Embed Size (px)
DESCRIPTION
Trata-se de um guia de laboratório de Métodos instumentais de análise para o uso didáctico de Farmacêuticos e Químicos e outras áreas afins.
Citation preview

INSTITUTO SUPERIOR DE CIÊNCIAS E TECNOLOGIA DE MOÇAMBIQUE
ISCTEM DEPARTAMENTO DE FARMÁCIA
GUIA DE LABORATÓRIO DE
MÉTODOS INSTRUMENTAIS DE ANÁLISE
Paulo José Cumbane 2009

1
ÍNDICE
1. RESUMO DAS NORMAS DE SEGURANÇA NO LABORATÓRIO DE QUÍMICA ................................... 4
1.0. AMOSTRAGEM ............................................................................................................................................ 7
1.1.0. Amostragem de Produto em processo e a Granel ............................................................................ 7
1.1.1. Amostragem de Produto Acabado ...................................................................................................... 7
1.2. CÁLCULO DE AMOSTRAGEM ................................................................................................................... 8
1.3. TRATAMENTO DAS AMOSTRAS PARA ANÁLISE................................................................................... 8
2.0. ENSAIOS ANALÍTICOS ............................................................................................................................ 11
2.0.1. Ensaios Organolépticos ........................................................................................................................ 11
2.1. Critério de Avaliação .............................................................................................................................. 12
2.3. ENSAIOS FÍSICO-QUÍMICOS ................................................................................................................. 12
2.3.1. Determinação de pH ............................................................................................................................ 12
2.3.2. Determinação de Viscosidade............................................................................................................ 13
2.3.3. Determinação de Densidade .............................................................................................................. 14
2.3.4. Determinação de Materiais Voláteis e Resíduo Seco .................................................................... 16
2.3.5. Determinação do Teor de Água/ Humidade .................................................................................. 16
2.3.6. Granulometria ....................................................................................................................................... 17
2.3.7. Teste de Centrífuga .............................................................................................................................. 17
2.4. ENSAIOS QUÍMICOS ............................................................................................................................... 17
3.0. MODELO DE RELATÓRIO ........................................................................................................................ 20
4.0. MÉTODO DE CÁLCULO E ERROS EM QUÍMICA ANALÍTICA ........................................................... 23
4.1. Métodos de Cálculo de Concentrações .............................................................................................. 23
4.3. Precisão, Exactidão, Sensibilidade e Limite de Detecção ............................................................... 26
4.3.1. Análise estatística dos Erros ............................................................................................................... 26
5. PARTE EXPERIMENTAL ................................................................................................................................. 30
5.0. ESPECTROSCOPIA DE ABSORÇÃO MOLECULAR (MAS) .................................................................. 30
5.1. Determinação Quantitativa de Aspirina, Paracetamol e Cafeína por MAS ................................ 30
5.1.1. Análise Multicomponente de um Comprimido ................................................................................. 30
5.1.2. Análise Quantitativa de Anadin-extra ............................................................................................. 30
5.2. TÉCNICA EXPERIMENTAL ........................................................................................................................ 31
5.3. PARTE INSTRUMENTAL ............................................................................................................................ 32

2
5.5. ESPECTROS DE ABSORÇÃO .................................................................................................................. 33
5.6. BIBLIOGRAGIA ......................................................................................................................................... 34
6.0. ANÁLISE DE ASPIRINA OU PARACETAMOL NUM COMPRIMIDO .................................................. 35
6.1. Análise quantitativa de um comprimido contendo apenas um componente ................................. 35
6.2. TÉCNICA EXPERIMENTAL ........................................................................................................................ 36
6.2.1. PARTE INSTRUMENTAL ......................................................................................................................... 37
6.3. BIBLIOGRAFIA ........................................................................................................................................... 38
7.0. ESPECTROSCOPIA DE ABSORÇÃO ATÓMICA (AAS) ....................................................................... 39
7.1. DETERMINAÇÃO DE CÁLCIO NO SUMO POR AAS .......................................................................... 39
7.2. PARTE EXPERIMENTAL ............................................................................................................................. 39
7.3. BIBLIOGRAFIA ........................................................................................................................................... 41
1.0. TITULAÇÕES CONDUCTIMÉTRICAS E POTENCIOMÉTRICAS ........................................................... 43
3. PARTE EXPERIMENTAL ................................................................................................................................. 44
3.2 TÉCNICA EXPERIMENTAL ......................................................................................................................... 45
4. QUESTIONÁRIO ........................................................................................................................................... 49
5. BIBLIOGRAFIA .............................................................................................................................................. 50

3
Segurança no Laboratório de Química

4
1. RESUMO DAS NORMAS DE SEGURANÇA NO LABORATÓRIO DE QUÍMICA
A prática da Química, seja ao nível profissional ou de aprendizado, exige que regras de segurança sejam rigorosamente seguidas para evitar acidentes e prejuízos de ordem humana ou material. Os acidentes podem ser minimizados ou até mesmo evitados se tomadas às devidas precauções. A seguir estão relacionadas algumas regras de segurança que você deverá colocar em prática para sua segurança e de seus colegas:
• Use sempre a bata de algodão de mangas compridas, na altura dos joelhos e fechados;
• Use calçado fechado de couro ou similar;
• Não use relógios, pulseiras, anéis ou quaisquer ornamentos durante o trabalho no laboratório;
• Não beba e não coma no laboratório;
• Nunca use material de laboratório para beber ou comer;
• É proibido fumar no laboratório ou em qualquer outro lugar que possa por em risco a
segurança ou saúde das pessoas;
• Caminhe com atenção e nunca corra no laboratório;
• Nunca teste amostras ou reagentes pelo sabor e os odores devem ser verificados com muito
cuidado;
• Não leve a mão à boca ou aos olhos quando estiver manuseando produtos químicos;
• Aventais de laboratório, luvas, óculos de protecção ou outras vestimentas não devem ser
usados fora do laboratório;
• Em caso de acidentes, mantenha a calma e chame o professor ou técnico responsável;
• Objectos pessoais como bolsas, blusas... devem ser guardados em armários de preferência em
áreas externas aos laboratórios;
• Brincadeiras são absolutamente proibidas nos laboratórios;
• Use o nicho sempre que trabalhar com solventes voláteis, tóxicos e reacções perigosas,
explosivas ou tóxicas;
• As substâncias inflamáveis devem ser manipuladas em locais distantes de fontes de
aquecimentos;
• O uso de pipetadores é requerido em qualquer circunstância ao utilizar pipetas;
• Lentes de contacto não devem ser usadas em laboratórios, pois podem absorver produtos
químicos e causar lesões nos olhos;

5
• Óculos protectores de segurança são requeridos durante todo o período de trabalho no
laboratório;
• Nunca jogue reagente ou resíduo de reacções na pia, procure o frasco de descarte;
• Ao final de cada aula, as vidrarias utilizadas durante o trabalho de laboratório devem ser
esvaziadas nos frascos de descarte e enxaguadas com água antes de serem enviadas para
limpeza;
• Vidrarias trincadas, lascadas ou quebradas devem ser descartadas e o técnico ou responsável
deve ser avisado;
• Antes de manipular qualquer reagente deve-se ter conhecimento de suas características com
relação à toxicidade, inflamabilidade e explosividade;
• Devem-se tomar cuidados especiais quando manipular substâncias com potencial cancerígena;
• Os reagentes e soluções devem ser claramente identificados e às soluções apresentar data de
preparo, validade e o nome do analista que a preparou;
• Todo acidente com reagentes deve ser limpo imediatamente protegendo-se se necessário. No
caso de ácidos e bases devem ser neutralizados antes da limpeza;
• Siga correctamente o roteiro de aula e não improvise, pois improvisações podem causar
acidentes, use sempre materiais e equipamentos adequados;
• Todas as substâncias são tóxicas, dependendo de sua concentração. Nunca confie no aspecto
de uma droga, deve-se conhecer suas propriedades para manipulá-la;
• Receber visitas apenas fora do laboratório, pois elas não conhecem as normas de segurança
e não estão adequadamente vestidas.
• Essas são algumas regras gerais que devemos seguir durante um trabalho no Laboratório.

