Embed Size (px)
Citation preview

Estudio de fundamentos y técnicas de simulaciónab-initio.
Aplicaciones al fluoruro de Aluminio α−AlF3
Lic. Jorge Luis Navarro SánchezDirector: Dr. Eduardo A. Albanesi
Seminario, Instituto de Física del LitoralIFIS CONICET-UNL
Octubre 10, 2014

Introducción Resumen
Resumen de la Charla
IntroducciónFundamentos Teóricos
Aproximaciones utilizadasMétodos computacionales.
Caracterización estructural de α−AlF3
Propiedades electrónicas- Corrección GWPropiedades ópticas- Corrección BSEPerspectivas de trabajo.Conclusiones
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 2 / 42

Introducción Objetivos
Objetivos
Como objetivos del trabajo se plantean los siguientes:
Estudiar los fundamentos y los esquemas de modelización ycaracterización ab-initio a nivel de detalle necesario en lananoescala incluyendo las correcciones por las interacciones demuchos cuerpos.Conocer los diferentes métodos con los cuales se implementanlas técnicas ab-initio por medio de dos de los programas másdifundidos al efecto, Wien2k y ABINIT.Aplicar los estudios anteriores para caracterizar y modelar pormedio de herramientas computacionales ab-initio el αAlF3.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 3 / 42

Introducción Objetivos
Objetivos
Como objetivos del trabajo se plantean los siguientes:Estudiar los fundamentos y los esquemas de modelización ycaracterización ab-initio a nivel de detalle necesario en lananoescala incluyendo las correcciones por las interacciones demuchos cuerpos.
Conocer los diferentes métodos con los cuales se implementanlas técnicas ab-initio por medio de dos de los programas másdifundidos al efecto, Wien2k y ABINIT.Aplicar los estudios anteriores para caracterizar y modelar pormedio de herramientas computacionales ab-initio el αAlF3.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 3 / 42

Introducción Objetivos
Objetivos
Como objetivos del trabajo se plantean los siguientes:Estudiar los fundamentos y los esquemas de modelización ycaracterización ab-initio a nivel de detalle necesario en lananoescala incluyendo las correcciones por las interacciones demuchos cuerpos.Conocer los diferentes métodos con los cuales se implementanlas técnicas ab-initio por medio de dos de los programas másdifundidos al efecto, Wien2k y ABINIT.
Aplicar los estudios anteriores para caracterizar y modelar pormedio de herramientas computacionales ab-initio el αAlF3.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 3 / 42

Introducción Objetivos
Objetivos
Como objetivos del trabajo se plantean los siguientes:Estudiar los fundamentos y los esquemas de modelización ycaracterización ab-initio a nivel de detalle necesario en lananoescala incluyendo las correcciones por las interacciones demuchos cuerpos.Conocer los diferentes métodos con los cuales se implementanlas técnicas ab-initio por medio de dos de los programas másdifundidos al efecto, Wien2k y ABINIT.Aplicar los estudios anteriores para caracterizar y modelar pormedio de herramientas computacionales ab-initio el αAlF3.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 3 / 42
Introducción Contexto
Calculos Ab-initio
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 4 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
Hamiltoniano para sistemas de muchos cuerpos. α y β son los núcleos, i y j son loselectrones.
H = −~2
2
∑α
1
mα∇2α −
~2
2me
∑i
∇2i +
∑α
∑β>α
ZαZβe2
~rαβ−∑α
∑i
Zαe2
~riα+∑i
∑i>j
e2
~rij
Energía cinética de los nucleos
Energía cinética de los electrones
Energía potencial debida a la repulsipon nuclear
Energía potencial debida a la atracción electrón-nucleo
Energía potencial debida a la repulsipon electrónica
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 5 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
Hamiltoniano para sistemas de muchos cuerpos. α y β son los núcleos, i y j son loselectrones.
H = −~2
2
∑α
1
mα∇2α −
~2
2me
∑i
∇2i +
∑α
∑β>α
ZαZβe2
~rαβ−∑α
∑i
Zαe2
~riα+∑i
∑i>j
e2
~rij
Energía cinética de los nucleos
Energía cinética de los electrones
Energía potencial debida a la repulsipon nuclear
Energía potencial debida a la atracción electrón-nucleo
Energía potencial debida a la repulsipon electrónica
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 5 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
Hamiltoniano para sistemas de muchos cuerpos. α y β son los núcleos, i y j son loselectrones.
H = −~2
2
∑α
1
mα∇2α −
~2
2me
∑i
∇2i +
∑α
∑β>α
ZαZβe2
~rαβ−∑α
∑i
Zαe2
~riα+∑i
∑i>j
e2
~rij
Energía cinética de los nucleos
Energía cinética de los electrones
Energía potencial debida a la repulsipon nuclear
Energía potencial debida a la atracción electrón-nucleo
Energía potencial debida a la repulsipon electrónica
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 5 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
Hamiltoniano para sistemas de muchos cuerpos. α y β son los núcleos, i y j son loselectrones.
H = −~2
2
∑α
1
mα∇2α −
~2
2me
∑i
∇2i +
∑α
∑β>α
ZαZβe2
~rαβ−∑α
∑i
Zαe2
~riα+∑i
∑i>j
e2
~rij
Energía cinética de los nucleos
Energía cinética de los electrones
Energía potencial debida a la repulsipon nuclear
Energía potencial debida a la atracción electrón-nucleo
Energía potencial debida a la repulsipon electrónica
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 5 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
Hamiltoniano para sistemas de muchos cuerpos. α y β son los núcleos, i y j son loselectrones.
H = −~2
2
∑α
1
mα∇2α −
~2
2me
∑i
∇2i +
∑α
∑β>α
ZαZβe2
~rαβ−∑α
∑i
Zαe2
~riα+∑i
∑i>j
e2
~rij
Energía cinética de los nucleos
Energía cinética de los electrones
Energía potencial debida a la repulsipon nuclear
Energía potencial debida a la atracción electrón-nucleo
Energía potencial debida a la repulsipon electrónica
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 5 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
HΨ(~qi, ~qα) = EΨ(~qi, ~qα)
Considerando que mα me
Hel = −~2
2me
∑i
∇2i −
∑α
∑i
Zαe2
~riα+∑j
∑i>j
e2
~rij
La ecuación de Schrödinger a resolver ahora es:
(Hel − VNN )Ψel = EelΨel
Donde
VNN =∑α
∑β>α
ZαZβe2
~Rαβ⇒ Vext
Born-Oppenheimer, permite desacoplar los movimientos electrónicos y nucleares.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 6 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
HΨ(~qi, ~qα) = EΨ(~qi, ~qα)
Considerando que mα me
Hel = −~2
2me
∑i
∇2i −
∑α
∑i
Zαe2
~riα+∑j
∑i>j
e2
~rij
La ecuación de Schrödinger a resolver ahora es:
(Hel − VNN )Ψel = EelΨel
Donde
VNN =∑α
∑β>α
ZαZβe2
~Rαβ⇒ Vext
Born-Oppenheimer, permite desacoplar los movimientos electrónicos y nucleares.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 6 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
HΨ(~qi, ~qα) = EΨ(~qi, ~qα)
Considerando que mα me
Hel = −~2
2me
∑i
∇2i −
∑α
∑i
Zαe2
~riα+∑j
∑i>j
e2
~rij
La ecuación de Schrödinger a resolver ahora es:
(Hel − VNN )Ψel = EelΨel
Donde
VNN =∑α
∑β>α
ZαZβe2
~Rαβ⇒ Vext
Born-Oppenheimer, permite desacoplar los movimientos electrónicos y nucleares.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 6 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
HΨ(~qi, ~qα) = EΨ(~qi, ~qα)
Considerando que mα me
Hel = −~2
2me
∑i
∇2i −
∑α
∑i
Zαe2
~riα+∑j
∑i>j
e2
~rij
La ecuación de Schrödinger a resolver ahora es:
(Hel − VNN )Ψel = EelΨel
Donde
VNN =∑α
∑β>α
ZαZβe2
~Rαβ⇒ Vext
Born-Oppenheimer, permite desacoplar los movimientos electrónicos y nucleares.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 6 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
HΨ(~qi, ~qα) = EΨ(~qi, ~qα)
Considerando que mα me
Hel = −~2
2me
∑i
∇2i −
∑α
∑i
Zαe2
~riα+∑j
∑i>j
e2
~rij
La ecuación de Schrödinger a resolver ahora es:
(Hel − VNN )Ψel = EelΨel
Donde
VNN =∑α
∑β>α
ZαZβe2
~Rαβ⇒ Vext
Born-Oppenheimer, permite desacoplar los movimientos electrónicos y nucleares.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 6 / 42

Fundamentos teóricos Aproximaciones utilizadas
Aproximación de Born Oppenheimer
HΨ(~qi, ~qα) = EΨ(~qi, ~qα)
Considerando que mα me
Hel = −~2
2me
∑i
∇2i −
∑α
∑i
Zαe2
~riα+∑j
∑i>j
e2
~rij
La ecuación de Schrödinger a resolver ahora es:
(Hel − VNN )Ψel = EelΨel
Donde
VNN =∑α
∑β>α
ZαZβe2
~Rαβ⇒ Vext
Born-Oppenheimer, permite desacoplar los movimientos electrónicos y nucleares.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 6 / 42

Fundamentos teóricos DFT
Ecuaciones de Kohn y Sham
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.1965 W. Kohn y L. Sham plantean el formalismo matemático de la DFT.Se separa la ecuación de
Schrödinger en N ecuaciones deuna sola partícula
[− ~2
2m∇2 + Veff (~r)
]ψi(~r) = εiψi(~r)
Donde la densidad total estadefinida por
ρ(~r) =
N∑i=1
|ψi(~r)|2
Se define el potencial efectivoVeff (~r) = Vext(r) + Vc(~r) + Vxc(~r)
El único término que se debe aproximar es el potencial de intercambioy correlación Vxc(~r)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 7 / 42

Fundamentos teóricos DFT
Ecuaciones de Kohn y Sham
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.
1965 W. Kohn y L. Sham plantean el formalismo matemático de la DFT.Se separa la ecuación de
Schrödinger en N ecuaciones deuna sola partícula
[− ~2
2m∇2 + Veff (~r)
]ψi(~r) = εiψi(~r)
Donde la densidad total estadefinida por
ρ(~r) =
N∑i=1
|ψi(~r)|2
Se define el potencial efectivoVeff (~r) = Vext(r) + Vc(~r) + Vxc(~r)
El único término que se debe aproximar es el potencial de intercambioy correlación Vxc(~r)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 7 / 42

Fundamentos teóricos DFT
Ecuaciones de Kohn y Sham
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.1965 W. Kohn y L. Sham plantean el formalismo matemático de la DFT.
Se separa la ecuación deSchrödinger en N ecuaciones deuna sola partícula
[− ~2
2m∇2 + Veff (~r)
]ψi(~r) = εiψi(~r)
Donde la densidad total estadefinida por
ρ(~r) =
N∑i=1
|ψi(~r)|2
Se define el potencial efectivoVeff (~r) = Vext(r) + Vc(~r) + Vxc(~r)
El único término que se debe aproximar es el potencial de intercambioy correlación Vxc(~r)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 7 / 42

