Embed Size (px)
Citation preview

i
UNIVERSIDAD DE CÓRDOBA
INSTITUTO DE ESTUDIOS DE POSTGRADO
MÁSTER EN BIOTECNOLOGÍA
Trabajo Fin de Máster
“Optimización de técnicas de proteómica ‘shotgun’
para la caracterización del proteoma de encina
(Quercus ilex)”
Isabel María Gómez Gálvez
Departamento de Bioquímica y Biología Molecular
Director: Jesús Valentín Jorrín Novo
Codirectora: M Ángeles Castillejo Sánchez
Córdoba, 06/2017


iii
Propuesta de Revisores: Jesús Valentín Jorrín Novo y M. Ángeles Castillejo Sánchez, directores del trabajo de fin de
máster titulado “Optimización de técnicas de proteómica ‘shotgun’ para la caracterización del
proteoma de encina (Quercus ilex)” proponen como posibles revisores de este a:
- Luis Valledor González. Biología de Organismos y Sistemas. Universidad de Oviedo.
- Jose Valero Galván. Departamento de Ciencias Básicas. Universidad Autónoma de
Ciudad Juárez, México. ([email protected])
- María Cristina Romero Rodríguez. Fitoquímica (Facultad de Ciencias Químicas).
Universidad Nacional de Asunción, Paraguay. ([email protected])
Jesús V. Jorrín Novo M. Ángeles Castillejo Sánchez
Fdo.: Fdo.:


v
Agradecimientos
Este Trabajo Fin de Máster, ha supuesto un esfuerzo, en el cual directa o
indirectamente han participado un gran número de personas ya sea opinando, corrigiendo,
dando ánimo o acompañando en los momentos en los que parecía que todo salía mal.
Además, este trabajo me ha permitido aprender, no solo en el campo profesional, sino
también a nivel personal y por eso agradezco a todas aquellas personas que de una u otra
forma han formado parte de este período.
En primer lugar, me gustaría agradecer a mi familia. Su gran esfuerzo y dedicación ha
permitido formarme y poder llegar a realizar este Trabajo Fin de Máster. Siempre han sido un
gran punto de apoyo, estando en todo momento a mi lado y ayudándome con todo lo que
estaba al alcance de su mano para facilitarme el camino.
A mis tutores, Jesús y M. Ángeles. Aunque el tiempo no ha estado especialmente a
nuestro favor, ellos han estado ahí en todo momento apoyándome y ayudándome a mejorar.
Resaltar su gran paciencia, y su disponibilidad frente a cualquier problema o duda que surgiera
durante el transcurso del trabajo, siempre dando palabras de apoyo.
A Maricarmen, por asegurarse de que todo estuviera en las mejores condiciones,
brindando toda la ayuda que fuera necesaria para que pudiéramos conseguir los mejores
resultados en nuestro trabajo. También me gustaría agradecer a Ana y Manolo, por todos los
conocimientos que me han aportado durante mi tiempo en el grupo y por estar siempre
dispuestos a ayudar.
Por último, no me puedo olvidar de mis compañeros. Cristina, gracias por estar
siempre dispuesta a echar una mano. Incluso cuando no íbamos muy bien de tiempo, siempre
has hecho un hueco para mí, y mis miles de análisis. A Kamilla, María Eugenia, Víctor, Rosa,
Conchi, Patricia, Fabiola, David y a muchas personas más que han formado parte de esta
familia. Gracias por ser un gran punto de apoyo, no solo a nivel académico, sino también a
nivel personal.
Esto supone el cierre de una etapa más, ha sido un placer vivirla con vosotros. Me llevo
grandes recuerdos de cada uno, y me siento muy afortunada por haber podido convivir con
esta gran familia, de la que estoy segura de que nunca me olvidaré. ¡GRACIAS POR TODO!


1
ÍNDICE
Índice de figuras ......................................................................................................................... 3
Índice de tablas ........................................................................................................................... 3
Resumen ..................................................................................................................................... 5
Objetivos ..................................................................................................................................... 7
Introducción ............................................................................................................................... 9
Materiales y métodos ............................................................................................................... 14
Material vegetal .................................................................................................................... 14
Diseño experimental ............................................................................................................. 15
Extracción y cuantificación de proteínas .............................................................................. 15
Espectrometría de masas...................................................................................................... 16
Identificación y cuantificación de proteínas ......................................................................... 18
Resultados y discusión .............................................................................................................. 19
Cuantificación de proteínas en extracto y en gel ................................................................. 19
Identificación de proteínas y agrupamiento funcional ......................................................... 20
Calidad de la identificación ................................................................................................... 23
Cuantificación de péptidos y proteínas ................................................................................ 25
Conclusiones ............................................................................................................................. 27
Referencias ............................................................................................................................... 28


3
Índice de figuras Figura 1. Flujo de trabajo en un experimento proteómico. ..................................................... 12
Figura 2. Material vegetal empleado ....................................................................................... 15
Figura 3. Cuantificación de proteínas en extracto y en gel ...................................................... 20
Figura 4. Árbol filogenético de familias botánicos para los que se obtuvo al menos una
identificación de ortólogos ....................................................................................................... 21
Figura 5. Número de péptidos y proteínas identificadas, usando las bases de datos de
UniprotKB-Viridiplantae y Quercus ilex. ................................................................................... 22
Figura 6. Diagrama de Venn mostrando el número de péptidos y proteínas identificadas a
partir de las dos bases de datos empleadas. ............................................................................ 22
Figura 7. Agrupación funcional de las proteínas identificadas................................................. 23
Figura 8. Parámetros de confianza en la identificación de proteínas, incluyendo número de
péptidos, % de cobertura y score. ............................................................................................ 24
Figura 9. Cuantificación relativa de proteínas, límite de detección y rango dinámico. ........... 25
Índice de tablas Tabla 1. Rendimiento de la extracción de proteínas para diferentes órganos ........................ 19
Tabla 2. Número de péptidos y proteínas identificados ......................................................... 21


5
Resumen
El presente trabajo fin de máster, titulado “Optimización de técnicas de proteómica
‘shotgun’ para la caracterización del proteoma de encina (Quercus ilex)” ha tenido como
objetivo la puesta a punto y optimización de técnicas de proteómica “shotgun” para su
posterior empleo en el estudio de diferentes aspectos de la biología de la encina (Quercus
ilex). Como cualquier técnica analítica, la proteómica ha de ser caracterizada, optimizada y
validada para cada sistema experimental. Antes de dar una interpretación biológica a los
resultados obtenidos se deben de plantear una serie de preguntas, entre otras: ¿Qué estamos
identificando y cómo de fiable es la identificación? ¿Cómo de válida es la cuantificación? ¿Cuál
es el límite de detección y el rango dinámico?
Se utilizó una muestra biológica resultante de la mezcla de extractos de diferentes
órganos de la planta (embrión, cotiledón, raíz y hoja), obtenidos mediante el método TCA-
acetona/fenol. La muestra se sometió a electroforesis 1-D (SDS-PAGE), llevándose a cabo la
cuantificación de proteínas a nivel de extracto y de gel. La banda única de proteínas se digirió
con tripsina y los péptidos resultantes se analizaron por nLC-MS/MS (Orbitrap, Q-OT-qIT). Se
llevó a cabo un ensayo de curva dilución-respuesta, en el rango 10-1000 ng equivalente de
seroalbúmina bovina (BSA). Para cada dilución se determinó el número de péptidos y
proteínas identificadas, los valores de confianza (número de péptidos, % de cobertura y score)
y los valores de cuantificación, límite de detección y rango dinámico. Para la identificación se
utilizó el programa Proteome Discoverer y dos bases de datos, la de UniProtKB-Viridiplantae
y otra construida a partir del transcriptoma de Quercus ilex (Guerrero-Sanchez et al., 2017).
El número de hits, ortólogos o productos génicos identificados dependió de la
concentración de proteínas y de la base de datos empleada. Los valores de confianza
obtenidos para las diferentes identificaciones fueron de 1-35 péptidos por proteína, 1-93 %
de cobertura y 1-355 de score. Los mejores resultados en cuanto a número de proteínas
identificadas y parámetros de confianza se obtuvieron con la concentración de 600 ng
equivalentes de BSA. La cuantificación relativa de proteínas se obtuvo a partir del área de pico,
encontrándose el límite de detección por debajo de 100 ng y el rango dinámico entre 100 y
1000 ng equivalente de BSA.
Palabras clave: Proteómica “shotgun”; Quercus ilex; parámetros de confianza, optimización
metodológica


7
Objetivos
El objetivo general del trabajo fin de máster fue la optimización de técnicas de proteómica
“shotgun” en la caracterización del proteoma de la encina, complementado así trabajos
previos del Grupo de Investigación “Bioquímica, Proteómica y Biología de Sistemas Vegetal y
Agroforestal” (ARG-164) en los que se emplearon técnicas de proteómica basadas en
electroforesis en gel.
Los objetivos específicos haciendo referencia a las distintas etapas de flujo de trabajo
fueron los siguientes:
i. Establecer el diseño experimental. Empleo de extractos resultado de la mezcla
obtenida a partir de diferentes órganos de la planta (embrión, cotiledón, raíz y
hoja).
ii. Analizar las distintas diluciones del extracto de proteínas en el rango 10-1000 ng
equivalente de BSA por espectrometría de masas nLC-MS/MS (Orbitrap Fusion, Q-
OT-qIT).
iii. Determinar el efecto de la cantidad de proteína y de la base de datos (UniprotKB-
Viridiplantae y Quercus ilex) sobre el número de proteínas identificadas y los
parámetros de confianza (número de péptidos, % de cobertura y valor de score).
iv. Establecer parámetros de cuantificación relativa, límite de detección y rango
dinámico.