6
AMOSTREGEM

7
1.0. AMOSTRAGEM
Amostragem é o processo definido de colecta que seja representativa de um todo, de acordo com um plano definido pelo tipo e pela quantidade de um determinado material ou produto. A rigor, é uma metodologia estatística sistemática para obter informações sobre alguma característica de uma população, através de estudo de uma fracção representativa ( i.e. amostra ) da população. Existem várias técnicas de amostragem que podem ser empregadas ( tais como amostragem aleatória simples, amostragem estratificada, amostragem sistemática, amostragem sequencial, amostragem por lote ), sendo a escolha de técnica determinada pelo propósito da amostragem e pelas condições sob as quais ela deve ser conduzida.
Amostra é a fracção representativa de um todo, seleccionado de tal modo que possua as características essenciais do conjunto que ela representa. 1.1. PROCESSO DE AMOSTRAGEM
1.1.0. Amostragem de Produto em processo e a Granel
Procedimento: i) O processo deverá ser realizado por pessoal devidamente treinado nos aspectos
operacionais e de segurança.
ii) A amostragem deverá ser executada em etapas definidas do processo.
iii) Devem ser utilizados acessórios e recipientes previamente definidos e devidamente
limpos para a colecta das amostras, com o produto em quantidade suficiente para
realização de todos os ensaios necessários.
iv) A amostra do produto deverá ser devidamente rotulada para garantir a identificação
e a restreabilidade do mesmo ( ex. Nome do Produto/Lote/Número da Ordem de
Fabricação/ Número de Tanque de Armazenamento/ Data de Fabricação).
v) As amostras deverão ser devidamente disponibilizadas para análise e retenção,
conforme procedimento interno da instituição.
1.1.1. Amostragem de Produto Acabado
Procedimentos: i) O processo deverá ser realizado por pessoal devidamente treinado nos aspectos
operacionais e de segurança.
ii) A colecta de produto acabado deverá ser realizado após o envase, em quantidade e
periodicidade suficientes para atender às necessidades de controle.

8
iii) O produto amostrado acabado deverá ser devidamente identificado, com informações
suficientes para sua restreabilidade ( ex. Nome do produto/Lote/Número da Ordem
de Fabricação/Número de Tanque de Armazenamento/Data de A fabricação )
1.2. CÁLCULO DE AMOSTRAGEM
O número de amostras a ser tomado pode ser equacionado pelas ferramentas da estatística, de modo a representar um valor amostral do total do produto. Seu cálculo pode ser feito de várias formas, sendo a equação a seguir a mais utilizada:
Número de amostras xn += Onde :
n = total de produtos x = normalmente aplica-se valor igual a 1
1.3. TRATAMENTO DAS AMOSTRAS PARA ANÁLISE
Os produtos analíticos podem apresentarem-se nos estados líquido, semi-sólido e sólido. Após seleccionar a forma de amostragem, a amostra deve ser tomada e tratada de seguinte forma, conforme o estado do produto: 1.3.1. Amostra em Estado Líquido
Neste estado encontram-se produtos tais como perfumes, soluções, loções, óleos, leites, aerossóis, entre outros.
Tomada a amostra para o ensaio : depois de homogeneizada, ela deve ser tratada de acordo com as condições especificadas. Alguns mililitros do líquido devem ser transferidos para um recipiente adequado, afim de realizar os ensaios pertinentes. 1.3.2. Amostra no estado Semi-Sólido a) Produtos acondicionados em embalagens com abertura estreita ( ex. Bisnagas, frascos
flexíveis ) : descarta-se a primeira alíquota do produto e retira-se a amostra para o ensaio.
b) Produtos acondicionados em embalagens com abertura larga ( ex. Potes ) : a camada superficial deve ser eliminada, homogeneizando-se o restante, para então ser efectuada a tomada da amostra para ensaio.
1.2.3. Tomada de amostra para ensaio. Há dois casos possíveis a) Pós : a embalagem deve ser agitada antes de ser aberta, a fim de garantir a
homogeneização da amostra. A seguir, deve ser efectuada a tomada de amostra para ensaio.

9
b) Pós compactados: camada superficial do sólido deve ser eliminado por meio de uma leve raspagem, para então ser efectuada a tomada de amostras para ensaio.
Obs. : Cada uma das amostras pode ser tratada individualmente ou em conjunto, na forma de pool, desde que originadas de um mesmo lote.

10
ENSAIOS ANALÍTICOS

11
2.0. ENSAIOS ANALÍTICOS
Os ensaios analíticos têm como objectivo verificar a conformidade dos materiais ou produtos frente às especificações estabelecidas. A execução desses ensaios deve ser realizada por pessoas qualificadas.
2.0.1. Ensaios Organolépticos
Ensaios organolépticos são procedimentos utilizados para avaliar as características de um produto, detectáveis pelos órgãos dos sentidos: aspecto, cor, odor, sabor e tacto. Fornecem parâmetros que permitem avaliar, de imediato, o estado da amostra em estudo por meio de análises comparativas, com o objectivo de verificar alterações como, separação de fases, precipitação e turvação, possibilitando o reconhecimento primário do produto. Deve-se utilizar uma amostra de referência ( ou padrão ) mantida em condições ambientais controladas, para evitar modificações nas propriedades organolépticas.
Para a execução dos ensaios organolépticos devem ser consideradas a forma física e as características de cada produto, tais como líquidos voláteis, não voláteis, semi-sólidos e sólidos. 2.0.2. Aspecto Método de Ensaio:
a análise da cor ( calorimetria ) pode ser realizado por meio visual ou instrumental. Na análise visual ( calorimetria visual ) compara-se visualmente a cor da amostra com a cor de um padrão armazenado em frasco da mesma especificação. Pode se efectuar essa análise sob condições de luz “branca”natural ou artificial ou ainda em câmaras especiais, com várias fontes de luz ( ou seja, vários comprimentos de onda ).
A análise instrumental substitui o olho humano como detector e pode ser feita por meio da calorimetria fotoeléctrica ou calorimetria espectro fotométrica. A calorimetria fotoeléctrica é o método que utiliza uma célula fotoeléctrica com o detector. É usualmente empregado com luz contida em intervalo relativamente estreito de comprimento de onda obtida pela passagem da luz branca através de filtros. Os aparelhos utilizados nesse método são conhecidos como colorímetros ou fotómetros de filtro. A calorimetria espectro fotométrica é o método que utiliza uma fonte de radiação em vários comprimentos de onda na região espectral do visível. O aparelho utilizado nesse método é conhecido como espectro fotómetro. 2.0.3. Odor
Método de Ensaio: A amostra e o padrão de referência, acondicionado no mesmo material de embalagem,
devem ter odor comparado directamente através do olfacto. 2.0.4. Sabor

