
s
Dra. Muriel
TUMORES SÓLIDOS INFANTILES Dra. M. MURIEL

Características de los tumores infantiles
Las células tumorales son embrionarias o sarcomas
Las localizaciones anatómicas son más profundas, los diagnósticos son accidentales (palpación de masa) o tardíos (evolucionados)

¿Por qué se produce?
Todas las células del organismo están sometidas a un complejo control de
división → diferenciación → muerte
↓ Célula cancerosa: Crecimiento indefinido → tumor Invasión tisular → metástasis

Tumores sólidos más frecuentes en niños
T.del SNC ( 15-25%) Neuroblastoma (7-10%) T. de Wilms (6%) Retinoblastoma (3%) Sarcomas:
Rabdomiosarcoma (5%) Osteosarcoma (4%) Sarcoma de Ewing (3%)

NEUROBLASTOMA

Neuroblastoma
T. Sólido extracraneal más frecuente en la infancia tiene su origen en la cresta neural (ganglios simpáticos pararaquideos y suprarrenales) Asienta sobre células embrionarias que pierden su diferenciación y continuan su división produciendo neuroblastos inmaduros T. maduros y benignos: Ganglioneuroma y
Feocromocitoma T. inmaduro y malignos: Neuroblastoma y
ganglioneuroblastoma

Neuroblastoma Epidemiologia
Frecuencia: 7-10% de tumores infantiles Incidencia: 10 casos / millón niños/ año Edad (al diagnóstico): < 5 años (75%) Alta tasa de remisiones espontáneas en los niños menores de un año

Neuroblastoma Localización
Localización Abdominal :75% Torácica: en mediastino posterior, 20%
En el periodo embrionario las simpatogonias emigran desde la cresta neural a lo que serán en el feto los ganglios simpáticos y la médula adrenal, por éste motivo los tumores derivados de ellas pueden asentar en las glándulas suprarrenales o bien en los ganglios simpáticos a lo largo de todo el eje espinal desde el cuello hasta el sacro

Neuroblastoma Clínica (I)
Manifestación más frecuente: Masa abdominal indolora, suele rebasar la línea media
Localización cervico-torácica: Pocos síntomas, compresión raquídea o VCS, Sindrome Claude-Bernard-Horner
Extendido: S. constitucional Nódulos azulados, frecuente en lactante Ojos de mapache: Infiltración retrobulbar

Neuroblastoma Clinica (II)
Area paraespinal: Los pacientes con NB en reloj de arena que se extienden al espacio
intramedular y comprimen medula, pueden referir dolor de espalda, cojera, hipotonía, arreflexia, atrofia muscular, paraplejia, escoliosis o alteración de esfínteres
En la piel: Las metastasis subcutaneas (estadio 4S) se manifiestan como
nodulos duros, no dolorosos, azulados, frecuentes en lactantes Síntomas paraneoplásicos Al ser tumores productores de catecolaminas pueden presentar
sofocos, cefaleas, palpitaciones, diarrea con fallo de medro asociada a distensión abdominal, hipertensión por aumento de renina en los que existe compresión renovascular. También pueden producir Parathormona y producir hipercalcemia
Puede secretar péptido intestinal vasoactivo dando lugar a diarrea acuosa intratable.

Neuroblastoma Clinica (III)
Sindrome Opsocerebelomioclónico (2%)
Consiste en descargas bruscas de movimientos rápidos involuntarios de los ojos en todas direcciones de la mirada que no fija (Opsosclonus). Asociadas a ellas presenta también mioclonias de los músculos del tronco y de las piernas. Estos síntomas son paraneoplásicos y se pueden resolver o no con la resección tumoral. Van asociadas a buen pronóstico vital, pero la mejoría puede ser lenta y parcial; con frecuencia es necesario tratar los síntomas

Neuroblastoma Clínica
Sd Claude Bernard Horner Neuroblastoma toracico
Ojos de mapache
Neuroblastoma torácico
Nódulos azulados

Forma 4S del lactante
Tumor primario localizado
Diseminacion a higado, piel y MO < 10% de cls tumorales
Niños < 1 año de edad MIBG negativa en MO