9
Introducción
Tres son los elementos que definen un proyecto de investigación, el sistema experimental,
la hipótesis u objetivos y la metodología. El presente trabajo se centra en el aspecto
metodológico y tiene como objetivo la puesta a punto, optimización y validación de técnicas
de proteómica “shotgun”1 para el análisis del proteoma de la encina y su posterior aplicación
a estudios de diferentes aspectos de la biología de dicha especie (variabilidad poblacional,
germinación de semillas y respuesta a estreses).
El trabajo queda encuadrado dentro de la línea de investigación sobre aproximaciones
moleculares al estudio de especies forestales, que se lleva a cabo en el Grupo de Investigación
“Bioquímica, Proteómica y Biología de Sistemas Vegetal y Agroforestal” (ARG-164). Este
presenta como novedad el empleo de las técnicas “shotgun” o libre de geles, las cuales
muestran una serie de ventajas sobre las técnicas basadas en gel utilizadas hasta el momento
(Romero-Rodríguez, 2015), como son: un mayor rendimiento en la identificación, mayor
rapidez de procesamiento y menor manipulación de la muestra. Cabe destacar, que ambas
técnicas se han descrito como complementarias en estudios proteómicos de sistemas
biológicos (Jorrín-Novo, 2014).
Un aspecto importante a tener en cuenta es que antes de realizar una interpretación
biológica de las proteínas identificadas, debemos de plantearnos una serie de preguntas: ¿Qué
estamos identificando y qué tan segura es la identificación? La calidad de la identificación en
cualquier análisis proteómico de espectrometría de masas es un factor crítico para obtener
tantas identificaciones de alta calidad como sea posible. Por tanto, es importante diferenciar
entre un hit y la identificación de ortólogos o productos génicos, así como establecer unos
valores mínimos en los parámetros de confianza obtenidos (el valor de score, el % de
cobertura y el número de péptidos identificados), dependiendo todo esto principalmente de
la base de datos utilizada (general o específica de especie). Si la respuesta a estas preguntas
no es suficientemente clara y la aproximación no es validada, nuestros resultados sólo serán
especulativos y nuestras conclusiones probablemente erróneas. Por este motivo, es necesaria
una optimización de los métodos y protocolos empleados para cada sistema biológico.
1 El término “shotgun” no está registrado en el diccionario de la RAE, pudiendo traducirse al castellano como “análisis masivo” o “análisis a gran escala”. Al ser el término en inglés bien entendido por la comunidad científica se ha decidido no traducirlo, a pesar de estar cometiendo un error gramatical o lingüístico.

La encina (Quercus ilex subsp. ballota [Desf.] Samp.) es la especie predominante en los
ecosistemas forestales de la cuenca del Mediterráneo occidental (Valero-Galván et al., 2014).
Presenta un interés tanto ecológico como económico. Su producto, la bellota, es el principal
componente de la dieta de un gran número de animales, algunos con un alto interés desde el
punto de vista del consumo humano, como es el caso del cerdo. Además, desde el punto de
vista climático, destaca su adaptación a climas de altas temperaturas (Simova-Stoilova et al.,
2015), siendo de gran interés ecológico en nuestra zona. Por otro lado, su biología, en
particular a nivel molecular, es en gran medida desconocida (Jorrín-Novo et al., 2014).
Estudios previos han demostrado la existencia de una elevada variabilidad poblacional y
polimorfismo en Quercus spp. Entre ellos se han realizado estudios anatómicos y
morfométricos (Castro-Díez et al., 1997), químicos (Valero-Galván et al., 2010), de
marcadores de ADN (Lumaret et al., 2009) y proteínas (Valero-Galván et al., 2011) entre
otros.
El bosque mediterráneo en general y la dehesa en particular se enfrenta a diversos
problemas que requieren solución. Los problemas históricos de sobreexplotación, así como
los actuales de pérdida de masa forestal por enfermedades y estreses ambientales (como el
síndrome de la seca) y los asociados a un escenario de futuro cambio climático, hacen que el
estudio de su biología pase a ser una prioridad de cara a diseñar programas de manejo y
conservación que aseguren su valor medioambiental (Jorrín-Novo et al., 2014).
El área de la biología y bioquímica molecular tiene algo que decir en cuanto a estos
objetivos. De todas las actuaciones biotecnológicas que se pueden utilizar en el ámbito
agroforestal, la caracterización de la biodiversidad y la selección de genotipos “élite” o “plus”
son las vías más adecuadas o viables. Esto es debido a las características de las especies
forestales (ciclos de vida muy largos y no domesticadas), lo que dificulta los programas de
mejora clásica. En dicho marco, la selección de individuos con determinadas características
fenotípicas, como las de adaptación y tolerancia a condiciones ambientales adversas (sequía
y patógenos, fundamentalmente) (Simova-Stoilova et al., 2015), sería un logro importante.
Por su parte, el estudio proteico puede llevarnos a la identificación de proteínas que, por su
actividad biológica y potencial, pueden darle valor añadido a la especie de estudio.
Este trabajo fin de máster se encuadra dentro de estos objetivos. La aproximación
biotecnológica requiere basarse en el estudio y conocimiento a nivel molecular. Para ello se

11
plantea el uso de técnicas de última generación como son las -ómicas, para caracterizar la
variabilidad existente y entender la respuesta a estreses ambientales. El análisis holístico
global, nos llevará a identificar marcadores, en nuestro caso proteínas, que se puedan utilizar
en la identificación y selección de individuos “plus”. El uso de la proteómica se justifica en que
es complementaria a otras disciplinas y aporta la información que no puede ser revelada por
otras técnicas como la genómica o transcriptómica. Es decir, la genómica nos da la idea de la
información contenida en un genoma, pero no toda esta información se expresa, y no siempre
existe una buena correlación entre transcritos y proteínas (Jorrín-Novo et al., 2014).
La proteómica, término acuñado en 1995 por Wilkins, tiene como objetivo el estudio del
proteoma, y se definió originalmente como “PROTein complement expressed by a genOME”,
es decir, el conjunto de proteínas expresadas por un genoma. A diferencia del genoma, el
proteoma es dinámico, y por tanto específico del tipo celular, estado de desarrollo y
condiciones ambientales (Wilkins et al, 1996).
En un sentido más amplio, el proteoma puede definirse como el conjunto total de especies
proteicas presentes en una unidad biológica (orgánulo, célula, tejido, órgano, individuo,
especie o ecosistema) en cualquier etapa de desarrollo y bajo condiciones ambientales
específicas. Mediante el uso de la proteómica, se espera saber cómo, dónde, cuándo y para
qué son los varios cientos de miles de especies de proteínas individuales producidas en un
organismo vivo, cómo interactúan entre sí y con otras moléculas, y cómo trabajan unas con
otras para encajar en el crecimiento programado y el desarrollo, e interactuar con su entorno
biótico y abiótico (Abril et al., 2011).
En general, el flujo de trabajo de un análisis típico de proteómica (Figura 1) consta de una
serie de etapas fundamentales; (1) diseño experimental y muestreo, (2) extracción de
proteínas, (3) separación de proteínas mediante técnicas de electroforesis en gel, (4) digestión
de las proteínas con el uso de tripsina, (5) análisis de los péptidos resultantes mediante
espectrometría de masas, (6) identificación y cuantificación de proteínas, empleando
diferentes algoritmos y bases de datos y (7) validación e interpretación biológica de los
resultados.
Cabe destacar, que la proteómica está cambiando en escala y enfoque, desde su objetivo
inicial de identificar tantas proteínas individuales como fuera posible en una muestra biológica

