Embed Size (px)
Citation preview

1
PARTE I
Muestreo y Evaluación Sensorial

2
PRACTICA No. 1
MUESTREO Y TRATAMIENTO ESTADÍSTICO
1. OBJETIVO
El alumno identificará y aplicará las diferentes técnicas de muestreo estadístico en
diversos tipos de alimentos y aprenderá a elaborar un marco muestral.
2. INTRODUCCIÓN
La toma de muestras es el acto de separar de un lote determinado, una muestra
representativa, a efectos de determinar mediante análisis de laboratorio la aptitud o no del
alimento puesto a consideración y los resultados obtenidos reflejarán las características del
lote del cual se tomo la muestra. En la práctica, es difícil lograr un muestro aleatorio
perfecto, sin embargo a través de las diferentes técnicas se puede obtener una muestra
representativa de un sistema como un todo. Si el sistema es homogéneo, cualquier muestra
sería aceptable, sin embargo, la mayoría de los sistemas son heterogéneos, por lo que es
necesario prepararlas (moler, pulverizar, agitar, etc.) para obtener una muestra uniforme.
Las muestras obtenidas para su análisis deben de cubrir los siguientes requisitos: ser
de tamaño suficiente para cubrir los requisitos de todas las determinaciones a las que se va
a someter; conservarlas para evitar cambios significativos de la misma; etiquetarla y
registrarla conteniendo toda la información necesaria para su identificación.
Existen diferentes métodos de muestreo para el análisis de alimentos, por lo que la
selección del mismo dependerá de: El propósito del análisis (aceptar o rechazar un
alimento, evaluar su calidad promedio o determinar su uniformidad); la naturaleza del lote a
analizar (tamaño; división en sub-lotes; carga o apilamiento); la naturaleza del material que
se va a analizar (homogeneidad, tamaño de la unidad; antecedentes; costos); la naturaleza
de los procedimientos de análisis (significancia de los resultados, ensayos destructivos o no
destructivos, tiempo de duración y costo de los análisis).
Antes de realizar un muestro, es necesario seleccionar el tipo de muestreo más
adecuado que se va a utilizar. Para ello se debe generar un “Marco Muestral” el cual

3
consiste en una lista, ordenación o cualquier otro método que permita visualizar en su
conjunto a todas y cada una de las unidades que componen la población, no siempre es
posible elaborar a detalle una lista, por lo que una visión de conjunto de la población sirve
para realizar un muestreo al azar. Otro aspecto importante es la variable a considerar, la
cual se debe elegir antes de hacer el estudio, es decir aquella que nos permita realizar los
análisis con la finalidad de cumplir con los objetivos propuestos, asimismo debe
seleccionarse la unidad en que se va a medir dicha variable.
3. MATERIALES, REACTIVOS Y EQUIPOS
Una bolsa con fresas, limones, naranjas o manzanas
21 paquetes chicos de cacahuates (de la misma marca)
1 vernier
Papel milimétrico (3 hojas)
Regla
Calculadora
Balanza analítica
4. METODOLOGÍA
I. Primeramente se debe seleccionar qué variable se va a medir a los productos que se
encuentran en la bolsa, posteriormente colocar estos productos en recipientes
grandes para poder extraer una muestra aleatoria, asegurándose de obtener
productos de todas las partes del recipiente; de la parte superficial, intermedia e
inferior, de un extremo, del centro y del otro extremo a fin de tener una buena
representación de los productos seleccionados. A cada producto seleccionado
realice la(s) mediciones que haya determinado hacer. Para este fin coloque sobre la
mesa una retícula que le permita identificar “lotes” o porciones y usando una tabla
de números aleatorios obtenga cuales productos se deben seleccionar. Si se
devuelve cada producto al recipiente la población no se alterará y, por tanto, la
probabilidad de seleccionar cualquier muestra permanecerá inalterada; a este
sistema se le denomina “Muestreo con Reemplazo”. Pero si no reintegra el
producto una vez medida, gradualmente se alterarán las probabilidades de selección

4
de la muestra (primero imperceptiblemente y poco a poco más grande) con lo que
la medición cambiará. A este método se le conoce como “Muestreo sin Reemplazo”
y debe usarse sólo en poblaciones grandes o cuando las mediciones a hacer
repercuten en la destrucción de la muestra.
Una vez obtenida la muestra obtenga las siguientes estadísticas descriptivas básicas:
Total
Media o Promedio
Varianza
Desviación estándar
Rango de variación
En donde la Varianza se obtiene por la fórmula:
S2 = (1 / n – 1) [ ∑x
2 – (∑x
2) / n] donde: n 0 tamaño de la muestra; x = valores
obtenidos.
y S = s2 corresponde a la desviación estándar
II. Ordene los paquetes de cacahuates en una línea, simulando una línea de producción,
formando lotes de 3 paquetes cada uno, simulando, cada uno, un lote de embarque.
De cada lote se obtendrá un paquete, el cual se abrirá y se contará el número de
cacahuates que contiene. Se procederá entonces a:
1) Obtener una muestra piloto, la cual tiene por objeto darnos un conocimiento de las
características estadísticas de la población; esto se realiza de la siguiente forma: se
obtiene el número de cacahuates del primer y segundo paquete, introduzca los datos
en la calculadora y obtenga la media y la varianza, en el papel milimétrico realice
una gráfica como la siguiente:
Varianza
Acumulada
1 2 3 4 5 6 7 8 9 10
Unidades Muestrales

5
Repita lo mismo con la tercera unidad y así sucesivamente hasta completar las diez
unidades o bien hasta que la gráfica se estabilice. De manera esquemática la gráfica
quedará de la siguiente forma:
Varianza
Acumulada
1 2 3 4 5 6 7 8 9 10
Unidades Muestrales
En el número de unidades en que se estabilice la gráfica (en el ejemplo) será la
muestra piloto, pues en ese momento se habrá eliminado el componente de varianza
debido al tamaño de muestra.
2) Para obtener el tamaño de muestra óptimo (o definitivo) se aplicará la siguiente
fórmula:
n’ = [(1.96)2 (C.V.)
2] / d
2
Donde:
n’ es una primera aproximación al tamaño de muestra; 1.96 es el valor de las tablas
de la distribución normal estándar para una significancia de 0.05; C.V. es el
coeficiente de variación de la población expresado como el porcentaje de la media
de acuerdo a la fórmula:
C.V. = (S) (100) / ˉx
En la cual:
ˉx y s son la media y la desviación estándar obtenidas a partir de la muestra piloto.
d’ es el error máximo de estimación que se desea tolerar expresado también como
porcentaje de la media:
d’ = (d x 100) / ˉx
En la cual:

6
d’ es el máximo error que se pueda tolerar expresado en las unidades en las que se
mide la variable de interés.
5. REPORTE DEL ALUMNO
Obtener la Media o Promedio
Varianza
Desviación estándar
Rango de variación
Realizar las gráficas arriba mencionadas
El reporte del alumno deberá incluir las siguientes secciones como se indica en el
apéndice 10.
6. BIBLIOGRAFÍA
Cochran, W.C. 1982. Técnicas de Muestreo. Ed. C.E.C.S.A. México, 513pp.
Méndez, R., I., 1976. Conceptos muy Elementales de Muestreo con Énfasis en la
Determinación del Tamaño de Muestra. Comunicaciones Técnicas. Serie Azul No.
6 I.I.M.A.S., U.N.A.M. 24pp.
Raj, D., 1972. Sampling Theory. Ed. McGraw Hill, U.S.A. 122pp.
Raj, D. 1979. La Estructura de las Encuestas por Muestreo. Fondo de Cultura
Económica, México, 234pp.
Scheaffer, R. L., W. Mendehalland, T. Ott, 1987. Elementos de Muestreo. Ed.
Grupo Editorial Iberoamericano, 325pp.
Zar, J. H. 1974. Biostatistical Analysis. Ed. Prentice Hall. Ingelwood, U.S.A.
620pp.

7
PRÁCTICA No. 2
SELECCION DE JUECES PARA PRUEBAS DE EVALUACIÓN DE
SABOR
1. OBJETIVO
Determinar la habilidad de los candidatos a jueces para detectar los cuatro sabores básicos,
mediante el uso de una prueba de ordenamiento con soluciones de azúcar, sal, ácido cítrico
y café, en diversas concentraciones.
2. INTRODUCCIÓN
Las pruebas de ordenamiento son muy sencillas, las cuales consisten en darles a los jueces
tres o más muestras que difieren en alguna propiedad, y se les pide que las pongan en orden
creciente o decreciente de dicha propiedad. Tiene la ventaja de ser rápida y su desventaja
es que la evaluación realizada es únicamente válida para el conjunto de muestras estudiado,
y no pueden compararse los resultados de un conjunto con los de otro. Sin embargo, su
aplicación en la industria alimentaria es muy común dada su sencillez, facilidad y rapidez.
El gusto o sabor de un alimento puede ser ácido (agrio), dulce, salado o amargo; o
bien puede haber una combinación de dos o más de estos cuatro. Esta propiedad sensorial
es detectada por medio de la lengua. Hay personas que pueden percibir un determinado
gusto, pero para los otros gustos, o sabores básicos, su percepción es muy pobre. Es
necesario determinar que sabores básicos puede detectar cada juez para después dejarles
participar en pruebas de sabor.
Si se van a probar caramelos u otros alimentos dulces, se deben emplear jueces con
habilidad para determinar el gusto dulce, mientras que para probar café o cerveza, los
jueces deben tener sensibilidad para el gusto amargo, si la muestra a probar es una fruta,
entonces se requieren jueces que tengan habilidad tanto para la detección del gusto dulce
como para percibir acidez. Y así, para los demás alimentos, es necesario considerar que
sabores básicos, tienen estos para seleccionar a los jueces que puedan apreciarlos y
medirlos adecuadamente.

8
3. MATERIALES, REACTIVOS Y EQUIPO
Soluciones de:
Azúcar: 10; 5; 2; 1 y 0.5 %
Sal: 5; 2; 1 y 0.5 %
Acido cítrico: 10; 5; 2; 1 y 0.5 %
Café: 0.1; 0.05; 0.02; 0.01 y 0.005 %
Vasos desechables de 50 mL
Agua purificada
4. METODOLOGÍA
Colocar 25 mL de cada solución en vasos desechables marcados con claves de tres o cuatro
números aleatorios y darlas a probar a cada uno de los candidatos a juez, proporcionándoles
una hoja para respuestas como la que se observa en la figura 1, asimismo darles un vaso
con agua purificada para enjuagarse la boca después de probar cada muestra. Para la
interpretación de los resultados puede recurrirse a dos métodos. El primero es mediante el
uso directo de tablas de totales de rangos o de Kramer, y el segundo es la aplicación del
análisis de varianza de datos transformados usando las tablas de Fisher y Yates.
MÉTODO 1. Uso de tablas de totales de rangos o de Kramer.
Supongamos que se realiza la prueba de salado en que participan 8 jueces y que tras la
ordenación se asignan cuatro puntos a la primera, tres a la segunda, dos a la tercera y una a
la cuarta. Las sumas de puntuaciones asignadas a cada muestra son:
MUESTRA 312 436 680 799
SUMA TOTAL 30 15 11 24
Aplicando la tabla de Kramer (Apéndice 1), para cuatro muestras o tratamientos y 8
repeticiones o replicas, obtienen los números 13-27 y 15-25 esto significa que los valores
inferiores a 13 y superiores a 27 presentan diferencias significativas. Por ello se puede
afirmar que las muestras 312 y 680 son diferentes con una probabilidad del 95%, o si se
prefiere expresarlo de otro modo, de que sólo hay una probabilidad del 5% de que la

9
diferencia entre ellas sea debida al azar. En el segundo renglón aparece el intervalo [15-25],
que significa que la muestra 680 es la que tiene la intensidad mínima de sabor salado
porque el total de 680 es inferior al valor de 15 de la suma de rangos de la tabla. El límite
superior del intervalo es de 25 por lo que la muestra 312 es la que tienen el valor salado
más alto.
Por lo tanto, usando la notación convencional, puede expresarse la significancia de los
datos como:
TOTALES: 312a 436a 680b 799a
METODO 2. Análisis de varianza de datos transformados
En este método se aplica el análisis de varianza pero transformando los datos a valores
numéricos según la tabla de Fisher y Yates (Apéndice 2). Dicha tabla se consulta con el
número de tratamientos y se obtienen uno o más números, los cuales se asignan a los
rangos dados por los jueces, de manera que le total para cada juez sea cero. Por ejemplo si
fueran 4 tratamientos, la tabla da los valores de 1.03 y 0.30, lo cual significa que a la
muestra a la que le fue asignada la intensidad mínima del atributo se le va a dar el valor -
1.03, a la siguiente -0.30, a la tercera 0.30, y a la de máxima intensidad +1.03. Por ejemplo
la tabla 1 muestra los resultados del análisis sensorial mediante la prueba de ordenamiento
de la intensidad de sabor dulce en galletas.
Tabla 1. Resultados del análisis sensorial mediante la prueba de ordenamiento la intensidad
de sabor dulce de galletas.
MUESTRAS
JUECES A B C D E
1 3 2 5 4 1
2 2 3 4 5 1
3 3 4 5 2 1
4 1 2 5 4 3
5 3 2 4 5 1
6 3 1 5 4 2
TOTALES 15 14 28 24 9
Nombre:______________________________________ Fecha:___________________

10
Se le han dado a usted 20 muestras con sabores dulce, amargo, salado y agrio, pruébelas y
sepárelas en cuatro grupos, dependiendo del sabor, y después, para cada sabor, ordénelas de
menor a mayor intensidad de sabor.
Indique sus respuestas usando la clave señalada en cada vaso. Enjuáguese la boca con agua
pura después de probar cada muestra. NO SE TRAGUE LAS MUESTRAS.
DULCE
Indique las claves de las muestras, de menor a mayor intensidad
___________ ____________ ______________ _____________ _____________
SALADO
Indique las claves de las muestras, de menor a mayor intensidad
____________ ____________ ______________ _____________ _____________
AMARGO
Indique las claves de las muestras, de menor a mayor intensidad
____________ ____________ ______________ _____________ _____________
AGRIO
Indique las claves de las muestras, de menor a mayor intensidad
____________ ____________ ______________ _____________ _____________
Figura 1. Cuestionario para pruebas de selección de jueces para evaluación de sabor.
Son 5 tratamientos y los números correspondientes de la tabla de Fisher y Yates son: 1.16 y
0.5; por lo tanto a las muestras se les asignan los valores numéricos de la siguiente manera:
1 (menor sabor dulce): -1.16
2: -0.50
3: 0.0
4: +0.50
5: +1.16
Por lo tanto, la tabla 1 de resultados queda modificada de manera que se muestra en la
tabla 2.
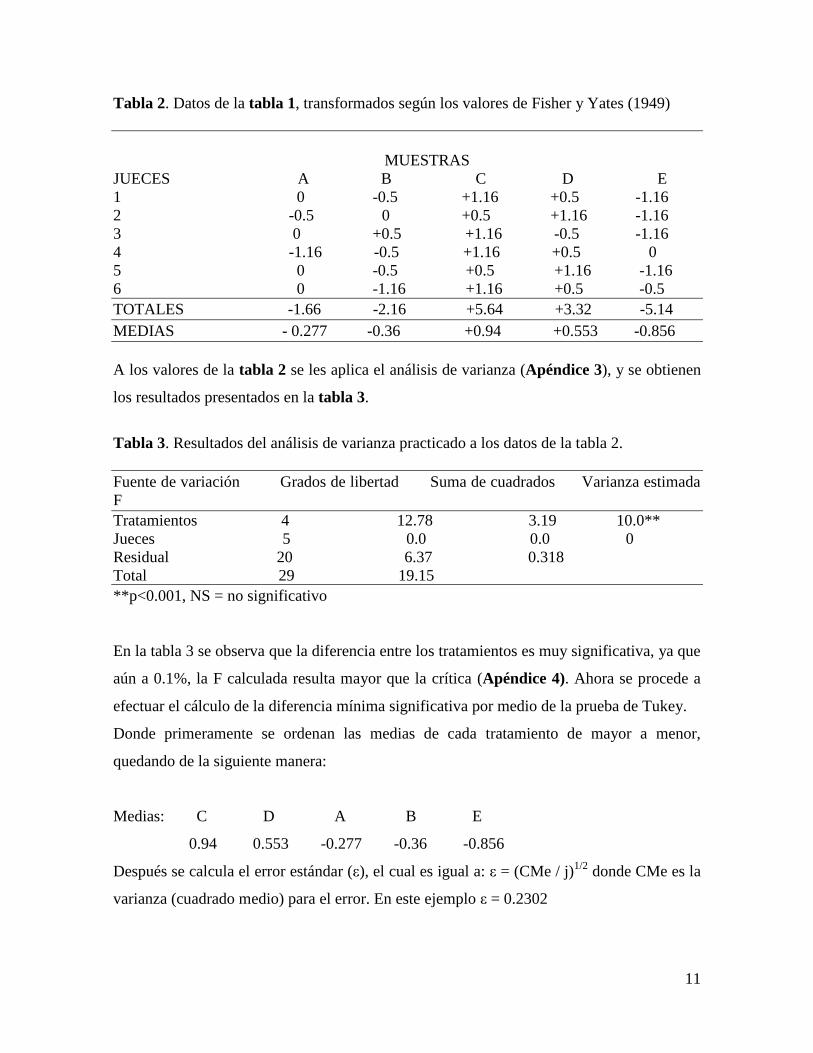
11
Tabla 2. Datos de la tabla 1, transformados según los valores de Fisher y Yates (1949)
MUESTRAS
JUECES A B C D E
1 0 -0.5 +1.16 +0.5 -1.16
2 -0.5 0 +0.5 +1.16 -1.16
3 0 +0.5 +1.16 -0.5 -1.16
4 -1.16 -0.5 +1.16 +0.5 0
5 0 -0.5 +0.5 +1.16 -1.16
6 0 -1.16 +1.16 +0.5 -0.5
TOTALES -1.66 -2.16 +5.64 +3.32 -5.14
MEDIAS - 0.277 -0.36 +0.94 +0.553 -0.856
A los valores de la tabla 2 se les aplica el análisis de varianza (Apéndice 3), y se obtienen
los resultados presentados en la tabla 3.
Tabla 3. Resultados del análisis de varianza practicado a los datos de la tabla 2.
Fuente de variación Grados de libertad Suma de cuadrados Varianza estimada
F
Tratamientos 4 12.78 3.19 10.0**
Jueces 5 0.0 0.0 0
Residual 20 6.37 0.318
Total 29 19.15
**p<0.001, NS = no significativo
En la tabla 3 se observa que la diferencia entre los tratamientos es muy significativa, ya que
aún a 0.1%, la F calculada resulta mayor que la crítica (Apéndice 4). Ahora se procede a
efectuar el cálculo de la diferencia mínima significativa por medio de la prueba de Tukey.
Donde primeramente se ordenan las medias de cada tratamiento de mayor a menor,
quedando de la siguiente manera:
Medias: C D A B E
0.94 0.553 -0.277 -0.36 -0.856
Después se calcula el error estándar (ε), el cual es igual a: ε = (CMe / j)1/2
donde CMe es la
varianza (cuadrado medio) para el error. En este ejemplo ε = 0.2302

12
Luego se consulta la tabla de rangos estudentizados significativos (Apéndice 5), con el
número de tratamientos (en este caso son 5) y los grados de libertad en el error (en este
ejemplo son 20), y el valor que se obtiene (RES) se multiplica por ε para obtener la
diferencia mínima significativa (D.M.S.)
D.M.S. = ε (RES) = 0.2302 (4.24) = 0.976
Después se comparan las diferencias entre las medias, y aquellas diferencias que sean
mayores a D.M.S. se consideran significativas.
C – E = 0.64 – (-0.856) = 1.796 > 0.976 Sí hay diferencia
C – B = 0.94 – (-0.36) = 1.3 > 0.976 Sí hay diferencia
C – A = 0.94 – (-0.277) = 1.217 > 0.976 Sí hay diferencia
C – D = 0.94 – ( 0.553) = 0.387 < 0.976 NO HAY DIFERENCIA
D – E = 0.553 – (-0.856) = 1.409 > 0.976 Sí hay diferencia
D – B = 0.553 – (-0.36) = 0.913 < 0.976 NO HAY DIFERENCIA
A – E = -0.277 – (-0.856 ) = 0.576 < 0.976 NO HAY DIFERENCIA
Por lo tanto, la significancia de los resultados puede expresarse así:
Medias: C D A B E
0.94a 0.553ab -0.277bc -0.36bc -0.856c
5. REPORTE DE ALUMNO
Presentar los resultados obtenidos por los dos métodos anteriormente descritos con la
finalidad de saber que jueces tienen la habilidad para realizar pruebas de sabor (ácido,
dulce, salado y amargo).
6. BIBLIOGRAFÍA Anzaldúa, M. A. 1994. “La Evaluación Sensorial de los Alimentos en la Teoría y en
la Práctica”. Editorial Acribia, S. A. Zragoza (España).
Sancho, J.; Bota, E. y de Castro, J.J. 2002. “ Introducción al Análisis Sensorial de
los Alimentos. ALFAOMEGA GRUPO EDITOR, S.A. de C.V.
Pedrero, D.L. y Pangborn, R.M. 1997. “Evaluación sensorial de los Alimentos”.
Métodos Analíticos. LONGMAN DE MEXICO EDITORES, S.A. de C.V.

13
PRACTICA No. 3
PRUEBAS DE PREFERENCIA
1. OBJETIVO
Ordenar, según las opiniones de un grupo de jueces, un par o una serie de muestras de
acuerdo con su aprecio personal o de su preferencia.
2. INTRODUCCCIÓN
El análisis sensorial de los alimentos se lleva a cabo con diferentes pruebas, según sea la
finalidad para la que se efectúe. Existen tres tipos principales de pruebas: Las pruebas
afectivas, las discriminativas y las descriptivas. Las pruebas afectivas son aquellas en las
que el juez expresa su reacción subjetiva ante el producto indicando si le gusta o disgusta.
Estas pruebas son las que presentan mayor variabilidad en los resultados, ya que se trata de
apreciaciones completamente personales. Para este tipo de pruebas es necesario contar con
un mínimo de 30 jueces no entrenados y estos deben ser consumidores habituales y
compradores del tipo de alimento en cuestión. Las pruebas afectivas pueden clasificarse en
tres tipos: pruebas de preferencia, de grado de satisfacción y de aceptación.
Las pruebas de preferencia, son sencillas en las cuales se desea conocer si los jueces
prefieren una muestra sobre otra, en este tipo de pruebas no se busca si los jueces pueden
distinguir entre dos muestras sino que se quiere evaluar si realmente prefieren determinada
muestra.
3. MATERIALES, REACTIVOS Y EQUIPO Cuatro sobres con polvo para preparar bebidas instantáneas (Kool-Aid, Zuko, etc.)
de diferente marca comercial pero del mismo sabor.
Agua purificada
Vasos desechables
Azúcar
Recipientes de 1 L
Cucharas
Refrigerador

14
4. METODOLOGÍA
Preparar las bebidas en recipiente de 1 L, de acuerdo a las indicaciones del fabricante, y
colocarlas en el refrigerador durante un tiempo aproximado de 2 a 3 horas antes de la
prueba. Después se coloca la bebida en vasos desechables, aproximadamente de 50 mL de
muestra en cada uno, codificados con números aleatorios (de 3 a 4 números). Lleve a cabo
la prueba a los jueces, los cuales deben enjuagarse la boca con agua purificada cada vez que
pruebe una muestra, además deberá presentar a los jueces las hojas para que indiquen las
respuestas (figura 2). Los datos se tabulan y se analizan de acuerdo a un ordenamiento por
rangos (o escalas de rangos ordinales)
Nombre:______________________________ Fecha:___________________________
Producto: Bebidas instantáneas
INSTRUCCIONES: Indique su aceptación al probar cada una de las muestras que se le
presentan (1 = menor preferencia, 4 = Mayor preferencia).
MUESTRA
8456 _____________
7913 _____________
7914 _____________
7915 _____________
COMENTARIOS:_______________________________________________________
Figura 2. Cuestionario para la prueba de preferencia
Para ilustrar la mecánica de este análisis, a continuación se presenta un ejemplo:
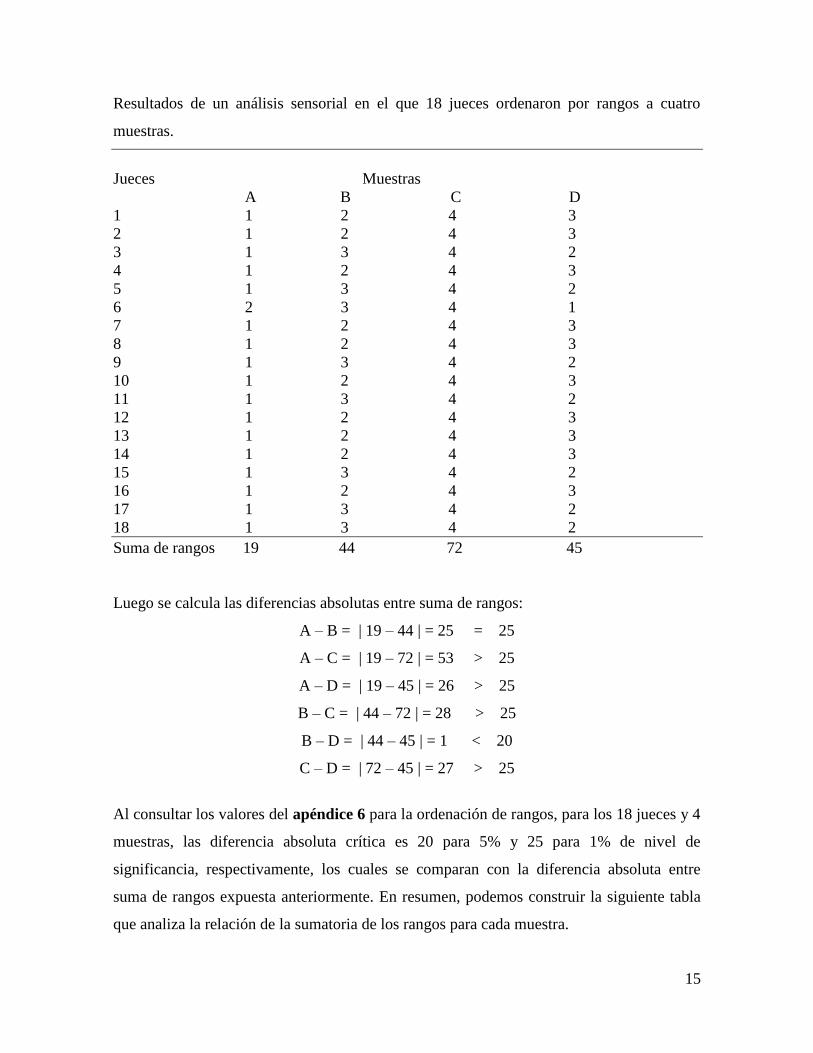
15
Resultados de un análisis sensorial en el que 18 jueces ordenaron por rangos a cuatro
muestras.
Jueces Muestras
A B C D
1 1 2 4 3
2 1 2 4 3
3 1 3 4 2
4 1 2 4 3
5 1 3 4 2
6 2 3 4 1
7 1 2 4 3
8 1 2 4 3
9 1 3 4 2
10 1 2 4 3
11 1 3 4 2
12 1 2 4 3
13 1 2 4 3
14 1 2 4 3
15 1 3 4 2
16 1 2 4 3
17 1 3 4 2
18 1 3 4 2
Suma de rangos 19 44 72 45
Luego se calcula las diferencias absolutas entre suma de rangos:
A – B = | 19 – 44 | = 25 = 25
A – C = | 19 – 72 | = 53 > 25
A – D = | 19 – 45 | = 26 > 25
B – C = | 44 – 72 | = 28 > 25
B – D = | 44 – 45 | = 1 < 20
C – D = | 72 – 45 | = 27 > 25
Al consultar los valores del apéndice 6 para la ordenación de rangos, para los 18 jueces y 4
muestras, las diferencia absoluta crítica es 20 para 5% y 25 para 1% de nivel de
significancia, respectivamente, los cuales se comparan con la diferencia absoluta entre
suma de rangos expuesta anteriormente. En resumen, podemos construir la siguiente tabla
que analiza la relación de la sumatoria de los rangos para cada muestra.

16
Muestras A B C D
Suma de rangos 19c 44
b 72ª 45
b
a,b,c = suma de rangos con distintos supraíndices indican diferencia significativa (p<0.01)
Mediante esta información se concluye que la muestra B y D no son diferentes entre sí de
manera significativa, pero si lo son con respecto a las muestras A y C, y a su vez entre sí;
todas las diferencias con un nivel de significancia menor igual a 0.01.
5. RESULTADOS
Tabular los datos y analizarlos de acuerdo a un ordenamiento por rangos (o escalas de
rangos ordinales)
6. BIBLIOGRAFÍA
Anzaldúa, M. A. 1994. “La Evaluación Sensorial de los Alimentos en la Teoría y en
la Práctica”. Editorial Acribia, S. A. Zaragoza (España).
Sancho, J.; Bota, E. y de Castro, J.J. 2002. “ Introducción al Análisis Sensorial de
los Alimentos. ALFAOMEGA GRUPO EDITOR, S.A. de C.V.
Pedrero, D.L. y Pangborn, R.M. 1997. “Evaluación sensorial de los Alimentos”.
Métodos Analíticos. LONGMAN DE MEXICO EDITORES, S.A. de C.V.

17
PRACTICA No. 4
PRUEBAS DE NIVEL DE AGRADO DE SATISFACCIÓN CON
ESCALAS HEDONICAS VERBALES.
1. OBJETIVO
Aplicar las escalas hedónicas verbales para determinar el grado de satisfacción que provoca
una muestra específica.
2. INTRODUCCCIÓN
Cuando se evalúan más de dos muestras a la vez, o cuando se desea obtener mayor
información acerca de un producto, se recurre a este tipo de pruebas, con la finalidad de
analizar objetivamente los datos de los jueces acerca de cuando les disgusta o les gusta un
alimento. Para realizar estas pruebas se utilizan las escalas hedónicas verbales o graficas y
la elección del tipo de escala depende de la edad de los jueces y del número de muestras a
evaluar.
Las escalas hedónicas verbales presentan a los jueces una descripción verbal de la
sensación que les produce la muestra. Deben de contener siempre un número impar de
puntos, y se debe de incluir siempre el punto central “ni me gusta ni me disgusta” al cual se
le asigna la calificación de cero. A los puntos de la escala por encima de este valor se le
otorgan valores numéricos positivos, indicando que las muestras son agradables; en
cambio, a los puntos por debajo del valor de indiferencia se les asignan los valores
negativos, correspondiendo a calificaciones de disgusto. Esta forma de asignar el valor
numérico tiene la ventaja de que facilita mucho los cálculos, y es posible reconocer al
primer vistazo si una muestra es agradable o desagradable.
3. MATERIALES, REACTIVOS Y EQUIPO
Cuatro bebidas refrescantes sabor (naranja, manzana, etc.) de diferente marca
comercial pero del mismo sabor.

18
Agua purificada
Vasos desechables
Refrigerador
4. METODOLOGÍA
Colocar las bebidas refrescantes de diferente sabor en el refrigerador durante un tiempo
aproximado de 2 a 3 horas antes de la prueba. Después se coloca el jugo en vasos
desechables, aproximadamente de 50 mL de muestra en cada uno, codificados con números
aleatorios (de 3 a 4 números). Lleve a cabo la prueba con los jueces (30 o más), los cuales
deben enjuagarse la boca con agua purificada cada vez que pruebe una muestra, además
deberá presentar a los jueces las hojas para que indiquen las respuestas (figura 3.).
Interpretación de resultados: Las calificaciones de la prueba hedónica se tabulan por juez-
consumidor (filas) y por producto (columnas), como se realizó en el ejemplo de la práctica
número 3, totalizando la sumatoria de cada columna y de cada fila para obtener un gran
total y después se aplica el análisis de varianza.
Nombre:__________________________________ Fecha: _____________________
PRODUCTO: JUGO DE NARANJA
Pruebe las muestras de jugo que se le presentan e indique,
Según la escala, su opinión sobre ellas.
Marque con una X el renglón que corresponda
a la calificación para cada muestra
MUESTRAS
ESCALA 5423 1978 2010 2050
Me gusta mucho ______ _______ ________ ________
Me gusta _______ ________ ________ _________
Me gusta poco ________ ________ _________ ________
Ni me gusta ni me disgusta ________ _________ _________ _________
Me disgusta poco ________ _________ _________ _________
Me disgusta _________ _________ _________ _________
Me disgusta mucho _________ __________ __________ _________
Comentarios: ___________________________________________________________
MUCHAS GRACIAS
Figura 3. Cuestionario de evaluación del grado de satisfacción por medio de una escala
hedónica verbal de siete puntos.

