Embed Size (px)
Citation preview

THESETHESE
En vue de l'obtention du
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSEDOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Cancérologie
JURY
Pr. Christian RECHER, Examinateur Pr. Didier BOUSCARY, Rapporteur Dr. Filippo ROSSELLI, Rapporteur
Dr. Jean-Sébastien HOFFMAN, Examinateur Dr. Hélène TOURRIERE, Examinateur
Pr. Bernard DUCOMMUN, Directeur de thèse Dr. Cécile DEMUR, Membre invité
Ecole doctorale : Biologie Santé Biotechnologies TOULOUSE III
Unité de recherche : Laboratoire de Biologie Cellulaire et Moléculaire du contrôle de la Prolifération Directeur(s) de Thèse : Pr. Bernard DUCOMMUN, Dr. Cécile DEMUR
Rapporteurs : Pr. Didier BOUSCARY, Dr. Filippo ROSSELLI
Présentée et soutenue par Cindy CAVELIER Le 4 mai 2010
Titre : Etude du point de contrôle des dommages à l'ADN
dans les Leucémies Aiguës Myéloïdes
et ciblage pharmacologique


1
REMERCIEMENTS Merci beaucoup à chaque membre du jury d’avoir accepté d’évaluer mon travail de thèse, à la fois médicale pour les scientifiques mais scientifique pour le monde médical…Ce n’était donc pas chose facile !! Merci donc à Christian Récher d’avoir présidé mon jury mais également de nous avoir apporter régulièrement des coups de pouce dans nos recherches et dans la compréhension des leucémies aiguës myéloïdes. Merci à Didier Bouscray, Filippo Rosselli, Hélène Tourrière et Jean-Sébastien Hoffmann, j’ai vraiment été très touchée par vos compliments et vos félicitations sur mes travaux de thèse. Il est maintenant temps de remercier mes directeurs de thèse. Merci Bernard tout d’abord pour cette thèse qui était pour moi presque tombée du ciel (presque !!) à des moments de découragement. J’étais vraiment ravie de votre proposition et j’ai été ravie de travailler à vos côtés pendant ces quelques années. Merci pour votre écoute, votre considération et votre rigueur scientifique. Et oui, la barre est très haute maintenant pour les suivants ;)). Merci Cécile d’avoir accepté de m’encadrer pendant la thèse au sein du service hématologie. Merci pour votre œil expert des LAM, pour votre bonne humeur et votre optimisme à toute épreuve. Les filles du bureau….Merci Christine pour ton aide et ton suivi quotidien qui m’ont permis de relever le challenge de la thèse !! Merci pour ta bonne humeur que tu savais si bien répandre dans notre bureau, je te souhaite tout plein de bonne chose pour la suite dans ta nouvelle équipe de recherche. Merci Fanny pour ton soutien et ton aide quotidiens que tu as su m’apporter en cette fin de thèse pas toujours facile moralement et physiquement. Bon courage et bonne chance à toi aussi pour la suite. Mes supers copines de la pause…Merci Amandine et Naïs pour votre soutien sans faille pendant ces 3 années et demi de thèse !! Vous avez réussi à rendre ma thèse beaucoup plus rigolote et à faire en sorte que je garde le moral même à des moments où je me sentais un peu seule dans le service (et dans le bureau)!! Je vous souhaite bien sûr le meilleur pour la suite, sur tous les plans. Tu verras Amandine la vie est parfois presque rose, même pour toi j’en suis sûre !! Merci aux différents membres du service hématologie, Véronique et Eric pour votre aide au cours de ma thèse et de l’après thèse !!, aux techniciennes d’hémato, Nicole, Monique et Aurore, de m’avoir accepter dans votre laboratoire et de toujours avoir pris du temps pour moi, même lorsque je vous détournais un peu de la routine !!, aux techniciennes (et technicien) de biologie moléculaire pour m’avoir permis de manipuler dans votre laboratoire. Merci à l’équipe de recherche de Bernard, Valérie, Odile, Martine, Denis, Béa, Christine, Thomas, Jennifer, Céline, Annie et Corinne, pour votre aide apportée dans mon projet de recherche. Merci aux différentes équipes du CPTP, les GD, BP (avec une mention toute spéciale quand même pour Stéphane qui nous a vraiment aidé à accomplir ce travail, merci pour tes remarques toujours pertinentes !!) et tout particulièrement les GL bien sûr…Merci Anne, Chris et Chris pour l’aide que vous avez su m’apporter très volontiers dès que j’ai eu besoin pas seulement depuis ma thèse puisque cela avait commencé un peu avant pour nous !!

2
Enfin, merci à mes proches, maman et mes frères et sœur, pour tous vos encourageants pendant ce long parcours !! Promis je vous embête plus avec mes histoires de stress lors d’examen (enfin peut être !). Merci enfin à ma petite famille, Tristan et Junon. Voilà l’aventure de la thèse est terminée pour nous !! Tristan, merci de m’avoir aussi bien accompagné jusqu’au bout!! Merci à ma petite puce, tellement adorable, tu m’as permis de conjuguer thèse et bébé.

3
ABREVIATIONS
ADN : Acide Désoxyribonucléique
AKT/ PKB : Protein Kinase B
ALDH : Aldéhyde Déshydrogénase
AML : Acute Myeloid Leukemia
Ang-1: Angiopoïétine-1
APC /C : Anaphase Promoting Complex/Cyclosome
Ara-C : cytarabine
ARN : Acide Ribonucléique
ARNi : ARN interferant
ARNm : ARN messager
ATM : Ataxia-Telangiectasia Mutated
ATP : Adénosine Tri-Phosphate
ATR : ATM and Rad3 related
ATRA : Acide Tout Trans rétinoïque
ATRIP : ATR Interacting Protein
BAALC: Brain And Acute Leukemia Cytoplasmic
BFU-E : Burst Forming Unit-Erythroid
BMP : Bone Morphogenic Proteins
BRCA1 : Breast Cancer susceptibility gene 1
BRCT : Breast Cancer c-Terminal domain
CAK : CDK-Activating Kinase
CAM-DR : Cell Adhesion Mediated-Drug Resistance
CBF-1 : C-promoter Binding Factor-1
CBP : CREB Binding Protein
CD34 : Cluster of Differentiation 34
CDC : Cell Division Cycle
CDK : Cyclin-Dependent Kinase
CEBP : CAAT/Enhancer Binding Protein
CFU : Colony Forming Unit
CFU-E : Colony Forming Unit-Erythrocyte
CFU-G : Colony Forming Unit-Granulocyte
CFU-GEMM : Colony Forming Unit-Granulocyte, Erythrocyte, Macrophage and
Megakaryocyte
CFU-GM : Colony Forming Unit-Granulocyte and Macrophage

4
CFU-L : Colony Forming Unit-Leukemia
CFU-M : Colony Forming Unit-Monocyte
CFU-mega : Colony Forming Unit-megakaryocyte
CHK1 : Checkpoint Kinase 1
CHK2 : Checkpoint Kinase 2
CKI : Cyclin Dependent Kinase Inhibitor
CLP : Common Lymphoïd Progenitor
CMP : Common Myeloid Progénitor
CS : Cellules Souches
CSH : Cellules Souches Hématopoïétiques
CSH-CT : CSH à Court Terme
CSH-LT : CSH à Long Terme
CSM : Cellules Souches Mésenchymateuses
CSL : Cellules Souches Leucémiques
DDR : DNA Damage Response
DNA-PKcs : DNA-dependent Protein Kinase catalytic subunit
DSB : Double Strand Breack
DSPases : Dual-Specifity Phosphatases
EGF : Epidermal Growth Factor
Eosino : Eosinophile
EPO : Erythropoïétine
ERG : Ets Related Gene
ERK : Extracellular-signal-Regulated Kinase
Erythro: Erythroblaste
FAB : French-American-British
FAK : Focal Adhesion Kinase
FGF : Fibroblast Growth Factor
FKHRL1 : Forkead in Rhabdomyosarcoma-Like 1
FLT3: Fms-Like Tyrosine kinase 3
FLT3-ITD : Fms-Like Tyrosine kinase 3- Internal Tandem Duplication
FT : Facteur de Transcription
G1/2 : Gap 1/2
G-CSF : Granulocyte-Colony Stimulating Factor
GM-CSF : Granulo-Macrophage- Colony Stimulating Factor
GSK3β : Glycogen Synthase Kinase-3β
HSC : Hematopoietic Stem Cell

5
ICAM : Intercellular Cell Adhesion Molecule
IL : Interleukine
INK4 : Inhibitors of CDK4
IR : Ionising radiation
JAK : Janus Kinase or Just Another Kinase
JNK : c-Jun NH2-terminal protein Kinase
KTLS : c-Kit+, Thy-1low, Lin –, Sca1+
LAM : Leucémie Aiguë Myéloïde
LEF/TCF : Lymphocyte Enhancer Factor/T Cell Factor
LIF : Leukemia Inhibitory Factor
LMC : Leucémie Myéloïde Chronique
LRP5/6 : Lipoprotein Related-Receptor Proteins 5/6
LSC : Leukemic Stem Cell
LTC-IC : Long-Term Culture Initiating Cell
LT-HSC : Long Term Hematopoïetic stem cell
Lympho T et B : Lymphocytes T et B
M : Mitosis
MAPK : Mitogenic-Activated Protein Kinase
MCM : Minichromosome Maintenance
M-CSF : Monocyte-Colony Stimulating Factor
MDM2 : Murine Double Minute 2
MEC : Matrice Extracellulaire
MEP : Megacaryocyte Erythrocyte Precursor
MGF : Mast cell Growth Factor
mKL : membrane-bound Kit Ligand
MLL : Mixed Lineage Leukemia
MN1 : Menigioma 1 gene
MO : Moelle Osseuse
Mono : Monocyte
MPF : Maturation Promoting Factor
Mrc1 : Mediator of Replication Checkpoint 1
MRN : MRE11-RAD50-NBS1
mTOR : mammalian Target Of Rapamycin
NCoR : complexe co-répresseur nucléaire
NES : Nuclear Export Signal
NFκB: Nuclear Factor-kappa B

6
Neutro : Neutrophile
NICD : Notch Intracellular Domain
NK : Natural Killer
NLS : Nuclear Localisation Sequence
NOD-SCID : Non Obese Diabetic-SCID
NPM : Nucléophosmine
PARP :Poly(ADP-ribose) polymérase
PDGF : Platelet-Derived growth Factor
PDK1/2 : 3-Phosphoinositide-Dependent Protein Kinase 1/2
Pgp : P-glycoprotéine
PI-3K : Phosphatidylinositol 3’-Kinase
PIKK : Phosphoinositide 3-Kinase Related Kinase
PKC : Protein Kinase C
PKA : cAMP-dependent Protein Kinase A
Plaq : Plaquettes
PLK : Polo Like Kinase
PML : Promyelocytic Leukemia
PP2A : Protein Phosphatase 2A
Pro B : Pro-lymphocyte B
Pro T : Pro-lymphocyte T
PS/TPase : Protéine Sérine/Thréonine Phosphatase
Ptc : Patched
PTEN : Phosphatase and Tensin homolog
PTPase : Protéine Tyrosine Phosphatase
PYK2 : Proline-rich tyrosine kinase 2
RAR : Retinoic Acid receptor
Rb : Rétinoblastome
RC : Rémission complète
RPA : Replication Protein A
RTK : Récepteur à activité Tyrosine Kinase
S : Synthesis
SAPK : Stress-Activated Protein Kinase
SCF : Skp1/Cul1/F-box
SCF : Stem Cell Factor
SCF-βTrCP : Skp-1–Cul1–F-box βTrCP
SCID : Severe Combined Immuno-Deficiency

7
Shh : Sonic Hedgehog
SHIP : Src Homology Inositol Phosphatase
SL-IC : SCID Leukemia Initiating Cell
SMD : syndrome myélodysplasique
SP : Side Population
SRC : SCID Repopulating Cells
STAT3 : Signal Transducer and Activator of Transcription 3
ST-HSC : Short Term-Hematopoietic stem cell
TGF-β : Transforming Growth Factor β
TK : Tyrosine Kinase
TopBP1 : DNA topoisomerase 2 binding protein 1
TPO : trombopoïétine
UV : Ultraviolet
VCAM : Vascular Cell Adhesion Molecule
VEGF : Vascular Endothelial Growth Factor
VLA : Very Late Antigen
WHO : World Health Organization
WT : Wilms Tumor protein

8

9
R E S U M ER E S U M E
Les Leucémies Aiguës Myéloïdes (LAM) représentent un groupe hétérogène de pathologies
développées aux dépend des progéniteurs myéloïdes immatures normaux, caractérisées sur
le plan moléculaire par des modifications de l'expression de facteurs transcriptionnels
aboutissant à un blocage de la maturation, et par une activation aberrante de molécules
de la transduction du signal conférant aux cellules leucémiques des avantages prolifératifs
et de survie.
Dans ce travail, nous avons montré que la protéine kinase CHK1, impliquée dans le
maintien de l’intégrité du génome en contrôlant l’arrêt du cycle cellulaire à la transition
G1/S et G2/M après dommages constitutifs à l’ADN, prolonge son activation après
dommages dans les cellules de LAM immatures permettant ainsi leur maintien en phase G2.
Cette meilleure efficacité du point de contrôle G2/M des cellules immatures contribue très
probablement à la chimiorésistance naturelle observée et largement documentée des
cellules leucémiques immatures. De plus, nous montrons que la kinase CHK1 est
anormalement activée dans les échantillons de LAM en raison de la présence de dommages
à l'ADN constitutifs dans ces cellules. L’étude des caractéristiques cytogénétiques des
patients nous a permis de mettre en évidence un haut niveau de dommages dans les LAM
de haut risque cytogénétique, en particulier celles avec des cellules à caryotype
complexe. L’inactivation de CHK1 par l’inhibiteur UCN-01 et par interférence à l'ARN
aboutit à une inhibition des capacités clonogènes des progéniteurs leucémiques et à une
chimiosensibilisation vis-à-vis de l’agent chimiothérapeutique ara-C. En revanche, les
cellules hématopoïétiques normales CD34+, qui ne présentent pas de dommages
constitutifs, sont beaucoup moins sensibles à l’UCN-01. Cet agent n’affecte pas non plus
les capacités clonogènes des progéniteurs granulo-monocytaires normaux aux doses
utilisées sur les cellules de LAM.
Ces résultats nous ont permis de montrer que la kinase CHK1 joue un rôle important à la
fois dans la chimiorésistance des cellules leucémiques immatures et dans l’établissement
de l’instabilité génétique observée dans les LAM à caryotype complexe. Enfin, ces travaux
ouvrent des perspectives intéressantes sur l’utilisation de composés inhibiteurs des kinases
du point de contrôle du cycle cellulaire dans les LAM et en particulier celles présentant le
plus mauvais pronostic, les LAM à caryotype complexe.

10
A B S T R A C TA B S T R A C T
Acute Myeloid Leukemia (AML) is a clonal hematopoietic disorder characterized by the
accumulation of malignant hematopoietic progenitor cells with an impaired myeloid
differentiation program. The molecular basis of AML is thought to be associated with the
acquisition of at least two types of critical cooperating mutations occurring at the
hematopoietic stem or committed progenitors level. Class I mutations, affecting tyrosine
kinases receptors and key components of cellular signalling pathways, confer growth and
proliferative advantages. They are associated with class II mutations, affecting
transcription factors thus leading to impaired normal differentiation program.
In this study, we were first interested in CHK1, a protein kinase involved in preserving
genome integrity by playing a critical role at the intra-S and G2/M cell cycle checkpoint
activated in DNA damage response. We have shown that activation of CHK1 was sustained
in immature cell lines, leading to a more stringent G2/M checkpoint in response to DNA
damage, thus impairing illegitimate entry into mitosis in presence of unrepaired DNA
damage and participating in their resistance to genotoxic agents. In a second study, we
have demonstrated an abnormal activation of the CHK1 kinase in a large panel of AML
patient samples, associated with the presence of constitutive DNA damage in absence of
genotoxic stress. Moreover, the level of CHK1 activation is significantly correlated with
unfavourable cytogenetic samples, particularly with complex karyotype phenotype. CHK1
inhibition by the pharmacological inhibitor UCN-01 or by RNA interference was found to
decrease the clonogenic capacity of the AML progenitors, and to induce a
chemosensitisation to ara-C. In contrast, growth of normal hematopoietic progenitors,
which do not display constitutive DNA damage, was not impaired by such treatment.
Overall, all these results underline the dual role of CHK1 kinase in AML pathology in the
chemoresistance of immature leukemic cells and in the establishment of the genomic
instability observed in complex karyotype AML. These findings could have major
pharmacologic consequences, because they open a therapeutic window for new compounds
targeting the cell cycle checkpoint machinery in AML and more particularly in the worst
prognostic group with complex karyotype.

11
S O M M A I R ES O M M A I R E
ABREVIATIONS ................................................................................ 1
RESUME .......................................................................................... 9
ABSTRACT..................................................................................... 10
SOMMAIRE .................................................................................... 11
INTRO DUCTION ............................................................................. 15
AVANT PROPOS ............................................................................. 16
I. CONTROLE DE L’INTEGRITE DU GENOME, ONCOGENESE ET THERAPEUTIQUE ........................................................................... 16
A. REGULATION DU CYCLE CELLULAIRE ET POINT DE CONTROLE .................................. 17 1. Le cycle cellulaire ............................................................................... 17
a) Les complexes CDK-Cycline ......................................................................................... 17 (1) Progression du cycle cellulaire.................................................................................. 17 (2) Régulation spatio-temporelle des complexes CDK-cyclines................................... 20 (3) Activation des complexes CDK-cycline .................................................................. 21 (4) Inhibition des complexes CDK-cyclines.................................................................. 22
b) Les phosphatases CDC25............................................................................................. 24 2. Points de contrôle du cycle cellulaire ................................................ 26
a) Les différents points de contrôle du cycle cellulaire.................................................... 26 b) Organisation de la voie de signalisation des dommages à l’ADN................................ 28
(1) La cinétique de réponse aux dommages à l’ADN ................................................... 29 (2) Les acteurs du point de contrôle des dommages à l’ADN....................................... 31
B. TUMORIGENESE ET BARRIERE ANTI-TUMORALE .................................................... 35 1. Dans les tumeurs solides..................................................................... 35 2. Dans les hémopathies ........................................................................ 36
a) Modèles de leucémogenèse ......................................................................................... 37 (1) Chez la souris......................................................................................................... 37 (2) Du syndrome myélodysplasique à la leucémie....................................................... 37
C. THERAPEUTIQUES CIBLANT LES ACTEURS DE LA VOIE DES DOMMAGES ........................ 38 1. Intérêt d’un ciblage du point de contrôle G2/M................................. 39 2. Inhibiteurs des acteurs de la voie des dommages............................... 40
a) Inhibiteurs des kinases ATM et ATR............................................................................. 40 b) Inhibiteurs de DNA-PK ................................................................................................ 41 c) Inhibiteurs de la kinase CHK2..................................................................................... 42 d) Inhibiteurs de la kinase CHK1 ..................................................................................... 42
(1) L’UCN-01 ............................................................................................................... 43 (2) Inhibiteurs spécifiques de CHK1 ............................................................................ 44
II. HEMATOPOÏESE & MICRO-ENVIRONNEMENT............................... 46 A. HEMATOPOÏESE NORMALE OU PHYSIOLOGIQUE ................................................... 46
1. Les cellules hématopoïétiques ........................................................... 47 a) Les cellules souches hématopoïétiques ....................................................................... 48
(1) Capacité de reconstitution de l’hématopoïèse...................................................... 48 (2) Capacité d’auto-renouvellement des CSH.............................................................. 51 (3) Pluripotence ou potentiel de différenciation multi-lignage ................................. 54
b) Les progéniteurs hématopoïétiques............................................................................ 56 c) Les précurseurs hématopoïétiques .............................................................................. 57
2. Le micro-environnement médullaire ................................................. 57 a) Interaction directe CSH-niche ..................................................................................... 59

12
b) Facteurs de croissance et cytokines de la niche hématopoïétique .............................. 60 c) Les composants de la matrice extra-cellulaire ............................................................ 61
B. HEMATOPOÏESE LEUCEMIQUE.......................................................................... 63 1. Caractéristiques ................................................................................. 63 2. Classification FAB des LAM.................................................................. 65 3. Pronostic et traitements .................................................................... 66
a) Enjeux thérapeutiques ................................................................................................ 66 b) Traitements ................................................................................................................ 67 c) Impact de l’âge dans le pronostic des LAM .................................................................. 67 d) Impact des caractéristiques cytogénétiques dans le pronostic des LAM...................... 68
(1) Pronostic favorable............................................................................................... 69 (2) Pronostic intermédiaire ........................................................................................ 71 (3) Pronostic défavorable........................................................................................... 73 (4) Les LAM3 ou leucémie aiguë promyélocytaire....................................................... 75
4. Concept de cellules souches leucémiques .......................................... 76 a) Origine des CSL............................................................................................................ 77 b) Caractérisation des CSL .............................................................................................. 78 c) Anomalies des voies de signalisation .......................................................................... 79
(1) Rôle des STATs ....................................................................................................... 79 (2) Rôle des ERK/MAPK ............................................................................................... 80 (3) Rôle de la voie PI3-kinase ...................................................................................... 81 (4) Les voies apoptotiques .......................................................................................... 82
5. Rôle du micro-environnement et de l’adhésion dans la leucémogenèse ......................................................................................... 83
a) Impact de l’intégrine α4β1 sur la leucémogenèse......................................................... 84 b) Impact de l’intégrine α5β1 sur la leucémogenèse ....................................................... 85
III. HEMATOPOÏESE ET CYCLE CELLULAIRE...................................... 87 A. CELLULES HEMATOPOÏETIQUES & CYCLE CELLULAIRE ............................................ 87
1. Les CSH................................................................................................ 87 a) Statut du cycle cellulaire dans les CSH ........................................................................ 87 b) Régulateurs du cycle cellulaire dans les CSH ............................................................... 88
(1) p21WAF1 & CSH........................................................................................................ 88 (2) p18INK4C ................................................................................................................. 89 (3) p16INK4A, p19ARF ..................................................................................................... 89 (4) Rb.......................................................................................................................... 89 (5) Les cyclines et CDK................................................................................................. 90
c) Niche hématopoïétique et cycle cellulaire .................................................................. 90 2. Au cours de la différenciation hématopoïétique................................ 91
a) Cycle cellulaire & différenciation................................................................................ 91 b) Régulateurs du cycle cellulaire & différenciation ....................................................... 92
(1) p21WAF1 .................................................................................................................. 92 (2) p27KIP1 ................................................................................................................... 92 (3) Autres régulateurs du cycle cellulaire ................................................................... 93
B. DEREGULATION DES ACTEURS DU CYCLE CELLULAIRE DANS LES LAM ........................ 94 1. Cycle cellulaire & LAM ........................................................................ 94 2. Régulateurs du cycle cellulaire & LAM................................................ 94
a) Cyclines & LAM ............................................................................................................ 94 b) CDKI & LAM................................................................................................................. 95 c) Kinases du cycle cellulaire & LAM................................................................................ 96 d) Rb & LAM..................................................................................................................... 97
3. Microenvironnement médullaire & LAM ............................................ 97
RESULTATS ET DISCUSSION ............................................................ 99
I. ANALYSE DU POINT DE CONTROLE G2/M DANS DES LIGNEES CELLULAIRES DE LAM ................................................................... 102
A. INTRODUCTION ........................................................................................ 102 B. RESULTATS.............................................................................................. 104

13
C. DISCUSSION ............................................................................................. 105
II. ACTIVATION CONSTITUTIVE DE LA VO IE DE SIGNALISATION DES DOMMAGES DANS LES ECHANTILLONS DE LAM DE CARYO TYPE COMPLEXE : INTERET THERAPEUTIQUE D’UN CIBLAGE DES POINTS DE CONTROLE .................................................................................. 107
A. INTRODUCTION ........................................................................................ 107 B. RESULTATS.............................................................................................. 108 C. DISCUSSION ............................................................................................. 109
DISCUSSION GENERALE ET PERSPECTIVES ...................................... 111
REFERENCES BIBLIOGRAPHIQUES .................................................. 119
ANNEXES ..................................................................................... 143

14

15
INTRODUCTIONINTRODUCTION

16
AVANT PROPOS
Nous développerons dans cette introduction les connaissances générales actuelles sur le
contrôle de l’intégrité du génome des cellules eucaryotes, puis ses dérégulations lors du
processus de tumorigenèse. Nous terminerons la première partie en présentant les
thérapies ciblant les acteurs du cycle cellulaire. Nous aborderons ensuite l’importance de
la régulation de la prolifération cellulaire dans l’hématopoïèse physiologique ainsi que ses
dérégulations dans le cas d’hématopoïèse leucémique. Enfin, nous terminerons cette
introduction en présentant les données bibliographiques rapportées sur le contrôle du
cycle cellulaire dans l’hématopoïèse physiologique et ses dérégulations dans les Leucémies
Aiguës Myéloïdes (LAM).
I. Contrôle de l’intégrité du génome, oncogenèse et
thérapeutique Chaque cellule d’un organisme se divise pour donner naissance à deux cellules filles au
cours du cycle cellulaire. Afin de garantir une transmission de l’information génétique la
plus fidèle possible, ce processus met en jeu de nombreux mécanismes de contrôle. La
compréhension et la connaissance des acteurs du cycle cellulaire ont permis et permettent
toujours de mettre en évidence les multiples dérégulations observées dans les cellules
cancéreuses favorisant une prolifération incontrôlée et ouvrent des perspectives
thérapeutiques nouvelles.
Dans cette première partie de l'introduction, nous présenterons l'état actuel des
connaissances sur les bases moléculaires du contrôle du cycle cellulaire en focalisant
principalement notre attention sur les mécanismes de préservation de l'intégrité
génomique, leur place dans la tumorigenèse et leur ciblage pharmacologique.

17
A. Régulation du cycle cellulaire et point de contrôle
1. Le cycle cellulaire
La cellule eucaryote prépare la duplication du matériel génétique porté par l’ADN en
phase G1 (Gap 1) et permet sa réplication semi-conservative en phase S (Synthesis). En
phase G2 (Gap 2), la cellule prépare la séparation du matériel génétique et la division
cellulaire qui auront lieu en mitose ou phase M. Les phases G1, S et G2 sont regroupées
sous le terme d’interphase. Cette vision classique et très descriptive du cycle cellulaire a
laissé place au cours des 20 dernières années à une caractérisation moléculaire des acteurs
et des voies de signalisation impliquées dans son contrôle et dans leur intégration aux
mécanismes qui contrôlent la prolifération et l'homéostasie tissulaire.
Figure 1 : Le cycle cellulaire eucaryote
Les deux événements majeurs du cycle cellulaire sont la réplication fidèle de l’information génétique en phase S et la ségrégation précise de ce matériel entre les deux cellules filles en mitose. Les phases G1, S et G2 constituent l’interphase. (D’après la thèse d’Anne Vidal).
a) Les complexes CDK-Cycline
(1) Progression du cycle cellulaire
La progression du cycle cellulaire est contrôlée par des kinases dépendantes des cyclines
(ou CDK pour Cyclin-Dependent-Kinase) qui se lient à des cyclines régulatrices spécifiques
permettant la succession des différentes phases du cycle cellulaire (Morgan, 1997).
Initialement, la première CDK a été mise en évidence chez les levures Saccharomyces

18
cerevisiae (Cdc28) et Saccharomyces Pombe (Cdc2). Le nombre de CDK et de cyclines
augmente considérablement au cours de l’évolution, cependant, seuls certains complexes
CDK-cyclines semblent contrôler la progression du cycle cellulaire (Malumbres and
Barbacid, 2009).
Selon, le modèle classique du cycle cellulaire chez les mammifères, les signaux
mitogéniques permettent, dans un premier temps, l’expression des cyclines de type D (D1,
D2 et D3) qui activent préférentiellement les CDK4 et 6 durant la phase G1 (Malumbres and
Barbacid, 2001). L’activation de ces complexes aboutit à l’inactivation partielle des
protéines de la famille du rétinoblastome pRB, p107 et p130 pour permettre l’expression
de la cycline E (E1 et E2) qui va lier et activer CDK2 (Harbour et al., 1999). Le complexe
CDK2-cycline E, en phosphorylant les protéines pRB, p107 et p130, permet leur
inactivation complète et ainsi la transition en phage S (Lundberg and Weinberg, 1998).
L’expression de la cycline E est finement contrôlée permettant de limiter l’accès des
cellules aux stades précoces de la synthèse d’ADN. En effet, l’utilisation de dominant-
négatif et d’anticorps dirigés contre CDK2 a permis d’inhiber la progression dans le cycle
de certaines cellules tumorales (Hochegger et al., 2008). CDK2 est ensuite activée par la
cycline A2 (ou A1 dans les cellules germinales) durant les phases tardives de la réplication,
permettant ainsi le passage vers la mitose (au cours de la phase G2). Pour finir, CDK1 est
activée par la cycline A en toute fin d’interphase pour permettre l’entrée en mitose
(Malumbres and Barbacid, 2005).
Figure 2 : Complexes CDK-Cyclines et progression dans le cycle cellulaire humain
Plusieurs CDK associées à différentes cyclines régulent le cycle cellulaire humain. Les travaux d’invalidation génétique dans la souris remettent en cause aujourd’hui ce schéma classique. (D’après la thèse d’Anne Vidal).

19
Ce modèle classique, selon lequel chaque passage d’une phase à l’autre du cycle cellulaire
serait gouverné par une CDK, a été considérablement remis en question grâce à des études
génétiques réalisées chez la souris. En effet, si l’invalidation du gène cdk1 est létale, les
pertes des loci cdk2, 4 ou 6 n’impactent pas sur le cycle cellulaire de la plupart des tissus
(Santamaria et al., 2007). Par exemple, la perte de cdk6 induit seulement un défaut
mineur dans la lignée érythroïde avec une réduction du nombre de globules rouges
périphériques (Malumbres et al., 2004).
L’absence de défauts majeurs du cycle cellulaire dans la plupart des cellules invalidées
pour une CDK interphasique n’est pas simplement due à la redondance des CDK en général.
En effet, les pertes concomitantes de plusieurs CDK permettent d’obtenir un phénotype
plus marqué, mais ne donnent pas non plus de défaut majeur du cycle cellulaire dans la
plupart des tissus. Par exemple, l’invalidation des Cdk4 et 6 induit des défauts de
prolifération seulement pour les précurseurs hématopoïétiques, pour les cellules non
hématopoïétiques, aucun défaut de phase G1 ou d’entrée dans le cycle n’a été observé
(Malumbres et al., 2004). L’effet de compensation par la troisième CDK interphasique,
CDK2, a pu être exclu avec les double-mutants (CDK2 et 6, CDK2 et 4) qui ne donnent
également que des défauts dans des tissus spécialisés (Barriere et al., 2007). Tout cela
suggère que les cyclines pourraient s’apparier avec d’autres partenaires que ceux
initialement décrits. Illustrant cette hypothèse, Aleem et ses collaborateurs ont mis en
évidence en 2005 une association entre CDK1 et la cycline E, ce complexe CDK1-cycline E
pourrait compenser l’absence de CDK2 à la transition G1/S dans les souris cdk2-/- (Aleem
et al., 2005). De manière surprenante, l’invalidation de l’ensemble des CDK interphasiques
chez la souris permet un développement embryonnaire jusqu’au stade E16.5-E17.5. Cela
signifie que CDK1 est suffisante pour permettre la prolifération et la division cellulaire de
la plupart des tissus du développement embryonnaire précoce (Santamaria et al., 2007).
Ainsi, le nombre important de CDK des organismes multi-cellulaires permet probablement
de mettre en jeu des niveaux complexes de régulation du contrôle de la prolifération dans
un très grand nombre de types cellulaires.
Initialement, seuls quelques substrats des complexes CDK-cycline étaient décrits tels que
les lamines nucléaires phosphorylées par CDK1, ou pRb qui pourrait être phosphorylée par
les cinq CDK décrites dans le paragraphe précédent. La liste s’est considérablement
étoffée ces dernières années (Malumbres and Barbacid, 2005), mais semble encore
probablement incomplète. La caractérisation des substrats des différents complexes CDK-
Cycline reste un enjeu majeur pour la compréhension des mécanismes qui contrôlent la
progression du cycle cellulaire.

20
CDK4 et CDK6 associées aux cyclines D phosphorylent et inactivent les protéines de
la famille du rétinoblastome pRB, p107 et p130. Ces protéines sont des répresseurs de la
transcription de gènes tels que ceux codant pour les histones déacétylases, les complexes
de remodelage de la chromatine ou les cyclines E.
Le complexe CDK2-cycline E permet l’initiation de la réplication notamment en
phosphorylant les protéines MCM (pour Minichromosome Maintenance) ainsi que plusieurs
protéines impliquées dans la réparation de l’ADN (BRCA1 : Breast Cancer susceptibility
gene 1) et de la modification des histones (p220, CBP pour CREB Binding Protein).
Les complexes CDK2-cycline A et CDK1-cycline A ont des cibles communes
vraisemblablement impliquées dans la sortie de phase S telles que pRB et p53.
Enfin, le plus grand nombre de cibles répertoriées concerne CDK-cycline B : plus de
70 protéines ciblées par ce complexe sont référencées, permettant la régulation de la
transition G2/M et la progression en mitose : les histones (condensation de la chromatine),
les phosphatases CDC25 (Cell Division Cycle 25) (entrée en mitose), les lamines A, B et C
(désassemblage de l’enveloppe nucléaire), la séparase (ségrégation du matériel
génétique), APC/C (Anaphase Promoting Complex/Cyclosome) (sortie de mitose)…
(Malumbres and Barbacid, 2005)
Par ailleurs, en plus des CDK interphasiques (CDK2, CDK4 et CDK6) et mitotique (CDK1),
d’autres membres de la famille ont été impliqués dans la régulation du cycle cellulaire. La
protéine CDK5 semble essentielle pour l’arrêt du cycle et la différenciation neuronale, le
complexe CDK7-cycline H active les CDK2 et CDK1, la CDK8 serait impliquée dans les
mécanismes d’amplification de gènes retrouvés dans les cancers du colon… (Malumbres and
Barbacid, 2009).
(2) Régulation spatio-temporelle des complexes CDK-
cyclines
L’activation des CDK nécessite l’association avec une cycline. La liaison se fait
principalement entre le domaine PSTAIRE de CDK1 ou 2, et le domaine CYCLIN BOX de la
cycline (une centaine d’acides aminés). Le complexe CDK-cycline change alors de
configuration au niveau d’une séquence T-loop, rendant accessible le site catalytique de la
CDK. La liaison CDK2-cycline A rend accessible à la phosphorylation trois acides aminés : la
thréonine 14, la tyrosine 15 et la thréonine 160 (Jeffrey et al., 1995).
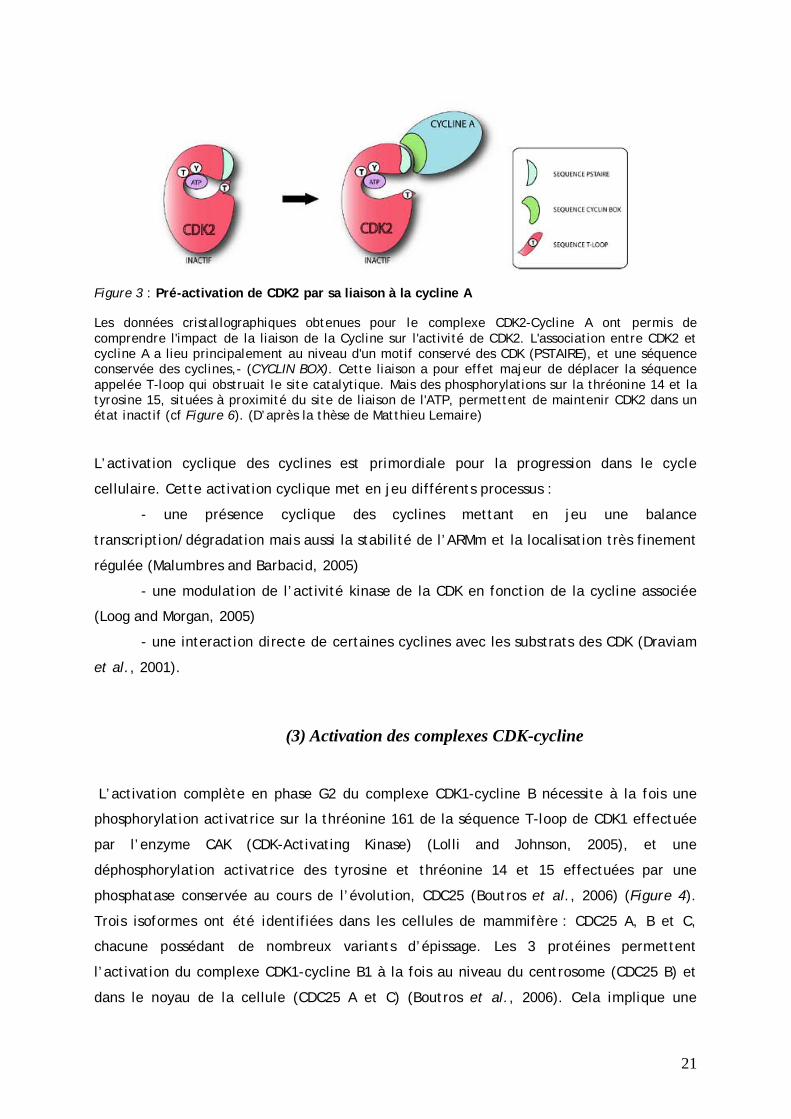
21
Figure 3 : Pré-activation de CDK2 par sa liaison à la cycline A
Les données cristallographiques obtenues pour le complexe CDK2-Cycline A ont permis de comprendre l'impact de la liaison de la Cycline sur l'activité de CDK2. L'association entre CDK2 et cycline A a lieu principalement au niveau d'un motif conservé des CDK (PSTAIRE), et une séquence conservée des cyclines,- (CYCLIN BOX). Cette liaison a pour effet majeur de déplacer la séquence appelée T-loop qui obstruait le site catalytique. Mais des phosphorylations sur la thréonine 14 et la tyrosine 15, situées à proximité du site de liaison de l'ATP, permettent de maintenir CDK2 dans un état inactif (cf Figure 6). (D’après la thèse de Matthieu Lemaire)
L’activation cyclique des cyclines est primordiale pour la progression dans le cycle
cellulaire. Cette activation cyclique met en jeu différents processus :
- une présence cyclique des cyclines mettant en jeu une balance
transcription/dégradation mais aussi la stabilité de l’ARMm et la localisation très finement
régulée (Malumbres and Barbacid, 2005)
- une modulation de l’activité kinase de la CDK en fonction de la cycline associée
(Loog and Morgan, 2005)
- une interaction directe de certaines cyclines avec les substrats des CDK (Draviam
et al., 2001).
(3) Activation des complexes CDK-cycline
L’activation complète en phase G2 du complexe CDK1-cycline B nécessite à la fois une
phosphorylation activatrice sur la thréonine 161 de la séquence T-loop de CDK1 effectuée
par l’enzyme CAK (CDK-Activating Kinase) (Lolli and Johnson, 2005), et une
déphosphorylation activatrice des tyrosine et thréonine 14 et 15 effectuées par une
phosphatase conservée au cours de l’évolution, CDC25 (Boutros et al., 2006) (Figure 4).
Trois isoformes ont été identifiées dans les cellules de mammifère : CDC25 A, B et C,
chacune possédant de nombreux variants d’épissage. Les 3 protéines permettent
l’activation du complexe CDK1-cycline B1 à la fois au niveau du centrosome (CDC25 B) et
dans le noyau de la cellule (CDC25 A et C) (Boutros et al., 2006). Cela implique une

22
régulation fine de la localisation des phosphatases mettant en jeu des séquences d’import
et d’export nucléaire, une interaction avec la protéine 14-3-3 permettant leur
séquestration cytoplasmique (Takizawa and Morgan, 2000), et une régulation de leur
stabilité notamment par la kinase CDK1 elle-même ((Hoffmann et al., 1993) ; (Baldin et
al., 2002)).
Figure 4 : Activation complète du complexe CDK1-cycline B
L'activation complète du complexe CDK1-cycline B nécessite la déphosphorylation des résidus thréonine 14 et tyrosine 15 par les phosphatases de la famille CDC25, ainsi que la phosphorylation par CAK de la Thréonine 161 située dans la séquence T-loop. Les phosphorylations de la thréonine 14 par MYT1 et de la tyrosine 15 par WEE1 inactivent le complexe, empêchant ainsi la fixation de l'ATP et/ou la reconnaissance des substrats. (D’après la thèse de Matthieu Lemaire)
(4) Inhibition des complexes CDK-cyclines
L’inactivation du complexe CDK1-cycline B est obtenue par phosphorylation de la tyrosine
15 de CDK1 par la kinase WEE1 localisée dans le noyau en interphase (Russell and Nurse,
1987). La phosphorylation de la thréonine 14 par MYT1, localisée aux membranes du
réticulum endoplasmique et de l’appareil de Golgi, semble uniquement venir renforcer
l’inactivation de CDK1 (Liu et al., 1997). La phosphorylation de ces 2 résidus empêcherait
la fixation de l’ATP ou la reconnaissance de substrats par le complexe CDK-cycline (Berry
and Gould, 1996) (Figure 4).
Par ailleurs, l’activité des CDK est régulée par deux familles d’inhibiteurs appelées CKI
(pour Cyclin-dependent Kinase Inhibitors) : les familles Cip/Kip et INK4 (Inhibitors of
CDK4) (Harper and Elledge, 1996) (Figure 5).

23
Les protéines de la famille Cip/Kip, p21CIP1, p27KIP1 et p57KIP2, inhibent toutes les complexes
CDK-cycline en modifiant la structure des CDK, empêchant ainsi la fixation de l’ATP (Russo
et al., 1998).
Les protéines de la famille INK4, p16INK4a, p15INK4b, p18INK4c, p19INK4d, inhibent les complexes
CDK4/6-cycline D, en s’associant vraisemblablement aux CDK4 et 6, empêchant leur
interaction avec les cyclines (Russo et al., 1998).
Figure 5 : Les protéines CKI (Cyclin Dependent Kinase Inhibitors)
Les études cristallographiques ont confirmé que les mécanismes d'inhibition des deux familles sont différents: p27Cip1 inhibe le complexe CDK2-Cycline A en empêchant la fixation de l'ATP et en obstruant le site catalytique, tandis que p16INK4a empêche l'association CDK6-Cycline D en provoquant une rotation du domaine PSTAIRE. (D’après la thèse de Matthieu Lemaire)
De manière paradoxale, les protéines p21CIP1 et p27KIP1 pourraient également participer à
l'assemblage et à l’activation des complexes CDK4/6-cycline D. Comme bon nombre
d’acteurs du cycle cellulaire, ces protéines inhibitrices des CDK possèdent d’autres rôles
tels que la régulation de l’apoptose, l’activation de la transcription (Coqueret, 2003), la
migration cellulaire (Besson et al., 2004) ou l’expansion des cellules souches (Besson et
al., 2007).

24
b) Les phosphatases CDC25
Les phosphatases CDC25 sont des Dual-Specificity-Phosphatases (DSPases), c’est-à-dire
qu’elles sont capables de déphosphoryler à la fois une tyrosine et une thréonine/sérine
situées sur la même molécule (Millar and Russell, 1992). Elles permettent l’activation des
CDK et ainsi la progression dans le cycle cellulaire. Dans les cellules humaines, 3 gènes
codent pour les phosphatases CDC25 : cdc25a, cdc25b et cdc25c. À ce jour, de nombreux
variants d’épissage ont été décrits : deux pour CDC25 A et cinq pour CDC25 B et C, mais on
connaît mal les spécificités de substrats et de fonctions propres à chaque isoforme (Figure
6). Des données récentes obtenues dans l’équipe du Pr. B. Ducommun mettent en évidence
qu’il existe d’autres formes de CDC25B, résultant non seulement d’épissage alternatif mais
également de transcription alternative par un promoteur interne à la séquence du gène
cdc25b. Ces autres formes de CDC25B possèdent des propriétés différentes en terme de
stabilité et de reprise en mitose après dommages à l’ADN (Julien en préparation).
Figure 6 : Les variants d’épissage des différentes isoformes des CDC25 Humaines
Le domaine régulateur situé en N-Terminal de CDC25 A, B et C est sujet à l’épissage alternatif (boîtes colorées) aboutissant à la génération de différentes isoformes. Pour CDC25 A, un seul exon est épissé dans cdc25a2 en comparaison avec cdc25a1. Pour CDC25 B, au moins quatre domaines différents codant pour des exons sont présents de façon différentielle dans chaque isoforme. Trois exons sont ciblés par l’épissage générant les divers variants codant pour CDC25 C. Les points d’interrogation indiquent la présence de deux domaines qui n’ont pas encore été confirmés dans CDC25B5. Dans CDC25C4 et CDC25C5, l’épissage alternatif conduit à un décalage du cadre de lecture aboutissant à la génération de nouveaux acides aminés (boîtes foncées). Dans CDC25C3, la délétion génère aussi un décalage du cadre de lecture menant à la formation d’une protéine tronquée entre autre pour le domaine catalytique donnant naissance à une protéine sans activité phosphatase. La forte conservation du domaine catalytique de CDC25 A, B et C est symbolisée par la couleur grise. La Cystéine catalytique est aussi mentionnée. (D’après (Boutros et al., 2007a))

25
Dans les cellules de mammifères, les trois isoformes de CDC25 sont impliquées dans le
contrôle des transitions du cycle cellulaire G1/S et G2/M :
CDC25 A apparaît dans la cellule en fin de phase G1, s’accumule jusqu’en mitose où elle va
être dégradée (Molinari et al., 2000) ; (Bernardi et al., 2000). La micro-injection
d’anticorps dirigés contre CDC25 A bloque les cellules en phase S (Hoffmann et al., 1993),
tandis que sa sur-expression accélère l’entrée en phase S et conduit à une activation
précoce des complexes CDK2-cycline E/CDK2-cycline A (Blomberg and Hoffmann, 1999).
Cependant, le rôle de CDC25 A ne se limite pas à la transition G1/S puisque l’inhibition de
son expression par interférence à l’ARN entraîne également une diminution du
pourcentage de cellules entrant en mitose, de même que sa sur-expression une entrée
prématurée en mitose par inactivation du complexe CDK1-cycline B1 (Mailand et al.,
2002).
CDC25 B et C ont été initialement décrites comme des phosphatases régulatrices de
l’entrée en mitose avec vraisemblablement des rôles différents. Alors que le niveau de
protéine CDC25 C reste relativement constant tout au long d’un cycle cellulaire normal
(Girard et al., 1992), CDC25 B s’accumule en phase S pour atteindre son niveau maximum
en mitose (Gabrielli et al., 1996). Si l’invalidation de CDC25 B ou CDC25 C conduit à un
blocage ou un retard de l’entrée en mitose, seule la sur-expression de CDC25 B conduit à
une entrée illégitime en phase M que la réplication soit achevée ou non (Lammer et al.,
1998). CDC25 B a été proposé comme l’activateur initial du pool de CDK1-cycline B localisé
aux centrosomes durant la transition G2/M (Lindqvist et al., 2005). L’activation résultante
de ce complexe induit ensuite la stabilisation et l’activation des CDC25 A et C, ce qui a
pour conséquence la pleine activation du MPF (Maturation Promoting Factor) dans le noyau
et donc l’entrée en mitose (Lindqvist et al., 2005). CDC25C déphosphoryle uniquement le
complexe CDK1-cycline B1 (Gabrielli et al., 1997), alors que CDC25 B pourrait
déphosphoryler les complexes CDK1-cycline B1 (Gabrielli et al., 1997) et CDK1-cycline A
(Sebastian et al., 1993) et CDK2-cycline A (Goldstone et al., 2001). La spécificité de
substrat de CDC25 B dépend de la phase du cycle cellulaire avec une affinité pour CDK2-
cycline A en phase S/G2 et pour CDK1-cycline B1 en phase G2/M (Lammer et al., 1998).
Les travaux récents de notre équipe suggèrent également que CDC25 B jouerait un rôle en
phase S dans le contrôle de la réplication (Bugler et al.) ainsi que dans le cycle de
duplication du centrosome ((Boutros et al., 2007b) ; Boutros en préparation).
Seule l'invalidation chez la souris du gène cdc25a est létale à un stade précoce de
l’embryogenèse (5-7ème jours) (Ferguson et al., 2005). Les souris invalidées pour les gènes
cdc25 b et/ou cdc25c ne présentent pas de phénotype particulier en dehors d’une stérilité
chez les femelles liée à l’incapacité des ovocytes cdc25b-/- à activer le MPF et à maturer

26
((Lincoln et al., 2002); (Chen et al., 2001)). Ces résultats surprenants sont en
contradiction avec ceux obtenus par l’équipe de Medema qui ont mis en évidence que seul
CDC25 B permet la reprise du cycle cellulaire au point de contrôle G2/M (van Vugt et al.,
2004), ainsi qu'avec les nombreux travaux décrivant les propriétés de ces trois
phosphatases. La capacité de CDC25 A à assurer une redondance fonctionnelle lors d'une
invalidation constitutive des deux autres phosphatases est une explication probable à cette
observation. Notre équipe vient d'obtenir des souris dans lesquelles l'expression de CDC25
B peut être invalidée de façon conditionnelle par la recombinase CRE. Ces animaux vont
être rapidement utilisés afin d’étudier de manière précise et de mieux comprendre le rôle
de CDC25 B dans de nombreux tissus et à différents stades du développement.
2. Points de contrôle du cycle cellulaire
Tous les organismes possèdent un certain nombre de mécanismes de contrôle assurant le
maintien de l’intégrité du génome. La division cellulaire et le maintien de l’intégrité du
génome nécessitent une distribution spatio-temporelle et une organisation précise des
différents acteurs du cycle. Chaque cellule d’un organisme est perpétuellement soumise à
des agressions intrinsèques (stress oxydant, erreur lors de la réplication…) et extrinsèques
(radiations, génotoxiques…) qui sont responsables de nombreuses altérations sur l’ADN. Ces
lésions sur l’ADN peuvent créer des adduits sur la molécule d’ADN empêchant ainsi
l’appariement des bases, la réplication ou la transcription, conduisant à la perte de bases
ou à des dommages simples ou double brins. Les points de contrôle du cycle cellulaire, qui
sont des voies de signalisation biochimiques, vont identifier les dommages sur l’ADN et
induire une réponse cellulaire en retardant la progression du cycle et activant les voies de
réparation des dommages. Lorsque les dommages sont trop importants, les acteurs des
points de contrôle pourront participer à l'induction de la mort cellulaire.
a) Les différents points de contrôle du cycle cellulaire
Au cours du cycle cellulaire des cellules eucaryotes, différents points de contrôle ont été
identifiés (Figure 7):
Le point de contrôle de la quiescence, avant l’entrée en phase G1 ; le point de contrôle
G1/S empêchant la réplication d’erreur en phase S ; le point de contrôle intra-S activé
lorsque la réplication est incomplète et lorsque l’ADN est endommagé ; le point de

27
contrôle G2, empêchant la dissémination des erreurs ; le point de contrôle mitotique
bloquant la séparation des chromatides sœurs ; le point de contrôle de la cytokinèse et le
point de contrôle des dommages à l’ADN permettant des arrêts du cycle en phase G1, G2,
S et en métaphase de la mitose.
Figure 7 : Les points de contrôle du cycle cellulaire
Les cellules dont l'ADN est endommagé en G1 déclenchent le point de contrôle G1/S, qui empêche l'initiation de la réplication et ainsi la copie des erreurs. Le point de contrôle intra-S ralentit la progression de la réplication en présence de dommages à l'ADN. Enfin le point de contrôle G2/M est activé par les dommages de l'ADN survenant en phase G2, et empêche l'entrée en mitose et la dissémination des erreurs. (D’après la thèse de Matthieu Lemaire).
Lorsque les points de contrôle vont être activés, deux réponses vont être mises en place :
une réponse rapide faisant intervenir des modifications post-traductionnelles des
différents acteurs et une réponse différée plus stable mettant en jeu l’activation de
nouveaux gènes.
- la réponse rapide résulte de nombreux événements de
phosphorylation/déphosphorylation mettant en jeu tous les acteurs du point de contrôle
concerné. Ces événements jouent sur la stabilité, la dégradation, l’activation ou
l’inhibition des différentes kinases/phosphatases. Nous développerons plus largement tous
ces événements dans les parties suivantes consacrées à la signalisation des dommages.
- la réponse différée résulte principalement de l’action du facteur de transcription
et gène suppresseur de tumeur p53. La protéine p53 joue un rôle majeur dans l’arrêt du
cycle en phase G1, mais son rôle en G2 est de mieux en mieux documenté (Taylor and
Stark, 2001). p53 agit tout d’abord sur la transcription de régulateurs des CDK tels que la
protéine 14-3-3, qui va maintenir le complexe CDK1-cycline B dans le cytoplasme, et la
protéine p21CIP1, qui va inhiber la phosphorylation activatrice de la CAK tout en inhibant

28
également les complexes CDK-cyclines. Enfin, p53 permet un arrêt durable du cycle en
réprimant indirectement la transcription de CDK1-cycline B1 (Taylor and Stark, 2001).
b) Organisation de la voie de signalisation des dommages
à l’ADN
Durant ma thèse, nous avons principalement porté notre attention sur les points de
contrôle activés par la voie de signalisation des dommages à l’ADN. Ainsi, dans la suite de
cette introduction, je focaliserai ma présentation sur la description des acteurs de cette
voie. Nous venons de voir que le point de contrôle des dommages à l’ADN peut induire des
arrêts du cycle en phase G1, S et G2. Pour conduire à cela, la signalisation des dommages
va être différente en fonction du type de lésions à l’ADN. Ainsi, selon qu’il s’agisse de
cassures simple ou double brin, de dommages induits par des rayonnements ultra-violets ou
des radiations ionisantes, la signalisation sera différente.
Pour décrire ces voies de signalisation induites lors de dommages, j’ai choisi l’exemple
d’une réponse à des cassures double brin (DSB pour Double Strand Breaks) puisque ce type
de dommage, très toxique, est plus difficile à réparer (Figure 8). La signalisation des DSB
met en jeu les trois kinases de la famille PIKK (phosphoinositide 3-kinase related kinases) :
ATM (Ataxia-Telangiectasia Mutated), ATR (ATM and Rad3 related) et DNA-PK (DNA-
dependent Protein Kinase catalytic subunit) (Bartek and Lukas, 2007).
Les acteurs de ce point de contrôle sont sub-divisés en 4 groupes : (i) les protéines
senseurs, qui vont détecter les dommages : le complexe MRN (MRE11-RAD50-NBS1), RAD17
et HUS1, (ii) les médiateurs, qui vont faciliter le recrutement des acteurs du point de
contrôle et des systèmes de réparation : les protéines à domaine BRCT (BRCA1 C-terminus
repeat), MDC1, 53BP1 (p53 Binding Protein 1), BRCA1, claspine, TopBP1 (Topoisomerase
binding protein 1) (iii) les transducteurs qui amplifient le signal reçu des senseurs : ATM,
ATR, ATRIP (ATR interacting protein), CHK1 (Checkpoint Kinase 1) et CHK2 et (iv) les
effecteurs qui permettent l’arrêt effectif du cycle cellulaire et la réparation des
dommages : p53, les phosphatases CDC25.
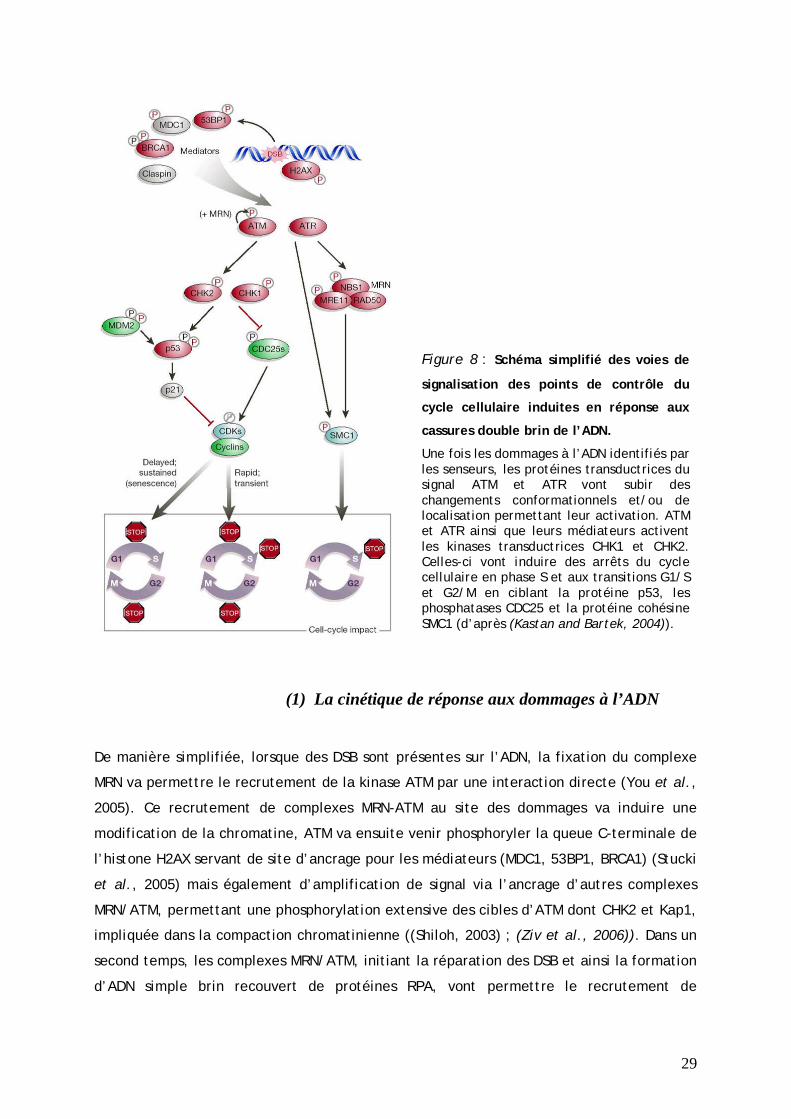
29
(1) La cinétique de réponse aux dommages à l’ADN
De manière simplifiée, lorsque des DSB sont présentes sur l’ADN, la fixation du complexe
MRN va permettre le recrutement de la kinase ATM par une interaction directe (You et al.,
2005). Ce recrutement de complexes MRN-ATM au site des dommages va induire une
modification de la chromatine, ATM va ensuite venir phosphoryler la queue C-terminale de
l’histone H2AX servant de site d’ancrage pour les médiateurs (MDC1, 53BP1, BRCA1) (Stucki
et al., 2005) mais également d’amplification de signal via l’ancrage d’autres complexes
MRN/ATM, permettant une phosphorylation extensive des cibles d’ATM dont CHK2 et Kap1,
impliquée dans la compaction chromatinienne ((Shiloh, 2003) ; (Ziv et al., 2006)). Dans un
second temps, les complexes MRN/ATM, initiant la réparation des DSB et ainsi la formation
d’ADN simple brin recouvert de protéines RPA, vont permettre le recrutement de
Figure 8 : Schéma simplifié des voies de
signalisation des points de contrôle du
cycle cellulaire induites en réponse aux
cassures double brin de l’ADN.
Une fois les dommages à l’ADN identifiés par les senseurs, les protéines transductrices du signal ATM et ATR vont subir des changements conformationnels et/ou de localisation permettant leur activation. ATM et ATR ainsi que leurs médiateurs activent les kinases transductrices CHK1 et CHK2. Celles-ci vont induire des arrêts du cycle cellulaire en phase S et aux transitions G1/S et G2/M en ciblant la protéine p53, les phosphatases CDC25 et la protéine cohésine SMC1 (d’après (Kastan and Bartek, 2004)).