dada, a analizar la dinámica del proteoma. Debido al gran avance de los equipos de
espectrometría de masas, los cuales cada vez son más precisos y generan mayor cantidad de
información, se requieren de potentes herramientas computacionales que sean capaces de
gestionar e integrar toda la información generada (Barbier-Brygoo et al., 2004).
Sin embargo, el potencial de la proteómica está lejos de ser totalmente explotado en la
investigación con especies vegetales. Sólo se ha investigado un bajo número de especies de
plantas a nivel proteico y, principalmente se han utilizado estrategias basadas en 2-DE
acopladas a espectrometría de masas, lo que resulta en una baja cobertura del proteoma
(Abril et al.,2011).
La cromatografía líquida (LC) acoplada a la espectrometría de masas, también denominada
“shotgun”, “gel-free” o libre de gel, es una alternativa emergente a la 2-DE para la separación
de proteínas, aunque ambas pueden combinarse en un solo experimento (Canovas et al.,
2004).
Los métodos “gel-free” proporcionan diseños experimentales más simples, requiriendo
menos manipulación de la muestra. Además, gracias al desarrollo de espectrómetros de
masas de alta resolución y alta precisión, se ha convertido en un enfoque importante en el
análisis cuantitativo de muestras biológicas. Estas técnicas, permiten una mayor cobertura del
proteoma, así como una estimación de la cantidad de proteínas en términos absolutos
(Gonzalez-Fernandez et al., 2013). Esta nueva herramienta tiene un gran potencial para llevar
Figura 1. Flujo de trabajo en un experimento proteómico. Se ilustran las diferentes etapas: (1) diseño experimental y muestreo, (2) extracción de proteínas, (3) separación de proteínas mediante técnicas de electroforesis en gel, (4) digestión de las proteínas con tripsina, (5) análisis de los péptidos resultantes mediante espectrometría de masas, (6) identificación y cuantificación de proteínas, mediante el empleo de algoritmos y bases de datos y (7) validación e interpretación biológica de los resultados.

13
a cabo el estudio de la expresión de proteínas a gran escala y la caracterización de sistemas
biológicos complejos (Davalieva et al., 2016).
Estos métodos obvian la etapa de separación de proteínas previa a la digestión con
proteasas (comúnmente tripsina), utilizándose extractos totales de proteínas. Los péptidos
resultantes se separan mediante cromatografía líquida (LC) acoplada a un equipo de
espectrometría de masas (LC-MS/MS). Se puede conseguir una mayor resolución incluyendo
etapas de pre-fraccionamiento antes de LC-MS/MS. Los métodos de pre-fraccionamiento
pueden incluir otros tipos de cromatografías, separación inicial por isoelectroenfoque (IEF), o
mediante geles 1-DE SDS PAGE (Davalieva et al, 2016).
Para la identificación de proteínas se utilizan software que requieren de una base de datos.
Si nuestro organismo no ha sido previamente anotado, nuestra búsqueda será por homología,
por lo que sólo nos permitirá hipotetizar o especular acerca de la identificación y función de
nuestras proteínas. Esto conlleva a que se obtenga un menor número de identificaciones, o
con parámetros de confianza de peor calidad debido a la presencia de proteínas específicas
de especie que no se encuentran representadas en esas bases de datos, o bien que a lo largo
de la evolución han sufrido diversas modificaciones en su secuencia (Romero-Rodríguez et al.,
2014). Para resolver este problema en especies no modelo, como es el caso de Quercus ilex,
la mejor opción es la creación de una base de datos específica a partir de transcritos de nuestra
especie. En este caso, las identificaciones corresponderán a productos génicos de la especie
en cuestión.
Además de la identificación, las técnicas “shotgun” están siendo aplicadas cada vez con
mayor frecuencia para la cuantificación de péptidos y proteínas en estudios de proteómica.
Podemos encontrar principalmente dos aproximaciones de cuantificación: por área de pico o
“peak area” y por conteo de espectros de masas o “spectral count” (Zhu et al., 2010; Xie et
al., 2011). En el caso del “Peak area”, un ion con una determinada masa/carga es detectado y
guardado con una intensidad concreta. Se ha observado que dicha intensidad se correlaciona
con la concentración del ion, aumentando la misma a medida que la cantidad de proteína
incrementa. Por su parte, la aproximación basada en “Spectral count”, consiste en un recuento
del número de espectros identificados del mismo péptido o proteína en cada uno de los
múltiples conjuntos de datos. Esto es posible ya que un aumento en la cantidad de proteína,
normalmente se relaciona con un aumento en el número de sus péptidos proteolíticos (Xie et

al., 2011). Este posible incremento de los sitios de corte tras la digestión supone un aumento
en la cobertura, el número de péptidos únicos identificados y el número total de espectros
MS/MS identificados (“spectral count”) para cada proteína.
Estas nuevas herramientas han sido ampliamente utilizadas en otros sistemas biológicos
como especies modelo y cultivos agronómicos (revisados en Jorrín et al., 2009), sin embargo,
existen pocos trabajos en los que se haya aplicado al estudio de especies forestales en general,
siendo en la encina casi inexistentes. Nuestro grupo ha iniciado un estudio dirigido a este fin,
siendo necesaria la optimización del flujo de trabajo con el objetivo de mejorar la calidad del
análisis y poder dar una interpretación correcta a nuestros resultados.
Materiales y métodos
Material vegetal
Los frutos de Q. ilex (bellotas) procedentes de la localidad de Aldea de Cuenca (Fuente
Obejuna, Córdoba; coordenadas UTM Huso 30 ETRS89, X:28857; Y:4238235.9), se
recolectaron en diciembre del 2015. Previo a su germinación, se seleccionaron aquellos que
no estaban dañados y de un tamaño homogéneo, siendo posteriormente limpiados y
esterilizados. Se siguió el protocolo establecido por Bonner et al. (1987).
Las bellotas, se sumergieron en agua, eliminando aquellas que flotaban, ya que estas
correspondían a frutos dañados (avellanados, con insectos). Para su esterilización, las bellotas
se transfirieron a una solución de hipoclorito de sodio al 2,5% (v/v) con unas gotas de Tween
20, y se dejaron en dicha solución durante 20 minutos. Finalmente, se lavaron
abundantemente con agua, se secaron y se almacenaron a 4°C en bolsas de polietileno
herméticamente cerradas (Caliskan, 2014; Corbineau et al., 2014).
Previamente a la germinación, y para que ésta fuera homogénea y sincronizada, se eliminó
el pericarpo y la porción distal (respecto al embrión) de las bellotas. Se sembraron en macetas
de 0,5 L conteniendo perlita y se llevaron, para su germinación y crecimiento, al invernadero
(Figura 2). Las condiciones durante este fueron: temperatura media, día/noche de 35/19⁰C, y
humedad relativa inferior al 43%. Las plantas se regaron periódicamente a capacidad de campo
con agua corriente, y una vez con solución nutritiva (macronutrientes FOLE 8-8-8 (Solem) y
micronutrientes microplus Fertiberia Jardín) (Hoagland et al., 1950).

15
Diseño experimental
Se preparó un extracto de proteínas que resultó de mezclar extractos obtenidos a partir
de diferentes órganos, incluyendo embrión (unos 200 mg), cotiledón, raíz y hojas (unos 500
mg de cada uno de ellos).
El embrión y los cotiledones se obtuvieron de semillas germinadas, y la raíz y las hojas de
plántulas de 4 meses (estadío de desarrollo de 10 hojas). Cada órgano (embrión, cotiledón,
raíz y hojas) fue congelado individualmente en nitrógeno líquido y almacenado a - 80⁰C hasta
su análisis.
Extracción y cuantificación de proteínas
Cada órgano fue homogeneizado independientemente con nitrógeno líquido en un
mortero, hasta obtener un polvo fino.
Se utilizó un método de precipitación basado en TCA-acetona-fenol, siguiendo el protocolo
de Wang et al. (2006), con algunas modificaciones. Al material previamente pulverizado en
nitrógeno líquido se le añadió una solución de 10% TCA en acetona. La suspensión se maceró
en mortero y posteriormente se sonicó (sonicador P Selecta Ultrasons) durante 10 min a la
máxima potencia. La mezcla se dejó a -20⁰C durante toda la noche. El precipitado se recuperó
por centrifugación (10000 g, 10 min, 4 ⁰C). Tras la eliminación del sobrenadante, el pellet se
Figura 2. Material vegetal empleado. Se obtuvo una muestra biológica resultado de la mezcla de extractos obtenidos a partir de diferentes órganos, incluyendo semillas germinadas (embriones y cotiledones), y raíces y hojas de plántulas. (1) Recogida de bellotas del árbol y transporte al laboratorio; (2) germinación; (3) siembra y crecimiento en invernadero; (4) muestreo.