12
Método de Ensaio: Compara-se o sabor da amostra com o do padrão de referência, directamente através do
paladar.
2.1. Critério de Avaliação
Após avaliação comparativa, a amostra tem que estar em conformidade com a amostra de referência ( padrão ) preestabelecida.
2.3. ENSAIOS FÍSICO-QUÍMICOS
Ensaios físico-químicos são operações técnicas que consistem em determinar uma ou mais características de um produto, processo ou serviço, de acordo com um procedimento especificado. Os equipamentos devem ser submetidos à manutenção e à calibração/aferição periódicas, de acordo com as especificações do fabricante, de forma a garantir que fornecem resultados válidos. Os métodos mais usuais são apresentados a seguir .
2.3.1. Determinação de pH
Definição
É o logaritmo negativo de concentração molar de iões de hidrogénios. Representa convencionalmente a acidez ou alcalinidade de uma solução. A escala de pH vai de 1 ( ácido) até 14 ( alcalino), sendo 7 o valor considerado pH neutro. 2.3.1.1. Princípios do método
O pH é determinado por potenciometria, pela determinação da diferença de potencial entre dois eléctrodos – imersos na amostra a ser analisada, e depende da actividade dos iões hidrogénios na solução. 2.3.1.2. Descrição do Método
Antes do uso, deve-se verificar a limpeza e determinar a sensibilidade do eléctrodo, utilizando-se soluções tampão de referência e, quando aplicável, ajustando-se o equipamento. Se o produto é um sólido ou semi-sólido, recomenda-se preparar uma solução/ dispersão/suspensão aquosa da amostra em uma concentração preestabelecida e determinar o pH da mistura com o eléctrodo apropriado. Em alguns casos, a medição pode ser feita directamente na amostra. Se o produto é uma loção ou solução, recomenda-se determinar o pH directamente sobre o líquido, imergindo-se o eléctrodo directamente nele.

13
Notas: 1) O modelo do equipamento e os tipos de eléctrodos a serem utilizados na medição do pH devem ser estabelecidos previamente, levando-se em consideração as características físico-químicas do produto. 2) Normalmente as medidas de pH são realizadas em meio aquoso. 3) Não tem significado medir pH em meio não-aquoso com eléctrodos convencionais. Para essa medida, devem ser utilizados eléctrodos específicos.
2.3.2. Determinação de Viscosidade
Definição
Viscosidade é a resistência que o produto oferece à deformação ou ao fluxo. A viscosidade depende das características físico-químicas e das condições de temperatura do material. A unidade fundamental da medida de viscosidade é o poise. Princípio
Consiste em medir a resistência de um material ao fluxo por meio da fricção ou do tempo de escoamento. Há vários métodos para se determinar a viscosidade. Os mais frequentes utilizam viscosímetros rotativos, de orifício e capilares.
• Determinação por viscosímetro rotativo: consiste na medição do torque requerido para rodar um fuso imerso em um dado fluido.
• Determinação por viscosímetro de orifício: consiste na medição do tempo de escoamento do material comparado com a água. Utiliza-se um copo na forma de cone (copo Ford), com um orifício na parte inferior por onde escoa o fluido. A escolha do diâmetro do orifício é feita em função da faixa de viscosidade a ser determinada.
• Determinação por viscosímetro capilar (Ostwald): consiste na medição do tempo de escoamento do material comparado com a água. A força hidrostática do líquido força-o a fluir através de um tubo capilar.
2.3.2.1. Descrição do Método
Vários são os métodos utilizados para a determinação da viscosidade de um fluido. Os métodos a seguir são os mais usuais em laboratórios:
1. Viscosímetro rotativo: dependendo da faixa de viscosidade da amostra, selecciona-se o fuso (sainete) adequado. A seguir, mergulha-se o fuso diagonalmente na amostra com temperatura estabilizada, conforme especificado, isenta de bolhas, até a marca (sulco) da haste do fuso, e nivela-se o aparelho. Verificada a ausência de bolhas junto ao fuso, procede-se à leitura da viscosidade, de acordo com o procedimento operacional do aparelho.
2. Viscosímetro de orifício: nivela-se o aparelho em superfície plana. Depois de se obstruir o orifício localizado na parte inferior do copo com o dedo e colocar lentamente a amostra até transbordar, com temperatura estabilizada, conforme especificado, nivela-se a superfície da amostra com uma espátula. Verifica-se então a presença de bolhas, que afectam a medida. Retira-se o dedo do orifício e, ao mesmo tempo, com a outra mão,

14
acciona-se o cronómetro. Imediatamente após o escoamento, pára-se o cronómetro e registra-se o tempo para fins de cálculo.
Cálculo:
VISCOSIDADE = A X T + B Onde: T = tempo expresso em segundos A e B = constantes definidas experimentalmente pelo fabricante, que variam para diferentes orifícios do copo.
Viscosímetro capilar : para determinar a viscosidade, deve-se transferir a amostra para o
viscosímetro e estabilizar o conjunto até a temperatura especificada. A seguir, aspira-se a amostra com o auxílio de um pipetador até a marca superior do menisco no viscosímetro e cronometra-se o tempo de escoamento entre a marca do menisco superior e do inferior. Repete-se esse procedimento três vezes e calcula-se a média.
Determinação da constante K: transfere-se a amostra para o viscosímetro e estabiliza-se o conjunto até a temperatura especificada. Aspira-se a amostra com o auxílio de um pipetador até a marca superior do menisco no viscosímetro e cronometra-se o tempo de escoamento entre a marca do menisco superior e do inferior. Repete-se esse procedimento cinco vezes e calcula-se a média. Cálculo da constante K:
K= 1/0,99823 X T Onde: 1 = 1 centipoise T = tempo de escoamento da água em segundos Cálculo da viscosidade:
V = T X K Onde: V = viscosidade da amostra em centipoises (cps) T = tempo de escoamento da amostra em segundos K = constante K De acordo com as características físicas do produto, podem ser utilizados diferentes tipos de viscosímetros.T
2.3.3. Determinação de Densidade
Definição
Densidade é a relação entre a massa e o volume. Existem várias formas de densidade: 1. Densidade absoluta é uma propriedade física de cada substância, cujo valor se calcula
pela relação entre certa massa da substância e o volume ocupado por essa massa (d=m/V), tomando por unidade geralmente o grama por centímetro cúbico (g/cm3). No sistema internacional, a unidade é o quilograma por metro cúbico (kg/m3).
2. Densidade relativa é a relação entre a densidade absoluta de uma substância e a densidade absoluta de outra substância estabelecida como padrão.

15
3. Densidade aparente é a relação directa entre a massa de uma amostra e seu volume específico, medido em proveta graduada.
4. Densidade específica é uma densidade relativa, sendo utilizada como padrão a densidade absoluta da água, que é igual a 1.000 kg/dm3 ou g/cm3 a 4°C temperatura em que a água é mais densa). No caso de gases, é tomada em relação ao ar ou ao hidrogénio.
Princípio
Baseia-se na razão entre a massa e o volume de uma dada amostra. Descrição do Método
A densidade pode ser medida utilizando-se picnómetro metálico, picnómetro de vidro, densímetro e densímetro digital.
Determinação da densidade aparente : Deve-se pesar uma quantidade da amostra e
introduzi-la na proveta, tampando-a e seguida. Para os produtos na forma de pó, é necessário
acomodar a amostra, eliminando o ar entre as partículas por meio de leves batidas em
movimentos verticais, padronizados, com altura fixa, sobre uma superfície lisa, até obter volume
constante. Deve-se anotar o volume.
Cálculo: Onde: dA = densidade aparente em g/ml m = massa da amostra em gramas v = volume final em mililitros
Determinação da densidade em picnómetro de vidro ou metálico: utiliza-se o de vidro para os produtos líquidos e o de metal para os produtos semi-sólidos e viscosos. Pesa-se o picnómetro vazio e anota-se o seu peso (M0). A seguir, deve-se enchê-lo completamente com água purificada, evitando-se a introdução de bolhas. Após secá-lo cuidadosamente, é necessário pesá-lo novamente e anotar seu peso (M1). O próximo passo é encher completamente o picnómetro (limpo e seco) com a amostra, evitando a formação de bolhas. Depois de secá-lo cuidadosamente, ele deve ser pesado mais uma vez e ter seu peso (M2) anotado. Cálculo: d=( M2 – M0)/M1 – M0 Onde: d = densidade M0 = massa do picnómetro vazio, em gramas M1 = massa do picnómetro com água purificada, em gramas M2 = massa do picnómetro com a amostra, em gramas
dA= m/v

