Embed Size (px)
Citation preview

LY333334 2.5 臨床に関する概括評価
フォルテオ皮下注キット 600 μg
フォルテオ皮下注カート 600 μg
2.5 臨床に関する概括評価
日本イーライリリー株式会社

LY333334 2.5 臨床に関する概括評価
目次
2.5 臨床に関する概括評価 .....................................................................................................12.5.1 製品開発の根拠 .........................................................................................................1
2.5.1.1 骨粗鬆症について .............................................................................................12.5.1.1.1 骨粗鬆症の定義及び概念 .........................................................................12.5.1.1.2 骨粗鬆症の成因 .........................................................................................32.5.1.1.3 骨粗鬆症の有病率 .....................................................................................3
2.5.1.2 現状の治療法と問題点 .....................................................................................42.5.1.2.1 ビスフォスフォネート製剤による治療 .................................................42.5.1.2.2 ホルモン補充療法 .....................................................................................52.5.1.2.3 選択的エストロゲン受容体モジュレーター(SERM)製剤によ
る治療 .........................................................................................................52.5.1.2.4 活性型ビタミン D3 製剤による治療 .......................................................52.5.1.2.5 現状の治療法と問題点のまとめ .............................................................5
2.5.1.3 臨床開発の科学的背景 .....................................................................................62.5.1.3.1 骨粗鬆症治療薬としてのテリパラチド .................................................62.5.1.3.2 テリパラチドの作用機序 .........................................................................6
2.5.1.4 臨床開発の概略 .................................................................................................72.5.1.4.1 外国における臨床開発の概略 .................................................................72.5.1.4.2 国内における臨床開発の概略 .................................................................7
2.5.1.4.2.1 初回治験相談(20 年 月 日) ...............................................82.5.1.4.2.2 国内における第 I 相単回投与試験(GHCO 試験)(20 年
月~20 年 月) ........................................................................82.5.1.4.2.3 後期第Ⅱ相試験開始前相談(20 年 月 日) .......................92.5.1.4.2.4 後期第 II 相試験(GHCS 試験)(2005 年 4 月~2006 年 3
月)........................................................................................................92.5.1.4.2.5 後期第 II 相試験終了後相談(20 年 月 日).....................92.5.1.4.2.6 第 III 相試験(GHDB 試験)(2007 年 2 月~2009 年 9 月).....10
2.5.1.5 外国臨床データの利用計画及び臨床データパッケージ............................112.5.2 生物薬剤学に関する概括評価 ...............................................................................13
2.5.2.1 バイオアベイラビリティ ...............................................................................132.5.2.2 投与部位の違いによる薬物動態への影響....................................................13
2.5.3 臨床薬理に関する概括評価 ...................................................................................142.5.3.1 健康成人による薬物動態 ...............................................................................142.5.3.2 骨粗鬆症患者における薬物動態 ...................................................................142.5.3.3 代謝及び排泄 ...................................................................................................142.5.3.4 カルシウム濃度に対する影響 .......................................................................152.5.3.5 特殊集団 ...........................................................................................................152.5.3.6 薬物相互作用 ...................................................................................................152.5.3.7 薬力学的解析 ...................................................................................................16
2.5.4 有効性の概括評価 ...................................................................................................172.5.4.1 有効性評価の概略 ...........................................................................................172.5.4.2 プラセボ対照二重盲検比較試験 ...................................................................17
2.5.4.2.1 試験デザインの概略 ...............................................................................172.5.4.2.2 有効性の評価項目 ...................................................................................182.5.4.2.3 有効性の評価の対象となった患者集団の特性....................................192.5.4.2.4 有効性の結果 ...........................................................................................20
2.5.4.2.4.1 国内プラセボ対照二重盲検比較試験の成績................................202.5.4.2.4.1.1 GHCS 試験(国内第 II 相試験)............................................202.5.4.2.4.1.2 GHDB 試験(国内第 III 相、ブリッジング試験)..............20

LY333334 2.5 臨床に関する概括評価
2.5.4.2.4.1.2.1 外国臨床データの日本人骨粗鬆症患者への外挿可
能性の検討 .......................................................................212.5.4.2.4.1.2.1.1 外挿可能性の評価方法............................................212.5.4.2.4.1.2.1.2 外国臨床データの外挿可能性の検討....................22
2.5.4.2.4.2 外国プラセボ対照二重盲検比較試験の成績................................242.5.4.2.4.2.1 GHAC 試験(外国第 III 相、ブリッジング対象試験)......242.5.4.2.4.2.2 GHAJ 試験(外国第 III 相試験)...........................................27
2.5.4.3 実薬対照試験 ...................................................................................................272.5.4.4 テリパラチド投与後にラロキシフェンの継続投与を検討した試験........292.5.4.5 テリパラチド投与中止後の追跡調査研究....................................................302.5.4.6 アジアで実施した試験 ...................................................................................322.5.4.7 骨吸収抑制剤の前治療又は併用を検討した試験........................................322.5.4.8 有効性評価のまとめ .......................................................................................33
2.5.5 安全性の概括評価 ...................................................................................................352.5.5.1 安全性評価の概略 ...........................................................................................352.5.5.2 安全性評価の対象となった被験者の人口統計学的及びその他の特性....362.5.5.3 テリパラチドの曝露及び使用経験................................................................372.5.5.4 有害事象 ...........................................................................................................37
2.5.5.4.1 比較的よく見られる有害事象 ...............................................................372.5.5.4.1.1 国内プラセボ対照二重盲検比較試験 ...........................................372.5.5.4.1.2 外国プラセボ対照二重盲検比較試験 ...........................................382.5.5.4.1.3 実薬対照試験 ...................................................................................392.5.5.4.1.4 テリパラチド投与後にラロキシフェンの継続投与を検討し
た試験 ...............................................................................................402.5.5.4.1.5 国内及び外国臨床試験の併合解析 ...............................................412.5.5.4.1.6 比較的よく見られる有害事象のまとめ........................................43
2.5.5.4.2 重篤及び他の重要な有害事象 ...............................................................432.5.5.4.3 テリパラチド投与中止後の追跡調査研究 ...........................................452.5.5.4.4 臨床薬理試験における安全性評価 .......................................................45
2.5.5.5 臨床検査 ...........................................................................................................462.5.5.6 バイタルサイン及び心電図 ...........................................................................472.5.5.7 テリパラチド投与において特に考慮すべき安全性....................................47
2.5.5.7.1 骨肉腫 .......................................................................................................472.5.5.7.2 高カルシウム血症 ...................................................................................482.5.5.7.3 起立性低血圧 ...........................................................................................492.5.5.7.4 腎障害 .......................................................................................................492.5.5.7.5 背部痛 .......................................................................................................49
2.5.5.8 薬物相互作用 ...................................................................................................502.5.5.9 市販後データ ...................................................................................................502.5.5.10 安全性評価のまとめ .......................................................................................51
2.5.6 ベネフィットとリスクに関する結論 ...................................................................522.5.6.1 骨折の危険性の高い骨粗鬆症の治療薬としてのテリパラチドの概略....522.5.6.2 テリパラチド投与によるベネフィット........................................................52
2.5.6.2.1 骨折防止効果 ...........................................................................................522.5.6.2.2 骨微細構造の再構築効果 .......................................................................532.5.6.2.3 骨密度増加効果 .......................................................................................542.5.6.2.4 骨形成マーカー先行優位の骨代謝マーカー反応................................562.5.6.2.5 腰背部痛に対する効果 ...........................................................................572.5.6.2.6 骨折の危険性の高い骨粗鬆症患者の治療におけるベネフィット....58
2.5.6.3 テリパラチド投与により予測されるリスク................................................582.5.6.3.1 骨肉腫 .......................................................................................................59
2.5.6.3.1.1 非臨床試験で認められた知見 .......................................................59

LY333334 2.5 臨床に関する概括評価
2.5.6.3.1.2 臨床試験及び市販後の安全性情報から得られた知見................592.5.6.3.2 有害事象 ...................................................................................................602.5.6.3.3 血清カルシウム及びそのほかの臨床検査に及ぼす影響....................612.5.6.3.4 骨折の危険性の高い骨粗鬆症患者に対するテリパラチド治療に
おけるリスク ...........................................................................................632.5.6.3.4.1 投与期間の設定 ...............................................................................632.5.6.3.4.2 在宅自己注射 ...................................................................................63
2.5.6.4 テリパラチドの臨床的意義のまとめ............................................................642.5.7 参考文献 ...................................................................................................................65

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
1
2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
ヒト副甲状腺ホルモン(以下、PTH とする)は間欠投与により骨形成促進作用を発揮
することが知られている。LY333334〔テリパラチド(遺伝子組換え)、以下、テリパラ
チドとする〕は、イーライリリー・アンド・カンパニーが開発した遺伝子組換え PTH
(1-34)製剤であり、PTH の活性部分である N 端側 34 個のアミノ酸で構成されている。
テリパラチドは、1 日 1 回皮下投与することにより骨形成を促進し、骨折の危険性の
高い骨粗鬆症患者における骨密度(以下、BMD とする)を速やかに増加させ、骨微細
構造を再構築することにより優れた骨折発生抑制効果を発揮する、新しい骨粗鬆症治療
剤である。
本項では、骨粗鬆症の概要、現状の治療法と問題点、テリパラチドの臨床開発の科学
的背景並びに臨床開発の概略からテリパラチドの製品開発の根拠について示す。
2.5.1.1 骨粗鬆症について
2.5.1.1.1 骨粗鬆症の定義及び概念
骨粗鬆症とは、骨強度の低下により骨の脆弱性が高まり骨折しやすくなった状態であ
る。骨粗鬆症は従来、BMD の減少及び骨微細構造の破綻により骨の脆弱化が起こり、
骨折の危険性が高まった状態と定義されてきた。しかし、骨強度の低下には BMD の低
下に加えて骨質の劣化も重要な役割を果たすことが明らかとなり、2000 年に米国国立衛
生研究所(以下、NIH とする)で開催されたコンセンサス会議において、「骨強度の低
下を特徴とし、骨折のリスクが増大しやすくなる骨格疾患」と定義された。また、骨強
度のほぼ 70%は BMD で、残りの 30%は骨質、すなわち骨微細構造、微細損傷(微細骨
折)の集積、骨代謝回転及び石灰化などにより説明されることが示され、骨粗鬆症にお
いて「骨強度の低下」と「骨折の危険性の増加」の 2 点が重視されるようになった
(NIH 2000)。
骨粗鬆症の診断基準は、1994 年に世界保健機構(以下、WHO とする)が提案した
BMD を指標とする診断基準が国際的に広く用いられているが、日本では原則として
BMD と骨折の両者を指標とした診断基準が用いられている。日本の原発性骨粗鬆症の
診断基準は、二重 X 線吸収法(以下、DXA 法とする)による BMD の測定値及び脆弱性
骨折の有無により設定されている。本診断基準によると、低 BMD を原因とする脆弱性
骨折があるものはすべて骨粗鬆症とし、脆弱性骨折のない場合は、BMD が若年成人平
均値(20~44 歳)(以下、YAM とする)の 70%未満を骨粗鬆症と診断する(折茂ほか
2001)。診断には BMD の値を用いるが、BMD の測定または評価が困難な場合には、脊
椎 X 線像を用いて評価する(表 2.5.1-1)。

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
2
表 2.5.1-1. 骨粗鬆症の診断基準
原発性骨粗鬆症の診断基準 (2000 年度改訂版)
低骨量をきたす骨粗鬆症以外の疾患または続発性骨粗鬆症を認めず、骨評価の結果が
下記の条件を満たす場合、原発性骨粗鬆症と診断する。
I. 脆弱性骨折 aあり
II. 脆弱性骨折なし
骨密度値 b 脊椎 X 線像での骨粗鬆化 c
正常 YAM の 80%以上 なし
骨量減少 YAM の 70%以上 80%未満 疑いあり
骨粗鬆症 YAM の 70%未満 あり
YAM: 若年成人平均値(20~44 歳)
a 脆弱性骨折:低骨量(骨密度が YAM の 80%未満、あるいは脊椎 X 線像で骨粗鬆化が
ある場合)が原因で、軽微な外力によって発生した非外傷性骨折。骨折部位は脊椎、
大腿骨頸部、橈骨遠位端、その他。
b 骨密度は原則として腰椎骨密度とする。ただし、高齢者において、脊椎変形などのた
めに腰椎骨密度の測定が適当でないと判断される場合には大腿骨頸部骨密度とする。
これらの測定が困難な場合は橈骨、第二中手骨、踵骨の骨密度を用いる。
c 脊椎 X 線像での骨粗鬆化の評価は、従来の骨萎縮度判定基準を参考にして行う。
脊椎 X 線像での骨粗鬆化 従来の骨萎縮度判定基準
なし 骨萎縮なし
疑いあり 骨萎縮度 I 度あり 骨萎縮度 II 度以上
骨粗鬆症の診断は、上記の通り BMD 及び脊椎 X 線像を基準に行われる。その一方で、
治療介入の基準として、臨床的危険因子を考慮して骨折の危険性の高い骨粗鬆症患者を
判定しようという検討は、世界的な動きとなりつつある。骨折の危険性の高い骨粗鬆症
患者を判別するための危険因子としては、低 BMD、年齢、既存骨折、ステロイド使用
などが知られている。すなわち、BMD が極端に低下している低 BMD の患者並びに同じ
BMD を示していても高齢者及び既存骨折などの危険因子を有する患者においては骨折
の危険性が高くなると報告されている(Kanis et al. 2005; Fujiwara et al. 2003)。
骨折は重篤度と頻度から最大の骨粗鬆症の合併症である。骨粗鬆症で最も高頻度に生
じる椎体骨折は慢性疼痛、脊椎変形による逆流性食道炎や呼吸機能低下などの機能障害
などにより生活の質(以下、QOL とする)のみならず生命予後をも悪化させることが明
らかとなってきている(骨粗鬆症の予防と治療ガイドライン 2006 年版; Kanis et al.
2004b)。一方、大腿骨頸部骨折は生命予後に最も影響する骨粗鬆症合併症であり、大
腿骨頸部骨折後の死亡率は高い(Cauley et al. 2000)。また、生存した患者においても自
立機能の障害を引き起こし、寝たきりなどを含め高齢者の QOL を著しく低下させる原
因となっている(骨粗鬆症の予防と治療ガイドライン 2006 年版; 厚生労働省 2008)。
骨粗鬆症治療の目的は骨折の予防である。骨折予防の治療に緊急性を要する骨折の危
険性が高い骨粗鬆症患者に対して、早期かつ有効な治療介入を行い効率的な骨折予防を
することは、骨粗鬆症患者の骨折後の生命、自立機能、QOL の維持につながり、ここに
骨粗鬆症の治療の目的と意義があると考えられている(藤原 2007; 骨粗鬆症検診・保健
指導マニュアル 2009)。

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
3
2.5.1.1.2 骨粗鬆症の成因
骨粗鬆症は、BMD の低下と骨質の劣化が大きく影響する(Raisz 2005; Seeman 2002)。
また、骨粗鬆症は、多因子疾患であり、遺伝要因と生活習慣(食事、運動、喫煙、アル
コールなど)が発症に大きく影響する。
骨粗鬆症は性別にかかわらず発症するが、閉経後の女性において最も多く認められる。
閉経後骨粗鬆症は、骨リモデリングサイクルが開始される頻度の増加の、いわゆる高代
謝回転の状態と、個々の骨改変部位における骨吸収と骨形成のアンバランスによって起
こる。閉経後女性では、内因性エストロゲン欠乏などにより異常に骨吸収が亢進し、骨
形成を上回ることによって、急速な BMD の減少を招く。また、骨梁に深い吸収窩が発
現することにより、連結性の低下や断裂から力学的強度が弱まり、脆弱性骨折のリスク
が高まるものと考えられる。また、高齢化が原因でおこる骨芽細胞とその前駆細胞の不
活化が、骨吸収と骨形成の不均衡に寄与していることも考えられる。これらの変化が原
因で、50 歳以上の女性の大部分で BMD の減少が進行し骨粗鬆症を発症する(骨粗鬆症
の予防と治療ガイドライン 2006 年版)。
男性においては、性腺機能低下症が骨粗鬆症の重要な要因だが、その他にもステロイ
ドの使用、甲状腺機能亢進症、並びに吸収不良障害などの他の基礎疾患も危険因子とな
る(Seeman 2002; Riggs et al. 1998)。加齢に伴うテストステロンの低下は、骨形成の低
下及び BMD の減少に寄与し、その結果骨の脆弱性に寄与している可能性がある。女性
と同様、男性における BMD の低下は、骨折発生率の増加の原因となる(Fatayerji et al.
1999)。
2.5.1.1.3 骨粗鬆症の有病率
骨粗鬆症は、米国、欧州、日本を含め 7,500 万人以上が罹患していると報告されてお
り、米国及び欧州を合わせると毎年 230 万件の骨折が骨粗鬆症によって生じている
(WHO 2003)。
日本における骨粗鬆症患者は、高齢化社会の進展とともに増加しその数は約 1,100 万
人と推定されている(骨粗鬆症の予防と治療ガイドライン 2006 年版)。骨粗鬆症に関
する疫学調査より、日本の骨粗鬆症の有病率は 50 歳以上の女性では 24%、50 歳以上の
男性では 4%であることが報告されている。また、年齢別の有病率としては、50 歳代 7%、
60 歳代 30%、70 歳代 37%、80 歳代 42%と、年齢が高くなるに従って有病率も上昇する
ことが確認されている(藤原ほか 1997)。
骨粗鬆症が進行すると骨折の危険性が上昇するが、中でも大腿骨頸部骨折は生命予後
に最も影響する骨粗鬆症合併症である。日本における大腿骨頸部骨折は、年間に 12 万
件を超えると推定され、このうち約 10%は 1 年以内に死亡し、約 30%は日常生活動作能
力(以下、ADL とする)が低下すると考えられている(骨粗鬆症の予防と治療ガイドラ
イン 2006 年版)。この大腿骨頸部骨折全体の治療費は 1 年間で 1,700 億円、介護費用は
4,400 億円にも上ると推測されており(吉村 2005)、高齢者人口の増加に伴い、大腿骨
頸部骨折による膨大な医療費及び介護費の出費は今後も増大することが懸念される
(Hagino et al. 2009)。

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
4
また、日本における椎体骨折については、70 歳代前半の 25%、80 歳代前半の 43%が
椎体骨折を有していると報告されている(Ross et al. 1995)。椎体骨折に関しても周囲
椎体の骨折連鎖を引き起こすことによる脊椎変形・姿勢異常、更にはこれに伴う消化器
系や呼吸器系の機能障害などにより、QOL の大幅な低下を引き起こすことが明らかとな
ってきている(骨粗鬆症の予防と治療ガイドライン 2006 年版)。
日本の総人口に占める 65 歳以上の高齢者の割合である高齢化率は上昇しつつあるこ
とから、骨粗鬆症の有病率が短期間で低下に転じるとは考えにくく、超高齢社会を前に
して、骨粗鬆症による骨折の防止、すなわち骨折の危険性の高い骨粗鬆症患者の治療は、
医療のみならず社会的にも急務の課題であることは明白である。
2.5.1.2 現状の治療法と問題点
日本における現在の骨粗鬆症の薬物治療としては、主に骨吸収抑制剤及びカルシウム
代謝改善剤が使用されている。骨吸収抑制剤としては、ビスフォスフォネート製剤、女
性ホルモン製剤、選択的エストロゲン受容体モジュレーター(以下、SERM とする)製
剤、カルシトニン製剤及びイプリフラボン製剤があり、カルシウム代謝改善剤として活
性型ビタミン D3製剤が挙げられる。以下に各薬剤の現状の治療法と問題点を示す。
2.5.1.2.1 ビスフォスフォネート製剤による治療
アミノ基を有する第 2、3 世代ビスフォスフォネート〔アレンドロン酸ナトリウム水
和物(以下、アレンドロネートとする)及びリセドロン酸ナトリウム水和物(以下、リ
セドロネートとする)〕は有効な骨粗鬆症の治療手段として普及している。ビスフォス
フォネート製剤の BMD 増加作用、骨折発生抑制作用に関しては多くの臨床試験結果が
報告されており、1 年間の投与により BMD は 3%~5%増加し、3 年間の投与により新規
椎体骨折発生率が 30%~50%低下することが示されている(Cummings et al. 1998; Harris
et al. 1999; 骨粗鬆症の予防と治療ガイドライン 2006 年版)。また、リセドロネートにお
いては 3 年間の投与により非椎体骨折の発生率が 40%減少することも報告されている
(McClung et al. 2001)。しかし、経口のビスフォスフォネート製剤では、他剤に比して
消化管障害の発現頻度が比較的高いことや、週1回投与製剤により緩和はされているも
のの、起床後の空腹での服薬や服薬後 30 分以内の食事や臥床が禁止されるために患者
の利便性が損なわれるなどの問題点も指摘されている(Adami et al. 1996; Kishimoto et al.
2006)。更に、ビスフォスフォネートは骨に長期間蓄積することが知られており、投与
後長期にわたり骨代謝回転を抑制するため、骨代謝過剰抑制による骨折治癒の遅延
(Odvina et al. 2005)や、最近では長期のアレンドロネート投与患者における大腿骨中
央部の骨折発生(Lenart et al. 2008)や顎骨壊死についても報告がなされている(Woo et
al. 2006)。米国骨粗鬆症財団(以下、NOF とする)のガイドラインによると、どのぐ
らい長期に投与すべきか又は長期投与により未知の副作用がないかなどが懸念事項とさ
れている(NOF 2008)。長期のビスフォスフォネート治療が骨粗鬆症患者にどのような
影響を与えるかについては現在のところ明らかではなく、今後更なる有効性及び安全性

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
5
の追跡並びにリスクベネフィットの解析が必要であることが示唆されている
(Armamento-Villareal et al. 2006)。
2.5.1.2.2 ホルモン補充療法
エストロゲン製剤を用いたホルモン補充療法は、閉経後骨粗鬆症の主病因であるエス
トロゲン欠乏を補充して是正するという生理的な治療法と考えられてきたが、子宮内膜
癌や乳癌の懸念及び性器出血などの問題により日本では以前から使用頻度が低かった。
長期の治療が必要な骨粗鬆症患者に対しては、エストロゲン製剤を用いたホルモン補充
療法以外の薬剤治療を選択することが薦められ、エストロゲン製剤は閉経後の更年期症
状などに対する短期的な治療に使用が限定されるものと推測される。
2.5.1.2.3 選択的エストロゲン受容体モジュレーター(SERM)製剤による治療
SERM の一種であるラロキシフェン塩酸塩(以下、ラロキシフェンとする)は、2004
年に日本において臨床応用された骨粗鬆症治療薬であり、閉経後骨粗鬆症に使用されて
いる。ラロキシフェン投与による BMD 増加はビスフォスフォネート製剤に比べ劣るも
のの、新規椎体骨折発生抑制効果はほぼ同等である(Delmas et al. 2002)。ラロキシフ
ェンの骨折発生抑制効果は、骨代謝回転を過度に抑制せず生理的な範囲に調節し、骨質
を改善する作用に起因するものと考えられている。ラロキシフェンに関しては、80 歳未
満の閉経後骨粗鬆症女性患者で BMD 増加効果、椎体骨折防止効果のエビデンスを有す
るが、非椎体骨折の防止効果に関するエビデンスは十分ではないと思われる(骨粗鬆症
の予防と治療ガイドライン 2006 年版)。
2.5.1.2.4 活性型ビタミン D3 製剤による治療
活性型ビタミン D3 製剤は骨粗鬆症治療薬として日本で最もよく処方されてきた薬剤
である。活性型ビタミン D3 製剤に関する臨床試験成績の報告は大部分が少数例の報告
であり、他の製剤に比較すると特に骨折発生抑制効果についてのエビデンスレベルは高
いとはいいがたく(骨粗鬆症の予防と治療ガイドライン 2006 年版)、欧米では骨粗鬆
症治療の適応を持っていない。
2.5.1.2.5 現状の治療法と問題点のまとめ
骨粗鬆症に対して使用されている主な薬剤である骨吸収抑制剤(ビスフォスフォネー
ト、SERM など)は、破骨細胞による骨吸収を抑制することにより BMD を増加あるい
は BMD の減少を防ぎ、骨折の発生を減少させる。既存の骨粗鬆症治療薬では 3 年間の
投与により椎体及び非椎体骨折の発生率が減少することなどが示されているが、短期間
での骨折発生抑制効果や骨微細構造の再構築作用はなく、骨折の防止を目的とした治療
を急務とする骨折の危険性の高い骨粗鬆症患者に対して不十分であると考えられる。
今後増加が見込まれる骨折の危険性の高い骨粗鬆症患者の骨折の予防を確実なものに
するためには、更に短期間で有効な骨折発生抑制効果を有する薬剤が必要と考えられる。

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
6
2.5.1.3 臨床開発の科学的背景
2.5.1.3.1 骨粗鬆症治療薬としてのテリパラチド
テリパラチドは 2002 年 11 月、骨折の危険性の高い骨粗鬆症を適応症として、1 日 1
回の皮下投与により骨形成を促進する初めての骨粗鬆症治療薬として米国で承認された。
2010 年 1 月 4 日現在では、テリパラチドは 83 ヵ国で承認されているが、日本ではこの
ような骨形成促進剤は上市されていない。
テリパラチドは、骨折の危険性の高い骨粗鬆症患者における骨形成促進作用により速
やかに BMD を増加させ、骨微細構造を再構築することにより短期間で優れた骨折発生
抑制効果を示す新しい骨粗鬆症治療剤である。また、日本における適応症は、外国での
対象患者と同様に「骨折の危険性の高い骨粗鬆症」を予定している。
テリパラチドの骨折の危険性の高い骨粗鬆症患者に対する有効性は臨床試験で示され
ている。テリパラチドの約 1.5 から 2 年間の短期間の投与により速やかに BMD を増加
させ骨微細構造を再構築することにより、65%の新規椎体骨折発生の抑制及び 53%の非
外傷性非椎体骨折発生の抑制効果が認められている。また、ビスフォスフォネート製剤
(アレンドロネート)に比して統計学的に有意な腰椎 BMD 増加効果も認められ、既存
の骨吸収抑制剤に対する優越性が認められている。
日本における骨粗鬆症患者は、高齢化社会の進展とともに増加し、その数は約 1,100
万人と推定されていることから、骨粗鬆症の治療及びそれに伴う骨折の予防は、高齢化
社会を迎えた日本において、重要な課題である。また、骨粗鬆症治療により骨折を如何
に効率よく防止し、患者の生命予後を改善するかということが今後の骨粗鬆症治療の鍵
となることから、骨折の危険性の高い骨粗鬆症患者において短期間で顕著な骨折防止効
果が期待できる骨粗鬆症治療薬の導入が必要である。
テリパラチドは既存薬にはない骨形成作用を有し、高い BMD 増加作用、骨微細構造
の再構築作用、その結果の優れた骨折発生抑制効果を発揮することから、従来の薬剤で
は十分な効果が得られないような、著しい BMD 低下症例や既に骨微細構造の破綻をき
たした症例など、特に骨折の危険性の高い骨粗鬆症患者に対して非常に有用な薬剤とな
ることが想定される。また、これらの効果が短期間で得られることから、骨折の危険性
の高い骨粗鬆症患者における第一選択薬になると考える。
2.5.1.3.2 テリパラチドの作用機序
テリパラチドは内因性 PTH の遺伝子組換え N-末端フラグメントであり、34 個の N-末
端アミノ酸で構成されている〔PTH(1-34)〕。84 個のアミノ酸からなる内因性 PTH は、
骨及び腎におけるカルシウム及びリン酸代謝の主要な調節因子である。PTH の生理的作
用としては、骨形成細胞(骨芽細胞)に対する直接作用による骨形成の促進、骨芽細胞
を介して間接的に破骨細胞分化・活性化の促進、カルシウムの尿細管再吸収及びリン酸
排泄の増大、活性型ビタミン D〔1α,25-(OH)2D〕産生作用を介したカルシウムの腸管吸
収の増大などがあげられる。特に、この骨形成促進作用は 1 日 1 回など間欠的に PTH を
投与した場合にのみ観察されると報告されている。PTH(1-34)は、内因性 PTH と同様