30
complexes ATR/ATRIP via les médiateurs TopBP1 et claspine (Kumagai et al., 2006). ATR
va alors phosphoryler l’ensemble de ces cibles dont CHK1 (Liu et al., 2006) (Figure 9).
L’activation des kinases CHK1 et CHK2 va permettre l’activation ou l’inhibition des
effecteurs (p53 et les CDC25) afin de ralentir ou d’arrêter le cycle cellulaire, de réparer
les lésions ou dans certains cas d’initier le processus apoptotique. L’inactivation des
phosphatases CDC25 par modification de leur localisation, modulation de leur activité
enzymatique, dégradation, va permettre de maintenir les complexes CDK-cyclines dans un
état inactif.
Figure 9 : Activation des kinases ATM et d’ATR en
réponse aux cassures doubles brin de l’ADN.
Les DSB reconnues par le complexe MRN vont recruter la kinase ATM. L’initiation du processus de réparation induit par ATM permet la formation d’un simple brin d’ADN recouvert de la protéine RPA permettant le recrutement du complexe ATR-ATRIP via TopBP1. ATR activée va phosphoryler ses cibles dont la kinase CHK1. (D’après (Bartek and Lukas, 2007))

31
(2) Les acteurs du point de contrôle des dommages à
l’ADN
Je focaliserai également la suite de cette présentation sur les acteurs principaux du point
de contrôle des dommages en réponse aux cassures double brin.
- La kinases ATM
ATM est une protéine de 350kDa, elle fait partie de la famille des PIKK mais ne présente
pas d’activité lipide kinase. La mutation d’ATM est responsable de l’Ataxia Telangiectasia,
une maladie autosomale récessive rare caractérisée par une dégénérescence cérébelleuse,
une immunodéficience, une instabilité génétique et une prédisposition au cancer (Shiloh,
1997). La perte d’ATM est non létale, suggérant qu’ATM ne soit pas essentielle au cycle
cellulaire normal et à la différenciation (Shiloh and Kastan, 2001). En l’absence de
dommage, ATM est maintenue inactive sous forme d’homodimères dans la cellule, les DSB
induisant un changement conformationnel de la protéine vont permettre son
autophosphorylation sur la sérine 1981 et ainsi sa monomérisation. ATM phosphoryle de
nombreux substrats parmi lesquels CHK2, BRCA1, p53, MDM2, H2AX, Rad17, CHK1.
- La kinase ATR
ATR a été découverte grâce aux bases de données du génome humain par homologie de
séquence avec ATM et Rad3. C’est une protéine de 303 kDa essentielle à la viabilité
cellulaire puisque lors de son invalidation les souris meurent à l’état embryonnaire, cela
suggère un rôle essentiel d’ATR en l’absence de dommages. Contrairement à ATM, ATR
semble constitutivement activée, d’où l’importance de sa localisation cellulaire. ATR en
complexe avec ATRIP est recrutée en présence d‘ADN simple brin par la protéine RPA. Le
rôle d’ATR ne se limite donc pas à la présence de DSB. ATR phosphoryle CHK1, p53, BRCA1,
H2AX, MDM2 et CHK2. Le modèle initial de phosphorylation exclusive de CHK2 par ATM et
de CHK1 par ATR semble assez remis en question dans différentes situations par exemple
lors de la réponse aux radiations ionisantes où CHK1 va être phosphorylée par ATM, on
parle de « crosstalk » entre ces protéines (Gatei et al., 2003).
- La protéine onco-suppressive p53
Cette protéine joue un rôle central dans le devenir cellulaire notamment lorsque les
cellules présentent des dommages sur leur ADN. En condition normale, le niveau de p53
reste faible grâce à la protéine MDM2 qui contrôle la dégradation protéasome-dépendante
de p53 mais aussi son export nucléaire (Liang and Clarke, 2001). Après détection de

32
dommages ADN, p53 est phosphorylée sur son domaine de trans-activation incluant les
sérines 15 et 20. La phosphorylation sur la sérine 15 par ATM et ATR permet la stabilisation
de p53 en inhibant son interaction avec MDM2 (Chehab et al., 1999). p53 est l’acteur clé
de l’arrêt du cycle cellulaire en phase G1. Sa cible transcriptionnelle essentielle est
p21WAF1 qui inhibe le complexe CDK2-cycline E et ainsi la transition G1/S (Harper et al.,
1993). p53 possède une centaine de gènes cibles lui permettant de réguler à la fois l’arrêt
du cycle cellulaire, la réparation des dommages, l’apoptose et la sénescence des cellules
(Riley et al., 2008).
- La protéine CHK2
CHK2 est une protéine exprimée de manière stable au cours du cycle cellulaire (Lukas et
al., 2001) mais inactive et sous forme monomérique, en absence de dommages. Cette
kinase est activée principalement par ATM en réponse à des DSB. La phosphorylation de la
thréonine 68 par ATM induit une dimérisation de la protéine, son autophosphorylation et
par conséquent son activation. Il semble que les kinases CHK1 et CHK2 aient partiellement
des rôles redondants, puisqu’un certain nombre de leurs cibles sont identiques : la kinase
TLK, la protéine PML, la kinase PLK3 et le facteur de transcription E2F1, permettant la
régulation de mécanismes cellulaires essentiels comme la réplication de l’ADN, la
progression dans le cycle, la restructuration de la chromatine ou l’apoptose (Bartek and
Lukas, 2003). Cependant, l’absence de mortalité lors de l'invalidation de chk2 chez la
souris , contrairement à ce qui est observé avec chk1, suggère des rôles indépendants et
propres de ces kinases (Takai et al., 2002). CHK2 permet une diminution rapide de CDC25
A indépendante de p53, par phosphorylation directe de la sérine 123 (Falck et al., 2001).
On retrouve de manière significative dans les tumeurs solides installées des anomalies au
niveau du gène chk2 avec soit des pertes d’hétérozygotie ou des mutations ponctuelles
(Bartkova et al., 2005).
- La protéine CHK1
La protéine de 56 kDa CHK1 est constituée de deux domaines d’environ 200 acides aminés :
le domaine kinase N-terminal et le domaine régulateur C-terminal. Le domaine kinase
présentant une activité constitutive va être inhibé par le domaine C-terminal,
probablement en cis par un mécanisme encore inconnu aujourd’hui. L’activation de la voie
des dommages permettant la phosphorylation du domaine C-terminal de CHK1 par
ATM/ATR permet la levée d’inhibition du domaine kinase (Kosoy and O'Connell, 2008). Des
travaux réalisés sur la levure ont mis en évidence des sites d’import et d’export nucléaire,
mais ils ne sont pas retrouvés dans la protéine humaine (Dunaway et al., 2005).

33
Contrairement à CHK2, l’expression de la kinase CHK1 est restreinte aux phases S et G2 du
cycle cellulaire (Lukas et al., 2001), elle est également active au cours d’un cycle
cellulaire non perturbé par des dommages (Jiang et al., 2003). La létalité précoce, au
stade embryonnaire, de l'invalidation de chk1 chez la souris met en évidence son rôle
essentiel au-delà du point de contrôle des dommages ADN, dans la surveillance du génome
durant la réplication de l’ADN (Takai et al., 2000). Des travaux récents portant sur les
formes mutées des différents sites de phosphorylation de CHK1 ont permis de grandes
avancées dans la compréhension de l’activation de la protéine. En l’absence de stress, une
phosphorylation faible de S345-CHK1 semble nécessaire pour empêcher l’entrée
prématurée des cellules en mitose. En revanche, en réponse aux dommages, l’activation
des protéines de la famille PIKK va permettre une activation directe de CHK1 5 à 10 fois
supérieure par phosphorylation de S345-CHK1 mais également de S317-CHK1 ((Liu et al.,
2000) ; (Zhao and Piwnica-Worms, 2001)) alors liée à la chromatine du noyau (Figure 10).
Cette forme phosphorylée semble indispensable au relargage chromatinien de
phosphoS345-CHK1 permettant alors son accumulation centrosomique ((Smits et al.,
2006) ; (Niida et al., 2007)). D’autre part la forme phosphoS345-CHK1 semble également
servir de site d’ancrage à la protéine 14-3-3, renforçant probablement l’export nucléaire
de CHK1 et donc l’inactivation du complexe CDK1-cycline B (Jiang et al., 2003),
permettant ainsi un arrêt du cycle durable avant la mitose.
Figure 10 : Modèle de régulation de la translocation de la kinase CHK1 vers le centrosome lors
de dommages à l’ADN.
La nécessité d’une phosphorylation de CHK1 sur la sérine 345 pour son association centrosomale n’est pas connue. (D’après (Niida et al., 2007)).
L’activation de CHK1 est maintenue tout au long de l’arrêt au point de contrôle. Des
travaux portant sur des mutants thermo-sensibles de CHK1 chez la levure ont permis de
mettre en évidence que l’inactivation de CHK1 durant le point de contrôle est suffisante

34
pour permettre une entrée illégitime en mitose en présence de dommages ADN encore
appelée catastrophe mitotique (Latif et al., 2004).
Lors de la reprise en mitose, son activité va diminuer mais reste supérieure à son activité
initiale, antérieure au stress. Dans les cellules humaines, deux phosphatases sont capables
de déphosphoryler CHK1 : la phosphatase Wip1 régulée par p53 et la protéine phosphatase
2A (PP2A). D’une part, Wip1 en déphosphorylant CHK1 sur la sérine 345 permet de
diminuer son activité à la fin du point de contrôle pour permettre un retour à son état
initiale après réparation des dommages (Lu et al., 2005b). D’autre part, l’équipe de
Piwnica-Worms a mis en évidence une régulation de l’activation de CHK1, elle-même
dépendante de son activité, mettant en jeu la kinase activatrice ATR et la phosphatase
PP2A (Leung-Pineda et al., 2006).
Par ailleurs, l’activation de la protéine CHK1 est régulée par de nombreuses autres
protéines telles que CDK1 elle-même (Shiromizu et al., 2006), ou PKB/AKT. De manière
intéressante, les équipes de Shtivelman et Parsons ont montré que les cellules invalidées
pour le gène suppresseur de tumeur pten vont phosphoryler CHK1 sur la sérine 280
conduisant à son ubiquitinylation sur la lysine 274 et ainsi à son inactivation par
translocation cytoplasmique ((King et al., 2004) ; (Puc and Parsons, 2005); (Puc et al.,
2005)). L’inhibition de CHK1 provoque ainsi une augmentation des CDC25 contribuant à la
mise en place de l’instabilité génétique observée dans les cellules cancéreuses
fréquemment mutées pour le gène pten (Shen et al., 2007).
In fine, l’activation de CHK1 va permettre l’inhibition indirecte des complexes CDK1-
cycline B en modulant l’activité des effecteurs de la voie des dommages. Différents
mécanismes ont pu être mis en évidence tel qu’une dégradation-protéasome dépendante
de CDC25 A consécutive à la phosphorylation de 3 sérines (76, 82 et 88), une séquestration
cytoplasmique de CDC25 C faisant intervenir la protéine 14-3-3 sur la sérine phosphorylée
216 (Sanchez et al., 1997), ou une activation de la protéine kinase WEE1 (O'Connell et al.,
1997).
Pour finir, il est intéressant de noter que contrairement à la kinase CHK2, CHK1 et ATR
sont très faiblement retrouvés mutées dans les cellules cancéreuses, suggérant
probablement que cette voie ATR-CHK1 est essentielle à la viabilité et à la prolifération
des cellules cancéreuses.
La meilleure connaissance des mécanismes d’action de ces acteurs clés de la voie des
dommages est essentielle pour permettre de comprendre les dérégulations observées dans
les cellules cancéreuses ainsi que pour permettre d’investir de nouveaux champs d’action
dans la thérapie des cancers.

35
B. Tumorigenèse et barrière anti-tumorale
1. Dans les tumeurs solides
Les études publiées par Gorgoulis et Bartkova en 2005 sont les premières à avoir mis en
évidence le rôle de la machinerie du point de contrôle des dommages ADN (ou DDR pour
DNA Damage Response) dans la progression tumorale ((Gorgoulis et al., 2005) ; (Bartkova
et al., 2005)). L’hypothèse initiale était que la croissance tumorale conduisait à
l’apparition d’un stress cellulaire mettant en jeu principalement l’attrition télomérique et
l’hypoxie menant à une activation de la voie des dommages à l’ADN. Dans ce modèle,
l’activation de la voie était une conséquence de l’initiation du processus tumoral. Les
études de 2005, menées sur un très large échantillonnage à la fois de tissu sain, pré-
tumoral et tumoral issus de diverses tumeurs solides (rein, sein, poumon, colon, peau), ont
permis de mettre en évidence une activation précoce des acteurs de la DDR :
phosphorylation de Thr68-CHK2, de Ser345 et 317-CHK1, et de H2AX, foci nucléaire de la
protéine 53BP1, niveau élevé de p53. L’activation maximale de cette voie est observée
dans les stades qui précédent ou très précoces du processus tumoral et retombe pour les
stades plus avancés, toutefois elle peut persister à des niveaux variables selon les tumeurs
examinées dans les phases plus avancées de la tumorigenèse. Les auteurs ont également
montré que l’activation de la voie des dommages précède les mutations et/ou pertes
d’expression de composants essentiels qui sont largement retrouvés dans les cancers
comme p53, ATM ou CHK2 (Figure 11).
Figure 11: Modèle d’activation du point de contrôle des dommages à l’ADN dans les cancers.
Une prolifération cellulaire anormalement stimulée aboutit à un stress réplicatif, à des cassures double brin de l’ADN (DSB), à une instabilité génétique, à l’activation du point de contrôle des dommages et à l’apoptose dépendante du gène suppresseur de tumeur p53. L’apoptose dépendante de p53 réprimant ainsi l’expansion des cellules cancéreuses, son inactivation constitue une pression de sélection. L’attrition télomérique et l’hypoxie peuvent aussi participer à la formation des DSB, à l’activation du point de contrôle et à l’instabilité génétique. (d’après (Gorgoulis et al., 2005)).

36
Dans ce modèle de progression tumorale, un stress réplicatif en amont serait responsable
de l’activation de la voie des dommages dans des cellules non-mutées impliquant des
dérégulations des acteurs du cycle comme des sur-expression de la cycline E, de CDC25 A,
ou du facteur de transcription E2F1, ou bien une sous-expression de la protéine RB. Cette
barrière qui implique la présence de points de contrôle encore valides devra être franchie
pour qu’une cellule devienne tumorale. Selon Bartek à la fois l’attrition télomérique et la
présence de radicaux libres oxygénés générés par le métabolisme cellulaire participeraient
également à l’activation de cette voie (Bartek and Lukas, 2007). Une étude réalisée par
l’équipe de Tanaka a d’ailleurs mis en évidence une diminution des dommages spontanés
mesurés par la phosphorylation de l’histone H2AX en cytométrie de flux lors d’utilisation
d’agents anti-oxydant ou d’analogues non-métabolisables du glucose (Tanaka et al., 2006).
Cependant, cette étude qui met en évidence la répartition des dommages au cours du
cycle cellulaire n’a été réalisée que sur des lignées cellulaires.
Ce modèle de progression tumoral permet d’expliquer de nombreuses caractéristiques des
cellules cancéreuses telles que la présence d’une grande instabilité génétique ou la haute
fréquence de mutations d’acteurs du point de contrôle des dommages ; cependant de
nombreuses questions restent non résolues: par quels mécanismes les oncogènes décrits
induisent-ils un stress réplicatif ? Quelle est la contribution des stress réplicatif et
oxydatif, et de l’attrition télomérique dans la génération de l’instabilité génétique ? Ce
modèle s’applique-t-il à l’ensemble des tumeurs ? Et enfin ce modèle ouvre-t-il de
nouvelles perspectives pour la prise en charge des cancers ?
2. Dans les hémopathies
La question de l’activation de la voie des dommages à l’ADN et la notion de barrière anti-
tumorale au cours du processus de leucémogenèse est beaucoup plus délicate à aborder
dans les hémopathies. En effet, contrairement aux tumeurs solides où la masse tumorale
localisée permet de travailler sur les tissus tumoraux et sur les tissus sains environnants,
de même que sur des stades plus ou moins avancés de la maladie, dans les leucémies on
ne détermine pas de stade évolutif de la maladie mais seulement un pourcentage
d’envahissement des cellules immatures (appelées blastes) dans la moelle osseuse des
patients.

37
a) Modèles de leucémogenèse
(1) Chez la souris
Un modèle murin de leucémogenèse a été mis au point par plusieurs équipes à partir de
différents constats argumentés dans la littérature. Tout d’abord, 30% des cas de syndromes
myélodysplasiques (SMD) progressent en Leucémie Aiguë Myéloïde (LAM), bien que les
mécanismes responsables de cette progression tumorale soient mal connus. Les SMD
représentent ainsi un modèle in vivo de leucémogenèse. Brièvement, les SMD sont un
groupe hétérogène de maladies clonales de la cellule souche hématopoïétique caractérisés
par une prolifération et une différenciation anormales des progéniteurs hématopoïétiques
conduisant à une dysplasie des trois lignées myéloïdes avec pour conséquence une
hématopoïèse inefficace et des cytopénies. Ces pathologies présentent une grande
instabilité génétique caractérisée par des anomalies caryotypiques dans 50% des cas, la
mutation activatrice de l’oncogène ras est présente dans 20% des cas. D’autre part, la
diminution observée du taux d’apoptose lors de la progression de la maladie corrèle avec
une augmentation intra-cellulaire des protéines de la famille BCL2 (Parker and Mufti,
2001). L’observation de l'altération importante de ces gènes a permis de suggérer qu’ils
puissent jouer un rôle dans la leucémogenèse, des modèles murins ont alors été générés.
Les souris exprimant NRAS activé présentent un phénotype léger avec une faible
augmentation du nombre de neutrophiles immatures (Kogan et al., 1998), et les souris sur-
exprimant BCL2 semblent avoir une augmentation du nombre de blastes dans la moelle
mais un nombre normal de cellules sanguines périphériques (Lagasse and Weissman, 1994).
En revanche, les souris NRAS activé et BCL2 sur-exprimé développent une maladie
analogue à une LAM sous 2 à 6 mois (Rassool et al., 2007). Ce modèle murin a permis de
mettre en évidence que l’activation des oncogènes ras et bcl2 est responsable de la
production de radicaux libres oxygénés, de DSB sur l’ADN et d’une activation de la voie des
dommages. Cependant, ce modèle utilisant les souris transgéniques ne rend pas compte de
l’évolution naturelle de la maladie et de sa progression vers les leucémies aiguës.
(2) Du syndrome myélodysplasique à la leucémie
Compte tenu de l’évolution naturelle des SMD vers les LAM, quelques études ont porté sur
l’activation de la voie des dommages lors de la leucémogenèse, faisant le pendant avec ce
qui est connu dans les tumeurs solides. L’étude menée par Horibe en 2007 a mis en

38
évidence qu’il existe une progression en terme d’activation de la voie des dommages dans
les différentes classifications des SMD. De manière simplifiée, les SMD de faible risque,
c’est-à-dire à plus faible risque de progression vers les LAM, présentent des niveaux plus
élevés de phosphorylation des acteurs de la voie des dommages (phospho-ATM, phospho-
CHK2, phospho-H2AX) que les SMD de haut risque ou les LAM secondaires (Horibe et al.,
2007). Une étude de Boehrer datant de 2009 a également appuyé ces résultats avec une
diminution de l’activation des kinases CHK1 et CHK2 lors de la progression des SMD vers les
LAM. Cependant, cela ne s’accompagne pas d’une modification de l’activation de la kinase
ATM ni du signal H2AX, qui ne diminuent pas, voire même augmentent lors de la
progression tumorale (Boehrer et al., 2009). Ces quelques résultats semblent suggérer que
la notion de barrière anti-tumorale largement décrite dans les tumeurs solides pourrait
également être valable au cours de la leucémogenèse. Cependant, des études
complémentaires semblent nécessaires pour que cette notion soit tout à fait admise :
d’une part, les études de leucémogenèse menées sur les SMD mettent en parallèle les SMD
et les LAM, or seul 30 à 40% de ces SMD évolueront en LAM ; de plus, ces LAM secondaires
constituent un type particulier de LAM dites de haut risque avec de nombreuses anomalies
du caryotype et souvent une résistance initiale au traitement, ce qui sous-entend que dans
ces conditions, on ne s’adresse qu’à un sous-type particulier de LAM.
Afin de répondre à ces questions, notre équipe à récemment mis en place une
collaboration avec des cliniciens du CHU Toulouse-Purpan pour étudier les échantillons de
SMD et réaliser une détection systématique à grande échelle de l’activation des acteurs de
la voie des dommages à l’ADN afin d’étudier plus largement ces profils au cours de
l'évolution de la maladie.
C. Thérapeutiques ciblant les acteurs de la voie des dommages
La grande majorité de nos connaissances dans le domaine de la prise en charge des
dommages à l’ADN vient de la détection de mutations présentes dans les maladies
héréditaires humaines prédisposant aux cancers. La liste est longue mais on peut prendre
pour exemple la transmission génétique de la mutation des gènes brca1 ou brac2, tous
deux impliqués dans la voie de réparation par recombinaison homologue, qui prédispose
aux cancers du sein et des ovaires (Bertwistle and Ashworth, 2000). Les progrès des
connaissances acquises au cours de la dernière décennie dans la compréhension des

39
mécanismes de contrôle du cycle cellulaire et dans la voie de réponse aux dommages à
l'ADN conduisent aujourd'hui à envisager de nouvelles stratégies thérapeutiques les ciblant
spécifiquement.
1. Intérêt d’un ciblage du point de contrôle G2/M
Plus de la moitié des tumeurs solides présente des anomalies du point de contrôle G1/S du
cycle cellulaire. Ces anomalies peuvent résulter de deux mécanismes : soit une levée du
point de contrôle G1/S, résultant principalement de la perte de fonction des gènes
suppresseurs de tumeurs p53 et rb, soit une accélération de la transition G1/S résultant de
mutations activatrices d’oncogènes tels que ras, cycline D, erbB, egfr…
Bien qu’il existe des variations entre les tumeurs, dépendant principalement de la capacité
de celles-ci à répondre aux agents radio- et chimiothérapeutiques, les cellules p53-/-
perdent leur capacité d’arrêt au point de contrôle G1/S en réponse aux radiations
ionisantes (Kuerbitz et al., 1992). Ce constat clinique a permis d’orienter de nouvelles
stratégies thérapeutiques visant à inactiver les acteurs du point de contrôle G2/M et en
particulier ceux de la voie de signalisation des dommages afin de cibler le maximum de
tumeurs y compris celles présentant des anomalies du point de contrôle G1/S.
Ainsi, il a été mis en évidence en 1995 que l’utilisation de l’inhbiteur caféine permet la
levée du point de contrôle G2/M en réponse aux radiations ionisantes. Cela permet une
radiosensibilisation supérieure des cellules déficientes pour p53 en comparaison avec leurs
homologues proficientes pour p53 (Powell et al., 1995). Malgré le fait que le mécanisme
d’action de la caféine n’était pas connu en 1995, son rôle dans la levée du point de
contrôle G2/M et dans la chimiosensilisation de nombreuses cellules tumorales a été
largement mis en évidence. Ce n’est quand 1999 que l’implication des kinases de la voie
des dommages ATM et ATR a été mise en évidence, validant ainsi l’utilisation d’inhibiteur
de la voie des dommages dans la sensibilisation des tumeurs p53 déficientes (Sarkaria et
al., 1999).
Ces premières découvertes ont ouvert le champ sur l’intérêt thérapeutique de l’utilisation
de ce type d’inhibiteur.

40
2. Inhibiteurs des acteurs de la voie des dommages
a) Inhibiteurs des kinases ATM et ATR
Comme nous l’avons vu précédemment, l’inhibition des kinases ATM/ATR par la caféine a
mis en évidence l’intérêt thérapeutique que représente la combinaison d’inhibiteurs de la
voie des dommages avec des agents endommageant l’ADN. Cependant, il a été mis en
évidence rapidement que la levée du point de contrôle G2/M par la caféine impliquait
d’autres voies de signalisation en dehors de celle des kinases ATM/ATR, nécessitant la
synthèse d’autres composés plus spécifiques (Cortez, 2003).
Les inhibiteurs de la famille des PIKK, LY294002 et wortmanine, inhibant
préférentiellement la protéine DNA-PK dans des cellules d’adénocarcinome (Sarkaria et
al., 1998), ont servi pour le criblage de composé spécifique des kinases ATM/ATR.
Une petite molécule, le KU55933, a pu ainsi être mise en évidence. Le KU55933 entre en
compétition avec l’ATP au niveau de la poche ATP de la kinase ATM, inhibant la kinase
avec une IC50 de 13 nM. Il permet une augmentation de la sensibilité aux radiations
ionisantes mais aussi à l’étoposide, à la doxorubicine et à la camptothécine dans des
lignées de tumeurs solides (Hickson et al., 2004).
Un autre composé inhibiteur d’ATM, le CP466722, a été découvert en 2008 par crible in
vitro de l’activité kinase d’ATM. Comme le KU55933, cet inhibiteur réversible permet une
radiosensibilisation des cellules HeLa (Rainey et al., 2008).
Malgré l’intérêt potentiel que représente l’utilisation de ces composés, aucun essai
clinique utilisant ces composés chez les patients n’a été publié, et aucun essai clinique en
cours n’est rapporté sur le site du ClinicalTrials.gov répertoriant les essais cliniques
achevés et en cours en cancérologie. Une explication possible est que la kinase ATM
possède de très nombreux substrats et est impliquée dans de nombreuses fonctions
cellulaires au-delà du point de contrôle G2/M : point de contrôle G1/S, trafic
intracellulaire, contrôle de la croissance cellulaire… Ainsi, les kinases ATM/ATR ne seraient
peut-être pas les meilleurs candidats pour la mise en place d’une stratégie ciblant le point
de contrôle G2/M.

41
b) Inhibiteurs de DNA-PK
La protéine DNA-PK est un médiateur dans le processus de réparation des cassures double
brin par recombinaison non homologue, son inhibition va donc diminuer ce processus de
réparation, augmentant ainsi la présence des dommages après traitement. L’intérêt de
diminuer l’activité de cette protéine résulte de deux observations : d’une part les souris
déficientes pour DNA-PK deviennent hypersensibles aux radiations ionisantes et aux autres
agents inducteurs de cassures double brin ; d’autre part, la sur-activation de DNA-PK dans
les cancers confère une radio- et une chimiorésistance aux cellules tumorales ((Collis et
al., 2005) ; (Muller et al., 1998)). Les résultats prometteurs obtenus en terme de radio-
sensibilisation avec les inhibiteurs de la famille PIKK, wortmannine et LY294002, ayant des
IC50 de l’ordre du nanomolaire et de 1,4 µM respectivement pour la protéine DNA-PK, ont
encouragé les recherches de nouveaux composés spécifiques de DNA-PK :
- L’IC87361 : permet une radiosensibiliation in vitro et in vivo chez les souris
xénogréffées (Shinohara et al., 2005).
- Le NU7026 : cinq fois plus sélectif pour DNA-PK que pour les kinases ATM/ATR,
avec une IC50 de 13 nM. Il permet une radio- et une chimiosensibilisation dans des lignées
tumorales et dans des modèles de souris xénogreffées en essai pré-clinique (Nutley et al.,
2005). Une étude de 2004 a mis en évidence que le NU7026 potentialise l’inhibition de
croissance des cellules de la lignée leucémique K562 en présence de poison de la
topisomérase II (idarubicine, daunorubicine, doxorubicine, étoposide) mais pas de la
camptothécine ou de l’Ara-C (Willmore et al., 2004).
- La vaniline : inhibe l’activité de la protéine DNA-PK et potentialise
significativement la chimiosensibilisation par le cisplatine dans une lignée de carcinome
ovarien (A2780), une lignée lymphoblastoïde B (TK6) ainsi qu’une lignée de lymphome
(GM00558). En revanche, ce composé ne permet pas d’induire une chimiosensibilité des
lignées de lymphome p53 déficientes (Raji et WT1) (Durant and Karran, 2003).
- Le SU11752 : inhibiteur compétitif de la sous-unité catalytique de DNA-PK. Il
permet une radio-sensibilisation des cellules de glioblastome humain (Ismail et al., 2004).
- La salvicine : inhibite naturellement la topoisomérase II et réprime également
l’activité kinase de DNA-PK (Lu et al., 2005a).

42
Malgré l’intérêt thérapeutique mise en évidence par de tels composés, il n’y a pas d’essai
clinique combinant des inhibiteurs de la DNA-PK rapporté pour des patients atteints de
cancer.
c) Inhibiteurs de la kinase CHK2
Comme pour les inhibiteurs des kinases ATM/ATR, conceptuellement l’inhibition des
protéines CHK1 et CHK2 devrait permettre une levée du point de contrôle des dommages
et une entrée illégitime en mitose. En réalité, les choses sont plus compliquées pour CHK2
compte tenu de son rôle dans l’induction de l’apoptose de manière dépendante et
indépendante de p53.
Il a été mis en évidence que la déplétion de CHK2 par un dominant négatif ou son
inhibition par le debromohymenialdesine induit une chimiosensibilisation à la
doxorubicine et une entrée illégitime en mitose suivie d’une apoptose dans les lignées
tumorales HeLa et HCT116 (Castedo et al., 2004). Cependant bien que cet effet
sensibilisateur est également été montré dans d’autres lignées (HEK293), contrairement à
CHK1, la déplétion de CHK2 par ARN interférence dans deux lignées cellulaires ne
potentialise pas les effets cytotoxiques des agents 5FU et gemcitabine (Morgan et al.,
2006). A cela, on peut ajouter que l’inhibition de l’activité kinase de CHK2 par le
VRX0466617 (IC50 pour CHK2 = 120 nM, pour CHK1 > 10 µM) atténue l’apoptose induite
par les radiations ionisantes et ne potentialise pas l’effet cytotoxique de la doxorubicine,
du taxol ou du cisplatine (Carlessi et al., 2007).
Les auteurs d’une revue publiée en 2007 rapportent que ces effets opposés obtenus sur la
sensibilité sont dus aux inhibiteurs qui ciblent la poche ATP de la kinase, et présentent
probablement des effets non spécifiques sur d’autres kinases. La question du ciblage de
CHK2 dans le traitement des tumeurs fait toujours débat (Antoni et al., 2007). Cependant,
un inhibiteur de CHK2, le LY2603618 est actuellement en cours d’évaluation dans des
essais cliniques de phase I et II sur des patients atteints de cancers pancréatiques (en
combinaison avec la gemcitabine) et du poumon (en combinaison avec le pemetrexed), et
permettra probablement une meilleure évaluation de ce type de composé (cf le lien
internet LY2603618).
d) Inhibiteurs de la kinase CHK1

43
(1) L’UCN-01
Les principaux travaux mettant en évidence l’intérêt d’une inhibition de la protéine CHK1
ont été réalisés ces dix dernières années avec un composé peu spécifique dérivant de la
staurosporine, l’UCN-01. Initialement, ce composé a été développé pour inhiber l’activité
de la protéine kinase C (PKC) (IC50 de 4,1 nM) (Akinaga et al., 1991), mais il a rapidement
été mis en évidence son rôle inhibiteur de la protéine CHK1 (IC50 de 7 nM) (Graves et al.,
2000) et plus récemment l’inactivation d’AKT via la protéine PDK1 (Phosphoinositide-
dependent protein kinase 1 ; IC50 = 33 nM) (Sato et al., 2002). L’UCN-01 en présence de
dommages à l’ADN induit une diminution de l’activité de CHK1 mise en évidence par
l’augmentation des phosphatases CDC25 C et A, conduisant à une reprise illégitime en
mitose (Graves et al., 2000). Le rôle spécifique de CHK1 dans la chimiosensibilisation a été
validé par des approches non-pharmacologiques en utilisant des formes de dominant-
négatif, ou d’interférence ADN (Lukas et al., 2004). Ainsi, l’effet bénéfique non spécifique
de l’UCN-01 combine ainsi l’inactivation des protéines CHK1 et PKC, un arrêt du cycle
cellulaire à la transition G1/S et la levée du point de contrôle des dommages. De manière
intéressante, l’UCN-01 induit une chimiosensibilité des cellules de lignées MCF-7
déficientes pour p53 au cisplatine, alors que leurs homologues proficientes pour p53 sont
chimiorésistantes à ce même traitement (Wang et al., 1996). L’UCN-01 permet la
potentialisation in vitro aux radiations ionisantes et à de multiples agents
chimiothérapeutiques (gemcitabine, ara-C, poisons de la topoisomérase I, cisplatine,
mytomicine et temozolomide) mais également in vivo sur des modèles de souris
xénogreffées (Tse et al., 2007). L’ensemble de ces résultats très encourageants a permis le
développement d’une quinzaine d’essais cliniques de phase I dans des tumeurs solides et
des hémopathies. Ces derniers combinant différents agents cytotoxiques : cisplatine (Lara
et al., 2005), 5FU (Kortmansky et al., 2005) et topotecan (Hotte et al., 2006), mais aussi
le carboplatine, le prednisone, l’irinotécan, la gemcitabine, la fludarabine, la
fluorouracile, ont permis de mettre en évidence une bonne tolérance de l’UCN-01 chez les
patients (pour les études publiées). Cependant, le seul essai clinique de phase II dont les
résultats ont été publiés combinant l’UCN-01 et le topotecan dans des cancers avancés des
ovaires montre que l’UCN-01 n’augmente pas l’activité cytotoxique du topotecan (Welch
et al., 2007). Les cinq essais cliniques de phase II en cours devraient permettre une
meilleure évaluation in vivo du potentiel chimiosensibilisateur de l’UCN-01.
Concernant, le rôle de l’UCN-01 dans les hémopathies, un essai pilote réalisait sur 12
patients atteints de LAM réfractaires a mis en évidence la pertinence du traitement
combinant l’UCN-01 avec l’ara-C. La combinaison induit une chimiosensibilité des blastes

44
en clonogénie, de même qu’une diminution de l’activation de CHK1 et d’AKT dans les
blastes de patients 24h après administration du co-traitement (Sampath et al., 2006).
D’autres travaux de ce type sont nécessaires pour valider l’intérêt thérapeutique de l’UCN-
01 dans les hémopathies. L’UCN-01 est toujours en cours d’évaluation dans 8 essais
cliniques de phase I (3) et II (5) seul ou en combinaison avec des agents cytotoxiques
(périfosine, topotécan, cytarabine, irinotécan, et fludarabine) dans des tumeurs solides et
des hémopathies, dont des lymphomes et des leucémies aiguës (cf le lien internet : UCN-
01). Cependant, les effets toxiques de l’UCN-01 déjà rapportés chez les patients ont
conduit les industriels à développer d’autres molécules inhibitrices spécifiques de la
protéine CHK1.
(2) Inhibiteurs spécifiques de CHK1
Voici une liste non exhaustive des composés spécifiques de CHK1 (parfois co-spécifique de
CHK2) rapportés dans la littérature :
- Le CHIR-124 : inhibiteur sélectif de CHK1 in vitro (IC50 = 0,3 nM). Il permet une
réduction de la prolifération de plusieurs lignées cellulaires mutées pour p53 (4 lignées
cellulaires de carcinome de sein et du colon) en combinaison avec des poisons de la
topisomérase (camptothécine et SN-38), une levée des points de contrôle S et G2/M en
combinaison avec le SN-38 dans une lignée tumorale de cancer du sein permettant ainsi
une augmentation de l’apoptose (MDA-MD-435), et enfin une potentialisation de l’effet
cytotoxique de l’irinotécan dans des modèles de souris xénogreffées (Tse et al., 2007).
- Le XL844 d’Exilixis : inhibiteur des kinases CHK1 et CHK2 (Ki = 2,2 nM pour CHK1
et 0,07 nM pour CHK2), il a permis d’augmenter la cytotoxicité in vitro et in vivo de la
gemcitabine (Matthews et al., 2007). Ces résultats prometteurs ont permis son évaluation
en essai clinique de phase I à partir de mai 2007 dans le traitement des lymphomes en
combinaison avec la gemcitabine. Le traitement s’est déroulé de la manière suivante :
administration bi-hebdomadaire d’XL844 en comprimé (5 à 100 mg) et hebdomadaire de
gemcitabine en intra-veineuse pendant 30 min (cf le lien XL-844). Cet essai clinique devait
se terminer en mai 2009, les résultats ne sont pas encore publiés.

45
- Les composés de Pfizer PF-00394691 et PF-00477736 : le PF-00394691 possède
une IC50 de 0,75 nM pour CHK1, il potentialise l’activité anti-tumorale de la gemcitabine,
de l’irinotecan et du cisplatine dans des souris xénogreffées. Celui-ci permet de diminuer
l’activation de CHK1 (perte de phospho-S317-CHK1) induite par l’irinotécan dans les
cellules tumorales des souris, par un mécanisme non identifié. Le PF-00477736 possède un
Ki de 0,49 nM pour CHK1, il permet la potentialisation de l’activité cytotoxique de la
gemcitabine de manière dose dépendante dans les souris xénogreffées (Blasina et al.,
2008). Ce composé est en cours d’évaluation dans un essai clinique de phase I en
combinaison avec la gemcitabine dans les tumeurs solides (cf le lien PF-00477736).
- L’AZD7762 d’ AstraZeneca est un inhibiteur équivalent des kinases CHK1 et CHK2
(IC50 = 5 nM pour CHK1). Il permet une potentialisation dose-dépendante in vitro et in vivo
des agents cytotoxiques gemcitabine et irinotécan (Zabludoff et al., 2008). Ces résultats
intéressants ont conduit à une évaluation en cours de ce composé en essai clinique de
phase I dans des tumeurs solides en combinaison avec la gemcitabine et l’irinotecan (cf le
lien AZD7762).
Cet état des lieux bibliographiques sur les composants pharmacologiques ciblant quelques-
uns des acteurs de la voie des dommages (il en existe bien sûr de nombreux autres parmi
lesquels : les phosphatases CDC25, WEE1, les protéines Polo-like Kinase (PLK), Aurora, la
Poly(ADP-ribose) polymérase 1…) permet de mettre en évidence l’importance actuelle des
thérapeutiques ciblées et combinées qui apportent de nouvelles solutions dans le domaine
de la cancérologie.

46
II. Hématopoïèse & micro-environnement
L’hématopoïèse est le processus par lequel une population minoritaire de cellules souches
hématopoïètiques (CSH) va donner naissance, à travers différentes étapes de
différenciation/expansion, à l’ensemble des éléments figurés du sang tout en conservant
l’intégrité de son pool de départ.
A. Hématopoïèse normale ou physiologique
L’hématopoïèse est définie comme l’ensemble des mécanismes qui assurent la formation
continue de cellules sanguines circulantes. Ce processus a lieu dès l’embryogenèse et se
poursuit durant toute la vie adulte d’un organisme. Il implique des mécanismes de
développement, d’auto-renouvellement et de différenciation des cellules primitives dites
CSH.
Compte tenu de la demi-vie très courte des cellules sanguines matures circulantes, les CSH
vont s’auto-renouveler continuellement pour donner :
- Deux CSH filles par division symétrique, préservant ainsi le groupe des CSH.
- Une CSH fille et une cellule qui va se différencier par division asymétrique,
alimentant ainsi les compartiments cellulaires plus différenciés.
- Eventuellement deux cellules différenciées mais cette option doit être minoritaire
pour préserver le pool des cellules souches.
Chez les mammifères, l’hématopoïèse réside dans le foie et la rate pour le fœtus, dans la
moelle osseuse (MO) des os plats et l’épiphyse des os longs pour l’adulte. La MO contenant
les cellules hématopoïétiques dans leur micro-environnement est appellée niche
hématopoïétique. Le rôle du micro-environnement médullaire est à la fois de soutenir
physiquement l’hématopoïèse mais aussi de réguler la quiescence, l’auto-renouvellement,
la survie et la différenciation des cellules hématopoïétiques grâce à des interactions
cellulaires directes et à la sécrétion de facteurs solubles comme les cytokines ou les
chimiokines.

47
1. Les cellules hématopoïétiques
Les cellules hématopoïétiques sont organisées de manière hiérarchisée avec au sommet de
la pyramide, les cellules souches primitives, et à la base les différentes cellules sanguines
matures (Figure 12).
Cette pyramide comprend quatre compartiments : (i) le compartiment des cellules
souches, (ii) le compartiment des progéniteurs multipotents MPP (Multipotent progenitors)
puis ceux propres à chaque lignage : avec les progéniteurs myéloïdes CMP (Common
Myeloid Progenitors) et lymphoïdes CLP (Common Lymphoïd Progenitors), donnant
naissance côté myéloïde : aux précurseurs MEP (Megacaryocyte Erythrocyte Precursor) et
GMP (Granulocyte Macrophage Precursors), (iii) le compartiment des précurseurs
hématopoïétiques, (iv) et enfin le compartiment des cellules sanguines matures avec : les
érythrocytes, moncytes, plaquettes, granulocytes, macrophages, cellules dendritiques,
lymphocytes B et T et les cellules NK (Natural Killer).
Figure 12: Schéma de l’hématopoïèse.
Les CSH (ou HSC pour Hematopoietic Stem Cell), comprenant les LT-HSC et ST-HSC (Long term et Short Term), se différencient en progéniteurs multipotents ou MPP (Multipotent progenitors) puis en progéniteurs myéloïdes/lymphoïdes communs (CMP/CLP pour Common Myeloid/Lymphoïd Progenitors) qui donneront à terme les cellules sanguines matures. MEP : Megacaryocyte Erythrocyte Precursor, GMP : Granulocyte Macrophage Precursors, Pro-DC : pro-dendritic cells, Pro-T : pro-T cells ; Pro-NK : pro-Natural Killer cells, Pro-B : pro-B cells. (d’après (Passegue et al., 2003)).

48
a) Les cellules souches hématopoïétiques
La première évidence expérimentale de l’existence des CSH remonte à 1961 avec la mise
en évidence par Till et McCulloch de l'existence d'une population clonogène de cellules de
la MO capable de générer des colonies hématopoïétiques multi-lignage dans la rate de
souris léthalement irradiées (Till and Mc, 1961). Cette population souche ne représente
pas plus de 0,01% des cellules de la MO.
Les CSH possèdent trois propriétés fondamentales :
- Une capacité de reconstitution de l’hématopoïèse myéloïde et lymphoïde chez
la souris.
- Une capacité d’auto-renouvellement permettant le maintien et l’amplification
de la population des CSH.
- Une pluripotence ou potentiel de différenciation multi-lignage permettant la
génération continue des cellules matures du sang.
(1) Capacité de reconstitution de l’hématopoïèse
Cette capacité de reconstitution de l’hématopoïèse chez la souris immuno-déficiente SCID
ou NOD-SCID représente le test absolu permettant de prouver que l’on a bien isolé des
CSH. Les tests de reconstitution consistent à mesurer la capacité des cellules
hématopoïétiques à repeupler et restaurer la MO de souris rendues aplasiques par
irradiation. L’évaluation de ce potentiel pour les cellules humaines a nécessité le
développement de systèmes de transplantations xénogéniques dans lesquels les réactions
immunitaires sont abolies (utilisation de souris immuno-déficientes SCID et NOD-SCID
comme hôte des CSH humaines). L’approche expérimentale consiste à injecter chez la
souris immuno-déficiente les CSH humaines par voie intraveineuse à des doses sub-létales.
Après plusieurs mois, les cellules hématopoïétiques humaines persistent dans la MO et la
rate de la souris. Ces xénogreffes permettent l’identification des cellules humaines
appelées SCID Repopulating Cells (SRC).
Ce test a permis de mettre en évidence deux sous-types de CSH humaines capables de
s’auto-renouveler et de se différencier mais également de définir leurs caractéristiques
phénotypiques (Figure 13):
- Le compartiment des CSH comprend : (i) les CSH à auto-renouvellement à long
terme ou LT-HSC (Long Term-Hematopoietic Stem Cells) qui s’auto-renouvellent de façon
illimitée tout au long de la vie de l’individu, reconstituant et pérennisant l’hématopoïèse

49
physiologique, (ii) les CSH à auto-renouvellement à court terme ou ST-HSC (Short Term-
Hematopoietic Stem Cells), plus différenciées que les précédentes, elles sont capables de
reconstituer l’hématopoïèse physiologique pendant une durée limitée d’environ 8
semaines. La population progénitrice multipotente est ainsi constituée de LT-HSC capable
de donner naissance aux ST-HSC donnant eux-mêmes naissance aux progéniteurs
multipotents qui ne possèdent plus de capacité d’auto-renouvellement (Morrison et al.,
1997).
Figure 13: Modèle de différenciation des CSH.
Le compartiment des CSH est composé de trois populations : les CSH à long terme (LT-HSC) possédant la plus forte capacité d’auto-renouvellement, les CSH à court terme (ST-HSC) à plus faible capacité d’auto-renouvellement et les progéniteurs multipotents (MPP) qui ne possèdent plus la capacité d’auto-renouvellement. Les MPP se différencient en progéniteurs myéloïdes/lymphoïdes communs (CMP et CLP). GMP : Granulocyte Macrophage Precursors, MEP : Megacaryocyte Erythrocyte Precursor, CTP : Cell T Precursors, CBP : Cell B Precursors, NKP : Natural Killer Precursors, Pro-T : Pro-T cells, Pro-B : Pro-B cells. (D’après (Reya et al., 2001)).
- Propriétés phénotypiques et cytochimiques des CSH
L’identification et la purification des CSH est basée sur deux approches expérimentales
réalisées par cytométrie de flux : le marquage membranaire des cellules et l’utilisation de
leurs propriétés cytochimiques.
Le marquage de protéines membranaires permet de définir des populations positives et
négatives pour le marqueur considéré (cf tableau annexe 1), d’autre part les protocoles de
marquage multi-couleur réalisés en cytométrie de flux permettent l’identification de
populations complexes. La majorité des CSH étant quiescente, en phase G0 ou G1 du cycle
cellulaire (Passegue et al., 2005) la méthode d’identification cytochimique consiste à
évaluer la résistance de ces cellules à des colorants vitaux (Rhodamine 123 ou Hoechst
33342). La Rhodamine 123, qui témoigne de l’activité cellulaire des mitochondries, et le
Hoechst 33342, qui s’intercale dans l’ADN, permettent l’identification des cellules en cycle

50
ou au métabolisme actif, les CSH ne sont alors pas marquées. Cette stratégie a permis
l’identification d’une population résistance au Hoechst 33342 appelée Side population (SP)
mille fois enrichie en LT-HSC. Ces méthodes seules ou couplées au marquage membranaire
permettent la purification de CSH capables de reconstituer la MO de souris irradiées avec
des rendements de plus en plus importants ((Wagers et al., 2002) ; (Camargo et al.,
2006)). Enfin, une autre méthode a été développée grâce à l’utilisation de l’enzyme
cytosolique Aldéhyde Déshydrogénase (ALDH) responsable de l’oxydation des aldéhydes
intracellulaires. L’ALDH est capable de dégrader les agents alkylants et participe ainsi à la
résistance des CSH aux agents toxiques (Hess et al., 2004). Elle est active dans certaines
cellules immatures et définit également une population capable de reconstitution
hématopoïétique à long terme (Hess et al., 2006).
L’identification des différentes populations par les marqueurs membranaires ou
l’utilisation des différents agents décrits ci-dessus, n’étant probablement pas dépourvu de
toxicité (les cellules identifiées conservent les marqueurs utilisés). Une autre manière
d’identifier les cellules peut être une sélection négative en éliminant les cellules
marquées, mais dans ce cas, il peut persister une population minoritaire de cellules non
souches (communication orale J.F. Mayol).
- Phénotypes des CSH murines : les CSH murines sont caractérisées par le
phénotype c-Kit+ Sca-1+ Lin- (KSL). La sélection d’une population exprimant faiblement ou
pas du tout la protéine membranaire CD34 (Cluster of Differentiation 34) dans la fraction
KSL (KSL-C34-/low) permet une augmentation du pourcentage de reconstitution de
l’hématopoïèse chez la souris irradiée (Osawa et al., 1996). Ainsi, la population KSL-C34-
/low, qui ne représente que 0,004% des cellules de la MO, est jusqu’à ce jour considérée
comme étant la plus proche des CSH murines.
L’analyse phénotypique des cellules SP murines confirme la négativité de la plupart des
marqueurs de lignage habituel, les SP ne représentent qu’une faible fraction des cellules
KSL+ mais aussi KSL-. D’autre part, même si une fraction significative de cellules SP est
KSL+ CD34-, la majorité est CD34+. Cela indique qu’il existe des CSH CD34+ et CD34-. Enfin,
les souris déficientes pour le transporteur bcrp-1 responsable de l’efflux du Hoechst 33342
ne présentent plus de population SP sans déficit hématopoïétique majeur (Zhou et al.,
2002).
- Phénotype des CSH humaines : chez l’homme, le marqueur membranaire le
plus utilisé pour la purification des cellules primitives est la glycoprotéine membranaire
CD34, car la plupart des cellules immatures l’expriment (Andrews et al., 1986). Cette
population étant très hétérogène, il a été mis en évidence que l’absence de marquage
CD38 (CD34+ CD38-) permet un enrichissement en cellules LT-HSC, alors que les cellules

51
CD34+ CD38+ permettent une reconstitution de MO à court terme seulement (Bhatia et al.,
1997). D’autres marqueurs ont été ajoutés afin de préciser le phénotype des CSH
humaines : CD34+ CD38- CD90+ CD33- HLA-DR- c-Kit+ Rho-123+.
(2) Capacité d’auto-renouvellement des CSH
L’une des perspectives les plus importantes dans le domaine de la biologie des cellules
souches vise à comprendre les mécanismes contrôlant l’auto-renouvellement des CSH.
L’auto-renouvellement est une caractéristique cruciale de la cellule souche qui permet
l’homéostasie de chaque tissu tout au long de la vie d’un individu. Alors que chaque cellule
souche possède ses propres capacités de développement, l’ensemble des cellules souches
assure leur auto-renouvellement et régulent la balance auto-renouvellement/
différenciation. L’élucidation des mécanismes de régulation de l’auto-renouvellement des
cellules souches normales est fondamentale pour appréhender les phénomènes de
dérégulation des cellules cancéreuses. Alors que les propriétés fonctionnelles et
phénotypiques des CSH ont été largement caractérisées, la question de la régulation de
l’auto-renouvellement n’est pas complètement résolue.
Dans la niche hématopoïétique, les CSH sont majoritairement en état de quiescence,
caractérisé par 2/3 des cellules en phase G0 du cycle cellulaire (Passegue et al., 2005).
Cet état permet à la fois de préserver les CSH des stress génotoxiques, notamment des
agents incorporés en phase S du cycle cellulaire, mais est également une caractéristique
déterminante du maintien du potentiel de reconstitution hématopoïétique.
La sortie de quiescence des CSH et donc l’entrée des cellules en cycle permet sans doute
en fonction des stimuli : (i) une reconstitution majoritaire du pool de cellules souches ou
du pool de cellules différenciées en raison des propriétés de division symmétrique ou
asymmétrique de ces cellules, (ii) une génération de deux cellules filles identiques par
division symétrique, (iii) une génération d’une cellule fille identique et d’une cellule fille
capable de se différencier par division asymétrique. Ces processus permettent à la fois le
maintien des propriétés et du nombre de CSH mais également l’expansion des cellules
hématopoïétiques qui s’engagent dans la différenciation myéloïde et lymphoïde
(Figure 14).

52
Figure 14 : Divisions symétrique et asymétrique des CSH dans le temps (t)
Les CSH (S sur cette figure) se divisent de façon symétrique pour se reproduire à l’identique, permettant ainsi le maintien du pool de cellules souches : c’est la capacité d’auto-renouvellement. Certaines CSH vont ensuite se diviser de façon asymétrique en s’engageant dans le processus de différenciation, donnant ainsi des cellules progénitrices capable de proliférer plus rapidement qui donneront elles-mêmes des cellules précurseurs. ((Ho and Wagner, 2007)).
Même si des progrès ont été réalisés dans la mise au point de conditions de culture
permettant le maintien de l’activité des CSH (Miller and Eaves, 1997), il est difficile de
conclure sur la combinaison exacte des facteurs de croissance impliqué dans l’auto-
renouvellement et l’expansion des CSH in vivo. Les voies de signalisation classiques
étudiées dans le contexte du développement embryonnaire – voies des morphogènes
Notch, Sonic hedgehog (Shh) et Wnt - sont de bons candidats de la régulation de l’auto-
renouvellement des CSH (Figure 15).
Figure 15: Les trois grandes voies morphogènes régulant l’auto-renouvellement des CSH.

53
L’activation de la voie Notch (bas) est assurée par l’interaction du récepteur Notch avec son ligand (Delta 1,2, 4 et Jagged 1, 2), ce qui entraîne le clivage et la libération du domaine intra-cellulaire notch 1 (NICD pour Notch Intracellular Domain) dans le noyau, qui en se liant au facteur de transcription CBF-1 (C-promoter Binding Factor-1) permet la transcription de gènes comme hes-1. Pour la voie Shh (milieu), la liaison du ligand hh sur le récepteur Patched lève l’inhibition que ce dernier exerçait sur le récepteur Smoothened et permet ainsi au facteur de transcription Gli d’induire la transcription de différents gènes parmi lesquels on retrouve celui du récepteur Patched. La voie Wnt (haut) est activée lorsqu’un ligand Wnt s’associe au récepteur frizzled couplé aux récepteurs LRP5/6 (lipoprotein related-Receptor Proteins 5/6), inhibant ainsi GSK3β qui ne peut plus exercer son rôle négatif sur le co-facteur transcriptionnel β-caténine, permettant alors la transcription avec LEF/TCF (Lymphocyte Enhancer Factor/T Cell Factor) de gènes impliqués dans l’auto-renouvellement des CSH comme la cycline D1. (D’après (Reya et al., 2001)).
Les morphogènes sont des molécules sécrétées durant l’embryogenèse pour définir le
développement embryonnaire, guider les ébauches tissulaires à partir de la cellule souche
pluripotente embryonnaire. Les ligands pro-mésodermes sont intéressants car ils sont les
précurseurs nécessaires à l’apparition du tissu hématopoïétique, et continuent à avoir un
rôle dans le maintien et la différenciation des CSH.
- La voie Notch : La voie Notch semble être impliquée dans la régulation de la
balance quiescence/auto-renouvellement des CSH. En effet, l’activité de Notch est forte
dans les CSH et décroît au cours de l’hématopoïèse. L’activation de Notch dans les CSH en
culture par son ligand Jagged-1 augmente la quantité des progéniteurs primitifs in vitro et
in vivo (Karanu et al., 2000). D’autre part, la transfection de la partie intra-cellulaire de
Notch immortalise les CSH et bloque la différenciation hématopoïétique. Le récepteur
Notch et ses ligands (Jagged et delta-like) ont été impliqués dans des translocations
dépistées chez des patients atteints de leucémie aiguë lymphoblastique. Récemment, une
étude a mis en évidence que l’expression ectopique de Notch-1 constitutivement actif
conduit à un arrêt du cycle cellulaire et à l’apoptose des cellules CD34+ (Chadwick et al.,
2007). D’autres investigations sont donc nécessaires pour préciser davantage la fonction de
la voie Notch dans cette balance.
- La voie Sonic-hedghog (Shh) : les ligands hh (sonic, indian, desert) sont reconnus
par un couple de récepteurs (Patched et Smoothened) qui sont présents à la surface des
CSH. Les populations hautement enrichies en CSH (CD34+ CD38- Lin-) augmentent leur
capacité d’auto-renouvellement lors d’une stimulation de la voie Shh in vitro grâce à la
signalisation en aval du morphogène BMP-4 (Bone Morphogenic Proteins) (Bhardwaj et al.,
2001). Cette signalisation est cruciale au cours de la stimulation des CSH, en effet, son
inhibition bloque la prolifération induite par Shh. Les BMP en tant que morphogènes
contrôlent également les CSH : BMP-2 et BMP-7 inhibent leur prolifération, alors que BMP-4
augmente la survie et la greffe des CSH dans la moelle des souris NOD/SCID (Ho and
Wagner, 2007).

