Embed Size (px)
Citation preview

Grupo n°3:
Joel Velez G
Catedra de Oftalmología Teórica.
8vo Ciclo C
Semestre A-2015.
Anomalías Congénitas Oculares.

Que son?
Son aquellas anomalías que se presentan desde el nacimiento.
Suelen ser producidas por efectos genéticos o teratógenos.
Entre los teratógenos no genéticos mas frecuentes están: tóxicos, deficiencias nutricionales, drogas, fallas al desarrollo, etc.
Aquellas lesiones antes de la organogénesis, suelen producir esbozo ocular y sistémico.
Aquellas lesiones que son después de la organogénesis, suelen producir esbozo ocular menos grave.

Anoftalmia.
Ausencia congénita del ojo.
Mencionada en 1557 y descrita 100 años después.
Según Mann, la ausencia del globo ocular se relaciona a la falta de formación de la foseta.
En una RM, se describen nervios ópticos de pequeño tamaño, por su posterior desarrollo.
Las anoftalmias verdaderas son raras, y se deben a malformaciones cerebrales graves.
Clínicamente el paciente presenta:
a. Parpados formados pero pequeños.
b. Hendidura palpebral pequeña.
c. Músculos extra oculares fijados al ebozo.
d. Orbita disminuida.
Como tratamiento se ha utilizado últimamente: prótesis cosméticas.

Anoftalmia

Microftalmia
Nanoftalmia.
Un globo ocular más pequeño que lo
normal.
Puede ser uni o bilateral .
Acompañada por otras malformaciones
o ser parte de un síndrome, trisomía
13.
Se debe generalmente a infecciones
intrauterinas como citomegalovirus y la
toxoplasmosis.
Según François:
a. Colobomatosa (asociada frecuentemente con un ojo mas pequeño). Pueden ser completo e incompleto.
b. Quiste colobomatoso (se da por un crecimiento excesivo de la capa interna). El tamaña del quiste depende del crecimiento de la capa interna.
Según su forma:
1. Pura: aquella que no se acompaña de otras malformaciones y sus características son:
Disminución del globo ocular (menor a 20 mm).
Aumento del espesor del cristalino (mayor 5mm).
2. No pura: se vincula a malformaciones oculares, siendo las mas frecuentes: esclerocornea, catarata congénita, etc.

Microftalmia

Nanoftalmia y criptoftalmia
Nanoftalmia:
Es un tipo de Microftalmia pura, bilateral.
Se describe por la reducción del tamaño ocular el cual no se relaciona a la del cristalino.
Criptoftalmia:
Es una malformación palpebral.
Generalmente es bilateral.
Existe una fusión de los parpados con la cornea y esta se asocia a Microftalmia.

Ciclopía
Es la presencia de un ojo único en
la parte medial de la cara.
Falta de separación de los ojos con
la falta de división del
prosencéfalo.
En esta rara anomalía hay un
desarrollo defectuoso del extremo
anterior del tubo neural, razón por
la cual siempre está asociada con
anormalidades del cerebro.
Clínicamente se caracteriza por:
a. Orbita central por encima de la probóscide.
b. El ojo único presenta: una esclera, dos corneas, dos iris, dos cristalinos, un vítreo y retina.
El trastorno cerebral que acompaña a la Ciclopía es holoprosencefalia.

Ciclopía

Coloboma
La fisura coroidea no se cierra
correctamente
Hendidura en el iris
Es un agujero o defecto del iris del
ojo. La mayoría de los colobomas
están presentes desde el nacimiento
Los colobomas del iris pueden lucir
como un agujero redondo y negro
localizado dentro o al lado de la parte
coloreada del ojo (iris)
Los colobomas generalmente se
diagnostican al nacer o poco después
El coloboma puede ocurrir debido a:
Afecciones hereditarias
Traumatismo de ojo
La mayoría de los casos de
coloboma son de causa
desconocida y no se relacionan
con otras anomalías. Un
pequeño porcentaje de
personas con coloboma tiene
otros problemas hereditarios del
desarrollo.

Coloboma

Megalocornea
Según algunos autores, es un glaucoma congénito de resolución espontanea.
Se le asocia a Huso de Krukerberg, hipoplasia del estroma iridiano.
La herencia es ligada al sexo en 90%.
De mayor presencia en herencia recesiva.
La clínica se presenta por:
a. Aumento del diámetro corneal (mayor o igual a 13 mm).
b. Es bilateral, simétrica y no progresiva.
c. La cámara anterior es profunda pero con aumento del espesor del cristalino.

Microcornea
Se piensa que el defecto es a nivel de los bordes anteriores de la copa óptica.
Se le puede asociar a coloboma, persistencia de la membrana pupilar, microfaquia y a síndromes como: Progeria, Norrie, Rieger, etc.
Su clínica se presenta por:
a. Cornea de disminuido tamaño (menos de 10 mm).
b. Tamaño ocular normal.
c. Puede presentarse uni o bilateral.
d. Puede presentar herencia autosómica o recesiva.

Megalo y Microcornea.

Embriotoxon Posterior.
Grupo de malformaciones frecuentemente autosómicas dominantes.
Se estima que se presenta en el 8-15% de la población.
Consiste en el engrosamiento de la línea Schwalbe y su posicionamiento anterior.
Se lo ve en el Síndrome de Alagille.
Clínicamente se presenta:
Anillo blanco retrocorneal completo o no paralelo al limbo.
Una forma de prevención y tratamiento es el control de la presión intraocular.

Embriotoxon Posterior

Anomalía de Axenfeld
Fue descrita en 1920 por Axenfeld.
Tiene herencia autosómica dominante.
Se presenta de manera bilateral.
Se le asocia a Embriotoxon posterior con adherencias iridocorneales entre la base del iris y el Embriotoxon posterior.
El 50% de los pacientes cursan con glaucoma tardío en la juventud.

Anomalía de Rieger
Descrita en 1934 por Rieger.
Cuando se le asocia a Axenfeld puede presentar trastornos oculares como:
a. Policoria por iris deshicente.
b. Degeneración macular.
c. Hipoplasia del nervio óptico.
Dependiendo a que anomalías se le asocia se puede presentar:
a. Dentarias: se presenta Rieger con microdontia ósea.
b. Faciales: hipoplasia de la rama ascendente del maxila superior.

Anomalía de Peters
Mencionada en 1887
Descrita por Peters en 1906.
Anomalía caracterizada por defecto de la membrana de Descemet en cornea central con ausencia del endotelio.
Forma parte de las disgenesias mesenquimatosas del segmento anterior.
El 80% de los casos son bilaterales.
El 50% de los casos se acompañan de glaucoma.
Clínicamente se presentan por:
a. Opacidad corneal central o paracentral.
b. Presentan sinequias para corneales.
c. Se presenta involucración del cristalino.

Anomalía de Peters

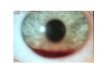
Aniridia
Malformación congénita bilateral caracterizada por la ausencia CASI total del Iris.
Se produce por Alteracion de las oleadas de las crestas neurales.
Puede presentarse de forma autosómica dominante, en donde el cromosoma 11 es el afecto.
Se relaciona con anomalías genitourinarias y tumor de Wilms.
Clínicamente Presenta:
a. Una variable agudeza visual.
b. Se acompaña de nistagmos y fotofobia.
c. Puede presentarse una catarata congénita de progreso lento.
d. A nivel corneal se presenta un trastorno en la membrana de Bowman.
e. Trastornos teratógenos pueden afectar de manera parcial o total dependiendo al periodo.

Aniridia