Embed Size (px)
Citation preview

Computational Skill for Modern Biology Research
Department of BiologyChungbuk National University
7th Lecture 2015.10.13
Ipython Notebook, Pandas, Matplotlib

Syllabus주 수업내용1 주차 Introduction : Why we need to learn this stuff?
2 주차 Basic of Unix and running BLAST in your PC
3 주차 Unix Command Prompt II and shell scripts
4 주차 Basic of programming (Python programming)
5 주차 Python Scripting II and sequence manipulations
6 주차 Ipython Notebook and Pandas
7 주차 Basic of Next Generation Sequencings and Tutorial
8 주차 Next Generation Sequencing Analysis I
9 주차10 주차 Next Generation Sequencing Analysis II
11 주차 R and statistical analysis
12 주차 Bioconductor I
13 주차 Bioconductor II
14 주차 Network analysis

DNA Sequencing
Watson & Crick, 1953
Double Strand Structure of DNA Determination of Nucleotide Sequence
Frederic Sanger, 1977

Sanger SequencingPrinciple
ACGTAGTAGAGAGATTAGTATATATATATAGGAGAGAGAACCGGCGTAT5’ 3’
TGCATCAT3’
dATPdGTPdCTPdTTP
DNA Polymerase
CTCT

OBaseCH2
OO
O
OP
H
OBaseCH2
OO
O
OP
H
OBaseCH2
OO
O
OP
H H
dideoxynucleotide
Chain termination!
ACGTAGTAGAGAGATTAGTATATATATATAGGTGCA
+ dATP, dCTP, dGTP, dTTP, ddATP
TGCATCA*TGCATCATCTCTCTA*TGCATCATCTCTCTAA*

dATPdGTPdCTPdTTP
ddATP
dATPdGTPdCTPdTTP
ddTTP
dATPdGTPdCTPdTTP
ddCTP
dATPdGTPdCTPdTTP
ddGTP
ATA*
A*
AT*
ATAA*ATAAG*ATAAGC*ATAAGCT*ATAAGCTA*

Original Sequencing
Istope and slab gel

Automatic Sanger SequencingCapillary electrophoresisDye-terminatorSequence Chromatogram

Sanger Sequencing
Read length : 800~1,000bp
Throughput : 936 samples/per day (Life technologies 3500xL genetic analyzer) 1kb*936 = 0.936Mbp/day/machine
1 대 : 50 일 가동 필요 , 10 대라면 5 일4Mb Bacterial Genome at 8X redundancy : we need 50Mbp

• Base calling : Analyze sequence chromatogram and convert as base pair
Accuracy of Sanger Sequencing
High Quality : Nonambiquous calling
Low Quality : very ambiquous calling

Phred Quality Score
Probability that base calling is WRONG
Def Q=-10 log10P P : probability of current base calling is wrong
만약 현재 염기서열 Calling 이 틀릴 확률이 1/10 이라면…Q= -10 log10 (10-1) = -10 * -1 = 10
Q=10 이면 약 90% 의 정확도를 지닌다 .
틀릴 확률이 1/100 이라면 ( 정확도 99%)
Q = -10 log10(10-2) = 20

Quality Trimming생거 시퀀싱의 경우 , 시퀀스 시작부분과 끝 부분의 퀄리티는 좋지 않다 .
이를 잘라낼 것인가 ? 이를 잘라내는 과정을 Quality Trimming 이라고 한다 .
Low quality region
Low quality region Low quality region

To cut or not to cut?
Sequence Trimming 을 강하게 하면 ..
- Sequence 에서 에러는 줄어든다- 그러나 유용한 데이터까지 버리는 것을 피할 수 없음 .

