Embed Size (px)
Citation preview

- 1 -
자료행렬과
반응변수
사전자료처리와
유전자선택
분류실시:
훈련집합
예측:
평가집합
생물학적특성
조정모수오분류율,
정분류율
제7장 분류분석
분류분석: 새로운 표본을 미리 정해진 특정 그룹으로 분류하는 분석
수집된 자료에 유전자 자료 및 그룹에 대한 정보를 함께 가지고 있는 경우에 적용
할 수 있는 방법
7.1 서론
마이크로어레이 자료의 분류분석
⋯ 번째 표본의 차원 mRNA 유전정보 ∈ 와 범주형 반응변수(outcome,
target 혹은 response) ∈ ⋯ 를 이용하여, 를 잘 구분해내는 들의 함수인
분류자(classifier)를 만든 후 새로운 값을 가진 자료에 대해서 를 예측하는 방법
마이크로어레이 자료의 분류분석의 절차
① 자료행렬과 반응변수로부터 사전자료처리를 통해 분류분석에 사용할 유전자 선별
② 분류자를 만들고 분류자의 성능을 평가하여 성능이 좋은 분류자 탐색
ⅰ 원자료를 훈련집합(training set)과 평가집합(test set)로 분할
ⅱ 훈련집합에서 분류함수를 구함
ⅲ 평가집합을 이용하여 분류함수의 성능을 평가
ⅳ 위의 결과를 분류조정인자에 반영하여 최종분류자를 만들 때까지 반복
마이크로어레이 자료의 분류분석의 목표
① 새로운 표본의 유전자의 정보로부터 반응범주를 정확하게 분류하고 예측
예) 새로운 환자의 유전자정보를 분류하고 암 판정 여부를 판단하기위한 예측작업
② 좋은 성능을 내는 소수의 유전자로 구성된 분류자를 개발
→ 중요한 소수의 유전자를 선별
예) 특정 질병 발병기작에 영향을 미치는 유전자들의 상호연관성 탐구
→ 신약 개발 등 질병치료에 직접적으로 관련된 제약산업으로 응용

- 2 -
7.2 분류분석
7.2.1 분류문제(Classification problem)
(1) 분류문제에 대한 자료구조와 가정
× 유전자 행렬 이외에 반응변수 가 함께 필요
⋯ ′ : 번째 표본 벡터
: 번째 범주형 반응변수
전체 표본 집합
⋯ ∈ ∈ ⋯ ⋯
∼
⋮
⋯
⋯
⋮ ⋮ ⋮ ⋮
⋯
: 와 번째 유전자 변수에 대한 공분산
: 번째 유전자 변수의 모분산
개의 범주에 속한 표본의 수 ⇒ 각각 ⋯
⋯ : 범주에 속하는 표본의 지수집합(index set)
반응범주 일 때, 표본평균벡터와 표본공분산행렬과 총평균벡터
∈
⋯
⋯
⋮ ⋮ ⋮ ⋮
⋯
∈
의 번째 원소
∈
:
범주 값을 갖는 표본자료 중 번째 변수에 대한 표본평균
의 ( ,)번째 원소
∈
:
범주 값을 갖는 표본자료 중 번째 변수에 대한 표본분산
의 ( ,)번째 원소
∈
:
범주 값을 갖는 표본자료 중 와 번째 변수에 대한 표본공분산

- 3 -
분류자를 구성할 때 사용하는 집합의 용도 구분
: 학습집합(learning set) 또는 훈련집합(training set)
: 평가집합(test set)
: 으로 구성한 분류자
, , : 표본 에 대한 예측범주
인 이범주 경우
각 범주에 속하는 표본의 수 : 각각
자료 행렬과 반응벡터 :
×
⋯
⋯
⋮ ⋮ ⋯ ⋮ ⋯
∈
훈련집합 로부터 최적의 분류자 를 구성
새로운 자료 를 의 부호에 따라 다음과 같이 분류
>0 이면 =1
<0 이면 =-1
=0 이면 의 분류 보류
인 다범주 경우
다범주 경우의 접근 전략
① 개의 범주에서 2개를 선택하여 판별하는 방법
개의 두 범주쌍에 대하여 분류분석을 하여 가장 많이 뽑힌 범주 선택
② 1대 -1 비교방법 : 한 범주와 나머지 -1개의 범주를 합친 범주를 비교
총 번 비교하고 난후 가장 많이 뽑힌 범주 선택
: 훈련집합 로부터 구한 의 예측값
Pr≠ Pr ≠ : 오분류율(misclassification error rate)
추정량 :
≠
(2) 분류분석의 단계
절차1 : 훈련집합 로부터 분류자를 구함
절차2 : 에서 예측을 하고 오분류율을 계산하여 분류자의 입력모수들을 조정
절차3 : 평가집합 로부터 최종분류자의 오분류율을 계산하여 그 성능을 평가

- 4 -
7.2.2 분류단계의 세부적 요소
(1) 여과(filtering)
신뢰성이 떨어지는 유전자나 정보가 많지 않은 자료를 제거하는 과정
① 비명시(non-specific) 여과 :
범주와 관계없이 실험적인 오류로 인해 신뢰성이 떨어지는 유전자를 제거하는 과정
예) 적당한 사전가지(유전자 발현 프로파일의 max/min과 max-min)를 통해 1000
개 정도의 유전자를 선택하고 나머지 유전자 제거
② 명시(specific) 여과 :
범주의 분류에 크게 기여하지 않은 유전자를 선택하여 제거하는 과정
예) 반응변수의 두 범주의 평균차이가 큰 유전자들만 선택하고 나머지 유전자 제거
(2) 자료의 변환
대부분의 마이크로어레이 실험의 특성상 단위는 일치하나 열과 행에 따라 척도가 차이가 나
는 경우가 많음 → 자료의 변환작업이 필요 → 행과 열에 따라 이차원으로 실시
변환에 관한 자세한 설명은 6장 참조
로그변환 log과 쌍곡탄젠트함수 tanh
<그림 7.3> log, tanh 변환효과

- 5 -
log : 가 음의 값을 가지면 변환 불가. 가 0근처로 갈수록 값이 급격히 작아짐
가 (0,1)구간에 있을 때 보다 (2,∞)구간에 있을 때에 변환값의 변화가 작음.
tanh exp exp exp exp
:
(-1,1)구간에서는 값과 변환값이 거의 동일
값이 아주 큰 값과 작은 값을 갖은 영역에서는 변환값이 -1과 1로 절단되는 효
과
행 중심화(centering) 변환
자료행렬 의 행 평균을 0 (각 유전자 에대해
)으로 만드는 변환
유전자마다 표본간 평균의 심각한 차이가 관찰된 경우 유전자 효과검증에 영향
열 중심화(centering) 변환
자료행렬 의 열 평균을 0 (각 표본 에대해
)으로 만드는 변환
같은 반응범주를 가지는 두 표본사이의 모든 유전자 발현값이 차이가 심할 경우 고려
척도(scale) 변환
자료의 분산을 1로 맞추는 변환
각 유전자 에 대해 혹은 각 표본 에 대해 로 맞추는 변환
유전자마다 변이가 심하게 날 경우에 필요한 변환
(2) 결측값(missing observation)
선형판별분석(LDA)과 이차판별분석(QDA)에서는 하나의 표본에서 특정유전자가 결측이 되
면 나머지 개의 관측된 정보를 쓸 수 없게 됨
결측값에 대한 처리 방법
① 대치법(imputation) :
-NN과 재귀 비정칙분해(recursive SVD)방법으로 결측값을 대치(impute)하여 자
료를 완전한 형태로 만들어 분석
② 결측값에 영향을 덜 받는 분류방법이나 거리 사용 :
분류회귀나무(CART)방법은 사전단계에서 자료를 완전한 형태로 만드는 과정이 필
요 없음. 상관계수를 거리로 사용하면 결측값에 덜 영향을 받음