Fundamentos teóricos DFT
Ecuaciones de Kohn y Sham
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.1965 W. Kohn y L. Sham plantean el formalismo matemático de la DFT.Se separa la ecuación de
Schrödinger en N ecuaciones deuna sola partícula
[− ~2
2m∇2 + Veff (~r)
]ψi(~r) = εiψi(~r)
Donde la densidad total estadefinida por
ρ(~r) =
N∑i=1
|ψi(~r)|2
Se define el potencial efectivoVeff (~r) = Vext(r) + Vc(~r) + Vxc(~r)
El único término que se debe aproximar es el potencial de intercambioy correlación Vxc(~r)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 7 / 42

Fundamentos teóricos DFT
Ecuaciones de Kohn y Sham
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.1965 W. Kohn y L. Sham plantean el formalismo matemático de la DFT.Se separa la ecuación de
Schrödinger en N ecuaciones deuna sola partícula
[− ~2
2m∇2 + Veff (~r)
]ψi(~r) = εiψi(~r)
Donde la densidad total estadefinida por
ρ(~r) =N∑i=1
|ψi(~r)|2
Se define el potencial efectivoVeff (~r) = Vext(r) + Vc(~r) + Vxc(~r)
El único término que se debe aproximar es el potencial de intercambioy correlación Vxc(~r)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 7 / 42

Fundamentos teóricos DFT
Ecuaciones de Kohn y Sham
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.1965 W. Kohn y L. Sham plantean el formalismo matemático de la DFT.Se separa la ecuación de
Schrödinger en N ecuaciones deuna sola partícula
[− ~2
2m∇2 + Veff (~r)
]ψi(~r) = εiψi(~r)
Donde la densidad total estadefinida por
ρ(~r) =N∑i=1
|ψi(~r)|2
Se define el potencial efectivoVeff (~r) = Vext(r) + Vc(~r) + Vxc(~r)
El único término que se debe aproximar es el potencial de intercambioy correlación Vxc(~r)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 7 / 42

Fundamentos teóricos DFT
Potencial de Intercambio y correlación
Existen diferentes aproximaciones para el potencial de intercambio ycorrelación
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 8 / 42

Fundamentos teóricos DFT
Potencial de Intercambio y correlación
Existen diferentes aproximaciones para el potencial de intercambio ycorrelación
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 8 / 42

Fundamentos teóricos DFT
Solución de las ecuaciones de Kohn y Sham
Se requiere resolver la llamada ecuación secular
H¯· Ci = εi · S
¯· Ci
Lo cual permite obtener la energía εi de Kohn y ShamDonde:
Sij = 〈Φj |Φk〉
Matriz de solapamiento
Hjk =
⟨Φj
∣∣∣∣− ~2m∇2 + VC + Vxc
∣∣∣∣Φk⟩Elementos de matriz del Hamiltoniano
Proceso iterativo, finaliza cuando los valores iniciales y finales de ρ(~r) son igualesdentro de un margen de tolerancia.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 9 / 42

Fundamentos teóricos DFT
Solución de las ecuaciones de Kohn y Sham
Se requiere resolver la llamada ecuación secular
H¯· Ci = εi · S
¯· Ci
Lo cual permite obtener la energía εi de Kohn y ShamDonde:
Sij = 〈Φj |Φk〉
Matriz de solapamiento
Hjk =
⟨Φj
∣∣∣∣− ~2m∇2 + VC + Vxc
∣∣∣∣Φk⟩Elementos de matriz del Hamiltoniano
Proceso iterativo, finaliza cuando los valores iniciales y finales de ρ(~r) son igualesdentro de un margen de tolerancia.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 9 / 42

Fundamentos teóricos DFT
Solución de las ecuaciones de Kohn y Sham
Se requiere resolver la llamada ecuación secular
H¯· Ci = εi · S
¯· Ci
Lo cual permite obtener la energía εi de Kohn y Sham
Donde:
Sij = 〈Φj |Φk〉
Matriz de solapamiento
Hjk =
⟨Φj
∣∣∣∣− ~2m∇2 + VC + Vxc
∣∣∣∣Φk⟩Elementos de matriz del Hamiltoniano
Proceso iterativo, finaliza cuando los valores iniciales y finales de ρ(~r) son igualesdentro de un margen de tolerancia.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 9 / 42

Fundamentos teóricos DFT
Solución de las ecuaciones de Kohn y Sham
Se requiere resolver la llamada ecuación secular
H¯· Ci = εi · S
¯· Ci
Lo cual permite obtener la energía εi de Kohn y ShamDonde:
Sij = 〈Φj |Φk〉
Matriz de solapamiento
Hjk =
⟨Φj
∣∣∣∣− ~2m∇2 + VC + Vxc
∣∣∣∣Φk⟩Elementos de matriz del Hamiltoniano
Proceso iterativo, finaliza cuando los valores iniciales y finales de ρ(~r) son igualesdentro de un margen de tolerancia.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 9 / 42

Fundamentos teóricos DFT
Solución de las ecuaciones de Kohn y Sham
Se requiere resolver la llamada ecuación secular
H¯· Ci = εi · S
¯· Ci
Lo cual permite obtener la energía εi de Kohn y ShamDonde:
Sij = 〈Φj |Φk〉
Matriz de solapamiento
Hjk =
⟨Φj
∣∣∣∣− ~2m∇2 + VC + Vxc
∣∣∣∣Φk⟩Elementos de matriz del Hamiltoniano
Proceso iterativo, finaliza cuando los valores iniciales y finales de ρ(~r) son igualesdentro de un margen de tolerancia.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 9 / 42

Fundamentos teóricos DFT
Solución de las ecuaciones de Kohn y Sham
Se requiere resolver la llamada ecuación secular
H¯· Ci = εi · S
¯· Ci
Lo cual permite obtener la energía εi de Kohn y ShamDonde:
Sij = 〈Φj |Φk〉
Matriz de solapamiento
Hjk =
⟨Φj
∣∣∣∣− ~2m∇2 + VC + Vxc
∣∣∣∣Φk⟩Elementos de matriz del Hamiltoniano
Proceso iterativo, finaliza cuando los valores iniciales y finales de ρ(~r) son igualesdentro de un margen de tolerancia.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 9 / 42

Fundamentos teóricos Correcciones Many Body
Extensiones a la DFT
Historicamente se ha logrado un cálculo adecuado de energías en el estadofundamental.
Subestimación de los band gaps en semiconductores y aisladores.La DOS calculada con LDA en sistemas con enlaces d y f (no llenos) no estande acuerdo con valores experimentales.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 10 / 42

Fundamentos teóricos Correcciones Many Body
Extensiones a la DFT
Historicamente se ha logrado un cálculo adecuado de energías en el estadofundamental.Subestimación de los band gaps en semiconductores y aisladores.
La DOS calculada con LDA en sistemas con enlaces d y f (no llenos) no estande acuerdo con valores experimentales.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 10 / 42

Fundamentos teóricos Correcciones Many Body
Extensiones a la DFT
Historicamente se ha logrado un cálculo adecuado de energías en el estadofundamental.Subestimación de los band gaps en semiconductores y aisladores.La DOS calculada con LDA en sistemas con enlaces d y f (no llenos) no estande acuerdo con valores experimentales.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 10 / 42

Fundamentos teóricos Correcciones Many Body
Extensiones a la DFT
Historicamente se ha logrado un cálculo adecuado de energías en el estadofundamental.Subestimación de los band gaps en semiconductores y aisladores.La DOS calculada con LDA en sistemas con enlaces d y f (no llenos) no estande acuerdo con valores experimentales.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 10 / 42

Fundamentos teóricos Correcciones Many Body
Extensiones a la DFT
Historicamente se ha logrado un cálculo adecuado de energías en el estadofundamental.Subestimación de los band gaps en semiconductores y aisladores.La DOS calculada con LDA en sistemas con enlaces d y f (no llenos) no estande acuerdo con valores experimentales.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 10 / 42

Fundamentos teóricos Correcciones Many Body
Corrección GWCuasipartícula: Combinación de una partícula real (Electron/hueco) y una nube depares virtuales de interacción electron-hueco que la rodean. Por esto se debe recurrira las llamadas teorias de muchos cuerpos, "Many body"
1965 Lars Hedin plantea la aproximación de apantallamiento dinámico GW
Donde W , se refiere al potencial de Coulomb apantallado.
W (~r1, ~r2) = ε−1v(~r1, ~r2)
Siendo v(~r1, ~r2) =e2
|~r1 − ~r2|el potencial de Coulomb estático.
Definiendo la evolución temporal del sistema por medio de la función de Green:
G(~r1, t1;~r2, t2) = −i〈N |Tψ(~r1, t1)ψ†(~r2, t2)|N〉
Con lo cual, se puede definir el operador autoenergía
Σ(~r1, ~r2, E) =i
2π
∫G(~r1, ~r2, E + E′)W (~r1, ~r2, E
′)dE′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 11 / 42

Fundamentos teóricos Correcciones Many Body
Corrección GWCuasipartícula: Combinación de una partícula real (Electron/hueco) y una nube depares virtuales de interacción electron-hueco que la rodean. Por esto se debe recurrira las llamadas teorias de muchos cuerpos, "Many body"
1965 Lars Hedin plantea la aproximación de apantallamiento dinámico GW
Donde W , se refiere al potencial de Coulomb apantallado.
W (~r1, ~r2) = ε−1v(~r1, ~r2)
Siendo v(~r1, ~r2) =e2
|~r1 − ~r2|el potencial de Coulomb estático.
Definiendo la evolución temporal del sistema por medio de la función de Green:
G(~r1, t1;~r2, t2) = −i〈N |Tψ(~r1, t1)ψ†(~r2, t2)|N〉
Con lo cual, se puede definir el operador autoenergía
Σ(~r1, ~r2, E) =i
2π
∫G(~r1, ~r2, E + E′)W (~r1, ~r2, E
′)dE′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 11 / 42

Fundamentos teóricos Correcciones Many Body
Corrección GWCuasipartícula: Combinación de una partícula real (Electron/hueco) y una nube depares virtuales de interacción electron-hueco que la rodean. Por esto se debe recurrira las llamadas teorias de muchos cuerpos, "Many body"
1965 Lars Hedin plantea la aproximación de apantallamiento dinámico GW
Donde W , se refiere al potencial de Coulomb apantallado.
W (~r1, ~r2) = ε−1v(~r1, ~r2)
Siendo v(~r1, ~r2) =e2
|~r1 − ~r2|el potencial de Coulomb estático.
Definiendo la evolución temporal del sistema por medio de la función de Green:
G(~r1, t1;~r2, t2) = −i〈N |Tψ(~r1, t1)ψ†(~r2, t2)|N〉
Con lo cual, se puede definir el operador autoenergía
Σ(~r1, ~r2, E) =i
2π
∫G(~r1, ~r2, E + E′)W (~r1, ~r2, E
′)dE′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 11 / 42

Fundamentos teóricos Correcciones Many Body
Corrección GWCuasipartícula: Combinación de una partícula real (Electron/hueco) y una nube depares virtuales de interacción electron-hueco que la rodean. Por esto se debe recurrira las llamadas teorias de muchos cuerpos, "Many body"
1965 Lars Hedin plantea la aproximación de apantallamiento dinámico GW
Donde W , se refiere al potencial de Coulomb apantallado.
W (~r1, ~r2) = ε−1v(~r1, ~r2)
Siendo v(~r1, ~r2) =e2
|~r1 − ~r2|el potencial de Coulomb estático.
Definiendo la evolución temporal del sistema por medio de la función de Green:
G(~r1, t1;~r2, t2) = −i〈N |Tψ(~r1, t1)ψ†(~r2, t2)|N〉
Con lo cual, se puede definir el operador autoenergía
Σ(~r1, ~r2, E) =i
2π
∫G(~r1, ~r2, E + E′)W (~r1, ~r2, E
′)dE′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 11 / 42

