Embed Size (px)
Citation preview

Hemofilia, Malaltia de Von Willebrand i altres
coagulopaties congènites
Dra. Cristina Marzo
Laboratori Clínic ICS Lleida
Secció d´hemostàsia
Hospital Universitari Arnau de
Vilanova de Lleida

COMPONENTES DE LA HEMOSTASIA

CONDICIONES PREANALíTICAS
• Información del paciente.
• Edad y sexo.
• Datos clínicos de interés en el momento de la
extracción:
– Hepatopatia o síndrome nefrótico
– Medicación: anticoagulantes orales, heparina
convencional o de bajo peso molecular,
aspirina.
– Información clínica del episodio.

CONDICIONES PREANALíTICAS
•Punción “limpia”.
•Tubo de recogida específico.
•Anticoagulante: citrato sódico 130 mM, 1 parte
de citrato y 9 de sangre. Concentración final: 13 mM.
•No de catéteres heparinizados.
•Se ha de mezclar inmediatamente.

CONDICIONES PREANALíTICAS
• Conservación de la sangre.
• Se conserva a temperatura ambiente.
• Ha de llegar al laboratorio en un tiempo
máximo de unas 2 horas.

CONDICIONES PREANALíTICAS
• Comprobaciones en el laboratorio:
identificación, etiquetaje…
• Tubo de recogida sea el correcto (Citrato).
• Volumen de sangre sea el correcto.
• Que la sangre no tenga coágulos.

CONDICIONES PREANALíTICAS
• Preparación del plasma.
• Centrifugar la sangre 1500 g (2500 rpm) 20
minutos a temperatura ambiente 18-25ºC.
• Separar el plasma.
• Observar el aspecto: lipémico, hemolizado, coagulado.
• Utilizar el plasma el mismo día o congelarlo.
• Descongelar una muestra, tecnicarla y no volver a congelarla.

ESTUDIO BÁSICO DE HEMOSTASIA
• Recuento plaquetar.
• T. de Protrombina (TP): Sensible a factores II,V, VII y X.
• T. de Tromboplastina (APTT): sensible a los factores: VIII, IX, XI, XII, precalicreina y Quininógeno.
• Fibrinógeno/ Tiempo de trombina.
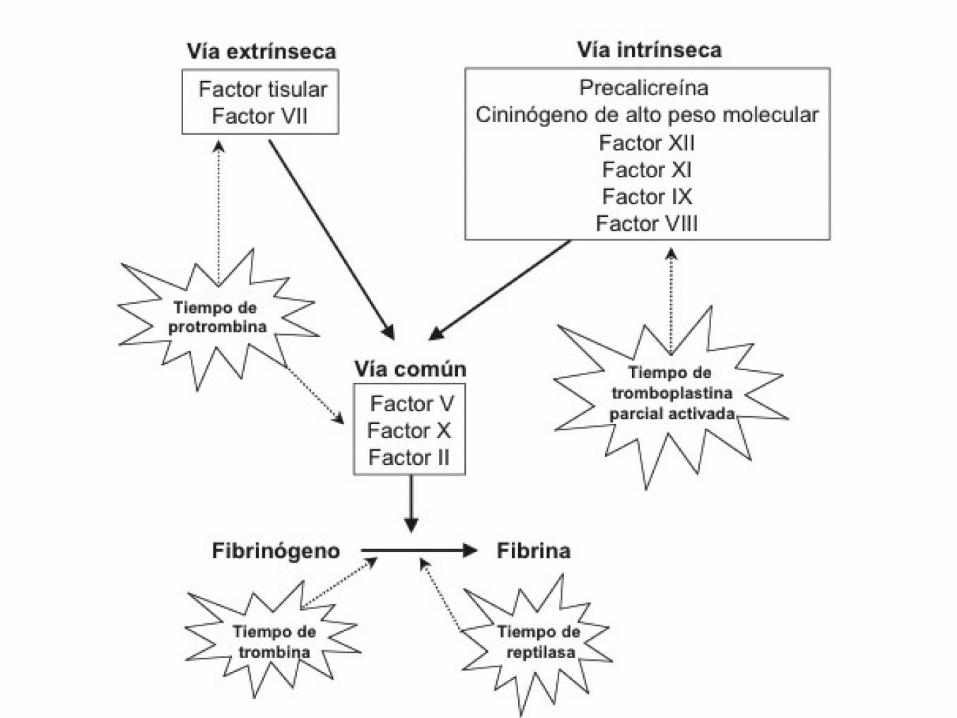