lavó tres veces con acetona previamente enfriada a -20⁰C, centrifugándose entre cada lavado
(4000 g, 15 min, 4⁰C). Al pellet final se le añadió buffer Fenol-SDS, dejándose a 4⁰C durante 5
min. Para la separación de las fases acuosa y fenólica, la mezcla se centrifugó (10000 g, 15
min, 4⁰C). Se recuperó la fase fenólica, a la que se le añadió una solución 0,1 M de acetato
amónico en metanol (-20⁰C), dejándolo precipitar toda la noche a -20⁰C. El pellet obtenido
tras la centrifugación (10000 g, 15 min, 4⁰C) se lavó una vez con metanol (100%) y otra con
acetona previamente enfriada (-20⁰C). El pellet final se secó a temperatura ambiente durante
2 h. Una vez seco, éste se solubilizó en solución (7 M urea, 2 M tiourea, 4% (w/v) CHAPS, 0,5%
(v/v) Triton X-100, 20 mM DTT) y se determinó la cantidad de proteínas mediante el método
Bradford, utilizando seroalbúmina bovina (BSA) como estándar (Bradford, 1975). La cantidad
de proteína de cada extracto se calculó a partir de los valores de absorbancia utilizando el
coeficiente ε= 0,0483 obtenido de la curva de calibrado.
Para el análisis proteómico se utilizó un extracto resultante de mezclar 300 µg de proteína
(equivalente de BSA) de cada una de las preparaciones de los diferentes órganos (embrión,
cotiledón, raíz y hoja). Dicho extracto se sometió a electroforesis SDS-PAGE al 12% de
acrilamida, tal y como se ha descrito en Valledor et al. (2014). Se llevó a cabo la electroforesis
a diferentes cantidades de proteína: 1, 5, 10, 20, 40, 80, 100 y 200 µg equivalente de BSA. La
electroforesis se detuvo cuando la muestra había atravesado el gel separador, resultando en
una banda única de 0,5 cm aproximadamente, tal y como se observó en la tinción del gel con
azul brillante de Coomassie G-250 (Neuhoff et al. 1988). Las imágenes del gel fueron
capturadas con un densitómetro (GS-900TM Calibrated densitometer, Bio-Rad),
determinándose la intensidad de banda con el programa Image Lab™ (versión 5.2.1, Bio-Rad).
La cantidad de proteína en la banda electroforética se estimó a partir de los valores de
intensidad (densidad óptica) obtenidos en una curva de calibrado con BSA como proteína de
referencia. Los valores de intensidad de banda se transformaron en cantidad de proteína
utilizando el coeficiente ε= 38,22 obtenido de la curva de calibrado.
Espectrometría de masas
Previamente al análisis por espectrometría de masas, las bandas se cortaron manualmente
con un escalpelo, troceándose el gel en fragmentos pequeños (1mm) para favorecer
posteriormente la digestión de las proteínas con tripsina.

17
Para el desteñido del gel, éste se sumergió en 100 µL de bicarbonato amónico 1
mM/Acetonitrilo 50%, dejándose en agitación a 37⁰C durante 30 minutos. Este paso se repitió
una vez más. Se eliminó la solución de bicarbonato y se añadió 100 µL de acetonitrilo (AcN),
dejándose a temperatura ambiente durante 5 min. Para la reducción y alquilación de las
proteínas se eliminó el AcN y se añadió, en primer lugar, DTT 20 mM en bicarbonato amónico
100 mM y posteriormente iodoacetamida 55 mM en bicarbonato amónico 100 Mm,
dejándose 30 min en cada solución. Finalmente, los trozos de gel se lavaron con bicarbonato
amónico 25 mM, dos veces, y otras dos veces con bicarbonato amónico 25 mM/AcN 50%.
Para la digestión, se utilizó una solución de tripsina (sequencing grade; Promega) a una
concentración final de 12,5 ng/µl en solución de bicarbonato amónico 25 mM, 10% AcN y 5
mM CaCl2, incubándose toda la noche a 37⁰C en agitación (aproximadamente 16 h).
Los péptidos resultantes de la hidrólisis fueron desalados con columnas de fase reversa C18
(Scharlau, C18100-01C). Las columnas fueron activadas con 70% AcN/0,1% TFA, lavadas con
0,1% TFA y a continuación se pasaron las muestras por las columnas. Los péptidos fueron
eluidos con AcN al 70%, secándose el eluido en un SpeedVac.
El análisis de espectrometría de masas se realizó en los equipos disponibles en el servicio
de la Universidad de Córdoba (Servicio Central de Apoyo a la Investigación-SCAI). Atendiendo
a la cuantificación de proteínas realizada anteriormente las siguientes cantidades de proteína
fueron analizadas mediante nLC-MS/MS (Orbitrap, Q-OT-qIT): 10, 50, 100, 200, 400, 600, 800
y 1000 ng (equivalente de BSA), cubriendo así un rango que iría desde un valor mínimo
detectable por el equipo de masas hasta un valor máximo correspondiente al de saturación
de la columna cromatográfica. Para ello se utilizó un equipo de cromatografía líquida nano-LC
(Dionex Ultimate 3000 nano UPLC, Thermo Scientific) utilizando una columna C18 75 µm x 50
Acclaim Pepmam (Thermo Scientific) acoplado a un espectrómetro de masas Orbitrap Fusion
(Q-OT-qIT, Thermo Scientific). La mezcla de péptidos fue previamente pasada por una
precolumna 300 um x 5 mm Acclaim Pepmap (Thermo Scientific) en 2% AcN/0.05% TFA
durante 5 min a un flujo de 5 µl/min. La separación de péptidos se realizó a 40 ⁰C. La fase
móvil A estaba compuesta por 0.1% acido fórmico y la fase móvil B por 20% AcN/0.1% acido
fórmico. La elución se llevó a cabo con un flujo de 300 nL/min, utilizando el siguiente gradiente
de la solución B en A: 4-35% (120 min); 35-55% (6 min); 55-90% (3 min); finalmente se llevó a
cabo un lavado con 90% B (8 min) y reequilibrado al 4% B (15 min). El tiempo total de la

cromatografía fue de 150 min. Los iones eluidos fueron convertidos a fase gaseosa por
ionización nano-electro espray y analizados en el equipo de espectrometría de masas
operando en modo positivo. El escaneo de péptidos precursores de rango 400 a 1500 m/z se
realizó a una resolución de 120K con un conteo de iones de 4 × 105. El análisis de masas en
tándem se realizó por aislamiento a 1,2 Da con el cuadrupolo, fragmentación CID con una
energía de colisión de 35 eV, y análisis de MS de exploración rápida en la trampa de iones.
Identificación y cuantificación de proteínas
Los datos generados por espectrometría de masas se procesaron usando el software
Proteome Discoverer (versión 2.1., Thermo Scientific). Los espectros MS2 se utilizaron para
realizar la búsqueda usando el algoritmo SEQUEST, fijándose los siguientes parámetros: la
tolerancia de masa para iones de 10 ppm y para los fragmentos de 0,8 Da. Solo se usaron
estados de carga +2 o superiores. Para la identificación, se estableció un FDR del 5% y las
modificaciones variables se establecieron en: acetilación del extremo N-terminal, oxidación
de la metionina y formación de carbamidometil cisteína. No se establecieron modificaciones
fijas, y se impuso un máximo de omisión de dos sitios de corte por la tripsina. (Jorrín-Novo,
2014)
La búsqueda se realizó utilizando dos bases de datos. Por una parte, la base de datos
Viridiplantae obtenida de UniProtKB (17/11/2017, http://www.uniprot.org/) y, además, se
hizo una segunda búsqueda empleando una base de datos específica de Quercus ilex, obtenida
a partir de su transcriptoma (Guerrero-Sanchez et al., 2017). Las proteínas identificadas,
fueron categorizadas funcionalmente utilizando el software MERCATOR
(http://www.plabipd.de/portal/mercator-sequence-annotation/) en 35 grupos funcionales,
los cuales se reagruparon en 16 grupos mayoritarios con el uso de la base de datos KEGG
(https://www.genome.jp/kegg/pathway.html).
La cuantificación relativa de las proteínas se realizó usando los valores área de pico
obtenidos con el software Proteome Discoverer (versión 2.1., Thermo Scientific). Estos
valores, son considerados como la media de la intensidad de señal MS de los tres péptidos
más representativos de cada proteína (los más intensos) (Silvia et al., 2006). Los datos fueron
normalizados por la suma total de estos valores en cada muestra.

