. Stemmer, W. P. DNA shuffling by random fragmentation and reassembly: in vitro recombination for molecular evolution. Proc. Natl. Acad. Sci. USA 91, 10747–51 (1994).
110. Iffland, A., Gendreizig, S., Tafelmeyer, P. & Johnsson, K. Changing the Substrate Specificity of Cytochrome c Peroxidase Using Directed Evolution. Biochem. Biophys. Res. Commun. 286, 126–32 (2001).
111. Eckert, K. A. & Kunkel, T. A. High fidelity DNA synthesis by the Thermus aquaticus DNA polymerase. Nucleic Acids Res. 18, 3739–44 (1990).
112. Tindall, K. R. & Kunkel, T. A. Fidelity of DNA Synthesis by the Thermus aquaticus DNA Polymerase. Biochemistry 27, 6008–13 (1988).
113. Zhou, Y. H., Zhang, X. P. & Ebright, R. H. Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase. Nucleic Acids Res. 19, 6052 (1991).
114. Cadwell, R. C. & Joyce, G. F. Randomization of genes by PCR mutagenesis. Genome Res. 2, 28–33 (1992).
115. Zaccolo, M., Williams, D. M., Brown, D. M. & Gherardi, E. An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 255, 589–603 (1996).
116. Reetz, M. T. & Krebs, G. P. L. Challenges in the directed evolution of stereoselective enzymes for use in organic chemistry. C. R. Chim. 14, 811–8 (2011).
117. Cline, J. & Hogrefe, H. H. Randomize gene sequence with new PCR mutagenesis kit. Strategies 13, 157–61 (2000).

Literaturverzeichnis
123
118. Wong, T. S., Tee, K. L., Hauer, B. & Schwaneberg, U. Sequence saturation mutagenesis (SeSaM): a novel method for directed evolution. Nucleic Acids Res. 32, e26 (2004).
119. Zhao, H., Giver, L., Shao, Z., Affholter, J. A. & Arnold, F. H. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat. Biotechnol. 16, 258–61 (1998).
120. Shao, Z., Zhao, H., Giver, L. & Arnold, F. H. Random-priming in vitro recombination: an effective tool for directed evolution. Nucleic Acids Res. 26, 681–3 (1998).
121. Müller, K. M., Stebel, S. C., Knall, S., Zipf, G., Bernauer, H. S. & Arndt, K. M. Nucleotide exchange and excision technology (NExT) DNA shuffling: a robust method for DNA fragmentation and directed evolution. Nucleic Acids Res. 33, e117 (2005).
122. Ostermeier, M., Shim, J. H. & Benkovic, S. J. A combinatorial approach to hybrid enzymes independent of DNA homology. Nat. Biotechnol. 17, 1205–9 (1999).
123. Lutz, S., Ostermeier, M., Moore, G. L., Maranas, C. D. & Benkovic, S. J. Creating multiple-crossover DNA libraries independent of sequence identity. Proc. Natl. Acad. Sci. USA 98, 11248–53 (2001).
124. O’Maille, P. E., Bakhtina, M. & Tsai, M.-D. Structure-based Combinatorial Protein Engineering (SCOPE). J. Mol. Biol. 321, 677–91 (2002).
125. Zhang, J. H., Dawes, G. & Stemmer, W. P. Directed evolution of a fucosidase from a galactosidase by DNA shuffling and screening. Proc. Natl. Acad. Sci. USA 94, 4504–9 (1997).
126. Stinson, S. C. Chiral Chemistry. Chem. Eng. News 79, 45–57 (2001).
127. Ohno, M. & Otsuka, M. in Org. React. (John Wiley & Sons, Inc., 2004).
128. Guis, H.-J., Lukas, K. L., Ball, W. A., Braun, S. & Lindner, H. J. Synthese homochiraler Bausteine fur die enantioselektive Totalsynthese von Cyclopentanoiden mit Esterase- katalysierter asymmetrischer Schliisselreaktion. Liebigs Ann. Chem. 687–716 (1986).
129. Klomp, D., Maschmeyer, T., Hanefeld, U. & Peters, J. A. Mechanism of homogeneously and heterogeneously catalysed Meerwein–Ponndorf–Verley–Oppenauer reactions for the racemisation of secondary alcohols. Chem. Eur. J. 10, 2088–93 (2004).
130. De Graauw, C. F., Peters, J. A., van Bekkum, H. & Huskens, J. Meerwein-Ponndorf-Verley Reductions and Oppenauer Oxidations: An Integrated Approach. Synth. 10, 1007–17 (1994).
