Embed Size (px)
Citation preview

150 ©2015 The Society of Polymer Science, Japan 高分子 64巻 3月号 (2015年)
展 望COVER STORY: Highlight Reviews
1.GROMACSとは?1.1 はじめに
GROMACS(Groningen Machine for Chemical Simulations)は、オランダ・フロニンゲン大で開発された分子動力学(MD)計算ソフトウェアである1)。最初から並列計算機利用を前提に開発された第二世代の草分け的なフリーなMDソフトウェアで、ほかの第二世代ソフトウェアであるLAMMPS、NAMD等と同様に、thread、MPI、GPU等での並列計算が売りとなっている。LAMMPSがEAMなどの無機系多体ポテンシャルや、連続体モデルとの接続等、より間口が広い印象があるのに対し、GROMACSは生体高分子を中心とした有機分子の保守本流的なソフトウェアで、有機材料系では、AMBER、CHARMM等の第一世代ソフトウェアに近い実績・ユーザー数をもつ印象がある。
上記のようにGROMACSは、AMBER等と同様、タンパクなどの生体高分子のシミュレーションを中心に開発されていることから、それ以外の非生体高分子への適用サポートは手薄で、関連する情報も相対的に少ない。
本稿では、ほかのフリーソフトウェアをGROMACSと連携して用いることにより、非生体高分子・一般分子へ適用する方法を、筆者らが最近取り組んでいる、Metal-organic framework(MOF)と呼ばれる配位高分子のMDシミュレーションを例に解説する。具体的には、MOFのモノマー(有機配位分子)の代表例である、4,4’-bipyridine(4,4’-bpy)のモデリングと、そのMD計算までの流れを説明する。GROMACSの最新メジャーバージョンは、2014年夏にリリースされた5.xであるが
(http://gromacs.org/を参照)、使用方法が前バージョンの4.xまでと多少異なるため、本稿では実績のある4.x
を前提に説明する。1.2 GROMACSによるMD計算の流れ図1に、以下で説明する手順全体の流れを示す。
まずMD計算に必要となるファイルをいくつかのフリーソフトを組み合わせて作製する。次にこれらを用いてGROMACSを用いた多分子集合体の計算を行う。以下ではソフトウェア間の連携を中心に簡単に紹介するが、Windows PC上での使用ソフトのインストール、コマンド入力等のより具体的な手順の詳細 は、WEBページ(http://staff.aist.go.jp/makoto-yoneya/ MDforKOUBUNSHI/)に記述したのでそちらを参照願いたい。
フリーな分子動力学シミュレーションソフトウェアGROMACSを、ほかのソフトウェアと連携して用いる方法を解説した。また、GROMACSの適用例として、筆者らが最近取り組んでいるMetal-organic frameworkと 呼 ばれる 配 位 高 分 子 ネットワーク自発形成過程の分子動力学シミュレーションを紹介した。
図1 本稿で説明する手順全体の流れ
特集 研究開発で役立つ高分子のソフトウェア
GROMACSと配位高分子の分子動力学計算
米谷 慎(独)産業技術総合研究所[305-8568]つくば市梅園1-1-1研究グループ長,博士(工学).専門はソフトマター科学.[email protected]/makoto-yoneya/

151©2015 The Society of Polymer Science, Japan高分子 64巻 3月号 (2015年)
展 望 COVER STORY: Highlight Reviews
1.2.1 一分子モデリングGROMACSの通常のターゲットである生体高分子系
の場合は、まずはその三次元構造のprotein database(PDB)ファイルから出発するのが一般的であるが、一般分子の場合は自分で化学構造を入力・作製する必要が ある。そのためには分子構造エディタソフトウェアを使うのが一般的と思われるが、ここでは当該ソフトウェアとしてフリー版ChemSketch(http://www.acdlabs.com/ resources/freeware/chemsketch/)を紹介する。図2に前出の4,4’-bipyridineをChemSketchを用いて描画した画面イメージを例示した。
ChemSketchは、図2のように描画した二次元の化学構造から、水素を自動付加し、簡易エネルギー最適化により三次元座標ファイルを簡単に作製できる点が大きな利点である。ChemSketchで水素付加・三次元化した1分子座標は、MDL-molファイル形式で保存する。
生 体 高 分 子 系 の 場 合 は、 次 にGROMACS同 包 のpdb2gmxモジュールで、GROMACSでのMD計算に必要なトポロジーファイル(*.top 力場パラメーターの割り当てなどが記述されたファイル)と座標ファイル(*.gro)をPDBファイルから生成するが、任意の一般分子構造ファイルから上記のファイルを生成するモジュールはGROMACSに同包されていない。この機能を一般分子に関して行うフリーソフトはいくつか開発されているが、ここではスタンドアローンで動くtopolbuild(http://www.gromacs.org/Downloads/User_contributions/Other_software)を紹介する。
topolbuildは、Sybyl-mol2形式の分子構造ファイルを入力として、上述のpdb2gmxと同様にトポロジーファイル(*.top)と座標ファイル(*.gro)を生成するフリーソフトウェアであり、力場としてTRIPOS、AMBER、general AMBER、OPLS-AA、GROMOS等を選択してGROMACS用トポロジーファイルを作製可能である。このtopolbuildに入力するSybyl-mol2形式と、前述の
ChemSketchが出力可能なMDL-mol形式の間のファイル形式変換が必要となるが、ここでは、化学構造ファイル形式変換を行うフリーソフトとして、OpenBabel
(http://openbabel.org/)を紹介する。OpenBabelはオリジナルのBabelから重用されてきた
きわめて多種の化学関連ファイル形式変換が可能なフリーソフトで、併せてGasteiger、charge equilibration
(QEq)法等の簡易手法による点電荷計算機能なども有する。MD計算のための点電荷計算には、GaussianなどのMO計算プログラムの結果から、electro static potential(ESP)電荷として得られたものを使うのが主流となっているが、ここではOpenBabelによるMDL-mol ファイルからSybil-mol2ファイル変換時に付加されるGasteiger電荷を用いる。
以上の手順により、ChemSketchで入力した分子構造から、GROMACSによるMD計算に必要となる一分子三次元座標ファイル(*.gro)とトポロジーファイル
(*.top)が得られる。1.2.2 分子集合体シミュレーション
液体・固体状態等の分子集合体のMD計算を行うためには、初期構造となる多分子座標ファイルが必要となる。通常MD計算では固体・液体状態の計算を、MDセルと呼ばれる単位セルを中心とした三次元周期境界条件下で計算することにより、仮想的なバルク状態を計算する。液体状態のMD計算の初期構造として、MDセル中にランダムに多数分子配置した座標ファイルは、GROMACSのgenboxモジュールによりtopolbuildで得られた一分子座標(*.gro)ファイルを基に作製可能である。genboxにより4,4’-bpy分 子 をMDセル 中 に128分子ランダムに配置した構造を、後述する分子構造ビューア-gOpenMolで表示したものを図3に示す。
図3 genboxで作製した初期構造(赤線:MDセル)
図2 ChemSketchによる分子構造描画例