19
Resultados y discusión
En este estudio se pretendió optimizar la técnica de proteómica “shotgun” para su
utilización en la caracterización del proteoma de la encina, así como para estudios de
variabilidad poblacional, de germinación y respuesta a estreses en esta especie.
Se utilizó una muestra de proteínas resultado de la mezcla equimolecular de extractos
obtenidos a partir de diferentes órganos de la planta, incluyendo embriones, cotiledones,
raíces y hojas. Dicho diseño experimental se planteó con el propósito de abarcar el mayor
número posible de proteínas. Las muestras procedieron de semillas germinadas y plantones
obtenidos de un individuo localizado en Aldea de Cuenca, Córdoba (X:288570, Y:4238235.9).
Cuantificación de proteínas en extracto y en gel
Los extractos se obtuvieron a partir de los diferentes órganos utilizando el método de
precipitación TCA-acetona-fenol. En la Tabla 1 se indican los valores de rendimiento de
proteínas para cada uno de los órganos, cuantificados a partir de los valores de absorbancia
obtenidos mediante el método Bradford.
Tabla 1. Rendimiento de la extracción de proteínas para diferentes órganos. La cuantificación de proteínas se llevó a cabo por el método de Bradford, utilizando BSA como estándar. Los valores se expresan como µg equivalente de BSA/mg de peso fresco de tejido.
Órgano Media ± DS
Cotiledón 5,00 ± 0,44
Raíz 0,58 ± 0,07
Embrión 4,88 ± 0,59
Hoja 3,86 ± 0,15
A partir de dichos valores se llevó a cabo una dilución seriada en el rango 1-200 µg
equivalentes de BSA. Las diferentes diluciones se sometieron a electroforesis SDS-PAGE, como
etapa previa a la espectrometría de masas. La electroforesis se detuvo una vez que la muestra
entró en el gel separador. Esta etapa preliminar tuvo como objetivo, además de la
cuantificación mediante este método, la limpieza y concentración de la muestra, antes del
análisis “shotgun”.
La cantidad de proteína en gel de electroforesis SDS-PAGE se estimó por extrapolación de
los valores de intensidad de banda obtenidos de las muestras de encina con los valores de una
curva de calibrado, realizada con diferentes concentraciones de BSA cubriendo el rango de 1-
200 µg.

En la Figura 3 se presentan las dos curvas correspondientes a la cuantificación de proteínas
en los extractos (método de Bradford) y en gel (intensidad de banda tras la tinción con
Coomassie). Como se puede apreciar, existe una diferencia ambos métodos, viéndose como
el método Bradford subestima la cantidad de proteína con respecto a la cuantificación en gel.
Identificación de proteínas y agrupamiento funcional
La identificación de proteínas se llevó a cabo a partir de los espectros de masas (valor m/z),
tanto de los péptidos (ion parental) como de los correspondientes productos de
fragmentación. Para ello se utilizó el software Proteome Discoverer (versión 2.1., Thermo
Scientific), y el algoritmo de búsqueda SEQUEST. Se utilizaron dos bases de datos de proteínas,
la de UniProtKB-Viridiplantae y la obtenida a partir del transcriptoma de Quercus ilex
(Guerrero-Sanchez et al., 2017). El número de péptidos y proteínas identificadas para ambas
bases de datos, así como el número de ortólogos (en el caso de UniprotKB-Viridiplantae) y
productos génicos (en el caso de Quercus ilex) identificados se muestran en la Tabla 2,
considerándose como ortólogos aquellos hits identificados en familias filogenéticamente
próximas a la familia Fagaceae, dentro del orden Fagales (Figura 4).
µg
de
pro
teín
a
µg de proteína equivalente a BSA (Bradford)
Figura 3. Cuantificación de proteínas en extracto y en gel. La cuantificación en extractos se llevó a cabo mediante el método de Bradford. La cuantificación en gel se llevó a cabo a partir de los valores de intensidad de banda. En ambos casos se utilizó como proteína de referencia la BSA. Los valores de proteína se calcularon a partir de la curva de calibrado presentada en la figura y se expresaron como µg equivalentes de BSA.
00
50
100
150
200
250
300
0 50 100 150 200 250
SDS-PAGE
Bradford