Limitation of Sanger Sequencing- 최초의 지놈인 Hemophillus influenza (1995) 를 비롯하여 효모 , 초파리 , 사람 , 마우스등의 많은 모델생물의 지놈은 Sanger Sequencing 으로 진행됨- Sanger Sequencing 을 위해서는 ..
Library ConstructionDNA preparation Sequencing
- Throughput : less than 1Mb/day/machine- Cost
Sanger Sequencing 으로 진행된 Human Genome Project 의 비용 : 2.7 billion US$ = 3 조원

Next Generation Sequencing (NGS)
- Instead of reference genome of single organism, we want to check persona differences
Personal Genome Sequencing
- 모델생물이 아닌 각종 생물 ( 작물 , 가축 등 ) 의 지놈 시퀀싱 ?
- Functional Genomic Application
Sanger Sequencing 으로는 이런 용도로 지놈 시퀀싱을 하기 어렵다 . ( 비용 , 노력 )

$1000 Genome
- 개별 인간의 지놈을 $1,000 ( 약 100 만원 ) 에 시퀀싱할 수 있다면 ..
• 병원에서 일반적인 검사 (X-ray, CT..) 를 하는 것처럼 개인에 대한 지놈 정보를 얻고
• 이 지놈 정보에 의해 개별적인 맞춤치료를 할 수 있지 않을까 ?
- 예 : 암환자
• 인간의 건강에 미치는 상당수의 요인이 유전적인 요인
• 암은 대개 DNA 의 돌연변이에 의한 이상• 환자에 따라서 발생하는 돌연변이는 제각각 틀림• 만약 암환자에 따른 돌연변이를 파악할 수 있다면 ..

Sequencing cost is dropping

Various NGS technology
Sequencing-By-Synthesis : Reversible Terminator Technology
-illumina/Solexa
Pyrosequencing : Detection of Pyrophosphate (Ppi) released from DNA polymerization
-454- Ion Torrent : Variation of Pyrosequening.
Single-Molecule Real Time (SMRT) Sequencing
Nanopore sequencing
- Oxford Nanopore
- Pacific Biosciences
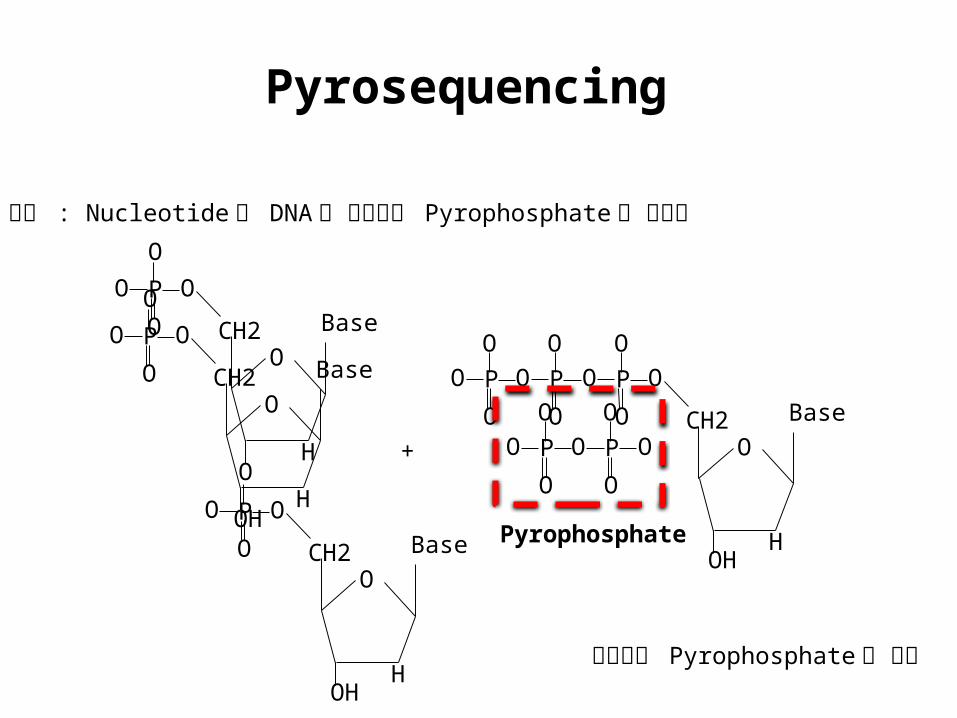
Pyrosequencing
원리 : Nucleotide 가 DNA 에 들어갈때 Pyrophosphate 가 방출됨
O
OBaseCH2
OO
OP
H
OBaseCH2
OO
O
OP
HOH
O
O
OPO
O
OP
OH
+
O
OBaseCH2
OO
OP
H OO
O
O
OPO
O
OP
OBaseCH2
O
O
O P
HOH
Pyrophosphate
방출되는 Pyrophosphate 를 정량