Fundamentos teóricos Correcciones Many Body
Corrección GWCuasipartícula: Combinación de una partícula real (Electron/hueco) y una nube depares virtuales de interacción electron-hueco que la rodean. Por esto se debe recurrira las llamadas teorias de muchos cuerpos, "Many body"
1965 Lars Hedin plantea la aproximación de apantallamiento dinámico GW
Donde W , se refiere al potencial de Coulomb apantallado.
W (~r1, ~r2) = ε−1v(~r1, ~r2)
Siendo v(~r1, ~r2) =e2
|~r1 − ~r2|el potencial de Coulomb estático.
Definiendo la evolución temporal del sistema por medio de la función de Green:
G(~r1, t1;~r2, t2) = −i〈N |Tψ(~r1, t1)ψ†(~r2, t2)|N〉
Con lo cual, se puede definir el operador autoenergía
Σ(~r1, ~r2, E) =i
2π
∫G(~r1, ~r2, E + E′)W (~r1, ~r2, E
′)dE′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 11 / 42

Fundamentos teóricos Correcciones Many Body
Corrección GW
Un calculo GW requiere en principio de la soluciónde la ecuación de cuasipartículas:
H0(~r1ψ(~r1)) +
∫Σ(~r1, ~r2, E)ψ(~r2)d3~r2 = Eψ(~r1)
Donde la interacción se incluye como unaperturbación en H0
H0(~r1) = T + VN (~r1) + vxc(~r1)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 12 / 42

Fundamentos teóricos Correcciones Many Body
Corrección GW
Un calculo GW requiere en principio de la soluciónde la ecuación de cuasipartículas:
H0(~r1ψ(~r1)) +
∫Σ(~r1, ~r2, E)ψ(~r2)d3~r2 = Eψ(~r1)
Donde la interacción se incluye como unaperturbación en H0
H0(~r1) = T + VN (~r1) + vxc(~r1)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 12 / 42

Fundamentos teóricos Correcciones Many Body
Corrección GW
Un calculo GW requiere en principio de la soluciónde la ecuación de cuasipartículas:
H0(~r1ψ(~r1)) +
∫Σ(~r1, ~r2, E)ψ(~r2)d3~r2 = Eψ(~r1)
Donde la interacción se incluye como unaperturbación en H0
H0(~r1) = T + VN (~r1) + vxc(~r1)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 12 / 42

Fundamentos teóricos Correcciones Many Body
Transiciones electrónicas
Parte imaginaría función dieléctrica
ε2(ω)αβ =4π2e2
m2ω2
∑i,f
∫〈f |pα|i〉〈f |pβ |i〉Wi(1−Wf )× δ(Ef − Ei − ~ω)d3k
Parte real función dieléctrica por medio de relaciones de Kramers y Kronig:
ε1(ω) = 1 +2
π
∫ ∞0
ε2(ω)ω′dω′
ω′2 − ω2
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 13 / 42

Fundamentos teóricos Correcciones Many Body
Transiciones electrónicas
Parte imaginaría función dieléctrica
ε2(ω)αβ =4π2e2
m2ω2
∑i,f
∫〈f |pα|i〉〈f |pβ |i〉Wi(1−Wf )× δ(Ef − Ei − ~ω)d3k
Parte real función dieléctrica por medio de relaciones de Kramers y Kronig:
ε1(ω) = 1 +2
π
∫ ∞0
ε2(ω)ω′dω′
ω′2 − ω2
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 13 / 42

Fundamentos teóricos Correcciones Many Body
Transiciones electrónicas
Parte imaginaría función dieléctrica
ε2(ω)αβ =4π2e2
m2ω2
∑i,f
∫〈f |pα|i〉〈f |pβ |i〉Wi(1−Wf )× δ(Ef − Ei − ~ω)d3k
Parte real función dieléctrica por medio de relaciones de Kramers y Kronig:
ε1(ω) = 1 +2
π
∫ ∞0
ε2(ω)ω′dω′
ω′2 − ω2
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 13 / 42

Fundamentos teóricos Correcciones Many Body
Transiciones electrónicas
Restantes funciones ópticas
Índice de refracción
n(ω) =
√|ε(ω)|+ ε1(ω)
2
Coeficiente de extinción:
K(ω) =
√|ε(ω)| − ε1(ω)
2
Reflectividad a incidencia normal:
R(ω) =(n− 1)2 + k2
(n+ 1)2 + k2
Coeficiente de absorción:
α(ω)j =2ω
c
(|ε(ω)j | − ε1(ω)j
2
) 12
Función de perdida de energíaelectrónica:
EELS(ω) = Im
− 1
ε(ω)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 14 / 42

Fundamentos teóricos Correcciones Many Body
Transiciones electrónicas
Restantes funciones ópticas
Índice de refracción
n(ω) =
√|ε(ω)|+ ε1(ω)
2
Coeficiente de extinción:
K(ω) =
√|ε(ω)| − ε1(ω)
2
Reflectividad a incidencia normal:
R(ω) =(n− 1)2 + k2
(n+ 1)2 + k2
Coeficiente de absorción:
α(ω)j =2ω
c
(|ε(ω)j | − ε1(ω)j
2
) 12
Función de perdida de energíaelectrónica:
EELS(ω) = Im
− 1
ε(ω)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 14 / 42

Fundamentos teóricos Correcciones Many Body
Transiciones electrónicas
Restantes funciones ópticas
Índice de refracción
n(ω) =
√|ε(ω)|+ ε1(ω)
2
Coeficiente de extinción:
K(ω) =
√|ε(ω)| − ε1(ω)
2
Reflectividad a incidencia normal:
R(ω) =(n− 1)2 + k2
(n+ 1)2 + k2
Coeficiente de absorción:
α(ω)j =2ω
c
(|ε(ω)j | − ε1(ω)j
2
) 12
Función de perdida de energíaelectrónica:
EELS(ω) = Im
− 1
ε(ω)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 14 / 42

Fundamentos teóricos Correcciones Many Body
Transiciones electrónicas
Restantes funciones ópticas
Índice de refracción
n(ω) =
√|ε(ω)|+ ε1(ω)
2
Coeficiente de extinción:
K(ω) =
√|ε(ω)| − ε1(ω)
2
Reflectividad a incidencia normal:
R(ω) =(n− 1)2 + k2
(n+ 1)2 + k2
Coeficiente de absorción:
α(ω)j =2ω
c
(|ε(ω)j | − ε1(ω)j
2
) 12
Función de perdida de energíaelectrónica:
EELS(ω) = Im
− 1
ε(ω)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 14 / 42

Fundamentos teóricos Correcciones Many Body
Transiciones electrónicas
Restantes funciones ópticas
Índice de refracción
n(ω) =
√|ε(ω)|+ ε1(ω)
2
Coeficiente de extinción:
K(ω) =
√|ε(ω)| − ε1(ω)
2
Reflectividad a incidencia normal:
R(ω) =(n− 1)2 + k2
(n+ 1)2 + k2
Coeficiente de absorción:
α(ω)j =2ω
c
(|ε(ω)j | − ε1(ω)j
2
) 12
Función de perdida de energíaelectrónica:
EELS(ω) = Im
− 1
ε(ω)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 14 / 42

Fundamentos teóricos Correcciones Many Body
Transiciones electrónicas
Restantes funciones ópticas
Índice de refracción
n(ω) =
√|ε(ω)|+ ε1(ω)
2
Coeficiente de extinción:
K(ω) =
√|ε(ω)| − ε1(ω)
2
Reflectividad a incidencia normal:
R(ω) =(n− 1)2 + k2
(n+ 1)2 + k2
Coeficiente de absorción:
α(ω)j =2ω
c
(|ε(ω)j | − ε1(ω)j
2
) 12
Función de perdida de energíaelectrónica:
EELS(ω) = Im
− 1
ε(ω)
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 14 / 42

Fundamentos teóricos Correcciones Many Body
Ecuación BSE
Ecuación de Bethe Salpeter: Corrección GW que incluye la estructura electrónica de Kohn ySham junto con la interacción de Coulomb apantallada incluyendo la interacción electrón-hueco,con la cual se puede mejorar la estimación de la función dielectrica ε2(ω)
ε2(ω) = 1− limq→0
(ω) =4πe2
q2
∑λ
|∑v,c〈v|eiq·r|c〉A
v,cλ |
2
ω − Eλ + iη
Eλ y Av,cλ son los autovalores y autovectores de:
Hexcvc,v′,c′A
v′,c′
λ = EλAv,cλ
Hexcvck,v′,c′k′ = Hdiag
vck;v′c′k′ +Hexchvc−fk;c′v′k′ +Hscr
vck;v′c′k′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 15 / 42

Fundamentos teóricos Correcciones Many Body
Ecuación BSE
Ecuación de Bethe Salpeter: Corrección GW que incluye la estructura electrónica de Kohn ySham junto con la interacción de Coulomb apantallada incluyendo la interacción electrón-hueco,con la cual se puede mejorar la estimación de la función dielectrica ε2(ω)
ε2(ω) = 1− limq→0
(ω) =4πe2
q2
∑λ
|∑v,c〈v|eiq·r|c〉A
v,cλ |
2
ω − Eλ + iη
Eλ y Av,cλ son los autovalores y autovectores de:
Hexcvc,v′,c′A
v′,c′
λ = EλAv,cλ
Hexcvck,v′,c′k′ = Hdiag
vck;v′c′k′ +Hexchvc−fk;c′v′k′ +Hscr
vck;v′c′k′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 15 / 42

Fundamentos teóricos Correcciones Many Body
Ecuación BSE
Ecuación de Bethe Salpeter: Corrección GW que incluye la estructura electrónica de Kohn ySham junto con la interacción de Coulomb apantallada incluyendo la interacción electrón-hueco,con la cual se puede mejorar la estimación de la función dielectrica ε2(ω)
ε2(ω) = 1− limq→0
(ω) =4πe2
q2
∑λ
|∑v,c〈v|eiq·r|c〉A
v,cλ |
2
ω − Eλ + iη
Eλ y Av,cλ son los autovalores y autovectores de:
Hexcvc,v′,c′A
v′,c′
λ = EλAv,cλ
Hexcvck,v′,c′k′ = Hdiag
vck;v′c′k′ +Hexchvc−fk;c′v′k′ +Hscr
vck;v′c′k′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 15 / 42

Fundamentos teóricos Correcciones Many Body
Ecuación BSE
Ecuación de Bethe Salpeter: Corrección GW que incluye la estructura electrónica de Kohn ySham junto con la interacción de Coulomb apantallada incluyendo la interacción electrón-hueco,con la cual se puede mejorar la estimación de la función dielectrica ε2(ω)
ε2(ω) = 1− limq→0
(ω) =4πe2
q2
∑λ
|∑v,c〈v|eiq·r|c〉A
v,cλ |
2
ω − Eλ + iη
Eλ y Av,cλ son los autovalores y autovectores de:
Hexcvc,v′,c′A
v′,c′
λ = EλAv,cλ
Hexcvck,v′,c′k′ = Hdiag
vck;v′c′k′ +Hexchvc−fk;c′v′k′ +Hscr
vck;v′c′k′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 15 / 42

