Embed Size (px)
Citation preview

OTWARCIE PRZEWODU DOKTORSKIEGO
Proponowany temat pracy doktorskiej:
Badania mikrostruktury kopolimerów metakrylowo–akrylowych
przy uŜyciu spektroskopii NMR
Marcin Pasich
Opiekun pracy:
Prof. UŚ dr hab. inŜ. Marek Matlengiewicz
Uniwersytet Śląski
Wydział Matematyki, Fizyki i Chemii
Instytut Chemii
Zakład Chemii i Technologii Środowiska
Katowice 2010

2
SPIS TREŚCI:
WSTĘP……………………………………………………………………………………...3
I. Mikrostruktura łańcucha polimerowego……………………………………………….3
1. Regioregularność, czyli izomeria pozycyjna…………………………………….....3
2. Taktyczność łańcucha polimerowego……………………………………………...4
II. Podstawy statystyki polimerów……………………………………………………….6
1. Homopolimery………………………..……………………………………………6
2. Kopolimery………………………………………………………………………...7
III. Wpływ mikrostruktury na właściwości fizykochemiczne polimeru………………….9
IV. Badania mikrostruktury kopolimerów za pomocą spektroskopii NMR……………...9
CEL I ZAKRES PRACY………………………………………………………………...14
BADANIA WŁASNE……………………………………………………………………15
I. Metodyka badań……………………………………………………………………..15
1. Otrzymywanie polimerów………………………………………………………..15
2. Rejestracja widm 13
C NMR polimerów………………………………………….16
3. Badania kalorymetryczne homopolimerów………………………………………16
II. Przedstawienie wyników badań.…………………………………………………….17
1. Otrzymywanie polimerów……………………………………………………….17
2. Widma 13
C NMR polimerów…………………………………………………….18
3. Porównanie zakresu karbonylowego układów metakrylowo–akrylowych……....28
4. Rozkład sekwencji homopolimeru poli(akrylanu metylu)………………………30
5. Rozkład sekwencji homopolimeru poli(akrylanu n-propylu)…………………...31
6. Badania kalorymetryczne homopolimerów……………………………………..32
PODSUMOWANIE I WNIOSKI………………….……………………………………34
LITERATURA CYTOWANA………………………………………………………….35
DOROBEK NAUKOWY……………………………………………………………….37
śYCIORYS……………………………………………………………………………..38

3
WSTĘP
I. Mikrostruktura łańcucha polimerowego
ZróŜnicowanie budowy przestrzennej łańcucha polimerowego, zwane
mikrostrukturą, jest jednym z istotnych czynników wpływających na właściwości
makroskopowe polimeru. Metody wykorzystywane do wyznaczania mikrostruktury moŜna
podzielić na chemiczne i fizyczne. Do metod chemicznych zaliczamy metody degradacyjne,
prowadzące do rozrywania długich łańcuchów na fragmenty o mniejszej masie molowej,
które następnie poddawane są analizie fizykochemicznej oraz metody chemicznej
transformacji w inne polimery, które moŜna następnie zbadać metodami fizycznymi.
Do metod fizycznych zaliczamy przede wszystkim metody spektroskopowe:
spektrofotometria UV–Vis (słuŜy tylko do badania polimerów zawierających ugrupowania
chromoforów sprzęŜonych), spektroskopia IR i Ramana, spektroskopia elektronowa
wykorzystująca promieniowanie Rentgena oraz spektroskopia magnetycznego rezonansu
jądrowego. Spośród wymienionych metod określania mikrostruktury tylko spektroskopia
NMR pozwala na szczegółowe przeprowadzenie badań strukturalnych polimerów [1].
Z chemicznego punktu widzenia mikrostruktura jest zdefiniowana jako wewnętrzne
ułoŜenie róŜnych sekwencji jednostek monomerycznych w łańcuchu polimerowym.
Elementami mikrostruktury są:
1. Regioregularność, czyli izomeria pozycyjna
Poprzez ten rodzaj izomerii rozumie się róŜnice wynikające z połoŜenia fragmentów
jednostki monomerycznej względem poprzedzającego i następującego fragmentu łańcucha.
Przyczyną istnienia tego rodzaju izomerii jest asymetryczna budowa monomeru.
Gdy w monomerze wyodrębnimy jego początek „głowę” i koniec „ogon”:
to istnieją trzy sposoby łączenia się takich jednostek monomerycznych:
a) „głowa–ogon”
CH CH2
R„głowa”
„ogon”
CH CH2
R
CH CH2
R

4
b) „głowa–głowa”
c) „ogon–ogon”
Połączeń „ogon–głowa” zazwyczaj nie da się odróŜnić od połączeń „głowa–ogon”.
W przypadków polimerów akrylowych, ze względu na trzy czynniki: stabilizację
rezonansową utworzonych form, efekt steryczny, czyli zawadę przestrzenną oraz polarność,
w większości przypadków zdecydowanie przewaŜa przyłączenie „głowa–ogon” i tylko takie
połączenia będą uwzględnione w prowadzonych badaniach mikrostruktury polimeru [1–5].
2. Taktyczność łańcucha polimerowego
Polimer, który posiada jednostki stereoizomeryczne rozmieszczone regularnie
nazywamy polimerem taktycznym. Pojęcie stereoregularności jest węŜsze od terminu
taktyczności, poniewaŜ do tego, aby polimer był taktyczny wystarczy, by w powtarzalnej
jednostce konfiguracyjnej była określona konfiguracja względna, co najmniej jednego
centrum chiralnego lub prochiralnego, a nie koniecznie wszystkich, jak to jest wymagane
w przypadku polimeru stereoregularnego [1].
W przypadku polimerów syntezowanych z monomerów winylowych istnieją co najmniej dwa
moŜliwe układy jednostek stereoregularnych:
� pierwszy, w którym kaŜdy asymetryczny atom węgla ma tę samą konfigurację – układ
izotaktyczny:
� drugi, gdy kolejne, asymetryczne atomy mają przeciwną konfigurację – układ
syndiotaktyczny:
CH2 CH CH CH2
RR
CH CH2 CH2 CH
R R
CH2 C CH2 C CH2 C CH2 C CH2 C
R R R R R
CH2
H H H H H
CH2 C CH2 C CH2 C CH2 C CH2 C
R H R H R
CH2
H R H R H

5
Istnieje równieŜ trzeci rodzaj połączeń monomerów winylowych – układ ataktyczny,
w którym kolejne asymetryczne atomy węgla mogą mieć dowolną konfigurację [5, 6]:
Aby bardziej szczegółowo przedstawić mikrostrukturę łańcucha moŜna wykorzystać
do tego konfiguracją względną. Gdy podstawowa jednostka konfiguracyjna polimeru zawiera
tylko jedno centrum prochiralne, to do jej określenia moŜna zastosować następującą
nomenklaturę:
� jeśli dwa sąsiednie centra stereogeniczne mają identyczną konfigurację, czyli podstawniki
R w uŜytej projekcji znajdują się po tej samej stronie płaszczyzny, to taki układ dwóch merów
(diada) zwany jest mezo (m):
� gdy dwa sąsiednie centra stereogeniczne mają konfigurację przeciwną, czyli podstawniki
R obecne są po przeciwnych stronach płaszczyzny, to wtedy diada nosi miano racemicznej (r)
[1, 5]:
Polimer idealnie izotaktyczny zawiera, więc tylko diady mezo (…mmmmmmmm…),
a polimer doskonale syndiotaktyczny zawiera wyłącznie diady racemiczne (…rrrrrrrrrrr…)
[1, 5]. Sekwencje zawierające trzy centra stereoizomeryczne nazywają się triadami
i są układami zbudowanymi z dwóch kolejnych stereodiad: mm, mr, rm i rr; w przypadku
dłuŜszych sekwencji, na przykład tetrad czy pentad, następuje połączenie odpowiednio trzech
i czterech centrów stereoizomerycznych [5].
CH2 C CH2 C CH2 C CH2 C CH2 C
R R H H R
CH2
H H R R H
CH2 C CH2 C CH2
R R
H H
CH2 C CH2 C CH2
R H
H R

6
II. Podstawy statystyki polimerów
W wyniku przeprowadzonych badań dotyczących kinetyki reakcji i załoŜeń
odnoszących się do mechanizmu polimeryzacji, został opracowany schemat rozkładu
prawdopodobieństwa występowania sekwencji, poniewaŜ matematyczny opis powstawania
łańcucha jest toŜsamy ze statystycznym opisem rozkładu sekwencji juŜ uformowanego
łańcucha [5].
1. Homopolimery
Prawdopodobieństwo przyłączenia jednostki konfiguracyjnej racemicznej (r)
moŜna wyrazić, jako P(r), a jednostki mezo (m), jako P(m):
)N(
)N()P(
mr
rr
+
= (1)
)N(
)N()P(
mr
mm
+
= (2)
N(r), N(m) – odpowiednio liczby jednostek racemicznych i mezo,
N(r + m) – całkowita liczba jednostek racemicznych i mezo.
W przypadku rozpatrywania segmentów łańcucha składającego się z triad (na przykład rr)
lub tetrad (na przykład rmr) prawdopodobieństwo występowania wymienionych sekwencji
jest zdefiniowane w podobny sposób:
)N(
)N()P(
rrrmmrmm
rrrr
+++
= (3)
) N(
)N()P(
rrrrmrmrmmrrrrmrmmmmrmmm
rmrrmr
+++++++
= (4)
Dla równań (1 – 4) moŜna napisać warunki normalizacji, dla kaŜdej sekwencji jednostek
konfiguracyjnych:
P(r) + P(m) = 1 (5)
P(rr) + P(mm) + P(rm) + P(mr) = 1 (6)
W statystyce Bernoulliego prawdopodobieństwo występowania diady, na przykład
mezo, moŜna wyrazić, jako funkcję tylko jednego parametru P(r), gdyŜ:
P(m) = 1 – P(r) (7)
to samo dotyczy prawdopodobieństwa utworzenia diady racemicznej.
Prawdopodobieństwo powstania triady rr wynosi P(r)2:
P(rr) = P(r)2 = [1 – P(m)]
2 (8)

7
W związku z tym, Ŝe spektroskopia NMR nie jest w stanie określić kierunku wzrostu
łańcucha (tzn. z której strony przyłączały się kolejne mery) lub rozróŜnić jego końców,
wprowadza się następujący zapis:
)(P rm + )(P mr = )(P rm = )(P mr (9)
Dla triady mr moŜna wtedy zapisać następujące równanie:
P( mr ) = 2·P(m)·P(r) = 2·P(m)·[1-P(m)] = 2·[1-P(r)]·P(r) (10)
a zatem liczba rozróŜnialnych dłuŜszych sekwencji, w porównaniu do liczby wszystkich
moŜliwych połączeń, ulega znacznemu zmniejszeniu.
Jeśli prawdopodobieństwo powstania kolejnej diady zaleŜy od rodzaju poprzedniej
diady (tzn. przyłączenie kolejnego monomeru o określonej konfiguracji jest uzaleŜnione
od konfiguracji końca łańcucha), to naleŜy zastosować prawdopodobieństwo warunkowe,
wyraŜone poprzez rozkład Markowa pierwszego rzędu:
)P(2
)P()/P(
r
rmmr
⋅
= (11)
)P(2
)P()/P(
m
mrrm
⋅
= (12)
)/P( mr – odpowiada prawdopodobieństwu występowania jednostki mezo zaraz po jednostce
racemicznej,
)/P( rm – odpowiada prawdopodobieństwu występowania jednostki racemicznej zaraz
po jednostce mezo.
W analogiczny sposób, jak dla statystyki Bernoulliego, moŜna przedstawić równania
na warunki normalizacji dla rozkładu Markowa pierwszego rzędu:
P(r/r) + P(r/m) = 1 (13)
P(m/m) + P(m/r) = 1 (14)
Natomiast prawdopodobieństwo P(r) naleŜy obliczyć z prawdopodobieństwa warunkowego
wyraŜonego poniŜszym wzorem [6, 7]:
P(r)·P(r/m) = P(m)·P(m/r) (15)
2. Kopolimery
W kopolimerach pojawia się dodatkowo moŜliwość róŜnego ułoŜenia kolejnych
komonomerów, co daje sekwencje kompozycyjne. Kolejne komonomery (na przykład A i M)
moŜna ułoŜyć na róŜne sposoby:
b) regularnie:
� blokowo – …AAAAAMMMAAAAAMMMAAAAA…

8
� przemiennie – …AMAMAMAMAMAMAMAMAM…
c) nieregularnie:
� przypadkowo – …AAMAMMMMAAMAMAMMMA…
Jeśli konfigurację łańcucha moŜna przedstawić, dla przykładu, następująco:
…rmmrmrmmr… to, gdy połączymy powyŜsze obydwa zapisy otrzymamy sekwencję
konfiguracyjno–kompozycyjną kopolimeru, która moŜe wyglądać następująco:
…ArMmMmArMmArMmMmArA…
W praktyce, w kopolimerach moŜe występować kilka szczególnych przypadków:
� gdy obydwa komonomery A i M nie dają efektów konfiguracyjnych lub efekty te są zbyt
słabe i nie da się ich zaobserwować, to zróŜnicowanie budowy łańcucha sprowadza się
do rozkładu komonomerów A i M (tylko sekwencje kompozycyjne);
� jesli izomeria konfiguracyjna jest obecna tylko w przypadku jednego komonomeru
(na przykład A), to sekwencje konfiguracyjne będą występować tylko pomiędzy jednostkami
tego samego komonomeru, na przykład:
…MAmAmAMMMAM…
� gdy w komonomerach A i M znajdują się róŜne podstawniki, czyli obydwie jednostki
komonomeryczne wniosą do łańcucha zróŜnicowanie konfiguracyjne, to dla kaŜdej sekwencji
kompozycyjnej naleŜy uwzględnić wszystkie moŜliwe sekwencje konfiguracyjne [4, 6, 7]:
)AM(P)MA(P rr ≠ (16)
)AAM(P)MAA(P mmmm ≠ (17)
)AAB(P)BAA(P rmmr ≠ (18)
)AM(P m = )MA(P m = )AM(P)MA(P mm + (19)
)AAM(P mr = )MAA(P rm = )AAM(P)MAA(P mrrm + (20)
Gdy rodzaj przyłączanego komonomeru nie zaleŜy od rodzaju komonomeru
znajdującego się na końcu wzrastającego łańcucha, a konfiguracja nowo powstałej diady
zaleŜy od końca łańcucha, to naleŜy połączyć obie statystyki, stosując rozkład Bernoulliego
dla sekwencji kompozycyjnych i rozkład Markowa pierwszego rzędu dla sekwencji
konfiguracyjnych, gdyŜ zaleŜności te są iloczynem prawdopodobieństwa przyłączenia danego
komonomeru i prawdopodobieństwa powstania określonej konfiguracji dwóch ostatnich
komonomerów [7].