Genomic Pyrosequencing

http://www.youtube.com/watch?v=nFfgWGFe0aA

Pyrosequencing
장점최초로 상용화된 Next Generation Sequencing Platforms(Relatively) Long Read Length : 800-1000bp
단점Low Throughput (400~700Mb) compared with other system Errors in Homopolymer
Roche GS-FLX
주 용도 De novo sequencing of bacteria16s rRNA

Semiconductor sequencing
원리Variation of PyrosequencingInstead of pyrophoshate, it detects H+ release from DNA polymerization

장점CostSpeed
단점ThroughputHomopolymer
Semiconductor sequencing
Ion Torrent 사 (Life Technology) 를 통해 다음의 기기로 상업화됨 .
PGM (Personal Genome Machine) Ion Proton
100-2Gb for PGM, 7.5-10Gb for Proton

Sequencing by Synthesis (SBS)
illumina sequencing
De facto Standard of Next Generation Sequencing차세대 시퀀싱의 ‘사실상 표준’
~ 90% of registered sequences in Genebank was sequenced by illumina/Solexa

Principle of illumina Sequencing
Flow cell지노믹 DNA 를 random 하게 조각을 낸 후 양 말단에 어댑터를 붙임
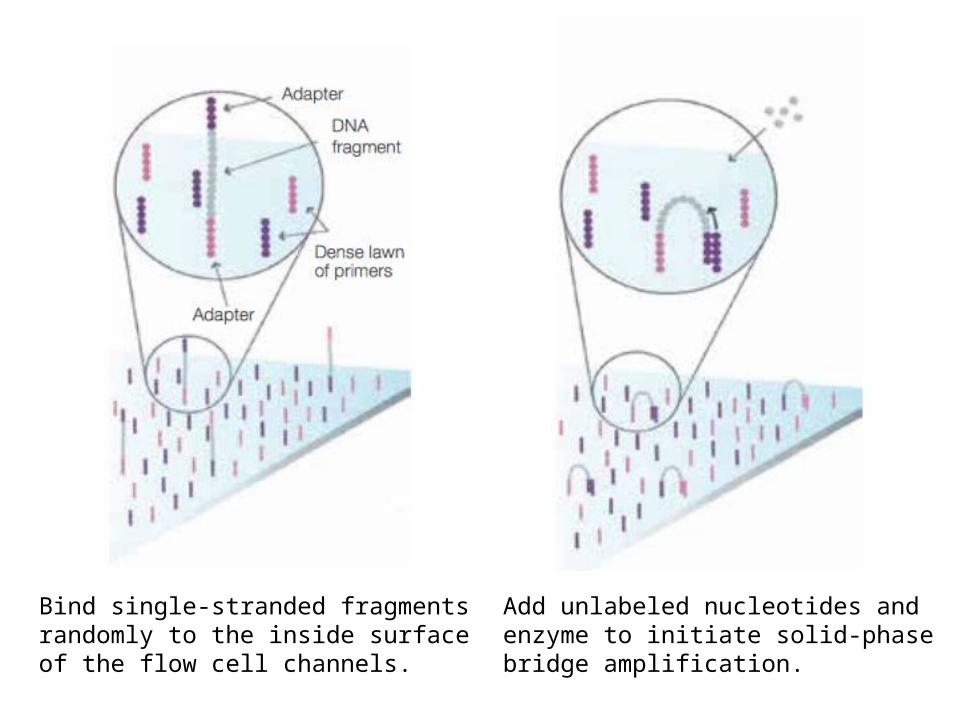
Bind single-stranded fragments randomly to the inside surface of the flow cell channels.
Add unlabeled nucleotides and enzyme to initiate solid-phase bridge amplification.

