Embed Size (px)
Citation preview

Boron trihalide adducts of 1,l-bis(piperidiny1)ethylene. 13C and "B nmr studies of a model system for fJ-carbon donation in enamine adducts
J. STEPHEN HARTMAN' AND ERIC C. KELUSKY Department of Chemistry, Brock University, St. Catharines. Ont., Canada L2S 3A1
Received September 9, 1980
J. STEPHEN HARTMAN and ERIC C. KELUSKY. Can. J. Chern. 59, 1284 (1981). 1,1-Bis(piperidiny1)ethylene coordinates to the boron trihalides through the CH, carbon, but the covalent adducts D.BX3 undergo
rearrangement to give predominantly the ionic adducts D,BX,+.BX,-. Striking variations in "B and 13C nmr peak widths support our proposed structures. The disappearance of peaks due to excessive broadening can be misleading in the interpretation of such systems.
J. STEPHEN HARTMAN et ERIC C. KELUSKY. Can. J. Chem. 59, 1284 (1981). Le bis(pipkridiny1)-1,1 ethyltne se coordonne aux trihalogknures de bore au moyen de I'atorne de carbone du CH,, mais les
adduits covalents obtenus, D.BX3, se transposent en donnant rnajoritairement les adduits ioniques D,BX,+.BX,-. Les fortes varia- tions observees dans les spectres de rmn du 13C et du "B sernblent confirmer les structures que nous proposons. La disparition des pics, due a un etalement excessif, peut conduire a une interprktation erronke de ces systemes.
[Traduit par le journal]
Introduction systems, in particular confirmation that P-carbon
The well-known stability of the amidinium ion donation occurs, has been hampered by the scar-
structure 1 (I) leads to the expectation of exclusive city of nmr data On carbon-donor boron trihalide I P-carbon protonation and complexation in 1, l-ene- adducts (7, 8). We now report 13C and lLB nmr
diamines (2). This is well established for protons- studies of boron trihalide coordination of 1,l-bis- (piperidiny1)ethylene (4), a 1,l-enediamine and a
I I model compound for P-carbon donation to Lewis /"\ /N\ acids. + ;C-R C=CH2 \N4' \N/a Results and Discussion
I I 13C Spectra 1 2 13C nmr parameters of 4 and its boron trihalide
tion (2). The few available studies of complexation adducts are given in Table 1- The P-carbon of 1,l-enediamines to simple Lewis acids (boron uncom~lexed 4 absorbs exce~t ional l~ far to high trihalides (3) and Group IV tetrahalides (4)) also field for an olefinic carbon, and this can be attrib- show that complexation is at the P-carbon site. In uted to the importance of the mesomeric forms 4b the boron trihalide adducts of 1,l-bis(dimethy1- and 4c, with the buildup of appreciable negative amino)ethylene (3), there is restricted rotation charge on this carbon. Similar but less pronounced about the central C-N bonds as a result of the effects occur in enamines (5,9). Changes in chemi- amidinium-ion partial double bond character (3). cal shift on complex formation are consistent with
In our current nmr studies of enamine adducts the P-carbon being the donor atom. Structures 4b ( 9 , we are attempting to detect ambidentate behav- and 4c should be the only mes0me1-i~ forms con- iour of the enamine ligands, in which both nitrogen tributing to the adducts, due to the localization of a and the P-carbon are potential donor sites (6). lone pair at the donor position on adduct formation. Interpretation of results in the enamine adduct The pronounced shift to high field of the P-carbon
Me on complexation to BF3 is consistent with the
I (2 required hybridization change to sp3. The shift ~e'~\ \ back to lower field in the heavier-halogen boron
+ \C-CH~BX~ trihalide adducts is consistent with the increasing ~e ,~* / cJ'c=cH2 Lewis acid strength of these compounds and the
I nearness of the BX, moiety. The 6ppm chemical Me shift difference between the BF, and BCl, adduct
3 4 P-carbon resonances is consistent with the 2.6 ppm difference between the 13C resonances of Me3N.BF3
'Author to whom correspondence should be addressed. and Me3N.BC13 (lo), and the 3-7ppm between 0008-404218 1108 1284-06$0 1.00/0
01981 National Research Council of CanadaIConseil national de recherches du Canada
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CO
NC
OR
DIA
UN
IV o
n 11
/12/
14Fo
r pe
rson
al u
se o
nly.