- 6 -
● 적합성(relevance) 유전자와 반응 범주사이에 과학적증거가 필요하다.
즉, Pr ≠ Pr 를 만족하는 유전자 변수
가 존재한다. 단Pr
● 희소성(sparsity) 같은 분류성능을 내더라도 유전자의 수가 작으면 작을수록 좋다.
● 최적성(optimality) 유전자수가 같더라도 분류성능이 더 좋아야 한다.
<표 7.1> 유전자 선택의 원칙
7.2.3 분류분석에서 유전자 선택하기(Feature Selection)
● 표본의 수에 비해 유전자의 수가 큰 마이크로어레이 자료의 분석에서는 중요한 소수의
유전자 (candidate gene)를 뽑아서 구성하는 것이 바람직
● 후보유전자를 찾는 방법 또한 분류방법의 선택 못지않게 중요
<표 7.1>은 유전자 선택의 세 가지 원칙인 희소성(sparsity), 최적성(optimality), 적합성
(relevance)을 설명. 적합성은 생물학적인 특성과 관련되어 있는 중요한 개념
(1) 유전자들 간의 상관성 반영여부에 따른 분류: 단변량 방법과 다변량 방법
단변량(univariate) 통계량에 기초한 방법들
가장 기본적인 방법. 각 유전자에 대하여 특정통계량 혹은 측도(measure)로 순위를 매겨
적당한 비율(5%)의 상위 유전자를 선택하여 평가하는 방식
Golub et al. (1999) : 각 유전자 마다 AML과 ALL의 가중치
를 계산
범주간의 평균의 차이가 크면서 산포의 값이 작은 유전자 탐색
한계점: 각 유전자의 개별적인 효과만 고려. 서로 상관성 있는 유전자자료의 특성을 잘 반
영하지 못함.

- 7 -
> temp <- data.frame(x1=a[5061,],x2=a[2230,]
+ ,group=as.factor(c(rep(1,27),rep(2,11))))
> t.test(temp[1:27,1],temp[28:38,1])$p.v
[1] 0.1214719
> t.test(temp[1:27,2],temp[28:38,2])$p.v
[1] 0.9761973
> summary(glm(factor(group)~x1,data=temp,family="binomial"))$coeff[,4]
(Intercept) x1
0.3077611 0.1427241
> summary(glm(factor(group)~x2,data=temp,family="binomial"))$coeff[,4]
(Intercept) x2
0.732290 0.976173
> summary(glm(factor(group)~x1+x2,data=temp,family="binomial"))$coeff[,4]
(Intercept) x1 x2
0.50344036 0.03107609 0.09334473
예) leukemia data에서 2230번째 유전자와 5061번째 유전자 조합

- 8 -
> temp <- data.frame(x1=a[2135,],x2=a[6504,]
+ ,group=as.factor(c(rep(1,27),rep(2,11))))
> t.test(temp[1:27,1],temp[28:38,1])$p.v
[1] 0.5714494
> t.test(temp[1:27,2],temp[28:38,2])$p.v
[1] 0.6874682
> summary(glm(factor(group)~x1,data=temp,family="binomial"))$coeff[,4]
(Intercept) x1
0.2010270 0.6869672
> summary(glm(factor(group)~x2,data=temp,family="binomial"))$coeff[,4]
(Intercept) x2
0.7722301 0.7686502
> summary(glm(factor(group)~x1+x2,data=temp,family="binomial"))$coeff[,4]
(Intercept) x1 x2
0.6736198 0.7513730 0.9907523
> summary(glm(factor(group)~x1+x2+x1:x2,data=temp,family="binomial"))$coeff[,4]
(Intercept) x1 x2 x1:x2
0.02759189 0.01998971 0.02660145 0.02003157
예) leukemia data에서 2135번째 유전자와 6504번째 유전자 조합

- 9 -
다변량(multivariate) 통계량에 기초한 방법들
대표적인 다변량 분석 방법: SVM, LDA, QDA
LDA, QDA: 표본의 수가 변수의 수보다 많아야 분석이 가능
→ 마이크로어레이 자료에는 직접적인 적용 불가
SVM: 표본의 수가 변수의 수보다 작더라도 분류분석 가능
(2) 유전자들을 선택하는 방향에 따른 분류 : 후진제거, 전진선택, 단계적 선택
후진 제거(backward elimination)
전체 개의 유전자집합에서 매 단계마다 반응범주 와 상대적으로 연관성이 적은 유전자를
제거하는 방식. 계산시간이 비교적 적게 걸림.
전진 선택(forward selection)
하나의 유전자로부터 시작하여 분류자로 사용될 유전자를 하나씩 추가적으로 선택하는 방식
단계적 선택(stepwise selection)
전진 선택 방법을 보완한 방법. 전진 선택방식과는 달리 새로운 유전자가 선택된 이후에 그
전 단계에서 미리 선택된 유전자의 제거여부를 추가적으로 결정하는 방식. 회귀분석에서 가
장 널리 쓰이는 방식.
Bo et al. (2002)
-검정으로 상위 유전자들을 뽑은 뒤, 가능한 모든 유전자 쌍 조합에 대해 분류분석을 실
시. 모든 유전자조합과 여러 방법들을 비교평가. 오분류율을 기준으로 비교했을 때에 전진
선택방식이 나머지 두 방식들보다 분류 성능면에서 크게 차이 나지 않음을 보임.
7.2.4 분류분석의 방법 결정
(1) 마이크로어레이 자료의 분류시 발생하는 과적합(overfitting) 문제점
분류분석에서의 과적합(overfiting) 문제 :
훈련집합에서 적합된 분류자가 평가집합에서는 훈련집합만큼의 성능을 보이지 않는 문제
과적합 문제 해결방안
① 조정화(regularization) : 사용자가 제어할 수 있는 조정모수(tuning parameter)를 사용
예) 역행렬을 계산하기 힘들 때 대각원소에 적당한 함수를 더함.
분류자가 복잡해지면 벌점을 주어서 간단한 분류자를 선택하도록 함.
② 차원 축소 : 전체 유전자변수를 몇 개의 주성분 변수에 사영
새로 생성된 변수에서 생물학적인 의미를 찾기 어려움.
원래의 변수를 그대로 사용하기 때문에 희소성의 원칙에도 위배.

- 10 -
(2) 분류자의 성능 평가 측도
각각 오분류들이 같은 위험을 가진다고 할 수 없음 → 손실함수(loss function) 사용
손실함수 × →
예) : 범주 를 로 예측 → 정분류 → 손실은 0
≠ : 범주 를 로 예측 → 양의 손실
손실 행렬 (loss matrix)
⋯
⋯
⋮ ⋮ ⋯ ⋮ ⋯
⋯ ⋯ ⋮ ⋮ ⋯ ⋮ ⋯
일 경우
⋅⋅ -1로 예측 1로 예측
<표 7.2> 에서 손실 행렬
⋅⋅ -1로 예측 1로 예측
<표 7.2> 에서 일반적인 손실 행렬
위험도(risk) : 손실함수의 기대값, , 일반적인 오차의 개념
예측 범주
-1 1
실제
범주
<표 7.4> 실제범주와 예측범주의 분류표
정분류율 :
오분류율 :
민감도(sensitivity) :
특이도(specificity) :
Receiver Operating Characteristic (ROC) 곡선
축: 1-특이도 축: 민감도
동일한 값에 대하여 위에 있는 곡선이 분류성능이 뛰어남.
일반적으로 곡선아래에 있는 전체면적을 계산하여 분류자의 성능을 비교.