16
Determinação da densidade em soluções alcoólicas: transfere-se a amostra para uma proveta adequada, ajustando-se a temperatura da amostra de acordo com a especificação do alcoómetro. A seguir, deve-se introduzir o densímetro (também chamado de Alcoómetro Gay-Lussac) na amostra e proceder à leitura na escala do densímetro.
Determinação da densidade por densímetro digital: depois de esperar o aparelho atingir a temperatura determinada pela calibração, injecta-se a amostra com uma seringa, lentamente, tendo o cuidado de não deixar formar bolhas no tubo de vidro. O aparelho, então, realizará a leitura.
2.3.4. Determinação de Materiais Voláteis e Resíduo Seco
Determinada quantidade da amostra, pesada analiticamente, é submetida à secagem em estufa aquecida a uma temperatura preestabelecida (de acordo com as características da amostra), até atingir peso constante. A diferença entre a massa da amostra, antes e depois da secagem, revela a massa dos componentes da formulação que volatilizam ou não naquelas condições. O material remanescente é denominado resíduo seco. Este método fornece resultados numéricos facilmente interpretados, normalmente expressos em percentagem. Cálculo de materiais voláteis:
Onde: MV = materiais voláteis em percentagem mi = massa inicial da amostra em gramas mf = massa final da amostra em gramas
Cálculo do resíduo seco: Onde: RS = resíduo seco em percentagem mi = massa inicial da amostra em gramas mf = massa final da amostra em gramas
2.3.5. Determinação do Teor de Água/ Humidade
Vários são os métodos utilizados para a determinação quantitativa de água em um produto acabado. Os mais usuais são: Método Gravimétrico, Método Destilação em aparelho de Dean & Stark e Método Titulométrico de Karl-Fischer. Esses métodos fornecem resultados numéricos, facilmente interpretados. O método de ensaio dependerá da escolha do equipamento utilizado.

17
2.3.6. Granulometria
Os produtos em forma de pós são constituídos de partículas de diâmetros variados. A proporção de partículas fora dos limites especificados poderá influenciar na aparência, na performance e na cor do produto. Para esse tipo de ensaio, podem ser utilizados os seguintes métodos:
• Tamisação: são utilizados tamis com malhas padronizadas para especificar o tamanho das partículas.
• Análise granulométrica por difracção a laser: utilizada para avaliar partículas de tamanho reduzido.
2.3.7. Teste de Centrífuga
A força da gravidade actua sobre os produtos, fazendo com que suas partículas se movam no seu interior. A centrifugação produz stress na amostra, simulando um aumento na força de gravidade, aumentando a mobilidade das partículas e antecipando possíveis instabilidades. Estas poderão ser observadas na forma de precipitação, separação de fases, formação de sedimento compacto (caking) e coalescência, entre outras. As amostras são centrifugadas em temperatura, tempo e velocidade padronizados. Em seguida, procede-se à avaliação visual. Geralmente, executa-se esse teste em estudos de estabilidade, podendo ser estendido ao controle de processo.
2.4. ENSAIOS QUÍMICOS
Determinação Química – Análise Qualitativa e Quantitativa A análise química é caracterizada como a aplicação de um processo ou de uma série de processos para qualificar e/ou quantificar uma substância ou componentes de uma mistura, ou para determinar a estrutura de compostos químicos. Os métodos de ensaios são diversos, porém no presente guia importa abordar os que tem aplicação corrente em farmácia (Métodos Ópticos e Métodos Electroquímicos). Avaliação dos Resultados Os resultados serão considerados satisfatórios quando as amostras apresentarem valor dentro da especificação estabelecida para o produto. Alguns produtos, em função do risco que podem apresentar, têm limites estabelecidos por regulamentação específica

18

19
MODELO DE RELATÓRIO

20
3.0. MODELO DE RELATÓRIO
I. RESUMO Determinação de ….. existente em (amostra - referir a origem) pela técnica de ……. .
Obteve-se …..± (erro) de produto por …. de amostra.
II. INTRODUÇÃO (1pag)
a) Fundamento do método
b) Expressão que relaciona a grandeza medida com a concentração. Definição dos parâmetros
envolvidos na equação.
c) Limite de detecção da técnica.
III. PARTE EXPERIMENTAL
Alterações introduzidas à técnica; referir as massas e aos volumes utilizados na
preparação dos padrões e das amostras; referência a todas as condições experimentais (por
exemplo: comprimento de onda da determinação, caudal de aspiração em absorção atómica;
tipo de aparelho utilizados e a respectivas marcas.
IV. RESULTADOS
a) Tabelas com resultados (por exemplo grandezas medidas versus volume ou concentração).
b) Gráficos (por exemplo rectas de calibração).
c) Determinação da quantidade de produto referido à amostra (em % sempre que possível)
d) Cálculo dos erros.
V. DISCUSSÃO E CRÍTICA
a)Dos resultados
b) Da técnica
VI. REFERÊNCIAS BIBLIOGRÁFICAS Boletim da Análise Efectuada
Título:
Amostra analisada:
Método/Aparelho:
Resultado Final:
Tabela dos Resultados:
Cp(ppm)

21
Amostra
Gráfico A vs S: ver folha em anexo
Cálculos da concentração na amostra inicial (com erros):
a) Equação da recta de calibração:
b) Concentração da amostra tirada da recta
c) Concentração na amostra inicial
Comentários ao trabalho (resultado obtido):
Nomes