131. Ponndorf, W. Der reversible Austausch der Oxydationsstufen zwischen Aldehyden oder Ketonen einerseits und primaren oder sekundaren Alkoholen anderseits. Zeitschrift für angew. Chemie 39, 138–43 (1926).

Literaturverzeichnis
124
132. Meerwein, H. & Schmidt, R. Ein neues Verfahren zur Reduktion von Aldehyden und Ketonen. Ann. der Chemie 444, 221–38 (1925).
133. Raja, M. U., Ramesh, R. & Ahn, K. H. Rhodium(III) NCN pincer complexes catalyzed transfer hydrogenation of ketones. Tetrahedron Lett. 50, 7014–7 (2009).
134. Pfaller, R. & Schneider, C. Production of (S)-2-Butanol by oxidative racemate resolution. US 2007/02207529 A1 (2007).
135. Martín-Matute, B., Edin, M., Bogár, K., Kaynak, F. B. & Bäckvall, J.-E. Combined ruthenium (II) and lipase catalysis for efficient dynamic kinetic resolution of secondary alcohols. Insight into the racemization mechanism. J. Am. Chem. Soc. 127, 8817–25 (2005).
136. Woodcock, D. M., Crowther, P. J., Doherty, J., Jefferson, S., DeCruz, E., Noyer-Weidner, M., Smith, S. S., Michael, M. Z. & Graham, M. W. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 17, 3469–78 (1989).
137. Studier, F. W. & Moffatt, B. A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–30 (1986).
138. Sambrook, J., Fritsch, E. F. & Maniatis, T. in Mol. Cloning (Cold Spring Harbor, New York, 1989).
139. Tartof, K. D. & Hobbs, C. A. Improved media for growing plasmid and cosmid clones. BRL Focus 9, (1987).
140. Birnboim, H. C. & Doly, J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res 7, 1513–23 (1979).
141. Saiki, R. K., Gelfand, D. H., Stoffel, S., Scharf, S. J., Higuchi, R., Horn, G. T., Mullis, K. B. & Erlich, H. A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 239, 487–91 (1988).
142. Kunkel, T. A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA 82, 488–92 (1985).
143. Nelson, M. & McClelland, M. Use of DNA methyltransferase/endonuclease enzyme combinations for megabase mapping of chromosomes. Methods Enzymol. 216, 279–303 (1992).
144. Hanahan, D. Studies on Transformation of Escherichia coli with Plasmids. J. Mol. Biol. 166, 557–80 (1983).
145. Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–5 (1970).
146. Ogino, H. & Ishikawa, H. Enzymes which are stable in the presence of organic solvents. J. Biosci. Bioeng. 91, 109–16 (2001).

Literaturverzeichnis
125
147. Irwin, A. J. & Jones, J. B. Asymmetric syntheses via enantiotopically selective horse liver alcohol dehydrogenase catalyzed oxidations of diols containing a prochiral center. J. Am. Chem. Soc. 99, 556–61 (1977).
148. Irwin, A. J. & Jones, J. B. Regiospecific and enantioselective horse liver alcohol dehydrogenase catalyzed oxidations of some hydroxycyclopentanes. J. Am. Chem. Soc. 99, 1625–30 (1977).
149. Wolberg, M., Filho, M. V, Bode, S., Geilenkirchen, P., Feldmann, R., Liese, A., Hummel, W. & Müller, M. Chemoenzymatic synthesis of the chiral side-chain of statins: application of an alcohol dehydrogenase catalysed ketone reduction on a large scale. Bioprocess Biosys. Eng. 31, 183–91 (2008).
150. Eckstein, M., Filho, M. V, Liese, A. & Kragl, U. Use of an ionic liquid in a two-phase system to improve an alcohol dehydrogenase catalysed reduction. Chem. Commun. 1084–5 (2004).
151. Hollmann, F., Arends, I. W. C. E. & Buehler, K. Biocatalytic redox reactions for organic synthesis: nonconventional regeneration methods. ChemCatChem 2, 762–82 (2010).
152. Daußmann, T., Rosen, T. C. & Dünkelmann, P. Oxidoreductases and Hydroxynitrilase Lyases: Complementary Enzymatic Technologies for Chiral Alcohols. Eng. Life Sci. 6, 125–9 (2006).
153. Schleheck, D. & Cook, A. M. ω-oxygenation of the alkyl sidechain of linear alkylbenzenesulfonate (LAS) surfactant in Parvibaculum lavamentivorans. Arch. Microbiol. 183, 369–77 (2005).