T.Tromboplastina / T. Cefalina/ APTT
Factores VIII, IX, XI , XII
• Plasma.
• Activador: sílica, ácido elágico, caolín etc
reactivo.
• Fosfolípidos (Cefalina).
• Calcio.
• Expresión de los resultados ratio: segundos
paciente/segundos control.

T DE PROTROMBINA
Factores II, V, VII, X
• Plasma.
• Factor tisular.
• Fosfolípidos tromboplastina.
• Calcio.
• Expresión de los resultados: ratio vs
INR:(segundos paciente / segundos control)ISI.

TIEMPO DE TROMBINA
•Plasma + trombina.
•Es el tiempo de coagulación obtenido al añadir
una concentración baja de trombina al plasma (1
U NIH/ml).
•Es sensible a la heparina (Tiempo de reptilase).
•Se alarga en las hepatopatias y en les hipo o
disfibrinogenemias.

TIEMPO DE REPTILASE
•Reptilase: veneno de serpiente que transforma el fibrinógeno en fibrina pero no es sensible a heparina.
•Se alarga en las hepatopatias y en les disfibrinogenemias.
•Es útil para descartar la presencia de heparina en la muestra (contaminación).

FIBRINÓGENO
•Plasma + Trombina
•Al añadir una concentración elevada de trombina
a un plasma, coagula en un tiempo que es
inversamente proporcional a la concentración de
fibrinógeno en la muestra.
•Fibrinógeno Clauss.
•Fibrinógeno derivado (calculado).
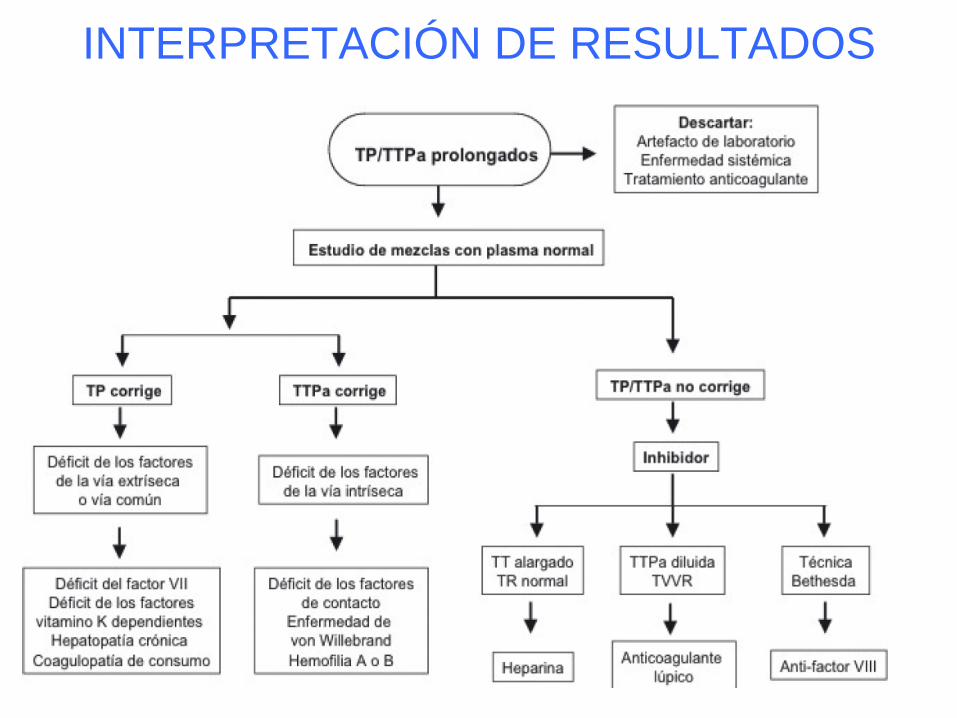
INTERPRETACIÓN DE RESULTADOS