19
5. RESULTADOS
Las calificaciones de la prueba hedónica se tabulan por juez-consumidor y por producto
(ejemplo de la práctica número 3), totalizando la sumatoria de cada columna y de cada fila
para obtener un gran total y después se aplica el análisis de varianza.
6. BIBLIOGRAFÍA
Anzaldúa, M. A. 1994. “La Evaluación Sensorial de los Alimentos en la Teoría y en
la Práctica”. Editorial Acribia, S. A. Zragoza (España).
Sancho, J.; Bota, E. y de Castro, J.J. 2002. “Introducción al Análisis Sensorial de los
Alimentos. ALFAOMEGA GRUPO EDITOR, S.A. de C.V.
Pedrero, D.L. y Pangborn, R.M. 1997. “Evaluación sensorial de los Alimentos”.
Métodos Analíticos. LONGMAN DE MEXICO EDITORES, S.A. de C.V.

20

21
PRACTICA No. 5
PRUEBA DE COMPARACIÓN POR PARES
1. OBJETIVO
Determinar si existente diferencia perceptible entre dos muestras
2. INTRODUCCCIÓN
Las pruebas discriminativas son aquellas en las que no se requiere conocer la sensación
subjetiva que produce un alimento en una persona, sino que se desea establecer si hay
diferencia significativa entre dos muestras o entre ellas y un patrón. Estas pruebas son
ampliamente utilizadas en el control de calidad para evaluar si las muestras de un lote están
siendo producidas con una calidad uniforme. Asimismo, por medio de ellas se puede
determinar el efecto de modificaciones en las condiciones del proceso sobre la calidad
sensorial del producto, las alteraciones introducidas por la sustitución de un ingrediente por
otro. Las pruebas discriminativas más comúnmente utilizadas son las siguientes: Prueba de
comparación apareada simple, Triangular, dúo-trío, de comparación apareada de Scheffé,
de comparaciones múltiples y de ordenamiento.
En la prueba de comparación por pares se presentan solamente dos muestras al juez y se
le pide que las compare en cuanto a alguna característica sensorial (dulzor, amargor,
dureza, olor, color, etc.) e indique cual de las dos tiene mayor intensidad de dicha
propiedad. En el tratamiento estadístico hay que tener en cuenta si la prueba es de tipo uni-
o bilateral. Se considera un ensayo unilateral cuando el director del panel sabe que hay
diferencia entre las muestras y desea averiguar si es percibida o no por el panel de jueces.
En la prueba bilateral no se sabe si hay diferencia entre las muestras, o bien hay razones
objetivas para creer que una será preferida a la otra.
3. MATERIALES, REACTIVOS Y EQUIPO Limones
Azúcar
Fructosa
Agua purificada

22
Vasos desechables
Azúcar
Recipientes de 1 L
Cucharas
Refrigerador
Potenciómetro
Refractómetro de Abbe
4. METODOLOGÍA
Elabore dos limonadas con el mismo contenido de sólidos solubles (°Bx) y pH, pero
endulzada con dos edulcorantes diferentes (azúcar y fructosa). Colocar las limonadas en el
refrigerador durante un tiempo aproximado de 2 a 3 horas antes de la prueba. Después se
colocan las limonadas en vasos desechables, aproximadamente de 50 mL de muestra en
cada uno, codificados con números aleatorios (de 3 a 4 números). Lleve a cabo la prueba
con los jueces, los cuales deben enjuagarse la boca con agua purificada cada vez que pruebe
una muestra, además deberá presentar a los jueces las hojas para que indiquen las
respuestas (figura 4.). La interpretación de los resultados se efectúa consultando el apéndice
7, para “prueba de una cola”, y se obtiene para el número de jueces que participaron en la
prueba y con el nivel de significancia escogido de antemano, el número mínimo de
respuestas correctas para que haya diferencia significativa entre las dos muestras.
Nombre:_________________________________________ Fecha: ________________
PRODUCTO: LIMONADA
Pruebe las dos muestras de limonada e indique cuál es la más dulce
Marque con una X la muestra más dulce
4513 2897
______ ______
Comentarios:
_________________________________________________________________________
MUCHAS GRACIAS
Figura 4. Cuestionario para prueba de comparación apareada simple.

23
5. RESULTADOS
Los resultados se obtienen consultando el apéndice 7, para “prueba de una cola”, con el
número de jueces y con el nivel de significancia, el número mínimo de respuestas correctas
para que haya diferencia significativa entre las dos muestras.
6. BIBLIOGRAFIA
Anzaldúa, M. A. 1994. “La Evaluación Sensorial de los Alimentos en la Teoría y en
la Práctica”. Editorial Acribia, S. A. Zaragoza (España).
Sancho, J.; Bota, E. y de Castro, J.J. 2002. “Introducción al Análisis Sensorial de los
Alimentos. ALFAOMEGA GRUPO EDITOR, S.A. de C.V.
Pedrero, D.L. y Pangborn, R.M. 1997. “Evaluación sensorial de los Alimentos”.
Métodos Analíticos. LONGMAN DE MEXICO EDITORES, S.A. de C.V.

24

25
PRACTICA No. 6
PRUEBA TRIANGULAR
1. OBJETIVO
Determinar si existe diferencia sensorialmente perceptible entre dos muestras, comparando
tres muestras a la vez, de las cuales dos son iguales entre sí y la otra diferente.
2. INTRODUCCIÓN
La prueba triangular consiste en presentar al catador tres muestras codificadas
convenientemente, de las cuales dos son iguales y sólo la tercera es diferente. El catador
debe indicar cual es diferente. La prueba recibe su nombre de la forma de presentarla:
generalmente cada muestra ocupa el vértice de un triángulo y se indica al catador que inicie
la degustación por uno de ellos y siga en orden. Aunque es una prueba sencilla y de fácil
interpretación esta sometida a muchas tendencias, sesgos, predisposiciones y prejuicios.
Uno de los sistemas de minimizarlos es presentar cada muestra un número igual de veces
en cada una de las posiciones del triángulo, pero esto complica la prueba. Por ejemplo,
partiendo de dos muestras B y C, deberán presentarse en las siguientes combinaciones:
BCC, BBC, BCB, CBB, CCB, CBC, donde la misma letra indica muestras iguales y la letra
diferente la posición de la muestra desigual. Dependiendo de la naturaleza del estímulo,
estas combinaciones aleatorias se ofrecen al juez en una o varias sesiones.
Esta prueba al igual que las demás pruebas de diferenciación, requiere que la variable
motivo de observación sensorial sea la única causa de variabilidad (por ejemplo: al
observar “sabor a chocolate”, que la atención del juez no se distraiga con el color o la forma
de la muestra). Por otra parte, de las muestras en estudio también se puede desconocer la
variable sensorial, por lo que simplemente esta prueba permitirá detectar si existe o no
diferencia entre las muestras, sin saber en que atributo. Resulta muy útil en el control de
calidad para asegurar que los distintos lotes de un producto mantengan las características o
para detectar si el cambio de un ingrediente provoca diferencias apreciables. Se emplea
muy a menudo en la industria alimentaria, cuando una empresa de esta naturaleza tenga
problemas para conseguir saborizantes que se utilizan para elaborar un producto, ya sea por
problemas del proveedor o por aumentos de precio, etc. En este caso es necesario

26
determinar cuál de las marcas existentes en el mercado puede ser eligida para sustituir este
saborizante sin que los consumidores detecten el cambio.
3. MATERIALES, REACTIVOS Y EQUIPO
1 kg de papas
Dos litros de aceite de marca comercial diferente (uno de maíz y otro de canola)
Freidora de papas
Pela papas
Sal
Agua purificada
4. METODOLOGÍA
Lavar, pelar y cortar las papas, después del corte debe volver a lavar las papas para eliminar
el exceso de almidón y sumergirlas en una solución de agua con sal (5%) para evitar el
oscurecimiento de las mismas. Sacar y secar las rodajas de papas y freír ½ kg estas en un
tipo de aceite (maíz) y el otro ½ kg en el otro tipo de aceite. Las muestras se darán a probar
a 12 jueces (pueden ser los que usted elija). Al aceite de maíz lo denominaremos X y al otro
Y. Los tríos a preparar sería los siguientes: XYY, XXY, XYX, YYX, YXX Y YXY. Si a la
muestra X la denominamos 545; a la segunda muestra X, la denominamos 875; a la muestra
Y, 321 y a la segunda muestra Y, 369, se deben preparar 12 muestras con el 545, 12
muestras con el 875 y seis muestras con el 321 y seis con el 369. Resultando los siguientes
tríos:
545-321-369 (Diferente-Igual-Igual)
545-875-321 (Igual-Igual-diferente)
545-321-875 (Igual-Diferente-Igual)
321-545-369 (Igual-Diferente-Igual)
321-369-545 (Igual-Igual-Diferente)
321-545-875 (Diferente-igual-igual)

27
Y como son 12 catadores se repetirá la misma combinación para cada uno de la docena.
Lleve a cabo la prueba con los jueces, los cuales deben enjuagarse la boca con agua
purificada cada vez que pruebe una muestra, además deberá presentar a los jueces las hojas
para que indiquen las respuestas (figura 5.).
La interpretación de los resultados se efectúa consultando el apéndice 8, en la cual
se reporta el número mínimo de respuestas correctas para establecer diferencias
significativas, al nivel de significancia que se haya elegido. Si los resultados obtenidos en la
cata dan que 9 de los 12 catadores identifican la muestra diferente se puede afirmar, con
una probabilidad de error del 1%, que las muestras son diferentes. Si hubiera un número de
catadores (por ejemplo un par) que no den respuesta, se restan al número total de jueces
(12-2=10) y se vuelve al apéndice 8 con el nuevo valor.
Nombre: ____________________________________________ Fecha: ____________
PRODUCTO: PAPAS FRITAS
Ante usted hay tres muestras, donde dos son idénticas y la tercera es diferente
Pruebe las tres muestras tantas veces como desee, empezando por la muestra situada a su
derecha. Cuando esté seguro, indique cuál es diferente
MARQUE CON UNA X LA CLAVE DE LA MUESTRA DIFERENTE
545 321 369
Comentarios:
_________________________________________________________________________
___________________________________________________________________
MUCHAS GRACIAS
Figura 5. Cuestionario para una prueba triangular.

28
5. RESULTADOS
Los resultados se obtienen consultando el apéndice 8, en el que se reporta el número
mínimo de respuestas correctas para establecer diferencia significativa, trabajar con un
nivel de significancia del 1%.
6. BIBLIOGRAFÍA
Anzaldúa, M. A. 1994. “La Evaluación Sensorial de los Alimentos en la Teoría y en
la Práctica”. Editorial Acribia, S. A. Zragoza (España).
Sancho, J.; Bota, E. y de Castro, J.J. 2002. “Introducción al Análisis Sensorial de los
Alimentos. ALFAOMEGA GRUPO EDITOR, S.A. de C.V.
Pedrero, D.L. y Pangborn, R.M. 1997. “Evaluación sensorial de los Alimentos”.
Métodos Analíticos. LONGMAN DE MEXICO EDITORES, S.A. de C.V.

29
PRACTICA No. 7
PRUEBA DÚO-TRÍO
1. OBJETIVO
Determinar si existe diferencia sensorialmente perceptible entre dos muestras, comparando
dos muestras desconocidas contra una tercera llamada de referencia, para indicar cuál de las
desconocidas es igual a la de referencia dada.
2. INTRODUCCIÓN
En esta prueba se le presentan al juez tres muestras, una identificada como referencia (R),
y las otras dos marcadas con una clave para ocultar su identidad; una de estas dos muestras
deberá ser necesariamente igual a la de referencia. La aplicación de esta prueba es similar a
la triangular, pero su eficiencia es menor ya que 50% de probabilidad de acierto por
casualidad, como en el caso de la prueba de comparación apareada, sin embargo, en esta
última se especifica que hay que tomar en cuenta un cierto atributo para establecer la
diferencia, mientras que en la prueba dúo-trío no es necesario especificarlo, sino sólo decir
que muestra es diferente. Esta prueba se utiliza para reducir el número de pruebas a probar,
por ejemplo cuando el sabor de las muestras es muy fuerte o picante, o cuando el alimento
tiene una textura desagradable.
3. MATERIALES, REACTIVOS Y EQUIPO
¼ de queso seco para rayar (marca comercial 1)
¼ de queso seco para rayar (marca comercial 2)
Cuchillos
Platos chicos desechables
Agua purificada

30
4. METODOLOGÍA
Cortar trocitos de aproximadamente 5 g, de queso seco para rayar cada uno y colocarlos en
platos desechables, codificarlos con números aleatorios de (3 a 4 números) y con la letra R.
Las muestras se deberán presentar a los jueces una codificada con la letra R y dos con los
números aleatorios de las cuales una debe ser igual a la de referencia (R). Lleve a cabo la
prueba con los jueces, los cuales deben enjuagarse la boca con agua purificada cada vez que
pruebe una muestra, además se presentará a los jueces las hojas para que indiquen la
respuesta (figura 6). La prueba dúo-trío se analiza de igual manera que la prueba de
comparación apareada simple, ya que la probabilidad de escoger la muestra correcta por
casualidad es también del 50 % (p = ½), y los resultados, tomado como número de aciertos,
pueden analizarse por Ji- cuadrada (apéndice 9) o la tabla para pruebas de significación de
pruebas pareadas (apéndice 7) de una sola cola.
Nombre: ________________________________ Fecha: _________________
PRODUCTO: QUESO SECO PARA RASPAR
Ante usted hay una muestra de referencia marcada con R y otras dos muestras marcadas
con claves
Una de estas muestras es idéntica a R y la otra es diferente
¿Cuál de las dos muestras es diferente de R?
Márquela con una X
7523 1985
Comentarios:______________________________________________________________
_____________________________________________________________
MUCHAS GRACIAS
Figura 6. Cuestionario para prueba dúo-trío.

31
La ji-cuadrada se utiliza para determinar si las comparaciones entre muestras que generan
las pruebas de comparación por pares, dúo-trío y triangular son significativamente
diferentes o no. La fórmula adecuada para estas pruebas sensoriales que involucran un
grado de libertad (g.l. = 1), es la llamada Ji-cuadrada ajustada.
Χ2 = ( │x1 - np│- 0.5)2 / np (1 – p)
Donde:
X = Número de opiniones acertadas
n = número total de ensayos practicados o número de jueces por repeticiones
efectuadas.
P = Probabilidad de éxito en un ensayo único
q = (1 – p) = Probabilidad de la falla en un ensayo único
0.5 o factor de corrección por continuidad en Ji-cuadrada ajustada. Este factor se
aplica sólo para un grado de libertad en el cual los resultados se consignan como
“acierto” o “falta”.
Ejemplo: en una prueba dúo-trío (p = 0.5) en la que 12 jueces participaron con tres
degustaciones (repeticiones), un total de 20 respuestas indicaron un fallo correcto ( se
detecto la muestra que era igual a la de referencia).
x = 20 respuestas correctas
n = 12 jueces por 3 repeticiones = 36 ensayos
p = ½ = 0.5
q = (1 – p) = 0.5
np = 36 x 0.5 = 18
Χ2 = (│20 - 18│- 0.5)2 / 18 (0.5) = 0.25

32
Consultando el valor de la tabla del apéndice 9 para Χ2 una cola, para grados de libertad
(g.l.) = 1 y p = 0.05 (o 5%) es 2.71, para p = 0.01 (1%) es 5.41. Mediante estos valores
podemos observar que la Χ2 calculada es menor que el valor de p = 0.05 y menor que el de
p = 0.01, por lo que declaramos que los jueces detectan de manera significativa (p < 0.05)
la igualdad entre las muestras codificadas y el control (R).
5. RESULTADOS
Determinar si los jueces detectan de manera significativa la diferencia entre las muestras y
el control.
6. BIBLIOGRAFÍA
Anzaldúa, M. A. 1994. “La Evaluación Sensorial de los Alimentos en la Teoría y en
la Práctica”. Editorial Acribia, S. A. Zragoza (España).
Sancho, J.; Bota, E. y de Castro, J.J. 2002. “ Introducción al Análisis Sensorial de
los Alimentos. ALFAOMEGA GRUPO EDITOR, S.A. de C.V.
Pedrero, D.L. y Pangborn, R.M. 1997. “Evaluación sensorial de los Alimentos”.
Métodos Analíticos. LONGMAN DE MEXICO EDITORES, S.A. de C.V.

33
PARTE II
Análisis de la Composición Proximal

34

35
PRACTICA No. 8
DETERMINACIÓN DE HUMEDAD
1. OBJETIVO
Determinar el contenido de humedad de un alimento
2. INTRODUCCIÓN
Todos los alimentos, cualquiera que sea el método de industrialización a que hayan sido
sometidos, contienen agua en mayor o menor proporción. Las cifras de contenido en agua
varían en los alimentos naturales. En los tejidos vegetales y animales, puede decirse que
existe en dos formas generales: “agua libre” y “agua ligada”. El agua libre o absorbida, que
es la forma predominante, se libera con gran facilidad. El agua ligada se halla combinada o
absorbida. Se encuentra en los alimentos como agua de cristalización (en los hidratos) o
ligada a las proteínas y a las moléculas de sacáridos y absorbida sobre la superficie de las
partículas coloidales.
Existen diferentes razones por las cuales, la mayoría de las industrias de alimentos
determinan la humedad en los alimentos, las principales son las siguientes:
a) El comprador de materias primas no desea adquirir agua en exceso.
b) El agua, si esta presente por encima de ciertos niveles, facilita el desarrollo de
microorganismos.
c) Para mantequilla, margarina, leche en polvo y queso esta señalada el máximo legal.
d) Los materiales pulverulentos se aglomeran en presencia de agua, por ejemplo azúcar
y sal.
e) La humedad de trigo debe ajustarse adecuadamente para facilitar la molienda.
f) La cantidad de agua presente puede afectar la textura.
g) La determinación del contenido en agua representa una vía sencilla para el control
de la concentración en las distintas etapas de los procesos de producción de
alimentos.

36
Métodos de secado
Los métodos de secado son los más comunes para valorar el contenido de humedad en los
alimentos; se calcula el porcentaje en agua por la perdida en peso debida a su eliminación
por calentamiento bajo condiciones normalizadas. Aunque estos métodos dan buenos
resultados que pueden interpretarse sobre bases de comparación, es preciso tener presente
que:
Algunas veces es difícil eliminar por secado toda la humedad presente;
A cierta temperatura el alimento es susceptible de descomponerse, con lo que se
volatilizan otras sustancias además de agua, y
También pueden perderse otras materias volátiles aparte de agua.
Método por secado de estufa
La determinación de secado en estufa se basa en la pérdida de peso de la muestra por
evaporación del agua. Para esto se requiere que la muestra sea térmicamente estable y que
no contenga una cantidad significativa de compuestos volátiles. El principio operacional del
método de determinación de humedad utilizando estufa y balanza analítica, incluye la
preparación de la muestra, pesado, secado, enfriado y pesado nuevamente de la muestra.
Precauciones que se deben tener en las determinaciones de humedad en estufa.
1. Los productos con un elevado contenido en azúcares y las carnes con un contenido
alto de grasa deben deshidratarse en estufa a vacío a temperaturas que no excedan
los 70ºC.
2. Los métodos de deshidratación en estufa son inadecuados para productos, como
las especias, ricas en sustancias volátiles distintas del agua.
3. La eliminación del agua de una muestra requiere que la presión parcial de agua en la
fase de vapor sea inferior a la que alcanza en la muestra; de ahí que sea necesario
cierto movimiento del aire; en una estufa de aire se logra abriendo parcialmente la
ventilación y en las estufas de vacío dando entrada a una lenta corriente de aire
seco.
4. La temperatura no es igual en los distintos puntos de la estufa, de ahí la
conveniencia de colocar el bulbo del termómetro en las proximidades de la muestra.

37
Las variaciones pueden alcanzar hasta más de tres grados en los tipos antiguos, en
los que el aire se mueve por convección. Las estufas más modernas de este tipo
están equipadas con eficaces sistemas, que la temperatura no varia un grado en las
distintas zonas.
5. Muchos productos son, tras su deshidratación, bastante higroscópicos; es preciso
por ello colocar la tapa de manera que ajuste tanto como sea posible
inmediatamente después de abrir la estufa y es necesario también pesar la cápsula
tan pronto como alcance la temperatura ambiente; para esto puede precisarse hasta
una hora si se utiliza un desecador de vidrio.
6. La reacción de pardeamiento que se produce por interacción entre los
aminoácidos y los azúcares reductores libera agua durante la deshidratación y se
acelera a temperaturas elevadas. Los alimentos ricos en proteínas y azúcares
reductores deben, por ello, desecarse con precaución, de preferencia en una estufa
de vacío a 60°C.
Método por secado en estufa de vacío
Se basa en el principio fisicoquímico que relaciona la presión de vapor con la presión del
sistema a una temperatura dada. Si se abate la presión del sistema, se abate la presión de
vapor y necesariamente se reduce su punto de ebullición. Si se sustrae aire de una estufa
por medio de vacío se incrementa la velocidad del secado.
Es necesario que la estufa tenga una salida de aire constante y que la presión no exceda
los 100 mm Hg. y 70°C, de manera que la muestra no se descomponga y que no se
evaporen los compuestos volátiles de la misma, cuya presión de vapor también a sido
modificada.
Método de secado en termobalanza
Este método se basa en evaporar de manera continua la humedad de la muestra y el registro
continuo de la perdida de peso, hasta que la muestra se sitúe a peso constante. El error de
pesada en este método se minimiza cuando la muestra no se expone constantemente al
ambiente.

38
Método de destilación azeotrópica
El método se basa en la destilación simultánea del agua con un líquido inmiscible en
proporciones constantes. El agua es destilada en un líquido inmiscible de alto punto de
ebullición, como son tolueno y xileno. El agua destilada y condensada se recolecta en una
trampa Bidwell para medir el volumen.
Precauciones que se deben tener con los procedimientos de destilación con disolvente.
1. Se recomienda usar los siguientes disolventes: Tolueno (Pe. = 112 ºC) y xileno (Pe.
= 137ºC).
2. La Asociación Americana de Comercio de Especias, en sus métodos oficiales
analíticos, recomienda el uso de benceno (Pe. = 80ºC) en lugar de tolueno, con
productos tales como pimientos rojos, cebollas deshidratadas, ajos deshidratados,
etc., que son ricos en azúcares y otras sustancias que pueden descomponerse,
liberando agua, a la temperatura de ebullición del tolueno.
3. Es preciso limpiar la totalidad del aparato, cada vez que se utilice, con ácido
sulfúrico-dicromato, enjuagarlo primero con agua y luego con alcohol y, finalmente,
secarlo.
4. Debe calibrarse el colector por sucesivas destilaciones con tolueno de cantidades de
agua medidas con precisión. Las lecturas deben aproximarse en centésimas de
mililitro.
5. La elección del colector depende del volumen de agua que se espera recoger, del
grado de precisión requerido y de la facilidad con que el disolvente refluya.
Método de Karl Fischer.
Es el único método químico comúnmente usado para la determinación de agua en alimentos
que precisamente se basa en su reactivo. Este reactivo fue descubierto en 1936 y consta de
yodo, dióxido de azufre, una amina (originalmente se empleaba piridina sin embargo por
cuestiones de seguridad y toxicidad se está reemplazando por imidazol) en un alcohol
(ejemplo metanol).

39
Inicialmente, el dióxido de azufre reacciona con el metanol para formar el éster el
cual es neutralizado por la base (1). El éster es oxidado por el yodo a metil sulfato en una
reacción que involucra al agua (2).
Las reacciones son las siguientes:
CH3OH + SO2 + RN → [RNH]SO3CH3 (1)
H2O + I2 + [RNH]SO3CH3 +2RN → [RNH] (SO4)CH3 + 2[RNH]I (2)
Habitualmente se utiliza un exceso de dióxido de azufre, piridina y metanol de
manera que la fuerza del reactivo venga determinada por la concentración de yodo. Este
reactivo es un poderoso deshidratante, por lo que tanto la muestra como el reactivo deben
protegerse contra la humedad del aire, cualquiera que sea la técnica usada. Se hace por
titulación y estas pueden ser visuales o potenciométricas. En su forma más simple el mismo
reactivo funciona como indicador. La disolución muestra mantiene un color amarillo
canario mientras haya agua, que cambia luego a amarillo cromato y después a pardo en el
momento del vire.
En su forma mas simple el método potenciométrico consta de una fuente de
corriente directa, un reóstato, un galvanómetro o microamperímetro y electrodos de platino,
dos cosas son necesarias para la determinación: una diferencia de potencial que nos de una
corriente y el contacto del titulante con el analito. Este método se aplica a alimentos con
bajo contenido de humedad por ejemplo frutas y vegetales deshidratados, aceite y café
tostado, no es recomendable para alimentos con alto contenido de humedad.
La tabla 1 muestra una comparación de los métodos para determinar la humedad de
los alimentos, as como sus ventajas y desventajas de cada uno de ellos.
Tabla 1. Comparación entre métodos para determinar humedad en alimentos.
Método Ventajas Desventajas
Secado en estufa
tamaño de la partícula, peso de la
muestra, posición de la muestra en
el horno, etc.
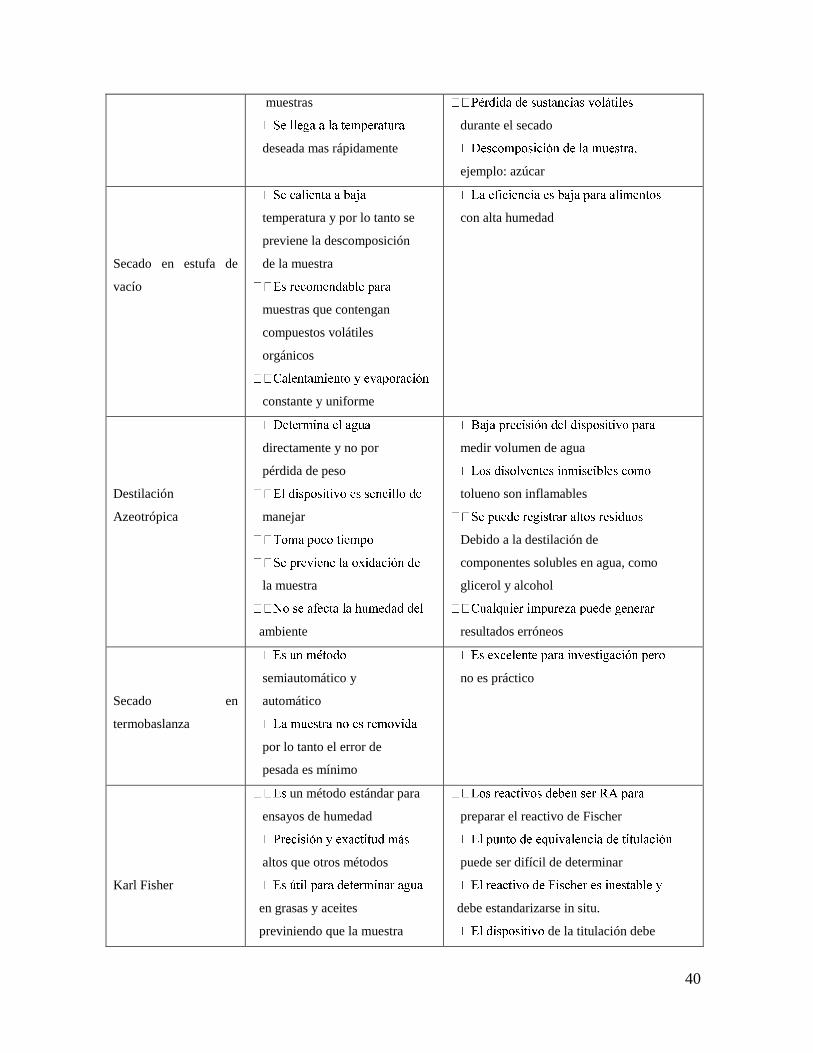
40
muestras
deseada mas rápidamente
durante el secado
ejemplo: azúcar
Secado en estufa de
vacío
temperatura y por lo tanto se
previene la descomposición
de la muestra
muestras que contengan
compuestos volátiles
orgánicos
constante y uniforme
con alta humedad
Destilación
Azeotrópica
directamente y no por
pérdida de peso
manejar
la muestra
ambiente
medir volumen de agua
tolueno son inflamables
Debido a la destilación de
componentes solubles en agua, como
glicerol y alcohol
resultados erróneos
Secado en
termobaslanza
semiautomático y
automático
por lo tanto el error de
pesada es mínimo
no es práctico
Karl Fisher
un método estándar para
ensayos de humedad
altos que otros métodos
en grasas y aceites
previniendo que la muestra
preparar el reactivo de Fischer
puede ser difícil de determinar
debe estandarizarse in situ.
de la titulación debe

41
se oxide
monta la determinación toma
pocos minutos
protegerse de la humedad atmosférica
debido a la excesiva sensibilidad del
reactivo a la humedad.
reactiva
A.- DETERMINACIÓN DE HUMEDAD POR MÉTODO DE SECADO
EN ESTUFA
a) Material y Equipo
Capsula de porcelana, previamente tarada (peso constante)
Pinzas para capsulas
Desecador
Balanza analítica
Estufa con temperatura controlada
b) Metodología
Primeramente se debe lavar la cápsula y llevarla a masa constante, colocándola en una
estufa a una temperatura de 130 C durante 2 hrs aproximadamente. Pesar con exactitud
entre 2-3 g de muestra, sobre la cápsula y colocarla en la estufa la cual debe estar a una
temperatura entre 90-100C. Retirarla de la estufa, dejarla enfriar en el desecador y
pesar tan pronto como se equilibre con la temperatura ambiente. Repetir hasta masa
constante. La pérdida de masa corresponde al contenido humedad de la misma. El
porcentaje de humedad se calcula mediante la formula (1).
% de humedad = [(B- C) 100] / A ( 1 )
Donde:
A = peso de muestra húmeda (g)
B = peso de crisol + muestra húmeda (g)
C= peso de crisol + muestra seca (g)

42
B.- DETERMINACIÓN DE HUMEDAD POR MÉTODO DE SECADO
EN ESTUFA DE VACÍO
a) Materiales y Equipo
Estufa de vacío
Capsula de porcelana, previamente tarada (peso constante)
Pinzas para capsula
Desecador
Balanza analítica
b) Metodología
Pesar de 2 a 3 g de muestra en una capsula de porcelana (previamente pesado después
de tenerlo a peso constante 2 hrs. a 130°C aprox.). Secar la muestra al menos por 24
hrs, en la estufa conectada a vacío a una temperatura de 70°C como máximo. Retirar de
la estufa, la capsula con la muestra seca, dejar enfriar en desecador y pesar tan pronto
como se equilibre con la temperatura ambiente. Repetir la operación hasta peso
constante. El porcentaje de humedad se calcula utilizando la formula (1 ).
C.- DETERMINACIÓN DE HUMEDAD POR METODO DE
DESTILACIÓN AZEOTRÓPICA.
a) Materiales y Equipo
Parrilla eléctrica
Condensador
Mangueras
Trampa de Bidwell
Matraz bola de 250 mL
Balanza analítica
Benceno, Tolueno o Xileno.

43
b) Metodología
Pesar 2-5 g de muestra en un matraz bola de 250 mL con junta esmerilada. Cubrir la
muestra con tolueno, Xileno o Benceno (100 mL aprox.). Acople al matraz un colector
para destilación azeotrópica y un refrigerante a este último en posición de reflujo
conectado al flujo de agua. Llene el vástago graduado del colector con el mismo
solvente desde la parte superior del refrigerante. Destile lentamente al principio e
incrementando la velocidad hasta que toda el agua haya sido destilada. Poco antes del
final de la destilación, lave el refrigerante con un poco de solvente desde la parte
superior. Continúe la destilación hasta que ya no varíe la cantidad de agua destilada en
el tubo colector. Lea el volumen directamente del tubo colector y calcule el porcentaje
de humedad considerando la densidad del agua. El porcentaje de humedad se calcula
mediante la siguiente formula.
% de Humedad = (mL de agua obtenidos / peso de la muestra) (100)
D.- DETERMINACIÓN DE HUMEDAD POR METODO DE SECADO
EN TERMOBALANZA
a) Materiales y Equipo
Temobalanza
Espátula
b) Metodología
Pesar de 8 a 10 g de muestra y colocarlos en una charola de aluminio formando una
capa lo más homogénea posible. Colocar la charola con la muestra en el espacio
destinado para ello en la termobalanza y encender el equipo. Registrar la pérdida de
peso o en su caso, el porcentaje de humedad (según el equipo) después de 10-15 min o
bien cuando ya no haya variación en la lectura.

