Embed Size (px)
Citation preview

Receptor−Ligand Interaction at 5‑HT3 Serotonin Receptors: A ClusterApproachBijan K. Rao,† Devleena Samanta,‡ Shawn Joshi,†,§ Kinjal Basu,∥ Sheryl D. Baldwin,⊥ Amrita Jha,#
Malgorzata Dukat,¶ Richard A. Glennon,¶ and Puru Jena*,†
†Department of Physics, §Department of Biomedical Engineering, ⊥Center for the Study of Biological Complexity, #Department ofChemistry, and ¶Department of Medicinal Chemistry, Virginia Commonwealth University, Richmond, Virginia 23284, United States‡Department of Chemistry and ∥Department of Statistics, Stanford University, Stanford, California 94305, United States
*S Supporting Information
ABSTRACT: A fundamental understanding of the interaction of ligands with biologicalreceptors is important because many drugs exert their influence via receptors. Using acluster approach, we have studied the role of structural and electronic parameters onreceptor−ligand binding by carrying out density functional theory based calculations. Asmodel systems, we have studied substituted arylguanidines, which activate 5-HT3receptors in a manner similar to that of serotonin. The geometries of the arylguanidinederivatives were fully optimized to obtain the lowest energy structures. Electronicproperties such as binding energies, dipole moments, polarizabilities, and electronaffinities, as well as geometric properties, such as molecular volume and dihedral angleswere calculated, and their relationship with binding affinity was evaluated. Resultsobtained were compared to experimental ligand−receptor binding affinity data available.These fundamental studies show that though both electronic and geometric properties ofthe ligands are important for binding, the electron affinities of the substituent species playa dominant role. Potential new fundamental indices for ligand−receptor affinity are alsodiscussed.
I. INTRODUCTION
Design of drugs to effectively treat various disorders can besignificantly improved if a fundamental understanding ofbiological processes at the atomic level can be achieved. Inspite of tremendous progress made in the development ofcomputer hardware and software as well as physical techniques,this understanding is difficult due to the complex architectureof biological molecules. Simple semiempirical methods havebeen traditionally used for this purpose as methods based onfirst-principles techniques cannot deal with molecules thatcontain thousands of atoms. Because for most of the reactionsonly the local environment of the reacting site of a molecule isof primary importance, it is possible to understand the keyparameters that control the reactivity of biological systems byusing a cluster approach that models the central reacting sitesurrounded by near neighbor atoms. We recall that atomicclusters are a new phase of matter intermediate between theatoms and bulk. At the dawn of this field, it was expected that astudy of the structure and properties of atomic clusters canillustrate how bulk properties evolve, one atom at a time. Inspite of considerable progress in this field1 an unambiguousanswer to the question “when does a cluster becomes a crystal?”has been difficult to find because the answer depends not onlyon the chemistry of the clusters but also on the property beinginvestigated. On the other hand, atomic clusters have beenfound to bridge many disciplines2−4 often addressingfundamental questions in physics, chemistry, biology, medicine,
and the environment. In this paper we use the cluster approachto focus on the design of drugs used for the treatment ofdisorders in the central nervous system (CNS), and show thatclusters can be used as a bridge between physics, chemistry, andmedicine.An effective strategy in drug design for the CNS has been to
identify specific receptors for important neurotransmitters.Serotonin (5-hydroxytryptamine or 5-HT) is one of the mostbasic neurotransmitters. There are seven different families ofserotonin receptors,5−9 5-HT1 through 5-HT7. Serotonin (5-HT) interacts with each receptor type7 and each receptor typecan be related to different physiological or pharmacologicalfunctions.9 Thus, it would be very useful to have therapeuticagents that selectively activate one receptor population, but notthe other 5-HT receptors. In some instances this has beenachieved.10,11 The challenge is to develop agents that haveselectivity as well as optimal binding properties with thereceptor of interest. In spite of considerable work on many ofthese receptors7−11 an atomic level first-principles under-standing of receptor−ligand is still lacking.