22
MÉTODOS DE CÁLCULO E ERROS EM QUÍMICA
ANALÍTICA
23
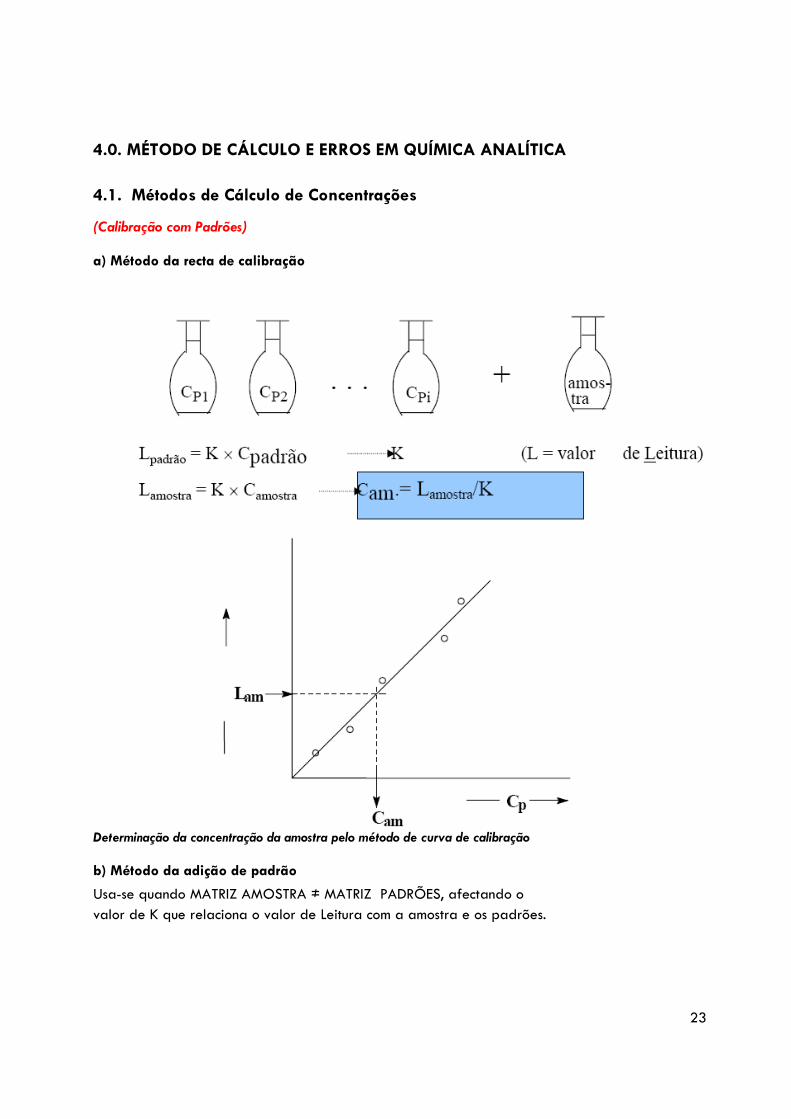
4.0. MÉTODO DE CÁLCULO E ERROS EM QUÍMICA ANALÍTICA
4.1. Métodos de Cálculo de Concentrações
(Calibração com Padrões) a) Método da recta de calibração
Determinação da concentração da amostra pelo método de curva de calibração b) Método da adição de padrão
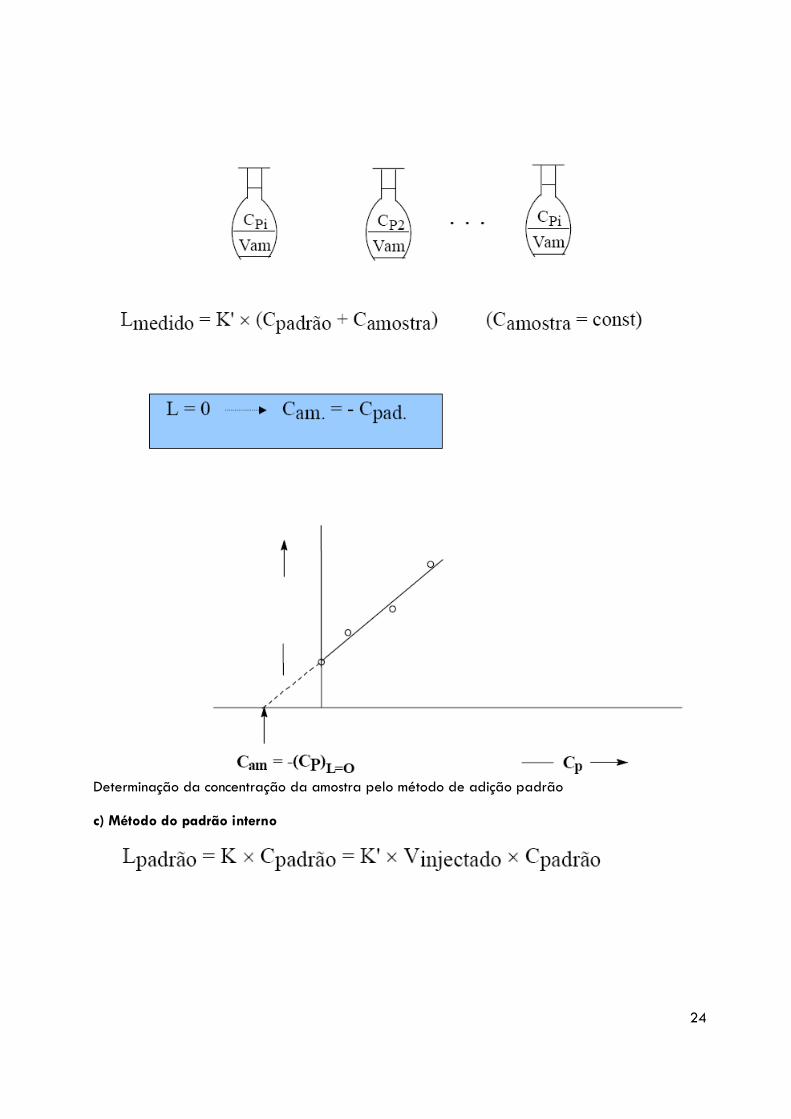
Usa-se quando MATRIZ AMOSTRA ≠ MATRIZ PADRÕES, afectando o valor de K que relaciona o valor de Leitura com a amostra e os padrões.
24
Determinação da concentração da amostra pelo método de adição padrão c) Método do padrão interno

25
Determinação da concentração da amostra pelo método de padrão interno

26
4.3. Precisão, Exactidão, Sensibilidade e Limite de Detecção
a) Precisão
Mede a dispersão dos resultados experimentais em relação à média.
(⇔ ∃ Erros aleatórios)
b) Exactidão
Mede o desvio em relação ao valor real.
(⇔ ∃ Erros sistemáticos)
c) Sensibilidade
Razão entre o acréscimo do valor lido, ΔL, e a variação da concentração ∆C,
correspondente aquele acréscimo (ΔL/ΔC). Se L = F(c) ⇔ relação linear ⇒ (ΔL/ΔC = declive da recta de calibração) c) Limite de Detecção
Menor valor que se pode distinguir do zero
⇓ Menor valor de concentração que um dado método consegue medir.
Avaliação: limite de detecção operacional (L.D.)
4.3.1. Análise estatística dos Erros
a) Recta dos mínimos quadrados e erros associados (programa Excel)
a: declive; σa erro associado. b: ordenada na origem; σb erro associado. Qualquer destes parâmetros é calculado pelo Excel
b) Cálculo do erro na concentração da amostra σx0, a partir da recta dos mínimos quadrados:

27
→→→→Método da recta de calibração
σy/x2 : desvio padrão da estimativa (standard error dado na folha de cálculo do Excel), n = número de pontos experimentais L = número de leituras para cada padrão ou amostra; x = média dos valores de xi; y = média dos valores de yi xi = cada valor de x
→→→→Método de adição de padrão

28
PARTE EXPERIMENTAL

29
MÉTODOS ÓPTICOS

30
5.0. ESPECTROSCOPIA DE ABSORÇÃO MOLECULAR (MAS)
5.1. Determinação Quantitativa de Aspirina, Paracetamol e Cafeína por MAS
Esta experiência tem como objectivo, a determinação quantitativa de aspirina, paracetamol e cafeína nos comprimidos de Anadin-extra, um analgésico à venda nas farmácias, por espectroscopia de absorção molecular (MAS) em UV-Vis. A análise destas drogas por métodos clássicos é quase impossível devido a vários factores com destaque, a fenómeno de hidrólise (aspirina) e a estabilidade dependente do pH ( no caso do paracetamol).
5.1.1. Análise Multicomponentes de um Comprimido
5.1.2. Análise Quantitativa de Anadin-extra
Uma vez que a aspirina e o seu produto de hidrólise (ácido salicílico) têm espectros de absorção do ultravioleta diferentes, a solução do comprimido de Anadin-extra deve ser fortemente básica para assegurar que toda a aspirina se hidrolise e apenas o ácido salicílico está a ser analisado. Do mesmo modo, o paracetamol é sensível ao pH e as formas ácida e básica têm diferentes espectros de absorção no UV, se a solução estiver fortemente básica, o paracetamol está todo na forma básica. Portanto, a pH elevado, a solução do comprimido de Anadin-extra contém ácido salicílico (forma básica), a forma básica do paracetamol e a cafeína.