154. Dong, W., Eichhorn, P., Radajewski, S., Schleheck, D., Denger, K., Knepper, T. P., Murrell, J. C. & Cook, A. M. Parvibaculum lavamentivorans converts linear alkylbenzenesulphonate surfactant to sulphophenylcarboxylates, α,β-unsaturated sulphophenylcarboxylates and sulphophenyldicarboxylates, which are degraded in communities. J. Appl. Microbiol. 96, 630–640 (2004).
155. Müller, M., Wolberg, M., Schubert, T. & Hummel, W. Enzyme-Catalyzed Regio- and Enantioselective Ketone Reductions. Adv. Biochem. Engin./Biotechnol. 92, 261–87 (2005).
156. Könst, P., Kara, S., Kochius, S., Holtmann, D., Arends, I. W. C. E., Ludwig, R. & Hollmann, F. Expanding the Scope of Laccase-Mediator Systems. ChemCatChem 5, 3027–32 (2013).
157. Broussy, S., Cheloha, R. W. & Berkowitz, D. B. Enantioselective, ketoreductase-based entry into pharmaceutical building blocks: ethanol as tunable nicotinamide reductant. Org. Lett. 11, 305–8 (2009).
158. Chou, C.-Y., Ko, T.-P., Wu, K.-J., Huang, K.-F., Lin, C.-H., Wong, C.-H. & Wang, A. H.-J. Modulation of substrate specificities of D-sialic acid aldolase through single mutations of Val-251. J. Biol. Chem. 286, 14057–64 (2011).

Literaturverzeichnis
126
159. Klibanov, A. M. Improving enzymes by using them in organic solvents. Nature 409, 241–6 (2001).
160. Klibanov, A. M. Why are enzymes less active in organic solvents than in water? Trends Biotechnol. 15, 97–101 (1997).
161. Yang, H., Jönsson, Å., Wehtje, E., Adlercreutz, P. & Mattiasson, B. The enantiomeric purity of alcohols formed by enzymatic reduction of ketones can be improved by optimisation of the temperature and by using a high co-substrate concentration. Biochim. Biophys. Acta 1336, 51–8 (1997).
162. Zaks, A. & Klibanov, A. M. Enzyme-catalyzed processes in organic solvents. Proc. Natl. Acad. Sci. USA 82, 3192–6 (1985).
163. Stepankova, V., Bidmanova, S., Koudelakova, T., Prokop, Z., Chaloupkova, R. & Damborsky, J. Strategies for stabilization of enzymes in organic solvents. ACS Catal. 3, 2823–36 (2013).
164. Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 42, 6406–36 (2013).
165. Rodrigues, R. C. & Fernandez-Lafuente, R. Lipase from Rhizomucor miehei as an industrial biocatalyst in chemical process. J. Mol. Catal. B Enzym. 64, 1–22 (2010).
166. Hong, S., Kim, H. S. & Kim, J. Highly Stabilized Lipase in Polyaniline Nanofibers for Surfactant- Mediated Esterification of Ibuprofen. Langmuir 911–5 (2014).
167. Kumar, D., Parshad, R. & Gupta, V. K. Application of a statistically enhanced, novel, organic solvent stable lipase from Bacillus safensis DVL-43. Int. J. Biol. Macromol. 66, 97–107 (2014).
168. László, K., Szava, A. & Simon, L. M. Stabilization of various alpha-chymotrypsin forms in aqueous-organic media by additives. J. Mol. Catal. B Enzym. 16, 141–6 (2001).
169. Adlercreutz, P. Activation of enzymes in organic media at low water activity by polyols and saccharides. Biochim. Biophys. Acta 1163, 144–8 (1993).
170. Konieczny, S., Krumm, C., Doert, D., Neufeld, K. & Tiller, J. C. Investigations on the activity of poly(2-oxazoline) enzyme conjugates dissolved in organic solvents. J. Biotechnol. 181, 55–63 (2014).
171. Schmid, R. D. Stabilized soluble enzymes. Adv. Biochem. Engin. 12, 41–118 (1979).
172. Ó’Fágáin, C. Enzyme stabilization—recent experimental progress. Enzym. Microb. Technol. 33, 137–49 (2003).
173. Bertoldi, M., Cellini, B., Laurents, D. V & Borri Voltattorni, C. Folding pathway of the pyridoxal 5’-phosphate C-S lyase MalY from Escherichia coli. Biochem. J. 389, 885–98 (2005).

Literaturverzeichnis
127
174. Van den Heuvel, R. H. H., Tahallah, N., Kamerbeek, N. M., Fraaije, M. W., van Berkel, W. J. H., Janssen, D. B. & Heck, A. J. R. Coenzyme binding during catalysis is beneficial for the stability of 4-hydroxyacetophenone monooxygenase. J. Biol. Chem. 280, 32115–21 (2005).