Neuroblastoma Diagnóstico
Enolasa neuronal especifica elevada Ac. VM, Ac HV, catecolaminas, dopamina elevadas en orina. Su
grado de elevación esta asociada a supervivencia acortada Tirosin-hidroxilasa elevada (marcador de. enfermedad residual y
recidivas muy precoz.) Aumento LDH y ferritina Rx tórax: masa mediastino posterior, masa retrocardiaca-
abdominal en reloj de arena Ecografía/ RMN Gammagrafia con MIBG Gammagrafia con Tc Biopsia-reseccion quirúrgica: histología, (N-Myc, citogenética,
Índice de ADN) Medula Osea bicrestal

Neuroblastoma Diagnóstico imagen (I)
Ecografia NB suprarrenal
Rx/TAC - NB torácico RM- NB paravertebral

Neuroblastoma Diagnóstico imagen (II)
a) Escintigrafia con tc99 que muestra una captación difusa en las metafisis de fémures y tibias
b) Ganmagrafia con MIBG de las extremidades inferiores que demuestra multiples areas de aumento de captación del contraste en fémures y tibias
MIBG:Método muy sensible, al diagnóstico. Positiva en el 95% de los casos. Sirve también para el seguimiento

Imagen retrocardiaca. NB toracoabdominal
Gammagrafia con MIBG
Neuroblastoma Diagnóstico imagen (III)
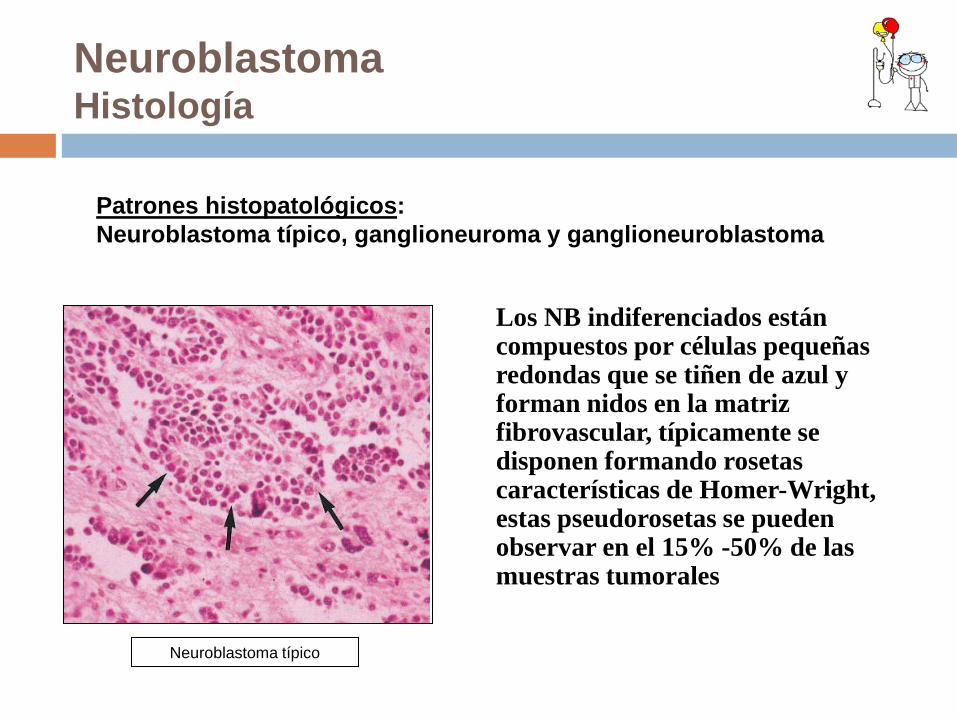
Neuroblastoma Histología
Los NB indiferenciados están compuestos por células pequeñas redondas que se tiñen de azul y forman nidos en la matriz fibrovascular, típicamente se disponen formando rosetas características de Homer-Wright, estas pseudorosetas se pueden observar en el 15% -50% de las muestras tumorales
Patrones histopatológicos: Neuroblastoma típico, ganglioneuroma y ganglioneuroblastoma
Neuroblastoma típico

Ganglioneuroma Histología
Tumor completamente diferenciado compuesto de células ganglionares maduras, estroma y células de Schwan

Ganglioneuroblastoma Histologia
En el GNB se encuentra todo el espectro de diferenciación celular entre el NB y el GN.
Se subclasifica en difuso Nodular (peor
pronóstico)
Tejido ganglionar maduro y estroma swaniano (Flecha curva) y células ganglionares maduras con abundante citoplasma (Flecha recta)en un GN.