HARTMAN AND KELUSKY
TABLE 1 . 13C nmr parametersa of adducts of 1 ,1-bis(piperidiny1)ethylene
Chemical shifts T Width at half height
(K) C-a c-8 C-2,6 C-3,5 C-4 of the C-B ~ e a k (Hz)b
Free 4
4/BF3
Sharp Sharp
Sharp Sharp Sharp
Ambient Ambient
"Chemical shifts are in ppm to low field of internal tetrameth Isilane. 20% solutions of 4 in deuterochloroform. 'Spectra were transformed with 5 Hz of line broadening. peais desciibed as "sharp" were not appreciably broader than the TMS peak under these conditions. <Not vicihle . - - . . -. - . - . *Also present in many adduct samples 'Reference 25.
a-carbon resonances of BF, and BCl, adducts of ethers (1 1).
The a-carbon resonance shifts to low field in the adducts and in protonaled 4 (Table 1). The chemi- cal shift of 168.7ppm for protonated 4 is consistent with values for other amidinium ions ([(Me2N)2CII]+, 156.4ppm (12), [(Me2N)(C6H5NH)CCl]+, 159ppm (13)). However correlations of C=N+ 13C chemi- cal shifts are generally poor, the nature of the anion having a strong influence, which can be as much as lOppm (12). The a-carbon shifts in the adducts (172- 178 ppm, and temperature-dependent) are close to the amidinium ion range. Doubling of the adduct
a-carbon resonance at low temperatures in some cases indicates additional complexity in the adduct systems, as is discussed below. The small shift to low field of the piperidinyl C-2 and C-6 carbons is consistent with adduct formation at a donor site remote from these carbons.
Peak widths of the resonances assigned to the P-carbons are given in Table 1. The resonances are broad or even unobservable at ambient tempera- ture, but become sharper as the temperature is lowered. This is consistent with coupling to "B which is undergoing quadrupole relaxation. Qua- drupole relaxation is more effective at lower tern- peratures, due to slower molecular tumbling. This causes a more complete collapse of the splittings due to "B-13C coupling, and of any residual peak broadening due to such coupling, at lower tempera- tures in the spectrum of the spin-112 nucleus coupled to the quadrupolar nucleus (14). Thus spectra of spin-112 nuclei coupled to boron are often simplified at low temperatures (7). In a
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CO
NC
OR
DIA
UN
IV o
n 11
/12/
14Fo
r pe
rson
al u
se o
nly.

1286 CAN. J . CHEM. VOL. 59, 1981
TABLE 2. I1B nmr parameters of 4/BX3 solutionsa
T (K) B&- D.BX, DZBX,+ Other
4 + BI, 253 -145.3 b b
273 - 146.0 b b
323 - 146.2 -74c -38' - 15'
4 + BI, + Me4N+I- 293 - 146.2 b b
323 - 146.2 -78(320 HZ) -36(>500HzC) - 16(>500 Hzc)
4 + BBr, 243 -42.4 -29.1(35 Hz) b
273 -42.9 -29.4(30 HZ) d
323 -42.7 -29.4(15 Hz) -28.1(280Hz)
4 + BCI, 253 -11.3 d
273 -11.3 - 12(-500 Hz)' 303 -11.3 - 12( 270 Hz)' 323 I I
4 + BF, 243 - 19.5 d
263 - 19.6 - 17.8(220 HZ)' 293 -19.7 - 17.0(210H~)~ 303 - 19.6 - 16.4(160Hz)' 323 - 19.5 o
"Chenlical shifts are in ppm to low field of external trimethox boron, (To converl to ppm to low field of boron trifluoride etherate, add 18.1 ppm.) Values In brackets are peak widths at half-height. The BX,- peai remalns quite sharp at all temperatures.
bNot visible. <Broad and badly overlapping peaks. dlnsufficienc resolution. eBroad resonance. likely a composite of D.BX, and DIBXIC. 'Speclrum is complex, apparently due to decomposition. 'Obscured by the BF,- peak and by poorly-resolved fine structure which is probably due to boron-fluorine coupling.
number of cases a carbon directly bonded to boron is undetectable at ambient temperature due to excessive broadening from coupling to llB, but does give a peak at low temperatures (15). Thus our I3C peak widths support our P-carbon peak as- signments and the proposed adduct structure.