175. Faber, K. Biotransformations in Organic Chemistry. Book (Springer, 2011).
176. Kara, S., Spickermann, D., Weckbecker, A., Leggewie, C., Arends, I. W. C. E. & Hollmann, F. Bioreductions Catalyzed by an Alcohol Dehydrogenase in Non-aqueous Media. ChemCatChem 6, 973–6 (2014).
177. Kroutil, W., Mang, H., Edegger, K. & Faber, K. Recent advances in the biocatalytic reduction of ketones and oxidation of sec-alcohols. Curr. Opin. Chem. Biol. 8, 120–6 (2004).
178. Hollmann, F., Arends, I. W. C. E., Buehler, K., Schallmey, A. & Bühler, B. Enzyme-mediated oxidations for the chemist. Green Chem. 13, 226–65 (2011).
179. Chenault, H. K. & Whitesides, G. M. Regeneration of nicotinamide cofactors for use in organic synthesis. Appl. Biochem. Biotechnol. 14, 147–97 (1987).
180. Jakoblinnert, A., Mladenov, R., Paul, A., Sibilla, F., Schwaneberg, U., Ansorge-Schumacher, M. B. & Domínguez de María, P. Asymmetric reduction of ketones with recombinant E. coli whole cells in neat substrates. Chem. Commun. 47, 12230–2 (2011).
181. Lavandera, I., Kern, A., Resch, V., Ferreira-Silva, B., Glieder, A., Fabian, W. M. F., DeWildeman, S. & Kroutil, W. One-way biohydrogen transfer for oxidation of sec-alcohols. Org. Lett. 10, 2155–8 (2008).
182. Kara, S., Spickermann, D., Schrittwieser, J. H., Leggewie, C., van Berkel, W. J. H., Arends, I. W. C. E. & Hollmann, F. More efficient redox biocatalysis by utilising 1,4-butanediol as a ‘smart cosubstrate’. Green Chem. 15, 330–5 (2013).
183. Galletti, P., Emer, E., Gucciardo, G., Quintavalla, A., Pori, M. & Giacomini, D. Chemoenzymatic synthesis of (2S)-2-arylpropanols through a dynamic kinetic resolution of 2-arylpropanals with alcohol dehydrogenases. Org. Biomol. Chem. 8, 4117–23 (2010).
184. Spickermann, D., Kara, S., Barackov, I., Hollmann, F., Schwaneberg, U., Duenkelmann, P. & Leggewie, C. Alcohol dehydrogenase stabilization by additives under industrially relevant reaction conditions. J. Mol. Catal. B Enzym. 103, 24–8 (2014).
185. Dombkowski, A. A. Disulfide by Design: A computational method for the rational design of disulfide bonds in proteins. Bioinformatics 19, 1852–3 (2003).
186. Guex, N. & Peitsch, M. C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 18, 2714–23 (1997).

Literaturverzeichnis
128
187. Richardson, T. I., Clarke, C. A., Ying, K. Y., Bleisch, T. J., Lopez, J. E., Jones, S. A., Hughes, N. E., Muehl, B. S., Lugar, C. W., Moore, T. L., Shetler, P. K., Zink, R. W., Osborne, J. J., Montrose-Rafizadeh, C., Patel, N., Geiser, A. G., Galvin, R. J. S. & Dodge, J. A. Novel 3-aryl indoles as progesterone receptor antagonists for uterine fibroids. ACS Med. Chem. Lett. 2, 148–53 (2011).
188. Bleisch, J. B., Clarke, C. A., Dodge, J. A., Jones, S. A., Lopez, J. E., Iii, C. W. L., Muehl, B. S., Richardson, T. I., Yee, Y. K. & Yu, K.-L. Indole sulfonamide modulators of progesterone receptors. WO2007087488 A9. (2007).
189. Kuhl, A., Kolkhof, P., Heckroth, H., Schlemmer, K.-H., Flamme, I., Perez, S. G., Gielen-Haertwig, H., Grosser, R., Ergüden, J.-K. & Lang, D. 4-Chromenonyl-1,4-dihydropyridines and their use. US 2010/0022484 A1. (2010).
190. Murali, C. & Creaser, E. H. Protein engineering of alcohol dehydrogenase -1. Effects of two amino acid changes in the active site of yeast ADH-1. Protein Eng. 1, 55–7 (1986).
191. Pham, V. T. & Phillip, R. S. Effects of substrate structure and temperature on the stereospecificity of secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus. J. Am. Chem. Soc. 112, 3629–32 (1990).