- 11 -
<그림 7.5> ROC 곡선
(3) 분류자의 성능 전략
방법 절차 특징
재대입를 사용하여 분류자를 만들고
다시 를 사용하여 평가오차를 과소 추정.
- CV를 사용하여 분류자를 만들고
를 사용하여 평가
인위적으로 와로 나누므
로 적당한 기준이 없음.
-fold CV
전체집합을 개의 부그룹으로 나
누어 (-1)개 와 1개의를
사용하여 평가. 개의 오차의 산
술평균으로 평가오차 추정
모든 자료가 (-1)번 와
에 속함.
LOOCV
(leave-out-one CV)=인 -fold CV
일반적으로 추정치의 분산은
크나 실제오차에 대한 편이는
작은 것으로 알려져 있음.
<표 7.5> 오차의 종류, 절차 및 그 특성
앙상블 방법(ensemble) 방법
표본의 크기가 일 때 개의 표본을 복원 추출하여 붓스트랩 표본을 만든 후에 오분류율을
구함(개의 붓스트랩 표본에 대해 반복).
평가 오분류율: 전체 개의 오분류율의 산술평균
붓스트랩 표본 자체의 가중치를 구하여 오분류율의 가중합

- 12 -
마진(margin)
Golub et al. (1999) : prediction strength(PS)라고 정의 → 각 표본의 예측력
예) 일 때 마진=, 일 때 마진=-
분류분석의 목표
해석이 용이하고 평가 집합의 새로운 표본에 대해 오분류가 적은 분류자를 만듦.
특이한 자료에 영향을 덜 받는 강건(robust)한 분류자를 만듦.
(4) 벌점효과(penalized effect)
● 분류함수 구성시 특정 표본이 오분류되면 그 손실을 많이 반영하도록 하여 특정 표본을
오분류하지 않은 쪽으로 분류자를 유도.
● 사전 경험과 사전 지식 필요 → 벌점과 관련된 입력모수는 특별한 주의가 필요
벌점효과의 의미
① 모집단의 분포의 변이보다 각 개체가 더 큰 변이를 갖도록 함
← 5.3절의 수정인자(fudge factor)와도 관련
② 오분류율과 분류자 차원사이의 균형(trade-off)을 맞추는 역할
7.3 모수적 분류분석
○ 모수적 방법 : 선형판별분석(LDA), 이차판별분석(QDA), 로지스틱 회귀분석
○ 비모수적 방법 : Support Vector Machines(SVM),
분류회귀나무(Classification And Regression Tree,CART)
7.3.1 Fisher의 Linear Discriminant Analysis(LDA)
□ Reference "Analysis of Microarray gene expressoin data" page 278
○ We shall consider the case where the training samples can be divided in two
known categories, denoted by and . Two groups and are said to be
linearly separable if there exists a constant and a linear function ′ such
that
′
, i f ∈
′
, i f ∈

- 13 -
The set of points ′ is called a separating hyperplane.
○ For simplicity of notation, we omit the specimen index in the remainder of this
selection. Assume that groups and have means and
, respectively. Also, assume that the covariance matrix is the same
for both group, I.e,
′ for
○ Fisher's LDA is based on finding the linear transformation ′ which maximizes
the ration of the between-groups sum of squares and the within-groups sum of
squares. This ratio can be expressed in the following form
′
′ ′
which equals the squared distance between linear combinations of means divided
by the variance of the linear combinations.
○ The maximum of the above ratio is attained at ′ ′ with
max′
′ ′
′
○ The linear transformation
′ ′
is called Fisher's linear discriminant function.
○ Let
′ ′
′
denote the midpoint between the means ′ and ′ of the two group
and .
It can be easily shown that ′ and ′ when
.
If an expression vector is from group , then the linear transformation ′
is , on the average, expected to be larger than the midpoint .
○ Similarly, if is from group , then the linear transformation ′ is expected
to be smaller than the midpoint

- 14 -
○ Hence Fisher's discrimination function can be used a classification device for the
test set as follows.
○ An expression vector is the test set is allocated to group if ′≥ , and
the group otherwise. Or, equivalently, expression vector in the test set is
allocated to group if
′
≥
and to group otherwise.
○ In practice, mean can be replaced by the sample mean for group
.
○ The covariance matrix can also be replaced by the sample covariance matrix
pooled from both groups and .
○ An observation in the test set is then allocated to group if
′
≥
and to group otherwise.
* 교재 내용 p246
○ learning data set을 p×n 라 가정하고, 여기서 n은 array p는 유전자라고 하고, k는 집
단의 수라고 가정.
○ 유전자 p에 대한 분산 행렬은 × 임.
○ 각 범주(집단)내에서의 변동들의 합 × 분산행렬 :
∊
→ 집단내 제곱합
범주(집단)간의 차이를 나타내는 × 변동행렬 :
∊
→ 집단간 제곱합
○ 와
그리고 의 추정량으로
각 범주의 표본평균 :
표본분산 :
공통분산(pooled variance) :

- 15 -
○ Fisher의 LDA는 Rayleigh 계수
를 사용하여 판별분석 하는 방법.
여기서 는 × 인 방향 벡터
○ 이라고 하면
― 각 변수의 선형결합 로 두고 은 들의 분산이라고 할 때
′ ′
∈
′∝′
′ ′
′ ′ ∈
′
∈
′
∝ ′
○ Rayleigh 계수 : ′
′
― 최대가 되는 경우는 선형결합 변수 에서의 각 범주의 평균차이를 크게 하면서 동
시에 분산이 작아야 하며
― 적절한 방향의 를 찾기만 하면 됨.
→ 적절한 방향 을 찾기 위해 (집단내분산분의 집단간 분산비) 고유치와 고
유벡터를 이용하여 구함.
○ 는 min 개의 영이 아닌 고유치들
≥ ≥⋯≥ 과 이에 상응하는 고유벡터 ⋯ 가짐.
○ 최대값 을 갖는 방향 은 첫 번째 분류방향
′
′ max′′
⋮ ⋮
′
′ max⊥ ′′ ≤
○ 다범주인 경우 관찰벡터 ⋯ ′
′ 들은 차원의 좌표공간에서 유클리안 거리.
예측판별 범주는
- 16 -
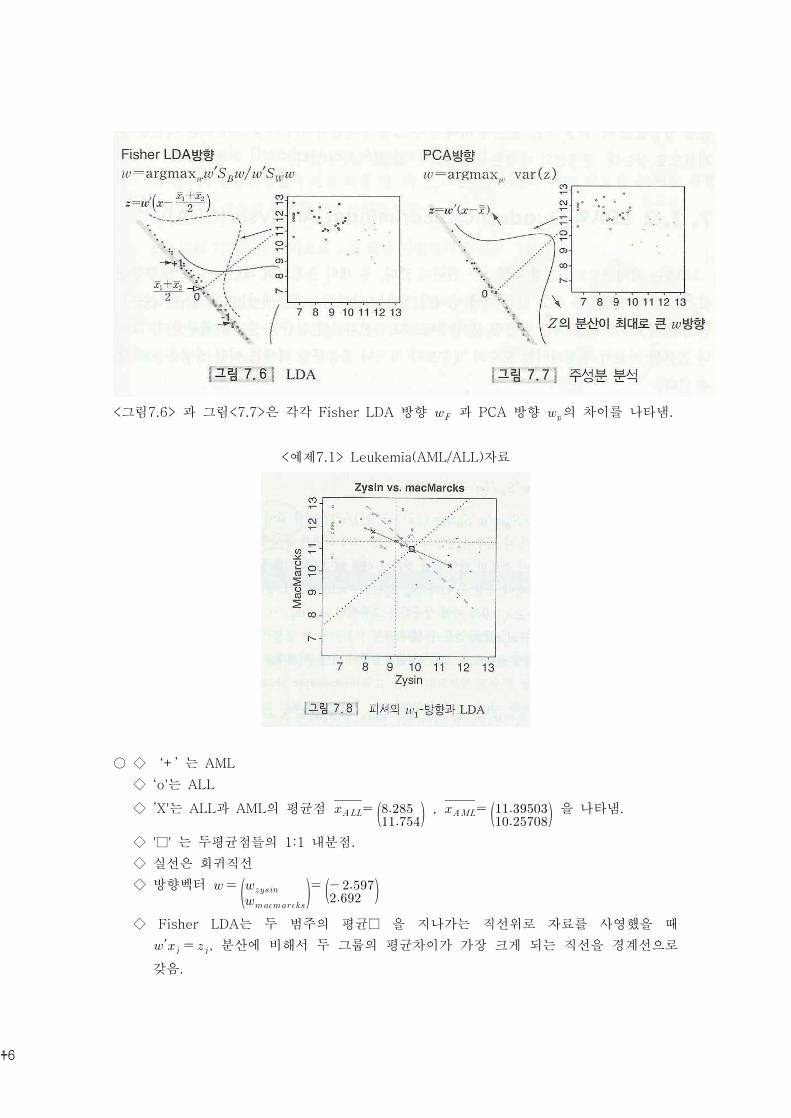
<그림7.6> 과 그림<7.7>은 각각 Fisher LDA 방향 과 PCA 방향 의 차이를 나타냄.
<예제7.1> Leukemia(AML/ALL)자료
○ ◇ ‘+’ 는 AML
◇ ‘o'는 ALL
◇ ’X'는 ALL과 AML의 평균점 , 을 나타냄.
◇ '□' 는 두평균점들의 1:1 내분점.
◇ 실선은 회귀직선
◇ 방향벡터
◇ Fisher LDA는 두 범주의 평균□ 을 지나가는 직선위로 자료를 사영했을 때
′ , 분산에 비해서 두 그룹의 평균차이가 가장 크게 되는 직선을 경계선으로
갖음.