APTT alargado
• Comprobar que el/la paciente no reciba
heparina ni anticoagulantes orales.
• Comprobar el alargamiento con una segunda
determinación (misma/nueva muestra).
• Estudio de APTT alargado:
– Anticoagulante lúpico,
– factores de la via intrínseca: VIII, IX, XI, XII,
– si procede estudio de inhibidores específicos.

Tiempo de protrombina alargado
•Comprobar que el paciente no reciba heparina ni
anticoagulantes orales.
•Estudio de TP alargado:
– factores de la via extrínseca: II, V, VII, X.
– si procede estudio de inhibidores específicos
•Comprobar que no tenga un AL.

Anticoagulante Lúpico
• No se asocia a sangrado sino a trombofilia.
• Se han descrito acciones sobre:
– Plasma: inhibición de la via de la Proteina C.
– Endotelio y monocitos: incremento del factor
tisular y disminución de síntesis de
prostaciclina.
– Plaquetas: incremento de síntesis de
tromboxano.

FACTORES VIA INTRÍNSECA
• Factores de la via intrínseca: VIII, IX, XI, XII.
• Plasma del paciente + plasma deficiente en el factor a determinar
• Cefalina (activador y fosfolípidos)
• Ca+2

FACTORES VIA EXTRÍNSECA
• Factores de la via extrínseca: II, V, VII, X
• Plasma del paciente + plasma deficiente
en el factor a determinar.
• Tromboplastina Ca+2

Factor Von Willebrand
• Se sintetiza en las células endoteliales y en los
megacariocitos y se acumula en los gránulos alfa
de las plaquetas.
• Forma puentes entre las plaquetas y el
subendotelio iniciando la agregación plaquetar.
• Forma un complejo con el factor VIII y lo
estabiliza.

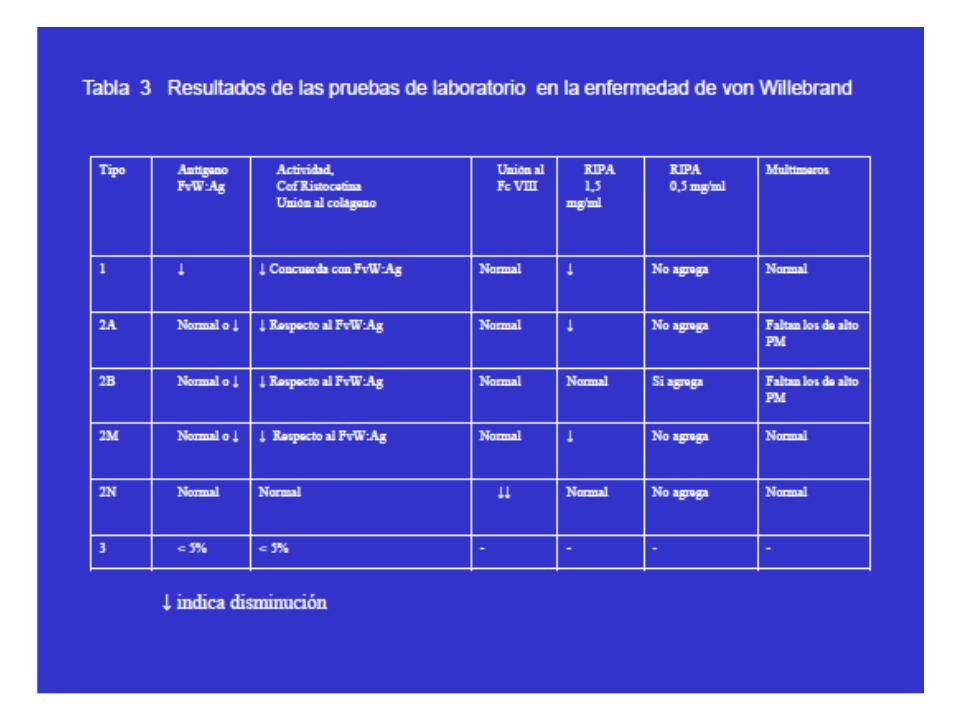
Valoración Factor von Willebrand
1) Valoración Immunológica (FvWAg): immunoturbidimetria
(latex).
2) Valoración funcional:
– Determinación cofactor ristocetina.
– Activitat FvW (receptor de la GPIb)
– Cocientes FvW:RCo/FvW:Ag; FVIII:C/FvW:Ag
– Pruebas diagnósticas avanzadas o de confirmación