Neuroblastoma Biología Molecular
• Amplificación del gen N-Myc (NMA). Se asocia a mal pronóstico por rápida progresión tumoral • Deleccion del 1p. Se asocia casi siempre con NMA. Si presentacion aislada mayor riesgo recidiva local • Alteraciones de 11q. Se encuentra en niños mayores y no se asocia a NMA. Pronóstico desfavorable • Índice de DNA. Hipodiploidia mal pronóstico.

International Neuroblastoma Staging System
Estadio 1. Tumor localizado con excisión macroscópica completa, con enfermedad residual microscópica o sin esta; ganglios linfáticos ipsilaterales microscópicamente negativos para el tumor (los ganglios adheridos al tumor primario y extirpados pueden ser positivos)
Estadioa 2ª.Tumor localizado sin extirpación macroscópica completa; ganglios linfáticos
ipsilaterales negativos microscópicamente para el tumor Estadio 2B.Tumor localizado con extirpación macroscópica completa o sin esta; ganglios linfáticos
ipsilaterales no adherentes positivos para el tumor. Los ganglios linfaticos contralaterales deben ser negativos microscopicamente.
Estadio3.Tumor irresecable unilateral, infiltrante que pasa la línea media , con afectación de los
ganglios linfáticos regionales o sin ésta, o tumor unilateral localizado con compromiso de los ganglios linfáticos contralaterales, o tumor en la línea media con extensión infiltrativa bilateral (irresecable) o por afectación de ganglios linfáticos. La línea media esta determinada por la columna vertebral. Los tumores que se originan en un lado y cruzan la línea media deben infiltrarse sobre esta o hacia el lado opuesto de la columna vertebral.
Estadio 4. Todo tumor primario diseminado a ganglios linfáticos distales, huesos, médula ósea,
hígado o piel a excepción del definido como estadio 4s Estadio4s.Tumor primario localizado, como se define para el estadio 1, 2Ay 2B con diseminación
limitada a la piel, hígado y/o médula ósea circunscrito a lactantes menores de 12meses de edad. La MIBG debe ser negativa para la MO y el nº de células tumorales debe ser < 10% en biopsia o aspirado de MO

Neuroblastoma Grupo pronóstico y tratamiento
Grupo de Bajo Riesgo: Niños con NB, estadio 1-2, cualquier edad y estadio 4-S sin
amplificación gen N-Myc Tratamiento: Cirugía Si hepatomegalia y/o compresión medular quimioterapia
Grupo Riesgo Intermedio: Niños con NB, estadio 3, sin amplificación gen N-Myc, mayores 18 meses y estadio 4 sin amplificación N-Myc en lactantes.
Tratamiento: Cirugía y QT intensidad moderada
Grupo Alto Riesgo: Niños mayores 18 meses en estadio 4 y cualquier estadio con
amplificación N-Myc, excepto estadio 1. Es el grupo mas frecuente
Tratamiento: QT inducción (Cojet) + Cirugia + Megaterapia (trasplante autólogo) + RT local + acido 13 cisretinoico + Ac monoclonales antigangliosidos.
SUPERVIVENCIA
Bajo riesgo 98%
Riesgo intermedio
90-95%
Alto riesgo 40-50%
Estadio IV-S >90%
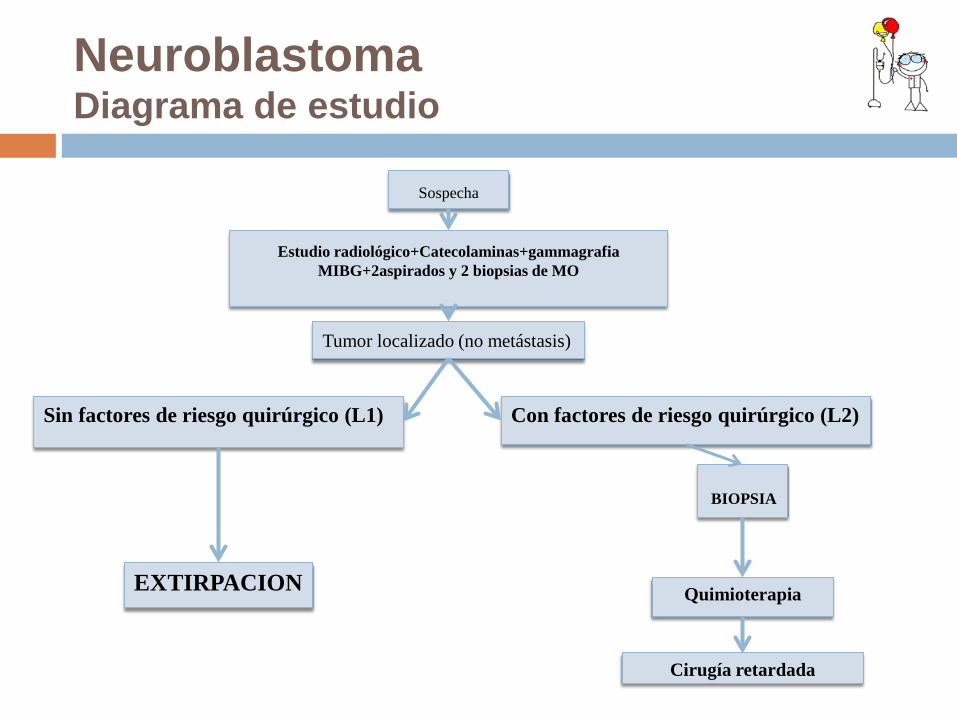
Neuroblastoma Diagrama de estudio
Sin factores de riesgo quirúrgico (L1) Con factores de riesgo quirúrgico (L2)
BIOPSIA
Quimioterapia
Cirugía retardada
EXTIRPACION
Sospecha
Estudio radiológico+Catecolaminas+gammagrafia MIBG+2aspirados y 2 biopsias de MO
Tumor localizado (no metástasis)