9
III. Wpływ mikrostruktury na właściwości fizykochemiczne polimeru
Właściwości fizykochemiczne polimerów akrylowych i metakrylowych w stanie
skondensowanym mogą zmieniać się w szerokim zakresie, co jest zdeterminowane wartością
temperatury zeszklenia danego polimeru oraz jego średnią masą molową.
Temperatura zeszklenia (Tg, ang. glass transition temperature) jest zdefiniowana,
jako temperatura przejścia polimeru ze stanu elastycznego (kauczukopodobnego) w stan
szklisty. PoniŜej temperatury Tg polimery są sztywne, kruche i podobne do szkła, natomiast
powyŜej tej wartości miękną, stają się ciągliwe, elastyczne, a w przypadku dalszego
ogrzewania zaczynają płynąć. Polimetakrylany i poliakrylany cechuje zaleŜność między
długością i stopniem rozgałęzienia bocznej alkilowej reszty estrowej a wartością temperatury
zeszklenia, to znaczy polimetakrylany mają wyŜsze wartości Tg niŜ poliakrylany.
Ze wzrostem długości podstawnika n-alkilowego następuje początkowo obniŜenie się
wartości Tg w obu grupach polimerów, lecz dalszy wzrost liczby atomów węgla
w podstawniku (>12 atomów C) powoduje ponowne podwyŜszenie temperatury zeszklenia;
natomiast rozgałęzienie podstawnika alkilowego przyczynia się do wzrostu Tg w porównaniu
z n-alkilowym łańcuchem o identycznej liczbie atomów węgla [8].
Tabela nr 1. Temperatura zeszklenia wybranych homopolimerów
Homopolimer: Temperatura zeszklenia, Tg [°C]:
poli(akrylan n-propylu) –45
poli(akrylan metylu) 6
poli(metakrylan metylu) izotaktyczny 45
poli(metakrylan metylu) ataktyczny 105
poli(metakrylan metylu) syndiotaktyczny 115
IV. Badania mikrostruktury kopolimerów za pomocą spektroskopii NMR
Kopolimery metakrylowo–akrylowe to układy, którym od wielu lat poświęca się
wiele uwagi i zainteresowania. W tabeli nr 2 przedstawiono listę kopolimerów akrylowych
i metakrylowych, które były przedmiotem badań w latach siedemdziesiątych,
osiemdziesiątych i na początku lat dziewięćdziesiątych, natomiast bardziej szczegółowy
przegląd publikacji obejmujący późniejsze lata zostanie przedstawiony w dalszej części tego
rozdziału.
10
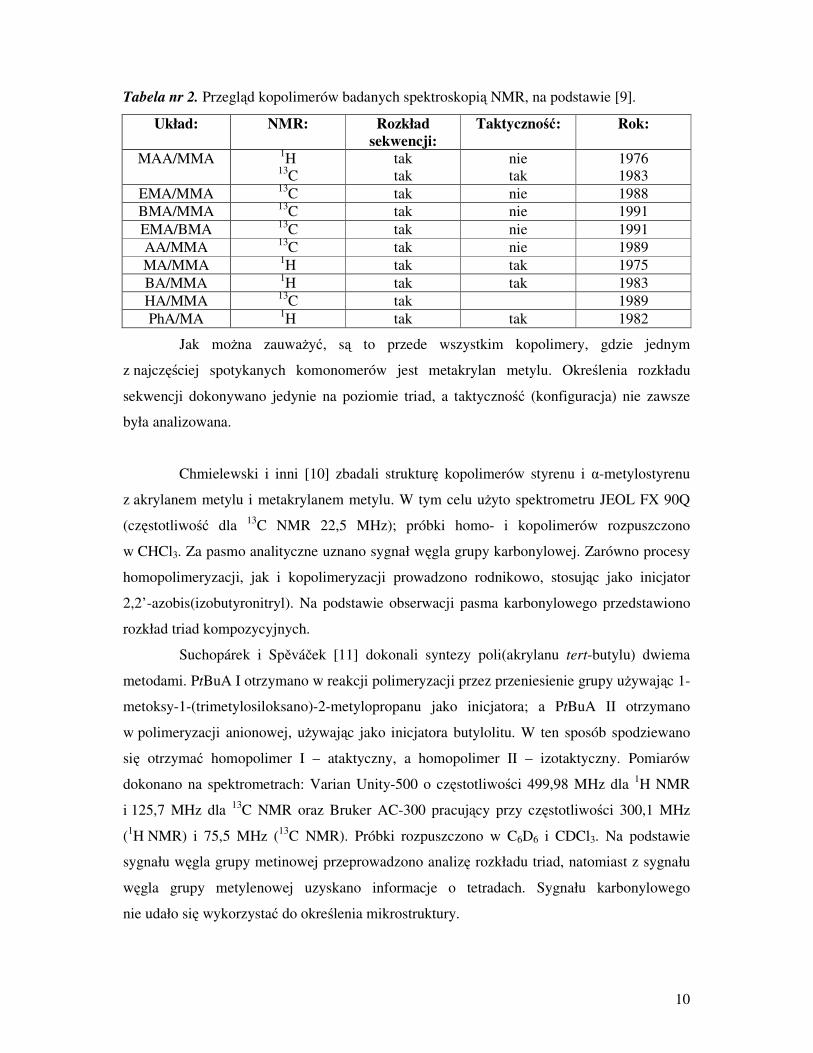
Tabela nr 2. Przegląd kopolimerów badanych spektroskopią NMR, na podstawie [9].
Układ: NMR: Rozkład
sekwencji:
Taktyczność: Rok:
MAA/MMA 1H
13C
tak
tak
nie
tak
1976
1983
EMA/MMA 13
C tak nie 1988
BMA/MMA 13
C tak nie 1991
EMA/BMA 13
C
tak nie 1991
AA/MMA 13
C tak nie 1989
MA/MMA 1H tak tak 1975
BA/MMA 1H tak tak 1983
HA/MMA 13
C tak 1989
PhA/MA 1H tak tak 1982
Jak moŜna zauwaŜyć, są to przede wszystkim kopolimery, gdzie jednym
z najczęściej spotykanych komonomerów jest metakrylan metylu. Określenia rozkładu
sekwencji dokonywano jedynie na poziomie triad, a taktyczność (konfiguracja) nie zawsze
była analizowana.
Chmielewski i inni [10] zbadali strukturę kopolimerów styrenu i α-metylostyrenu
z akrylanem metylu i metakrylanem metylu. W tym celu uŜyto spektrometru JEOL FX 90Q
(częstotliwość dla 13
C NMR 22,5 MHz); próbki homo- i kopolimerów rozpuszczono
w CHCl3. Za pasmo analityczne uznano sygnał węgla grupy karbonylowej. Zarówno procesy
homopolimeryzacji, jak i kopolimeryzacji prowadzono rodnikowo, stosując jako inicjator
2,2’-azobis(izobutyronitryl). Na podstawie obserwacji pasma karbonylowego przedstawiono
rozkład triad kompozycyjnych.
Suchopárek i Spěváček [11] dokonali syntezy poli(akrylanu tert-butylu) dwiema
metodami. PtBuA I otrzymano w reakcji polimeryzacji przez przeniesienie grupy uŜywając 1-
metoksy-1-(trimetylosiloksano)-2-metylopropanu jako inicjatora; a PtBuA II otrzymano
w polimeryzacji anionowej, uŜywając jako inicjatora butylolitu. W ten sposób spodziewano
się otrzymać homopolimer I – ataktyczny, a homopolimer II – izotaktyczny. Pomiarów
dokonano na spektrometrach: Varian Unity-500 o częstotliwości 499,98 MHz dla 1H NMR
i 125,7 MHz dla 13
C NMR oraz Bruker AC-300 pracujący przy częstotliwości 300,1 MHz
(1H NMR) i 75,5 MHz (
13C NMR). Próbki rozpuszczono w C6D6 i CDCl3. Na podstawie
sygnału węgla grupy metinowej przeprowadzono analizę rozkładu triad, natomiast z sygnału
węgla grupy metylenowej uzyskano informacje o tetradach. Sygnału karbonylowego
nie udało się wykorzystać do określenia mikrostruktury.

11
De la Fuente i inni [12] przeprowadzili badania statystycznego kopolimeru
metakrylanu metylu z akrylanem butylu, który otrzymano na drodze kontrolowanej
polimeryzacji rodnikowej, ATRP, w masie. Do analizy spektroskopowej uŜyto trzech próbek
o następujących składach komonomerów: 75:25, 50:50, 25:75; które rozpuszczono w CDCl3.
Pomiarów dokonano na spektrometrze Varian Oxford 300, a analiza sygnału węgla grupy
karbonylowej pozwoliła na przedstawienie rozkładu sekwencji na poziomie triad.
Kim i Harwood [13] zbadali mikrostrukturę kopolimeru metakrylanu metylu
z akrylanem metylu. PMMA, PMA i serię kopolimerów o róŜnym składzie zsyntetyzowano
stosując polimeryzację w masie, a jako inicjatora uŜyto AIBN. Do rejestracji widm 13
C NMR
wykorzystano spektrometr Varian Unity Plus 600, pracujący przy częstotliwości 150 MHz.
Opierając się na sygnale grupy α-metylowej i metinowej przeprowadzono analizę rozkładu
sekwencji kompozycyjnych na poziomie triad i częściowo na poziomie pentad. Nie podjęto
się wnioskowania na temat mikrostruktury polimeru na podstawie pasma karbonylowego,
ze względu na nakładanie się sygnałów grupy C=O, pochodzących zarówno od jednostek
metakrylanu metylu jak i akrylanu metylu.
Dhal i współpracownicy [14] zajęli się badaniem mikrostruktury kopolimeru
metakrylanu glicydylu, GMA, z akrylanem izo-butylu i n-butylu. Widma 13
C NMR wykonano
na spektrometrze JEOL JNM-FX100 pracującym przy częstotliwości 25,05 MHz, a jako
rozpuszczalnika uŜyto CDCl3. Na podstawie sygnału rezonansowego węgla karbonylowego
i węgla grupy metylowej dokonano analizy sekwencji PGMA na poziomie pentad,
natomiast piki z obszaru węgla karbonylowego dla PnBA i PiBA dostarczyły informacji
jedynie o diadach, co wskazuje, Ŝe otrzymane w tej pracy homopolimery akrylowe były
ataktyczne. W przypadku kopolimerów, na podstawie zakresu karbonylowego, udało się
przedstawić rozkład sekwencji na poziomie triad.
W roku 2000 ukazała się publikacja Matlengiewicza i współpracowników [15],
którzy przeprowadzili analizę sygnału β-CH2 w kopolimerze metakrylanu metylu i akrylanu
etylu. Homopolimery: PMMA, PEA i kopolimer zostały otrzymane w wyniku polimeryzacji
rodnikowej z zastosowaniem AIBN, a drugą próbkę PMMA otrzymano anionowo stosując
fluorenolit, jako inicjator. Widma 13
C NMR zostały wykonane na spektrometrze Bruker DRX
400 przy częstotliwości 100 MHz, z zastosowaniem techniki DEPT, a jako rozpuszczalnika
uŜyto CDCl3. Na podstawie pasma rezonansowego grupy metylenowej dokonano analizy
na poziomie diad i tetrad oraz przypisano wartości inkrementów α i β na podstawie
przesunięcia chemicznego.