31
Estes três componentes têm espectros de UV diferentes e por isso eles podem ser analisados simultaneamente, através da medição da absorvância da solução a três comprimentos de onda diferentes e compor-se o sistema de 3 equações. Com efeito a absorvância total da solução comprimido a cada comprimento de onda é devida à soma das contribuições dos três componentes presentes. (propriedade aditiva da Lei de Beer)
em que 1, 2 e 3 são os três comprimentos de onda aos quais se fazem as medições; CA, CP, e Cc são as concentrações de aspirina, paracetamol e cafeína, respectivamente; os e são as nove absortividades das três componentes aos três comprimentos de onda.
O sistema das três equações pode ser resolvido com a calculadora ou usando matemática de matrizes para resolver uma equação matricial.
5.2. TÉCNICA EXPERIMENTAL
A- Preparação das Soluções
1) Solução 0.1M NaOH (500 mL) 2) Soluções de aspirina (padrão) (a) Solução de Aspirina 1,000 g / L
Pese rigorosamente cerca de 1,000 g de ácido acetilsalicílico (aspirina) num copo, adicione alguns mL de metanol (cerca de 10mL) para dissolver o sólido, transfira para um balão aferido de 1 L, e dilua com água destilada até ao traço e agite bem. Solução de Trabalho A
Prepare uma solução em que toda a aspirina tenha sido hidrolisada a ácido salicílico pipetando 5 mL da solução (a) para um balão volumétrico de 100 mL, adicionando 40 mL de solução 0.1M NaOH e diluindo até ao traço com água destilada. Agite bem e deixe em repouso 10 min. para assegurar que toda a aspirina é hidrolisada a ácido salicílico. (b) Solução de paracetamol 1,000 g / L
Pese rigorosamente cerca de 1,000 g de paracetamol (4-acetamidofenol) para um copo, adicione alguns mL de metanol (cerca de 10 mL)até dissolver e transfira para o balão de 1L, diluindo com água (agite bem). Solução de trabalho B
Prepare uma solução em que todo o paracetamol esteja na forma básica, pipetando 2 ml de solução (b) para um balão volumétrico de 100 ml, adicionando 40 ml de solução 0.1M NaOH, perfazendo o volume com água destilada.

32
(c) Solução de cafeína a 1,000 g / L
Proceda como nas soluções a e b Solução de trabalho C
Proceda como nas soluções a e b (d) Soluções de Anadin-Extra
Solução inicial (100mg / 100 mL)
Esmague um comprimido num almofariz até obter um pó muito fino. Pese rigorosamente cerca de 100 mg do pó num copo, adicione alguns mL de metanol e agite durante alguns minutos. Transfira então para o balão volumétrico e dilua até ao traço com água. Solução de trabalho D
Filtre a solução inicial, pipete 2 mL desta solução para um balão de 100 mL, perfazendo o volume com água destilada. Esta solução é instável e deve ser usada apenas até 1-2 h depois de ser preparada. Calcule a média da massa dos comprimidos de Anadin-extra
Pese 7 comprimidos e calcule a média. Solução padrão
Prepare em balões volumétricos de 50 mL, 4 soluções padrão para a aspirina, paracetamol e cafeína, a partir das correspondentes soluções de trabalho, pipetando 5, 10, 20 e 25 mL para balões volumétricos de 50 mL e diluindo até ao traço com água destilada.
5.3. PARTE INSTRUMENTAL
1. Ligue o aparelho e faça acerto do zero, do aparelho com a solução do branco nas 2 células (fazer o varrimento de comprimento de onda na gama 230-340 nm e absorvância 0-0.5).
2. Para um resto das medições deixar a solução de branco na célula de referência (posição posterior).
3. Para cada um dos componentes, traçar o espectro da solução mais concentrada dos padrões. Determine o comprimento de onda de máxima absorção para cada um dos componentes o qual deverá ser cerca de 296 nm (aspirina, na verdade é ácido salicílico), 257 nm (paracetamol na forma básica) e 273 nm (cafeína).
4. Para cada componente meça as absorvâncias dos padrões e amostra aos três comprimentos de onda determinados anteriormente. A absorvância amostra deverá cair dentro da gama de calibração, diluindo se necessário.
33
5.4. CÁLCULOS
1)Trace as 3 curvas de calibração para cada um dos três componentes (use
concentrações em mg/L) e calcule as correspondentes 3×3 absortividades (ε). Escreva o sistema das 3 equações lineares a três incógnitas. Para o resolver, construa a matrix A das absortividades correspondentes ao sistema , calcule as concentrações dos três componentes na solução do comprimido. Calcule a concentração (mg/comprimido) dos três componentes no comprimido.
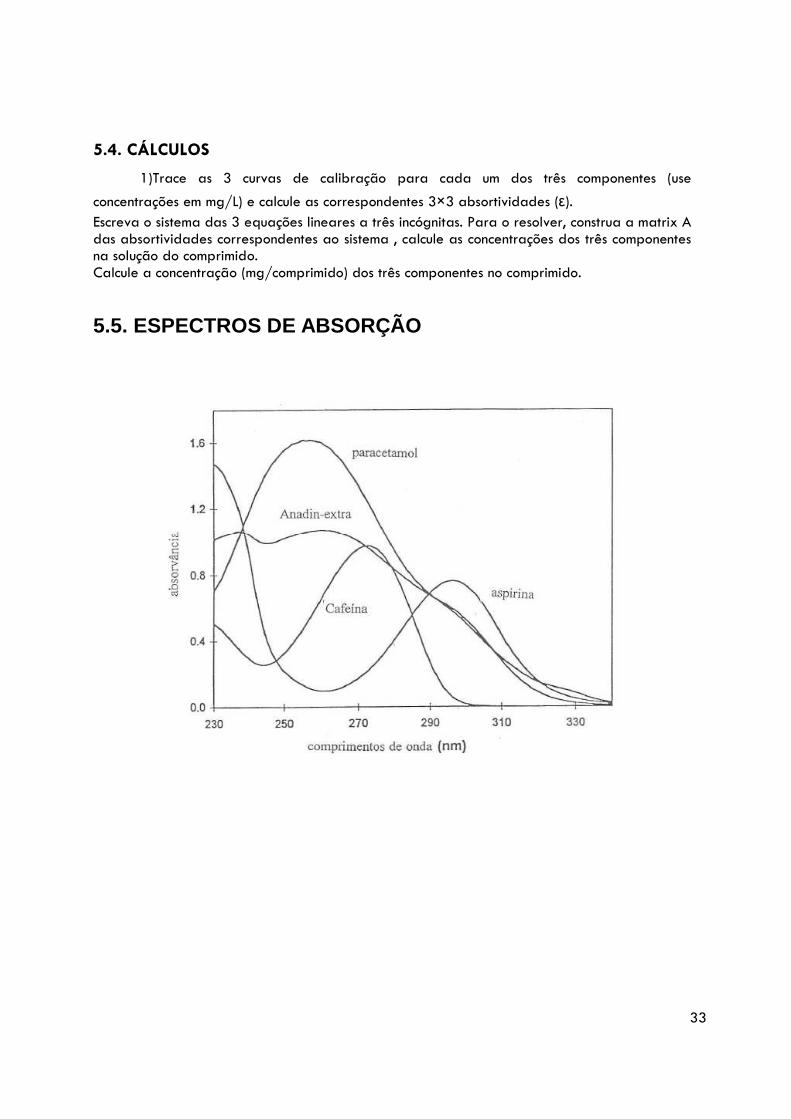
5.5. ESPECTROS DE ABSORÇÃO

34
5.6. BIBLIOGRAGIA
1) M.L. Simões Gonçalves, Métodos Instrumentais de Análise de Soluções, Fundação Calouste Gulbenkian,1889, cap.2. 2) D. A. Skoog and J.J. Leary, Principles of Instrumental Analysis, 4th Edition, Saunders, 1992, cap. 7 (pág. 123-149) e cap. 8 pág. 150-175. 3) J. Kerkay, J. Chem. Ed., 1979, 56, 331-333.