21
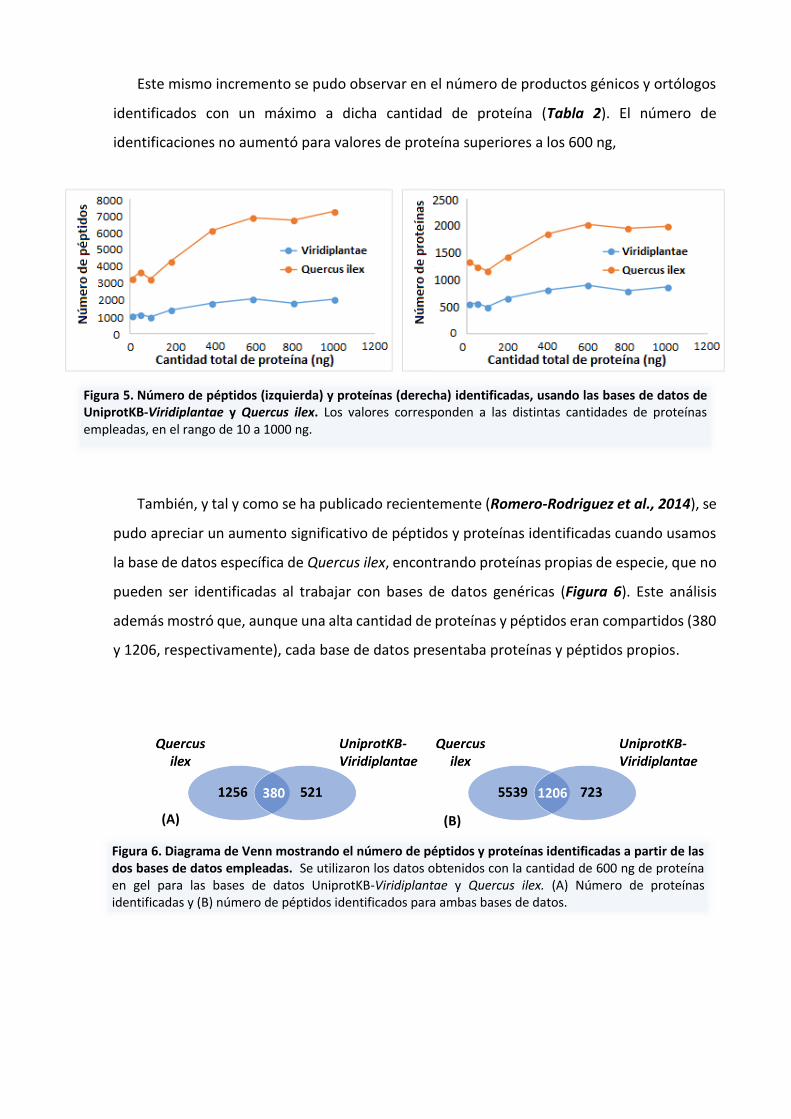
En base a estos resultados pudimos observar que el número de péptidos y proteínas
identificadas incrementa con la cantidad de proteína analizada, con valores máximos a 600ng
(Tabla 2). Estos valores se han representado en la Figura 5, en la que podemos apreciar que
el número de identificaciones depende de la cantidad de proteína utilizada, incrementando
de forma lineal de 100 a 600 ng.
Tabla 2. Número de péptidos y proteínas identificados. Los valores corresponden a las diferentes diluciones empleadas, en el rango de 10-1000 ng. Se utilizaron las bases de datos UniprotKB (Viridiplantae) y Quercus ilex. Se indican ortólogos y productos génicos, señalando número de péptidos o proteínas totales y únicas.
Cantidad de proteínas
(ng)
UniprotKB-Viridiplantae Quercus ilex
Péptidos (totales/únicos)
Proteínas (totales/únicas)
Productos ortólogos
Péptidos (totales/únicos)
Proteínas (totales/únicas)
Productos génicos
10 1071/996 556/556 85 3266/3206 1337/1141 1141
50 1134/1110 563/563 95 3663/3643 1244/1058 1244
100 1001/987 495/495 73 3245/3234 1168/993 1168
200 1410/1363 659/659 100 4301/2455 1425/1180 1425
400 1806/1678 812/812 116 6126/6001 1851/1511 1851
600 2068/1929 901/901 135 6885/6745 2028/1636 2028
800 1821/1678 793/793 122 6743/6589 1955/1587 1955
1000 2047/1821 869/869 124 7264/7034 1992/1604 1992
Consistentes 211 152 124 375
Figura 4. Árbol filogenético de familias botánicos para los que se obtuvo al menos una identificación de ortólogos. Solo aparece una parte de los órdenes correspondientes a la clasificación de eudicotiledóneas, donde se agrupa la encina (Q. ilex). Se ha rodeado los órdenes de especies consideradas como ortólogos a Quercus ilex.
Este mismo incremento se pudo observar en el número de productos génicos y ortólogos
identificados con un máximo a dicha cantidad de proteína (Tabla 2). El número de
identificaciones no aumentó para valores de proteína superiores a los 600 ng,
También, y tal y como se ha publicado recientemente (Romero-Rodriguez et al., 2014), se
pudo apreciar un aumento significativo de péptidos y proteínas identificadas cuando usamos
la base de datos específica de Quercus ilex, encontrando proteínas propias de especie, que no
pueden ser identificadas al trabajar con bases de datos genéricas (Figura 6). Este análisis
además mostró que, aunque una alta cantidad de proteínas y péptidos eran compartidos (380
y 1206, respectivamente), cada base de datos presentaba proteínas y péptidos propios.
Figura 5. Número de péptidos (izquierda) y proteínas (derecha) identificadas, usando las bases de datos de UniprotKB-Viridiplantae y Quercus ilex. Los valores corresponden a las distintas cantidades de proteínas empleadas, en el rango de 10 a 1000 ng.
(A) (B)
Figura 6. Diagrama de Venn mostrando el número de péptidos y proteínas identificadas a partir de las dos bases de datos empleadas. Se utilizaron los datos obtenidos con la cantidad de 600 ng de proteína en gel para las bases de datos UniprotKB-Viridiplantae y Quercus ilex. (A) Número de proteínas identificadas y (B) número de péptidos identificados para ambas bases de datos.
23
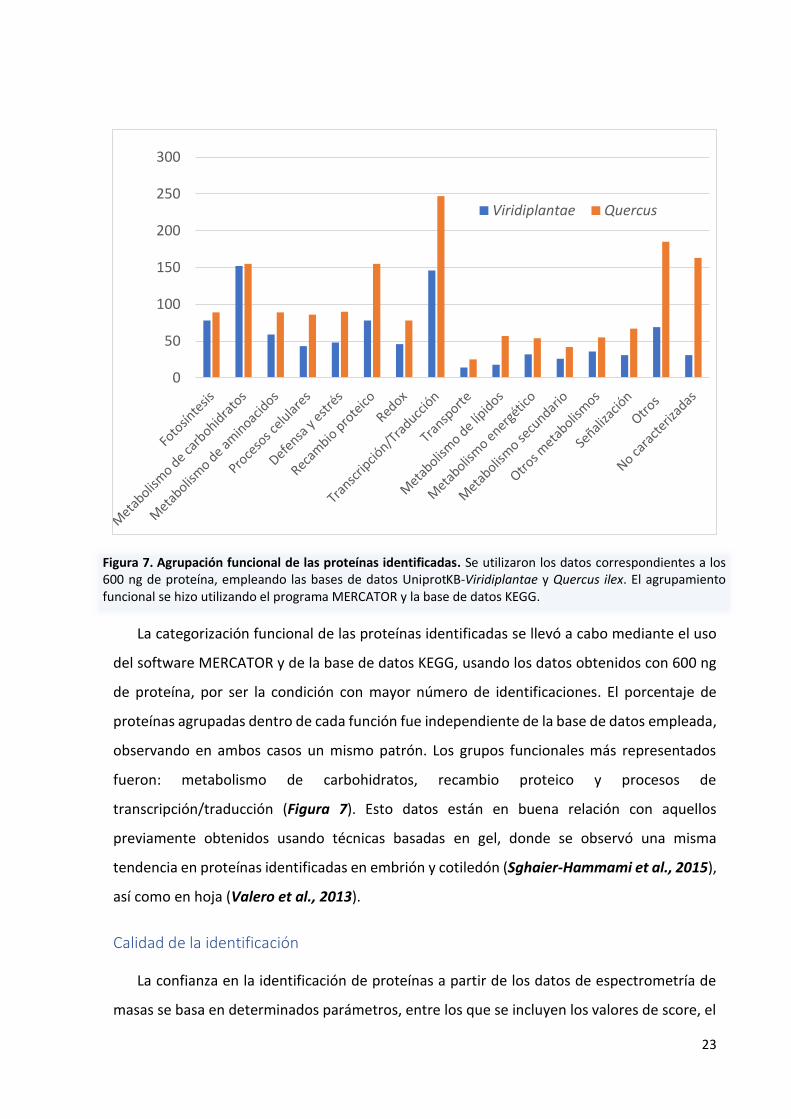
La categorización funcional de las proteínas identificadas se llevó a cabo mediante el uso
del software MERCATOR y de la base de datos KEGG, usando los datos obtenidos con 600 ng
de proteína, por ser la condición con mayor número de identificaciones. El porcentaje de
proteínas agrupadas dentro de cada función fue independiente de la base de datos empleada,
observando en ambos casos un mismo patrón. Los grupos funcionales más representados
fueron: metabolismo de carbohidratos, recambio proteico y procesos de
transcripción/traducción (Figura 7). Esto datos están en buena relación con aquellos
previamente obtenidos usando técnicas basadas en gel, donde se observó una misma
tendencia en proteínas identificadas en embrión y cotiledón (Sghaier-Hammami et al., 2015),
así como en hoja (Valero et al., 2013).
Calidad de la identificación
La confianza en la identificación de proteínas a partir de los datos de espectrometría de
masas se basa en determinados parámetros, entre los que se incluyen los valores de score, el
Figura 7. Agrupación funcional de las proteínas identificadas. Se utilizaron los datos correspondientes a los 600 ng de proteína, empleando las bases de datos UniprotKB-Viridiplantae y Quercus ilex. El agrupamiento funcional se hizo utilizando el programa MERCATOR y la base de datos KEGG.
0
50
100
150
200
250
300
Viridiplantae Quercus
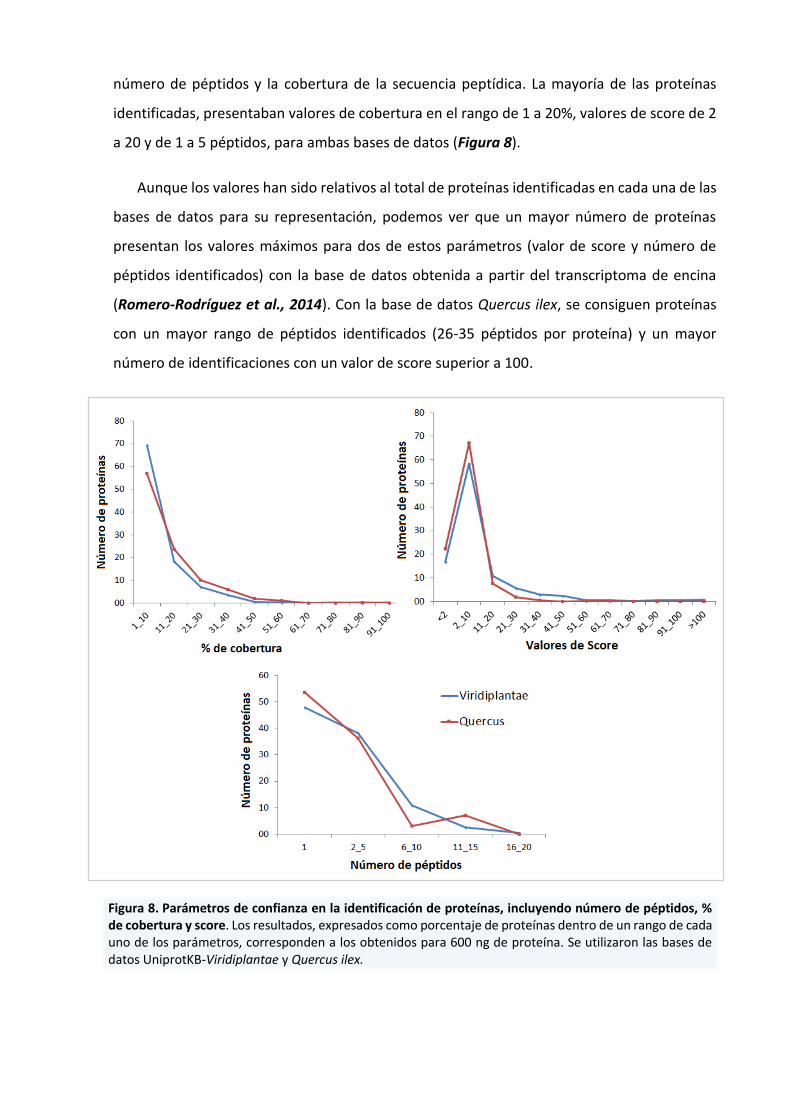
número de péptidos y la cobertura de la secuencia peptídica. La mayoría de las proteínas
identificadas, presentaban valores de cobertura en el rango de 1 a 20%, valores de score de 2
a 20 y de 1 a 5 péptidos, para ambas bases de datos (Figura 8).
Aunque los valores han sido relativos al total de proteínas identificadas en cada una de las
bases de datos para su representación, podemos ver que un mayor número de proteínas
presentan los valores máximos para dos de estos parámetros (valor de score y número de
péptidos identificados) con la base de datos obtenida a partir del transcriptoma de encina
(Romero-Rodríguez et al., 2014). Con la base de datos Quercus ilex, se consiguen proteínas
con un mayor rango de péptidos identificados (26-35 péptidos por proteína) y un mayor
número de identificaciones con un valor de score superior a 100.
Figura 8. Parámetros de confianza en la identificación de proteínas, incluyendo número de péptidos, % de cobertura y score. Los resultados, expresados como porcentaje de proteínas dentro de un rango de cada uno de los parámetros, corresponden a los obtenidos para 600 ng de proteína. Se utilizaron las bases de datos UniprotKB-Viridiplantae y Quercus ilex.
25
Uno de los propósitos principales de los estudios proteómicos mediante espectrometría
de masas es identificar tantas proteínas como sea posible. Sin embargo, la calidad de estas
identificaciones también es de gran importancia. Ya vimos que, con el uso de bases de datos
específicas, el número de identificaciones aumenta (Figura 5). Atendiendo a estos últimos
resultados, hemos comprobado además que los parámetros de confianza también mejoran
con la base de datos creada a partir del transcriptoma de Quercus ilex (Figura 8). Así pues, con
el uso de bases de datos específicas se consiguen alcanzar los objetivos planteados en
cualquier estudio de proteómica “shotgun”, mejorando tanto en el número de
identificaciones como los parámetros de confianza y, por tanto, la fiabilidad de los resultados
obtenidos.
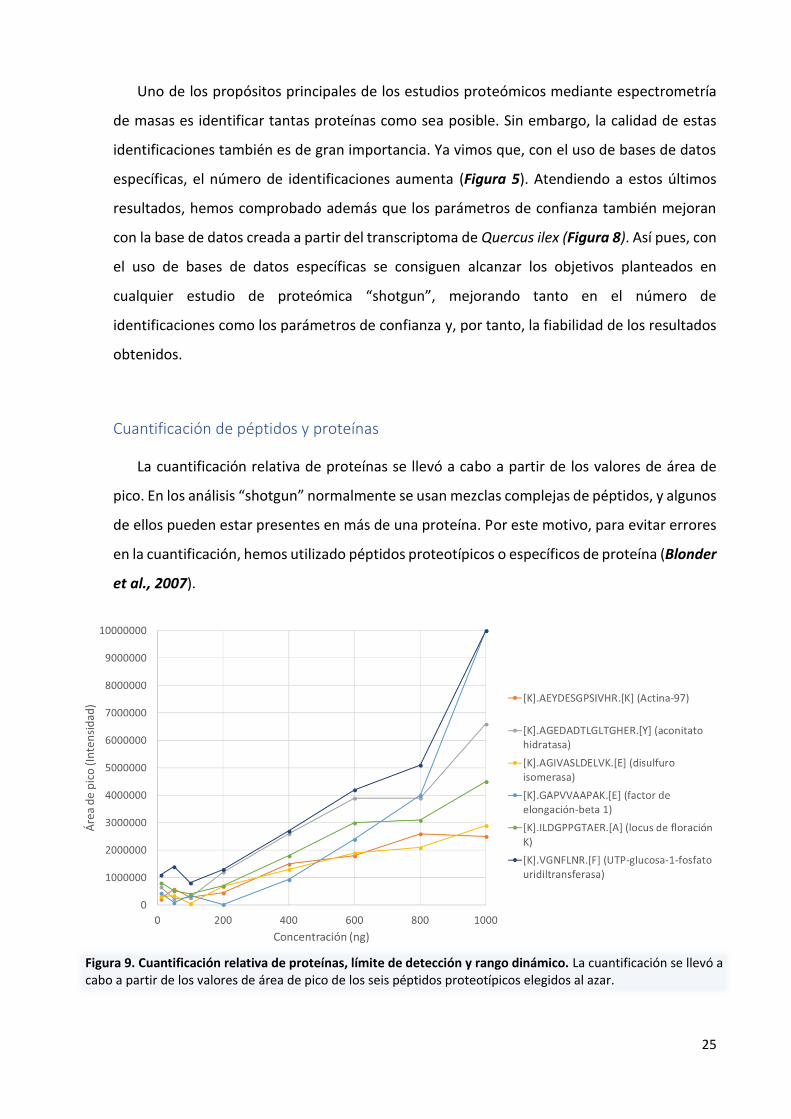
Cuantificación de péptidos y proteínas
La cuantificación relativa de proteínas se llevó a cabo a partir de los valores de área de
pico. En los análisis “shotgun” normalmente se usan mezclas complejas de péptidos, y algunos
de ellos pueden estar presentes en más de una proteína. Por este motivo, para evitar errores
en la cuantificación, hemos utilizado péptidos proteotípicos o específicos de proteína (Blonder
et al., 2007).
Figura 9. Cuantificación relativa de proteínas, límite de detección y rango dinámico. La cuantificación se llevó a cabo a partir de los valores de área de pico de los seis péptidos proteotípicos elegidos al azar.