54
- La voie Wnt : les protéines Wnt sont des molécules de signalisation intra-cellulaire
qui régulent le développement de différents organismes et sont dérégulées dans plusieurs
cancers. La stimulation des progéniteurs hématopoïétiques et des CSH avec des protéines
Wnt solubles ou avec les activateurs en aval de la voie Wnt (β-caténine) induit leur
expansion lors de culture à long terme (Reya, 2003). De plus, l’expression ectopique
d’inhibiteur de la voie Wnt (Axin) induit une inhibition de la prolifération des CSH,
augmente leur apoptose in vitro et diminue la capacité de reconstitution hématopoïétique
chez la souris.
D’autres voies de signalisation faisant intervenir par exemple JAK/STAT, PTEN ou bcl-2
jouent également un rôle dans la régulation de la balance quiescence/auto-
renouvellement. D’autre part, les facteurs de transcription (FT) sont aussi importants dans
cette régulation. En effet, quelle que soit la voie de transduction du signal, elle aboutit à
une régulation génétique des FT qui vont gouverner la réponse cellulaire, en terme de
renouvellement, différenciation, apoptose, sénescence, migration ou adhésion. Ces choix
cruciaux se font de manière intégrée, leur dérégulation étant la base du processus de
leucémogenèse. Les FT PU.1 et ceux de la famille Homeobox (Hox) sont particulièrement
impliqués dans l’auto-renouvellement des CSH.
- Les facteurs de transcription PU.1 : les CSH PU.1-/- présentent une altération de
l’auto-renouvellement et de la différenciation initiale vers les progéniteurs immatures
lymphoïdes et myéloïdes. Même si ces FT sont exprimés à faible taux dans les CSH (leur
expression dépend de la différenciation), ils contrôlent néanmoins la synthèse de
récepteurs à des cytokines, ce qui participe à la survie et à l’expansion du groupe CSH.
- Les facteurs de transcription de la famille Hox : ils ont d’abord été impliqués dans
la leucémogenèse et ont ensuite montré leur rôle critique au niveau de la CSH normale,
notamment HoxB3 et B4. HoxB4 est un FT qui peut être sécrété, une manipulation
génétique induisant sa synthèse par les cellules stromales entraîne une nette augmentation
des CSH in vitro lors de co-culture avec des cellules CD34+ CD38-.
(3) Pluripotence ou potentiel de différenciation multi-
lignage
Que ce soit pour maintenir l’homéostasie hématopoïétique ou pour faire face à un stress
hématologique, les CSH sont capables d’alimenter les différents lignages hématopoïétiques
de l’organisme permettant la génération de l’ensemble des cellules du sang. Pour cela, les

55
CSH doivent sortir de leur état de quiescence, s’expandre et se différencier. Des
expériences de vidéo-microscopie ont permis de mettre en évidence que la division
asymétrique est beaucoup plus fréquente dans les cellules CD34+ CD38- que dans les CD34+
CD38+ (Huang et al., 1999), ainsi le potentiel d’auto-renouvellement est perdu
progressivement au cours des stades initiaux de la différenciation. Même si, ce taux de
division asymétrique des cellules immatures reste constant en présence de molécules
régulatrices qui stimulent par exemple le pourcentage mitotique ou la génération de
différents types de colonies (Punzel et al., 2002).
Parmi les facteurs extrinsèques qui peuvent jouer sur la quantité de division asymétrique,
le contact direct des CSH avec les cellules de la niche hématopoïétique augmente
significativement le nombre de cellules qui entrent en division asymétrique, leur
polarisation cellulaire et l’orientation du fuseau mitotique (Punzel et al., 2003). D’autre
part, il est bien établi que la différenciation hématopoïétique implique également d’autres
signaux extrinsèques tels que des facteurs solubles, cytokines présentes dans le micro-
environnement. In vitro, il est possible d’orienter la différenciation des cellules immatures
vers un lignage particulier : par exemple vers une différenciation myéloïde par ajout de
MGF (Mast cell Growth factor), interleukine-3 (IL-3), GM-CSF (Granulo-Macrophage-Colony
Stimulating Factor), M-CSF (Monocyte-CSF), et G-CSF (Granulocyte-CSF). Les cytokines
sont classées en 3 groupes :
- Les cytokines synergiques : SCF, LIF (Leukemia Inhibitory Factor), IL-1, 6, 11,
permettant l’augmentation du nombre de CSH en cycle à partir des CSH quiescentes,
induisant la sensibilisation des cellules aux autres cytokines en permettant notamment
l’expression des récepteurs membranaires spécifiques.
- Les cytokines multipotentes : IL-3, 7, GM-CSF, agissant à la fois sur les CSH et les
progéniteurs en cycle, stimulant la prolifération, la survie, et la différenciation.
- Les cytokines restreintes : G-CSF, M-CSF, EPO (érythropoïétine), TPO
(trombopoïétine), IL-5, agissant sur la différenciation terminale des progéniteurs engagés,
et la maturation.
D’un point de vue moléculaire, c-myc, un des acteurs clés impliqué dans la régulation du
cycle cellulaire, peut aussi jouer un rôle important dans l’induction du processus de
différenciation. c-myc est capable de réprimer l’expression de molécules d’adhésion
comme les intégrines α2β1 et α5β1, et la N-cadhérine. Ainsi, Wilson et ses collaborateurs
ont proposé un modèle de régulation des CSH par l’adhésion à la niche médullaire
contrôlé par c-myc. Ainsi, lorsque c-myc serait fortement exprimé, les CSH, en réponse à

56
un signal mitogène, génèreraient des progéniteurs engagés sortant de la niche grâce à la
répression des molécules d’adhésion contrôlées par c-myc. Au contraire, quand c-myc
serait inactif, en présence d’un signal mitogénique, ces molécules resteraient fortement
exprimées, retenant ainsi les cellules dans la niche pour permettre leur expansion.
Figure 16: Modèle de régulation du devenir des CSH par l’adhésion à la niche
hématopoïétique contrôlée par le niveau de c-myc.
Les CSH quiescentes (haut), exprimant peu de c-myc, sont ancrées à la niche par différentes molécules (N-cadhérine, LFA-1, intégrines α2β1 et α5β1). En réponse à une stimulation mitogénique, les CSH vont générer deux cellules filles, qui en absence d’induction de c-myc (a) restent ancrées à la niche (expansion des CSH), qui lors d’une induction de c-myc dans une seule cellule fille (b) ou dans les deux (c), répriment les molécules d’adhésion permettant ainsi une sortie de la niche hématopoïétique et la différenciation des CSH. (D’après (Wilson et al., 2004)).
b) Les progéniteurs hématopoïétiques
Ce compartiment comprend des progéniteurs immatures qui sont multipotents et
possèdent une grande capacité de prolifération et des progéniteurs matures qui ont un
potentiel de prolifération et de différenciation plus restreint. Ce compartiment regroupe
ainsi une population hétérogène, avec des cellules perdant progressivement leur pouvoir
d’auto-renouvellement, et présentant un pouvoir de prolifération accru.
L’identification des progéniteurs hématopoïétiques est possible par cytométrie de flux, en
effet, ces cellules possèdent à la fois des marqueurs de maturité (CD38, HLA-DR, CD71) et
des marqueurs propres à chaque lignage (CD33, CD13, CD15 pour la lignée myéloïde) ; mais
également par des cultures clonogéniques permettant une analyse des cellules matures
générées. La culture clonogénique consiste en la mise en culture en milieu semi-solide (par
exemple la méthylcellulose) des progéniteurs hématopoïétiques, qui donneront naissance à

57
des colonies morphologiquement reconnaissables au bout de 7 à 14 jours d’incubation.
Pour la branche myéloïde, on distingue les progéniteurs suivants :
- Les CFU-GM (Colony Forming Unit- Granulocyte and Macrophage), progéniteurs
granulo-monocytaires qui donneront naissance aux CFU-G (Granulocyte) granulocytaires et
CFU-M (Monocyte) monocytaires.
- Les BFU-E (Burst Forming Unit-Erythroid) qui sont les progéniteurs érythroïdes se
différenciant en CFU-E (Erythrocyte) erythrocytaires.
- Les CFU-Meg (Megacaryocyte), progéniteurs de la lignée mégacaryocytaire.
- Les CFU-GEMM (Granulocyte Erythrocyte Macrophage and Megacaryocyte) qui sont
des progéniteurs plus primitifs donnant naissance à des colonies des différents lignages
CFU-GM, CFU-E, CFU-Meg.
c) Les précurseurs hématopoïétiques
Contrairement aux progéniteurs hématopoïétiques, les précurseurs sont des cellules
identifiables morphologiquement, elles possèdent un phénotype et des propriétés
fonctionnelles propres à chaque lignage. Elles représentent la majorité (99%) des cellules
médullaires. Ces cellules n’ont plus de capacité d’auto-renouvellement et ont un faible
pouvoir prolifératif. Une fois que le processus de maturation est achevé, ces cellules vont
passer dans la circulation périphérique pour y assurer leur fonction et ont alors une durée
de vie limitée.
2. Le micro-environnement médullaire
La quiescence des CSH, l’auto-renouvellement et la différenciation sont régulés par des
mécanismes intrinsèques (tels que la régulation épigénètique des CSH contrôlée par
exemple par les protéines de la famille polycomb impliquées dans le remodelage de la
chromatine) et extrinsèques. Ces mécanismes extrinsèques impliquent des changements du
devenir des CS dictés par le micro-environnement. L’effet du micro-environnement sur les
CSH peut se faire directement, grâce à des interactions physiques mettant en jeu des
molécules d’adhésion (intégrines, cadhérines) ou indirectement grâce à la sécrétion de
nombreuses molécules (cytokines, facteurs de croissance).
Quatre grandes caractéristiques sont applicables à la niche des CS quelque soit leur type
tissulaire :

58
- Sa taille est limitée, ainsi que le nombre de CS. La niche hématopoïétique est
située au niveau de l’endosteum, sous l’os trabéculaire des os plats et/ou longs chez
l’adulte.
- Les contacts CS-cellules du micro-environnement sont indispensables pour le
maintien de la quiescence cellulaire.
- Les produits sécrétés (morphogènes, cytokines, facteurs de croissance) sont à
l’origine des contacts inter-cellulaires et de la création d’un équilibre
activateur/inhibiteur du nombre et de la fonction des CS.
- La présence d’une cellule non souche dans la niche peut lui conférer des
« propriétés de CS ».
La niche des CSH se situe dans un endroit précis de la MO, au niveau de la région
endostéale de l’os trabéculaire, zone dynamique à l’interface de la MO et de l’os, où celui-
ci subit continuellement des remodelages. Le micro-environnement est composé de
populations cellulaires hétérogènes (ostéoblastes, fibroblastes, adipocytes, cellules
endothéliales) dérivant d’un précurseur commun, la CS mésenchymateuse (CSM). Les CSM
vont sécréter de nombreux facteurs solubles impliqués dans la quiescence, la survie ou la
différenciation des CSH, elles vont également se différencier pour donner naissance à
divers types cellulaires comme les cellules réticulaires, les adipocytes, et surtout les
ostéoblastes qui représentent l’ossature de la niche hématopoïétique.
Figure 17: Niche hématopoïétique au sein de la moelle osseuse.

59
Les ostéoblastes exprimant la N-cadhérine (SNO pour Spindle-shaped N-cadherin-expressing osteoblats) forment la niche endostéale quiescente qui maintient la quiescence des CSH (HSC). L’auto-renouvellement des CSH permet la production de cellules progénitrices multipotentes (MPP) par division asymétrique et/ou par une stimulation de l’environnement médullaire. Ces MPP sont capables de se différencier pour donner toutes les cellules hématopoïétiques de chaque lignage. En réponse à un stress, les CSH quiescentes pourraient être activées et recrutées au niveau de la niche vasculaire. L’auto-renouvellement des CSH dans la niche vasculaire reste à être déterminé. (D’après (Wilson and Trumpp, 2006)).
a) Interaction directe CSH-niche
Comme nous l’avons vu précédemment, l’interaction entre les cellules hématopoïétiques
et la niche hématopoïétique est un composant majeur de la régulation de l’hématopoïèse.
Différentes études ont mis en évidence que les ostéoblastes constituent un composant
majeur de la niche régulant la localisation, la quiescence, l’auto-renouvellement, et la
différenciation des CSH. En effet, leur déplétion chez la souris provoque une diminution du
nombre de cellules dans la MO s’accompagnant de la perte des progéniteurs myéloïdes,
lymphoïdes et érythroïdes ainsi que d’une hématopoïèse extra-médullaire localisée dans la
rate (Visnjic et al., 2004). De plus, les ostéoblastes soutiennent les cellules
hématopoïétiques in vitro et maintiennent les cellules responsables de la culture à long
terme (LTC-IC : Long Terme Culture-Initiating Cell) (Taichman et al., 1996), en assurant
un véritable contrôle du nombre de CSH de la niche qui pourrait mettre en jeu la voie du
morphogène Notch (Calvi et al., 2003) ou l’interaction N-cadhérine-CD45 (Zhang et al.,
2003).
L’interaction CSH-ostéoblastes fait intervenir de nombreuses molécules d’adhésion parmi
lesquelles les intégrines α4β1 et α5β1 semblent jouer un rôle clé. Il a été montré
récemment que les intégrines α4β1 sont cruciales dans le maintien de la survie et des
capacités clonogènes des précurseurs immatures. En effet, l’ajout de ligands solubles des
intégrines α4β1 (VCAM-1 et fibronectine) dans le milieu permet de maintenir ces capacités
(Jung et al., 2005). D’autre part, l’utilisation d’anticorps bloquant a permis de valider
l’implication de ces intégrines α4β1 et α5β1.
L’importance de ces molécules d’adhésion dans la régulation des CSH implique que leur
niveau d’expression soit très finement contrôlé. Ainsi, différentes études révèlent
l’existence d’un dialogue sous forme de boucle paracrine entre les CSH et les cellules de la
niche, les CSH moduleraient ainsi la composition de leur micro-environnement (Jung et al.,
2008). Une illustration de cette propriété est l’implication de c-myc dans l’induction du
processus de différenciation et de sortie de la niche hématopoïétique (Wilson et al., 2004)
(cf II. A. 1. A) (3) Pluripotence ou potentiel de différenciation multilignage).
L’interface CSH/ostéoblaste ne se résume pas à la simple association intégrine/ligand, il
existe une véritable synapse complexe impliquant de nombreuses autres molécules

60
d’adhésion inter-cellulaire (Figure 18). Différentes études d’invalidation ont montré
l’importance des intégrines α4β1 et du récepteur au ligand c-kit (mKL pour membrane-
bound Kit Ligand) dans les propriétés d’auto-renouvellement des CSH et leur maintien
((Priestley et al., 2006); (Barker, 1997)). D’autre part, la signalisation calcique et
probablement impliqu ée comme le suggère le fait que la liaison MUC1/ligand ICAM-1 joue
un rôle dans le phénomène de migration trans-endothéliale des cellules du cancer du sein,
mettant en évidence l’importance que pourrait avoir les remaniement du cytosquelette
dépendants des interactions CS/niche dans le phénomène de tumorigenèse (Rahn et al.,
2004).
Figure 18: La synapse ostéoblaste/CSH. (D’après (Wilson and Trumpp, 2006)).
b) Facteurs de croissance et cytokines de la niche hématopoïétique
Les cytokines et les facteurs de croissance sécrétés localement peuvent moduler le devenir
des CS grâce à l’activation de signaux de transduction spécifiques. Le TGF-β est l’un des
quelques régulateurs négatifs connus des CSH. Il maintient la quiescence des CSH en
bloquant notamment l’expression de récepteurs aux cytokines comme celui de c-Kit, du
FLT3, de MPL ou à l’IL-6 (Fortunel et al., 2003). Cependant, le rôle in vivo du TGF-β reste

61
à élucider, puisque la déplétion du récepteur n’induit pas de changement dans la
répartition du cycle cellulaire des cellules (Larsson et al., 2001). D’autre part,
l’angiopoïétine-1 (ANG-1) produite par les cellules stromales, en interagissant avec le
récepteur à activité tyrosine kinase TIE2 induit une augmentation de la quiescence des CSH
(Arai et al., 2004). Bien que les mécanismes de maintien des cellules en quiescence ne
soient pas précisément détaillés dans ces deux cas, il est tout à fait possible d’envisager
qu’il s’agisse de p21WAF1 qui est directement réprimé par c-myc, régulateur clé de la niche
hématopoïétique. Les CSH quiescentes sont caractérisées par un niveau élevé de p21WAF1 et
une absence d’expression de c-myc, alors que les CSH plus actives présentent une
expression réduite de p21WAF1 associée à une activité augmentée de c-myc.
Le FGF-1 (Fibroblast Growth Factor-1) pourrait aussi être impliqué dans l’auto-
renouvellement (de Haan et al., 2003).
c) Les composants de la matrice extra-cellulaire
La niche hématopoïétique ne comprend pas seulement des éléments cellulaires, mais
également un grand nombre de facteurs solubles et insolubles constituant le micro-
environnement moléculaire. Ces molécules sont synthétisées principalement par les
cellules stromales puis sécrétées, exprimées à la surface ou assemblées pour former la
matrice extra-cellulaire (MEC). Les protéines insolubles regroupent notamment les
protéines constituant la MEC, les molécules d’adhérence, et les enzymes matricielles ;
tandis que les médiateurs solubles peuvent être de deux types : les facteurs de croissance
(ou cytokines) et les chimiokines. Les collagènes, la fibronectine, les laminines, la
thrombospondine, l’ostéopontine et les glycosaminoglycanes sont les constituants
principaux de la MEC. Ces molécules favorisent l’adhérence des CSH et régulent la
concentration locale de chimiokines et de cytokines en les présentant aux CSH (Long et
al., 1992).
- La fibronectine : elle est considérée par beaucoup d’auteurs comme la protéine
majeure de l’adhérence cellulaire (Dufour et al., 1986). Dans la MO, elle est produite
principalement par les cellules endothéliales et les fibroblastes. Molécule ubiquitaire
d’adhérence, elle participe à la régulation de l’ensemble des fonctions cellulaires comme
la survie, la prolifération, la différenciation et la motilité. C’est une glycoprotéine de 450
à 500 kDa qui interagit avec les intégrines α4β1 ou α5β1 et le CD44. La fibronectine
stimule la production de BFU-E, CFU-GM, CFu-Mix (progéniteurs de plusieurs lignages) en
culture, régule la prolifération des progéniteurs CD34+ CD38- et joue un rôle dans la

62
reconstitution hématopoïétique des souris immuno-déficientes ((Schofield et al., 1998) ;
(van der Loo et al., 1998) ; (Yokota et al., 1998)). Les cellules hématopoïétiques
interagissent avec différents domaines de la fibronectine en fonction de leur stade de
différenciation : les BFU-E et les CFU-GM se lient à un fragment situé au centre de la
molécule contenant la séquence RGDS qui est le ligand de l’intégrine α5β1 (VLA-5), les
progéniteurs plus immatures se fixent sur un site de l’extrémité C-terminale contenant le
peptide CS-1 reconnu par l’intégrine α4β1 (VLA-4) ((Verfaillie et al., 1991) ; (Oostendorp
and Dormer, 1997)).
- L’ostéopontine : elle peut être cytokine ou molécule d’adhésion, sécrétée par les
ostéoblastes, elle peut se lier avec des récepteurs présents à la surface des CSH comme le
CD44 (récepteur à l’acide hyaluronique) et aux intégrines α4β1 et α5β1. Les modèles
animaux d'invalidation démontrent son rôle direct dans l’homéostasie des CSH. En effet,
les CSH ostéopontine -/- sont beaucoup moins quiescentes que des CSH normales (Stier et
al., 2005). Elle jouerait le rôle de régulateur négatif des CSH, restreignant le nombre de
CSH dans la niche.
- L’acide hyaluronique participe également à l’attraction des CSH dans la niche, à
leur ancrage, à leur survie et au maintien de leur quiescence (Nilsson et al., 2003).
Enfin, cette zone trabéculaire de l’os étant une zone hypoxique, les CSH développent des
propriétés liées à ces conditions comme l’expression de la protéine nucléophosmine par
exemple qui va influencer la prolifération des CSH ainsi que leur résistance aux stress
oxydatifs et génotoxiques tout en maintenant leurs propriétés d’auto-renouvellement (Li
et al., 2004).
Un travail récent a mis en évidence l’existence d’une autre niche dite vasculaire (Kiel et
al., 2005). Cette niche située au centre de la MO est caractérisée par la présence de CSH à
proximité de vaisseaux spécifiques, pouvant permettre aux cellules de gagner la
circulation veineuse, ou de gagner la MO dans le cas d’une reconstitution hématopoïétique
par exemple. On pense que cette zone serait plutôt une zone de transit des CSH, la niche
ostéoblastique étant le véritable réservoir des CSH capable d’assurer le maintien de la
quiescence et de l’auto-renouvellement des CSH.
Ainsi, la niche hématopoïétique est un compartiment très complexe et très dynamique,
dans lequel le maintien des CSH fait intervenir de nombreux types cellulaires, comme les
cellules endothéliales, les progéniteurs mésenchymateux, les mégacaryocytes, les cellules

63
immunitaires, les adipocytes, les cellules réticulaires, les ostéoblastes et les ostéoclastes,
ainsi que des interactions inter-cellulaires directes et indirectes.
B. Hématopoïèse leucémique
1. Caractéristiques
Les Leucémies Aiguës Myéloïdes (LAM) sont caractérisées par l’expansion clonale de
cellules malignes issues d’un des différents lignages myéloïdes bloqués à des stades
variables de la différenciation granulo-monocytaire. Cette population clonale leucémique
dérive de la transformation maligne d’une cellule souche ou d’un progéniteur myéloïde
plus différencié ayant ré-acquis des propriétés d’auto-renouvellement. L’hématopoïèse
leucémique conserve des caractéristiques comparables à celle de l’hématopoïèse normale
(Figure 19). Le clone leucémique est en effet organisé de façon hiérarchique en trois
compartiments distincts :
- Un compartiment minoritaire de cellules souches leucémiques (CSL) de phénotype
immature (Lin- CD34+ CD38- CD33+ CD96+ CD123+) pour la plupart quiescentes mais
capables d’auto-renouvellement ou d’engagement dans un processus de prolifération/
différenciation.
- Un compartiment plus mature de progéniteurs leucémiques (CFU-L pour Colony
Forming Unit-Leukemia) ayant perdu la capacité d’auto-renouvellement mais hautement
proliférant et ayant des propriétés clonogènes et de différenciation limitée.
- Un compartiment majoritaire de cellules leucémiques appelé blastes ayant des
capacités de différenciation limitée bloqué à un stade donné de différenciation granulo-
monocytaire ((Bonnet and Dick, 1997); (Passegue et al., 2003) ;(Taussig et al.,
2005) ;(Hosen et al., 2007)).

64
Figure 19: Similarités entre les cellules souches hématopoïétiques (CSH) et les cellules souches
leucémiques (CSL).
Comme dans le système hématopoïétique normal, les cellules souches leucémiques (LSC pour leukaemia stem cells) dérivent des CSH CD34+ CD38-. Au cours de l’hématopoïèse normale (droite), les CSH (HSC) donnent les cellules capables de repeupler la moelle osseuse de souris aplasiques (SRC pour SCID repopulating cell) douées d’auto-renouvellement et capables de donner naissance à tous les lignages hématopoïétiques. Durant la leucémogenèse (gauche), les CSL (ou SL-IC pour SCID leukaemia-initiating cells) expriment certains marqueurs communs aux CSH, elles sont capables de donner naissance aux progéniteurs leucémiques clonogènes et aux cellules blastiques non clonogènes (bulk). (D’après (Huntly and Gilliland, 2005)).
L’hématopoïèse leucémique est associée à une inhibition de l’hématopoïèse normale par
des mécanismes mal connus conduisant à un état d’insuffisance médullaire majeure qui
fait la gravité de cette maladie. D’autre part, cette maladie est aggravée par une
dissémination sanguine des blastes qui vont envahir d’autres organes hématopoïétiques
(rate, ganglions, foie) et non-hématopoïétiques (peau, système nerveux central, testicule).
D’un point de vue moléculaire, deux événements mutationnels seraient nécessaires et
suffisants pour induire la leucémie selon la théorie du « two hits model » (Warner et al.,
2005). Dans un premier temps, des mutations de classe II consistant en un remaniement de
facteurs de transcription dans des translocations chromosomiques récurrentes, comme
AML1-ETO, CBFβ/MYH11, PML-RARα ou MLL/AF9, vont bloquer le processus de
différenciation myéloïde en réprimant la transcription de gènes cibles, constituant ainsi un
événement nécessaire à la leucémogénèse. Dans un second temps, des mutations de classe
I consistant en la mutation de gènes impliqués dans les voies de signalisation intra-

65
cellulaire, comme FLT3, Ras ou c-Kit, vont conférer aux cellules des avantages de survie et
de prolifération (Gilliland et al., 2004). Toutefois, ce modèle n’intègre pas d’autres
caractéristiques des processus néoplasiques comme l’instabilité génétique, l’échappement
à la réponse immune, la néo-angiogénèse ou les interactions avec le micro-environnement
médullaire qui jouent également un rôle important dans la leucémogénèse et la réponse
aux agents génotoxiques.
2. Classification FAB des LAM
Etant donné que les LAM sont issues de différents lignages myéloïdes bloqués à des stades
variable de la différenciation, elles constituent une pathologie hétérogène. Une
classification dite FAB (French-American-British) a été mise en place basée sur des critères
morphologiques et cytochimiques de différenciation, accompagnés pour certaines
catégories d’un phénotypage immunologique (McKenna, 2000). Cette classification
comprend 8 groupes de M0 à M7, chacun correspondant à un niveau de blocage dans la
différenciation hématopoïétique. Bien que cette classification tende actuellement à
disparaître au profit de la classification de l’Organisation Mondiale de la Santé
(classification WHO : World Health Organization) intégrant à la fois des caractéristiques
cytochimiques, cytogénétiques, morphologiques et les antécedents, elle est toujours
utilisée pour la caractérisation rapide des nouveaux cas diagnostiqués de LAM (Vardiman et
al., 2002) (Figure 20).
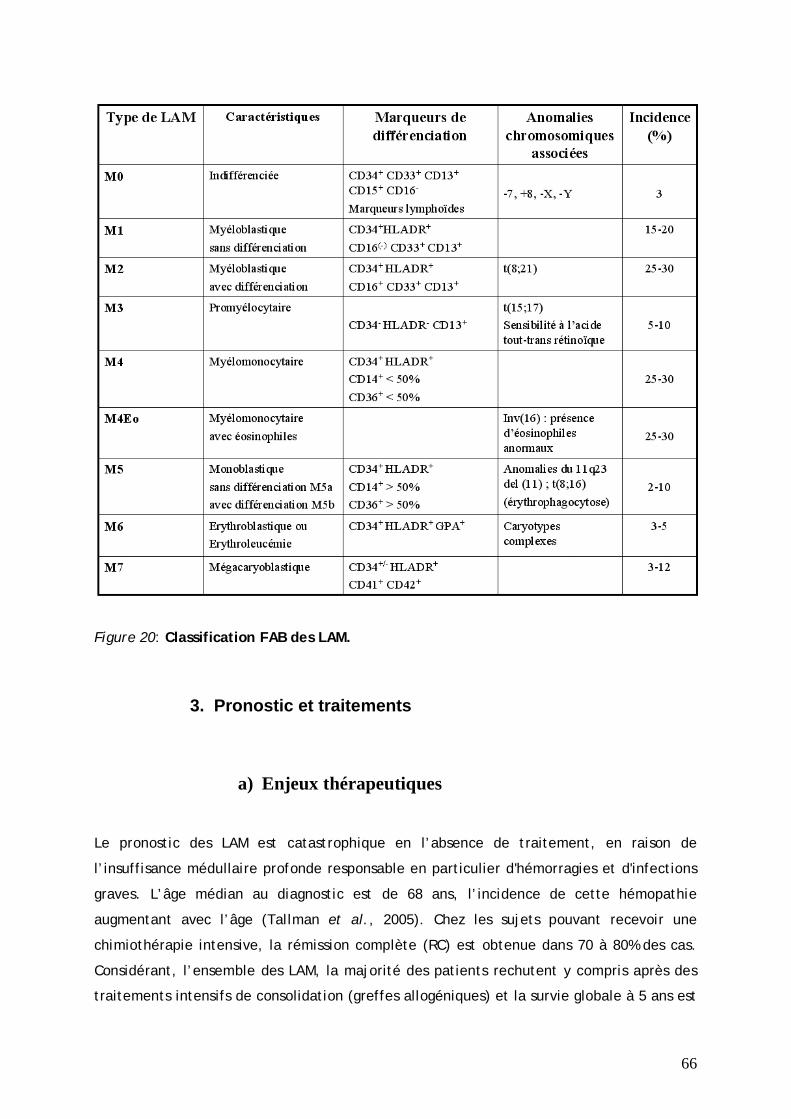
66
Figure 20: Classification FAB des LAM.
3. Pronostic et traitements
a) Enjeux thérapeutiques
Le pronostic des LAM est catastrophique en l’absence de traitement, en raison de
l’insuffisance médullaire profonde responsable en particulier d'hémorragies et d'infections
graves. L’âge médian au diagnostic est de 68 ans, l’incidence de cette hémopathie
augmentant avec l’âge (Tallman et al., 2005). Chez les sujets pouvant recevoir une
chimiothérapie intensive, la rémission complète (RC) est obtenue dans 70 à 80% des cas.
Considérant, l’ensemble des LAM, la majorité des patients rechutent y compris après des
traitements intensifs de consolidation (greffes allogéniques) et la survie globale à 5 ans est

67
de l’ordre de 30 à 40% chez les sujets de moins de 60 ans. Pour les sujets plus âgés, la
survie globale est encore plus mauvaise (moins de 20%). Ceci étant lié d’une part à la
toxicité des traitements intensifs souvent non applicables aux sujets âgés et à la résistance
primaire plus fréquente aux traitements.
Néanmoins, les résultats thérapeutiques varient énormément selon que l’on considère les
deux facteurs pronostiques les plus discriminants qui sont l’âge et les profils
cytogénétiques.
b) Traitements
Le traitement actuel des LAM du sujet jeune repose dans un premier temps sur un
traitement d’induction associant une anthracycline et de l’aracytine. Ce traitement est
suivi d’un traitement de consolidation qui peut être de 2 types selon les caractéristiques
des patients et l’existence d’un donneur : (i) intensification par chimiothérapie seule
(essentiellement avec de l’aracytine à haute dose) suivi ou non d’autogreffe de CSH ; (ii)
chimiothérapie suivie d’une allogreffe de CSH. A l’heure actuelle, on opte parfois pour un
traitement continu avec des molécules comparables à celles utilisées pour les SMD
(Azacytidine).
c) Impact de l’âge dans le pronostic des LAM
L’âge est déterminant car les traitements intensifs de consolidation ne sont pas
applicables aux patients les plus âgés en raison de leur toxicité. De plus, ces patients
développent des maladies sensiblement différentes de celles des sujets plus jeunes avec
des maladies plus résistantes aux agents cytotoxiques (LAM secondaires, cytogénétique
complexe, forte expression de la P-glycoprotéine impliquée dans l’efflux des drogues…).
Compte tenu du vieillissement de la population dans notre pays, le traitement de ce
groupe de malades devient un enjeu majeur de santé publique.

68
Figure 21: Proportion et caractéristiques des LAM chez les sujets jeunes et âgés.
Les sujets âgés ont un âge supérieur ou égal à 60 ans, les sujets jeunes un âge inférieur à 60. L’incidence est donnée pour 100 000 habitants américans/an. Le taux de rémission complète est rapporté après administration du traitement classique par une anthracycline. (Azacytidine ) (D’après (Stone et al., 2004)).
d) Impact des caractéristiques cytogénétiques dans le
pronostic des LAM
A l’exception des LAM3 qui est un cas particulier, des anomalies chromosomiques
récurrentes permettent de classer les LAM en trois sous-groupes de pronostic très
différents.
Figure 22: Impact des caractéristiques cytogénétiques sur le survie globale des patients.

69
La survie globale est mesurée de la date d’entrée des patients dans l’étude jusqu’à leur mort.
(D’après (Jourdan et al., 2005)).
Figure 23: Impact de la cytogénétique dans les LAM des patients de moins de 60 ans. (D’après
(Slovak et al., 2000)).
(1) Pronostic favorable
Le groupe cytogénétique de bon pronostic est défini par la présence de la translocation
t(8;21)(q22;q22) ou des remaniements du chromosome 16 inv16(p13;q22)/
t(16;16)(p13;q22). Ces deux anomalies sont présentes dans environ 10 à 15% des LAM du
sujet jeune (< 60 ans), 5% des LAM du sujet âgé et sont communément regroupés sous le
terme de Core Binding Factor Leukemia (CBF). Ces deux anomalies chromosomiques
affectent les deux sous-unités du facteur de transcription CBF, interférant ainsi sur le
processus de différenciation hématopoïétique. CBF est un facteur de transcription
hétérodimérique composé d’une sous-unité CBFα liant l’ADN au niveau des régions
régulatrices de type « enhancer » et d’une sous-unité unique ne liant pas l’ADN, CBFβ.
Chez l’homme, il existe quatre gènes codant pour les différentes sous-unités du facteur de
transcription : trois codant pour la sous-unité α (CBFA1 à 3) et un codant pour la sous-unité
β CBFB (Speck et al., 1999). CBFα2 n’est exprimé que dans les cellules myéloïdes et
lymphoïdes, la perte complète d’expression des sous-unités α2 et β est incompatible avec
le développement d’une hématopoïèse normale. Les remaniements t(8;21) et inv(16)
impliquant les gènes CBFA2 et CBFB génèrent des protéines chimériques.

70
D’un point de vue moléculaire, la translocation t(8;21) fusionne la partie amino-terminale
du gène AML1 (Acute Myeloid Leukemia 1 autrement appelé RUNX1/CBFα/PEBP2αβ) du
chromosome 21 avec la majeure partie du gène ETO (Eight-Twenty One ou MTG8) du
chromosome 8. Cette protéine chimérique AML1-ETO induit un blocage de la
différenciation des LAM, et est caractéristique de certaines LAM 2. L’inversion du
chromosome 16 génère la fusion de la partie amino-terminale de CBFβ avec la majeure
partie de la protéine MYH11. Cette protéine de fusion conduit à un blocage de la
différenciation myéloïde à travers la séquestration cytoplasmique de CBFβ, et est
caractéristique des LAM 4 à éosinophiles.
Ces maladies sont chimiosensibles, le taux de RC est très élevé (90 à 95%), et le taux de
rechute est de l’ordre de 40%. Mais, une proportion non négligeable de patient est ainsi
guérie uniquement par chimiothérapie en épargnant aux patients une éventuelle greffe
allogénique beaucoup plus toxique. De plus, certains facteurs pronostiques moléculaires,
comme les mutations du récepteur à activité tyrosine kinase c-Kit, peuvent permettre
d’identifier des patients à haut risque au sein de ce groupe de pronostic favorable.
Figure 24: Le complexe CBF et son association dans les leucémies.
Le facteur de transcription CBF est formé de l’association des deux sous-unités CBFα et CBFβ, celle-ci augmentant l’affinité de CBFα pour l’ADN. Trois gènes codent pour la sous-unité CBFα, l’un de ces gènes, CBFA2, codé par CBFB et le plus fréquemment muté dans les leucémies aiguës. (D’après (Speck et al., 1999)).
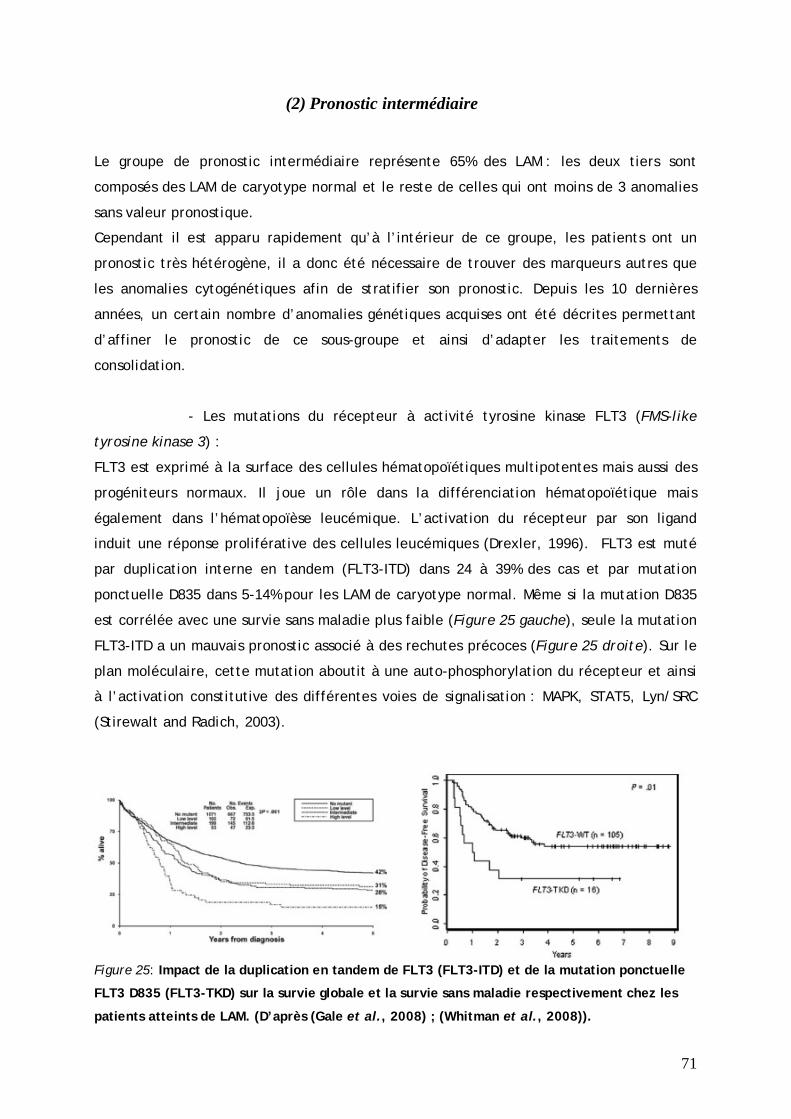
71
(2) Pronostic intermédiaire
Le groupe de pronostic intermédiaire représente 65% des LAM : les deux tiers sont
composés des LAM de caryotype normal et le reste de celles qui ont moins de 3 anomalies
sans valeur pronostique.
Cependant il est apparu rapidement qu’à l’intérieur de ce groupe, les patients ont un
pronostic très hétérogène, il a donc été nécessaire de trouver des marqueurs autres que
les anomalies cytogénétiques afin de stratifier son pronostic. Depuis les 10 dernières
années, un certain nombre d’anomalies génétiques acquises ont été décrites permettant
d’affiner le pronostic de ce sous-groupe et ainsi d’adapter les traitements de
consolidation.
- Les mutations du récepteur à activité tyrosine kinase FLT3 (FMS-like
tyrosine kinase 3) :
FLT3 est exprimé à la surface des cellules hématopoïétiques multipotentes mais aussi des
progéniteurs normaux. Il joue un rôle dans la différenciation hématopoïétique mais
également dans l’hématopoïèse leucémique. L’activation du récepteur par son ligand
induit une réponse proliférative des cellules leucémiques (Drexler, 1996). FLT3 est muté
par duplication interne en tandem (FLT3-ITD) dans 24 à 39% des cas et par mutation
ponctuelle D835 dans 5-14% pour les LAM de caryotype normal. Même si la mutation D835
est corrélée avec une survie sans maladie plus faible (Figure 25 gauche), seule la mutation
FLT3-ITD a un mauvais pronostic associé à des rechutes précoces (Figure 25 droite). Sur le
plan moléculaire, cette mutation aboutit à une auto-phosphorylation du récepteur et ainsi
à l’activation constitutive des différentes voies de signalisation : MAPK, STAT5, Lyn/SRC
(Stirewalt and Radich, 2003).
Figure 25: Impact de la duplication en tandem de FLT3 (FLT3-ITD) et de la mutation ponctuelle
FLT3 D835 (FLT3-TKD) sur la survie globale et la survie sans maladie respectivement chez les
patients atteints de LAM. (D’après (Gale et al., 2008) ; (Whitman et al., 2008)).
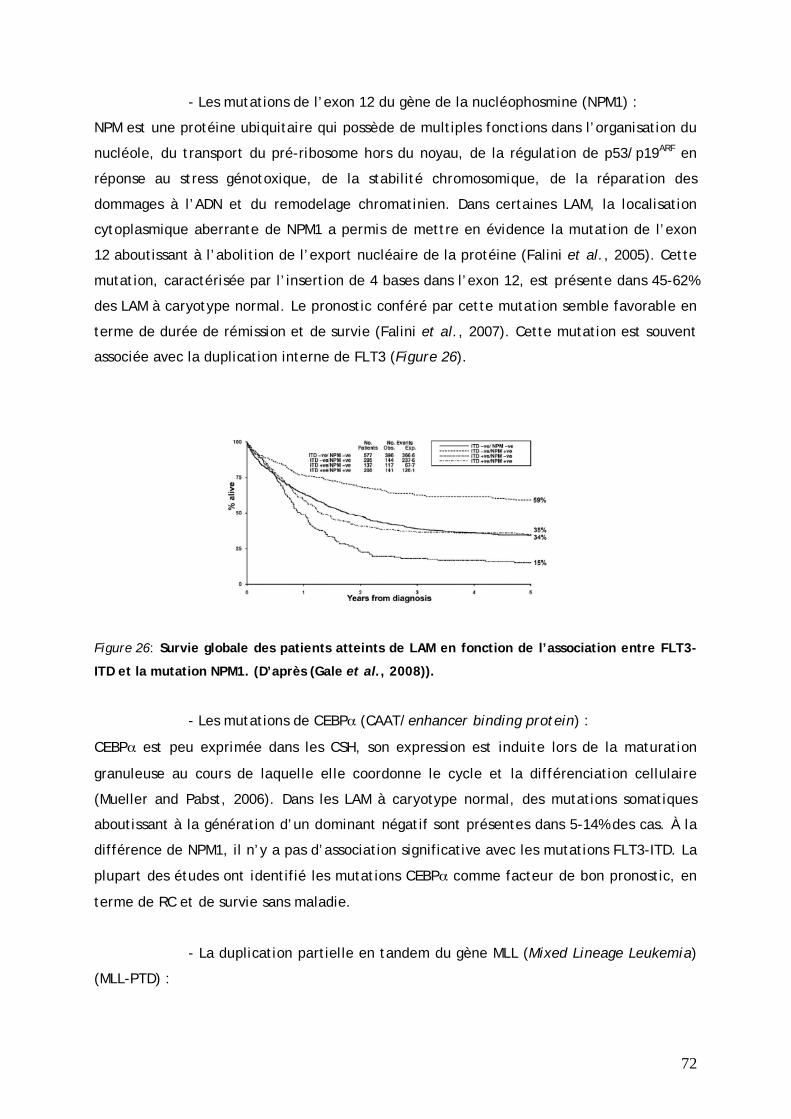
72
- Les mutations de l’exon 12 du gène de la nucléophosmine (NPM1) :
NPM est une protéine ubiquitaire qui possède de multiples fonctions dans l’organisation du
nucléole, du transport du pré-ribosome hors du noyau, de la régulation de p53/p19ARF en
réponse au stress génotoxique, de la stabilité chromosomique, de la réparation des
dommages à l’ADN et du remodelage chromatinien. Dans certaines LAM, la localisation
cytoplasmique aberrante de NPM1 a permis de mettre en évidence la mutation de l’exon
12 aboutissant à l’abolition de l’export nucléaire de la protéine (Falini et al., 2005). Cette
mutation, caractérisée par l’insertion de 4 bases dans l’exon 12, est présente dans 45-62%
des LAM à caryotype normal. Le pronostic conféré par cette mutation semble favorable en
terme de durée de rémission et de survie (Falini et al., 2007). Cette mutation est souvent
associée avec la duplication interne de FLT3 (Figure 26).
Figure 26: Survie globale des patients atteints de LAM en fonction de l’association entre FLT3-
ITD et la mutation NPM1. (D’après (Gale et al., 2008)).
- Les mutations de CEBPα (CAAT/enhancer binding protein) :
CEBPα est peu exprimée dans les CSH, son expression est induite lors de la maturation
granuleuse au cours de laquelle elle coordonne le cycle et la différenciation cellulaire
(Mueller and Pabst, 2006). Dans les LAM à caryotype normal, des mutations somatiques
aboutissant à la génération d’un dominant négatif sont présentes dans 5-14% des cas. À la
différence de NPM1, il n’y a pas d’association significative avec les mutations FLT3-ITD. La
plupart des études ont identifié les mutations CEBPα comme facteur de bon pronostic, en
terme de RC et de survie sans maladie.
- La duplication partielle en tandem du gène MLL (Mixed Lineage Leukemia)
(MLL-PTD) :

73
Elle est présente dans 5-11% des LAM à caryotype normal, la valeur pronostique péjorative
reste controversée, en effet, une étude récente n’a pas retrouvée d’impact sur la survie,
le pronostic serait plutôt lié aux autres marqueurs moléculaires (NPM1, FLT3-ITD, BAALC et
ERG) (Whitman et al., 2007).
- La sur-expression de BAALC (Brain And Acute Leukemia Cytoplasmic), de
ERG (Ets Remated Gene), et de MN1 (Menigioma 1 gene) :
Ces événements sont également des facteurs péjoratifs dans les LAM à caryotype normal
associé à une augmentation de l’incidence des rechutes et à une diminution de la survie.
Ces anomalies sont beaucoup moins utilisées dans la classification pronostique du fait de la
difficulté d’interprétation de la sur-expression.
Les taux de rémission complète du groupe de pronostic intermédiaire sont de l’ordre de
85% et la survie sans maladie à 3 ans est de 40 à 60% en fonction des séries et des
traitements. Actuellement, le traitement comporte une chimiothérapie d’induction et une
intensification thérapeutique orientée par la présence ou l’absence des marqueurs que
nous venons de citer. En effet, l’allogreffe donne de meilleurs résultats en terme de
prévention des rechutes mais sa toxicité ne permet pas toujours de transformer cet
avantage en gain de survie par rapport à la chimiothérapie intensive.
En ce qui concerne les malades FLT3 non mutés (FLT3-) et NPM1 muté (NPM1+) ils reçoivent
à l’heure actuelle un traitement d’induction suivi d’une intensification par chimiothérapie
sans allogreffe. Dans tous les autres cas (FLT3+ NPM1- ; FLT3+ NPM1+ ; FLT3- NPM1-) les
patients sont candidats à une intensification par allogreffe idéalement par un donneur
familial ou s’il n’existe pas par un donneur choisi sur le fichier national ou sur un fichier
international des donneurs de moelle.
.
(3) Pronostic défavorable
Le groupe de pronostic défavorable représente 15% des sujets jeunes et 30% des sujets de
plus de 60 ans. Les anomalies cytogénétiques reconnues par la majorité des groupes de
traitement des LAM sont les suivantes : caryotypes complexes (présentant au moins trois
anomalies cytogénétiques non apparentées), -5/5q-, -7/7q-, t(9;22), t(6;9), anomalie
11q23 en excluant la t(9;11), anomalie 3q26(evi1).

74
Toutefois, certains groupes anglais (UK-MRC) ne parlent de complexité qu’à partir de cinq
anomalies. Récemment, l’impact péjoratif de la perte d’autosomes sur le pronostic a été
reconnu : la monosomal caryotype classification classe les patients dans le groupe de
mauvais pronostic s’ils sont porteurs d’au moins deux monosomies ou bien d’une
monosomie et d’une anomalie de structure (Breems et al., 2008).
Ces anomalies (gains et pertes chromosomiques) s’intègrent dans un groupe d’expression
en micro-arrays où il est décrit une surexpression des gènes régulant la réparation de
l’ADN et la ségrégation chromosomiques (Schoch et al., 2005). Il existe dans ces maladies
une plus grande instabilité des micro-satelliltes, une plus grande activité télomérase que
dans les autres sous-groupes de LAM et surtout une plus grande inactivation du gène
suppresseur de tumeur p53 (jusqu’à 80% des cas) expliquant probablement en grande
partie la chimiorésistance observée en clinique (Haferlach et al., 2008). Le taux de RC est
significativement plus faible (30-50) et la durée de RC plus courte (6-8 mois) aboutissant à
un pronostic catastrophique quel que soit l’âge et les modalités thérapeutiques, en effet
même l’allogreffe n’a pas démontré de supériorité dans ce sous-groupe.
- Les LAM à caryotype complexe :
Les LAM à caryotype complexe sont définies par la présence de plus de trois anomalies
cytogénétiques non apparentées, mais dans 94% des cas, au moins cinq anomalies sont
observées. L’incidence de ce sous-type augmente lorsqu’il existe des maladies
hématologiques ou des traitements antérieurs notamment par des agents alkylants
(Christiansen et al., 2001) mais également avec l’âge des patients (l’incidence est 25 fois
augmentée entre les 21-30 ans par rapport aux 61-70 ans). L’approche thérapeutique est
alors restreinte car trop toxique pour les sujets âgés (Schoch et al., 2005). La présence des
cellules immatures CD34+ est également plus fréquente dans ce sous-type. Même si tous les
mécanismes impliqués dans l’instabilité génétique observée dans les LAM à caryotypes
complexes ne sont pas complètement élucidés, Schoch et ses collaborateurs ont mis en
évidence des profils transcriptionnels différents entre les LAM à caryotypes normaux et
complexes, incluant notamment des gènes impliqués dans la réparation des dommages
(rad50, xrcc4), dans la transduction du signal (nr3c1), et dans le contrôle du cycle
cellulaire (ppp2ca, cdc23) (Schoch et al., 2005). Les mutations de p53, très fréquentes
dans les LAM à caryotype complexe (80% contre 14% dans les autres sous-types), jouent
probablement un grand rôle dans l’instabilité génétique observée et dans la
chimiorésistance (Haferlach et al., 2008). Compte tenu des nombreux mécanismes de
résistance supplémentaires décrits pour ce sous-type de LAM, la sensibilité au traitement
quand il est possible est très faible et en conséquence, la médiane de survie est très
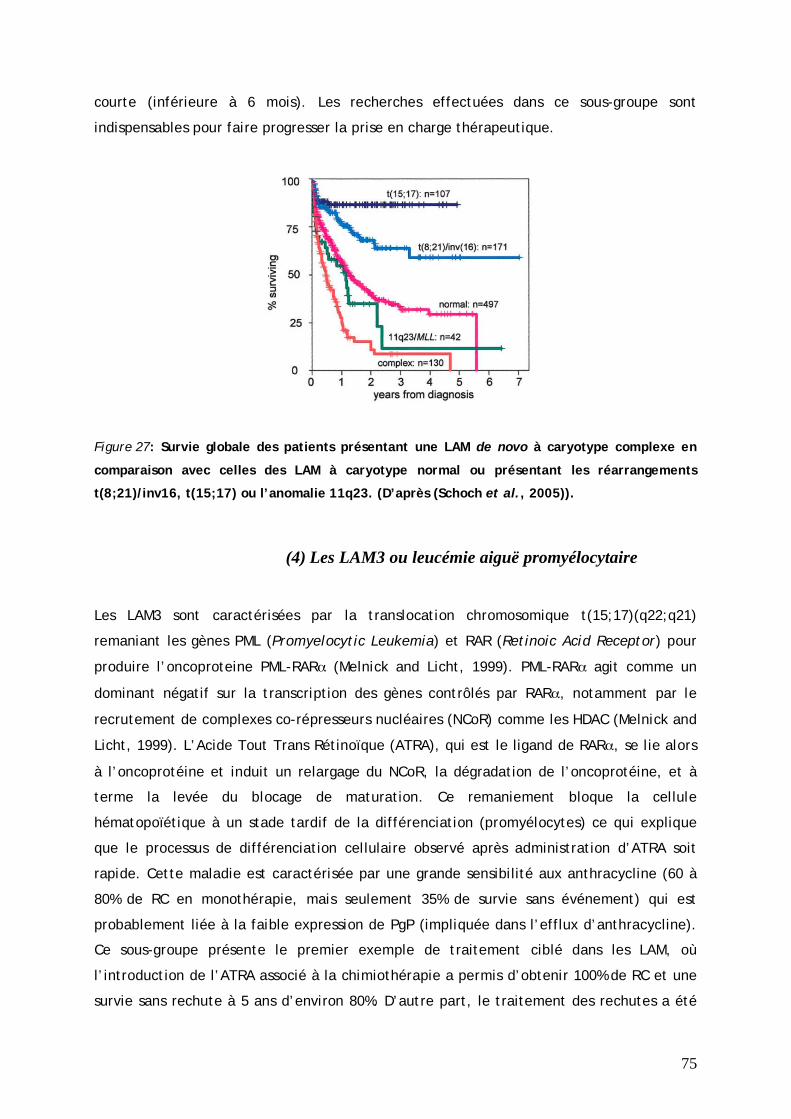
75
courte (inférieure à 6 mois). Les recherches effectuées dans ce sous-groupe sont
indispensables pour faire progresser la prise en charge thérapeutique.
Figure 27: Survie globale des patients présentant une LAM de novo à caryotype complexe en
comparaison avec celles des LAM à caryotype normal ou présentant les réarrangements
t(8;21)/inv16, t(15;17) ou l’anomalie 11q23. (D’après (Schoch et al., 2005)).
(4) Les LAM3 ou leucémie aiguë promyélocytaire
Les LAM3 sont caractérisées par la translocation chromosomique t(15;17)(q22;q21)
remaniant les gènes PML (Promyelocytic Leukemia) et RAR (Retinoic Acid Receptor) pour
produire l’oncoproteine PML-RARα (Melnick and Licht, 1999). PML-RARα agit comme un
dominant négatif sur la transcription des gènes contrôlés par RARα, notamment par le
recrutement de complexes co-répresseurs nucléaires (NCoR) comme les HDAC (Melnick and
Licht, 1999). L’Acide Tout Trans Rétinoïque (ATRA), qui est le ligand de RARα, se lie alors
à l’oncoprotéine et induit un relargage du NCoR, la dégradation de l’oncoprotéine, et à
terme la levée du blocage de maturation. Ce remaniement bloque la cellule
hématopoïétique à un stade tardif de la différenciation (promyélocytes) ce qui explique
que le processus de différenciation cellulaire observé après administration d’ATRA soit
rapide. Cette maladie est caractérisée par une grande sensibilité aux anthracycline (60 à
80% de RC en monothérapie, mais seulement 35% de survie sans événement) qui est
probablement liée à la faible expression de PgP (impliquée dans l’efflux d’anthracycline).
Ce sous-groupe présente le premier exemple de traitement ciblé dans les LAM, où
l’introduction de l’ATRA associé à la chimiothérapie a permis d’obtenir 100% de RC et une
survie sans rechute à 5 ans d’environ 80%. D’autre part, le traitement des rechutes a été

76
grandement amélioré par l’introduction du trioxyde d’arsenic qui permet également la
dégradation de l’oncogène PML-RARα. Ce qui fait que le pronostic de cette maladie est à
l’heure actuelle presque exclusivement conditionnée par la prise en charge initiale car,
cette hémopathie est caractérisée par la fréquence de la gravité des accidents aiguës
secondaires à la CIVD (coagulation intra-vasculaire disséminée) et à la fibrinolyse.
Le schéma ci-dessous résume les différentes étapes nécessaires pour la caractérisation des
LAM. Cette caractérisation consiste en une analyse cytogénétique et moléculaire très vaste
permettant une adaptation du traitement des patients (Figure 28).
Figure 28: Représentation schématique des analyses cellulaires, moléculaires et génétiques
nécessaires pour l’établissement du diagnostic des LAM. (D’après (Renneville et al., 2008)).
4. Concept de cellules souches leucémiques
L’existence d’une population de cellules souches leucémiques (CSL) possédant des
propriétés d’auto-renouvellement, capable d’initier la leucémie et de la maintenir à long
terme, a été démontrée clairement en 1997. Ces travaux ont permis de mettre en
évidence que seulement une très petite fraction des cellules leucémiques (0,2-1%) de
phénotype immature (Thy- (CD90) CD34+ CD38-) était capable d’induire une leucémie chez

77
l’animal immuno-déficient ((Blair et al., 1997) ; (Bonnet and Dick, 1997)). L’hématopoïèse
leucémique garde ainsi une organisation hiérarchisée sur le modèle des cellules
hématopoïétiques normales (cf II. B. 1. Caractérisques). Le concept général de CSL est
maintenant bien établi, il sert de référence pour l’identification de cellules souches
cancéreuses dans les tumeurs solides.
Ces compartiments ont une sensibilité différente à la thérapie, les chimiothérapies
actuelles sont très efficaces dans l’éradication des populations leucémiques plus
différenciées (CFU-L et blastes) mais moins efficaces sur le compartiment des cellules
souches comme le montre la fréquence des rechutes.
a) Origine des CSL
Dans les LAM, comme dans la plupart des cellules cancéreuses, il est difficile d’identifier
les cellules responsables du phénotype tumoral. Plusieurs hypothèses peuvent être
formulées pour expliquer l’émergence des CSL (Passegue et al., 2003) :
- Les CSL, qui possédent des propriétés d’auto-renouvellement, de reconstitution
de l’hématopoïèse chez l’animal immuno-déficient, de différenciation et de résistance à
l’apoptose qui leurs sont propres, pourraient provenir de CSH ayant accumulé un certain
nombre de mutations.
.
- Les CSL pourraient provenir de progéniteurs plus matures où l’événement
oncogénique conférerait aux CSL des caractéristiques de CS (parmi lesquelles l’auto-
renouvellement). Dans les LAM3, la translocation t(15;17) est absente dans les cellules plus
différenciées CD34+ CD38+ contrairement aux cellules CD34+ CD38- (Turhan et al., 1995).
Ainsi, les événements oncogéniques pourraient survenir tout au long du processus de
différenciation mais la LAM ne s’exprimerait qu’à des stades de différenciation plus
tardifs. Ainsi dans la grande majorité des cas, les caractéristiques de la LAM induite
dépendraient strictement du blocage de différenciation de la population transformée
- L’événement oncogénique aurait lieu dans les CSH, mais ne créerait un phénotype
tumoral qu’à des stades de différenciation plus tardifs. Dans les LAM t(8;21) en RC, les
patients présentent des CSH non leucémiques portant la translocation et capables de se
différencier normalement in vitro. Il est donc nécessaire qu’il y ait une mutation
supplémentaire plus tardive pour conduire au phénotype tumoral.