Splittings due to llB-13C coupling can usually be seen in 13C nmr spectra of tetrahedral boron compounds with C-B bonds, but these splittings are often not visible in three-coordinate boron compounds (7). This is in accord with the expecta- tion that quadrupole relaxation of boron should be less effective in tetrahedral boron compounds due to their higher symmetry (14). The absence of such splittings in the 13C spectra of the present series of adducts is thus somewhat unusual. llB chemical shifts are not consistent with three-coordinate boron. The effect of the bulky enediamine donor in slowing molecular tumbling is probably responsi- ble for the effectiveness of "B quadrupole relaxa- tion in these adducts, leading to I3C and l lB peak widths more typical of trigonal boron than of tetrahedral boron.
I1B Spectra Solutions of 4 with the boron trihalides in a 1:l
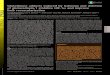
molar ratio or with excess 4 give more than one I1B nmr signal, and the signals have a very wide range of peak widths (Table 2 and Fig. 1). Uncomplexed boron trihalide peaks, which would absorb at low
-30 -40 -30 -40 - 30 -LO ppm
FIG. 1. I1B nmr spectra of 4.BBr3 at various temperatures. The spectra were transformed with 5Hz of line broadening, slightly increasing the apparent width of the BBr4- signal.
field, are absent, as are resonances in the RBX2 region which is at even lower field (8). The spectra are consistent with adduct formation followed by redistribution of donor for halogen to give the ionic form of the adducts, D2BX2+.BX4-.
[l] 2D.BX3 * D2BX2+ + BX4-
Comparison with literature values (16-18)2 of l lB chemical shifts of adducts and related species
3 e e also Table IV of ref. 8.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CO
NC
OR
DIA
UN
IV o
n 11
/12/
14Fo
r pe
rson
al u
se o
nly.

HARTMAN AND KELUSKY
TABLE 3. "B chemical shifts of model compoundsa
X F C1 Br I Reference
BX4- - 19.9 -11.6 -42.4 - 146.0 16 Me3N.BX3 - 15.9 -8.6 -20.9 -7 1.4 17
I Me2S.BX3 - 15.2 -11.2 -29.5 -87.0 18 . . . . BX3 -8.6- -6.5 28-30 20-22 . . . . . . . . . . . . . .
-24- -26 . i
See ref. 8, Table 4 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
'In ppm to low field of trimethoxyboron.
(Table 3) shows that the sharpest peak in the l lB spectra of the 4/BX3 samples corresponds to the tetrahaloborate anion BX4-. From the size of the peak, BX4- is a major species. Boron cations such as D2BX2+ are well-known species (19,20) but llB chemical shifts have been reported for few halo- boron cations (8).
In the 4/BBr3 and 4/B13 systems, one broad resonance has a chemical shift consistent with the values for known simple boron trihalide adducts (Table 3) and is assigned to 4.BX3. An additional, even broader resonance occurs at lower field, and we have assigned this to D2BX2+. One further very broad resonance is present in the 4.B13 system and might arise from a species such as D3B12+. Prece- dent exists for such species, which are most easily formed when iodines, having the weakest boron- halogen bonds, are available to be displaced (19). Supporting evidence for our assignments comes from addition of tetramethylammonium iodide to solutions of 4 and BI,. The size of the initially-small -78ppm peak, assigned to 4.B13, is greatly in- creased relative to both of the lower-field peaks, in accord with reactions such as [2].
[2] D2BIZt + I- = D.B13 + D
In the lB spectrum of the 4/BBr3 system (Fig. 1) the resonance of intermediate chemical shift and peak width that is assigned to D.BBr3 is small, consistent with equilibrium [I] being far to the right. The very slight chemical shift difference between the broad resonances assigned to D.BBr3 and D2BBr2+, together with the small range of chemical shifts of B-Br compounds when com- pared to analogous B-I compounds (Table 3), makes it unlikely that any further broad resonance signals, such as might arise from D3BBr2+, would be detectable. The broadest resonance could be a composite signal, rather than solely due to D2BBr2+.
"B peak areas are not consistent with D2BX2+ and BX,- as the only ions in solution. The peaks assigned to cations are considerably larger than those assigned to BX4- ions. Halide ion is another possible counterion for D2BX2+ (19,20) and would not be detected in this study.