44
Nota: Dependiendo del equipo, es necesario regular la intensidad de la lámpara para
evitar que la muestra se queme y el resultado sea erróneo.
4. REPORTE DEL ALUMNO
Calcular el porcentaje de humedad, reportándolo como pérdida por secado a 90-
100°C.
Calcular el porcentaje de humedad, reportándolo como pérdida por secado en estufa
de vacío a 70±1°C.
Calcular el porcentaje de humedad de la muestra
Realizar un reporte como se indica en el apéndice 10.

45
PRACTICA No. 9
DETERMINACIÓN DE CENIZAS
1. OBJETIVO
Determinar el contenido de cenizas que corresponden a las sales minerales presentes en la
muestra
2. INTRODUCCIÓN
Las cenizas de un alimento son un término analítico equivalente al residuo inorgánico que
queda después de calcinar la materia orgánica. Las cenizas normalmente, no son las mismas
sustancias inorgánicas presentes en el alimento original, debido a las perdidas por
volatilización o a las interacciones químicas entre los constituyentes.
El valor principal de la determinación de cenizas (y también de las cenizas solubles
en agua, la alcalinidad de las cenizas y las cenizas insolubles en ácido) es que supone un
método sencillo para determinar la calidad de ciertos alimentos, por ejemplo en las especias
y en la gelatina es un inconveniente un alto contenido en cenizas. Las cenizas de los
alimentos deberán estar comprendidas entre ciertos valores, lo cual facilitará en parte su
identificación.
a. Los productos que contienen mucha agua se secan primero sobre un plato eléctrico
caliente o al baño María.
b. La consideración principal es que el producto no desprenda humos.
c. En general, la temperatura adecuada de la mufla son 500°C. Sin embargo, los
cloruros, pueden volatilizarse a esta temperatura.
d. Las cenizas se utilizan muchas veces para la determinación de constituyentes
individuales, por ejemplo cloruros, fosfatos, calcio y hierro.
Para la determinación de cenizas se siguen dos métodos; en seco y vía húmeda.
Método de cenizas totales
La determinación en seco es el método más común para cuantificar la totalidad de

46
minerales en alimentos y se basa en la descomposición de la materia orgánica quedando
solamente materia inorgánica en la muestra, es eficiente ya que determina tanto cenizas
solubles en agua, insolubles y solubles en medio ácido.
En este método toda la materia orgánica se oxida en ausencia de flama a una
temperatura que fluctúa entre los 550 - 600°C; el material inorgánico que no se volatiliza a
esta temperatura se conoce como ceniza.
Determinación de cenizas en húmedo.
La determinación húmeda se basa en la descomposición de la materia orgánica en medio
ácido por lo que la materia inorgánica puede ser determinada por gravimetría de las sales
que precipiten, y también por algún otro método analítico para las sales que permanezcan
en disolución acuosa o ácida. Para la determinación húmeda se dan cenizas alcalinas, ácidas
y neutras y esto se basa en el tipo de anión o catión ya sea metálico o complejo de tal forma
hay minerales como tartratos, citratos que producirán cenizas con un carácter alcalino. Es
necesario tomar en cuenta que también un índice de alcalinidad de cenizas es muestra del
contenido de carbonatos en disolución acuosa. Las ventajas y desventajas de estos métodos
se muestran en la tabla 2
Tabla 2. Comparación entre métodos para determinación de cenizas totales
Método Ventajas desventajas
Seco 1.- Simple
2.- No se requiere atención durante
la generación de cenizas
3.- No se requieren reactivos
4.- Se pueden manejar muchas
Muestras
5.- Es un método estándar para la
determinación de cenizas
1.- Se requiere alta temperatura
2.- El equipo es caro
3.- Hay perdidas por volatización
4.- Hay interacciones entre minerales y
recipientes.
5.-Absorción de elementos traza por
recipientes de porcelana o sílice
6.- Poca utilidad para análisis de Hg, As, P
y Se
7.- Calentamiento excesivo puede hacer
ciertos componentes insolubles.
8.- Hay una dificultad de manejo de cenizas
por ser higroscópicas, sensibles a la luz,

47
etc.
Húmedo 1.- Relativamente no se requiere
alta temperatura
2.- El dispositivo es simple
3.- La oxidación es rápida
4.- Se mantiene la disolución
acuosa lo cual es bueno para
análisis mineral.
5.- El equipo no es caro
6.- no hay volatilización de
minerales
1.- se requieren altas cantidades de
materiales corrosivos.
2.- Se requieren ácidos explosivos
3.- Se requiere estandarizar los reactivos
4.- Las reacciones son fumantes
5.- Manejar sistemáticamente varias
muestras no se puede
6.- El procedimiento es tedioso y gasta mucho
tiempo.
A.- DETERMINACIÓN DE CENIZAS POR EL MÉTODO DE
CENIZAS TOTALES
a) Materiales y Equipo
Crisol de porcelana o platina
Mechero de Bunsen
Mufla con temperatura controlada
Desecador
Balanza analítica
Pinzas para crisol
b) Metodología
Llevar el crisol a masa constante, colocándolo en la mufla a 500-600 C durante 2 hrs
aproximadamente.. Colocar en el mismo de 2 a 5 g de muestra. Carbonizar lentamente
con el mechero para evitar pérdidas por arrastre en el humo, hasta que cese su
desprendimiento. Calcinar en la mufla a una temperatura de 550-600 ºC (2 ó 3 horas
aproximadamente). Repetir la operación anterior si es necesario, hasta conseguir unas

48
cenizas blancas o ligeramente grises, homogéneas. Enfriar en el desecador y pesar. El
porcentaje de cenizas se calcula de acuerdo a la siguiente formula.
% de cenizas = [A-B) 100] / C
A = peso del crisol con la ceniza (g)
B = peso del crisol vacío (g)
C = peso de la muestra (g)
Nota: Elévese lentamente la temperatura de la mufla hasta alcanzar la de incineración
sin que se formen llamas. Una combustión demasiado activa puede ocasionar pérdidas
de cenizas o conducir a que se fundan o formen inclusiones de carbono que no se
incineren, Llevase cuidado en evitar la pérdida de cenizas ligeras; manténgase la
cápsula cubierta por un pequeño vidrio de reloj incluso dentro del desecador.
B.- DETERMINACIÓN DE CENIZAS POR EL MÉTODO DE
CENIZAS EN HUMEDO
a) Materiales y Equipo
Vasos de precipitado de 250 mL
Ácido nítrico concentrado
Placa de calentamiento eléctrica
Matraz aforado de 100 mL
Balanza analítica
Estufa con temperatura controlada
b) Metodología
Pesar 5 g de muestra en un vaso de precipitados, adicionar 10 mL de ácido nítrico
concentrado, calentar durante una hora hasta la obtención de color traslúcido, enfriar,
recuperar, filtrar en matraz aforado de 100 mL, aforar con agua. Tomar una alícuota de
10 mL y colocarlo en un vaso de precipitado de 250 mL, colocado previamente a peso

49
constante, evaporar a sequedad, colocar en estufa hasta peso constante. Calcular por
diferencia de peso la cantidad de minerales en la alícuota y relacionarlo con el aforo.
3. REPORTE DEL ALUMNO
Calcular el porcentaje de cenizas por los dos métodos
Realizar un reporte como se indica en el apéndice 10.

50
PRACTICA No. 10
DETERMINACIÓN DE PROTEÍNAS POR EL MÉTODO KJELDAHL
1. OBJETIVO
Determinar el contenido de proteína de un alimento por el método Kjeldahl
2. INTRODUCCIÓN
El nitrógeno presente en los alimentos lo podemos encontrar formando parte de la proteína
y otros compuestos. Las proteínas se encuentran formadas por cadenas de aminoácidos.
Algunos aminoácidos son esenciales y otros no esenciales. Los aminoácidos no esenciales
pueden ser sintetizados en el cuerpo, pero los aminoácidos esenciales deben estar presentes
en la dieta porque el cuerpo no los puede sintetizar. Existen diversos métodos para la
cuantificación de las proteínas, todos ellos basados en algunas de sus propiedades típicas,
como pueden ser los patrones de adsorción de las radiaciones electromagnéticas de los
grupos aromáticos, la reactividad del enlace peptídico, su contenido de nitrógeno, etc.
El método de Kjeldahl es el que más se utiliza e incluso se toma como referencia o
comparación cuando se usan otras técnicas; con este procedimiento se mide el nitrógeno
total de un alimento sin hacer distinción entre aquel que proviene de las proteínas y el no
proteínico; esto puede dar lugar a errores de cálculo. Entre los compuestos que contienen
nitrógeno, pero que nos son proteínas y que se encuentran en los alimentos, se tiene la urea,
ácidos nucleicos, dopamina, colina). El factor de conversión de N2 a proteína es específico
en cada caso y proviene de dividir 100 entre el porcentaje de N2 (que es ya conocido) del
polímero; por ejemplo, en el caso de la leche, los polipéptidos presentan 16% de N2 en
forma pura, por lo que su factor se conversión será 100/16=6.25.
El método se basa en la determinación de la cantidad de nitrógeno orgánico contenido
en productos alimentarios, compromete dos pasos consecutivos:
a) La descomposición de la materia orgánica bajo calentamiento en presencia de ácido
sulfúrico.
b) El registro de la cantidad de amoniaco obtenida de la muestra.

51
Durante el proceso de descomposición ocurre la deshidratación y carbonización de la
materia orgánica combinada con la oxidación de carbono a dióxido de carbono. El
nitrógeno orgánico es transformado a amoniaco que se retiene en la disolución como
sulfato de amonio. La recuperación del nitrógeno y velocidad del proceso pueden ser
incrementados adicionando sales que abaten la temperatura de descomposición (sulfato de
potasio) y por un catalizador.
El método de Kjeldahl consta de las siguientes etapas:
a) Digestión Proteína + H2SO4 → CO2 + (NH4)2SO4 + SO2
b) Destilación (NH4)2SO4 + 2NaOH → Na2SO4 + NH3 ↑+ H2O
NH3 + H3BO3 →NH4H2BO3
c) Titulación
NH4H2BO3 + HCl → H3BO3 + NH4Cl
En la mezcla de digestión se incluye sulfato sódico para aumentar el punto de ebullición y
un catalizador para acelerar la reacción, tal como sulfato de cobre. El amoniaco en el
destilado se retiene en ácido bórico y y se titula con HCL en presencia de un indicar.
3. MATERIALES, EQUIPO Y REACTIVOS
Equipo kjeldahl
Matraces Kjeldahl de 800 mL
Probeta de 50 mL
H2SO4 concentrado p.a. (98%)
K2SO4 o Na2SO4 anhidro
CuSO4.5H2O
NaOH 40%
Solución de HBO3 al 4%
HCl 0.1 N

52
Indicador rojo de metilo
Bureta de 25 mL
Probeta de 100 mL
Soporte y pinzas para bureta
Matraz erlenmeyer de 250 mL
Balanza analítica
4. METODOLOGÍA
Digestión: Pesar una muestra de alimento entre 2 y 4 g y colocarla en un matraz Kjeldahl de
500 mL. Agregar 20 g de CuSO4.5H2O y 5 g de K2SO4 o Na2SO4 anhidro, unas perlas de
vidrio y 25 mL de H2SO4 concentrado, todo el material debe estar sumergido en el ácido para
que no haya pérdidas de nitrógeno. Colocar el matraz en el digestor de equipo kjeldahl y
calentar la mezcla hasta que se haya digerido toda la materia orgánica (no se observan
partículas carbonosas sin oxidar y el líquido queda translúcido y de color débilmente verdoso
o azul-verdoso). No se debe retirar el matraz del digestor si este sigue desprendiendo humos,
ya que estos son muy tóxicos. La digestión demanda entre 1 y 2 hs. Enfriar y agregar
cuidadosamente 200 mL de agua destilada.
Destilación: Colocar el matraz con la muestra digerida a un refrigerante por medio de una
trampa adecuada, además de colocar un matraz erlenmeyer con 50 ml de H3BO3 al 4% (sobre
el cual se va a recoger el NH3 destilado) y unas gotas de indicador (rojo de metilo), y ponerlo a
la salida del refrigerante, cuidando que el extremo del mismo quede sumergido en la solución
ácida. Antes de conectar completamente el matraz al destilador se va agregando con cuidado
de 80-100 mL de solución de NaOH 40% para neutralizar el ácido sulfúrico, primero sin agitar
para que se ubique en el fondo, y una vez agregado todo, conectar bien el matraz, agitar para
lograr la mezcla (el medio se hace fuertemente alcalino que se detecta por formación de un
precipitado pardo oscuro, dispersado por efecto de la ebullición) y simultáneamente se
comienza el calentamiento a ebullición del contenido del matraz. El indicador vira a amarillo
cuando empieza a destilarse el NH3 por arrastre en corriente de vapor. Se sigue destilando
hasta obtener aproximadamente 125 mL de destilado en el matraz erlenmeyer colector (los

53
primeros 100 mL de destilado contienen generalmente la totalidad del NH3). Una vez
alcanzado dicho volumen, se retira el matraz erlenmeyer y luego se suspende el calentamiento.
Valoración: El destilado se valora con solución de HCl 0.1 N, hasta lograr el viraje del
indicador al color inicial rojo.
Cálculos:
% Proteínas = ( mL gastados x N x 0.014 x F x 100 ) / g de muestra.
Donde:
F = factor de conversión de N2 a proteína (de acuerdo al alimento analizado)
N = normalidad de HCl
0.014 = mili-equivalente del nitrógeno
La tabla 1 muestra los diferentes factores de conversión que se utilizan en diversos tipos de
alimentos para calcular el porcentaje de proteínas.
Tabla 1. Factores de conversión de N2 a proteína de algunos alimentos
Alimento % de nitrógeno Factor de conversión
Vegetales
Productos de soya
Harina de trigo
Harina de maíz
Harina de avena
Carne y derivados
Cebada
Gelatina
Huevo
Leche y prods.
lácteos
Proteína (General)
Legumbres
17.35
17.35
17.54
16.00
17.15
16.00
17.15
18.02
15.97
15.67
16.00
16.00
5.70
5.70
5.70
6.25
5.83
6.25
5.83
5.55
6.25
6.38
6.25
6.25

54
5. REPORTE DEL ALUMNO
Calcular el porcentaje de nitrógeno y en base al tipo de alimento que utilizo en su
práctica, utilizar el factor adecuado para determinar el porcentaje de proteínas que
tienen el mismo.
Realizar un reporte como se indica en el apéndice 10.

55
PRACTICA No. 11
EXTRACCIÓN Y CUANTIFICACIÓN DE EXTRACTO ETÉREO O
GRASA CRUDA POR EL MÉTODO SOXHLET
1. OBJETIVO
Cuantificar el contenido de extracto etéreo o grasa cruda de un alimento.
2. INTRODUCCIÓN
Los lípidos, junto con las proteínas y carbohidratos, constituyen los principales
componentes estructurales de los alimentos. Se definen como un grupo heterogéneo de
compuestos que son insolubles en agua pero solubles en disolventes orgánicos tales como
éter, cloroformo, benceno o acetona. Todos los lípidos contienen carbón, hidrógeno y
oxigeno, y algunos también contienen fósforo y nitrógeno. Los lípidos comprenden un
grupo de sustancias que tienen propiedades comunes y similitudes en la composición, sin
embargo algunos, tales como los triacilgliceroles son muy hidrofóbicos. Otros, tales como
los di y monoacilgliceroles tienen movilidad hidrofóbica e hidrofílica en su molécula por lo
que pueden ser solubles en disolventes relativamente polares
Los constituyentes grasos de los alimentos consisten en diversas sustancias
lípidicas. El contenido en grasa (algunas veces llamado extracto etéreo o grasa cruda), el
cual se puede considerar que consiste de constituyentes lípidicos libres o sea aquellos que
pueden ser extraídos por los disolventes menos polares como las fracciones ligeras del
petróleo y el éter dietílico, mientras que los constituyentes lípidos “combinados” necesitan
disolventes más polares tales como alcoholes para ser extraídos. Las uniones de los lípidos
pueden romperse por hidrólisis o algún otro tratamiento químico para producir lípidos
libres. Por esto la cantidad de lípidos que se extraen en los alimentos dependerá del método
de análisis que se haya usado.
Se llama grasa cruda a la fracción separada del material seco por extracción en
forma directa con solventes orgánicos (éter de petróleo, éter etílico, acetona, cloroformo,
etc.). Se habla de extracto etéreo y no de grasa debido a que en este método el solvente

56
recomendado extrae además de los triglicéridos otros tipos de sustancias lipidicas solubles
en el solvente.
El contenido total de lípidos se determina comúnmente por métodos de extracción
con disolventes orgánicos (por ejemplo Soxhlet, Goldfish, Mojonnier), sin embargo
también puede cuantificarse por métodos de extracción que no incluyen disolventes (por
ejemplo, Babcock, Gerber) y por métodos instrumentales que se basan en propiedades
físicas o químicas de los lípidos (por ejemplo, infrarrojo, densidad y absorción es rayos X).
El método de extracción soxhlet, consiste en una extracción semicontinua con un
disolvente orgánico. En este método el disolvente se calienta, se volatiliza y condensa
goteando sobre la muestra la cual queda sumergida en el disolvente. Posteriormente éste es
sifoneado al matraz de calentamiento para empezar de nuevo el proceso. El contenido de
grasa se cuantifica por diferencia de peso
3. MATERIALES, EQUIPO Y REACTIVOS
Aparato de extracción Soxhlet
Parrilla eléctrica
Mangueras
Soporte universal
Pinzas para soporte
Desecador
Balanza analítica
Éter etílico o de petróleo, hexano, etc.
Cartucho de celulosa
Estufa con control de temperatura
4. METODOLOGÍA
Poner a peso constante un matraz bola de fondo plano (soxhlet) en la estufa a 100ºC,
aproximadamente 2 hrs. Colocar de 4 a 5 g de muestra (seca) sobre un papel, enrollarlo y
colocarlo en un cartucho de celulosa, tapar con un algodón (No apretar el algodón contra la
muestra) y colocar el cartucho en el extractor.

57
Conectar el matraz al extractor, en el que se debe encontrar el cartucho con la muestra,
agregar dos cargas del disolvente (generalmente hexano) y posteriormente conectar éste al
refrigerante (no poner grasa en las juntas). Y posteriormente conectar este al refrigerante y
calentar el matraz con parrilla a ebullición suave durante cuatro horas aproximadamente.
Una vez extraída toda la grasa, se recupera el disolvente antes de que se descargue
(evitando el sifoneo), quitar el cartucho con la muestra desengrasada, seguir calentando
hasta la casi total eliminación del disolvente, quitar el matraz y secar el extracto en la estufa
a 100ºC por 30 min., enfriar y pesar. Calcular el porcentaje de grasa de acuerdo a la
siguiente formula.
.
Cálculos:
% Extracto etéreo = [( MG - M )/ W] X 100
Donde:
MG = peso del matraz con grasa
M = peso del matraz
W = peso de la muestra
5. REPORTE DEL ALUMNO
Calcular el porcentaje de extracto etéreo o grasa cruda de la muestra utilizada
Realizar un reporte como se indica en el apéndice 10.

58
PRACTICA No. 12
DETERMINACIÓN DE FIBRA CRUDA
1. OBJETIVO
Cuantificar la fibra presente en el alimento a analizar
2. INTRODUCCIÓN
La fibra cruda es el residuo orgánico combustible e insoluble que queda después de que la
muestra se ha tratado en ciertas condiciones y representa la porción no digerible de los
alimentos. Esta constituida de celulosa, lignina y hemicelulosa.
La celulosa es el principal polisacárido estructural de los vegetales; está presente, en
forma de fibra, en los tallos y hojas y en las paredes de todas las células, asociado a
hemicelulosas y pectinas. No es digerible por el hombre, pero participa en la formación del
contenido hidratado que facilita la evaluación de otras materias no digeribles.
La celulosa es responsable, en gran parte de la textura de los alimentos vegetales,
pues su dureza parece estar ligada al carácter cristalino de la celulosa. La pre-cocción
atenúa estas características, mientras que el almacenamiento ya sea por congelación o
deshidratado tiende a acentuarlas.
Existen tres tipos de fibra: fibra cruda, fibra dietética y fibra vegetal. La fibra cruda
es, por definición, el residuo obtenido tras el tratamiento de los vegetales con ácidos y
álcalis; la fibra dietética incluye celulosa, hemicelulosa, lignina, pectinas, gomas y tejidos
animales no degradables como mucopolisacáridos. La fibra vegetal se refiere
fundamentalmente a los elementos fibrosos de la pared de la célula vegetal. Una
clasificación mas es aquella que parte de su grado de solubilidad en agua y se clasifica en:
Fibra soluble: las fibras solubles, como las pectinas, polisacáridos vegetales, goma guar,
mucílago y algunas hemicelulosas, se encuentran principalmente en las frutas y vegetales,
especialmente en naranjas, manzanas y zanahorias. Se encuentran también en las hojuelas
del salvado de avena, cebada y legumbres. Las fibras solubles forman mezclas de
consistencia viscosa cuyo grado depende de la fuente de vegetal o fruta utilizado.

59
Fibra insoluble: las fibras insolubles, como la celulosa, la mayoría de las hemicelulosas y la
lignina, forman con el agua mezclas de baja viscosidad. Los cereales son especialmente
ricos en fibra insoluble (salvado de trigo).
El método para determinar la fibra cruda se basa en la extracción de la misma con ácido y
base de la fracción insoluble presente en el material.
3. MATERIALES, EQUIPO Y REACTIVOS
Digestor de fibra cruda
Embudo de Bushener
Crisol de Gooch
Bomba de vacío
Rejilla de asbesto
Asbesto preparado
Mechero
Antiespumante (vaselina liquida)
Pinzas para crisol
Probeta de 100 mL
Papel filtro sin cenizas
Pipetas
Vasos de berzelius
Matraz kitazato
H2SO4 al 1.25%
NaOH al 1.25%
Alcohol etílico al 96%
Hexano (Éter de petróleo o etílico)
4. METODOLOGÍA
Digestión ácida:
1. Se pesan de 1 a 2 g de muestra desengrasada y se colocan en el vaso de berzelius,
agregan 200 mL de H2SO4 al 1.25% en peso caliente, una pizca de asbesto

60
preparado (lavado con HNO3, H2O y calcinado) y unas gotas de antiespumante,
colocar el vaso en el digestor-condensador para fibra cruda
2. Se hierve durante 30 minutos.
Digestión alcalina:
3. Después de la digestión ácida, enfriar el vaso y agregar 200 mL de NaOH al 1.25%
en peso caliente, y
4. Se hierve durante 30 min.
Después de realizar estos dos procesos, se deja enfriar y se filtra sobre un papel filtro (sin
cenizas) el cual fue puesto a peso constante. Se lava el residuo con agua caliente, hasta la
neutralidad (3 o 4 porciones de 25 mL de agua caliente), y por último con una porción de
25 mL de alcohol. Posteriormente secar el residuo con el papel filtro durante 1 hora a 95ºC,
enfriar en desecador y pesar. Registrar el peso del residuo. Después se calcina el residuo
seco y pesado con el papel filtro en un crisol para determinación de cenizas y se calcula el
peso de las cenizas. Se utiliza la siguiente formula para determinar el porcentaje de fibra
cruda.
% Fibra cruda = W1 - W2 x 100
W 0
Donde:
W0 = Peso de la muestra
W1 = Peso del crisol + muestra digerida y seca - peso del papel filtro
W2 = Peso del crisol + cenizas
5. REPORTE DEL ALUMNO
Calcular el porcentaje de fibra cruda del alimento utilizado
Realizar un reporte como se indica en el apéndice 10.

61
DETERMINACIÓN DE CARBOHIDRATOS (POR DIFERENCIA)
% de carbohidratos = 100 – ( % humedad + % de cenizas + % de grasa + % de proteína +
% fibra cruda).
DETERMINACIÓN DEL VALOR ENERGETICO DE LA MUESTRA
La unidad de energía contenido en los alimentos, así como la que interviene con mayor
frecuencia en los procesos metabólicos, se expresa como una unidad de calor. La caloría.
Una caloría se define como la cantidad de calor requerida para elevar la temperatura de 1 g
de agua en 1ºC (de 15 a 16ºC). Esta cantidad de calor es excesivamente pequeña de manera
que en nutrición es común usar en su lugar, a una gran caloría o kilocaloría (Kcal), la cual
es 1000 veces mayor.
Valores calóricos de los componentes de los alimentos
Componente Kcal por gramo
Carbohidratos 4
Grasas 9
Proteínas 4
Determinación del valor energético del alimento (VEA)
VEA = % de proteínas X 4 + % de grasa X 9 + % de carbohidratos X 4 = Kcal /100 g
de muestra.
6. BIBLIOGRAFIA
A.O.A.C. 1980. Association of Official Agricultural Chemists. Official Methods of
Analysis. Washington, D.C.

62

63
PARTE III
ANALISIS FISICOQUÍMICO DE LECHES Y PRODUCTOS
LACTEOS

64

65
PRACTICA No. 13
I. ANALISIS FISICOQUIMICO DE LECHES
El examen de la leche incluye diversos análisis físico-químicos y microbiológicos, al llegar
está a la factoría a granel o en recipientes, normalmente se inspecciona para determinar su
calidad, en base a: olor, temperatura, densidad, contenido en grasas, acidez y por la prueba
de la resazurina. Las muestras con los niveles más bajos de extracto seco magro también se
examinan por el agua añadida mediante el ensayo del punto de congelación. Después del
proceso de pasteurización, se realiza la prueba de la fosfatasa.
A) PRUEBA DEL ALCOHOL
1. OBJETIVO
Determinar la estabilidad de la leche al calor
2. INTRODUCCIÓN
La prueba del alcohol en la leche es un método práctico para evaluar la estabilidad
de la leche al calor. Este método se basa en el hecho de que el alcohol afecta las
proteínas de la leche deshidratándolas y desnaturalizándolos. Las leches normales
son estables al alcohol y al calor, sin embargo, la leche acidificada y con balance
salino incorrecto es inestable en dichas condiciones. Esta se debe efectuar al llegar
la leche a la factoría.
3. MATERIALES, REACTIVOS Y EQUIPO
Leche fresca
Tubos de ensayo
Termómetro
Alcohol etílico al 68%

66
4. METODOLOGÍA
Colocar 10 mL de leche cruda en un tubo de ensayo y agregar 10 mL de alcohol etílico
al 68%. Invertir el tubo tres veces y observar si se forman floculos, la temperatura de la
leche debe ser de 20ºC.
5. RESULTADOS
Reportar si hay formación de floculos lo cual indica una anormalidad de la leche.
Realizar un reporte de acuerdo al apéndice 10
B) DENSIDAD
1. OBJETIVO
Determinar la densidad de la leche de vaca por el método de lactodensímetro de
Quévenne.
2. INTRODUCCIÓN
La leche es una emulsión grasa-agua; consecuentemente su densidad es una función
de la densidad de la grasa y del agua, así como de las proporciones de estos
componentes. La densidad de la grasa es de aproximadamente 0.93 y la de los
sólidos no grasos1.5; cuando el contenido de grasa en la leche aumenta la densidad
disminuye; cuando los sólidos no grasos de la leche aumentan, la densidad también
se incrementa.
Este método se basa en la determinación de la densidad de la leche
utilizando el lactodensímetro de Quevenne, haciendo la lectura a 288 ºK (15 ºC),
aunque también puede efectuarse a otras temperaturas pero corrigiendo la lectura a
288 ºK (15 ªC).
Un tipo difundido de lactodensímetro, es el Quevenne, cuyo vástago con
escala graduada comprende valores entre 15 y 40 que corresponden a las milésimas
de densidad por encima de la unidad, es decir que el número 32 del lactodensímetro
indica la densidad 1,032. El instrumento esta calibrado a 15º C y a esa temperatura;

67
por lo tanto; el número leído representa la densidad de la leche. A temperaturas
diferentes, debe recurrirse a tablas especiales de corrección. Cuando la discrepancia
con respecto a los 15 ºC no es mucha (no más de ± 5ºC), se puede obtener la
corrección sumando o restando 0,0002 a la densidad hallada, o bien 0,2 a los grados
leídos en el lactodensímetro, por cada grado de temperatura respectivamente
superior o inferior a 15º.
La densidad de la leche es bastante variable. La densidad de la leche fresca
entera es de aproximadamente 1.030 Kg/m3 si la materia grasa está completamente
liquida. Durante la refrigeración, la grasa solidificada y la densidad de la leche
aumenta aproximadamente 1.2 Kg / m3 a 10ºC. La densidad de la leche se eleva
cuando incrementa el contenido en extracto seco magro, pero disminuye conforme
aumenta el contenido en materia grasa.
3. MATERIALES, REACTIVOS Y EQUIPO
Leche fresca o pasteurizada
Lactodensímetro de Quévenne con termómetro integrado, escala ºC.
Probeta graduada de 500 mL
Dos vasos pp. de 500 mL
4. METODOLOGÍA
Homogenizar la muestra, haciendo pasar de un vaso a otro varias veces, y verterla
en la probeta hasta la marca, evitando la formación de espuma, introducir
cuidadosamente el lactodensímetro, dejándolo que flote libremente sin tocar las
paredes hasta que de un nivel constante, leer en la parte de menisco, anotando la
temperatura del termómetro interno del lactodensímetro. Si la leche es fresca, se
debe precalentar a 40ºC y después enfriarla a 20ºC. Esto es necesario, ya que la
densidad cambia durante las primeras horas (fenómeno de Recknagel). Las leches
viejas no necesitan esta preparación.
Hacer la corrección al valor de la densidad, aumentando o restando el valor
de la leche aparente, el valor dado de multiplicar la diferencia que existe entre la
temperatura dada y la normal (15ºC), es decir los grados centígrados por arriba o

68
por debajo de dicha temperatura por el factor de corrección .0002 y después
sumando o restando dicha cantidad.
Por ejemplo: si la lectura en la escala indica 32 y la temperatura fue de 16 ºC, la
densidad en este casa será: 1.032 + 0.0002 = 1.0322
Si la lectura en la escala indica 31 y la temperatura de 10 ºC, la densidad sería:
1.031 – (0.0002 X 5) = 1.031 – 0.001 = 1.030
5. RESULTADOS
Determinar la densidad de la leche
Realizar un reporte como se indica en el apéndice 10
C) ACIDEZ
1. OBJETIVO
Determinar el grado de acidez de la leche de vaca por el método volumétrico
2. INTRODUCCIÓN
La leche de vaca presenta un pH comprendido entre 6.6 y 6.8, siendo la acidez total
debida a una suma de tres reacciones fundamentales y a una cuarta de carácter eventual.
Las cuales son las siguientes:
1. Acidez debido a su contenido de caseína.
2. Acidez debida a las sustancias minerales y a la presencia de ácidos orgánicos.
3. Reacciones secundarias debidas a los fosfatos presentes en la leche.
4. “Acidez desarrollada”, debida al ácido láctico y a otros ácidos procedentes de la
degradación microbiana de la lactosa en las leches en proceso de alteración. Las
tres primeras representan la “acidez natural” de la leche. La cuarta puede existir
debido a condiciones higiénico-sanitarias no adecuadas.
En general, la determinación de la acidez de la leche es una medida indirecta de su
calidad sanitaria. Este análisis es aplicado de forma habitual a la leche cruda, así como

69
también a la leche tratada térmicamente. El primer caso, reviste particular importancia
económica, puesto que la tendencia a nivel mundial es fijar el precio de la compra de
leche a los productores por su calidad, valorando no solo el volumen o masa de leche,
sino también la calidad fisicoquímica y sanitaria de la misma.
La acidez se mide con base a una titulación alcalimétrica con hidróxido de sodio 0.1
N utilizando fenolftaleína como indicador o, en su caso, utilizando un potenciómetro
para detectar el pH de 8.3 que corresponde al fin de la titulación. La leche generalmente
tiene una acidez de 1.3 a 1.7 g/L expresada en acido láctico.
La acidez normal de la leche se debe principalmente a su contenido de caseína
(0.05- 0.08%) y de fosfatos. También contribuyen a la acidez el dióxido de carbono
(0.01 -0.02 %), los citratos (0.01%) y la albumina (menos de 0.001%).
3. MATERIALES, EQUIPOS Y REACTIVOS Leche fresca o pasteurizada
Pipeta graduada de 10 mL
Pipeta volumétrica de 20 mL
Matraz erlenmeyer de 125 mL
Bureta de 25 mL
NaOH 0.1 N
Fenolftaleína al 1%
4. METODOLOGÍA
Medir 20 mL de muestra y colocarlos en un matraz y diluir con dos veces su
volumen con agua libre de CO2. Añadir 2-3 gotas de fenolftaleína y titular con
NaOH 0.1 N hasta la aparición de un color rosa persistente cuando menos por un
minuto. Se recomienda hacer esto por lo menos dos veces.
Calculos:
Acidez g/L (ácido láctico) = (V x N x 90 ) / M
Donde:
V = mL de NaOH 0.1 N gastados en la titulación
N = Normalidad de la solución de NaOH
M = Volumen de la muestra

70
5. RESULTADOS
Reportar la acidez en g/L o como porcentaje de ácido láctico
Realizar un reporte como se indica en el apéndice 10
D) DETERMINACIÓN DE CLORUROS
1. OBJETIVO
Determinar el contenido de cloruros que se encuentran en la leche de vaca
2. INTRODUCCIÓN
Los tejidos de la ubre de la vaca permiten que el cloruro de sodio del plasma
sanguíneo pase a la leche secretada. Un descenso o aumento de la calidad normal de
cloruros en la leche fresca indica una condición anormal de la ubre (mastitis); pero
también puede indicar que la leche procede de una fuente pobre o bien que ésta ha
sido adulterada con un posible reconstituyente. La cantidad normal de cloruros
presentes en leche es de 0.85 a 1.25 g/L expresados en cloruros.
El fundamento de método se basa en que a una muestra acidificada de leche
se le añade un exceso de AgNO3, que con los cloruros forma AgCl. El exceso de
AgNO3 se valora con una solución de sulfocianuro de amonio usando como
indicador sulfato férrico amónico, el cual toma un color pardo rojizo con el
sulfocianuro en exceso.
3. MATERIALES, EQUIPO Y REACTIVOS Leche cruda
Matraz erlenmeyer de 250 mL
Pipetas volumétricas de 10 mL
Pipetas graduadas de 10 mL
Bureta de 25 mL

71
Placa de calentamiento
Ácido nítrico concentrado
Sulfocianuro de amonio 0.1 N (1.0 mL es equivalente a 1.0 mL de AgNO3 0.1 N)
Sulfato férrico-amónico, solución saturada, aproximadamente al 14%, con 5% de
HNO3.
4. METODOLOGÍA
Colocar 10 mL de leche en el matraz erlenmeyer, agregar 10 mL de AgNO3 0.1 N t
10 mL de HNO3 concentrado (libre de halógenos) y calentar hasta ebullición
durante 3-5 minutos, dejar de calentar cuando su contenido aparezca cristalino y de
color amarillo; agregar 100 mL de agua y 2 mL de la solución de sulfato férrico-
amónico. Titular con la solución 0.1 N de sulfocianuro de amonio el exceso de
AgNO3 hasta obtener un cambio de color pardo rojizo que indica el final de la
reacción.
Cálculos:
Cloruros (Cl) g/L = [(V1 x N1) – (V2 x N2)] (3.545)
Donde:
V1 = mL de AgNO3 0.1
N1 = Normalidad de la solución de AgNO3
V2 = mL de NH4SCN 0.1 N
N2 = Normalidad de la solución de NH4SCN
5. RESULTADOS
Reportar el contenido de cloruros en la leche.
Realizar un reporte de acuerdo al apéndice 10

72
E) GRADO REFRACTOMÉTRICO
1. OBJETIVO
Determinar el grado refractométrico de la leche por el método del sulfato de
cobre
2. INTRODUCCIÓN
El grado refractométrico del suero de la leche se utiliza para determinar la presencia
de agua agregada a la misma. Si se ha añadido agua la porción de las sales solubles
de la leche disminuirá en el suero por lo que el grado refractométrico disminuirá
también Las leches que dan lecturas por debajo de 1.337 a 20 ºC, se consideran
añadidas de agua.
Se usa una solución de sulfato de cobre para desproteínizar y separar el suero, el
cual una vez filtrado y ajustado a 20 ºC, se le determina el grado refractemétrico,
previamente a este debe ajustarse el refractómetro de inmersión con agua destilada.
3. MATERIALES, EQUIPO Y REACTIVOS Matraz erlenmeyer de 250 mL
Pipetas volumétricas de 20 mL
Pipetas volumétricas de 5 mL
Termómetro
Embudo
Papel filtro
Refractómetro de ABBE
Solución de sulfato de cobre (pesar 72.5 g de CuSO4.5H2O y diluir a 1 L con agua
destilada)
4. METODOLOGÍA Colocar en un matraz erlenmeyer 20 mL de leche, añadir 5 mL de sulfato de cobre
pentahidratado, agitar y filtrar. Ajustar la temperatura a 20 ºC del filtrado y hacer la
lectura del mismo en el refractómetro.