78
Figure 29: Modèles de leucémogenèse à partir de cellules souches hématopoïétiques.
Trois scénarios sont possibles : a) La CSH est la cible de l’événement oncogénique, conduisant à une cellule pré-leucémique qui donnera une CSL (ou LSC) après acquisition de mutations secondaires. b) La CSH est la cible de l’événement oncogénique conduisant après différenciation à un progéniteur pré-leucémique qui donnera une CSL après acquisition de mutations secondaires. c) Le progéniteur myéloïde commun (CMP) subit les mutations initiales donnant naissance à un progéniteur pré-leucémique qui donnera une CSL après mutations additionnelles. (D’après (Jordan and Guzman, 2004)).
b) Caractérisation des CSL
Elle repose sur les mêmes approches que celles qui sont utilisées pour caractériser les CSH,
c’est-à-dire principalement de la cytométrie en flux et du tri cellulaire. Les CSL
présentent des caractéristiques phénotypiques communes avec les CSH normales : CD34+
CD38- HLADR- (le plus souvent), mais d’autres marqueurs membranaires ont pu être
identifiés permettant de discriminer les CSL des CSH, comme l’expression du CD123,
l’antigène CLL1 (C type-like molecule 1) et le CD96. Le CD123 (IL-3-Rα) est exprimé sur les
cellules leucémiques CD34+ CD38- et relativement peu sur les CSH normales (Jordan and
Guzman, 2004). Plus récemment, l’antigène CLL1 a été mis en évidence à la surface des
CSL et ne serait pas retrouvé au niveau des CSH normales (van Rhenen et al., 2007). En
revanche, les CSL sont également caractérisées par l’absence d’expression des marqueurs
CD117 (c-Kit) et CD90.

79
c) Anomalies des voies de signalisation
Les études des voies de signalisation ont été principalement réalisées sur l’ensemble de la
population leucémique, et les données portant sur l’activation de ces voies dans les CSL
commencent à être décrites de même que quelques notions pharmacologiques (Bardet et
al., 2006). La plupart des modèles de leucémogenèse ont montré que l’introduction de
tyrosines kinases (TK) activées seules (FLT3-ITD, TEL-PDGFRα, BCR-ABL) dans des CSH de
souris ne permet pas l’induction d’un phénotype leucémique. La transformation
leucémique n’est observée que lorsque ces mutations des TK (dites mutations de classe I)
sont associées à des mutations de facteurs de transcription (mutations de classe II). Les
mutations des TK aboutissent à une activation constitutive des voies de signalisation d’aval
de survie/prolifération comme les MAPK, PI3K/AKT, STAT. Des inhibiteurs
pharmacologiques ciblant ces voies sont en cours d’évaluation dans des protocoles
cliniques, seule l’association de ces inhibiteurs avec des agents chimiothérapeutiques
semble prometteuse.
(1) Rôle des STATs
Les STATs sont activées par les récepteurs aux cytokines et aux facteurs de croissance à
activité tyrosine kinase (TK). Ces récepteurs peuvent activer directement les STATs par
phosphorylation ou par l’intermédiaire de tyrosines kinases cytoplasmiques comme KAK ou
SRC. Après phosphorylation, les STATs forment des dimères immédiatement relocalisés au
noyau et reconnaissent les régions promotrices de nombreux gènes. Dans un contexte
oncogénique, les récepteurs à activité TK constitutivement activés comme BRC-ABL ou
FLT3-ITD sont capables d’activer les STATs indépendamment des cytokines ou des facteurs
de croissance ((Sternberg and Gilliland, 2004); (Benekli et al., 2003)). En effet,
l’activation constitutive de STAT 5 est retrouvée lors d’autophosphorylation de FLT3
dupliqué ou non. Dans les LAM, STAT 3 et STAT 5 sont activés dans respectivement 28 et
22% de cas ((Xia et al., 1998) ; (Spiekermann et al., 2001) ; (Spiekermann et al., 2002)).
De plus l’activation de STAT 3 représente un facteur pronostique péjoratif. Les patients
ayant une activation constitutive de STAT 3 ont une survie sans maladie significativement
plus courte, de façon indépendante de l’âge, du caractère secondaire de la leucémie et du
caryotype (Benekli et al., 2002). De plus, les blastes de certains patients expriment STAT
3β, qui est une isoforme tronquée en c-terminal, et a un rôle de dominant négatif vis-à-vis
de STAT 3α. La dérégulation de la voie STAT peut également découler de leur activation

80
constitutive mais aussi de la production de cette isoforme tronquée STAT 3β qui conserve
des capacités de dimérisation et de liaison à l’ADN (Benekli et al., 2002). Enfin,
l’activation sélective de STAT 5 dans les CSH, mais pas dans les progéniteurs multipotents,
induit une importante myéloprolifération, suggérant l’importance de STAT 5 phosphorylée
dans l’auto-renouvellement des CSL (Kato et al., 2005).
Les cibles des STATs ne sont pas connues dans les LAM. Toutefois, la surexpression de STAT
5 ou l’expression de TEL-JAK, qui active constitutivement les STATs, peuvent générer un
syndrome myéloprolifératif (Schwaller et al., 1998). Dans ces cellules, les protéines STAT
contribuent à l’activation de la cyline D, de p21WAF1, BCL-xL, Pim1 et c-myc ((Dumon et
al., 1999) ; (Matsumura et al., 1999) ; (Nosaka and Kitamura, 2002) ; (Takahashi et al.,
2004) ; (George et al., 2004)). Par ailleurs, FLT3-ITD, exprimé dans les cellules murines
32D ou Ba/F3, induit l’activation de STAT 5 et contrôle l’expression des kinases Pim-1 et
Pim-2 au niveau transcriptionnel et protéique ((Mizuki et al., 2003) ; (Zhang et al., 2000);
(Adam et al., 2006)).
L’ensemble de ces résultats met en évidence le rôle important des protéines STATs dans la
stimulation de la prolifération des cellules leucémiques de LAM.
(2) Rôle des ERK/MAPK
La voie ERK est impliquée dans les phénomènes mitogéniques et anti-apoptotiques.
L’activation aberrante des MAPK est un phénomène fréquent dans les LAM, elle est estimée
entre 50 et 83% des cas selon les séries de patients étudiées ((Towatari et al., 1997) ;
(Ricciardi et al., 2005) ; (Milella et al., 2001)). Dans la série la plus large (138 patients), la
phosphorylation de MAPK est détectée dans 74% des cas. Cette activité anormale n’est pas
corrélée à des mutations de l’oncogène ras. Il semble que l’activation constitutive de la
voie ERK soit liée à une activation de MEK, une hyper-expression de ERK et une diminution
de l’expression de la phosphatase PAC1 qui régule négativement ERK (Kim et al., 1999). Le
pronostic des patients ayant une phosphorylation constitutive de MAPK est mauvais
((Milella et al., 2003); (Kornblau et al., 2006)). Dans les échantillons où une activation
constitutive est détectée, l’inhibition de ERK/MAPK par le PD98059 ou PD184352 induit une
inhibition de prolifération par un blocage du cycle cellulaire associé à une stabilisation de
p27KIP1 et une inhibition de la transcription de p21WAF1 (Milella et al., 2001). Ces résultats
sont en accord avec d’autres travaux décrivant que des bas niveaux de p27KIP1 et des hauts
niveaux de p21WAF1 confèrent un mauvais pronostic dans les LAM ((Yokozawa et al., 2000) ;
(Zhang et al., 1995)). De plus, l’activation conditionnelle de cette voie par l’introduction

81
de formes actives de Raf dans des cellules hématopoïétiques murines, induit un phénotype
leucémique chez la souris (Konopleva et al., 2005). Enfin, dans les cellules
hématopoïétiques CD34+ normales, on ne détecte pas ou peu de phosphorylation de ERK à
moins de les stimuler avec du G-CSF. De fait, le PD98059 induit peu toxicité sur les
progéniteurs hématopoïétiques normaux (Milella et al., 2001).
(3) Rôle de la voie PI3-kinase
Plusieurs études récentes ont montré que la voie PI3-Kinase/AKT est activée dans les
blastes leucémiques dans 50 à 91% des cas (Min et al., 2003). Les sous-unités p110α, -β, et
–γ sont exprimées dans 50 à 70% des LAM mais à des niveaux variables alors que p110δ est
constamment exprimée à des niveaux élevés ((Sujobert et al., 2005) ; (Billottet et al.,
2006)). Ainsi, un inhibiteur spécifique de cette isoforme, l’IC87114, empêche l’activation
d’AKT dans les blastes au même titre que le LY294002, qui lui n’est pas spécifique d’une
isoforme particulière. Le traitement avec ce composé inhibe la prolifération des cellules
leucémiques avec une faible toxicité sans affecter celle des progéniteurs normaux
contrairement au LY294002. L’activation de la voie PI3-Kinase n’est pas liée à des
mutations de la sous-unité p110δ (Cornillet-Lefebvre et al., 2006). D’autre part, la
phosphorylation constitutive d’AKT sur la thréonine 308 et la sérine 473 est rapportée dans
50 à 70% des échantillons frais issus de patients atteints de LAM, de plus il a été mis en
évidence par nos collaborateurs que la phosphorylation du résidu thréonine 308 est
retrouvée dans les LAM de haut risque cytogénétique (Gallay et al., 2009). Cette
phosphorylation précoce dans la différenciation hématopoïétique (cellules CD34+ CD38-
CD123+) n’est pas retrouvée dans les cellules CD34+ normales. Plusieurs études ont établi
que la phosphorylation d’AKT est un facteur pronostique péjoratif dans les LAM pour la
survie globale ((Kornblau et al., 2006) ; (Min et al., 2003) ; (Gallay et al., 2009)), ceci
pourrait en parti être du à l’induction de la délocalisation de p27KIP1 dans le cytoplasme
loin de ses cibles conventionnelles (Min et al., 2004). Récemment la protéine PTEN,
régulateur négatif de la voie PI3-Kinase/AKT, a été identifiée comme impliquée dans le
maintien des CSH en contrôlant la balance quiescence/auto-renouvellement. En effet,
l’inactivation de PTEN dans les CSH médullaires diminue leur expansion à long terme et
altère leur capacité de reconstitution (Zhang et al., 2006). Ces effets peuvent être
réversés par un traitement à la rapamycine, un inhibiteur de la kinase mTOR situé en aval
d’AKT, qui rétablit l’hématopoïèse normale et empêche la leucémogenèse (Yilmaz et al.,
2006). Dans les LAM, une phosphorylation de PTEN a été mise en évidence dans 74% des cas

82
et est corrélée avec une diminution de la survie globale des patients (Cheong et al., 2003).
Cette phosphorylation stabilise la molécule mais la rend aussi moins active, ce qui résulte
d’une phosphorylation d’AKT signant l’activation de la voie PI3-Kinase.
(4) Les voies apoptotiques
Une des grandes propriétés des CSL est leur grande résistance au stress ; cet avantage de
survie pourrait être mis à profit pour développer des mutations oncogéniques
supplémentaires, voire même être à l’origine des nombreuses rechutes observées chez les
patients atteints de LAM.
De manière intéressante, le marqueur membranaire CD123 exprimé préférentiellement sur
les CSL par rapport aux CSH, peut activer des membres de la famille Bcl2 mais aussi NF-κB
(Guzman et al., 2001). Cette différence d’activation entre les CSH et les CSL a permis
d’apporter de nouvelles perspectives thérapeutiques. Par contre, il existe d’autres modes
possibles d’activation de NF-κB, par les voies PI3K et Ras notamment, activées par des
mutations de récepteurs à activité TK, fréquentes dans les LAM, FLT3 notamment. Les
membres de la famille Bcl-2 sont aussi dérégulés dans le contexte des LAM. En effet, les
blastes de LAM expriment plus de Bcl-2 que leurs équivalents normaux et la sur-expression
de Bcl-xL chez la souris participe au développement de LAM. D’autres études montrent que
la transformation dans le lignage myéloïde impliquerait l’altération concomitante des deux
voies d’apoptose, Fas et Bcl-2 (Traver et al., 1998).
L’ensemble de ces recherches portant sur l’étude de la dérégulation des voies de
signalisation cellulaire dans les LAM a permis la génération de diverses molécules
pharmacologiques permettant un meilleur ciblage des cellules leucémiques en fonction de
leurs propriétés (Figure 30).

83
Figure 30: Ciblage pharmacologique des marqueurs de surface cellulaire et des voies de
signalisation activées dans les CSL. (D’après (Krause and Van Etten, 2007)).
5. Rôle du micro-environnement et de l’adhésion dans la leucémogenèse
Parallèlement à l'altération des voies de signalisation, les propriétés d'adhérence et de
migration, de prolifération et de survie des cellules leucémiques dépendent aussi de leur
micro-environnement médullaire. Ces propriétés résultent de l'intégration cellulaire des
signaux intrinsèques et extrinsèques à la cellule. L’impact du stroma médullaire dans les
LAM est assez mal connu.
En effet, il y a peu d’informations sur l’état des cellules stromales dans les LAM.
Cependant, les perturbations fonctionnelles de la niche semblent évidentes. En effet,
plusieurs groupes ont démontré, par exemple, que la néo-vascularisation est augmentée
dans la MO des patients atteints de LAM ((Hussong et al., 2000) ; (Padro et al., 2000)).
Ceci corrèle avec le fait que l’expression du VEGF (Vascular Endothelial Growth Factor)
(ainsi que de l’angiopoïétine-2 et de son récepteur Tie-2 (Schliemann et al., 2006)) est
significativement augmentée dans la MO des patients (Bellamy et al., 1999) et représente
un facteur de mauvais pronostic. De plus, les modifications de la niche avec
principalement une augmentation de la production de cytokines, vont avoir pour
conséquence l’existence d’un foyer inflammatoire présentant une concentration élevée de
cellules immunitaires et de protéines matricielles. Plusieurs études proposent que des
anomalies des cellules de l’immunité innée expliqueraient en partie la tolérance
immunitaire aux blastes leucémiques ((Costello et al., 2002) ; (Fauriat et al., 2005)). De

84
plus, une différenciation de cellules de patients atteints de LAM in vitro peut aboutir à la
génération de cellules leucémiques développant des caractéristiques de cellules
dendritiques produisant une grande quantité de fibronectine, pouvant être à l’origine de
l’amplification dérégulée des cellules leucémiques (Vialle-Castellano et al., 2004).
Nous avons évoqué plus haut le rôle potentiel d’une dérégulation de c-Myc sur l’expression
des intégrines/cadhérines, et sur l’ancrage des CSH dans leur niche. Dans le contexte
leucémique, les CSL expriment les intégrines α4β1 et α5β1, cependant il a été mis en
évidence une sur-expression de celle-ci dans les cellules leucémiques CD34+ (De Waele et
al., 1999). Ces intégrines ont un rôle crucial dans le processus d’adhérence des blastes au
micro-environnement médullaire dont la fibronectine (Stucki et al., 2001). De plus, les
blastes pourraient secréter et exprimer à leur surface davantage de fibronectine (Vialle-
Castellano et al., 2004).
Ainsi, la transformation leucémique des CSH s’accompagne certainement d’anomalies
adhésives pouvant contribuer à la leucémogenèse. Cependant, très peu d’informations
existent concernant l’influence de telles anomalies sur le plan fonctionnel.
a) Impact de l’intégrine α4β1 sur la leucémogenèse
L’intégrine α4β1 est exprimée à la surface des blastes et a été impliquée principalement
dans la migration et l’adhésion au stroma médullaire (Burger et al., 2003).
Son expression est associée à un mauvais pronostic, dont l’hyperleucocytose (Vila et al.,
1995) et de résistance aux drogues. En effet, l’utilisation in vitro d’anticorps spécifiques
dirigés contre VLA-4 a mis en évidence que l’interaction entre VLA-4, à la surface des
cellules leucémiques, et la fibronectine sur les cellules stromales, aurait un rôle majeur
dans le pronostic des LAM (Matsunaga et al., 2003). De plus, Matsunaga et ses
collaborateurs ont montré que les cellules leucémiques exprimant l’intégrine α4β1
acquièrent une résistance à l’anoïkis et à l’apoptose induite par un agent
chimiothérapeutique, via la voie de signalisation PI3-Kinase/AKT/Bcl-2 activée par
l’interaction entre l’intégrine α4β1 et la fibronectine. Ils ont ensuite mis en évidence que
la combinaison d’un anticorps dirigé contre cette intégrine et d’une anthracyline
permettait d’obtenir 100 % de survie des souris atteintes de maladie minimale résiduelle
(Matsunaga et al., 2003). Cependant, ils n’ont pas pris en compte la forte expression de
l’intégrine α5β1 à la surface des cellules leucémiques pouvant être responsable de
rechutes et de résistantes à la chimiothérapie.

85
b) Impact de l’intégrine α5β1 sur la leucémogenèse
L’intégrine α5β1 est exprimée à la surface des cellules leucémiques et sa sur-expression a
été déterminée en comparaison avec les cellules CD34+ normales (De Waele et al., 1999).
Sa sur-surpression semble conférer une résistance à l’apoptose ainsi qu’un avantage
prolifératif des blastes. En effet, la stimulation de cette intégrine par le SCF dans les
cellules de LAM induit une augmentation de l’adhésion sur fibronectine, conduisant à une
inhibition de l’apoptose et à une stimulation de leur prolifération, a contrario des cellules
CD34+ normales (Bendall et al., 1998). Par ailleurs, une étude portant sur les mécanismes
de résistance aux drogues des cellules leucémiques induite par l’adhésion (ou CAM-DR pour
Cell Adhesion Mediated-Drug Resistance) montre l’implication de l’intégrine α5β1
(Damiano et al., 2001).
Enfin, dans le cas de Leucémie Myéloïde Chronique (LMC), l’intégrine α5β1 est aussi sur-
exprimée sur les cellules progénitrices leucémiques du fait de la présence de l’oncogène
BCR-ABL provoquant l’adhésion à la fibronectine puis la prolifération cellulaire via la
dégradation de p27KIP1 (Kramer et al., 1999). Ce résultat est d’autant plus intéressant que
suite à la phase chronique de la LMC, les patients peuvent entrer dans une phase accélérée
puis une crise blastique de LAM. Donc le processus d’acutisation de la LMC peut nous aider
à mieux comprendre les mécanismes spécifiques de signalisation mis en jeu dans les LAM.
Cependant, les études portant sur l’influence de l’oncogène sur l’adhésion à la
fibronectine ou aux cellules stromales donnent des résultats contradictoires : tandis que
certaines équipes montrent que la translocation BCR-ABL induit l’adhésion des cellules à la
fibronectine ((Kramer et al., 1999) ; (Bazzoni et al., 1996)), d’autres suggèrent le
contraire (Ramaraj et al., 2004). Par ailleurs, un travail récent soutient la première
hypothèse selon laquelle les cellules exprimant l’oncogène BCR-ABL adhèrent plus
fortement aux cellules stromales et cela via une augmentation de l’expression de
l’intégrine β1 (Fierro et al., 2008).
D’autres investigations seront donc nécessaires afin de déterminer précisément comment
cet oncogène régule l’adhésion des cellules leucémiques à la matrice extra-cellulaire et
quel impact fonctionnel cela leur confère ; ce qui sera d’un grand intérêt pour la
compréhension des mécanismes de transformation cellulaire induits par cet oncogène.
L’adhésion des CSL à la fibronectine par l’intermédiaire des intégrines α4β1 et α5β1 joue
donc un rôle majeur dans la rétention des cellules leucémiques dans la niche, la régulation
de la prolifération et de la survie de ces cellules. Pour ces raisons, il est intéressant de
noter qu’il existe un peptide chimérique, appelé la RetronectinTM (Takara), qui possède, à

86
la fois, le domaine CS-1 (site de liaison de l’intégrine α4β1) et le domaine CBD (site de
liaison de l’intégrine α5β1) de la fibronectine. L’utilisation de ce peptide permettrait donc
de cibler les cellules exprimant fortement ces deux intégrines, comme les cellules
leucémiques, et d’augmenter l’action cytotoxique d’un agent chimiothérapeutique en
épargnant les cellules saines.

87
III. Hématopoïèse et cycle cellulaire
A. Cellules hématopoïétiques & Cycle cellulaire
Les différents mécanismes de régulation mis en jeu au cours de l’hématopoïèse
permettent de répondre rapidement et de manière très efficace à des stress
hématologiques, comme une infection, une hémorragie, ou une exposition à des agents
génotoxiques, grâce à la double capacité d’auto-renouvellement et de différenciation de
la population des CSH. Le statut de prolifération des CSH doit ainsi être finement contrôlé
pour assurer à la fois une production continue de progéniteurs hématopoïétiques en
absence de stress et leur différenciation massive en condition de stress, sans bien sûr qu’il
y ait épuisement du pool de CSH. Un tel contrôle nécessite une régulation appropriée de
l’entrée et de la progression dans le cycle cellulaire, qui joue un rôle fondamental dans le
devenir des CSH. L’activité des régulateurs du cycle cellulaire varie selon la population
hématopoïétique considérée et par conséquent selon son niveau de différenciation
hématopoïétique.
1. Les CSH
a) Statut du cycle cellulaire dans les CSH
Le potentiel de reconstitution hématopoïétique, chez la souris immunodéficiente irradiée,
dépend largement de la position des cellules hématopoïétiques dans le cycle cellulaire. En
effet, ce potentiel est maximal pour les CSH quiescentes en phase G0 du cycle, et il
diminue considérablement lorsque les CSH entrent en phase G1 ((Gothot et al., 1997);
(Passegue et al., 2005)). Un travail récent a étudié plus finement la position des LT-CSH
dans le cycle, et mis en évidence que les 2/3 sont quiescentes (phase G0) alors que le 1/3
restant est en phase G1 et S-G2/M du cycle. Ces LT-CSH non quiescentes seraient déjà en
cycle et/ou engagées dans le processus de différenciation, ainsi capables de se
différencier rapidement en populations proliférantes ST-CSH et progénitrices
multipotentes (Passegue et al., 2005). D’autre part, des expériences d’administration
continue de BrdU (24 heures) réalisées chez la souris ont mis en évidence la faible

88
proportion de LT-CSH capables d’entrer en cycle au cours du temps. Cela sous-entendant,
qu’une autre population, peut être les LT-CSH non quiescentes, soit capable d’entrer en
cycle beaucoup plus rapidement, en cas de stress notamment. Seulement 24% des LT-CSH
incorporent le BrdU en 24 heures, alors que plus de 50% des ST-CSH et progéniteurs
multipotents sont BrdU positifs après 24 heures. ((Cheshier et al., 1999) ; (Passegue et al.,
2005)).
Historiquement, les LT-CSH ont été identifiées et isolées uniquement sur la base de leur
capacité à reconstituer l’hématopoïèse d’un hôte irradié, cependant l’évolution des
techniques d’identification et de purification cellulaire permet de préciser le phénotype
des LT-CSH et de faire avancer les connaissances dans le domaine de l’hématopoïèse et du
cycle cellulaire.
b) Régulateurs du cycle cellulaire dans les CSH
L’entrée restreinte des CSH en cycle nécessite la régulation fine des différents activateurs
et inhibiteurs du cycle cellulaire tant au niveau de leur expression que de leur activité. Les
CDKI sont les protéines qui s’opposent à la progression du cycle cellulaire, elles sont donc
impliquées dans le maintien de la quiescence des CSH chez l’homme et la souris.
(1) p21WAF1 & CSH
Dans les souris déficientes en p21WAF1 (p21-/-), la population de CSH en phase G0 diminue,
alors que celle des CSH en cycle augmente. Ces dernières perdent leurs propriétés de
cellules souches en devenant sensibles au 5-FU et incapables de reconstituer
l’hématopoïèse des souris immunodéficientes irradiées (Cheng et al., 2000b). Cela suggère
que p21WAF1 restreigne l’entrée des CSH en cycle, préservant ainsi ces cellules de la
toxicité des agents s’incorporant dans les cellules en cycle. Le maintien de la quiescence
cellulaire par p21WAF1 consiste principalement à empêcher l’interaction entre CDK2 et le
complexe p130-E2F4, maintenant ainsi les cellules en phase G0/G1 du cycle.
Différentes protéines présentes dans les cellules hématopoïétiques sont capables de
moduler l’expression de p21WAF1 : le facteur de croissance TGF-β1 naturellement produit
par les CSH (Hatzfeld et al., 1991), les facteurs de transcription WT (wilms tumor
protein) (Ellisen et al., 2001), MLL (Caslini et al., 2000), RB (Decesse et al., 2001).

89
Néanmoins, l’implication de ces derniers facteurs dans le maintien de la quiescence
cellulaire n’est pas établie.
(2) p18INK4C
La déplétion de p18INK4C entraîne une amélioration significative du potentiel de
reconstitution hématopoïétique chez la souris due à une augmentation de l’auto-
renouvellement des CSH p18-/- (Yuan et al., 2004). p18INK4C semble donc jouer un rôle
important en limitant l’auto-renouvellement des CSH in vivo. Dans les souris doublement
invalidées pour p18INK4C et p21WAF1, la délétion de p18INKC s’oppose à la perte accélérée des
CSH observée lors d’une reconstitution hématopoïétique par des CSH délétées pour
p21CIP1/WAF1 (Yu et al., 2006). p18INK4C exerce son rôle en interagissant spécifiquement avec
les CDK4 et 6, empêchant leur liaison avec la cycline D, en phase G1 précoce du cycle
cellulaire. D’autre part, l'expression de la protéine p18INK4C est considérablement induite
(de 12 à 50 fois) au cours de la myogénèse et de l’adipogénèse, mettant en évidence son
rôle important dans l’induction et/ou le maintien de l’arrêt du cycle cellulaire lors du
processus de différenciation (Phelps et al., 1998).
(3) p16INK4A, p19ARF
Les protéines p16INK4A et p19ARF sont impliquées dans l’auto-renouvellement des CSH via
une régulation épigénétique par le répresseur transcriptionnel bmi-1, appartenant aux
gènes de la famille polycomb. Cette famille de gènes a pour fonction le remodelage de la
structure de la chromatine à travers la modification des histones. L’expression de bmi-1
est restreinte à la MO chez l’homme et la souris, elle est élevée dans les CSH et diminue
au cours de la différenciation hématopoïétique. Les souris bmi-1-/- meurent précocement
(2 mois) d’une aplasie médullaire ((Jacobs et al., 1999) ;(Park et al., 2003)). p16INK4A et
p19ARF inhibent l’interaction de la cycline D avec les CDK4 et 6, induisant ainsi un arrêt du
cycle en phase G0/G1 du cycle cellulaire.
(4) Rb
Afin de s’affranchir de la redondance fonctionnelle des différents membres de la famille
Rb, qui n’avait pas permis jusqu’alors de mettre en évidence un défaut phénotypique clair

90
au niveau de l’hématopoïèse, une équipe américaine a montré récemment que la délétion
simultanée des trois gènes de la famille (pRB, p130 et p107) induit une perte de la
quiescence des CSH. Des dérégulations comme l’activation du complexe CDK4-cycline D ou
la perte de fonction de p16INK4A, conduisant à l’inactivation du suppresseur de tumeur Rb,
ont été fréquemment décrites dans de nombreux cancers mais également dans les
hémopathies telles que les leucémies et les lymphomes ((Leoncini et al., 2006) ; (Spike
and Macleod, 2005)).
(5) Les cyclines et CDK
Les souris doublement mutées pour les gènes cdk4 et cdk6 meurent d’anémie sévère au
stade embryonnaire (Malumbres et al., 2004). Malgré cette mortalité embryonnaire,
l’analyse phénotypique révèle une prolifération cellulaire et une apoptose normales dans
la plupart des tissus analysés, sauf pour le tissu hématopoïétique. Ces résultats mettent en
évidence que les protéines Cdk4 et Cdk6 qui lient et sont activées par les cyclines de la
famille D (D1, D2, D3) ne sont pas essentielles pour réguler l’entrée des cellules en cycle
après une stimulation mitogénique dans la plupart des tissus, mais elles sont
indispensables pour la mise en place d’une hématopoïèse normale. Une étude de Kozar
portant sur l’analyse de souris délétées pour les 3 cyclines de la famille D, confirme que
les cyclines D sont essentielles pour l’hématopoïèse, alors qu’elles ne sont pas essentielles
pour la prolifération de nombreux tissus (Kozar et al., 2004).
c) Niche hématopoïétique et cycle cellulaire
Les interactions entre les CSH et leur microenvironnement médullaire appelé niche ont
permis de faire avancer les connaissances dans le maintien de la quiescence des CSH. En
effet, les CSH ont besoin de se détacher de leur niche dans laquelle elles sont maintenues
pour entrer en division asymétrique et en expansion pour maintenir l’hématopoïèse. Il a
été montré que le microenvironnement médullaire influence l’état de quiescence des CSH
grâce à des interactions directes comme la formation de complexes β-caténine/N-
cadhérine entre les ostéoblastes et les LT-CSH (Zhang et al., 2003), ou bien Tie-
2/angiopoïétine-1 entre les ostéoblastes et les CSH quiescentes (Arai et al., 2004).
Cependant, les acteurs du cycle cellulaire mis en jeu après stimulation par le
microenvironnement ont été peu étudiés. Une étude rapporte que l’engagement de la β1-

91
intégrine, constituant majeur de la fibonectine, inhibe l’entrée et la progression en phase
S des cellules hématopoïétiques immatures CD34+ grâce à l’augmentation de p27KIP1,
inhibiteur du complexe CDK2-cycline E et de l’activité de CDK2 (Jiang et al., 2000).
Les analyses des différents mutants des acteurs du cycle (p21WAF1, p27KIP1, p16INK4A, p18INK4C,
p19ARF, RB), précédemment mentionnés, ont permis de mettre en évidence leur influence
sur le devenir des CSH. Néanmoins, l’utilisation de délétions conditionnelles restreintes au
compartiment des CSH permet de préciser le rôle de ces acteurs dans l’hématopoïèse. En
effet, la délétion conditionnelle de la protéine RB entraîne un désordre myéloprolifératif
caractérisé par une hématopoïèse extra-médullaire et la mobilisation des cellules
primitives dans la périphérie (Walkley et al., 2007). Ce phénotype n’est pas observé
lorsque les CSH rb-/- sont maintenues dans un microenvironnement sain, démontrant ainsi
que RB contrôle les CSH de manière extrinsèque en maintenant la compétence du
microenvironnement à supporter une hématopoïèse normale. Ce phénomène ne semble pas
restreint à RB puisque cela est aussi le cas pour p27KIP1. En effet, l’hyperplasie des cellules
hématopoïétiques du thymus et de la rate n’est observable qu’à condition que les souris
receveurs soient aussi p27-/-.
Ce résultat soulève donc l’importance du contexte environnemental dans la régulation des
fonctions des CSH et le rôle majeur que semblent jouer les acteurs du cycle cellulaire dans
la régulation de ce microenvironnement.
2. Au cours de la différenciation hématopoïétique
a) Cycle cellulaire & différenciation
Le processus de différenciation hématopoïétique s’accompagne d’une réduction
significative du pourcentage de cellules en phase G0 du cycle cellulaire : 67% pour les LT-
CSH, 59% pour les ST-CSH, 50% pour les progéniteurs multipotents, 49% pour les
progéniteurs myéloïdes communs, et 4% pour les progéniteurs myélomonocytiques
(Passegue et al., 2005). De plus, les progéniteurs myéloïdes communs et les progéniteurs
myélomonocytiques sont capables de cycler en 24 heures comme le montre l’incorporation
du BrdU après injection intrapéritonéale chez la souris.

92
b) Régulateurs du cycle cellulaire & différenciation
(1) p21WAF1
De manière assez surprenante, alors que p21WAF1 semble indispensable à l’inhibition de
l’entrée des CSH en cycle, cette protéine est fortement exprimée dans les populations en
cycle plus différenciées : les progéniteurs et précurseurs hématopoïétiques (Steinman et
al., 1998). La déplétion de p21WAF1 chez la souris induit une diminution du nombre de
progéniteurs myéloïdes, mettant en évidence son rôle dans la réplication et la survie des
progéniteurs. Ce rôle atypique de p21WAF1 dans l’expansion du pool de progéniteurs
refléterait sa capacité à augmenter la liaison du complexe CDK-cycline, facilitant ainsi
l’entrée des cellules en phase S (LaBaer et al., 1997). D’autres études portant sur la
différenciation des cellules hématopoïétiques ont mis évidence le rôle direct de p21WAF1
dans ce processus, écartant ainsi une action indirecte à travers l’arrêt du cycle cellulaire
associé au processus de différenciation ((Steinman et al., 1998) ; (Freemerman et al.,
1997)). Toutefois, ce rôle semble être redondant puisque d’autres CDKI comme p19ARF ou
p27KIP1, ou les membres de la famille RB, participent aussi à la différenciation
hématopoïétique (Mori et al., 1999).
De nombreux facteurs influencent le rôle de p21WAF1 au cours de la différenciation : le
répresseur AML1 (Lutterbach et al., 2000), les activateurs hox A10 et E2F1 ((Bromleigh and
Freedman, 2000) ; (Gartel et al., 1998)) pour l’expansion des progéniteurs, et plus
tardivement les protéines CEBPα et STAT ((Harris et al., 2001); (Coqueret and Gascan,
2000)) pour le processus de différenciation.
(2) p27KIP1
Les souris déficientes pour p27KIP1 présentent une augmentation du nombre de progéniteurs
alors que le nombre de CSH reste constant (Cheng et al., 2000a). Ce phénotype met en
évidence le rôle de p27KIP1 dans le contrôle de la division cellulaire au niveau des
progéniteurs. En condition normale de croissance, l’expression importante de p27KIP1
restreint le statut de prolifération des progéniteurs, en revanche en présence de certaines
cytokines, la répression de p27KIP1 va permettre une augmentation de l’entrée en cycle des
progéniteurs (Hoshikawa et al., 1998). Bien que les mécanismes d’action des différentes
cytokines ne soient pas complètement élucidés, un certain nombre ont été décrits. La

93
régulation négative de p27KIP1 dépendante des cytokines peut faire intervenir le répresseur
transcriptionnel STAT3, l’activateur transcriptionnel forhead protein (FKHR-L1), le
protéasome, mais également des mécanismes de relocalisation cytoplasmique de la
protéine (Yaroslavskiy et al., 1999).
Comme pour p21WAF1, p27KIP1 ne semble pas strictement nécessaire pour la différenciation
hématopoïétique. Toutefois, contrairement à p21WAF1, la déplétion de p27KIP1 entraîne un
déséquilibre de la différenciation hématopoïétique, avec une augmentation de la
population progénitrice et une diminution des neutrophiles (de Koning et al., 2000).
Le concours simultané des régulateurs p21WAF1 et p27KIP1 semble important pour permettre
la sortie du cycle cellulaire et la différenciation terminale mégacaryocytaire (Taniguchi et
al., 1999).
(3) Autres régulateurs du cycle cellulaire
Le fait que chez la souris le double mutant p21-/- et p27-/- soit viable suggère que
d’autres régulateurs du cycle cellulaire soient nécessairement mis en jeu durant la
différenciation cellulaire. Cependant, peu de données bibliographiques rapportent
l’importance d’autres régulateurs du cycle cellulaire dans la différenciation
hématopoïétique. Les cyclines sont impliquées dans ce processus, une diminution de leur
expression a été mise en évidence lors de la différenciation. Cependant, l’importance
d’une cycline particulière dans ce processus varie selon le lignage hématopoïétique
considéré. La cycline D1 est plus impliquée dans la différenciation des LT-CSH en ST-CSH
et progéniteurs multipotents (Passegue et al., 2005), alors que les cyclines D2 et D3
seraient impliquées dans la différenciation granulocytaire (Kato and Sherr, 1993).
Les acteurs du cycle cellulaire possèdent des fonctions importantes dans la régulation de
l’entrée en cycle au cours de la différenciation des cellules hématopoïétiques. Les études
qui se sont intéressées à cette problématique soulèvent d’autres questions notamment
celles de la régulation et de la fonction des CKI dans d’autres processus que l’arrêt du
cycle cellulaire comme la différenciation, l’apoptose et la prolifération.

94
B. Dérégulation des acteurs du cycle cellulaire dans les LAM
1. Cycle cellulaire & LAM
Nous venons de voir les nombreux acteurs mis en jeu dans la régulation très fine du cycle
cellulaire permettant le maintien de la balance quiescence/auto-
renouvellement/différenciation hématopoïétique. La moindre anomalie d’un de ces
acteurs peut impacter sur cette balance et contribuer à l’initiation du processus de
leucémogénèse, cependant, ces anomalies peuvent aussi être la résultante de ce
processus.
Il a été constaté depuis quelques années que des dérégulations du cycle cellulaire peuvent
constituer de nouveaux mécanismes de résistance sélectionnés par les cellules
leucémiques pour échapper à la mort cellulaire induite par les agents
chimiothérapeutiques. C’est ainsi que les cellules leucémiques immatures ont développé la
capacité de prolonger l’arrêt en phase G2/M du cycle, en présence d’agents
endommageant l’ADN ((Bruno et al., 1998) ; (Clave et al., 1997)). Ce mécanisme peu
documenté dans la bibliographie nécessite probablement différentes dérégulations des
acteurs de la phase G2/M du cycle cellulaire. Ce point a fait l’objet de nos publications et
sera abordé dans la partie Résultats et Discussion.
Dans les hémopathies, contrairement aux cancers solides, la mutation du gène suppresseur
de tumeur p53 est peu fréquente, cependant cette fréquence augmente considérablement
dans certains groupes tels que les patients de haut risque cytogénétique présentant un
caryotype complexe. Compte tenu de la faible fréquence des mutations de p53 dans les
LAM, le point de contrôle G1/S est à priori opérationnel, ce qui constitue une grande
différence avec les cancers solides.
2. Régulateurs du cycle cellulaire & LAM
a) Cyclines & LAM
Les cyclines étant des régulateurs positifs du cycle cellulaire, leurs surexpressions ont été
retrouvées dans de nombreux cancers, d’autre part, des dérégulations de la voie de
signalisation CDK4/6-cycline D-Rb contrôlant la transition G1/S ont également été mises en
évidence dans de nombreux cancers. Une surexpression de la cycline A1 a été mise en
évidence dans des hémopathies à différenciation myéloïde avec une plus forte incidence

95
dans des échantillons de LAM ((Kramer et al., 1998) ; (Yang et al., 1999)). De manière
intéressante, la surexpression spécifique de la cycline A1 dans le lignage myéloïde chez la
souris conduit à une augmentation du nombre de cellules immatures et induit un
phénotype leucémique dans 15% des cas (Liao et al., 2001). D’autre part, la cycline A1
surexprimée dans les souris transgéniques présente une localisation anormalement
cytoplasmique, ces anomalies de localisation ont par ailleurs été trouvées dans les lignées
cellulaires de LAM et dans les échantillons de LAM (Ekberg et al., 2004). Ainsi, l’expression
anormale et/ou la localisation cytoplasmique de la cycline A1 dans les cellules myéloïdes
immatures pourraient perturber la transition G1/S et ainsi leur différenciation.
Quelques études portant sur les LAM ont mis en évidence des anomalies d’expression et/ou
de localisation des autres cyclines, en particulier une localisation cytoplasmique anormale
de la cycline B1 dans 76% d’échantillons de LAM (Ersvaer et al., 2007) et une surexpression
de la cycline E dans 27% d’échantillons de LAM (Iida et al., 1997).
b) CDKI & LAM
Les CDKI étant des régulateurs négatifs de la progression dans le cycle cellulaire,
différents mécanismes conduisant à leur inactivation ont été décrits dans les cancers.
Dans les tumeurs, l’expression et/ou l’activité de p27KIP1 est souvent diminuée, faisant
intervenir différents mécanismes tels qu’une séquestration cytoplasmique ou une
augmentation de sa dégradation (Blain et al., 2003). La localisation cytoplasmique de
p27KIP1 est d’ailleurs associée à des cancers de grades avancés et de mauvais pronostic
((Singh et al., 1998) ; (Viglietto et al., 2002)). La voie de signalisation AKT/PKB est
impliquée dans la régulation négative de p27KIP1, les mécanismes mis en jeu impliquent la
protéolyse (Sun et al., 1999), la répression transcriptionnelle via le facteur
transcriptionnel FKHR-L1 (forhead protein) (Medema et al., 2000), ou bien la séquestration
cytoplasmique de p27KIP1 ((Viglietto et al., 2002) ; (Shin et al., 2002)). Dans les LAM, il été
mis en évidence que la suractivation de la voie AKT/PKB, fréquente dans les LAM,
augmente la localisation anormalement cytoplasmique de p27KIP1. Cette localisation
anormale est associée à une diminution de la survie chez les patients atteints de LAM (Min
et al., 2004).
De même que pour p27KIP1, l’expression de p15INK4b est très souvent diminuée voire perdue
dans les hémopathies myéloïdes et lymphoïdes. Le mécanisme le plus courant conduisant à
l’inactivation de p15INK4b est l’hyperméthylation de son promoteur dans 80% des cas de

96
LAM. Chez la souris, la perte d’un allèle du gène INK4b induit une augmentation de la
susceptibilité à développer une leucémie (Rosu-Myles and Wolff, 2008).
Un article a mis en évidence une surexpression des autres protéines de la famille INK4,
p18INK4c et p19INK4d, dans des échantillons de LAM ainsi que dans les progéniteurs normaux
par rapport aux populations plus différenciées granulocytaires et monocytaires. Ce résultat
suggère que le maintien de l’expression de ces CDKI dans les LAM pourrait contribuer au
blocage de la différenciation caractéristique de ces cellules (Tschan et al., 1999).
Très récemment, une étude publiée dans le journal Nature a montré le rôle critique de
p21WAF1 dans l’initiation et le maintien de la leucémogénèse (Viale et al., 2009). Les
résultats de l’étude mettent en évidence que l’expression des protéines de fusion
oncogéniques telles que PML-RAR ou AML1-ETO dans les CSH induit des dommages sur
l’ADN et un ralentissement du cycle cellulaire dépendant de p21WAF1, permettant la
réparation des dommages. p21WAF1 joue donc un rôle important dans l’accumulation de CSH
génétiquement instables.
c) Kinases du cycle cellulaire & LAM
Depuis peu, des études portant sur différentes kinases du cycle cellulaire telles que Aurora
A et B ou PLK1, ont mis en évidence qu’elles aussi peuvent être exprimées de façon
aberrante dans les LAM. Les kinases Aurora A ou B, impliquées dans la régulation de la
mitose, sont exprimées de manière variable dans des lignées cellulaires de LAM, et
surexprimées dans des échantillons de LAM en comparaison avec des cellules normales.
L’utilisation d’inhibiteur spécifique de ces kinases (ZM447439) provoque un arrêt de
croissance et l’apoptose des cellules de LAM, suggérant que ces kinases contribuent aussi
de façon importante au développement de la maladie (Ikezoe et al., 2007).
D’autre part, une étude plus récente menée par le même groupe vient de mettre en
évidence la surexpression aberrante de la kinase PLK1 dans des lignées cellulaires de LAM
et dans des échantillons de LAM, cette kinase est impliquée dans l’entrée des cellules en
mitose, la formation du fuseau mitotique, ou encore la cytokinèse. L’invalidation de PLK1
par interférence à l’ARN ou l’utilisation d’inhibiteur pharmacologique (GW843682X)
permettent également une inhibition de la prolifération des cellules de LAM (Ikezoe et al.,
2009). Des résultats similaires ont été obtenus par nos collaborateurs qui ont mis en
évidence une perte des capacités clonogènes des échantillons de LAM en présence de
l’inhibiteur de PLK1 BI2536. Celui-ci a peu d’effet sur les cellules progénitrices myélïdes
normales (Renner et al., 2009).

97
Enfin, une stratégie d’inhibition de la kinase CHK1, impliquée dans le point de contrôle
G2/M du cycle cellulaire, a été utilisée en combinaison avec l’agent chimiothérapeutique
ara-C dans des échantillons de LAM. L’utilisation de cet inhibiteur après un traitement à
l’ara-C chez les patients induit une diminution de l’activation des protéines CHK1 et AKT.
Il permet également de diminuer la capacité clonogène des cellules de LAM (Sampath et
al., 2006). Jusqu’à présent, aucune étude n’a mis en évidence de dérégulation de la kinase
CHK1 dans les LAM, ceci a fait l’objet d’une partie de mon travail de thèse et sera
développé dans la partie Résultats et Discussion.
d) Rb & LAM
La protéine RB semble aussi être dérégulée dans les LAM. Sa perte d’expression ou son
hyperméthylation ont été constatées dans 27% et 17% des LAM respectivement.
Les différentes dérégulations constatées des acteurs du cycle cellulaire mettent en
évidence la place importante qu’ils jouent dans les LAM. En effet, chaque acteur
contribuant à moduler la balance quiescence/auto-renouvellement/différenciation peut
potentiellement être impliqué ans le processus de leucémogenèse. Ce constat souligne
l’importance de l’étude de ces acteurs dans cette pathologie dans le but d’apporter de
nouvelles solutions thérapeutiques en manipulant pharmacologiquement ces voies.
3. Microenvironnement médullaire & LAM
Dans les LAM, le rôle de l’adhésion à la MEC sur la prolifération cellulaire reste peu
documenté. Une étude assez ancienne a mis en évidence qu’une exposition des cellules de
LAM au facteur de croissance SCF augmente leur adhésion à la fibronectine. Ce mécanisme
s’accompagne d’une inhibition de l’apoptose et d’une augmentation de la prolifération des
cellules de LAM (Bendall et al., 1998). Plus récemment, une étude de nos collaborateurs a
mis en évidence l’implication de la phosphatase CDC25 A dans la prolifération induite par
l’adhésion à la fibronectine dans différentes lignées cellulaires de LAM et dans des
échantillons de LAM (Fernandez-Vidal et al., 2006). L’expression augmentée de CDC25A
implique la voie de signalisation PI3K/AKT/mTor, la protéine CHK1 et la machinerie de
dégradation dépendante du protéasome.

98
Bien que quelques articles de la littérature décrivent des dérégulations de plusieurs
acteurs du cycle cellulaire dans les LAM, peu d’études se sont intéressées aux mécanismes
moléculaires conduisant à ces dérégulations ainsi qu’aux conséquences fonctionnelles que
cela peut engendrer sur le développement de la pathologie.

99
RESU LTAT S ET RESU LTAT S ET
DISCUSSIONDISCUSSION

100
PRESENTATION DES ETUDES
Mon projet de thèse a été réalisé sous la co-direction du Pr. Bernard Ducommun (LBCMCP-
CNRS UMR5088) et du Dr Cécile Demur (praticien hospitalier au laboratoire d’hématologie
et INSERM U563). Il s’inscrit dans le cadre d’une collaboration étroite mise en place depuis
trois ans entre les laboratoires des deux co-directeurs. Il s’appuie sur la création d’une
localisation secondaire de l’UMR5088 en milieu hospitalier et le développement d’un
projet de recherche sur le cycle cellulaire dans les hémopathies soutenu par l’INCa. Au
cours de ma thèse, nous avons publié trois articles soulignant l’importance du cycle
cellulaire dans la chimiorésistance des LAM d’une part et sa contribution dans l’instabilité
génétique des LAM d’autre part. Nos deux publications majeures sont citées ci-après, elles
seront reprises en détail dans les parties I et II du chapitre Résultats et Discussion, notre
troisième article a fait l’objet d’une publication dans le journal European Journal of
Pharmacology (2008 Sep 4 ;591(1-3) :102-5) et est rapporté dans l’annexe 2:
J’ai présenté dans l’introduction générale quelques données bibliographiques concernant
le rôle peu décrit du cycle cellulaire dans la chimiorésistance des Leucémies Aiguës
Myéloïdes (LAM). La première partie de mes travaux a donc eu pour but d’évaluer la
fonctionnalité et l'implication du point de contrôle G2/M du cycle cellulaire dans deux
lignées cellulaires de LAM (KG1a CD34+ et U937 CD34-) en réponse un l’agent responsable
de cassures double brin, l'étoposide. L’efficacité de ce point de contrôle étant différente
selon le degré de maturation des cellules, nous avons analysé les profils d’expression de
cinq acteurs majeurs du point de contrôle G2/M et la reprise en mitose après dommages.
Nous avons porté notre attention sur la kinase CHK1 qui est essentielle pour l’arrêt en
phase G2 du cycle après dommages et dont l’invalidation induit une chimiosensibilisation à
la fois des cellules KG1a mais également des cellules issues de patients atteints de LAM.
Article paru dans le journal Oncogene : 2008 Jun 19;27(27):3811-20.

101
Dans la deuxième partie de mes travaux, je me suis intéressée au statut d’activation de la
protéine CHK1, acteur majeur de la voie des dommages à l’ADN dans les cellules de
patients atteints de LAM. L’activation spontanée et variable détectée dans 50 échantillons
de LAM m’a amenée d’une part à établir des corrélations entre ces profils d’activation et
les caractéristiques moléculaires et caryotypiques des échantillons ; et d’autre part à
évaluer la présence de dommages spontanés dans ces cellules. J’ai poursuivi cette étude
en faisant des comparaisons de sensibilité et de chimiosensibilisation lors de l’invalidation
ou de l’inhibition de la kinase CHK1 dans des cellules de LAM de niveaux de dommages
variables.
Article paru dans le journal Cancer Research. 2009 Nov 15;69(22):8652-61.

102
I. Analyse du point de contrôle G2/M dans des lignées cellulaires de LAM
A. Introduction
La prise en charge des patients atteints de LAM repose sur l’utilisation d’agents
chimiothérapeutiques, la cytarabine (ara-C) et une anthracycline (habituellement la
daunorubicine). Ces traitements permettent la mise en rémission complète de plus de 70%
des cas de LAM. Ce pourcentage est cependant très variable selon le type, les antécédents,
et l’âge du patient. Dans tous les cas, les rechutes sont très fréquentes, de l’ordre de 50 à
80%. Cette observation clinique suggère que la chimiothérapie est très efficace dans
l’éradication des cellules progénitrices leucémiques plus différenciées mais beaucoup
moins dans le compartiment des cellules immatures, dont les cellules souches
leucémiques, responsables des rechutes, font partie. De nombreux mécanismes impliqués
dans la résistance des cellules leucémiques immatures ont été mis en évidence parmi
lesquels l’efflux des drogues du type anthracycline par la protéine Pgp, des capacités de
réparation supérieures notamment par le système MMR, la quiescence des cellules,
l’inhibition de la réponse apoptotique… L’équipe du Pr. Guy Laurent a largement contribué
à décrire ces différents mécanismes de résistance dans la lignée KG1a (Laurent and
Jaffrezou, 2001).
Depuis une dizaine d’années, quelques publications ont envisagé la contribution des
acteurs du cycle cellulaire dans la résistance des cellules leucémiques aux agents
chimiothérapeutiques. L’une des premières études datant de 1998 a pu mettre en évidence
qu’un prolongement du point de contrôle G2/M du cycle en réponse aux radiations
ionisantes induit un échappement des cellules immatures à l’apoptose, même si le message
principal de la publication ne traitait pas ce sujet directement (Bruno et al., 1998). De
même, des profils de réponses variables ont été obtenus lors de l’utilisation de la
calicheamycine sur différentes lignées cellulaires de LAM, avec pour certaines des arrêts
en phase G2 permettant un échappement à l’apoptose (Amico et al., 2003). Dans une
étude plus récente datant de 2006, Sampath et ses collaborateurs ont mis en évidence une
activation variable de la kinase CHK1 dans une série de 12 patients atteints de LAM
réfractaires au traitement (Sampath et al., 2006).
Dans ce contexte, et compte tenu du peu de données bibliographiques concernant le
véritable impact du cycle cellulaire sur la réponse aux agents endommageant l’ADN, il nous
est apparu pertinent d’évaluer de manière beaucoup plus précise la fonctionnalité et

103
l'implication du point de contrôle G2/M dans des lignées cellulaires de LAM en réponse à un
agent responsable de cassures double brin comme l'étoposide.