192. Pham, V. T., Phillips, R. S. & Ljungdahl, L. G. Temperature-dependent enantiospecificity of secondary alcohol dehydrogenase from Thermoanaerobacter ethanolicus. J. Am. Chem. Soc. 111, 1935–1936 (1989).
193. Ke, T. & Klibanov, A. M. On enzymatic activity in organic solvents as a function of enzyme history. Biotechnol. Bioeng. 57, 746–50 (1998).
194. Klibanov, A. M. Enzymatic catalysis in anhydrous organic solvents. Trends Biochem. Sci. 14, 141–4 (1989).
195. Dai, L. & Klibanov, A. M. Striking activation of oxidative enzymes suspended in nonaqueous media. Proc. Natl. Acad. Sci. USA 96, 9475–8 (1999).
196. Ståhl, M., Jeppson-Wistrand, U., Månsson, M.-O. & Mosbach, K. Induced stereoselectivity and substrate selectivity of bio-imprinted alpha-chymotrypsin in anhydrous organic media. J. Am. Chem. Soc. 113, 9366–8 (1991).
197. Debulis, K. & Klibanov, A. M. Dramatic enhancement of enzymatic activity in organic solvents by lyoprotectants. Biotechnol. Bioeng. 41, 566–71 (1993).
198. Adolph, H. W., Maurer, P., Schneider-Bernlöhr, H., Sartorius, C. & Zeppezauer, M. Substrate specificity and stereoselectivity of horse liver alcohol dehydrogenase. Eur. J. Biochem. 201, 615–25 (1991).
199. Van Osselaer, T. A., Lemiere, G. L., Lepoivre, J. A. & Alderweireldt, F. C. Enzymatic in vitro reduction of ketones. Part 2. Study of coenzyme recycling in an ethanol-cyclohexanone system with HLADH (Horse Liver Alcohol Dehydrogenase). J. C. S. Perkin II 1181–8 (1978).

Literaturverzeichnis
129
200. Phillips, R. S. Temperature effects on stereochemistry of enzymatic reactions. Enzyme Microb. Technol. 14, 417–419 (1992).
201. Phillips, R. S. Temperature modulation of the stereochemistry of enzymatic catalysis: Prospects for exploitation. Trends Biotechnol. 14, 13–6 (1996).
202. Arrhenius, S. . Z Phys Chem 4, 226–48 (1889).
203. Henricus van’t Hoff, J. Études de dynamique chimique. (Frederik Muller & Co., 1884).
204. Iyer, P. V & Ananthanarayan, L. Enzyme stability and stabilization-aqueous and non-aqueous environment. Process Biochem. 43, 1019–32 (2008).
205. Valivety, R. H., Halling, P. J. & Macrae, A. R. Water as a competitive inhibitor of lipase-catalysed esterification in organic media. Biotechnol. Lett. 15, 1133–1138 (1993).
206. Reetz, M. T. Laboratory evolution of stereoselective enzymes: a prolific source of catalysts for asymmetric reactions. Angew. Chem. Int. Ed. 50, 138–74 (2011).
207. Matsuura, T., Miyai, K., Trakulnaleamsai, S., Yomo, T., Shima, Y., Miki, S., Yamamoto, K. & Urabe, I. Evolutionary molecular engineering by random elongation mutagenesis. Nat. Biotechnol. 17, 58–61 (1999).
208. Mateo, C., Palomo, J. M., Fernandez-Lorente, G., Guisan, J. M. & Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzym. Microb. Technol. 40, 1451–63 (2007).
209. Yadav, J. K. & Prakash, V. Stabilization of alpha-amylase, the key enzyme in carbohydrates properties alterations, at low pH. Int. J. Food Prop. 14, 1182–96 (2011).
210. Filatova, L. Y., Becker, S. C., Donovan, D. M., Gladilin, A. K. & Klyachko, N. L. LysK, the enzyme lysing Staphylococcus aureus cells: Specific kinetic features and approaches towards stabilization. Biochimie 92, 507–13 (2010).
211. Morgan, J. A. & Clark, D. S. Salt-activation of nonhydrolase enzymes for use in organic solvents. Biotechnol. Bioeng. 85, 456–9 (2004).
212. Andersson, M. M. & Hatti-Kaul, R. Protein stabilising effect of polyethyleneimine. J Biotechnol 72, 21–31 (1999).
213. Rocha, J. M. S., Gil, M. H. & Garcia, F. A. P. Effects of additives on the activity of a covalently immobilised lipase in organic media. J. Biotechnol. 66, 61–7 (1998).