Special Issue: A. W. Castleman, Jr. Festschrift
Received: February 20, 2014Revised: May 29, 2014Published: May 30, 2014
Article
pubs.acs.org/JPCA
© 2014 American Chemical Society 8471 dx.doi.org/10.1021/jp5017906 | J. Phys. Chem. A 2014, 118, 8471−8476

Multiple receptor types can exist for a single neuro-transmitter and, indeed, multiple receptors for serotonin havebeen identified.11,12 Further, there are different types ofreceptor families, e.g., G-protein coupled receptors (GPCRs)and ligand gated ion channel receptors (LGICRs).10,11 Forthese receptors, the neurotransmitter ligand binds to thereceptor and elicits a response. Although neurotransmitters arethe natural or endogenous ligands for these receptors, syntheticligands can also bind to, and activate, the receptors.10
Receptors are traditionally viewed as “nano-locks” of specificthree-dimensional structure. The ligands are considered as“nano-keys” that must be “docked”, fitting into the receptor,like a key in a lock.13−16 Further, there must be mechanismsthat bind the “docked” ligand to the receptor. Conventionalwisdom is that the most important factors relate to alteration ofthe protein receptors, and the achievement of a stableconformation for the “nano-lock/nano-key” complex. Suchmechanisms arise from conformational changes in the receptorand the hydrophilic or lipophilic nature of the ligands, and fromother electronic and physicochemical parameters. For drugdesign, it is essential to gain an understanding of howreceptor−ligand binding can be modulated. The motivationof our work is that quantum mechanical studies based on first-principles cluster approach can, perhaps, shed light on thescience underlying these interactions.This study involves a specific family of serotonin receptors,
the 5-HT3 receptors, which are the only ion channel receptorsin the 5-HT receptor family17,18 and are involved in painperception, anxiety, and mental disorders. Dukat and Glennonhave previously identified a set of synthetic ligands,arylguanidines, that show activity at this receptor.19−21
Arylguanidines (AG) have been shown to activate 5-HT3receptors in a manner similar to that for serotonin. Therefore,arylguanidine derivatives have been synthesized and studiedextensively for their binding with these receptors.19−21 It wasfound that with different substituents at different positions inthe aryl ring, binding affinities vary over a very large range.19,21
Quantitative structure−activity relationship (QSAR) studieshave indicated that affinity is dependent upon the electronwithdrawing nature of the substituents at specific positions aswell as a relationship to molecular polarizability.21 However, astriking inconsistency (to be discussed later) in binding dataremained unexplained and no parameter or property had beenidentified to explain these apparent anomalies.In this work, we have conducted quantum mechanical
calculations from first-principles using density functional theoryto investigate both structural and electronic parameters onwhich the binding affinities of arylguanidine derivatives mightdepend.
II. METHODSExperimental Procedure. To assess the ability of a ligand
to bind to a receptor, we focused on receptor “affinity”, whichdefines its ability to bind to a specific receptor. The measure ofaffinity, Ki, is the dissociation constant. Because Ki is anequilibrium dissociation constant, larger Ki values mean poorerbinding and vice versa; e.g., a Ki = 1 nM represents high affinity,whereas a Ki = 10 000 nM represents 10 000-fold lower affinity.Using −log Ki or pKi (where Ki is in units of molarity), receptoraffinity varies directly with pKi. It might be noted that it iscustomary to use the pKi for statistical correlation analysis. Theaffinity can be measured using radioligand binding techniqueswhere receptors (in cell homogenates expressing the 5-HT3
receptor) are labeled with radioligand and the ability of agentsto displace the radioligand is measured. The procedures usedfor these measurements and the synthesis of all arylguanidine(AG) compounds were detailed in an earlier work.19 A total of14 molecules spanning a broad range of binding affinities werestudied and the values are given in Table 1.