Tumor de Wilms

T. de Wilms (Nefroblastoma)
EPIDEMIOLOGIA: Tumor renal más frecuente en la infancia Frecuencia 6% cancer infantil Incidencia: 8-10 casos / millon niños <15 años /año Pico incidencia: Entre 3-4 años Igual ambos sexos
Los nefroblastomas son tumores derivados del blastoma renal embrionario

T. de Wilms Tipo I:
Presencia de restos nefrogénicos intralobares Edad presentación precoz Histologia favorable. Predominio de estroma Anomalías genito-urinarias: WAGR y Denys-Drash Aniridia
T. de Wilms Tipo II: Presencia de restos nefrogénicos perilobares Edad presentación tardía Histologia con anaplasia o favorable con blastema Síndromes con gigantismo: Beckwith-Wiedeman /hemihipertrofias
T. de Wilms Biología molecular
Causado por alteración de genes responsables desarrollo genito-urinario Las alteraciones moleculares que producen inactivación del gen WT1 o la perdida de marcaje del gen WT2 pueden provocar la persistencia de restos nefrogénicos que se pueden transformar en tumor de Wilms

Tumor Wilms Aniridia Malformación Genitourinaria Retraso mental
Sindromes asociados T. Wilms I Sindrome WAGR

Sindromes asociados T. Wilms I Malformacion Denys-Drash
Pseudohermafroditismo Enfermedad renal degenerativa Tumor Wilms

Aniridia

Sindromes asociados T. Wilms II Síndrome de Beckwith-Wiedeman
Exoglosia Gigantismo Onfalocele Hemihipertrofia Visceromegalias Tumor Wilms

T. de Wilms Clínica
Masa abdominal palpable, indolora, lisa, no suele pasar la línea media
Dolor abdominal, vómitos Hematuria macroscopica, alteraciones sedimento
urinario Hipertensión (60%). ↑ actividad renina Fiebre, hipercalcemia, Poliglobulia Al ser un tumor retroperitoneal no suele causar
obstrucción intestinal

T. de Wilms Clínica
Masa abdominal

T. de Wilms Diagnóstico
Estudios radiológicos objetivos: Confirmar la presencia de tumor intrarrenal Orientar al cirujano en tratamiento Evaluar riñón contralateral y presencia de invasión vascular por el tumor
Estudios a realizar: Ecografía abdominal con doppler TC abdominal RM abdominal Radiografía tórax /TC tórax

Efecto masa
Pielografia: Cálices distorsionados
Ecografia: Masa heterogénica e hiperecogénica renal
T. De Wilms Diagnóstico

T. De Wilms Diagnóstico
TC contraste:
Tumor riñón izdo., menos frecuentes las calcificaciones y zonas de hemorragia que en el neuroblastoma
TC: T. Wilms en RD

T. De Wilms Diagnóstico
RM: Corte coronal T Wilms dcho: desplaza aorta, hígado y cava inferior
RM: Corte axial T Wilms dcho: rebasa línea media