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
7
に特異的 G 蛋白共役 PTH/PTHrP 受容体に結合し、骨及び腎に対する内因性 PTH の既知
の in vivo における生体内作用である骨形成促進作用を発揮する。
PTH(1-34)の骨への作用は投与スケジュールによって左右され、PTH(1-34)を 1
日 1 回の皮下投与として間欠的に投与すると、骨梁並びに皮質骨の内膜及び外膜面にお
いて骨芽細胞機能が活性化され、破骨細胞機能を上回るため、骨新生が誘発される。
2.5.1.4 臨床開発の概略
2.5.1.4.1 外国における臨床開発の概略
テリパラチドは、イーライリリー・アンド・カンパニーにより骨粗鬆症の治療薬とし
て開発された。外国における第 III 相試験を実施中の 1998 年 12 月に、ラットがん原性試
験において骨肉腫を含む骨腫瘍性病変所見が認められたため、進行中であったすべての
テリパラチドの臨床試験を自主的に中止した。その後、イーライリリー・アンド・カン
パニーは、サルにおける長期投与試験及び中止した臨床試験の追跡調査を実施し、米国
食品医薬品局(以下、FDA とする)との議論を重ねた結果、ラットで発生した骨肉腫が
ヒトにおいて発症する可能性は低いとされ、中止した臨床試験成績を用いて承認申請す
ることが可能であるとの合意に至った。
米国においては、2000 年 11 月 29 日、テリパラチドの新薬承認申請を行い、骨折の危
険性の高い閉経後骨粗鬆症女性の治療及び原発性又は性腺機能低下による骨粗鬆症を有
する男性の骨量増加を適応症として、2002 年 11 月 26 日に承認された。FDA の審査過程
においてテリパラチドの投与期間についても検討され、外国で実施した閉経後骨粗鬆症
女性を対象としたプラセボ対照二重盲検比較試験(GHAC 試験)のテリパラチドの最大
投与期間を考慮し 24 ヵ月を超えて本剤の使用を推奨しないこととされた。なお、ステ
ロイド性骨粗鬆症の効能追加申請を 2007 年 2 月 8 日に行い、2009 年 7 月 22 日に承認さ
れた。
一方、欧州においては、2001 年 5 月 31 日、欧州医薬品委員会(以下、CPMP とす
る)に対し、本剤の販売承認申請を行い、2003 年 6 月 10 日、骨折の危険性の高い閉経
後骨粗鬆症女性の治療を適応症として承認された。CPMP においても投与期間の上限に
ついて検討がなされ、GHAC 試験のテリパラチド投与期間の中央値を考慮し、投与期間
上限は 18 ヵ月間と設定された。骨折の危険性の高い骨粗鬆症男性の治療に対する適応
症は 2007 年 6 月に承認、ステロイド性骨粗鬆症については、2008 年 4 月に承認された。
なお、欧州においても、近年実施された GHCA 試験などの臨床試験データを基に、2008
年 10 月 15 日、投与期間上限を 24 ヵ月に延長する申請を行い、2009 年 2 月 25 日に承認
された。
2.5.1.4.2 国内における臨床開発の概略
外国におけるテリパラチドの承認及び承認申請以降の安全性に関する情報を受けて、
本剤の薬剤特性、骨粗鬆症に関する診断方法・治療方法及び既に得られていたテリパラ
チドの外国臨床データ等を考慮して、日本ではブリッジング開発を選択し、外国臨床試
験成績を外挿して承認申請することを計画した。

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
8
第 I 相試験である GHCO 試験は、日本人及び外国人の閉経後女性を対象に行った。
GHCO 試験において、民族的内因性要因が薬物動態に及ぼす影響について検討した結果、
テリパラチド 10 µg~60 µg までの薬物動態は線形性を示し、日本人へのテリパラチドの
投与は良好な忍容性を示した。また、日本人の忍容性は外国人と比較して、臨床的に問
題となるような大きな違いはないことが示唆された。この結果を受けて、国内における
第 II 相試験である日本人の閉経後骨粗鬆症女性を対象とした用量反応試験を実施し、腰
椎 BMD 変化率を指標とした場合の用量反応関係を評価した。その結果、日本人の臨床
推奨用量は 20 μg と考えられ、外国での臨床推奨用量と同じであることが確認された。
その後、国内第 III 相試験はブリッジング試験として実施し、テリパラチド 20 μg を
12 ヵ月間投与したときのプラセボに対する優越性が認められ、日本人骨粗鬆症患者に対
するテリパラチド 20 μg の有効性及び安全性が確認された。また、ブリッジング対象試
験(GHAC 試験)及び他の外国で実施されたプラセボ対照二重盲検比較試験の有効性を
比較した結果、ブリッジングの成立要件を満たしたことから、外国臨床データの日本人
骨粗鬆症患者への外挿を考慮した承認申請が可能であると考えた。
なお、国内での第 I 相臨床試験、第 II 相臨床試験及び第 III 相臨床試験の開始前に治験
相談を行い機構より助言を得た。
以下に、各治験相談と国内臨床試験の概略を記載する。
2.5.1.4.2.1 初回治験相談(20 年 月 日)
非臨床試験のラットがん原性試験において骨肉腫を含む骨腫瘍性病変所見が認められ
たが、その後のデータからヒトに骨肉腫を発生させる可能性は低いと判断し、日本にお
いても臨床試験を実施することは適切であると判断した。
について機構の意見を
聴くため治験相談を行った。その結果、ラットがん原性試験結果、外国臨床試験成績等
を勘案し、「
」との助言を得て、
日本での開発を開始することを決定した(第 1.13 項参照)。
2.5.1.4.2.2 国内における第 I 相単回投与試験(GHCO 試験)(20 年 月~20
年 月)
テリパラチドを単回投与したときの安全性、薬物動態を評価することを目的に、日本
人及び外国人の閉経後健康女性を対象とした、テリパラチド 10 g~60 g までの単回投
与試験を米国ハワイ州にて実施した。日本人閉経後女性でテリパラチド 10 µg~60 µg ま
で、外国人閉経後女性でテリパラチド 20 µg~60 µg までの用量範囲で薬物動態は線形性
を示し、年齢、性別及び食事による影響をほとんど受けないと考えられた。体重には日
本人と外国人間で違いが認められたが、この違いは有効性及び安全性に影響を与えるも
のではないと考えられた。また、日本人と外国人で、投与量調節を必要とするような、
明らかな薬物動態の差は認められなかった。

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
9
2.5.1.4.2.3 後期第Ⅱ相試験開始前相談(20 年 月 日)
後期第 II 相試験開始前相談を実施し、
について相談した。
との助言を
受けた。また、
との助言を受けた(第 1.13 項参照)。
2.5.1.4.2.4 後期第 II 相試験(GHCS 試験) (2005 年 4 月~2006 年 3 月)
骨折の危険性の高い閉経後骨粗鬆症女性を対象に、テリパラチド 10 μg、20 μg 及び
40 μg を 6 ヵ月間投与した際の用量反応関係を検討することを目的として、第 II 相プラ
セボ対照二重盲検試験を実施した。被験者は治験薬投与開始前に自己注射練習用資材を
用いて自己注射の手技を学習し、治験責任医師又は分担医師により自己注射手技の習得
が十分であると判断された被験者を治験薬割付の対象とした。試験の結果、有効性の主
要評価項目である腰椎(L2-L4)BMD 変化率において、プラセボ群と比較し 10 μg 群、
20 μg 群及び 40 μg 群は投与量の増加に伴って統計学的に有意な増加を示した。骨代謝マ
ーカーの変化について、テリパラチド 20 μg 群及び 40 μg 群ではいずれの骨形成マーカ
ー及び骨吸収マーカーにも上昇が認められたが、10 μg 群では骨形成マーカーである血
清骨型アルカリフォスファターゼ(以下、BAP とする)及び骨吸収マーカーである血清
I 型コラーゲン架橋 C-テロペプチド(以下、CTX とする)の上昇は認められず、テリパ
ラチド投与群間に違いが認められた。プラセボ群ではいずれの骨代謝マーカーの上昇も
認められなかった。また、安全性の結果から、40 μg 群では因果関係を否定できない有
害事象及び有害事象による中止が、10 μg 群及び 20 μg 群に比べて多く認められた。以上
の結果から、主要評価項目である腰椎 BMD については、テリパラチドの用量反応関係
が示されたが、副次的評価項目である骨代謝マーカーの 10 μg 群の結果及び安全性にお
ける 40 μg 群の結果を考慮し、日本人骨粗鬆症患者の臨床推奨用量は 20 μg が妥当であ
ると考えられた。
2.5.1.4.2.5 後期第 II 相試験終了後相談(20 年 月 日)
国内第Ⅱ相試験(GHCS 試験)から、本剤の 10 μg~40 μg までの用量反応性及び安全
性が確認され、日本人骨粗鬆症患者の臨床推奨用量として 20 μg が適切と考えられた。
また、テリパラチドは外因性及び内因性の民族的要因を受けにくいと考えられたことか
ら、本剤 20 μg 及びプラセボを二重盲検下で 12 ヵ月間投与したプラセボ比較対照試験を
ブリッジング試験として実施することは適切であると考えた。
後期第 II 相試験終了後相談では、
について相談を行った。

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
10
相談の結果、
との助言を得た。
との助言を得た(第 1.13 項参照)。
2.5.1.4.2.6 第 III 相試験(GHDB 試験)(2007 年 2 月~2009 年 9 月)
骨折の危険性の高い骨粗鬆症患者に対する有効性及び安全性を評価するために、テリ
パラチド 20 g を 12 ヵ月間投与した際の本剤の有効性及び 18 ヵ月間投与した際の安全
性等を評価するため、プラセボ対照二重盲検比較期間である第 1 期とオープン期間であ
る第 2 期からなる第 III 相試験を開始した。
ブリッジングが成立したと判断するための要件及び評価方法をあらかじめ設定するた
めに、GHAC 試験におけるベースラインの腰椎 BMD の値による部分集団解析の結果を
検討したところ、テリパラチド投与後の腰椎 BMD 変化率は、外国試験に比べて国内試
験において高くなることが予想された。したがって、有効性のブリッジングが成立した
と判断するための要件及び評価方法を 12 ヵ月後の腰椎(L1-L4)BMD 変化率について
テリパラチド 20 µg 群のプラセボ群に対する優越性が示され、ブリッジング対象試験
(GHAC 試験)と同様の結果が得られることとして設定した。
また、試験開始後、欧州における投与期間上限の延長計画を受けて、日本人骨粗鬆症
患者における本剤 24 ヵ月間投与の使用経験を得るために、進行中の GHDB 試験におけ
る治験薬投与期間を 24 ヵ月に変更した(20 年 月 日治験計画変更届書提出)。
GHDB 試験実施中、 について医薬品追加相談を
行った(20 年 月 日相談申込、 月 日までに書面にて実施)。総合機構より、
「
」との助言を受けた。
本助言内容に基づき、申請者は
こととして本剤の開発を進めた。また、
予定である。
第 1 期の二重盲検比較期間である 12 ヵ月時点での有効性の解析の結果、主要評価項
目である腰椎(L2-L4)BMD の平均変化率について、テリパラチド 20 μg のプラセボに
対する優越性が認められ、日本人骨粗鬆症患者に対する有効性が確認された。また、第
1 期二重盲検比較期間に新規椎体骨折が認められた被験者の割合は、プラセボ群で 6.0%
(4/67)に認められたのに対し、テリパラチド群では 3.7%(5/136)であった。安全性
に関しては、テリパラチド 20 μg を 12 ヵ月間投与した時の忍容性は良好であり、外国臨
床試験と国内第 II 相試験の安全性プロファイルに大きな違いがないことが示された。ブ

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
11
リッジング対象試験(GHAC 試験)及び他の外国で実施されたプラセボ対照二重盲検比
較試験の有効性を比較した結果、ブリッジングの成立要件を満たしたことから、外国臨
床データの日本人骨粗鬆症患者への外挿を考慮した承認申請が可能であると考えた。
第 2 期のオープン期間において、テリパラチド 20 g の 1 日 1 回 18 ヵ月間投与の結果、
有効かつ安全であることが認められた。テリパラチド 20 g の投与による腰椎 BMD の
増加は、12 ヵ月間の二重盲検比較期間に、時間依存的かつプラセボ群に比し統計学的に
有意であることが確認され、その後 6 ヵ月間にも継続して観察された。また、プラセボ
を 12 ヵ月間投与後、テリパラチド 20 g を 6 ヵ月間投与した被験者における腰椎 BMD
の増加は、試験期間を通じてテリパラチド 20 g を投与した被験者において投与 6 ヵ月
後に観察された結果と同様であった。また、試験期間を通じてテリパラチド 20 g を投
与した被験者において、投与期間を 12 ヵ月間から 18 ヵ月間まで延長した結果、さらな
る安全性上の懸念や重要な安全性の所見の増加は認められず、テリパラチド 20 g を骨
折の危険性の高い日本人骨粗鬆症患者に 1 日 1 回 18 ヵ月間投与した際の忍容性は良好で
あることが示された。
日本における申請効能効果は、骨折の危険性の高い骨粗鬆症であり、用法用量は 1 日
1 回テリパラチド(遺伝子組換え)として 20 μg を皮下注射する。なお、本剤の投与は
18 ヵ月間までとすることとする。
2.5.1.5 外国臨床データの利用計画及び臨床データパッケージ
本剤の製造販売承認申請に含まれる、有効性評価及び安全性評価に用いた臨床試験一
覧を以下の表に示す(表 2.5.1-2)。国内プラセボ対照二重盲検比較試験である GHDB
試験をブリッジング試験、外国プラセボ対照二重盲検比較試験である GHAC 試験をブリ
ッジング対象試験として承認申請を実施する。

LY333334 2.5 臨床に関する概括評価
2.5.1 製品開発の根拠
12
表 2.5.1-2. 承認申請に用いる臨床試験の一覧
試験の種類 評価資料 参考資料
国内プラセボ対照二重盲検
比較試験
GHCS、GHDB(ブリッジ
ング試験)-
外国プラセボ対照二重盲検
比較試験
GHAC(ブリッジング対象
試験)、GHAJGHAA
テリパラチド投与中止後の
追跡調査研究GHBJ -
実薬対照試験 GHBM GHBZ、GHAH、GHCXテリパラチド投与後にラロ
キシフェンの継続投与を検
討した試験
GHCA GHBQ
日本人及び外国人を対象と
した臨床薬理試験GHCO -
外国人を対象とした臨床薬
理試験
GHBO、GHAD、GHAE、GHAW、GHBA、GHBC、GHBI、GHBR
GHAB、GHAK、GHAM、
GHAN、GHAS、GHAT、GHBF、GHCE
アジアで実施した試験 - GHCB、GHCC、GHCF骨吸収抑制剤の前治療又は
併用を検討した試験- GHCM、GHCK、GHCD
ホルモン補充療法の併用を
検討した試験- GHAF
ペン型注入器のコンプライ
アンスを検討した試験- GHCP
早期に中止された試験(有
効性の解析対象とならなか
った試験)
-GHAL、GHAV、GHAU、GHCU

LY333334 2.5 臨床に関する概括評価
2.5.2 生物薬剤学に関する概括評価
13
2.5.2 生物薬剤学に関する概括評価
2.5.2.1 バイオアベイラビリティ
外国人健康成人を対象にテリパラチドを皮下及び静脈内に投与し、皮下投与時の絶対
的バイオアベイラビリティの評価を行った(GHBI 試験)。
母集団薬物動態解析によるテリパラチド皮下投与時の絶対的バイオアベイラビリティ
の推定値は約 95%であり、テリパラチドは皮下投与によりほぼ完全に吸収されると考え
られた(第 2.7.1.2.1 項参照)。また、テリパラチドは皮下へ投与するため、テリパラチ
ドの吸収に食事のタイミングや胃内 pH が影響を及ぼさないと考えられる。
2.5.2.2 投与部位の違いによる薬物動態への影響
外国人女性骨粗鬆症患者(GHAC 試験)及び男性骨粗鬆症患者(GHAJ 試験)を対象
とした第 III 相試験において、母集団薬物動態解析によりテリパラチドの薬物動態の評
価を行った。両試験において、テリパラチドの薬物動態に影響を及ぼす可能性がある因
子の 1 つとして、投与部位の違い(大腿部及び腹壁)についても検討を行った。
GHAC 試験及び GHAJ 試験における母集団薬物動態解析の結果、大腿部へ皮下注射し
たときには腹壁への投与時に比べて見かけの分布容積(V/F)がそれぞれ 21%及び 30%
増加すると推定された(第 2.7.1.2.2 項参照)。母集団薬物動態パラメータを用いたシミ
ュレーションの結果、大腿部への投与時には腹壁への投与時に比べて最高血清中テリパ
ラチド濃度(Cmax)がわずかに低下すると予測された。血清中テリパラチド濃度-時間曲
線下面積(AUC)に投与部位による違いは認められなかった。投与部位の違いによる
Cmax の差は小さく、腹壁及び大腿部のいずれに皮下注射しても、テリパラチドの有効性
及び安全性に関して臨床上問題とはならないと考えられた。

LY333334 2.5 臨床に関する概括評価
2.5.3 臨床薬理に関する概括評価
14
2.5.3 臨床薬理に関する概括評価
2.5.3.1 健康成人による薬物動態
テリパラチドを単回皮下投与したとき、テリパラチドは投与後すぐに最高血清中濃度
(Cmax)に到達した後、速やかに 1 相性に消失した(GHCO 試験)。日本人及び外国人
のいずれにおいても、テリパラチド皮下投与後の血清中テリパラチド濃度-時間曲線下面
積(AUC)及び Cmaxは投与量の増加にしたがって増加した。また、テリパラチドを 1 日
1 回 14 日間反復投与したとき蓄積性は認められなかった(GHAD 試験)。
日本人にテリパラチド 40 µg を単回投与したときの Cmax 及び AUC の平均値は、外国
人の値と比べてそれぞれ約 30%及び 40%高値であった。それぞれのパラメータを体重で
補正することにより、Cmax 及び AUC の平均値は日本人と外国人ほぼ同様となったこと
から、日本人と外国人における薬物動態の違いは体重の違いにより説明できると考えら
れた(第 2.7.2.2.2.1 項参照)。また、日本人と外国人において安全性の結果に差は認め
られなかったことから(第 2.7.4.2.1.1.11.1 項参照)、体重の違いによる投与量調節は必
要ないと考えられた。
2.5.3.2 骨粗鬆症患者における薬物動態
日本人女性骨粗鬆症患者を対象とした第 II 相試験(GHCS 試験)並びに外国人女性骨
粗鬆症患者及び外国人男性骨粗鬆症患者を対象とした第 III 相試験(それぞれ GHAC 試
験及び GHAJ 試験)において、母集団薬物動態解析によりテリパラチドの薬物動態の評
価を行った(第 2.7.2.2.3 項参照)。
いずれの試験においても、母集団薬物動態解析により体重が見かけの分布容積
(V/F)に対する有意な共変量であった。体重の増加するにしたがって V/F が増加し、
その結果、Cmax が低下すると推定された。また、GHAJ 試験においてクレアチニンクリ
アランスが見かけのクリアランス(CL/F)に対する有意な共変量であった。クレアチニ
ンクリアランスが低下するにしたがって CL/F が低下すると推定されたが、その影響の
程度は小さく、臨床的には問題とならないと考えられた。
骨粗鬆症患者と健康成人におけるテリパラチドの薬物動態は同様であった(第
2.7.2.3.1 項参照)。
2.5.3.3 代謝及び排泄
PTH(1-34)の分解には、肝細胞や他の組織中のタンパク分解酵素のほか、クッパー
細胞やその他のマクロファージなどいくつもの因子が関わっていると思われる(Segre et
al. 1981, Bringhurst et al. 1988, Daugaard 1996, Murray et al. 2005)。また、PTH(1-34)は
腎、肝、肺などで分解されることが報告されている(Liao et al. In press)。テリパラチ
ドは PTH(1-34)とアミノ酸配列が同じであり、PTH(1-34)と同様に分解されて排泄
されると考えられる。

LY333334 2.5 臨床に関する概括評価
2.5.3 臨床薬理に関する概括評価
15
2.5.3.4 カルシウム濃度に対する影響
テリパラチドを 1 日 1 回反復投与したとき、投与の約 2 時間後に血清中カルシウム濃
度が上昇し始め、約 4~6 時間後に最高濃度に達した。その後、投与 16~24 時間後には
ベースラインまで低下した。いずれの試験においてもカルシウム濃度の上昇は一過性の
ものであった。また、尿中へのカルシウム排出に影響を及ぼす利尿薬をテリパラチドと
併用したとき、血清中及び尿中カルシウム濃度の変化に臨床的に問題となるような変化
は認められなかった。
2.5.3.5 特殊集団
外国人の軽度及び中等度の腎障害を有する患者(クレアチニンクリアランス:31~
75 mL/min)にテリパラチド 40 µg を単回皮下投与したとき、腎機能が正常な被験者(ク
レアチニンクリアランス:90 mL/min 以上)と比べて薬物動態パラメータに有意な違い
は認められなかった。しかし、重度の腎障害を有する患者(クレアチニンクリアラン
ス:30 mL/min 以下)にテリパラチド 40 µg を単回皮下投与したとき、腎機能が正常な
被験者と比べて AUC0-∞が 89%高く、t1/2が 77%延長した。一方、Cmaxや投与 3 時間後ま
での AUC(AUC0-3)に有意な差は認められなかった。重度の腎障害を有する患者では血
中からのテリパラチドの消失に遅延が認められているため、慎重に投与する必要がある
(GHAW 試験)。
NYHA(New York Heart Association)心機能分類でクラス 1~3 に分類された心不全患
者にテリパラチド 20 µg を単回皮下投与したところ、健康成人における薬物動態(GHBI
試験)とほぼ同様の結果が得られた。また、血圧や心拍数にも臨床上問題となるような
差は認められず、心不全患者にテリパラチドを投与するときに投与量の調整は必要ない
と考えられた(GHBC 試験)。
軽度又は中等度の高血圧を有する患者にテリパラチド 40 µg を β 遮断薬又はカルシウ
ム拮抗薬と併用投与した。β 遮断薬又はカルシウム拮抗薬併用の有無に関わらず、テリ
パラチドを投与することにより、血圧がわずかに低下する傾向が認められたが、β 遮断
薬又はカルシウム拮抗薬により血圧低下が増強されることはなかった。したがって、軽
度又は中等度の高血圧を有する患者にテリパラチドを投与するときに投与量の調整は必
要ないと考えられ、また、テリパラチドと β 遮断薬又はカルシウム拮抗薬を併用投与す
ることが可能であると考えられた(GHAE 試験)。
2.5.3.6 薬物相互作用
テリパラチドは PTH(1-34)と同一のアミノ酸配列である。PTH(1-34)は肝細胞や
他の組織中のタンパク分解酵素のほか、クッパー細胞やその他のマクロファージなどで
速やかに分解されることが知られており、テリパラチドはチトクロム P450(CYP)酵素
による代謝を受けないと考えられる。また、カニクイザルの肝ミクロソームを用いた in
vitro 試験において、テリパラチドは CYP 酵素に影響を与えないことが明らかになって
いる。これらのことから、テリパラチドと他の薬剤を併用したとき、CYP 酵素を介した
薬物動態学的相互作用を生じる可能性は低いと考えられる。

LY333334 2.5 臨床に関する概括評価
2.5.3 臨床薬理に関する概括評価
16
また、テリパラチドは血清カルシウム濃度を一過性に増加させる薬理作用を有してい
る。また、海外における臨床薬理試験では、有害事象のひとつとして起立性低血圧が認
められている。これらのことから、カルシウム拮抗薬、アテノロール(β 遮断薬)、フ
ロセミド(ループ利尿薬)、ヒドロクロロチアジド(利尿薬)及びジゴキシンとの薬物
相互作用の有無を検討した。
いずれの薬剤においてもテリパラチドとの薬物動態学的又は薬力学的な薬物相互作用
は認められなかった。したがって、これらの薬剤と併用するとき、投与量調整の必要は
ないと考えられた。
2.5.3.7 薬力学的解析
外国人女性骨粗鬆症患者を対象とした第 III 相試験(GHAC 試験)において、腰椎及
び大腿骨頸部の骨密度(BMD)の変化を指標として母集団薬力学解析を行った。
テリパラチド投与量が 40 μg のとき、20 μg 投与時に比べ各時点における腰椎 BMD 変
化量が 52%上昇すると推定された。また、高齢の患者は若齢者に比べ、ベースラインの
腰椎 BMD が低い患者は高い患者に比べ、尿中 N-テロペプチドのベースラインが高い患
者は低い患者に比べ腰椎 BMD 変化量の増加が大きいと推定された。
テリパラチド投与量が 40 μg のとき、20 μg 投与時に比べ各時点における大腿骨頸部
BMD 変化量が 69%上昇すると推定された。また、高齢の患者は若齢の患者に比べ、ベ
ースラインの尿中 N-テロペプチドが高い患者は低い患者に比べ、体重が重い患者は軽い
患者に比べ大腿骨頸部 BMD 変化量の増加が大きいと推定された。

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
17
2.5.4 有効性の概括評価
2.5.4.1 有効性評価の概略
テリパラチドに関する外国臨床データの日本人骨粗鬆症患者への外挿可能性について、
「外国臨床データを受け入れる際に考慮すべき民族的要因について(医薬審第 672 号、
1998 年 8 月 11 日)」(厚生省 1998)を参照して、日本人と外国人における外因性要因、
内因性要因及び薬物動態を比較した結果、テリパラチドは民族的要因の影響を受けにく
い薬剤であると考えられた。更に、外国臨床データとの類似性を検討するため、国内に
おいてプラセボ対照二重盲検比較試験(国内第 III 相 GHDB 試験)をブリッジング試験
として実施し、ブリッジング対象試験(外国第 III 相 GHAC 試験)及び他のプラセボ対
照二重盲検比較試験(国内第 II 相 GHCS 試験及び外国第 III 相 GHAJ 試験)の試験成績
との類似性を検討した。ブリッジング対象試験である GHAC 試験結果などを考察するこ
とにより、あらかじめブリッジングの成立要件を設定し、それに基づき外国臨床データ
の外挿可能性を判断した上で、外国臨床データを用いた有効性評価を行った。
骨粗鬆症患者に対するテリパラチドの有効性は、国内で実施した試験 2 試験及び外国
で実施した試験 17 試験の計 19 試験において検討した(表 2.5.4-1)。19 試験中 7 試験を
有効性評価の評価資料とし、12 試験を参考資料とした。
表 2.5.4-1. 有効性評価のための評価資料及び参考資料
試験の種類 評価資料 参考資料
国内プラセボ対照二重盲検比較試
験
GHCSGHDB(ブリッジング試験)
-
外国プラセボ対照二重盲検比較試
験GHAC(ブリッジング対象試験)GHAJ GHAA
テリパラチド投与中止後の追跡調
査研究GHBJ -
実薬対照試験 GHBM GHBZ、GHAH、GHCXテリパラチド投与後にラロキシフ
ェンの継続投与を検討した試験GHCA GHBQ
アジアで実施した試験 - GHCB、GHCC、GHCF骨吸収抑制剤の前治療又は併用を
検討した試験- GHCM、GHCK、GHCD
ホルモン補充療法の併用を検討し
た試験- GHAF
2.5.4.2 プラセボ対照二重盲検比較試験
2.5.4.2.1 試験デザインの概略
国内及び外国で実施したプラセボ対照二重盲検比較試験の対象は「骨折の危険性の高
い骨粗鬆症患者」であり、GHDB 試験は原発性骨粗鬆症患者(閉経後女性及び男性)、
GHCS 試験及び GHAC 試験は閉経後骨粗鬆症女性患者、GHAJ 試験は原発性骨粗鬆症男
性患者を対象とした。
国内で実施した GHDB 試験では、原発性骨粗鬆症の診断基準(折茂ほか 2001)、並
びに骨折の危険因子である年齢及び低 BMD を考慮して、以下の 3 基準のいずれかを満
たす 55 歳以上の閉経後女性及び男性を組み入れた。

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
18
1. 腰椎 BMD が YAM の 80%1未満であり、かつ脆弱性椎体骨折を 1 個以上有する患
者
2. 腰椎 BMD が YAM の 70%1未満であり、かつ年齢が 65 歳以上の患者
3. 腰椎 BMD が YAM の 65%1未満であり、かつ年齢が 55 歳以上の患者
外国で実施した GHAC 試験では、30 歳から 85 歳の既存椎体骨折を有する閉経後女性
を対象としており、GHDB 試験及び GHAC 試験の選択基準には違いがあった。しかしな
がら、既存骨折がない場合でも、BMD が約 1 標準偏差(以下、SD とする)低下した場
合の骨折の危険性は、既存骨折を有する場合とほぼ同程度であること(藤原 2005)、年
齢が 65 歳で T スコアが-2.5 である白人女性が 5 年間に骨折する危険性は、年齢が 55 歳
で T スコアが-3.0 である白人女性とほぼ同程度であること(Cummings et al. 2002)が報
告されている。そのため、既存椎体骨折に加え、年齢及び低 BMD で規定した GHDB 試
験の 3 基準における骨折の危険性は、基準間で同程度と考えられた。また、既存椎体骨
折、年齢及び低 BMD は男性においても骨折の危険因子であり、既存椎体骨折、年齢及
び BMD で調整すると、椎体骨折の発生率について男女間に大きな違いは認められなか
った(Fujiwara et al. 2003)。以上のことから、骨折の危険性の観点では、GHDB 試験と
GHAC 試験の対象患者との間に、大きな違いはないと考えられ、両試験間の比較を行う
ことは妥当であると判断した。GHCS 試験では GHDB 試験と同様に、既存椎体骨折、年
齢及び低 BMD で規定した選択基準を用い、55 歳以上の閉経後女性を組み入れた。
GHAJ 試験では、30 歳から 85 歳の T スコアが-2.0 以下である男性を組み入れた。
治験薬は、ペン型注入器を用いた自己注射により、1 日 1 回皮下投与した。また、基
礎治療として、全被験者にカルシウム及びビタミン D を投与した。
用量については、国内第 II 相試験である GHCS 試験では、日本人患者における用量反
応関係を検討するために、プラセボ群、テリパラチド 10 g 群、20 g 群及び 40 g 群を
設定した。国内第 III 相試験である GHDB 試験では、GHCS 試験の結果からテリパラチ
ド 20 g 群を設定し、プラセボ群と比較した。外国で実施した GHAC 試験及び GHAJ 試
験では、プラセボ群、テリパラチド 20 g 群及び 40 g 群を設定した。
2.5.4.2.2 有効性の評価項目
国内で実施した GHCS 試験及び GHDB 試験、並びに外国で実施した GHAJ 試験では、
有効性の主要評価項目を腰椎 BMD とした。一方、外国で実施した GHAC 試験の主要評
価項目は、新規椎体骨折であった。ブリッジング試験(GHDB 試験)、ブリッジング対
象試験(GHAC 試験)及び他のプラセボ対照二重盲検比較試験間の有効性の類似性は、
腰椎(L1-L4)BMD を指標として検討した。
骨粗鬆症治療の第一の目的は骨折の抑制であるが、BMD が治療効果を判定するサロ
ゲートマーカーとして広く用いられている。テリパラチドにおける椎体骨折抑制効果に
対する BMD の寄与率は 30%から 41%であり、骨吸収抑制剤における寄与率と比較して
1 YAM の 80%は T スコア-1.7、70%は T スコア-2.6、65%は T スコア-3.0 程度に相当すると考えら
れる(Orimo et al. 1998)。