12
Brar i współpracownicy [16] podjęli się wyznaczenia mikrostruktury łańcucha
polimerowego dla układu metakrylan metylu – akrylan etylu stosując techniki jedno-
i dwuwymiarowe. Kopolimer MMA/EA otrzymano w polimeryzacji rodnikowej w masie,
a jako inicjatora uŜyto AIBN. Widma 1D i 2D NMR zarejestrowano w CDCl3,
na spektrometrze 300 MHz Bruker DPX przy częstotliwości odpowiednio 300,13 MHz
i 75,5MHz; równieŜ z uŜyciem techniki DEPT. Wykorzystując sygnał węgla grupy metylowej
i grupy –CH dokonano analizy na poziomie pentad, natomiast stosując metodę DEPT
i technikę dwuwymiarowego przedstawiania widm 13
C NMR, na podstawie pasma
rezonansowego grupy metylenowej opisano rozkład tetrad, a dla obszaru węgla
karbonylowego udało się określić rozkład triad.
Kolejną swoją publikację [17] Matlengiewicz poświęcił wnikliwej analizie
kopolimerów akrylowych. Kopolimeryzacji rodnikowej (z uŜyciem AIBN, jako inicjatora)
poddano takie układy, jak: metakrylan metylu – akrylan etylu (50/50), metakrylan metylu –
akrylan butylu (40/60), metakrylan nonylu – metakrylanu metylu (70/30) i akrylan nonylu –
akrylan dimetyloaminy (50/50). Widma 13
C NMR zostały wykonane w CDCl3, przy pomocy
spektrometru Bruker AM 400 przy częstotliwości 100 MHz. Obserwowano pasmo
rezonansowe grupy karbonylowej oraz z pomocą statystyki Bernoulliego i Markowa
pierwszego rzędu przedstawiono rozkład pentad konfiguracyjno–kompozycyjnych.
Nguyen i współpracownicy [18], przedstawili mikrostrukturę łańcucha kopolimeru
metakrylanu metylu z akrylanem etylu. Kopolimer zsyntetyzowano rodnikowo uŜywając,
AIBN jako inicjatora. Widma 13
C NMR zostały wykonane w CDCl3, na spektrometrze Bruker
AM 400 przy częstotliwości 100 MHz. Obiektem zainteresowania był zakres karbonylowy
widma. Na jego podstawie przypisano triady i pentady, następnie zasymulowano widma
PMMA/EA o składach 70/30, 50/50 i 30/70 oraz przypisano wartości inkrementów
α na podstawie przesunięć chemicznych.
Nguyen wraz ze współpracownikami [19] przedstawiła publikację omawiającą
zastosowanie inkrementalnej metody wyznaczania rozkładu sekwencji homopolimerów.
W tym celu zsyntezowano poli(metakrylan metylu) dwiema róŜnymi metodami: rodnikowo
i anionowo (z uŜyciem fluorenolitu, jako inicjatora). Otrzymano w ten sposób odpowiednio
homopolimer syndiotaktyczny i izotaktyczny. Widma 13
C NMR zostały zarejestrowane
w CDCl3, na spektrometrze Bruker DRX 400 przy częstotliwości 100 MHz. Przeanalizowano
część widma dotyczącą zakresu karbonylowego, przypisano wartości inkrementów α, α1, β, β1
i γ na podstawie przesunięć chemicznych sekwencji konfiguracyjnych w syndiotaktycznym

13
i izotaktycznym PMMA oraz dla triad, pentad i heptad porównano ze sobą rozkład Markowa
pierwszego i drugiego rzędu.
Z kolei Matlengiewicz [20] dokonał dokładnej analizy syndiotaktycznego
poli(metakrylanu metylu), który otrzymano na drodze polimeryzacji rodnikowej stosując,
jako inicjator AIBN. Widma 13
C NMR zarejestrowano na spektrometrze Bruker AM 400,
przy częstotliwości 100 MHz uŜywając, jako rozpuszczalnika CDCl3. Skupiono się głównie
na obszarze węgla grupy karbonylowej i dokonano analizy rozkładu sekwencji na poziomie
heptad. Następnie porównano ze sobą statystyki: Bernoulliego i Markowa pierwszego rzędu
dla triad, pentad i heptad.
W roku 2007 ukazały się dwie publikacje Bujaka i współpracowników
przedstawiające charakterystykę mikrostrukturalną homopolimerów akrylanu butylu.
W pierwszej z nich [21] omówiono syntezę poli(akrylanu n-butylu), PnBA, z zastosowaniem
inicjatora rodnikowego AIBN. Zarejestrowano widma 100 MHz 13
C NMR otrzymanych
próbek na spektrometrze Bruker Avance 400, stosując jako rozpuszczalnik deuterowany
chloroform i deuterowany benzen. Całkując obszar pod pikami grupy α-CH określono rozkład
triad konfiguracyjnych (rr, rm i mm) dla PnBA. Analizę mikrostrukturalną tego
homopolimeru przeprowadzono w oparciu o sygnał grupy karbonylowej obejmujący zakres
od 174 do 174,7 ppm. Stosując statystykę Bernoulliego i Markowa pierwszego rzędu
oraz inkrementalną metodę obliczania przesunięcia chemicznego dokonano wyznaczenia
mikrostruktury poli(akrylanu n-butylu) na poziomie pentad konfiguracyjnych. Wykorzystując
środowisko Matlab (MatchWorks, Inc.) i dane uzyskane z obliczeń statystycznych
przeprowadzono symulację sygnału C=O PnBA stosując szerokość połówkową linii 3 i 5 Hz.
Druga publikacja Bujaka [22] została poświęcona poli(akrylanowi tert-butylu).
Próbki tego homopolimeru uzyskano dwiema metodami: PtBA I – zsyntezowano
w polimeryzacji jonowej z uŜyciem n-butylolitu, jako inicjatora, a PtBA II otrzymano
rodnikowo (jako inicjatora uŜyto AIBN). 100 MHz widma 13
C NMR otrzymanych próbek
zarejestrowano na spektrometrze Bruker Avance 400 stosując trzy róŜne deuterowane
rozpuszczalniki: chloroform, benzen i aceton. Następnie zebrano i porównano między sobą
sygnały karbonylowe PtBA I i PtBA II w róŜnych rozpuszczalnikach, wybierając do dalszej
analizy widma zarejestrowane w CDCl3 i C6D6. Na podstawie sygnału grupy α-CH obliczono
zawartość triad konfiguracyjnych. Wykorzystując analogiczną metodę obliczeń,
jak dla PnBA, przedstawiono mikrostrukturę PtBA I i PtBA II na poziomie pentad
konfiguracyjnych oraz przeprowadzono symulację sygnału karbonylowego stosując szerokość
połówkową linii 1, 5 i 10 Hz.

14
CEL I ZAKRES PRACY
Celem pracy jest określenie mikrostruktury wybranych kopolimerów metakrylowo–
akrylowych oraz odpowiednich homopolimerów akrylowych. Jako komonomer metakrylowy
wybrano metakrylan metylu, a jako komonomery akrylowe wytypowano akrylan metylu
oraz akrylany propylu. Badania mikrostrukturalne polimerów zostaną przeprowadzone przy
uŜyciu spektroskopii 13
C NMR z wykorzystaniem sygnału węgli karbonylowych (sekwencje
nieparzyste: triady, pentady, itd.) i metylenowych (sekwencje parzyste: diady, tetrady, itd.).
Przewidywany zakres prac będzie obejmować syntezę homopolimerów akrylanów
metylu i propylu oraz odpowiednich kopolimerów metakrylowo–akrylowych o zmiennym
udziale molowym komonomerów. W kolejnym etapie prac przewidziano rejestrację widm
NMR, wytypowanie sygnałów rezonansowych czułych na mikrostrukturę, a następnie
szczegółową analizę sygnału karbonylowego homo- i kopolimerów. Informacje uzyskane
z badań mikrostrukturalnych tych polimerów posłuŜą do poszukiwania wpływu rozkładu
sekwencji na ich właściwości termochemiczne, badane przy uŜyciu róŜnicowej kalorymetrii
skaningowej, DSC.

15
BADANIA WŁASNE
Biorąc pod uwagę dotychczasowe wyniki badań nad układami metakrylowo–
akrylowymi podjąłem się charakteryzowania mikrostruktury kopolimerów metakrylanu
metylu z akrylanami metylu i propylu.
Z danych literaturowych wiadomo, Ŝe Kim i Harwood [13] przeprowadzili
charakterystykę mikrostrukturalną poli(metakrylanu metylu-co-akrylanu metylu),
PMMA/MA, w oparciu o sygnały grupy α-metylowej i metinowej, jednak sygnał grup
karbonylowych nie został przez nich poddany analizie. W związku z powyŜszym
postanowiłem uzupełnić badania mikrostruktury kopolimeru PMMA/MA o analizę sygnału
grup C=O.
W trakcie studiów literaturowych dotyczących mikrostruktury kopolimerów
metakrylowo–akrylowych nie spotkałem się z charakterystyką homopolimerów akrylanu
propylu oraz ich kopolimerów z metakrylanem metylu. Biorąc pod uwagę brak informacji
o rozkładzie sekwencji konfiguracyjnych w homopolimerach, jak i sekwencji
konfiguracyjno–kompozycyjnych w kopolimerach zawierających akrylany propylu uznałem
za interesujące zbadanie pełnej mikrostruktury tychŜe polimerów. Jako pierwszy, do badań
został wytypowany homopolimer akrylanu n-propylu i jego kopolimer z metakrylanem
metylu. W następnej kolejności zaplanowano prace badawcze nad poli(akrylanem izopropylu)
oraz poli(metakrylanem metylu-co-akrylanem izopropylu), PMMA/iPrA.
I. Metodyka badań
1. Otrzymywanie polimerów
Synteza homopolimerów
Badane homopolimery: poli(metakrylan metylu), PMMA, poli(akrylan metylu),
PMA, oraz poli(akrylan n-propylu), PnPrA, otrzymano na drodze polimeryzacji rodnikowej,
spodziewając się uzyskać homopolimery syndiotaktyczny [13, 20].
Synteza kopolimerów poli(metakrylanu metylu-co-akrylanu metylu), PMMA/MA
i poli(metakrylanu metylu-co-akrylanu n-propylu), PMMA/nPrA
Na podstawie równania kopolimeryzacji:
+
+=
[M][A]r
[A][M]r
[A]
]M[
d[A]
d[M]
2
1