- 17 -
7.3.2 LDA와 QDA
― LDA는 최대가능도 추정량과 관련. 두개의 유전자의 패턴이 같은 방향성을 갖거나
반대 방향성을 갖고 있을 때에 이러한 정보는 단변량 t검정에서는 고려되지 않음
― LDA와 QDA는 다변량 분석 방법으로 유전자간의 상관성 정보를 활용.
문제점 : 표본의 자료가 추정해야 할 모수의 개수보다 작거나 불충분할 때에는 사용할 수
없음.
□ Maximum likelihood(ML) discriminant rule
k 범주에서 Y=k 일때 확률벡터 가 다변량 정규분포 를 따른다고 가정.
- 확률밀도함수
Pr
′
만약 k 범주일때 x가 나올 확률이 k 가 아닌 다른 범주의 경우보다 크다면 x에 예측범
주는 k라고 함.
이 규칙에 해당되는 분류자를 최대가능도(Maximum likelihood,ML) 이라 하며 다음과
같음.
C(x) = argmaxk pr(x|y = k)
= argmaxk
= argmaxk
′
□ 실제적으로 을 아는 경우가 드물기 때문에 다음과 같이 변환해서 사용함.
C(x) = argmaxk
= argmaxk log
= argmaxk
log
′
′ log
□ ML 분류자의 일반형은 아래와 같음.
′ log
(1) Quadratic Discriminant Analysis (QDA)
각 범주의 공분산 행렬이 서로 다를때, 즉 ≠ 일 적용
(2) Linear Discriminant Analysis (LDA)
공분산 행렬이 ⋯ 인 경우에 적용

- 18 -
log
′
(3) Diagonalized Quadratic Discriminant Analysis (DQDA)
-모든 에 대해 ≠ 이 만족될 때 적용할 수 있는 분석.
-이 경우 분산행렬은 대각행렬이 되고 log 는 간단히
log 로 표현.
(4) Diagonalized Linear Discriminant Analysis (DLDA)
- ≠ 이고 i 번째 변수에 대한 분산이 에 상관없이 동일한 경우, 즉
⋯ 인 경우에 적용할수 있는 분석. 공통분산행렬을 라고 할때
인 경우에 분류함수.
log
′
log 는 간단히
log 로 표현.
■ DLDA와 Golub et.al(1999) 비교
- DLDA 분류자
′
′
여기서 ⋯
, 는 i 번째 표본분산
- Golub et.al (1999)에서 사용한 분류자
Reference paper "Comparison of Discrimination Methods for the Classification of Tumors
Using Gene Expression Data" page 7-9

- 19 -
□ In a situation where the class conditional densities are known, the
maximum likelihood(ML) discriminant rule predicts the class of an observation
… by that which gives the largest likelihood to x, i.e., by C(x) =
argmaxk pr(x|y = k). (When the class conditional densities are fully known, a
learning set is not needed and the classier is simply C(x).)
□ In practice, however, even if the forms of the class conditional densities are
known, their parameters must be estimated from a learning set. Using parameter
estimates in place of the unknown parameters yields the sample maximum
likelihood discriminant rule.
□ For multivariate normal class densities, i.e., for ∼ , the ML
discriminant rule is
′ log
□ In general, this is a quadratic discriminant rule. Interesting special cases include:
1. When the class densities have the same covariance matrix, , the
discriminant rule is based on the square of the Mahalanobis distance and
is linear
′
2. When the class densities have diagonal covariance matrices,
∆ …
the discriminant rule is given by additive quadratic
contributions from each variable
log
3. In this simplest case, when the class densities have the same diagonal
covariance matrix
∆ …
, the discriminant rule is linear and given by
□ We refer to special cases 2 and 3 as diagonal quadratic (DQDA) and linear
(DLDA)
discriminant analysis, respectively. For the corresponding sample ML discriminant
rules,

- 20 -
the population mean vectors and covariance matrices are estimated from a
learning set L,
by the sample mean vectors and covariance matrices, respectively: and
.
□ For the constant covariance matrix case, the pooled estimate of the common
covariance
matrix is used:
.
□ Similar discriminant rules as above arise in a Bayesian context, where the
predicted class is
chosen to maximize the posterior class probabilities pr(y = k|x) [20].
□ In one of the 1st applications of a discrimination method to gene expression
data, Golub et
al. [17] proposed a "weighted voting scheme" for binary classification.
□ This method turns out to be a minor variant of the sample ML rule
corresponding to
special case 3. For two classes k = 1 and 2, the sample ML rule classifies an
observation
… as 1 iff
≥
that is, iff
≥
□ The discriminant function can be rewritten as , where ,
and
.
□ This is almost the same function as used in Golub et al., except for which
Golub et al. dene as
. → p(g,c) page526 column3 line7
□ Not only is an unusual way to calculate the standard error of a
difference, but having standard deviations instead of variances in the
denominator of produces the wrong units.

- 21 -
□ For each prediction made by the classifier, Golub et al. also dene a prediction
strength, PS,
which indicates the "margin of victory"
max min
max min page526 note 20
where max and max . Golub et al. choose a
conservative
prediction strength threshold of .3 below which no predictions are made.
7.3.3 로지스틱회귀분석
○ 일반화 선형모형의 일종으로 이항분포 가정하에서 최대가능도 추정법으로 모수를 추정
○ Iterative Reweighted least squares(IRWLS)라고 불리는 Newton-Raphson 알고리즘
을 이용하여 반복적인 근사해를 찾음.
Newton-Raphson 방법
-우도함수의 2차미분, 변곡정도를 활용
-근사해는 초기 입력값에 영향을 받기 때문에 찾는 해가 반드시 최대값인 ML 추정치가
된다는 보장이 없음.
○ K=2 인 경우, 특정 유전자 의 발현 정도에 따라 암세포에 속하는지 정상세포에 속하는
지 모형.
범주를 -1(암세포), +1(정상세포)
logPr Pr
단, Pr exp exp
○ 가능도 함수를 구성하여 와 을 추정.
Pr exp exp
○ 이 예측확률을 이용하여 분류자를 다음과 같이 구성
Pr ⇔
○ LDA 분류함수룰 이용하여 절편 와 기울기 표시
′
○ 기울기를 나타내는 미분계수
× exp ′
exp ′

- 22 -
Reference " The Elements of Statistcal Learning Data Mining" page 95
□ The logistic regression model arises from the desire to model the posterior
probabilities of the classes via linear functions in , while at the same time
ensuring that they sum to one and remain in [0,1]. The model has the form
logPr Pr
logPr Pr
⋮ ⋮
logPr Pr
□ The model is specified in terms of log-odds or logit transformations
(reflecting the constraint that the probabilities sum to one).
□ Although the model uses the last class as the denominator in the odds-rations,
the choice of denominator is arbitrary in that the estimates are equivariant under
this choice.
□ A simple calculation shows that
Pr
exp
exp
…
Pr
exp
and they clearly sum to one.
□ To emphasize the dependence on the entire parameter set
…
, we denote the probabilities
Pr .
□ When , this model is especially simple, since there is only a single linear
function. It is widely used in biostatistical applications where binary
response(two classes) occur quite frequently.
■ Fitting Logistic Regression Models
□ Logistic regression models are usually fit by maximum likelihood, using the
conditional likelihood of G given X.
□ Since Pr completely specifies the conditional distribution, the multinomial
distribution is appropriate.
□ The log-likelihood for N observations is

- 23 -
log
where Pr
□ We discuss in detail the two-class case, since the algorithms simplify
considerably.
□ It is convenient to code the two-class via a 0/1 response , where
when , and when .
□ The log-likelihood can be written
log log
log
Here , and we assume that the vector of inputs includes the
constant term 1 to accommodate the intercept.
□ To maximize the log-likelihood, we set its derivatives to zero.
□ The score equations are
------(1)
which are equations nonlinear in .
□ Notice that since the first component of is 1. the first score equation
specifies that
the expected number of class ones matches the
observed number ( and hence also class twos.)
□ To solve the score equations (1), we use the Newton-Raphson algorithm, which
requires the second-derivative or Hessian matrix
□ Starting with , a single Newton-Raphson update is
where the derivatives are evaluated at
□ It is convenient to write the score and Hessian in matrix notation.
□ Let denote the vector of values, the × matrix of values, the
vector of fitted probabilities with th element and a × diagonal
matrix of weights with th diagonal element
.
□ Then
and