35
6.0. ANÁLISE DE ASPIRINA OU PARACETAMOL NUM COMPRIMIDO
6.1. Análise quantitativa de um comprimido contendo apenas um componente
Uma vez que a aspirina e o seu produto de hidrólise (ácido salicílico) têm espectros de absorção do ultravioleta diferentes, a solução do comprimido deve ser fortemente básica para assegurar que toda a aspirina se hidrolise e apenas o ácido salicílico seja objecto de análise. Do mesmo modo, o paracetamol é sensível ao pH e as formas ácida e básica têm diferentes espectros de absorção no UV, o facto da solução estar fortemente básica, assegura que este esteja todo na forma básica. Portanto, a pH elevado, a solução do comprimido em estudo contém ou o ácido salicílico (forma básica) ou a forma básica do paracetamol. O método espectrofotométrico que vamos utilizar para a determinação de um destes componentes (acido acetilsalicílico ou paracetamol) baseia-se no facto destes apresentarem absorção de radiação electromagnética UV, devendo a absorvância medida seguir a lei de Lambert-Beer:
A = εbc A - absorvância
ε - absortividade molar b - espessura da célula c - concentração molar da espécie
É importante realçar que, neste tipo de determinação, em que apenas se pretende dosear um componente, a amostra a utilizar não poderá conter simultaneamente os dois componentes acima referidos (nem outros constituintes que apresentem absorções de radiação na gama do espectro de absorção do componente em estudo, como ex. a cafeína, ver fig.). Neste caso ter-se-ia que optar por uma determinação simultânea, tendo em conta a aditividade da lei de Beer.

36
6.2. TÉCNICA EXPERIMENTAL
A- Preparação das Soluções
A-1- Soluções para a determinação da aspirina
1) Solução 0.1 M NaOH (500 mL) 2) Soluções de aspirina 2-1- Solução inicial (1,000 g/ L)
Pese rigorosamente cerca de 1,000 g de ácido acetilsalicílico (aspirina) num copo, adicione alguns mL de metanol (cerca de 10 mL) para dissolver o sólido, transfira para um balão aferido de 1 L, e dilua com água destilada até ao traço e agite bem. 2-2 – Solução de trabalho
Prepare uma solução em que toda a aspirina tenha sido hidrolisada a ácido salicílico pipetando 5 mL da solução de partida para um balão volumétrico de 100 mL, adicionando 40 mL de solução 0.1 M NaOH e diluindo até ao traço com água destilada. Agite bem e deixe em repouso 10 min. para assegurar que toda a aspirina é hidrolisada a ácido salicílico. A-2 - Soluções para a determinação do paracetamol
1) Solução 0.1 M NaOH (500 mL) 2) Soluções de paracetamol 2-1 – Solução inicial (1g/L)
Pese rigorosamente cerca de 1 g de paracetamol (4-acetamidofenol) para um copo, adicione alguns mL de metanol (cerca de 10 mL)até dissolver e transfira para o balão de 1L, diluindo com água (agite bem). 2-2 – Solução de trabalho
Prepare uma solução em que todo o paracetamol esteja na forma básica, pipetando 2 mL de solução inicial de paracetamol para um balão volumétrico de 100 mL, adicionando 40 mL de solução 0.1 M NaOH, e depois diluindo até ao traço com água. A-3– Soluções de Comprimido
1) Solução inicial (100 mg/100 mL) Esmague um comprimido num almofariz até obter um pó muito fino. Pese rigorosamente cerca de 100 mg do pó num copo, adicione alguns mL de metanol e agite durante alguns minutos. Transfira então para o balão volumétrico e dilua até ao traço com água. 1-2 - Solução de trabalho
Filtre a solução inicial, pipete 2 mL desta solução para um balão de 100 mL, e proceda como em 2-2. Esta solução é instável e deve ser usada apenas até 1-2 h depois de ser preparada.

37
A-4– Soluções Padrão
Em balões volumétricos de 50 mL, prepare 5 soluções padrão para a aspirina e paracetamol, a partir das correspondentes soluções de trabalho, pipetando 5, 10, 15, 20 e 25 mL para balões volumétricos de 50 mL e dilua até ao traço com água destilada.
6.2.1. Parte Instrumental
1. Ligue o aparelho e faça o acerto do zero do aparelho com a solução do branco nas duas células. O aparelho traça uma linha de base recta (fazer o varrimento de comprimento de onda na gama 230-340 nm e absorvância 0.0-0.5). Para o resto das medições deixar a solução de branco na célula de referência (posição posterior).
2. Trace o espectro da solução mais concentrada dos padrões. Determine o comprimento de onda de absorção máxima do componente em estudo, o qual deverá ser cerca de 296 nm (aspirina, na verdade é ácido salicílico) e 257 nm (paracetamol na forma básica).
3. Para o componente a dosear, meça as absorvâncias dos padrões e da amostra ao comprimento de onda determinado anteriormente. A absorvância da amostra deverá cair dentro da gama de calibração, diluindo se necessário.
4. Para a determinação da concentração na amostra, calcule a massa dos comprimidos (pese 7 comprimidos e calcule a média).

38
6.3. BIBLIOGRAFIA
1) M.L. Simões Gonçalves, Métodos Instrumentais de Análise de Soluções, Fundação Calouste Gulbenkian,1989, cap.2. 2) D.A. Skoog and J.J. Leary, Principles of Instrumental Analysis, 4th Edition, Saunders, 1992, cap. 7 (pág. 123-149) e Cap. 8, pág. 150-175. 3) J. Kerkay, J. Chem. Ed., 1979, 56, 331-333.

39
7.0. ESPECTROSCOPIA DE ABSORÇÃO ATÓMICA (AAS)
7.1. Determinação de Cálcio no sumo por AAS
A absorção atómica é um método de chama em que esta é usada para atomizar uma solução, mas não para excitar os átomos a um estado de energia mais elevado, ao invés do que sucede na fotometria de chama. O facto de a chama ser apenas utilizada para atomizar a solução elimina parte das interferências presentes nesta última técnica.
A excitação é, no caso da absorção atómica, produzida por uma lâmpada cujo cátodo emissor é feito do elemento a determinar e emite radiações com as frequências características desse elemento. Os átomos neutros, livres, no estado de mais baixa energia existentes na chama podem absorvê-las, incluindo a frequência de ressonância correspondente à transição do estado fundamental para o nível excitado de mais baixa energia. Estes átomos excitados voltam depois ao estado fundamental, podendo emitir energia radiante com a frequência de ressonância, mas esta é transmitida em todas as direcções e a fracção detectada pelo espectro fotómetro não produz perturbação notável na medida da radiação não absorvida. A radiação que passou através da chama, com o elemento a determinar e com o solvente, vai em seguida a um sistema de selecção de comprimentos de onda e depois a sua intensidade é medida num detector, determinando-se qual a percentagem que foi absorvida. Num espectro fotómetro de absorção atómica, munido dum registador, mede-se a absorção em relação à linha de base traçada com o solvente. A absorvância e a concentração estão relacionadas pela lei de Lambert-Beer.
em que I - intensidade de radiação transmitida I - intensidade de radiação incidente T - transmitância A - absorvância a - absortividade (constante para cada espécie absorvente) b - espessura do meio absorvente (neste caso a chama) c - concentração
7.2. PARTE EXPERIMENTAL
7.2.1. Material e aparelhagem
- material corrente de laboratório - espectro fotómetro de absorção atómica - lâmpada de cátodo oco de cálcio - centrífuga 7.2.3. Soluções a preparar
• 1000ml de solução 500 μg/ml em Ca, a partir de carbonato de cálcio (adicione HCl gota a gota até dissolver).