Fundamentos teóricos Correcciones Many Body
Ecuación BSE
Ecuación de Bethe Salpeter: Corrección GW que incluye la estructura electrónica de Kohn ySham junto con la interacción de Coulomb apantallada incluyendo la interacción electrón-hueco,con la cual se puede mejorar la estimación de la función dielectrica ε2(ω)
ε2(ω) = 1− limq→0
(ω) =4πe2
q2
∑λ
|∑v,c〈v|eiq·r|c〉A
v,cλ |
2
ω − Eλ + iη
Eλ y Av,cλ son los autovalores y autovectores de:
Hexcvc,v′,c′A
v′,c′
λ = EλAv,cλ
Hexcvck,v′,c′k′ = Hdiag
vck;v′c′k′ +Hexchvc−fk;c′v′k′ +Hscr
vck;v′c′k′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 15 / 42

Fundamentos teóricos Correcciones Many Body
Ecuación BSE
Ecuación de Bethe Salpeter: Corrección GW que incluye la estructura electrónica de Kohn ySham junto con la interacción de Coulomb apantallada incluyendo la interacción electrón-hueco,con la cual se puede mejorar la estimación de la función dielectrica ε2(ω)
ε2(ω) = 1− limq→0
(ω) =4πe2
q2
∑λ
|∑v,c〈v|eiq·r|c〉A
v,cλ |
2
ω − Eλ + iη
Eλ y Av,cλ son los autovalores y autovectores de:
Hexcvc,v′,c′A
v′,c′
λ = EλAv,cλ
Hexcvck,v′,c′k′ = Hdiag
vck;v′c′k′ +Hexchvc−fk;c′v′k′ +Hscr
vck;v′c′k′
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 15 / 42

Métodos computacionales
Método LAPW
LAPW Linearized Augmented PlaneWavesSe divide el sistema en dos regiones
1 Esféras atómicas centradasalrededor de los sitios atómicos
2 Región intersticial
Wien2k
Este método es utilizado por elprograma Wien2k.Página web:http://www.wien2k.at
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 16 / 42

Métodos computacionales
Método LAPWLAPW Linearized Augmented PlaneWaves
Se divide el sistema en dos regiones
1 Esféras atómicas centradasalrededor de los sitios atómicos
2 Región intersticial
Wien2k
Este método es utilizado por elprograma Wien2k.Página web:http://www.wien2k.at
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 16 / 42

Métodos computacionales
Método LAPWLAPW Linearized Augmented PlaneWavesSe divide el sistema en dos regiones
1 Esféras atómicas centradasalrededor de los sitios atómicos
2 Región intersticial
Wien2k
Este método es utilizado por elprograma Wien2k.Página web:http://www.wien2k.at
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 16 / 42

Métodos computacionales
Método LAPWLAPW Linearized Augmented PlaneWavesSe divide el sistema en dos regiones
1 Esféras atómicas centradasalrededor de los sitios atómicos
2 Región intersticial
Wien2k
Este método es utilizado por elprograma Wien2k.Página web:http://www.wien2k.at
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 16 / 42

Métodos computacionales
Método LAPWLAPW Linearized Augmented PlaneWavesSe divide el sistema en dos regiones
1 Esféras atómicas centradasalrededor de los sitios atómicos
2 Región intersticial
Wien2k
Este método es utilizado por elprograma Wien2k.Página web:http://www.wien2k.at
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 16 / 42

Métodos computacionales
Método LAPWLAPW Linearized Augmented PlaneWavesSe divide el sistema en dos regiones
1 Esféras atómicas centradasalrededor de los sitios atómicos
2 Región intersticial
Wien2k
Este método es utilizado por elprograma Wien2k.Página web:http://www.wien2k.at
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 16 / 42

Métodos computacionales
Método de Pseudopotenciales
Generalidades
Este método es utilizado por el programa ABINIT.Página web: http://www.abinit.org.
1 Reemplaza el potencial nuclear por unpseudopotencial que tiene en cuenta solo losefectos ocasionados por los electrónes devalencia.
2 Reduce el número de orbitales que seincluyen en el cálculo al incluir menoselectrones.
3 Se usa una base de ondas planas pararepresentar las funciones de ondaψ(~r) = ei(
~k+ ~K)·~r
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 17 / 42

Métodos computacionales
Método de Pseudopotenciales
Generalidades
Este método es utilizado por el programa ABINIT.Página web: http://www.abinit.org.
1 Reemplaza el potencial nuclear por unpseudopotencial que tiene en cuenta solo losefectos ocasionados por los electrónes devalencia.
2 Reduce el número de orbitales que seincluyen en el cálculo al incluir menoselectrones.
3 Se usa una base de ondas planas pararepresentar las funciones de ondaψ(~r) = ei(
~k+ ~K)·~r
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 17 / 42

Métodos computacionales
Método de Pseudopotenciales
Generalidades
Este método es utilizado por el programa ABINIT.Página web: http://www.abinit.org.
1 Reemplaza el potencial nuclear por unpseudopotencial que tiene en cuenta solo losefectos ocasionados por los electrónes devalencia.
2 Reduce el número de orbitales que seincluyen en el cálculo al incluir menoselectrones.
3 Se usa una base de ondas planas pararepresentar las funciones de ondaψ(~r) = ei(
~k+ ~K)·~r
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 17 / 42

Métodos computacionales
Método de Pseudopotenciales
Generalidades
Este método es utilizado por el programa ABINIT.Página web: http://www.abinit.org.
1 Reemplaza el potencial nuclear por unpseudopotencial que tiene en cuenta solo losefectos ocasionados por los electrónes devalencia.
2 Reduce el número de orbitales que seincluyen en el cálculo al incluir menoselectrones.
3 Se usa una base de ondas planas pararepresentar las funciones de ondaψ(~r) = ei(
~k+ ~K)·~r
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 17 / 42

Métodos computacionales
Método de Pseudopotenciales
Generalidades
Este método es utilizado por el programa ABINIT.Página web: http://www.abinit.org.
1 Reemplaza el potencial nuclear por unpseudopotencial que tiene en cuenta solo losefectos ocasionados por los electrónes devalencia.
2 Reduce el número de orbitales que seincluyen en el cálculo al incluir menoselectrones.
3 Se usa una base de ondas planas pararepresentar las funciones de ondaψ(~r) = ei(
~k+ ~K)·~r
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 17 / 42

Resultados Caracterización estructural α− AlF3
Polimorfismos AlF3
Fase estable a temperatura ambiente
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 18 / 42

Resultados Caracterización estructural α− AlF3
Polimorfismos AlF3
Fase estable a temperatura ambiente
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 18 / 42

Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42

Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42

Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42

Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42

Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42
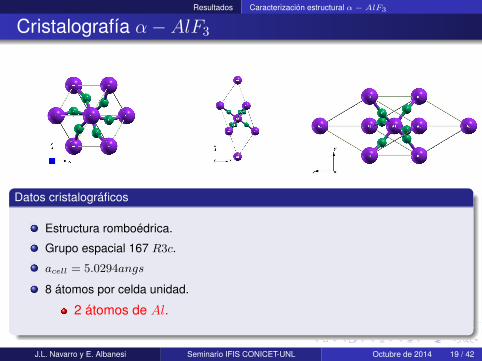
Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.
6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42

Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42

Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42

Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42

Resultados Caracterización estructural α− AlF3
Cristalografía α− AlF3
Datos cristalográficos
Estructura romboédrica.
Grupo espacial 167 R3c.
acell = 5.0294angs
8 átomos por celda unidad.
2 átomos de Al.6 átomos de F .
Aislante iónico.
Un calculo All-electron incluye 80electrones (26Al, 54F ).
Un calculo con pseudopotencialesincluye 48 electrones (18Al, 30F ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 19 / 42

Resultados Caracterización estructural α− AlF3
Propiedades estructurales
Por medio de las ecuaciones de estado de Birch-Murnagham,la presión en función del volumense define como:
P (V ) =3
2B0
[(V0
V
)7/3
−(V0
V
)5/3]
1 +3
4(B′0 − 4)
[(V0
v
)2/3
− 1
]
mientras que la energía en función de volumen se define como:
E(V ) = E0 +9V0B0
16
[(
V0
V
)2/3
− 1
]3B′0 +
[(V0
V
)2/3
− 1
]2 [6− 4
(V0
V
)2/3]
B0 es el modulo de Bulk del material.
B′0 su derivada.
V0 es el volumen de equilibrio
E0 es la enegía mínima
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 20 / 42

Resultados Caracterización estructural α− AlF3
Propiedades estructurales
Por medio de las ecuaciones de estado de Birch-Murnagham,la presión en función del volumense define como:
P (V ) =3
2B0
[(V0
V
)7/3
−(V0
V
)5/3]
1 +3
4(B′0 − 4)
[(V0
v
)2/3
− 1
]
mientras que la energía en función de volumen se define como:
E(V ) = E0 +9V0B0
16
[(
V0
V
)2/3
− 1
]3B′0 +
[(V0
V
)2/3
− 1
]2 [6− 4
(V0
V
)2/3]
B0 es el modulo de Bulk del material.
B′0 su derivada.
V0 es el volumen de equilibrio
E0 es la enegía mínima
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 20 / 42

Resultados Caracterización estructural α− AlF3
Propiedades estructurales
Por medio de las ecuaciones de estado de Birch-Murnagham,la presión en función del volumense define como:
P (V ) =3
2B0
[(V0
V
)7/3
−(V0
V
)5/3]
1 +3
4(B′0 − 4)
[(V0
v
)2/3
− 1
]
mientras que la energía en función de volumen se define como:
E(V ) = E0 +9V0B0
16
[(
V0
V
)2/3
− 1
]3B′0 +
[(V0
V
)2/3
− 1
]2 [6− 4
(V0
V
)2/3]
B0 es el modulo de Bulk del material.
B′0 su derivada.
V0 es el volumen de equilibrio
E0 es la enegía mínima
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 20 / 42

Resultados Caracterización estructural α− AlF3
Popiedades estructuralesCurvas obtenidas por fiteo de Birch Murnagham
800 1000Volumen (Bohr^3)
-150,9
-150,85
-150,8
-150,75
En
erg
ia (
Ha)
Datos calculados ABINITBirch-Murnag
Energia Vs.Volumen
alfa-AlF3
800 1000Volumen (Bohr^3)
-20
0
20
40
60
Pre
sion (
GP
a)
Datos Calculados ABINITBirch-Murnag
Presion Vs Volumenalfa-AlF3
E0 = −150.88Ha
V0 = 943.52Bohr3
B0 = 137GPa
B′0 = 3.47
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 21 / 42

Resultados Caracterización estructural α− AlF3
Popiedades estructuralesCurvas obtenidas por fiteo de Birch Murnagham
800 1000Volumen (Bohr^3)
-150,9
-150,85
-150,8
-150,75
En
erg
ia (
Ha)
Datos calculados ABINITBirch-Murnag
Energia Vs.Volumen
alfa-AlF3
800 1000Volumen (Bohr^3)
-20
0
20
40
60
Pre
sion (
GP
a)
Datos Calculados ABINITBirch-Murnag
Presion Vs Volumenalfa-AlF3
E0 = −150.88Ha
V0 = 943.52Bohr3
B0 = 137GPa
B′0 = 3.47
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 21 / 42