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
19
高いと考えられている(Chen et al. 2006)。また、テリパラチドは骨微細構造を改善し
(第 2.5.4.2.4.2.1 項、Jiang et al. 2003)、この海綿骨微細構造の改善は、腰椎及び大腿骨
頸部 BMD の増加と相関していることが報告されている(Chen et al. 2007)。更に、腰椎
の定量的コンピュータ断層撮影法(以下、QCT とする)を用いた有限要素解析の結果、
アレンドロネートに比べてテリパラチドにおいてより高い腰椎骨強度の増加が認められ
ること、アレンドロネートでは DXA で測定した腰椎 BMD と有限要素解析で得られた腰
椎骨強度との間にほとんど相関関係がなかったが、テリパラチドではある程度の相関が
認められることが報告された(Keaveny et al. 2007)。以上のことから、骨形成促進剤で
あるテリパラチドは BMD 増加を介した骨微細構造改善及び骨強度増加効果を有するた
めに、骨吸収抑制剤に比べて BMD 増加と骨折抑制効果との関連性が高いと考えられる。
そのため、テリパラチドによる治療において、BMD は骨粗鬆症における骨折抑制効果
のサロゲートマーカーとして有効であると考えられた。
腰椎 BMD 及び椎体骨折のほか、有効性の副次的評価項目として、非椎体骨折、大腿
骨頸部及び大腿骨近位部を含む他の部位の BMD、骨形成マーカー〔血清 I 型プロコラー
ゲン-N-プロペプチド(以下、PINP とする)、血清 I 型プロコラーゲン-C-プロペプチド
(以下、PICP とする)、血清骨型アルカリフォスファターゼ(以下、BAP とする)〕、
骨吸収マーカー〔尿中 I型コラーゲン架橋 N-テロペプチド(以下、NTX とする)、血清
I 型コラーゲン架橋 C-テロペプチド(以下、CTX とする)、尿中遊離デオキシピリジノ
リン(以下、DPD とする)〕、骨微細構造、並びに腰背部痛への効果を評価し、骨粗鬆
症治療におけるテリパラチドの有効性を多面的に検討した。
2.5.4.2.3 有効性の評価の対象となった患者集団の特性
55 歳以上の閉経後骨粗鬆症女性患者を対象とした国内第 II 相 GHCS 試験では、154 例
(プラセボ群 38 例、テリパラチド 10 μg 群 38 例、20 μg 群 39 例及び 40 μg 群 39 例)を
解析対象とした。全被験者の平均年齢は 71.0 歳、平均体重は 48.18 kg、平均腰椎(L1-
L4)BMD は 0.6005 g/cm2であり、既存骨折なしの被験者の割合は 57.1%であった。
55 歳以上の原発性骨粗鬆症患者を対象とした国内第 III 相 GHDB 試験では、203 例
(プラセボ群 67 例及びテリパラチド 20 μg 群 136 例)を解析対象とした。203 例中 14 例
(プラセボ群 5 例及び 20 μg 群 9 例)は男性であった。その他の 189 例の被験者は閉経
後女性であった。全被験者の平均年齢は 69.6 歳、平均体重は 49.24 kg、平均腰椎(L1-
L4)BMD は 0.6143 g/cm2であり、既存骨折なしの被験者の割合は 59.1%であった。国内
で実施した GHCS 試験及び GHDB 試験の人口統計学的及びその他のベースラインの特
性は、試験間で同様であった。国内試験では、骨折の危険性の高い骨粗鬆症患者として、
原発性骨粗鬆症の診断基準、並びに骨折の危険因子である年齢及び低 BMD を考慮して
被験者を組み入れており、申請効能効果を骨折の危険性の高い骨粗鬆症としているため、
市販後に実際にテリパラチドが投与されると予想される集団と試験対象集団との間に大
きな差異はないと考えられた。
30 歳から 85 歳の閉経後骨粗鬆症女性患者を対象とした外国第 III 相 GHAC 試験では、
1637 例(プラセボ群 544 例、テリパラチド 20 μg 群 541 例及び 40 μg 群 552 例)を解析

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
20
対象とした。全被験者の平均年齢は 69.5 歳、平均体重は 65.80 kg、平均腰椎(L1-L4)
BMD は 0.8204 g/cm2 であり、既存骨折なしの被験者の割合は 9.3%であった。国内で実
施した GHCS 試験及び GHDB 試験と比べて、外国で実施した GHAC 試験ではベースラ
インにおける体重及び腰椎 BMD の値が高く、既存骨折なしの被験者の割合が低かった。
30 歳から 85 歳の原発性骨粗鬆症男性患者を対象とした外国第 III 相 GHAJ 試験では、
437 例(プラセボ群 147 例、テリパラチド 20 μg 群 151 例及び 40 μg 群 139 例)を解析対
象とした。全被験者の平均年齢は 58.7 歳、平均体重は 75.71 kg、平均腰椎(L1-L4)
BMD は 0.8724 g/cm2 であった。女性を対象とした GHAC 試験と比べて、男性を対象と
した GHAJ 試験ではベースラインにおける年齢が低く、体重及び腰椎 BMD の値が高か
った。
2.5.4.2.4 有効性の結果
2.5.4.2.4.1 国内プラセボ対照二重盲検比較試験の成績
2.5.4.2.4.1.1 GHCS 試験(国内第 II 相試験)
GHCS 試験の治験薬投与期間は 6 ヵ月(24 週)であった。主要評価項目である腰椎
(L2-L4)BMD のベースラインから最終観察時までの平均変化率は、プラセボ群で
0.66%、テリパラチド 10 g 群で 5.80%、20 μg 群で 6.40%、40 μg 群で 11.47%であった。
Williams の検定による主要解析の結果、10 μg 群、20 μg 群及び 40 μg 群は、プラセボ群
に比べ投与量の増加に伴って統計学的に有意な増加を示した(すべての投与群について
p<0.001)。
副次的評価項目である骨代謝マーカー(血清 PINP、血清 PICP、血清 BAP 及び血清
CTX)の変化を評価した結果、20 μg 群及び 40 μg 群ではいずれの骨代謝マーカーにも上
昇が認められたが、10 μg 群では骨形成マーカーである血清 BAP 及び骨吸収マーカーで
ある血清 CTX の上昇は認められなかった。安全性の結果から、40 μg 群では因果関係を
否定できない有害事象及び有害事象による中止が、他の投与群に比べて多く認められた。
以上の結果から、主要評価項目である腰椎 BMD について、テリパラチド 10 g 群、
20 g 群及び 40 μg 群は、プラセボ群に比べ投与量の増加に伴った統計学的に有意な増加
を示し、用量反応関係が示された。副次的評価項目である骨代謝マーカーにおける
10 g 群の結果及び安全性における 40 μg 群の結果を考慮すると、日本人骨粗鬆症患者に
おける臨床推奨用量は、20 μg と考えられた。
2.5.4.2.4.1.2 GHDB 試験(国内第 III 相、ブリッジング試験)
GHDB 試験の治験薬投与期間は、24 ヵ月(104 週)である。GHDB 試験の治験薬投与
期間は 3 期からなり、第 1 期の二重盲検比較期間 12 ヵ月(52 週)のデータを、主要解
析及びブリッジング対象試験との類似性の検討に用いた。主要評価項目である腰椎
(L2-L4)BMD のベースラインから第 1 期二重盲検比較期間の最終観察時までの平均変
化率は、プラセボ群で 0.04%、テリパラチド 20 g 群で 9.82%であった。2 標本 t 検定に
よる主要解析の結果、投与群間に統計学的に有意な差が認められた(p<0.001)。

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
21
副次的評価項目であるベースラインから第 1 期二重盲検比較期間の最終観察時までの
大腿骨頸部及び大腿骨近位部 BMD の平均変化率は、プラセボ群ではそれぞれ 0.46%及
び-0.22%、20 g 群ではそれぞれ 2.24%及び 2.66%であった。骨代謝マーカーは、20 g
群で投与開始 1 ヵ月後から血清 PINP の上昇が認められ、3 ヵ月後から血清 CTX の上昇
が認められた。血清 BAP の上昇は認められなかった。第 1 期二重盲検比較期間に新規椎
体骨折が認められた被験者の割合は、プラセボ群及び 20 μg 群でそれぞれ 6.0%(4/67)
及び 3.7%(5/136)、脆弱性非椎体骨折が認められた被験者の割合はそれぞれ 1.5%
(1/67)及び 0.7%(1/136)であった。骨折抑制の検証を主要目的にしていないため統
計学的な仮説検定は実施していないが、いずれの骨折が認められた被験者の割合も、プ
ラセボ群に比べて 20 μg 群において低かった。腰背部痛の程度の変化に対する効果を検
討した結果、テリパラチドの明らかな影響は認められなかった。
更に、第 1 期二重盲検比較期間の後に第 2 期オープン期間〔6 ヵ月(24 週)〕を実施
した結果、テリパラチド 20 g を 18 ヵ月間投与した群において、BMD の増加は投与期
間中継続して観察された。また、骨形成マーカーである血清 PINP 及び骨吸収マーカー
である血清 CTX の上昇も継続して認められた。更に、テリパラチド 20 g を 18 ヵ月間
投与した群において、第 2 期オープン期間に新規椎体骨折又は椎体骨折の悪化は認めら
れず、治験担当医師の判定による椎体骨折及び非椎体骨折も認められなかった。
以上の結果から、主要評価項目である腰椎 BMD において、テリパラチド 20 g のプ
ラセボに対する優越性が認められ、日本人骨粗鬆症患者に対する有効性が確認された。
2.5.4.2.4.1.2.1 外国臨床データの日本人骨粗鬆症患者への外挿可能性の検討
2.5.4.2.4.1.2.1.1 外挿可能性の評価方法
GHDB 試験はブリッジング試験として実施した試験であり、ブリッジング対象試験
(GHAC 試験)及び他のプラセボ対照二重盲検比較試験(GHCS 試験及び GHAJ 試験)
の試験成績との類似性を検討した。ブリッジングが成立したと判断するための要件及び
類似性の検討方法をあらかじめ設定するために、GHAC 試験におけるベースラインの腰
椎 BMD の値による部分集団解析の結果を検討したところ、テリパラチド投与後の腰椎
BMD 変化率は、ベースラインにおける腰椎 BMD の値が低いほど高くなることが示唆さ
れた。ベースラインにおける腰椎 BMD の値は、各民族における YAM の値の違いから、
外国人に比べて日本人において低いと考えられた。そのため、テリパラチド投与後の腰
椎 BMD 変化率は、外国人に比べて日本人において高くなることが予想された。以上の
考察に基づき、有効性における類似性の検討において、有効性のブリッジングが成立し
たと判断するための要件及び検討方法を、あらかじめ以下のように設定した。
ベースラインから 12 ヵ月後の腰椎(L1-L4)BMD 変化率のテリパラチド 20 μg 群と
プラセボ群との差は、外国人の結果に比べて日本人において大きいが、日本人の結
果から、12 ヵ月後の腰椎(L1-L4)BMD 変化率についてテリパラチド 20 g 群のプ
ラセボ群に対する優越性が示され、外国人と同様の結果が得られる。

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
22
有効性のブリッジング成立要件の可否は、ベースラインから 12 ヵ月後の腰椎(L1-
L4)BMD 変化率について、テリパラチド 20 μg 群とプラセボ群との差の 95%信頼区間
の下限値が 0 を超えることが、いずれの試験においても確認できるかどうかにより判断
する。変化量についても、同様の検討を行う。用量反応性の比較は、GHCS 試験及び
GHAC 試験の結果を用いて行い、各テリパラチド用量に対するベースラインから 6 ヵ月
後の腰椎(L1-L4)BMD 変化率及び変化量の平均値及び標準偏差を図示し、試験間の用
量反応の類似性を確認する。また、GHDB 試験及び GHAC 試験において、被験者ごとの
ベースラインにおける腰椎(L1-L4)BMD の値に対する 12 ヵ月時点の腰椎(L1-L4)
BMD 変化率又は変化量を散布図として示し、両試験におけるテリパラチド投与による
反応とベースラインの値との関連性を検討する。各民族集団の YAM を基準とし、骨粗
鬆症の診断基準に用いられる T スコアは、骨粗鬆症の診断の観点から両試験で同様に扱
うことができると考えられることから、両試験の被験者ごとのベースラインにおける T
スコアに対する 12 ヵ月時点の腰椎(L1-L4)BMD 変化率又は変化量についても、散布
図を用いて検討する。更に、GHAJ 試験、GHAC 試験及び GHDB 試験における腰椎
BMD の結果に基づき、性別ごとの結果を比較する。また、骨代謝マーカーは、各試験
において測定した種類ごとに、経時的変化を試験間で比較する。
これらの検討結果に加えて、安全性に関する成績を比較した上で、総合的に外国臨床
データ、特に骨折抑制効果の日本人骨粗鬆症患者への外挿可能性を評価することとした。
2.5.4.2.4.1.2.1.2 外国臨床データの外挿可能性の検討
ブリッジング試験(GHDB 試験)、ブリッジング対象試験(GHAC 試験)及び他のプ
ラセボ対照二重盲検比較試験の有効性における類似性を検討したところ、以下の結果が
得られ、有効性のブリッジング成立要件を満たすことが確認された。
GHDB 試験及び GHAC 試験において、ベースラインから 12 ヵ月後の腰椎(L1-L4)
BMD 変化率のテリパラチド群とプラセボ群との差〔平均値(95%信頼区間)〕は、
GHDB 試験で 10.20%(8.57%, 11.84%)及び GHAC 試験で 7.41%(6.70%, 8.12%)で
あり、いずれの試験においても、テリパラチド 20 g 群のプラセボ群に対する優越
性が認められた(表 2.5.4-2、図 2.5.4-1)。ベースラインから 12 ヵ月後の腰椎(L1-
L4)BMD 変化率のテリパラチド群とプラセボ群との差の平均値は、GHAC 試験に
比べて GHDB 試験において高かった。
腰椎(L1-L4)BMD 変化量についても、いずれの試験においてもテリパラチド 20 μg
群のプラセボ群に対する優越性が示された(表 2.5.4-2、図 2.5.4-1)。ベースラインから
12 ヵ月後の腰椎(L1-L4)BMD 変化量のテリパラチド群とプラセボ群との差の平均値は、
両試験間で同程度であった。
GHCS 試験及び GHAC 試験のいずれにおいても、テリパラチド投与量の増加に伴って
6 ヵ月後の腰椎(L1-L4)BMD 変化率及び変化量が増加し、ほぼ同様の用量反応性が示
された(図 2.5.4-2)。

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
23
テリパラチド投与による反応とベースラインの腰椎 BMD の値との関連性を散布図を
用いて検討した結果、GHDB 試験の被験者は、GHAC 試験の被験者に比べてベースライ
ンの腰椎(L1-L4)BMD が低い範囲に分布を示し、いずれの試験においても、ベースラ
インにおける腰椎(L1-L4)BMD の値が低いほど、12 ヵ月時点の腰椎(L1-L4)BMD の
変化率が高くなる傾向が認められた。また、腰椎 BMD(L1-L4)変化量は、変化率に比
べるとベースラインの値の影響を受けにくいことが示された。ベースラインにおける T
スコアとの関連性を検討した結果からも、骨粗鬆症の診断の観点からは GHDB 試験と
GHAC 試験の被験者集団は類似した集団であると考えられ、テリパラチド投与による反
応とベースラインの T スコアとの関連性が、両試験間で類似していることが認められた。
そのため、テリパラチド投与による腰椎 BMD の反応性について、両試験間で本質的な
違いがないことが示された。
更に、テリパラチド投与による腰椎 BMD の反応性に男女差は認められなかった。ま
た、骨代謝マーカーは、いずれの試験においても同様の経時的変化を示した。安全性に
関する成績についても、日本人に特徴的な有害事象の発現は認められなかった。以上の
ことから、外国臨床データの日本人骨粗鬆症患者への外挿は可能であると判断した。
表 2.5.4-2. ベースラインから投与後 12 ヵ月時点の腰椎(L1-L4)骨密度(BMD)
変化率(%)及び変化量(g/cm2)の比較(GHDB 試験及び GHAC 試験)
Study Treatment Group N Mean SD SE 95% CIPercent Change
GHACPlacebo 467 0.84 4.87 0.23 -
LY20 466 8.25 6.10 0.28 -Difference (LY20-Placebo) - 7.41 5.52 0.36 (6.70, 8.12)
GHDBPlacebo 60 0.23 4.44 0.57 -
LY20 121 10.43 5.61 0.51 -Difference (LY20-Placebo) - 10.20 5.25 0.83 (8.57, 11.84)
Change
GHACPlacebo 467 0.0065 0.0373 0.0017 -
LY20 466 0.0632 0.0426 0.0020 -Difference (LY20-Placebo) - 0.0567 0.0400 0.0026 (0.0515, 0.0618)
GHDBPlacebo 60 0.0008 0.0256 0.0033 -
LY20 121 0.0630 0.0313 0.0028 -Difference (LY20-Placebo) - 0.0623 0.0296 0.0047 (0.0530, 0.0715)
SD = 標準偏差、SE = 標準誤差、95% CI = 95%信頼区間
Placebo = プラセボ、LY20 = テリパラチド 20 μg、Difference (LY20-Placebo) = テリパラチド 20 μg群とプラセボ
群との差

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
24
Mean with 95% Confidence IntervalBMD = Bone Mineral Density(骨密度)
LY20 = テリパラチド 20 μg、Placebo = プラセボ
LY20-Placeboは、テリパラチド 20 μg群とプラセボ群との差を示す。
図 2.5.4-1. ベースラインから投与後 12 ヵ月時点の腰椎(L1-L4)骨密度(BMD)
変化率(左図)及び変化量(右図)の比較(GHDB 試験及び GHAC 試験)
Mean with Standard DeviationBMD = Bone Mineral Density(骨密度)
Placebo = プラセボ、LY10 = テリパラチド 10 μg,、LY20 = テリパラチド 20 μg,、LY40 = テリパラチド 40 μg
図 2.5.4-2. 投与後 6 ヵ月時点の腰椎(L1-L4)骨密度(BMD)変化率(左図)及び
変化量(右図)における用量反応関係(GHCS 試験及び GHAC 試験)
2.5.4.2.4.2 外国プラセボ対照二重盲検比較試験の成績
2.5.4.2.4.2.1 GHAC 試験(外国第 III 相、ブリッジング対象試験)
GHAC 試験の治験薬投与期間は 3 年の予定であったが、治験依頼者の判断により途中
で試験を中止したため(第 2.7.3.1.1 項参照)、実際の治験薬投与期間(中央値)は 19
ヵ月であった。以下に評価項目ごとに結果を記載する。
(1) 椎体骨折
主要評価項目である新規椎体骨折が認められた被験者の割合は、プラセボ群 14.3%
(64/448)、テリパラチド 20 μg 群 5.0%(22/444)、40 μg 群 4.4%(19/434)であった

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
25
(表 2.5.4-3)。カイ二乗検定の結果、いずれのテリパラチド群においても、プラセボ群
との間に統計学的に有意な差が認められた(いずれも p<0.001)。プラセボ群に対する
新規椎体骨折発生の相対的なリスクの減少は、20 μg 群で 65%、40 μg 群で 69%であった。
複数、重度、並びに中等度又は重度の新規椎体骨折が認められた被験者の割合もテリパ
ラチド群において低下し、プラセボ群との間に統計学的に有意な差が認められた(いず
れも p<0.05)。プラセボ群に対する相対的なリスクの減少は、複数の新規椎体骨折では、
20 μg 群及び 40 μg 群でそれぞれ 77%及び 86%、重度の新規椎体骨折ではそれぞれ 100%
及び 77%、中等度又は重度の新規椎体骨折ではそれぞれ 90%及び 78%であった。
表 2.5.4-3. 新規椎体骨折の要約(GHAC 試験)
Placebon=448
PTH20n=444
PTH40n=434
Number of patients with ≥1 new fracture (%) 64 (14.3%) 22 (5.0%) 19 (4.4%)Relative risk reduction compared with placebo — 65% 69%Relative risk (95% CI) compared with placebo — 0.347 (0.218, 0.553) 0.306 (0.187, 0.503)Comparison with placebo [a] — p<0.001 p<0.001n = number of patients with evaluable baseline and endpoint x-ray films, CI = confidence interval(信頼区間)
Placebo = プラセボ、PTH20 = テリパラチド 20 μg、PTH40 = テリパラチド 40 μg[a] Pearson’s chi-square test.CSR Table GHAC.11.7から抜粋
(2) 非椎体骨折
副次的評価項目である非椎体骨折が認められた被験者の割合は、プラセボ群 9.7%
(53/544)、テリパラチド 20 μg 群 6.3%(34/541)、40 μg 群 5.8%(32/552)であり、い
ずれのテリパラチド群においても、プラセボ群との間に統計学的な有意差が認められた
(20 μg 群 p=0.036、40 μg 群 p=0.015、カイ二乗検定)。プラセボ群に対する非椎体骨折
発生の相対的なリスクの減少は、20 μg 群で 35%、40 μg 群で 40%であった。非外傷性非
椎体骨折(健康人では通常は骨折を起こさない軽微な外力によって発生した骨折とす
る)が認められた被験者の割合は、プラセボ群 5.5%(30/544)、20 μg 群 2.6%
(14/541)、40 μg 群 2.5%(14/552)であり、いずれのテリパラチド群においても、プ
ラセボ群との間に統計学的な有意差が認められた(20 μg 群 p=0.015、40 μg 群 p=0.012、
カイ二乗検定)。プラセボ群に対する非外傷性非椎体骨折発生の相対的なリスクの減少
は、20 μg 群で 53%、40 μg 群で 54%であった。骨折の非発生持続割合に関する Kaplan-
Meier 曲線において、非椎体骨折では投与後約 12 ヵ月、非外傷性非椎体骨折では投与後
約 9 ヵ月に、プラセボ群の曲線は両テリパラチド群の曲線から乖離し始めた。
(3) 骨密度
副次的評価項目であるベースラインから 18 ヵ月時点までの腰椎 BMD の平均変化率は、
プラセボ群で 1.06%、テリパラチド 20 μg 群で 10.31%、40 μg 群で 14.76%であった。ま
た、ベースラインから 12 ヵ月時点までの大腿骨頸部 BMD の平均変化率は、プラセボ群、
20 μg 群及び 40 μg 群でそれぞれ-0.00%、1.54%及び 3.06%、大腿骨近位部 BMD の平均
変化率はそれぞれ-0.53%、1.70%及び 2.55%であった。腰椎、大腿骨頸部及び大腿骨近
位部 BMD の変化率について、いずれのテリパラチド群においてもプラセボ群との間に

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
26
統計学的に有意な差が認められた(いずれも p<0.001、分散分析)。また、ベースライ
ンから最終観察時までの橈骨超遠位端 BMD の平均変化率は、プラセボ群、20 μg 群及び
40 μg 群で、それぞれ-1.64%、-0.11%及び-1.49%、橈骨遠位 1/3BMD では、それぞれ
-1.28%、-2.07%及び-3.21%であった。橈骨 BMD はテリパラチド群で減少したが、
これは比較的石灰化度の低い新生骨の添加により骨外径が増加し、投影面積が増加した
ための見かけ上の変化と考えられた(Zanchetta et al. 2003)。非外傷性橈骨骨折の発生
した被験者数も、プラセボ群で 544 例中 7 例、20 μg 群で 541 例中 2 例、40 μg 群で 552
例中 3 例であり、プラセボ群に比べてテリパラチド群で増加しなかった。
(4) 骨代謝マーカー
副次的評価項目である骨代謝マーカーについて、テリパラチド投与により骨形成マー
カー(血清 PICP 及び血清 BAP)の上昇が投与開始後 1 ヵ月時点から認められ、その後
遅れて骨吸収マーカー(尿中 NTX 及び尿中 DPD)が上昇した。
(5) 骨微細構造
骨生検は、試験参加前に骨生検の実施について同意の得られた被験者において実施し、
骨生検を実施する被験者は 3 投与群に無作為に割り付けられた。ベースライン及び投与
開始後の両方において評価可能であった 51 例(プラセボ群 19 例、20 μg 群 18 例、40 μg
群 14 例)の腸骨稜の骨生検標本について、2 次元組織形態計測及びマイクロコンピュー
タ断層撮影法による 3 次元組織形態計測を用いた骨微細構造の再解析を実施した(Jiang
et al. 2003)。プラセボ群とテリパラチド 20 μg 群及び 40 μg 群併合群(以下、併合群と
する)との比較は、必要に応じて順位に変換し、t 検定を用いて行った。2 次元組織形態
計測パラメータにおけるベースラインから最終観察時までの変化率(中央値)について、
以下のパラメータで投与群間に統計学的に有意な差が認められた:骨量の増加(併合群
14.3%、プラセボ群-24.0%、p=0.001)及び骨髄腔体積の減少(併合群-15.9%、プラセ
ボ群 112.2%、p=0.004)。3 次元組織形態計測パラメータにおけるベースラインからの変
化率(中央値)については、以下のパラメータで投与群間に統計学的に有意な差が認め
られた:ストラクチャーモデルインデックスの減少(併合群-12.2%、プラセボ群 7.1%、
p=0.025)、連結密度の増加(併合群 19.1%、プラセボ群-14.0%、p=0.034)及び皮質骨
幅の増加(併合群 22.0%、プラセボ群 2.9%、p=0.012)。一方、3 次元組織形態計測パラ
メータにおいて、骨量(併合群 7.2%、プラセボ群-5.0%)及び皮質骨多孔性(併合群
0.69%、プラセボ群-21.0%)の変化について、投与群間に統計学的に有意な差は認めら
れなかった(それぞれ p=0.098 及び p=0.457)。以上のことから、テリパラチド投与によ
り、海綿骨量及び連結密度の増加、骨梁形状のより板状構造へのシフト、並びに皮質骨
幅の増加が認められ、骨粗鬆症により悪化した骨微細構造がより正常な状態に改善され
ることが示された。
以上の結果から、主要評価項目である新規椎体骨折が認められた被験者の割合におい
て、テリパラチド 20 g 及び 40 g のプラセボに対する優越性が示され、テリパラチド