73
5. RESULTADOS
Reportar el grado refractómétrico de la leche
Realizar un reporte de acuerdo al apéndice 10
F) PUNTO DE CONGELACIÓN
1. OBJETIVO
Determinar el punto de congelación con el crióscopo de Hotvet en leche
2. INTRODUCCIÓN
A través de los años, la calidad de la leche se ha visto disminuida debida a la
adición de agua. La leche se congela por debajo de los 0ºC ya que las sustancias
disueltas bajan el punto de congelación de los disolventes puros. El punto de
congelación de la leche, es una de sus propiedades más constantes, y por lo tanto,
efectuando su determinación se puede detectar la adición de agua. Cuando a la leche
se le añade agua, hay una disminución de l a cantidad de lactosa y de las sales
disueltas, ocasionando que el punto de congelación se aproxime al del agua.
El principio en el cuál se basa la técnica de la crioscopía es la Ley de Raoult,
que señala tanto el descenso crióscopico, como el ascenso ebulloscopico, están
determinados por la concentración molecular de las sustancias disueltas. Al enfriar
una solución diluida se alcanza eventualmente una temperatura, en la cual, es
disolvente sólido (soluto) comienza a separarse. La temperatura a la cual comienza
la separación se conoce como punto de congelación de la solución. Las lecturas
normales deben ser de -530 a -560 ºC, cuando la lectura es mayor a -530 ºC se
presume de la adición de agua.
3. MATERIALES, EQUIPO Y REACTIVOS Ácido sulfúrico concentrado
Éter etílico

74
Alcohol etílico
Agua destilada
Solución A (sacarosa al 7%)
Solución B (Sacarosa al 10%)
Matraz kitasato
Termómetro estándar de 1 – 2ºC subdividido en decimas y centésimas, lo que hace
posible las lecturas de 0.001 grados
Crióscopo de Hotvet
Bomba de vacío
4. METODOLOGÍA
Colocar en el tubo de congelación una pequeña cantidad de alcohol etílico, verter en
el frasco de Dewar 400 mL de éter etílico a menos de -2ºC, conectar la bomba de
vacío a flujo moderado conforme se juzga por la agitación del ácido sulfúrico en el
matraz kitasato, colocar de 30-35 mL de agua a -10ºC en el tubo de ensayo a
continuación colocar el termómetro y el agitador dentro. Mover el agitador de arriba
abajo una vez cada segundo o dos, mantener el flujo de aire y cuando el refrigerante
alcanza -2.5 ºC empieza a congelar el agua subiendo rápidamente el mercurio.
Golpear lentamente el termómetro hasta que el nivel del mercurio permanezca
estable, efectuar al lectura. El tubo se ensayo se enjuaga con la solución en estudio
antes de llenarlo nuevamente con 30-35 mL a menos de -10ºC, repetir la lectura con
otra alícuota. Repetir todo el procedimiento con la solución A de sacarosa, solución
B y leche. Al repetir las determinaciones debe comprobarse que el volumen del éter
sea de 400 mL. Generalmente se requiere de añadir de 10-14 mL en cada
determinación.
Cálculos:
A = [(T – T”) / T] x 100
Donde:
A = Por ciento en peso agua añadida
T = 0.545 que es el punto de congelación promedio en ºC en leches normales
T” = Punto de congelación de la muestra.

75
5. RESULTADOS
Reportar el por ciento en peso de agua añadida de la leche
Realizar un reporte de acuerdo al apéndice 10
G. SÓLIDOS TOTALES
1. OBJETIVO
Determinar la cantidad de sólidos totales
2. INTRODUCCIÓN
Los sólidos de la leche incluyen todos los compuestos presentes en ésta, excepto el
agua. Sus límites son estrechos y característicos y su determinación es útil para
revelar algunas adulteraciones. El valor normal es de 115 a 125 g/L. este método
consiste en que a un volumen definido de leche se le evapora el agua por
calentamiento, primero con vapor de agua y después en una estufa a temperatura de
98-100 ºC.
3. MATERIALES, EQUIPOS Y REACTIVOS Leche de vaca
Pipetas volumétricas de 2 mL
Cápsulas de níquel o porcelana de 5 cm de diámetro
Grasa
Baño de vapor
Estufa con temperatura controlada
Desecador
Balanza analítica

76
4. METODOLOGÍA
Medir 2 mL de leche y colocarlos en la cápsula de níquel o porcelana, previamente
puestos a peso constante, con una cama de grasa, colocar la cápsula sobre un baño
de agua a sequedad. Transferir la cápsula a la estufa y secar durante 3 horas a 98-
100 ºC. Enfriar en el desecador y pesar el residuo.
Cálculos:
Sólidos totales (g/L) = [(P2 – P1) x 100] / M
Donde:
P2 = Peso de la cápsula con el residuo seco
P1 = Peso de la cápsula con la cama de grasa
M = Volumen de la muestra en mL.
6. RESULTADOS
Reportar el contenido de sólidos totales de la muestra.
Realizar un reporte de acuerdo al apéndice 10
H) DETERMINACIÓN DE GRASA (METODO DE GERBER)
1. OBJETIVO
Determinar el contenido de grasa en leche por el método Gerber
2. INTRODUCCIÓN
La grasa es el constituyente más importante de la leche y es el que fija el precio de
la leche en el comercio; a él se recurre para el control de materias primas para la
fabricación de mantequilla, queso y otros productos lácteos para verificar el
rendimiento de las vacas y para el control del descremado premeditado de la leche.
Este método se basa en la disolución de todos los componentes de la leche, excepto
la grasa, en ácido sulfúrico. Emplea alcohol iso-amílico para ayudar a romper la
emulsión de la leche y evitar que se queme la capa de grasa. El alcohol iso-amílico

77
reacciona con el ácido sulfúrico formando un éster que es completamente soluble en
dicho ácido.
3. MATERIALES, EQUIPOS Y REACTIVOS Leche de vaca
Butirometro de gerber con una sola abertura, graduado de modo que cada marca
corresponda al 1% de grasa de leche (Butirométro de 1-8%)
Centrifuga para butirómetro Gerber
Tapones para butirómetro
Pipetas volumétricas de 11 mL
Ácido sulfúrico al 90%
Alcohol iso-amílico
4. METODOLOGÍA
Colocar 10 mL de ácido sulfúrico en el butirómetro evitando bañar las paredes
internas del cuello; añadir lentamente resbalando por las paredes y sin mezclar, 11
mL de leche de modo que se forme un estrato de leche sobre el ácido,
inmediatamente agregar 1 mL de alcohol iso-amílico. Cerrar con el tapón y agitar
enérgicamente con el que se produce un fuerte calentamiento y la disolución en
ácido de los albuminoides de la leche; colocar el butirómetro en un baño de agua
caliente y mantenerlo a 65-70 ºC por 10 minutos. Centrifugar por 2 minutos a 1100
rpm y colocarlo por último en el baño de agua caliente durante 4-5 minutos. Leer el
espesor de la capa de grasa acumulada en la parte superior; como el tapón queda
hacia abajo, éste debe movilizarse cuidadosamente hasta colocar los límites de la
capa de grasa dentro de la escala, la cual expresa directamente la cantidad en
porcentaje de la grasa contenida en la leche.
5. RESULTADOS
Reportar el porcentaje de grasa en leche
Realizar un reporte de acuerdo al apéndice 10

78
I) DETERMINACIÓN DE GRASA (METODO DE ROESE-GOTTLIEB)
1. OBJETIVO
Determinar el contenido de grasa de grasa en leche por el método de Rose-
Gottlieb.
2. INTRODUCCIÓN
Este método fue propuesto por la Federación Internacional de Lacticinios; la
Organización Internacional de Estandarización y las Publicaciones del A.O.A.C.,
informe que fue publicado como Norma Internacional por FAO/WHO en el Código
de Principios concernientes con la leche, productos de la leche y las Normas
Asociadas.
Este método consiste en someter a la leche a un procedimiento de extracción de la
grasa utilizando como disolvente orgánico una mezcla de alcohol etílico, éter etílico
y éter de petróleo. Posteriormente se evaporan los disolventes y se pesa el residuo
calculándolo como grasa por litro en peso. Se utiliza como método de arbitraje
especialmente enlas leches homogeneizadas y pasteurizadas.
3. MATERIALES, EQUIPO Y REACTIVOS Leche de vaca
Tubos de extracción de roehring o mojonnier
Vasos o cápsulas de vidrio o níquel
Probetas
Balanza analítica
Estufa con control de temperatura
Desecador
Baño de agua con sistema eléctrico para control de temperatura o placa de
calentamiento
Hidróxido de amonio (NH4OH)
Éter de petróleo

79
Éter etílico libre de peróxido
Alcohol etílico al 95 %.
4. METODOLOGÍA
En un tubo de Roehring o Mojonnier colocar 10 mL de la muestra y agregar 1.25
mL de NH4OH (si la muestra esta agria emplear 2 mL). Mezclar perfectamente,
agregar 10 mL de alcohol etílico y volver a mezclar; adicionar 25 mL de éter etílico,
tapar el tubo con tapón de corcho, hule sintético o vidrio esmerilado y agitar
vigorosamente por un minuto; agregar 25 mL de éter de petróleo y repetir la
agitación vigorosa; centrifugar el tubo, si es de Mojonnier, a una velocidad de 600
rpm hasta que se aclare la porción superior del líquido o bien se le deja en reposo, si
el tubo es de Roehring, hasta que se obtenga el mismo resultado. Si se decanta la
solución etérea a un vaso, o cápsula de metal o vidrio lavando el labio y el tapón del
tubo con una mezcla de los dos éteres. Estos lavados se agregan al vaso o cápsula;
se repite la extracción del líquido sobrante en el tubo dos veces más, utilizando 15
mL de cada disolvente en cada ocasión y agregando agua si fuese necesario, pero
sin hacer los lavados con la mezcla de disolventes. Evaporar los disolventes de la
cápsula en placa o baño de vapor, a una temperatura apropiada que permita la
eficiente evaporación; desecar la grasa extraída a la temperatura del agua a
ebullición cuando se utiliza un baño y después en la estufa a 102 ± 2 ºC o al vacío a
70-75 ºC con presión menor de 50 mm de Hg. Pesar los vasos o las cápsulas ya frías
utilizando un testigo preparado en forma similar. Extraer la grasa del vaso o cápsula
ya pesada utilizando éter de petróleo caliente; volver a secar la cápsula o vaso, como
se indicó antes y pesar después cuando esté fría.
La diferencia, en peso multiplicado por 100 indica la cantidad por litro de grasa en
la muestra. En todos los casos es necesaria una corrección para conocer el peso
exacto de la grasa, lo que se logra restando el peso obtenido en el testigo. Cuando se
emplean los mismos reactivos si el testigo es mayor de 0.5 mg purificar o
reemplazar estos reactivos. Las diferencias en las determinaciones por duplicado

80
practicadas por el mismo analista debe ser más o menos variables en 0.03 g de
grasa/1000 mL de producto.
5. RESULTADOS
Reportar el contenido de grasa en leche
Realizar un reporte de acuerdo al apéndice 10
J) DETECCIÓN DE FOSFATASA EN LECHE, CREMA Y
MANTEQUILLA
1. OBJETIVO
Determinar la fosfatasa residual en leche pasteurizada
2. INTRODUCCIÓN
La fosfatasa es una enzima normalmente presente en la leche cruda. En las
condiciones ordinarias de pasteurización (lenta, rápida o ultra-rápida) la enzima se
inactiva. Se ha demostrado que esta enzima es más difícil de destruir que la mayoría
de los organismos patogénicos termo-resistentes que pudieran estar presentes en la
leche, como por ejemplo el bacilo tuberculoso. La prueba es de gran utilidad para
decidir si la leche ha sido o no pasteurizada, si la leche pasteurizada se ha mezclado
con leche cruda, o incluso si la pasteurización ha sido deficiente.
Este método se basa en incubar la leche con fenilfosfato en solución
reguladora de hidróxido de bario. Si la fosfatasa activa esta presente, el fenilfosfato
se hidroliza y forma fenol.
C6H5OPO3H2 + H2O → C6H5OH + H3PO4
En la leche que ha sido pasteurizada eficientemente, la fosfatasa se destruye
y no hay hidrólisis. El fenol formado se determina colorimétricamente haciéndolo
reaccionar con 2,6-Dibromoquininacloimida (B.C.Q:) obteniéndose un color azul.
Esta es la forma estable del reactivo cualitativo para fosfatasa de Scharer. Al añadir
estos reactivos a la muestra dan una reacción de color la cual puede ser comparada

81
con las pruebas control y en la tabla de colores localizando el color más cercano o
igual por simple inspección visual, lo cual es de gran valor en la supervisión
ambulante de plantas pasteurizadoras de leche.
3. MATERIALES, REACTIVOS Y EQUIPO
Leche pasteurizada
Balanza analítica
Estufa para incubar
Cronómetro
Tubos de ensayo
Pipetas de capacidad necesarias
LACTO – ZIMA I (Fenilfosfato de sodio y buffer alcalino)
LACTO – ZIMA II (Reactivo desarrollador de color)
4. METODOLOGÍA
Para la realización de esta prueba se utilizan dos tubos de ensayo, uno se marca
como problema y otro como control; se les añade a cada uno 10 mL de agua
destilada a 37 – 39ºC y 250 mg de polvo de LACTO – ZIMA I y se mezcla hasta
que se disuelva. Al tubo problema se le añade 1 mL de leche por analizar y se
mezcla. Al tubo control, se procede de la misma manera, pero con leche caliente en
la cual la fosfatasa ha sido previamente destruida por calentamiento a 85 ºC. Pa
tener mayor seguridad es necesario incubar la mezcla de ambos tubos en una estufa
a 45 ºC, durante 10 minutos. Agregar 250 mg de polvo de LACTO – ZIMA II a
ambos tubos, se dejan en reposo durante 10 minutos y se agitan, a los 5 minutos
comparar los colores de los dos tubos con la tabla de colores (está tabla de colores
es proporcionada por el fabricante de los reactivos de LACTO – ZIMA I y II), la
cual facilita la interpretación en forma esquemática. La intensidad del color varía
con la leche analizada. Cuando se obtiene un tinte gris o café rojizo, la prueba de la
fosfatasa en negativa. Cuando el tinte es ligeramente azul, la reacción es débilmente
positiva y cuando el tinte es azul intenso, la reacción es fuertemente positiva.

82
La técnica de la prueba de la fosfatasa en crema es la misma que en la leche. Para
aplicarla a la mantequilla, se funden 10 g en baño de agua a 40 ºC y se centrifugan;
la capa acuosa se separa y se trabaja como se describe para leche. Las pequeñas
cantidades de fenol interfieren en la prueba, por lo que se requiere usar una pipeta
deferente por cada prueba y, no usar plástico ni tapones de hule.
5. RESULTADOS
Reportar si la prueba es positiva o negativa
Realizar un reporte de acuerdo al apéndice 10
K) LACTOSA
1. OBJETIVO
Determinación del contenido de lactosa en leche
2. INTRODUCCIÓN
La lactosa es el principal carbohidrato de la leche y está considerado por algunos
como el único; sin embargo, también se encuentran pequeñas cantidades de glucosa,
galactosa, sacarosa, cerebrósidos y algunos aminoazúcares dereivados de la
hexosamina. A pesar de que estos azúcares están en concentraciones muy bajas,
pueden ejercer una influencia muy importante en la estabilidad de la leche, sobre
todo en aquella que ha sido sujeta a tratamientos térmicos. La lactosa tiene
aproximadamente 15% de la dulzura de la sacarosa, y contribuye, junto con las
sales, al sabor global de la leche. Su determinación se efectúa con la finalidad de
descubrir si la leche ha sido adulterada con agua, o si ésta proviene de vacas
enfermas de mastitis.
Este método se fundamenta en que la muestra primeramente se defeca para
precipitar las proteínas utilizando soluciones de acetato de plomo y sulfato de sodio.
Se filtra, y en el filtrado se determina la lactosa aprovechando su propiedad de ser

83
un azúcar reductor directo que reduce el cobre de sus sales alcalinas, mediante una
valoración volumétrica, según el método de Lane y Eynon.
3. MATERIALES, REACTIVOS Y EQUIPO
Lana de vidrio
Matraces volumétricos de 100 mL
Matraces erlenmeyer de 125 mL
Matraces erlenmeyer de 300 mL
Pipetas volumétricas de 10 mL
Pipetas volumétricas de 5 mL
Pipetas tipo Mohr de 10 mL graduadas en 0.1 mL
Bureta de 25 mL graduadas en 0.1 mL
Placa de calentamiento
Solución acuosa saturada de sub-acetato de plomo
Solución ascuosa saturada de sulfato de sodio
Ácido acético glacial
Solución acuosa de azul de metileno al 0.2%
Solución acuosa de KI al 20%
Solución de tiosulfato de sodio 0.1N.
Solución de Fehling A: Disolver 34.639 g de CuSO4.5H2O en agu destilada, y diluir a
500 mL; filtrar a tavesde lana de vidrio. Ajustar la solución, determinando el contenido
de cobre en una alicota con tiosulfato de sodio 0.1 N y KI al 20% hasta obtener 440.9
mg de cobre por cada 25 mL.
Solución de Fehling B: Disolver 173 g de tartrato de sodio y potacio y 50 g de NaOH
en agua y diluir a 500 mL; dejar reposar 2 días y filtrar a través de lana de vidrio.
Solución patrón de lactosa: Disolver 10 g de lactosa anhidra y pura y diluir a 1 L
con solución acuosa al 0.2% de ácido benzoico.

84
Titulación de la solución de Fehling: colocar 5 mL de la solución Fehling A y 5 mL
de la solución Fehling B en un matraz erlenmeyer de 300 mL y agregar 50 mL de agua,
calentar en una placa de calentamiento a ebullición y agregar poco a poco, con una
bureta, solución patrón de lactosa hasta la casi reducción total del cobre. Añadir 1 mL
de azul de metileno y continuar la titulación hasta la desaparición del color azul.
Calcular los mg de lactosa que se necesitan para titular la solución de Fehling A y B.
Este valor corresponde al factor (F) del reactivo.
4. METODOLOGÍA
Defecación de la leche: Colocar 10 mL de leche en un matraz volumétrico de 100
mL con 25 mL de agua. Añadir 6 mL de solución saturada de sub-acetato de plomo,
10 mL de sulfato de sodio y 1 mL de ácido acético glacial. Dejar reposar media
hora. Diluir a la marca con agua, filtrar, y el filtrado colocarlo en una bureta.
Determinación de la lactosa: colocar 5 mL de la solución Fehling A y 5 mL de la
solución Fehling B en un matraz erlenmeyer de 300 mL y agregar 50 mL de agua,
calentar en una placa de calentamiento a ebullición y agregar poco a poco, con una
bureta, la solución filtrada hasta la casi reducción total del cobre. Añadir 1 mL de
azul de metileno y continuar la titulación hasta la desaparición del color azul.
Cálculos:
Lactosa g/L = (F / V) x 100
F = Factor del reactivo en mg de lactosa
V = mL del filtrado que se necesitan para titular la solución de Fehling A y B.
5. RESULTADOS
Reportar el contenido de lactosa en la muestra utilizada.
Realizar un reporte de acuerdo al apéndice 10

85
L) DETERMINACIÓN DE PROTEÍNAS (TITULACIÓN DE
FORMALDEHÍDO)
1. OBJETIVO
Determinar el porcentaje de proteínas por el método de titulación de formaldehido
2. INTRODUCCIÓN
La titulación con formaldehido es un método rápido para la determinación de proteínas
en leche fresca. Cuando se agrega formaldehido a la leche neutralizada, se liberan ácidos
libres en proporción a la cantidad de proteínas presentes. Esta acidez producida puede ser
titulada con un álcali.
Cuando se agrega el formaldehido, el grupo amino (-NH2) es reemplazado por el
grupo imino-metileno (-N=CH2) y el grupo carboxilo (-COOH) se encuentra disponible
para ser utilizado. La reacción se describe de la siguiente manera:
R·COOH·NH2 + H·CH=O → H2O + R·COOH·N=CH2
3. MATERIAL, REACTIVOS Y EQUIPO
Matraces erlenmeyer de 125 mL
Pipetas volumétricas de 2 y 10 mL
Pipetas de 1 mL graduadas en décimas
Bureta de 10 mL o microbureta
Solución de oxalato de potasio al 4%
Solución alcohólica de fenolftaleína al 0.5%
Solución de NaOH 0.1 N
Solución de formaldehido G. R. (40%)

86
4. METODOLOGÍA
A 10 mL de muestra agregar 0.4 mL de solución saturada de oxalato de potasio y
0.5 mL de fenolftaleína. Agitar y dejar reposar por dos minutos. Neutralizar con
NaOH 0.1 N hasta obtener un color rosa pálido. Agregar 2 mL de formaldehido.
Dejar reposar dos minutos y titular la nueva acidez producida con el NaOH 0.1 N
hasta obtener el color rosa pálido que indica el punto final. Titular simultáneamente
un blanco con 10 mL de agua destilada, 2 mL de formaldehido y 0.5 mL de
fenolftaleína.
Cálculos:
g/L de proteína = (V1 – V2) X 1.74 X 10
Donde:
V1 = Volumen de NaOH 0.1 N gastado para titular la muestra
V2 = Volumen de NaOH 0.1 N gastado para titular el blanco
1.74 = Factor empírico
5. RESULTADOS
Reportar El contenido de proteínas en leche
Realizar un reporte de acuerdo al apéndice 10

87
II. QUESOS
Preparación de la muestra: Pasar la muestra sin corteza 3 veces a través de un cortador de
alimentos o cortar o desmenuzar la muestra finamente y mezclar muy bien. Con los quesos,
crema cottage y quesos similares, proceder de la siguiente manera: poner de 300-600 g de
muestra, que este a una temperatura aproximada de 15 ºC en el vaso de una licuadora de 1
L y mezclar por unos minutos (2 – 5) para obtener una mezcla homogénea. La temperatura
final debe ser de 25 ºC aproximadamente.
A) GRASA
1. OBJETIVO
Determinación de la grasa butírica en quesos por el método de Gerber-Van Gulik
2. INTRODUCCIÓN
Este método se basa en la digestión parcial de los componentes del queso, excepto
la grasa, butírica en ácido sulfúrico. Emplea alcohol isoamílico para disminuir la
tensión en la interface entre la grasa y la mezcla en reacción (ácido sulfúrico-leche),
lo que facilita el ascenso de los glóbulos pequeños de grasa por centrifugación. El
alcohol isoamílico reacciona con el ácido sulfúrico formando un éster que es
completamente soluble en dicho ácido.
3. MATERIALES, REACTIVOS Y EQUIPO
Ácido sulfúrico concentrado (90%)
Alcohol isoamílico
Embudo con llave de paso para liberar 1 mL.
Embudo con llave de paso para liberar 10 mL
Butirómetro de Gerber-Van Gulik para quesos (graduado hasta 40%)
Tapones para butirómetro
Centrífuga para butirómetro Gerber
Baño de agua con temperatura regulada (65ºC)

88
4. METODOLOGÍA
Pesar directamente sobre el embudo con tapón previamente tararos 3 ± 0.001 g de
muestra. Colocar la muestra dentro del butirómetro y con una pipeta se vierten 10
mL de ácido sulfúrico (90%) de tal manera que cubra todo el queso , tapar el
butirómetro y colocarlo en baño de agua (65ºC, 30 minutos), agitar cuidadosamente
de dos a tres veces durante ese lapso de tiempo para disolver todas las partículas de
queso. Agregar 1 mL de alcohol isoamílico y agitar. Terminar de llenar el
butirómetro con ácido sulfúrico, hasta que el volumen llegue aproximadamente ¾
partes de la columna graduada, taparlo y volverlo a meter al baño de agua por 5
minutos, mezclar y centrifugar a 12000 rpm durante 5 minutos, volver a meter el
butirómetro en el baño durante 10 minutos, hacer la lectura.
5. RESULTADOS
Reportar el porcentaje de grasa obtenido en el queso
Realizar un reporte de acuerdo al apéndice 10
B) ACIDEZ
Se añade agua templada (40 ºC) a 10 g de muestra hasta tener un volumen total de 105 mL.
Se agita la muestra vigorosamente, se filtra y se valora en porciones de 25 mL
(aproximadamente 2.5 g de muestra) con NaOH 0.1 N utilizando fenolftaleína como
indicador. Se calcula la acidez como ácido láctico.
1 mL de NaOH 0.1 N = 0.0009 g de ácido láctico
Para la determinación de pH del queso blando, l os electrodos se pueden introducir
directamente a la muestra. Los quesos duros se deben ablandar previamente mezclándolos
con un volumen mínimo de agua destilada. (VER PRÁCTICA III)

89
C) DETERMINACIÓN DE SAL EN QUESO
Se pesan 2 g de muestra preparada en un matraz erlenmeyer de 250 mL y se añaden 10 mL
de agua y 25 mL de AgNO3 0.05 N. Se calienta la mezcla a 80 ºC, se agita vigorosamente
para dispersar la muestra, se añaden 10 mL de HNO3 concentrado y se digiere la cuajada
por ebullición suave durante 10 minutos. En este punto el cloruro de plata deberá ser
granular, el líquido de color limón claro y la capa de grasa exc enta de sólidos. Se añaden
0.3 g de urea a la disolución caliente, se mezcla, se enfría, y se agrega 1 mL de
nitrobenceno y se mezcla de nuevo. Se añaden 2 mL del indicador de nitrato de plata con
KCNS 0.05 N hasta un tinte naranja persistente durante 15 segundos (VER PRACTICA
IV).
5. BIBLIOGRAFÍA
PEARSON. D; Técnicas de laboratorio para el análisis de alimentos; Acribia, S.A.
Zaragoza (España) 1993.
HART F. L; Análisis moderno de los alimentos; Acribia. Zaragoza (España), 1991.
Association of Official Analytical Chemistry. Official Methods of Analysis
(A.O.A.C.) 1980.
NMX-F-443-1983.ALIMENTOS. Leche fluida. Punto de congelación. Crióscopo
Hotvet. Normas Mexicanas. Dirección General de Normas.
NMX-F-100-1984. ALIMENTOS. LACTEOS. Determinación de la grasa butírica
en quesos.
PARRILLA, C.C., SALDATE, C.O. Y NICOLI, T. M., 1989. Manual de Técnicas y
Procedimientos de Laboratorio para Análisis Microbiológico de Leches; Secretaría
de Salud.