Resultados Caracterización estructural α− AlF3
Popiedades estructuralesCurvas obtenidas por fiteo de Birch Murnagham
800 1000Volumen (Bohr^3)
-150,9
-150,85
-150,8
-150,75
En
erg
ia (
Ha)
Datos calculados ABINITBirch-Murnag
Energia Vs.Volumen
alfa-AlF3
800 1000Volumen (Bohr^3)
-20
0
20
40
60
Pre
sion (
GP
a)
Datos Calculados ABINITBirch-Murnag
Presion Vs Volumenalfa-AlF3
E0 = −150.88Ha
V0 = 943.52Bohr3
B0 = 137GPa
B′0 = 3.47
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 21 / 42

Resultados Estructura electrónica
Densidad de estados (total)
DOS Total ABINIT
Band Gap DFT 7.79eV
DOS Total Wien2k
-20 -10 0 10 20 30Energy (eV)
0
5
10
15
20
25
DO
S (
Sta
tes
Vo
l-1 e
V-1
)
DOS total αAlF3 Wien2k
Band Gap del mismo orden demagnitud.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 22 / 42
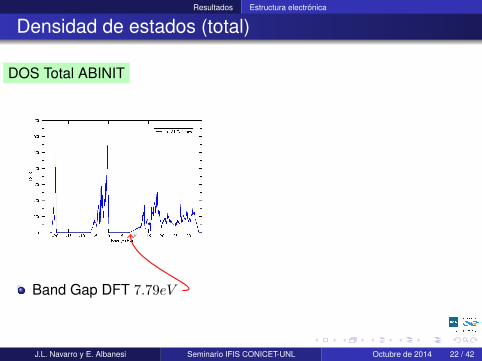
Resultados Estructura electrónica
Densidad de estados (total)
DOS Total ABINIT
Band Gap DFT 7.79eV
DOS Total Wien2k
-20 -10 0 10 20 30Energy (eV)
0
5
10
15
20
25
DO
S (
Sta
tes
Vo
l-1 e
V-1
)
DOS total αAlF3 Wien2k
Band Gap del mismo orden demagnitud.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 22 / 42

Resultados Estructura electrónica
Densidad de estados (total)
DOS Total ABINIT
Band Gap DFT 7.79eV
DOS Total Wien2k
-20 -10 0 10 20 30Energy (eV)
0
5
10
15
20
25
DO
S (
Sta
tes
Vol-1
eV
-1)
DOS total αAlF3 Wien2k
Band Gap del mismo orden demagnitud.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 22 / 42

Resultados Estructura electrónica
Densidad de estados (total)
DOS Total ABINIT
Band Gap DFT 7.79eV
DOS Total Wien2k
-20 -10 0 10 20 30Energy (eV)
0
5
10
15
20
25
DO
S (
Sta
tes
Vol-1
eV
-1)
DOS total αAlF3 Wien2k
Band Gap del mismo orden demagnitud.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 22 / 42

Resultados Estructura electrónica
Densidad de estados parcial (PDOS)
Principales picos calculados a partir de DOS en (eV) y sus estados aportantes para el α−AlF3.Los orbitales se muestran en orden decreciente de contribución.
0
50
100
150
200
F-s StatesF-p States
F-d States
0
5
10
15
20
25
30
DO
S (
Sta
tes
Vol-1
eV
-1)
Al-s StatesAl-p States
Al-d States
-20 -10 0 10 20 30
Energy (eV)
0
100
200
300
400
500
600
700
Total DOS α−AlF3
0
F C
on
du
ctio
n D
OS
VBM CBM
c1
c2
c3
c4 c
5
c6
c7
c8
c9
c10
V1
v2
v2
v3
v4
v5
v6
v7v
8
VBM Max Zona valencia
CMB Mín zona conducción
Pico DOS EnergíaDOS
Estadosapor-tantes.
Mayor MenorV8 −5.02 F-p Al-s Al-pV7 −3.85 F-p Al-pV6 −3.19 F-p Al-p Al-dV5 −2.82 F-p Al-p Al-dV4 −2.16 F-p Al-dV3 −1.64 F-p Al-dV2 −0.98 F-pV1 −0.32 F-pV BM 0CMB 10.81C1 14.15 Al-s Al-p F-pC2 15.7 Al-s Al-p F-p F-sC3 16.14 Al-s Al-p F-pC4 16.43 Al-s F-p Al-pC5 17.39 Al-p Al-sC6 18.27 Al-p Al-s F-p F-sC7 18.93 F-p Al-s Al-sC8 19.74 Al-d Al-p F-pC9 20.11 Al-p Al-d F-pC10 21.09 Al-d Al-p F-p
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 23 / 42

Resultados Estructura electrónica
Densidad de estados parcial (PDOS)
Principales picos calculados a partir de DOS en (eV) y sus estados aportantes para el α−AlF3.Los orbitales se muestran en orden decreciente de contribución.
0
50
100
150
200
F-s StatesF-p States
F-d States
0
5
10
15
20
25
30
DO
S (
Sta
tes
Vol-1
eV
-1)
Al-s StatesAl-p States
Al-d States
-20 -10 0 10 20 30
Energy (eV)
0
100
200
300
400
500
600
700
Total DOS α−AlF3
0
F C
on
du
ctio
n D
OS
VBM CBM
c1
c2
c3
c4 c
5
c6
c7
c8
c9
c10
V1
v2
v2
v3
v4
v5
v6
v7v
8
VBM Max Zona valencia
CMB Mín zona conducción
Pico DOS EnergíaDOS
Estadosapor-tantes.
Mayor MenorV8 −5.02 F-p Al-s Al-pV7 −3.85 F-p Al-pV6 −3.19 F-p Al-p Al-dV5 −2.82 F-p Al-p Al-dV4 −2.16 F-p Al-dV3 −1.64 F-p Al-dV2 −0.98 F-pV1 −0.32 F-pV BM 0CMB 10.81C1 14.15 Al-s Al-p F-pC2 15.7 Al-s Al-p F-p F-sC3 16.14 Al-s Al-p F-pC4 16.43 Al-s F-p Al-pC5 17.39 Al-p Al-sC6 18.27 Al-p Al-s F-p F-sC7 18.93 F-p Al-s Al-sC8 19.74 Al-d Al-p F-pC9 20.11 Al-p Al-d F-pC10 21.09 Al-d Al-p F-p
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 23 / 42

Resultados Estructura electrónica
Estructura de Bandas α− AlF3
L U X U Gamma L U WWave vector (k)
-20
-15
-10
-5
0
5
10
15
20
En
erg
y (
eV)
εF
Estructura de bandas del α−AlF3 donde se observa el Gapdirecto en el punto Γ de 7.79eV
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 24 / 42

Resultados Estructura electrónica
Estructura de Bandas α− AlF3
L U X U Gamma L U WWave vector (k)
-20
-15
-10
-5
0
5
10
15
20
En
erg
y (
eV)
εF
Estructura de bandas del α−AlF3 donde se observa el Gapdirecto en el punto Γ de 7.79eV
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 24 / 42

Resultados Estructura electrónica
Corección GW a la estructura de bandas
Estructura de bandas del α−AlF3 con gap corregido con GW.Gap obtenido 10.81 eV en el punto Γ.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 25 / 42

Resultados Estructura electrónica
Corección GW a la estructura de bandas
Estructura de bandas del α−AlF3 con gap corregido con GW.Gap obtenido 10.81 eV en el punto Γ.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 25 / 42

Resultados Propiedades ópticas
Función dielectríca εi(ω)
0
0,5
1
1,5
2ε
2 (
ω)
ε2(ω) Dir xx
ε2(ω) Dir yy
ε2(ω) Dir zz
0 10 20 30Energy (eV)
0
0,5
1
1,5
2
2,5
3
ε1 (
ω)
ε1(ω) Dir xx
ε1(ω) Dir yy
ε1(ω) Dir zz
E0
E1
E2
E3
E4
E5
E6
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 26 / 42
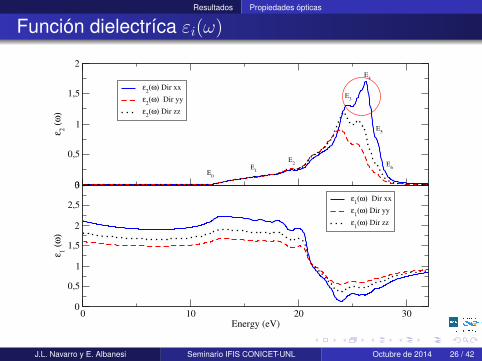
Resultados Propiedades ópticas
Función dielectríca εi(ω)
0
0,5
1
1,5
2ε
2 (
ω)
ε2(ω) Dir xx
ε2(ω) Dir yy
ε2(ω) Dir zz
0 10 20 30Energy (eV)
0
0,5
1
1,5
2
2,5
3
ε1 (
ω)
ε1(ω) Dir xx
ε1(ω) Dir yy
ε1(ω) Dir zz
E0
E1
E2
E3
E4
E5
E6
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 26 / 42

Resultados Propiedades ópticas
Funciones ópticas
RefracciónCoeficiente deExtinciónReflectividad
Grilla de puntos k de25x25x25 convergidaincluyendo 36 bandas.ABINIT
0,6
0,8
1
1,2
1,4
1,6
n(ω
)
n(ω) Dir xx
n( ω) Dir yy
n(ω) Dir zz
0
0,2
0,4
0,6
0,8
K(ω
)
K(ω) Dir xx
K(ω) Dir yy
K(ω) Dir zz
0 5 10 15 20 25 30
Energy (eV)
0
0,05
0,1
0,15
R(ω
)
R(ω) Dir xx
R(ω) Dir yy
R(ω) Dir zz
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 27 / 42

Resultados Propiedades ópticas
Funciones ópticas
RefracciónCoeficiente deExtinciónReflectividad
Grilla de puntos k de25x25x25 convergidaincluyendo 36 bandas.ABINIT
0,6
0,8
1
1,2
1,4
1,6
n(ω
)
n(ω) Dir xx
n( ω) Dir yy
n(ω) Dir zz
0
0,2
0,4
0,6
0,8
K(ω
)
K(ω) Dir xx
K(ω) Dir yy
K(ω) Dir zz
0 5 10 15 20 25 30
Energy (eV)
0
0,05
0,1
0,15
R(ω
)
R(ω) Dir xx
R(ω) Dir yy
R(ω) Dir zz
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 27 / 42

Resultados Propiedades ópticas
Funciones ópticas
RefracciónCoeficiente deExtinciónReflectividad
Grilla de puntos k de25x25x25 convergidaincluyendo 36 bandas.ABINIT
0,6
0,8
1
1,2
1,4
1,6
n(ω
)
n(ω) Dir xx
n( ω) Dir yy
n(ω) Dir zz
0
0,2
0,4
0,6
0,8
K(ω
)
K(ω) Dir xx
K(ω) Dir yy
K(ω) Dir zz
0 5 10 15 20 25 30
Energy (eV)
0
0,05
0,1
0,15
R(ω
)
R(ω) Dir xx
R(ω) Dir yy
R(ω) Dir zz
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 27 / 42