TC: Mtx pulmonares t. Wilms
T. De Wilms Diagnóstico
Mx de T. de Wilms (numular con bulla alrededor

Cuadros histológicos desfavorables: Anaplasia T. rabdoide Sarcoma de cls claras
Metástasis locales: por invasión de la
cápsula renal y por vía linfática a los ganglios regionales
Metástasis distales: Hematógenas. pulmonares 85% y hepáticas 15%
La histologia presenta un patrón compuesto por cls epiteliales blastema y elementos estromales. Aproximadamente 90% de todos tienen una histologia favorable
T. De Wilms Anatomia Patológica

T. De Wilms Estadiaje
Estadio I. Tumor limitado al riñón y completamente resecado, capsula intacta Estadio II Tumor completamente resecado con extension a la capsula o seno
renal Estadio III Tumor residual tras cirugía confinado al abdomen incluyendo márgenes
positivos Ganglios positivos locoregionales Infiltración peritoneal o implantes Rotura tumoral operatoria o resección en más de una pieza Estadio IV Metástasis hematógenas (pulmón, hígado huesos o ganglios a distancia) Estadio V Tumores renales bilaterales

Quimioterapia preoperatoria reductora Extirpación quirúrgica + QT postoperatoria +/- RT Pronóstico: Supervivencia largo plazo: 90%
DD: Nefroma mesoblástico: Tumor renal congénito más frecuente Predominio en varones. Aparece antes del año de edad (promedio a los 2 meses) . Se relaciona con polihidrapnios en embarazo. Leiomioma o leiomiosarcoma de bajo grado con nefronas
atrapadas. Puede producir renina. Tratamiento: resección quirúrgica
T. De Wilms Tratamiento
PROTOCOLO SIOP

Retinoblastoma

Retinoblastoma Concepto y epidemiologia
• Es un tumor que se origina en la célula embrionaria de la retina. • Se debe a defectos del gen del retinoblastoma (RB1) • Tumor intraocular mas frecuente en pediatría • Frecuencia: 3% de las neoplasia infantiles • Incidencia: 1 / 18.000 RN / año

Retinoblastoma Formas clínicas
RETINOBLASTOMA ESPORADICO (60%)
RETINOBLASTOMA HEREDITARIO (40%)
Número tumores Tumor único Múltiples tumores
Lateralidad Unilateral Uní (15%) o bilateral
Antecedentes familiares
No 40%
Biología molecular Perdida funcional de ambos alelos de RB1
Perdida Primer alelo En una célula de la retina (somático)
En una célula de línea germinal
Perdida 2º alelo Somático Somático
Edad media aparición 24 meses 12 meses
Riesgo 2ª neoplasia No Elevado