90
PARTE IV
ANÁLISIS FISICOQUÍMICOS DE CARNE Y PRODUCTOS CARNICOS

91

92
PRÁCTICA No. 14
A) DETERMINACIÓN DE FECULA O ALMIDÓN POR HIDRÓLISIS ACIDA
1. OBJETIVO
Determinar en porcentaje de fécula o almidón por hidrólisis ácida en muestras de
embutidos cárnicos
2. INTRODUCCIÓN
Para Determinar la fécula o almidón presente en un embutido, este método incluye dos
pruebas: la prueba cualitativa que se menciona, debe preceder al ensayo cuantitativo, el
cual debe aplicarse en los casos de positividad o cuando se sospeche el empleo de harina
de soya para la cual la prueba de yodo no es aplicable. Mediante el método cuantitativo
para fécula, se detecta la presencia de otros carbohidratos, como glucógeno del hígado u
otras vísceras o aquellos empleados en la manufactura del alimento.
3. MATERIALES, REACTIVOS Y EQUIPO
Materiales para la prueba cualitativa
Capsula de porcelana o vasos de precipitado de 50 mL
Agitador de vidrio
Tijeras o cuchillos
Baño de agua con termostato y termómetro
Mortero
Matraz aforado de 100 mL
Vasos de precipitado de 100 mL
Parrilla eléctrica
Materiales para la prueba cuantitativa
Matraces aforados de: 100, 250, 500 y 1000 mL
Matraces erlenmeyer de 250 y 500 mL
Embudo
Papel filtro

93
Bureta de 50 mL
Columna refrigerante
Pipetas de 10 mL.
Pipetas volumétricas de 5 mL
Balanza analítica
Reactivos para la prueba cualitativa
Solución de Lugol: Disolver 10 g de yoduro de potasio (KI) en 70 mL de agua
destilada, cuando éste completamente disuelto agregar 5 g de yodo sublimado,
disolver y completar el volumen a 100 mL con agua destilada.
Reactivos para la prueba cuantitativa
Solución A: Solución de sulfato de cobre: Disolver 34.639 g de CuSO4.5H2O en
agua destilada y diluir a 500 mL utilizando un matraz volumétrico, filtrar a través de
lana de vidrio o papel filtro.
Solución B: Tartrato de sodio y potasio: Disolver 173 g de tartrado doble de sodio y
potasio (Sal de Rochelle) (KNaC4H4O6.$H2O) y 50 g de NaOH en agua y diluir a
500 mL, dejar reposar dos días y filtrar.
Solución patrón de sacarosa invertida al 1%. Pesar 9.5 g de sacarosa (Q.P.) y
disolver en 50 mL de agua, agregar 5 mL de HCl concentrado y diluir a 100 mL.
Guardar al algunos días a temperatura ambiente (aproximadamente 7 días a 12-
15ºC; 3 días a 20-25ºC o 15 minutos a 67ºC) después de esta inversión, diluir a un
litro (esta solución es estable durante algunos meses en refrigeración.
Solución acuosa de azul de metileno al 0.2%
Solución de NaOH 1:1 m/v
Acido clorhídrico concentrado
Solución alcohólica de fenolftaleína al 1%
Solución diluida de sacarosa Diluir 10 mL de la solución patrón previamente
neutralizada a 100 mL con agua (1 mL = mg de sacarosa)
(*) Titulación de la solución A-B
Medir con pipeta volumétrica 5 mL de la solución A y 5 mL de la solución B en un matraz
erlenmeyer de 500 mL, agregar 100 mL de agua y unas perlas de cristal, calentar en parrilla
eléctrica a ebullición y agregar poco a poco, con bureta, la solución patrón de sacarosa

94
diluida, hasta casi reducción total del cobre. Agregar 1 mL de azul de metileno y continuar
con la titulación hasta la desaparición del color azul (mantener continua la emisión de vapor
para prevenir la reoxidación del cobre o del indicador). Si el gasto es menor de 15 mL o
mayor de 50 mL de la solución de azúcar invertida, hacer la disolución apropiada o
reformulación para que quede dentro de este rango. Calcular los mg de sacarosa que se
necesitan para titular la solución A-B, este valor corresponde al factor (F) del reactivo.
Estos mg se calculan multiplicando los mL de sacarosa invertida requeridos para la
titulación, por los mg/mL de la misma. Este titulo equivale a loa mg totales de azúcar
invertido para reducir el cobre de las alícuotas.
4. METODOLOGÍA
PRUEBA CUALITATIVA
Pesar 5 g de muestra previamente molida y transferirla a una capsula de porcela o vaso de
precipitados de 50 mL, agregar 10 mL de agua destilada caliente o una cantidad suficiente
para que la muestra se disgregue perfectamente. Agregar unas gotas de la solución de lugol
y mezclar con la ayuda de un agitador de vidrio. La aparición de un color azul indica la
presencia de almidón.
PRUEBA CUANTITATIVA
En un matraz erlenmeyer de 500 mL colocar de 1-5 g de muestra molida y agregar 150 mL
de agua y 25 mL de HCl concentrado, colocar el matraz en el refrigerante y reflujar de 60-
90 minutos, enfriar, y agregar unas gotas de fenolftaleína, neutralizar con NaOH 1:1 y
enfriar. Pasar a un matraz volumétrico de 250 mL, llevar a la marca con agua, filtar, colocar
el filtrado en la bureta y proceder a la titulación como se indico anteriormente (*).
Cálculos:
% de almidón= [[ (250 X F) /V] X 100 X 0.9] / PM
Donde:
V = mL gastados para titular la solución A-B
F = Factor del reactivo en g de sacarosa
0.9 = Factor de conversión de glucosa a almidón
PM = Peso de la muestra.

95
Al valor obtenido para fécula se debe restar la cantidad correspondiente a los azucares
reductores totales.
5. RESULTADOS
Reportar el porcentaje de almidón en la muestra
Realizar un reporte de acuerdo al apéndice 10.
B). DETERMINACIÓN DE AZUCARES REDUCTORES TOTALES
1. OBJETIVO
Determinar la cantidad de azucares reductores totales en una muestra de embutidos
cárnicos.
2. INTRODUCCIÓN
Este método se basa en que la muestra primeramente se defeca con la finalidad de
precipitar las proteínas utilizando una solución de acetato de plomo, filtrando e
hidrolizando con ácido clorhídrico a una temperatura de 67ºC para invertir los azucares no
reductores y aprovechar la propiedad que tienen los azúcares reductores de reducir el cobre
de sus sales alcalinas, mediante una valoración volumétrica, según el método de Lane y
Eynon.
3. MATERIALES, REACTIVOS Y EQUIPO
Solución acuosa saturada de acetato de plomo de plomo neutro
Solución acuosa saturada de oxalato de sodio o potasio
Todos los materiales, reactivos y equipos de la práctica para determinar el
porcentaje de fécula por hidrolisis ácida en muestras de embutidos cárnicos.
4. METODOLOGÍA
Pesar de 5-10 g de muestra previamente molida y colocarla en un vaso de precipitado de
400 mL, agregar 100 mL de agua, agitar para suspender todo el material y agregar de 2-10

96
mL de la solución saturada de acetato de plomo neutro, agitar y dejar sedimentar. Agregar
poco a poco solución saturada de oxalato de sodio o potasio hasta la total precipitación del
acetato en exceso, filtrar, recibiendo el filtrado en un matraz volumétrico de 250 mL.
Agregar 10 mL de HCl concentrado al matraz volumétrico que contienen el filtrado y
calentar 15 minutos en baño de agua a 67ºC, enfriar, agregar unas gotas de fenolftaleína,
neutralizar con NaOH 1:1, enfriar, diluir a la marca con agua y proceder a la titulación,
como se indica en el punto de la titulación de la solución A-B (*) de la práctica para
determinar el porcentaje de fécula por hidrolisis ácida en muestras de embutidos cárnicos.
Cálculos:
% de azúcares reductores = [[ (250 X F) /V] X 100] / PM
Donde:
V = mL gastados para titular la solución A-B
F = Factor del reactivo en g de sacarosa
PM = Peso de la muestra.
5. RESULTADOS
Reportar el porcentaje de reductores en la muestra
Realizar un reporte de acuerdo al apéndice 10.
C) DETERMINACIÓN DE FOSFATO (MÉTODO COLORIMÉTRICO)
1. OBJETIVO
Determinar el porcentaje de fosfatos en un producto cárnico
2. INTRODUCCIÓN
Cuando una solución ácida de ortofosfatos es tratada con una solución de ácido molíbdico y
ácido vanádico en medio ácido, se forma un complejo de color amarillo-naranja de ácido
vanadomolibdifosfórico (H3PO4. VO3. 11MoO3.n H2O) cuya absorción se lee en un
espectrofotómetro a 420-480 nm (Reacción de Misson`s)

97
3. MATERIALES, REACTIVOS Y EQUIPO
Capsulas o crisoles de porcelana
Matraces volumétricos de 100, 250 y 1000 mL
Pipetas volumétricas de 5, 10 y 25 mL
Vasos de precipitados de 500 mL
Agitador de vidrio
Bureta de 50 mL
Mechero de Bunsen.
Triángulo de porcelana.
Pinzas para crisol.
Embudo.
Soporte universal y anillo.
Papel filtro.
Papel indicador de pH.
Balanza analítica
Espectrofotómetro
Mufla
Hidróxido de amonio
Acido nítrico 1:2 v/v
Acido clorhídrico 5 N
Mezcla de vanadio-molibdato: Disolver 20 g de molibdato de amonio en 400 mL de agua a
la temperatura de 50°C y enfriar. Disolver por separado un g de vanadato de amonio en 300
ml de agua hirviendo, enfriar y agregar gradualmente 140 mL de ácido nítrico concentrado
agitando ocasionalmente, en este momento agregar gradualmente la solución de molibdato
de amonio, previamente preparada y completar el volumen con agua a un litro.
Solución de Nitrato de Magnesio: Disolver 950 g de Mg (N03)2 · 6H2O libre de fósforo, en
agua y completar el volumen con agua a un litro.

98
Solución Patrón de Fósforo: Preparar una solución que contenga 3.834 g de KH2P04 en un
litro de agua. Colocar una alícuota de 25 mL en un matraz volumétrico de 250 mL y
completar el volumen con agua. Un ml de esta solución patrón equivale a 0.2 mg de P2O5.
PREPARACIÓN DE LA CURVA PATRÓN
Poner alícuotas de 0.0, 2.5, 5, 10, 20, 30, 40 y 50 mL de la solución patrón de fósforo, en
matraces aforados de 100 mL y diluir con agua a un volumen entre 50 y 60 mL. Agregar a
cada matraz unas gotas de NH4OH (0.88 mL) y con HNO3 1:2 llevarlos a medio ácido.
Agregar 25 mL de reactivo de vanadio-molibdato, completar el volumen con agua a 100
mL y mezclar. Dejar reposar durante 10 minutos. Transferir las soluciones a las celdas del
espectrofotómetro y medir la absorbancia a 470 nanómetros. Trazar en papel
semilogarítmico de un ciclo, una curva de absorbancia contra concentración de P2O5.
4. METODOLOGÍA
Tomar una muestra de 2 a 2.5 g en una cápsula o crisol de porcelana, humedecerla con unos
mililitros de solución de Mg (NO3)2 · 6H2O y evaporar, incinerar a una temperatura de
600°C, disolver las cenizas con 10 mL de HCl 5 N, calentar hasta ebullición, enfriar y
transferir a un matraz volumétrico de 100 mL con ayuda de unos mililitros de agua. Si
todas las cenizas no se solubilizaron, filtrar para pasar al matraz volumétrico, neutralizar
agregando NH4OH gota a gota (pH = 7.2)
El volumen de la solución después de la neutralización debe ser entre 50 y 60 mL,
acidificar ligeramente con HNO3 1:2, utilizando el papel indicador de pH, agregar 25 ml del
reactivo de molibdato de vanadio, completar el volumen a 100 mL con agua, mezclar y
dejar reposar 10 minutos.
Transferir la solución problema a las celdas del espectrofotómetro y medir la
absorbancia a 470 nanómetros y comparar con la curva patrón.
Cálculos:
% P2O5 = (L X 100) / m

99
Donde:
L = Lectura del problema en mg de P2O5 al comparar con la curva.
m = Masa de la muestra en gramos.
5. RESULTADOS
Reportar el porcentaje de fosfatos en la muestra
Realizar un reporte de acuerdo al apéndice 10
D) DETERMINACIÓN DE NITRATOS (METODO COLORIMETRICO)
1. OBJETIVO
Determinar las ppm de nitratos en embutidos.
2. INTRODUCCIÓN
Este método colorimétrico se fundamenta en la formación de un compuesto coloreado del
ácido nitrofenildisulfónico a partir del ácido fenoldisulfónico por cualquier nitrato presente
y en medio alcalino.
3. MATERIALES, REACTIVOS Y EQUIPO
Fenol
Acido sulfúrico concentrado
Sulfato de potasio y aluminio dodecahidratado
Hidróxido de amonio
Cloruro de bario
Nitrato de sodio seco
Solución saturada de acetato de plomo
Solución saturada de sulfato de plata libre de nitratos
Solución de fenoldisulfónico: Calentar en baño maría 6 g de fenol con 37 mL de ácido
sulfúrico concentrado, enfriar y añadir 3 mL de agua.

100
Crema de alúmina: Preparar una solución saturada de sulfato de potasio y aluminio
dodecahidratado. Añadir hidróxido de amonio con constante agitación hasta que la solución
sea alcalina al papel tornasol. Dejar que se asiente el precipitado y lavar por decantación
con agua hasta que el agua de lavado de ligeramente la reacción para sulfatos con cloruro
de bario. Tirar el exceso de agua y guardar la crema residual en un frasco cerrado.
Solución patrón de nitrato de sodio: Disolver 1 g de nitrato de sodio seco en agua, pasarlo
a un matraz volumétrico de 1000 ml y completar el volumen con agua. Pasar una alícuota
de 10 ml de esta solución a un vaso de precipitados de 50 mL y evaporarla en baño maría
de 70 a 80°C a sequedad; añadir 2 mL de la misma solución y mezclar rápidamente por
medio de un agitador de vidrio. Calentar un minuto en baño maría de 70 a 80°C, pasar a un
matraz volumétrico de 100 mL y completar el volumen con agua. Esta solución tiene una
concentración de 0.1 mg de nitrato de sodio por mililitro. Con esta concentración preparar
una serie de 20 tubos.
Tomar en un vaso de precipitados de 50 ml una alícuota de 10 ml de la solución original (1
g de nitrato de sodio en 1000 ml de agua) y evaporarla a sequedad en baño maría de 70 a
80°C, añadir 2 mL de la misma solución y mezclar rápidamente por medio de un agitador
de vidrio. Calentar durante un minuto en el baño maría de 70 a 80°C, pasar a un matraz
volumétrico de 1000 mL y completar el volumen con agua. Esta solución tiene una
concentración de 0.01 mg de nitrato de sodio por mililitro. Con esta concentración preparar
una segunda serie de 20 tubos.
Vasos de precipitados de 50, 100 y 250 mL
Matraces volumétricos de 100 y 1000 mL
Pipetas volumétricas 1, 2, 5, 10 y 25 mL
Pipetas graduadas en décimas de ml de 5, 10 y 20 mL
Agitar de vidrio
Tubos de Nessler de 50 ml o tubos de ensayo de 60 a 70 mL
Embudo

101
Papel filtro
Papel tornasol
Frascos para guardar los reactivos
Papel milimétrico para graficar
Baño maría o de vapor
Soporte universal y anillo
Balanza analítica con ± 0.1 mg de sensibilidad.
Espectrofotómetro
PREPARACIÓN DE LAS CURVAS DE COMPARACIÓN O PATRONES
Medir en dos series de tubos de Nessler de 50 mL o en tubos de ensayo de 60 a 70 mL,
alícuotas de 1 a 20 mL de las dos soluciones diluídas de nitrato de sodio, añadir 5 mL de
hidróxido de amonio 0.88 a cada tubo diluir hasta completar 50 mL con agua.
Los tubos patrones así preparados son estables por algunas semanas si se guardan bien
tapados, leer en el espectrofotómetro a una longitud de onda de 420 nanómetros y trazar
las dos curvas graficando absorbancia contra concentración.
4. METODOLOGÍA
Pasar de 2 a 5 g de muestra finamente molida y homogeneizar a un matraz aforado de 100
mL, añadir de 20 a 30 mL de agua y calentar a baño maría a la temperatura de 70 a 80°C
por 20 minutos, agitando, añadir de 2 a 3 mL de solución saturada de acetato de plomo
agitar y dejar sedimentar, si el líquido sobrenadante queda turbio, añadir de 5 a 8 mL de
crema de alúmina agitar y dejar sedimentar; si el líquido sobrenadante queda turbio, añadir
3.5 mL de solución saturada de sulfato de plata libre de nitratos, agitar, completar el
volumen con agua a 100 mL, volver a agitar y filtrar a través de papel, regresando el
filtrado hasta que pase claro.
Evaporar a sequedad en baño maría a una temperatura de 70 a 80°C, una alícuota de 25 mL
de filtrado. Retirar y añadir 1 mL de solución de ácido fenoldisulfónico, mezclar
rápidamente con el agitador de vidrio, añadir 1 mL de agua y 3 a 4 gotas de ácido sulfúrico
y calentar en baño maría de 70 a 80°C de 2 a 3 minutos, teniendo cuidado de no secar la
muestra, añadir aproximadamente 25 ml de agua y un exceso de hidróxido de amonio 0.88,

102
transferir a un tubo de Nessler o de ensayo (con aforo de 50 ml), añadir 1 ó 2 ml de crema
de alúmina si no está completamente claro, completar el volumen con agua y filtrar si es
necesario.
El aparato se ajusta a cero de transmitancia con un blanco preparado evaporando 25 mL del
filtrado clarificado, añadir 1 mL de ácido sulfúrico concentrado, mezclar rápidamente con
un agitador de vidrio, añadir 1 mL de agua y calentar a baño maría a la temperatura de 70 a
80°C de 2 a 3 minutos, teniendo cuidado de no secar la muestra, añadir 25 mL de agua y un
exceso de hidróxido de amonio 0.88, transferir a un tubo con aforo de 50 mL y completar el
volumen con agua, leer a 420 namómetros para comparar con la curva patrón.
Cálculos:
ppm NaNO3 = (L X 4 X 1000) / m
Donde:
L= Lectura de la solución problema en mg de nitrato de sodio al comparar con la
curva.
m= Masa de la muestra en gramos
5. RESULTADOS
Reportar el porcentaje de nitratos en ppm de la muestra
Realizar un reporte de acuerdo al apéndice 10.
E) DETERMINACIÓN DE NITRITOS (METODO COLORIMETRICO)
1. OBJETIVO
Determinar las ppm de nitritos en productos cárnicos.
2. INTRODUCCIÓN
Este método de prueba se fundamenta en la reacción colorida entre los nitritos y el
colorante con grupo funcional azo a un pH entre 2.0 y 2.5, por la copulación del ácido

103
sulfanílico diazoado con clorhidrato de naftilamina, resultando una coloración roja.
3. MATERIALES, REACTIVOS Y EQUIPO
Acido sulfanílico.
Acido acético glacial.
Alfanaftilamina (naftilamina 1).
Zinc en polvo.
Sulfato de potasio y aluminio dodecahidratado.
Hidróxido de amonio 0.88.
Cloruro de bario.
Nitrito de sodio.
Cloruro mercúrico.
Carbón vegetal activado.
Vasos de precipitados de 50, 100 y 250 mL
Matraces volumétricos de 250 y 1000 mL
Pipetas volumétricas de 1, 2 y 10 mL
Agitador de vidrio
Tubos de Nessler de 50 mL o tubos de ensayo de 60 a 70 mL
Pipetas de 5, y 20 mL graduadas en décimos de mL
Embudo
Papel filtro
Papel tornasol
Frascos para guardar los reactivos
Baño María o de vapor con termostato y termómetro
Soporte universal y anillo
Papel milimétrico para graficar
Mortero
Balanza analítica con = 0.1 mg de sensibilidad.
Espectrofotómetro

104
Solución de ácido sulfanílico: Disolver en caliente 0.5 g de ácido sulfanílico, 30 mL de
ácido acético glacial y 120 mL de agua libre de nitritos y filtrar. Guardar en refrigeración.
Solución de alfanaftilimina: Disolver por calentamiento 0.1 g de alfanaftilamina
(naftilamina 1) en 120 mL de agua libre de nitritos, enfriar, agregar 30 mL de ácido acético
glacial y filtrar. Guardar en refrigeración.
Si cualquiera de las dos soluciones anteriores se torna colorida, agitar con 0.5 g de zinc en
polvo y filtrar.
Crema de alúmina: Preparar una solución saturada de sulfato de potasio y aluminio
dodecahidratado, añadir hidróxido de amonio 0.88 con agitación constante hasta que la
solución sea alcalina al papel tornasol. Dejar sedimentar el precipitado y lavar por
decantación con agua hasta que el agua de lavado dé ligeramente la reacción para sulfatos
con cloruro de bario. Quitar el exceso de agua y guardar la crema residual en un frasco
cerrado.
Solución patrón de nitrito de sodio: Disolver 0.5000 g de nitrito de sodio en agua libre de
nitritos hasta completar 1000 mL, Diluir una alícuota de 10 mL de esta solución hasta
completar un volumen de 1000 mL. Un mililitro de esta solución contiene 0.005 mg de
nitrito de sodio.
PREPARACIÓN DE LA CURVA DE COMPARACIÓN O PATRÓN
Medir en tubos de Nessler de 50 mL o en tubos de ensayo de 60 a 70 mL los siguientes
volúmenes de solución patrón de nitrito de sodio: 0.0, 0.5, 2.0, 4.0, 6.0, 8.0, 10.0, 12.0,
14.0, 16.0, 18.0 mL, añadir a cada tubo 2 mL de solución de ácido sulfanílico y 2 mL de
solución de alfanaftilamina. Mezclar perfectamente y dejar reposar 20 minutos. Leer en
espectrofotómetro a una longitud de onda de 520 nanómetros, y trazar una curva graficando
concentración contra absorbencia.

105
4. METODOLOGÍA
Colocar 2 g de muestra finamente molida y homogeneizada en un vaso de precipitados de
50 mL, añadir aproximadamente 40 mL de agua a la temperatura de 80°C, mezclar
perfectamente con el agitador teniendo cuidado de romper todos los grumos, transferir todo
el contenido a un matraz volumétrico de 250 mL, lavar el vaso y el agitador con porciones
de agua caliente (160 mL aproximadamente), colocar el matraz en baño maría o de vapor a
la temperatura de 70 a 80° C durante dos horas, agitando.
Añadir 10 mL de solución saturada de cloruro mercúrico y mezclar para aclarar
completamente, añadir 5 mL de crema de alúmina y si hay color añadir menos de 0.5 g de
carbón vegetal activado. Enfriar a temperatura ambiente, completar el volumen con agua
libre de nitritos y mezclar otra vez, filtrar y determinar nitritos. Tomando una alícuota de 50
mL del filtrado y colocarla en un tubo y agregarle 2 mL de la solución de ácido sulfanílico
y 2 mL de la de alfanaftilamina.
Mezclar y dejar reposar 20 minutos para que desarrolle el color rosa, leer en
espectrofotómetro transfiriendo una porción adecuada de la solución problema a la celda
del espectrofotómetro y determinar la absorbencia a una longitud de onda de 520 nm.
El aparato se ajusta a cero de transmitancia con un blanco de 50 mL de agua libre de
nitritos, más 2 mL de la solución de ácido sulfanílico y 2 mL de la de alfanaftilamina.
Cálculos:
ppm NaNO2 = (L X 5 X 1000) / m
Donde:
L = Lectura del problema en mg de nitritos al comparar con la curva.
m = Masa de la muestra en gramos

106
5. RESULTADOS
Reportar el contenido de nitritos en ppm de la muestra
Realizar un reporte de acuerdo al apéndice 10
6. BIBLIOGRAFÍA
Dirección General de Investigación en Salud Pública Secretaría de Salubridad y
Asistencia.
Técnicas para el Análisis Fisicoquímico de Alimentos. 1976.
Association of Official Analytical Chemists. Official Methods of Analysis. Nitrites,
in Cured Meats 24.037; Twelfth Edition 1975.
Pearson, D. 1993. Técnicas de Laboratorio para el Análisis de Alimentos. 6ª
Edición. Editorial Acribia, S.A. Zaragoza (España).
MNX-F-097-S-1978. Determinación de nitritos en embutidos
NMX-F-321-S-1978. Determinación de fécula por hidrólisis acida en embutidos.
Association of Official Analytical Chemists. Official Methods of Analysis. Part
25.012-1975.
MNX-F-318-S-1978. Determinación de nitratos en embutidos
MNX-F-320-S-1978. Determinación de fosfatos en embutidos

107
PARTE VI
ANALISIS FISICOQUIMICOS DE GRANOS Y HARINAS DE CEREALES

108

109
PRACTICA No. 15
I. DENSIDAD EN GRANOS ENTEROS DE CEREALES
1. OBJETIVO
Determinar la densidad de granos enteros de cereales
2. INTRODUCCIÓN
El término de peso hectolítrico se refiere al concepto de "densidad aparente" del grano Se
habla de densidad aparente porque es una relación entre la masa y el volumen de una
muestra de grano, considerando dentro de este volumen no solamente el espacio ocupado
por los granos propiamente dichos sino también el volumen de los espacios intergranulares.
Por hacer referencia a un volumen fácilmente comprensible para los usuarios, se
acostumbra expresar los resultados de este análisis en libras por bushel en el sistema de
USA y en kilogramos por hectolitro o por metro cúbico en el sistema métrico decimal
Existen varios sistemas que dictaminan la calidad del grano por medio del estudio
de la densidad. Indudablemente, el más importante y práctico es la determinación del peso
hectolitrico o volumétrico realizado con el medidor WINCHESTER BUSHEL METER
(figura1). El sistema consiste en la determinación del peso en lb o kg de un cierto volumen
de grano expresado en búshels (2150.42 plg3 o 36.37 L) o hectolitros llenado y, o empacado
bajo condiciones estandarizadas.
Los valores de peso hectolítrico o densidad están relacionados con la calidad del
grano de cereal. Los granos masa densos son menos susceptibles al ataque por insectos. En
el caso del trigo el peso hectolitrico es un parámetro que se usa para la determinación del
grado para su comercialización y venta.
En trigos se prefieren granos con pesos hectolítrico o densidades altas ya que son
más rendidores mientras que en maíz esto no es importante debido que se quiere obtener
almidón (y para ello hay que remojar el grano con sulfito de sodio). Un maíz con menor
densidad es mas fácil de remojar y posteriormente procesar para obtener almidón.

110
Figura 1. Winchester Bushel Meter
3. MATERIALES, REACTIVOS Y EQUIPO
Granos de cereales
Winchester Bushel Meter
4. METODOLOGIA
Colocar la muestra en el recipiente cónico del equipo, el cual tiene un volumen
especificado y posteriormente pesarla con la finalidad de conocer el peso del grano
contenido en dicho volumen.
5. RESULTADOS
Reportar la densidad del grano obtenida
Realizar un reporte como se indica en el apéndice
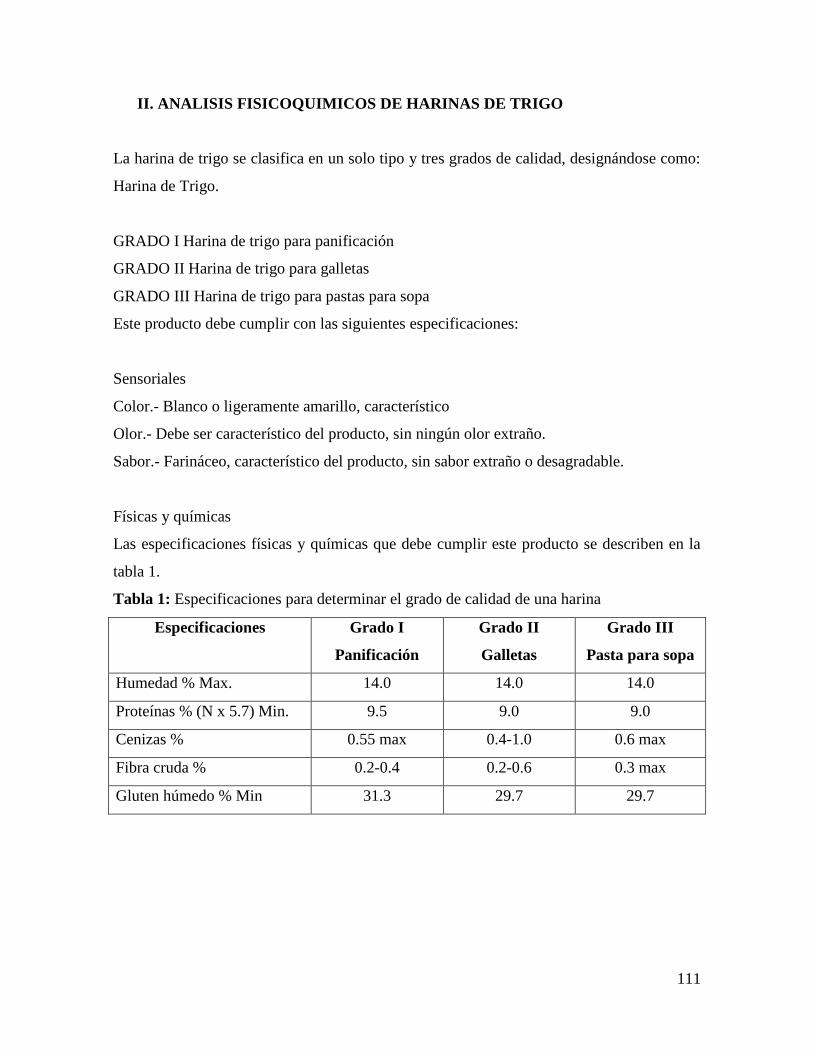
111
II. ANALISIS FISICOQUIMICOS DE HARINAS DE TRIGO
La harina de trigo se clasifica en un solo tipo y tres grados de calidad, designándose como:
Harina de Trigo.
GRADO I Harina de trigo para panificación
GRADO II Harina de trigo para galletas
GRADO III Harina de trigo para pastas para sopa
Este producto debe cumplir con las siguientes especificaciones:
Sensoriales
Color.- Blanco o ligeramente amarillo, característico
Olor.- Debe ser característico del producto, sin ningún olor extraño.
Sabor.- Farináceo, característico del producto, sin sabor extraño o desagradable.
Físicas y químicas
Las especificaciones físicas y químicas que debe cumplir este producto se describen en la
tabla 1.
Tabla 1: Especificaciones para determinar el grado de calidad de una harina
Especificaciones Grado I
Panificación
Grado II
Galletas
Grado III
Pasta para sopa
Humedad % Max. 14.0 14.0 14.0
Proteínas % (N x 5.7) Min. 9.5 9.0 9.0
Cenizas % 0.55 max 0.4-1.0 0.6 max
Fibra cruda % 0.2-0.4 0.2-0.6 0.3 max
Gluten húmedo % Min 31.3 29.7 29.7

112
A) DETERMINACIÓN DE GLUTEN (MÉTODO: LAVADO A MANO)
1. OBJETIVO
Determinar el contenido de gluten en una muestra de harina de trigo
2. INTRODUCCIÓN
La fuerza de una harina depende en gran parte de la naturaleza y cantidad de gluten
presente. Tales propiedades se pueden apreciar formando una pasta con agua o agua salina
de la cual se puede separar el almidón y la proteína soluble (albumina y parte de las
globulinas), por lavado quedando un residuo húmedo con propiedades cohesivas y elásticas
llamado gluten vital (principalmente, glutelinas y prolaminas).
3. MATERIALES, REACTIVOS Y EQUIPO
Harina de trigo
Tamiz o manta de cielo
Capsula de porcelana
Balanza analítica
Estufa de calentamiento
Probeta de 50 mL
Lugol
4. METODOLOGÍA
Pesar 25 g de muestra de harina y colocarla en una capsula de porcelana, agregar 15 mL de
agua y amasar para formar una masa, evitando que se adhieran partículas de masa a las
paredes, dejar reposar durante una hora. Colocar la masa sobre un trozo de manta de cielo o
tamiz y llevarla al chorro de agua con el fin de eliminar el almidón que contienen la masa,
lavar con agua hasta que ésta no de coloración azul con el reactivo de lugol. Dejar el
residuo en agua durante una hora y hacer una bola con las manos, eliminándole la mayor
cantidad de agua posible por compresión, entonces pasarla a una capsula previamente
tarada y pesar.

113
Cálculos:
% de gluten húmedo = (R / M) X 100
Donde:
R = Residuo de la muestra en gramos
M = Peso de la muestra en gramos
Levar a la estufa a 90ºC el gluten húmedo, hasta masa constante y pesar. Reportar el
resultado en % de gluten seco.
5. RESULTADOS
Reportar el porcentaje de gluten Húmedo y seco
Realizar un reporte como se indica en el apéndice 10
B) DETERMINACIÓN DE pH
1. OBJETIVO
Determinar el pH de una muestra de harina de trigo
2. INTRODUCCIÓN
La acidez medida por el valor de pH es un importante factor para el control de muchos
procesos, tanto naturales como de fabricación. Es un considerando de gran importancia en
la conservación y almacenamiento de alimentos, por su efecto inhibidor de
microorganismos y enzimas. En general las bacterias son más sensibles a los iones de
hidrogeno que los fermentos y los mohos. La mayor parte de los organismos tienen límites
de pH máximo y mínimo para su desarrollo y un rango óptimo para un crecimiento más
rápido. Los iones de hidrogeno también influyen en la cantidad de calor que se debe aplicar
a las conservas para alcanzar su esterilización comercial, por ejemplo, los vegetales y las
carnes se someten a temperaturas más elevadas y durante mayor tiempo que las frutas que
son más ácidas. El valor de pH afecta a diversas propiedades físicas de algunos alimentos,
por ejemplo, la textura y la estabilidad o resistencia de los geles de gelatina y del de

114
pectina-azúcar-ácido de las mermeladas. Junto con la humedad, la determinación del pH es,
probablemente, la que más frecuentemente se realiza en la industria de los alimentos.
3. MATERIALES, REACTIVOS Y EQUIPO
Harina de trigo
Vasos de precipitado de 100 mL
Agitador de vidrio
Balanza analítica
Potenciómetro
4. METODOLOGÍA
Pesar 10 g de muestra de harina y suspenderla en 100 mL de agua destilada, recientemente
hervida y a 25ºC, se mezcla hasta obtener una suspensión homogénea que se deja macerar
durante 30 minutos, agitando de vez en cuando; después se deja reposar, decantar el
líquido, se filtra y se mide el pH.
5. RESULTADOS
Reportar el pH obtenido de la muestra de harina
Realizar un reporte como se indica en el apéndice 10
.
C) DETERMINACIÓN DE ACIDEZ
1. OBJETIVO
Determinar la acidez de una muestra de harina
2. INTRODUCCIÓN
La acidez de las harinas aumenta durante el almacenamiento, sobre todo si están a
temperaturas demasiado altas, con la consiguiente debilitación de la fermentabilidad, por
otra parte, la acidez de una harina morena es mayor que la de una blanca preparadas ambas
a partir del mismo grano.