104
B. Résultats
Dans cette première étude menée en collaboration avec C. Didier, nous avons comparé
deux lignées cellulaires de LAM qui présentent de nombreuses anomalies cytogénétiques:
les cellules de lignée cellulaire KGIa myéloblastiques immatures CD34+ et les cellules de la
lignée U937 monoblastiques CD34-. Sur ces lignées, nous avons évalué l’efficacité du point
du contrôle G2/M en présence d’étoposide.
Cette étude nous a permis de mettre en évidence que les cellules KG1a possèdent un point
de contrôle G2/M beaucoup plus efficace que les cellules U937 puisqu’elles ne reprennent
leur progression en mitose qu’une fois les dommages réparés ; la phosphatase CDC25B
jouant un rôle clé dans l’entrée en mitose après dommage. D’autre part, l’invalidation de
la protéine CHK1, par interférence à l’ARN ou par l’agent pharmacologique UCN-01, inhibe
le point de contrôle G2/M et induit une catastrophe mitotique permettant une
sensibilisation à l’étoposide à la fois des cellules KG1a mais également des cellules
d’échantillons myéloïdes issus de patients atteints de LAM.
Ces résultats mettent en évidence que l’efficacité du point de contrôle G2/M varie dans
les cellules de LAM et probablement au cours de la différenciation hématopoïétique. Ce
mécanisme confère à la cellule immature un mécanisme de protection supplémentaire
pouvant être responsable de la résistance aux traitements. Ce travail montre également la
pertinence de l'utilisation des traitements combinant des inhibiteurs des acteurs du point
de contrôle G2/M et des agents endommageant l’ADN.
Ce travail est présenté en détail dans la publication suivante.
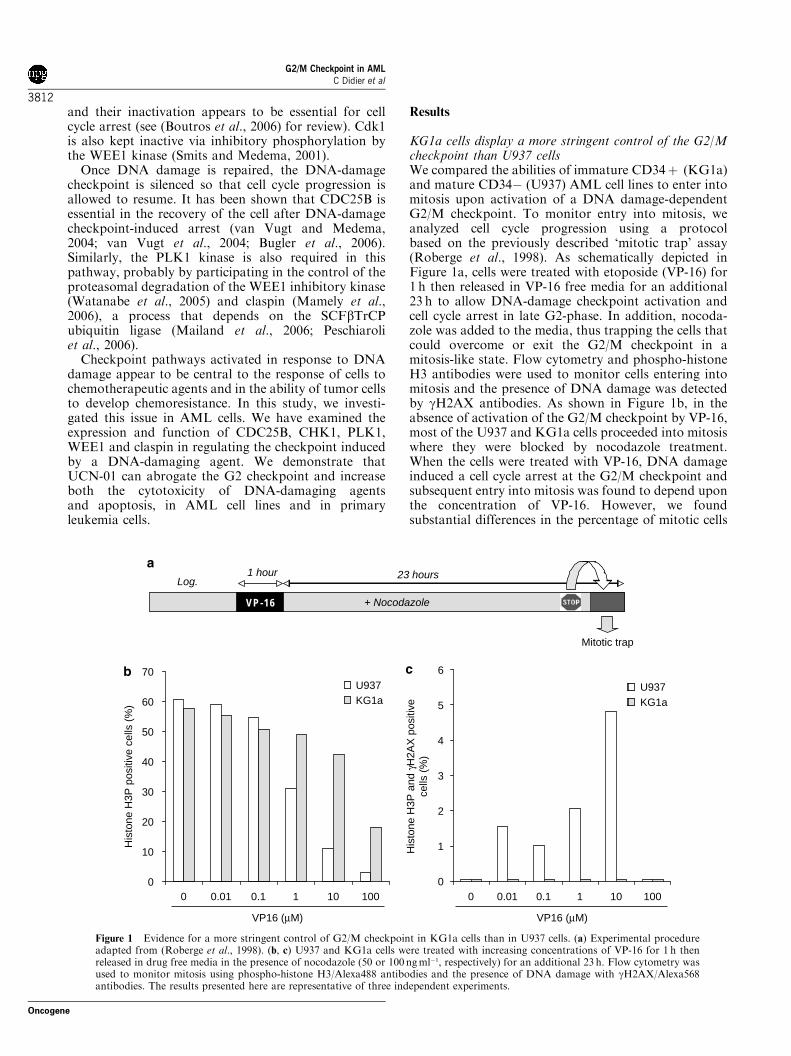
and their inactivation appears to be essential for cellcycle arrest (see (Boutros et al., 2006) for review). Cdk1is also kept inactive via inhibitory phosphorylation bythe WEE1 kinase (Smits and Medema, 2001).
Once DNA damage is repaired, the DNA-damagecheckpoint is silenced so that cell cycle progression isallowed to resume. It has been shown that CDC25B isessential in the recovery of the cell after DNA-damagecheckpoint-induced arrest (van Vugt and Medema,2004; van Vugt et al., 2004; Bugler et al., 2006).Similarly, the PLK1 kinase is also required in thispathway, probably by participating in the control of theproteasomal degradation of the WEE1 inhibitory kinase(Watanabe et al., 2005) and claspin (Mamely et al.,2006), a process that depends on the SCFbTrCPubiquitin ligase (Mailand et al., 2006; Peschiaroliet al., 2006).
Checkpoint pathways activated in response to DNAdamage appear to be central to the response of cells tochemotherapeutic agents and in the ability of tumor cellsto develop chemoresistance. In this study, we investi-gated this issue in AML cells. We have examined theexpression and function of CDC25B, CHK1, PLK1,WEE1 and claspin in regulating the checkpoint inducedby a DNA-damaging agent. We demonstrate thatUCN-01 can abrogate the G2 checkpoint and increaseboth the cytotoxicity of DNA-damaging agentsand apoptosis, in AML cell lines and in primaryleukemia cells.
Results
KG1a cells display a more stringent control of the G2/Mcheckpoint than U937 cellsWe compared the abilities of immature CD34þ (KG1a)and mature CD34� (U937) AML cell lines to enter intomitosis upon activation of a DNA damage-dependentG2/M checkpoint. To monitor entry into mitosis, weanalyzed cell cycle progression using a protocolbased on the previously described ‘mitotic trap’ assay(Roberge et al., 1998). As schematically depicted inFigure 1a, cells were treated with etoposide (VP-16) for1 h then released in VP-16 free media for an additional23 h to allow DNA-damage checkpoint activation andcell cycle arrest in late G2-phase. In addition, nocoda-zole was added to the media, thus trapping the cells thatcould overcome or exit the G2/M checkpoint in amitosis-like state. Flow cytometry and phospho-histoneH3 antibodies were used to monitor cells entering intomitosis and the presence of DNA damage was detectedby gH2AX antibodies. As shown in Figure 1b, in theabsence of activation of the G2/M checkpoint by VP-16,most of the U937 and KG1a cells proceeded into mitosiswhere they were blocked by nocodazole treatment.When the cells were treated with VP-16, DNA damageinduced a cell cycle arrest at the G2/M checkpoint andsubsequent entry into mitosis was found to depend uponthe concentration of VP-16. However, we foundsubstantial differences in the percentage of mitotic cells
VP16 (µM)
VP-16
23 hoursLog.
+ Nocodazole
1 hour
Mitotic trap
His
tone
H3P
pos
itive
cel
ls (
%)
U937KG1a
0
1
2
3
4
5
6
U937KG1a
0
10
20
30
40
50
60
70
0 0.01 0.1 1 10 100
VP16 (µM)
0 0.01 0.1 1 10 100
His
tone
H3P
and
γH
2AX
pos
itive
cells
(%
)
Figure 1 Evidence for a more stringent control of G2/M checkpoint in KG1a cells than in U937 cells. (a) Experimental procedureadapted from (Roberge et al., 1998). (b, c) U937 and KG1a cells were treated with increasing concentrations of VP-16 for 1 h thenreleased in drug free media in the presence of nocodazole (50 or 100 ngml�1, respectively) for an additional 23 h. Flow cytometry wasused to monitor mitosis using phospho-histone H3/Alexa488 antibodies and the presence of DNA damage with gH2AX/Alexa568antibodies. The results presented here are representative of three independent experiments.
G2/M Checkpoint in AMLC Didier et al
3812
Oncogene
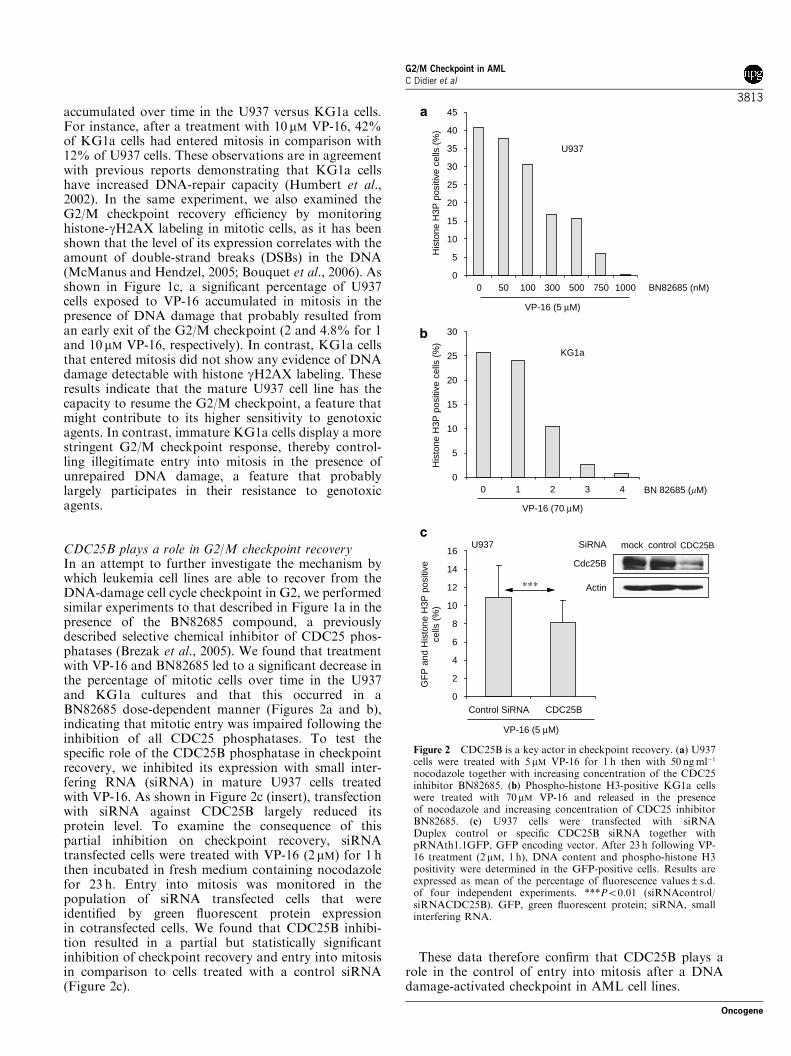
accumulated over time in the U937 versus KG1a cells.For instance, after a treatment with 10 mM VP-16, 42%of KG1a cells had entered mitosis in comparison with12% of U937 cells. These observations are in agreementwith previous reports demonstrating that KG1a cellshave increased DNA-repair capacity (Humbert et al.,2002). In the same experiment, we also examined theG2/M checkpoint recovery efficiency by monitoringhistone-gH2AX labeling in mitotic cells, as it has beenshown that the level of its expression correlates with theamount of double-strand breaks (DSBs) in the DNA(McManus and Hendzel, 2005; Bouquet et al., 2006). Asshown in Figure 1c, a significant percentage of U937cells exposed to VP-16 accumulated in mitosis in thepresence of DNA damage that probably resulted froman early exit of the G2/M checkpoint (2 and 4.8% for 1and 10 mM VP-16, respectively). In contrast, KG1a cellsthat entered mitosis did not show any evidence of DNAdamage detectable with histone gH2AX labeling. Theseresults indicate that the mature U937 cell line has thecapacity to resume the G2/M checkpoint, a feature thatmight contribute to its higher sensitivity to genotoxicagents. In contrast, immature KG1a cells display a morestringent G2/M checkpoint response, thereby control-ling illegitimate entry into mitosis in the presence ofunrepaired DNA damage, a feature that probablylargely participates in their resistance to genotoxicagents.
CDC25B plays a role in G2/M checkpoint recoveryIn an attempt to further investigate the mechanism bywhich leukemia cell lines are able to recover from theDNA-damage cell cycle checkpoint in G2, we performedsimilar experiments to that described in Figure 1a in thepresence of the BN82685 compound, a previouslydescribed selective chemical inhibitor of CDC25 phos-phatases (Brezak et al., 2005). We found that treatmentwith VP-16 and BN82685 led to a significant decrease inthe percentage of mitotic cells over time in the U937and KG1a cultures and that this occurred in aBN82685 dose-dependent manner (Figures 2a and b),indicating that mitotic entry was impaired following theinhibition of all CDC25 phosphatases. To test thespecific role of the CDC25B phosphatase in checkpointrecovery, we inhibited its expression with small inter-fering RNA (siRNA) in mature U937 cells treatedwith VP-16. As shown in Figure 2c (insert), transfectionwith siRNA against CDC25B largely reduced itsprotein level. To examine the consequence of thispartial inhibition on checkpoint recovery, siRNAtransfected cells were treated with VP-16 (2 mM) for 1 hthen incubated in fresh medium containing nocodazolefor 23 h. Entry into mitosis was monitored in thepopulation of siRNA transfected cells that wereidentified by green fluorescent protein expressionin cotransfected cells. We found that CDC25B inhibi-tion resulted in a partial but statistically significantinhibition of checkpoint recovery and entry into mitosisin comparison to cells treated with a control siRNA(Figure 2c).
These data therefore confirm that CDC25B plays arole in the control of entry into mitosis after a DNAdamage-activated checkpoint in AML cell lines.
0
5
10
15
20
25
30
0 1 2 3 4
VP-16 (70 µM)
BN 82685 (�M)
Cdc25B
control CDC25B
Actin
mockSiRNA
0
2
4
6
8
10
12
14
16
Control SiRNA CDC25B
VP-16 (5 µM)
U937
His
tone
H3P
pos
itive
cel
ls (
%)
GF
P a
nd H
isto
ne H
3P p
ositi
vece
lls (
%)
∗∗∗
VP-16 (5 µM)
His
tone
H3P
pos
itive
cel
ls (
%)
0
5
10
15
20
25
30
35
40
45
0 50 100 300 500 750 1000
KG1a
U937
BN82685 (nM)
Figure 2 CDC25B is a key actor in checkpoint recovery. (a) U937cells were treated with 5 mM VP-16 for 1 h then with 50 ngml�1
nocodazole together with increasing concentration of the CDC25inhibitor BN82685. (b) Phospho-histone H3-positive KG1a cellswere treated with 70mM VP-16 and released in the presenceof nocodazole and increasing concentration of CDC25 inhibitorBN82685. (c) U937 cells were transfected with siRNADuplex control or specific CDC25B siRNA together withpRNAth1.1GFP, GFP encoding vector. After 23 h following VP-16 treatment (2mM, 1 h), DNA content and phospho-histone H3positivity were determined in the GFP-positive cells. Results areexpressed as mean of the percentage of fluorescence values±s.d.of four independent experiments. ***Po0.01 (siRNAcontrol/siRNACDC25B). GFP, green fluorescent protein; siRNA, smallinterfering RNA.
G2/M Checkpoint in AMLC Didier et al
3813
Oncogene
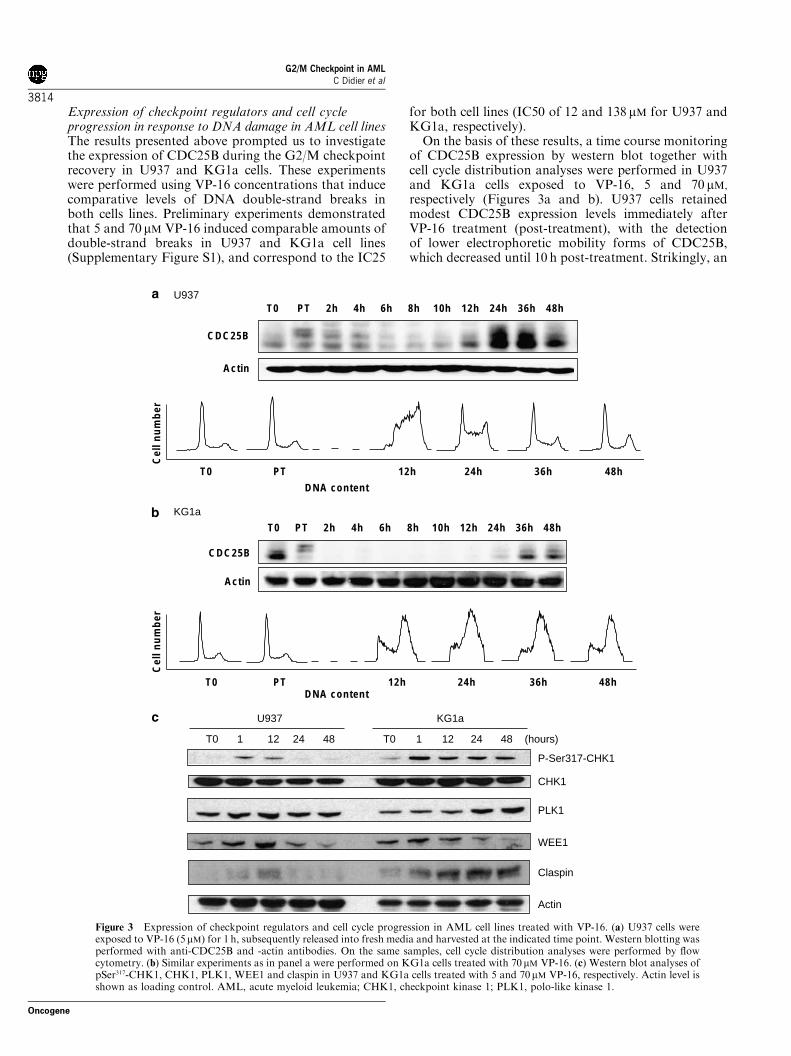
Expression of checkpoint regulators and cell cycleprogression in response to DNA damage in AML cell linesThe results presented above prompted us to investigatethe expression of CDC25B during the G2/M checkpointrecovery in U937 and KG1a cells. These experimentswere performed using VP-16 concentrations that inducecomparative levels of DNA double-strand breaks inboth cells lines. Preliminary experiments demonstratedthat 5 and 70 mM VP-16 induced comparable amounts ofdouble-strand breaks in U937 and KG1a cell lines(Supplementary Figure S1), and correspond to the IC25
for both cell lines (IC50 of 12 and 138 mM for U937 andKG1a, respectively).
On the basis of these results, a time course monitoringof CDC25B expression by western blot together withcell cycle distribution analyses were performed in U937and KG1a cells exposed to VP-16, 5 and 70 mM,
respectively (Figures 3a and b). U937 cells retainedmodest CDC25B expression levels immediately afterVP-16 treatment (post-treatment), with the detectionof lower electrophoretic mobility forms of CDC25B,which decreased until 10 h post-treatment. Strikingly, an
CDC25B
T0 PT 48h36h24h12h10h8h6h4h2h
Actin
DNA content
Cel
l nu
mb
er
T0 PT 12h 24h 36h 48h
T0 PT 2h 4h 48h36h24h12h10h8h6h
CDC25B
Actin
DNA contentT0 PT 12h 24h 36h 48h
KG1a
U937
Cel
l nu
mb
er
WEE1
Claspin
T0 1 12 4824 T0 1 12 24 48
PLK1
P-Ser317-CHK1
(hours)
Actin
CHK1
U937 KG1a
Figure 3 Expression of checkpoint regulators and cell cycle progression in AML cell lines treated with VP-16. (a) U937 cells wereexposed to VP-16 (5mM) for 1 h, subsequently released into fresh media and harvested at the indicated time point. Western blotting wasperformed with anti-CDC25B and -actin antibodies. On the same samples, cell cycle distribution analyses were performed by flowcytometry. (b) Similar experiments as in panel a were performed on KG1a cells treated with 70 mM VP-16. (c) Western blot analyses ofpSer317-CHK1, CHK1, PLK1, WEE1 and claspin in U937 and KG1a cells treated with 5 and 70mM VP-16, respectively. Actin level isshown as loading control. AML, acute myeloid leukemia; CHK1, checkpoint kinase 1; PLK1, polo-like kinase 1.
G2/M Checkpoint in AMLC Didier et al
3814
Oncogene

increase in the level of CDC25B was observed after 24 h,concomitant with a progressive exit of the cells from theG2/M checkpoint as shown by flow cytometry analysis(Figure 3a). Similar results were observed in additionalexperiments with VP-16 concentration up to 10 mM (datanot shown). Similar experiments performed on KG1acells revealed that the CDC25B protein levels alsoprogressively decreased after treatment, then recoveredto a high level at 36 and 48 h post-treatment. However,in these cells, analyses of the DNA content showed anintense and prolonged cell cycle arrest with a G2 DNAcontent, but in the absence of significant cell death, asshown by lack of cells with sub-G1 DNA content(Figure 3b). A similar accumulation of CDC25B wasobserved when the experiment was performed in thepresence of VP-16 concentrations ranging from 20 to200 mM (data not shown).
These observations indicate that a strong increase inthe level of CDC25B is observed in U937 and KG1aafter DNA damage. However, only in U937 cells is thisincrease associated with an ability to exit the G2/Mcheckpoint. KG1a cells remain predominantly arrestedat the checkpoint and entry into mitosis is very limited.Thus, from these experiments we can conclude that inKG1a cells, the weak increase in CDC25B levels afterDNA damage does not seem to be sufficient to allowcheckpoint recovery.
To gain more insight into the molecular mechanismsof the G2 checkpoint recovery, we monitored CHK1and pSer317-CHK1 expression level by western blotanalyses in U937 and KG1a cells exposed to VP-16(Figure 3c). No significant change in total CHK1protein levels could be observed. However, in both celllines, a striking CHK1 activation was detected byphosphorylation on S317 in response to genotoxicstress. Interestingly, CHK1 phosphorylation appearsto decrease more rapidly in U937 than in KG1a. Wealso examined PLK1 level and observed a minorincrease 24 h after VP-16 treatment in U937 cells,concomitant with a progressive exit of the cells fromthe G2/M checkpoint (Figure 3c). The PLK1 level wasalso found to be significantly increased in KG1a cells,but this was not correlated with exit from the G2 block.
We therefore addressed the question of PLK1 activityby monitoring the levels of claspin and WEE1. Asshown in Figure 3c, claspin level initially increased inU937 cells exposed to VP-16, then decreased at 24 and48 h after treatment. By contrast, in KG1a cells, claspinprotein level rapidly increased and remained elevatedin response to VP-16. WEE1 protein level was found todecrease at late time point in response to genotoxictreatment in both cells lines.
Taken together, these observations are in favor of amore intense and prolonged checkpoint activation in theKG1a cell line than in U937 cells.
Combined exposure of KG1a immature AML cells to VP-16 and UCN-01 results in dramatic checkpoint recoveryWe next sought to overcome the G2/M checkpointmechanism in the KG1a cells using the CHK1 kinase
inhibitor UCN-01 (Graves et al., 2000). Recovery fromthe G2/M checkpoint was examined by monitoringthe percentage of mitotic cells (Figure 4a) and by theprogressive entry into an apoptotic process (Figure 4b).As shown, treatment of KG1a cells with 100 nM
UCN-01 alone already had a mitotic and proapoptoticinducing effect, suggesting that CHK1 indeed exerts astrong inhibitory effect on cell cycle progression.Combination of UCN-01 with VP-16 treatment resultedin a more significant increase in the percentage ofmitotic and apoptotic cells (Figures 4a and b).
To assess the specific role of CHK1, KG1a cells weretransfected with CHK1 siRNA, leading to an importantdecrease in CHK1 protein level (Figure 4c). A significantincrease in mitotic population of cells transfected withCHK1 siRNA was observed following VP-16 (10 mM)exposure (Po0.001). Taken together, these findingsindicate that CHK1 siRNA abolishes the strong G2checkpoint and sensitizes the immature KG1a cells to aDNA-damaging agent.
Checkpoint recovery in AML blasts from patientsTo examine the role of CHK1 in resistance of AMLblasts from patients, we evaluated whether the effect ofUCN-01 could sensitize the colony formation efficiencyof primary leukemic cells from patient samples totreatment with VP-16. The colony formation efficiencyof AML blasts treated with VP-16 and/or UCN-01, wasmonitored and the IC50 for VP-16 was determined(Figure 5a). We found that combination with UCN-01had a strong potentialization effect with an importantdecrease of the IC50 for VP-16. The sensitization factor,which is the ratio between the IC50 for VP-16 alone andVP-16 in combination with UCN-01, was derived fromthese data.
Based on these findings, we compared the sensitiza-tion factors from 17 AML patients belonging to the M1and M2 classes with those of AML patients belonging tothe M4 and M5 classes of the French–American–Britishclassification. As shown in Figure 5b, combination ofVP-16 with UCN-01 had a significant potentializingeffect on M1 and M2 (mean 2.1±0.4) compared to M4and M5 groups (mean 1.0±0.1) (Po0.05, Figure 5b).To elucidate the mechanism(s) by which M1 and M2patients are more readily sensitized to UCN-01, weevaluated in six patients from each group the totalCHK1 level and the phosphorylation of Ser345, anindicator of its activation level. For this purpose, AMLblasts from patients were grown in EGM2 mediumin the presence of SCF, GM-CSF, IL-3 and Flt3 ligandfor 48 h before analyses. Cell cycle distribution wasmonitored and no significant difference was observedbetween the two groups (Supplementary Figure S2). Asshown in Figure 5c, we observed variability in CHK1expression and activation level in the two AML patientgroups. However, these results could not be correlatedwith sensibilization status.
In conclusion, these results indicate that in bothKG1a immature cells and leukemic progenitors cellsfrom AML patients, G2/M checkpoint recovery and
G2/M Checkpoint in AMLC Didier et al
3815
Oncogene
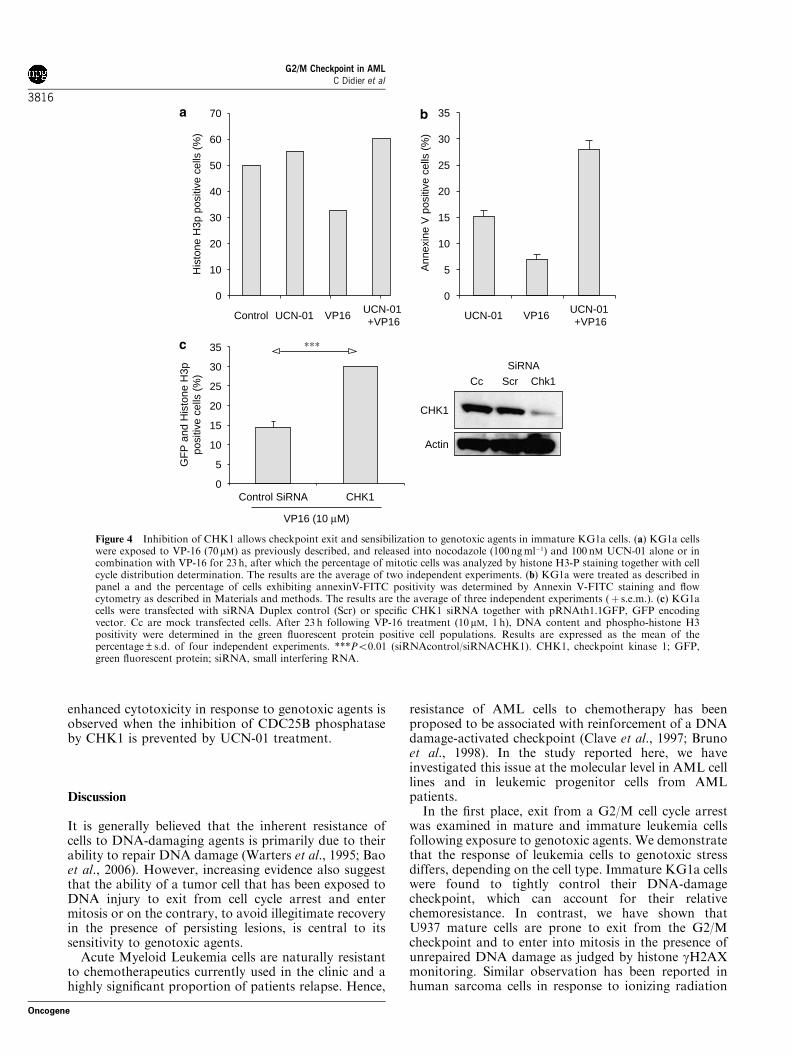
enhanced cytotoxicity in response to genotoxic agents isobserved when the inhibition of CDC25B phosphataseby CHK1 is prevented by UCN-01 treatment.
Discussion
It is generally believed that the inherent resistance ofcells to DNA-damaging agents is primarily due to theirability to repair DNA damage (Warters et al., 1995; Baoet al., 2006). However, increasing evidence also suggestthat the ability of a tumor cell that has been exposed toDNA injury to exit from cell cycle arrest and entermitosis or on the contrary, to avoid illegitimate recoveryin the presence of persisting lesions, is central to itssensitivity to genotoxic agents.
Acute Myeloid Leukemia cells are naturally resistantto chemotherapeutics currently used in the clinic and ahighly significant proportion of patients relapse. Hence,
resistance of AML cells to chemotherapy has beenproposed to be associated with reinforcement of a DNAdamage-activated checkpoint (Clave et al., 1997; Brunoet al., 1998). In the study reported here, we haveinvestigated this issue at the molecular level in AML celllines and in leukemic progenitor cells from AMLpatients.
In the first place, exit from a G2/M cell cycle arrestwas examined in mature and immature leukemia cellsfollowing exposure to genotoxic agents. We demonstratethat the response of leukemia cells to genotoxic stressdiffers, depending on the cell type. Immature KG1a cellswere found to tightly control their DNA-damagecheckpoint, which can account for their relativechemoresistance. In contrast, we have shown thatU937 mature cells are prone to exit from the G2/Mcheckpoint and to enter into mitosis in the presence ofunrepaired DNA damage as judged by histone gH2AXmonitoring. Similar observation has been reported inhuman sarcoma cells in response to ionizing radiation
Cc Scr Chk1
CHK1
Actin
SiRNA
0
5
10
15
20
25
30
35
Control SiRNA CHK1
VP16 (10 µM)
GF
P a
nd H
isto
ne H
3ppo
sitiv
e ce
lls (
%)
∗∗∗
Ann
exin
e V
pos
itive
cel
ls (
%)
0
5
10
15
20
25
30
35
UCN-01 VP16UCN-01 +VP16
His
tone
H3p
pos
itive
cel
ls (
%)
0
10
20
30
40
50
60
70
Control UCN-01 VP16UCN-01 +VP16
Figure 4 Inhibition of CHK1 allows checkpoint exit and sensibilization to genotoxic agents in immature KG1a cells. (a) KG1a cellswere exposed to VP-16 (70mM) as previously described, and released into nocodazole (100 ngml�1) and 100 nM UCN-01 alone or incombination with VP-16 for 23 h, after which the percentage of mitotic cells was analyzed by histone H3-P staining together with cellcycle distribution determination. The results are the average of two independent experiments. (b) KG1a were treated as described inpanel a and the percentage of cells exhibiting annexinV-FITC positivity was determined by Annexin V-FITC staining and flowcytometry as described in Materials and methods. The results are the average of three independent experiments (þ s.e.m.). (c) KG1acells were transfected with siRNA Duplex control (Scr) or specific CHK1 siRNA together with pRNAth1.1GFP, GFP encodingvector. Cc are mock transfected cells. After 23 h following VP-16 treatment (10 mM, 1 h), DNA content and phospho-histone H3positivity were determined in the green fluorescent protein positive cell populations. Results are expressed as the mean of thepercentage±s.d. of four independent experiments. ***Po0.01 (siRNAcontrol/siRNACHK1). CHK1, checkpoint kinase 1; GFP,green fluorescent protein; siRNA, small interfering RNA.
G2/M Checkpoint in AMLC Didier et al
3816
Oncogene
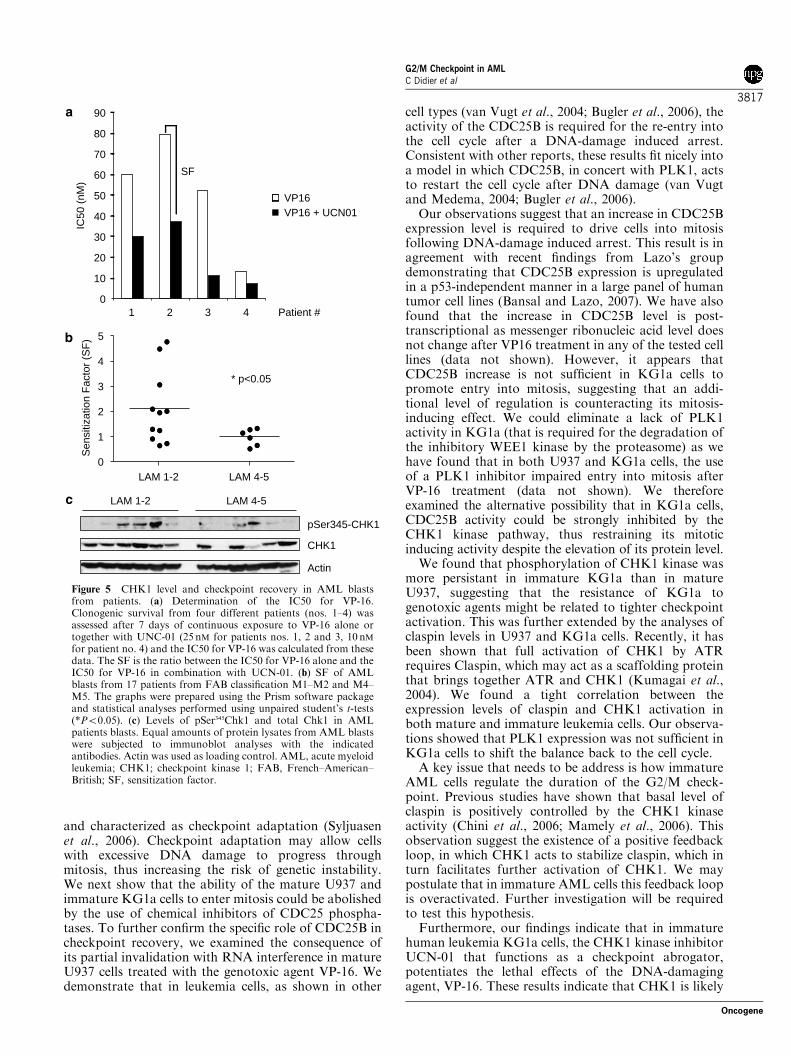
and characterized as checkpoint adaptation (Syljuasenet al., 2006). Checkpoint adaptation may allow cellswith excessive DNA damage to progress throughmitosis, thus increasing the risk of genetic instability.We next show that the ability of the mature U937 andimmature KG1a cells to enter mitosis could be abolishedby the use of chemical inhibitors of CDC25 phospha-tases. To further confirm the specific role of CDC25B incheckpoint recovery, we examined the consequence ofits partial invalidation with RNA interference in matureU937 cells treated with the genotoxic agent VP-16. Wedemonstrate that in leukemia cells, as shown in other
cell types (van Vugt et al., 2004; Bugler et al., 2006), theactivity of the CDC25B is required for the re-entry intothe cell cycle after a DNA-damage induced arrest.Consistent with other reports, these results fit nicely intoa model in which CDC25B, in concert with PLK1, actsto restart the cell cycle after DNA damage (van Vugtand Medema, 2004; Bugler et al., 2006).
Our observations suggest that an increase in CDC25Bexpression level is required to drive cells into mitosisfollowing DNA-damage induced arrest. This result is inagreement with recent findings from Lazo’s groupdemonstrating that CDC25B expression is upregulatedin a p53-independent manner in a large panel of humantumor cell lines (Bansal and Lazo, 2007). We have alsofound that the increase in CDC25B level is post-transcriptional as messenger ribonucleic acid level doesnot change after VP16 treatment in any of the tested celllines (data not shown). However, it appears thatCDC25B increase is not sufficient in KG1a cells topromote entry into mitosis, suggesting that an addi-tional level of regulation is counteracting its mitosis-inducing effect. We could eliminate a lack of PLK1activity in KG1a (that is required for the degradation ofthe inhibitory WEE1 kinase by the proteasome) as wehave found that in both U937 and KG1a cells, the useof a PLK1 inhibitor impaired entry into mitosis afterVP-16 treatment (data not shown). We thereforeexamined the alternative possibility that in KG1a cells,CDC25B activity could be strongly inhibited by theCHK1 kinase pathway, thus restraining its mitoticinducing activity despite the elevation of its protein level.
We found that phosphorylation of CHK1 kinase wasmore persistant in immature KG1a than in matureU937, suggesting that the resistance of KG1a togenotoxic agents might be related to tighter checkpointactivation. This was further extended by the analyses ofclaspin levels in U937 and KG1a cells. Recently, it hasbeen shown that full activation of CHK1 by ATRrequires Claspin, which may act as a scaffolding proteinthat brings together ATR and CHK1 (Kumagai et al.,2004). We found a tight correlation between theexpression levels of claspin and CHK1 activation inboth mature and immature leukemia cells. Our observa-tions showed that PLK1 expression was not sufficient inKG1a cells to shift the balance back to the cell cycle.
A key issue that needs to be address is how immatureAML cells regulate the duration of the G2/M check-point. Previous studies have shown that basal level ofclaspin is positively controlled by the CHK1 kinaseactivity (Chini et al., 2006; Mamely et al., 2006). Thisobservation suggest the existence of a positive feedbackloop, in which CHK1 acts to stabilize claspin, which inturn facilitates further activation of CHK1. We maypostulate that in immature AML cells this feedback loopis overactivated. Further investigation will be requiredto test this hypothesis.
Furthermore, our findings indicate that in immaturehuman leukemia KG1a cells, the CHK1 kinase inhibitorUCN-01 that functions as a checkpoint abrogator,potentiates the lethal effects of the DNA-damagingagent, VP-16. These results indicate that CHK1 is likely
0
1
2
3
4
5
LAM 1-2 LAM 4-5
Sen
sitiz
atio
n F
acto
r (S
F)
Actin
pSer345-CHK1
CHK1
0
10
20
30
40
50
60
70
80
90
1 2 3 4
VP16VP16 + UCN01
Patient #
LAM 1-2 LAM 4-5
* p<0.05
IC50
(nM
)
SF
Figure 5 CHK1 level and checkpoint recovery in AML blastsfrom patients. (a) Determination of the IC50 for VP-16.Clonogenic survival from four different patients (nos. 1–4) wasassessed after 7 days of continuous exposure to VP-16 alone ortogether with UNC-01 (25 nM for patients nos. 1, 2 and 3, 10 nM
for patient no. 4) and the IC50 for VP-16 was calculated from thesedata. The SF is the ratio between the IC50 for VP-16 alone and theIC50 for VP-16 in combination with UCN-01. (b) SF of AMLblasts from 17 patients from FAB classification M1–M2 and M4–M5. The graphs were prepared using the Prism software packageand statistical analyses performed using unpaired student’s t-tests(*Po0.05). (c) Levels of pSer345Chk1 and total Chk1 in AMLpatients blasts. Equal amounts of protein lysates from AML blastswere subjected to immunoblot analyses with the indicatedantibodies. Actin was used as loading control. AML, acute myeloidleukemia; CHK1; checkpoint kinase 1; FAB, French–American–British; SF, sensitization factor.
G2/M Checkpoint in AMLC Didier et al
3817
Oncogene

to play a key role in the G2/M checkpoint activation ofAML cells, probably through the phosphorylation andthe inhibition of CDC25 phosphatases. It is alsotherefore very likely, as schematically illustrated inFigure 6, that a robust activation of the ATR/claspin/CHK1 pathway (Bartek and Lukas, 2007), which leadsto CDC25B phosphorylation and inactivation, is actingin KG1a cells thus rendering their checkpoint responseextremely efficient and decreasing the efficiency ofgenotoxic treatment.
Finally, we examined whether the checkpoint abro-gating effect of UCN-01 could sensitize primaryleukemic cells from AML patients to genotoxic stress.We have found that the colony formation efficiency ofprimary leukemic cells from AML patients M1 and M2(French-American-British classification (FAB)) exposedto UCN-01 in combination with VP-16, was stronglyinhibited compared to the M4 and M5 groups.However, we could not detect any correlated variationin CHK1 level or activity to explain these resultsat the molecular level. Further studies will be requiredto investigate and compare in more detail theDNA-damage checkpoint response pathway betweenthese two groups of patients.
Our observations, together with a recent reportshowing loss of cloning potential when blasts frompatients where exposed to Ara-C in combination withUCN-01 (Sampath et al., 2006), further reinforces a
model in which a very active checkpoint pathway mightbe a common characteristic of immature leukemic cells.
Checkpoint abrogation can potentiate the efficacy ofDNA damaging agents in cancer cells, and is thereforethought to represent an attractive therapeutic target forcancer treatment. The relevance of this strategy isfurther reinforced by the increasing experimentalevidence for a G2/M checkpoint reinforcement in cancerstem cells, associated with increased CHK activity(Bao et al., 2006).
Our findings suggest that combined treatment withUCN-01 and DNA damaging agents may warrantfurther examination as a novel therapeutic strategy inthe treatment of AML. Further studies with blastsfrom a larger set of AML patients will be required toinvestigate and evaluate in detail the therapeuticpotential for checkpoint inhibitors.
Materials and methods
Cell culturesU937 and KG1a were cultured as described (Fernandez-Vidalet al., 2006). Leukemic blasts from patients were obtainedupon informed consent from bone marrow aspirates by ficoll-Hypaque density gradient centrifugation. Frozen cells werethawed in EGM2 medium (Fernandez-Vidal et al., 2006)supplemented with stem cell factor (100 ng ml�1), IL-3(5 ng ml�1), Flt3 ligand (100 ngml�1), and GM-CSF(10 ng ml�1). Cells were left at 1� 106 ml�1 for 48 h inthese conditions during which moderate cell death occurred(10–40% of the cells depending of the patient) then furtherprocessed.
Cytokines, pharmacologic inhibitors and reagentsVP-16 was from Sigma (St Louis, MO, USA). UCN-01 wasprovided by the National Cancer Institute (Rockville, MD,USA). BN82685 was provided by the Institut Henri Beaufour(IPSEN). SCF, interleukin (IL)-3, Flt3 ligand (FL), andgranulocyte-macrophage colony-stimulating factor (GM-CSF) were from R&D Systems (Minneapolis, MN). Otherproducts were purchased from Sigma (Saint Quentin-Fallavier,France).
Transfection of small interfering RNA2� 106 cells were resuspended in 100 ml Nucleofector Kit V forU937 and kit L for KG1a according to manufacturer’sinstructions (Amaxa, Cologne, Germany). CDC25B siRNA(Kramer et al., 2004; Lindqvist et al., 2004), CHK1 siRNA(Schmitt et al., 2006) or control siRNA (Eurogentec, Belgium)together with pRNATH1.1 green fluorescent protein expres-sing vector (Genescript) (Bugler et al., 2006) were added andcells were immediately transfected using the Amaxa nucleo-fector device (program V-001). Cells were subsequentlyresuspended in warm medium, cultured at 106 cells/ml andprocessed after 24 h for experiments.
Analysis of cell cycle progression, apoptosis and gH2AX stainingCells were harvested and fixed in ice-cold 70% ethanol.Cells were permeabilized with PBS-0.25% Triton X-100and subsequently stained with rabbit anti-phospho-histoneH3 (Upstate Biotechnology, Lake Placid, NY, USA) andpropidium iodide. To select the transfected (green fluorescentprotein-positive) cells, a paraformaldehyde fixation was
CHK1
CDC25B
G2 M
WEE1
PLK1
BN82685SiRNA
UCN-01SiRNA
DNA injuryATR
Claspin
Figure 6 Working model. Checkpoint recovery and entry intomitosis is dependent on the activity of the CDC25B phosphataseand on the PLK1 kinase that trigger proteasome-dependentdegradation of the inhibitory WEE1 kinase and the adaptorprotein, claspin. We propose that the reinforcement of thecheckpoint in KG1a cells is linked to the strong inhibitory actionof the CHK1 pathway on CDC25B. As demonstrated in thismanuscript, inhibition of CDC25B (by BN82685 or siRNA) furtherinhibits entry into mitosis, while pharmacological inhibition ofCHK1 by UCN-01 abrogates the checkpoint and favors illegiti-mate entry into mitosis. (See Discussion for further details). CHK1;checkpoint kinase 1; siRNA, small interfering RNA
G2/M Checkpoint in AMLC Didier et al
3818
Oncogene

performed with the intrastain kit from DakoCytomation, andthen the staining was performed as above. Data were collectedon a BD FACSCalibur cytometer and analyzed using the CellQuest (Becton Dickinson, Franklin Lakes, NJ, USA) andModfit software (Verity Software). Apoptotic cells weredetected with Annexin V-FITC detection kit from BDPharMingen (San Diego, CA, USA). Staining for gH2AXwas conducted as described by the manufacturer (Euromedex/Upstate Biotechnology; mouse monoclonal anti-phospho-histone gH2AX antibody; 1:500 dilution). DNA content forblast cells was processed using a coulter DNA Prep reagent kit(Beckman Coulter, Fullerton, CA, USA).
Colony formation analysis with AML patient cellsColony formation efficiency of primary leukemic cells wereobtained from bone marrow aspirates from AML patients atdiagnosis. Colony formation efficiency of primary leukemiccells were resuspended in H441000 methyl-cellulose mediumcomplemented with 10% 5637-conditioned medium. UCN-01,VP-16 or DMSO was added at the indicated concentration,and the cells were incubated at 37 1C for 7 days. Colonies withmore than 20 cells were scored.
Western blot analysesCells were harvested and lysed as described previously(Dutertre et al., 2004). Protein Samples were separated on
4–12% bis-tris-gels, transferred onto nitrocellulose membrane,and immmunostained. Antibodies were: anti-CDC25B (1:1000)Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-
pSer345CHK1 and anti-pSer317CHK1 (1:1000) Cell SignalingTechnology (Beverly, MA, USA), anti-CHK1 (1:1000) fromSanta Cruz Biotechnology (Santa Cruz, CA, USA), anti-PLK1(1:1000) from Zymed laboratories, anti-Wee1 (1:1000) CellSignaling Technology (Beverly, MA, USA) and anti-actin(1:10 000) from Chemicon (Temecula, CA, USA).
StatisticsResults are expressed as mean values±standard deviationsor standard error of the mean as indicated. Statistical analysesof the data were performed by unpaired student t-test.Differences were considered as significant for P-values lessthan 0.05.
Acknowledgements
We gratefully acknowledge R Boutros for corrections andcomments on the manuscript. CD and CC are supported by agrant from the Institut National du Cancer (PL103). This workwas also supported by CNRS, l’Universite Paul Sabatier andla Ligue Nationale Contre le Cancer (Equipe labellisee 2005)grants to BD.
References
Amico D, Barbui AM, Erba E, Rambaldi A, Introna M, Golay J.(2003). Differential response of human acute myeloid leukemiacells to gemtuzumab ozogamicin in vitro: role of Chk1 and Chk2phosphorylation and caspase 3. Blood 101: 4589–4597.
Bansal P, Lazo JS. (2007). Induction of Cdc25B regulates cell cycleresumption after genotoxic stress. Cancer Res 67: 3356–3363.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al.(2006). Glioma stem cells promote radioresistance by preferentialactivation of the DNA damage response. Nature 444: 756–760.
Bartek J, Lukas J. (2003). Chk1 and Chk2 kinases in checkpointcontrol and cancer. Cancer Cell 3: 421–429.
Bartek J, Lukas J. (2007). DNA damage checkpoints: from initiationto recovery or adaptation. Curr Opin Cell Biol 19: 238–245.
Blagosklonny MV. (2006). Target for cancer therapy: proliferatingcells or stem cells. Leukemia 20: 385–391.
Bouquet F, Muller C, Salles B. (2006). The loss of gammaH2AX signalis a marker of DNA double strand breaks repair only at low levels ofDNA damage. Cell Cycle 5: 1116–1122.
Boutros R, Dozier C, Ducommun B. (2006). The when and wheres ofCDC25 phosphatases. Curr Opin Cell Biol 18: 185–191.
Brezak MC, Quaranta M, Contour-Galcera MO, Lavergne O,Mondesert O, Auvray P et al. (2005). Inhibition of human tumorcell growth in vivo by an orally bioavailable inhibitor of CDC25phosphatases. Mol Cancer Ther 4: 1378–1387.
Bruno AP, Laurent G, Averbeck D, Demur C, Bonnet J, Bettaieb Aet al. (1998). Lack of ceramide generation in TF-1 human myeloidleukemic cells resistant to ionizing radiation. Cell Death Differ 5:172–182.
Bugler B, Quaranta M, Aressy B, Brezak MC, Prevost G, DucommunB. (2006). Genotoxic-activated G2-M checkpoint exit is dependenton CDC25B phosphatase expression. Mol Cancer Ther 5:1446–1451.
Chini CC, Chen J. (2003). Human claspin is required for replicationcheckpoint control. J Biol Chem 278: 30057–30062.
Chini CC, Chen J. (2006). Repeated phosphopeptide motifs in humanClaspin are phosphorylated by Chk1 and mediate Claspin function.J Biol Chem 281: 33276–33282.
Chini CC, Wood J, Chen J. (2006). Chk1 is required to maintainclaspin stability. Oncogene 25: 4165–4171.
Clarke CA, Clarke PR. (2005). DNA-dependent phosphorylationof Chk1 and Claspin in a human cell-free system. Biochem J 388:705–712.
Clave E, Carosella ED, Gluckman E, Socie G. (1997). Radiation-enhanced expression of interferon-inducible genes in the KG1aprimitive hematopoietic cell line. Leukemia 11: 114–119.
Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier Cet al. (2004). Phosphorylation of CDC25B by Aurora-A at thecentrosome contributes to the G2/M transition. J Cell Sci 117:2523–2531.
Fernandez-Vidal A, Ysebaert L, Didier C, Betous R, De Toni F,Prade-Houdellier N et al. (2006). Cell adhesion regulates CDC25Aexpression and proliferation in acute myeloid leukemia. Cancer Res
66: 7128–7135.Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O’Connor PM
et al. (2000). The Chk1 protein kinase and the Cdc25C regulatorypathways are targets of the anticancer agent UCN-01. J Biol Chem 275:5600–5605.
Humbert O, Hermine T, Hernandez H, Bouget T, Selves J, Laurent Get al. (2002). Implication of protein kinase C in the regulation ofDNA mismatch repair protein expression and function. J Biol Chem
277: 18061–18068.Kramer A, Mailand N, Lukas C, Syljuasen RG, Wilkinson CJ, Nigg
EA et al. (2004). Centrosome-associated Chk1 prevents prematureactivation of cyclin-B-Cdk1 kinase. Nat Cell Biol 6: 884–891.
Kumagai A, Dunphy WG. (2000). Claspin, a novel protein requiredfor the activation of Chk1 during a DNA replication checkpointresponse in Xenopus egg extracts. Mol Cell 6: 839–849.
Kumagai A, Kim SM, Dunphy WG. (2004). Claspin and the activatedform of ATR-ATRIP collaborate in the activation of Chk1. J Biol
Chem 279: 49599–49608.Lindqvist A, Kallstrom H, Karlsson Rosenthal C. (2004). Character-
isation of Cdc25B localisation and nuclear export during the cellcycle and in response to stress. J Cell Sci 117: 4979–4990.
Mailand N, Bekker-Jensen S, Bartek J, Lukas J. (2006). Destruction ofClaspin by SCFbetaTrCP restrains Chk1 activation and facilitatesrecovery from genotoxic stress. Mol Cell 23: 307–318.
Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, PerrakisA et al. (2006). Polo-like kinase-1 controls proteasome-dependent
G2/M Checkpoint in AMLC Didier et al
3819
Oncogene

degradation of Claspin during checkpoint recovery. Curr Biol 16:1950–1955.
McManus KJ, Hendzel MJ. (2005). ATM-dependent DNA damage-independent mitotic phosphorylation of H2AX in normally growingmammalian cells. Mol Biol Cell 16: 5013–5025.
Nyberg KA, Michelson RJ, Putnam CW, Weinert TA. (2002). Towardmaintaining the genome: DNA damage and replication checkpoints.Annu Rev Genet 36: 617–656.
Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, HalazonetisT, Sherman NE et al. (2006). SCFbetaTrCP-mediated degradationof Claspin regulates recovery from the DNA replication checkpointresponse. Mol Cell 23: 319–329.
Roberge M, Berlinck RG, Xu L, Anderson HJ, Lim LY, Curman Det al. (1998). High-throughput assay for G2 checkpoint inhibitorsand identification of the structurally novel compound isogranulati-mide. Cancer Res 58: 5701–5706.
Sampath D, Cortes J, Estrov Z, Du M, Shi Z, Andreeff M et al. (2006).Pharmacodynamics of cytarabine alone and in combination with7-hydroxystaurosporine (UCN-01) in AML blasts in vitro andduring a clinical trial. Blood 107: 2517–2524.
Schmitt E, Boutros R, Froment C, Monsarrat B, Ducommun B,Dozier C. (2006). CHK1 phosphorylates CDC25B during the cellcycle in the absence of DNA damage. J Cell Sci 119: 4269–4275.
Shiloh Y. (2003). ATM: ready, set, go. Cell Cycle 2: 116–117.Smits VA, Medema RH. (2001). Checking out the G(2)/M transition.
Biochim Biophys Acta 1519: 1–12.Syljuasen RG, Jensen S, Bartek J, Lukas J. (2006). Adaptation to the
ionizing radiation-induced G2 checkpoint occurs in human cells anddepends on checkpoint kinase 1 and Polo-like kinase 1 kinases.Cancer Res 66: 10253–10257.
van Vugt MA, Bras A, Medema RH. (2004). Polo-like kinase-1controls recovery from a G2 DNA damage-induced arrest inmammalian cells. Mol Cell 15: 799–811.
van Vugt MA, Medema RH. (2004). Checkpoint adaptationand recovery: back with Polo after the break. Cell Cycle 3:1383–1386.
Warters RL, Barrows LR, Chen DJ. (1995). DNA double-strand breakrepair in two radiation-sensitive mouse mammary carcinoma celllines. Mutat Res 336: 1–7.
Watanabe N, Arai H, Iwasaki J, Shiina M, Ogata K, Hunter T et al.(2005). Cyclin-dependent kinase (CDK) phosphorylation destabi-lizes somatic Wee1 via multiple pathways. Proc Natl Acad Sci USA
102: 11663–11668.Xu B, Kim ST, Lim DS, Kastan MB. (2002). Two molecularly distinct
G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell
Biol 22: 1049–1059.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
G2/M Checkpoint in AMLC Didier et al
3820
Oncogene

105
C. Discussion
L’ensemble de ces résultats nous permet d’établir le schéma suivant qui met en évidence
l’état d’activation du point de contrôle G2/M du cycle cellulaire à 48h post-traitement
dans les cellules KG1a CD34+ et U937 CD34-.
De manière comparable avec ce qui a été montré dans les tumeurs solides de glioblastome,
les cellules immatures répondent de manière plus intense et durable au traitement par
l’étoposide (Bao et al., 2006). En effet, ces cellules ont la capacité de maintenir
l’activation de la protéine CHK1 jusqu’à 48h et probablement au-delà, alors que les
cellules plus différenciées éteignent cette activation dès 24h. De plus et de manière
surprenante, la seule activation de CHK1 permet le maintien du point de contrôle G2/M
alors que toute la machinerie de reprise en mitose est déjà mise en place dès 24h après
traitement dans les cellules KG1a avec une augmentation de la quantité des protéines
CDC25 B et PLK1, et une dégradation de WEE1. Ainsi, la protéine CHK1 semble jouer un
rôle majeur dans le maintien en phase G2 après traitement des cellules leucémiques
immatures, avec probablement des différences en terme d’activation, de localisation ou
de stabilité de la protéine. Ces modifications font appel à des événements post-
traductionnels puisque le niveau d’ARNm CHK1 évalué dans un panel de lignées de LAM
n’est pas modifié (résultats non montrés).
D’autre part, nous montrons que l’efficacité du point de contrôle G2/M varie dans les deux
lignées cellulaires, allant dans le sens d’une diminution au cours de la différenciation
hématopoïétique. Néanmoins cette étude menée sur deux lignées distinctes n’est pas
suffisante pour conclure car chacune des deux lignées possède des caractéristiques
propres. Il nous a alors semblé intéressant d’évaluer l’efficacité du point de contrôle G2/M
dans des cellules de LAM issues de patients. Les résultats que nous avons pu obtenir sur des
cellules CD34+ et CD34- mises en culture dans un cocktail de cytokines ont permis de
confirmer nos résultats précédents en montrant que les cellules CD34+ maintiennent
davantage l’arrêt en phase G2 du cycle après traitement par l’étoposide. Cependant, il est
délicat d’aborder cette question sur des cellules primaires qui cyclent très peu in vivo dans
la moelle des patients et qui entrent massivement en cycle cellulaire en présence d'un
milieu plus riche en cytokines que le plasma normal. Une étude complémentaire menée sur
le même échantillon CD34+ normal avant et après différenciation granulo-monocytaire
serait nécessaire pour valider cette notion. Nous essayons de répondre à cette question en
évaluant de manière large l’expression et l’activation des acteurs du cycle cellulaire au

106
cours de la différenciation des cellules immatures normales CD34+. Cependant, l’accès aux
cellules CD34+ normales est difficile mais surtout les quantités obtenues après purification
ne nous permettent pas de répondre à l’ensemble des questions proposées sur le même
échantillon. Néanmoins, des travaux réalisés par nos collaborateurs de l’INSERM U563
viennent de mettre en évidence que les cellules CD34+ normales, fraîchement isolées de la
moelle osseuse de fémur, expriment les phosphatases CDC25 A et CDC25 B alors qu’elles
sont en phase G0/G1 du cycle cellulaire (V. De Mas communication personnelle). Ces
expressions diminuent au cours de la différenciation érythroïde, ainsi qu'au cours de
l’entrée massive en cycle des cellules. Ces résultats préliminaires semblent très
intéressants et pourraient nous faire suspecter d’autres rôles des acteurs du cycle
cellulaire, en particulier pour les phosphatases CDC25, au cours de la différenciation
hématopoïétique. Le statut de CHK1 restera à évaluer dans cette étude.
Compte tenu du rôle clé que semble jouer la protéine CHK1 dans le maintien de l’arrêt du
cycle cellulaire préférentiellement dans les cellules immatures et ainsi dans la
chimiorésistance des cellules de LAM, de même que l’intérêt potentiel d’une thérapie
visant à invalider la protéine, nous avons poursuivi notre étude en portant notre intérêt sur
le statut d’activation de CHK1 dans des cellules de LAM issues de patients.

107
II. Activation constitutive de la voie de signalisation des dommages dans les échantillons de LAM de caryotype complexe : intérêt thérapeutique d’un ciblage des points de contrôle
A. Introduction
Notre précédent travail mettant en évidence le rôle critique de la protéine CHK1 dans la
chimiorésistance des cellules immatures de LAM, nous a conduit à évaluer son statut
d’activation avant et après dommages à l'ADN dans des échantillons de patients atteints de
LAM. Nous avons rapidement constaté que la protéine CHK1 présente une activation basale
et variable dans nos échantillons en l’absence de dommages à l’ADN. Ce qui met en
évidence le double rôle de CHK1 dans la chimiorésistance des cellules de LAM en présence
de dommages à l’ADN et dans l’établissement de l’instabilité génétique.
Comme cela a été décrit dans l'introduction, l’activation de la voie de signalisation des
dommages dépendante des oncogènes semble jouer un rôle crucial dans l’établissement du
processus tumoral. En ce qui concerne l'oncogenèse des tumeurs solides, il est maintenant
bien établi que, dans les stades précoces qui précédent le développement de la tumeur,
les principaux acteurs de la voie de réponse aux dommages sont activés : γH2AX, ATM/ATR,
CHK1/CHK2, et qu’ils contribuent à l’établissement de l’instabilité génétique ((Gorgoulis
et al., 2005) ; (Bartkova et al., 2005)). Par contre, dans les stades plus avancés et lorsque
la tumeur est installée, la voie des dommages est moins activée voire éteinte selon les
tissus observés
Cependant, la question de la participation de cette voie au processus de tumorigenèse est
beaucoup moins documentée dans les hémopathies ; c’est pourquoi notre étude est l’une
des premières à adresser cette problématique dans un large échantillonnage de cellules de
patients atteints de LAM.

108
B. Résultats
Ce travail a pu être mené exclusivement sur des cellules de patients grâce à la mise en
place d’une collection hémopathies INSERM Midi-Pyrénées (HIMIP) (N° d’autorisation de
cession AC-2008-129, N° de déclaration DC2008-307 collection N°1). Cette collection a été
constituée il y a de nombreuses années par les hématologues appartenant à la fois au CHU
et à l’INSERM U563. Ces échantillons documentés nous permettent de connaître les
caractéristiques propres de chaque échantillon. Ces informations sont particulièrement
importantes dans le cas des LAM qui sont des maladies très hétérogènes.
Ce travail m’a permis de mettre en évidence une activation basale de la protéine CHK1 en
absence d’agents endommageant l’ADN, dans des échantillons de LAM prélevés au
diagnostic avant tout traitement. Cette activation basale de CHK1 résulte de la présence
de dommages spontanés sur l’ADN analysés par la mesure de la phosphorylation de
l’histone H2AX. L’étude d’un large panel d’échantillons (n = 50) m’a permis de montrer
que les LAM de caryotype complexe (de mauvais pronostic) présentent davantage de
dommages spontanés que les LAM de caryotype normal ou simple, de risque intermédiaire
selon la classification cytogénétique pronostique. Et en conséquence, l’invalidation de
CHK1 par interférence à l’ARN et utilisation d’inhibiteur UCN-01 est plus efficace dans les
échantillons de caryotype complexe présentant plus de dommages à l'ADN. En revanche,
l’inhibition de CHK1 par l’UCN-01 aux doses utilisées sur les cellules de LAM n’a pas d’effet
sur l’hématopoïèse normale. L’ensemble de ces résultats ouvre une réelle fenêtre
thérapeutique sur l’utilisation d’inhibiteurs de CHK1 dans la prise en charge des LAM de
caryotype complexe dont le pronostic est catastrophique quel que soit l’âge et les
modalités thérapeutiques puisque même l’allogreffe n’a pas démontrée de supériorité
dans ce sous-groupe.
Ce travail est présenté en détail dans la publication suivante.