214. Triantafyllou, A. Ö., Wehtje, E., Adlercreutz, P. & Mattiasson, B. How do additives affect enzyme activity and stability in nonaqueous media? Biotechnol. Bioeng. 54, 67–76 (1997).
215. Klibanov, A. in Adv. Appl. Microbiol. (Laskin, A. I.) 29, 1–28 (Academic Press, 1983).

Literaturverzeichnis
130
216. Qu, Y., Bolen, C. L. & Bolen, D. W. Osmolyte-driven contraction of a random coil protein. Proc. Natl. Acad. Sci. USA 95, 9268–73 (1998).
217. Kaushik, J. K. & Bhat, R. Thermal Stability of Proteins in Aqueous Polyol Solutions : Role of the Surface Tension of Water in the Stabilizing Effect of Polyols. J. Phys. Chem. B 102, 7058–66 (1998).
218. Combes, D., Yoovidhya, T., Girbal, E., Willemot, R.-M. & Monsan, P. Mechanism of Enzyme Stabilization. Ann. N. Y. Acad. Sci. 501, 59–62 (1987).
219. Mozhaev, V. V, Sergeeva, M. V, Belova, A. B. & Khmelnitsky, Y. L. Multipoint attachment to a support protects enzyme from inactivation by organic solvents: alpha-Chymotrypsin in aqueous solutions of alcohols and diols. Biotechnol. Bioeng. 35, 653–9 (1990).
220. Raza, S., Fransson, L. & Hult, K. Enantioselectivity in Candida antarctica lipase B: a molecular dynamics study. Protein Sci. 10, 329–38 (2001).
221. Wang, Y., Wang, R., Li, Q., Zhang, Z. & Feng, Y. Kinetic resolution of rac-alkyl alcohols via lipase-catalyzed enantioselective acylation using succinic anhydride as acylating agent. J. Mol. Catal. B Enzym. 56, 142–45 (2009).
222. Overbeeke, P. L., Ottosson, J., Hult, K., Jongejan, J. & Duine, J. The Temperature Dependence of Enzymatic Kinetic Resolutions Reveals the Relative Contribution of Enthalpy and Entropy to Enzymatic Enantioselectivity. Biocatal. Biotransform. 17, 61–79 (1999).
223. Schroer, K., Tacha, E., Bringer-Meyer, S., Hummel, W., Daußmann, T., Pfaller, R. & Lütz, S. Continous asymmetric ketone reduction processes with tailor-made microorganisms. J. Biotechnol. 131, S95–6 (2007).
224. Wolfson, A., Dlugy, C., Tavor, D., Blumenfeld, J. & Shotland, Y. Baker’s yeast catalyzed asymmetric reduction in glycerol. Tetrahedron Asymmetry 17, 2043–5 (2006).
225. Fonseca, A. M., Monte, F. J. Q., de Oliveira, M. D. C. F., de Mattos, M. C., Cordell, G., Braz-Filho, R. & Lemos, T. L. G. Coconut water (Cocos nucifera L.)-A new biocatalyst system for organic synthesis. J. Mol. Catal. B Enzym. 57, 78–82 (2009).
226. Mozhaev, V. V, Khmelnitsky, Y. L., Sergeeva, M. V, Belova, A. B., Klyachko, N. L., Levashov, A. V & Martinek, K. Catalytic activity and denaturation of enzymes in water/organic cosolvent mixtures. Eur. J. Biochem. 184, 597–602 (1989).
227. Kuntz Jr, I. D. & Kauzmann, W. in Adv. Protein Chem. (Anfinsen, C. B., Edsall, J. T., Richards, F. M. & Eisenber, B. T.) 28, 239–345 (Academic Press, 1974).
228. Rupley, J. A. & Careri, G. Protein Hydration and Function. Adv. Protein Chem. 41, 37–172 (1991).
229. Gregory, R. B. Protein-solvent Interactions. Recl. des Trav. Chim. des Pays-Bas 115, 305 (1996).

Literaturverzeichnis
131
230. Griebenow, K. & Klibanov, A. M. On protein denaturation in aqueous-organic mixtures but not in pure organic solvents. J. Am. Chem. Soc. 118, 11695–700 (1996).
231. Hansch, C., Leo, A. & Hoekman, D. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology. (American Chemical Society, 1995).
232. Kara, S., Spickermann, D., Schrittwieser, J. H., Weckbecker, A., Leggewie, C., Arends, I. W. C. E. & Hollmann, F. Access to lactone building blocks via horse liver alcohol dehydrogenase-catalyzed oxidative lactonization. ACS Catal. 3, 2436–9 (2013).