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
27
の 19 ヵ月間(中央値)の投与は、新規椎体骨折の抑制に有効であった。更に、テリパ
ラチド投与により、非椎体骨折の抑制、腰椎、大腿骨頸部及び大腿骨近位部 BMD の増
加、骨代謝マーカーの上昇、並びに骨微細構造の改善効果が示された。
2.5.4.2.4.2.2 GHAJ 試験(外国第 III 相試験)
GHAJ 試験の治験薬投与期間は 2 年の予定であったが、治験依頼者の判断により途中
で試験を中止したため(第 2.7.3.1.1 項参照)、実際の治験薬投与期間(中央値)は 11
ヵ月であった。主要評価項目である腰椎 BMD のベースラインから最終観察時(最大 12
ヵ月)までの平均変化率は、プラセボ群で 0.54%、テリパラチド 20 g 群で 5.73%、
40 g 群で 8.75%であった。また、ベースラインから 12 ヵ月時点までの平均変化率は、
プラセボ群で 0.58%、テリパラチド 20 μg 群で 6.07%、40 μg 群で 9.41%であった。分散
分析を用いて解析した結果、いずれのテリパラチド群においても、プラセボ群との間に
統計学的に有意な差が認められた(いずれも p<0.001)。
副次的評価項目である大腿骨頸部及び大腿骨近位部 BMD のベースラインから 12 ヵ月
時点までの平均変化率は、大腿骨頸部 BMD ではプラセボ群、20 μg 群及び 40 μg 群でそ
れぞれ 0.27%、1.40%及び 3.15%、大腿骨近位部 BMD ではそれぞれ 0.41%、1.28%及び
2.61%であり、いずれのテリパラチド群においてもプラセボ群との間に統計学的に有意
な差が認められた(いずれも p<0.05)。橈骨 BMD はテリパラチド群で減少したが、
GHAC 試験と同じ理由によると考えられた。骨代謝マーカーについて、骨形成マーカー
(血清 BAP 及び血清 PICP)の上昇は、投与開始後 1 ヵ月時点から認められ、その後遅
れて骨吸収マーカー(尿中 NTX 及び尿中 DPD)が上昇した。
以上の結果から、主要評価項目である腰椎 BMD において、テリパラチド 20 g 及び
40 g のプラセボに対する優越性が認められ、男性患者に対する有効性が確認された。
2.5.4.3 実薬対照試験
GHBM 試験はアレンドロネートを対照とする二重盲検比較試験である。主要目的は、
テリパラチド 20 μg 又はアレンドロネート 10 mg を 1 日 1 回 18 ヵ月間投与したときの腰
椎 BMD 変化率を比較することであった。対象は、BMD が T スコア-2.5 以下-4.0 以上
の閉経後骨粗鬆症女性患者であった。基礎治療として全被験者にカルシウム及びビタミ
ン D を投与した。解析対象は 203 例(テリパラチド群 102 例、アレンドロネート群 101
例)であった。全被験者の平均年齢は 65.9 歳、平均体重は 62.37 kg、平均腰椎 BMD は
0.7493 g/cm2であった。
主要評価項目である腰椎 BMD のベースラインから投与後 18 ヵ月までの平均変化量及
び平均変化率は、テリパラチド群で 0.08 g/cm2 及び 10.92%、アレンドロネート群で
0.04 g/cm2及び 5.51%であった。また、腰椎 BMD のベースラインから最終観察時までの
平均変化量及び平均変化率は、テリパラチド群で 0.07 g/cm2 及び 8.69%、アレンドロネ
ート群で 0.04 g/cm2 及び 5.18%であった。分散分析を用いて解析した結果、投与群間に
統計学的な有意差が認められた(いずれも p<0.001)。繰り返し測定値に関する混合効
果モデル(mixed effects model with repeated measurements:以下、MMRM とする)を用い

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
28
て解析した結果、腰椎 BMD 変化量及び変化率は、投与後 6 ヵ月以降のすべての時点に
おいてアレンドロネート群に比べてテリパラチド群で高く、投与群間に統計学的に有意
な差が認められた(いずれも p<0.05)。
副次的評価項目であるベースラインから最終観察時における大腿骨頸部及び大腿骨近
位部 BMD の平均変化率は、大腿骨頸部 BMD ではテリパラチド群及びアレンドロネー
ト群でそれぞれ 3.37%及び 2.98%、大腿骨近位部 BMD ではそれぞれ 2.74%及び 2.98%で
あった。最終観察時において投与群間に統計学的に有意な差は認められなかった。骨形
成マーカー(血清 BAP、血清 PICP 及び血清 PINP)及び骨吸収マーカー(尿中 NTX)
は、テリパラチド群では上昇したのに対し、アレンドロネート群では低下した。質問票
を用いて腰背部痛を評価した結果、投与群間に明らかな違いは認められなかった。
また、参考資料とした GHBZ 試験は、ステロイド性骨粗鬆症患者を対象とし、主要目
的として、ベースラインから投与後 18 ヵ月までの腰椎 BMD の増加に関して、テリパラ
チドがアレンドロネートを上回ることを検討した。治験薬投与期間は、18 ヵ月の二重盲
検主要期及び 18 ヵ月の二重盲検継続期の計 36 ヵ月間であった。解析対象は 428 例(ア
レンドロネート 10 mg 群 214 例、テリパラチド 20 μg 群 214 例)であった。
ベースラインから二重盲検主要期(18 ヵ月)の最終観察時までの腰椎 BMD 変化量の
最小二乗平均値は、テリパラチド群で 0.059 g/cm2、アレンドロネート群で 0.028 g/cm2で
あり、投与群間に統計学的な有意差が認められた(p<0.001、分散分析)。ベースライン
から二重盲検継続期(36 ヵ月)の最終観察時までの BMD 変化量の最小二乗平均値は、
腰椎 BMD では、テリパラチド群及びアレンドロネート群でそれぞれ 0.073 g/cm2 及び
0.034 g/cm2、大腿骨頸部 BMD ではそれぞれ 0.033 g/cm2及び 0.017 g/cm2、大腿骨近位部
BMD ではそれぞれ 0.032 g/cm2及び 0.017 g/cm2であり、投与群間に統計学的な有意差が
認められた(いずれも p<0.001、分散分析)。腰椎 BMD では 3 ヵ月時点以降、大腿骨頸
部及び大腿骨近位部 BMD では 12 ヵ月時点以降のすべての測定時点において、いずれの
BMD の変化率もテリパラチド群において高く、投与群間に統計学的に有意な差が認め
られた(いずれも p<0.05、MMRM)。骨折については、18 ヵ月時点において X 線撮影
の集中判定により新規椎体骨折が認められた被験者は、テリパラチド群で 171 例中 1 例
(0.6%)、アレンドロネート群では 165 例中 10 例(6.1%)であり、投与群間に統計学
的に有意な差が認められた(p=0.004、Cochran-Mantel-Haenszel 検定)。非椎体骨折を発
生した被験者は、テリパラチド群で 214 例中 12 例(5.6%)、アレンドロネート群で 214
例中 8 例(3.7%)であり、投与群間に統計学的に有意な差は認められなかった。骨代謝
マーカーは、GHBM 試験と同様にテリパラチド群では上昇し、アレンドロネート群では
低下した。
以上の結果から、腰椎 BMD において、テリパラチド群のアレンドロネート群に対す
る優越性が認められた。更に、骨代謝マーカーについては、骨形成促進剤と骨吸収抑制
剤の作用機序の差異を反映し、テリパラチド群で上昇し、アレンドロネート群で低下す
ることが示された。

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
29
2.5.4.4 テリパラチド投与後にラロキシフェンの継続投与を検討した試験
GHCA 試験は、2 つのサブスタディによって構成された非盲検試験であった。主要目
的は 2 つあり、1 つはテリパラチド 20 μg を 24 ヵ月間持続投与したとき(以下、テリパ
ラチド/テリパラチド群とする)の腰椎 BMD の変化を、テリパラチド 20 μg を 12 ヵ月
間投与後、カルシウム及びビタミン D のみ 12 ヵ月間投与したとき(以下、テリパラチ
ド/無治療群とする)と比較することであった。もう 1 つの主要目的は、テリパラチド
20 μg を 12 ヵ月間投与後、ラロキシフェン 60 mg を 12 ヵ月間投与したとき(以下、テリ
パラチド/ラロキシフェン群とする)の腰椎 BMD の変化を、テリパラチド/無治療群
と比較することであった。対象は、1 個以上の脆弱性椎体又は非椎体骨折の既往があり、
BMD が T スコア-2.5 以下の閉経後骨粗鬆症女性患者であった。基礎治療として全被験
者にカルシウム及びビタミン D を投与した。
サブスタディ 1 では、3 群を比較検討した。解析対象は全体で 632 例であった。第 2
期の投与群ごとの解析対象は 503 例(テリパラチド/テリパラチド群 304 例、テリパラ
チド/ラロキシフェン群 97 例、テリパラチド/無治療群 102 例)であった。被験者の
平均年齢は 69.9 歳であり、平均腰椎 BMD は 0.743 g/cm2であった。
主要評価項目である腰椎 BMD について、ベースラインから 24 ヵ月までの腰椎 BMD
変化量の最小二乗平均値は、テリパラチド/テリパラチド群 0.079 g/cm2、テリパラチド
/ラロキシフェン群 0.058 g/cm2、テリパラチド/無治療群 0.028 g/cm2であり、いずれの
群においても、統計学的に有意な増加が認められた(p<0.001、MMRM)。24 ヵ月まで
の変化量について、テリパラチド/テリパラチド群とテリパラチド/無治療群との差、
テリパラチド/ラロキシフェン群とテリパラチド/無治療群との差、及びテリパラチド
/テリパラチド群とテリパラチド/ラロキシフェン群との差は、いずれも統計学的に有
意であった(p<0.001、MMRM)。テリパラチド/テリパラチド群では 24 ヵ月間の投与
期間を通じて腰椎 BMD が持続的に増加し、テリパラチド/ラロキシフェン群では第 1
期の腰椎 BMD の増加を第 2 期において維持したものの、テリパラチド/無治療群では
12 ヵ月以降腰椎 BMD が減少した。
サブスタディ 2 では、骨吸収抑制剤無効例へのテリパラチド 20 g の 24 ヵ月間投与の
影響を検討した。骨吸収抑制剤無効例は以下のいずれかを満たす場合とした:a) 骨吸収
抑制剤治療開始 1 年後以降の脆弱性の椎体骨折又は非椎体骨折の既往、b) 2 年間以上の
骨吸収抑制剤治療にもかかわらず腰椎、大腿骨近位部又は大腿骨頸部の BMD が T スコ
ア-3 以下、c) 過去 2 年間の骨吸収抑制剤治療にもかかわらず腰椎、大腿骨近位部又は
大腿骨頸部の BMD が 3.5%以上の低下。解析対象は 234 例であった。全被験者の平均年
齢は 70.2 歳であり、平均腰椎 BMD は 0.715 g/cm2であった。サブスタディ 2 において、
腰椎、大腿骨近位部及び大腿骨頸部 BMD は持続的に増加し、ベースラインから 24 ヵ月
までの変化は統計学的に有意であった(いずれも p<0.001、MMRM)。
視覚的アナログスケール(Visual Analogue Scale;以下、VAS とする)により腰背部痛
を検討した結果、サブスタディ 1 のすべての投与群とサブスタディ 2 において、VAS の
スケールにベースラインからの統計学的に有意な低下が認められた(いずれも p<0.05、
1 標本 t 検定)。

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
30
また、参考資料とした GHBQ 試験においても、テリパラチド 20 µg を 1 年間投与後、
ラロキシフェン 60 mg を 1 年間継続投与すると、テリパラチド投与により増加した腰椎
BMD の大部分が維持されることが確認された。
以上の結果から、テリパラチド 12 ヵ月間投与後、ラロキシフェン 12 ヵ月投与に切り
替えた被験者では、1 年目の BMD が 2 年目において維持されることが示された。更に、
テリパラチド 24 ヵ月間投与により、腰椎、大腿骨頸部及び大腿骨近位部 BMD がテリパ
ラチド投与期間中持続的に増加した。この持続的な増加は、骨吸収抑制剤無効例におい
ても同様に認められた。
2.5.4.5 テリパラチド投与中止後の追跡調査研究
GHBJ 試験は、先行して実施した臨床試験 7 試験(GHAC、GHAF、GHAH、GHAJ、
GHAL、GHAU 及び GHAV 試験)のいずれかに参加し、テリパラチド 20 μg、40 μg 又は
プラセボ投与を受けた被験者を対象とした、テリパラチド投与中止後の追跡調査研究で
ある。追跡調査中、医師は他の骨粗鬆症治療薬を自由に処方することができた。GHBJ
試験は、安全性と効果の持続性を調査する 24 ヵ月の追跡調査期間と、重篤な有害事象
を観察する 30 ヵ月間の追加観察期間の合計 4.5 年間の試験として構成された。先行する
試験ごとに割り付けられた投与群別に、有効性の結果を評価した。最も調査例数が多か
った GHAC 試験集団と GHAJ 試験集団における結果を以下に記載する。GHAC 試験から
は 1263 例(閉経後骨粗鬆症女性)、GHAJ 試験からは 355 例(原発性骨粗鬆症男性)が
GHBJ 試験に参加した。以下に先行する試験及び評価項目ごとに結果を記載する。
(1) GHAC 試験集団における椎体骨折
GHAC 試験の最終観察時から 18 ヵ月(中央値)時点までに新規椎体骨折が発生した
被験者の割合は、プラセボ群 19.5%(67/344)、20 μg 群 11.6%(42/361)、40 μg 群
10.7%(36/338)であった。カイ二乗検定の結果、プラセボ群との間に統計学的に有意な
差が認められた(20 μg 群 p=0.004、40 μg 群 p=0.001)。プラセボ群に対する相対的なリ
スクの減少は、20 μg 群では 40%、40 μg 群では 45%であった。
(2) GHAC 試験集団における非椎体骨折
GHAC 試験の最終観察時から 30 ヵ月(中央値)時点までに非椎体骨折が認められた
被験者の割合は、プラセボ群 14.5%(60/414)、20 μg 群 12.4%(54/436)、40 μg 群
10.4%(43/412)であり、プラセボ群に比較しテリパラチド群で減少していたが、投与群
間に統計学的に有意な差は認められなかった(20 μg 群 p=0.367、40 μg 群 p=0.078、カイ
二乗検定)。GHAC 試験のベースラインから最終観察後 30 ヵ月(中央値)時点までに
非椎体骨折が認められた被験者の割合は、プラセボ群 22.5%(93/414)、20 μg 群 17.7%
(77/436)、40 μg 群 15.3%(63/412)とプラセボ群に比較しテリパラチド群で減少し、
40 μg 群ではプラセボ群との間に統計学的に有意な差が認められた(20 μg 群 p=0.080、
40 μg 群 p=0.008)。GHAC 試験の最終観察時から 30 ヵ月(中央値)時点までに非外傷
性非椎体骨折が認められた被験者の割合は、プラセボ群 8.5%(35/414)、20 μg 群 6.2%

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
31
(27/436)、40 μg 群 5.1%(21/412)であり、プラセボ群と比較し減少していたが、統
計学的に有意な差は認められなかった(20 μg 群 p=0.205、40 μg 群 p=0.055)。GHAC 試
験のベースラインから最終観察後 30 ヵ月(中央値)時点までに非外傷性非椎体骨折が
認められた被験者の割合は、プラセボ群 13.3%(55/414)、20 μg 群 8.5%(37/436)、
40 μg 群 7.3%(30/412)とプラセボ群に比べてテリパラチド群で減少し、いずれのテリ
パラチド群においてもプラセボ群との間に統計学的に有意な差が認められた(20 μg 群
p=0.024、40 μg 群 p=0.005)。
(3) GHAC 試験集団における腰椎骨密度
GHAC 試験のベースラインから最終観察後 30 ヵ月(中央値)時点までの腰椎 BMD の
平均変化率は、プラセボ群 5.10%、20 μg 群 7.38%、40 μg 群 9.11%であった。いずれの
テリパラチド群においても、プラセボ群との間に統計学的に有意な差が認められた
(20 μg 群 p=0.002、40 μg 群 p<0.001、分散分析)。テリパラチドの投与によって増加し
た腰椎 BMD は、最終観察後次第に低下したが、GHAC 試験の最終観察時から 30 ヵ月経
過時点においても、テリパラチド投与開始時点と比べると高い BMD を維持していた。
(4) GHAJ 試験集団における椎体骨折
GHAJ 試験のベースラインから最終観察後 18 ヵ月(中央値)時点までに 1 個以上の新
規椎体骨折が発生した被験者の割合は、プラセボ群 11.9%(12/101)、20 μg 群 5.7%
(5/87)、40 μg 群 6.2%(5/81)であり、プラセボ群と比較しテリパラチド群で減少し
ていたが、投与群間に統計学的に有意な差は認められなかった(20 μg 群 p=0.144、40 μg
群 p=0.188、カイ二乗検定)。プラセボ群に対する相対的なリスクの減少は、20 μg 群で
は 52%、40 μg 群では 48%であった。
(5) GHAJ 試験集団における腰椎骨密度
GHAJ 試験のベースラインから最終観察後 30 ヵ月(中央値)時点までの腰椎 BMD の
平均変化率は、プラセボ群 2.50%、20 μg 群 4.13%、40 μg 群 5.23%であった。分散分析
で比較した結果、40 μg 群ではプラセボ群との間に統計学的に有意な差が認められた
(p=0.003)が、20 μg 群では有意差は認められなかった(p=0.099)。
以上の結果から、テリパラチド投与中止後に他の骨粗鬆症治療薬の使用が制限されて
いない状況下において、GHAC 試験でテリパラチドの投与を 19 ヵ月間(中央値)受け
た被験者では、最終観察後 18 ヵ月時点においてプラセボ群と比較した新規椎体骨折の
抑制効果の持続、最終観察後 30 ヵ月時点において非外傷性非椎体骨折の抑制効果及び
腰椎 BMD 増加効果の持続が認められた。GHAJ 試験においてテリパラチドの投与を 11
ヵ月間(中央値)受けた被験者では、GHAJ 試験のベースラインから最終観察後 18 ヵ月
時点において新規椎体骨折を発生した被験者の割合はプラセボに比べて減少していたが、
統計学的に有意な差は認められなかった。また、GHAJ 試験のベースラインから最終観

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
32
察後 30 ヵ月時点において腰椎 BMD 変化率はプラセボに比べて高かったが、統計学的に
有意な差は認められなかった。
2.5.4.6 アジアで実施した試験
参考資料とした、アジアで実施した実薬対照試験である GHCB 試験、GHCC 試験及び
GHCF 試験では、閉経後骨粗鬆症女性に対し、テリパラチド 20 µg 又はカルシトニン
100 IU を 6 ヵ月間投与した時の腰椎 BMD の変化を比較した。解析対象は、GHCB 試験、
GHCC 試験及び GHCF 試験でそれぞれ 60 例、70 例及び 104 例であった。テリパラチド
群の最終観察時における腰椎 BMD の変化量及び変化率の平均値は、GHCB 試験でそれ
ぞれ 0.033 g/cm2及び 4.55%、GHCC 試験でそれぞれ 0.027 g/cm2及び 4.16%、GHCF試験
でそれぞれ 0.036 g/cm2及び 5.03%であった。いずれの試験においても、最終観察時にお
ける腰椎 BMD の変化量はテリパラチド群において高く、投与群間に統計学的に有意な
差が認められた(GHCB 試験:p=0.003、2 標本 t 検定、GHCC 試験:p=0.012、共分散分
析、GHCF 試験:p<0.001、共分散分析)。国内で実施した 6 ヵ月投与試験である GHCS
試験では、テリパラチド 20 g 群の最終観察時における腰椎(L1-L4)BMD の変化量及
び変化率の平均値は、それぞれ 0.037 g/cm2及び 6.19%であり、アジアで実施した試験の
結果と比べて大きな違いは認められなかった。
2.5.4.7 骨吸収抑制剤の前治療又は併用を検討した試験
参考資料とした GHCM 試験では、アレンドロネートで治療されていた(以下、アレ
ンドロネート前投与層とする)又はラロキシフェンで治療されていた(以下、ラロキシ
フェン前投与層とする)閉経後骨粗鬆症女性患者に対して、前治療を継続しテリパラチ
ドを追加した場合(以下、併用群とする)と前治療を中止しテリパラチドに切り替えた
場合(以下、切り替え群とする)における腰椎、大腿骨頸部及び大腿骨近位部 BMD の
変化を検討した。投与期間は 18 ヵ月であった。解析対象は、アレンドロネート前投与
層 102 例(切り替え群 50 例、併用群 52 例)、ラロキシフェン前投与層 96 例(切り替え
群 49 例、併用群 47 例)であった。最終観察時において、いずれの部位の BMD もすべ
ての投与群で増加した(腰椎 BMD 変化率の最小二乗平均値:アレンドロネート前投与
層切り替え群 4.7%及び併用群 8.3%、ラロキシフェン前投与層切り替え群 7.9%及び併用
群 8.3%)。アレンドロネート前投与層及びラロキシフェン前投与層共に、最終観察時に
おける BMD の変化率は、切り替え群に比べて併用群の方が高かった。骨代謝マーカー
では、血清 PINP、血清 BAP 及び血清 CTX がすべての投与群で上昇した。アレンドロネ
ート前投与層では、切り替え群の方が併用群よりも大きく上昇した。ラロキシフェン前
投与層では、切り替え群の方が併用群よりも大きく上昇したが、その投与群間差はアレ
ンドロネート前投与層よりも小さかった。
以上の結果から、骨吸収抑制剤による前治療からテリパラチドへの切り替え及び骨吸
収抑制剤とテリパラチドとの併用のいずれにおいても、BMD の増加が認められた。骨
吸収抑制剤による前治療を受けていた被験者において、テリパラチドとの併用は、テリ
パラチドへの切り替えと同等又は同等以上の BMD の増加を示した。骨代謝マーカーの

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
33
評価からも、骨吸収抑制剤による前治療からテリパラチドへの切り替え及び骨吸収抑制
剤とテリパラチドとの併用のいずれにおいても、テリパラチドの骨形成促進作用が示さ
れた。
2.5.4.8 有効性評価のまとめ
国内第 II 相プラセボ対照二重盲検比較試験である GHCS 試験の結果、主要評価項目で
ある腰椎 BMD 変化率について、テリパラチド 10 g 群、20 g 群及び 40 μg 群は、プラ
セボ群に比べ、投与量の増加に伴って統計学的に有意な増加を示した。副次的評価項目
である骨代謝マーカーにおける 10 g 群の結果及び安全性における 40 μg 群の結果を考
慮すると、日本人骨粗鬆症患者における臨床推奨用量は、20 μg と考えられた。その後
に実施した国内第 III 相プラセボ対照二重盲検比較試験である GHDB 試験では、主要評
価項目である腰椎 BMD 変化率について、テリパラチド 20 g のプラセボに対する優越
性が認められ、日本人骨粗鬆症患者に対するテリパラチド 20 g の有効性が確認された。
ブリッジング試験(GHDB 試験)とブリッジング対象試験(GHAC 試験)及び他のプ
ラセボ対照二重盲検比較試験の有効性の類似性を検討した結果、あらかじめ設定した評
価方法によりブリッジングの成立要件を満たし、試験間の類似性が確認できた。更に、
腰椎 BMD の反応性に男女差は認められず、骨代謝マーカーはいずれの試験においても
同様の経時的変化を示した。安全性に関する成績についても、日本人に特徴的な有害事
象の発現は認められなかったことから、外国臨床データの日本人骨粗鬆症患者への外挿
は可能であると判断し、外国臨床データを用いて有効性を評価した。
外国プラセボ対照二重盲検比較試験である GHAC 試験の結果、主要評価項目である新
規椎体骨折において、テリパラチドのプラセボに対する優越性が認められ、テリパラチ
ド 20 g が、19 ヵ月間(中央値)の投与により新規椎体骨折抑制効果を示すことが明ら
かとなった。更に、非椎体骨折の抑制、腰椎、大腿骨頸部及び大腿骨近位部 BMD の増
加、骨代謝マーカーの上昇、並びに骨微細構造の改善効果が示された。また、男性患者
を対象とした外国プラセボ対照二重盲検比較試験である GHAJ 試験の結果、主要評価項
目である腰椎 BMD において、テリパラチド 20 g のプラセボに対する優越性が認めら
れた。
実薬対照試験の結果、主要評価項目である腰椎 BMD において、18 ヵ月間の投与によ
り、テリパラチド 20 g 群のアレンドロネート 10 mg 群に対する優越性が認められた。
骨代謝マーカーの経時的変化の違いから、両薬剤は異なる作用機序を有することが明ら
かとなった。
テリパラチド投与後にラロキシフェンの継続投与を検討した試験の結果、テリパラチ
ド 20 g の 12 ヵ月間投与による BMD 増加は、ラロキシフェン 60 mg の 12 ヵ月投与に
切り替え後維持されることが示された。更に、テリパラチド 20 g の 24 ヵ月間投与によ
り、腰椎、大腿骨頸部及び大腿骨近位部 BMD が持続的に増加することが示された。こ
の BMD の持続的な増加は、骨吸収抑制剤無効例においても認められた。
テリパラチド投与中止後の追跡調査研究の結果、他の骨粗鬆症治療薬使用の制限がな
い状況下において、テリパラチド投与中止後の効果の持続が示された。特に、19 ヵ月間

LY333334 2.5 臨床に関する概括評価
2.5.4 有効性の概括評価
34
(中央値)テリパラチドが投与された GHAC 試験後では、最終観察後 18 ヵ月時点にお
いて新規椎体骨折の抑制効果の持続、最終観察後 30 ヵ月時点において非外傷性非椎体
骨折の抑制効果及び腰椎 BMD 増加効果の持続が認められた。
以上のことから、骨形成促進剤であるテリパラチド 20 g の有効性が、骨折の危険性
の高い日本人骨粗鬆症患者で示された。テリパラチドには投与期間の上限が設定される
予定であるが、テリパラチド 20 g の 18 ヵ月間の投与により、椎体骨折及び非椎体骨折
の抑制効果、アレンドロネートよりも高い BMD 増加、骨形成促進作用を反映する骨代
謝マーカー上昇、並びに骨粗鬆症により悪化した骨微細構造の正常な状態への改善が認
められる。更に、テリパラチドの投与により持続的に BMD が増加し、投与終了後にお
いても、他の骨粗鬆症治療薬使用の制限がない状況下において、骨折抑制及び骨量増加
の効果の持続が認められる。

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
35
2.5.5 安全性の概括評価
2.5.5.1 安全性評価の概略
安全性の評価に用いた試験一覧を表 2.5.5-1 に示す。安全性評価は、臨床試験 6 試験及
び追跡調査研究 1 試験と、臨床薬理試験 9 試験の成績に基づいて行った。臨床試験 17 試
験及び臨床薬理試験 8 試験を参考資料とした。評価資料において、国内及び外国におけ
る安全性プロファイルの評価として、個々の試験間の比較と、国内と外国で実施した臨
床試験の併合解析による比較を行った。更に、2008 年 11 月 26 日をデータ締切日として、
進行中の臨床試験を含むすべての臨床試験、臨床薬理試験及び市販後臨床試験の死亡及
び重篤な有害事象、並びに外国市販後の自発報告における有害事象、死亡及び重篤な有
害事象について評価した。
ブリッジング試験の GHDB 試験とブリッジング対象試験の GHAC 試験を含む評価資
料において、国内及び外国の臨床試験の結果を比較したところ、あらかじめ規定した成
立要件を満たし、テリパラチド投与時の日本人に対する安全性は良好で、外国データと
類似していたことから、外国臨床試験の安全性データを日本人に外挿して、評価した
(第 2.7.3.3.2.1 項参照)。
表 2.5.5-1. 安全性評価のための評価資料及び参考資料
試験の種類 評価資料 参考資料
国内プラセボ対照二重盲検比較
試験
GHCS、GHDB(ブリッジング
試験)-
外国プラセボ対照二重盲検比較
試験
GHAC(ブリッジング対象試
験)、GHAJGHAA
テリパラチド投与中止後の追跡
調査研究GHBJ -
実薬対照試験 GHBM GHBZ、GHAH、GHCXテリパラチド投与後にラロキシ
フェンの継続投与を検討した試
験
GHCA GHBQ
日本人及び外国人を対象とした
臨床薬理試験GHCO -
外国人を対象とした臨床薬理試
験
GHBO、GHAD、GHAE、GHAW、GHBA、GHBC、GHBI、GHBR
GHAB、GHAK、GHAM、
GHAN、GHAS、GHAT、GHBF、GHCE
アジアで実施した試験 - GHCB、GHCC、GHCF骨吸収抑制剤の前治療又は併用
を検討した試験- GHCM、GHCK、GHCD
ホルモン補充療法の併用を検討
した試験- GHAF
ペン型注入器のコンプライアン
スを検討した試験- GHCP
早期に中止された試験(有効性
の解析対象とならなかった試
験)
-GHAL、GHAV、GHAU、GHCU
国内臨床試験 GHDB 試験では、日本人における長期投与の安全性を検討するために、
12 ヵ月間の第 1 期二重盲検比較期間終了後に 6 ヵ月間の第 2 期オープン期間及び 6 ヵ月
間の第 3 期長期投与延長期間の合計 12 ヵ月間の継続投与(最大投与期間:24 ヵ月間)