115
3. MATERIALES, REACTIVOS Y EQUIPO
Harina de trigo
Frasco con tapón esmerilado
Bureta de 25 mL
Soporte y pinzas para bureta
Etanol al 96 % (Neutralizado a la fenolftaleína)
Hidróxido de sodio 0.02 N
Fenolftaleína 0.1% en etanol neutralizado
Balanza analítica
4. METODOLOGÍA
Pesar 5 g de harina y colocarla en un frasco con tapón esmerilado, adicionar 25 mL de de
etanol al 96% (neutralizado exactamente a la fenolftaleína) y dejarlo reposar durante 36
horas (agitar cada 8 horas). Tomar 10 mL del líquido separadoy titular con NaOH 0.02 N.
Cálculos:
Acidez = ( A x N x meq x 1000) / ( M x a)
Donde:
A = mL de NaOH empleados
N = Normalidad del NaOH
M = Peso de la muestra en gramos
A = Alicuota tomada (mL)
meq = Miliequivalentes del ácido predominante en la muestra
5. RESULTADOS
Reportar la acidez obtenida de la muestra de harina
Realizar un reporte como se indica en el apéndice 10
.

116
D) ACTIVIDAD DIATASICA
1. OBJETIVO
Determinar el contenido de maltosa de una harina por el método de Blish y Sandstedt
2. INTRODUCCIÓN
Cuando una harina de trigo para pan se mezcla con agua, sal y levadura, la masa producida
debe fermentar dando suficiente azúcar para la adecuada producción de gas y debe
hincharse debido a la presión del mismo. Por lo tanto, las harinas para producir pan deben
contener adecuadas cantidades de proteínas, tener condiciones físicas idóneas y ser capaces
de convertir el almidón en azúcar, de lo cual es una medida la actividad diastasica. Las
principales enzimas diastásicas presentes son la α-amilasa y la β-amilasa.
3. MATERIALES, REACTIVOS Y EQUIPO
Potenciómetro
Balanza analítica
Ferricianuro de potasio
Carbonato de sodio anhidro
Matraces aforados de 100 mL y de 1 L
Pipetas volumétricas de 10 mL
Acido acético glacial
Sulfato de zinc
Cloruro de potasio
Acetato de sodio anhidro
Acido sulfúrico
Yoduro de potasio
Hidróxido de sodio
Almidón al 1%
Tiosulfato de sodio 0.1N
Bureta de 25 mL
Matraces erlenmeyer de 250 mL

117
Tubos de ensayo grandes
Disolución alcalina de ferricianuro de potasio (0.1 N). Se disuelven 33 g de ferricianuro de
potasio y 44 g de carbonato de sodio anhidro en agua y se diluye la solución a un litro.
Reactivo de ácido acético. Se disuelven 40 g de sulfato de zinc y 70 g de cloruro de potasio
en 600 mL de agua, se añaden lentamente 200 mL de ácido acético glacial y se diluye la
solución con agua a un litro.
Disolución tampón (pH = 4.6 – 4.8). se disuelven 4.1 g de acetato sódico anhidro en agua,
se añaden 3 mL de ácido acético glacial y se diluye la solución con agua a un litro.
Acido sulfúrico (3.7 N). Se añaden cuidadosamente, con agitación, 10 mL de ácido
sulfúrico concentrado sobre unos 80 mL de agua y se diluye la solución con agua a 100 mL.
Disolución de Yoduro de potasio. Se añaden 50 g de KI a 100 mL de agua fría y se añade
una gota de NaoH 10 N. La disolución debe usarse recién preparada.
4. METODOLOGIA
Se introducen 5 g de harina y una cucharadita de arena calcinada, un matraz erlenmeyer de
250 mL y se mezclan por rotación. Se calienta la mezcla a 30ºC, se añaden 46 mL de
disolución tampón (también a 30ºC) y se agita hasta que toda la harina este en suspensión.
Se coloca el matraz en un baño de agua a 30ºC durante una hora, rotándolo cada 15
minutos. Al final de la hora se añaden 2 mL de H2SO4 3.7 N, se mezcla, se añaden 2 mL de
disolución de wolframato de sodio al 12%, se mezcla de nuevo, se deja reposar durante 2
minutos y se filtra, desechando las 10 primeras gotas. Inmediatamente se pipetean 5 mL del
filtrado dentro de un tubo de ensayo grande, se añaden 10 mL de disolución de ferricianuro
de potasio y se sumerge el tubo de ensayo en agua hirviendo durante 20 minutos con la
superficie del líquido en el tubo unos 4 cm por debajo del nivel del agua hirviendo. Se
enfría el tubo con agua corriente y se pasa el contenido a un erlenmeyer de 100 mL,
lavando con 25 mL del reactivo de ácido acético. Se añade 1 mL de la disolución de KI y 2
mL de disolución de almidón al 1%., se mezcla bien y se valora por retroceso con tiosulfato
de sodio 0.1N (x mL) hasta desaparecer completamente el color azul. (Si el material del
tubo es incoloro en vez de amarillo luego del tratamiento en el baño de agua hirviendo y no

118
se vuelve azul después de añadir KI, se repite la determinación usando menos extracto, por
ejemplo; 1 o 2.5 mL. En tales casos se diluye la mezcla hasta 5 mL con agua y se modifican
los cálculos correpondientes).
y = mL de ferricianuro 0.1 N reducido = (10 – x) mL
El índice de maltosa, expresado en porcentaje de maltosa formada, se puede obtener con la
ayuda de la tabla 2. Al interpretar los valores de la literatura hay que tener presente, que en
muchos países incluyendo U.S.A., el contenido de maltosa se expresa en mg de maltosa
producidos por 10 g de harina (100 x el índice Reino Unido). Para que una harina pueda
utilizarse para pan debe contener un porcentaje de maltosa del 2.0-3.5. Por debajo de este
rango hay una inadecuada actividad diastásica de azúcar en la masa y el pan obtenido es
probable que tenga una corteza descolorida. La adición de harina de malta incrementa
notablemente el contenido de maltosa. Un valor por encima de 3.5 indica una excesiva
actividad de la α-amilasa y se obtiene una miga pegajosa. La mayor parte de las harinas
para pan tienen la adecuada β-amilasa, la cual no obstante actúa sobre almidón deteriorado,
cuya cantidad varía considerablemente.
5. RESULTADOS
Reportar índice de maltosa obtenido de la muestra de harina
Realizar un reporte como se indica en el apéndice 10
119
Table 2. Índices de maltosa obtenidos usando el método de Blish y Sandstedt
y mL ferricianuro
0.1 N reducido
Indice de maltosa
%
y mL ferricianuro
0.1 N reducido
Indice de maltosa
%
y mL ferricianuro
0.1 N reducido
Indice de maltosa
%
0.1 0.05 3.4 1.71 6.7 3.79
0.2 0.10 3.5 1.76 6.8 3.85
0.3 0.15 3.6 1.82 6.9 3.02
0.4 0.20 3.7 1.88 7.0 3.98
0.5 0.25 3.8 1.95 7.1 4.06
0.6 0.31 3.9 2.01 7.2 4.12
0.7 0.36 4.0 2.07 7.3 4.18
0.8 0.41 4.1 2.13 7.4 4.25
0.9 0.46 4.2 2.18 7.5 4.31
1.0 0.51 4.3 2.25 7.6 4.38
1.1 0.56 4.4 2.31 7.7 4.45
1.2 0.60 4.5 2.37 7.8 4.51
1.3 0.65 4.6 2.44 7.9 4.58
1.4 0.71 4.7 2.51 8.0 4.65
1.5 0.76 4.8 2.57 8.1 4.72
1.6 0.80 4.9 2.64 8.2 4.78
1.7 0.85 5.0 2.70 8.3 4.85
1.8 0.90 5.1 2.76 8.4 4.92
1.9 0.96 5.2 2.82 8.5 4.99
2.0 1.01 5.3 2.88 8.6 5.05
2.1 1.06 5.4 2.95 8.7 5.12
2.2 1.11 5.5 3.02 8.8 5.19
2.3 1.16 5.6 3.08 8.9 5.27
2.4 1.21 5.7 3.15 9.0 5.34
2.5 1.26 5.8 3.22 9.1 5.42
2.6 1.30 5.9 3.28 9.2 5.50
2.7 1.35 6.0 3.34 9.3 5.58
2.8 1.40 6.1 3.41 9.4 5.68
2.9 1.45 6.2 3.47 9.5 5.78
3.0 1.51 6.3 3.53 9.6 5.88
3.1 1.56 6.4 3.60 9.7 5.98
3.2 1.61 6.5 3.67 9.8 6.08
3.3 1.66 6.6 3.73 9.9 6.18

120
D) PRUEBAS DE SEDIMENTACIÓN DE HARINAS (INDICE DE PELSHENKE)
1. OBJETIVO
Determinar el valor e índice de Pelshenke.de la harina de trigo
2. INTRODUCCIÓN
Estas pruebas están diseñadas para predecir la funcionalidad y, o potencial de las muestras
de trigo, especialmente las destinadas para panificación. Existen varias pruebas que están
basadas en el mismo principio: la de Pelshenke y las de Sedimentación. La prueba de
Pelshenke utiliza harina de trigo entero mezclado con agua y levadura. Después de hidratar
y amasar, la masa resultante es moldeada en forma de bola y sumergida en una probeta, la
cual es colocada en un gabinete de fermentación a 30ºC, el tiempo que demora la harina en
desintegrarse es el valor de Pelshenke, indicando la calidad del gluten, entre más fuerte sea
el gluten más tiempo tarda en desintegrarse el amasado anterior. Los valores observados en
harinas suaves son de 20-30 hasta 100-175 minutos, mientras que las harinas panaderas
demoran hasta 400 minutos en desintegrarse. La división del valor de la prueba entre el
contenido proteico da el índice de Pelshenke. Los trigos panaderos tienen un índice mayor
que los trigos suaves o galleteros.
3. MATERIALES, REACTIVOS Y EQUIPO
Capsula de porcelana
Harina de trigo
Probetas de 250 mL
Balanza analítica
Levadura
4. METODOLOGIA
Colocar 5 g de harina en una capsula de porcelana y agregar 2,5 mL de agua y 0,25 g de
levadura, amasar y formar de una bola, la cual se coloca en una probeta que contiene 175
mL de agua a 32 ºC. La bola, que al comienzo se va al fondo de la probeta, pronto emerge a

121
la superficie, donde permanece un tiempo variable, hasta que se desintegra. Se llama cifra
de Pelshenke o tiempo de fermentación los minutos transcurridos desde que se coloca la
bola de masa en la probeta hasta la ruptura y varía de 25 minutos a 5 horas, según la fuerza
panadera del trigo. Dividiendo el tiempo de fermentación por el porcentaje de proteína se
obtiene una constante específica para cada variedad de trigo, llamado índice de Pelshenke.
5. RESULTADOS
Reportar el valor e índice de Pelshenke.
Realizar un reporte como se indica en el apéndice 10
6. BIBLIOGRAFÍA
Pearson, D. 1993. Técnicas de Laboratorio para el Análisis de Alimentos. 2ª
Edición. Editorial ACRIBIA, S. A. Zaragoza, España.
Serna-Saldivar, S. 1996. Química, Almacenamiento e Industrialización de los
Cereales. 1ª Edición. A.G.T. Editor, S. A.
INSTRUCTIVO DE ALIMENTOS VOL. II. (1991). IPN, Escuela Nacional de
Ciencias Biológicas. Departamento de Ingeniería Bioquímica

122

123
PARTE VII
ANALISIS FISICO-QUIMICO DE FRUTAS Y HORTALIZAS

124

125
PRACTICA 16
1. DETERMINACIÓN DE pH
1. OBJETIVO
Determinar el pH de un producto hortofrutícola
2. INTRODUCCIÓN
El pH es el logaritmo común del número de litros de disolución que contienen un
equivalente gramo de iones hidrógeno. Así pues:
pH = - log10 [ H+ ]
El rango de pH va de 0 a 14; un valor por debajo de 7 representa una disolución ácida, 7
una disolución neutra y por encima de 7 una disolución alcalina. Los valores del pH de
algunos alimentos y productos son: ácido cítrico, 2.0; limón, 2.2-2.4; vinagre, 2.4-3.4;
manzana, 2.9-3.3; pan, 5.0-6.0; col, 5.2-5.4; harina de 6.0-6.5; leche, 6.4-6.8.
El pH se puede determinar colorimétricamente utilizando los indicadores
adecuados, pero se determina con mayor exactitud por métodos eléctricos. La mayoría de
los potenciómetros miden la diferencia de potencial entre un electrodo patrón de
calomelanos y un electrodo de vidrio por balanceo.
La acidez medida por el valor del pH es un importante factor para el control de
muchos procesos, tanto naturales como de fabricación. El pH es de gran importancia en la
conservación y almacenamiento de alimentos, por su efecto inhibidor del desarrollo de
microorganismos y enzimas. La mayor parte de los organismos tienen límites de pH
máximo y mínimo para su desarrollo y un rango optimo para su crecimiento más rápido.
Los iones hidrogeno también influyen en la cantidad de calor que se debe aplicar a
las conservas para alcanzar su esterilización comercial, por ejemplo, los vegetales y las
carnes se someten a temperaturas más elevadas y durante mayor tiempo que las frutas que
son más acidas.
El valor del pH afecta además a diversas propiedades físicas de algunos alimentos,
por ejemplo, la textura y la estabilidad o resistencia de los geles de la gelatina y del de

126
pectina/azúcar/ácido de las mermeladas. Junto con la humedad, la determinación del pH es,
probablemente, la que más frecuentemente se realiza en la industria de los alimentos.
3. MATERIALES, REEACTIVOS Y EQUIPO
Potenciómetro
Vasos de precipitado
Termómetro
Matraz aforado de 100 mL
4. METODOLOGÍA
Pesar 10 g de muestra y agitar con 100 mL de agua destilada hervida y fría, a 25 ºC, hasta
obtener una suspensión homogénea, macerar durante 30 minutos y agitar ocasionalmente,
decantando después de reposar 10 minutos, llevar el líquido a un vaso de precipitado de 100
mL y determinar el pH utilizando el potenciómetro. Tener especial cuidado en el manejo de
los electrodos y recordar que estos deberán estar perfectamente secos y calibrados con una
solución de pH conocido, para obtener así un dato confiable.
5. RESULTADOS
Reportar el pH obtenido en la muestra utilizada
Realizar un reporte de acuerdo al apéndice 10
II. DETERMINACIÓN DE LA ACIDEZ TITULABLE
1. OBJETIVO
Determinar la acidez titulable de una muestra
2. INTRODUCCIÓN
Además de que el pH es un factor importante para la conservación y estabilización de
ciertos geles, el contenido total de ácido de un alimento es un ensayo de los más sencillos

127
para el control de una formulación. La mayoría de los ácidos comúnmente usados en la
industria de los alimentos (por ejemplo: cítrico, tartárico) son objeto de valoración. La
acidez valorable total se determina casi siempre con NaOH 0.1 N o 0.5 N e indicador de
fenolftaleína (pH 8.3-10.0) o azul de bromotimol (pH 6.0-7.6).
La acidez valorable total se suele indicar en términos del ácido predominante entre
los existentes, por ejemplo: en la leche como ácido láctico, en la mayor parte de las frutas
como ácido cítrico, en las manzanas como ácido málico y en el vinagre como ácido acético.
En el control industrial es frecuente dar la acidez como “número de mL de álcali 0.1 N
consumido por 10g”, dado que los resultados de las valoraciones son tan interesantes por si
mismos como por comparación entre ellos. En ciertos casos se expresa la acidez en
términos de equivalencia de peso de un álcali determinado. Por ejemplo: los fosfatos ácidos
utilizados en la levadura en polvo se expresan normalmente en términos de bicarbonato de
sodio.
3. MATERIALES, EQUIPO Y REACTIVOS
Matraces erlenmeyer de 100 mL
Vasos de precipitado de 100 mL
Bureta de 25 mL
Soporte y pinzas para buretas
NaOH 0.1 N
Alcohol neutralizado
Fenolftaleína al 0.1 % en etanol
4. METODOLOGÍA
Pesar aproximadamente 10 g de muestra y añadir 50 mL de agua destilada o alcohol
neutralizado si la muestra es insoluble en agua, titular con NaOH utilizando fenolftaleína
como indicador hasta observar un cambio de color que persista por lo menos un minuto. Si
las muestras son de color oscuro, seguir la neutralización midiendo los cambios de pH en
un potenciómetro.

128
Cálculos:
Expresar los resultados en gramos del ácido predominante en la muestra, por 100 mL de
muestra.
Acidez = [ (A x N x meq) / m] x 100
Donde:
A = mL de NaOH 0.1 N empleados en la titulación
N = Normalidad del NaOH
meq = Miliequivalente del ácido predominante , en gramos.
m = Masa de la muestra en gramos
5. RESULTADOS
Reportar la acidez obtenida en la muestra utilizada
Realizar un reporte de acuerdo al apéndice 10
III. VITAMINA C (Ácido ascórbico)
1. OBJETIVO
Determinar el contenido de Vitamina C
2. INTRODUCCIÓN
El contenido de vitamina C en las frutas y verduras varía dependiendo del grado de
madurez, es menor cuando están verdes, aumenta su cantidad cuando alcanza su madurez y
luego vuelve a disminuir; por lo que la fruta madura pierde parte de su contenido. Lo más
recomendable es comer las frutas y verduras frescas puesto que la acción del calor destruye
esta vitamina, además cuando se pone en contacto con el aire se oxida y pierde su actividad.
El déficit de vitamina C produce Escorbuto, que se caracteriza por hinchamientos,
hemorragias en las encías y caída de los dientes. Algunos otros efectos atribuidos a esta
vitamina son: mejor cicatrización de heridas, alivio de encías sangrantes, reducción de

129
alergias, prevención del resfriado común, y en general fortalecimiento del organismo. Las
principales fuentes de vitamina C son: Hortalizas y Frutas
3. MATERIALES, REACTIVOS Y EQUIPO
Matraz aforado de 250 y 100 mL
Matraz erlenmeyer de 250 mL
Dos pipetas graduadas de 10 mL
Acido ascórbico (solución tipo)
Acido oxálico al 0.4 %
Éter etílico
2,6 diclorofenol indofenol
Bicarbonato de sodio
Licuadora
PREPARACIÓN DE LOS REACTIVOS
Solución tipo de ácido ascórbico; Disolver 50 mg del reactivo en 60 mL de ácido oxálico
al 0.4% y aforar con agua destilada a 250 mL. Un mililitro de esta solución equivale a 0.2
mg de ácido ascórbico.
Soluciuón de 2,6 diclorofenol indofenol; Disolver 50 mg del reactivo en agua destilada,
llevar a 200 mL con agua y filtrar. Esta solución es estable en refrigeración durante dos
semanas. Adicionar 40 mg de bicarbonato de sodio para aumentar su estabilidad.
Titulación del ácido azcorbico; Colocar en un matraz erlenmeyer de 125 mL, 10 mL de la
solución tipo de ácido ascórbico. Adicionar con bureta el colorante de 2,6 diclorofenol
indofenol hasta la aparición de un color ligeramente rosado. Repetir la valoración para tener
un resultado más exacto.
PREPARACIÓN DE LA MUESTRA
En aquellas muestras donde pueda extraerse fácilmente el jugo, por ejemplo en cítricos,
presionar y filtrar o bien centrifugar. Medir de 10 a 20 mL de la muestra y añadir una
cantidad igual de ácido oxálico al 0.4% y aforar a 250 mL con agua destilada.

130
En caso de que la muestra sea sólida, moler en una licuadora 50 g, de la misma con 100 mL
de solución de ácido oxálico durante 2 minutos y aforar a 250 mL con ácido, filtrar y
centrifugar.
La cantidad de muestra así como de ácido oxálico a utilizar, dependerá de la riqueza en
vitamina C dela muestra que se este trabajando.
4. METODOLOGIA
Tomar una alícuota de la muestra, cuando el color de la misma pueda enmascarar el vire del
colorante, agregar 20 mL de éter etílico, agitar y titular con el 2,6 diclorofenol indofenol
hasta la aparición de un color rosa pálido persistente durante un minuto. El vire se
observará en la capa de éter etílico.
CALCULOS:
Calculo del equivalente “E”, del 2,6 diclorofenol indofenol. La cantidad del colorante
utilizada en la valoración de la solución tipo de ácido ascórbico es equivalente a 2 mg de
ácido ascórbico, por lo que el valor de “E” será:
E = [mL de 2,6 diclorofenol indofenol] / [2 mg de ácido ascórbico]
Calculo del contenido de ácido ascórbico en la muestra
A. A. = [G x V] / [a x m x E]
Donde:
A. A. = Contenido de ácido ascórbico en la muestra (mg/g)
G = mL de colorante utilizados en la valoración
E = Equivalente de 2,6 diclorofenol indofenol (mg de ácido ascórbico que reaccionan con 1
mL de colorante)
V = volumen al que se aforo
a = Alícuota utilizada
m = Masa de la muestra en gramos

131
5. RESULTADOS
Reportar el contenido de vitamina C (mg / g) en la muestra utilizada
Realizar un reporte de acuerdo al apéndice 10.
IV. DETERMINACIÓN DE CLORUROS (Método de Volhart)
1. OBJETIVO
Determinar el contenido de cloruros en salmueras utilizadas en el enlatado de hortalizas
2. INTRODUCCIÓN
El fundamento de método se basa en que a una muestra acidificada de salmuera se le añade
un exceso de AgNO3, que con los cloruros forma AgCl. El exceso de AgNO3 se valora con
una solución de tiocianato de potasio usando como indicador sulfato férrico amónico, el
cual toma un color naranja pálido rosa permanente.
3. MATERIALES, EQUIPO Y REACTIVOS
Matraz erlenmeyer de 250 mL
Pipetas volumétricas de 10 mL
Pipetas graduadas de 10 mL
Bureta de 25 mL
Ácido nítrico concentrado
Nitrato de plata 0.1 N
Tiocianato de potasio 0.1 N
Sulfato férrico-amónico al 5 %,
4. METODOLOGIA
Colocar en un matraz aforado de 100 mL, en forma cuantitativa, 10 mL de salmuera y
llevar al aforo con agua destilada, agitar, pasar una alícuota de 10 mL a un matraz

132
erlenmeyer de 250 mL, agregar 10 mL de AgNO3 0.1 N (deberá estar en exceso) y 1 mL de
HNO3 concentrado, agregar 1 mL de la solución de sulfato férrico-amónico al 5 % y agitar
vigorosamente para coagular el precipitado formado de AgCl; titular con la solución 0.1 N
de tiocianato de potasio el exceso de AgNO3 hasta obtener un color naranja pálido
permanente.
Cálculos:
% Cloruros = [(A x N1) – (B x N2)] x [meq x 100 ] / [m x a]
Donde:
A = mL de AgNO3 0.1
N1 = Normalidad de la solución de AgNO3
B = mL de KSCN 0.1 N
N2 = Normalidad de la solución de KSCN
m = Masa de muestra en gramos
a = Alícuota
5. RESULTADOS
Reportar el contenido de cloruros en la salmuera
Realizar un reporte de acuerdo al apéndice 10
V. DETERMINACIÓN DE CARBOHIDRATOS SOLUBLES
TOTALES (Índice de refracción)
1. OBJETIVO
Determinar el contenido de carbohidratos solubles totales
2. INTRODUCCIÓN
Cuando la radiación electromagnética pasa de un medio a otro, cambia de dirección, se
dobla o se refracta. La relación entre el ángulo de incidencia al seno del ángulo de

133
refracción se llama índice de refracción (RI), el cual varía con la naturaleza del compuesto,
la temperatura, la longitud de onda de la luz y la concentración del compuesto. Si las tres
primeras variables se hacen constantes la concentración del compuesto se puede determinar
midiendo el RI, de tal forma que este, se utiliza para determinar sólidos totales en
disolución. El uso del RI para determinar concentraciones es preciso solamente para
sacarosa pura u otras disoluciones puras, también se utiliza para obtener concentraciones
aproximadas de azucares para productos líquidos en cuyo caso la solución debe ser clara.
Los refractómetros pueden leer directamente en unidades de sacarosa. (Nielsen, 1998).
En general los azucares se determinan por métodos físicos indirectos
(refractómetrico, polarímetrico o aerómetrico) o por métodos químicos semiempíricos
(volumétricos o gravimétricos). La determinación por métodos refractométricos se usan
para el control rápido en la factoría. Los azucares a menudo se expresan en equivalentes de
sacarosa.
3. MATERIALES, EQUIPO Y REACTIVOS
Refractómetro ABBE
Papel Watman No. 1
Embudos
Matraces aforado de 100 mL
Solución de acetato de plomo al 10 %
Oxalato de sodio o potasio
PREPARACIÓN DE LA MUESTRA
Si las soluciones de azúcares tienen partículas en suspensión se requiere eliminarlas por
filtración en papel Whatman No. 1. Para muestras sólidas se recomienda disolver una
cantidad conocida en un volumen exacto de agua y filtrar, en caso de presentarse material
insoluble. Cuando las muestras son turbias o muy coloridas se requiere de clarificación,
para lo cual tomar una alícuota (entre 10 y 50 mL, dependiendo de la cantidad de azúcares
que contienen), y colocar en un matraz aforado de 100 mL. Diluir aproximadamente 50 mL
y tratar con 1 mL de solución de acetato de plomo, diluir al volumen y filtrar sobre papel
Whatman Nº 1, recuperando una solución clara. Para eliminar el exceso de plomo adicionar

134
oxalato de sodio o potasio sólido, mezclar y filtrar nuevamente, descartando los primeros
mL.
4. METODOLOGÍA
Colocar una o dos gotas de la solución en el prisma del refractómetro de campo,
adecuadamente calibrado. Cierre la tapa, suavemente, la muestra debe cubrir
completamente la superficie del prisma. Mirar la escala a través de la “mirilla”. Leer en la
escala, en la intersección de los campos. En caso de que la separación de los campos no sea
clara, ajustar moviendo la base del objetivo. Eliminar la muestra del prisma, utilizando un
papel suave húmedo. Los refractómetros ABBE se calibran a 0%, colocando unas gotas de
agua destilada en el prisma. Si la separación de los campos no marca 0%, en la escala,
ajustar girando el tornillo que se encuentra en la parte superior de la base del prisma.
5. RESULTADOS
Reportar el contenido de carbohidratos solubles totales en la muestra (% de
sacarosa)
Realizar un reporte de acuerdo al apéndice 10
VI. DETERMINACIÓN DE CARBOHIDRATOS REDUCTORES (Método de Lane
y Eynon)
1. OBJETIVO
Determinar el contenido de carbohidratos reductores
2. INTRODUCCIÓN
Cuando un azúcar reductor se calienta en condiciones básicas se degrada y algunos de los
productos de degradación reducen los iones cúpricos para formar oxido cuproso. (Pomeranz
y Meloan, 2000). Una amplia variedad de métodos se han utilizado para determinar

135
azucares reductores, todos estos han sido variaciones del método de Fehling. Estas
variaciones han sido para cada tipo de alimento, en cualquier caso el principal logro de
modificaciones ha sido mejorar la precisión de reducción y eliminar cualquier factor que
interfiera con la producción de oxido de cobre, de los factores estudiados, la alcalinidad del
reactivo, la proporción, el tiempo de calentamiento, la concentración del azúcar, parecen ser
los mas importantes. A partir de esto, diferentes técnicas han sido utilizadas para
determinar el oxido cuproso que se forma y se han obtenido datos de calibración; de estos
métodos los mas usados son el método de Lane-Eynon y el método Munson y Walker.
El método Lane-Eynon consiste en determinar el volumen de disolución de azúcar
que se necesita para reducir 10 o 25 mL de disolución de Fehling en presencia de azul de
metileno como indicador. El aire se elimina de la mezcla reactante manteniendo el líquido
hirviendo durante la valoración. Naturalmente la sacarosa debe de hidrolizarse a azúcar
invertido (glucosa + fructosa) antes de su valoración.
3. MATERIALES, EQUIPO Y REACTIVOS
Bureta de 25 mL
Matraces aforados de 1000 y de 500 mL
Perlas de vidrio
Placa de calentamiento
Soporte universal
Pinzas para bureta
Fenolftaleína
HCl 1 N
MaOH al 10%
Sacarosa Q. P.
Solución A de Fehling: Pesar 69.278 g CuSO4.5H2O y disolverlo en 1000 mL de agua
destilada. Se filtra.
Solución B de Fehling: Pesar 346 g de sal de Rochelle ( KNaC4H4O8. 4H2O) + 100 g de
NaOH por litro de agua. Se filtra.
Azul de metileno al 1 %.
136
NORMALIZACIÓN DE LA DISOLUCIÓN DE FEHLING
En un vaso de 300 mL se disuelven 2.375 g de sacarosa Q. P. (desecada a 100 ºC) en 130
mL de agua, se añaden 15 mL de HCl 1 N y se hierve la mezclas durante 2 minutos, con la
finalidad de obtener el azúcar invertido. Se enfría, se añaden 2 gotas de fenolftaleína, se
neutraliza con NaOH al 10 %, se pasa a un matraz aforado de 500 mL y se enrasa con agua
destilada. La disolución se valora según el método de “valoración exacta”, empleando 10
mL de la disolución de Fehling.
1 mL de disolución patrón de azúcar invertido = 0.00475 g de sacarosa = 0.005 g de azúcar
invertido.
10 mL de disolución de Fehling = 10.5 mL de disolución patrón de azúcar invertido.
Calculo del contenido de azúcar por el método de Lane y Eynon
La proporción de azúcares reductores en la disolución se puede obtener a partir del factor
apropiado de la siguiente tabla, cuando se usan 10 o 25 mL de disolución de Fehling.
Usando 10 mL de disolución de Fehling Usando 25 mL de disolución de Fehling
Titulo Azúcar invertido no sacarosa Titulo Azúcar invertido no sacarosa
15 50.6 15 123.6
16 50.6 16 123.6
17 50.7 17 123.6
18 50.8 18 123.7
19 50.8 19 123.7
20 50.8 20 123.8
21 51.0 21 123.8
22 51.0 22 123.9
23 51.1 23 123.9
24 51.2 24 124.0
25 51.2 25 124.0
26 51.3 26 124.0
27 51.4 27 124.1
28 51.4 28 124.2
29 51.5 29 124.2
30 51.5 30 124.3

137
31 51.6 31 124.3
32 51.6 32 124.4
33 51.7 33 124.4
34 51.7 34 124.5
35 51.8 35 124.5
36 51.8 36 124.6
37 51.9 37 124.6
38 51.9 38 124.7
39 52.0 39 124.7
40 52.0 40 124.8
41 52.1 41 124.8
42 52.1 42 124.9
43 52.2 43 124.9
44 52.2 44 125.0
45 52.3 45 125.0
46 52.3 46 125.1
47 52.4 47 125.1
48 52.4 48 125.2
49 52.5 49 125.2
50 52.5 50 125.3
PREPARACIÓN DE LA MUESTRA
Se sigue el mismo procedimiento de la práctica 3 (Determinación de carbohidratos solubles
totales)
4. METODOLOGÍA
Mezclar en un matraz erlenmeyer 2.5 mL de solución A y 2.5 mL de la solución B y
agregar 50 mL de agua destilada. Calentar a ebullición (con un mechero o una placa de
calentamiento) y sin quitar de la fuente de calentamiento, añadir con bureta la solución con
los carbohidratos reductores, de tal manera que sólo falte agregar de 0.5 a 1 mL para
terminar la titulación, para lo que deberá realizarse una prueba inicial y agregue 0.5 mL de
indicador de azul de metileno. Todo el proceso debe realizarse en menos de 3 min. Las
soluciones deberán tener una concentración tal, que se requiera más de 10 mL y menos de
40 mL para reducir todo el cobre del reactivo de Fehling, si se usa una bureta de 50 mL