The enzyme incorporates nucleotides to build double-stranded bridges on the solid-phase substrate.
Denaturation leaves single-stranded templates anchored to the substrate.
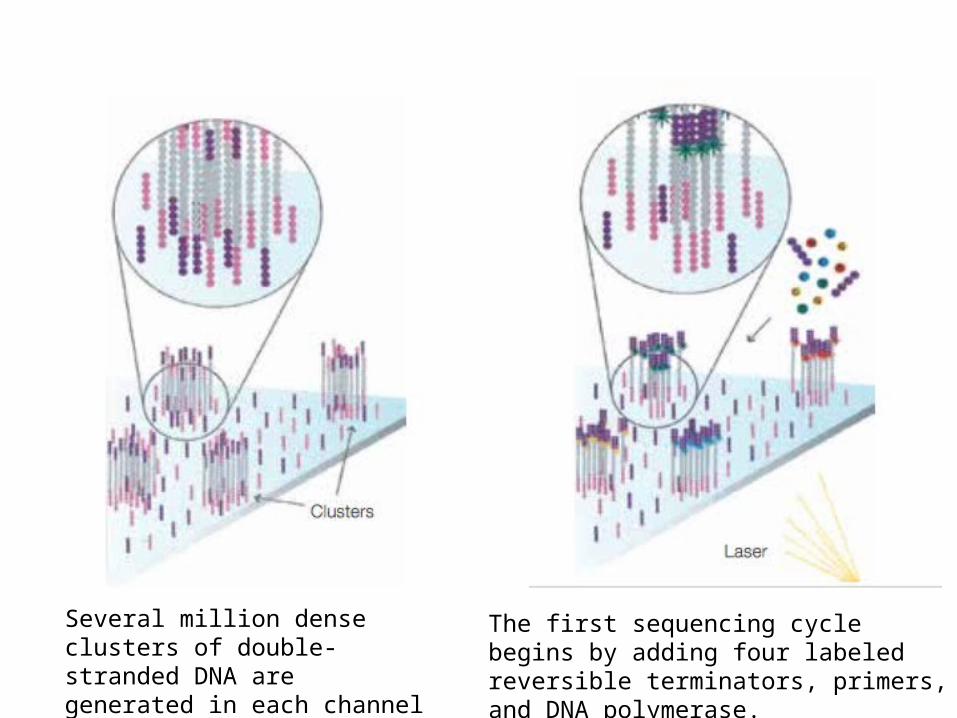
Several million dense clusters of double-stranded DNA are generated in each channel of the flow cell.
The first sequencing cycle begins by adding four labeled reversible terminators, primers, and DNA polymerase.

After laser excitation, the emitted fluorescence from each cluster is captured and the first base is identified.
Sequential Read of Bases

Reversible Terminators

Sequencing Throughput
HiSeq 2500
MiSeq
900-1,000Gb/in 6 days
13-15Gb/in 55hrs (2x300bp)
(2x125bp)
Human Whole Genome : 3Gb * 50 = 150bp
HiSeq 2500 의 경우 하루에 Human Whole Genome 1 개를 50x 배수로 시퀀싱할 용량 !

Single Molecule Real Time Sequencing (SMRT)
지금까지의 시퀀싱은 근본적으로 ‘한번에 하나의 베이스’ 만을 읽는 방법이었음 .
Single Molecule Real Time Sequencing : DNA 중합효소의 단일분자를 관찰하여 , 단일분자의DNA 중합효소에 의한 DNA 합성을 실시간으로 관찰

PacBio RS II (Pacific Bioscience)
http://www.youtube.com/watch?v=v8p4ph2MAvI

장점- Very Loooooooooooooong (~more than 15kb) Read is possible
- Fast (120 minitue)
(Advantages in Genome Assembly)
Single Molecule Real Time Sequencing (SMRT)

http://www.slideshare.net/flxlex/combining-pacbio-with-short-read-technology-for-improved-de-novo-genome-
assemblyRepeats
Repeat copy 1 Repeat copy 2
Collapsed repeat consensus
고등동물의 지놈에는 Repeat 가 다수 존재Repeat 는 Gap 을 만드는 주범
Why long sequencing reads are needed?