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
36
を実施した。本項には第 1 期二重盲検比較期間の投与 12 ヵ月間及びその後の第 2 期オー
プン期間を含めた投与 18 ヵ月間の安全性成績を示す。第 1 期開始時にプラセボに割り
付けられた群をプラセボ群、第 1 期開始時にテリパラチド 20 g に割り付けられた群を
20 g 群とした。なお、いずれの被験者も第 2 期においてはテリパラチド 20 g を投与し
たため、第 2 期の結果を含む記述では、投与薬剤を明確にする目的で、必要に応じて、
プラセボ群(プラセボ→20 g)及び 20 g 群(20 g→20 g)と記載した。
2.5.5.2 安全性評価の対象となった被験者の人口統計学的及びその他の特性
国内臨床試験において、対象となった患者は、GHCS 試験では骨折の危険性の高い閉
経後骨粗鬆症女性患者、並びに GHDB 試験では骨折の危険性の高い原発性骨粗鬆症患者
(閉経後骨粗鬆症女性及び男性)であった。外国臨床試験において対象となった患者は、
GHAC 試験では椎体骨折を有する閉経後骨粗鬆症女性患者であり、GHAJ 試験では原発
性骨粗鬆症男性患者、GHBM 試験及び GHCA 試験では閉経後骨粗鬆症女性患者であっ
た。これらの試験における投与群ごとの年齢の平均値は 58.06 歳から 72.5 歳であった。
全ての被験者は、試験期間を通じて、基礎治療としてカルシウムとビタミン D の投与を
受けた。骨粗鬆症に関する選択基準は、既存骨折、YAM 又は T スコアに基づく若年成
人の平均 BMD に対する低下により規定した。また、主な除外基準を以下のとおり設定
した。
続発性骨粗鬆症及び骨ページェット病などの原発性骨粗鬆症以外の代謝性骨疾患
の既往を有する患者、又は合併している患者
副甲状腺機能亢進症又は副甲状腺機能低下症を有する患者
甲状腺機能異常を有する患者(慢性甲状腺機能低下症で、十分な補充療法を受け
ている患者は除く)
腎結石又は尿路結石の既往を有する患者、又は合併している患者
悪性腫瘍の既往を有する患者、又は合併している患者
テリパラチドの添付文書(案)では、臨床試験で除外した上記の主な患者集団につい
て制限を設けているため、市販後のテリパラチドの適正使用下においては、予想外の患
者集団に投与される可能性は低いと考えられる。しかし、以下のような安全性データベ
ースの限界も考えられる。
国内臨床試験の GHDB 試験において、骨粗鬆症疾患の実態を反映して、男性骨粗
鬆症患者は 14 例で、女性骨粗鬆症患者に比較して少なかった。
腎障害を有する患者を対象にした臨床薬理試験の GHAW 試験で、重度の腎障害
を有する患者は 5 例であり、当該疾患を有する患者への曝露は限られていた。
参考資料ではあるが、ステロイド性骨粗鬆症患者を対象とした第 III/IV 相臨床試
験の GHBZ 試験で、テリパラチド投与を受けた閉経前の患者は 37 例であり、閉
経前患者への曝露は限られていた。

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
37
2.5.5.3 テリパラチドの曝露及び使用経験
国内臨床試験においてテリパラチドの投与を受けた被験者数は、GHCS 試験 116 例、
GHDB 試験 195 例(第 1 期にテリパラチドの投与を受けた被験者 136 例及び第 2 期にプ
ラセボからテリパラチドに切り替えた被験者 59 例)であった。投与期間の中央値は、
GHCS 試験ではテリパラチド 10 g 群、20 g 群及び 40 g 群ともに 169.0 日であり、
GHDB 試験の第 1 期二重盲検比較期間(12 ヵ月)ではテリパラチド 20 g 群で 365.0 日、
第 2 期オープン期間終了時(18 ヵ月)では 20 g 群(20 g→20 g)で 533.0 日であった。
治療の遵守状況について、全試験期間にわたって、服薬遵守状況が 70%以上で良好とさ
れた被験者の各投与群における割合は、GHCS 試験で 92.3%以上、GHDB 試験の第 1 期
で 96.3%以上及び第 2 期で 96.6%以上であった。国内臨床試験において、患者は自己注
射により本剤を投与しており、両試験共に服薬遵守状況が良好であった患者数は 90%以
上であり、日本人の自己注射手技による本剤投与は、安全性上の懸念もなく、服薬遵守
状況が良好であることが示された。
外国で当時進行中であった GHAC 試験及び GHAJ 試験を含む 7 臨床試験は、ラットが
ん原性試験において骨肉腫を含む骨腫瘍性病変が認められたことを受け、治験依頼者の
判断により 1998 年に試験を中止した。そのため、評価資料のうち GHAC 試験及び
GHAJ 試験の 2 試験では、予定投与期間を完了した被験者はいなかった。GHAC 試験に
おける投与期間の中央値は 19 ヵ月であり、18~23 ヵ月間の投与を受けた被験者数は、
20 g 群では 375 例(69.3%)、40 g 群では 362 例(65.6%)であった。GHAJ 試験にお
ける投与期間の中央値は 11 ヵ月であり、9 ヵ月以上の投与を受けた被験者数は、20 g
群で 106 例(70.2%)と 40 g 群で 91 例(65.5%)であった。アレンドロネートとの比較
を行った GHBM 試験において、テリパラチドの投与を受けた 102 例の投与期間の中央
値は 537.0 日であった。非盲検下でテリパラチド 20 g の 24 ヵ月間投与を行った GHCA
試験において、2 年目もテリパラチド 20 g の投与を継続した被験者 504 例のうち、478
例が 18 ヵ月以上の投与を受け、24 ヵ月間のテリパラチド投与の試験期間を完了した被
験者数は 476 例であった。参考資料のステロイド性骨粗鬆症患者を対象とした GHBZ 試
験において、テリパラチド 20 g の投与を受けた被験者 214 例の曝露期間の中央値は
1086 日であり、そのうち 36 ヵ月間の試験を完了した被験者数は 123 例であった。
2.5.5.4 有害事象
2.5.5.4.1 比較的よく見られる有害事象
2.5.5.4.1.1 国内プラセボ対照二重盲検比較試験
1 件以上の有害事象を発現した被験者数は、GHCS 試験では、プラセボ群で 38 例中 29
例(76.3%)、10 g 群で 38 例中 30 例(78.9%)、20 g 群で 39 例中 33 例(84.6%)、
40 g 群で 39 例中 32 例(82.1%)であった。GHDB 試験の第 1 期二重盲検比較期間にお
いては、プラセボ群で 67 例中 59 例(88.1%)、20 g 群で 136 例中 116 例(85.3%)で
あった。両試験共に、各投与群における 1 件以上の有害事象の発現率はほぼ同程度であ
り、それぞれの試験において投与群間における統計学的に有意な差は認められなかった
(それぞれ p=0.791 及び p=0.670;Fisher の直接確率計算法)。

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
38
GHCS 試験において、全テリパラチド群の 5%以上の被験者に発現し、プラセボ群よ
りも発現率の高かった有害事象は、悪心(プラセボ群 10.5%、全テリパラチド群 11.2%、
以下同様)、湿疹(2.6%、6.9%)、頭痛(2.6%、6.9%)、転倒(2.6%、6.0%)、筋痙
縮(0%、6.0%)及び血中尿酸増加(0%、5.2%)であり、このうち悪心、頭痛、筋痙縮
及び血中尿酸増加は、2%以上の被験者で治験薬との因果関係が否定できないと判断され
た。有害事象の重症度は、概して軽度又は中等度であった。悪心及び頭痛の発現率は、
他の 3 投与群に比べて 40 g 群において高かった。悪心と頭痛が最初に発現するまでの
時間を検討した Kaplan-Meier 曲線から、40 g 群における最初の発現は投与開始後初期
に多く認められた。
GHDB 試験の第 1 期二重盲検比較期間において、テリパラチド群の 5%以上の被験者
に発現し、プラセボ群よりも発現率の高かった有害事象は、便秘(プラセボ群 3.0%、テ
リパラチド群 7.4%、以下同様)、頭痛(4.5%、6.6%)、骨関節炎(6.0%、6.6%)、浮
動性めまい(4.5%、5.9%)、上気道の炎症(4.5%、5.9%)及び膀胱炎(3.0%、5.1%)
であり、このうち頭痛の 1 例は治験薬との因果関係が否定できないと判断された。悪心
の発現はテリパラチド群で 1 例(0.7%)であり、他の試験に比較して少なかった。重症
度は、概して軽度又は中等度であった。
また、GHDB 試験の第 1 期二重盲検比較期間後に第 2 期オープン期間においてテリパ
ラチド 20 μg の投与を 6 ヵ月継続したとき、合計 18 ヵ月の投与期間に 1 件以上の有害事
象を発現した被験者数は、テリパラチド 20 g 群(20 g→20 g)では 136 例中 123 例
(90.4%)、プラセボ群(プラセボ→20 g)では 67 例中 60 例(89.6%)であった。
20 μg 群において投与 12 ヵ月間に被験者の 5%超に認められた有害事象は鼻咽頭炎、背
部痛、便秘、転倒・転落、頭痛、骨関節炎、関節痛、挫傷、浮動性めまい、季節性アレ
ルギー、上気道の炎症及び膀胱炎であったが、その後テリパラチド 20 μg の投与を継続
して計 18 ヵ月間投与した後は鼻咽頭炎、背部痛、便秘、転倒・転落、挫傷、浮動性め
まい、頭痛、骨関節炎、上気道の炎症、関節痛、季節性アレルギー、膀胱炎、下痢、接
触性皮膚炎及び不眠症であった。また、第 2 期で高度と判定された有害事象はなかった。
2.5.5.4.1.2 外国プラセボ対照二重盲検比較試験
GHAC 試験において、1 件以上の有害事象を発現した被験者数は、プラセボ群で 544
例中 473 例(86.9%)、20 g 群で 541 例中 447 例(82.6%)、40 g 群で 552 例中 476 例
(86.2%)であった。GHAJ 試験においては、プラセボ群で 147 例中 112 例(76.2%)、
20 g 群で 151 例中 121 例(80.1%)、40 g 群で 139 例中 112 例(80.6%)であった。い
ずれの試験においても有害事象の発現率は投与群間で同程度であった。
GHAC 試験において、テリパラチド 20 g 群及び 40 g 群の発現率が 5%以上で、プラ
セボ群よりも両投与群の発現率が高かった有害事象は、悪心(プラセボ群 7.5%、20 g
群 9.4%、40 g 群 17.8%、以下同様)、無力症(7.2%、8.9%、10.1%)、浮動性めまい
(6.1%、9.2%、8.0%)、咳嗽増加(5.5%、6.7%、6.3%)、発疹(5.3%、5.7%、6.0%)
及び下痢(5.3%、5.9%、5.6%)であった。また、全投与群間で統計学的有意差の認めら
れた有害事象のうち、40 g 群で発現頻度の高かったものは頭痛(8.3%、8.1%、13.0%)

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
39
及び悪心であり、背部痛はテリパラチド群の方が発現率は低く、40 g 群で最も低かっ
た(22.6%、16.8%、15.8%)。悪心と頭痛が最初に発現するまでの時間を検討した
Kaplan-Meier 曲線から、40 g 群における最初の発現は、投与開始後初期に多く認められ
た。
GHAJ 試験において、テリパラチド 20 g 群及び 40 g 群の発現率が 5%以上で、プラ
セボ群よりも発現率の高かった有害事象は、疼痛(プラセボ群 12.9%、20 g 群 17.9%、
40 g 群 15.8%、以下同様)、鼻炎(9.5%、9.9%、11.5%)、関節痛(6.1%、9.3%、
6.5%)、無力症(5.4%、7.9%、11.5%)、悪心(3.4%、5.3%、18.7%)及び頭痛(4.1%、
5.3%、10.8%)であった。このうち、鼻炎、無力症、悪心及び頭痛の発現率は 20 g 群
よりも 40 g 群の方が高かった。頭痛が最初に発現するまでの時間の検討から、40 g 群
における最初の発現は、投与開始後初期に多く認められた。
外国で実施したプラセボ対照試験 2 試験において認められた有害事象の重症度は、概
して軽度又は中等度であった。悪心及び頭痛の 40 g 群における最初の発現は、投与開
始後初期に多く認められた。
2.5.5.4.1.3 実薬対照試験
GHBM 試験において、アレンドロネート 10 mg を対照薬として、テリパラチド 20 g
の評価を行った。1 件以上の有害事象を発現した被験者数は、アレンドロネート群では
101 例中 80 例(79.2%)及びテリパラチド群では 102 例中 87 例(85.3%)であった。テ
リパラチド群における発現率が 5%以上で、アレンドロネート群よりもテリパラチド群
の方が発現率の高かった有害事象は、頭痛(アレンドロネート群 5.9%、テリパラチド群
11.8%、以下同様)、鼻咽頭炎(5.9%、11.8%)、悪心(6.9%、10.8%)、筋痙攣(4.0%、
8.8%)、浮動性めまい(5.9%、8.8%)、関節痛(6.9%、8.8%)、四肢痛(6.9%、
7.8%)、消化不良(4.0%、7.8%)、便秘(3.0%、5.9%)、下痢(2.0%、5.9%)及びう
つ病(3.0%、5.9%)であった。一方、テリパラチド群における発現率が 5%以上で、テ
リパラチド群よりもアレンドロネート群の方が発現率の高かった有害事象は、背部痛で
あり、テリパラチド群 25.5%に対してアレンドロネート群 38.6%であった。本試験にお
いて、良性、悪性又は詳細不明の腫瘍の有害事象の発現は、テリパラチド群では認めら
れなかったが、アレンドロネート群では 5 例(5.0%)に認められた。
重症度別では、軽度、中等度及び高度の有害事象を 1 件以上発現した被験者数は、ア
レンドロネート群でそれぞれ 11 例(10.9%)、41 例(40.6%)及び 28 例(27.7%)、テ
リパラチド群でそれぞれ 23 例(22.5%)、43 例(42.2%)及び 21 例(20.6%)であった。
テリパラチド群はアレンドロネート群に比べて軽度の有害事象を発現した割合が高く、
高度の有害事象を発現した被験者の割合が低いという結果が示され、重症度別の有害事
象発現の割合において投与群間に統計学的な有意差が認められた(p=0.035;Mantel-
Haenszel exact test)。
参考資料の GHBZ 試験において、ステロイド性骨粗鬆症患者を対象に、アレンドロネ
ート 10 mg とテリパラチド 20 g の比較を行った。1 件以上の有害事象を発現した被験

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
40
者数は、アレンドロネート群では 214 例中 184 例(86.0%)及びテリパラチド群では 214
例中 194 例(90.7%)であった。
テリパラチド群においてアレンドロネート群より多く発現し、投与群間で有意差が認
められた有害事象は、悪心(アレンドロネート群 8.4%、テリパラチド群 16.8%、以下同
様)、呼吸困難(2.8%、7.5%)、不眠症(1.4%、5.6%)及びウイルス感染(0%、
2.3%)であった(第 2.7.6.3.5 項、表 2.7.6.3.5-33 参照)。
アレンドロネート群においてテリパラチド群より多く発現し、投与群間で統計学的な
有意差が認められた有害事象は、体重減少、多発性関節炎及び肝酵素上昇であった。体
重減少、多発性関節炎及び肝酵素上昇の発現率は、アレンドロネート群でそれぞれ 4.2%、
1.9%及び 1.9%であり、テリパラチド群ではいずれも認められなかった。
GHBZ 試験の対象は、原発性でなくステロイド誘発性骨粗鬆症患者である点で他の試
験と異なっており、両投与群において発現頻度が最も高かった器官別大分類(SOC)は、
共に「感染症および寄生虫症」であったことは特徴として考えられた。この結果は、被
験者が継続的なステロイドの投与を受けているために想定内の結果であり、また投与群
間に統計学的な有意差は認められなかった。
2.5.5.4.1.4 テリパラチド投与後にラロキシフェンの継続投与を検討した試験
GHCA 試験は、サブスタディ 1 とサブスタディ 2 の 2 つのサブスタディで構成された。
サブスタディ 1 では、被験者 634 例は 1 年目にテリパラチド 20 g の投与を受けた後、2
年目に各被験者をテリパラチド群、ラロキシフェン群又は無治療群に割り付けた。サブ
スタディ 2 では、骨吸収抑制剤無効例を対象とし、全被験者 234 例がテリパラチド 20
g の投与を 24 ヵ月間受けた。GHCA 試験でテリパラチド投与を受けていた期間、すな
わち、サブスタディ 1 の 1 年目(1 年目の全被験者)とテリパラチド群の 2 年目(テリ
パラチド/テリパラチド群)、及びサブスタディ 2 の 24 ヵ月間において、1 件以上の有
害事象を発現した被験者数は、866 例中 631 例(72.9%)であった。テリパラチド投与を
受けた被験者において 1 件以上の有害事象を発現した被験者の割合(初発)を 6 ヵ月ご
とに見た場合、最初の 6 ヵ月間では 58.7%、7-12 ヵ月では 41.2%、13-18 ヵ月では 38.3%、
19-24 ヵ月では 34.2%であった。5%以上の被験者に見られた有害事象(0-24 ヵ月)は、
悪心(13.3%)、関節痛(10.0%)、頭痛(7.9%)、高血圧(6.7%)、四肢痛(6.1%)、
筋痙攣(5.7%)及び下痢(5.3%)であった。このうち、高血圧以外は、1%以上の被験
者において治験薬との因果関係が否定できないと判断された。
本剤投与中に、全被験者 866 例のうち 190 例(21.9%)が軽度、245 例(28.3%)が中
等度、196 例(22.6%)が高度の有害事象を発現した。テリパラチドを投与した被験者の
0.5%以上に見られた高度の有害事象は、悪心(1.8%)、高血圧(1.2%)、背部痛
(1.0%)、頭痛(0.8%)、関節痛、浮動性めまい及び失神(それぞれ 0.7%)、下痢、
四肢痛、肺炎及び回転性眩暈(それぞれ 0.6%)であった。
本試験では投与期間に伴い、継続投与例数の減少も認められるために、投与期間と有
害事象の発現の関連性を厳密に議論することは容易ではないが、少なくとも長期投与に

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
41
伴い有害事象の発現率が増加する傾向は認められなかった。また、長期投与に伴い、特
定の有害事象の発現が増加する傾向や重症度が悪化する傾向は認められなかった。
2.5.5.4.1.5 国内及び外国臨床試験の併合解析
国内プラセボ対照二重盲検比較試験の GHCS 試験及び GHDB 試験、並びに外国プラ
セボ対照二重盲検比較試験の GHAC 試験及び GHAJ 試験と実薬対照試験の GHBM 試験
についてそれぞれ併合解析を行い安全性プロファイルの比較を行った。
国内臨床試験の GHCS 試験及び GHDB 試験(投与 12 ヵ月間)の併合解析で、20 g
群の併合群(N=175)における発現率が 5%以上で、プラセボ併合群よりも発現率の高
かった有害事象は、便秘(プラセボ併合群 2.9%、テリパラチド 20 g 併合群 6.9%、以
下同様)、浮動性めまい(3.8%、6.3%)、頭痛(3.8%、5.7%)、骨関節炎(3.8%、
5.7%)、関節痛(3.8%、5.1%)、挫傷(3.8%、5.1%)及び上気道の炎症(2.9%、
5.1%)であった(表 2.5.5-2)。テリパラチド 20 g 併合群の 1%以上の被験者で治験薬
との因果関係が否定できないと判断されたものは、血中アルカリホスファターゼ増加、
血中尿酸増加、上腹部痛、頭痛、高尿酸血症及び悪心であった。
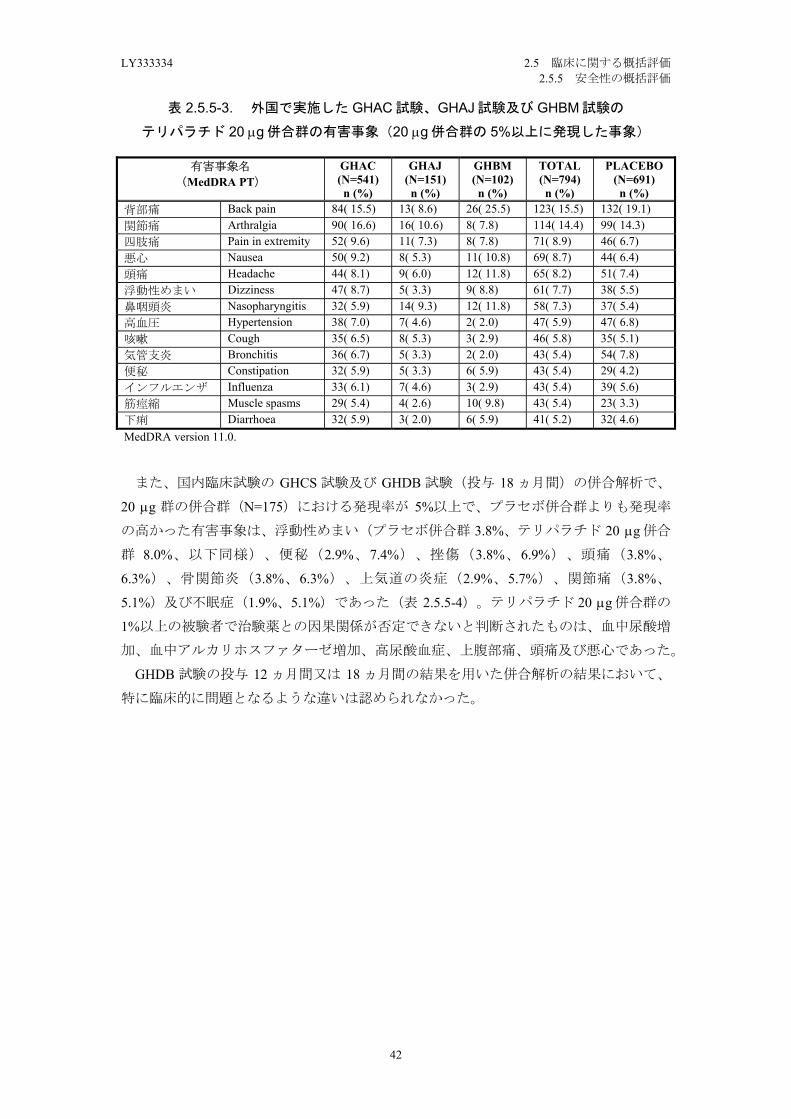
外国臨床試験の 3 試験の併合解析で、テリパラチド 20 g 併合群(N=794)における
発現率が 5%以上で、プラセボ併合群よりも発現率の高かった有害事象は、関節痛(プ
ラセボ併合群 14.3%、テリパラチド 20 g 併合群 14.4%、以下同様)、四肢痛(6.7%、
8.9%)、悪心(6.4%、8.7%)、頭痛(7.4%、8.2%)、浮動性めまい(5.5%、7.7%)、
鼻咽頭炎(5.4%、7.3%)、咳嗽(5.1%、5.8%)、便秘(4.2%、5.4%)、筋痙縮(3.3%、
5.4%)及び下痢(4.6%、5.2%)であった(表 2.5.5-3)。テリパラチド 20 g 併合群の
1%以上の被験者で治験薬との因果関係が否定できないと判断されたものは、悪心、浮動
性めまい、関節痛、頭痛、消化不良、筋痙縮、四肢痛、下痢、腹痛、無力症、便秘、疲
労、回転性めまい及び高血圧であった。
表 2.5.5-2. 国内で実施した GHCS 試験及び GHDB 試験(投与 12 ヵ月間)の
テリパラチド 20 g 併合群の有害事象(20 g 併合群の 5%以上に発現した事象)
有害事象名
(MedDRA PT)GHCS(N=39)n (%)
GHDB(N=136)n (%)
TOTAL(N=175)n (%)
PLACEBO(N=105)n (%)
鼻咽頭炎 Nasopharyngitis 9( 23.1) 38( 27.9) 47( 26.9) 37( 35.2)背部痛 Back pain 0( 0.0) 17( 12.5) 17( 9.7) 14( 13.3)便秘 Constipation 2( 5.1) 10( 7.4) 12( 6.9) 3( 2.9)浮動性めまい Dizziness 3( 7.7) 8( 5.9) 11( 6.3) 4( 3.8)頭痛 Headache 1( 2.6) 9( 6.6) 10( 5.7) 4( 3.8)骨関節炎 Osteoarthritis 1( 2.6) 9( 6.6) 10( 5.7) 4( 3.8)関節痛 Arthralgia 1( 2.6) 8( 5.9) 9( 5.1) 4( 3.8)挫傷 Contusion 1( 2.6) 8( 5.9) 9( 5.1) 4( 3.8)転倒・転落 Fall 0( 0.0) 9( 6.6) 9( 5.1) 7( 6.7)上気道の炎症 Upper respiratory
tract inflammation1( 2.6) 8( 5.9) 9( 5.1) 3( 2.9)
MedDRA version 11.0.
LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
42
表 2.5.5-3. 外国で実施した GHAC 試験、GHAJ 試験及び GHBM 試験の
テリパラチド 20 g 併合群の有害事象(20 g 併合群の 5%以上に発現した事象)
有害事象名
(MedDRA PT)GHAC(N=541)n (%)
GHAJ(N=151)n (%)
GHBM(N=102)n (%)
TOTAL(N=794)n (%)
PLACEBO(N=691)n (%)
背部痛 Back pain 84( 15.5) 13( 8.6) 26( 25.5) 123( 15.5) 132( 19.1)関節痛 Arthralgia 90( 16.6) 16( 10.6) 8( 7.8) 114( 14.4) 99( 14.3)四肢痛 Pain in extremity 52( 9.6) 11( 7.3) 8( 7.8) 71( 8.9) 46( 6.7)悪心 Nausea 50( 9.2) 8( 5.3) 11( 10.8) 69( 8.7) 44( 6.4)頭痛 Headache 44( 8.1) 9( 6.0) 12( 11.8) 65( 8.2) 51( 7.4)浮動性めまい Dizziness 47( 8.7) 5( 3.3) 9( 8.8) 61( 7.7) 38( 5.5)鼻咽頭炎 Nasopharyngitis 32( 5.9) 14( 9.3) 12( 11.8) 58( 7.3) 37( 5.4)高血圧 Hypertension 38( 7.0) 7( 4.6) 2( 2.0) 47( 5.9) 47( 6.8)咳嗽 Cough 35( 6.5) 8( 5.3) 3( 2.9) 46( 5.8) 35( 5.1)気管支炎 Bronchitis 36( 6.7) 5( 3.3) 2( 2.0) 43( 5.4) 54( 7.8)便秘 Constipation 32( 5.9) 5( 3.3) 6( 5.9) 43( 5.4) 29( 4.2)インフルエンザ Influenza 33( 6.1) 7( 4.6) 3( 2.9) 43( 5.4) 39( 5.6)筋痙縮 Muscle spasms 29( 5.4) 4( 2.6) 10( 9.8) 43( 5.4) 23( 3.3)下痢 Diarrhoea 32( 5.9) 3( 2.0) 6( 5.9) 41( 5.2) 32( 4.6)MedDRA version 11.0.
また、国内臨床試験の GHCS 試験及び GHDB 試験(投与 18 ヵ月間)の併合解析で、
20 g 群の併合群(N=175)における発現率が 5%以上で、プラセボ併合群よりも発現率
の高かった有害事象は、浮動性めまい(プラセボ併合群 3.8%、テリパラチド 20 g 併合
群 8.0%、以下同様)、便秘(2.9%、7.4%)、挫傷(3.8%、6.9%)、頭痛(3.8%、
6.3%)、骨関節炎(3.8%、6.3%)、上気道の炎症(2.9%、5.7%)、関節痛(3.8%、
5.1%)及び不眠症(1.9%、5.1%)であった(表 2.5.5-4)。テリパラチド 20 g 併合群の
1%以上の被験者で治験薬との因果関係が否定できないと判断されたものは、血中尿酸増
加、血中アルカリホスファターゼ増加、高尿酸血症、上腹部痛、頭痛及び悪心であった。
GHDB 試験の投与 12 ヵ月間又は 18 ヵ月間の結果を用いた併合解析の結果において、
特に臨床的に問題となるような違いは認められなかった。