Figure 1 shows the structures of serotonin and thearylguanidines examined. The “R” indicates various substituents
on the arylguanidine ring. The positions where the substitutionof the H atom was made are marked by their ring position. Thesubstituent, or R group, in each different molecule was varied toeffect possible binding behavior toward the serotonin 5-HT3receptor. Specifically, these compounds provided an ideal studyfor they were developed by substitution of a variety of R groupsat the 3-, 4-, and/or 5-positions of the aryl ring. They weresynthesized and experimentally evaluated for binding affinity atthe 5-HT3 receptor using whole cell preparations andradioligand binding.19−21
Computational Procedure. Earlier studies have shownthat, by itself, phenylguanidine (2 in Table 1) does not bindwith high affinity to 5-HT3 receptors.19,21 However, onsubstitution, some substituents increased binding affinitywhereas others decreased it. Although these affinities aretraditionally understood from quantitative structure−activityrelationship (QSAR) studies that are empirically based, therewas an apparent inconsistency in the previous QSAR thatremained unexplained. That is, high affinity could be (partially)explained by the presence of electron withdrawing substituentsat the ring 3-position (e.g., −Cl) but it was difficult to accountfor the lower than expected affinity of the stronger electronwithdrawing −CF3 analogues. This led to investigation of
Table 1. Binding Affinity Data for the SubstitutedArylguanidines (AGs)21
AG compound (R) Ki (nM) pKi
1 3-CF3 2440 5.612 H 2340 5.633 3-OCH3 1600 5.804 3-Cl 32 7.495 3-Cl, 5-OCH3 18 7.746 3,5-diCl 5 8.307 3,4,5-triCl 0.7 9.158 4-Cl 325 6.499 4-CF3 230 6.6410 3-CF3, 4-Cl 36 7.4411 3-NO2 85 7.0712 3-CN 123 6.9113 3-OH 2020 5.6914 3-CH3 6520 5.19
Figure 1. Chemical structures of serotonin and the arylguanidines.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5017906 | J. Phys. Chem. A 2014, 118, 8471−84768472

molecular properties such as polarizability,21 but −CF3-substituted analogues continued to appear as outliers.With a view to obtaining first-principles explanation of
binding, calculations using density functional theory wereperformed on these substituted arylguanidines. The geometriesof these were optimized to obtain the lowest energy structurefor each compound. We used B3LYP22−24 hybrid functional forthe exchange−correlation potential and 6-311++G**25,26 basisset for C, H, N, O, Cl, and F atoms. All structures wereoptimized without any symmetry constraint. No imaginaryfrequencies were found, indicating that the structures aredynamically stable. All calculations were performed usingGaussian 03 software.27
Several electronic and geometric properties of varioussubstituted arylguanidines were investigated to determine theeffects of the electronic structure as well as topology onreceptor binding affinity (pKi). These included (1) dipolemoments (DM), (2) polarization (dipole moment/volume),(3) molecular volume (intended to show the effect of sterichindrance), (4) dihedral angle between the guanidyl moietyand the benzene ring as the substituent was changed, (5)binding energy (atomization energy), (6) ionization potential(IP), (7) electron affinity (EA), (8) charge on the guanidylmoiety, (9) magnitude of exact polarizabilities, (10) totalcharge associated with the atom(s) and group(s) in positions 3,4, and 5, (11) relative EA, (12) relative IP of the substituent(13) binding energy per atom, and (14) relative substituentbinding energy. The relative EA (IP) was computed by takingthe difference between the EA (IP) of the substituted speciesand the H atom they replace. Similarly, the relative substituentbinding energy was computed by taking the difference betweenbinding energy of the substituent to the phenylguanidinemoiety and the binding energy of the H replaced. The chargeon an atom was computed using the natural bond orbital(NBO) analysis.The calculations in this study did not include the structure of
the receptor. Instead, the intent here was to discern whichmolecular attributes of the ligands themselves influence bindingto the 5-HT3 receptor. That is, the structure of the receptor(although possibly existing in multiple conformations) was heldconstant. Routine QSAR studies have provided someinformation but have not yet adequately accounted forunexpected differences in receptor binding.21 Some of theproblems were attributed to the possibility of rotamericbinding,21 but a persistent problem was their inability toaccount for the binding of −CF3 arylguanidines. In our work,we took a cluster approach; i.e., we computed the electronicand geometric properties of arylguanidine molecules asopposed to bulk. In this way, we tried to discern molecularparameters that are important for the receptor−ligand binding.Because the chemical nature of the substituent and its