CASO CLÍNICO
• Niño de 14 meses derivado a CCEE de
Hemostasia para estudio de r-PTTA alargada.
• Hematomas ante mínimos traumatismos desde
el nacimiento. No sangrados mucosos ni
hemartros.
• No antecedentes familiares de diátesis
hemorrágica (++++).

Analítica sanguínea
Hemograma

Estudio de hemostasia básico

Dosificación de factores

Estudio de hemostasia: estudio de inhibidores

Dosificación de factores: estudio de inhibidores

HEMOFILIA A : diagnóstico genético

HEMOFILIA: ENFERMEDAD DE REYES

SOSPECHA CLÍNICA
Varón Edad temprana de la vida
Historia de diátesis hemorrágica familiar
Clínica de sangrado inusual

DATOS DE LABORATORIO
• TP normal
• PTTA alargada
• Dosificación de factores: VIII, IX, XI y XII
Hemofilia A: F VIII
Hemofilia B: F IX
• Descartar presencia de inhibidores

HEMOFILIA CONGÉNITA
• Diátesis hemorrágica hereditaria recesiva ligada al
cromosoma X.
• Hemofilia A (F VIII) > Hemofilia B (F IX)
• Varones afectos, mujeres portadoras
(excepciones)
• Frecuencia: 1/10000 nacimientos
– HA: 1/5000 varones
– HB: 1/30000 varones
– Afectos mundialmente +/- 400000 (0.05%)

FACTOR VIII y FACTOR vW
• En plasma como precursor inactivo unido a
FvW.
• El FvW protege al FVIII de su proteolisis y
aclaramiento, evita su unión prematura a los
fosfolípidos asegurando la correcta
localización del FVIII en las zonas donde debe
ejercer su función.