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
43
表 2.5.5-4. 国内で実施した GHCS 試験及び GHDB 試験(投与 18 ヵ月間)の
テリパラチド 20 g 併合群の有害事象(20 g 併合群の 5%以上に発現した事象)
有害事象名
(MedDRA PT)
GHCS GHDB TOTAL PLACEBO PLACEBO→20 μg
(N=39) (N=136) (N=175) (N=105) (N=67)n (%) n (%) n (%) n (%) n (%)
鼻咽頭炎 Nasopharyngitis 9( 23.1) 45( 33.1) 54( 30.9) 37( 35.2) 29( 43.3)背部痛 Back pain 0( 0.0) 22( 16.2) 22( 12.6) 14( 13.3) 16( 23.9)浮動性めまい Dizziness 3( 7.7) 11( 8.1) 14( 8.0) 4( 3.8) 3( 4.5)便秘 Constipation 2( 5.1) 11( 8.1) 13( 7.4) 3( 2.9) 4( 6.0)挫傷 Contusion 1( 2.6) 11( 8.1) 12( 6.9) 4( 3.8) 5( 7.5)転倒・転落 Fall 0( 0.0) 11( 8.1) 11( 6.3) 7( 6.7) 7( 10.4)頭痛 Headache 1( 2.6) 10( 7.4) 11( 6.3) 4( 3.8) 4( 6.0)骨関節炎 Osteoarthritis 1( 2.6) 10( 7.4) 11( 6.3) 4( 3.8) 5( 7.5)
上気道の炎症Upper respiratory tract inflammation 1( 2.6) 9( 6.6) 10( 5.7) 3( 2.9) 3( 4.5)
関節痛 Arthralgia 1( 2.6) 8( 5.9) 9( 5.1) 4( 3.8) 7( 10.4)不眠症 Insomnia 2( 5.1) 7( 5.1) 9( 5.1) 2( 1.9) 2( 3.0)MedDRA version 12.0.「PLACEBO」は、GHCS 試験のプラセボ群のデータと GHDB 試験のプラセボ群の二重盲検比較期間 12ヵ月間におけるデータの併合を示す。
「PLACEBO→20 g」は、GHDB 試験のプラセボ群の二重盲検比較期間 12 ヵ月間(プラセボ投与)及
びオープン期間 6 ヵ月間(テリパラチド 20 g 投与)の合計 18 ヵ月間のデータを示す。
2.5.5.4.1.6 比較的よく見られる有害事象のまとめ
国内で実施した GHCS 試験及び GHDB 試験において認められた有害事象の発現状況
は、外国臨床試験の結果と類似していた。よく見られた有害事象である悪心及び頭痛の
最初の発現は、投与開始後初期に多く認められた。報告された有害事象の重症度は、ほ
とんどが軽度又は中等度であった。また、薬物動態における外国試験と国内試験の類似
性の検討の結果、日本人と外国人の体重の違いにより薬物動態に差が認められたが、安
全性の観点から問題となることはないと考えられた。以上より、臨床推奨用量であるテ
リパラチド 20 g 投与の忍容性は良好であり、国内において特徴的な有害事象の発現傾
向は認められなかった。
2.5.5.4.2 重篤及び他の重要な有害事象
国内で実施した GHCS 試験及び GHDB 試験において、死亡例は認められなかった。
GHCS 試験において、重篤な有害事象は、手骨折(10 μg 群)、一過性脳虚血発作(20
μg 群)及び食欲減退(40 μg 群)がそれぞれ 1 例ずつ認められた。いずれも、治験薬、
治験手順又はペン型注入器との因果関係はないと判断された。治験薬投与中に発現した
重篤な有害事象を理由に試験を中止した被験者は認められなかった。有害事象による中
止は、20 g 群が 2 例(血中カリウム増加 1 例及び骨関節炎 1 例)、40 g 群が 4 例(悪
心 2 例、呼吸困難 1 例及び倦怠感 1 例)の 6 例であった。GHDB 試験の第 1 期二重盲検
比較期間において、重篤な有害事象はプラセボ群で 7 例(10.4%)、テリパラチド 20 μg
群で 7 例(5.1%)に認められた。このうち、テリパラチド群の血圧低下及び乳癌は治験
薬との因果関係が否定できないと判断され、テリパラチド群の腹痛は治験手順との因果

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
44
関係が否定できないと判断された。重篤な有害事象による中止は、テリパラチド群では
血圧低下、肺腺癌及び乳癌の各 1 例ずつの合計 3 例、プラセボ群ではアレルギー性気管
支肺アスペルギルス症の 1 例であった。また、第 2 期移行後に重篤な有害事象を発現し
た被験者は、プラセボ群(プラセボ→20 g)2 例、20 g 群(20 g→20 g)3 例であっ
た。第 2 期に認められたいずれの重篤な有害事象も、治験薬又は治験手順との因果関係
はないと判断された。第 2 期における重篤な有害事象による中止は、プラセボ群(プラ
セボ→20 g)の動脈解離の 1 例であった。有害事象による中止は、第 1 期二重盲検比較
期間においてプラセボ群 2 例とテリパラチド群 6 例の合計 8 例であり、そのうち 4 例は
重篤な有害事象による中止であった。また、第 2 期における有害事象による中止は、プ
ラセボ群(プラセボ→20 g)1 例と 20 g 群(20 g→20 g)3 例の合計 4 例であり、そ
のうち 1 例が重篤な有害事象による中止であった。国内臨床試験において、テリパラチ
ド投与における重篤な有害事象の発現に関して、一定の傾向は認められなかった。
外国臨床試験の GHAC 試験において、死亡は 16 例でいずれも治験薬との因果関係は
ないと判断された。1 件以上の重篤な有害事象が認められた被験者数は、プラセボ群で
544 例中 113 例(20.8%)、テリパラチド 20 g 群で 541 例中 93 例(17.2%)、40 g 群
では 552 例中 109 例(19.7%)であり、投与群間に統計学的な有意な差は認められなか
った。全体で 1%以上の被験者で報告された重篤な有害事象は、外科手術手技 125 例
(7.6%)、事故による外傷 37 例(2.3%)及び腹痛 22 例(1.3%)であったが、これらの
投与群における発現率は同程度であった。有害事象により本試験を中止した被験者数は
126 例(7.7%)であった。有害事象による中止率は、プラセボ群 5.9%、20 g 群 6.5%、
40 g 群 10.7%であり、40 g 群で高く(全投与群間で p=0.005)、試験中止に至った有
害事象のうち最も多く見られたものは悪心(12 例、0.7%)であった。
GHAJ 試験において、死亡は 2 例でいずれも治験薬との因果関係はないと判断された。
1 件以上の重篤な有害事象が認められた被験者数は、プラセボ群で 147 例中 16 例
(10.9%)、テリパラチド 20 g 群で 151 例中 15 例(9.9%)、40 g 群では 139 例中 14
例(10.1%)であり、投与群間に統計学的な有意差は認められなかった。全体で 4 例
(0.9%)以上に報告された重篤な有害事象は、外科手術手技(2.1%)、事故による外傷、
咳嗽増加及び発熱(それぞれ 0.9%)であった。重篤な有害事象の発現について、プラセ
ボ群よりもテリパラチド投与群で顕著に多く認められた事象はなく、多くの事象の発現
は 2 例以下であり、本剤投与による影響が考えられる特定の重篤な有害事象が発現する
傾向は認められなかった。有害事象による中止は、プラセボ投与群 7 例(4.8%)、20 μg
群 14 例(9.3%)及び 40 μg 群 18 例(12.9%)の合計 39 例(8.9%)であった。最も多く
見られた試験中止に至った有害事象は悪心であり、悪心による中止例は 40 μg 群のみに
5 例(3.6%)認められた。
以上のように、臨床試験で報告された重篤な有害事象の発現は、一定の傾向を示すも
のではなかった。また、プラセボ群に比べて、テリパラチド投与により重篤な有害事象
の発現リスクが増大する結果は認められなかった。

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
45
2.5.5.4.3 テリパラチド投与中止後の追跡調査研究
GHBJ 試験において、GHAC 試験及び GHAJ 試験を含む外国で実施した臨床試験 7 試
験について、投与中止後から 5 年間(GHBJ 試験の観察期間は 4.5 年間)の追跡調査を実
施した。有害事象は 24 ヵ月、重篤な有害事象は 5 年間評価した。GHBJ 試験では、テリ
パラチドの投与は行っていないが、カルシウムとビタミン D の補給に加えて、医師の判
断により他の骨粗鬆症治療薬を投与できた。安全性の評価は、先行する試験集団ごとに
行った。
概して、24 ヵ月までの有害事象の安全性評価では、先行する試験でのテリパラチドの
投与期間及び追跡調査期間のいずれにおいても、本剤の良好な忍容性が認められ、安全
性に関する大きな懸念は認められなかった。本剤の投与期間中、背部痛の発現は有意に
低く、この背部痛の発現低下は GHBJ 試験の追跡調査期間中も維持された。40 g 群の
悪心及び頭痛は、本剤と関連があると考えられたが、追跡調査期間中、これらの有害事
象の発現は、テリパラチド群とプラセボ群の両方において発現頻度は低かった。
重篤な有害事象は、先行する試験でのテリパラチド投与中止から最大 5 年間にわたり、
追跡調査した。1943 例の被験者のうち、69%には重篤な有害事象が認められなかった。
GHAC 試験、GHAJ 試験及び GHAH 試験の試験集団において、死亡に関して投与群間で
統計学的な有意な差は認められなかった。これらの試験集団の 1.0%以上に認められた重
篤な有害事象の発現に関して、投与群間に統計学的な有意差は認められなかった。先行
する試験の全試験集団において、テリパラチド投与を受けた被験者で骨肉腫又は原発性
骨悪性腫瘍の発現は認められなかった。
概して、テリパラチド投与中止後に、被験者の安全性に関して問題と考えられるリス
クは見られなかった。先行する試験でテリパラチド投与を受けた被験者において、骨癌、
心血管系疾患及び椎体骨折の発現増加は認められず、骨肉腫の発現も認められなかった。
2.5.5.4.4 臨床薬理試験における安全性評価
日本人及び外国人の閉経後健康女性被験者を対象に実施した GHCO 試験において、日
本人において最もよく見られた因果関係を否定できない有害事象は頭痛であり、続いて
嘔吐、悪心、浮動性めまいであった。これらの有害事象は外国人においても同様に認め
られており、重症度は、日本人、外国人共にすべて軽度又は中等度であり、高度なもの
は認められなかった。多くの有害事象(99 件中 84 件)は 40 µg 又は 60 µg 投与時に発現
し、用量の増加とともに有害事象の発現が増加する傾向が認められた。日本人及び外国
人健康被験者に対するテリパラチド 60 µg までの単回投与の忍容性は良好であり、安全
性に重大な問題はないものと考えられた。
外国の 11 試験の併合解析において、健康成人男性及び女性並びに高血圧患者及び慢
性腎障害患者を対象に、5~100 µg の範囲のテリパラチドを投与した。ほとんどの有害
事象は投与後 4 時間以内に発現し、用量の増加に伴い因果関係を否定できない有害事象
が増加する傾向が認められた。5~15 µg 投与では因果関係を否定できない有害事象は発
現しなかった。20 µg 投与では 42 例中 7 例において因果関係を否定できない有害事象が

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
46
発現した。臨床的に最も問題となる有害事象は 1 例に見られた頭部ふらふら感を伴う一
過性の起立性低血圧であった。
30~40 μg 群と 60~100 μg 群において最もよく見られた有害事象は頭痛であり、次に
よく見られた有害事象は浮動性めまいであった。浮動性めまいはすべて投与後 4 時間以
内に発現し、しばしば立位血圧の臨床的に問題と考えられる変化を伴った。その他のよ
く見られた有害事象は悪心、嘔吐及び起立性低血圧であった。
反復投与時に認められた有害事象は、単回投与時と同様の発現傾向であった。高血圧
又は慢性腎障害を有する患者のテリパラチド 40 µg の投与に対する忍容性は良好であり、
健康被験者と同様であった。
重篤な有害事象は 2 例に認められた。そのうちテリパラチドを投与した被験者におけ
る重篤な有害事象は、外国人で白血球増加症が 1 例に認められた。
以上のように、テリパラチドは男女健康成人被験者、高血圧患者及び慢性腎障害患者
を対象とする第 I 相試験において、良好な忍容性及び安全性を示した。臨床的に問題と
考えられる有害事象として起立性低血圧があり、その多くは 30 µg 以上の用量で認めら
れた。
2.5.5.5 臨床検査
国内及び外国で実施した臨床試験において、測定した臨床検査値の経時的変化につい
て臨床的に問題となるような事象は認められなかった。PTH の生理作用として血清カル
シウムの増加が予測されたため、GHAC 試験、GHAJ 試験及び GHCS 試験においては、
投与後 4~6 時間及び各来院時の治験薬投与前(投与後約 24 時間)に血清カルシウムの
測定を行った。GHAC 試験、GHAJ 試験において、投与後 4~6 時間の血清カルシウムは、
特にテリパラチド 40 g 投与群で増加が認められたが、この増加は一過性であり、投与
前値においてはベースラインとほぼ同程度のレベルまで戻っていた。また投与期間に伴
って増加する傾向も認められなかった。国内臨床試験の GHCS 試験における投与後 4~
6 時間及び投与前血清カルシウムの結果は、変化量はわずかであり臨床的に問題となら
ないと考えられた。GHDB 試験では、投与前血清カルシウムの測定のみ実施し、他の試
験で認められた結果と同様の傾向であった。以上より、テリパラチド投与による血清カ
ルシウムの増加は、一過性であり、臨床的に問題となるものではないと考えた。
その他の項目では、外国及び国内で実施した臨床試験において、血清尿酸の増加が認
められた。血清尿酸の増加は、本事象に関連して発現する可能性のある痛風や尿路結石
症などの臨床症状の発現を伴わなかった。国内臨床試験では因果関係を否定できない有
害事象として血中尿酸増加が多く報告され、GHCS 試験の全テリパラチド群で 6 例
(5.2%)と多く見られた事象であり、GHDB 試験では投与 12 ヵ月間にテリパラチド群
で高尿酸血症 2 例(1.5%)及び血中尿酸増加 1 例(0.7%)、投与 18 ヵ月間に 20 g 群
(20 g→20 g)で高尿酸血症が 3 例(2.2%)及び血中尿酸増加が 2 例(1.5%)、プラ
セボ群(プラセボ→20 g)で高尿酸血症が 2 例(3.0%)に認められた。尿路結石を有す
る患者でのテリパラチド投与の臨床試験結果がないことから、尿路結石を有する患者及
びその既往のある患者には、テリパラチドを慎重に投与する必要があると考える。肝機

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
47
能検査において、肝機能異常を示唆する臨床検査値の変動は認められなかった。腎機能
検査においてベースラインに対して、血清尿素窒素の増加、血清マグネシウムの低下な
どの所見が認められ、血液学検査において赤血球及び白血球関連の項目にわずかな変化
が認められたが、これらの変化は極めて小さく、また多くは可逆的で投与終了時点にお
いてはプラセボと同程度であり、臨床的に問題となるものではないと考えられた。
2.5.5.6 バイタルサイン及び心電図
国内で実施した GHCS 試験において、12 週時点で 20 g 群の心拍数がベースラインと
比較して統計学的に有意な増加が認められ、GHDB 試験において、52 週時点で 20 g 群
の心拍数、収縮期血圧及び拡張期血圧にベースラインと比較して統計学的に有意な減少
が、76 週時点でプラセボ群(プラセボ→20 g)及び 20 g 群(20 g→20 g)において、
心拍数の平均値に、ベースラインからの統計学的に有意な減少が認められたが、これら
の変化は小さく臨床的に問題となるものではないと考えられた。GHDB 試験の第 1 期に
20 g 群で血圧低下の重篤な有害事象が 1 例発現し、試験を中止した。外国で実施した
GHAC 試験及び GHAJ 試験において、テリパラチド投与群における心拍数、収縮期血圧
及び拡張期血圧のベースラインからの変化は極めて小さく、投与群間で差は認められな
かった。以上のように、バイタルサインについて、20 g 投与により臨床的に問題とな
る変化は認められなかった。臨床薬理試験において、主に 30 g 以上の投与群で、投与
後 3~5 時間に起立性低血圧が認められた。
心電図に関しては、GHCS 試験におけるいずれの投与群でもベースラインから 24 週時
までに臨床的に問題となる変化は認められなかった。GHDB 試験では、テリパラチド投
与群において、ベースラインから 24 週及び 52 週時までの変化について投与群間に統計
学的な有意差は認められなかった。更に、ベースラインから 76 週時までについても、
臨床的に問題となる変化は認められなかった。参考資料の外国で実施した GHBQ 試験で
は、一部の被験者について心電図への影響を検討した結果、20 g の投与による QTc 間
隔に対する短期的又は長期的に臨床上問題となる影響は認められなかった。以上より心
電図所見において、臨床的に問題となる変化は認められなかった。
2.5.5.7 テリパラチド投与において特に考慮すべき安全性
2.5.5.7.1 骨肉腫
非臨床試験で Fischer 344 系ラットを用いた 2 年間がん原性試験において、骨肉腫を含
む骨腫瘍性病変が認められたことにより、当時外国で進行中であった GHAC 試験及び
GHAJ 試験を含む実施中であった臨床試験 7 試験が治験依頼者の判断で途中中止となっ
た。ラットで見られた骨に対する増殖性病変は、サルにおける 18 ヵ月間投与試験では
認められず、サルにおいては骨肉腫の発現も認められなかった。ラットでは、ヒトやサ
ルと異なり、ほぼ生涯にわたり骨の長軸方向への成長が継続することや、骨代謝回転が
速いことなど、ラットとヒトの骨の生理は異なっている。ラットがん原性試験では骨量
の過剰な増加が観察されたが、これは上記のラットにおける特有の骨の生理が関連して
いると考えられた。

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
48
現在までに、テリパラチド投与を行った全臨床試験及び市販後臨床試験において、本
剤の投与期間中に骨肉腫の発現は認められておらず、更に追跡調査研究である GHBJ 試
験で、投与終了後 5 年間(GHBJ 試験の観察期間は 4.5 年)で、骨癌の発現率の上昇は認
められず、骨肉腫の発現も報告されなかった。
市販後の自発報告において、これまでに 5 例の骨肉腫が報告されている。2008 年 11
月 26 日時点で、全世界でテリパラチド投与を受けた患者数は 65.1 万人と推定されてお
り、テリパラチド投与開始後の年数を勘案すると約 150 万人年に相当する。一方、60 歳
以上の一般集団における骨肉腫の自然発症率は 100 万人年あたり約 4 件であり、これは
テリパラチドの自発報告における発症率とほぼ同程度と考えられた。
イーライリリー・アンド・カンパニーは、GHBX 研究として、40 歳以上の男性及び女
性で骨肉腫を発症した症例について、テリパラチドの投与歴を調査している。2008 年
12 月 15 日までに、米国で 1025 例、欧州の 5 ヵ国で 89 例の骨肉腫の発症が報告されて
おり、そのうち詳細調査が実施できた症例(それぞれ 461 例、62 例)において、本剤の
投与歴のある症例は認められていない。イーライリリー・アンド・カンパニーは、テリ
パラチド投与に関連する骨肉腫発症例を更に調査するため、GHBX 研究の観察期間は承
認後 10 年間から 15 年間に延長することを決定し、今後も継続して本剤と骨肉腫発症の
関連性について注意深く観察することとしている。
以上より、ラットで認められた骨肉腫がヒトにおいても発症する可能性は低いと考え
られた。しかしながら、ヒトにおける骨肉腫の発症に関しては不明である。よって、本
剤の投与期間の上限を、それまでに実施した臨床試験の投与期間を基に設定することと
し、更に骨肉腫発生リスクの高いと考えられる患者には投与しないこととした。
2.5.5.7.2 高カルシウム血症
PTH はカルシウムの恒常性を調節するホルモンであるため、テリパラチド投与により
PTH の薬理作用に基づく血清カルシウムの増加が予測された。国内で実施した GHCS 試
験及び外国で実施した GHAC 試験と GHAJ 試験においては、本剤投与後 4~6 時間に一
過性の血清カルシウムの増加が認められたが、この増加は投与前値ではベースライン値
まで戻っており、持続的な増加は認められず、臨床的に問題となるものでないと考えら
れた。GHDB 試験では、プラセボ群及び 20 μg 群において、血清カルシウム及び補正血
清カルシウムの中央値にベースラインからの統計学的に有意な上昇が認められたが、こ
の上昇は小さく、他の試験における結果と同様であった。外国及び国内のプラセボ対照
試験における高カルシウム血症の発現は、プラセボ群では認められず、テリパラチド投
与群では GHDB 試験、GHAC 試験及び GHAJ 試験でそれぞれ1例ずつと、GHBM 試験
で 3 例及び GHCA 試験 32 例に認められた。なお、GHCA 試験では各治験実施施設での
基準範囲上限を超えた場合に高カルシウム血症として報告された。
PTH の薬理作用として血清カルシウム増加作用があり、一過性ではあるものの本剤投
与により血清カルシウムの増加が認められ、低頻度ではあるが高カルシウム血症の報告
があることから、血清カルシウム上昇に関しては、患者に十分な説明を行い、持続性の
血清カルシウム上昇が疑われる症状が認められた場合は、テリパラチドの投与を中止し

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
49
て速やかに診察を受けるよう指導する必要がある。また、本剤と活性型ビタミン D 製剤
の併用により、相加的に血清カルシウムが増加することが懸念されるため、両薬剤の併
用投与は避けることが望ましい。
2.5.5.7.3 起立性低血圧
起立性低血圧は、臨床薬理試験で、主に 30 g 以上の投与群において報告されたが、
骨粗鬆症患者を対象とした評価資料の臨床試験では 2 例のみが報告された。
臨床薬理試験では、起立性低血圧は、日本人と外国人を比較した GHCO 試験において
33 例中 4 例(6 件)に認められ、20 μg 投与後に発現した 1 例を除き、他はすべて 30 μg
よりも高用量の投与群であった。外国臨床薬理試験 11 試験の併合解析の因果関係を否
定できない有害事象において、体位性低血圧として 20 μg 群で 1 例、30~40 μg で 3 例、
60~100 μg で 1 例に認められた。多くの症状は、立位血圧測定時に頭がふらふらして立
位を保てなかったもので、投与後 3~5 時間に発現し、5~15 分間持続した。
一方、臨床試験では、テリパラチド投与による起立性低血圧の報告は国内で実施した
GHDB 試験と外国で実施した GHCA 試験における 20 μg 群の 1 例ずつ(発現率:それぞ
れ 0.7%、0.1%)であった。
起立性低血圧は、臨床薬理試験では臨床的に問題と考えられる影響が認められ、その
多くは 30 μg よりも高用量において発現していた。本剤投与後の起立性低血圧によるリ
スクを回避するために、高所での作業、自動車の運転など危険を伴う作業に従事する場
合には注意する必要がある。
2.5.5.7.4 腎障害
腎機能正常者と軽度から重度の腎障害を有する患者(クレアチニンクリアランス値:
正常;≥90 mL/min、軽度~中等度;31-75 mL/min、重度;≤30 mL/min)において、テリ
パラチド 40 μg の血清カルシウム及び尿中カルシウムに対する影響を検討した(GHAW
試験)。重度の腎障害を有する患者 5 例において、治験薬との因果関係を否定できない
有害事象の発現は、頭痛 1 例のみであった。腎機能正常者と腎障害を有する患者のいず
れにも血中総カルシウムに対する影響は認められなかったが、腎障害を有する患者では
腎機能正常者に比べて血中イオン化カルシウムが有意に低下した。24 時間尿中カルシウ
ム排泄量は、腎障害を有する患者では 60~65%減少したが、この差は腎障害の重症度の
程度により、カルシウム分画排泄率に差がでたためであると推定された。
以上のように、腎障害を有する被験者においても、本剤の忍容性は良好であった。し
かし、重度腎障害を有する被験者の薬物動態パラメーターは、腎機能正常の被験者に対
して、AUC0-∞は 89%高く、T1/2は 77%の延長が認められたことなどを勘案し、腎障害を
有する患者に対しては慎重に投与する必要がある。
2.5.5.7.5 背部痛
背部痛は高齢者や骨粗鬆症患者において発症頻度が高くなることが予測される。国内
の GHCS 試験及び GHDB 試験(投与 12 ヵ月間)の併合解析では、プラセボ群 13.3%、

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
50
全テリパラチド投与群 8.7%、外国の GHAC 試験、GHAJ 試験及び GHBM 試験の併合解
析では、プラセボ群 19.1%、全テリパラチド群 14.5%であった。背部痛は、国内及び外
国臨床試験の併合解析では共に 2 番目に多く見られた有害事象であったが、概してテリ
パラチド群において背部痛の発現は低かった。
個々の試験においては、国内臨床試験の GHCS 試験では、プラセボ群 10.5%、10 g
群 10.5%、20 g 群 0%及び 40 g 群 2.6%であり(p=0.082)、GHDB 試験の第 1 期二重盲
検比較期間では、プラセボ群 14.9%及びテリパラチド群 12.5%であった(p=0.663)。外
国臨床試験の GHAC 試験では、プラセボ群 22.6%、20 g 群 16.8%及び 40 g 群 15.8%で
あり(p=0.007)、GHAJ 試験では、プラセボ群 12.9%、20 g 群 9.3%及び 40 g 群 7.9%
であり(p=0.342)、GHBM 試験では、アレンドロネート群 38.6%及びテリパラチド群
25.5%であった(p=0.05)。重症度は、テリパラチド投与群において、GHCS 試験及び
GHDB 試験では高度の背部痛はなく、その他の試験でもほとんどが軽度又は中等度であ
った。
以上のように、背部痛は骨粗鬆症患者の被験者特性を反映して、国内及び外国におい
て発現率が高かったため、背部痛の発現には注意を要するが、テリパラチド群において
発現率が低かったことから、テリパラチド投与により背部痛発現のリスクが低下する可
能性が考えられた。
2.5.5.8 薬物相互作用
臨床薬理試験において、ヒドロクロロチアジド、フロセミド、カルシウム拮抗薬又は
遮断薬、ジゴキシン、及びホルモン補充療法又はラロキシフェンとの薬物相互作用を
検討した。これらの薬剤との併用により、臨床的に問題となる薬物相互作用は認められ
なかった。
参考資料の臨床試験において、骨粗鬆症治療薬のラロキシフェン(GHCD 試験)、ホ
ルモン補充療法(GHAF 試験)及びアレンドロネート又はラロキシフェン(GHCM 試
験)とテリパラチドの併用の影響について検討した。テリパラチドとこれらの薬剤の併
用による臨床的に問題となる安全性上の問題は認められなかった。
2.5.5.9 市販後データ
テリパラチドは、2002 年 11 月に米国にて承認されて以来、2010 年 1 月 4 日現在 83 の
国又は地域で承認されている。2008 年 11 月 26 日時点におけるテリパラチド投与を受け
た患者数は約 65.1 万人と推定されている。2008 年 11 月 26 日時点において、市販後の自
発報告にて 541 件の死亡例が報告されている。重篤な有害事象は 8165 件報告されてお
り、そのうち報告された件数の中で 2%以上であったよく見られた事象は、転倒・転落
(3.97%)、肺炎(2.35%)及び股関節部骨折(2.02%)であった。非重篤な有害事象は
30920 件報告されており、そのうち報告された件数の中で 2%以上であった事象は、悪心
(4.27%)、浮動性めまい(3.53%)、頭痛(2.62%)、筋痙縮(2.55%)、四肢痛
(2.21%)及び関節痛(2.07%)であった。自発報告において報告された重篤な有害事象
は臨床試験における重篤な有害事象の中でも発現率の高い事象であり、また股関節部骨

LY333334 2.5 臨床に関する概括評価
2.5.5 安全性の概括評価
51
折は高齢者及び骨粗鬆症患者ではよく見られる事象でもある。非重篤な有害事象の安全
性プロファイルは、臨床試験においてよく見られた有害事象と類似しており、市販後に
おいて予測できない有害事象や特定の有害事象が発現する傾向は認められなかった。
2.5.5.10 安全性評価のまとめ
国内及び外国で実施した臨床試験及び臨床薬理試験で報告された安全性に関する成績
から、テリパラチド 20 μg の忍容性及び安全性プロファイルは良好であった。国内で実
施した臨床試験における有害事象の発現は、外国で実施した試験における有害事象と類
似した傾向を示し、日本人で特徴的に発現する有害事象は認められず、外国試験成績か
ら予測可能なものであった。評価資料とした臨床試験の投与期間は最長 24 ヵ月であっ
たが、有害事象の発現率及び発現傾向は試験間で特に差は認められなかった。
また、非臨床試験の成績から懸念された骨肉腫の発現については、自発報告で 5 例の
報告があるが、この発症率は自然発症率と同程度であった。臨床試験では、投与期間中
及び投与中止後の追跡調査期間ともに骨肉腫の発現は認められなかった。動物種の違い
から、ラットで認められた骨肉腫がヒトで発現する可能性は低いと考えられる。しかし
ながら、ヒトにおける骨肉腫の発症に関しては不明である。よって、市販後における安
全性情報を観察するために GHBX 研究を実施している。
市販後の自発報告における有害事象及び重篤な有害事象は、概して臨床試験で認めら
れた事象と類似しており、臨床試験の安全性プロファイルから予測できるものであった。
しかし、市販後において、日本人の骨粗鬆症患者に予測しない安全性上の所見が認めら
れる可能性も否定できないため、継続して安全性情報について注意を払っていく。
以上より、テリパラチド 20 μg の 1 日 1 回皮下投与は、適切な患者に対して適正に使
用される場合、忍容性が良好で安全な薬剤であると考えた。