Para ello seis péptidos proteotípicos pertenecientes a distintas proteínas se eligieron al
azar: Actina-97 ([K].AEYDESGPSIVHR.[K]), aconitato hidratasa ([K].AGEDADTLGLTGHER.[Y]),
disulfuro isomerasa ([K].AGIVASLDELVK.[E]), factor de elongación-beta 1
([K].GAPVVAAPAK.[E]), locus de floración K ([K].ILDGPPGTAER.[A]) y UTP-glucosa-1-fosfato
uridiltransferasa ([K].VGNFLNR.[F]) (Figura 9).
Los valores de área de pico (variable dependiente) se representaron frente a la cantidad
de proteína analizada (variable independiente), observándose que el límite de detección del
equipo de masas está en torno a los 100 ng de proteína y que el rango dinámico lineal se sitúa
entre los 100 y 800 ng para los seis péptidos analizados.

27
Conclusiones
Se ha puesto a punto una estrategia de proteómica “shotgun” para encina. De los
resultados obtenidos se puede concluir que:
i. El número de péptidos y proteínas identificadas máximo fue de 6885 y 1851,
respectivamente; correspondientes a 1511 proteínas únicas.
ii. El número de proteínas identificadas dependió de la cantidad de proteínas
analizadas y de la base de datos empleada.
iii. Los rangos de los parámetros de confianza obtenidos en la identificación fueron de
1-35 péptidos por proteína, 1-93 % de cobertura y 1-355 valor de score.
iv. El número de proteínas identificadas, así como los parámetros de confianza
obtenidos en la identificación fueron mayores cuando se utilizó la base de datos
específica de Quercus ilex que cuando se usó la base de datos genética UniprotKB-
Viridiplantae.
v. Las proteínas identificadas se agruparon en 16 categorías funcionales mayoritarias,
siendo las más representadas las del metabolismo de carbohidratos, de recambio
proteico y de procesos de transcripción/traducción.
vi. La cuantificación relativa a partir de los datos de área de pico es válida para
cantidades de proteína comprendidas entre 100 y 800 ng, el cual constituye el
rango dinámico para el sistema estudiado, la encina.