Resultados Propiedades ópticas
Funciones ópticas
RefracciónCoeficiente deExtinciónReflectividad
Grilla de puntos k de25x25x25 convergidaincluyendo 36 bandas.ABINIT
0,6
0,8
1
1,2
1,4
1,6
n(ω
)
n(ω) Dir xx
n( ω) Dir yy
n(ω) Dir zz
0
0,2
0,4
0,6
0,8
K(ω
)
K(ω) Dir xx
K(ω) Dir yy
K(ω) Dir zz
0 5 10 15 20 25 30
Energy (eV)
0
0,05
0,1
0,15
R(ω
)
R(ω) Dir xx
R(ω) Dir yy
R(ω) Dir zz
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 27 / 42
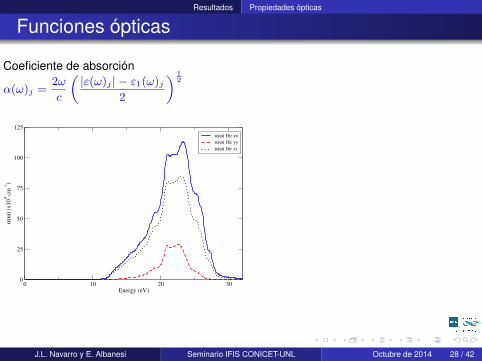
Resultados Propiedades ópticas
Funciones ópticas
Coeficiente de absorción
α(ω)j =2ω
c
(|ε(ω)j | − ε1(ω)j
2
) 12
0 10 20 30Energy (eV)
0
25
50
75
100
125
α(ω
) (x
10
4 c
m-1
)
α(ω) Dir xx
α(ω) Dir yy
α(ω) Dir zz
Función de perdida de energía electrónica
EELS(ω) = Im
− 1
ε(ω)
0 10 20 30Energy (eV)
0
0,5
1
1,5
2
- Im
ε (
ω)
EELS(ω) Dir xx
EELS(ω) Dir yy
EELS(ω) Dir zz
Cálculos realizados con una grilla de puntos k de 25x25x25 totalmente convergidaincluyendo 36 bandas, por medio del Programa ABINIT
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 28 / 42

Resultados Propiedades ópticas
Funciones ópticas
Coeficiente de absorción
α(ω)j =2ω
c
(|ε(ω)j | − ε1(ω)j
2
) 12
0 10 20 30Energy (eV)
0
25
50
75
100
125
α(ω
) (x
10
4 c
m-1
)
α(ω) Dir xx
α(ω) Dir yy
α(ω) Dir zz
Función de perdida de energía electrónica
EELS(ω) = Im
− 1
ε(ω)
0 10 20 30Energy (eV)
0
0,5
1
1,5
2
- Im
ε (
ω)
EELS(ω) Dir xx
EELS(ω) Dir yy
EELS(ω) Dir zz
Cálculos realizados con una grilla de puntos k de 25x25x25 totalmente convergidaincluyendo 36 bandas, por medio del Programa ABINIT
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 28 / 42

Resultados Propiedades ópticas
Corrección BSE
Función dielectríca ε2(ω)
ExcitonesGrilla de puntos k de14x14x14 convergidaimplementando BSE.ABINIT
0 10 20 30Energy (eV)
0
1
2
3
4
ε2(ω
)
ε2(ω) Dir xx DFT
ε2(ω) Dir yy DFT
ε2(ω) Dir zz DFT
ε2(ω) BSE
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 29 / 42

Resultados Propiedades ópticas
Corrección BSE
Función dielectríca ε2(ω)
ExcitonesGrilla de puntos k de14x14x14 convergidaimplementando BSE.ABINIT
0 10 20 30Energy (eV)
0
1
2
3
4
ε2(ω
)
ε2(ω) Dir xx DFT
ε2(ω) Dir yy DFT
ε2(ω) Dir zz DFT
ε2(ω) BSE
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 29 / 42

Resultados Propiedades ópticas
Corrección BSE
Función dielectríca ε2(ω)
ExcitonesGrilla de puntos k de14x14x14 convergidaimplementando BSE.ABINIT
0 10 20 30Energy (eV)
0
1
2
3
4
ε2(ω
)
ε2(ω) Dir xx DFT
ε2(ω) Dir yy DFT
ε2(ω) Dir zz DFT
ε2(ω) BSE
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 29 / 42

Resultados Propiedades ópticas
Corrección BSE
Función dielectríca ε2(ω)
ExcitonesGrilla de puntos k de14x14x14 convergidaimplementando BSE.ABINIT
0 10 20 30Energy (eV)
0
1
2
3
4
ε2(ω
)
ε2(ω) Dir xx DFT
ε2(ω) Dir yy DFT
ε2(ω) Dir zz DFT
ε2(ω) BSE
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 29 / 42

Resultados Propiedades ópticas
Corrección BSE
Función de perdida de energía electronica EELS(ω)
ExcitonesGrilla de puntos k de14x14x14 convergidaimplementando BSE.ABINIT
0 10 20 30Energy (eV)
0
0,5
1
1,5
2
-Im
ε(ω
)
EELS(ω) Dir xx DFT
EELS(ω) Dir yy DFT
EELS(ω) Dir yy DFT
EELS(ω) BSE
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 30 / 42

Resultados Propiedades ópticas
Corrección BSE
Función de perdida de energía electronica EELS(ω)
ExcitonesGrilla de puntos k de14x14x14 convergidaimplementando BSE.ABINIT
0 10 20 30Energy (eV)
0
0,5
1
1,5
2
-Im
ε(ω
)
EELS(ω) Dir xx DFT
EELS(ω) Dir yy DFT
EELS(ω) Dir yy DFT
EELS(ω) BSE
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 30 / 42

Resultados Propiedades ópticas
Corrección BSE
Función de perdida de energía electronica EELS(ω)
ExcitonesGrilla de puntos k de14x14x14 convergidaimplementando BSE.ABINIT
0 10 20 30Energy (eV)
0
0,5
1
1,5
2
-Im
ε(ω
)
EELS(ω) Dir xx DFT
EELS(ω) Dir yy DFT
EELS(ω) Dir yy DFT
EELS(ω) BSE
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 30 / 42

Resultados Comparación experimental
EELS(ω) Exp Vs Teoría
10 20 30Energy (eV)
0,5
1
1,5-I
m ε
EELS(ω) BSE
EELS(ω) Peak 1 Exp
EELS(ω) DFT
EELS(ω) exp 2 Peak
Técnicas XPS y UPSEin = 100eV
Ángulo incidencia: 30
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 31 / 42

Para terminar...
Perspectivas
Modelado de interfaces
Modelado de interface α−AlF3 − Cu(100)
Modelado de interface α−AlF3 − Cu(111)
Calulo de fenómenos de transporte en dichas interfases,obtención de curvas I-VImplementación de otros progamas de cálculo (Ej. OpenMX,SIESTA).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 32 / 42

Para terminar...
Perspectivas
Modelado de interfaces
Modelado de interface α−AlF3 − Cu(100)
Modelado de interface α−AlF3 − Cu(111)
Calulo de fenómenos de transporte en dichas interfases,obtención de curvas I-VImplementación de otros progamas de cálculo (Ej. OpenMX,SIESTA).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 32 / 42

Para terminar...
Perspectivas
Modelado de interfaces
Modelado de interface α−AlF3 − Cu(100)
Modelado de interface α−AlF3 − Cu(111)
Calulo de fenómenos de transporte en dichas interfases,obtención de curvas I-V
Implementación de otros progamas de cálculo (Ej. OpenMX,SIESTA).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 32 / 42

Para terminar...
Perspectivas
Modelado de interfaces
Modelado de interface α−AlF3 − Cu(100)
Modelado de interface α−AlF3 − Cu(111)
Calulo de fenómenos de transporte en dichas interfases,obtención de curvas I-VImplementación de otros progamas de cálculo (Ej. OpenMX,SIESTA).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 32 / 42

Para terminar...
Conclusiones
Resultados:
El α−AlF3 es un material del grupo espacial R3C del grupo de simetría 167. Es unmaterial que en su celda unitaria cuenta con 8 átomos, 2 de Al y 6 de F .
Modulo de bulk 137GPa, derivada del módulo de bulk respecto a la presión 3.47, volumende equilibrio 943.53Bohr3, energía mínima 150.88Ha.
Band-gap de 7.793 eV directo en el punto Γ calculado con DFT.
Band-gap GW de 10.81eV corregido en el band gap del material.
Constante dieléctrica ε0 de 1.92 y de 2.0 en presencia de campos locales.
El α−AlF3 presenta una gran absorción en una energía que va desde los 19 eV hasta 24eV.
Corrección BSE. Cálculo de las propiedades ópticas que incluyendo efectos excitónicosregión del band-gap (10− 14eV ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 33 / 42

Para terminar...
Conclusiones
Resultados:
El α−AlF3 es un material del grupo espacial R3C del grupo de simetría 167. Es unmaterial que en su celda unitaria cuenta con 8 átomos, 2 de Al y 6 de F .
Modulo de bulk 137GPa, derivada del módulo de bulk respecto a la presión 3.47, volumende equilibrio 943.53Bohr3, energía mínima 150.88Ha.
Band-gap de 7.793 eV directo en el punto Γ calculado con DFT.
Band-gap GW de 10.81eV corregido en el band gap del material.
Constante dieléctrica ε0 de 1.92 y de 2.0 en presencia de campos locales.
El α−AlF3 presenta una gran absorción en una energía que va desde los 19 eV hasta 24eV.
Corrección BSE. Cálculo de las propiedades ópticas que incluyendo efectos excitónicosregión del band-gap (10− 14eV ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 33 / 42

Para terminar...
Conclusiones
Resultados:
El α−AlF3 es un material del grupo espacial R3C del grupo de simetría 167. Es unmaterial que en su celda unitaria cuenta con 8 átomos, 2 de Al y 6 de F .
Modulo de bulk 137GPa, derivada del módulo de bulk respecto a la presión 3.47, volumende equilibrio 943.53Bohr3, energía mínima 150.88Ha.
Band-gap de 7.793 eV directo en el punto Γ calculado con DFT.
Band-gap GW de 10.81eV corregido en el band gap del material.
Constante dieléctrica ε0 de 1.92 y de 2.0 en presencia de campos locales.
El α−AlF3 presenta una gran absorción en una energía que va desde los 19 eV hasta 24eV.
Corrección BSE. Cálculo de las propiedades ópticas que incluyendo efectos excitónicosregión del band-gap (10− 14eV ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 33 / 42

Para terminar...
Conclusiones
Resultados:
El α−AlF3 es un material del grupo espacial R3C del grupo de simetría 167. Es unmaterial que en su celda unitaria cuenta con 8 átomos, 2 de Al y 6 de F .
Modulo de bulk 137GPa, derivada del módulo de bulk respecto a la presión 3.47, volumende equilibrio 943.53Bohr3, energía mínima 150.88Ha.
Band-gap de 7.793 eV directo en el punto Γ calculado con DFT.
Band-gap GW de 10.81eV corregido en el band gap del material.
Constante dieléctrica ε0 de 1.92 y de 2.0 en presencia de campos locales.
El α−AlF3 presenta una gran absorción en una energía que va desde los 19 eV hasta 24eV.
Corrección BSE. Cálculo de las propiedades ópticas que incluyendo efectos excitónicosregión del band-gap (10− 14eV ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 33 / 42