233. Sih, C. J. & Chen, C.-S. Mikrobielle asymmetrische Katalyse - enantioselektive Reduktion von Ketonen. Angew. Chem. 96, 556–65 (1984).
234. Webpage. SeSaM-Biotech GmbH. at <www.sesam-biotech.com>
235. Reetz, M. T., Wilensek, S., Zha, D. & Jaeger, K.-E. Directed Evolution of an Enantioselective Enzyme through Combinatorial Multiple-Cassette Mutagenesis. Angew. Chem. Int. Ed. 40, 3589–91 (2001).
236. Martínez, R. & Schwaneberg, U. A roadmap to directed enzyme evolution and screening systems for biotechnological applications. Biol. Res. 46, 395–405 (2013).
237. Wong, T. S., Zhurina, D. & Schwaneberg, U. The diversity challenge in directed protein evolution. Comb. Chem. High Throughput Screen. 9, 271–88 (2006).
238. Arnold, F. H. Combinatorial and computational challenges for biocatalyst design. Nature 409, 253–7 (2001).
239. Silva, C., Da, S., Silva, N., Matamá, T., Araújo, R., Martins, M., Chen, S., Chen, J., Wu, J., Casal, M. & Cavaco-Paulo, A. Engineered Thermobifida fusca cutinase with increased activity on polyester substrates. Biotechnol. J. 6, 1230–9 (2011).
240. Araújo, R., Silva, C., O’Neill, A., Micaelo, N., Guebitz, G., Soares, C. M., Casal, M. & Cavaco-Paulo, A. Tailoring cutinase activity towards polyethylene terephthalate and polyamide 6,6 fibers. J. Bacteriol. 128, 849–57 (2007).
241. Shivange, A. V, Dennig, A. & Schwaneberg, U. Multi-site saturation by OmniChange yields a pH- and thermally improved phytase. J. Biotechnol. 170, 68–72 (2014).
242. Reetz, M. T. & Wu, S. Laboratory evolution of robust and enantioselective Baeyer−Villiger monooxygenases for asymmetric catalysis. J. Am. Chem. Soc. 131, 15424–32 (2009).
243. Morel, N., Bon, S., Greenblatt, H. M., van Belle, D., Wodak, S. J., Sussman, J. L., Massoulié, J. & Silman, I. Effect of mutations within the peripheral anionic site on the stability of acetylcholinesterase. Mol. Pharmacol. 55, 982–92 (1999).
244. Floor, R. J., Wijma, H. J., Colpa, D. I., Ramos-Silva, A., Jekel, P. A., Szymański, W., Feringa, B. L., Marrink, S. J. & Janssen, D. B. Computational Library Design for Increasing Haloalkane Dehalogenase Stability. ChemBioChem 15, 1660–72 (2014).

Literaturverzeichnis
132
245. Ansorge-Schumacher, M. B., Slusarczyk, H., Schümers, J. & Hirtz, D. Directed evolution of formate dehydrogenase from Candida boidinii for improved stability during entrapment in polyacrylamide. FEBS J. 273, 3938–45 (2006).
246. Reetz, M. T., Wang, L.-W. & Bocola, M. Directed Evolution of Enantioselective Enzymes: Iterative Cycles of CASTing for Probing Protein-Sequence Space. Angew. Chem. 118, 1258–63 (2006).
247. Robinson, C. R. & Sauer, R. T. Striking stabilization of Arc repressor by an engineered disulfide bond. Biochemistry 39, 12494–502 (2000).
248. Yu, X. W., Tan, N. J., Xiao, R. & Xu, Y. Engineering a Disulfide Bond in the Lid Hinge Region of Rhizopus chinensis Lipase: Increased Thermostability and Altered Acyl Chain Length Specificity. PLoS One 7, 1–7 (2012).
249. Prodanovic, R., Ostafe, R., Scacioc, A. & Schwaneberg, U. Ultrahigh-throughput screening system for directed glucose oxidase evolution in yeast cells. Comb. Chem. High Throughput Screen. 14, 55–60 (2011).
250. Li, Q.-S., Schwaneberg, U., Fischer, M., Schmitt, J., Pleiss, J., Lutz-Wahl, S. & Schmid, R. D. Rational evolution of a medium chain-specific cytochrome P-450 BM-3 variant. Biochim. Biophys. Acta 1545, 114–21 (2001).
251. Wang, T.-W., Zhu, H., Ma, X.-Y., Zhang, T., Ma, Y.-S. & Wei, D.-Z. Methods for Mutant Library Construction Casting a Wider Net. Mol. Biotechnol. 34, 55–68 (2006).