- 24 -
□ The Newton-Raphson step is thus
□ In the second and third line we have re-expressed the Newton-Raphson step as
a weighted least squares step, with the response
sometimes known as the adjusted response.
□ These equations get solved re-peatedly, since at each iteration p change, and
hence so does W and z.
□ This algorithm is referred to as iteratively re-weighted least squares or IRLS,
since each iteration solves the weighted least squares problem:
←
□ It seems that is a good starting value for the iterative procedure, although
convergence is never guaranteed.
□ Typically the algorithm does converge, since the log-likelihood is concave, but
overshooting can occur.
□ In the rare cases that the log-likelihood decreases, step size halving will
guarantee convergence.
7.4 비모수적 분류분석
7.4.1 Support Vector Machine
SVM 방법: 두 그룹을 잘 분리시키는 분류 초평면을 찾는 방법
기존의 선형 분류방법보다 확장성이 좋고 모집단의 가정이 없음
최종 분류규칙을 일부 표본에만 의존
SVM의 본격적인 적용: 기계를 이용한 우편번호의 자동분류 문제
SVM의 핵심적인 요소
① 초평면 분류자와 Support Vectors(SVs)
② 커널 (Kernel function)
이범주 문제에서의 SVM
∈ 인 분류에서 범주를 가장 잘 구분하는 분류 초평면 ⟨ ⟩ 을 찾음.
여기서 ⟨⋅⋅⟩는 벡터간의 내적, 는 평면의 법선벡터(tangent vector), 는 절편

- 25 -
<그림 7.13> 초평면과 마진 (Smola et al., 2002)
margin
: 분류 초평면과 가장 가까운 표본까지의 거리
가 클수록 일반적 분류성능관점에서 더 나은 분류기준
를 기준으로 ( , )를 결정
차원에서 비선형규칙 만들기
① 큰 차원인 ( )으로 확장
② 확장된 공간에서 선형분류 실시
③ 원래 공간 에서 해석 → 에 대해서 비선형 규칙
Support Vectors(SVs)
분류기준을 결정짓는 경계에 놓여 있거나 경계와 밀접한 연관이 있는 표본
SVM에서의 분류문제
① 분류 가능한 경우 (separable case) → 교재에서 언급
분류규칙에 의해 완벽히 분류
SVs: 분류규칙에서 가장 가까운 표본
② 분류 불가능한 경우 (nonseparable case) → Smora et al. (2002) 참조
완벽하게 오분류율이 0이 되지 않는 경우
SVs: 오분류된 표본 + 마진 안에 있는 표본
(1) 분류 가능한 모형
⟨ ⟩ : -1 차원 초평면
원점으로부터 초평면까지의 거리 =

- 26 -
점 에서 초평면까지의 거리=
⟨ ⟩
점 과 : 초평면에서 가장 가까운 점
점 나 점 에서 초평면까지의 거리 (margin)≡
점 에서 점 까지의 방향상의 거리=
SVM의 시도
를 최대화 하여 두 그룹을 가장 멀리 떨어지게 하는 초평면을 선택
핵심 문제 (primal problem)
목적함수 min
∈
subj ect to ⟨⟩ ≥ i f
⟨⟩ ≤ i f ⋯
↓
또 다른 표현 : ⟨⟩ ≥
→ 제한조건이 있는 최적화 문제 (constrained optimization problem)
→ 라그랑지안(Lagrangian)을 이용하여 다음의 LP를 유도
LP: min min
⟨ ⟩
with
⟨ ⟩ ≥ ∀
≥ ∀
⟨ ⟩ ∀(7.2)
● LP의 해는 반드시 조건 (7.2) (feasible region)을 만족.
● (7.2)의 조건으로부터 해의 쌍 들은 볼록집합(convex set)을 이룸.
● 볼록집합 위에서 볼록함수(convex function) 을 최소화
→항상 최소값을 가지는 유일해가 존재
Kurush-Kuhn-Tucker (KKT) 조건
조건 (7.2)와 를 미분하여 구한 두 개의 방정식

- 27 -
① ∂∂
② ∂∂
③
⟨ ⟩ ≥ ∀
≥ ∀
⟨ ⟩ ∀
<표 7.6> KKT 조건
LP문제를 컴퓨터가 이해하기 쉬운 쌍대문제(dual problem)로 재구성
<표 7.6>의 ①
와 ②
를 LP에 대입
→ 다음의 쌍대(dual)문제 DLP가 유도
DLP: max ≥ max ≥
subj ect to
≥ ∀
∀(7.3)
DLP의 행렬표현식
max ≥
subject to. ≥
여기서 ⋯ ′ , ⋯ ′ , .
Quadratic programming(QP) : 에 대한 2차식을 컴퓨터로 푸는 과정
● 절편 는 에 해당한 들로부터 구할 수 있음. (Keerthi et al., 1999)
, 로부터 구한 최종 분류함수와 분류자
⟨ ⟩ ⟨
⟩
⟨ ⟩
SVs: 에 해당하는 들
(2) 마이크로어레이 자료 분석: SVM을 통한 유전자 선택 절차

- 28 -
⋯에 대해서 변수 인덱스(index) 저장집합을 Ranked로, 참가변수집합을 Var로 표기
● 절차1 : Var = ⋯에 변수가 하나도 없을 때까지 반복
(a) 인덱스 집합 Var의 변수들로 훈련용 집합 에서 SVM을 적합
(b) Var에 있는 각 변수 에 대해, 기준 를 계산
(는 변수 를 제거하고 SVM 적합한 경우를 의미)
(c) argmin 가 되는 유전자 변수를 선택
(d) 변수 를 인덱스 저장 집합 Ranked=[]에 저장
(e) Var에서 를 제거
● 절차2 : Ranked 집합에서 저장된 역순으로 일정한 수의 유전자들을 선택하고, 로
SVM 모형에 적합하고 로 평가
<표 7.7> SVM 재귀적 후진제거 변수 선택 알고리즘 (SVM-RFE)
Western et al. (2002)
평가오차의 상한을 자료의 총 변동영역 과 마진 로서 표현하고 이 상한을 최소
화하여 특정한 유전자를 선택
Guyon et al. (2002)
SVM을 수행하면서 특정한 유전자를 제거한 효과를 계산
순차적으로 암과의 관련성이 상대적으로 작은 유전자를 제거
후진제거법을 반복적으로 적용하여 순차적인 유전자 수열을 구성
← 재귀 변수 제거법 (Recursive Feature Elimination, RFE)
Leukemia자료에 SVM-RFE를 적용
→ 분류에 상당히 기여한다고 알려진 Zysin등의 유전자 발견
신장암자료에 SVM-RFE를 적용
→ 순수하게 신장암 정보와 관련이 있는 유전자를 찾아냄
(3) 마이크로어레이 자료분석에서 SVM의 장점
● 정확성
QP알고리즘 - 표본수가 적은 마이크로어레이 자료에서는 최적해를 정확히 구할 수 있음.
최적해 근사 알고리즘
Keerthi et al. (1999)가 제시한 sequential minimization optimization (SMO)
● 추가적인 정보
: 분류함수에 기여 못하는 표본
: 오분류가 일어났거나 분류의 경계근처의 있는 표본
→ 표본 의 특성을 살피는데 유용하게 사용될 수 있음
● 확장성(flexibility)