16
gdzie:
r1 – współczynnik reaktywności komonomeru M (metakrylan metylu, MMA),
r2 – współczynnik reaktywności komonomeru A (akrylan: metylu (MA), n-propylu (nPrA)),
[M], [A] – odpowiednio stęŜenia molowe komonomerów M i A,
d[A]
d[M]– stosunek molowy komonomerów M i A w kopolimerze,
wyznaczonego niezaleŜnie przez Alfreya i Goldfingera, Mayo i Lewisa oraz Walla [1, 6, 23]
obliczono ilości reagentów niezbędnych do reakcji kopolimeryzacji. Wykorzystano
współczynniki reaktywności zamieszczone w publikacji [13], które dla układu PMMA/MA
wynoszą r1(MMA) = 2,60 i r2(MA) = 0,27. W przypadku kopolimeryzacji poli(metakrylanu
metylu-co-akrylanu n-propylu) zastosowano analogiczne warunki, jak w przypadku
kopolimeru PMMA/MA. Wartości współczynników reaktywności dla PMMA/nPrA, zostały
wyznaczone metodą Finemana–Rossa i wynoszą odpowiednio: r1(MMA) = 2,08, r2(nPrA) = 0,49.
Analogicznie, jak w przypadku homopolimerów kopolimery metakrylowo–akrylowe
otrzymano na drodze rodnikowej, a dobierając warunki syntezy skorzystano w publikacji
Kima i Harwooda [13].
2. Rejestracja widm 13C NMR polimerów
Widma 13
C NMR zarejestrowano przy częstotliwości 100 MHz na spektrometrze
Bruker Avance 400. Sporządzono próbki homo- i kopolimerów o stęŜeniu około 10% w C6D6
i CDCl3. Rejestrację 12000 skanów (NS) przeprowadzono w temperaturze T = 313 K
zarówno dla deuterowanego benzenu, jak i deuterowanego chloroformu stosując odstępy
między kolejnymi impulsami wynoszące D1 = 4 s.
3. Badania kalorymetryczne homopolimerów
Badania DSC przeprowadzono na kalorymetrze Mettler Toledo DSC 822e. OdwaŜki
homopolimerów umieszczano w aluminiowych naczyńkach, które zamykano mechaniczne.
Próbki ogrzewano z szybkością 20,00 °C/min, potem temperaturę utrzymywano na stałym
poziomie przez trzy minuty, następnie chłodzono ze stałą szybkością – 20,00 °C/min,
aby ponownie stabilizować przez trzy minuty w – 65 °C; następnie próbka była ponownie
poddawana ogrzewaniu. Przedstawione krzywe pochodzą z trzeciego przebiegu grzanie–
chłodzenie. Badania wykonano zgodnie z normami: PN-EN ISO 11357-1:2009
„Tworzywa sztuczne - RóŜnicowa kalorymetria skaningowa (DSC) - Część 1: Zasady ogólne”
17
i ISO 11357-2:1999 “Plastics - Differential scanning calorimetry (DSC) - Part 2:
Determination of glass transition temperature”.
II. Przedstawienie wyników badań
1. Otrzymywanie polimerów
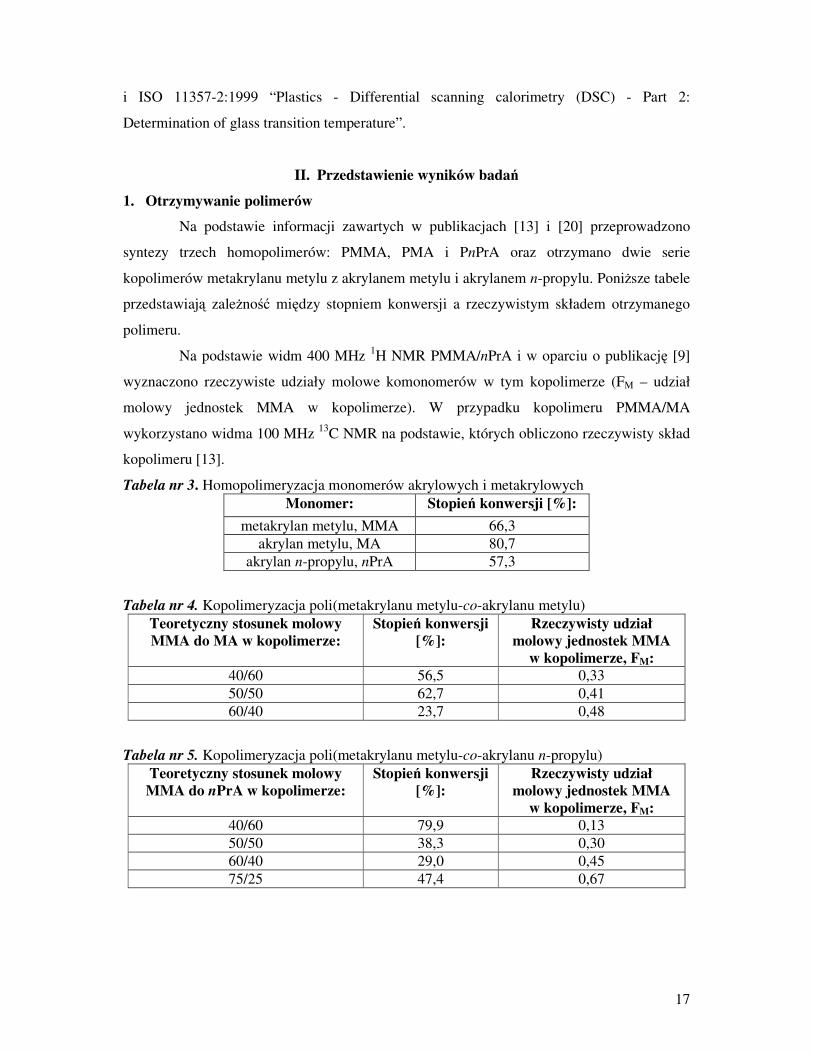
Na podstawie informacji zawartych w publikacjach [13] i [20] przeprowadzono
syntezy trzech homopolimerów: PMMA, PMA i PnPrA oraz otrzymano dwie serie
kopolimerów metakrylanu metylu z akrylanem metylu i akrylanem n-propylu. PoniŜsze tabele
przedstawiają zaleŜność między stopniem konwersji a rzeczywistym składem otrzymanego
polimeru.
Na podstawie widm 400 MHz 1H NMR PMMA/nPrA i w oparciu o publikację [9]
wyznaczono rzeczywiste udziały molowe komonomerów w tym kopolimerze (FM – udział
molowy jednostek MMA w kopolimerze). W przypadku kopolimeru PMMA/MA
wykorzystano widma 100 MHz 13
C NMR na podstawie, których obliczono rzeczywisty skład
kopolimeru [13].
Tabela nr 3. Homopolimeryzacja monomerów akrylowych i metakrylowych
Monomer: Stopień konwersji [%]:
metakrylan metylu, MMA 66,3
akrylan metylu, MA 80,7
akrylan n-propylu, nPrA 57,3
Tabela nr 4. Kopolimeryzacja poli(metakrylanu metylu-co-akrylanu metylu)
Teoretyczny stosunek molowy
MMA do MA w kopolimerze:
Stopień konwersji
[%]:
Rzeczywisty udział
molowy jednostek MMA
w kopolimerze, FM:
40/60 56,5 0,33
50/50 62,7 0,41
60/40 23,7 0,48
Tabela nr 5. Kopolimeryzacja poli(metakrylanu metylu-co-akrylanu n-propylu)
Teoretyczny stosunek molowy
MMA do nPrA w kopolimerze:
Stopień konwersji
[%]:
Rzeczywisty udział
molowy jednostek MMA
w kopolimerze, FM:
40/60 79,9 0,13
50/50 38,3 0,30
60/40 29,0 0,45
75/25 47,4 0,67
18
-OCH3
180 160 140 120 100 80 60 40 20 0Chemical Shift (ppm)
TMS
C6D6
C=O
α-C
α-CH3 β-CH2
2. Widma 13C NMR polimerów
Otrzymane próbki polimerów posłuŜyły do przygotowania ok. 10% roztworów,
które następnie poddano badaniom magnetycznego rezonansu jądrowego. Na podstawie
uzyskanych widm NMR przypisano sygnały rezonansowe odpowiednim węglom z łańcucha
polimerowego i poddano wnikliwej analizie zakres karbonylowy.
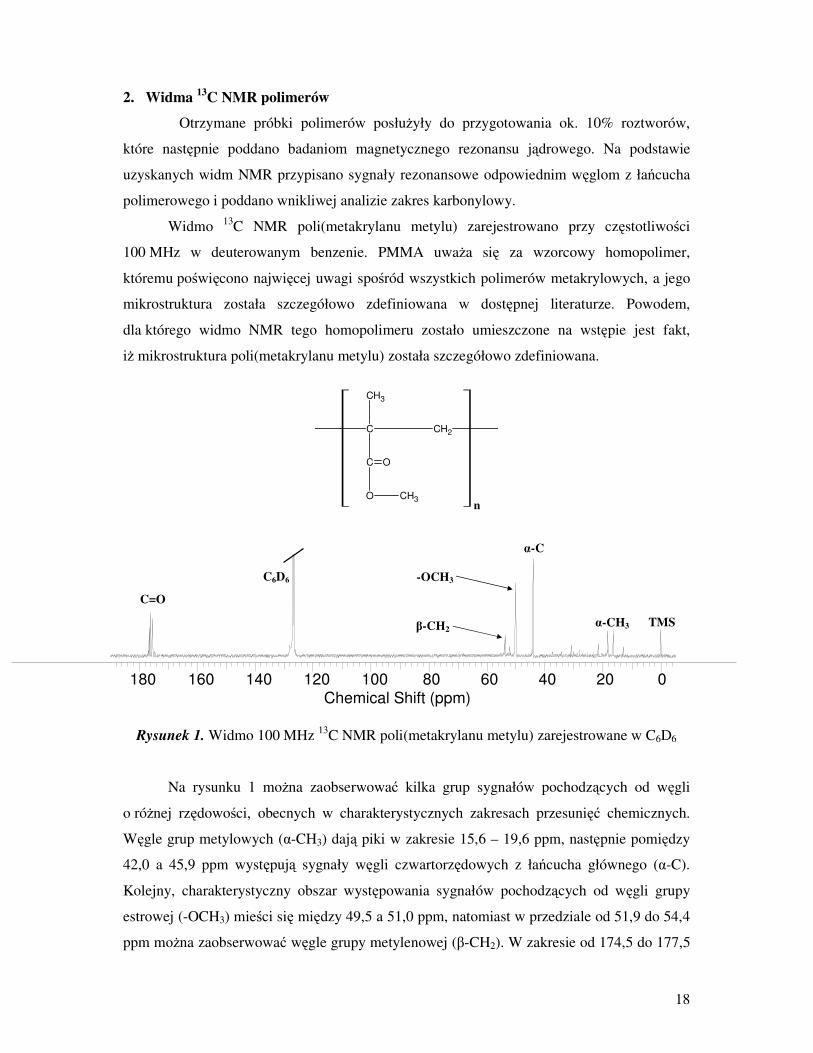
Widmo 13
C NMR poli(metakrylanu metylu) zarejestrowano przy częstotliwości
100 MHz w deuterowanym benzenie. PMMA uwaŜa się za wzorcowy homopolimer,
któremu poświęcono najwięcej uwagi spośród wszystkich polimerów metakrylowych, a jego
mikrostruktura została szczegółowo zdefiniowana w dostępnej literaturze. Powodem,
dla którego widmo NMR tego homopolimeru zostało umieszczone na wstępie jest fakt,
iŜ mikrostruktura poli(metakrylanu metylu) została szczegółowo zdefiniowana.
Rysunek 1. Widmo 100 MHz 13
C NMR poli(metakrylanu metylu) zarejestrowane w C6D6
Na rysunku 1 moŜna zaobserwować kilka grup sygnałów pochodzących od węgli
o róŜnej rzędowości, obecnych w charakterystycznych zakresach przesunięć chemicznych.
Węgle grup metylowych (α-CH3) dają piki w zakresie 15,6 – 19,6 ppm, następnie pomiędzy
42,0 a 45,9 ppm występują sygnały węgli czwartorzędowych z łańcucha głównego (α-C).
Kolejny, charakterystyczny obszar występowania sygnałów pochodzących od węgli grupy
estrowej (-OCH3) mieści się między 49,5 a 51,0 ppm, natomiast w przedziale od 51,9 do 54,4
ppm moŜna zaobserwować węgle grupy metylenowej (β-CH2). W zakresie od 174,5 do 177,5
C CH2
CH3
C
O CH3
O
n

19
55 50 45 40 35 30 25 20 15Chemical Shift (ppm) ppm
-OCH3
α-CH3
β-CH2
α-C
177.5 177.0 176.5 176.0 175.5 175.0 174.5 174.0Chemical Shift (ppm) ppm
ppm są obecne sygnały węgli karbonylowych (C=O). Przypisania sygnałów dokonano
na podstawie publikacji [5] i uwzględniono róŜnice w przesunięciach chemicznych związane
z uŜytym rozpuszczalnikiem.
Rysunek 2. Zakres alifatyczny widma 100 MHz 13
C NMR PMMA zarejestrowanego w C6D6
Rysunek 3. Zakres karbonylowy widma 100 MHz 13
C NMR PMMA zarejestrowanego
w C6D6
Kolejnym homopolimerem, badanym w niniejszej pracy był poli(akrylan metylu),
PMA. Warunki rejestracji były analogiczne, jak w przypadku poli(metakrylanu metylu).
CH CH2
C
O CH3
O
n

20
180 160 140 120 100 80 60 40 20 0ppm
TMS
C=O
C6D6 -OCH3
α-CH
β-CH2
50 48 46 44 42 40 38 36 34 32 30Chemical Shift (ppm)
ppm
β-CH2
α-CH
-OCH3
Rysunek 3. Widmo 100 MHz 13
C NMR poli(akrylanu metylu) zarejestrowane w C6D6
Na widmie 100 MHz 13
C NMR PMA moŜna wyróŜnić kilka grup sygnałów,
analogicznie, jak w przypadku PMMA. Sygnały węgli grup metylenowych (β-CH2) moŜna
zaobserwować w granicach 33,3 – 35,1 ppm, a w przedziale od 39,4 do 41,2 ppm obecne
są piki pochodzące od sygnałów węgli α-CH, z łańcucha polimerowego. Charakterystyczny,
pojedynczy pik przy 50,17 ppm pochodzi od węgli grup -OCH3, a odosobniony sygnał
w zakresie od 173,0 – 174,0 ppm naleŜy do węgli grup karbonylowych homopolimeru.
Przypisania sygnałów dokonano na podstawie publikacji [13] i odniesiono do wzorca – TMS.
Rysunek 4. Zakres alifatyczny widma 100 MHz 13
C NMR PMA zarejestrowanego w C6D6
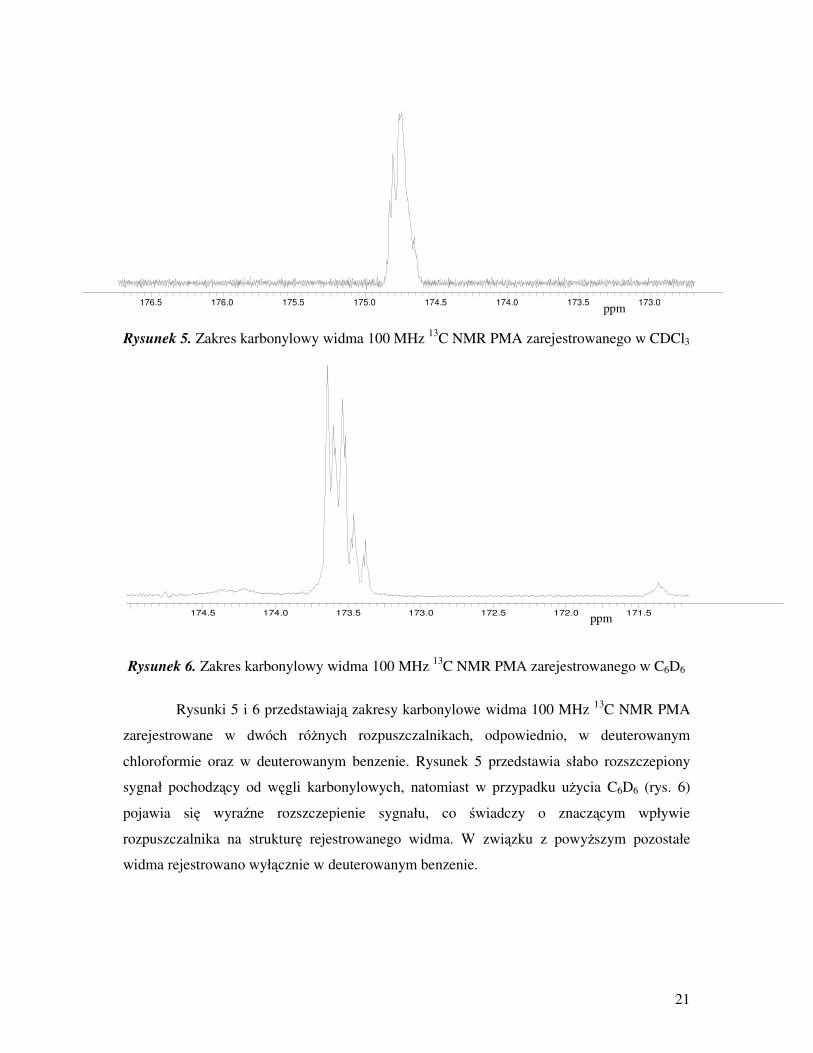
21
174.5 174.0 173.5 173.0 172.5 172.0 171.5Chemical Shift (ppm) ppm
Rysunek 5. Zakres karbonylowy widma 100 MHz 13
C NMR PMA zarejestrowanego w CDCl3
Rysunek 6. Zakres karbonylowy widma 100 MHz 13
C NMR PMA zarejestrowanego w C6D6
Rysunki 5 i 6 przedstawiają zakresy karbonylowe widma 100 MHz 13
C NMR PMA
zarejestrowane w dwóch róŜnych rozpuszczalnikach, odpowiednio, w deuterowanym
chloroformie oraz w deuterowanym benzenie. Rysunek 5 przedstawia słabo rozszczepiony
sygnał pochodzący od węgli karbonylowych, natomiast w przypadku uŜycia C6D6 (rys. 6)
pojawia się wyraźne rozszczepienie sygnału, co świadczy o znaczącym wpływie
rozpuszczalnika na strukturę rejestrowanego widma. W związku z powyŜszym pozostałe
widma rejestrowano wyłącznie w deuterowanym benzenie.
176.5 176.0 175.5 175.0 174.5 174.0 173.5 173.0Chemical Shift (ppm) ppm