DIAGNÓSTICO HEMOFILIA A
ALTERACIONES MOLECULARES MÁS FRECUENTES: - 45-50% inversión intrón 22 - 2-5% inversión intrón 1 - 5-10% grandes deleciones (1 o > exones) - Mutaciones puntuales
Base de datos de las mutaciones: http://www.factorviii-db.org/
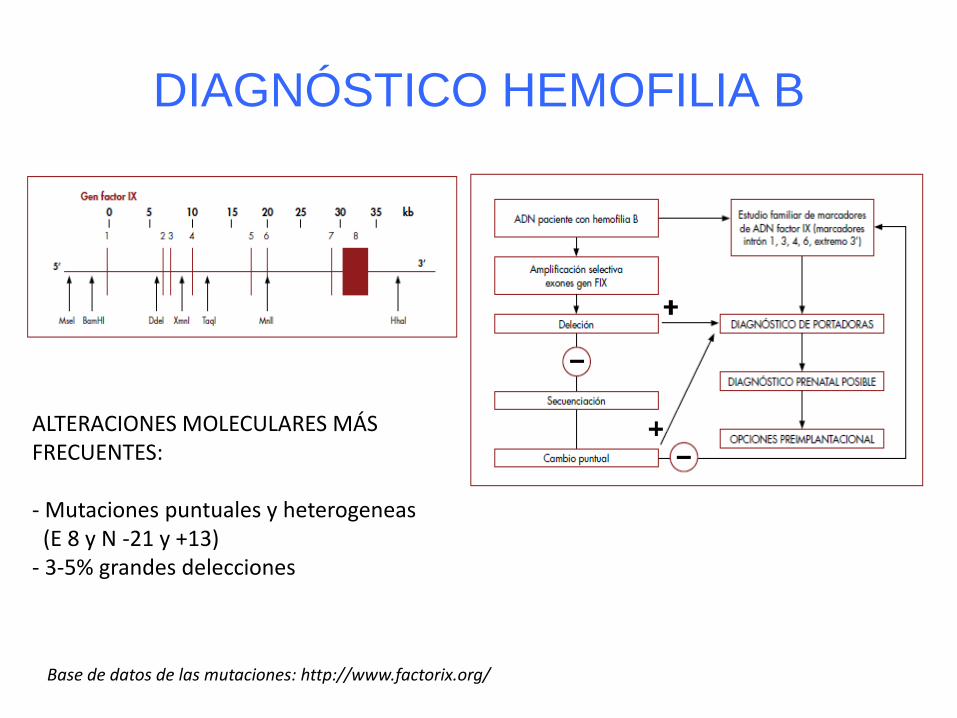
DIAGNÓSTICO HEMOFILIA B
ALTERACIONES MOLECULARES MÁS FRECUENTES: - Mutaciones puntuales y heterogeneas (E 8 y N -21 y +13) - 3-5% grandes delecciones
Base de datos de las mutaciones: http://www.factorix.org/

HERENCIA DE LA HEMOFILIA

TIPOS DE PORTADORAS
PORTADORAS SEGURAS POSIBLES PORTADORAS
• Mujer con un solo hijo
hemofílico sin
antecedentes familiares
de hemofilia
• Hijas de mujer portadora
segura
• Mujer con historia familiar
de varón hemofílico por
via materna
• Hijas de varón hemofílico
• Mujer con hijo hemofílico
y antecedente de varón
hemofílico por vía
materna
• Mujer con más de un hijo
hemofílico (excepto
embarazo gemelar)

TIPOS Y GRADOS DE HEMOFILIA
• A: Factor VIII
• B: Factor IX
• Grave <1%
• Moderada 1-5%
• Leve 5-30%

CLASIFICACIÓN DE LA HEMOFILIA

HEMORRAGIAS
• Grave • espontáneas
• mínimos traumatismos
• Moderada • traumatismos leves o moderados
• Leve • asintomática
• estudio familiar
• estudio de hemostasia
• sangrado postquirúrgico
Las manifestaciones hemorrágicas en la hemofilia, dependientes de la gravedad de
la deficiencia, se caracterizan por sangrado espontáneo (principalmente articular y
muscular) y sangrado grave tras cirugía o traumatismo.

CARACTERÍSTICAS DE LOS SANGRADOS

LOCALIZACIONES RELEVANTES
- Hemorragia en suelo de boca y laringe: obstrucción
aerea.
- Cadera: necrosis aséptica.
- SNC: vigilar recaidas.
- Hematuria: descartar otras causas.
- Músculo psoas: sd. compartimentales.

RECOMENDACIONES BÁSICAS:
- No AINES ni AAS.
- Utilización de casco protector.
- Vacunas VHA y VHB sc (basal, 1m, 6m).
- Estudio genético familiar.
- Terapia sustitutiva: dosis de factor para
situación urgente.