The small llB chemical shift ranges in the 4/BC13 and 4/BF3 systems do not allow adequate resolu- tion of peaks in addition to BX4-. However, there is definitely a large broad resonance underlying the BX4- peak in each system. It becomes less broad at higher temperatures, but resolution into separate signals has not been possible. The existence of the broad, poorly resolved resonance does indicate that reactions analogous to those in the heavier boron trihalide adduct systems are occurring.
Peak widths and their variation with temperature are the most striking feature of the "B spectra. In contrast to the 13C spectra, some peaks became too broad to detect at the lowest temperatures, in keeping with very short lifetimes of the "B spin states due to more effective quadrupole relaxation at these temperatures (14). The variations of llB peak widths support our assignments. The tetra- haloborate ions, with full tetrahedral symmetry, give quite sharp peaks to the lowest temperatures studied, because the high electric field symmetry about boron decreases the effectiveness of quad- rupole relaxation. The adducts D.BX3, with only C3, symmetry about boron and also with molecular tumbling slowed due to the attachment of a bulky donor group, undergo more effective quadrupole relaxation and hence give much broader reso- nances. Since the boron is in a somewhat-flexible C-CHI-BX3 unit, considerable local tumbling should still be possible. D2BX2+ ions, with only C2, symmetry, should give the broadest peaks. The effect should be accentuated by the much slower tumbling of the boron-containing part of the mole- cule due to anchoring between two very bulky donor groups. These are indeed the broadest peaks.
Boron trihalide adduct systems can be far more complex than is frequently assumed; reaction [I] is one of several possible complications (21). llB chemical shifts of the tetrahaloborates confirm reaction [I], in accord with a recent suggestion (8). Nuclear magnetic resonance studies involving only donor-molecule nuclei can be misleading in that they give no direct evidence of this reaction. llB chemical shift and peak width studies should have
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CO
NC
OR
DIA
UN
IV o
n 11
/12/
14Fo
r pe
rson
al u
se o
nly.

1288 CAN. J . CHEM. VOL. 59, 1981
similar value in other adduct systems in which such complications may occur.
Further Aspects 1,l-Enediamines have low stabilities, especially
in the presence of Lewis acids, and this severely limits their usefulness as ligands. The present study has been limited by the tendency of many nrnr samples to give precipitates on standing, and a few features of the spectra have not been fully ex- plained. Thus 13C spectra show that protonated 4 is present in variable amounts in our samples. Possi- ble reactions of the initially-formed adducts include loss of an acidic P-hydrogen to give an organoboron enediamine, 5 or 6:
I
I /N\ ,~x3
\N/c=c\H + base .H+
I
I /N\ 7 x 2
\,/c=c\H + base .H+ X-
6
When the base in eqs. [3] and [4] is excess uncomplexed enediamine, protonated 4 would be formed. Such reactions have been reported for enediamine-group IV Lewis acid complexes by Weingarten and Wagar (4), and some evidence for similar reactions has been found for the adduct 3 (X = F) by 'H and 19F nrnr (3). Such reactions apparently occur to a significant extent on standing in methylene chloride solution at ambient tempera- ture for a few hours (22), consistent with the behaviour of carbon acids which often undergo relatively slow proton transfer (23). However, absence of the olefinic-carbon resonances expec- ted for 5 or 6 argues against this reaction being the source of protonated 4. Protonation might also
arise from traces of hydrogen halide present in the boron trihalide (24) or from DC1 which is often present in deuterochloroform, but this normally gives only traces of the protonated species.
Doubling of the 13C a-carbon resonances in some of the adduct spectra at low temperatures could be the result of ion pairing effects, since C=N+ shifts are quite anion-dependent (12). The absence of an equivalent doubling of the P-carbon resonances, together with the coalescence of the a-carbon resonances as the temperature is raised, excludes the possibility that the donor environments in D.BX, and D2BX2+ give rise to the separate a-carbon resonances. The boron-carbon bonds must be long-lived to account for the broadening of the adduct P-carbon resonances at higher tempera- tures, and hence a-carbon resonances from D.BX, and D2BX2+ would not coalesce. Ion pairing, on the other hand, would have the greatest effects on the a-carbon, and changes involving the counterion would be fast, consistent with the data. However, confirmation of such ion pairing effects awaits further work.