Para terminar...
Conclusiones
Resultados:
El α−AlF3 es un material del grupo espacial R3C del grupo de simetría 167. Es unmaterial que en su celda unitaria cuenta con 8 átomos, 2 de Al y 6 de F .
Modulo de bulk 137GPa, derivada del módulo de bulk respecto a la presión 3.47, volumende equilibrio 943.53Bohr3, energía mínima 150.88Ha.
Band-gap de 7.793 eV directo en el punto Γ calculado con DFT.
Band-gap GW de 10.81eV corregido en el band gap del material.
Constante dieléctrica ε0 de 1.92 y de 2.0 en presencia de campos locales.
El α−AlF3 presenta una gran absorción en una energía que va desde los 19 eV hasta 24eV.
Corrección BSE. Cálculo de las propiedades ópticas que incluyendo efectos excitónicosregión del band-gap (10− 14eV ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 33 / 42

Para terminar...
Conclusiones
Resultados:
El α−AlF3 es un material del grupo espacial R3C del grupo de simetría 167. Es unmaterial que en su celda unitaria cuenta con 8 átomos, 2 de Al y 6 de F .
Modulo de bulk 137GPa, derivada del módulo de bulk respecto a la presión 3.47, volumende equilibrio 943.53Bohr3, energía mínima 150.88Ha.
Band-gap de 7.793 eV directo en el punto Γ calculado con DFT.
Band-gap GW de 10.81eV corregido en el band gap del material.
Constante dieléctrica ε0 de 1.92 y de 2.0 en presencia de campos locales.
El α−AlF3 presenta una gran absorción en una energía que va desde los 19 eV hasta 24eV.
Corrección BSE. Cálculo de las propiedades ópticas que incluyendo efectos excitónicosregión del band-gap (10− 14eV ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 33 / 42

Para terminar...
Conclusiones
Resultados:
El α−AlF3 es un material del grupo espacial R3C del grupo de simetría 167. Es unmaterial que en su celda unitaria cuenta con 8 átomos, 2 de Al y 6 de F .
Modulo de bulk 137GPa, derivada del módulo de bulk respecto a la presión 3.47, volumende equilibrio 943.53Bohr3, energía mínima 150.88Ha.
Band-gap de 7.793 eV directo en el punto Γ calculado con DFT.
Band-gap GW de 10.81eV corregido en el band gap del material.
Constante dieléctrica ε0 de 1.92 y de 2.0 en presencia de campos locales.
El α−AlF3 presenta una gran absorción en una energía que va desde los 19 eV hasta 24eV.
Corrección BSE. Cálculo de las propiedades ópticas que incluyendo efectos excitónicosregión del band-gap (10− 14eV ).
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 33 / 42

Para terminar...
Final
Gracias por su atención
Hasta la próxima...
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 34 / 42

Para terminar...
Teoréma de Block
Electrones de Block[− ~2
2m∇2 + U(~r)
]ψ = Eψ
Asumiendo la periodicidad de la red
V (~r + ~R) = V (~r)
“Las autofunciones o funciones propias ψ de la ecuación de onda para un potencialperiódico V (~r + ~R) = V (~r) son el producto de una onda plana de la forma ei
~k·~r poruna función u~k(~r) que posee la periodicidad de la red cristalina que conforma elsólido”
ψ~k = ei~k·~ru~k(~r)
u~k(~r + ~R) = u~k(~r) para todo vector ~R de la red de Bravais
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 35 / 42

Para terminar...
Teoréma de Block
Electrones de Block[− ~2
2m∇2 + U(~r)
]ψ = Eψ
Asumiendo la periodicidad de la red
V (~r + ~R) = V (~r)
“Las autofunciones o funciones propias ψ de la ecuación de onda para un potencialperiódico V (~r + ~R) = V (~r) son el producto de una onda plana de la forma ei
~k·~r poruna función u~k(~r) que posee la periodicidad de la red cristalina que conforma elsólido”
ψ~k = ei~k·~ru~k(~r)
u~k(~r + ~R) = u~k(~r) para todo vector ~R de la red de Bravais
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 35 / 42

Para terminar...
Teoréma de Block
Electrones de Block[− ~2
2m∇2 + U(~r)
]ψ = Eψ
Asumiendo la periodicidad de la red
V (~r + ~R) = V (~r)
“Las autofunciones o funciones propias ψ de la ecuación de onda para un potencialperiódico V (~r + ~R) = V (~r) son el producto de una onda plana de la forma ei
~k·~r poruna función u~k(~r) que posee la periodicidad de la red cristalina que conforma elsólido”
ψ~k = ei~k·~ru~k(~r)
u~k(~r + ~R) = u~k(~r) para todo vector ~R de la red de Bravais
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 35 / 42

Para terminar...
Teoréma de Block
Electrones de Block[− ~2
2m∇2 + U(~r)
]ψ = Eψ
Asumiendo la periodicidad de la red
V (~r + ~R) = V (~r)
“Las autofunciones o funciones propias ψ de la ecuación de onda para un potencialperiódico V (~r + ~R) = V (~r) son el producto de una onda plana de la forma ei
~k·~r poruna función u~k(~r) que posee la periodicidad de la red cristalina que conforma elsólido”
ψ~k = ei~k·~ru~k(~r)
u~k(~r + ~R) = u~k(~r) para todo vector ~R de la red de Bravais
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 35 / 42

Para terminar...
Teoréma de Block
Electrones de Block[− ~2
2m∇2 + U(~r)
]ψ = Eψ
Asumiendo la periodicidad de la red
V (~r + ~R) = V (~r)
“Las autofunciones o funciones propias ψ de la ecuación de onda para un potencialperiódico V (~r + ~R) = V (~r) son el producto de una onda plana de la forma ei
~k·~r poruna función u~k(~r) que posee la periodicidad de la red cristalina que conforma elsólido”
ψ~k = ei~k·~ru~k(~r)
u~k(~r + ~R) = u~k(~r) para todo vector ~R de la red de BravaisJ.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 35 / 42

Para terminar...
Teoremas de Hohenberg y Kohn
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.
Theorem1. La densidad electrónica ρ(~r) del estado fundamental determina univocamente elpotencial externo Vext(~r), a menos de una constante. ρ(~r) es necesario perosuficiente para construir el Hamiltoniano H. Por lo que al conocer H, se conoceríaψ0(~r1, ...., ~rN ).
Theorem2 .Se puede definir un funcional universal de la energía E[ρ], en términos de ladensidad de carga ρ(~r), valida para cualquier potencial externo Vext(~r).Para un dado Vext(~r), la energía exacta del estado fundamental es el mínimo de lafuncional E[ρ], y la densidad que minimiza la funcional es la densidad de carga delestado fundamental.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 36 / 42

Para terminar...
Teoremas de Hohenberg y Kohn
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.
Theorem1. La densidad electrónica ρ(~r) del estado fundamental determina univocamente elpotencial externo Vext(~r), a menos de una constante. ρ(~r) es necesario perosuficiente para construir el Hamiltoniano H. Por lo que al conocer H, se conoceríaψ0(~r1, ...., ~rN ).
Theorem2 .Se puede definir un funcional universal de la energía E[ρ], en términos de ladensidad de carga ρ(~r), valida para cualquier potencial externo Vext(~r).Para un dado Vext(~r), la energía exacta del estado fundamental es el mínimo de lafuncional E[ρ], y la densidad que minimiza la funcional es la densidad de carga delestado fundamental.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 36 / 42

Para terminar...
Teoremas de Hohenberg y Kohn
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.
Theorem1. La densidad electrónica ρ(~r) del estado fundamental determina univocamente elpotencial externo Vext(~r), a menos de una constante. ρ(~r) es necesario perosuficiente para construir el Hamiltoniano H. Por lo que al conocer H, se conoceríaψ0(~r1, ...., ~rN ).
Theorem2 .Se puede definir un funcional universal de la energía E[ρ], en términos de ladensidad de carga ρ(~r), valida para cualquier potencial externo Vext(~r).Para un dado Vext(~r), la energía exacta del estado fundamental es el mínimo de lafuncional E[ρ], y la densidad que minimiza la funcional es la densidad de carga delestado fundamental.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 36 / 42

Para terminar...
Teoremas de Hohenberg y Kohn
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.
Theorem1. La densidad electrónica ρ(~r) del estado fundamental determina univocamente elpotencial externo Vext(~r), a menos de una constante. ρ(~r) es necesario perosuficiente para construir el Hamiltoniano H. Por lo que al conocer H, se conoceríaψ0(~r1, ...., ~rN ).
Theorem2 .Se puede definir un funcional universal de la energía E[ρ], en términos de ladensidad de carga ρ(~r), valida para cualquier potencial externo Vext(~r).Para un dado Vext(~r), la energía exacta del estado fundamental es el mínimo de lafuncional E[ρ], y la densidad que minimiza la funcional es la densidad de carga delestado fundamental.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 36 / 42

Para terminar...
Teoremas de Hohenberg y Kohn
N∑i=1
[− ~2
2m∇2i + Vext(~ri)
]+
1
2
N∑i6=j=1
e2
|~ri − ~rj |
ψ(~ri, ....~rN ) = Eψ(~ri, ....~rN )
1964 P. Hohenberg Y W. Kohn plantean los teoremas que dan fundamento a la DFT.
Theorem1. La densidad electrónica ρ(~r) del estado fundamental determina univocamente elpotencial externo Vext(~r), a menos de una constante. ρ(~r) es necesario perosuficiente para construir el Hamiltoniano H. Por lo que al conocer H, se conoceríaψ0(~r1, ...., ~rN ).
Theorem2 .Se puede definir un funcional universal de la energía E[ρ], en términos de ladensidad de carga ρ(~r), valida para cualquier potencial externo Vext(~r).Para un dado Vext(~r), la energía exacta del estado fundamental es el mínimo de lafuncional E[ρ], y la densidad que minimiza la funcional es la densidad de carga delestado fundamental.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 36 / 42

Para terminar...
Herramientas para resolver la ecuación deSchrödinger.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 37 / 42

Para terminar...
Comaparación con DOS Teorica
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 38 / 42

Para terminar...
Densidad de estados parcial Al en α− AlF3
DOS parciales obtenidas con ABINIT y con Wien2k
0
0,1
0,2
0,3
0,4
0,5
DO
S (
Sta
tes/
eV)
Al s-states
Al p-states
Al d-states
Al f-states
-20 -10 0 10 20 30
Energy (eV)
0
5
10
15
20
25
30
DO
S (
Sta
tes/
eV)
DOS Total α−AlF3
0
0,5
1
1,5
DO
S (
Sta
tes/
eV)
Al s-states
Al p-states
Al d-states
Al f-states
Al-Dos
-20 -10 0 10 20 30
Energy (eV)
0
5
10
15
20
25
30
DO
S (
Sta
tes/
eV)
DOS Total α−AlF3
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 39 / 42

Para terminar...
Densidad de estados parcial Al en α− AlF3
DOS parciales obtenidas con ABINIT y con Wien2k
0
0,1
0,2
0,3
0,4
0,5
DO
S (
Sta
tes/
eV)
Al s-states
Al p-states
Al d-states
Al f-states
-20 -10 0 10 20 30
Energy (eV)
0
5
10
15
20
25
30
DO
S (
Sta
tes/
eV)
DOS Total α−AlF3
0
0,5
1
1,5
DO
S (
Sta
tes/
eV)
Al s-states
Al p-states
Al d-states
Al f-states
Al-Dos
-20 -10 0 10 20 30
Energy (eV)
0
5
10
15
20
25
30
DO
S (
Sta
tes/
eV)
DOS Total α−AlF3
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 39 / 42