Retinoblastoma Histologia

Retinoblastoma Manifestaciones clinicas
Leucocoria (60 %) Reflejo (“ojo gato”) Estrabismo (20 %)
Ojo rojo poco frecuente) Heterocromia iris (poco frecuente)
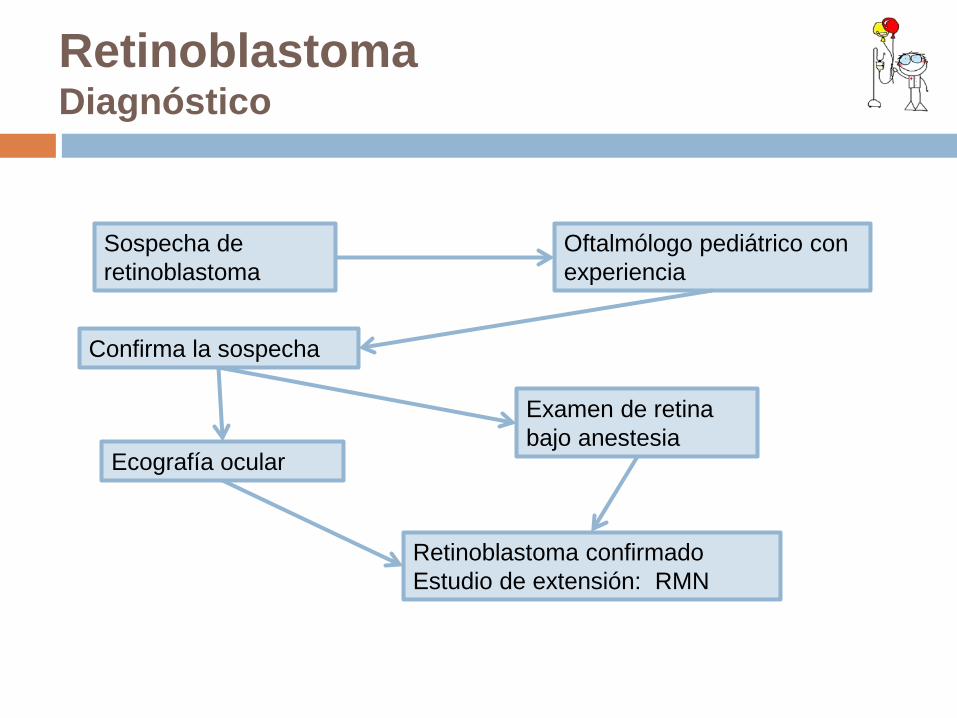
Retinoblastoma Diagnóstico
Sospecha de retinoblastoma
Oftalmólogo pediátrico con experiencia
Examen de retina bajo anestesia
Confirma la sospecha
Ecografía ocular
Retinoblastoma confirmado Estudio de extensión: RMN
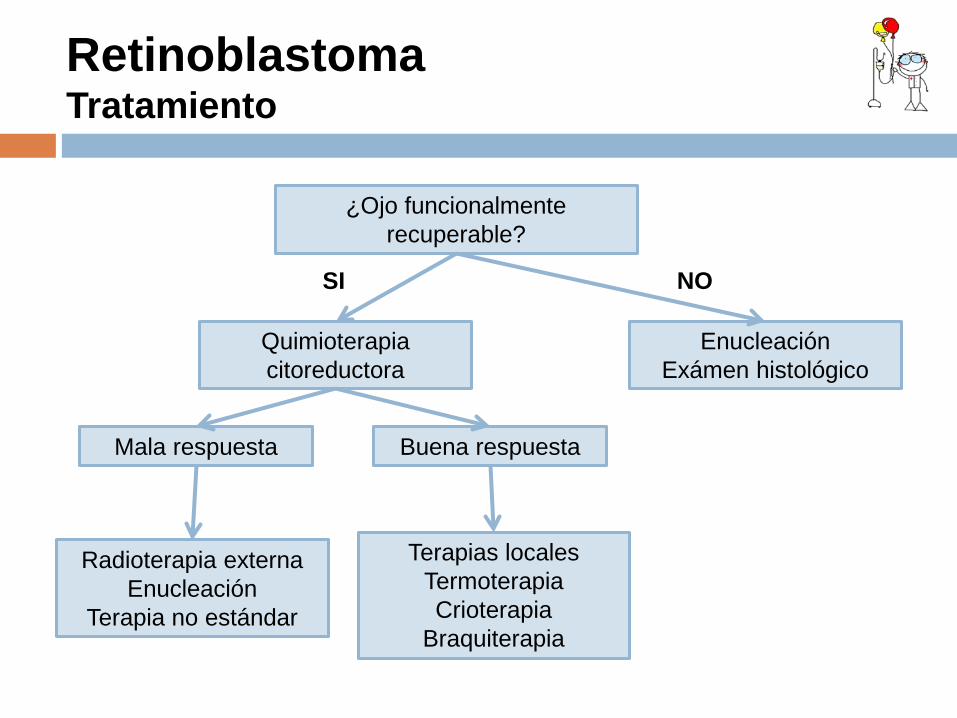
Retinoblastoma Tratamiento
¿Ojo funcionalmente recuperable?
Terapias locales Termoterapia Crioterapia
Braquiterapia
Radioterapia externa Enucleación
Terapia no estándar
Buena respuesta Mala respuesta
Enucleación Exámen histológico
Quimioterapia citoreductora
SI NO

RABDOMIOSARCOMA

Rabdomiosarcoma Concepto y epidemiologia
• Tumor maligno embrionario derivado de células mesenquimales (rabdomioblastos) cuya diferenciación a células de musculo estriado maduro es incompleta
• Puede localizarse en cualquier parte del organismo, incluyendo lugares donde no hay musculo esquelético
• Tumor de tejidos blandos mas frecuente en el niño
• Frecuencia: 5 % del cáncer pediátrico
• Asociación a anomalías congénitas del SNC y genitourinarias
• Mayor incidencia en: Síndrome Wideman Bekwith
Síndrome Gorlin Neurofibromatosis tipo I Síndrome Li-Fraumeni Síndrome alcohólico fetal