- 29 -
분류함수를 만들 때 비선형함수로 확장 가능
(4) 마이크로어레이 자료분석에서 SVM의 단점
● SVM을 이용하여 표본을 분류할 수 있지만 유전자변수를 선택하는 방법이 취약
→ RFE등과 같은 별도의 유전자 변수선택 알고리즘과의 연동이 필수
● 커널과 같이 복잡한 함수를 도입
→ 최종분류자의 설명력이 LDA등 모수적인 방법에 비해 떨어짐
7.4.2 Classification and Regression Tree (CART)
CART: Beiman(1984)이 고안
의사결정나무(decision tree)에 기초
현재노드의 분할여부를 판단, 이분할(binary split)을 실시
<그림 7.15> 현재 노드와 작은 노드들
: 현재노드 : 왼쪽의 자식노드 : 오른쪽의 자식노드
: 전체나무 : 분리규칙(splitting rule)
● 나무 구성후 가지치기(pruning)를 실시하여 더 나은 나무 구조로 만듬.
● 만들어진 나무에 새로운 나무를 대입하여 말단노드(terminal node)에서 구한 오분류율로
나무를 평가
: 번째 범주에 속하는 표본의 수
: ⋯번째 범주에 속할 확률의 추정량
: 노드에서 범주 를 갖는 표본의 수
: 한 표본이 노드 에 속하면서 동시에 범주 에 속할 확률의 재대입
추정치

- 30 -
: 한 표본이 노드 에 속할 확률의 재대입 추정치
: 노드 에 속한 한 표본이 범주 에 속할 확률의 재대입 추정치
노드에서의 불순도 함수 ⋯ , ≥ ,
① 는 ( , , ⋯ ,) 에서만 최대
② 는 (1, 0, ⋯ , 0), (0, 1, ⋯ , 0), ⋯ ,(0, 0, ⋯ , 1)에서만 최소
③ 는 ⋯ 의 대칭함수
⋯ : 불순도 함수를 에 대한 함수로 표현
∈
∈
: 나무 전체의 불순도 함수
분리규칙 의 효과
하나의 노드와 그 자식노드의 불순도의 차이
나무 전체에서의 불순도의 차이
불순도함수의 종류
① 지니 인덱스 (gini index)
② 엔트로피 (entropy)
log
● 일반적으로 설명력이 뛰어나고 이질적인(non-homogeneous) 자료를 잘 분리한다고
알려져 있음(Breiman, 1998).
마이크로어레이 분석에서의 적용
① 유전자를 특정기준에 의해 뽑아서 10~100개의 사이로 크기를 축소하여 사용
② 최상위 루트 노드에서 분리기준으로 가장 먼저 선택되는 유전자 상위
10~100개를 선택
7.5 앙상블방법 (Ensemble approach)
- 한 개의 분류자 대신 한 개 이상의 분류자를 구성하고, 이들을 고려하여 최종 분류자를
선택.
장점 : 한 번의 잘못된 결정으로 생기는 위험을 최소화.
앙상블(ensemble) 방법 : 여러번의 의사결정들에서 다수결 원칙으로 반응 범주를 예측하는

- 31 -
전략. 보통 여러 개의 개별 분류자를 결합(combine)하는 기법.
앙상블 방법의 대표적인 예 : 투표(voting) 방식, 개별분류자(weak learner)들의 마진까지
고려한 부스팅(boosting)
투표 방식 : R 개의 개별분류자가 주어졌을 시, 새로운 표본에 대하여 투표 방식을 개별
분류자에 의해 예측되는 범주들 중 다수의 범주로 할당하는 방식
부스팅 : 개별 분류자들의 가중합을 이용하여, 최종 예측을 하는 방식. 가중투표(weighted
voting) 방식.
투표 방법
- 소수의 유의한 유전자 탐색의 목적보다는 보다 정확한 분류에 더 목적을 둠.
- 투표방법에 쓰이는 여러 개의 분류자들에는 임의의 분류방법이 적용 가능하나 주로
CART 분류방법을 중심으로 발전
투표 방법의 핵심 요소
(1) 투표 방식
(2) 정보 획득 방법
- 표본집단이 약간 변하더라도 분류결과가 잘 변하지 않는 분류자 혹은 분류결과가 잘 변
하는 분류자가 있으며 이러한 성질을 분류자의 불안정성(instability)라고 함. 따라서 투
표방법은 의사결정 시행마다 분류자의 결과의 변동 여부에 주목해야함.
- 일반적으로 CART와 k-NN은 불안정하며, LDA는 안정적.
7.5.1 k-Nearest Neighborhood method(k-NN)
- CART는 이분법적 사고의 관점에서 가장 직관적인 방법이고, k-NN은 거리 개념의 관점
에서 볼 때 가장 직관적인 방법
- Simple nonparametric methods for classifying specimens that can capture
nonlinearities in the true boundary between classes when the number of
specimens is sufficient.
- Does not involve estimation of numerous parameters, it often is an effective
method even when the number of specimens is relatively small
- 거리를 기준으로 가장 가까이 있으면 이웃으로 표한하고, 이웃사이에는 비슷한 정보나 여
건을 공유하고 있다는 가정을 함.

- 32 -
절차1 : TS에 속한 x와 가장 가까운 k개의 이웃 관찰치를 LS에서 찾는다.
절차2 : x의 범주는 절차 1에서 찾은 k개 이웃의 y중에서, 투표를 통해 다수가 되는
y로 예측한다.
- Common distance measure : Euclidean, Mahalanobis and 1-correlation coefficient
- k-NN 방법은 결측치를 보정하거나 확률밀도함수추정 방법으로 보다 널리 쓰임.
k-NN 알고리즘
- LS에 속하는 표본 ∈ 와 TS에 속하는 표본들과의 관계를 거리 행렬을 만들어 가장
유사한 k개의 표본을 찾고, 이 표본 중에서 가장 다수를 차지하는 y값으로 범주를 예측.
여기서 k는 사용자가 임의로 지정해도 되나 CV과정을 통해 최선의 k를 택하는 것이 바
람직하다.
- For small sample sizes, smaller values of k such as 1 or 3 are most appropriate.
- Optimized values of k were generally less than 7 in the studies of Dudoit et al.
(2002).
- 범주를 구분하는 함수가 명시적으로 제시되지 않는 단점이 있어 설명력이 떨어지나, 다른
방법과 비교할 때에 기준이 되는 방법으로 유용하게 사용.

- 33 -
집합 ⋯ 과 r번째 분류자의 중요도는 ⋯에 대하여
절차1 : For ⋯
- 휸련집합 생성
- 분류자 구성
절차2 : 분류자들의 집합열, 구축하고 새로운 표본 에 대해서
예측범주 집합열, 구성
절차3 : 에 대한 예측 범주는 다음과 같이 투표로 예측
arg
arg
7.5.2 배깅(Bagging)
- Breiman(1996, 1998)이 제안한 투표방법으로 주로 CART를 이용하여 여러 개의 분류자
를 만들고 투표를 통하여 통합 분류자를 구하는 방법
(1) 집합 ⋯ 으로부터 bootstrap을 이용하여, 총 R개의 bootstrap 표
본 집합열 을 만듬.
(2) ⋯번째 bootstrap 표본집합으로 구성한 분류자의 가중치를 로 두고,
을 집합 L에서의 분류자라 하면, 각 집합 마다 분류자 를 구함.
(3) R개의 가중치와 분류자들의 집합열 을 형성.
배깅 알고리즘
- CART와 같이 불안정한 분류자에 대하여 배깅을 적용하게 되면 분류자의 성능을 많이 향
상시킬 수 있음.
- LDA와 같이 자료에 적합시킬 모형이 제한적(자료의 평균과 분산에만 의존하는 의미에서)
이라서 분류방법이 안정적인 경우에는 배깅을 적용하더라도 그 성능이 별로 향상되지
않음
- 투표 전략을 사용하는 것은 결국 분류성능의 증가를 꾀하기 위한 것이므로 투표의 효과
가 극대화되지 못하는 경우에는 바람직한 전략이라 할 수 없음.
7.5.3 부스팅(Boosting)
- 배깅은 번째 bootstrap을 발생시킬 때, 직전 단계인 번째의 분류결과는 영향을