152 ©2015 The Society of Polymer Science, Japan 高分子 64巻 3月号 (2015年)
展 望COVER STORY: Highlight Reviews
GROMACSによるMD計算のために最後に必要なのが、時間積分のステップ幅や、温度、圧力制御の方法などのMD計算の計算条件を指定するための計算パラメーターファイル(*.mdp)で、マニュアルや サンプルを見ながら作製することとなる(詳細は、前出のWEBページhttp://staff.aist.go.jp/makoto-yoneya/MDforKOUBUNSHI/を参照)。
具 体 的 なMD計 算 は、GROMACSの モジ ュ ー ルgromppにより上述の、初期座標ファイル(*.gro)、トポロジーファイル(*.top)、MD計算パラメーターファイル(*.mdp)ファイルを統合して、バイナリトポロジーファイル(*.tpr)とし、それを用いて、MD計算モジュールmdrunでMD計算を行う形となり、出力として各種のエネルギーや温度・圧力等の時系列出力ファイル(*.edr)、トラジェクトリファイル(*.trr、*.xtc)等が得られる。GROMACSには、これらの出力ファイルを入力として、各種の解析(動径分布関数、平均二乗変位、各種の相関関数等)を行うモジュールがある程度整備されているのも特徴である。GROMACSにはトラジェクトリをアニメーションとして表示するモジュールngmxも同包されてはいるが、ここでは、gOpenMol
(http://www.csc.fi/english/pages/g0penMol)を 紹 介する。gOpenMolはすでに開発が停止した古参フリーソフトであるが、ほかの描画ソフトでは多少面倒なMDセルが簡単に表示可能であるなど、使い方が簡単なのが利点である。
2.配位高分子のMD計算ポーラスネットワーク構造をとる配位高分子は、近
年、Metal-organic framework(MOF)と呼ばれ、ガス貯蔵材料などの応用が期待され、活発な研究開発が行われている2)。
前節ではGROMACSによるMD計算の流れを、MOFの 有機配位子の代表例である、4,4’-bipyridine(bpy)を例にして説明した。実際の系では、適当な溶媒中で4,4’-bpy と、Cd(NO3)2を混合することにより、二次元スクエア グリッドからなる2-D MOF、{[Cd(4,4’-bpy)2](NO3)2}∞
等が自発的に形成される3)。このようなモノマーからの配位高分子ネットワークの自己組織化プロセスのモデリング・シミュレーションは、MOF形成プロセスの理解に有用であり、チャレンジングな課題である。
ほかの多くの自己組織化過程と同様、上記のMOF自発形成プロセスの時間スケールは、通常のMD計算が扱える時間スケールの上限(現状でマイクロ秒程度)
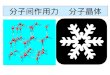
を超えており、時間スケールのギャップを埋める何らかのモデリングが必須となる。筆者らは、溶媒を陽に扱わずに、いわゆるimplicit solventとして扱う4)ことにより、ランダムに配置したモノマーと金属イオンからの配位高分子ネットワークの自発形成のシミュレーションに成功した5),6)。図4に、上記モデルにより自発的に得られた三次元
キュービックネットワークを示す。同様のモデルにより実現されている配位超分子の自発形成7)と同様に、当該配位結合が、結合・解離の双方向性を有する弱い相互作用(cationic dummy-atom method8)でモデル化)である点が自己組織化の観点からキーとなっていることがわかる。このようなMOFの自己組織化プロセスのモデリング・シミュレーションは、新規なMOF系の開発に役立つと考え研究を進めている。本稿が、少しでもGROMACSを用いた分子シミュレーションに興味をおもちいただくきっかけになれば幸いである。
文 献1) B. Hess, et al., J. Chem. Theo. Comp., 4, 435 (2008)2) H. Furukawa, et al., Science, 341, 1230444 (2013)3) M. Fujita, et al., J. Am. Chem. Soc., 116, 1151 (1994)4) M. Yoneya, et al., J. Am. Chem. Soc., 134, 14401 (2012)5) M. Yoneya, et al., “Proc. MOF2014”, P1-083, Kobe (2014)6) 米谷ほか, 分子シミュレーション討論会予稿集, S112 (2014)7) M. Yoneya, et al., ACS Nano, 8, 1290 (2014)8) Y. Pang, et al., J. Mol. Modeling, 5, 196 (1999)
図4 自発形成により得られたMOFネットワーク