Experimental Therapeutics, Molecular Targets, and Chemical Biology
Constitutive Activation of the DNA Damage Signaling Pathway inAcute Myeloid Leukemia with Complex Karyotype: PotentialImportance for Checkpoint Targeting Therapy
Cindy Cavelier,1,2 Christine Didier,1,2 Naïs Prade,3 Véronique Mansat-De Mas,3,4
Stéphane Manenti,3 Christian Recher,3,4 Cécile Demur,3,4 and Bernard Ducommun1,2,4
1Université de Toulouse, LBCMCP, 2Centre National de la Recherche Scientifique, LBCMCP-UMR5088, 3Institut Nationalde la Sante et de la Recherche Medicale, Centre de Physiopathologie Toulouse Purpan-U563, and4Laboratoire d'Hématologie-CHU Toulouse-Purpan, TSA 40031, Toulouse, France
AbstractGenomic instability in solid tumors participates in the onco-genetic process and is associated with the activation of theDNA damage response pathway. Here, we report the activa-tion of the constitutive DNA damage and checkpoint pathwayassociated with complex karyotypes in samples from patientswith acute myeloid leukemia (AML). We show that antagoniz-ing CHK1 kinase with a small inhibitory compound or by RNAinterference strongly reduces the clonogenic properties ofhigh–DNA damage level AML samples, particularly those withcomplex karyotypes. Moreover, we observe a beneficial effectof CHK1 inhibition in high–DNA damage level AML samplestreated with 1-β-D-arabinofuranosylcytosine. In contrast,CHK1 inhibition has no effect on the clonogenic propertiesof normal hematopoietic progenitors. All together, our resultsindicate that CHK1 inhibition may represent an attractivetherapeutic opportunity in AML with complex karyotype.[Cancer Res 2009;69(22):8652–61]
IntroductionAcute myeloid leukemia (AML), the most frequent acute leu-
kemia in adults, is a clonal disorder characterized by the accu-mulation of malignant hematopoietic progenitor cells (HPC)with an impaired differentiation program. Despite considerableprogress in the therapy of AML and a high rate of completeremission after induction chemotherapy, most patients relapseand succumb to the disease (1). This clinical observation high-lights the difficulties involved in eradicating the leukemic stemcells that are responsible for the emergence of new leukemicprogenitor cells leading to relapse. AML is a heterogeneous dis-ease and patients differ strongly in their response to therapy, inthe occurrence of relapse and in overall survival, according toprognosis factors such as age and presence of molecular and/or cytogenetic abnormalities. Many different genetic defectsare thought to underlie the heterogeneity of this disease (2).On the basis of cytogenetic analysis, the classification of Grim-wade and Schlenk (3, 4) defines three prognosis groups (favor-able, intermediate, and high risk). Complex karyotype AML,
belonging to cytogenetic high-risk AML, is characterized by thepresence of at least three unrelated cytogenetic abnormalitiesand accounts for 10% to 20% of AML cases. The percentage in-creases with age and this group is considered to have the worstprognosis, despite intensive therapy (5).Our understanding of the molecular basis of the emergence of
genomic instability in solid tumors has benefited considerablyfrom recent work demonstrating the involvement of a replica-tion stress activating the DNA damage response pathway (6).At an early stage of tumorigenesis, oncogene-induced DNAdamage, associated with replicative and oxidative stresses, is re-sponsible for creating double-strand breaks, thus leading to activa-tion of the DNA damage signaling pathway, with phosphorylationof histone H2AX and ATM and activation of checkpoint kinases(CHK1 and CHK2; refs. 7, 8). Activation of the DNA signalingpathway in precancerous lesions has been suggested to constitutean anticancer barrier (7). A limited number of studies have ad-dressed the question of the presence of DNA double-strand breakin hematologic cancers. Constitutive DNA damage was observedin two myeloid leukemia cell lines (K562 and HL60; ref. 9) and,more recently, an increase in detectable DNA damage was re-ported in a mouse model of progression from myelodysplasiato leukemia (10). However, as concern samples from AML pa-tients, the status of the DNA damage signaling pathway hasnot been documented.The activation of the DNA damage pathway, through check-
point kinase phosphorylation, leads to cell cycle arrest in S-phaseor at the G2-M checkpoint (11, 12). CDC25 phosphatases havebeen shown to be the main effectors of active CHK1 (13), andthe abrogation of the activity of this kinase using small inhibitorycompounds or small interfering rna (siRNA) knockdown strate-gies has been reported to result in checkpoint bypass and in sen-sitization of the cells to DNA-damaging agents (14–17). Forinstance, the use of 7-hydroxystaurosporine (UCN-01), a CHK1 in-hibitor, induces sensitization of AML progenitors to the nucleo-side analogue 1-!-D-arabinofuranosylcytosine (ara-C; ref. 18).Moreover, several phase-1 and -2 trials in solid tumors have beenreported, for UCN-01, either alone (19–21) or in combination withestablished cytotoxic agents (22, 23). These observations have ledto the recent development of checkpoint kinase-inhibitory com-pounds, some of which, including UCN-01, are currently beingevaluated in early clinical trials.In the study reported here, we have investigated the existence
of constitutive DNA damage and activation of the DNA damagepathway in a large panel of AML patient samples. Our results re-veal high levels of DNA damage in complex karyotype AML andshow the high sensitivity of these cells to UCN-01. Together with
Note: Supplementary data for this article are available at Cancer Research Online(http://cancerres.aacrjournals.org/).
Requests for reprints: Bernard Ducommun and Cécile Demur, 118 Route deNarbonne, 31062 Toulouse Cedex, France. Phone: 33-5-61-55-62-10; Fax: 33-5-61-55-81-09; E-mail: [email protected].
©2009 American Association for Cancer Research.doi:10.1158/0008-5472.CAN-09-0939
8652Cancer Res 2009; 69: (22). November 15, 2009 www.aacrjournals.org

the demonstration that CHK1 inhibition has no negative effect onthe growth of normal hematopoietic progenitors, our resultshighlight the interest of this kinase for checkpoint targetingtherapy in hematologic malignancies.
Materials and MethodsCells. Fresh AML samples were obtained, after informed consent, from
patients diagnosed at the Hematology Department of Toulouse UniversityMedical Centre (France). AML cells were isolated from bone marrow byFicoll-Hypaque density-gradient centrifugation and were cryopreserved inIscove's modified Dulbecco medium (IMDM) with DMSO (10% final con-centration) and FCS (50% final concentration). All samples were keptfrozen in liquid nitrogen by the HIMIP collection of the U563 InstitutNational de la Sante et de la Recherche Medicale department (n°DC-2008-307-CPTP1 HIMIP). Frozen cells were thawed in IMDM mediumand immediately processed for clonogenic assays or cultured in IMDM,10% FCS for 72 h for viability testing. For the DNA damaging treatment,cells were cultured in IMDM, 10% BIT (StemCell Technologies) supple-mented with SCF (100 ng/mL), IL3 (5 ng/mL), FLT3 (100 ng/mL) andgranulocyte macrophage colony-stimulating factor (10 ng/mL) for 24 h,then treated with etoposide (VP-16; 17 "mol/L) for 30 min, released indrug-free media, and finally analyzed for their H2AX phosphorylation pro-file. Normal bone marrow CD34+ HPCs were obtained after informed con-sent from discarded fragments from hematologically healthy patients whohad undergone hip surgery. Mononuclear cells from bone marrow wereobtained by Ficoll-Hypaque density-gradient centrifugation, after whichisolation of HPCs was performed by positive selection of CD34-expressingcells using magnetic separation and EasySep procedure (StemCellTechnologies).
Pharmacologic inhibitors and antibodies. UCN-01 was provided bythe National Cancer Institute. Ara-C and other products used werepurchased from Sigma-Aldrich. Antibodies were monoclonal and poly-clonal anti–phospho-S139-H2AX (1:100 and 1:25, respectively; UpstateBiotechnology and Cell Signaling), anti–phospho-S317-CHK1 (1:75; signal-ing), anti-CHK1 (1:75; Santa Cruz Biotechnology), PE/Cy7-conjugated anti-CD34 (1:100; Beckman Coulter), PE-conjugated anti-CD123 (1:50; BD) andAPC-conjugated anti-CD38 (1:10; Biolegend), and Alexa 488–conjugatedanti-rabbit and anti-mouse (1:500; Invitrogen).
Flow cytometry. Immunostaining was performed according to the pre-viously described protocol (24). Samples were analyzed in an FC500 flowcytometer (Beckman Coulter) and subsequently processed using the CXPsoftware. Four-color immunostaining was carried out according to thepreviously described protocol (25). Samples were analyzed with LSRII flowcytometer (BD), followed by data treatment using the BD FACSDiva soft-ware. All flow cytometry results are expressed as the ratio between meanfluorescence intensity of the stained sample and that of its control iso-typic antibody.
Immunofluorescence microscopy. Immunofluorescence studies wereperformed as previously described (26). Cellular DNA was counterstainedwith 4!,6-diamidino-2-phenylindole. Images were acquired using a DM6000microscope (Leica microsystems) fitted with a Roper COOLsnap ES CCDcamera and subsequently processed using the MetaMorph and ImageJsoftwares.
Clonogenic assays. The percentage of leukemic cells present in pa-tients' bone marrow is reported in Table 1. Clonogenic assays on AML cellsand on fresh human bone marrow CD34+ HPC cells were made accordingto a method previously described (27).
siRNA experiments. AML cells were transfected using the Amaxanucleofection technology (Amaxa). Leukemic cells (5 ! 106) were sus-pended in 100 "L of either kit V or human monocyte or human CD34+cell nucleofector solution mixed with 30 pmol control and CHK1 siRNAoligonucleotide sequences. These experiments were performed either withsingle control and CHK1 siRNA (Quiagen and Dharmacon, as described inref. 28) or with a pool of siRNA (control and CHK1) as described (Dhar-macon control and CHK1 SMART pool reagents #001820 and #003255,
respectively; ref. 29). Cells were immediately nucleofected with an AmaxaNucleofector apparatus (program U-15 or Y-001 or U-008). They werethen transferred to culture prewarmed medium and divided into twosamples. One was used immediately for clonogenic assay and the secondwas cultured in IMDM 10% FCS for 24 h to evaluate the CHK1 knock-down efficiency by Western blot.
Viability test. AML cells (1 ! 105/well) were incubated in 100 "L ofIMDM 10% FCS with increasing UCN-01 concentrations for 72 h. To deter-mine the percentage viability, 10 "L of Proliferation Reagent WST-1(Roche) were added and the cells were incubated for 3 h. The absorbanceat 420 nm (Mithras LB 940, Berthold technologies) of samples corrected fora background control allowed us to calculate the percentage viability ofAML samples and to determine the UCN-01 IC50.
Statistics. Results are expressed as means ± SEM. Statistical analyseswere performed by the unpaired Student's t test and Pearson correlationtest with Prism 4 software. Differences were considered as significant forP values of !0.05.
ResultsHigh levels of constitutive DNA damage in complex karyo-
type AML patient samples. We first examined the existence ofdetectable constitutive DNA damage in samples of blasts fromAML patient, using flow cytometry to quantify H2AX phosphory-lation (30). As indicated in Fig. 1A, obtained from two AML blastcell samples (patients #20 and #44, Table 1 and SupplementaryData 1) and one normal hematopoietic progenitor CD34-positivecell (HPC) sample, flow cytometry histograms reveal a variablelevel of phospho-H2AX labeling in malignant samples and con-firm the absence of detectable labeling in the normal HPC sam-ple. A series of 49 patient samples (see Table 1) was thenexamined using the same assay and the phospho-H2AX level ofeach was calculated (see Materials and Methods). As depicted onthe left side of Fig. 1B, normal HPC did not display any detect-able DNA damage and their phospho-H2AX level was about 1(mean, 1.31 ± 0.13; n = 7). In contrast, AML blasts from patientsamples expressed a higher and very variable level of phospho-H2AX (mean, 3.90 ± 0.36; n = 49; **, P = 0.007), indicating avariable level of constitutive DNA damage. On the right side ofFig. 1B, the phospho-H2AX level of the same patients is dividedinto two groups according to their cytogenetic profiles (see Table 1).The high-risk group (mean, 5.07 ± 0.86; n = 16, including 11complex karyotype samples) displayed a significantly higher levelof phospho-H2AX than the intermediate one (mean, 3.08 ± 0.21;n = 31; **, P = 0.007).We next tested the variability of these AML samples with
respect to constitutive phospho-H2AX labeling as a physiologicresponse to DNA injury and examined their capacity to inducean additional level of phospho-H2AX after treatment with a gen-otoxic agent. To this end, we evaluated phospho-H2AX levels intwo AML samples, before and 1 h after treatment with the DNA-damaging agent VP-16. We observed in both samples a rapid in-crease of phospho-H2AX level (data not shown), reaching a peak,1 hour after treatment (Fig. 1C), in the same range as the consti-tutive damage levels observed in some AML samples (Fig. 1B).Thus, AML blasts samples with a certain level of constitutiveDNA damage retained the ability to increase this level inresponse to a DNA-damaging agent.Damage was not induced by freezing and thawing of the patient
material, as we found similar phospho-H2AX levels in threesamples of fresh and thawed AML cells and in one sample ofnormal HPC (Supplementary Data 2).
Checkpoint Targeting in Acute Myeloid Leukemia
8653 Cancer Res 2009; 69: (22). November 15, 2009www.aacrjournals.org
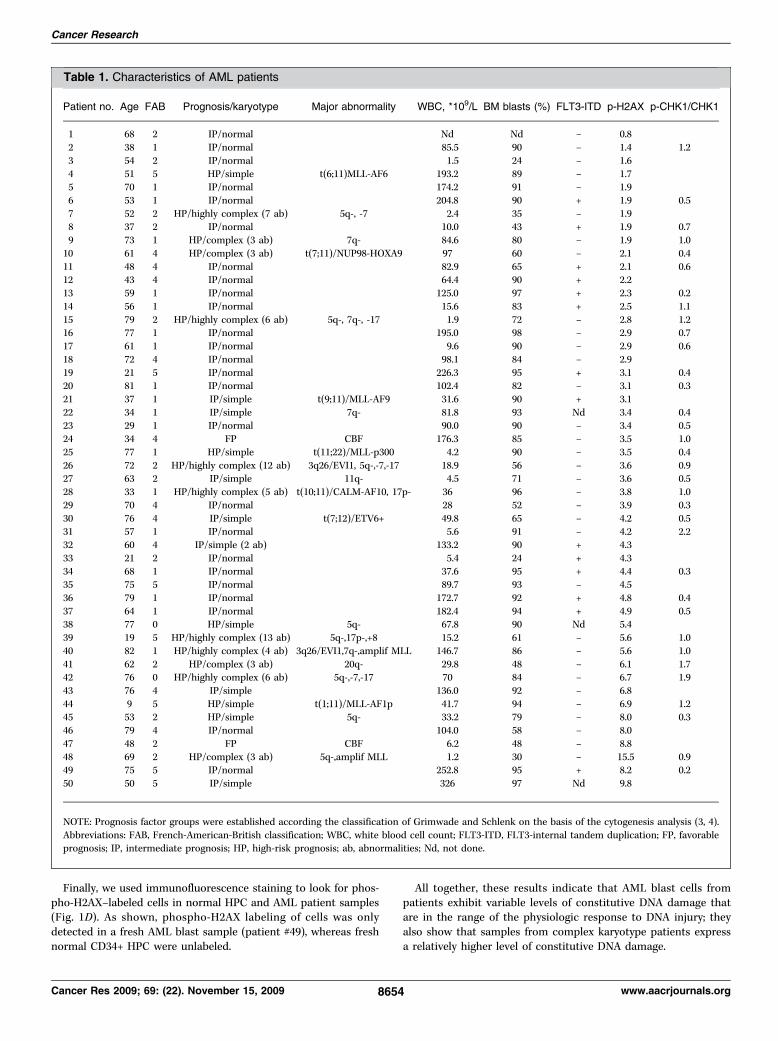
Finally, we used immunofluorescence staining to look for phos-pho-H2AX–labeled cells in normal HPC and AML patient samples(Fig. 1D). As shown, phospho-H2AX labeling of cells was onlydetected in a fresh AML blast sample (patient #49), whereas freshnormal CD34+ HPC were unlabeled.
All together, these results indicate that AML blast cells frompatients exhibit variable levels of constitutive DNA damage thatare in the range of the physiologic response to DNA injury; theyalso show that samples from complex karyotype patients expressa relatively higher level of constitutive DNA damage.
Table 1. Characteristics of AML patients
Patient no. Age FAB Prognosis/karyotype Major abnormality WBC, *109/L BM blasts (%) FLT3-ITD p-H2AX p-CHK1/CHK1
1 68 2 IP/normal Nd Nd " 0.82 38 1 IP/normal 85.5 90 " 1.4 1.23 54 2 IP/normal 1.5 24 " 1.64 51 5 HP/simple t(6;11)MLL-AF6 193.2 89 " 1.75 70 1 IP/normal 174.2 91 " 1.96 53 1 IP/normal 204.8 90 + 1.9 0.57 52 2 HP/highly complex (7 ab) 5q-, -7 2.4 35 " 1.98 37 2 IP/normal 10.0 43 + 1.9 0.79 73 1 HP/complex (3 ab) 7q- 84.6 80 " 1.9 1.010 61 4 HP/complex (3 ab) t(7;11)/NUP98-HOXA9 97 60 " 2.1 0.411 48 4 IP/normal 82.9 65 + 2.1 0.612 43 4 IP/normal 64.4 90 + 2.213 59 1 IP/normal 125.0 97 + 2.3 0.214 56 1 IP/normal 15.6 83 + 2.5 1.115 79 2 HP/highly complex (6 ab) 5q-, 7q-, -17 1.9 72 " 2.8 1.216 77 1 IP/normal 195.0 98 " 2.9 0.717 61 1 IP/normal 9.6 90 " 2.9 0.618 72 4 IP/normal 98.1 84 " 2.919 21 5 IP/normal 226.3 95 + 3.1 0.420 81 1 IP/normal 102.4 82 " 3.1 0.321 37 1 IP/simple t(9;11)/MLL-AF9 31.6 90 + 3.122 34 1 IP/simple 7q- 81.8 93 Nd 3.4 0.423 29 1 IP/normal 90.0 90 " 3.4 0.524 34 4 FP CBF 176.3 85 " 3.5 1.025 77 1 HP/simple t(11;22)/MLL-p300 4.2 90 " 3.5 0.426 72 2 HP/highly complex (12 ab) 3q26/EVI1, 5q-,-7,-17 18.9 56 " 3.6 0.927 63 2 IP/simple 11q- 4.5 71 " 3.6 0.528 33 1 HP/highly complex (5 ab) t(10;11)/CALM-AF10, 17p- 36 96 " 3.8 1.029 70 4 IP/normal 28 52 " 3.9 0.330 76 4 IP/simple t(7;12)/ETV6+ 49.8 65 " 4.2 0.531 57 1 IP/normal 5.6 91 " 4.2 2.232 60 4 IP/simple (2 ab) 133.2 90 + 4.333 21 2 IP/normal 5.4 24 + 4.334 68 1 IP/normal 37.6 95 + 4.4 0.335 75 5 IP/normal 89.7 93 " 4.536 79 1 IP/normal 172.7 92 + 4.8 0.437 64 1 IP/normal 182.4 94 + 4.9 0.538 77 0 HP/simple 5q- 67.8 90 Nd 5.439 19 5 HP/highly complex (13 ab) 5q-,17p-,+8 15.2 61 " 5.6 1.040 82 1 HP/highly complex (4 ab) 3q26/EVI1,7q-,amplif MLL 146.7 86 " 5.6 1.041 62 2 HP/complex (3 ab) 20q- 29.8 48 " 6.1 1.742 76 0 HP/highly complex (6 ab) 5q-,-7,-17 70 84 " 6.7 1.943 76 4 IP/simple 136.0 92 " 6.844 9 5 HP/simple t(1;11)/MLL-AF1p 41.7 94 " 6.9 1.245 53 2 HP/simple 5q- 33.2 79 " 8.0 0.346 79 4 IP/normal 104.0 58 " 8.047 48 2 FP CBF 6.2 48 " 8.848 69 2 HP/complex (3 ab) 5q-,amplif MLL 1.2 30 " 15.5 0.949 75 5 IP/normal 252.8 95 + 8.2 0.250 50 5 IP/simple 326 97 Nd 9.8
NOTE: Prognosis factor groups were established according the classification of Grimwade and Schlenk on the basis of the cytogenesis analysis (3, 4).Abbreviations: FAB, French-American-British classification; WBC, white blood cell count; FLT3-ITD, FLT3-internal tandem duplication; FP, favorableprognosis; IP, intermediate prognosis; HP, high-risk prognosis; ab, abnormalities; Nd, not done.
Cancer Research
8654Cancer Res 2009; 69: (22). November 15, 2009 www.aacrjournals.org
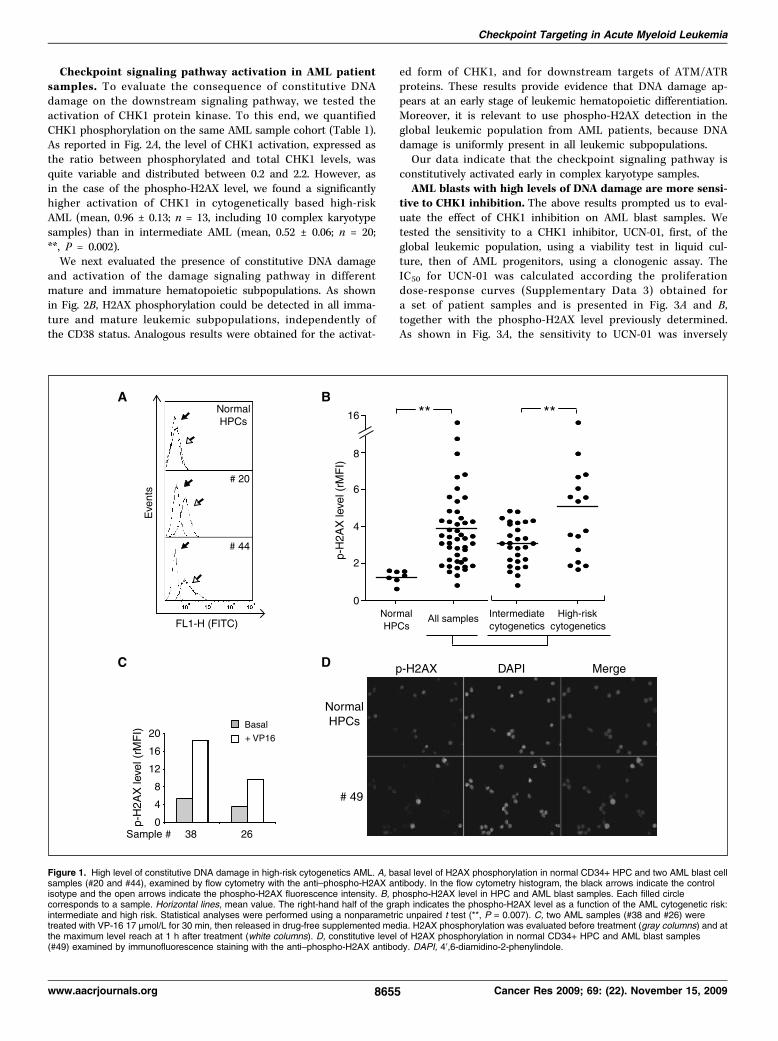
Checkpoint signaling pathway activation in AML patientsamples. To evaluate the consequence of constitutive DNAdamage on the downstream signaling pathway, we tested theactivation of CHK1 protein kinase. To this end, we quantifiedCHK1 phosphorylation on the same AML sample cohort (Table 1).As reported in Fig. 2A, the level of CHK1 activation, expressed asthe ratio between phosphorylated and total CHK1 levels, wasquite variable and distributed between 0.2 and 2.2. However, asin the case of the phospho-H2AX level, we found a significantlyhigher activation of CHK1 in cytogenetically based high-riskAML (mean, 0.96 ± 0.13; n = 13, including 10 complex karyotypesamples) than in intermediate AML (mean, 0.52 ± 0.06; n = 20;**, P = 0.002).We next evaluated the presence of constitutive DNA damage
and activation of the damage signaling pathway in differentmature and immature hematopoietic subpopulations. As shownin Fig. 2B, H2AX phosphorylation could be detected in all imma-ture and mature leukemic subpopulations, independently ofthe CD38 status. Analogous results were obtained for the activat-
ed form of CHK1, and for downstream targets of ATM/ATRproteins. These results provide evidence that DNA damage ap-pears at an early stage of leukemic hematopoietic differentiation.Moreover, it is relevant to use phospho-H2AX detection in theglobal leukemic population from AML patients, because DNAdamage is uniformly present in all leukemic subpopulations.Our data indicate that the checkpoint signaling pathway is
constitutively activated early in complex karyotype samples.AML blasts with high levels of DNA damage are more sensi-
tive to CHK1 inhibition. The above results prompted us to eval-uate the effect of CHK1 inhibition on AML blast samples. Wetested the sensitivity to a CHK1 inhibitor, UCN-01, first, of theglobal leukemic population, using a viability test in liquid cul-ture, then of AML progenitors, using a clonogenic assay. TheIC50 for UCN-01 was calculated according the proliferationdose-response curves (Supplementary Data 3) obtained fora set of patient samples and is presented in Fig. 3A and B,together with the phospho-H2AX level previously determined.As shown in Fig. 3A, the sensitivity to UCN-01 was inversely
Figure 1. High level of constitutive DNA damage in high-risk cytogenetics AML. A, basal level of H2AX phosphorylation in normal CD34+ HPC and two AML blast cellsamples (#20 and #44), examined by flow cytometry with the anti–phospho-H2AX antibody. In the flow cytometry histogram, the black arrows indicate the controlisotype and the open arrows indicate the phospho-H2AX fluorescence intensity. B, phospho-H2AX level in HPC and AML blast samples. Each filled circlecorresponds to a sample. Horizontal lines, mean value. The right-hand half of the graph indicates the phospho-H2AX level as a function of the AML cytogenetic risk:intermediate and high risk. Statistical analyses were performed using a nonparametric unpaired t test (**, P = 0.007). C, two AML samples (#38 and #26) weretreated with VP-16 17 !mol/L for 30 min, then released in drug-free supplemented media. H2AX phosphorylation was evaluated before treatment (gray columns) and atthe maximum level reach at 1 h after treatment (white columns). D, constitutive level of H2AX phosphorylation in normal CD34+ HPC and AML blast samples(#49) examined by immunofluorescence staining with the anti–phospho-H2AX antibody. DAPI, 4′,6-diamidino-2-phenylindole.
Checkpoint Targeting in Acute Myeloid Leukemia
8655 Cancer Res 2009; 69: (22). November 15, 2009www.aacrjournals.org
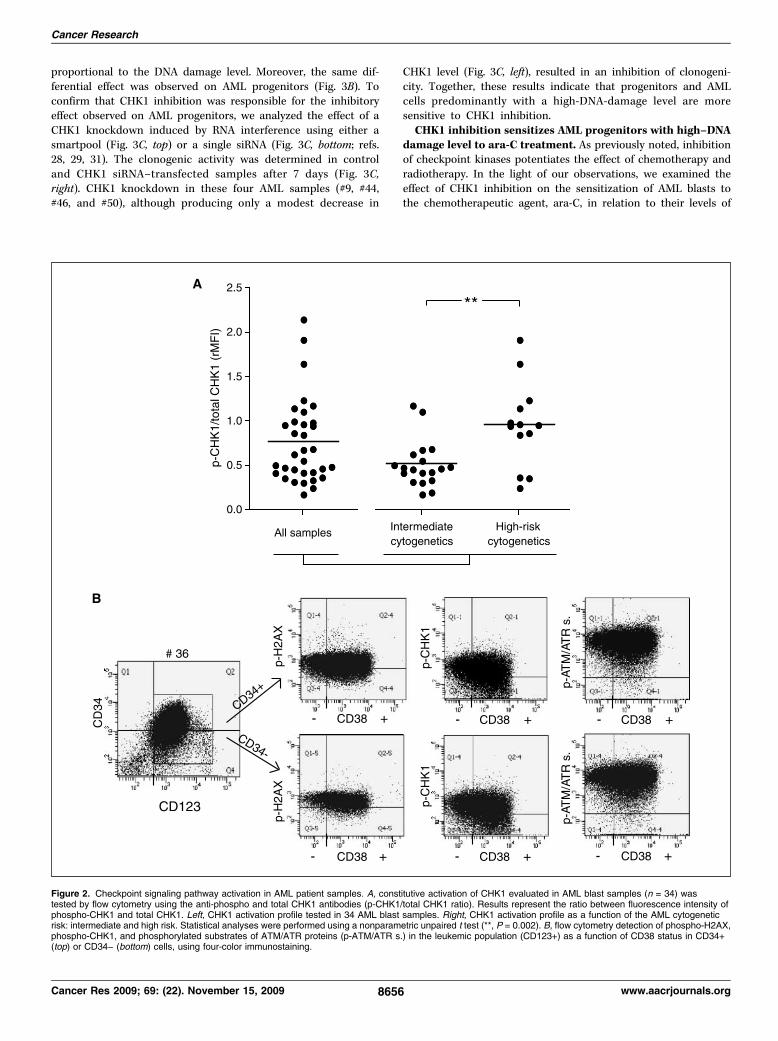
proportional to the DNA damage level. Moreover, the same dif-ferential effect was observed on AML progenitors (Fig. 3B). Toconfirm that CHK1 inhibition was responsible for the inhibitoryeffect observed on AML progenitors, we analyzed the effect of aCHK1 knockdown induced by RNA interference using either asmartpool (Fig. 3C, top) or a single siRNA (Fig. 3C, bottom; refs.28, 29, 31). The clonogenic activity was determined in controland CHK1 siRNA–transfected samples after 7 days (Fig. 3C,right). CHK1 knockdown in these four AML samples (#9, #44,#46, and #50), although producing only a modest decrease in
CHK1 level (Fig. 3C, left), resulted in an inhibition of clonogeni-city. Together, these results indicate that progenitors and AMLcells predominantly with a high-DNA-damage level are moresensitive to CHK1 inhibition.CHK1 inhibition sensitizes AML progenitors with high–DNA
damage level to ara-C treatment. As previously noted, inhibitionof checkpoint kinases potentiates the effect of chemotherapy andradiotherapy. In the light of our observations, we examined theeffect of CHK1 inhibition on the sensitization of AML blasts tothe chemotherapeutic agent, ara-C, in relation to their levels of
Figure 2. Checkpoint signaling pathway activation in AML patient samples. A, constitutive activation of CHK1 evaluated in AML blast samples (n = 34) wastested by flow cytometry using the anti-phospho and total CHK1 antibodies (p-CHK1/total CHK1 ratio). Results represent the ratio between fluorescence intensity ofphospho-CHK1 and total CHK1. Left, CHK1 activation profile tested in 34 AML blast samples. Right, CHK1 activation profile as a function of the AML cytogeneticrisk: intermediate and high risk. Statistical analyses were performed using a nonparametric unpaired t test (**, P = 0.002). B, flow cytometry detection of phospho-H2AX,phospho-CHK1, and phosphorylated substrates of ATM/ATR proteins (p-ATM/ATR s.) in the leukemic population (CD123+) as a function of CD38 status in CD34+(top) or CD34! (bottom) cells, using four-color immunostaining.
Cancer Research
8656Cancer Res 2009; 69: (22). November 15, 2009 www.aacrjournals.org
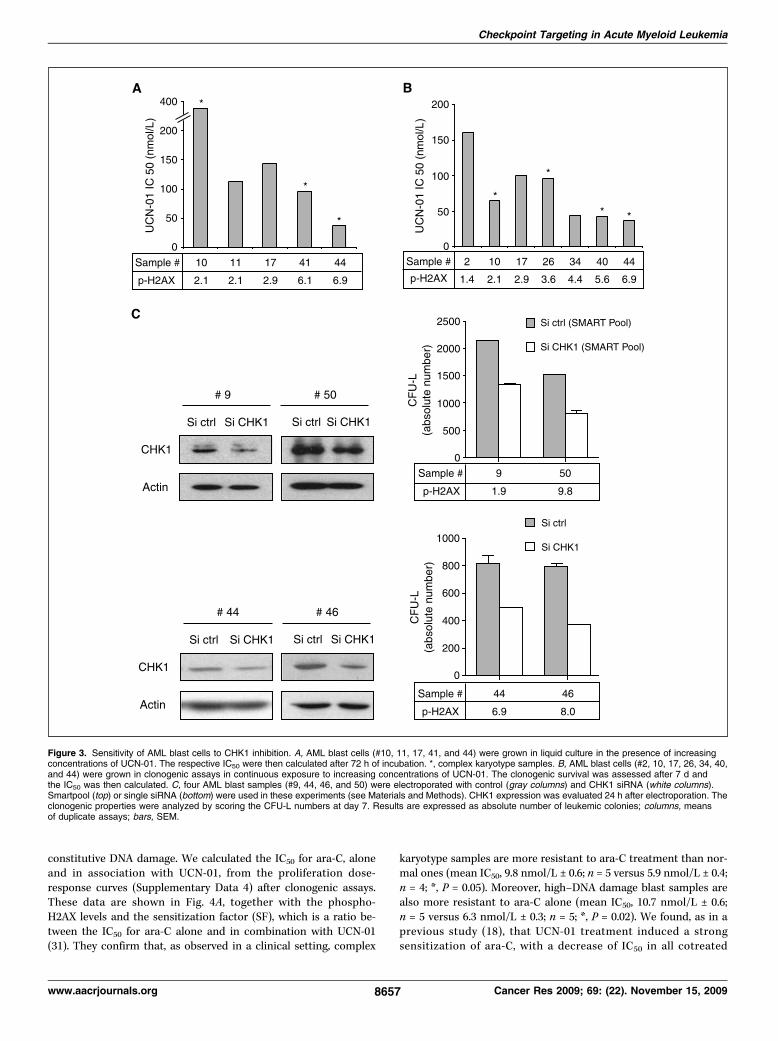
constitutive DNA damage. We calculated the IC50 for ara-C, aloneand in association with UCN-01, from the proliferation dose-response curves (Supplementary Data 4) after clonogenic assays.These data are shown in Fig. 4A, together with the phospho-H2AX levels and the sensitization factor (SF), which is a ratio be-tween the IC50 for ara-C alone and in combination with UCN-01(31). They confirm that, as observed in a clinical setting, complex
karyotype samples are more resistant to ara-C treatment than nor-mal ones (mean IC50, 9.8 nmol/L ± 0.6; n = 5 versus 5.9 nmol/L ± 0.4;n = 4; *, P = 0.05). Moreover, high–DNA damage blast samples arealso more resistant to ara-C alone (mean IC50, 10.7 nmol/L ± 0.6;n = 5 versus 6.3 nmol/L ± 0.3; n = 5; *, P = 0.02). We found, as in aprevious study (18), that UCN-01 treatment induced a strongsensitization of ara-C, with a decrease of IC50 in all cotreated
Figure 3. Sensitivity of AML blast cells to CHK1 inhibition. A, AML blast cells (#10, 11, 17, 41, and 44) were grown in liquid culture in the presence of increasingconcentrations of UCN-01. The respective IC50 were then calculated after 72 h of incubation. *, complex karyotype samples. B, AML blast cells (#2, 10, 17, 26, 34, 40,and 44) were grown in clonogenic assays in continuous exposure to increasing concentrations of UCN-01. The clonogenic survival was assessed after 7 d andthe IC50 was then calculated. C, four AML blast samples (#9, 44, 46, and 50) were electroporated with control (gray columns) and CHK1 siRNA (white columns).Smartpool (top) or single siRNA (bottom) were used in these experiments (see Materials and Methods). CHK1 expression was evaluated 24 h after electroporation. Theclonogenic properties were analyzed by scoring the CFU-L numbers at day 7. Results are expressed as absolute number of leukemic colonies; columns, meansof duplicate assays; bars, SEM.
Checkpoint Targeting in Acute Myeloid Leukemia
8657 Cancer Res 2009; 69: (22). November 15, 2009www.aacrjournals.org
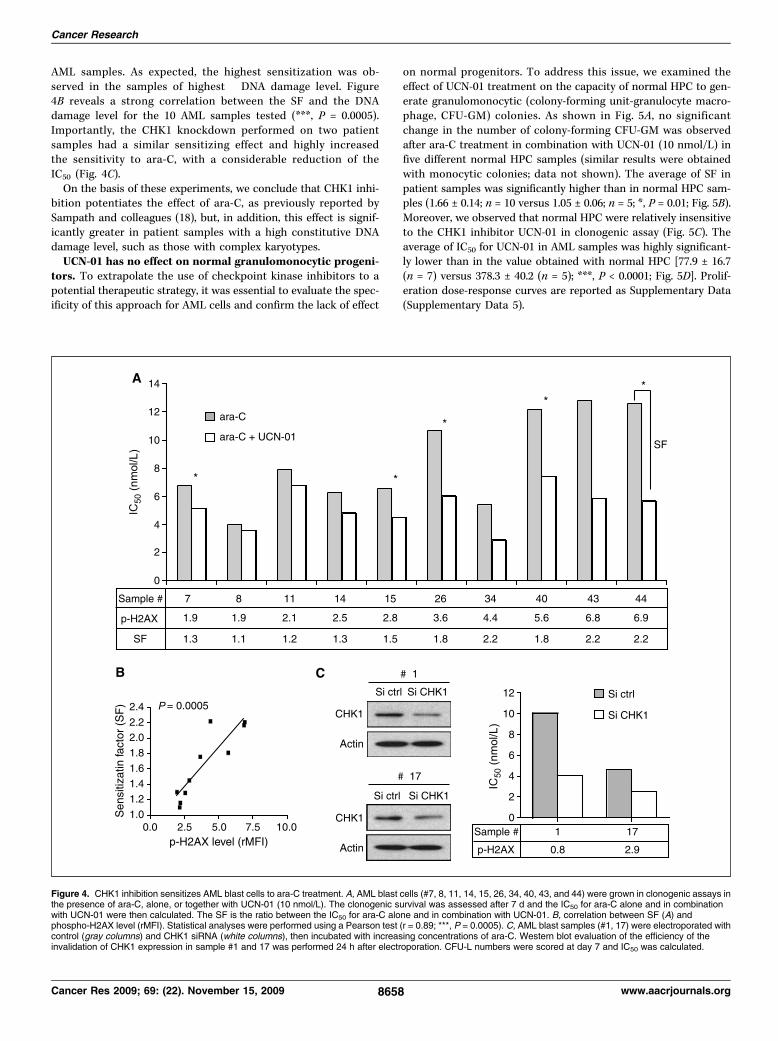
AML samples. As expected, the highest sensitization was ob-served in the samples of highest DNA damage level. Figure4B reveals a strong correlation between the SF and the DNAdamage level for the 10 AML samples tested (***, P = 0.0005).Importantly, the CHK1 knockdown performed on two patientsamples had a similar sensitizing effect and highly increasedthe sensitivity to ara-C, with a considerable reduction of theIC50 (Fig. 4C).On the basis of these experiments, we conclude that CHK1 inhi-
bition potentiates the effect of ara-C, as previously reported bySampath and colleagues (18), but, in addition, this effect is signif-icantly greater in patient samples with a high constitutive DNAdamage level, such as those with complex karyotypes.UCN-01 has no effect on normal granulomonocytic progeni-
tors. To extrapolate the use of checkpoint kinase inhibitors to apotential therapeutic strategy, it was essential to evaluate the spec-ificity of this approach for AML cells and confirm the lack of effect
on normal progenitors. To address this issue, we examined theeffect of UCN-01 treatment on the capacity of normal HPC to gen-erate granulomonocytic (colony-forming unit-granulocyte macro-phage, CFU-GM) colonies. As shown in Fig. 5A, no significantchange in the number of colony-forming CFU-GM was observedafter ara-C treatment in combination with UCN-01 (10 nmol/L) infive different normal HPC samples (similar results were obtainedwith monocytic colonies; data not shown). The average of SF inpatient samples was significantly higher than in normal HPC sam-ples (1.66 ± 0.14; n = 10 versus 1.05 ± 0.06; n = 5; *, P = 0.01; Fig. 5B).Moreover, we observed that normal HPC were relatively insensitiveto the CHK1 inhibitor UCN-01 in clonogenic assay (Fig. 5C). Theaverage of IC50 for UCN-01 in AML samples was highly significant-ly lower than in the value obtained with normal HPC [77.9 ± 16.7(n = 7) versus 378.3 ± 40.2 (n = 5); ***, P < 0.0001; Fig. 5D]. Prolif-eration dose-response curves are reported as Supplementary Data(Supplementary Data 5).
Figure 4. CHK1 inhibition sensitizes AML blast cells to ara-C treatment. A, AML blast cells (#7, 8, 11, 14, 15, 26, 34, 40, 43, and 44) were grown in clonogenic assays inthe presence of ara-C, alone, or together with UCN-01 (10 nmol/L). The clonogenic survival was assessed after 7 d and the IC50 for ara-C alone and in combinationwith UCN-01 were then calculated. The SF is the ratio between the IC50 for ara-C alone and in combination with UCN-01. B, correlation between SF (A) andphospho-H2AX level (rMFI). Statistical analyses were performed using a Pearson test (r = 0.89; ***, P = 0.0005). C, AML blast samples (#1, 17) were electroporated withcontrol (gray columns) and CHK1 siRNA (white columns), then incubated with increasing concentrations of ara-C. Western blot evaluation of the efficiency of theinvalidation of CHK1 expression in sample #1 and 17 was performed 24 h after electroporation. CFU-L numbers were scored at day 7 and IC50 was calculated.
Cancer Research
8658Cancer Res 2009; 69: (22). November 15, 2009 www.aacrjournals.org
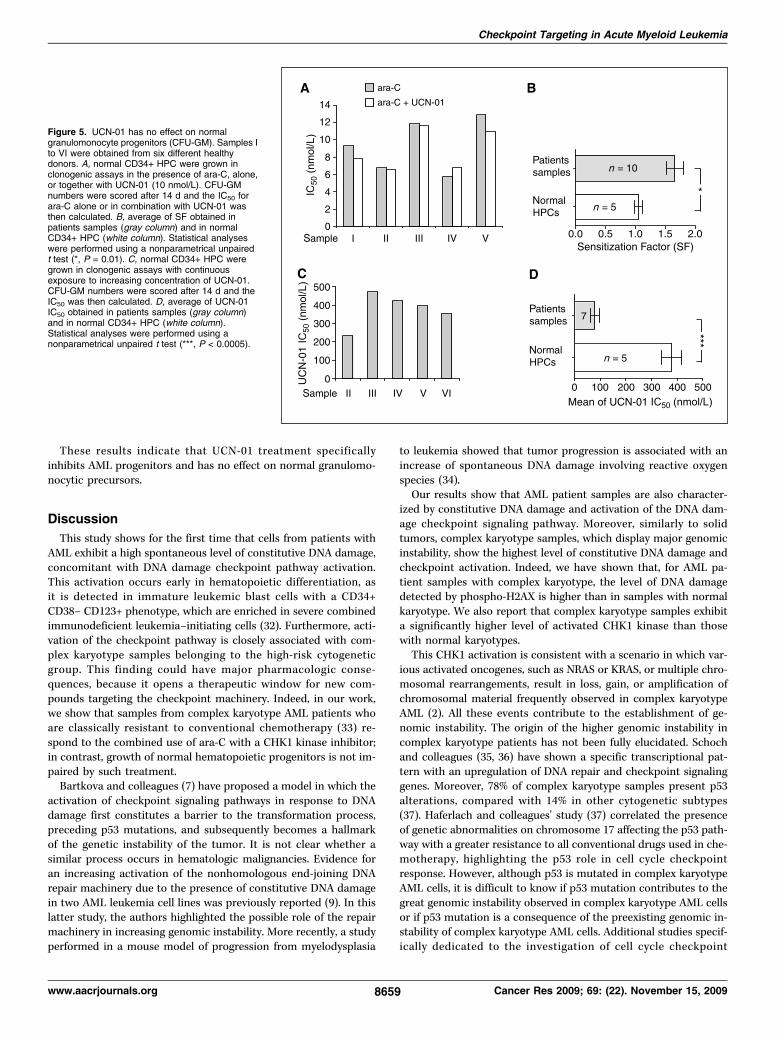
These results indicate that UCN-01 treatment specificallyinhibits AML progenitors and has no effect on normal granulomo-nocytic precursors.
DiscussionThis study shows for the first time that cells from patients with
AML exhibit a high spontaneous level of constitutive DNA damage,concomitant with DNA damage checkpoint pathway activation.This activation occurs early in hematopoietic differentiation, asit is detected in immature leukemic blast cells with a CD34+CD38" CD123+ phenotype, which are enriched in severe combinedimmunodeficient leukemia–initiating cells (32). Furthermore, acti-vation of the checkpoint pathway is closely associated with com-plex karyotype samples belonging to the high-risk cytogeneticgroup. This finding could have major pharmacologic conse-quences, because it opens a therapeutic window for new com-pounds targeting the checkpoint machinery. Indeed, in our work,we show that samples from complex karyotype AML patients whoare classically resistant to conventional chemotherapy (33) re-spond to the combined use of ara-C with a CHK1 kinase inhibitor;in contrast, growth of normal hematopoietic progenitors is not im-paired by such treatment.Bartkova and colleagues (7) have proposed a model in which the
activation of checkpoint signaling pathways in response to DNAdamage first constitutes a barrier to the transformation process,preceding p53 mutations, and subsequently becomes a hallmarkof the genetic instability of the tumor. It is not clear whether asimilar process occurs in hematologic malignancies. Evidence foran increasing activation of the nonhomologous end-joining DNArepair machinery due to the presence of constitutive DNA damagein two AML leukemia cell lines was previously reported (9). In thislatter study, the authors highlighted the possible role of the repairmachinery in increasing genomic instability. More recently, a studyperformed in a mouse model of progression from myelodysplasia
to leukemia showed that tumor progression is associated with anincrease of spontaneous DNA damage involving reactive oxygenspecies (34).Our results show that AML patient samples are also character-
ized by constitutive DNA damage and activation of the DNA dam-age checkpoint signaling pathway. Moreover, similarly to solidtumors, complex karyotype samples, which display major genomicinstability, show the highest level of constitutive DNA damage andcheckpoint activation. Indeed, we have shown that, for AML pa-tient samples with complex karyotype, the level of DNA damagedetected by phospho-H2AX is higher than in samples with normalkaryotype. We also report that complex karyotype samples exhibita significantly higher level of activated CHK1 kinase than thosewith normal karyotypes.This CHK1 activation is consistent with a scenario in which var-
ious activated oncogenes, such as NRAS or KRAS, or multiple chro-mosomal rearrangements, result in loss, gain, or amplification ofchromosomal material frequently observed in complex karyotypeAML (2). All these events contribute to the establishment of ge-nomic instability. The origin of the higher genomic instability incomplex karyotype patients has not been fully elucidated. Schochand colleagues (35, 36) have shown a specific transcriptional pat-tern with an upregulation of DNA repair and checkpoint signalinggenes. Moreover, 78% of complex karyotype samples present p53alterations, compared with 14% in other cytogenetic subtypes(37). Haferlach and colleagues' study (37) correlated the presenceof genetic abnormalities on chromosome 17 affecting the p53 path-way with a greater resistance to all conventional drugs used in che-motherapy, highlighting the p53 role in cell cycle checkpointresponse. However, although p53 is mutated in complex karyotypeAML cells, it is difficult to know if p53 mutation contributes to thegreat genomic instability observed in complex karyotype AML cellsor if p53 mutation is a consequence of the preexisting genomic in-stability of complex karyotype AML cells. Additional studies specif-ically dedicated to the investigation of cell cycle checkpoint
Figure 5. UCN-01 has no effect on normalgranulomonocyte progenitors (CFU-GM). Samples Ito VI were obtained from six different healthydonors. A, normal CD34+ HPC were grown inclonogenic assays in the presence of ara-C, alone,or together with UCN-01 (10 nmol/L). CFU-GMnumbers were scored after 14 d and the IC50 forara-C alone or in combination with UCN-01 wasthen calculated. B, average of SF obtained inpatients samples (gray column) and in normalCD34+ HPC (white column). Statistical analyseswere performed using a nonparametrical unpairedt test (*, P = 0.01). C, normal CD34+ HPC weregrown in clonogenic assays with continuousexposure to increasing concentration of UCN-01.CFU-GM numbers were scored after 14 d and theIC50 was then calculated. D, average of UCN-01IC50 obtained in patients samples (gray column)and in normal CD34+ HPC (white column).Statistical analyses were performed using anonparametrical unpaired t test (***, P < 0.0005).
Checkpoint Targeting in Acute Myeloid Leukemia
8659 Cancer Res 2009; 69: (22). November 15, 2009www.aacrjournals.org

response and AML p53 mutation will be necessary to evaluateprecisely its impact.In Bartkova's model, activated oncogenes, associated with repli-
cative and oxidative stresses, are responsible for DNA injury, lead-ing to the activation of the DNA damage signaling pathway (7).Considering the high genetic instability of complex karyotype sam-ples, it is more than likely that similar mechanisms operate inAML. Among various mechanisms of resistance to chemotherapy,it has been shown that complex karyotype AML displays a com-bined activity both of multidrug resistance proteins P-gp andMDR-related protein 1, which contributes to the chemoresistance(38). In this study, we confirm that complex karyotype AMLsamples display the highest resistance to chemotherapy, even thatinvolving the widely used antimetabolite, ara-C, which is not aP-gp substrate. Moreover, this poor sensitivity is also likely to becorrelated with their lower proliferative activity in comparisonwith the favorable and intermediate prognosis group (39). Theweak proliferative activity explains the low incorporation ofara-C into DNA and, consequently, contributes to the poor effectof this treatment, which precludes complete clinical remission. Inview of the global resistance mechanisms to chemotherapy used bycomplex karyotype cells, the use of checkpoint inhibitors mayrepresent an attractive pharmacologic strategy to increase theefficacy of ara-C treatment in the group of complex karyotypeAML patients. Our findings strongly support this idea. First,we show that complex karyotype samples are particularly sensitiveto CHK1 inhibition by UCN-01 and by RNA interference. Moreimportantly, we have shown that low concentrations of UCN-01induce a sensitization to ara-C treatment that is particularlymarked in the high–DNA damage sample. Similar results wereobtained when UCN-01 and VP-16 were used together (data not
shown). Moreover, Sampath and colleagues (18) have shown thatUCN-01, in combination with ara-C, induces a rapid decrease ofthe constitutive activation CHK1 in 8 AML samples.Above all, we have clearly shown that the same concentration
UCN-01 has no effect on the growth and clonogenic capacity ofnormal HPCs, thus indicating that such an association betweena checkpoint targeting agent and an antimetabolite has a specificeffect on abnormal hematopoiesis.Finally, our study shows that evaluation of the DNA damage
level by flow cytometry detection of histone H2AX phosphorylationis a very easy and direct assay to routinely select the patients forwhom such a combinative treatment may be beneficial. The use ofthis simple technology could be of real interest to identify AMLpatients with complex karyotype AML and high DNA damageand to constitute the large cohort of such patients that are nowrequired to evaluate in detail the therapeutic potential of check-point inhibitors.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
AcknowledgmentsReceived 3/11/09; revised 7/27/09; accepted 8/24/09; published OnlineFirst 10/20/09.
Grant support: C.N.R.S, l'Université Paul Sabatier, l'Institut National du Cancer(Programme libre 2005 et 2008), la région Midi-Pyrénées, and la Ligue NationaleContre le Cancer (Equipe labellisée 2008; B. Ducommun).
The costs of publication of this article were defrayed in part by the paymentof page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank J. Smith for corrections and comments on the manuscript, N.Dastugue for karyotype analyses of each patient, and P. Bourin for the generousgift of hematopoietic cells.
References1. Tallman MS, Gilliland DG, Rowe JM. Drug therapy foracute myeloid leukemia. Blood 2005;106:1154–63.
2. Kelly LM, Gilliland DG. Genetics of myeloid leukemias.Annu Rev Genomics Hum Genet 2002;3:179–98.
3. Grimwade D, Walker H, Oliver F, et al. The importanceof diagnostic cytogenetics on outcome in AML: analysisof 1,612 patients entered into the MRC AML 10 trial.The Medical Research Council Adult and Children'sLeukaemia Working Parties. Blood 1998;92:2322–33.
4. Schlenk RF, Benner A, Hartmann F, et al. Risk-adaptedpostremission therapy in acute myeloid leukemia: re-sults of the German multicenter AML HD93 treatmenttrial. Leukemia 2003;17:1521–8.
5. Schoch C, Haferlach T, Haase D, et al. Patients withde novo acute myeloid leukaemia and complex karyo-type aberrations show a poor prognosis despite inten-sive treatment: a study of 90 patients. Br J Haematol2001;112:118–26.
6. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development.Science 2008;319:1352–5.
7. Bartkova J, Horejsi Z, Koed K, et al. DNA damage re-sponse as a candidate anti-cancer barrier in early hu-man tumorigenesis. Nature 2005;434:864–70.
8. Tanaka T, Halicka HD, Huang X, Traganos F, Darzyn-kiewicz Z. Constitutive histone H2AX phosphorylationand ATM activation, the reporters of DNA damage byendogenous oxidants. Cell Cycle 2006;5:1940–5.
9. Brady N, Gaymes TJ, Cheung M, Mufti GJ, Rassool FV.Increased error-prone NHEJ activity in myeloid leuke-mias is associated with DNA damage at sites that re-cruit key nonhomologous end-joining proteins. CancerRes 2003;63:1798–805.
10. Rassool FV, Gaymes TJ, Omidvar N, et al. Reactiveoxygen species, DNA damage, and error-prone repair:a model for genomic instability with progression inmyeloid leukemia? Cancer Res 2007;67:8762–71.
11. Bartek J, Lukas J. Chk1 and Chk2 kinases in check-point control and cancer. Cancer Cell 2003;3:421–9.
12. Shiloh Y. ATM: ready, set, go. Cell Cycle 2003;2:116–7.13. Boutros R, Dozier C, Ducommun B. The when andwheres of CDC25 phosphatases. Curr Opin Cell Biol2006;18:185–91.
14. Sampath D, Shi Z, Plunkett W. Inhibition of cyclin-dependent kinase 2 by the Chk1–25A pathway duringthe S-phase checkpoint activated by fludarabine: dysre-gulation by 7-hydroxystaurosporine. Mol Pharmacol2002;62:680–8.
15. Shao RG, Cao CX, Pommier Y. Abrogation of Chk1-mediated S/G2 checkpoint by UCN-01 enhances ara-C-induced cytotoxicity in human colon cancer cells. ActaPharmacol Sin 2004;25:756–62.
16. Shi Z, Azuma A, Sampath D, Li YX, Huang P, PlunkettW. S-Phase arrest by nucleoside analogues and abroga-tion of survival without cell cycle progression by 7-hy-droxystaurosporine. Cancer Res 2001;61:1065–72.
17. Wang S, Wang Z, Grant S. Bryostatin 1 and UCN-01potentiate 1-!-D-arabinofuranosylcytosine-induced ap-optosis in human myeloid leukemia cells through dispa-rate mechanisms. Mol Pharmacol 2003;63:232–42.
18. Sampath D, Cortes J, Estrov Z, et al. Pharmacody-namics of cytarabine alone and in combination with7-hydroxystaurosporine (UCN-01) in AML blasts in vitroand during a clinical trial. Blood 2006;107:2517–24.
19. Sausville EA, Arbuck SG, Messmann R, et al. PhaseI trial of 72-hour continuous infusion UCN-01 in pa-tients with refractory neoplasms. J Clin Oncol 2001;19:2319–33.
20. Fuse E, Kuwabara T, Sparreboom A, Sausville EA,Figg WD. Review of UCN-01 development: a lesson inthe importance of clinical pharmacology. J Clin Pharma-col 2005;45:394–403.
21. Dees EC, Baker SD, O'Reilly S, et al. A phase I andpharmacokinetic study of short infusions of UCN-01in patients with refractory solid tumors. Clin CancerRes 2005;11:664–71.
22. Kortmansky J, Shah MA, Kaubisch A, et al. Phase Itrial of the cyclin-dependent kinase inhibitor and pro-tein kinase C inhibitor 7-hydroxystaurosporine in com-bination with Fluorouracil in patients with advancedsolid tumors. J Clin Oncol 2005;23:1875–84.
23. Lara PN, Jr., Mack PC, Synold T, et al. The cyclin-dependent kinase inhibitor UCN-01 plus cisplatin inadvanced solid tumors: a California cancer consortiumphase I pharmacokinetic and molecular correlative trial.Clin Cancer Res 2005;11:4444–50.
24. Gallay N, Dos Santos C, Cuzin L, et al. The level ofAKT phosphorylation on threonine 308 but not on ser-ine 473 is associated with high-risk cytogenetics andpredicts poor overall survival in acute myeloid leukae-mia. Leukemia 2009;23:1029–38.
25. Bardet V, Tamburini J, Ifrah N, et al. Single cell anal-ysis of phosphoinositide 3-kinase/Akt and ERK activa-tion in acute myeloid leukemia by flow cytometry.Haematologica 2006;91:757–64.
26. Boutros R, Lobjois V, Ducommun B. CDC25B in-volvement in the centrosome duplication cycle and inmicrotubule nucleation. Cancer Res 2007;67:11557–64.
27. Recher C, Beyne-Rauzy O, Demur C, et al. Antileuke-mic activity of rapamycin in acute myeloid leukemia.Blood 2005;105:2527–34.
28. Schmitt E, Boutros R, Froment C, Monsarrat B,Ducommun B, Dozier C. CHK1 phosphorylates CDC25B
Cancer Research
8660Cancer Res 2009; 69: (22). November 15, 2009 www.aacrjournals.org

during the cell cycle in the absence of DNA damage. JCell Sci 2006;119:4269–75.
29. Syljuasen RG, Sorensen CS, Hansen LT, et al. Inhibi-tion of human Chk1 causes increased initiation of DNAreplication, phosphorylation of ATR targets, and DNAbreakage. Mol Cell Biol 2005;25:3553–62.
30. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, BonnerWM. DNA double-stranded breaks induce histone H2AXphosphorylation on serine 139. J Biol Chem 1998;273:5858–68.
31. Didier C, Cavelier C, Quaranta M, et al. G2/M check-point stringency is a key parameter in the sensitivity ofAML cells to genotoxic stress. Oncogene 2008;27:3811–20.
32. Jordan CT, Guzman ML. Mechanisms controllingpathogenesis and survival of leukemic stem cells. Onco-gene 2004;23:7178–87.
33. Chen CC, Yang CF, Lee KD, et al. Complex karyotypesconfer a poor survival in adult acute myeloid leukemiawith unfavorable cytogenetic abnormalities. CancerGenet Cytogenet 2007;174:138–46.
34. Rassool FV. DNA double strand breaks (DSB) andnon-homologous end joining (NHEJ) pathways in hu-man leukemia. Cancer Lett 2003;193:1–9.
35. Lindvall C, Furge K, Bjorkholm M, et al. Combinedgenetic and transcriptional profiling of acute myeloidleukemia with normal and complex karyotypes. Haema-tologica 2004;89:1072–81.
36. Schoch C, Kohlmann A, Dugas M, et al. Genomicgains and losses influence expression levels of genes lo-cated within the affected regions: a study on acute my-eloid leukemias with trisomy 8, 11, or 13, monosomy 7,or deletion 5q. Leukemia 2005;19:1224–8.
37. Haferlach C, Dicker F, Herholz H, Schnittger S,Kern W, Haferlach T. Mutations of the TP53 genein acute myeloid leukemia are strongly associatedwith a complex aberrant karyotype. Leukemia 2008;22:1539–41.
38. Legrand O, Simonin G, Beauchamp-Nicoud A,Zittoun R, Marie JP. Simultaneous activity of MRP1and Pgp is correlated with in vitro resistance to dauno-rubicin and with in vivo resistance in adult acute mye-loid leukemia. Blood 1999;94:1046–56.
39. Braess J, Jahns-Streubel G, Schoch C, et al. Pro-liferative activity of leukaemic blasts and cytosinearabinoside pharmacodynamics are associated withcytogenetically defined prognostic subgroups inacute myeloid leukaemia. Br J Haematol 2001;113:975–82.
Checkpoint Targeting in Acute Myeloid Leukemia
8661 Cancer Res 2009; 69: (22). November 15, 2009www.aacrjournals.org

109
C. Discussion
Lorsque nous avons initié ce travail, notre idée de départ était d’évaluer le statut
d’activation de CHK1 dans les LAM en la corrélant avec le degré de maturation des cellules
(CD34+ vs CD34-), en raison des résultats de notre précédente étude. De fait, nous avons
trouvé des activations variables de la protéine allant dans le sens d’une activation
spontanée majoritaire dans les LAM CD34+. Conceptuellement, ce résultat signifierait que
les cellules immatures verrouilleraient davantage leur point de contrôle G2/M en l’absence
d’agents génotoxiques, ce qui en présence de dommages leur permettraient d’arrêter
probablement plus facilement et plus rapidement le cycle cellulaire, contribuant ainsi à
leur chimiorésistance naturelle. Toutefois, ce résultat doit être nuancé car je n’ai pas
comparé directement la quantité de dommages spontanés entre les cellules CD34+ et
CD34-. Cependant le marqueur CD34 n’étant pas un marqueur absolu de la différenciation,
en effet son niveau d’expression au cours de la différenciation peut varier selon les
lignages hématopoïétiques. Nous avons donc recherché à établir une corrélation avec ce
qui est considéré à l’heure actuelle comme les marqueurs pronostiques les plus solides
dans les LAM, c’est à dire les caractéristiques cytogénétiques. Et nous avons donc montré
que l’activation de la protéine CHK1 est supérieure dans les LAM de mauvais pronostic
(selon la classification cytogénétique) en particulier les LAM à caryotype complexe.
Dans les tumeurs solides, la présence de dommages spontanés semble résulter
essentiellement d’un stress réplicatif lié à une activité oncogénique en amont (Bartkova et
al., 2005), écartant ainsi la contribution d’autres stress et notamment le stress oxydatif.
Cette notion est toutefois discutée puisque dans certaines publications l’utilisation d’anti-
oxydant permet de réduire la quantité de dommages spontanés sur l’ADN (Tanaka et al.,
2006). Compte tenu de la quantité massive de radicaux libres oxygénés évalués grâce à une
sonde fluorescente dans nos cellules de LAM, nous avons évalué la contribution du stress
oxydatif dans la génération de dommages spontanés. Parmi les nombreux anti-oxydants
que nous avons testés (N-acéthyl cystéine, NDGA, trolox, GSHE) seul un analogue non-
métabolisable du glucose, le 2-DG, nous a permis de diminuer significativement la quantité
de radicaux libres oxygénés et ainsi les dommages spontanés dans les cellules (résultats
non montrés). Cependant, comme on ne pouvait pas exclure que nos conditions de culture,
de décongélation… induisaient elles-mêmes un stress oxydatif, nous avons testé l’impact
de plusieurs conditions (échantillons frais, décongelés, milieux de culture…) sans obtenir
de variation significative de la quantité de dommages (résultats supplémentaires de notre
publication). L’ensemble de ces résultats signifie que le métabolisme de base des cellules

110
de LAM génère un stress oxydatif qui contribue probablement à la génération des cassures
de l’ADN.
D’autre part, le modèle de tumorigenèse met en évidence une perte de la signalisation des
dommages lorsque la tumeur est installée, par conséquent en présence d’une forte
instabilité génétique, avec majoritairement la perte d’une copie du gène p53 ou sa
mutation, et dans quelques cas la perte ou la mutation des gènes atm ou chk2. Sur ce
point, les LAM sont très différentes des tumeurs solides, puisque la perte du gène p53 est
minoritaire avec seulement 14% des cas de LAM (Haferlach et al., 2008). Ce n’est que dans
les LAM de caryotype complexe fortement instables que l’on trouve une perte fréquente du
gène p53 (78%). Cependant, la perte d’activité de p53 ne dépend pas seulement de la
perte du gène. En effet, de nombreuses mutations, insertions/délétions du gène sont
décrites dont la plupart ont pour résultat une diminution de l’activité de la protéine. Afin
de définir de façon plus complète le rôle de p53 dans l’activation de la voie de
signalisation des dommages dans les hémopathies, nous avons identifié les différentes
mutations du gène dans nos 50 échantillons de LAM afin d’établir d’éventuelles
corrélations avec notre profil de dommages spontanés. De plus, nous avons élargi notre
recherche aux différentes mutations récurrentes qui ont un impact sur le pronostic des
LAM. Cette question sera développée dans la discussion générale de ce travail.
Pour finir, notre étude a permis de mettre en évidence une corrélation entre l’activation
de la protéine CHK1 et la sensibilisation par l’UCN-01. En effet dans notre travail, plus les
cellules présentent des dommages spontanés plus elles activent les acteurs de la voie des
dommages, établissant ainsi de nombreuses autres dérégulations. Dans ces cas,
l’inactivation de la voie est davantage délétère conduisant à une sensibilisation accrue à
l’agent chimiothérapeutique ara-C. Bien que sa spécificité sur CHK1 ne soit absolu,
l’intérêt thérapeutique de l’UCN-01 est toujours d’actualité. En effet, il est évalué en
essai clinique de phase I et II dans les tumeurs du sein, les leucémies aiguës, les SMD et les
lymphomes. D’autres composés plus spécifiques de CHK1 ont été développés notamment
l’AZD7762 par le groupe Astrazeneca. Une collaboration toute récente entre C. Didier et le
groupe nous a permis d’obtenir cet inhibiteur pour lequel nous sommes en train de tester
l’efficacité sur les cellules de LAM.
L’ensemble de ces résultats ouvre des perspectives thérapeutiques intéressantes visant à
utiliser des inhibiteurs de la voie des dommages et en particulier de la protéine CHK1.