40
• 250 ml solução 100 μg/ml em Ca, a partir da solução anterior. • 100 ml solução 5 % em La, a partir de óxido de lantâneo (La2O3 , adicione gota a gota
até se dissolver). - HCl (6 M). 7.2.4. Técnica
Preparação da amostra
Filtre cerca de 50 ml de sumo por um filtro de poros apertados. Se necessário centrifugue previamente a amostra de modo a obter um líquido límpido. Determinação do cálcio
Em balões de 100 ml prepare uma gama de 5 soluções-padrão de concentração compreendida
entre 0 e 7 ppm em Ca a partir da solução 100μg/ /ml. Adicione a todas elas 10 ml da solução de La, 20 ml da solução de HCl e 5 ml de sumo. Prepare ainda um branco para calibrar o aparelho. Determine o teor em Ca no sumo pelo método de adição de padrão.

41
7.3. BIBLIOGRAFIA
o N. Strohl, J. Chem. Educ., vol. 62, nº 4, pág. 343 (1985). o M. Lurdes S. S. Gonçalves, Métodos Instrumentais para Análise de Soluções,
Ed.Fundação Calouste Gulbenkian, Lisboa (1983).

42
MÉTODOS ELECTROQUÍMICOS

43
1.0. TITULAÇÕES CONDUTIMÉTRICAS E POTENCIOMÉTRICAS
APLICADAS A REACÇÕES ÁCIDO-BASE
1.1. OBJECTIVO DO TRABALHO
a) Traçado de curvas de titulação ácido-base utilizando o método conductimétrico e o método potenciométrico. b) Determinação do teor em ácido acetilsalicilico de um comprimido de aspirina. c) Titulação de um aminoácido utilizando simultaneamente o método conductimétrico e o método potenciométrico.
Solução padrão é uma solução cuja concentração é conhecida com rigor. Esta pode ser preparada a partir de uma substância primária. Na aferição de soluções básicas, utiliza-se com frequência o ácido ftalalato de potássio, que é uma substância primária.
Os comprimidos de aspirina são constituídos por ácido acetilsalicílico, um ácido monoprótico com a seguinte fórmula molecular:
O seu teor por comprimido será determinado por titulações com base forte - NaOH -
usando para detecção do ponto final de titulação uma mistura de indicadores - azul de bromotimol e vermelho de fenol Indicador pH Viragem de cor - azul de bromotimol 6.0 - 7.6 amarelo para azul
- vermelho de fenol 6.8 - 8.4 amarelo para vermelho
- mistura dos dois indicadores 7.5 amarelo para roxo
- fenolftaleina 8.3 – 10 incolor para carmim

44
Na segunda parte deste trabalho será efectuada uma titulação conductimétrica- glicina.
A glicina é um aminoácido não polar, com um ponto isoeléctrico de 5.97.
A combinação de dois métodos (conductimetria e potenciometria) na titulação da glicina com hidróxido de sódio permite obter mais informação acerca do aminoácido, além de ilustrar as vantagens de ambos os métodos. Em solução aquosa a glicina existe em três formas: a forma dibásica, completamente protonada, +NH3CH2COOH; a forma monoprótica, zwiteriónica, +NH3CH2COO- e a forma aniónica, NH2CH2COO- .A concentração relativa destas espécies depende do valor de pH da solução. A titulação da glicina completamente protonada com NaOH produz uma curva de titulação de pH típica de um ácido diprótico. Através desta curva é possível calcular as duas constantes de dissociação (K1 e K2) e a concentração original de glicina. A titulação conductimétrica origina igualmente uma curva com dois pontos de equivalência. A combinação da informação obtida de ambas as curvas permite a determinação dos valores de conductividade equivalente das espécies de glicina carregada.
3. PARTE EXPERIMENTAL 3.1. Material e reagentes
• Conductímetro • Célula conductimétrica • Medidor de pH e eléctrodo de pH • Agitador electromagnético • Comprimido de aspirina • Água destilada • Soluções já preparadas: - Glicina / HCl 0,03 M; a preparar na altura - Indicadores azul de bromotimol e vermelho de fenol
Soluções a preparar:
• Hidróxido de sódio 0,1 M • **Ftalato ácido de potássio 0,5 M • Ácido clorídrico 0.01M ** Solução rigorosa, para isso utilizar a balança analítica para a pesagem do produto sólido se necessário. Tome nota da quantidade de produto pesado.

45
3.2 Técnica experimental
3.2.1. Padronização da solução de NaOH
Use a solução de ftalato ácido de potássio para padronizar a solução de hidróxido de sódio 0.1M, titulando potenciometricamente 20 ml da solução de ftalato ácido 0,05 M com a solução de NaOH. Trace a curva potenciométrica e determine o ponto de equivalência. 3.2.2. Titulação conductimétrica HCl – NaOH
Num copo de 250 ml, adicione 100 ml de ácido clorídrico 0,01 M e 50 ml de água destilada. Titule com hidróxido de sódio 0,1 M. Trace a curva de titulação conductimétrica e determine o ponto de equivalência. 3.2.3. Titulação do ácido acetilsalicílico
a) Dissolva, a quente, um comprimido de aspirina com cerca de 100 ml de água destilada; deixe arrefecer e filtre para um balão aferido de 500 ml, fazendo repetidas lavagens do resíduo com água destilada. Perfaça o volume.
b) Em seguida, titule em duplicado 100 ml da solução de aspirina, com uma solução de hidróxido de sódio ~0,05 M (título rigoroso determinado anteriormente). Use 1 ml da mistura de indicadores, azul de bromotimol + vermelho de fenol, cuja viragem se dá de amarelo para roxo. 3.2.4. Titulação da glicina
Num copo de 250 ml, adicione 50 ml de água destilada a 50 ml de solução de glicina 0,03 M. Titule com NaOH 0,1 M. Esta titulação deve ser efectuada utilizando simultaneamente ambos os métodos, potenciométrico e conductimétrico.

46
Titulação : Hidrogenoftalato de potássio/ NaOH
V.Titulante
pH
dpH
dV
dpH/dV

47
Titulação : HCl / NaOH
V.Titulante Condutância mhocm -1

48
Titulação : Glicina / NaOH
V titulante
pH
Condutância mhocm -1

49
4. QUESTIONÁRIO
4.1. Traçar em papel milimétrico, escolhendo uma escala apropriada, a curva "pH/Volume de titulante" para a titulação potenciométricas efectuada (titulação hidróxido sódio/ftalato ácido de potássio). Determinar o ponto de equivalência pelo método da primeira derivada (máximo da curva que representa dpH/dV em função de V, sendo V o volume de titulante adicionado). Para isso, calcular para cada par de valores da curva e representar este valor em função do volume. Sobreponha a curva da derivada à curva de titulação. 4.2. Cálculo do pH ao longo da curva de titulação e comparação com os valores experimentais. Apresente os cálculos que efectuar para o preenchimento da tabela seguinte.
4.3. Determine o título exacto das soluções:
• Ftalato ácido de potássio • Hidróxido de sódio
4.4. Traçar em papel milimétrico, escolhendo uma escala apropriada, a curva
"condutividade/Volume de titulante" para a titulação conductimétrica efectuada (titulação HCl / NaOH). Determinar o ponto de equivalência. 4.5. Aspirina
• Peso do comprimido: • Teor em aspirina (% p/p):
4.6 Traçar em papel milimétrico as curvas de titulação da glicina com NaOH, em função do pH e da condutividade. 4.6.1. Calcular a concentração inicial de glicina e os valores das constantes de dissociação. Comparar com os valores da literatura. 4.6.2. Calcular a condutividade molar para as espécies GlyH2+ e Gly-. Comparar com os valores da literatura.

50
5. BIBLIOGRAFIA
[1] M.L.Gonçalves, "Métodos Instrumentais para Análise de Soluções", Edição da Fundação Calouste Gulbenkian, Lisboa. [2] Rosenthal, L.C. and Nathan, L.C. (1981) J.Chem.Ed. 58, 656-658. [3] Calhorda, M.J. (1993) Boletim da Sociedade Portuguesa de Química, 51, 56-58.





