- 34 -
집합 ⋯ 에 대하여
절차1 : 자료 들에 대한 분포 Pr ⋯
(분포 Pr ⋯ Pr 에 따라 자료 들이 복원추출 됨.)
절차2 : For ⋯
- 분포 Pr ⋯ Pr 을 이용하여 훈련집합에서 분류실시
- 분류자 → 에 대하여 오차 ≠ 를 계산
- 번째 분류자 의 중요도 ln
(단, ≥ i f ≤ 이며, 이 줄어들면 는 증가한다.)
- 각 자료의 분포 Pr 를 갱신 :
Pr
Pr ×
i f
i f ≠
Pr exp
( ≥ or 이면 Pr 로 설정, 는 확률의 합이 1이
받지 않고 독립적으로 표본을 뽑으나 부스팅은 직전 시점의 오분류율을 반영하여 번째
표본을 뽑음.
- 따라서 부스팅은 번째 분류자에 대한 중요도 이 다름.
- 가장 널리 알려져 있는 부스팅 방법은 AdaBoost(Adaptive Boosting).
- 회수 에 따라 달라지는 가중치는 두 가지가 있음.
(1) 매 시행마다 추출하는 자료의 가중치의 분포 Pr ⋯Pr
(2) 개별 분류자에 대한 가충치
- 단계에서 주어진 자료의 분포 Pr ⋯Pr 에 따라 복원추출하고 분류자
를 구하고 오분류율 을 구해서 이를 바탕으로 번째의
Pr ⋯Pr 을 갱신한다.
- 번의 개별 분류자들을 구한 뒤, 새로운 표본에 대한 최종분류함수는
로 예측
AdaBoost 알고리즘

- 35 -
되도록 하는 항)
절차3 : 최종 투표 분류자
- 배깅보다 우수한 성능을 보이며, 훈련집합에서 추정된 오분류율이 0이 되어도 계속해서
마진을 증가시키려고 노력을 함. 이점이 배깅보다 나은 요소가 되어 평가 오차를
감소시킴.
- SVM에서 살펴본 마진과의 차이점은 부스팅은 bootstrap 표본열 개를 사용하므로 개
에 대한 가중 합으로 표시. 자료 에 대해서 마진은
로 정의.
- Philip et al. (2003)는 AdaBoost 등에서 추정방법을 개선하고, 각 bootstrap 표본열
마다 적용되는 CART에서 오직 하나의 유전자 값만 분리기준으로 사용하기를 제안.
대신에
ln 을 사용하여 자료의 수가 작은 데서 기인하는 오차
의 불확정성을 조정. 여기서 ∈ 는 유전자 군집의 개수와 관련된 입력 모수로서 CV
로 최적의 d 값을 찾음.
오분류율 관점에서 Leukemia(AML/ALL) 자료 등과 같은 분류가 비교적 잘 되는 자료에
대해서 분류자의 성능을 향상.
7.6 분류분석의 정리와 토의
(1) 변수선택의 중요성
- 통계적인 분류분석은 많은 수의 표본을 관찰할 수 있다는 가정을 전제.
- 마이크로어레이 자료는 유전자의 수는 많고 표본 수는 적기 때문에 이러한 가정을 만족
하기가 어려움.
- 이미 알려진 기존의 방법을 적용하려면 변수(유전자)의 수를 줄여야 가능하며 반응범주와
밀접한 연관을 갖는 변수를 선택하여야 좋은 분류분석이 가능.
(2) 비선형 분류자의 필요성
- 선형 분류자는 비선형 분류자에 비해 해석이 쉽고 사용하기도 편한 장점을 지님. 하지만
마이크로어레이 자료의 경우 유전자의 발현현상이 일방적으로 선형패턴을 보일 것으로
기대하는 것은 무리.

- 36 -
분류방법 유전자 선택 전략 입력 모수
LDA, QDA
고정된 하나의 유전자(혹은 두 세 개의
유전자 조합)에 대해, F-검정 (K=2는
t-검정)으로
상위유전자(상위유전자조합)선택
DLDA, DQDA를 사용할 지 결정
로지스틱1. LDA와 같은 방법을 할 수 있다.
2. 전진선택 혹은 후진선택
SVM
1. LDA와 같은 방법을 할 수 있다.
2. 전진선택 혹은 후진선택
1. C값 지정
2. 커널종류와 커널에 해당하는 d,
를 지정(흔히 사용하는 값은
와 배깅, 부스팅 LDA와 같은 방법을 적용할 수 있다. 반복 회수
- 따라서 비선형 분류자는 선형 분류자 보다 정확하게 반응 그룹을 구분할 것으로 기대되
기 때문에 해석이 난해하고 사용하기도 쉽지 않음에도 불구하고 비선형 분류자에 대한
연구는 활발하게 진행될 것으로 기대.
(3) 조정모수의 해석
- 분류분석은 반응범주를 구별하는 규칙을 찾는 것이며, 이렇게 구성된 규칙은 재사용
가능한 것이 바람직.
- 현실적으로 분류성능을 향상시켰음에도 불구하고 원인을 규명하기가 힘들 때가 많음.
- 분석자가 선택한 입력한 조정모수가 많으면 해석이 어렵고 CV에 의해 선택할 최적의
모수 값들의 조합을 찾는 것 자체가 커다란 문제를 야기시킬 수 있으나, 오로지 표본의
분류에만 목적이 있다면 조정모수들의 다양함은 최고의 분류자를 찾는 여러 가지 해법의
가능성을 제시해줌.
각 분류방법에서 사용자 입력이 필요한 조정모수
7.7 R 프로그램 응용 - Classification
1. Data 설명
- 훈련용 집합 (Golub_Train) : 총 38명 (ALL 27명; AML 11명), 7,129개의 gene.
- 평가용 집합 (Golub_Test) : 총 34명 (ALL 20명; AML 14명), 7,129개의 gene.
- 통합용 집합 (Golub_Merge) : 총 72명 (ALL 47명; AML 25명), 7,129개의 gene.
2. Golub 자료 불러오기
library(golubEsets)
data(Golub_Train); data(Golub_Test); data(Golub_Merge);

- 37 -
###자료의 여과 변환 ###
### 변환 함수 ###
library(genefilter)
GolubTrans <-function(eSet) {
X<-exprs(eSet)
X[X<100]<-100 ##휘도값이 100 미만은 100으로 고정
X[X>16000]<-16000 ##휘도값이 16000 초과는 16000으로 고정
X <- log2(X) ## 로그 2 변환
exprs(eSet) <- X
eSet
}
### 여과 함수 ###
mmfilt <- function(r=5, d=500, na.rm=TRUE) {
function(x) {
minval <- min(2^x, na.rm=na.rm)
maxval <- max(2^x, na.rm=na.rm)
(maxval/minval > r) && (maxval-minval > d) ## 최대값과 최소값의 차이와 비
## 율을 이용한 여과
}
}
gTrn <- GolubTrans(Golub_Train)
gTest <- GolubTrans(Golub_Test)
gMerge <- GolubTrans(Golub_Merge)
ffun<-mmfilt()
#ffun <- filterfun(mmfilt)
sub <- genefilter(gTrn, ffun)
sub[c(2401,3398,4168)] <- FALSE
sum(sub) ## 3051
gTrnS <- gTrn[sub,]
gTestS <- gTest[sub,]
gMergeS <- gMerge[sub,]
Ytr <-Golub_Train$ALL.AML
Ytest <- Golub_Test$ALL.AML

- 38 -
Ymerge <- Golub_Merge$ALL.AML
3. ANOVA를 이용한 유전자 선택
af <- Anova(c(rep(1,27),rep(2,11)), .00001) ## single argument function으로 변환
anovasub <- sub[sub==TRUE]
#for( i in 1 : sum(anovasub) )
#anovasub[i] <- af(exprs(gTrnS)[i,])
anovasub<-apply(exprs(gTrnS),1,af) ## gene 별로 분산분석후 유의한 gene을 TRUE로.
sum(anovasub) ##68
gTrA <- gTrnS[anovasub, ]
gTeA <- gTestS[anovasub, ]
gMeA <- gMergeS[anovasub, ]
whBad1 <- (apply(exprs(gTrA), 1, mad) == 0)
whBad2 <- (apply(exprs(gTeA), 1, mad) == 0)
whBad <- whBad1 | whBad2
sum(whBad) #11
gTrA <- gTrA[!whBad, ]
gTeA <- gTeA[!whBad, ]
gMeA <- gMeA[!whBad, ]
4. 표준화
star <- function(x) (x-median(x))/mad(x)
TrExprs <- t(apply(exprs(gTrA), 1, star))
TeExprs <- t(apply(exprs(gTeA), 1, star))
MeExprs <- t(apply(exprs(gMeA), 1, star))
5. LDA 프로그램
library(MASS)
gTr.lda <- lda(t(TrExprs), Ytr)
plot(gTr.lda)
preds.lda <- predict(gTr.lda, t(TeExprs))
table(preds.lda$class, Ytest)