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
52
2.5.6 ベネフィットとリスクに関する結論
2.5.6.1 骨折の危険性の高い骨粗鬆症の治療薬としてのテリパラチドの概略
日本における骨粗鬆症患者は、高齢化社会の進展とともに増加し、その数は約 1,100
万人(2000 年)と推定されていることから、骨粗鬆症の治療及びそれに伴う骨折の予防
は、高齢化社会を迎えた日本において、重要な課題である。最近の報告では大腿骨近位
部骨折と椎体骨折を合わせた総医療費は 2,382~3,218 億円、医療・介護費用は 7,974~
9,895 億円と推計されている(原田ほか 2005)。したがって、骨粗鬆症治療により骨折
を如何に効率よく防止できるか、ということが骨折により低下する患者の QOL の改善、
寝たきりの防止、更には医療費抑制の鍵となることから、特に骨折の危険性の高い骨粗
鬆症患者において顕著な骨折防止効果の期待できる骨粗鬆症治療薬が必要である。骨粗
鬆症と診断された患者の中でも、既に脆弱性骨折を有している場合はもちろんのこと、
脆弱性骨折を有していない場合でも高齢や低骨密度などの骨折の危険因子を有している
場合、骨折の危険性は高まった状態であると考えられる。テリパラチドの皮下投与療法
は、既に骨微細構造が破綻をきたした骨折の危険性の高い骨粗鬆症患者において、速や
かに骨微細構造を再構築し、骨粗鬆症を回復方向へ転換する。このような効果は他の骨
吸収抑制剤にはなく新規性を示すものである。この新規的なテリパラチドの骨形成促進
作用に伴い、約 1.5 から 2 年間の短期間の治療で他剤と比して顕著な椎体及び大腿骨骨
密度増加作用、更には椎体骨折及び非椎体骨折の発生抑制効果が発揮されると考えられ
る。日本ではこのような新規性を持つ骨形成促進剤は未だ上市されておらず、既に骨微
細構造が破綻をきたした骨折の危険性の高い骨粗鬆症患者の治療において必要である。
2.5.6.2 テリパラチド投与によるベネフィット
2.5.6.2.1 骨折防止効果
骨形成促進剤であるテリパラチドは、骨折の危険性の高い骨粗鬆症患者の骨密度を有
意に増加させ、骨微細構造を改善、再構築することにより、結果として新規椎体骨折及
び非椎体骨折の発生を抑制することが確認されている。
外国で実施した大規模な骨折抑制試験である GHAC 試験は、1 個以上の中等度又は 2
個以上の軽度椎体骨折を有する閉経後骨粗鬆症女性患者 1,637 例を対象とし、プラセボ
投与を対照として、テリパラチド 20 μg 又は 40 μg の 1 日 1 回皮下投与による新規椎体
骨折発生率の抑制効果を主要評価項目として実施された。本試験の投与期間は計画時点
では 3 年であったが、試験実施中にラットがん原性試験において骨肉腫が認められたた
め早期に中止した。このため、被験者におけるテリパラチドの投与期間は、19 ヵ月(中
央値)であった。GHAC 試験では、テリパラチド 20 μg 及び 40 μg の 19 ヵ月(中央値)
の投与により、プラセボ群と比較して椎体及び非椎体骨折の発生を有意に抑制した。プ
ラセボ群に対する新規椎体骨折の相対的なリスクの減少は、テリパラチド 20 μg 群で
65%、40 μg 群で 69%であった。更に、複数の新規椎体骨折の発生の場合、プラセボ群
に対する相対的なリスクの減少は、テリパラチド 20 μg 群で 77%、40 μg 群で 86%であ
り、中等度又は重度の新規椎体骨折の発生の場合、テリパラチド 20 μg 群で 90%、40 μg
群で 78%であった。プラセボ群に対する非椎体骨折の相対的なリスクの減少は、テリパ

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
53
ラチド 20 μg 群で 35%、40 μg 群で 40%であった。更に非外傷性非椎体骨折の発生にお
ける相対的な骨折リスクの減少は、テリパラチド 20 μg 群で 53%、40 μg 群で 54%であ
った(第 2.5.4.2.4.2.1 項参照)。また、1 人の患者における骨折を防止するために必要な
患者数(Number needed to treat; NNT)はテリパラチド 20 g の 19 ヵ月(中央値)投与に
おいて椎体骨折で 11、非椎体骨折で 34 という値を示している(Hodsman et al. 2005)。
外国で実施したテリパラチド投与中止後の追跡調査研究である GHBJ 試験の結果から、
GHAC 試験においてテリパラチドの投与を 19 ヵ月(中央値)受けた被験者では、GHAC
試験の最終観察時から 18 ヵ月時点においてプラセボ群と比較した新規椎体骨折の抑制
効果の持続が認められた(プラセボ群に対する相対的なリスクの減少:20 μg 群で 40%、
40 μg 群で 45%)。GHBJ 試験中にいずれの群でも 60%前後の被験者に他の骨粗鬆症薬の
投与が認められてはいるが、テリパラチド投与群においては投与終了後にも非椎体骨折
抑制効果の持続は認められていた(第 2.5.4.5 項参照)。GHAC 試験の最終観察時から
30 ヵ月経過時点までの、非椎体骨折の非発生持続割合に関する Kaplan-Meier 曲線では、
プラセボ群の曲線は両テリパラチド投与群の曲線から乖離し続けたままであった(第
2.7.3.5.1.1.1.1.2 項参照)。
ステロイド使用の骨折に対する影響は骨密度や既存骨折とは独立していて、ステロイ
ド使用時の骨折リスクは約 2~4 倍になることが知られており(Kanis et al. 2004a)、ス
テロイド性骨粗鬆症は骨折の危険性の高い骨粗鬆症の代表的なものとしてあげられる。
ステロイド性骨粗鬆症患者を対象とした外国臨床試験(GHBZ 試験)において、テリパ
ラチド群はアレンドロネート群と比較して統計学的に有意な椎体骨折発生抑制を認めた
〔18 ヵ月での新規椎体骨折発生率:テリパラチド群で 171 例中 1 例(0.6%)、アレンド
ロネート群では 165 例中 10 例(6.1%)、p=0.004〕(第 2.5.4.3 項参照)。
現在日本では、骨粗鬆症治療薬としてビスフォスフォネート製剤や SERM 製剤などの
骨吸収抑制剤が主に用いられている。それぞれの臨床試験において対象被験者、試験デ
ザインなどが異なるため単純な比較は必ずしも適切ではないが、テリパラチドの 20 μg
による治療は、椎体骨折、更には非椎体骨折においても、既存の骨吸収抑制剤ではみら
れなかった 50%を越える相対的なリスクの減少を示した。特に非椎体骨折では、投与後
36 ヵ月後において唯一相対的な非椎体骨折リスクの減少が認められているリセドロネー
ト〔プラセボに対して 40%の相対的な骨折リスクの減少(Harris et al. 1999)〕より高い
抑制効果がより短い期間〔19 ヵ月間(中央値)〕の投与後において示された。
このように、テリパラチドは、約 1.5 から 2 年という比較的短期間の投与において、
従来の骨粗鬆症治療薬を上回る椎体骨折及び非椎体骨折の抑制効果を有することが明ら
かにされた。
2.5.6.2.2 骨微細構造の再構築効果
テリパラチドは新規の骨形成促進剤であり、骨折の危険性の高い骨粗鬆症患者におい
て骨微細構造を再構築する作用が認められている。
GHAC 試験では、一部の被験者において骨生検を実施することにより、テリパラチド
を投与した場合の骨質の変化を組織学的に評価した。テリパラチド投与により、海綿骨

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
54
量及び連結性の増加、骨梁形状の板状構造へのシフト、並びに皮質骨の多孔性
(porosity)の増加を伴わない皮質骨幅の増加が認められた(Jiang et al. 2003)。これら
の海綿骨及び皮質骨の形態学的な変化、すなわち骨微細構造再構築作用は、力学的健全
性を増加させ、新たな椎体骨折及び非椎体骨折の発現を減少させることが示唆されてい
る。
日本で用いられているビスフォスフォネート製剤、SERM 製剤などの骨吸収抑制剤は、
骨吸収を抑制して BMD を増加し、椎体骨折の危険性を低減する効果を示すことは認め
られてはいるが、骨形成促進により骨微細構造を再構築する作用はなく、骨折の危険性
の高い骨粗鬆症患者に対して十分な効果を発揮しているとは言い切れない(第 2.5.1.2 項
参照)。これらの結果からも、テリパラチドは骨形成を促進し、骨の強度を増強し、短
い期間で優れた骨折抑制効果を示すことが可能であり、その作用機序は既存の骨粗鬆症
治療薬にはない新規性を示している(Riggs and Parfitt 2005)。
2.5.6.2.3 骨密度増加効果
国内第 II 相試験(GHCS 試験)及び第 III 相試験(GHDB 試験)においては、骨折の
危険性の高い日本人原発性骨粗鬆症患者(第 II 相試験は閉経後女性患者が対象)におけ
るテリパラチド投与後の腰椎 BMD 変化率を評価した。GHCS 試験においてテリパラチ
ドを 24 週間連日皮下投与した際の腰椎 BMD 変化率は、10 μg、20 μg 及び 40 μg 群でプ
ラセボ群に比べ投与量の増加に従って統計学的に有意な増加を示した(すべての投与群
について p<0.001)。ベースラインから最終観察時までの平均変化率は、20 μg 群で
6.4%であった。GHDB 試験の第 1 期二重盲検比較期間 52 週間(12 ヵ月間)においては、
テリパラチド 20 μg 群において投与期間に応じて腰椎 BMD が増加し、ベースラインか
らの平均変化率は 6 ヵ月(24 週)時点で 7.35%、12 ヵ月(52 週)時点で 10.04%であっ
た(第 2.7.6.3.2.6.2.1.1 項参照)。その後第 2 期オープン期間 24 週間(6 ヵ月間)を実施
した結果、腰椎 BMD は持続的に増加し、テリパラチド 20 μg を 18 ヵ月間投与した被験
者における腰椎 BMD のベースラインからの平均変化率は、11.93%であった(第
2.7.6.4.1.6.1.1 項参照)。外国で実施したプラセボ対照二重盲検比較試験(GHAC 及び
GHAJ 試験)においても、腰椎 BMD のベースラインからの変化率は、テリパラチド
20 μg 群(GHAC 試験:12 ヵ月時点 8.26%及び 18 ヵ月時点 10.31%、GHAJ 試験:12 ヵ月
時点 6.07%)及び 40 μg 群(GHAC 試験:12 ヵ月時点 11.87%及び 18 ヵ月時点 14.76%、
GHAJ 試験:12 ヵ月時点 9.41%)において、プラセボ群との間に統計学的に有意な差が
認められた(第 2.7.3.3.2.2.2.1.1 項参照)。
更にテリパラチドにおいては、ビスフォスフォネート製剤(アレンドロネート)に比
して統計学的に有意な腰椎 BMD 増加効果が認められている(第 2.5.4.3 項参照)。アレ
ンドロネート 10 mg を実薬対照とした、テリパラチド第 IV 相臨床試験(GHBM 試験)
では 203 例の閉経後骨粗鬆症女性患者が割り付けられ、テリパラチド 20 μg 又はアレン
ドロネート 10 mg を 18 ヵ月投与した場合の腰椎 BMD 変化率を比較した。投与開始後
18 ヵ月時点において、テリパラチド投与群における腰椎 BMD のベースラインからの平

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
55
均変化率は 10.92%、アレンドロネート群は 5.51%であり、6 ヵ月以降のすべての腰椎
BMD の測定時点において両投与群間に統計学的な有意差が認められた。
また、一部の被験者では QCT を用いた有限要素解析法により、テリパラチドはアレ
ンドロネートに比較して 18 ヵ月投与後 5 倍の骨密度増加に応じた骨強度(bone
strength/density ratio)の増加を示すことが報告された(Keaveny et al. 2007)。これは前
述のテリパラチドによる微細構造の再構築に基づく骨強度の増加によるものと考えられ
た。
大腿骨 BMD は最も鋭敏に大腿骨近位部骨折を予測することが、最近の検討より明ら
かになっている(Kanis et al. 2008)。国内第 III 相試験(GHDB 試験)において大腿骨
BMD はテリパラチド 20 μg 群で投与期間に応じて増加し、第 1 期二重盲検比較期間(12
ヵ月間)では、大腿骨近位部 BMD のベースラインから最終観察時までの平均変化率は
2.66%(プラセボ群-0.22%)であり、大腿骨頸部 BMD のベースラインからの平均変化
率は 2.24%(プラセボ群 0.46%)であった。外国で実施したプラセボ対照二重盲検比較
試験(GHAC 試験及び GHAJ 試験)では、投与後 12 ヵ月までの大腿骨頸部及び大腿骨
近位部 BMD 変化率について、テリパラチド 20 μg 群及び 40 μg 群においてプラセボ群と
の間に有意な差が認められた〔大腿骨頸部 BMD:20 μg 群 1.54%(GHAC 試験)及び
1.40%(GHAJ 試験)、40 μg 群 3.06%(GHAC 試験)及び 3.15%(GHAJ 試験)、大腿骨
近位部 BMD:20 μg 群 1.70%(GHAC 試験)及び 1.28%(GHAJ 試験)、40 μg 群 2.55%
(GHAC 試験)及び 2.61%(GHAJ 試験)〕(第 2.7.3.3.2.2.2.1.2 項及び第 2.7.3.3.2.2.2.1.3
項参照)。
テリパラチドの腰椎及び大腿骨 BMD に対する 2 年間の臨床試験の結果(GHCA 試
験)、2 年間のテリパラチド投与期間中にわたり持続的な大腿骨頸部及び近位部 BMD
の増加が見られることが明らかになった(第 2.5.4.4 項参照)。
GHCA 試験のサブスタディ 1 では 1 個以上の脆弱性椎体又は非椎体骨折の既往があり
BMD が T スコア-2.5 以下の閉経後骨粗鬆症女性患者を対象とし、テリパラチド 20 μg
を全被験者に 12 ヵ月間投与(第 1 期)した後、次の 12 ヵ月(第 2 期)にテリパラチド
20 μg 投与、ラロキシフェン 60 mg 投与又は無治療を実施した。テリパラチド 24 ヵ月投
与群において腰椎 BMD は 12 ヵ月 9.3%、18 ヵ月 11.1% 、24 ヵ月 13.1%と持続して増加
を示した。更に大腿骨近位部 BMD は 12 ヵ月 1.8%、18 ヵ月 2.7%、24 ヵ月 3.8%、大腿
骨頸部 BMD は 12 ヵ月 2.2%、18 ヵ月 3.1% 、24 ヵ月 4.8%の増加を示した。特に大腿骨
の BMD に関しては、18 ヵ月から 24 ヵ月における増加が最も大きくみられた
(Obermayer-Pietsch et al. 2008)。GHBM 試験の結果からも、大腿骨頸部及び近位部
BMD について最終観察時においてベースラインより統計学的に有意に増加することが
認められた。しかしながら、これらの変化は腰椎 BMD の結果とは異なり、テリパラチ
ド群とアレンドロネート群との間に統計学的に有意な差はみられなかった。なおステロ
イド性骨粗鬆症を対象とした外国臨床試験(GHBZ 試験)においては、テリパラチド投
与によりアレンドロネートと比較して統計学的に有意な腰椎及び大腿骨 BMD 増加効果
が認められた(第 2.5.4.3 項参照)。

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
56
これらの結果より、外国大規模臨床試験において認められた腰椎及び大腿骨 BMD に
おける有効性が、骨折の危険性の高い日本人患者におけるテリパラチド投与においても
示された。更に、テリパラチドは既存の骨吸収抑制剤のうち最も BMD 増加効果が高い
と考えられるアレンドロネートと比較しても、顕著な骨密度増加効果を示すことが明ら
かになった。
2.5.6.2.4 骨形成マーカー先行優位の骨代謝マーカー反応
骨代謝マーカーは、骨形成と骨吸収のバランス、つまり骨における代謝回転の状態を
反映し、その測定結果により、将来の骨質を予測するものである。近年、骨代謝マーカ
ーが骨折の危険性を予測できるということを示唆するデータが集積されつつある(Szulc
and Delmas 2008)。また GHAC 試験の事後解析により、骨形成マーカーである PICP の
1 ヵ月での上昇及び PINP の 3 ヵ月での上昇が投与後 18 ヵ月時における BMD 変化を予
測すること(Chen et al. 2005)、骨形成マーカーである BAP の 1 ヵ月での上昇及び PICP
の 1 ヵ月での上昇が投与後 22 ヵ月時での骨微細構造の改善と相関することが報告され
ている(Dobnig et al. 2005)。
GHBM 試験では、テリパラチドとアレンドロネート投与開始後の骨代謝マーカーの経
時的変化を評価した。その結果、テリパラチドは、骨吸収抑制剤であるアレンドロネー
トとは全く異なった骨代謝マーカーの経時的変化を示すことが明らかとなった。すなわ
ち、テリパラチド 20 μg 群では、投与後 1 ヵ月で血清 PINP に代表される骨形成マーカー
が上昇し、その後遅れて NTX に代表される骨吸収マーカーが上昇した。対照的にアレ
ンドロネート群では、骨形成マーカー及び骨吸収マーカーの両方が、投与開始後 1 ヵ月
から 12 ヵ月のすべての測定時期において有意に減少した(第 2.7.3.3.2.2.3.2 項参照)。
これは、テリパラチドとアレンドロネートの作用機序の違いを明確に示すものである。
すなわち、テリパラチドが投与後速やかに骨形成を促進し、骨吸収面のみならず骨静止
面における骨形成(modeling-based formation)を促進することにより、正常な骨質を保
ちつつ新生骨の形成を誘導して anabolic window と呼ばれる骨形成先行優位の骨代謝回転
を促進することを示唆しているのに対し、アレンドロネートは骨吸収を抑制することに
伴い骨形成も抑制し、主に石灰化の亢進結果として骨密度増加が認められることを示唆
している。
国内第 II 相試験(GHCS 試験)及び第 III 相試験(GHDB 試験)においても、骨折の
危険性の高い日本人原発性骨粗鬆症患者(第 II 相試験は閉経後女性患者が対象)におけ
るテリパラチド投与後の骨代謝マーカーの変化が、副次的評価項目として評価された。
GHCS 試験において、テリパラチド 20 μg、40 μg 投与により骨形成マーカー(血清 PINP、
PICP、BAP)の速やかな上昇(12 週時、24 週時)、それに遅れて骨吸収マーカー(血
清 CTX)の上昇がみられた。GHDB 試験においてもテリパラチド 20 μg 投与により、血
清 PINP の速やかな上昇(12 週時、24 週時)、それに遅れて血清 CTX の上昇がみられ
た。GHDB 試験において投与後 12 ヵ月にわたり血清 BAP の上昇はみられなかった(第
2.5.4.2.4.1 項参照)。PTH 投与は比較的分化段階初期の骨芽細胞に作用することが知ら
れており、これらの細胞群は I 型コラーゲンを発現しているが、分化後期に発現する

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
57
BAP は発現していない(Szulc and Delmas 2008)。よって、一般に PTH 投与による骨形
成マーカーBAP の上昇は PINP の動きに比して顕著ではなく、遅れて上昇すると考えら
れる。しかしながら、国内で実施した GHCS 試験では 20 μg 群において血清 BAP の上昇
が認められており、GHDB 試験において投与後 12 ヵ月にわたり血清 BAP の上昇が認め
られなかった理由は不明である。
これらの結果を総合すると、テリパラチドが投与後速やかに anabolic window と呼ばれ
る骨形成先行優位の骨代謝回転を促進し、その結果、正常な骨質を保ちつつ骨微細構造
を再構築することが外国及び国内臨床試験における骨代謝マーカーの結果からも示唆さ
れた。
2.5.6.2.5 腰背部痛に対する効果
日本における「骨粗鬆症の予防と治療ガイドライン 2006 年版」では、疼痛に対して
は、カルシトニン製剤の筋肉注射とアレンドロネート製剤が推奨されている。カルシト
ニン製剤は骨折防止効果に関するエビデンスは少なく、骨粗鬆症における疼痛を適応症
としている。アレンドロネートは、外国大規模試験において腰背部痛に伴う活動制限
(腰痛に伴う臥床日数、活動制限日数の減少)に対する効果が示されている(Nevitt et
al. 2000)。
GHAC 試験において、有害事象として腰背部痛を発現した被験者数は、プラセボ群
123 例(22.6%)、テリパラチド 20 μg 群 91 例(16.8%)及び 40 μg 群 87 例(15.8%)で
あり、テリパラチド投与群に比べてプラセボ群で発現率が高く、3 投与群間で統計学的
な有意差が認められた(p=0.008)。GHBM 試験においても、有害事象として腰背部痛
が新たに発現(又は悪化)した患者は、アレンドロネート群の 38.6%に比べ、テリパラ
チド 20 μg 群では 25.5%と少なかった(p=0.05)。更に重症度ごとでは、中等度の腰背
部痛が発現した患者は、テリパラチド 20 μg 群で 15%に、アレンドロネート群で 33%に
認められ、高度の腰背部痛が発現した患者は、テリパラチド 20 μg 群で 4%に、アレンド
ロネート群で 12%に認められた。このように、アレンドロネートとの比較による臨床試
験においても、テリパラチド投与により、骨粗鬆症患者が有する中等度から高度の腰背
部痛を抑制することが確認された。外国臨床試験 5 試験を併合した総被験者数 2670 例
の Meta-analysis によると、テリパラチド併合群において、腰背部痛発現リスクは低下し、
相対リスクはすべての腰背部痛では 0.66、中等度又は高度の腰背部痛では 0.60、高度の
腰背部痛のみでは 0.44 であった(第 2.7.4.2.1.1.13.1.9 項参照、Nevitt et al. 2006)。更に
GHCA 試験においては VAS による腰背部痛が検討された。サブスタディ 1 及び骨吸収
抑制剤無効例への投与の影響を検討したサブスタディ 2 の両方において、ベースライン
からの VAS のスケールに統計学的に有意な低下が認められた(第 2.7.3.3.2.2.5 項参照)。
国内臨床試験において、GHCS 試験における腰背部痛の発現は、プラセボ群 4 例
(10.5%)、10 μg 群 4 例(10.5%)、20 μg 群 0 例(0.0%)及び 40 μg 群 1 例(2.6%)で
あった。国内第 III 相試験(GHDB 試験)においては、探索的にテリパラチド及びプラ
セボ投与患者における痛みの程度を 4 段階の被験者申告により評価したが、腰背部痛の
増悪あるいは軽減した患者の割合の相違の傾向をみることはできなかった。この結果に

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
58
関しては、ベースラインでの中等度以上の痛みを示した患者数が両群ともに 10%以下と
少なかった影響やプラセボ効果の影響などが考えられる(第 2.7.3.3.2.2.5.1 項参照)。
GHDB 試験の第 1 期二重盲検比較期間における有害事象としての腰背部痛の発現はプラ
セボ群 10 例(14.9%)、20 μg 群 17 例(12.5%)であり、投与群間で顕著な差はなく、
20 μg 群の方がわずかに低かった(第 2.7.4.2.1.1.13.1.9 項参照)。
以上よりテリパラチドによる腰背部痛に対する効果は外国においてエビデンスが構築
されてきているが、現在まで国内臨床試験で得られた結果も外国臨床試験のエビデンス
と矛盾したものではないと考える。
2.5.6.2.6 骨折の危険性の高い骨粗鬆症患者の治療におけるベネフィット
前項までに述べたとおり、テリパラチドは、その強力な骨形成促進作用により有意に
腰椎及び大腿骨 BMD を増加させ、約 1.5 から 2 年という限られた投与期間において、
従来の骨粗鬆症治療薬を上回る骨量増加効果や椎体骨折及び非椎体骨折抑制効果を示し
た。更に、テリパラチドはすでに破綻をきたした骨の微細構造を再構築し、骨粗鬆症の
回復方向への転換を可能とするこれまでの骨粗鬆症治療薬とは全く異なった作用機構及
び治療効果を有する薬剤である。骨代謝マーカーの経時的変化からも、投与後速やかに
anabolic window と呼ばれる骨形成先行優位の骨代謝回転を促進し、その結果、正常な骨
質を保ちつつ骨微細構造を再構築するというテリパラチドの作用機序の新規性が示唆さ
れている。更にはすでに腰背部痛に対するエビデンスを有する既存薬(アレンドロネー
ト)と同等以上に腰背部痛を軽減する結果も、外国における実薬対照臨床試験により示
されている。現在日本において、骨折の危険性の高い骨粗鬆症には経口ビスフォスフォ
ネート製剤が主として使用されている。しかしながら、ビスフォスフォネート製剤は骨
に長期間蓄積することが知られており、長期投与により過度に骨代謝回転を抑制した場
合の骨質の低下による骨折治癒の遅延及び障害(Armamento-Villareal et al. 2006; Lenart et
al. 2008)や顎骨壊死についてのリスクも指摘されている。更に、週 1 回製剤により緩和
はされているものの、起床時の空腹での服用や服用後 30 分以内の食事や臥床が禁止さ
れているために、患者の利便性が失われるなどの問題もある(第 2.5.1.2.1 項参照)。テ
リパラチドは在宅自己注射による投与が行われる薬剤であるが、国内及び外国臨床試験
において優れたコンプライアンスを示し、ビスフォスフォネート製剤に見られるような
服用時の制限などもなく、安全性プロファイルに関してもビスフォスフォネート製剤と
比して優れていると考えられた。
以上の結果から、本剤は直ちに治療を必要とする骨粗鬆症患者、すなわち、骨折の危
険性の高い骨粗鬆症患者において、既存治療薬より高い有用性が期待できると考えられ
る。
2.5.6.3 テリパラチド投与により予測されるリスク
テリパラチドの非臨床試験及び臨床試験の結果、並びに市販後の自発報告から、安全
性所見として骨肉腫、有害事象及び臨床検査値への影響に関して、以下に記載する。

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
59
2.5.6.3.1 骨肉腫
2.5.6.3.1.1 非臨床試験で認められた知見
Fischer344 系ラットを用いて実施された 2 年間がん原性試験において、骨肉腫を含む
骨増殖性病変が認められた。
ラットでは、ヒトと異なり皮質骨内に骨内を縦走する血管を中心に同心円状の層板を
形成するハバース系がないとされており、テリパラチドは海綿骨表面に加えて皮質骨内
面及び骨膜表面にも新生骨を広範に被覆することによって、皮質骨の骨量を過剰に増大
させる。本がん原性試験で認められたように、ヒトに対する推奨用量を上回る量のテリ
パラチドを、ほぼ一生涯に相当する期間にわたりラットに皮下投与すると、骨量の著明
な増加、骨の変形、及び骨髄腔の消失又は縮小が生じた。また、限局性骨芽細胞過形成、
骨腫、骨芽細胞腫及び骨肉腫など、過形成性及び腫瘍性の骨病変が観察された。これら
は用量に相関して発現し、約 17 ヵ月間の投与後に初めて検出されたものであった。一
方、ラットにテリパラチドを長期間投与しても、非骨組織の悪性新生物の発生率増加は
認められなかった。その後実施された 2 回目のラット 2 年間がん原性試験から、ラット
のテリパラチド誘発性骨肉腫は投与期間かつ用量に依存することが明らかになった。観
察されたラット骨増殖性病変の分子/細胞レベルでの発生機構は現在不明であるが、こ
れまでに得られた結果は骨の長期刺激に関与する機構と一致する。その後、霊長類にお
けるテリパラチドの骨組織に対する増殖性病変の発生について検討するために、カニク
イザルにテリパラチドを 18 ヵ月間投与し、投与終了後 3 年間観察する試験を実施した。
その結果、18 ヵ月の投与期間及び 3 年間の投与終了後の観察期間を通して、テリパラチ
ド投与に起因すると考えられる骨組織における増殖性病変は認められなかった。ラット
ではほぼ生涯にわたり骨の長軸方向への成長が継続し、骨代謝回転速度が速いなど、根
本的にラットとヒトの骨の生理は異なっていること、更には霊長類における観察結果よ
り、骨粗鬆症の治療のためにテリパラチドを投与されたヒトにおいて、骨肉腫発症の危
険性が増大することを示唆するものではないと考えている(第 2.5.5.7.1 項及び第 2.6.6.1
項参照)。
2.5.6.3.1.2 臨床試験及び市販後の安全性情報から得られた知見
イーライリリー・アンド・カンパニーは、2002 年 11 月に世界で初めてテリパラチド
が承認されて以来、テリパラチドの安全性に関する監視プログラムを継続している。
2008 年 11 月 26 日時点においてテリパラチドは 65.1 万人の患者に投与されたと推定され
ており、テリパラチド投与開始後の年数を勘案すると、観察規模は 150 万人年に相当す
る。2008 年 11 月 26 日までの自発報告において、骨肉腫として報告されたものは 5 例あ
る(第 2.5.5.7.1 項参照)。一方、60 歳以上の一般集団における骨肉腫の基礎発症率は
100 万人あたり 4 件程度と考えられているため(Harper et al. 2007)、テリパラチドの自
発報告による骨肉腫の発症率は、ほぼ自然発生頻度のレベルであると考えられる。更に、
テリパラチド投与による骨肉腫発現のリスクを評価する目的で、イーライリリー・アン
ド・カンパニーは米国及び欧州 5 ヵ国での骨肉腫新規発症例につき、発症前にテリパラ
チドが投与されたことがあるかについての調査を実施している(GHBX 研究)。本研究