Heterozygosity
http://commons.wikimedia.org/wiki/File:Chromosome_1.svg
*
*
*
서로 다른 상동염색체 간의 차이

Polymorphic contig 2
Polymorphic contig 3
Contig 4Contig 1

Repeat copy 1 Repeat copy 2
Long reads can span repeats
Polymorphic contig 2
Polymorphic contig 3
Contig 4Contig 1
and heterozygous regions

SMRT: ProsReference 가 없는 De novo sequencing/assembly 에 유리함
Methylation 연구
Full length cDNA 의 시퀀싱이 가능

Systemic Error 가 아닌 Random Error 인 관계로 Coverage 를 높이면 극복 가능
SMRT : Cons
- Sequencing Accuracy
단일 Read 의 정확도는 80-85% 수준- Systemic Error ( 틀린 부분은 계속 틀리는 ) 가 아닌 Random Error
AGGGGGGGATGAGACCCATGAGATCCTTAAAGGGGGG-ATGAGACCCATGAGATCCTTAAAGGGGGGGGATGAGACCCATGAGATCCTTAAAGGGG---ATGAGACCCATGAGATCCTTAAAGGGGG--ATGAGACCCATGAGATCCTTAAAGGGGCGGGATGAGACCCATGAGATCCTTAA
Systemic Error
AGGGGGGGATGAGACCCATGAGATCCTTAAAGGGGGGGTTGAGACCCATGGGATCCTTAAAGGGGGGGAGGAGACCCATGAGTTCCATAAAGGGGGGAATGAGACCCATCAGATACTTAA
Random Error
AGCGGGGGATGAGACCCATGAGATCCTATAAGGGGGGGATGAGTCCGATGAGATCCTTAA
- Throughput : 0.5/Gb per run

Nanopore SequencingSequencing for the Future?
- Nanopore : 생체막 (Membrane) 에 존재하는미세공- Pore 내부로 DNA 를 흘려보냄으로써 전위차가생성
- 해당하는 전위차를 이용하여 DNA 서열을 결정
- Experimental Stages

Oxford Nanopore : MiniOn
USB-Connected Sequencer
Sequence Accuracu : 60-70% (now)

Data Processing of NGS Data
+ Sequence Quality Data (Phred Like Score)
>GBUDRX201C2TIH length=489 xy=1145_0599 region=1 run=R_2010_02_05_07_58_35_ACAAGATGCCATTGTCCCCCGGCCTCCTGCTGCTGCTGCTCTCCGGGGCCACGGCCACCGCTGCCCTGCCCCTGGAGGGTGGCCCCACCGGCCGAGACAGCGAGCATATGCAGGAAGCGGCAGGAATAAGGAAAAGCAGCCTCCTGACTTTCCTCGCTTGGTGGTTTGAGTGGACCTCCCAGGCCAGTGCCGGGCCCCTCATAGGAGAGGAAGCTCGGGAGGTGGCCAGGCGGCAGGAAGGCGCACCCCCCCAGCAATCCGCGCGCCGGGACAGAATGCCCTGCAGGAACTTCTTCTGGAAGACCTTCTCCTCCTGCAAATAAAACCTCACCCATGAATGCTCACGCAAGTTTAATTACAGACCTGAA
NGS 의 결과물은 결국 Sequence Data (A,C,G,T)
>GBUDRX201C2TIH length=489 xy=1145_0599 region=1 run=R_2010_02_05_07_58_35_40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 4040 40 40 40 40 40 34 34 34 34 40 40 40 40 40 40 40 40 40 40 40 40 40 40 4040 39 39 39 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 4040 40 34 34 34 40 40 40 40 40 40 40 40 40 40 40 40 40 40 39 39 39 40 40 4040 39 39 39 40 39 39 39 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 4040 30 30 30 30 39 40 40 40 40 36 36 36 36 38 40 40 18 18 18 18 18 34 39 4040 40 40 40 40 40 40 40 40 40 35 35 26 26 26 35 40 40 40 40 40 40 40 40 4040 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 4040 40 40 40 40 40 40 40 39 39 39 40 40 40 40 40 40 40 40 40 40 40 40 40 3939 39 28 28 28 28 28 39 39 23 40 26 26 26 26 39 39 37 40 40 40 40 40 40 4040 40 40 40 40 40 40 30 30 30 30 40 40 40 40 40 40 40 40 40 40 40 40 40 3939 39 40 40 40 40 40 40 40 40 40 40 40 40 40 40 39 39 39 40 40 40 40 40 4040 40 40 40 40 40 40 40 40 40 40 40 39 39 39 40 40 40 40 40 40 40 40 40 4040 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 40 39 39 39 40 40 40
FASTA
Quality