- 39 -
Ytest
ALL AML
ALL 19 12
AML 1 2
정분류율 = 0.618
오분류율 = 0.382
6. k-NN 프로그램
library(class)
knn3 <- knn(t(TrExprs), t(TeExprs), factor(Ytr), k=3)
table(knn3, Ytest)
knn5.cvpreds <- knn.cv(t(MeExprs), factor(Ymerge), k=5)# leave one-out CV
table(knn5.cvpreds, Ymerge)
Ytest
knn3 ALL AML
ALL 17 6
AML 3 8
정분류율 = 0.735
오분류율 = 0.265
Ymerge
knn5.cvpreds ALL AML
ALL 43 4
AML 4 21
정분류율 = 0.889
오분류율 = 0.111
7. SVM 프로그램
library(e1071)
svm1 <- svm(t(exprs(gTrA)), Golub_Train$ALL, type="C-classification",
kernel="linear")
trpred <- predict(svm1,t(exprs(gTrA)))
sum( trpred != Golub_Train$ALL)
table(trpred, Golub_Train$ALL)
#trcv <- svm(t(exprs(gTrA)), Golub_Train$ALL, type="C-classification",
#kernel="linear", cross=10)
#summary(trcv)

- 40 -
trcv <- svm(t(exprs(gMeA)), Golub_Merge$ALL, type="C-classification",
kernel="linear", cross=10)
summary(trcv)
tepred <- predict(svm1, t(exprs(gTeA)))
sum(tepred != Golub_Test$ALL)
table(tepred, Golub_Test$ALL)
trpred ALL AML
ALL 27 0
AML 0 11
정분류율 = 1
오분류율 = 0
10-fold cross-validation on training data:
Total Accuracy: 98.61111
Single Accuracies:
100 100 100 100 100 100 85.71429 100 100 100
tepred ALL AML
ALL 20 2
AML 0 12
정분류율 = 0.941
오분류율 = 0.059
8. CART 프로그램
library(tree)
ngTrA <- data.frame(t(exprs(gTrA)), Ytr )
ngTeA <- data.frame(t(exprs(gTeA)), Ytest )
ngTrA.tree <- tree(Ytr~., data=ngTrA)
ngTrA.tree
summary(ngTrA.tree)
plot(ngTrA.tree)
text(ngTrA.tree)
tepred <- predict.tree(ngTrA.tree, ngTeA)
tepred <-tepred[,1]; tepred[tepred==1] <-"ALL"
tepred[tepred==0] <-"AML"
sum(tepred != Ytest)
table(tepred, Ytest)

- 41 -
Ytest
tepred ALL AML
ALL 18 1
AML 2 13
정분류율 = 0.912
오분류율 = 0.088
9. 발견된 유전자 정보 구하는 함수
library(golubEsets)
library(hu6800)
library(annotate)
affyID <- ls(env=hu6800CHR)
(1:7129)[affyID=="X95735_at"]
mygene <- affyID[6614]
get(mygene, env = hu6800ACCNUM)
get(mygene, env = hu6800ENTREZID)
get(mygene, env = hu6800SYMBOL)
get(mygene, env = hu6800GENENAME)
#get(mygene, env = hu6800SUMFUNC)
get(mygene, env = hu6800UNIGENE)
get(mygene, env = hu6800CHR)
get(mygene, env = hu6800CHRLOC)
get(mygene, env = hu6800MAP)
get(mygene, env = hu6800PMID)
get(mygene, env = hu6800GO)
pmids <- getPMID(mygene, data = "hu6800")
pubmed(pmids, disp = "browser")
get(mygene, env = hu6800)
data<-exprs(gTrA)
pval<-numeric()

- 42 -
for(i in 1:length(data[,1])){
lma<-lm(data[i,]~Ytr)
pval[i]<-summary(lma)$coef[2,4]
}
mis_e <- function(x) {
len<-length(x[1,])
tot<-sum(x)
right.c=0
for(i in 1:len){
right.c<-right.c+x[i,i]
}
mis.c <- tot-right.c
mis.r <- round(mis.c/tot,3)
mis.r
}
TrExprs1<-TrExprs[1:20,]
TeExprs1<-TeExprs[1:20,]
gTrA1<-exprs(gTrA)[1:20,]
gTeA1<-exprs(gTeA)[1:20,]
for(i in 1:20){
TrExprs1[i,]<-TrExprs[order(pval)==i,]
TeExprs1[i,]<-TeExprs[order(pval)==i,]
gTrA1[i,]<-exprs(gTrA)[order(pval)==i,]
gTeA1[i,]<-exprs(gTeA)[order(pval)==i,]
}
tr_lda1<-te_lda1<-tr_svm1<-te_svm1<-tr_svm2<-te_svm2<-tr_svm3<-te_svm3<-num
eric()
for (i in 1:19){
# LDA
gTr.lda <- lda(t(TrExprs1[1:(i+1),]), Ytr)
preds.lda1<-predict(gTr.lda, t(TrExprs1[1:(i+1),]))
preds.lda2 <- predict(gTr.lda, t(TeExprs[1:(i+1),]))
tr<-table(preds.lda1$class, Ytr)
tr_lda1[i]<-mis_e(tr)
te<-table(preds.lda2$class, Ytest)
te_lda1[i]<-mis_e(te)

- 43 -
#SVM-linear
svm1 <- svm(t(gTrA1[1:(i+1),]), Ytr, type="C-classification", kernel="linear")
trpred <- predict(svm1,t(gTrA1[1:(i+1),]))
svm_tr<-table(trpred, Ytr)
tr_svm1[i]<-mis_e(svm_tr)
tepred <- predict(svm1, t(gTeA1[1:(i+1),]))
svm_te<-table(tepred, Ytest)
te_svm1[i]<-mis_e(svm_te)
#SVM-sigmoid
svm1 <- svm(t(gTrA1[1:(i+1),]), Ytr, type="C-classification", kernel="sigmoid")
trpred <- predict(svm1,t(gTrA1[1:(i+1),]))
svm_tr<-table(trpred, Ytr)
tr_svm2[i]<-mis_e(svm_tr)
tepred <- predict(svm1, t(gTeA1[1:(i+1),]))
svm_te<-table(tepred, Ytest)
te_svm2[i]<-mis_e(svm_te)
#SVM-polynomial
svm1 <- svm(t(gTrA1[1:(i+1),]), Ytr, type="C-classification", kernel="polynomial")
trpred <- predict(svm1,t(gTrA1[1:(i+1),]))
svm_tr<-table(trpred, Ytr)
tr_svm3[i]<-mis_e(svm_tr)
tepred <- predict(svm1, t(gTeA1[1:(i+1),]))
svm_te<-table(tepred, Ytest)
te_svm3[i]<-mis_e(svm_te)
}
plot(2:20,tr_lda1,lty=2,type="o",col=1,ylim=c(0,0.5))
lines(2:20,tr_svm1,lty=3,type="o",col=2,pch=17)
lines(2:20,tr_svm2,lty=4,type="o",col=3,pch=18)
lines(2:20,tr_svm3,lty=5,type="o",col=4,pch=19)
plot(2:20,te_lda1,lty=2,type="o",col=1,ylim=c(0,0.5))
lines(2:20,te_svm1,lty=3,type="o",col=2,pch=17)
lines(2:20,te_svm2,lty=4,type="o",col=3,pch=18)
lines(2:20,te_svm3,lty=5,type="o",col=4,pch=19)

- 44 -



















![제7장 포트폴리오 분석 [호환 모드]contents.kocw.or.kr/document/week07_1.pdf•효율적포트폴리오(efficient portfolio ) : 지배원리 를만족시키는포트폴리오](https://img.pdfslide.tips/doc/110x75/602901a044af2c59bd26ff95/oe7-e-e-eeoe-aoeeefficient.jpg)


![제7장 포트폴리오 분석 [호환 모드]elearning.kocw.net/document/week07_1.pdf · 제7장포트폴리오분석 1.불확실성, 위험 2.효용이론 3.평균․분산기준포트폴리오이론](https://img.pdfslide.tips/doc/110x75/5a80b4307f8b9a9d308c8bc2/7-7-1.jpg)