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
60
は 2002 年に開始し、2008 年 12 月 15 日時点で米国での骨肉腫発生例の 69%(1025 例)
が登録され、そのうち 461 例が調査完了している。欧州 5 ヵ国においても同様の調査が
行われており、89 例の発生例のうち 62 例が調査完了している。いずれの調査において
もテリパラチド投与歴が確定している骨肉腫例は認められていない。本研究はテリパラ
チド投与患者における骨肉腫発生リスクが増加するか否かにつき検討する目的で、調査
を継続中である。2008 年 3 月 15 日までイーライリリー・アンド・カンパニーが実施し
たテリパラチドの臨床試験は 46 試験、継続中のものは 7 試験あり、7598 例の患者がテ
リパラチド投与をうけており、これは 37000 人年に相当するが、骨肉腫の発生は認めら
れていない(第 2.7.4.6.6.3 項参照)。更に、追跡調査研究である GHBJ 試験で、外国の
第 III 相試験 7 試験に参加した患者の投与終了後 5 年間(GHBJ 試験の観察期間は 4.5
年)の安全性を評価した結果、先行する試験で本剤を投与した患者において、癌の発現
率の上昇は認められず、骨肉腫の発現も報告されなかった(第 2.5.5.4.3 項参照)。
以上より、ラットで発生した骨肉腫がヒトにおいても発症する可能性は低いと考えら
れた。しかしながら、ヒトにおける骨肉腫の発症に関しては不明である。よって本剤の
投与期間の上限を、それまでに実施した臨床試験の投与期間を基に設定することし、さ
らに骨肉腫発生リスクの高いと考えられる患者には投与しないこととした(第 2.5.5.7.1
項参照)。
2.5.6.3.2 有害事象
国内第 II 相試験(GHCS 試験、テリパラチド投与期間 6 ヵ月)においてプラセボ群と
比較してテリパラチド投与群で発現率が高く、かつテリパラチド(10 μg、20 μg 及び
40 μg)投与群のいずれかにおいて 5%以上の発現率が認められた有害事象は、「悪心」、
「湿疹」、「頭痛」、「転倒」、「筋痙縮」及び「血中尿酸増加」であった。有害事象
の発現率は 40 μg 群において高かった(第 2.5.5.4.1.1 項参照)。国内第 III 相試験
(GHDB 試験、第 1 期二重盲検比較期間 12 ヵ月及び第 2 期オープン期間 6 ヵ月間)にお
いて死亡例はなく、重篤な有害事象は、第 1 期二重盲検比較期間においてプラセボ群 7
例(10.4%)及びテリパラチド投与群 7 例(5.1%)、その後の第 2 期オープン期間にお
いてすべての被験者にテリパラチド 20 μg を投与中に 5 例に認められた。このうちテリ
パラチド投与と因果関係の否定できないものは血圧低下、乳癌であった(第 2.5.5.4.2 項
参照)。血圧低下の症例は、テリパラチド投与後 24 週時の規定来院時においてテリパ
ラチド投与後 15 分に血圧が 60/40 まで低下したと報告されたが、2 時間半後に 100/64 ま
で回復した症例で、投与中止となった。乳癌の症例は、テリパラチド投与後約 1 年にて
乳癌にて入院となった症例である。投薬開始前に乳癌の存在は確認できなかったとのこ
とから、因果関係を否定できないと判断された(第 2.7.6.3.2.7 項参照)。本試験にて第
1 期二重盲検比較期間の 52 週間のプラセボ(n=67)又はテリパラチド 20 μg(n=136)投
与を受けた被験者のうち、有害事象を 1 件以上発現した被験者はプラセボ群 59 例
(88.1%)、テリパラチド 20 μg 群 116 例(85.3%)であり、この発現率はほぼ外国第 III
相試験(GHAC 試験)と同様であった。プラセボ群と比較してテリパラチド投与群で発
現率が高く、かつテリパラチド投与群のいずれかにおいて 5%以上の発現率が認められ

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
61
た有害事象は「便秘」、「頭痛」、「骨関節炎」、「浮動性めまい」、「上気道の炎症」
及び「膀胱炎」であったが、いずれも発現率はプラセボ投与群と統計学的有意差を認め
なかった(第 2.5.5.4.1.1 項参照)。その後、第 2 期オープン期間を実施し、投与期間を
12 ヵ月から 18 ヵ月まで延長した結果、さらなる安全性上の懸念や重要な安全性の所見
の増加は認められなかった。
外国大規模臨床試験(GHAC 試験)において認められた主な有害事象(プラセボ及び
20 μg 群)の発現率を国内第 III 相試験と比較検討したところ、「頭痛」、「浮動性めま
い」、「筋痙縮」及び「嘔気」はほぼ外国及び国内で同様の出現傾向がみられたが、
「悪心」の発現は国内第 III 相試験では少なかった〔GHAC 試験プラセボ群 41 例
(7.5%)、20 μg 群 51 例(9.4%)、40 μg 群 98 例(17.8%)、GHDB 試験〔第 1 期二重
盲検比較期間(投与 12 ヵ月間)〕プラセボ群 1 例(1.5%)、20 μg 群 1 例(0.7%)〕。
日本人骨粗鬆症患者で認められた有害事象は、外国臨床試験で認められた有害事象に含
まれており、日本人特有の有害事象は認められなかった(第 2.7.4.2.1.1.13.1.1 項参照)。
国内で実施した 2 試験〔GHCS 試験及び GHDB 試験(投与 12 ヵ月間)〕と外国で実施
した 3 試験(GHAC 試験、GHAJ 試験及び GHBM 試験)の統合解析を比較した場合、全
テリパラチド投与群及びテリパラチド 20 μg 群においてよく見られた因果関係を否定で
きない有害事象の発現傾向は、国内外で類似していた(第 2.5.5.4.1.6 項参照)。国内に
おいては血中尿酸増加と血中アルカリフォスファターゼ増加の発現が高い傾向が認めら
れたが、これらは外国で実施した試験の臨床検査結果でよく見られた変化であり、国内
で特徴的に発現が多く見られた現象ではなかった(第 2.5.5.5 項参照)。
更に国内第 II 相及び第 III 相試験(投与 12 ヵ月間)の成績をあわせて因果関係あり、
又は不明の有害事象の解析を行うと、テリパラチド 10 μg から 40 μg を投与した安全性
評価対象 252 例中 44 例(17.5%)に因果関係あり、又は不明の有害事象(臨床検査値異
常を含む)が認められた。主な因果関係あり、又は不明の有害事象は、血中尿酸増加 7
例(2.8%)、頭痛 7 例(2.8%)、悪心 7 例(2.8%)、血中アルカリフォスファターゼ増
加 4 例(1.6%)、筋痙縮 3 例(1.2%)であった。なお、プラセボの投与を受けた 105 例
中 11 例(10.5%)に因果関係あり、又は不明の有害事象(臨床検査値異常を含む)が認
められた(第 2.7.4.2.1.1.12.2.2 項参照)。
以上よりテリパラチドの安全性プロファイルに問題はなく、忍容性は良好であった。
骨粗鬆症患者を対象とした安全性の評価資料とした試験においては 2 例のみの報告で頻
度は稀であるが、外国臨床薬理試験(GHCO 試験)において有害事象として 4 例(6
件)報告された起立性低血圧、及び国内外国臨床試験において頻度は高くないものの認
められている浮動性めまいに関しては注意が必要と考え、高所での作業、自動車の運転
など危険を伴う作業に従事する場合は注意させることとした(第 2.5.5.7.3 項参照)。
2.5.6.3.3 血清カルシウム及びそのほかの臨床検査に及ぼす影響
テリパラチドの薬理作用により、投与後一過性の血清カルシウム上昇がおこることが
知られている。

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
62
外国大規模臨床試験(GHAC 試験)では、テリパラチド 20 μg 群及 40 μg 群において、
投与後 4~6 時間時点で、基準範囲を超えた血清カルシウム濃度を示す被験者が認めら
れた。この基準値上限を超えた血清カルシウム濃度を示した被験者数は、プラセボ群と
比較してテリパラチド 20 μg 群及び 40 μg 群で多かったが、これらの変動は臨床的に問
題となるものではなかった。なお、投与後 24 時間時点で得られた血清カルシウム濃度
(以下、投与前血清カルシウムとする)は、すべての投与群においてベースライン値ま
で戻っており、持続的な増加は認められなかった。
国内第 II 相試験(GHCS 試験、テリパラチド投与期間 6 ヵ月)においてもテリパラチ
ド 20 μg 群及び 40 μg 群において投与後 4~6 時間時点で、ベースライン値に比して有意
な補正血清カルシウム濃度の上昇がみられたが、その上昇の中央値は最大 0.3 mg/dL に
とどまった。治験中に、統計学的に有意な投与前補正カルシウム値の上昇がみられたが、
臨床的に問題となるものではなかった。治験薬投与前の補正血清カルシウム値が
11 mg/dL を超えた被験者はプラセボ群の 1 例のみ(24 週時)で、テリパラチド投与によ
る投与前の補正血清カルシウムの変化量は小さく、最大でも平均 0.3 mg/dL の上昇にと
どまり臨床的に問題となるものではないと考えられた(第 2.7.4.3.1.1.1.1 項参照)。
国内第 III 相試験(GHDB 試験、二重盲検比較期間 12 ヵ月及びオープン期間 6 ヵ月)
においても、ベースライン値に対してテリパラチド投与後 4~76 週にわたって統計学的
に有意な補正カルシウム値の上昇がみられたが、その中央値は最大でも平均 0.3 mg/dL
の上昇にとどまった。また、テリパラチド投与群で 1 例一過性の高カルシウム血症
(12.3 mg/dL)が報告されたが、治験薬投与を継続したまま無処置で 8 日後に回復した。
高カルシウム血症による臨床症状を呈した症例はみられず、また高カルシウム血症によ
り中止になった症例もみられなかった。国内臨床試験における高カルシウム血症の発現
は、この 1 例(0.7%)のみであった(第 2.7.4.3.1.1.2.1 項参照)。
外国臨床試験における高カルシウム血症の発現は、GHAC 試験の 40 μg 群で 1 例
(0.2%)、GHAJ 試験の 40 μg 群で 1 例(0.7%)、GHBM 試験のテリパラチド群で 3 例
(2.9%)であった。GHCA 試験ではプラセボ投与群がなく、更に各治験実施施設での基
準範囲上限を超えた場合に高カルシウム血症として報告していたため、テリパラチド群
で 32 例(3.7%)の報告があった(第 2.5.5.7.2 項参照)。
これらの知見から、テリパラチド投与による血清カルシウムの変化量は小さく、臨床
的に問題となるものではないと考えられた。しかし、PTH の生理作用として血清カルシ
ウム増加作用があり、一過性であるものの本剤投与により血清カルシウムの増加が認め
られたことから、ベースラインでの高カルシウム血症の患者はテリパラチド投与により
高カルシウム増悪のリスクがあると考えられるため禁忌とした。更に重要な基本的注意
として、血清カルシウム値上昇が認められた場合の対応について設定した。日本で広く
用いられている活性型ビタミン D 製剤もテリパラチドと併用した場合相加作用により血
清カルシウムが高くなるおそれがあるため、できるだけ併用は避けることが望ましいと
した(第 2.5.5.7.2 項参照)。
また国内及び外国臨床試験において血清尿酸値の上昇が指摘されている。外国臨床試
験では血清尿酸の増加は、本事象に関連して発現する可能性のある痛風や尿路結石症の

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
63
発現を伴わなかった。しかし、尿路結石を有する患者でのテリパラチド投与の臨床試験
結果がないことから尿路結石を有する患者及びその既往のある患者は慎重投与と規定し
た(第 2.5.5.5 項参照)。更に外国の臨床薬理試験において、重度の腎障害を有する患者
では血中からのテリパラチドの消失に遅延が認められていることより、腎障害を有する
患者は慎重投与と規定した。
2.5.6.3.4 骨折の危険性の高い骨粗鬆症患者に対するテリパラチド治療におけるリス
ク
2.5.6.3.4.1 投与期間の設定
テリパラチドは開発途中である 1998 年に、ラットがん原性試験において骨肉腫を含
む骨腫瘍性病変所見が認められたため、イーライリリー・アンド・カンパニーは自主的
に本剤の全臨床試験を中止した。その後、サルにおける長期投与試験において骨肉腫は
認められなかったこと、本剤投与の臨床試験及び中止した臨床試験の追跡調査において
も本剤投与に起因すると思われる骨肉腫が認められなかったことから、ラットで発生し
た骨肉腫がヒトにおいても発症する可能性は低いと考えられた。しかしながら、ヒトに
おける骨肉腫の発症に関しては不明である。よって本剤の投与期間の上限を、それまで
に実施した臨床試験の投与期間を基に設定することとなった。すなわち、米国における
本剤の投与期間上限は、GHAC 試験の最大投与期間を考慮し 2 年間とされ、また欧州で
は、GHAC 試験で投与期間の中央値を考慮し 18 ヵ月と設定された。なお、欧州におい
ても近年実施された臨床試験成績より、投与期間上限の 24 ヵ月への延長が申請され承
認された(第 2.5.5.7.1 項参照)。
このようにテリパラチドは投与期間の上限が設定される薬剤であるが、その強力な骨
形成促進作用により速やかに腰椎及び大腿骨 BMD を増加させ、約 1.5 から 2 年という
限られた投与期間においても、従来の骨粗鬆症治療薬を上回る骨量増加効果や椎体骨折
及び非椎体骨折抑制効果を示す。
2.5.6.3.4.2 在宅自己注射
テリパラチドは 1 日 1 回の皮下注射を用法・用量として外国及び国内において開発さ
れた経緯をもつ薬剤である。骨折の危険性の高い骨粗鬆症患者の通院などに関する利便
性を鑑みて在宅自己注射による投与が外国・国内臨床試験、外国市販後において行われ
ている。一般に既存骨折がある、あるいは高齢などの骨折の危険性の高い骨粗鬆症患者
においては在宅自己注射の困難が想定される。しかしながら、国内第 II 相試験(GHCS
試験、テリパラチド投与期間 6 ヵ月)において骨折の危険性の高い閉経後女性患者への
テリパラチドの在宅自己注射による投与は、概して忍容性が良好であった。70%以上の
服薬遵守状況を示した被験者の割合は、いずれの投与群においても 92.3%以上と良好で
あった(第 2.7.4.1.2.1.1.1 項参照)。国内第 III 相試験(GHDB 試験、二重盲検比較期間
12 ヵ月及びオープン期間 6 ヵ月)においても骨折の危険性の高い日本人男性及び女性患
者へのテリパラチドの投与は、概して忍容性が良好であり、70%以上の遵守状況を示し
た被験者の割合は、第 1 期二重盲検比較期間においてプラセボ群 98.5%、テリパラチド

LY333334 2.5 臨床に関する概括評価
2.5.6 ベネフィットとリスクに関する結論
64
投与群 96.3%、第 2 期オープン期間において、第 1 期でプラセボの投与を受け、第 2 期
でテリパラチドの投与を受けた群で 96.6%、第 1 期及び第 2 期共にテリパラチドの投与
を受けた群で 100.0%と良好であった(第 2.7.4.1.2.1.2.1 項参照)。外国臨床試験及び市
販後においても比較的良好な遵守状況が得られている。なお外国及び国内臨床試験にお
いては、治験薬投与開始前にトレーニングを実施し、操作方法を習得した患者のみ対象
とした。よって十分な患者指導及びインフォームドコンセントを行うことにより、日本
において高い遵守状況で在宅自己注射によるテリパラチド投与を行うことは可能である。
2.5.6.4 テリパラチドの臨床的意義のまとめ
テリパラチドの皮下投与療法は、すでに骨微細構造が破綻をきたした骨折の危険性の
高い骨粗鬆症患者において、速やかに骨微細構造を再構築し、「生理学的に正常な骨」
の形成を促進し、骨粗鬆症の回復方向への転換をするという他の薬剤にはない新規的な
作用を有する。この新規的な骨形成促進作用に伴い、2 年以内の短期間の治療で他剤と
比して類を見ない椎体及び大腿骨の骨密度増加作用、更には 50%を超える椎体骨折及び
非椎体脆弱骨折の発生抑制効果が発揮されると考えられる。
テリパラチドの投与により問題となる副作用は認められず、安全かつ忍容性のある薬
剤である。外国及び国内臨床試験において発現率が高かった有害事象は「頭痛」、「悪
心」などの比較的軽度なものが主であった。またラットにおいて骨肉腫発現が報告され
たものの、サルにおける実験結果では認められず、臨床試験、臨床試験終了後の追跡調
査、外国における新規骨肉腫発生患者調査、外国市販後調査の結果などからも骨粗鬆症
の治療のためにテリパラチドを投与された患者において、骨肉腫発症の危険性が増大す
ることを示唆するエビデンスは得られていない。
以上より、ベネフィットとリスクのバランスに関する全般的な観点から、テリパラチ
ドが骨折の危険性の高い骨粗鬆症患者にもたらすベネフィットは、懸念されるリスクを
上回るものと考えられる。

LY333334 2.5 臨床に関する概括評価
2.5.7 参考文献
65
2.5.7 参考文献
Adami S, Zamberlan N. Adverse effects of bisphosphonates. A comparative review. Drug Saf.
1996;14(3):158-70.
Armamento-Villareal R, Napoli N, Panwar V, Novack D. Suppressed bone turnover during
alendronate therapy for high-turnover osteoporosis. N Engl J Med. 2006;335(19):2048-50.
Bringhurst FR, Stern AM, Yotts M, Mizrahi N, Segre GV, Potts JT Jr. Peripheral metabolism
of PTH: fate of biologically active amino terminus in vivo. Am J Physiol. 1988;255:886-93.
Cauley JA, Thompson DE, Ensrud KC, Scott JC, Black D. Risk of mortality following clinical
fractures. Osteoporos Int. 2000;11:556-61.
Chen P, Satterwhite JH, Licata AA, Lewiecki EM, Sipos AA, Misurski DM, et al. Early
changes in biochemical markers of bone formation predict BMD response to Teriparatide in
postmenopausal women with osteoporosis. J Bone Miner Res. 2005;20(6):962-70.
Chen P, Miller PD, Delmas PD, Misurski DA, Krege JH. Change in lumbar spine BMD and
vertebral fracture risk reduction in Teriparatide-treated postmenopausal women with osteoporosis.
J Bone Miner Res. 2006;21(11):1785-90.
Chen P, Miller PD, Recker R, Resch H, Rana A, Pavo I, et al. Increase in BMD correlate with
improvements in bone microarchitecture with Teriparatide treatment in postmenopausal women
with osteoporosis. J Bone Miner Res. 2007;22(8):1173-80.
Cummings SR, Black DM, Thompson DE, Applegate WB, Barrett-Connor E, Musliner TA, et
al. Effect of alendronate on risk of fracture in women with low bone density but without vertebral
fractures. JAMA. 1998;280(24):2077-82.
Cummings SR, Bates D, Black DM. Clinical use of bone densitometry. JAMA. 2002
Oct;288(15):1889-97.
Daugaard H. Peripheral metabolism of parathyroid hormone. Danish Medical Bulletin. 1996
Jun;43(3):203-15.
Delmas PD, Ensrud KE, Adachi JD, Harper KD, Sarkar S, Gennari C, et al. Efficacy of
raloxifene on vertebral fracture risk reduction in postmenopausal women with osteoporosis: four-
year results from a randomized clinical trial. J Clin Endocrinol Metab. 2002;87(8):3609-17.
Dobnig H, Sipos A, Jiang Y, Fahrleitner-Pammer A, Ste-Marie L, Gallagher JC, et al. Early
changes in biochemical markers of bone formation correlate with improvements in bone structure
during Teriparatide therapy. J Clin Endocrinol Metab. 2005;90(7):3970-7.
Fatayerji D, Cooper AM, Eastell R. Total body and regional bone mineral density in men:
effect of age. Osteoporos Int. 1999;10(1):59-65.
Fujiwara S, Kasagi F, Masunari N, Naito K, Suzuki G, Fukunaga M. Fracture prediction from
bone mineral density in Japanese men and women. J Bone Miner Res. 2003;18(8):1547-53.
Hagino H, Furukawa K, Fujiwara S, Okano T, Katagiri H, Yamamoto K, et al. Recent trends in
the incidence and lifetime risk of hip fracture in Tottori, Japan. Osteoporos Int. 2009;20(4):543-8.
Harper KD, Marcus R, Mitlak BH. Osteosarcoma and Teriparatide?. J Bone Miner Res.
2007;22(2):334.

LY333334 2.5 臨床に関する概括評価
2.5.7 参考文献
66
Harris ST, Watts NB, Genant HK, McKeever CD, Hangartner T, Keller M, et al. Effects of
risedronate treatment on vertebral and nonvertebral fractures in women with postmenopausal
osteoporosis. JAMA. 1999 Oct 13;282(14):1344-52.
Hodsman AB, Bauer DC, Dempster DW, Dian L, Hanley DA, Harris ST, et al. Parathyroid
hormone and Teriparatide for the treatment of osteoporosis: a review of the evidence and
suggested guidelines for its use. Endocrine Reviews. 2005;26(5):688-703.
Jiang Y, Zhao JJ, Mitlak BH, Wang O, Genant HK, Eriksen EF. Recombinant human
parathyroid hormone (1-34) [Teriparatide] improves both cortical and cancellous bone structure. J
Bone Miner Res. 2003;18(11):1932-41.
Kanis JA, Johansson H, Oden A, Johnell O, de Laet C, Melton III LJ, et al. A meta-analysis of
prior corticosteroid use and fracture risk. J Bone Miner Res. 2004a Jun 19(6): 893-9.
Kanis JA, Oden A, Johnell O, De Laet C, Jonsson B. Excess mortality after hospitalisation for
vertebral fracture. Osteoporos Int. 2004b;15(2):108-12.
Kanis JA, Borgstrom F, De Laet C, Johansson H, Johnell O, Jonsson B, et al. Assessment of
fracture risk. Osteoporos Int. 2005;16:581-9.
Kanis JA, McCloskey EV, Johansson H, Oden A, Melton III LJ, Khaltaev N. A reference
standard for the description of osteoporosis. Bone. 2008;42:467-75.
Keaveny TM, Donley DW, Hoffmann PF, Mitlak BH, Glass EV, San Martin JA. Effects of
Teriparatide and alendronate on vertebral strength as assessed by finite element modeling of QCT
scans in women with osteoporosis. J Bone Miner Res. 2007;22(1):149-57.
Kishimoto H, Fukunaga M, Kushida K, Shiraki M, Itabashi A, Nawata H, et al. Efficacy and
tolerability of once-weekly administration of 17.5 mg risedronate in Japanese patients. J Bone
Miner Metab. 2006;24:405-13.
Lenart BA, Lorich DG, Lane JM. Atypical fractures of the femoral diaphysis in
postmenopausal women taking Alendronate. N Engl J Med. 2008;358(12):1304-6.
Liao S, Qie JK, Xue M, Zhang ZQ, Liu KL, Ruan JX. Metabolic stability of human parathyroid
hormone peptide hPTH (1-34) in rat tissue homogenates: kinetics and products of proteolytic
degradation. Amino Acids; In press.
McClung MR, Geusens P, Miller PD, Zippel H, Bensen WG, Roux C et al.; Hip Intervention
Program Study Group. Effect of risedronate on the risk of hip fracture in elderly women. Hip
Intervention Program Study G. N Engl J Med. 2001 Feb 1;344(5):333-40.
Murray TM, Rao LG, Divieti P, Bringhurst FR. Parathyroid hormone secretion and action:
evidence for discrete receptors for the carboxyl-terminal region and related biological actions of
carboxyl-terminal ligands. Endocrine Reviews. 2005 Feb;26(1):78-113.
Nevitt MC, Thompson DE, Black DM, Rubin SR, Ensrud K, Yates AJ; Fracture Intervention
Trial Research Group. Effect of Alendronate on limited-activity days and bed-disability days
caused by back pain in postmenopausal women with existing vertebral fractures. Arch Intern Med.
2000 Jan 10;160:77-85.

LY333334 2.5 臨床に関する概括評価
2.5.7 参考文献
67
Nevitt MC, Chen P, Dore RK, Reginster J, Kiel DP, Zanchetta JR, et al. Reduced risk of back
pain following Teriparatide treatment: a meta-analysis. Osteoporos Int. 2006;17:273–80.
NIH: Osteoporosis Prevention, Diagnosis, and Therapy. NIH Consensus Statement.
2000;17(1): 1-45.
NOF: National Osteoporosis Foundation. Clinician's guide to prevention and treatment of
osteoporosis. Washington, DC: National Osteoporosis Foundation; 2008.
Obermayer-Pietsch B, Marin F, McCloskey EV, Hadji P, Farrerons J, Boonen S, et al. Effects
of two years of daily Teriparatide treatment on BMD in postmenopausal women with severe
osteoporosis with and without prior antiresorptive treatment. J Bone Miner Res.
2008;23(10):1591-600.
Odvina CV, Zerwekh JE, Rao DS, Maalouf N, Gottschalk FA, Pak CY. Severely suppressed
bone turnover: a potential complication of alendronate therapy. J Clin Endocrinol Metab.
2005;90(3):1294-301.
Orimo H, Sugioka Y, Fukunaga M, Muto Y, Hotokebuchi T, Gorai I, et al. Diagnostic criteria
of primary osteoporosis. J Bone Miner Metab. 1998;16:139-50.
Raisz LG. Pathogenesis of osteoporosis: concepts, conflicts, and prospects. J Clin Invest.
2005;115(12):3318-25.
Riggs BL, Khosla S, Melton III LJ. A unitary model for involutional osteoporosis: estrogen
deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes
to bone loss in aging men. J Bone Miner Res. 1998;13(5):763-73.
Riggs BL, Parfitt AM. Drugs used to treat osteoporosis: the critical need for a uniform
nomenclature based on their action on bone remodeling. J Bone Miner Res. 2005;20(2):177-84.
Ross PD, Fujiwara S, Huang C, Davis JW, Epstein RS, Wasnich RD, et al. Vertebral fracture
prevalence in women in Hiroshima compared to Caucasians or Japanese in the US. Int J
Epidemiol. 1995;24(6):1171-7.
Seeman E. Pathogenesis of bone fragility in women and men. Lancet. 2002 May 25;359:1841-
50.
Segre GV, Perkins AS, Witters LA, Potts JT Jr. Metabolism of parathyroid hormone by
isolated rat kupffer cells and hepatocytes. J Clin Invest. 1981 Feb;67:449-57.
Szulc P, Delmas PD. Biochemical markers of bone turnover: potential use in the investigation
and management of postmenopausal osteoporosis. Osteoporos Int. 2008;19:1683–704.
WHO: Prevention and management of osteoporosis. Report of a WHO Study Group. Geneva,
World Health Organization, 2003 (WHO Technical Report Series, No. 921).
Woo SB, Hellstein JW, Kalmar JR. Systematic review: bisphosphonates and osteonecrosis of
the jaws. Ann Intern Med. 2006;144(10):753-61.
Zanchetta JR, Bogado CE, Ferretti JL, Wang O, Wilson MG, Sato M, et al. Effects of
Teriparatide [recombinant human parathyroid hormone (1-34)] on cortical bone in
postmenopausal women with osteoporosis. J Bone Miner Res. 2003;18(3):539-43.

LY333334 2.5 臨床に関する概括評価
2.5.7 参考文献
68
折茂肇, 林泰史, 福永仁夫, 曽根照喜, 藤原佐枝子, 白木正孝, ほか. ; 日本骨代謝学会骨粗
鬆症診断基準検討委員会. 原発性骨粗鬆症の診断基準(2000 年度改訂版). 日本骨代謝学
会誌. 2001;18(3):76-82.
厚生省. 外国臨床データを受け入れる際に考慮すべき民族的要因について(医薬審第
672 号、平成 10 年 8 月 11 日). 1998.(添付せず)
厚生労働省. 平成 19 年国民生活基礎調査の概況. 2008.
骨粗鬆症検診・保健指導マニュアル. 細井孝之, 福永仁夫, editors. 東京: ライフサイエン
ス出版; 2009. p.2-3.
骨粗鬆症の予防と治療ガイドライン作成委員会, editor. 骨粗鬆症の予防と治療ガイドラ
イン 2006 年版. 東京: ライフサイエンス出版; 2006.(添付せず)
原田敦, 松井康素, 竹村真里枝, 伊藤全哉, 若尾典充, 太田壽城. 骨粗鬆症の医療経済-疫
学, 費用と介入法別費用・効用分析-. 日老医誌. 2005;42(6):596-608.
藤原佐枝子, 増成直美, 児玉和紀, 福永仁夫. 腰椎・大腿骨骨塩量カットオフ値を使った
骨粗鬆症有病率の検討. Osteoporosis Jpn. 1997;5(2):223-6.
藤原佐枝子 . 骨粗鬆症による椎体・非椎体骨折リスクの EBM. 医学のあゆみ .
2005;212(2):139-42.
藤原佐枝子. WHO の FRAT と骨折リスクの知見. 日本臨床. 2007;65(9):265-8.
吉村典子. 骨粗鬆症と骨折の医療・社会的影響. 日本内科学会雑誌. 2005;94(4):13-19.





















![認定看護師教育基準カリキュラム · 臨床解剖学、臨床病理学、臨床生理 学を学ぶ 1)臨床解剖学 2)臨床病理学 3)臨床生理学 [授業形態]](https://img.pdfslide.tips/doc/110x75/5f0ebd437e708231d440b3f0/ecoeeeffff-eeeccecc.jpg)