22
-OCH2CH2CH3
-OCH2CH2CH3 -OCH2CH2CH3
α-CH
180 160 140 120 100 80 60 40 20 0ppm
C=O
C6D6
β-CH2
TMS
Poli(akrylan n-propylu), PnPrA, zsyntezowano metodą polimeryzacji rodnikowej,
w roztworze, uŜywając jako inicjatora 2,2’-azobis(izobutyronitrylu), AIBN; z otrzymanej
próbki pobrano odwaŜkę i rozpuszczono w deuterowanym benzenie; tak przygotowany ok.
10% roztwór posłuŜył do rejestracji 100 MHz widma 13
C NMR.
Rysunek 7. Widmo 100 MHz 13
C NMR poli(akrylanu n-propylu) zarejestrowane w C6D6
Na rysunku 7 przedstawiono widmo homopolimeru poli(akrylanu n-propylu)
wraz z przypisaniem sygnałów rezonansowych odpowiednim atomom węgla. Sygnały węgli
pochodzące od grupy n-propylowej mają charakter pojedynczych pików znajdujących się
przy 9,2 ppm (-OCH2CH2CH3), 21,0 ppm (-OCH2CH2CH3) i 63.9 ppm (-OCH2CH2CH3).
Sygnał grupy metylenowej (β-CH2) obejmuje zakres od 32,7 do 36,3 ppm, natomiast sygnał
węgla α-CH występuje w przedziale 40,4 – 41,4 ppm. Węgle grup karbonylowych dają sygnał
w granicach od 172,5 do 174,0 ppm. PowyŜszego przypisania sygnałów karbonylowych
dokonano na podstawie publikacji [21], w której przedstawiono analogiczny homopolimer,
poli(akrylan n-butylu). Wartości przysunięć chemicznych odniesiono do bieŜących warunków
rejestracji widma 13
C NMR.
CH CH2
C
O
O
CH2 CH2 CH3
n
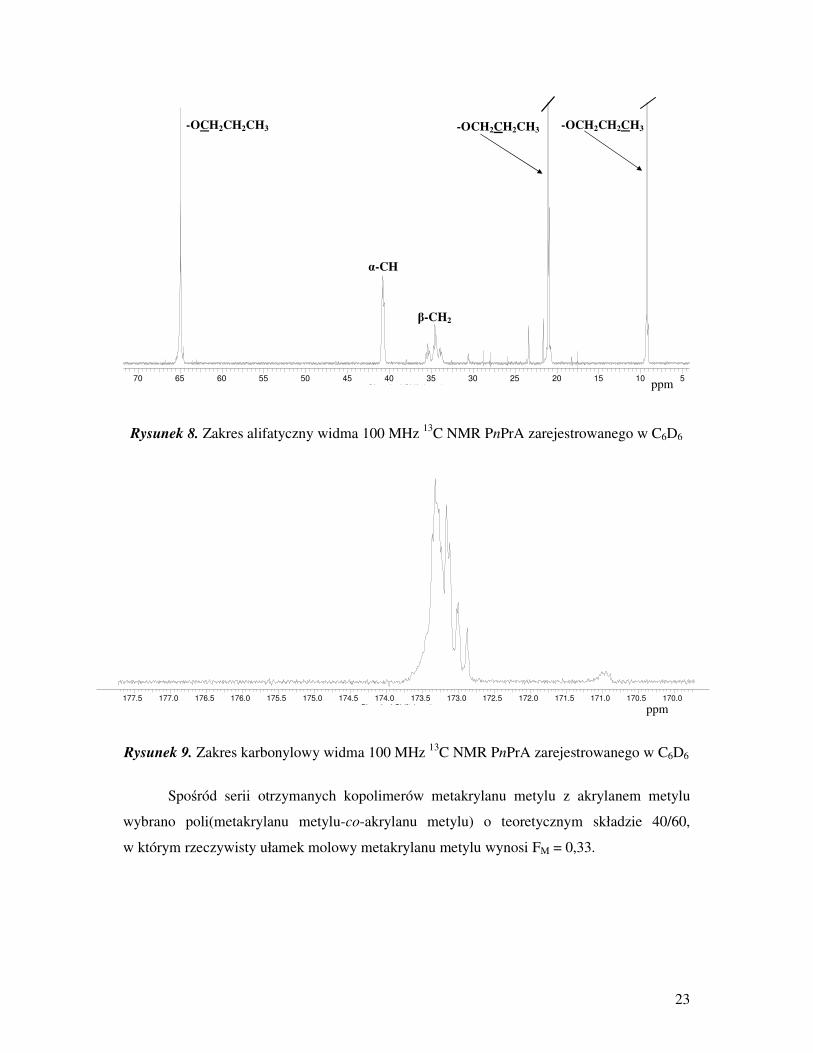
23
-OCH2CH2CH3
β-CH2
70 65 60 55 50 45 40 35 30 25 20 15 10 5Chemical Shift (ppm) ppm
α-CH
-OCH2CH2CH3 -OCH2CH2CH3
177.5 177.0 176.5 176.0 175.5 175.0 174.5 174.0 173.5 173.0 172.5 172.0 171.5 171.0 170.5 170.0Chemical Shift (ppm) ppm
Rysunek 8. Zakres alifatyczny widma 100 MHz 13
C NMR PnPrA zarejestrowanego w C6D6
Rysunek 9. Zakres karbonylowy widma 100 MHz 13
C NMR PnPrA zarejestrowanego w C6D6
Spośród serii otrzymanych kopolimerów metakrylanu metylu z akrylanem metylu
wybrano poli(metakrylanu metylu-co-akrylanu metylu) o teoretycznym składzie 40/60,
w którym rzeczywisty ułamek molowy metakrylanu metylu wynosi FM = 0,33.
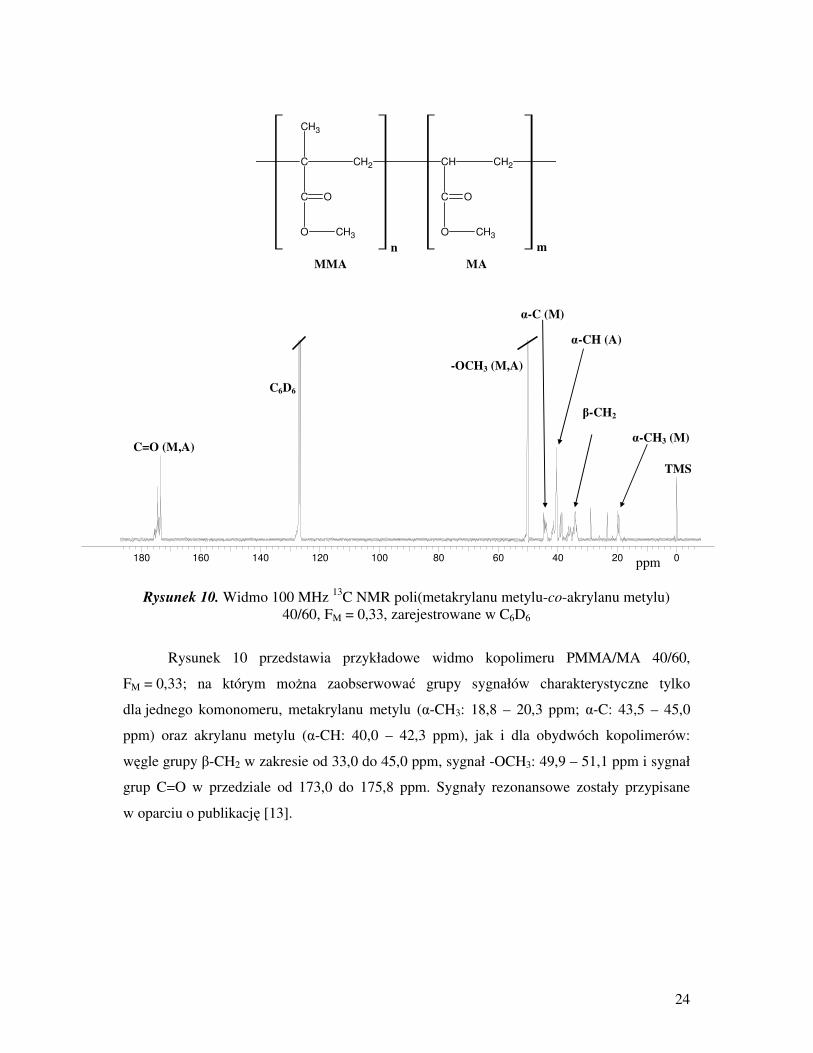
24
β-CH2
α-CH (A)
α-CH3 (M)
C6D6
180 160 140 120 100 80 60 40 20 0ppm
C=O (M,A)
-OCH3 (M,A)
TMS
α-C (M)
Rysunek 10. Widmo 100 MHz 13
C NMR poli(metakrylanu metylu-co-akrylanu metylu)
40/60, FM = 0,33, zarejestrowane w C6D6
Rysunek 10 przedstawia przykładowe widmo kopolimeru PMMA/MA 40/60,
FM = 0,33; na którym moŜna zaobserwować grupy sygnałów charakterystyczne tylko
dla jednego komonomeru, metakrylanu metylu (α-CH3: 18,8 – 20,3 ppm; α-C: 43,5 – 45,0
ppm) oraz akrylanu metylu (α-CH: 40,0 – 42,3 ppm), jak i dla obydwóch kopolimerów:
węgle grupy β-CH2 w zakresie od 33,0 do 45,0 ppm, sygnał -OCH3: 49,9 – 51,1 ppm i sygnał
grup C=O w przedziale od 173,0 do 175,8 ppm. Sygnały rezonansowe zostały przypisane
w oparciu o publikację [13].
C
CH3
C
O CH3
O
CH2 CH
C
O CH3
O
CH2
n m
MMA MA
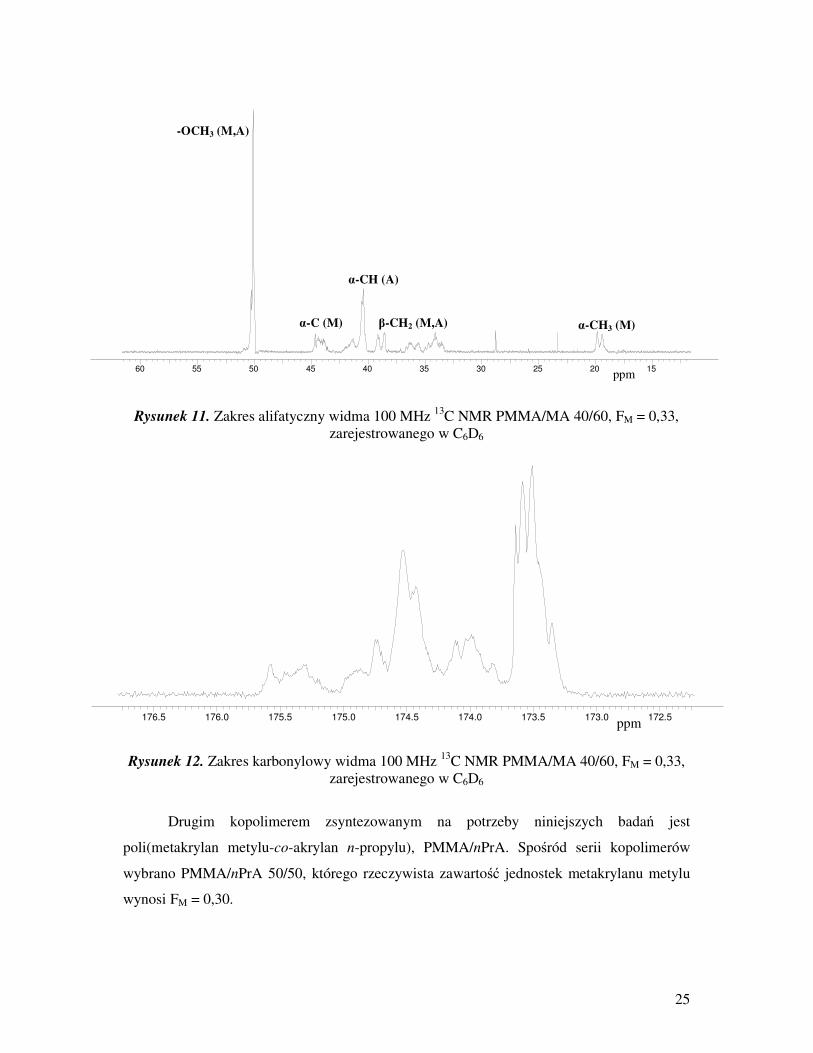
25
60 55 50 45 40 35 30 25 20 15Chemical Shift (ppm) ppm
-OCH3 (M,A)
α-C (M)
α-CH (A)
β-CH2 (M,A) α-CH3 (M)
176.5 176.0 175.5 175.0 174.5 174.0 173.5 173.0 172.5Chemical Shift (ppm) ppm
Rysunek 11. Zakres alifatyczny widma 100 MHz 13
C NMR PMMA/MA 40/60, FM = 0,33,
zarejestrowanego w C6D6
Rysunek 12. Zakres karbonylowy widma 100 MHz 13
C NMR PMMA/MA 40/60, FM = 0,33,
zarejestrowanego w C6D6
Drugim kopolimerem zsyntezowanym na potrzeby niniejszych badań jest
poli(metakrylan metylu-co-akrylan n-propylu), PMMA/nPrA. Spośród serii kopolimerów
wybrano PMMA/nPrA 50/50, którego rzeczywista zawartość jednostek metakrylanu metylu
wynosi FM = 0,30.
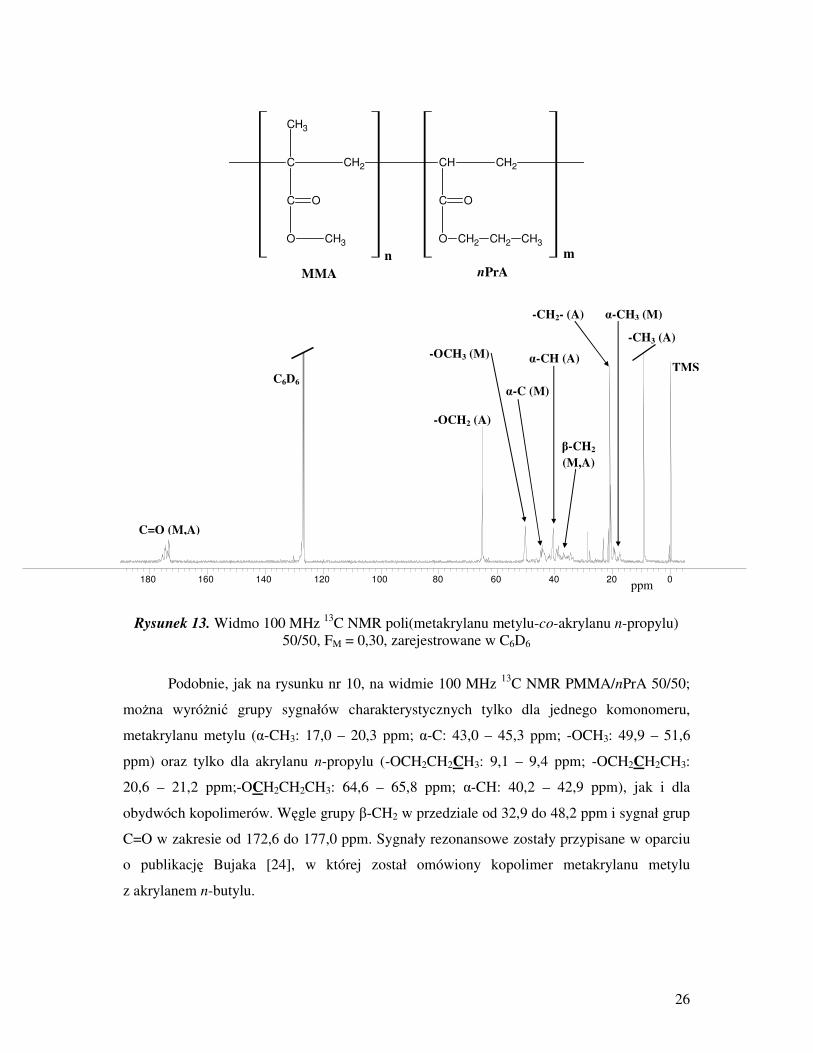
26
Rysunek 13. Widmo 100 MHz 13
C NMR poli(metakrylanu metylu-co-akrylanu n-propylu)
50/50, FM = 0,30, zarejestrowane w C6D6
Podobnie, jak na rysunku nr 10, na widmie 100 MHz 13
C NMR PMMA/nPrA 50/50;
moŜna wyróŜnić grupy sygnałów charakterystycznych tylko dla jednego komonomeru,
metakrylanu metylu (α-CH3: 17,0 – 20,3 ppm; α-C: 43,0 – 45,3 ppm; -OCH3: 49,9 – 51,6
ppm) oraz tylko dla akrylanu n-propylu (-OCH2CH2CH3: 9,1 – 9,4 ppm; -OCH2CH2CH3:
20,6 – 21,2 ppm;-OCH2CH2CH3: 64,6 – 65,8 ppm; α-CH: 40,2 – 42,9 ppm), jak i dla
obydwóch kopolimerów. Węgle grupy β-CH2 w przedziale od 32,9 do 48,2 ppm i sygnał grup
C=O w zakresie od 172,6 do 177,0 ppm. Sygnały rezonansowe zostały przypisane w oparciu
o publikację Bujaka [24], w której został omówiony kopolimer metakrylanu metylu
z akrylanem n-butylu.
β-CH2
(M,A)
-CH2- (A) α-CH3 (M)
-OCH3 (M)
-OCH2 (A)
TMS
180 160 140 120 100 80 60 40 20 0Chemical Shift (ppm) ppm
C=O (M,A)
C6D6
-CH3 (A)
α-C (M)
α-CH (A)
C
CH3
C
O CH3
O
CH2 CH
C O
CH2
O CH2 CH2 CH3
n m
MMA nPrA
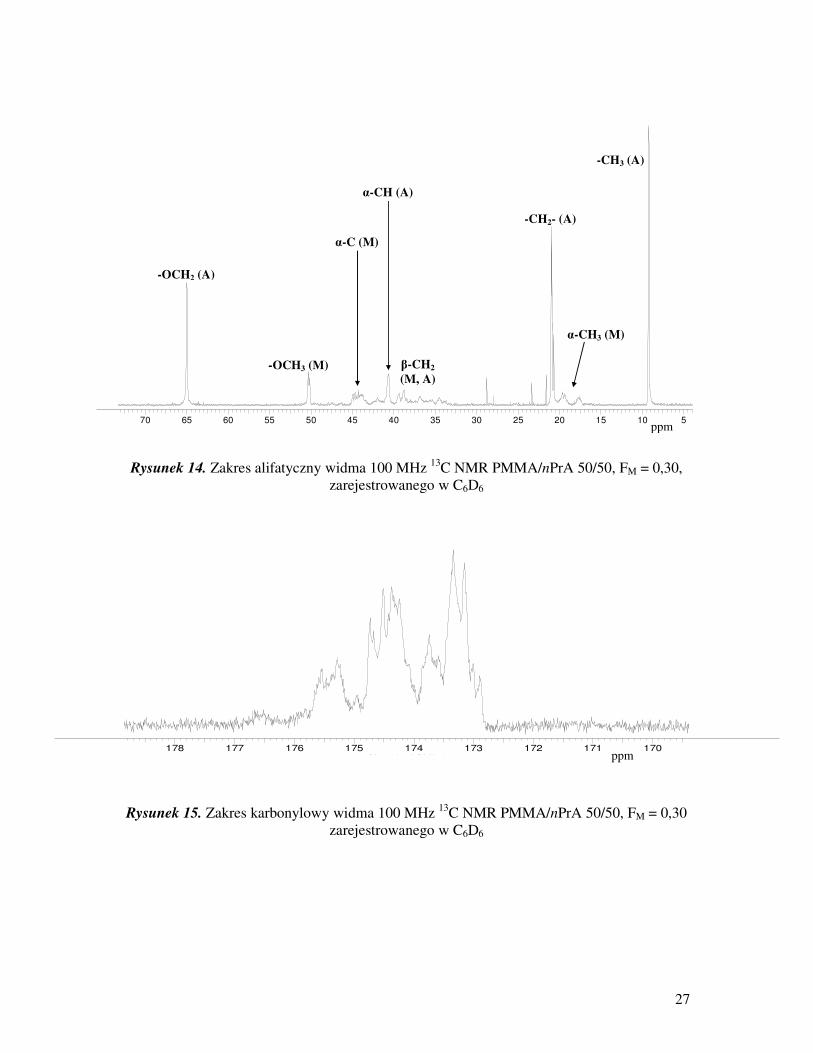
27
Rysunek 14. Zakres alifatyczny widma 100 MHz 13
C NMR PMMA/nPrA 50/50, FM = 0,30,
zarejestrowanego w C6D6
Rysunek 15. Zakres karbonylowy widma 100 MHz 13
C NMR PMMA/nPrA 50/50, FM = 0,30
zarejestrowanego w C6D6
α-CH3 (M)
β-CH2
(M, A)
α-CH (A)
70 65 60 55 50 45 40 35 30 25 20 15 10 5Chemical Shift (ppm) ppm
-CH3 (A)
-CH2- (A)
-OCH2 (A)
-OCH3 (M)
α-C (M)
178 177 176 175 174 173 172 171 170Chemical Shift (ppm) ppm
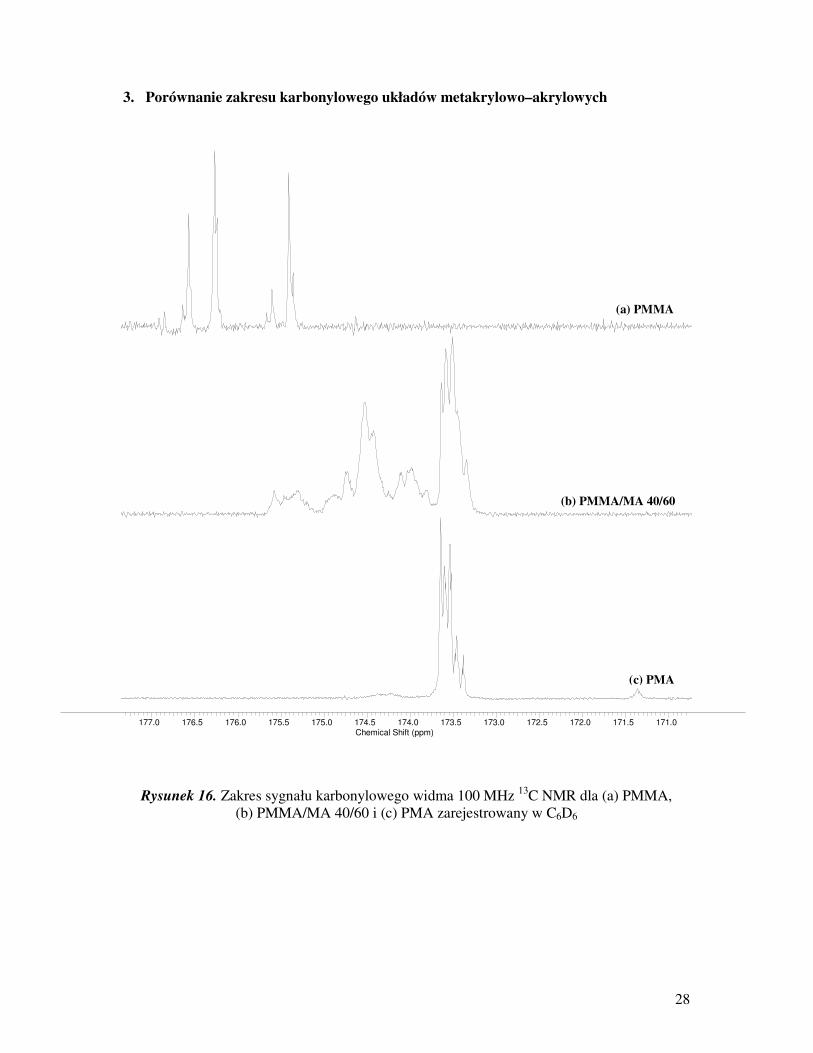
28
3. Porównanie zakresu karbonylowego układów metakrylowo–akrylowych
Rysunek 16. Zakres sygnału karbonylowego widma 100 MHz 13
C NMR dla (a) PMMA,
(b) PMMA/MA 40/60 i (c) PMA zarejestrowany w C6D6
177.0 176.5 176.0 175.5 175.0 174.5 174.0 173.5 173.0 172.5 172.0 171.5 171.0Chemical Shift (ppm)
(a) PMMA
(b) PMMA/MA 40/60
(c) PMA
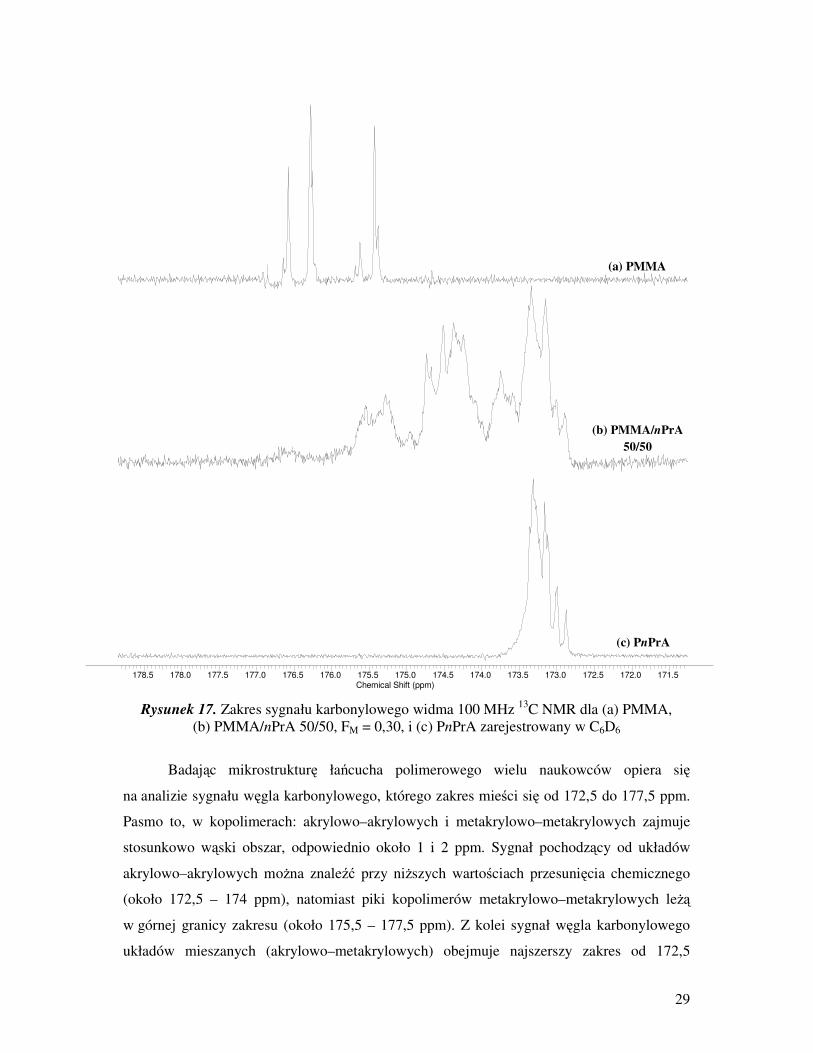