138
Titular el reactivo de Fehling de igual forma, empleando una solución estándar para obtener
el factor correspondiente, que debe ser expresado como gramos del carbohidrato que reduce
todo el cobre en las condiciones de trabajo.
Cálculos
La proporción de azúcares reductores en la disolución se puede obtener a partir del factor
apropiado obtenido de la tabla anterior.
Azúcar (mg) por 100 mL de disolución = [Factor x 100] / [Valoración (mL)]
Además la sacarosa puede convertirse a azúcar invertido antes de la valoración, y entonces;
Concentración de sacarosa = Azúcar invertido x 0.95
5. RESULTADOS
Reportar el azúcar (mg) por 100 mL de disolución y concentración de sacarosa
contenida en la muestra
Realizar un reporte como se indica en el apéndice 10
6. BIBLIOGRAFÍA
PEARSON. D; Técnicas de laboratorio para el análisis de alimentos; Acribia, S.A.
Zaragoza (España) 1993.
HART F. L; Análisis moderno de los alimentos; Acribia. Zaragoza (España), 1991.
Association of Official Analytical Chemistry. Official Methods of Analysis
(A.O.A.C.) 1980.
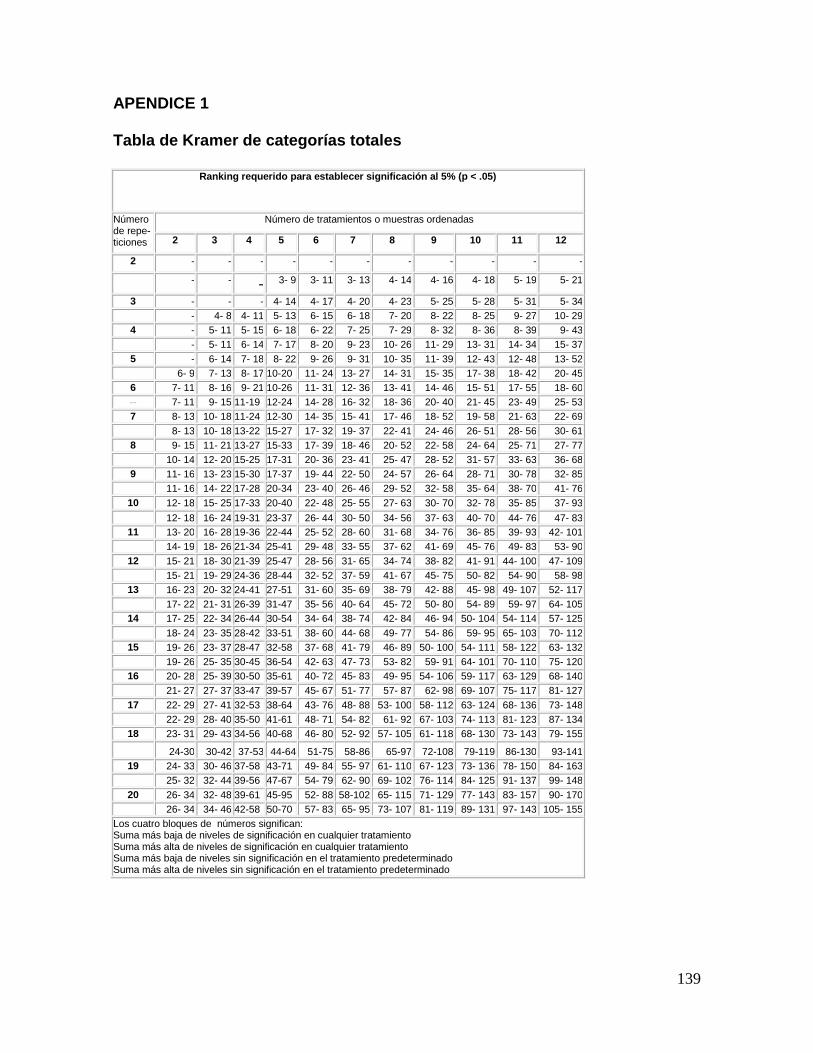
139
APENDICE 1
Tabla de Kramer de categorías totales
Ranking requerido para establecer significación al 5% (p < .05)
Número de repe- ticiones
Número de tratamientos o muestras ordenadas
2 3 4 5 6 7 8 9 10 11 12
2 - - - - - - - - - - -
- - - - 3- 9 3- 11 3- 13 4- 14 4- 16 4- 18 5- 19 5- 21
3 - - - 4- 14 4- 17 4- 20 4- 23 5- 25 5- 28 5- 31 5- 34 - - 4- 8 4- 11 5- 13 6- 15 6- 18 7- 20 8- 22 8- 25 9- 27 10- 29 4 - 5- 11 5- 15 6- 18 6- 22 7- 25 7- 29 8- 32 8- 36 8- 39 9- 43 - - 5- 11 6- 14 7- 17 8- 20 9- 23 10- 26 11- 29 13- 31 14- 34 15- 37 5 - 6- 14 7- 18 8- 22 9- 26 9- 31 10- 35 11- 39 12- 43 12- 48 13- 52 - 6- 9 7- 13 8- 17 10-20 11- 24 13- 27 14- 31 15- 35 17- 38 18- 42 20- 45 6 7- 11 8- 16 9- 21 10-26 11- 31 12- 36 13- 41 14- 46 15- 51 17- 55 18- 60 -- 7- 11 9- 15 11-19 12-24 14- 28 16- 32 18- 36 20- 40 21- 45 23- 49 25- 53 7 8- 13 10- 18 11-24 12-30 14- 35 15- 41 17- 46 18- 52 19- 58 21- 63 22- 69 - 8- 13 10- 18 13-22 15-27 17- 32 19- 37 22- 41 24- 46 26- 51 28- 56 30- 61 8 9- 15 11- 21 13-27 15-33 17- 39 18- 46 20- 52 22- 58 24- 64 25- 71 27- 77 - 10- 14 12- 20 15-25 17-31 20- 36 23- 41 25- 47 28- 52 31- 57 33- 63 36- 68 9 11- 16 13- 23 15-30 17-37 19- 44 22- 50 24- 57 26- 64 28- 71 30- 78 32- 85 - 11- 16 14- 22 17-28 20-34 23- 40 26- 46 29- 52 32- 58 35- 64 38- 70 41- 76
10 12- 18 15- 25 17-33 20-40 22- 48 25- 55 27- 63 30- 70 32- 78 35- 85 37- 93 - 12- 18 16- 24 19-31 23-37 26- 44 30- 50 34- 56 37- 63 40- 70 44- 76 47- 83
11 13- 20 16- 28 19-36 22-44 25- 52 28- 60 31- 68 34- 76 36- 85 39- 93 42- 101 - 14- 19 18- 26 21-34 25-41 29- 48 33- 55 37- 62 41- 69 45- 76 49- 83 53- 90
12 15- 21 18- 30 21-39 25-47 28- 56 31- 65 34- 74 38- 82 41- 91 44- 100 47- 109 - 15- 21 19- 29 24-36 28-44 32- 52 37- 59 41- 67 45- 75 50- 82 54- 90 58- 98
13 16- 23 20- 32 24-41 27-51 31- 60 35- 69 38- 79 42- 88 45- 98 49- 107 52- 117 - 17- 22 21- 31 26-39 31-47 35- 56 40- 64 45- 72 50- 80 54- 89 59- 97 64- 105
14 17- 25 22- 34 26-44 30-54 34- 64 38- 74 42- 84 46- 94 50- 104 54- 114 57- 125 - 18- 24 23- 35 28-42 33-51 38- 60 44- 68 49- 77 54- 86 59- 95 65- 103 70- 112
15 19- 26 23- 37 28-47 32-58 37- 68 41- 79 46- 89 50- 100 54- 111 58- 122 63- 132 - 19- 26 25- 35 30-45 36-54 42- 63 47- 73 53- 82 59- 91 64- 101 70- 110 75- 120
16 20- 28 25- 39 30-50 35-61 40- 72 45- 83 49- 95 54- 106 59- 117 63- 129 68- 140 -- 21- 27 27- 37 33-47 39-57 45- 67 51- 77 57- 87 62- 98 69- 107 75- 117 81- 127 17 22- 29 27- 41 32-53 38-64 43- 76 48- 88 53- 100 58- 112 63- 124 68- 136 73- 148 - 22- 29 28- 40 35-50 41-61 48- 71 54- 82 61- 92 67- 103 74- 113 81- 123 87- 134
18 23- 31 29- 43 34-56 40-68 46- 80 52- 92 57- 105 61- 118 68- 130 73- 143 79- 155 - 24-30 30-42 37-53 44-64 51-75 58-86 65-97 72-108 79-119 86-130 93-141
19 24- 33 30- 46 37-58 43-71 49- 84 55- 97 61- 110 67- 123 73- 136 78- 150 84- 163 - 25- 32 32- 44 39-56 47-67 54- 79 62- 90 69- 102 76- 114 84- 125 91- 137 99- 148
20 26- 34 32- 48 39-61 45-95 52- 88 58-102 65- 115 71- 129 77- 143 83- 157 90- 170 - 26- 34 34- 46 42-58 50-70 57- 83 65- 95 73- 107 81- 119 89- 131 97- 143 105- 155
Los cuatro bloques de números significan: Suma más baja de niveles de significación en cualquier tratamiento Suma más alta de niveles de significación en cualquier tratamiento Suma más baja de niveles sin significación en el tratamiento predeterminado Suma más alta de niveles sin significación en el tratamiento predeterminado
140
Tabla de Kramer de categorías totales
Ranking requerido para establecer significación al l% (p < .O1) Número de repeti- ciones
Número de tratamientos o muestras ordenadas 2 3 4 5 6 7 8 9 10 11 12
2 - - - - - - - - - - - - - - - - - - - - 3- 19 3- 21 3- 23
3 - - - - - - - - 4- 29 4- 32 4- 35 - - - - 4- 14 4- 17 4- 20 5- 22 5- 25 6- 27 6- 30 6- 33 4 - - - 5- 19 5- 23 5- 27 6- 30 6- 34 6- 38 6- 42 7- 45 - - - 5- 15 6- 18 6- 22 7- 25 8- 28 8- 32 9- 35 10- 38 10- 42 5 - 6- 19 7- 23 7- 28 8- 32 8- 37 9- 41 9- 46 10- 50 10- 55
- - 6-14 7-18 8-22 9-26 10-30 11-34 12-38 13-42 14-46 15-50 6 - 7- 17 8- 22 9- 27 9- 33 10- 38 11- 43 12- 48 13- 53 13- 59 14- 64 - - 8- 16 9- 21 10- 26 12- 30 13- 35 14- 40 16- 44 17- 49 18- 54 20- 58 7 - 8- 20 10- 25 11- 31 12- 37 13- 43 14- 49 15- 55 16- 61 17- 67 18- 73 - 8- 13 9- 19 11- 24 12- 30 14- 35 16- 40 18- 45 19- 51 21- 56 23- 61 25- 66 8 9- 15 10- 22 11- 29 13- 35 14- 42 16- 48 17- 55 19- 61 20- 68 21- 75 23- 81 - 9- 15 11- 21 13- 27 15- 33 17- 39 19- 45 21- 51 23- 57 25- 63 28- 68 30- 74 9 10- 17 12- 24 13- 32 15- 39 17- 46 19- 53 21- 60 22- 68 24- 75 26- 82 27- 90 - 10- 17 12- 24 15- 30 17- 37 20- 43 22- 50 25- 56 27- 63 30- 69 32- 76 35- 82
10 11- 19 13- 27 15- 35 18- 42 20- 50 22- 58 24- 66 26- 74 28- 82 30- 90 32- 98 - 11- 19 14- 26 17- 33 20- 40 23- 47 25- 55 28- 62 31- 69 34- 76 37- 83 40- 90
11 12- 21 15- 29 17- 38 20- 46 22- 55 25- 63 27- 72 30- 80 32- 89 34- 98 37- 106 - 13- 20 16- 28 19- 36 22- 44 25- 52 29- 59 32- 67 35- 75 39- 82 42- 90 45- 98
12 14- 22 17- 31 19- 41 22- 50 25- 59 28- 68 31- 77 33- 87 36- 96 39- 105 42- 114 --- 14- 22 18- 30 21- 39 25- 47 28- 56 32- 64 36- 72 39- 81 43- 89 47- 97 50- 106 13 15- 24 18- 34 21- 44 25- 53 28- 63 31- 73 34- 83 37- 93 40- 103 43- 113 46- 123 - 15- 24 19- 33 23- 42 27- 51 31- 60 35- 69 39- 78 44- 86 48- 95 52- 104 56- 113
14 16- 26 20- 36 24- 46 27- 57 31- 67 34- 78 38- 88 41- 98 45- 109 48- 120 51- 131 - 17- 25 21- 35 25- 45 30- 54 34- 64 39- 73 43- 83 48- 92 52- 102 57- 121 61- 121
15 18- 27 22- 38 26- 49 30- 60 34- 71 37- 83 41- 94 45- 105 49- 116 53- 127 56- 139 - 18- 27 23- 37 28- 47 32- 58 37- 68 42- 78 47- 88 52- 98 57- 108 62- 118 67- 128
16 19- 29 23- 41 28- 52 32- 64 36- 76 41- 87 45- 99 49- 111 53- 123 57- 135 62- 146 -- 19- 29 25- 39 30- 50 35- 61 40- 72 46- 82 51- 93 56- 104 61- 115 67- 125 72- 136 17 20- 31 25- 43 30- 55 35- 67 39- 80 44- 92 49- 104 53- 117 58- 129 62- 142 67- 154 - 21- 30 26- 42 32- 53 38- 64 43- 76 49- 87 55- 98 60- 110 66- 121 72- 132 78- 143
18 22- 32 27- 45 32- 58 37- 71 42- 84 47- 97 52- 110 57- 123 62- 136 67- 149 72- 162 - 22- 32 28- 44 34- 56 40- 68 46- 80 52- 92 57- 105 62- 118 68- 130 73- 143 79- 155
19 23- 34 29- 47 34- 61 40- 74 45- 88 50- 102 56- 115 61- 129 67- 142 72- 156 77- 170 - 24- 33 30- 46 36- 59 43- 71 49- 84 56- 96 62- 109 69- 121 76- 133 82- 146 89- 158
20 24- 36 30- 50 36- 64 42- 78 48- 92 54- 106 60- 120 65- 135 71- 149 77- 163 82- 178 - 25- 35 32- 48 38- 62 45- 75 52- 88 59- 101 66- 114 73- 127 80- 140 87- 153 94- 166
Estas Tablas corresponden a una reproducción de las Tablas de A. Kramer, publicadas en Food Technol. 17 (12), 124-125 (1963).
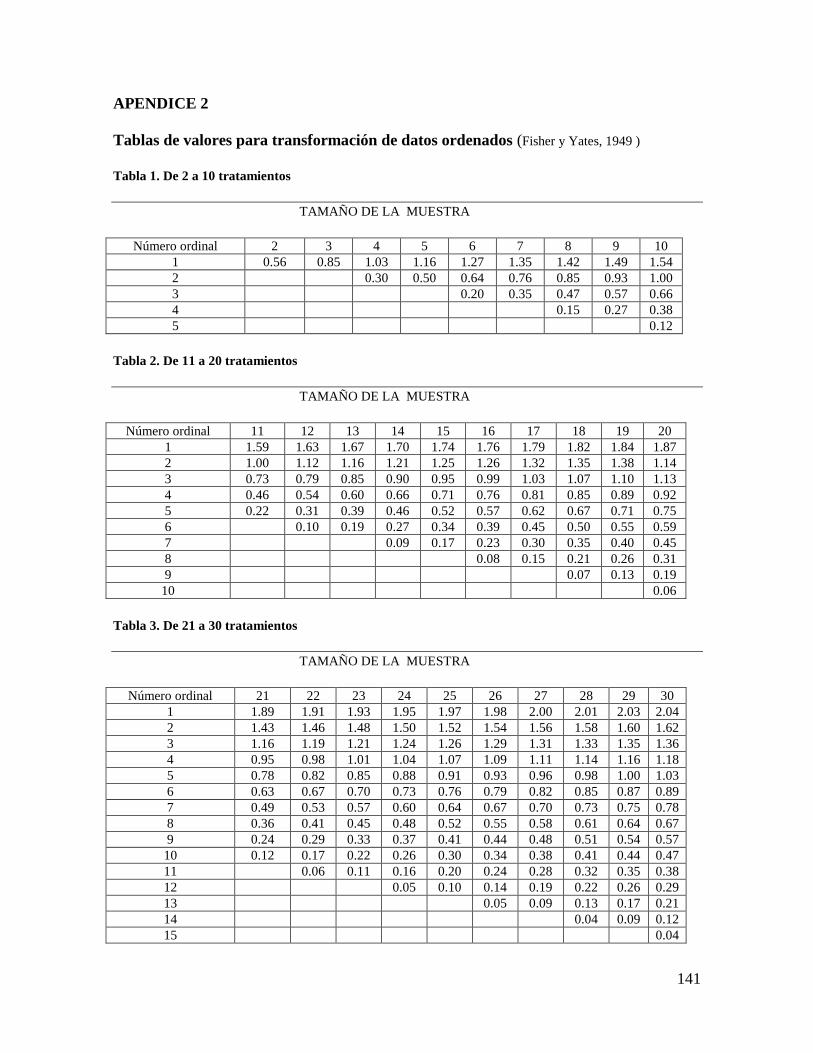
141
APENDICE 2
Tablas de valores para transformación de datos ordenados (Fisher y Yates, 1949 )
Tabla 1. De 2 a 10 tratamientos
TAMAÑO DE LA MUESTRA
Número ordinal 2 3 4 5 6 7 8 9 10
1 0.56 0.85 1.03 1.16 1.27 1.35 1.42 1.49 1.54
2 0.30 0.50 0.64 0.76 0.85 0.93 1.00
3 0.20 0.35 0.47 0.57 0.66
4 0.15 0.27 0.38
5 0.12
Tabla 2. De 11 a 20 tratamientos
TAMAÑO DE LA MUESTRA
Número ordinal 11 12 13 14 15 16 17 18 19 20
1 1.59 1.63 1.67 1.70 1.74 1.76 1.79 1.82 1.84 1.87
2 1.00 1.12 1.16 1.21 1.25 1.26 1.32 1.35 1.38 1.14
3 0.73 0.79 0.85 0.90 0.95 0.99 1.03 1.07 1.10 1.13
4 0.46 0.54 0.60 0.66 0.71 0.76 0.81 0.85 0.89 0.92
5 0.22 0.31 0.39 0.46 0.52 0.57 0.62 0.67 0.71 0.75
6 0.10 0.19 0.27 0.34 0.39 0.45 0.50 0.55 0.59
7 0.09 0.17 0.23 0.30 0.35 0.40 0.45
8 0.08 0.15 0.21 0.26 0.31
9 0.07 0.13 0.19
10 0.06
Tabla 3. De 21 a 30 tratamientos
TAMAÑO DE LA MUESTRA
Número ordinal 21 22 23 24 25 26 27 28 29 30
1 1.89 1.91 1.93 1.95 1.97 1.98 2.00 2.01 2.03 2.04
2 1.43 1.46 1.48 1.50 1.52 1.54 1.56 1.58 1.60 1.62
3 1.16 1.19 1.21 1.24 1.26 1.29 1.31 1.33 1.35 1.36
4 0.95 0.98 1.01 1.04 1.07 1.09 1.11 1.14 1.16 1.18
5 0.78 0.82 0.85 0.88 0.91 0.93 0.96 0.98 1.00 1.03
6 0.63 0.67 0.70 0.73 0.76 0.79 0.82 0.85 0.87 0.89
7 0.49 0.53 0.57 0.60 0.64 0.67 0.70 0.73 0.75 0.78
8 0.36 0.41 0.45 0.48 0.52 0.55 0.58 0.61 0.64 0.67
9 0.24 0.29 0.33 0.37 0.41 0.44 0.48 0.51 0.54 0.57
10 0.12 0.17 0.22 0.26 0.30 0.34 0.38 0.41 0.44 0.47
11 0.06 0.11 0.16 0.20 0.24 0.28 0.32 0.35 0.38
12 0.05 0.10 0.14 0.19 0.22 0.26 0.29
13 0.05 0.09 0.13 0.17 0.21
14 0.04 0.09 0.12
15 0.04

142
APENDICE 3
ANALISIS DE VARIANZA PARA EVALUACIONES SENSORIALES CON UNA
VARIABLE (PRODUCTO) Y REPETICIONES (CATADORES O JUECES)
Para analizar el efecto de varios niveles de una variable (dulzor, intensidad de color, etc.)
en un producto, se aplica el método estadístico de la varianza. En él se compara la variación
propia de la variable con la residual, o sea, la varianza debida al error experimental y la
debida al azar.
En primer lugar se determinan los grados de libertad:
GLv = grados de libertad de la variable = m – 1
en donde m son los niveles (dulzor, intensidad de color, etc.) de la variable en estudio
GLj = Grados de libertad de jueces = n – 1
En donde n es el número de jueces
GLt = grados de libertad totales = (n)(m) -1
GLr = Grados de libertad de residual = GLt – GLv – GLj
Luego se calcula el factor de corrección a partir de la suma de cuadrados
FC = Factor de corrección = TT2/ [(n)(m)]
En donde TT es la suma total de observaciones, es decir:
TT = ∑Xij
Y con este factor de corrección se pasa a obtener las sumas de cuadrados:
SCv = Suma de los cuadrados de la variable = [(Tc1)2
+ (Tc2)2+…+ (Tcm)
2] / [n - FC]
En donde Tcj son los totales de cada columna, j = 1,2,3,….,m
SCj = Suma de cuadrados de los jueces = [(Tr1)2 + (Tr2)
2+….+(Trm)
2] / [m - FC]
En donde Tri son los totales de cada fila, i = 1,2,3, ….., n
SCt = Suma de cuadrados totales
= Suma de cada observación al cuadrado – FC