ESTRATEGIAS EN LA TERAPIA
SUSTITUTIVA

TRATAMIENTO SUSTITUTIVO DE LOS
EPISODIOS HEMORRÁGICOS AGUDOS
- 1 UI de FVIII por Kg elevará el FVIII circulante
0,02 U/mL
- 1 UI de FIX por Kg elevará el FIX circulante
0,01 U/mL

TRATAMIENTO A DEMANDA EN
SITUACIONES AGUDAS
HEMORRAGIA FACTOR ÓPTIMO (%) F VIII (UI/Kg) FIX (UI/Kg)
Articular 30-50 12-25 30-50
Muscular 30-50 12-25 30-50
Hematuria 30-50 12-25 30-50
Mucosa oral 30-50 12-25 30-50
Epistaxis 30-50 12-25 30-50
Digestivo 40-60 30-40 40-60
SNC 80-100 50 80-100
Retroperitoneal 50-100 30-50 60-100

RIESGO QUIRÚRGICO
• Grave:
– de alto riesgo
• Moderada:
– de alto riesgo
• Leve:
– sangrado postquirúrgico

CIRUGÍA MENOR
• Adenoidectomía
• Implantación de catéter central
• Circuncisión
• Cirugía dental
• Biopsias


Extracción dentaria
• Nivel mínimo: 50%
• Antifibrinolíticos durante 7 días
• Considerar la aplicación de hemostático local


EL RETO: LA PROFILAXIS
- Niños con hemofilia severa
- Eficaz en la prevención de la enfermedad articular
- Recomendada por: WFH y US Hemophilia Foundation
Hemorragias articulares espontáneas
Sinovitis crónica
Artropatia crónica
Discapacidad
Interferencia con la actividad normal
Codos
Rodillas
Tobillos

ESQUEMAS DE PROFILAXIS
- Modelo sueco (dosis altas): Malmö protocol
25-40 UI/Kg administradas 3-2 días/semana
- Modelo holandés (dosis intermedias): Utrecht protocol
15-30 UI/Kg administradas 3-2 días/semana
- Modelo canadiense (dosis escalonadas)
50 UI/Kg/semana
30 UI/Kg/semana
25 UI/Kg/48h
- Modelo farmacocinético (individualizado)



ENF. DE VON WILLEBRAND: definición
• Trastorno hemorrágico hereditario más
frecuente.
• Anomalías cualitativas y/o cuantitativas de
la proteína factor de von Willebrand
(FvW).
• Herencia: autosómica dominante o, con
menos frecuencia, recesiva.

FACTOR VON WILLEBRAND
El FvW es una pieza clave en la interacción endotelio-plaqueta y desempeña
un papel relevante en la formación del trombo en la pared vascular

ENF DE VON WILLEBRAND: generalidades
• Grupo heterogéneo: variabilidad expresión
clínica (gravedad).
• Diversidad presentación en un mismo
enfermo (niveles de F VW reactante fase
aguda).
• Grupo sanguíneo: grupo 0 niveles 25 %
menos.

ENF VON WILLEBRAND: diagnóstico
• Historia clínica de diátesis hemorrágica
(personal y familiar): intensidad, gravedad,
frecuencia y duración.
• Realización del árbol genealógico.
• Pruebas de laboratorio: defectos
cuantitativos y/o cualitativos del F vW.

ENF VON WILLEBRAND: clínica hemorrágica
• Anamnesis sistémica
• Exploración física
• Puntuación de síntomas (baremo hemorrágico)

ENF VON WILLEBRAND: clínica hemorrágica
Petequias
Hematoma
Equimosis

ENF VON WILLEBRAND: clínica hemorrágica
-Hemorragias mucocutáneas:
•epistaxis,
•hematomas ,
•hemorragias cavidad oral,
•menorragias.
-Hemartros y
hematomas partes
blandas: deficiencias severas.

ENFERMEDAD DE VON WILLEBRAND

ENF DE VON WILLEBRAND: diagnóstico
• Pruebas de cribado:
– Recuento plaquetar
– Función plaquetar: TH/TO
– TTPa
– Factor VIII: C
– Factor vW: Ag
– Factor vW: Rco
– Cocientes FvW:RCo/FvW:Ag;FVIII:C/FvW:Ag
– Grupo sanguíneo

ENF VON WILLEBRAND: pruebas avanzadas
• Si está indicado repetir las pruebas
iniciales para detectar VW.
• Multímeros.
• Prueba de unión al colágeno.
• Prueba de unión al factor VIII.
• RIPA.
• Estudio genético.