Para terminar...
Densidad de estados parcial F en α− AlF3
DOS parciales obtenidas con ABINIT y con Wien2k
0
20
40
60
80
100
F D
OS
(Sta
tes
Vol-1
eV
-1) F-s States
F-p States
F-d States
-20 -15 -10 -5 0 5 10 15 20 25 30Energy (eV)
0
100
200
300
400
500
600
700
Tota
l D
OS
(S
tate
s V
ol-1
eV
-1)
Total DOS α−AlF3
0
2
4
6
8
10
F D
OS
(Sta
tes
Vol-1
eV
-1) Zoom Zona de conduccion
0
2.5
DO
S (
Sta
tes
Vo
l-1 e
V-1
)
F s-statesF p-states
F d-states
F f-states
-20 -10 0 10 20 30
Energy (eV)
0
5
10
15
20
25
30
DO
S (
Sta
tes
Vo
l-1 e
V-1
) DOS Total αAlF3
Energy (eV)0
DO
S (
Sta
tes
Vo
l-1 e
V-1
)
Zoom zona de Conduccion
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 40 / 42

Para terminar...
Densidad de estados parcial F en α− AlF3
DOS parciales obtenidas con ABINIT y con Wien2k
0
20
40
60
80
100
F D
OS
(Sta
tes
Vol-1
eV
-1) F-s States
F-p States
F-d States
-20 -15 -10 -5 0 5 10 15 20 25 30Energy (eV)
0
100
200
300
400
500
600
700
Tota
l D
OS
(S
tate
s V
ol-1
eV
-1)
Total DOS α−AlF3
0
2
4
6
8
10
F D
OS
(Sta
tes
Vol-1
eV
-1) Zoom Zona de conduccion
0
2.5
DO
S (
Sta
tes
Vo
l-1 e
V-1
)
F s-statesF p-states
F d-states
F f-states
-20 -10 0 10 20 30
Energy (eV)
0
5
10
15
20
25
30
DO
S (
Sta
tes
Vo
l-1 e
V-1
) DOS Total αAlF3
Energy (eV)0
DO
S (
Sta
tes
Vo
l-1 e
V-1
)
Zoom zona de Conduccion
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 40 / 42
Para terminar...
Propiedades ópticas con Wien2kFunción dielectrica εi(ω)
0
0,5
1
1,5
2
2,5
3
ε2(ω
)
ε2(ω) Dir xx
ε2(ω) Dir yy
ε2(ω) Dir zz
0 10 20 30 40Energy (eV)
0
0,5
1
1,5
2
2,5
3
ε1(ω
)
ε1(ω) Dir xx
ε1(ω) Dir yy
ε1(ω) Dir zz
Refracción n(ω), Extinción K(ω), Reflectividad R(ω)
0,60,8
1
1,21,41,61,8
2
n(ω
)
n(ω) Dir xx
n(ω) Dir yy
n(ω) Dir zz
0
0,5
1
1,5
2
K(ω
)
K(ω) Dir xx
K(ω) Dir yy
K(ω) Dir zz
0 10 20 30 40Energy (eV)
0
0,05
0,1
0,15
0,2
0,25
R(ω
)
R(ω) Dir xx
R(ω) Dir yy
R(ω) Dir zz
Coeficiente de absorción α(ω)
0 10 20 30 40Energy (eV)
0
50
100
150
200
250
300
α(ω
) (x
10
4 c
m-1
)
α(ω) Dir xx
α(ω) Dir yy
α(ω) Dir zz
Función perdida de energía electrónica EELS(ω)
0 10 20 30 40Energy (eV)
0
0,5
1
1,5
2
2,5
-Im
ε(ω
)
EELS (ω) Dir xx
EELS (ω) Dir yy
EELS (ω) Dir zz
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 41 / 42

Para terminar...
Propiedades ópticas con Wien2kFunción dielectrica εi(ω)
0
0,5
1
1,5
2
2,5
3
ε2(ω
)
ε2(ω) Dir xx
ε2(ω) Dir yy
ε2(ω) Dir zz
0 10 20 30 40Energy (eV)
0
0,5
1
1,5
2
2,5
3
ε1(ω
)
ε1(ω) Dir xx
ε1(ω) Dir yy
ε1(ω) Dir zz
Refracción n(ω), Extinción K(ω), Reflectividad R(ω)
0,60,8
1
1,21,41,61,8
2
n(ω
)
n(ω) Dir xx
n(ω) Dir yy
n(ω) Dir zz
0
0,5
1
1,5
2
K(ω
)
K(ω) Dir xx
K(ω) Dir yy
K(ω) Dir zz
0 10 20 30 40Energy (eV)
0
0,05
0,1
0,15
0,2
0,25
R(ω
)
R(ω) Dir xx
R(ω) Dir yy
R(ω) Dir zz
Coeficiente de absorción α(ω)
0 10 20 30 40Energy (eV)
0
50
100
150
200
250
300
α(ω
) (x
10
4 c
m-1
)
α(ω) Dir xx
α(ω) Dir yy
α(ω) Dir zz
Función perdida de energía electrónica EELS(ω)
0 10 20 30 40Energy (eV)
0
0,5
1
1,5
2
2,5
-Im
ε(ω
)
EELS (ω) Dir xx
EELS (ω) Dir yy
EELS (ω) Dir zz
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 41 / 42

Para terminar...
Propiedades ópticas con Wien2kFunción dielectrica εi(ω)
0
0,5
1
1,5
2
2,5
3
ε2(ω
)
ε2(ω) Dir xx
ε2(ω) Dir yy
ε2(ω) Dir zz
0 10 20 30 40Energy (eV)
0
0,5
1
1,5
2
2,5
3
ε1(ω
)
ε1(ω) Dir xx
ε1(ω) Dir yy
ε1(ω) Dir zz
Refracción n(ω), Extinción K(ω), Reflectividad R(ω)
0,60,8
1
1,21,41,61,8
2
n(ω
)
n(ω) Dir xx
n(ω) Dir yy
n(ω) Dir zz
0
0,5
1
1,5
2
K(ω
)
K(ω) Dir xx
K(ω) Dir yy
K(ω) Dir zz
0 10 20 30 40Energy (eV)
0
0,05
0,1
0,15
0,2
0,25
R(ω
)
R(ω) Dir xx
R(ω) Dir yy
R(ω) Dir zz
Coeficiente de absorción α(ω)
0 10 20 30 40Energy (eV)
0
50
100
150
200
250
300
α(ω
) (x
10
4 c
m-1
)
α(ω) Dir xx
α(ω) Dir yy
α(ω) Dir zz
Función perdida de energía electrónica EELS(ω)
0 10 20 30 40Energy (eV)
0
0,5
1
1,5
2
2,5
-Im
ε(ω
)
EELS (ω) Dir xx
EELS (ω) Dir yy
EELS (ω) Dir zz
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 41 / 42

Para terminar...
Propiedades ópticas con Wien2kFunción dielectrica εi(ω)
0
0,5
1
1,5
2
2,5
3
ε2(ω
)
ε2(ω) Dir xx
ε2(ω) Dir yy
ε2(ω) Dir zz
0 10 20 30 40Energy (eV)
0
0,5
1
1,5
2
2,5
3
ε1(ω
)
ε1(ω) Dir xx
ε1(ω) Dir yy
ε1(ω) Dir zz
Refracción n(ω), Extinción K(ω), Reflectividad R(ω)
0,60,8
1
1,21,41,61,8
2
n(ω
)
n(ω) Dir xx
n(ω) Dir yy
n(ω) Dir zz
0
0,5
1
1,5
2
K(ω
)
K(ω) Dir xx
K(ω) Dir yy
K(ω) Dir zz
0 10 20 30 40Energy (eV)
0
0,05
0,1
0,15
0,2
0,25
R(ω
)
R(ω) Dir xx
R(ω) Dir yy
R(ω) Dir zz
Coeficiente de absorción α(ω)
0 10 20 30 40Energy (eV)
0
50
100
150
200
250
300
α(ω
) (x
10
4 c
m-1
)
α(ω) Dir xx
α(ω) Dir yy
α(ω) Dir zz
Función perdida de energía electrónica EELS(ω)
0 10 20 30 40Energy (eV)
0
0,5
1
1,5
2
2,5
-Im
ε(ω
)
EELS (ω) Dir xx
EELS (ω) Dir yy
EELS (ω) Dir zz
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 41 / 42

Para terminar...
Ecuación BSE
El término diagonal representa transiciones fotoabsortivas
Hdiagvck,v′,c′k′ = (Eck − Evk)δvv′δcc′δkk′
El segundo término representa el intercambio electrón-hueco
Hexchvc~k,v′,c′~k′ = 2vv
′c′~k′
vc~k= 2
4π
Ω
∑~G6=0
1
|~G|2〈c~k|ei ~G~r|v~k〉 × 〈c′~k′|e−i ~G~r|v′~k′〉
El tercer término toma en cuenta el apantalamiento por medio de ε−1M
HSCRvc~k,v′,c′~k′ = W v′c′~k′
vc~k= −4π
Ω
∑~G~G′
ε−1~G~G′(~q)
|~q + ~G|2〈c~k|ei(~q+~G)~r|c′~k′〉 × 〈v′~k′|e−i(~q+~G)~r|v~k〉δ~q,~k−~k′
Donde ~G y ~G′ son vectores de la red recíproca.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 42 / 42

Para terminar...
Ecuación BSE
El término diagonal representa transiciones fotoabsortivas
Hdiagvck,v′,c′k′ = (Eck − Evk)δvv′δcc′δkk′
El segundo término representa el intercambio electrón-hueco
Hexchvc~k,v′,c′~k′ = 2vv
′c′~k′
vc~k= 2
4π
Ω
∑~G6=0
1
|~G|2〈c~k|ei ~G~r|v~k〉 × 〈c′~k′|e−i ~G~r|v′~k′〉
El tercer término toma en cuenta el apantalamiento por medio de ε−1M
HSCRvc~k,v′,c′~k′ = W v′c′~k′
vc~k= −4π
Ω
∑~G~G′
ε−1~G~G′(~q)
|~q + ~G|2〈c~k|ei(~q+~G)~r|c′~k′〉 × 〈v′~k′|e−i(~q+~G)~r|v~k〉δ~q,~k−~k′
Donde ~G y ~G′ son vectores de la red recíproca.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 42 / 42

Para terminar...
Ecuación BSE
El término diagonal representa transiciones fotoabsortivas
Hdiagvck,v′,c′k′ = (Eck − Evk)δvv′δcc′δkk′
El segundo término representa el intercambio electrón-hueco
Hexchvc~k,v′,c′~k′ = 2vv
′c′~k′
vc~k= 2
4π
Ω
∑~G6=0
1
|~G|2〈c~k|ei ~G~r|v~k〉 × 〈c′~k′|e−i ~G~r|v′~k′〉
El tercer término toma en cuenta el apantalamiento por medio de ε−1M
HSCRvc~k,v′,c′~k′ = W v′c′~k′
vc~k= −4π
Ω
∑~G~G′
ε−1~G~G′(~q)
|~q + ~G|2〈c~k|ei(~q+~G)~r|c′~k′〉 × 〈v′~k′|e−i(~q+~G)~r|v~k〉δ~q,~k−~k′
Donde ~G y ~G′ son vectores de la red recíproca.
J.L. Navarro y E. Albanesi Seminario IFIS CONICET-UNL Octubre de 2014 42 / 42





