111
DISCUSSION DISCUSSION
GENERALE ET GENERALE ET
PERSPECTIVESPERSPECTIVES

112
Ces deux études ont permis de mettre en évidence que les acteurs du cycle cellulaire et en
particulier la kinase CHK1 jouent des rôles clés en contribuant à la chimiorésistance dans
des lignées immatures de LAM KG1a mais également en participant à la forte instabilité
génétique observée dans les LAM à caryotypes complexes.
L’étude que nous avons réalisée sur l’efficacité du point de contrôle G2/M et qui a fait
l’objet d'une publication dans le journal Oncogene est à ce titre très informative. En effet,
elle permet d’impliquer pour la première fois les acteurs du cycle cellulaire dans le
maintien prolongé en phase G2 du cycle cellulaire après dommages, mettant en évidence
le rôle majeur de la kinase CHK1 qui contribue très probablement ainsi à la
chimiorésistance des cellules immatures. Cette étude a permis d’apporter de nouveaux
éléments de réflexion sur l’implication du cycle cellulaire dans les LAM et ainsi de proposer
de nouvelles pistes thérapeutiques consistant à combiner un inhibiteur de la kinase CHK1
avec un agent inducteur de dommages (ici l’étoposide). Toutefois, l’une des principales
difficultés à laquelle nous sommes confrontés dans le domaine des leucémies aiguës
myéloïdes est la très grande hétérogénéité de la maladie. Ainsi, les études réalisées sur
des lignées cellulaires bien qu’étant très représentatives des LAM (en particulier pour les
KG1a et KG1) ne rendent probablement compte que d’un faible échantillon peu
représentatif de la diversité de la pathologie. De plus, et c'est une des limites principales
de l'utilisation de lignées cellulaires modèles, il est très délicat d'extrapoler des
observations réalisées sur des lignées qui prolifèrent activement dans nos milieux de
culture cellulaire et des cellules qui cyclent très peu dans la moelle osseuse des patients.
Les analyses de profil de cycle cellulaire des blastes fraîchement extraits de la moelle
osseuse des patients, m'ont permis d'observer que le pourcentage de cellules en phase
G0/G1 du cycle varie entre 80 et 96%, contre environ 70% dans les KG1a en culture ; et de
1 à 10% en phase G2/M pour les blastes contre 20% pour des KG1a, la phase S étant
quasiment inexistante dans les blastes. Ainsi, la répartition et la cinétique du cycle est
radicalement différente, limitant ainsi les corrélations possibles entre les résultats obtenus
dans les lignées et les cellules de patients. Dans la même perspective d'une meilleure
connaissance de l'état prolifératif des blastes de patients, nous avons mené en
collaboration avec le Pr. C. Récher un travail qui a consisté à établir les profils de
répartition dans le cycle cellulaire des blastes au diagnostic, puis 12h et 36h après le début
de l’administration de leur chimiothérapie. Ces profils réalisés sur 6 patients ont révélé
une entrée progressive en phase sub-G1 (apoptose) sans que l’on puisse réellement voir de
modifications témoignant d'accumulation des cellules en phase G1 ou G2/M du cycle
cellulaire (résultats non montrés).

113
L’ensemble de ces résultats nous a poussé à mener un travail exclusivement réalisé sur des
cellules de patients afin de mieux rendre compte de l’hétérogénéité des LAM, de mettre
en évidence des mécanismes réellement impliqués dans les cellules de malades, et de
tester plus largement la pertinence thérapeutique d’un inhibiteur de la kinase CHK1
combiné à l’ara-C utilisée lors de la chimiothérapie des LAM.
L’étude que nous avons réalisée sur l’activation constitutive de CHK1 dans les LAM et qui a
fait l’objet d'une publication dans le journal Cancer Research soulève différents points de
discussion et des perspectives biologiques et thérapeutiques qui nous semblent
intéressantes. En effet, elle nous a permis de mettre en évidence pour la première fois
dans des échantillons de LAM issus de patients la présence de dommages constitutifs
(détectés par le marqueur γH2AX) responsables d’une activation basale de la protéine
CHK1 majoritairement retrouvées dans les LAM porteuses d'un caryotype complexe. Comme
cela a été rappelé dans l'introduction, la présence de dommages constitutifs dans les
cellules de LAM n’a jamais réellement été abordée dans la littérature. En effet, l’étude
publiée par Horibe en 2007 fait le constat de dommages constitutifs à l'ADN dans les LAM
secondaires aux syndromes myélodysplasiques (n = 10), faisant plutôt le lien avec les
données sur la progression du processus tumoral dans les tumeurs solides ((Horibe et al.,
2007) ; (Gorgoulis et al., 2005) ; (Bartkova et al., 2005)). L’étude publiée par Boherer en
2009 avait également pour but d’évaluer la présence de dommages à l'ADN constitutifs et
l’activation de la voie des dommages au cours de la progression du syndrome
myélodysplasique vers la leucémie (Boehrer et al., 2009). Les auteurs rapportent la
présence de dommages spontanés dans les cellules de LAM mais l’effectif considéré est
très limité (n = 3). Ainsi, à ce jour aucune étude menée sur un large échantillonnage
n’avait encore mis en évidence de dommages spontanés sur l’ADN des cellules de LAM ni
fait de corrélation entre la présence de dommages constitutifs et les caractéristiques
cytogénétiques des LAM. Les données rapportées sur les tumeurs solides ont
vraisemblablement influencé et précipité les recherches sur la progression tumorale dans
le domaine de l’hématologie.
De plus, nous avons montré que la quantité de dommages constitutifs présents est corrélée
de manière significative avec la chimiosensibilisation à l’ara-C par l’inhibiteur de la kinase
CHK1, l’UCN-01. L’inhibiteur seul permet également une sensibilisation très variable des
cellules de LAM, soulignant une « dépendance » différente vis-à-vis de la voie de
signalisation des dommages selon les échantillons et leurs caractéristiques propres. La
sensibilisation majoritaire des LAM à caryotypes complexes ouvre le champ sur une

114
nouvelle opportunité de traitement de ce sous-groupe de patients qui, comme nous l’avons
vu dans l’introduction, présente une médiane de survie de l’ordre de quelques mois
seulement. Bien que nous ayons confirmé l’implication directe de l’UCN-01 dans la
chimiosensibilisation des échantillons de LAM par interférence ARN, nous ne pouvons pas
exclure d’autres actions de l’inhibiteur, en particulier sur la voie de survie PI3K/AKT,
puisque nous avons vu dans l’introduction que l’UCN-01 permet une inactivation indirecte
d’AKT. Ces données sont d’autant plus intéressantes que des résultats publiés récemment
par le Pr. C. Récher, mettent en évidence une activation d’AKT sur la thréonine 308 dans
des échantillons de LAM à haut risque cytogénétique (Gallay et al., 2009). Ainsi le bénéfice
de l’UCN-01 dans les LAM de haut risque cytogénétique pourrait également impliquer
l’inactivation indirecte d’AKT par PDK1 responsable de sa déphosphorylation sur la
thréonine 308 (Sato et al., 2002). De plus, il a été mis en évidence que la sérine/thréonine
phosphatase PP2A impliquée dans l’inactivation in vitro d’AKT mais également de CHK1
((Millward et al., 1999) ; (Leung-Pineda et al., 2006)), présente une activité plus faible
dans les LAM de haut risque cytogénétique que dans les LAM de risque intermédiaire
(Gallay et al., 2009). Ainsi, cette activation plus faible de PP2A dans les LAM de haut
risque pourrait également jouer un rôle dans l’activation constitutive de la protéine CHK1
que nous rapportons. L’ensemble de ces résultats met en évidence l’intérêt que
représente l’utilisation d’inhibiteurs de la protéine CHK1 dans la prise en charge des LAM
de haut risque cytogénétique. Il est donc indispensable de valider les résultats obtenus
avec l'UCN-01 en utilisant d'autres composés inhibiteurs de CHK1 aux propriétés inhibitrices
plus spécifiques. Notre collaboration récente avec le groupe AstraZeneca nous a permis
d’obtenir un inhibiteur spécifique de la protéine CHK1, l’AZD7762, actuellement en cours
d’évaluation dans trois essais cliniques de phase I dans des tumeurs solides en combinaison
avec la gemcitabine et l’irinotécan. Des tests rapides de viabilité cellulaire en milieu
liquide et milieu semi-solide avec l'AZD7762 devraient nous permettre d'évaluer l’effet
chimiosensibilisateur de cet inhibiteur de CHK1 sur des échantillons de LAM et de valider
les hypothèses proposées à la suite de nos travaux réalisés avec l'UCN-01.
Compte tenu du rôle de l’activation d’oncogènes et/ou de la perte de gènes suppresseurs
de tumeurs dans l’établissement de l’instabilité génétique conduisant au développement
tumoral, il nous a semblé essentiel de répertorier les mutations présentes dans les 50
échantillons de LAM que nous avons examinés dans notre étude (Cancer Research).
Dans un premier temps, nous avons porté notre attention sur les mutations ponctuelles
et/ou les pertes de copie du gène p53 afin de tester l’impact réciproque de l’activation de
la voie des dommages et de la perte de fonction de ce gène. Nos travaux de recherche des
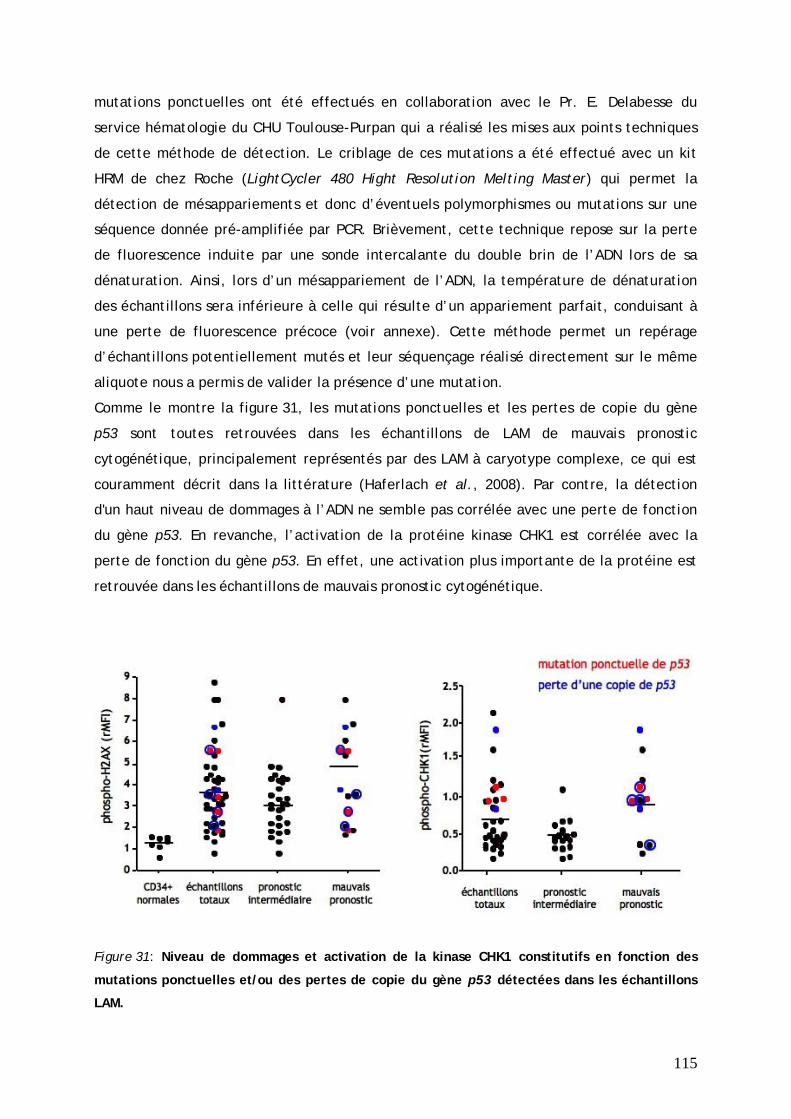
115
mutations ponctuelles ont été effectués en collaboration avec le Pr. E. Delabesse du
service hématologie du CHU Toulouse-Purpan qui a réalisé les mises aux points techniques
de cette méthode de détection. Le criblage de ces mutations a été effectué avec un kit
HRM de chez Roche (LightCycler 480 Hight Resolution Melting Master) qui permet la
détection de mésappariements et donc d’éventuels polymorphismes ou mutations sur une
séquence donnée pré-amplifiée par PCR. Brièvement, cette technique repose sur la perte
de fluorescence induite par une sonde intercalante du double brin de l’ADN lors de sa
dénaturation. Ainsi, lors d’un mésappariement de l’ADN, la température de dénaturation
des échantillons sera inférieure à celle qui résulte d’un appariement parfait, conduisant à
une perte de fluorescence précoce (voir annexe). Cette méthode permet un repérage
d’échantillons potentiellement mutés et leur séquençage réalisé directement sur le même
aliquote nous a permis de valider la présence d’une mutation.
Comme le montre la figure 31, les mutations ponctuelles et les pertes de copie du gène
p53 sont toutes retrouvées dans les échantillons de LAM de mauvais pronostic
cytogénétique, principalement représentés par des LAM à caryotype complexe, ce qui est
couramment décrit dans la littérature (Haferlach et al., 2008). Par contre, la détection
d'un haut niveau de dommages à l’ADN ne semble pas corrélée avec une perte de fonction
du gène p53. En revanche, l’activation de la protéine kinase CHK1 est corrélée avec la
perte de fonction du gène p53. En effet, une activation plus importante de la protéine est
retrouvée dans les échantillons de mauvais pronostic cytogénétique.
Figure 31: Niveau de dommages et activation de la kinase CHK1 constitutifs en fonction des
mutations ponctuelles et/ou des pertes de copie du gène p53 détectées dans les échantillons
LAM.

116
Les mutations et pertes de fonction du gène p53 sont majoritairement retrouvées dans les LAM de mauvais pronostic cytogénétique. Cette analyse moléculaire ne permet pas de mettre en évidence de corrélation entre le niveau de dommages et la perte de fonction du gène p53. En revanche, l’activation constitutive de la kinase CHK1 est corrélée (p = 0,0005) avec la perte de fonction du gène p53. Les niveaux de dommages et activation de CHK1 ont été évalués en cryométrie de flux, les valeurs représentent le ratio des intensités moyennes de fluorescence (Mean Fluorescence Intensity ratio) entre l’anticorps isotypique et l’anticorps dirigé contre la phosphorylation (D’après (Cavelier et al., 2009)).
Ces données semblent en contradiction avec les données de la littérature décrivant le
modèle de tumorigenèse dans les tumeurs solides (cf Introduction). En effet, dans les LAM,
la perte de fonction du gène p53 n’est pas accompagnée d’une inactivation de la voie de
signalisation des dommages. Néanmoins, l’évaluation de l’activation de CHK1 que nous
rapportons a été réalisée au diagnostic de la pathologie, ce qui ne préjuge en rien de
l’évolution de cette activation à des stades plus tardifs. De plus, la question de
l’implication de la voie des dommages dans l’établissement de l’instabilité génétique au
cours de la leucémogenèse reste ouverte. En effet, les études de leucémogenèse basées
sur l’évolution des SMD vers les LAM ne rendent pas compte de l'évolution des différents
marqueurs lors de l’apparition d’une LAM primitive et sont donc trop sélectives d’un sous-
groupe de LAM. L’utilisation de modèle murin développant progressivement une leucémie,
tel que ceux délété de manière conditionnelle pour le gène pten, permettrait d’adresser
et de documenter cette question (Yilmaz et al., 2006).
Dans un second temps, nous nous sommes intéressés plus largement aux mutations
fréquemment retrouvées dans les LAM qui induisent l’activation de voies de signalisation et
à ce titre qui pourraient avoir un impact sur l’instabilité génétique dans les LAM afin de
tester une éventuelle corrélation entre ces mutations et l’activation de la voie des
dommages. Nous avons donc évalué la présence de la duplication en tandem et des
mutations ponctuelles de flt3 (FLT3-ITD et FLT3-TKD respectivement), et des mutations
ponctuelles des gènes nras, kras et kit. En effet, la mutation FLT3-ITD est présente dans
24 à 39% des LAM et conduit à une activation constitutive du récepteur tyrosine kinase, il
en résulte une augmentation de la prolifération cellulaire, une inhibition de l’apoptose à
travers l’activation des voies de signalisation MAPK, STAT et Lyn/SRC. La mutation FLT3-
TKD est beaucoup moins fréquente avec seulement 5 à 14% des LAM. Même si son rôle dans
les LAM est mal connu, elle est associée à une survie sans maladie inférieure à celle des
patients non mutés dans le groupe des caryotypes normaux (Whitman et al., 2008). Comme
dans les tumeurs solides, le rôle des mutations du gène ras dans la dérégulation des voies
de signalisation notamment la voie MAPK et l’établissement de l’instabilité génétique, a
été rapporté dans les LAM et dans les modèles murins de leucémogenèse ((Braun and
Shannon, 2008) ; (Rassool et al., 2007)). Dans les LAM, nras est plus fréquemment muté
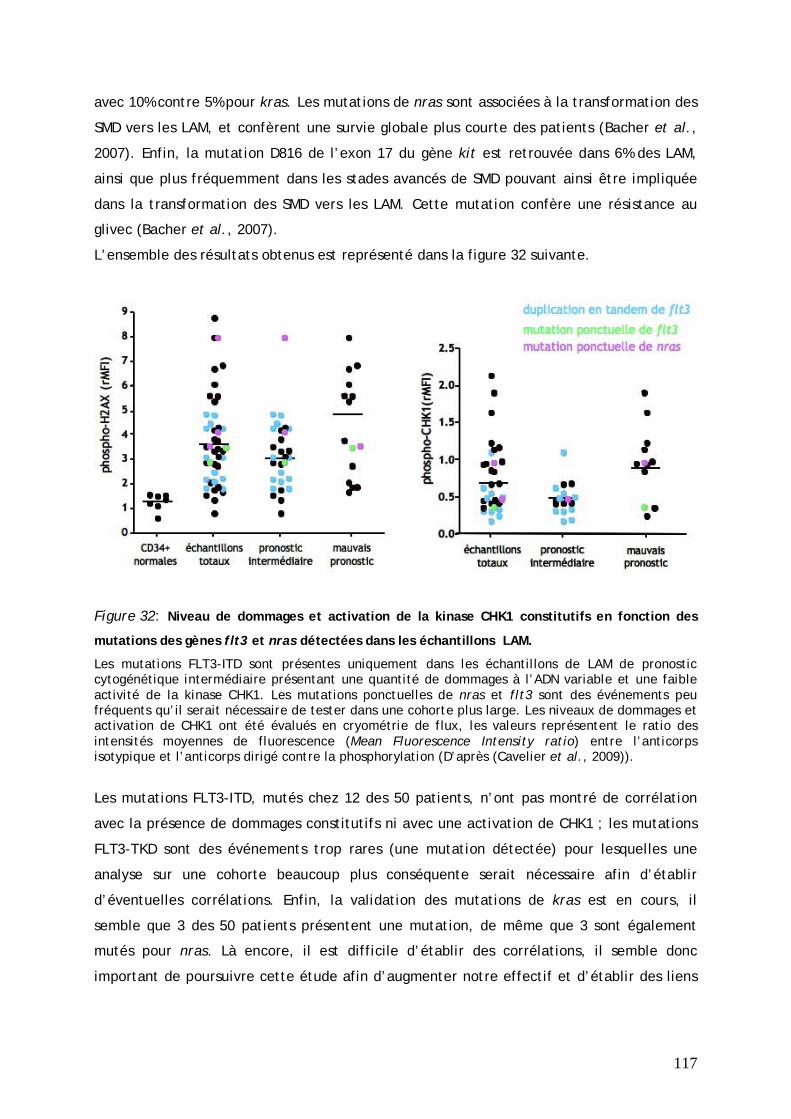
117
avec 10% contre 5% pour kras. Les mutations de nras sont associées à la transformation des
SMD vers les LAM, et confèrent une survie globale plus courte des patients (Bacher et al.,
2007). Enfin, la mutation D816 de l’exon 17 du gène kit est retrouvée dans 6% des LAM,
ainsi que plus fréquemment dans les stades avancés de SMD pouvant ainsi être impliquée
dans la transformation des SMD vers les LAM. Cette mutation confère une résistance au
glivec (Bacher et al., 2007).
L’ensemble des résultats obtenus est représenté dans la figure 32 suivante.
Figure 32: Niveau de dommages et activation de la kinase CHK1 constitutifs en fonction des
mutations des gènes flt3 et nras détectées dans les échantillons LAM.
Les mutations FLT3-ITD sont présentes uniquement dans les échantillons de LAM de pronostic cytogénétique intermédiaire présentant une quantité de dommages à l’ADN variable et une faible activité de la kinase CHK1. Les mutations ponctuelles de nras et flt3 sont des événements peu fréquents qu’il serait nécessaire de tester dans une cohorte plus large. Les niveaux de dommages et activation de CHK1 ont été évalués en cryométrie de flux, les valeurs représentent le ratio des intensités moyennes de fluorescence (Mean Fluorescence Intensity ratio) entre l’anticorps isotypique et l’anticorps dirigé contre la phosphorylation (D’après (Cavelier et al., 2009)).
Les mutations FLT3-ITD, mutés chez 12 des 50 patients, n’ont pas montré de corrélation
avec la présence de dommages constitutifs ni avec une activation de CHK1 ; les mutations
FLT3-TKD sont des événements trop rares (une mutation détectée) pour lesquelles une
analyse sur une cohorte beaucoup plus conséquente serait nécessaire afin d’établir
d’éventuelles corrélations. Enfin, la validation des mutations de kras est en cours, il
semble que 3 des 50 patients présentent une mutation, de même que 3 sont également
mutés pour nras. Là encore, il est difficile d’établir des corrélations, il semble donc
important de poursuivre cette étude afin d’augmenter notre effectif et d’établir des liens

118
entre l’activation de la voie des dommages à l’ADN et les mutations du gène ras chez les
patients atteints de LAM.
L’ensemble des travaux réalisés dans le cadre de cette thèse mettent une évidence une
corrélation entre l’activation de la kinase CHK1 et la perte de fonction du gène
suppresseur de tumeur p53. Il me semble particulièrement intéressant de poursuivre la
détection systématique de l’activation de cette protéine en cytomètrie de flux sur les
échantillons de LAM au diagnostic afin d’augmenter la cohorte de patients et de renforcer
nos résultats. En effet, cette activation majoritaire des cellules p53 déficientes permet
une sensibilité plus importante aux inhibiteurs de la kinase CHK1. C’est précisément dans
ce contexte que l’utilisation des inhibiteurs de la voie des dommages et plus
particulièrement de CHK1, qui joue un rôle majeur dans le point de contrôle G2/M, ont
émergé et ont montré leur véritable intérêt en terme de radio- et chimiosensibilisation.
L’effet chimiosensibilisateur de l’UCN-01 obtenu dans les LAM avec un caryotype complexe
est encourageant et pourrait représenter une perspective thérapeutique très pertinente
pour ce sous-groupe de patients. En effet, aucune thérapeutique dans ce sous-groupe n’a
fait ses preuves à l’heure actuelle. Nous pouvons également souligner que la détection de
l’activation de la kinase CHK1 et des mutations de p53 sont des tests (cytométrie en flux
et HRM) que l’on peut imaginer effectuer en routine dans des laboratoires spécialisés et
qui pourraient ainsi servir à caractériser au diagnostic les patients qui pourraient tirer le
maximum de bénéfice de ces traitements.
Pour finir, l’ensemble de mes travaux de thèse portant sur l’implication du cycle cellulaire
et de ses points de contrôle dans la pathologie des LAM nous a permis d’apporter de
nouveaux éléments de réflexion dans la connaissance des mécanismes de chimiorésistance
et d’instabilité génétique des LAM. Ces travaux montrent à quel point il est pertinent de
mener une recherche translationnelle entre les laboratoires et la clinique afin d’aller vers
une connaissance accrue de la pathologie sur le plan cellulaire et moléculaire permettant
de proposer des perspectives thérapeutiques de plus en plus ciblées et ainsi de plus en plus
efficaces.

119
REFERENCES REFERENCES
BIBLIOGRAPHIQUESBIBLIOGRAPHIQUES

120
Adam M, Pogacic V, Bendit M, Chappuis R, Nawijn MC, Duyster J et al (2006). Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res 66: 3828-35. Akinaga S, Gomi K, Morimoto M, Tamaoki T, Okabe M (1991). Antitumor activity of UCN-01, a selective inhibitor of protein kinase C, in murine and human tumor models. Cancer Res 51: 4888-92. Aleem E, Kiyokawa H, Kaldis P (2005). Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat Cell Biol 7: 831-6. Amico D, Barbui AM, Erba E, Rambaldi A, Introna M, Golay J (2003). Differential response of human acute myeloid leukemia cells to gemtuzumab ozogamicin in vitro: role of Chk1 and Chk2 phosphorylation and caspase 3. Blood 101: 4589-97. Andrews RG, Singer JW, Bernstein ID (1986). Monoclonal antibody 12-8 recognizes a 115-kd molecule present on both unipotent and multipotent hematopoietic colony-forming cells and their precursors. Blood 67: 842-5. Antoni L, Sodha N, Collins I, Garrett MD (2007). CHK2 kinase: cancer susceptibility and cancer therapy - two sides of the same coin? Nat Rev Cancer 7: 925-36. Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K et al (2004). Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118: 149-61. Bacher U, Haferlach T, Kern W, Haferlach C, Schnittger S (2007). A comparative study of molecular mutations in 381 patients with myelodysplastic syndrome and in 4130 patients with acute myeloid leukemia. Haematologica 92: 744-52. Baldin V, Pelpel K, Cazales M, Cans C, Ducommun B (2002). Nuclear localization of CDC25B1 and serine 146 integrity are required for induction of mitosis. J Biol Chem 277: 35176-82. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444: 756-60. Bardet V, Tamburini J, Ifrah N, Dreyfus F, Mayeux P, Bouscary D et al (2006). Single cell analysis of phosphoinositide 3-kinase/Akt and ERK activation in acute myeloid leukemia by flow cytometry. Haematologica 91: 757-64. Barker JE (1997). Early transplantation to a normal microenvironment prevents the development of Steel hematopoietic stem cell defects. Exp Hematol 25: 542-7. Barriere C, Santamaria D, Cerqueira A, Galan J, Martin A, Ortega S et al (2007). Mice thrive without Cdk4 and Cdk2. Mol Oncol 1: 72-83. Bartek J, Lukas J (2003). Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421-9. Bartek J, Lukas J (2007). DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 19: 238-45.

121
Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K et al (2005). DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 434: 864-70. Bazzoni G, Carlesso N, Griffin JD, Hemler ME (1996). Bcr/Abl expression stimulates integrin function in hematopoietic cell lines. J Clin Invest 98: 521-528. Bellamy WT, Richter L, Frutiger Y, Grogan TM (1999). Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Res 59: 728-33. Bendall LJ, Makrynikola V, Hutchinson A, Bianchi AC, Bradstock KF, Gottlieb DJ (1998). Stem cell factor enhances the adhesion of AML cells to fibronectin and augments fibronectin-mediated anti-apoptotic and proliferative signals. Leukemia 12: 1375-82. Benekli M, Baer MR, Baumann H, Wetzler M (2003). Signal transducer and activator of transcription proteins in leukemias. Blood 101: 2940-54. Benekli M, Xia Z, Donohue KA, Ford LA, Pixley LA, Baer MR et al (2002). Constitutive activity of signal transducer and activator of transcription 3 protein in acute myeloid leukemia blasts is associated with short disease-free survival. Blood 99: 252-7. Bernardi R, Liebermann DA, Hoffman B (2000). Cdc25A stability is controlled by the ubiquitin-proteasome pathway during cell cycle progression and terminal differentiation. Oncogene 19: 2447-54. Berry LD, Gould KL (1996). Regulation of Cdc2 activity by phosphorylation at T14/Y15. Prog Cell Cycle Res 2: 99-105. Bertwistle D, Ashworth A (2000). BRCA1 and BRCA2. Curr Biol 10: R582. Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM (2004). p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev 18: 862-76. Besson A, Hwang HC, Cicero S, Donovan SL, Gurian-West M, Johnson D et al (2007). Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev 21: 1731-46. Bhardwaj G, Murdoch B, Wu D, Baker DP, Williams KP, Chadwick K et al (2001). Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol 2: 172-80. Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE (1997). Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci U S A 94: 5320-5. Billottet C, Grandage VL, Gale RE, Quattropani A, Rommel C, Vanhaesebroeck B et al (2006). A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene 25: 6648-59. Blain SW, Scher HI, Cordon-Cardo C, Koff A (2003). p27 as a target for cancer therapeutics. Cancer Cell 3: 111-5.

122
Blair A, Hogge DE, Ailles LE, Lansdorp PM, Sutherland HJ (1997). Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood 89: 3104-12. Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J et al (2008). Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther 7: 2394-404. Blomberg I, Hoffmann I (1999). Ectopic expression of Cdc25A accelerates the G(1)/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Mol Cell Biol 19: 6183-94. Boehrer S, Ades L, Tajeddine N, Hofmann WK, Kriener S, Bug G et al (2009). Suppression of the DNA damage response in acute myeloid leukemia versus myelodysplastic syndrome. Oncogene 28: 2205-18. Bonnet D, Dick JE (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3: 730-7. Boutros R, Dozier C, Ducommun B (2006). The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol 18: 185-91. Boutros R, Lobjois V, Ducommun B (2007a). CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer 7: 495-507. Boutros R, Lobjois V, Ducommun B (2007b). CDC25B involvement in the centrosome duplication cycle and in microtubule nucleation. Cancer Res 67: 11557-64. Braun BS, Shannon K (2008). Targeting Ras in myeloid leukemias. Clin Cancer Res 14: 2249-52. Breems DA, Van Putten WL, De Greef GE, Van Zelderen-Bhola SL, Gerssen-Schoorl KB, Mellink CH et al (2008). Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol 26: 4791-7. Bromleigh VC, Freedman LP (2000). p21 is a transcriptional target of HOXA10 in differentiating myelomonocytic cells. Genes Dev 14: 2581-6. Bruno AP, Laurent G, Averbeck D, Demur C, Bonnet J, Bettaieb A et al (1998). Lack of ceramide generation in TF-1 human myeloid leukemic cells resistant to ionizing radiation. Cell Death Differ 5: 172-82. Bugler B, Schmitt E, Aressy B, Ducommun B Unscheduled expression of CDC25B in S-phase leads to replicative stress and DNA damage. Mol Cancer 9: 29. Burger JA, Spoo A, Dwenger A, Burger M, Behringer D (2003). CXCR4 chemokine receptors (CD184) and alpha4beta1 integrins mediate spontaneous migration of human CD34+ progenitors and acute myeloid leukaemia cells beneath marrow stromal cells (pseudoemperipolesis). Br J Haematol 122: 579-89. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC et al (2003). Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425: 841-6.

123
Camargo FD, Chambers SM, Drew E, McNagny KM, Goodell MA (2006). Hematopoietic stem cells do not engraft with absolute efficiencies. Blood 107: 501-7. Carlessi L, Buscemi G, Larson G, Hong Z, Wu JZ, Delia D (2007). Biochemical and cellular characterization of VRX0466617, a novel and selective inhibitor for the checkpoint kinase Chk2. Mol Cancer Ther 6: 935-44. Caslini C, Shilatifard A, Yang L, Hess JL (2000). The amino terminus of the mixed lineage leukemia protein (MLL) promotes cell cycle arrest and monocytic differentiation. Proc Natl Acad Sci U S A 97: 2797-802. Castedo M, Perfettini JL, Roumier T, Yakushijin K, Horne D, Medema R et al (2004). The cell cycle checkpoint kinase Chk2 is a negative regulator of mitotic catastrophe. Oncogene 23: 4353-61. Cavelier C, Didier C, Prade N, Mansat-De Mas V, Manenti S, Recher C et al (2009). Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: potential importance for checkpoint targeting therapy. Cancer Res 69: 8652-61. Chadwick N, Nostro MC, Baron M, Mottram R, Brady G, Buckle AM (2007). Notch signaling induces apoptosis in primary human CD34+ hematopoietic progenitor cells. Stem Cells 25: 203-10. Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD (1999). Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci U S A 96: 13777-82. Chen MS, Hurov J, White LS, Woodford-Thomas T, Piwnica-Worms H (2001). Absence of apparent phenotype in mice lacking Cdc25C protein phosphatase. Mol Cell Biol 21: 3853-61. Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden DT (2000a). Stem cell repopulation efficiency but not pool size is governed by p27(kip1). Nat Med 6: 1235-40. Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M et al (2000b). Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287: 1804-8. Cheong JW, Eom JI, Maeng HY, Lee ST, Hahn JS, Ko YW et al (2003). Phosphatase and tensin homologue phosphorylation in the C-terminal regulatory domain is frequently observed in acute myeloid leukaemia and associated with poor clinical outcome. Br J Haematol 122: 454-6. Cheshier SH, Morrison SJ, Liao X, Weissman IL (1999). In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc Natl Acad Sci U S A 96: 3120-5. Christiansen DH, Andersen MK, Pedersen-Bjergaard J (2001). Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol 19: 1405-13.

124
Clave E, Carosella ED, Gluckman E, Socie G (1997). Radiation-enhanced expression of interferon-inducible genes in the KG1a primitive hematopoietic cell line. Leukemia 11: 114-9. Collis SJ, DeWeese TL, Jeggo PA, Parker AR (2005). The life and death of DNA-PK. Oncogene 24: 949-61. Coqueret O (2003). New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol 13: 65-70. Coqueret O, Gascan H (2000). Functional interaction of STAT3 transcription factor with the cell cycle inhibitor p21WAF1/CIP1/SDI1. J Biol Chem 275: 18794-800. Cornillet-Lefebvre P, Cuccuini W, Bardet V, Tamburini J, Gillot L, Ifrah N et al (2006). Constitutive phosphoinositide 3-kinase activation in acute myeloid leukemia is not due to p110delta mutations. Leukemia 20: 374-6. Cortez D (2003). Caffeine inhibits checkpoint responses without inhibiting the ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) protein kinases. J Biol Chem 278: 37139-45. Costello RT, Sivori S, Marcenaro E, Lafage-Pochitaloff M, Mozziconacci MJ, Reviron D et al (2002). Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 99: 3661-7. Damiano JS, Hazlehurst LA, Dalton WS (2001). Cell adhesion-mediated drug resistance (CAM-DR) protects the K562 chronic myelogenous leukemia cell line from apoptosis induced by BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation. Leukemia 15: 1232-9. de Haan G, Weersing E, Dontje B, van Os R, Bystrykh LV, Vellenga E et al (2003). In vitro generation of long-term repopulating hematopoietic stem cells by fibroblast growth factor-1. Dev Cell 4: 241-51. de Koning JP, Soede-Bobok AA, Ward AC, Schelen AM, Antonissen C, van Leeuwen D et al (2000). STAT3-mediated differentiation and survival and of myeloid cells in response to granulocyte colony-stimulating factor: role for the cyclin-dependent kinase inhibitor p27(Kip1). Oncogene 19: 3290-8. De Waele M, Renmans W, Jochmans K, Schots R, Lacor P, Trullemans F et al (1999). Different expression of adhesion molecules on CD34+ cells in AML and B-lineage ALL and their normal bone marrow counterparts. Eur J Haematol 63: 192-201. Decesse JT, Medjkane S, Datto MB, Cremisi CE (2001). RB regulates transcription of the p21/WAF1/CIP1 gene. Oncogene 20: 962-71. Draviam VM, Orrechia S, Lowe M, Pardi R, Pines J (2001). The localization of human cyclins B1 and B2 determines CDK1 substrate specificity and neither enzyme requires MEK to disassemble the Golgi apparatus. J Cell Biol 152: 945-58. Drexler HG (1996). Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 10: 588-99. Dufour S, Duband JL, Thiery JP (1986). Role of a major cell-substratum adhesion system in cell behavior and morphogenesis. Biol Cell 58: 1-13.

125
Dumon S, Santos SC, Debierre-Grockiego F, Gouilleux-Gruart V, Cocault L, Boucheron C et al (1999). IL-3 dependent regulation of Bcl-xL gene expression by STAT5 in a bone marrow derived cell line. Oncogene 18: 4191-9. Dunaway S, Liu HY, Walworth NC (2005). Interaction of 14-3-3 protein with Chk1 affects localization and checkpoint function. J Cell Sci 118: 39-50. Durant S, Karran P (2003). Vanillins--a novel family of DNA-PK inhibitors. Nucleic Acids Res 31: 5501-12. Ekberg J, Landberg G, Holm C, Richter J, Wolgemuth DJ, Persson JL (2004). Regulation of the cyclin A1 protein is associated with its differential subcellular localization in hematopoietic and leukemic cells. Oncogene 23: 9082-9. Ellisen LW, Carlesso N, Cheng T, Scadden DT, Haber DA (2001). The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J 20: 1897-909. Ersvaer E, Zhang JY, McCormack E, Olsnes A, Anensen N, Tan EM et al (2007). Cyclin B1 is commonly expressed in the cytoplasm of primary human acute myelogenous leukemia cells and serves as a leukemia-associated antigen associated with autoantibody response in a subset of patients. Eur J Haematol 79: 210-25. Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J (2001). The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410: 842-7. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L et al (2005). Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352: 254-66. Falini B, Nicoletti I, Martelli MF, Mecucci C (2007). Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood 109: 874-85. Fauriat C, Moretta A, Olive D, Costello RT (2005). Defective killing of dendritic cells by autologous natural killer cells from acute myeloid leukemia patients. Blood 106: 2186-8. Ferguson AM, White LS, Donovan PJ, Piwnica-Worms H (2005). Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol Cell Biol 25: 2853-60. Fernandez-Vidal A, Ysebaert L, Didier C, Betous R, De Toni F, Prade-Houdellier N et al (2006). Cell adhesion regulates CDC25A expression and proliferation in acute myeloid leukemia. Cancer Res 66: 7128-35. Fierro FA, Taubenberger A, Puech PH, Ehninger G, Bornhauser M, Muller DJ et al (2008). BCR/ABL expression of myeloid progenitors increases beta1-integrin mediated adhesion to stromal cells. J Mol Biol 377: 1082-93. Fortunel NO, Hatzfeld JA, Monier MN, Hatzfeld A (2003). Control of hematopoietic stem/progenitor cell fate by transforming growth factor-beta. Oncol Res 13: 445-53.

126
Freemerman AJ, Vrana JA, Tombes RM, Jiang H, Chellappan SP, Fisher PB et al (1997). Effects of antisense p21 (WAF1/CIP1/MDA6) expression on the induction of differentiation and drug-mediated apoptosis in human myeloid leukemia cells (HL-60). Leukemia 11: 504-13. Gabrielli BG, Clark JM, McCormack AK, Ellem KA (1997). Hyperphosphorylation of the N-terminal domain of Cdc25 regulates activity toward cyclin B1/Cdc2 but not cyclin A/Cdk2. J Biol Chem 272: 28607-14. Gabrielli BG, De Souza CP, Tonks ID, Clark JM, Hayward NK, Ellem KA (1996). Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J Cell Sci 109 ( Pt 5): 1081-93. Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK et al (2008). The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 111: 2776-84. Gallay N, Dos Santos C, Cuzin L, Bousquet M, Simmonet Gouy V, Chaussade C et al (2009). The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia 23: 1029-38. Gartel AL, Goufman E, Tevosian SG, Shih H, Yee AS, Tyner AL (1998). Activation and repression of p21(WAF1/CIP1) transcription by RB binding proteins. Oncogene 17: 3463-9. Gatei M, Sloper K, Sorensen C, Syljuasen R, Falck J, Hobson K et al (2003). Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem 278: 14806-11. George P, Bali P, Cohen P, Tao J, Guo F, Sigua C et al (2004). Cotreatment with 17-allylamino-demethoxygeldanamycin and FLT-3 kinase inhibitor PKC412 is highly effective against human acute myelogenous leukemia cells with mutant FLT-3. Cancer Res 64: 3645-52. Gilliland DG, Jordan CT, Felix CA (2004). The molecular basis of leukemia. Hematology Am Soc Hematol Educ Program: 80-97. Girard F, Strausfeld U, Cavadore JC, Russell P, Fernandez A, Lamb NJ (1992). cdc25 is a nuclear protein expressed constitutively throughout the cell cycle in nontransformed mammalian cells. J Cell Biol 118: 785-94. Goldstone S, Pavey S, Forrest A, Sinnamon J, Gabrielli B (2001). Cdc25-dependent activation of cyclin A/cdk2 is blocked in G2 phase arrested cells independently of ATM/ATR. Oncogene 20: 921-32. Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T et al (2005). Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 434: 907-13. Gothot A, Pyatt R, McMahel J, Rice S, Srour EF (1997). Functional heterogeneity of human CD34(+) cells isolated in subcompartments of the G0 /G1 phase of the cell cycle. Blood 90: 4384-93.

127
Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O'Connor PM et al (2000). The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem 275: 5600-5. Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA et al (2001). Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 98: 2301-7. Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T (2008). Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 22: 1539-41. Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC (1999). Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98: 859-69. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ (1993). The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75: 805-16. Harper JW, Elledge SJ (1996). Cdk inhibitors in development and cancer. Curr Opin Genet Dev 6: 56-64. Harris TE, Albrecht JH, Nakanishi M, Darlington GJ (2001). CCAAT/enhancer-binding protein-alpha cooperates with p21 to inhibit cyclin-dependent kinase-2 activity and induces growth arrest independent of DNA binding. J Biol Chem 276: 29200-9. Hatzfeld J, Li ML, Brown EL, Sookdeo H, Levesque JP, O'Toole T et al (1991). Release of early human hematopoietic progenitors from quiescence by antisense transforming growth factor beta 1 or Rb oligonucleotides. J Exp Med 174: 925-9. Hess DA, Meyerrose TE, Wirthlin L, Craft TP, Herrbrich PE, Creer MH et al (2004). Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood 104: 1648-55. Hess DA, Wirthlin L, Craft TP, Herrbrich PE, Hohm SA, Lahey R et al (2006). Selection based on CD133 and high aldehyde dehydrogenase activity isolates long-term reconstituting human hematopoietic stem cells. Blood 107: 2162-9. Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI et al (2004). Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64: 9152-9. Ho AD, Wagner W (2007). The beauty of asymmetry: asymmetric divisions and self-renewal in the haematopoietic system. Curr Opin Hematol 14: 330-6. Hochegger H, Takeda S, Hunt T (2008). Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat Rev Mol Cell Biol 9: 910-6. Hoffmann I, Clarke PR, Marcote MJ, Karsenti E, Draetta G (1993). Phosphorylation and activation of human cdc25-C by cdc2--cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J 12: 53-63.

128
Horibe S, Takagi M, Unno J, Nagasawa M, Morio T, Arai A et al (2007). DNA damage check points prevent leukemic transformation in myelodysplastic syndrome. Leukemia 21: 2195-8. Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M et al (2007). CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proc Natl Acad Sci U S A 104: 11008-13. Hoshikawa Y, Amimoto K, Mizuguchi R, Hatakeyama M (1998). Highly controlled heterologous gene expression through combined utilization of the tetracycline-repressible transactivator and the lac repressor. Anal Biochem 261: 211-8. Hotte SJ, Oza A, Winquist EW, Moore M, Chen EX, Brown S et al (2006). Phase I trial of UCN-01 in combination with topotecan in patients with advanced solid cancers: a Princess Margaret Hospital Phase II Consortium study. Ann Oncol 17: 334-40. Huang S, Law P, Francis K, Palsson BO, Ho AD (1999). Symmetry of initial cell divisions among primitive hematopoietic progenitors is independent of ontogenic age and regulatory molecules. Blood 94: 2595-604. Huntly BJ, Gilliland DG (2005). Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer 5: 311-21. Hussong JW, Rodgers GM, Shami PJ (2000). Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood 95: 309-13. Iida H, Towatari M, Tanimoto M, Morishita Y, Kodera Y, Saito H (1997). Overexpression of cyclin E in acute myelogenous leukemia. Blood 90: 3707-13. Ikezoe T, Yang J, Nishioka C, Takezaki Y, Tasaka T, Togitani K et al (2009). A novel treatment strategy targeting polo-like kinase 1 in hematological malignancies. Leukemia 23: 1564-76. Ikezoe T, Yang J, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y et al (2007). A novel treatment strategy targeting Aurora kinases in acute myelogenous leukemia. Mol Cancer Ther 6: 1851-7. Ismail IH, Martensson S, Moshinsky D, Rice A, Tang C, Howlett A et al (2004). SU11752 inhibits the DNA-dependent protein kinase and DNA double-strand break repair resulting in ionizing radiation sensitization. Oncogene 23: 873-82. Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M (1999). The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397: 164-8. Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J et al (1995). Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376: 313-20. Jiang K, Pereira E, Maxfield M, Russell B, Goudelock DM, Sanchez Y (2003). Regulation of Chk1 includes chromatin association and 14-3-3 binding following phosphorylation on Ser-345. J Biol Chem 278: 25207-17.

129
Jiang Y, Prosper F, Verfaillie CM (2000). Opposing effects of engagement of integrins and stimulation of cytokine receptors on cell cycle progression of normal human hematopoietic progenitors. Blood 95: 846-54. Jordan CT, Guzman ML (2004). Mechanisms controlling pathogenesis and survival of leukemic stem cells. Oncogene 23: 7178-87. Jourdan E, Boiron JM, Dastugue N, Vey N, Marit G, Rigal-Huguet F et al (2005). Early allogeneic stem-cell transplantation for young adults with acute myeloblastic leukemia in first complete remission: an intent-to-treat long-term analysis of the BGMT experience. J Clin Oncol 23: 7676-84. Jung Y, Song J, Shiozawa Y, Wang J, Wang Z, Williams B et al (2008). Hematopoietic stem cells regulate mesenchymal stromal cell induction into osteoblasts thereby participating in the formation of the stem cell niche. Stem Cells 26: 2042-51. Jung Y, Wang J, Havens A, Sun Y, Jin T, Taichman RS (2005). Cell-to-cell contact is critical for the survival of hematopoietic progenitor cells on osteoblasts. Cytokine 32: 155-62. Karanu FN, Murdoch B, Gallacher L, Wu DM, Koremoto M, Sakano S et al (2000). The notch ligand jagged-1 represents a novel growth factor of human hematopoietic stem cells. J Exp Med 192: 1365-72. Kastan MB, Bartek J (2004). Cell-cycle checkpoints and cancer. Nature 432: 316-23. Kato JY, Sherr CJ (1993). Inhibition of granulocyte differentiation by G1 cyclins D2 and D3 but not D1. Proc Natl Acad Sci U S A 90: 11513-7. Kato Y, Iwama A, Tadokoro Y, Shimoda K, Minoguchi M, Akira S et al (2005). Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J Exp Med 202: 169-79. Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121: 1109-21. Kim SC, Hahn JS, Min YH, Yoo NC, Ko YW, Lee WJ (1999). Constitutive activation of extracellular signal-regulated kinase in human acute leukemias: combined role of activation of MEK, hyperexpression of extracellular signal-regulated kinase, and downregulation of a phosphatase, PAC1. Blood 93: 3893-9. King FW, Skeen J, Hay N, Shtivelman E (2004). Inhibition of Chk1 by activated PKB/Akt. Cell Cycle 3: 634-7. Kogan SC, Lagasse E, Atwater S, Bae SC, Weissman I, Ito Y et al (1998). The PEBP2betaMYH11 fusion created by Inv(16)(p13;q22) in myeloid leukemia impairs neutrophil maturation and contributes to granulocytic dysplasia. Proc Natl Acad Sci U S A 95: 11863-8. Konopleva M, Shi Y, Steelman LS, Shelton JG, Munsell M, Marini F et al (2005). Development of a conditional in vivo model to evaluate the efficacy of small molecule inhibitors for the treatment of Raf-transformed hematopoietic cells. Cancer Res 65: 9962-70.

130
Kornblau SM, Womble M, Qiu YH, Jackson CE, Chen W, Konopleva M et al (2006). Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood 108: 2358-65. Kortmansky J, Shah MA, Kaubisch A, Weyerbacher A, Yi S, Tong W et al (2005). Phase I trial of the cyclin-dependent kinase inhibitor and protein kinase C inhibitor 7-hydroxystaurosporine in combination with Fluorouracil in patients with advanced solid tumors. J Clin Oncol 23: 1875-84. Kosoy A, O'Connell MJ (2008). Regulation of Chk1 by its C-terminal domain. Mol Biol Cell 19: 4546-53. Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E et al (2004). Mouse development and cell proliferation in the absence of D-cyclins. Cell 118: 477-91. Kramer A, Hochhaus A, Saussele S, Reichert A, Willer A, Hehlmann R (1998). Cyclin A1 is predominantly expressed in hematological malignancies with myeloid differentiation. Leukemia 12: 893-8. Kramer A, Horner S, Willer A, Fruehauf S, Hochhaus A, Hallek M et al (1999). Adhesion to fibronectin stimulates proliferation of wild-type and bcr/abl-transfected murine hematopoietic cells. Proc Natl Acad Sci U S A 96: 2087-92. Krause DS, Van Etten RA (2007). Right on target: eradicating leukemic stem cells. Trends Mol Med 13: 470-81. Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB (1992). Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci U S A 89: 7491-5. Kumagai A, Lee J, Yoo HY, Dunphy WG (2006). TopBP1 activates the ATR-ATRIP complex. Cell 124: 943-55. LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS et al (1997). New functional activities for the p21 family of CDK inhibitors. Genes Dev 11: 847-62. Lagasse E, Weissman IL (1994). bcl-2 inhibits apoptosis of neutrophils but not their engulfment by macrophages. J Exp Med 179: 1047-52. Lammer C, Wagerer S, Saffrich R, Mertens D, Ansorge W, Hoffmann I (1998). The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J Cell Sci 111 ( Pt 16): 2445-53. Lara PN, Jr., Mack PC, Synold T, Frankel P, Longmate J, Gumerlock PH et al (2005). The cyclin-dependent kinase inhibitor UCN-01 plus cisplatin in advanced solid tumors: a California cancer consortium phase I pharmacokinetic and molecular correlative trial. Clin Cancer Res 11: 4444-50. Larsson J, Goumans MJ, Sjostrand LJ, van Rooijen MA, Ward D, Leveen P et al (2001). Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J 20: 1663-73. Latif C, den Elzen NR, O'Connell MJ (2004). DNA damage checkpoint maintenance through sustained Chk1 activity. J Cell Sci 117: 3489-98.

131
Laurent G, Jaffrezou JP (2001). Signaling pathways activated by daunorubicin. Blood 98: 913-24. Leoncini L, Bellan C, De Falco G (2006). Retinoblastoma gene family expression in lymphoid tissues. Oncogene 25: 5309-14. Leung-Pineda V, Ryan CE, Piwnica-Worms H (2006). Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol Cell Biol 26: 7529-38. Li J, Zhang X, Sejas DP, Bagby GC, Pang Q (2004). Hypoxia-induced nucleophosmin protects cell death through inhibition of p53. J Biol Chem 279: 41275-9. Liang SH, Clarke MF (2001). Regulation of p53 localization. Eur J Biochem 268: 2779-83. Liao C, Wang XY, Wei HQ, Li SQ, Merghoub T, Pandolfi PP et al (2001). Altered myelopoiesis and the development of acute myeloid leukemia in transgenic mice overexpressing cyclin A1. Proc Natl Acad Sci U S A 98: 6853-8. Lincoln AJ, Wickramasinghe D, Stein P, Schultz RM, Palko ME, De Miguel MP et al (2002). Cdc25b phosphatase is required for resumption of meiosis during oocyte maturation. Nat Genet 30: 446-9. Lindqvist A, Kallstrom H, Lundgren A, Barsoum E, Rosenthal CK (2005). Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J Cell Biol 171: 35-45. Liu F, Stanton JJ, Wu Z, Piwnica-Worms H (1997). The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol Cell Biol 17: 571-83. Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K et al (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14: 1448-59. Liu S, Bekker-Jensen S, Mailand N, Lukas C, Bartek J, Lukas J (2006). Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol 26: 6056-64. Lolli G, Johnson LN (2005). CAK-Cyclin-dependent Activating Kinase: a key kinase in cell cycle control and a target for drugs? Cell Cycle 4: 572-7. Long MW, Briddell R, Walter AW, Bruno E, Hoffman R (1992). Human hematopoietic stem cell adherence to cytokines and matrix molecules. J Clin Invest 90: 251-5. Loog M, Morgan DO (2005). Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature 434: 104-8. Lu HR, Zhu H, Huang M, Chen Y, Cai YJ, Miao ZH et al (2005a). Reactive oxygen species elicit apoptosis by concurrently disrupting topoisomerase II and DNA-dependent protein kinase. Mol Pharmacol 68: 983-94. Lu X, Nannenga B, Donehower LA (2005b). PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 19: 1162-74.