29
Rysunek 17. Zakres sygnału karbonylowego widma 100 MHz 13
C NMR dla (a) PMMA,
(b) PMMA/nPrA 50/50, FM = 0,30, i (c) PnPrA zarejestrowany w C6D6
Badając mikrostrukturę łańcucha polimerowego wielu naukowców opiera się
na analizie sygnału węgla karbonylowego, którego zakres mieści się od 172,5 do 177,5 ppm.
Pasmo to, w kopolimerach: akrylowo–akrylowych i metakrylowo–metakrylowych zajmuje
stosunkowo wąski obszar, odpowiednio około 1 i 2 ppm. Sygnał pochodzący od układów
akrylowo–akrylowych moŜna znaleźć przy niŜszych wartościach przesunięcia chemicznego
(około 172,5 – 174 ppm), natomiast piki kopolimerów metakrylowo–metakrylowych leŜą
w górnej granicy zakresu (około 175,5 – 177,5 ppm). Z kolei sygnał węgla karbonylowego
układów mieszanych (akrylowo–metakrylowych) obejmuje najszerszy zakres od 172,5
178.5 178.0 177.5 177.0 176.5 176.0 175.5 175.0 174.5 174.0 173.5 173.0 172.5 172.0 171.5Chemical Shift (ppm)
(a) PMMA
(b) PMMA/nPrA
50/50
(c) PnPrA
30
do 177,5 ppm, łącznie z zakresami wartości przesunięcia chemicznego dla kopolimerów
akrylowo–akrylowych i metakrylowo–metakrylowych [17].
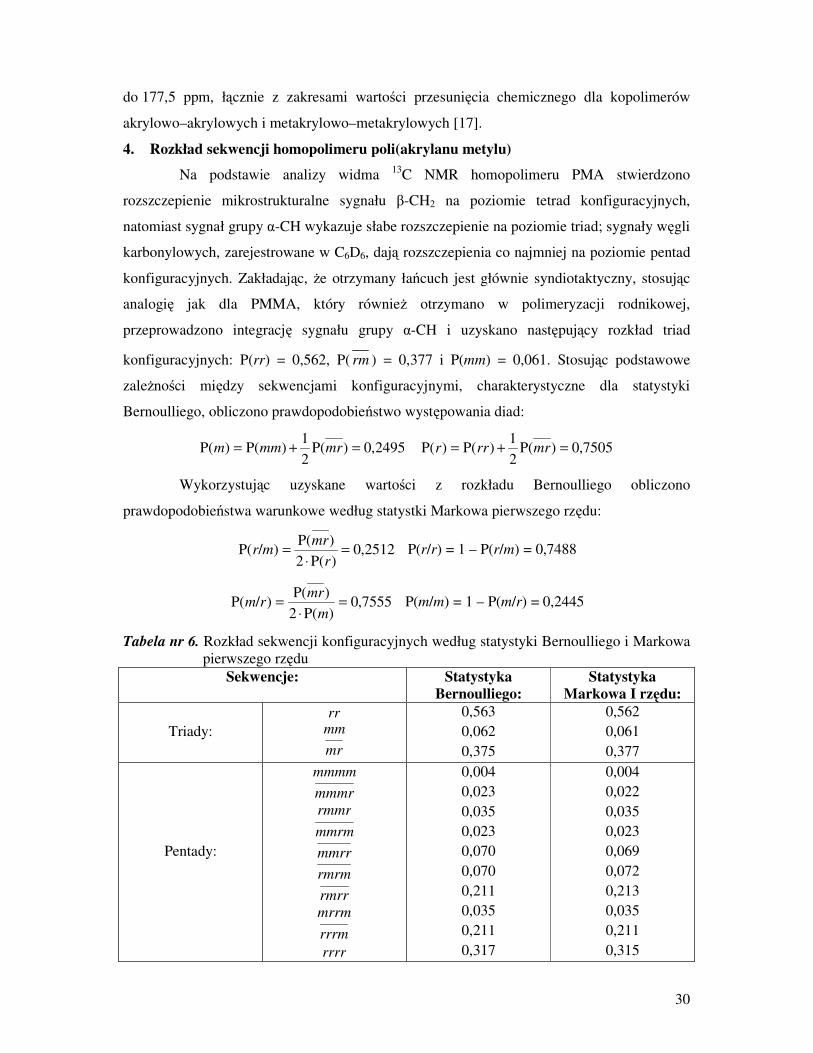
4. Rozkład sekwencji homopolimeru poli(akrylanu metylu)
Na podstawie analizy widma 13
C NMR homopolimeru PMA stwierdzono
rozszczepienie mikrostrukturalne sygnału β-CH2 na poziomie tetrad konfiguracyjnych,
natomiast sygnał grupy α-CH wykazuje słabe rozszczepienie na poziomie triad; sygnały węgli
karbonylowych, zarejestrowane w C6D6, dają rozszczepienia co najmniej na poziomie pentad
konfiguracyjnych. Zakładając, Ŝe otrzymany łańcuch jest głównie syndiotaktyczny, stosując
analogię jak dla PMMA, który równieŜ otrzymano w polimeryzacji rodnikowej,
przeprowadzono integrację sygnału grupy α-CH i uzyskano następujący rozkład triad
konfiguracyjnych: P(rr) = 0,562, P( rm ) = 0,377 i P(mm) = 0,061. Stosując podstawowe
zaleŜności między sekwencjami konfiguracyjnymi, charakterystyczne dla statystyki
Bernoulliego, obliczono prawdopodobieństwo występowania diad:
2495,0)P(2
1)P()P( =+= mrmmm 7505,0)P(
2
1)P()P( =+= mrrrr
Wykorzystując uzyskane wartości z rozkładu Bernoulliego obliczono
prawdopodobieństwa warunkowe według statystki Markowa pierwszego rzędu:
2512,0)P(2
)P()/P( =
⋅
=
r
mrmr P(r/r) = 1 – P(r/m) = 0,7488
7555,0)P(2
)P()/P( =
⋅
=
m
mrrm P(m/m) = 1 – P(m/r) = 0,2445
Tabela nr 6. Rozkład sekwencji konfiguracyjnych według statystyki Bernoulliego i Markowa
pierwszego rzędu
Sekwencje: Statystyka Bernoulliego:
Statystyka Markowa I rzędu:
Triady:
rr mm
mr
0,563
0,062
0,375
0,562
0,061
0,377
Pentady:
mmmm
mmmr
rmmr
mmrm
mmrr
rmrm
rmrr
mrrm
rrrm
rrrr
0,004
0,023
0,035
0,023
0,070
0,070
0,211
0,035
0,211
0,317
0,004
0,022
0,035
0,023
0,069
0,072
0,213
0,035
0,211
0,315
31
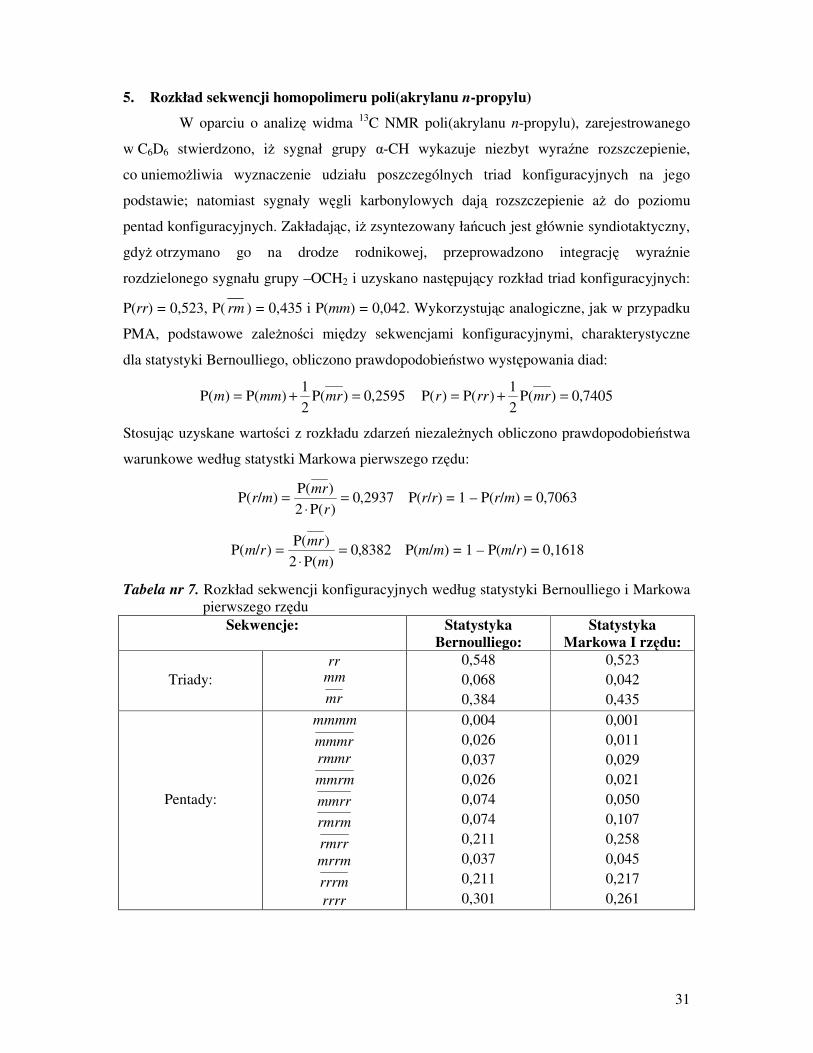
5. Rozkład sekwencji homopolimeru poli(akrylanu n-propylu)
W oparciu o analizę widma 13
C NMR poli(akrylanu n-propylu), zarejestrowanego
w C6D6 stwierdzono, iŜ sygnał grupy α-CH wykazuje niezbyt wyraźne rozszczepienie,
co uniemoŜliwia wyznaczenie udziału poszczególnych triad konfiguracyjnych na jego
podstawie; natomiast sygnały węgli karbonylowych dają rozszczepienie aŜ do poziomu
pentad konfiguracyjnych. Zakładając, iŜ zsyntezowany łańcuch jest głównie syndiotaktyczny,
gdyŜ otrzymano go na drodze rodnikowej, przeprowadzono integrację wyraźnie
rozdzielonego sygnału grupy –OCH2 i uzyskano następujący rozkład triad konfiguracyjnych:
P(rr) = 0,523, P( rm ) = 0,435 i P(mm) = 0,042. Wykorzystując analogiczne, jak w przypadku
PMA, podstawowe zaleŜności między sekwencjami konfiguracyjnymi, charakterystyczne
dla statystyki Bernoulliego, obliczono prawdopodobieństwo występowania diad:
2595,0)P(2
1)P()P( =+= mrmmm 7405,0)P(
2
1)P()P( =+= mrrrr
Stosując uzyskane wartości z rozkładu zdarzeń niezaleŜnych obliczono prawdopodobieństwa
warunkowe według statystki Markowa pierwszego rzędu:
2937,0)P(2
)P()/P( =
⋅
=
r
mrmr P(r/r) = 1 – P(r/m) = 0,7063
8382,0)P(2
)P()/P( =
⋅
=
m
mrrm P(m/m) = 1 – P(m/r) = 0,1618
Tabela nr 7. Rozkład sekwencji konfiguracyjnych według statystyki Bernoulliego i Markowa
pierwszego rzędu
Sekwencje: Statystyka
Bernoulliego:
Statystyka
Markowa I rzędu:
Triady:
rr mm
mr
0,548
0,068
0,384
0,523
0,042
0,435
Pentady:
mmmm
mmmr
rmmr
mmrm
mmrr
rmrm
rmrr
mrrm
rrrm
rrrr
0,004
0,026
0,037
0,026
0,074
0,074
0,211
0,037
0,211
0,301
0,001
0,011
0,029
0,021
0,050
0,107
0,258
0,045
0,217
0,261
32
Glass Transi tionOnset 117,28 °CMidpoint 122,89 °C
!]5[Marcin pr. PMMAMarcin pr. PMMA, 3,1600 mg
Wg^-1
0,5
°C-60 -40 -20 0 20 40 60 80 100 120 140 160 180 200
exoexoexoexo
SSSSTATATATARRRReeee SW 9.01 SW 9.01 SW 9.01 SW 9.01Lab: METTLERLab: METTLERLab: METTLERLab: METTLER
Glass Transi tionOnset -12,31 °CMidpoint -6,04 °C
!]5[Mar cin PMA powt.Marcin PMA powt., 18,3600 mg
Wg^-1
0,5
°C-60 -50 -40 -30 -20 -10 0 10 20 30 40 50 60 70
exoexoexoexo
SSSSTATATATARRRReeee SW 9.01 SW 9.01 SW 9.01 SW 9.01Lab: METTLERLab: METTLERLab: METTLERLab: METTLER
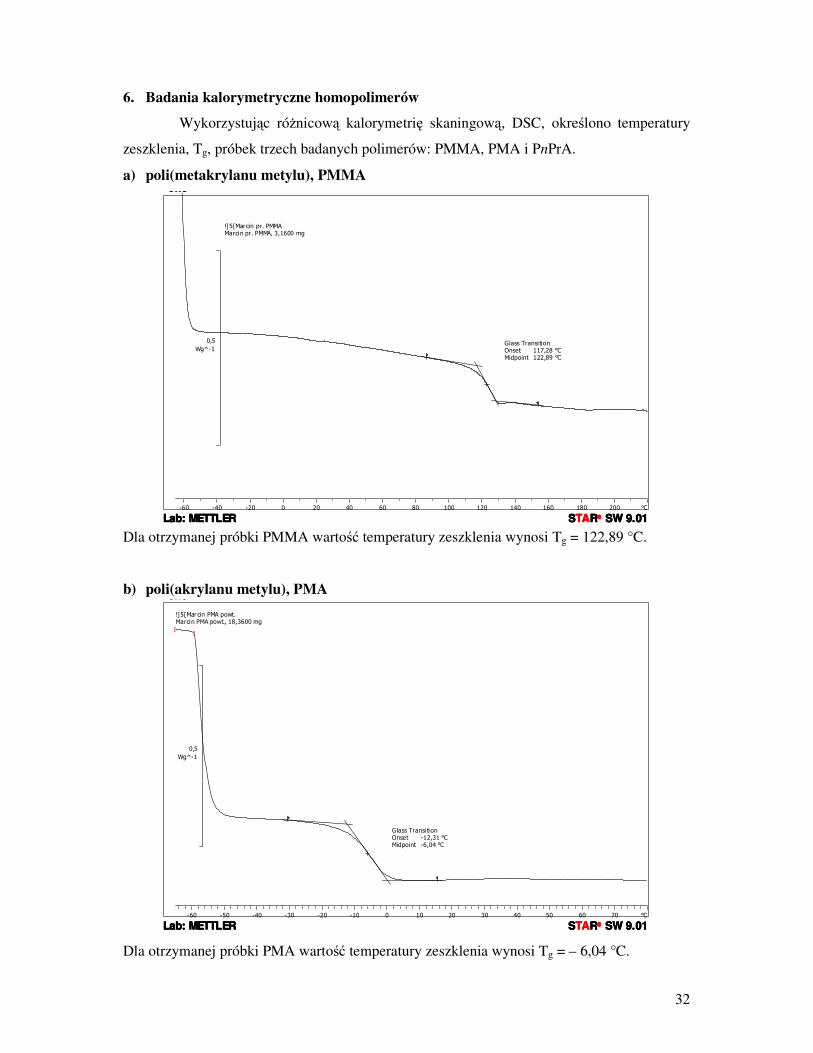
6. Badania kalorymetryczne homopolimerów
Wykorzystując róŜnicową kalorymetrię skaningową, DSC, określono temperatury
zeszklenia, Tg, próbek trzech badanych polimerów: PMMA, PMA i PnPrA.
a) poli(metakrylanu metylu), PMMA
Dla otrzymanej próbki PMMA wartość temperatury zeszklenia wynosi Tg = 122,89 °C.
b) poli(akrylanu metylu), PMA
Dla otrzymanej próbki PMA wartość temperatury zeszklenia wynosi Tg = – 6,04 °C.