252. Bornscheuer, U. T. Trends and challenges in enzyme technology. Adv. Biochem. Engin./Biotechnol. 100, 181–203 (2005).
253. Benkert, P., Biasini, M. & Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–50 (2011).
254. Petersen, M. T., Jonson, P. H. & Petersen, S. B. Amino acid neighbours and detailed conformational analysis of cysteines in proteins. Protein Eng. 12, 535–48 (1999).
255. Fiorentino, G., Cannio, R., Rossi, M. & Bartolucci, S. Decreasing the stability and changing the substrate specificity of the Bacillus stearothermophilus alcohol dehydrogenase by single amino acid replacements. Protein Eng. 11, 925–30 (1998).
256. Goihberg, E., Dym, O., Tel-Or, S., Levin, I., Peretz, M. & Burstein, Y. A Single Proline Substitution is Critical for the Thermostabilization of Clostridium beijerinckii Alcohol Dehydrogenase. Proteins: Struct., Funct., Bioinf. 204, 196–204 (2007).
257. Annaluru, N., Watanabe, S., Pack, S. P., Saleh, A. A., Kodaki, T. & Makino, K. Thermostabilization of Pichia stipitis xylitol dehydrogenase by mutation of structural zinc-binding loop. J. Biotechnol. 129, 717–22 (2007).
258. Pennacchio, A., Esposito, L., Zagari, A., Rossi, M. & Raia, C. A. Role of tryptophan 95 in substrate specificity and structural stability of Sulfolobus solfataricus alcohol dehydrogenase. Extremophiles 13, 751–61 (2009).

Literaturverzeichnis
133
259. Bogin, O., Levin, I., Hacham, Y., Tel-or, S., Peretz, M., Frolow, F. & Burstein, Y. Structural basis for the enhanced thermal stability of alcohol dehydrogenase mutants from the mesophilic bacterium Clostridium beijerinckii: contribution of salt bridging. Protein Sci. 11, 2561–74 (2002).
260. Bogin, O., Peretz, M., Hacham, Y., Korkhin, Y., Frolow, F., Kalb, A. J. & Burstein, Y. Enhanced thermal stability of Clostridium beijerinckii alcohol dehydrogenase after strategic substitution of amino acid residues with prolines from the homologous thermophilic Thermoanaerobacter brockii alcohol dehydrogenase. Protein Sci. 7, 1156–63 (1998).
261. De Bolle, X., Vinals, C., Prozzi, D., Paquet, J.-Y., Leplae, R., Depiereux, E., Vandenhaute, J. & Feytmans, E. Identification of residues potentially involved in the interactions between subunits in yeast alcohol dehydrogenases. Eur. J. Biochem. 231, 214–9 (1995).
262. Xie, Y., An, J., Yang, G., Wu, G., Zhang, Y., Cui, L. & Feng, Y. Enhanced Enzyme Kinetic Stability by Increasing Rigidity within the Active Site. J. Biol. Chem. 289, 7994–8006 (2014).
263. Hamnevik, E., Blikstad, C., Norrehed, S. & Widersten, M. Kinetic characterization of Rhodococcus ruber DSM 44541 alcohol dehydrogenase A. J. Mol. Catal. B Enzym. 99, 68–78 (2014).
264. Reetz, M. T., Bocola, M., Carballeira, J. D., Zha, D. & Vogel, A. Expanding the range of substrate acceptance of enzymes: combinatorial active-site saturation test. Angew. Chem. Int. Ed. 44, 4192–6 (2005).
265. Degering, C. Optimierung heterologer Proteinsekretion in Bacillus unter Verwendung wirtsfremder und künstlicher Signalpeptide. PhD-Thesis - Düsseld. Univ. (2010). at <http://docserv.uni-duesseldorf.de/servlets/DocumentServlet?id=18434>
266. Paetzel, M., Dalbey, R. E. & Strynadka, N. C. Crystal structure of a bacterial signal peptidase in complex with a beta-lactam inhibitor. Nature 396, 186–90 (1998).

Literaturverzeichnis
134

Anhang
135
Anhang
Hiermit versichere ich, dass ich die vorliegende Arbeit selbstständig verfasst und keine
anderen als die angegebenen Quellen und Hilfsmittel benutzt habe, dass alle Stellen der
Arbeit, die wörtlich oder sinngemäß aus anderen Quellen übernommen wurden, als solche
kenntlich gemacht sind und dass die Arbeit in gleicher oder ähnlicher Form noch keiner
Prüfungsbehörde vorgelegt wurde.
Aachen, den
Dominik Spickermann
Recommended

