A 1,l-enediamine complex with platinum(I1) has recently been reported (25). Two isomers, an q2 (metal-olefin) complex and an q (P-carbon-donor) complex analogous to our adducts, were postulated on the basis of lH and I3C nrnr data. 13C chemical shifts for the q 1 complex are very similar to ours (Table 1) and an especially large 13C-195Pt coupling constant provides further evidence for the struc- ture. The inherent interest of 1,l-enediamines as carbon-donor "ylide" ligands merits further stud- ies of their coordination behaviour, in spite of difficulties arising from their high reactivity.
The readiness of boron trihalide adducts of 1,l-enediamines to rearrange according to eq. [ I ] throws light on our current studies of possible ambidentate behaviour of enamines as ligands to the boron trihalides. Initially-formed nitrogen- donor adducts of certain enamines convert to a second species, which 13C nrnr indicates is the P-carbon-donor adduct (5). However, llB nmr, showing only BX,- resonances for the second species, seemed inconsistent with the 13C results. Our present study, showing that carbon-donor BX, adducts are especially susceptible to reaction [I], resolves the apparent inconsistency. We have apparently been unable to detect llB nrnr signals of the enamine D.BX3 and D2BX2+ species due to excessive peak broadening. We will report our enamine coordination studies in a further paper.
Experimental 1,l-Bis(piperidiny1)ethylene (4) was synthesized by reacting
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CO
NC
OR
DIA
UN
IV o
n 11
/12/
14Fo
r pe
rson
al u
se o
nly.

HARTMAN A N D KELUSKY 1289
1,1, I-triethoxyethane and piperidine in the presence of toluene- 4-sulfonyl chloride, according to the method of Baganz and Domaschke (26). Freshly distilled 4 is clear and colorless but it discolors on standing. The nmr samples were prepared in a nitrogen-filled glove box and made up as 20% solutions of the enediamine in deuterochloroform.
Boron tribromide and boron triiodide (Alfa Inorganics), which had been transferred and sealed into ampoules on a high-vacuum system, were opened in the glove box and added to the enediamine solutions which were cooled to -50°C. BF, and BCI, (Matheson) were added directly from the cylinder to some samples. For other samples, BF, and BCI, were purified on the high-vacuum system and condensed onto a solution of the enediamine at -196'C, and allowed to warm and react; the excess boron trihalide and the solvent were pumped off, leaving the adduct as a colorless oil.
"C and l lB nmr spectra were obtained on a Bruker WP-60 Fourier Transform nmr spectrometer operating at 15.08 MHz and at 19.25 MHz, respectively; 10 mm sample tubes were used. For 13C spectra a 3759 Hz sweep width was used, with a 1.09 s acquisition time for 8K of data memory, using quadrature detection. A 30" pulse was used, with the Phase Alternating Pulse Sequence, and up to 5000 pulses were accumulated. The FID's were transformed with 0.5Hz of line broadening. Off- resonance decoupled spectra were obtained to confirm peak assignments and used a proton frequency set at 250Hz to high field of tetramethylsilane. For I1B spectra various sweep widths and memory sizes were used in order to optimize spectra containing both sharp and broad resonances. Transformation of FID's with extreme line broadening (> 100Hz) was found to be helpful in confirming the presence of very broad peaks which were dimcult to detect above the noise when zero or small line broadening was applied.
Acknowledgements We thank the Natural Sciences and Engineering
Research Council of Canada for financial support of this work, and Mr. Arnold Fox and Mr. T. R. B. Jones for technical assistance.
1. W. KANTLEHNER. In Amidinium salts in organic chemis- try. Vol. 2. Edited b y H. Bohme. John Wiley and Sons, New York. 1979 ((Vol. 9, Part 2 of Advances in organic chemistry. Methods and results).
2. J . D. WILSON, C. F. HOBBS, and H. WEINGARTEN. J. Org. Chem. 35, 1542 (1970).
3. J. S. HARTMAN and R. R. YETMAN. Can. J. Chem. 54,1130 (1976).
4. H. WEINGARTEN and J. S. WAGER. Synth. Inorg. Met.-Org. Chem. 1, 123 (1971).
5. J . S . HARTMAN and B. D. MCGARVEY. Paper IN-28, presented at the 63rd Canadian Chemical Conference, Ottawa, Ontario, June 1980; J. S. HARTMAN, E. C. KELUSKY, and B. D. MCGARVEY. In preparation.