33
Glass TransitionOnset -35,45 °CMidpoint -33,45 °C
!]5[Pasich pr. Pn-PrAPasich pr. Pn-PrA, 9,5200 mg
Wg^-1
1
°C-60 -40 -20 0 20 40 60 80 100 120 140 160 180
exoexoexoexo
SSSSTATATATARRRReeee SW 9.01 SW 9.01 SW 9.01 SW 9.01Lab: METTLERLab: METTLERLab: METTLERLab: METTLER
c) poli(akrylanu n-propylu), PnPrA
Wartość temperatury zeszklenia PnPrA wynosi Tg = – 35,45 °C.

34
PODSUMOWANIE I WNIOSKI
1. Przeprowadzono syntezy wybranych kopolimerów metakrylanu metylu z akrylanem
metylu i akrylanem n-propylu oraz odpowiednich homopolimerów z uŜyciem inicjatora
rodnikowego, 2,2’-azobis(izobutyronitrylu). Zarejestrowano ich widma 13
C NMR;
wytypowano sygnały rezonansowe czułe na efekty mikrostrukturalne oraz dokonano
pomiarów temperatury zeszklenia, Tg, otrzymanych homopolimerów.
2. Na podstawie widm 1H NMR wyznaczono rzeczywiste udziały molowe komonomerów
dla poli(metakrylanu metylu-co-akrylanu n-propylu), natomiast wykorzystując widma 13
C NMR określono rzeczywiste składy dla poli(metakrylanu metylu-co-akrylanu metylu).
3. Wyznaczono współczynniki reaktywności (r1 i r2) dla nowo otrzymanego kopolimeru
metakrylanu metylu z akrylanem n-propylu uzyskując wartości zbliŜone do analogicznego
układu poli(metakrylanu metylu-co-akrylanu n-butylu).
4. Zastosowanie deuterowanego chloroformu będącego typowym rozpuszczalnikiem
w badaniach mikrostrukturalnych układów akrylowych pozwala na obserwacje rozszczepień konfiguracyjnych pochodzących tylko od metakrylanu metylu, natomiast nie widać takich
rozszczepień w sygnałach obu akrylanów. Zastosowanie deuterowanego benzenu pozwoliło
na obserwacje efektów konfiguracyjnych pochodzących równieŜ od jednostek akrylowych.
5. Widma 13
C NMR wybranych kopolimerów metakrylanu metylu z akrylanem metylu
i akrylanem n-propylu wykazują czułość na efekty kompozycyjne i konfiguracyjne, wywołane
zarówno przez jednostki metakrylanu metylu, jak i obu akrylanów.
6. Na podstawie sygnału α-CH określono rozkład triad konfiguracyjnych dla poli(akrylanu
metylu), a w przypadku poli(akrylanu n-propylu) wykorzystano sygnał grupy –OCH2.
Korzystając z relacji pomiędzy intensywnościami sekwencji róŜnej długości obliczono
rozkład diad oraz pentad, które posłuŜą do symulacji sygnałów karbonylowych otrzymanych
homopolimerów.