132
Lukas C, Bartkova J, Latella L, Falck J, Mailand N, Schroeder T et al (2001). DNA damage-activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res 61: 4990-3. Lukas J, Lukas C, Bartek J (2004). Mammalian cell cycle checkpoints: signalling pathways and their organization in space and time. DNA Repair (Amst) 3: 997-1007. Lundberg AS, Weinberg RA (1998). Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 18: 753-61. Lutterbach B, Westendorf JJ, Linggi B, Isaac S, Seto E, Hiebert SW (2000). A mechanism of repression by acute myeloid leukemia-1, the target of multiple chromosomal translocations in acute leukemia. J Biol Chem 275: 651-6. Mailand N, Podtelejnikov AV, Groth A, Mann M, Bartek J, Lukas J (2002). Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J 21: 5911-20. Malumbres M, Barbacid M (2001). To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1: 222-31. Malumbres M, Barbacid M (2005). Mammalian cyclin-dependent kinases. Trends Biochem Sci 30: 630-41. Malumbres M, Barbacid M (2009). Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9: 153-66. Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S et al (2004). Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118: 493-504. Matsumura I, Kitamura T, Wakao H, Tanaka H, Hashimoto K, Albanese C et al (1999). Transcriptional regulation of the cyclin D1 promoter by STAT5: its involvement in cytokine-dependent growth of hematopoietic cells. EMBO J 18: 1367-77. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A et al (2003). Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med 9: 1158-65. Matthews DJ, Yakes FM, Chen J, Tadano M, Bornheim L, Clary DO et al (2007). Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle 6: 104-10. McKenna RW (2000). Multifaceted approach to the diagnosis and classification of acute leukemias. Clin Chem 46: 1252-9. Medema RH, Kops GJ, Bos JL, Burgering BM (2000). AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404: 782-7. Melnick A, Licht JD (1999). Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93: 3167-215.

133
Milella M, Kornblau SM, Andreeff M (2003). The mitogen-activated protein kinase signaling module as a therapeutic target in hematologic malignancies. Rev Clin Exp Hematol 7: 160-90. Milella M, Kornblau SM, Estrov Z, Carter BZ, Lapillonne H, Harris D et al (2001). Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia. J Clin Invest 108: 851-9. Millar JB, Russell P (1992). The cdc25 M-phase inducer: an unconventional protein phosphatase. Cell 68: 407-10. Miller CL, Eaves CJ (1997). Expansion in vitro of adult murine hematopoietic stem cells with transplantable lympho-myeloid reconstituting ability. Proc Natl Acad Sci U S A 94: 13648-53. Millward TA, Zolnierowicz S, Hemmings BA (1999). Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci 24: 186-91. Min YH, Cheong JW, Kim JY, Eom JI, Lee ST, Hahn JS et al (2004). Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res 64: 5225-31. Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, Jeung HK et al (2003). Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia 17: 995-7. Mizuki M, Schwable J, Steur C, Choudhary C, Agrawal S, Sargin B et al (2003). Suppression of myeloid transcription factors and induction of STAT response genes by AML-specific Flt3 mutations. Blood 101: 3164-73. Molinari M, Mercurio C, Dominguez J, Goubin F, Draetta GF (2000). Human Cdc25 A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Rep 1: 71-9. Morgan DO (1997). Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 13: 261-91. Morgan MA, Parsels LA, Parsels JD, Lawrence TS, Maybaum J (2006). The relationship of premature mitosis to cytotoxicity in response to checkpoint abrogation and antimetabolite treatment. Cell Cycle 5: 1983-8. Mori A, Higashi H, Hoshikawa Y, Imamura M, Asaka M, Hatakeyama M (1999). Granulocytic differentiation of myeloid progenitor cells by p130, the retinoblastoma tumor suppressor homologue. Oncogene 18: 6209-21. Morrison SJ, Wandycz AM, Hemmati HD, Wright DE, Weissman IL (1997). Identification of a lineage of multipotent hematopoietic progenitors. Development 124: 1929-39. Mueller BU, Pabst T (2006). C/EBPalpha and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol 13: 7-14.

134
Muller C, Christodoulopoulos G, Salles B, Panasci L (1998). DNA-Dependent protein kinase activity correlates with clinical and in vitro sensitivity of chronic lymphocytic leukemia lymphocytes to nitrogen mustards. Blood 92: 2213-9. Niida H, Katsuno Y, Banerjee B, Hande MP, Nakanishi M (2007). Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol Cell Biol 27: 2572-81. Nilsson SK, Haylock DN, Johnston HM, Occhiodoro T, Brown TJ, Simmons PJ (2003). Hyaluronan is synthesized by primitive hemopoietic cells, participates in their lodgment at the endosteum following transplantation, and is involved in the regulation of their proliferation and differentiation in vitro. Blood 101: 856-62. Nosaka T, Kitamura T (2002). Pim-1 expression is sufficient to induce cytokine independence in murine hematopoietic cells, but is dispensable for BCR-ABL-mediated transformation. Exp Hematol 30: 697-702. Nutley BP, Smith NF, Hayes A, Kelland LR, Brunton L, Golding BT et al (2005). Preclinical pharmacokinetics and metabolism of a novel prototype DNA-PK inhibitor NU7026. Br J Cancer 93: 1011-8. O'Connell MJ, Raleigh JM, Verkade HM, Nurse P (1997). Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J 16: 545-54. Oostendorp RA, Dormer P (1997). VLA-4-mediated interactions between normal human hematopoietic progenitors and stromal cells. Leuk Lymphoma 24: 423-35. Osawa M, Hanada K, Hamada H, Nakauchi H (1996). Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 273: 242-5. Padro T, Ruiz S, Bieker R, Burger H, Steins M, Kienast J et al (2000). Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood 95: 2637-44. Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL et al (2003). Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423: 302-5. Parker JE, Mufti GJ (2001). The role of apoptosis in the pathogenesis of the myelodysplastic syndromes. Int J Hematol 73: 416-28. Passegue E, Jamieson CH, Ailles LE, Weissman IL (2003). Normal and leukemic hematopoiesis: are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc Natl Acad Sci U S A 100 Suppl 1: 11842-9. Passegue E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL (2005). Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med 202: 1599-611. Phelps DE, Hsiao KM, Li Y, Hu N, Franklin DS, Westphal E et al (1998). Coupled transcriptional and translational control of cyclin-dependent kinase inhibitor p18INK4c expression during myogenesis. Mol Cell Biol 18: 2334-43. Powell SN, DeFrank JS, Connell P, Eogan M, Preffer F, Dombkowski D et al (1995). Differential sensitivity of p53(-) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay. Cancer Res 55: 1643-8.

135
Priestley GV, Scott LM, Ulyanova T, Papayannopoulou T (2006). Lack of alpha4 integrin expression in stem cells restricts competitive function and self-renewal activity. Blood 107: 2959-67. Puc J, Keniry M, Li HS, Pandita TK, Choudhury AD, Memeo L et al (2005). Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 7: 193-204. Puc J, Parsons R (2005). PTEN loss inhibits CHK1 to cause double stranded-DNA breaks in cells. Cell Cycle 4: 927-9. Punzel M, Liu D, Zhang T, Eckstein V, Miesala K, Ho AD (2003). The symmetry of initial divisions of human hematopoietic progenitors is altered only by the cellular microenvironment. Exp Hematol 31: 339-47. Punzel M, Zhang T, Liu D, Eckstein V, Ho AD (2002). Functional analysis of initial cell divisions defines the subsequent fate of individual human CD34(+)CD38(-) cells. Exp Hematol 30: 464-72. Rahn JJ, Shen Q, Mah BK, Hugh JC (2004). MUC1 initiates a calcium signal after ligation by intercellular adhesion molecule-1. J Biol Chem 279: 29386-90. Rainey MD, Charlton ME, Stanton RV, Kastan MB (2008). Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res 68: 7466-74. Ramaraj P, Singh H, Niu N, Chu S, Holtz M, Yee JK et al (2004). Effect of mutational inactivation of tyrosine kinase activity on BCR/ABL-induced abnormalities in cell growth and adhesion in human hematopoietic progenitors. Cancer Res 64: 5322-31. Rassool FV, Gaymes TJ, Omidvar N, Brady N, Beurlet S, Pla M et al (2007). Reactive oxygen species, DNA damage, and error-prone repair: a model for genomic instability with progression in myeloid leukemia? Cancer Res 67: 8762-71. Renner AG, Dos Santos C, Recher C, Bailly C, Creancier L, Kruczynski A et al (2009). Polo-like kinase 1 is overexpressed in acute myeloid leukemia and its inhibition preferentially targets the proliferation of leukemic cells. Blood 114: 659-62. Renneville A, Roumier C, Biggio V, Nibourel O, Boissel N, Fenaux P et al (2008). Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia 22: 915-31. Reya T (2003). Regulation of hematopoietic stem cell self-renewal. Recent Prog Horm Res 58: 283-95. Reya T, Morrison SJ, Clarke MF, Weissman IL (2001). Stem cells, cancer, and cancer stem cells. Nature 414: 105-11. Ricciardi MR, McQueen T, Chism D, Milella M, Estey E, Kaldjian E et al (2005). Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia 19: 1543-9. Riley T, Sontag E, Chen P, Levine A (2008). Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 9: 402-12.

136
Rosu-Myles M, Wolff L (2008). p15Ink4b: dual function in myelopoiesis and inactivation in myeloid disease. Blood Cells Mol Dis 40: 406-9. Russell P, Nurse P (1987). Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 49: 559-67. Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP (1998). Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature 395: 237-43. Sampath D, Cortes J, Estrov Z, Du M, Shi Z, Andreeff M et al (2006). Pharmacodynamics of cytarabine alone and in combination with 7-hydroxystaurosporine (UCN-01) in AML blasts in vitro and during a clinical trial. Blood 107: 2517-24. Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H et al (1997). Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277: 1497-501. Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K et al (2007). Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448: 811-5. Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM et al (1999). Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res 59: 4375-82. Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT (1998). Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res 58: 4375-82. Sato S, Fujita N, Tsuruo T (2002). Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine). Oncogene 21: 1727-38. Schliemann C, Bieker R, Padro T, Kessler T, Hintelmann H, Buchner T et al (2006). Expression of angiopoietins and their receptor Tie2 in the bone marrow of patients with acute myeloid leukemia. Haematologica 91: 1203-11. Schoch C, Kern W, Kohlmann A, Hiddemann W, Schnittger S, Haferlach T (2005). Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer 43: 227-38. Schofield KP, Humphries MJ, de Wynter E, Testa N, Gallagher JT (1998). The effect of alpha4 beta1-integrin binding sequences of fibronectin on growth of cells from human hematopoietic progenitors. Blood 91: 3230-8. Schwaller J, Frantsve J, Aster J, Williams IR, Tomasson MH, Ross TS et al (1998). Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO J 17: 5321-33. Sebastian B, Kakizuka A, Hunter T (1993). Cdc25M2 activation of cyclin-dependent kinases by dephosphorylation of threonine-14 and tyrosine-15. Proc Natl Acad Sci U S A 90: 3521-4. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP et al (2007). Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128: 157-70.

137
Shiloh Y (1997). Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet 31: 635-62. Shiloh Y (2003). ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155-68. Shiloh Y, Kastan MB (2001). ATM: genome stability, neuronal development, and cancer cross paths. Adv Cancer Res 83: 209-54. Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J et al (2002). PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med 8: 1145-52. Shinohara ET, Geng L, Tan J, Chen H, Shir Y, Edwards E et al (2005). DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res 65: 4987-92. Shiromizu T, Goto H, Tomono Y, Bartek J, Totsukawa G, Inoko A et al (2006). Regulation of mitotic function of Chk1 through phosphorylation at novel sites by cyclin-dependent kinase 1 (Cdk1). Genes Cells 11: 477-85. Singh SP, Lipman J, Goldman H, Ellis FH, Jr., Aizenman L, Cangi MG et al (1998). Loss or altered subcellular localization of p27 in Barrett's associated adenocarcinoma. Cancer Res 58: 1730-5. Slovak ML, Kopecky KJ, Cassileth PA, Harrington DH, Theil KS, Mohamed A et al (2000). Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood 96: 4075-83. Smits VA, Reaper PM, Jackson SP (2006). Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol 16: 150-9. Speck NA, Stacy T, Wang Q, North T, Gu TL, Miller J et al (1999). Core-binding factor: a central player in hematopoiesis and leukemia. Cancer Res 59: 1789s-1793s. Spiekermann K, Biethahn S, Wilde S, Hiddemann W, Alves F (2001). Constitutive activation of STAT transcription factors in acute myelogenous leukemia. Eur J Haematol 67: 63-71. Spiekermann K, Pau M, Schwab R, Schmieja K, Franzrahe S, Hiddemann W (2002). Constitutive activation of STAT3 and STAT5 is induced by leukemic fusion proteins with protein tyrosine kinase activity and is sufficient for transformation of hematopoietic precursor cells. Exp Hematol 30: 262-71. Spike BT, Macleod KF (2005). The Rb tumor suppressor in stress responses and hematopoietic homeostasis. Cell Cycle 4: 42-5. Steinman RA, Huang J, Yaroslavskiy B, Goff JP, Ball ED, Nguyen A (1998). Regulation of p21(WAF1) expression during normal myeloid differentiation. Blood 91: 4531-42. Sternberg DW, Gilliland DG (2004). The role of signal transducer and activator of transcription factors in leukemogenesis. J Clin Oncol 22: 361-71.

138
Stier S, Ko Y, Forkert R, Lutz C, Neuhaus T, Grunewald E et al (2005). Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med 201: 1781-91. Stirewalt DL, Radich JP (2003). The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer 3: 650-65. Stone RM, O'Donnell MR, Sekeres MA (2004). Acute myeloid leukemia. Hematology Am Soc Hematol Educ Program: 98-117. Stucki A, Rivier AS, Gikic M, Monai N, Schapira M, Spertini O (2001). Endothelial cell activation by myeloblasts: molecular mechanisms of leukostasis and leukemic cell dissemination. Blood 97: 2121-9. Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP (2005). MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123: 1213-26. Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F et al (2005). Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood 106: 1063-6. Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J et al (1999). PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A 96: 6199-204. Taichman RS, Reilly MJ, Emerson SG (1996). Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood 87: 518-24. Takahashi S, Harigae H, Kaku M, Sasaki T, Licht JD (2004). Flt3 mutation activates p21WAF1/CIP1 gene expression through the action of STAT5. Biochem Biophys Res Commun 316: 85-92. Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S et al (2002). Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J 21: 5195-205. Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T et al (2000). Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev 14: 1439-47. Takizawa CG, Morgan DO (2000). Control of mitosis by changes in the subcellular location of cyclin-B1-Cdk1 and Cdc25C. Curr Opin Cell Biol 12: 658-65. Tallman MS, Gilliland DG, Rowe JM (2005). Drug therapy for acute myeloid leukemia. Blood 106: 1154-63. Tanaka T, Kurose A, Halicka HD, Traganos F, Darzynkiewicz Z (2006). 2-deoxy-D-glucose reduces the level of constitutive activation of ATM and phosphorylation of histone H2AX. Cell Cycle 5: 878-82. Taniguchi T, Endo H, Chikatsu N, Uchimaru K, Asano S, Fujita T et al (1999). Expression of p21(Cip1/Waf1/Sdi1) and p27(Kip1) cyclin-dependent kinase inhibitors during human hematopoiesis. Blood 93: 4167-78.

139
Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G et al (2005). Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood 106: 4086-92. Taylor WR, Stark GR (2001). Regulation of the G2/M transition by p53. Oncogene 20: 1803-15. Till JE, Mc CE (1961). A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat Res 14: 213-22. Towatari M, Iida H, Tanimoto M, Iwata H, Hamaguchi M, Saito H (1997). Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia 11: 479-84. Traver D, Akashi K, Weissman IL, Lagasse E (1998). Mice defective in two apoptosis pathways in the myeloid lineage develop acute myeloblastic leukemia. Immunity 9: 47-57. Tschan MP, Peters UR, Cajot JF, Betticher DC, Fey MF, Tobler A (1999). The cyclin-dependent kinase inhibitors p18INK4c and p19INK4d are highly expressed in CD34+ progenitor and acute myeloid leukaemic cells but not in normal differentiated myeloid cells. Br J Haematol 106: 644-51. Tse AN, Carvajal R, Schwartz GK (2007). Targeting checkpoint kinase 1 in cancer therapeutics. Clin Cancer Res 13: 1955-60. Turhan AG, Lemoine FM, Debert C, Bonnet ML, Baillou C, Picard F et al (1995). Highly purified primitive hematopoietic stem cells are PML-RARA negative and generate nonclonal progenitors in acute promyelocytic leukemia. Blood 85: 2154-61. van der Loo JC, Xiao X, McMillin D, Hashino K, Kato I, Williams DA (1998). VLA-5 is expressed by mouse and human long-term repopulating hematopoietic cells and mediates adhesion to extracellular matrix protein fibronectin. J Clin Invest 102: 1051-61. van Rhenen A, van Dongen GA, Kelder A, Rombouts EJ, Feller N, Moshaver B et al (2007). The novel AML stem cell associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 110: 2659-66. van Vugt MA, van de Weerdt BC, Vader G, Janssen H, Calafat J, Klompmaker R et al (2004). Polo-like kinase-1 is required for bipolar spindle formation but is dispensable for anaphase promoting complex/Cdc20 activation and initiation of cytokinesis. J Biol Chem 279: 36841-54. Vardiman JW, Harris NL, Brunning RD (2002). The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100: 2292-302. Verfaillie CM, McCarthy JB, McGlave PB (1991). Differentiation of primitive human multipotent hematopoietic progenitors into single lineage clonogenic progenitors is accompanied by alterations in their interaction with fibronectin. J Exp Med 174: 693-703. Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D et al (2009). Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 457: 51-6.

140
Vialle-Castellano A, Gaugler B, Mohty M, Isnardon D, van Baren N, Olive D (2004). Abundant expression of fibronectin is a major feature of leukemic dendritic cells differentiated from patients with acute myeloid leukemia. Leukemia 18: 426-33. Viglietto G, Motti ML, Bruni P, Melillo RM, D'Alessio A, Califano D et al (2002). Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med 8: 1136-44. Vila L, Thomas X, Campos L, Sabido O, Archimbaud E (1995). Expression of VLA molecules on acute leukemia cells: relationship with disease characteristics. Exp Hematol 23: 514-8. Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL (2004). Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood 103: 3258-64. Wagers AJ, Sherwood RI, Christensen JL, Weissman IL (2002). Little evidence for developmental plasticity of adult hematopoietic stem cells. Science 297: 2256-9. Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH (2007). Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 129: 1081-95. Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O'Connor PM (1996). UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J Natl Cancer Inst 88: 956-65. Warner JK, Wang JC, Takenaka K, Doulatov S, McKenzie JL, Harrington L et al (2005). Direct evidence for cooperating genetic events in the leukemic transformation of normal human hematopoietic cells. Leukemia 19: 1794-805. Welch S, Hirte HW, Carey MS, Hotte SJ, Tsao MS, Brown S et al (2007). UCN-01 in combination with topotecan in patients with advanced recurrent ovarian cancer: a study of the Princess Margaret Hospital Phase II consortium. Gynecol Oncol 106: 305-10. Whitman SP, Ruppert AS, Marcucci G, Mrozek K, Paschka P, Langer C et al (2007). Long-term disease-free survivors with cytogenetically normal acute myeloid leukemia and MLL partial tandem duplication: a Cancer and Leukemia Group B study. Blood 109: 5164-7. Whitman SP, Ruppert AS, Radmacher MD, Mrozek K, Paschka P, Langer C et al (2008). FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood 111: 1552-9. Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA et al (2004). A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 103: 4659-65. Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM et al (2004). c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 18: 2747-63. Wilson A, Trumpp A (2006). Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol 6: 93-106.

141
Xia Z, Baer MR, Block AW, Baumann H, Wetzler M (1998). Expression of signal transducers and activators of transcription proteins in acute myeloid leukemia blasts. Cancer Res 58: 3173-80. Yang R, Nakamaki T, Lubbert M, Said J, Sakashita A, Freyaldenhoven BS et al (1999). Cyclin A1 expression in leukemia and normal hematopoietic cells. Blood 93: 2067-74. Yaroslavskiy B, Watkins S, Donnenberg AD, Patton TJ, Steinman RA (1999). Subcellular and cell-cycle expression profiles of CDK-inhibitors in normal differentiating myeloid cells. Blood 93: 2907-17. Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H et al (2006). Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441: 475-82. Yokota T, Oritani K, Mitsui H, Aoyama K, Ishikawa J, Sugahara H et al (1998). Growth-supporting activities of fibronectin on hematopoietic stem/progenitor cells in vitro and in vivo: structural requirement for fibronectin activities of CS1 and cell-binding domains. Blood 91: 3263-72. Yokozawa T, Towatari M, Iida H, Takeyama K, Tanimoto M, Kiyoi H et al (2000). Prognostic significance of the cell cycle inhibitor p27Kip1 in acute myeloid leukemia. Leukemia 14: 28-33. You Z, Chahwan C, Bailis J, Hunter T, Russell P (2005). ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol 25: 5363-79. Yu H, Yuan Y, Shen H, Cheng T (2006). Hematopoietic stem cell exhaustion impacted by p18 INK4C and p21 Cip1/Waf1 in opposite manners. Blood 107: 1200-6. Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T (2004). In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol 6: 436-42. Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL et al (2008). AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther 7: 2955-66. Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT et al (2006). PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441: 518-22. Zhang J, Niu C, Ye L, Huang H, He X, Tong WG et al (2003). Identification of the haematopoietic stem cell niche and control of the niche size. Nature 425: 836-41. Zhang S, Fukuda S, Lee Y, Hangoc G, Cooper S, Spolski R et al (2000). Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J Exp Med 192: 719-28. Zhang W, Kornblau SM, Kobayashi T, Gambel A, Claxton D, Deisseroth AB (1995). High levels of constitutive WAF1/Cip1 protein are associated with chemoresistance in acute myelogenous leukemia. Clin Cancer Res 1: 1051-7.

142
Zhao H, Piwnica-Worms H (2001). ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21: 4129-39. Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP (2002). Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci U S A 99: 12339-44. Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC et al (2006). Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol 8: 870-6.

143
ANNEXESANNEXES
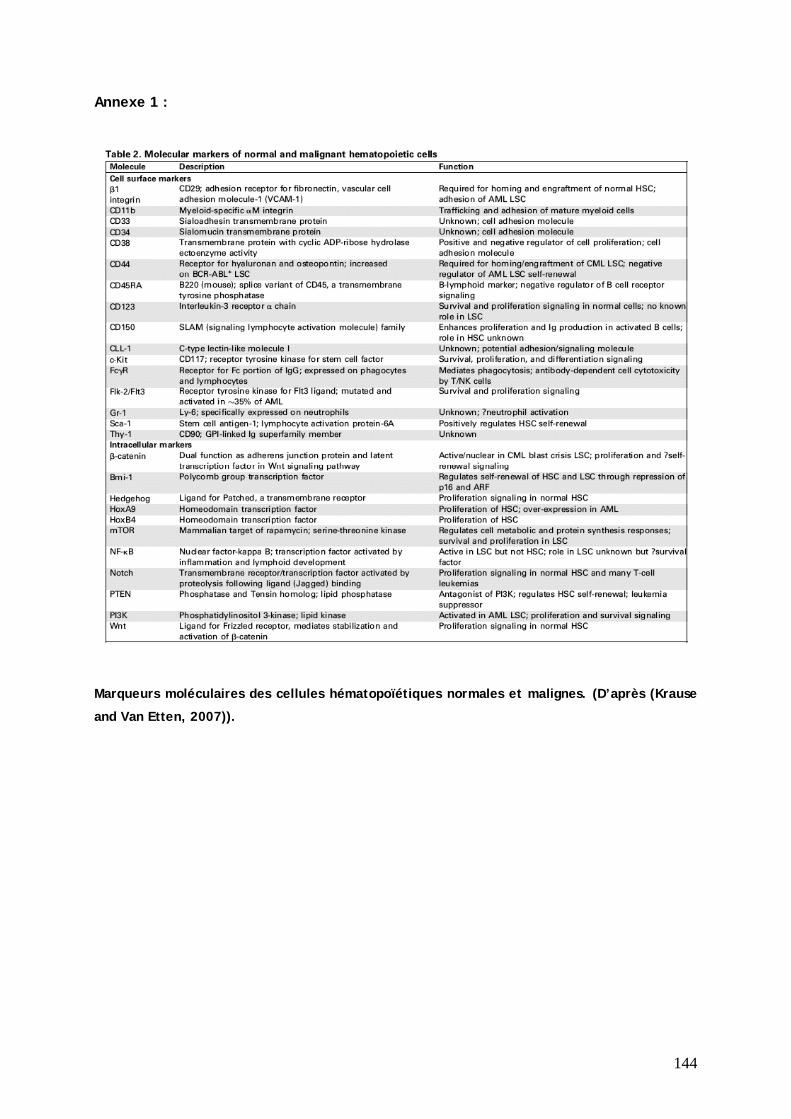
144
Annexe 1 :
Marqueurs moléculaires des cellules hématopoïétiques normales et malignes. (D’après (Krause
and Van Etten, 2007)).

European Journal of Pharmacology 591 (2008) 102–105
Contents lists available at ScienceDirect
European Journal of Pharmacology
j ourna l homepage: www.e lsev ie r.com/ locate /e jphar
Evaluation of Polo-like Kinase 1 inhibition on the G2/M checkpoint in AcuteMyelocytic Leukaemia
Christine Didier a,1, Cindy Cavelier a,1, Muriel Quaranta a, Cécile Demur b,c, Bernard Ducommun a,d,⁎a LBCMCP-CNRS UMR5088, IFR109 “Institut d'Exploration Fonctionnelle des Génomes”, University of Toulouse, 118 route de Narbonne, 31062 Toulouse, Franceb INSERM U563, Centre de Physiopathologie Toulouse Purpan, CHU Purpan, 31024 Toulouse Cedex 3, Francec Service d'Hématologie. Pavillon Lefebvre CHU Purpan, 31059 Toulouse Cedex, Franced UA4124. Pavillon Lefebvre CHU Purpan, TSA 40031, 31059 Toulouse Cedex, France
⁎ Corresponding author. LBCMCP-CNRS UMR5088,Fonctionnelle des Génomes”, University of Toulouse, 1Toulouse, France. Tel.: +33 5 61 55 62 10; fax: +33 5 61 5
E-mail address: [email protected] (B. Ducommun).1 These two authors equally contributed to this work
0014-2999/$ – see front matter © 2008 Elsevier B.V. Aldoi:10.1016/j.ejphar.2008.06.062
A B S T R A C T
A R T I C L E I N F OArticle history:
Polo-Kinase 1 (PLK1) is a key Received 31 January 2008Received in revised form 9 June 2008Accepted 12 June 2008Available online 19 June 2008Keywords:AML (Acute Myelocytic Leukaemia)PLK1 (Polo-Kinase 1)Cell cycleG2/M checkpoint
cell cycle regulator that is necessary for checkpoint recovery after DNA damage-induced G2 arrest. We have examined the effects of PLK inhibition in Acute Myelocytic Leukaemia (AML)cells, whose resistance to genotoxic agents is thought to be associated with checkpoint reinforcement. Wereport that in U937 AML cells, PLK1 participates in checkpoint recovery, and that inhibition of PLK by theGW843682X compound results in mitotic accumulation and apoptosis. We also found that when challengedwith VP-16, inhibition of PLK1 prevented U937 cells from checkpoint exit. Finally, we found that treatmentwith GW843682X slightly reduced genotoxic-induced inhibition of colony formation efficiency of primaryleukaemia cells (CFU-L) from AML patients.
© 2008 Elsevier B.V. All rights reserved.
1. Introduction
Polo-Kinase family members (PLK1–4) are key players in cell cycleregulation, especially during G2 phase and mitosis. The foundingmember Polo-Kinase 1 (PLK1) specifically localizes to centrosomes,the midzone of the mitotic spindle, and the post-mitotic bridge. PLK1promotes entry into mitosis and regulates mitotic progressionthrough the phosphorylation of proteins that control either theirlocalization or their proteasome-dependent degradation (van deWeerdt and Medema, 2006). Depletion of PLK1 by RNA interferenceresults in an accumulation of cells arrested in metaphase withabnormal chromatin morphologies. Knock-down of PLK1 also reducescell survival and increases the drug sensitivity of tumor cells, but notnormal cells (Liu et al., 2006; Spankuch et al., 2006). PLK1 isoverexpressed in a large variety of cancer types, and its over-expression often correlates with poor prognosis (Kneisel et al.,2002; Weichert et al., 2004; Yamada et al., 2004). Thus, inhibition ofPLK1 kinase activity would appear to represent an attractive approachfor the development of novel anticancer therapies (Barr et al., 2004). Anumber of promising compounds have been identified whose effectson cancer cells are currently under investigation (Strebhardt andUllrich, 2006). For instance, GW843682X, described as compound 1 in
IFR109 “Institut d'Exploration18 route de Narbonne, 310625 81 09.
.
l rights reserved.
Lansing et al. (2007), is an ATP-competitive inhibitor that hasnanomolar activity against purified PLK enzymes and that can inhibitthe growth of tumor cell lines in vitro. This compound has also beenshown to delay the G2/M transition, induce mitotic spindle defects,and cause multinucleation in the lung adenocarcinoma cell line NCI-H460 (Lansing et al., 2007). GW843682X was also shown in the samestudy to inhibit at a similar concentration PLK3, a member of the Polo-like kinase family that is proposed to be involved in the control of theG1/S transition and S-phase progression (Zimmerman and Erikson,2007a,b).
When DNA is damaged by genotoxic agents, the checkpointkinases are activated to either allow time for DNA repair or to initiateprogrammed cell death if the damage is irreparable (Bartek and Lukas,2003; Nyberg et al., 2002; Shiloh, 2003; Xu et al., 2002). Once DNA-damage is repaired, the checkpoint mechanism is silenced so as toallow the cell to resume cycling. In order to firemitosis-inducing CDK–Cyclin complexes, the activity of CDC25 phosphatases must beactivated, while inhibiting kinases such as WEE1, are inactivated.CDC25B has further been shown to be essential in the recovery of thecell from DNA damage-induced checkpoint arrest (Bugler et al., 2006;van Vugt et al., 2004; van Vugt and Medema, 2004). PLK1 has beensimilarly implicated in the recovery from checkpoint arrest and inpromoting mitotic entry through the degradation of the MYT1/WEE1kinases and of claspin by the proteasome pathway (Mamely et al.,2006; van Vugt et al., 2004; Watanabe et al., 2005). PLK3 has beenproposed to participate in the checkpoint response via the p53pathway, but whether PLK3 is also involved in checkpoint recoveryremains to be determined (van de Weerdt and Medema, 2006).

Fig. 1. Expression of PLK1 and cell cycle progression in U937 AML cells treated withVP-16. U937 cells were exposed to VP-16 (5 μM) for 1 h, released into fresh media andthen harvested at the indicated time points. (A) Cells were fixed and phospho-histoneH3-positive cells were analyzed by flow cytometry. (B) Western blotting analyseswere performed using anti-PLK1 and -actin antibodies, and cell cycle distributionsdetermined by flow cytometry analyses of DNA content.
103C. Didier et al. / European Journal of Pharmacology 591 (2008) 102–105
Disease relapse and resistance of Acute Myelocytic Leukaemia(AML) to chemotherapy is thought to be associated with increased G2/M checkpoint stringency and the inhibition of checkpoint recovery(Blagosklonny, 2006; Didier et al., 2008). In this study, we investigatedthe functional consequences of the pharmacological inhibition ofPLK1, in Acute Myelocytic Leukaemia. We have examined the role ofthe PLK1 kinase in regulating checkpoint activation induced by a DNA-damaging agent, and have characterized the effect of PLK1 inhibitionby GW843682X on the cytotoxicity associated with DNA-damage inleukaemic cell lines and primary leukaemia blasts from AML patients.
2. Materials and methods
2.1. Cell culture
The human leukaemia monocytic cell line U937 was obtained fromthe German Collection of Microorganisms and Cell Cultures(Braunschweig, Germany). Cells were grown in RPMI-1640 mediumsupplemented with 10% fetal bovine serum (U937) in the presence of2 mM L-glutamine and 100 units/ml penicillin/100 mg/ml streptomy-cin. The cells were passaged into fresh medium every 2 or 3 days andmaintained at 3×105 cells/ml in a humidified 5% CO2 incubator at37 °C.
2.2. Pharmacologic inhibitors and reagents
The PLK1 inhibitor GW843682X (5-(5,6-dimethoxy-1H-benzimi-dazol-1-yl)-3-{[2-(trifluoromethyl)-benzyl]oxy}thiophene-2-carbox-amide) was kindly provided by J. Jackson (GlaxoSmithKline). Thepolyclonal anti-PLK1 antibody was purchased from Cell Signalling(Berverly, MA), polyclonal anti-phospho-histone H3 was from UpstateBiotechnology (Lake Placid, NY) and the monoclonal anti-actinantibody was from Sigma (St. Louis, MO). Other products werepurchased from Sigma (Saint Quentin-Fallavier, France).
2.3. Analyses of cell cycle progression and apoptosis
Cells were harvested and fixed in ice-cold 70% ethanol. Cells werepermeabilized with PBS-0.25% Triton X-100 and subsequentlystained with anti-phospho-histone H3 and propidium iodide asdescribed previously (Bugler et al., 2006). Apoptotic cells weredetected with Annexin V-FITC/7-AAD detection kit from BeckmanCoulter (Fullerton, CA) following the manufacturer's instructions.Data were collected using a BD FACSCalibur cytometer and analyzedusing the Cell Quest (Becton Dickinson) and Modfit (Verity Software)software packages.
2.4. Cell proliferation assay
The inhibition of cell proliferation was determined using acolorimetric assay WST1 based on cleavage of the tetrazolium saltWST1 by mitochondrial dehydrogenases in viable cells, leading toformazan formation (Roche Diagnostic). U937 cell lines were seededin 96-well plates at day 0 in RPMI-1640 supplemented with 10% fetalbovine serum in the presence of 2 mM L-glutamine and 100 units/mlpenicillin/100 mg/ml streptomycin. At day 1, cells were treated for48 h with increasing concentrations of GW843682X. These experi-ments were performed in triplicate.
2.5. Western blot analyses
Cells were harvested and lysed as described previously (Dutertre etal., 2004). Samples were separated on a 7.5% SDS-PAGE gel, transferredonto nitrocellulose membrane, and immmunostained. Antibodieswere used at the following dilution: anti-PLK1 (1:500) and anti-actin(1:10,000).
2.6. Colony formation analyses on AML patients cells
CFU-L were obtained from bone marrow aspirates from AMLpatients at diagnosis. The cells were cryopreserved after informedconsent. CFU-L cells were resuspended in H 4230 methyl-cellulosemedium complemented with 10% 5637-conditioned medium.GW843682X, VP-16 or DMSO were added at the indicated concentra-tions, and the cells were incubated at 37 °C for 7 days. Colonies withmore than 20 cells were scored.
3. Results
3.1. Expression of PLK1 and cell cycle progression in response to DNA-damage in U937 cells
In order to monitor mitotic entry in U937 AML cells in response toDNA-damage, we examined phospho-Histone H3 positivity by flowcytometry (Fig. 1A). As expected, upon 1 h treatment with thegenotoxic agent etoposide (VP-16) checkpoint activation-mediatedinhibition of mitotic entry was observed. However, by 24 h post-treatment, a significant fraction of cells had recovered and enteredmitosis.
We then similarly treated U937 cells and investigated the variationof PLK1 expression bywestern blot analysis, togetherwith a study of thecell cycle distribution. U937 cells had modest PLK1 expression levelimmediately after VP-16 treatment, which was significantly increasedby 24 h after treatment (Fig. 1B), concomitantly with a progressive exitof the cells from the checkpoint arrest and their progression intomitosis (Fig. 1A and B). This observation indicated that, in agreementwith the previously demonstrated role for PLK1 in checkpoint recoveryand re-entry into mitosis, an increase in PLK1 level potentiates therecovery of leukaemia cells from checkpoint arrest.
3.2. The PLK inhibitor GW843682X inhibits U937 AML cell proliferation
In order to inhibit the activity of PLK1, we used the newly describedspecific compoundGW843682X (Lansing et al., 2007).We found using a
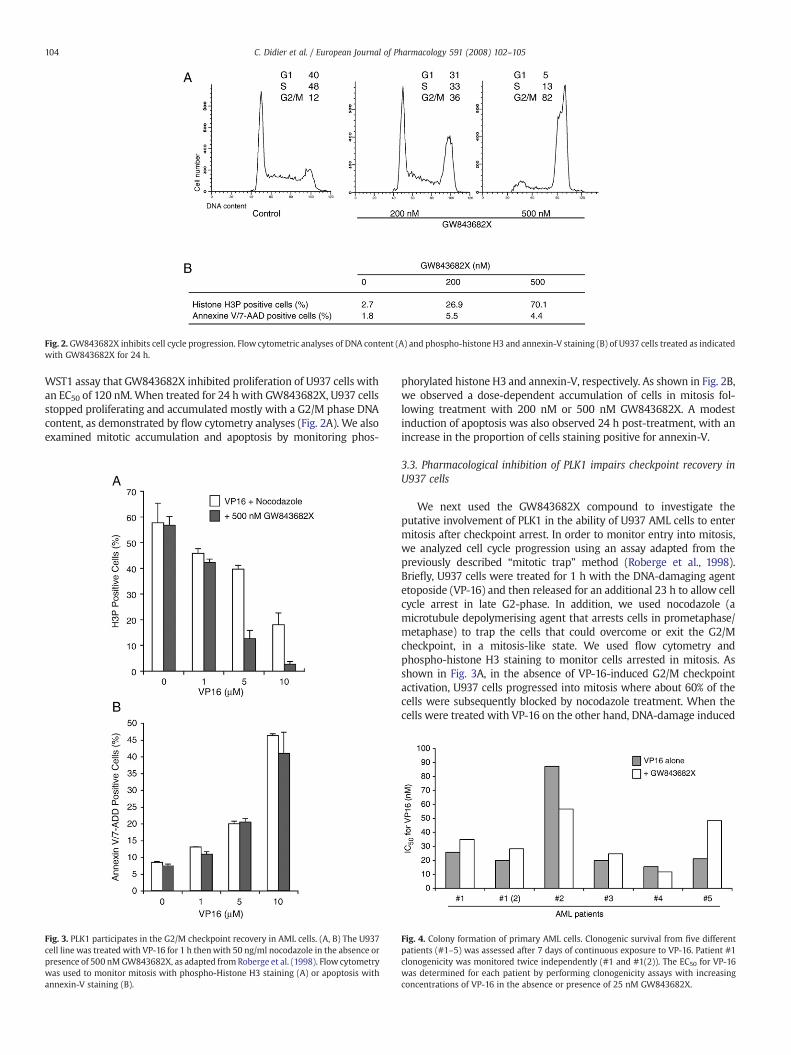
Fig. 2. GW843682X inhibits cell cycle progression. Flow cytometric analyses of DNA content (A) and phospho-histone H3 and annexin-V staining (B) of U937 cells treated as indicatedwith GW843682X for 24 h.
104 C. Didier et al. / European Journal of Pharmacology 591 (2008) 102–105
WST1 assay that GW843682X inhibited proliferation of U937 cells withan EC50 of 120 nM.When treated for 24 h with GW843682X, U937 cellsstopped proliferating and accumulated mostly with a G2/M phase DNAcontent, as demonstrated by flow cytometry analyses (Fig. 2A). We alsoexamined mitotic accumulation and apoptosis by monitoring phos-
Fig. 3. PLK1 participates in the G2/M checkpoint recovery in AML cells. (A, B) The U937cell line was treated with VP-16 for 1 h thenwith 50 ng/ml nocodazole in the absence orpresence of 500 nMGW843682X, as adapted from Roberge et al. (1998). Flow cytometrywas used to monitor mitosis with phospho-Histone H3 staining (A) or apoptosis withannexin-V staining (B).
phorylated histone H3 and annexin-V, respectively. As shown in Fig. 2B,we observed a dose-dependent accumulation of cells in mitosis fol-lowing treatment with 200 nM or 500 nM GW843682X. A modestinduction of apoptosis was also observed 24 h post-treatment, with anincrease in the proportion of cells staining positive for annexin-V.
3.3. Pharmacological inhibition of PLK1 impairs checkpoint recovery inU937 cells
We next used the GW843682X compound to investigate theputative involvement of PLK1 in the ability of U937 AML cells to entermitosis after checkpoint arrest. In order to monitor entry into mitosis,we analyzed cell cycle progression using an assay adapted from thepreviously described “mitotic trap” method (Roberge et al., 1998).Briefly, U937 cells were treated for 1 h with the DNA-damaging agentetoposide (VP-16) and then released for an additional 23 h to allow cellcycle arrest in late G2-phase. In addition, we used nocodazole (amicrotubule depolymerising agent that arrests cells in prometaphase/metaphase) to trap the cells that could overcome or exit the G2/Mcheckpoint, in a mitosis-like state. We used flow cytometry andphospho-histone H3 staining to monitor cells arrested in mitosis. Asshown in Fig. 3A, in the absence of VP-16-induced G2/M checkpointactivation, U937 cells progressed into mitosis where about 60% of thecells were subsequently blocked by nocodazole treatment. When thecells were treated with VP-16 on the other hand, DNA-damage induced
Fig. 4. Colony formation of primary AML cells. Clonogenic survival from five differentpatients (#1–5) was assessed after 7 days of continuous exposure to VP-16. Patient #1clonogenicity was monitored twice independently (#1 and #1(2)). The EC50 for VP-16was determined for each patient by performing clonogenicity assays with increasingconcentrations of VP-16 in the absence or presence of 25 nM GW843682X.

Fig. 5. Model for PLK1 involvement in the G2/M checkpoint pathway. Checkpointrecovery and entry into mitosis is dependent on the activity of the PLK1 kinase that(1) triggers proteasome-dependent degradation of the inhibitory WEE1 kinase and(2) presumably regulates the activity of the CDC25B phosphatase.
105C. Didier et al. / European Journal of Pharmacology 591 (2008) 102–105
a cell cycle arrest at the G2/M checkpoint and subsequent entry intomitosis was found to depend upon the concentration of VP-16.Interestingly, PLK1 inhibition by GW843682X further impaired theability of U937 cells to overcome the VP-16 induced cell cyclecheckpoint arrest and to enter into mitosis (Fig. 3A). For instance, weobserved more than 50% inhibition of entry into mitosis in cells treatedwith 5 µM VP-16 when combined with 500 nM GW843682X. Theseresults strongly suggest that, as described in other cell models, PLK1plays an important role during recovery from a G2 DNA-damage arrestin AML cell lines. However, no additive effect on apoptosiswas observedafter combining VP-16 and GW843682X (Fig. 3B).
3.4. Combined exposure of AML blast cells to VP-16 and GW843682Xresults in checkpoint reinforcement
We next examined whether the effect of PLK1 inhibition couldmodulate the survival of CFU-L primary leukaemic cells that had beenexposed to a treatment with VP-16. The EC50 values for VP-16 alone, or incombination with GW843682X (25 nM (Fig. 4) or 50 nM (data notshown)) were determined for five AML patients. These concentrations ofGW843682Xwereused in this experiment as theydisplayedno inhibitoryeffect, when used alone, on the clonogenicity efficiency. Of the 5 patientsamples examined, we observed a modest beneficial effect of associatingthe genotoxic agent VP-16with chemical inhibition of PLK1 in only 1 case,patient #2 (Fig. 4). In that case, the EC50 for VP-16 was reduced by about30% in the presence of GW843682X. In contrast, blast cells from patient#5 and to aminor extent, frompatients #1 and#3,weremore resistant toVP-16 treatment when associated with GW843682X than with VP-16alone. Thus, we conclude that, as far as CFU-L survival is concerned,inhibition of PLK appears to increase the EC50 and therefore modestlyreduces the efficiency of the genotoxic agent VP-16.
4. Discussion
On the basis of the already reported role for PLK1 in checkpoint exit(van Vugt et al., 2004) and of the results we obtained with the U937 cellline (Fig. 3), we speculated that PLK1 inhibition could also result incheckpoint reinforcement and increased resistance to genotoxictreatment. Indeed, we found this to be the case in leukaemic cellsfrom patients exposed to the PLK1 inhibitor GW843682X, in combina-tionwith the genotoxic agent VP-16 (Fig. 4). As it has been reported thatPLK3 performs opposite functions to PLK1 in the cellular response toDNA-damage (van deWeerdt andMedema, 2006; Xie et al., 2001a,b), itis unlikely that our observations result from the inhibition of PLK3. Wepropose that a consequence of PLK1 inhibition and subsequentperturbation of PLK1-dependent events, such as inhibition of WEE1degradation but persistent activation of the CDC25B phosphatase, maybe the emission of contradicting signals (see Fig. 5).
Acknowledgements
We thank R. Boutros for corrections and comments on the manu-script. We gratefully acknowledge GlaxoSmithKline for providing the
GW843682X compound. CD and CC are supported by a grant from theInstitut National du Cancer (PL103). This work was also supported by C.N.R.S, l'Université Paul Sabatier, la région Midi-Pyrénées and la LigueNationale Contre le Cancer (Equipe labellisée 2005) to BD.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.ejphar.2008.06.062.
References
Barr, F.A., Sillje, H.H., Nigg, E.A., 2004. Polo-like kinases and the orchestration of celldivision. Nat. Rev. Mol. Cell Biol. 5, 429–440.
Bartek, J., Lukas, J., 2003. Chk1 and Chk2 kinases in checkpoint control and cancer.Cancer Cell. 3, 421–429.
Blagosklonny, M.V., 2006. Target for cancer therapy: proliferating cells or stem cells.Leukemia 20, 385–391.
Bugler, B., Quaranta, M., Aressy, B., Brezak, M.C., Prevost, G., Ducommun, B., 2006.Genotoxic-activated G2-M checkpoint exit is dependent on CDC25B phosphataseexpression. Mol. Cancer Ther. 5, 1446–1451.
Didier, C., Cavelier, C., Quaranta, M., Galcera, M.O., Demur, C., Laurent, G., Manenti, S.,Ducommun, B., 2008. G2/M checkpoint stringency is a key parameter in thesensitivity of AML cells to genotoxic stress. Oncogene June 19;27 (27), 3811–20.
Dutertre, S., Cazales, M., Quaranta, M., Froment, C., Trabut, V., Dozier, C., Mirey, G.,Bouche, J., Theis-Febvre, N., Schmitt, E., Monsarrat, B., Prigent, C., Ducommun, B.,2004. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to theG2/M transition. J. Cell Sci. 117, 2523–2531.
Kneisel, L., Strebhardt, K., Bernd, A., Wolter, M., Binder, A., Kaufmann, R., 2002. Expressionof polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease.J. Cutan. Pathol. 29, 354–358.
Lansing, T.J., McConnell, R.T., Duckett, D.R., Spehar, G.M., Knick, V.B., Hassler, D.F., Noro,N., Furuta, M., Emmitte, K.A., Gilmer, T.M., Mook Jr., R.A., Cheung, M., 2007. In vitrobiological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol.Cancer Ther. 6, 450–459.
Liu, X., Lei, M., Erikson, R.L., 2006. Normal cells, but not cancer cells, survive severe Plk1depletion. Mol. Cell Biol. 26, 2093–2108.
Mamely, I., van Vugt, M.A., Smits, V.A., Semple, J.I., Lemmens, B., Perrakis, A., Medema, R.H.,Freire, R., 2006. Polo-like kinase-1 controls proteasome-dependent degradation ofClaspin during checkpoint recovery. Curr. Biol. 16, 1950–1955.
Nyberg, K.A., Michelson, R.J., Putnam, C.W., Weinert, T.A., 2002. Towardmaintaining thegenome: DNA damage and replication checkpoints. Annu. Rev. Genet. 36, 617–656.
Roberge, M., Berlinck, R.G., Xu, L., Anderson, H.J., Lim, L.Y., Curman, D., Stringer, C.M.,Friend, S.H., Davies, P., Vincent, I., Haggarty, S.J., Kelly, M.T., Britton, R., Piers, E.,Andersen, R.J., 1998. High-throughput assay for G2 checkpoint inhibitors andidentification of the structurally novel compound isogranulatimide. Cancer Res. 58,5701–5706.
Shiloh, Y., 2003. ATM: ready, set, go. Cell Cycle 2, 116–117.Spankuch, B., Heim, S., Kurunci-Csacsko, E., Lindenau, C., Yuan, J., Kaufmann, M.,
Strebhardt, K., 2006. Down-regulation of Polo-like kinase 1 elevates drug sensitivityof breast cancer cells in vitro and in vivo. Cancer Res. 66, 5836–5846.
Strebhardt, K., Ullrich, A., 2006. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev.Cancer 6, 321–330.
van de Weerdt, B.C., Medema, R.H., 2006. Polo-like kinases: a team in control of thedivision. Cell Cycle 5, 853–864.
van Vugt, M.A., Medema, R.H., 2004. Checkpoint adaptation and recovery: back withPolo after the break. Cell Cycle. 3, 1383–1386.
van Vugt, M.A., Bras, A., Medema, R.H., 2004. Polo-like kinase-1 controls recovery from aG2 DNA damage-induced arrest in mammalian cells. Mol. Cell 15, 799–811.
Watanabe, N., Arai, H., Iwasaki, J., Shiina, M., Ogata, K., Hunter, T., Osada, H., 2005.Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 viamultiple pathways. Proc. Natl. Acad. Sci. U.S.A. 102, 11663–11668.
Weichert, W., Schmidt, M., Gekeler, V., Denkert, C., Stephan, C., Jung, K., Loening, S.,Dietel, M., Kristiansen, G., 2004. Polo-like kinase 1 is overexpressed in prostatecancer and linked to higher tumor grades. Prostate 60, 240–245.
Xie, S., Wang, Q., Wu, H., Cogswell, J., Lu, L., Jhanwar-Uniyal, M., Dai, W., 2001a. Reactiveoxygen species-induced phosphorylation of p53 on serine 20 is mediated in part bypolo-like kinase-3. J. Biol. Chem. 276, 36194–36199.
Xie, S., Wu, H., Wang, Q., Cogswell, J.P., Husain, I., Conn, C., Stambrook, P., Jhanwar-Uniyal, M., Dai, W., 2001b. Plk3 functionally links DNA damage to cell cycle arrestand apoptosis at least in part via the p53 pathway. J. Biol. Chem. 276, 43305–43312.
Xu, B., Kim, S.T., Lim, D.S., Kastan, M.B., 2002. Two molecularly distinct G(2)/Mcheckpoints are induced by ionizing irradiation. Mol. Cell Biol. 22, 1049–1059.
Yamada, S., Ohira, M., Horie, H., Ando, K., Takayasu, H., Suzuki, Y., Sugano, S., Hirata, T.,Goto, T., Matsunaga, T., Hiyama, E., Hayashi, Y., Ando, H., Suita, S., Kaneko, M., Sasaki,F., Hashizume, K., Ohnuma, N., Nakagawara, A., 2004. Expression profiling anddifferential screening between hepatoblastomas and the corresponding normallivers: identification of high expression of the PLK1 oncogene as a poor-prognosticindicator of hepatoblastomas. Oncogene 23, 5901–5911.
Zimmerman, W.C., Erikson, R.L., 2007a. Finding Plk3. Cell Cycle 6, 1314–1318.Zimmerman, W.C., Erikson, R.L., 2007b. Polo-like kinase 3 is required for entry into S
phase. Proc. Natl. Acad. Sci. U.S.A. 104, 1847–1852.

145
Annexe 3 : Protocole HRM
Les ADN des 50 échantillons de LAM collectés au sein du laboratoire d’hématologie du CHU
Toulouse-Purpan ont été dosés à l’aide d’un spectrophotomètre NanoDrop (Thermo
Scientific) et ajustés à la concentration de 10ng/µL. Ces ADN ont été ensuite préparés
selon la technique HRM décrite dans le protocole Roche (LightCycler 480 Hight Resolution
Melting Master) en utilisant les proportions suivantes :
Le design de l’ensemble des couples d’amorces a été effectué par le Pr. E. Delabesse, et
nous a permis une amplification :
- des exons 2 à 11 du gène p53
- des exons 2 et 3 des gènes nras et kras
- de l’exon 20 de flt3
La technique HRM consiste en une pré-amplification par PCR comprenant une dénaturation
initiale de 10 minutes à 95°C, suivie de 50 cycles de PCR (10 secondes à 95°C, 15 secondes
entre 63 et 58°C puis 25 secondes à 72°C). L’amplicon amplifié va subir une dénaturation
par élévation de la température de 72 à 95°C, au cours de laquelle le Light Cycler 480
effectue 25 mesures de la fluorescence émise par la sonde intercalante de l’ADN double
brin (contenue dans le kit LC480 HRM Master MIX) par degré, cela permet d’établir une
courbe de fusion qui rend compte de l’état de dénaturation de l’échantillon.
Courbes de fusion obtenues pour l’exon 7 du gène p53
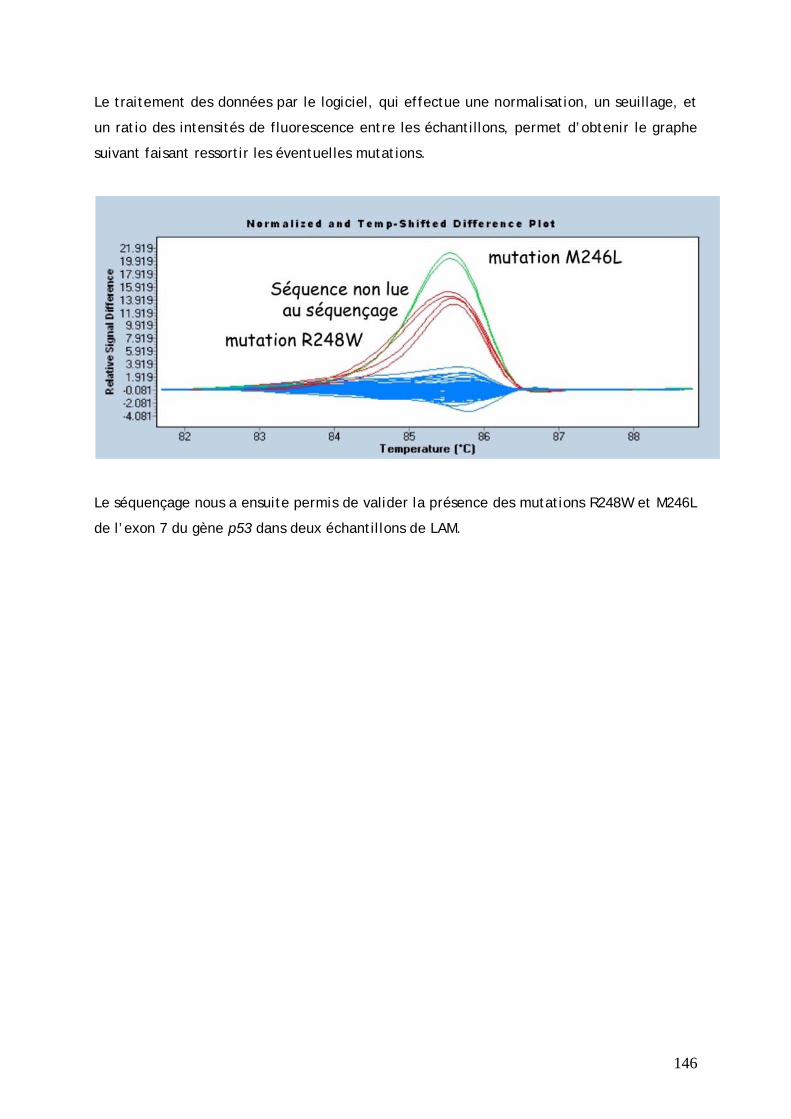
146
Le traitement des données par le logiciel, qui effectue une normalisation, un seuillage, et
un ratio des intensités de fluorescence entre les échantillons, permet d’obtenir le graphe
suivant faisant ressortir les éventuelles mutations.
Le séquençage nous a ensuite permis de valider la présence des mutations R248W et M246L
de l’exon 7 du gène p53 dans deux échantillons de LAM.

147

AUTEUR : Cindy Cavelier TITRE : Etude du point de contrôle des dommages à l’ADN dans les Leucémies Aiguës Myéloïdes et ciblage pharmacologique DIRECTEURS DE THESE : Pr Bernard Ducommun et Dr cécile Demur LIEU ET DATE DE SOUTENANCE : CHU Toulouse-Purpan, le 4 mai 2010 RESUME : Les Leucémies Aiguës Myéloïdes (LAM) représentent un groupe hétérogène de pathologies développées aux dépend des progéniteurs myéloïdes immatures normaux, caractérisées sur le plan moléculaire par des modifications de l'expression de facteurs transcriptionnels aboutissant à un blocage de la maturation, et par une activation aberrante de molécules de la transduction du signal conférant aux cellules leucémiques des avantages prolifératifs et de survie. Dans ce travail, nous avons montré que la protéine kinase CHK1, impliquée dans le maintien de l’intégrité du génome en contrôlant l’arrêt du cycle cellulaire à la transition G1/S et G2/M après dommages constitutifs à l’ADN, prolonge son activation après dommages dans les cellules de LAM immatures permettant ainsi leur maintien en phase G2. Cette meilleure efficacité du point de contrôle G2/M des cellules immatures contribue très probablement à la chimiorésistance naturelle observée et largement documentée des cellules leucémiques immatures. De plus, nous montrons que la kinase CHK1 est anormalement activée dans les échantillons de LAM en raison de la présence de dommages à l'ADN constitutifs dans ces cellules. L’étude des caractéristiques cytogénétiques des patients nous a permis de mettre en évidence un haut niveau de dommages dans les LAM de haut risque cytogénétique, en particulier celles avec des cellules à caryotype complexe. L’inactivation de CHK1 par l’inhibiteur UCN-01 et par interférence à l'ARN aboutit à une inhibition des capacités clonogènes des progéniteurs leucémiques et à une chimiosensibilisation vis-à-vis de l’agent chimiothérapeutique ara-C. En revanche, les cellules hématopoïétiques normales CD34+, qui ne présentent pas de dommages constitutifs, sont beaucoup moins sensibles à l’UCN-01. Cet agent n’affecte pas non plus les capacités clonogènes des progéniteurs granulo-monocytaires normaux aux doses utilisées sur les cellules de LAM. Ces résultats nous ont permis de montrer que la kinase CHK1 joue un rôle important à la fois dans la chimiorésistance des cellules leucémiques immatures et dans l’établissement de l’instabilité génétique observée dans les LAM à caryotype complexe. Enfin, ces travaux ouvrent des perspectives intéressantes sur l’utilisation de composés inhibiteurs des kinases du point de contrôle du cycle cellulaire dans les LAM et en particulier celles présentant le plus mauvais pronostic, les LAM à caryotype complexe. MOTS-CLES : leucémies aiguës myéloïdes, cycle cellulaire et points de contrôle, kinase CHK1, dommages à l’ADN, inhibiteurs pharmacologiques.





