Rabdomiosarcoma Histologia
•Buen pronóstico: RMS botroide (aspecto racimo uvas, crecimiento espacios abiertos. 6%
RMS células fusiformes
•Pronóstico intermedio: RMS embrionario. Las células asemejan músculo embrionario. 60%.
•Mal pronóstico: RMS alveolar. Células forman pequeños espacios huecos o alveolos 20%
•Tumor de células pequeñas, redondas y azules
•Positividad para marcadores inmuno-histoquimicos musculares: Miogenina, MyoD, actina musculoespecifica, mioglobina y/o desmina
RMS embrionario
RMS alveolar

Rabdomiosarcoma Biología molecular
RMS embrionario: Perdida heterocigosidad del cromosoma 11 Sobreexpresion del gen IGF-2 RMS alveolar: Traslocacion (2: 13) en el 55-85 % casos Traslocacion (1:13) en el 5- 15 % casos Como consecuencia de estas traslocaciones se generan proteínas de fusión que tienen efecto oncogénico y que inhiben la diferenciación miogénica y la apoptosis

Rabdomiosarcoma Clinica
El RMS puede presentarse en cualquier región anatómica. La clínica depende de su localización.
REGION ANATOMICA Y FRECUENCIA LOCALIZACION MANIFESTACION CLINICA
Genito urinaria 25%
•Vejiga y próstata 15% •Vagina y útero 10%
Obstrucción urinaria/hematuria Sangrado vaginal/T.vulva
Cabeza y cuello 25%
•Parameningeos 15%: senos paranasales, fosa nasal y fosa pterigomaxilar •No parameningeos 10%
Epistaxis /masa en fosa nasal. Masa palpable
Extremidades 20 %
•Extremidades Masa palpable
Orbita 10%
•Orbita Exoftalmos
Otras localizaciones 20%
•Vía biliar •Pared torácica
Masa abdominal + ictericia Masa torácica /Derrame pleural
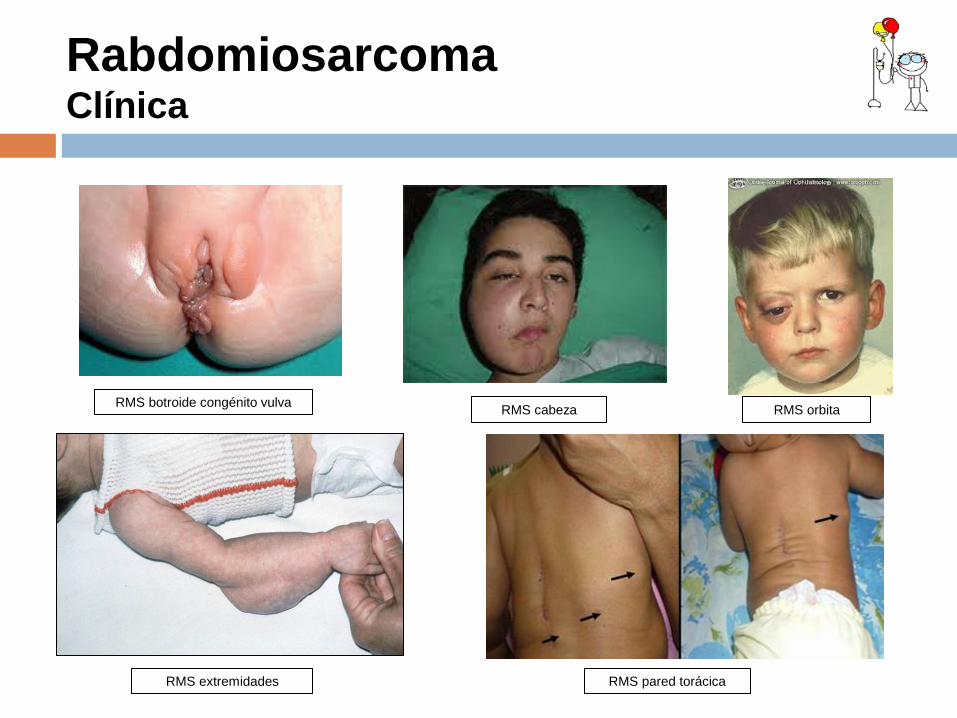
Rabdomiosarcoma Clínica
RMS botroide congénito vulva RMS cabeza RMS orbita
RMS extremidades RMS pared torácica