35
LITERATURA CYTOWANA
[1] Florjańczyk Z., Penczek S. (red.) praca zbiorowa „Chemia polimerów” tom I; Oficyna
Wydawnicza Politechniki Warszawskiej, Warszawa 2001,
[2] Koenig J.L. “Spectroscopy of Polymers”; American Chemical Society, Washington
1992,
[3] Bovey F.A., Mirau P.A. “NMR of Polymers”; Academic Press; San Diego 1996,
[4] Tonelli A.E. “NMR Spectroscopy and Polymer Microstructure: The Conformational
Connection”; VCH, New York 1989,
[5] Matlengiewicz M. „Zastosowanie spektroskopii 13
C NMR do charakteryzowania
wieloskładnikowych mieszanin petro- i karbochemicznych oraz mikrostruktury polimerów
akrylowych”; Wydawnictwo Fundacji im. Wojciecha Świętosławskiego, Gliwice 1997,
[6] Stevens P.M. „Wprowadzenie do chemii polimerów”; PWN, Warszawa 1983,
[7] Fischer H., Hendra P., Hummel D.O. i in. “Polymer Spectroscopy”; Verlag Chemie,
Weinheim 1974,
[8] Florjańczyk Z., Penczek S. (red.) praca zbiorowa „Chemia polimerów” tom II; Oficyna
Wydawnicza Politechniki Warszawskiej, Warszawa 2002,
[9] Aerdts A.M., German A.L., van der Velden G.P.M. “Determination of the Reactivity
Ratios, Sequence Distribution and Stereoregularity of Butyl Acrylate-Methyl Methacrylate
Copolymers by Means of Proton and Carbon-13 NMR”; Magn. Reson. Chem., 32, S80
(1994),
[10] Chmielewski A., śurakowska–Orszagh J., Makulski W. „Zastosowanie spektroskopii 13
C NMR z transformacją Fouriera do badania struktury kopolimerów winylowych”;
Polimery, 39, 142 (1994),
[11] Suchopárek M., Spěváček J. “Characterization of the Stereochemical Structure
of Poly(tert-butyl acrylate) by One- and Two Dimensional NMR Spectroscopy”;
Macromolecules, 26, 102 (1993),
[12] De la Fuente J.L., Fernandez-Garcia M., Fernandez-Sanz M., Madruga E.L.
“Sequence Distribution and Stereoregularity of Methyl Methacrylate and Butyl Acrylate
Statistical Copolymers Synthesized by Atom Transfer Radical Polymerization”;
Macromolecules, 34, 5833 (2001),
[13] Kim Y., Harwood J. “Analysis of sequence distribution in methyl methacrylate-methyl
acrylate copolymers by 13
C NMR spectroscopy”; Polymer, 43, 3229 (2002),
[14] Dhal P.K., Babu G.N., Nanda R.K. “Microstructure Elucidation of Glycidyl
Methacrylate-Alkyl Acrylate Copolymers by 13
C NMR Spectroscopy”; Macromolecules, 17,
1131 (1984)
[15] Matlengiewicz M., Nguyen G., Nicole D., Henzel N. “Analysis of β-CH2 Signals in
the 13
C NMR Spectra of the Methyl Methacrylate-Ethyl Acrylate Copolymer as a Tool for
Microstructure Determination”; J. Polym. Sci.: Part A: Polym. Chem., 38, 2147 (2000),
[16] Brar A.S., Hooda S., Kumar R. “Compositional and Configurational Sequence
Determintion of Methyl Methacrylate/Ethyl Acrylate Copolymers by One- And Two-
Dimensional Nuclear Magnetic Resonance Spectroscopy”; J. Polym. Sci.: Part A: Polym.
Chem., 41, 313 (2003),
[17] Matlengiewicz M. “Pentad Distribution of Acrylic Copolymers by 13
C NMR
Spectroscopy”; Polish J. Chem., 71, 947 (1997),
[18] Nguyen G., Nicole D., Swistek M., Matlengiewicz M., Wiegert B. “Sequence
distribution of the methyl methacrylate-ethyl acrylate copolymer by 13
C n.m.r. spectroscopy”;
Polymer, 38 (1997),

36
[19] Nguyen G., Matlengiewicz M., Nicole D. “Incremental method for determination
of sequence distribution of poly(methyl methacrylate) by 13
C NMR spectroscopy”; Analusis,
27, 847 (1999),
[20] Matlengiewicz M. “Heptad distribution of the syndiotactic poly(methyl methacrylate)
(PMMA)”; Analusis, 24, 60 (1996),
[21] Bujak P., Matlengiewicz M., Henzel N. “Incremental Calculation of Sequence
Distribution of Poly(Butyl Acrylate)”; International Journal of Polymer Anal. Charact.,12, 95
(2007),
[22] Bujak P., Henzel N., Matlengiewicz M. “Microstructure Study of Poly(tert-butyl
acrylate by 13
C NMR Spectroscopy”; International Journal of Polymer Anal. Charact.,12,
431 (2007),
[23] Pielichowski J.; Puszyński A. „Chemia polimerów”; Wydawnictwa Naukowo–
Techniczne „Teza”, Kraków 2004,
[24] Bujak P., Henzel N., Matlengiewicz M. “Microstructure Study of Methyl
Methacrylate/n-Butyl Acrylate Copolymer by 13
C NMR Spectroscopy”; International Journal
of Polymer Anal. Charact.,13, 149 (2008)

37
DOROBEK NAUKOWY:
� Publikacje:
1. P. Bujak, M. Matlengiewicz, M. Pasich, N. Henzel,
Microstructure of methyl methacrylate/tert-butyl acrylate copolymer characterized
by 13
C NMR spectroscopy,
Polym. Bull., 64(3), 259–273 (2010)
2. M. Matlengiewicz, M. Pasich, P. Bujak, N. Henzel,
Microstructure Analysis of Acrylate–Methacrylate Copolymers by 13
C NMR Spectroscopy,
Int. J. Polym. Anal. Charact., 14(8), 686–94 (2009)
3. M. Pasich, M. Matlengiewicz,
Zastosowanie spektroskopii magnetycznego rezonansu jądrowego do badania mikrostruktury
układów metakrylowo–akrylowych,
Zeszyty Naukowe Politechniki Rzeszowskiej – Chemia, 20, 111–115 (2009)
� Udział w konferencjach naukowych:
1. P. Bujak, M. Pasich, M. Maślankiewicz, M. Matlengiewicz,
Characterization of the Stereochemical Structure of Poly(tert-Butyl Acrylate) by 13
C NMR
Spectroscopy,
XLIX Zjazd Naukowy Polskiego Towarzystwa Chemicznego, Paper no. S7–P6
18 – 22 wrzesień 2006, Gdańsk
2. M. Matlengiewicz, M. Pasich,
Application of NMR Spectroscopy to the Analysis of Microstructure of Methyl
Methacrylate – Methyl Acrylate Copolymer”
8th
International Conference: Advances in Coatings Technology, ACT’08
Conference papers (ISBN 978-83-917693-8-6), Paper no. 51
25 – 27 November 2008, Warszawa
3. M. Pasich, M. Matlengiewicz,
Zastosowanie spektroskopii magnetycznego rezonansu jądrowego do badania mikrostruktury
układów metakrylowo–akrylowych,
XIV Profesorskie Warsztaty Naukowe: „Przetwórstwo Tworzyw Polimerowych”
15 – 17 czerwiec 2009, Krasiczyn
4. M. Matlengiewicz, M. Pasich, P. Bujak, N. Henzel
Microstructure Analysis of Acrylate – Methacrylate Copolymers by 13
C NMR Spectroscopy
22nd
International Symposium on Polymer Analysis and Characterization, ISPAC’09
Conference Paper no. 5
22 – 24 June 2009, Zlin, Czech Republic
5. M. Matlengiewicz, M. Pasich,
NMR Study of Poly(Propyl Acrylate),
8th
International Conferences: Advances in Plastics Technology, APT’09
Conference papers (ISBN 978-83-917693-9-3), Paper no. 30
03 – 05 November 2009, Katowice

38
śYCIORYS:
� Dane personalne:
Imię i nazwisko: Marcin PASICH
Data i miejsce urodzenia: 03.03.1982, Będzin
Adres: ul. Chopina 7/15
41-300 Dąbrowa Górnicza
� Wykształcenie:
2007 – obecnie Uniwersytet Śląski w Katowicach
Wydział Matematyki, Fizyki i Chemii – studia doktoranckie
2001 – 2006 Uniwersytet Śląski w Katowicach
Wydział Matematyki, Fizyki i Chemii
kierunek: chemia ogólna
temat pracy magisterskiej: „Badanie mikrostruktury
kopolimerów metakrylanu metylu i akrylanu tert-butylu
z wykorzystaniem spektroskopii 13
C NMR”
1997 – 2001 II Liceum Ogólnokształcące im. Stanisława Wyspiańskiego
w Będzinie
� Praca zawodowa:
2007 – obecnie Instytut InŜynierii Materiałów Polimerowych i Barwników
w Toruniu; Oddział Zamiejscowy Farb i Tworzyw
w Gliwicach
stanowisko – asystent
� Znajomość języków obcych:
– język angielski: praktyczny
– język francuski: podstawowy