FASTQ File
@SEQ_IDGATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT+!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
Sequence ID
Sequence
Quality
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
Lower HigherQuality Score
0 93
Quality Score = ASCII CODE-33
FASTQ = FASTA + Quality

Quality Check of Sequencing Data
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/

Examples of Good Quality Data

Examples of Bad Quality Data

NGS 데이터의 응용De novo Genome Sequencing
- Sequence Assembly
Resequencing
- Sequence Alignments to Reference Genome
RNA-Seq
- Sequencing of mRNA- Measurement of expression levels of each transcripts
CHiP-Seq (Chromatin-Immunoprecipitation Sequencing)

Resequencing
Human Whole Genome Sequence : 전체 지놈 서열을 대상으로 시퀀싱을 수행(30x-50x of Human Genome size)
Exome Sequencing: Exon 영역에 해당하는 DNA 영역만 선택적으로 시퀀싱을 수행(50-100x of Exon region)
이미 표준 서열이 존재하는 생물 ( 예 : Human) 을 대상으로 개체간의 변이를 알기 위하여시퀀싱을 수행하는 일
Resequencing 의 구분
Size of Sequencing Data
Human Whole Genome : 30-50x of human genome x 3.2Gb = 100-150GbyteHuman Whole Exome : 100x of Human Exome = 10Gbyte

Whole Genome sequencing
Hybridization with oligoCorrespond to exon

Data Analysis
De novo sequencing
Sequence Assembly
Resequencing, RNA-Seq, CHIP-Seq..
Align to Reference Sequence

Sequence Mapping to Reference Sequences
이미 완성된 특정 생물에 대한 표준서열이 있는 경우 , 이것을 기준으로 하여 시퀀스 데이터를Align 하여 표준서열과의 변이를 찾는다
NGS Data : Short Reads
Sanger Sequecing : 700-1000bpNGS (illumina) : 75-200bp
NGS 데이터는 Sanger Sequencing 에 비해서 개별 Read 의 길이가 매우 짧은 관계로 Assembly 를 수행하여 Contig 을 형성하기가 어렵다 .
같은 종의 생물일 경우에는 ‘대개의’ 지놈 구조는 유사함따라서 ‘지놈의 전체적인 구조는 거의 유사하다고 간주하고’ 차이만을 찾기 위하여 기존에밝혀진 지놈 구조를 참조하여 여기에 시퀀스 데이터를 align 하여 차이를 찾는다 .

Lander-Waterman StatisticsG : 시퀀싱하려는 지놈의 길이N : 우리가 시퀀싱한 낱개 시퀀스의 갯수 L : 각각의 시퀀스의 길이 c = nL/G : 커버리지 ( 지놈의 길이에 비해서 몇 배 시퀀스를 더했는가 ?) T: 검출가능한 최소의 시퀀스간 overlapσ = (L-t)/L
Contig 의 갯수 = Ne-cσ Contig 의 길이 = L((ecσ – 1) / c + 1 – σ)

- SNV (Single Nucleotide Variation)
서로 다른 개체간에 어느정도의 서열 변이를 가지고 있는가 ?
- CNV (Copy-Number Variation) / Structural Variation>
유전체의 특정 영역이 중복 혹은 삭제되었는가 ?
Resequencing 으로 얻을 수 있는 정보


Sequence Align to Reference Sequences
이전에 알아보았던 서열정렬과 근본적으로는 원리가 동일 ..
어마어마하게 큰 데이터 : 지금보다 속도가 빠른 Alignment 방식이 필요함
다음 시간에…
























![[아카데미X] 7강. 상표제도의 이해](https://img.pdfslide.tips/doc/110x75/58707f921a28ab57368b6097/x-7-58707f921a28ab57368b6097.jpg)




![19-A4.ppt [호환 모드] - 캐드앤그래픽스 · 2012-04-24 · 2 • Computer Aided Graphics • (범용) CAX 분야= 사용기술 논의의 방향-CAD : AutoCAD, CATIA, UG,](https://img.pdfslide.tips/doc/110x75/5f057c587e708231d41332e1/19-a4ppt-eeoe-eoeee-2012-04-24-2-a-computer-aided.jpg)