143
= [ (X11)2 + (X12)
2+ ….+ (Xnm)
2] - FC
SCr = Suma de cuadrados del residual = SCt – SCv – SCj
Con estos valores corregidos, ya se pueden calcular las varianzas, que se obtienen
dividiendo la suma de los cuadrados entre los grados de libertad correspondientes a cada
una de ellas:
Vv = Varianza debida a la variable = SCv / GLv
Vj = Varianza debida a los jueces = Scj / GLj
Vr = Varianza debida al azar o residual = SCr / GLr
Finalmente con las varianza se obtienen los valores F, de acuerdo a las formulas:
Fv = Vv / Vr
Fj = Vj / Vr
Estos valores de F se comparan con los de las tablas (Ft), con los grados de libertad de la
fuente de variación que se esté considerando, es decir, GLv o GLj como los grados de
libertad en el numerador (n1) y, siempre, con GLr, como grados de libertad en el
denominador (n2), y con un nivel de significancia escogido, 5% o 1% en la mayoría de los
casos. Si F<Ft no hay efecto significativo de la fuente de variación considerada sobre los
resultados; en cambio si es mayor o igual, sí hay diferencia significativa y en este caso es
conveniente obtener la diferencia mínima significativa con la prueba de Tukey.
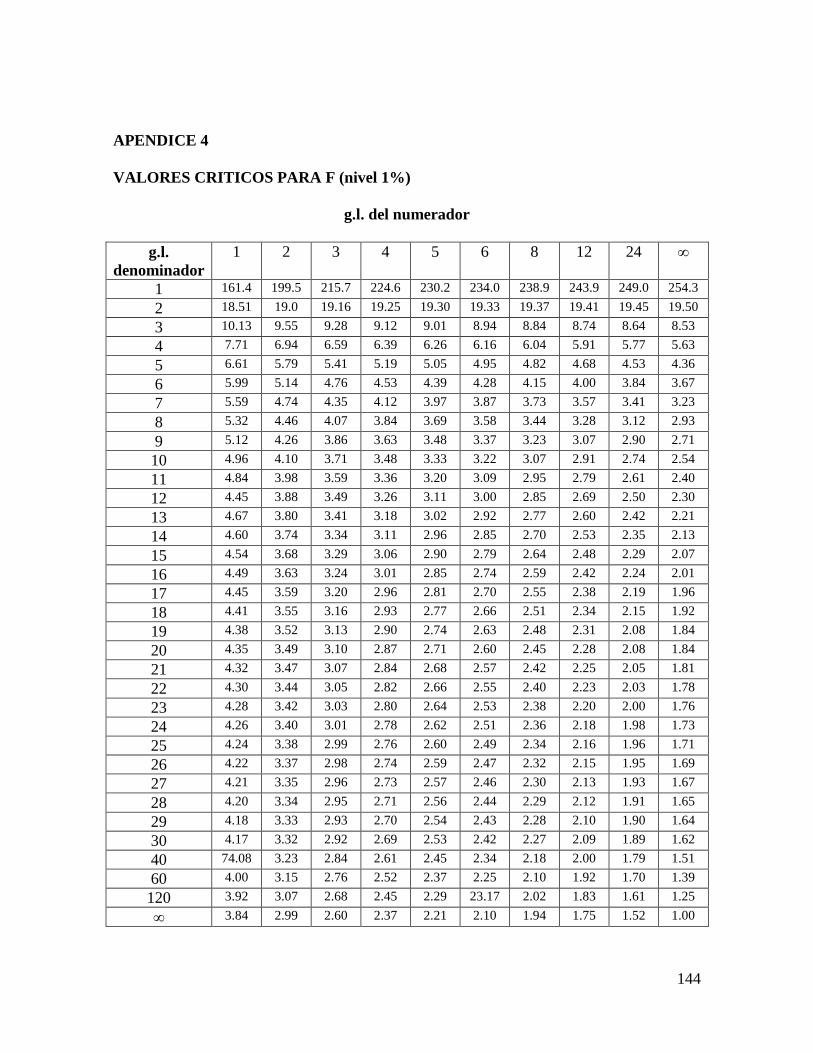
144
APENDICE 4
VALORES CRITICOS PARA F (nivel 1%)
g.l. del numerador
g.l.
denominador
1 2 3 4 5 6 8 12 24 ∞
1 161.4 199.5 215.7 224.6 230.2 234.0 238.9 243.9 249.0 254.3
2 18.51 19.0 19.16 19.25 19.30 19.33 19.37 19.41 19.45 19.50
3 10.13 9.55 9.28 9.12 9.01 8.94 8.84 8.74 8.64 8.53
4 7.71 6.94 6.59 6.39 6.26 6.16 6.04 5.91 5.77 5.63
5 6.61 5.79 5.41 5.19 5.05 4.95 4.82 4.68 4.53 4.36
6 5.99 5.14 4.76 4.53 4.39 4.28 4.15 4.00 3.84 3.67
7 5.59 4.74 4.35 4.12 3.97 3.87 3.73 3.57 3.41 3.23
8 5.32 4.46 4.07 3.84 3.69 3.58 3.44 3.28 3.12 2.93
9 5.12 4.26 3.86 3.63 3.48 3.37 3.23 3.07 2.90 2.71
10 4.96 4.10 3.71 3.48 3.33 3.22 3.07 2.91 2.74 2.54
11 4.84 3.98 3.59 3.36 3.20 3.09 2.95 2.79 2.61 2.40
12 4.45 3.88 3.49 3.26 3.11 3.00 2.85 2.69 2.50 2.30
13 4.67 3.80 3.41 3.18 3.02 2.92 2.77 2.60 2.42 2.21
14 4.60 3.74 3.34 3.11 2.96 2.85 2.70 2.53 2.35 2.13
15 4.54 3.68 3.29 3.06 2.90 2.79 2.64 2.48 2.29 2.07
16 4.49 3.63 3.24 3.01 2.85 2.74 2.59 2.42 2.24 2.01
17 4.45 3.59 3.20 2.96 2.81 2.70 2.55 2.38 2.19 1.96
18 4.41 3.55 3.16 2.93 2.77 2.66 2.51 2.34 2.15 1.92
19 4.38 3.52 3.13 2.90 2.74 2.63 2.48 2.31 2.08 1.84
20 4.35 3.49 3.10 2.87 2.71 2.60 2.45 2.28 2.08 1.84
21 4.32 3.47 3.07 2.84 2.68 2.57 2.42 2.25 2.05 1.81
22 4.30 3.44 3.05 2.82 2.66 2.55 2.40 2.23 2.03 1.78
23 4.28 3.42 3.03 2.80 2.64 2.53 2.38 2.20 2.00 1.76
24 4.26 3.40 3.01 2.78 2.62 2.51 2.36 2.18 1.98 1.73
25 4.24 3.38 2.99 2.76 2.60 2.49 2.34 2.16 1.96 1.71
26 4.22 3.37 2.98 2.74 2.59 2.47 2.32 2.15 1.95 1.69
27 4.21 3.35 2.96 2.73 2.57 2.46 2.30 2.13 1.93 1.67
28 4.20 3.34 2.95 2.71 2.56 2.44 2.29 2.12 1.91 1.65
29 4.18 3.33 2.93 2.70 2.54 2.43 2.28 2.10 1.90 1.64
30 4.17 3.32 2.92 2.69 2.53 2.42 2.27 2.09 1.89 1.62
40 74.08 3.23 2.84 2.61 2.45 2.34 2.18 2.00 1.79 1.51
60 4.00 3.15 2.76 2.52 2.37 2.25 2.10 1.92 1.70 1.39
120 3.92 3.07 2.68 2.45 2.29 23.17 2.02 1.83 1.61 1.25
∞ 3.84 2.99 2.60 2.37 2.21 2.10 1.94 1.75 1.52 1.00
145
VALORES CRITICOS PARA F (Nivel 5%).
g.l. del numerador
g.l.
denominador 1 2 3 4 5 6 8 12 24 ∞
1 4052 4999 5403 5625 5764 5859 5982 6106 6234 6366
2 68.50 99.00 99.17 99.25 99.30 99.33 99.37 99.42 99.46 99.50
3 34.12 30.82 29.46 28.71 28.24 27.91 27.49 27.05 26.60 26.12
4 21.2 18.00 16.69 15.98 15.52 15.21 14.80 14.37 13.93 13.46
5 16.26 13.27 12.06 11.39 10.97 10.67 10.29 9.89 9.47 9.02
6 13.74 10.92 9.78 9.15 8.75 8.47 8.10 7.72 7.31 6.88
7 12.25 9.55 8.45 7.85 7.46 7.16 6.84 6.47 6.07 5.65
8 11.26 8.65 7.59 7.01 6.63 6.37 6.03 5.67 5.28 4.86
9 10.56 8.02 6.99 6.42 6.06 5.80 5.47 5.11 4.73 4.31
10 10.04 7.56 6.55 5.99 5.64 5.39 5.06 4.71 4.33 3.91
11 9.65 7.20 6.22 5.67 5.32 5.07 4.74 4.40 4.02 3.60
12 9.33 6.83 5.95 5.41 5.06 4.82 4.50 4.16 3.78 3.36
13 9.07 6.70 5.74 5.20 4.86 4.62 4.30 3.96 3.59 3.16
14 8.86 6.51 5.56 5.03 4.69 4.46 4.14 3.80 3.43 3.00
15 8.68 6.36 5.42 4.89 4.56 4.32 4.00 3.67 3.29 2.87
16 8.53 6.23 5.29 4.77 4.44 4.20 3.89 3.55 3.18 2.75
17 8.40 6.11 5.18 4.67 4.34 4.10 3.79 3.45 3.08 2.65
18 8.28 6.01 5.09 4.58 4.25 4.01 3.71 3.37 3.00 2.57
19 8.18 5.93 5.01 4.50 4.17 3.94 3.63 3.30 2.92 2.49
20 8.10 5.85 4.94 4.43 4.10 3.87 3.56 3.23 2.86 2.452
21 8.02 5.78 4.87 4.37 4.04 3.81 3.51 3.17 2.80 2.36
22 7.94 5.72 4.82 4.31 3.99 3.76 3.45 3.12 2.75 2.31
23 7.88 5.66 4.76 4.26 3.94 3.71 3.41 3.07 2.70 2.26
24 7.82 5.61 4.72 4.22 3.90 3.67 3.36 3.03 2.66 2.21
25 7.77 5.57 4.68 4.18 3.86 3.63 3.32 2.99 2.62 2.17
26 7.72 5.53 4.46 4.14 3.82 3.59 3.29 2.96 2.58 2.13
27 7.68 5.49 4.60 4.11 3.78 3.56 3.26 2.93 2.55 2.10
28 7.64 5.45 4.57 4.07 3.75 3.53 3.23 2.90 2.52 2.06
29 7.60 5.42 4.54 4.04 3.73 3.50 3.20 2.87 2.49 2.03
30 7.56 5.39 4.51 4.02 3.70 3.47 3.17 2.84 2.47 2.01
40 7.31 5.18 4.31 3.83 3.51 3.29 2.99 2.66 2.29 1.80
60 7.08 4.98 4.13 3.65 3.34 3.12 2.82 2.50 2.12 1.60
120 6.85 4.79 3.95 3.48 3.17 2.96 2.66 2.34 1.95 1.38
∞ 6.64 4.60 3.78 3.32 3.02 2.80 2.51 2.18 1.79 1.00
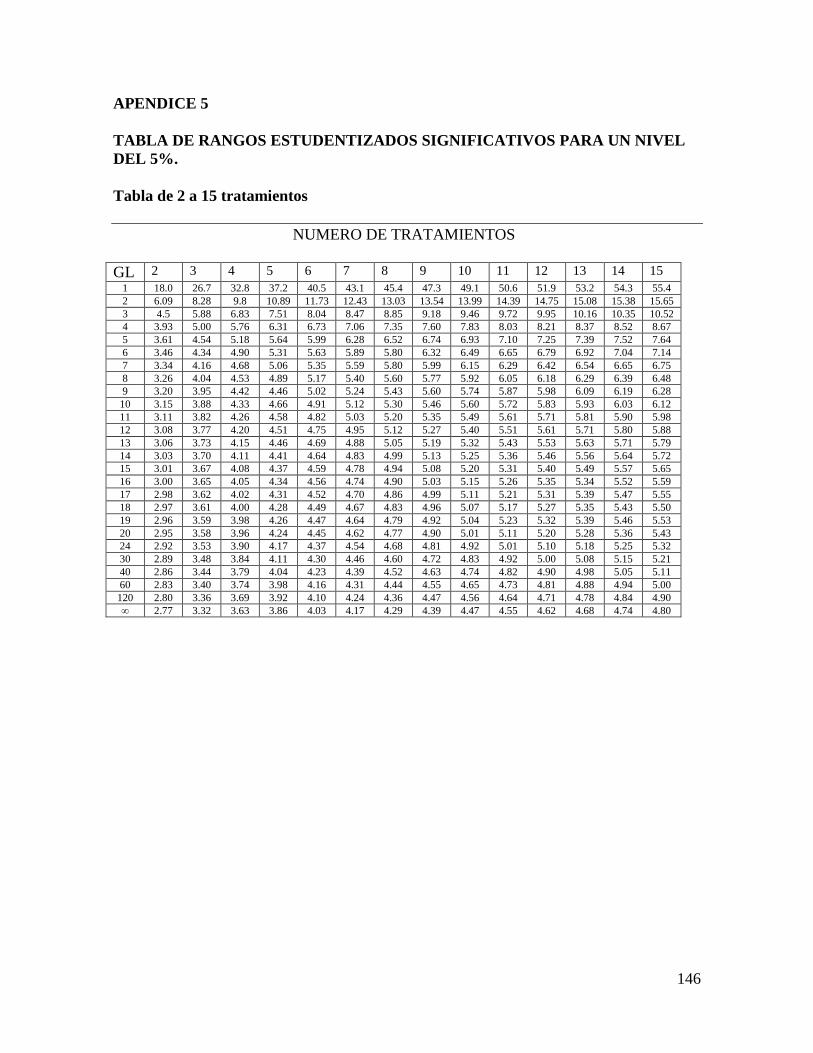
146
APENDICE 5
TABLA DE RANGOS ESTUDENTIZADOS SIGNIFICATIVOS PARA UN NIVEL
DEL 5%.
Tabla de 2 a 15 tratamientos
NUMERO DE TRATAMIENTOS
GL 2 3 4 5 6 7 8 9 10 11 12 13 14 15
1 18.0 26.7 32.8 37.2 40.5 43.1 45.4 47.3 49.1 50.6 51.9 53.2 54.3 55.4
2 6.09 8.28 9.8 10.89 11.73 12.43 13.03 13.54 13.99 14.39 14.75 15.08 15.38 15.65
3 4.5 5.88 6.83 7.51 8.04 8.47 8.85 9.18 9.46 9.72 9.95 10.16 10.35 10.52
4 3.93 5.00 5.76 6.31 6.73 7.06 7.35 7.60 7.83 8.03 8.21 8.37 8.52 8.67
5 3.61 4.54 5.18 5.64 5.99 6.28 6.52 6.74 6.93 7.10 7.25 7.39 7.52 7.64
6 3.46 4.34 4.90 5.31 5.63 5.89 5.80 6.32 6.49 6.65 6.79 6.92 7.04 7.14
7 3.34 4.16 4.68 5.06 5.35 5.59 5.80 5.99 6.15 6.29 6.42 6.54 6.65 6.75
8 3.26 4.04 4.53 4.89 5.17 5.40 5.60 5.77 5.92 6.05 6.18 6.29 6.39 6.48
9 3.20 3.95 4.42 4.46 5.02 5.24 5.43 5.60 5.74 5.87 5.98 6.09 6.19 6.28
10 3.15 3.88 4.33 4.66 4.91 5.12 5.30 5.46 5.60 5.72 5.83 5.93 6.03 6.12
11 3.11 3.82 4.26 4.58 4.82 5.03 5.20 5.35 5.49 5.61 5.71 5.81 5.90 5.98
12 3.08 3.77 4.20 4.51 4.75 4.95 5.12 5.27 5.40 5.51 5.61 5.71 5.80 5.88
13 3.06 3.73 4.15 4.46 4.69 4.88 5.05 5.19 5.32 5.43 5.53 5.63 5.71 5.79
14 3.03 3.70 4.11 4.41 4.64 4.83 4.99 5.13 5.25 5.36 5.46 5.56 5.64 5.72
15 3.01 3.67 4.08 4.37 4.59 4.78 4.94 5.08 5.20 5.31 5.40 5.49 5.57 5.65
16 3.00 3.65 4.05 4.34 4.56 4.74 4.90 5.03 5.15 5.26 5.35 5.34 5.52 5.59
17 2.98 3.62 4.02 4.31 4.52 4.70 4.86 4.99 5.11 5.21 5.31 5.39 5.47 5.55
18 2.97 3.61 4.00 4.28 4.49 4.67 4.83 4.96 5.07 5.17 5.27 5.35 5.43 5.50
19 2.96 3.59 3.98 4.26 4.47 4.64 4.79 4.92 5.04 5.23 5.32 5.39 5.46 5.53
20 2.95 3.58 3.96 4.24 4.45 4.62 4.77 4.90 5.01 5.11 5.20 5.28 5.36 5.43
24 2.92 3.53 3.90 4.17 4.37 4.54 4.68 4.81 4.92 5.01 5.10 5.18 5.25 5.32
30 2.89 3.48 3.84 4.11 4.30 4.46 4.60 4.72 4.83 4.92 5.00 5.08 5.15 5.21
40 2.86 3.44 3.79 4.04 4.23 4.39 4.52 4.63 4.74 4.82 4.90 4.98 5.05 5.11
60 2.83 3.40 3.74 3.98 4.16 4.31 4.44 4.55 4.65 4.73 4.81 4.88 4.94 5.00
120 2.80 3.36 3.69 3.92 4.10 4.24 4.36 4.47 4.56 4.64 4.71 4.78 4.84 4.90
∞ 2.77 3.32 3.63 3.86 4.03 4.17 4.29 4.39 4.47 4.55 4.62 4.68 4.74 4.80
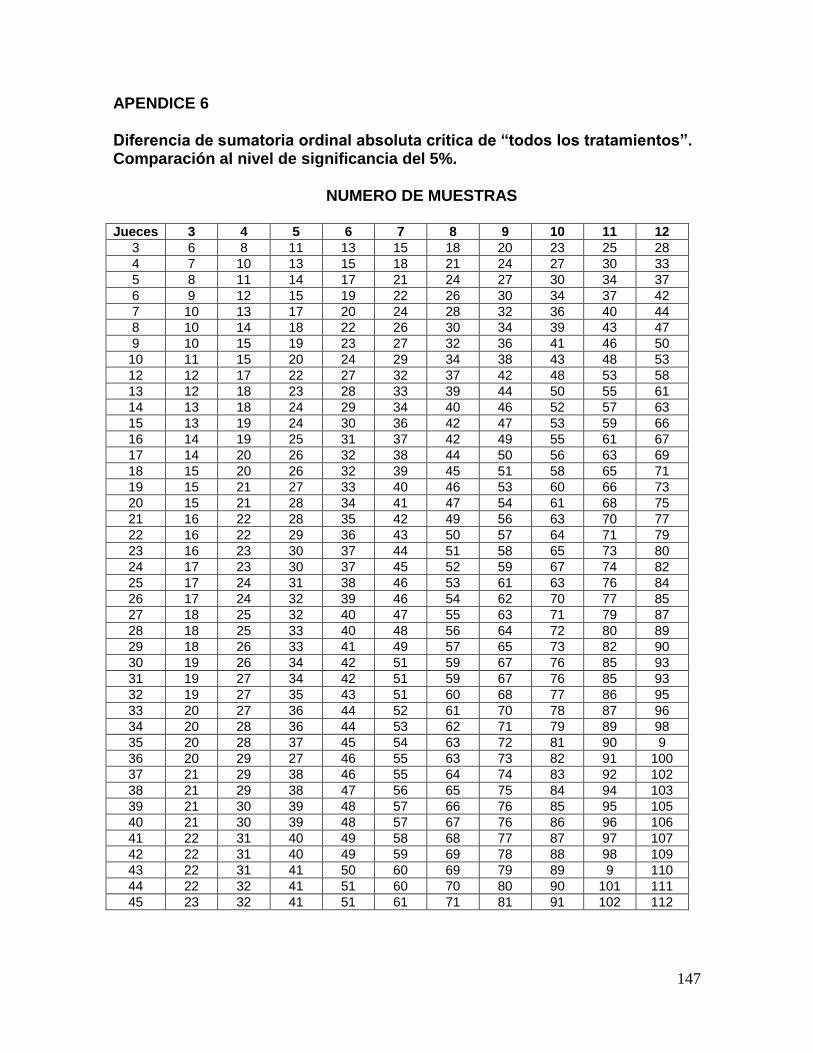
147
APENDICE 6
Diferencia de sumatoria ordinal absoluta crítica de “todos los tratamientos”. Comparación al nivel de significancia del 5%.
NUMERO DE MUESTRAS
Jueces 3 4 5 6 7 8 9 10 11 12
3 6 8 11 13 15 18 20 23 25 28
4 7 10 13 15 18 21 24 27 30 33
5 8 11 14 17 21 24 27 30 34 37
6 9 12 15 19 22 26 30 34 37 42
7 10 13 17 20 24 28 32 36 40 44
8 10 14 18 22 26 30 34 39 43 47
9 10 15 19 23 27 32 36 41 46 50
10 11 15 20 24 29 34 38 43 48 53
12 12 17 22 27 32 37 42 48 53 58
13 12 18 23 28 33 39 44 50 55 61
14 13 18 24 29 34 40 46 52 57 63
15 13 19 24 30 36 42 47 53 59 66
16 14 19 25 31 37 42 49 55 61 67
17 14 20 26 32 38 44 50 56 63 69
18 15 20 26 32 39 45 51 58 65 71
19 15 21 27 33 40 46 53 60 66 73
20 15 21 28 34 41 47 54 61 68 75
21 16 22 28 35 42 49 56 63 70 77
22 16 22 29 36 43 50 57 64 71 79
23 16 23 30 37 44 51 58 65 73 80
24 17 23 30 37 45 52 59 67 74 82
25 17 24 31 38 46 53 61 63 76 84
26 17 24 32 39 46 54 62 70 77 85
27 18 25 32 40 47 55 63 71 79 87
28 18 25 33 40 48 56 64 72 80 89
29 18 26 33 41 49 57 65 73 82 90
30 19 26 34 42 51 59 67 76 85 93
31 19 27 34 42 51 59 67 76 85 93
32 19 27 35 43 51 60 68 77 86 95
33 20 27 36 44 52 61 70 78 87 96
34 20 28 36 44 53 62 71 79 89 98
35 20 28 37 45 54 63 72 81 90 9
36 20 29 27 46 55 63 73 82 91 100
37 21 29 38 46 55 64 74 83 92 102
38 21 29 38 47 56 65 75 84 94 103
39 21 30 39 48 57 66 76 85 95 105
40 21 30 39 48 57 67 76 86 96 106
41 22 31 40 49 58 68 77 87 97 107
42 22 31 40 49 59 69 78 88 98 109
43 22 31 41 50 60 69 79 89 9 110
44 22 32 41 51 60 70 80 90 101 111
45 23 32 41 51 61 71 81 91 102 112

148
Diferencia de sumatoria ordinal absoluta crítica de “todos los tratamientos”. Comparación al nivel de significancia del 1%.
NUMERO DE MUESTRAS
Jueces 3 4 5 6 7 8 9 10 11 12
3 - 9 12 14 17 19 22 24 27 30
4 8 11 14 17 20 23 26 29 32 36
5 8 11 14 17 2 23 26 29 32 36
6 10 14 18 21 25 29 33 37 41 45
7 11 15 19 23 28 32 36 40 45 49
8 12 16 21 25 30 34 39 43 48 53
9 13 17 22 27 32 36 41 46 51 56
10 13 18 23 28 33 38 44 49 54 59
11 14 19 24 30 35 40 46 51 57 63
12 15 20 26 31 37 42 48 54 60 66
13 15 21 27 32 38 44 50 56 62 68
14 16 22 28 34 40 46 52 58 65 71
15 16 22 28 35 41 48 54 60 67 74
16 17 23 30 36 43 49 56 63 70 77
17 17 24 31 37 44 51 58 65 72 79
18 18 25 31 38 45 52 60 67 74 81
19 18 25 32 39 46 54 61 69 76 84
20 19 26 33 40 48 55 63 70 78 86
21 19 27 34 41 49 56 64 72 80 88
22 20 27 35 42 50 58 66 74 82 90
23 20 28 32 43 51 59 67 75 84 92
24 21 28 36 44 52 60 69 77 85 94
25 21 29 37 45 53 62 70 79 87 96
26 22 29 38 46 54 63 71 80 89 98
27 22 30 38 47 55 64 73 82 91 100
28 22 31 39 48 56 65 74 83 92 101
29 23 31 40 48 57 66 75 85 94 103
30 23 32 40 49 58 67 77 86 95 105
31 23 32 41 50 59 69 78 87 97 107
32 24 33 42 51 60 70 79 89 99 108
33 24 33 42 52 61 71 80 90 100 110
34 25 34 43 52 62 72 82 92 102 112
35 25 34 44 53 63 73 83 93 103 113
36 25 35 44 54 64 74 84 94 105 115
37 26 35 45 55 65 75 85 95 106 117
38 26 36 45 55 66 76 86 97 107 118
39 26 36 46 56 66 77 87 98 109 120
40 27 36 47 57 67 78 88 99 110 121
41 27 37 47 57 69 79 90 100 112 123
42 27 37 48 58 69 80 91 102 113 124
43 28 38 48 59 70 81 92 103 114 126
44 28 38 49 60 70 82 93 104 115 127
45 28 39 49 60 71 82 94 105 117 128
46 28 39 50 61 72 83 95 106 118 130
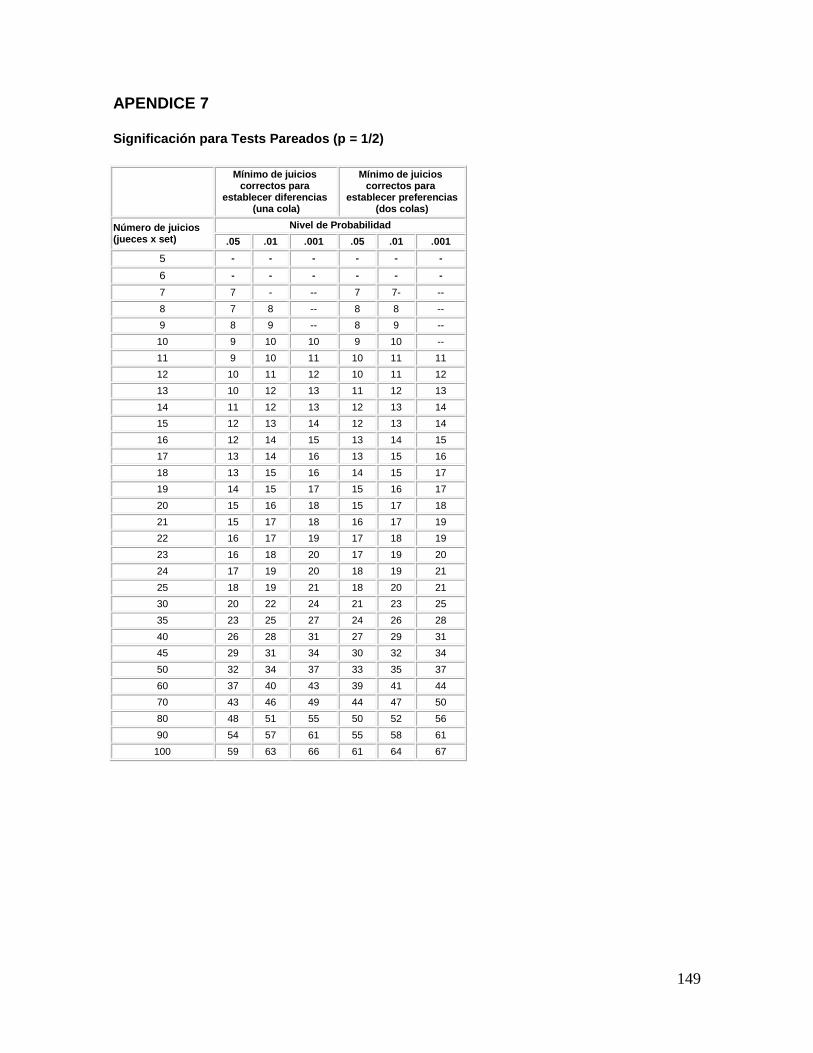
149
APENDICE 7
Significación para Tests Pareados (p = 1/2)
- Mínimo de juicios
correctos para establecer diferencias
(una cola)
Mínimo de juicios correctos para
establecer preferencias (dos colas)
Número de juicios (jueces x set)
Nivel de Probabilidad .05 .01 .001 .05 .01 .001
5 - - - - - -
6 - - - - - -
7 7 - -- 7 7- -- 8 7 8 -- 8 8 -- 9 8 9 -- 8 9 -- 10 9 10 10 9 10 -- 11 9 10 11 10 11 11 12 10 11 12 10 11 12 13 10 12 13 11 12 13 14 11 12 13 12 13 14 15 12 13 14 12 13 14 16 12 14 15 13 14 15 17 13 14 16 13 15 16 18 13 15 16 14 15 17 19 14 15 17 15 16 17 20 15 16 18 15 17 18 21 15 17 18 16 17 19 22 16 17 19 17 18 19 23 16 18 20 17 19 20 24 17 19 20 18 19 21 25 18 19 21 18 20 21 30 20 22 24 21 23 25 35 23 25 27 24 26 28 40 26 28 31 27 29 31 45 29 31 34 30 32 34 50 32 34 37 33 35 37 60 37 40 43 39 41 44 70 43 46 49 44 47 50 80 48 51 55 50 52 56 90 54 57 61 55 58 61
100 59 63 66 61 64 67
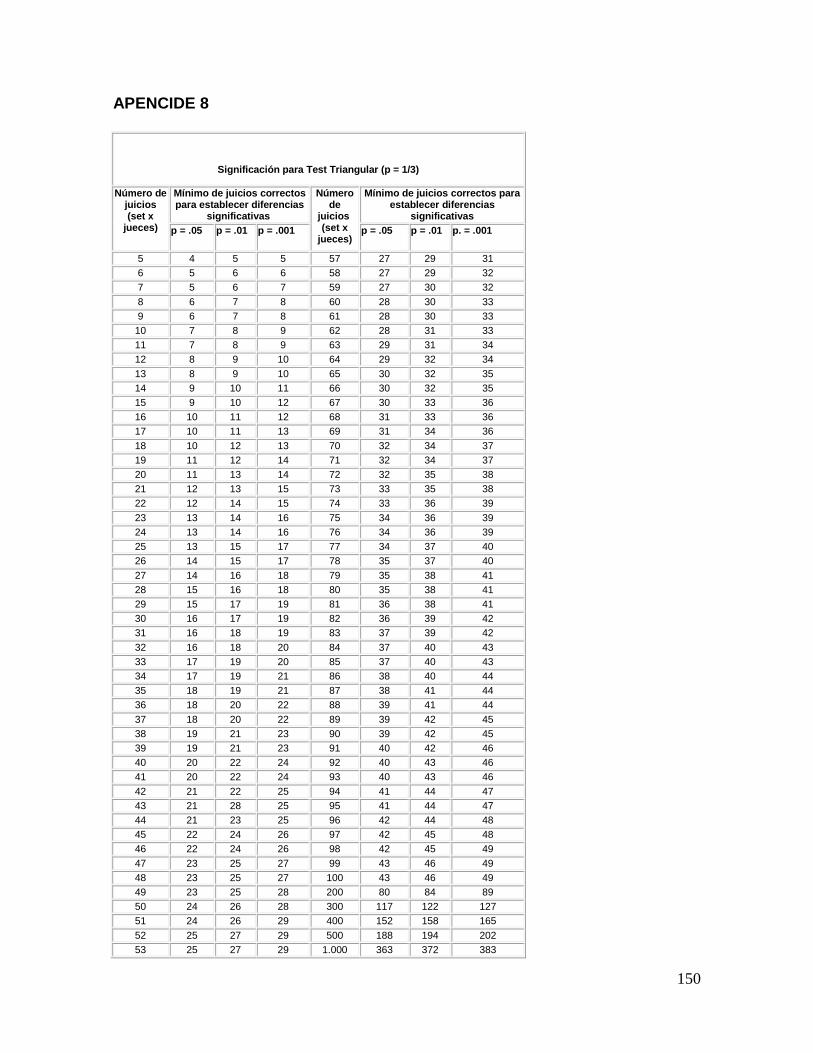
150
APENCIDE 8
Significación para Test Triangular (p = 1/3)
Número de juicios (set x
jueces)
Mínimo de juicios correctos para establecer diferencias
significativas
Número de
juicios (set x
jueces)
Mínimo de juicios correctos para establecer diferencias
significativas p = .05 p = .01 p = .001 p = .05 p = .01 p. = .001
5 4 5 5 57 27 29 31 6 5 6 6 58 27 29 32 7 5 6 7 59 27 30 32 8 6 7 8 60 28 30 33 9 6 7 8 61 28 30 33
10 7 8 9 62 28 31 33 11 7 8 9 63 29 31 34 12 8 9 10 64 29 32 34 13 8 9 10 65 30 32 35 14 9 10 11 66 30 32 35 15 9 10 12 67 30 33 36 16 10 11 12 68 31 33 36 17 10 11 13 69 31 34 36 18 10 12 13 70 32 34 37 19 11 12 14 71 32 34 37 20 11 13 14 72 32 35 38 21 12 13 15 73 33 35 38 22 12 14 15 74 33 36 39 23 13 14 16 75 34 36 39 24 13 14 16 76 34 36 39 25 13 15 17 77 34 37 40 26 14 15 17 78 35 37 40 27 14 16 18 79 35 38 41 28 15 16 18 80 35 38 41 29 15 17 19 81 36 38 41 30 16 17 19 82 36 39 42 31 16 18 19 83 37 39 42 32 16 18 20 84 37 40 43 33 17 19 20 85 37 40 43 34 17 19 21 86 38 40 44 35 18 19 21 87 38 41 44 36 18 20 22 88 39 41 44 37 18 20 22 89 39 42 45 38 19 21 23 90 39 42 45 39 19 21 23 91 40 42 46 40 20 22 24 92 40 43 46 41 20 22 24 93 40 43 46 42 21 22 25 94 41 44 47 43 21 28 25 95 41 44 47 44 21 23 25 96 42 44 48 45 22 24 26 97 42 45 48 46 22 24 26 98 42 45 49 47 23 25 27 99 43 46 49 48 23 25 27 100 43 46 49 49 23 25 28 200 80 84 89 50 24 26 28 300 117 122 127 51 24 26 29 400 152 158 165 52 25 27 29 500 188 194 202 53 25 27 29 1.000 363 372 383
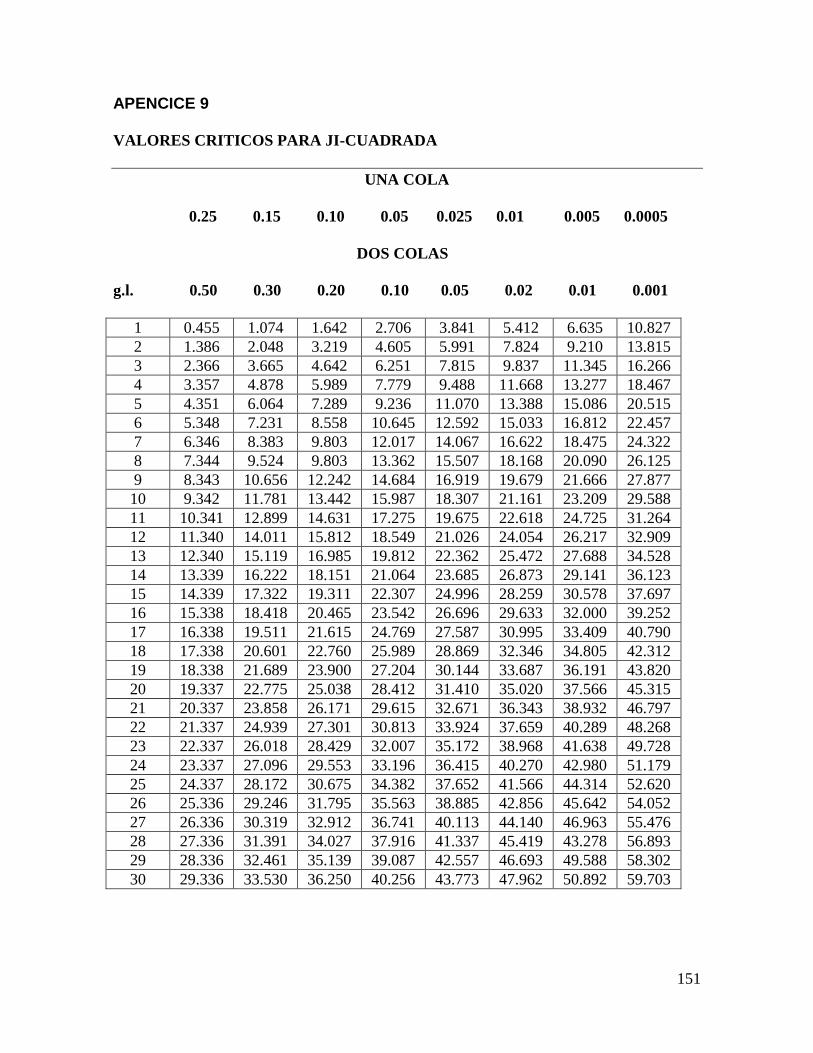
151
APENCICE 9
VALORES CRITICOS PARA JI-CUADRADA
UNA COLA
0.25 0.15 0.10 0.05 0.025 0.01 0.005 0.0005
DOS COLAS
g.l. 0.50 0.30 0.20 0.10 0.05 0.02 0.01 0.001
1 0.455 1.074 1.642 2.706 3.841 5.412 6.635 10.827
2 1.386 2.048 3.219 4.605 5.991 7.824 9.210 13.815
3 2.366 3.665 4.642 6.251 7.815 9.837 11.345 16.266
4 3.357 4.878 5.989 7.779 9.488 11.668 13.277 18.467
5 4.351 6.064 7.289 9.236 11.070 13.388 15.086 20.515
6 5.348 7.231 8.558 10.645 12.592 15.033 16.812 22.457
7 6.346 8.383 9.803 12.017 14.067 16.622 18.475 24.322
8 7.344 9.524 9.803 13.362 15.507 18.168 20.090 26.125
9 8.343 10.656 12.242 14.684 16.919 19.679 21.666 27.877
10 9.342 11.781 13.442 15.987 18.307 21.161 23.209 29.588
11 10.341 12.899 14.631 17.275 19.675 22.618 24.725 31.264
12 11.340 14.011 15.812 18.549 21.026 24.054 26.217 32.909
13 12.340 15.119 16.985 19.812 22.362 25.472 27.688 34.528
14 13.339 16.222 18.151 21.064 23.685 26.873 29.141 36.123
15 14.339 17.322 19.311 22.307 24.996 28.259 30.578 37.697
16 15.338 18.418 20.465 23.542 26.696 29.633 32.000 39.252
17 16.338 19.511 21.615 24.769 27.587 30.995 33.409 40.790
18 17.338 20.601 22.760 25.989 28.869 32.346 34.805 42.312
19 18.338 21.689 23.900 27.204 30.144 33.687 36.191 43.820
20 19.337 22.775 25.038 28.412 31.410 35.020 37.566 45.315
21 20.337 23.858 26.171 29.615 32.671 36.343 38.932 46.797
22 21.337 24.939 27.301 30.813 33.924 37.659 40.289 48.268
23 22.337 26.018 28.429 32.007 35.172 38.968 41.638 49.728
24 23.337 27.096 29.553 33.196 36.415 40.270 42.980 51.179
25 24.337 28.172 30.675 34.382 37.652 41.566 44.314 52.620
26 25.336 29.246 31.795 35.563 38.885 42.856 45.642 54.052
27 26.336 30.319 32.912 36.741 40.113 44.140 46.963 55.476
28 27.336 31.391 34.027 37.916 41.337 45.419 43.278 56.893
29 28.336 32.461 35.139 39.087 42.557 46.693 49.588 58.302
30 29.336 33.530 36.250 40.256 43.773 47.962 50.892 59.703

152
APÉNDICE 10
INFORME DE LAS PRÁCTICAS DE LABORATORIO
El informe de laboratorio debe incluir las siguientes secciones:
1. Portada
2. Introducción
3. Objetivos
4. Metodología
5. Resultados
6. Discusión
7. Referencias bibliográficas
PORTADA
La portada debe incluir lo siguiente:
Nombre de la institución con los logotipos correspondientes
Titulo de la práctica
Nombre del alumno (o de todos los alumnos que forman el equipo)
Fecha de realización del mismo
INTRODUCCIÓN
La introducción debe incluir información tocante al análisis que realizó en el laboratorio y
hacer comentarios breves de la importancia de la práctica, no debe ser exactamente la
misma que se presenta en cada práctica de este manual.
OBJETIVOS
Los objetivos deben ser concisos y explicar en una oración por qué se realiza la práctica.
Aunque en este manual se incluyen los objetivos de cada práctica, los alumnos deben
volver a expresarlos con sus propias palabras.
METODOLOGÍA
En vista de que este manual contiene una metodología detallada de cada práctica, esta debe
ser breve y anotar cada uno de los pasos que se llevan a cabo para realizar cada una de las
prácticas y además deberá incluir las características de muestra y de los equipos utilizados.
RESULTADOS
Esta sección debe presentar los datos en forma organizada como tablas y gráficas,
enumérelas y titúlelas. Incorpore al pie de las tablas para explicar las unidades y
abreviaturas o para explicar las irregularidades. Cuando los resultados requieran cálculos,

153
muestre un ejemplo únicamente de cada tipo de cálculo e incluya unidades. Aquí se deben
mostrar datos individuales y datos del equipo, es decir, datos obtenidos únicamente por el
estudiante y por el equipo (compañeros de clase). Se pueden incorporar comentarios sobre
los datos recolectados por todos los individuos o todos los equipos de la clase.
DISCUSIÓN
Aquí se explicarán los resultados, por lo que debe hacerse hincapié en lo referente a las
tablas y graficas y si estos son significativos en un determinado análisis. Además explicar si
estos son correctos o si hubo algún problema durante el desarrollo de la práctica o si esta
debe ser modificada para obtener resultados más confiables.
REFERENCIAS
Los estudiantes deben citar hechos o datos tomados de fuentes que no sean del manual o
notas de clase. Deben consultarse y citarse por menos dos referencias, incluidos libros,
artículos de investigación, artículos de revisión y sitios web.





