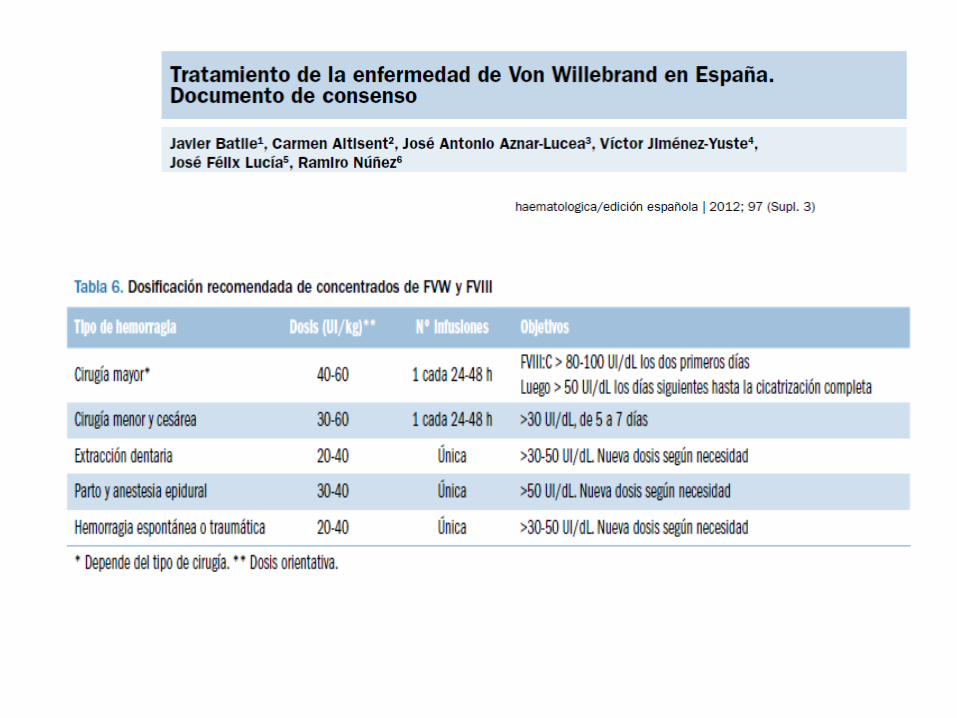
ENF VON WILLEBRAND: TRATAMIENTO
• Diagnóstico correcto
• Historia hemorrágica previa
• Niveles de Factor VIII y VW
• Gravedad y duración del episodio

ENF VON WILLEBRAND: TRATAMIENTO
• Antifibrinolíticos
• DDAVP: vía sc, intravenosa, intranasal.
• Ttº sustitutivo: concentrados de factores:
-Unidades necesarias=peso corporal (Kg) x
aumento deseado de f vW (% o UI/dl) x 0.5.
-En niños dosis un 20% superior a la del adulto,
teniendo en cuenta su mayor volumen
plasmático.

OTRAS COAGULOPATÍAS
• Las deficiencias de factor poco comunes son trastornos
de la coagulación hereditarios por un problema con uno
o varios factores de la coagulación diferentes al factor
VIII y/o IX.
• Las deficiencias de factor poco comunes son trastornos
en los que uno de los factores de la coagulación están
ausentes o no funcionan adecuadamente.
• Los conocimientos sobre estos trastornos son menores
porque se diagnostican muy rara vez.

Niveles descendidos ≠ coagulopatía

OTRAS COAGULOPATÍAS


Niveles y semivida plasmática
Deficiencia Niveles Semivida
Fibrinógeno >1 g/L 2-4 d
Protrombina >10% 3-4 d
V 10% 36 h
VII >20% 2-6 h
V+VIII 40% FV 36 h
F VIII 10-14h
X >40% 40-60 h
XI 15-20% 50 h
XII --- ---
XIII 30% 9-12 d

OTRAS COAGULOPATÍAS