Rabdomiosarcoma Estadiaje. Clasificacion Clinica IRS
Grupo I: Resección completa, márgenes negativos Grupo II: Resección macroscópica, evidencia extensión locoregional
Grupo IIa: Resección completa, márgenes positivos
Grupo IIb: Resección completa, márgenes negativos, ganglios linfáticos positivos
Grupo IIc: Resección completa, márgenes positivos, ganglios linfáticos extirpados positivos
Grupo III: Enfermedad residual macroscópica, incluyendo ganglios linfáticos no resecados Grupo IV: Metástasis a distancia presentes al diagnóstico

Rabdomiosarcoma Factores pronósticos
FACTORES RIESGO
FAVORABLES DESFAVORABLES
Histología embrionario alveolar
Estadio grupo I grupo II-III
Afectación Ganglionar
no afectación (N0)
afectación ganglionar (N1)
Localización orbita, cabeza, cuello (no parameníngeo), genitourinario (no vejiga/próstata)
resto localizaciones
Tamaño </= 5 centímetros > 5 centímetros
Edad < 10 años > 10 años

Rabdomiosarcoma Tratamiento
TRATAMIENTO
Local: Cirugía y/o radioterapia Quimioterapia: Citorreduccion previa a tratamiento local Control diseminación distancia
VAC (vincristina+actinomicina D+ciclofosfamida) IVA (ifosfamida+vincristina+actinomicina D)
Pronóstico: RMS localizado y resecable SLE: 60-70% RMS metastásico SLE: 20 %
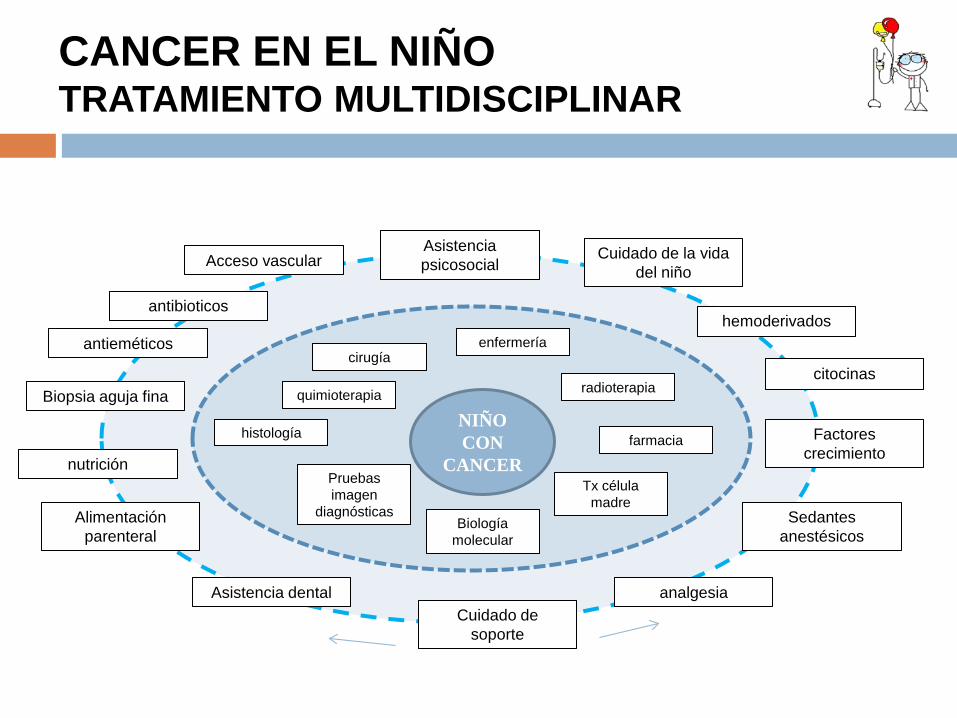
antibioticos
nutrición
Factores crecimiento
Biopsia aguja fina
antieméticos
citocinas
Sedantes anestésicos
analgesia
Alimentación parenteral
Asistencia dental
Cuidado de la vida del niño
Asistencia psicosocial Acceso vascular
hemoderivados
Cuidado de soporte
histología
Pruebas imagen
diagnósticas
Tx célula madre
enfermería
radioterapia
cirugía
Biología molecular
farmacia
quimioterapia
NIÑO CON
CANCER
CANCER EN EL NIÑO TRATAMIENTO MULTIDISCIPLINAR

La población infantil representa actualmente el 30% de toda la población mundial pero es el 100% de la población futura.
Recommended

