Referencias
Abril N.; Gion J.M.; Kerner R.; Müller-Starck G.; Navarro-Cerrillo R.M.; Plomion C.; Renaut J.; Valledor
L.; Jorrín-Novo J.V. (2011). Proteomics research on forest trees the most recalcitrant and orphan plant
species. Phytochemestry 71: 1219-1242.
Barbier-Brygoo H.; Jouard J. (2004). Focus on plant proteomics. Plant Physiology and Biochemistry 42:
913-917.
Blonder J.; Veenstra T.D. (2007). Computacional prediction of preoteotypic peptides. Nature
Biotechnology 25(1): 125-131
Bonner, F. T.; Vozzo, J. A. (1987) Seed Biology and Technology of Quercus. General Technical Report,
SO-66. New Orleans, LA: U.S. Dept. of Agriculture, Forest Service, Southern Forest Experiment Station.
21 p.
Bradford M.M. (1975). A Rapid and Sensitive Method for the Quantitation of Microgram Quantities
of Protein Utilizing the Principle of Protein-Dye Binding. Analytical Biochemestry 72: 248-254.
Caliskan, S. (2014) Germination and seedling growth of holm oak (Quercus ilex L.): effects of
provenance, temperature, and radicle pruning. iForest - Biogeosciences and Forestry, 7:103-109.
Cánovas F.M.; Dumas-Gaudot E.; Recorbet G.; Jorrín J.; Mock H.P.; Rossignol M. (2004). Plant proteome
analysis. Proteomics 4: 285-298.
Castro-Díez P.; Villar-Salvador P.; Pérez-Rontomé C.; Maestro-Martínez M.; Montserrat-Martí G.
(1997). Leaf morphology and leaf chemical composition in three Quercus (Fagaceae) species along a
rainfall gradient in NE Spain. Trees-Structure and Function 11:127–34.
Corbineau, F.; Xia, Q.; Bailly, C. and El-Maarouf-Bouteau, H. (2014) Ethylene, a key factor in the
regulation of seed dormancy. Frontiers in Plant Science, 5:539.
Davalieva K.; Kostovska I.M.; Dwork A.J. (2016). Proteomics Research in Schizophrenia. Frontiers in
Cellular Neuroscience 10: 18.

29
González-Fernández R.; Aloria K.; Arizmendi J.M.; Jorrín-Novo J.V. (2013). Application of Label-Free
Shotgun nUPLC-MS and 2-DE Approaches in the Study of Botrytis cinereal Mycelium. Journal of
Proteome Research 12: 3042-3056.
Guerrero-Sanchez V.M.; Maldonado-Alconada A.M.; Amil-Ruiz F.; Jorrin-Novo J. (2017). Holm Oak
(Quercus ilex) Transcriptome. De novo Sequencing and Assembly Analysis. Frontiers in Molecular
Biosciences 4:70
Hoagland D.R.; Arnon D. I. (1950). The Water-Culture Method for Growing Plants without Soil.
California Agricultural Experiment Station, Circular-347.
Jorrin-Novo J. V.; Maldonado A. M.; Echevarría-Zomeño S.; Valledor L.; Castillejo M. A.; Curto M.;
Valero J.; Sghaier B.; Donoso G.; Redondo I. (2009). Plant proteomics update (2007-2008). Second
generation proteomic techniques, an appropriate experimental design and data analysis to fulfill
MIAPE standards, increase plant proteome coverage and biological knowledge. Journal of
Proteomics 72: 285–314.
Jorrín-Novo J.; Navarro-Cerrillo R.M. (2014). Variabilidad y respuesta a distintos estreses en
poblaciones de encina (Quercus ilex L.) en Andalucía mediante una aproximación proteómica.
Ecosistemas 23(2):99-107.
Jorrín-Novo, J.V. (2014). Plant proteomics: methods and protocols. Methods in Molecular Biology,
1072: 3-13.
Lumaret R; Jabbour-Zahab R. (2009). Ancient and current gene flow between two distantly related
Mediterranean oak species, Quercus suber and Q. ilex. Annals of Botany 104:725–736.
Neuhoff V.; Arold N.; Taube D.; Ehrhardt W. (1988). Improved staining of proteins in polyacrylamide
gels including isoelectric focusing gels with clear background at nanogram sensitivity using Comassie
Brilliant Blue G-250 and R-250. Electrophoresis 9: 255-62.
Romero-Rodriguez M.C.; Pascual J.; Valledor L.; Jorrin-Novo J. (2014). Improving the quality of protein
identification in non-model species. Characterization of Quercus ilex seed and Pinus radiata needle
proteomes by using SEQUEST and custom databases. Journal of Proteomics 105: 85-91

Romero-Rodriguez, M.C.; Abril N.; Jorrín-Novo J. (2015). Aproximaciones -ómicas al estudio de la
germinación de semillas de especies recalcitrantes: el caso de la encina (Quercus ilex subsp. Ballota)
(Tesis Doctoral). Universidad de Córdoba
Sghaier-Hammami B.; Redondo-López I.; Valero-Galván J.; Jorrín-Novo J. V. (2016). Protein profile of
cotyledon, tegument, and embryonic axis of mature acorns from a non.orthodox plant species:
Quercus ilex. Planta, 243(2): 369-96
Silva J. C.; Gorenstein M. V.; Li G.-Z.; Vissers J. P. C.; Geromanos S. J. (2006). Absolute Quantification
of Proteins by LCMS. Molecular & Cellular Proteomics, 5(1): 144–156.
Simova-Stoilova L.P.; Romero-Rodríguez M. C.; Sánchez-Lucas R.; Navarro-Cerrillo R. M.; Medina-
Aunon J. A.; Jorrín-Novo J.V. (2015). 2-DE proteomics analysis of drought treated seedlings of Quercus
ilex supports a root active strategy for metabolic adaptation in response to water shortage. Frontier
in Plant Sciende 6: 627.
Valero-Galván J.; González-Fernandez R.; Navarro-Cerrillo R. M.; Gil-Pelegrín E.; Jorrín-Novo J. V.
(2013). Physiological and proteomic analyses of drought stress response in Holm oak provenances.
Journal of Proteome Research, 12(11): 5110-23
Valero-Galván J.; González-Fernández R.; Navarro-Cerrillo R.M.; Gil-Pelegrín E.; Jorrín-Novo J.V. (2014).
Physiological and Proteomics Analyses of Drought Stress Response in Holm Oak Provenances. Journal
of proteome Research 12(11):5110-23.
Valero-Galván J.; Jorrín-Novo J.; Cabrera A.; Ariza D.; García-Olmo J.; Cerrillo R. (2010). Population
variability based on the morphometry and chemical composition of the acorn in Holm oak (Quercus
ilex subsp. ballota [Desf.] Samp.). European Journal of Forest Research 131:893-904.
Valero-Galván J.; Valledor L.; Navarro Cerrillo R. M.; Gil Pelegrín E.; Jorrín-Novo J V. (2011). Studies of
variability in Holm oak (Quercus ilex subsp. ballota[Desf.] Samp.) through acorn protein profile
analysis. Journal of Proteomics 74:1244 – 1255.
Valledor L.; Wolfram W. (2014). Plants proteomics: methods and protocols. Methods in Molecular
Biology, 1072: 347-58

31
Wang W.; Vignani R.; Scali M.; Mauro C. (2006). A universal and rapid protocol for protein extraction
from recalcitrant plant tissues for proteomic analysis. Electrophoresis 27: 2782-2786.
Wilkins M.R., Sanchez J.C.; Gooley A.A.; Appel R.D.; Humphery-Smith I.; Hochstrasser D.F.; Williams
K.L. (1996). Progress with Proteome Projects: Why all Proteins Expressed by a Genome Should be
Identified and How to Do It. Biotechnology and Genetic Engineering Reviews 13: 19-50.
Xie F.; Liu T.; Qian W. J.; Petyuk V. A.; Smith R. D. (2011). Liquid Chromatography-Mass Spectrometry-
based Quantitative Proteomics. The Journal of biological Chemestry 286 (29): 25443-25449
Zhu W.; Smith J. W.; Huang C. M. (2010). Mass spectrometry-based label-free quantitative
proteomics. Journal of Biomedicine and Biotechnology, 1-6. doi:10.1155/2010/840518


33
Jesús V. Jorrín Novo M. Ángeles Castillejo Sánchez
Fdo.: Fdo.:
Isabel M. Gómez Gálvez
Fdo.:















![Dens Qit [Genesis] - GospelGogospelgo.com/q/Dong Bible - Portions.pdf · Dens Qit [Genesis] 1 Wangc Menl daengv menl daengv dih 1 Dens qit, Wangc Menl daengv menl daengv dih. 2 Xic](https://img.pdfslide.tips/doc/110x75/5a8baade7f8b9af27f8c272b/dens-qit-genesis-bible-portionspdfdens-qit-genesis-1-wangc-menl-daengv.jpg)