6. A. G. COOK (Editor). Enamines: synthesis, structure, and reactions. Marcel Dekker, New York. 1969.
7. B. WRACKMEYER. h o g . NucI. Magn. Reson. Spectrosc. 12,227 (1979).
8. H. NOTH and B. WRACKMEYER. NMR basic principles and progress. Vol. 14. Edited b y P. Diehl, E. Fluck, and R. Kosfeld. Springer-Verlag, Berlin. 1978.
9. D. T O U R W ~ , G. VAN BINST, S. A. G. DE GRAAF, and U. K. PANDIT. Org. Magn. Reson. 7, 433 (1975); M. G. AHMED and P. W. HICKMOTT. J. Chem. Soc. Perkin Trans. 11,838 (1977); M. G. AHMED, P. W. HICKMOTT, and R. D. SOELISTYOWATI. J. Chem. Soc. Perkin Trans. 11, 372 (1978); R. STRADI, P. TRIMARCO, and A. VIGEVANI. J . Chem. Soc. Perkin Trans. I, l(1978).
10. J. M. MILLER and T. R. B. JONES. Inorg. Chem. 15, 284 (1976).
11. A. FRATIELLO, R. KUBO, D. LIU, and G. VIDULICH. J. Chem. Soc. Perkin Trans. 11, 1415 (1975).
12. C. RABILLER, J. P. RENOU, and G. J. MARTIN. J. Chem. Soc. Perkin Trans. 11, 536 (1977).
13. R. MER~NYI . In Iminium salts inorganic chemistry. Vol. 1. Edited b y H. Bohme and H. G. Viehe. Wiley Interscience, 1976. pp. 25-105. (Vol. 9, Part 1 of Advances in organic chemistry. Methods and results).
14. M. SUZUKI and R. KUBO. Mol. Phys. 7, 201 (1963); J. BACON, R. J. GILLESPIE, and J. W. QUAIL. Can. J. Chem. 41,3063 (1963); J . BACON, R. J . GILLESPIE, J. S. HARTMAN, and U. R. K. RAO. MoI. Phys. 18,561 (1970); R. WEISS and R. N. GRIMES. J. Am. Chem. Soc. 100, 1401 (1978).
15. J . D. ODOM, T. F . MOORE, R. GOETZE, H. NOTH, and B. WRACKMEYER. J. Organomet. Chem. 173, 15 (1979); B. R. GRAGG, W. J. LAYTON, and K. NIEDENZU. J. Organomet. Chem. 132,29 (1977).
16. J. S. HARTMAN and G. J. SCHROBILGEN. Inorg. Chem. 11, 940 (1972).
17. H. BINDER and E. FLUCK. Z. Anorg. Allgem. Chem. 381, 116 (1971).
18. M. J. BULA and J. S . HARTMAN. J. Chem. Soc. Dalton Trans. 1047 (1973). . .
19. R. E. RYSCHKEWITSCH. In Boron hydride chemistry. Edited b y E. L. Muetterties. Academic Press, New York. 1975. Chapt. 6.
20. 0. P. SHITOV, S. L . IOFFE, V. A. TARTAKOVSKII, and S. S. NOVIKOV. Russ. Chem. Rev. 40, 905 (1970).
21. G. E. RYSCHKEWITSCH and W. H. MYERS. Synth. React. Inorg. Metal-Org. Chem. 5, 123 (1975).
22. R. R. YETMAN. M.Sc. thesis, Brock University, St. Catha- r i n e ~ , Ont. 1975.
23. A. G. KRESGE. ACC. Chem. Res. 8, 354 (1975). 24. J. S . HARTMAN and G. J. SCHROBILGEN. Can. J . Chem. 50,
713 (1972). 25. P. P. PONTI, J. C. BALDWIN, and W. C. KASKA. Inorg.
Chem. 18,873 (1979). 26. H. BAGANZ and L . DOMASCHKE. Chem. Ber. 95, 2095
(1962). Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
CO
NC
OR
DIA
UN
IV o
n 11
/12/
14Fo
r pe
rson
al u
se o
nly.