substitution position are the principal changes from the basicarylguanidine structure, we looked at the problem from theperspective of the nature of these changes. Consequently,computing properties for the substituents themselves seemedlogical and summing the changes in each position, we obtainedthe following results. We define the relative electron affinity(EA), ΔEA, as
∑ ∑Δ = −EA EA(S) EA(H)
where EA(S) is the electron affinity of the substituent andEA(H) is the electron affinity of the hydrogen atom replaced by
the substituent. Hence, we are taking into account the changein electron affinity of the substituent as we replace H by anyother substituent group. Considering this change to becumulative, we investigated the correlation between thisquantity and pKi. For example, in the case of 3-Cl, 5-OCH3-AG two hydrogen atoms have been replaced by substituents.Hence the total relative electron affinity is given by
Δ = + −EA EA(Cl) EA(OCH ) 2EA(H)3
Similarly, the relative IP is defined as ΔIP = ∑IP(S) −∑IP(H) and the relative substituent binding energy is definedas RSBE = BE(S) − BE(H).To provide a basis for using relative electron affinities to
quantify the binding affinity of substituent ligands, we note thatin classical organic chemistry these substituents are designated,respectively, as electron donating or electron withdrawinggroups. Here, however, we have actually computationallydetermined the fundamental physical factor involved in electrondonating or electron withdrawing groups, namely, theionization potential and electron affinity of those groups.
III. RESULTS AND DISCUSSION
The optimized geometries of phenylguanidine and itssubstituted derivatives are given in Figure 2. It was previouslysuggested that both “substituent” parameters and “wholemolecule” properties might account for the binding of AGsto 5-HT3 receptors.21 Here, we investigate both using first-principles techniques.Our task is to model the pKi (called Y hereafter) in terms of
14 different predictors, namely, DM, polarization, molecularvolume, dihedral angle, binding energy, IP, EA, NBO charge onthe guanidyl moiety, magnitude of exact polarizability, chargeon 3,4,5 substituents, relative EA, relative IP, binding energyper atom, and relative substituent binding energy (called X1, X2,..., X14 hereafter). A simple linear regression (SLR) between theY of the 14 molecules and each of the predictors wasperformed. The correlation coefficients obtained using thisbasic approach, are given in Table 2.According to Table 2, relative electron affinity and total
energy are the most important parameters that affect binding.We noticed an increase in binding with increased relative EA ofthe substituents. Figure 3 shows the variation of binding affinitywith relative EA for all 14 molecules studied.Although this method is fairly simple, it does not provide an
accurate description of the variation of Y with Xi. This isbecause it ignores the possibility of more than one predictorvariable affecting Y. The low magnitude of the correlationcoefficient (r) between Y and Xm attained through SLR doesnot necessarily imply that the variable Xm is insignificant. Ifmultivariate regression analysis were to be done, Xm might be asignificant predictor in conjunction with other variables.Moreover, correlation between the predictor variables (e.g.,Xm and Xn) is neglected in SLR.Because our goal is to find which of the predictor variables
are important and which are not, we have the problem of fittingpredictors X on a variable Y to come up with the best modelthat not only gives a good theoretical interpretation but alsohelps in future predictions with minimum error. Thisnecessitates multivariate regression analysis. To fit Y onto thedata matrix X, it was required to find β such that
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5017906 | J. Phys. Chem. A 2014, 118, 8471−84768473
∑ β β− ′ = || − ||=
Y X Y X( )i
n
i i1
22
2
is minimized, where β is the coefficient of the predictorvariables. Because all the variables are being used in such asimple model, we are clearly overfitting the data, and this modelwill surely have a huge variance for future predictions.Known literature tells us how to select the important
predictor variables through a procedure as generalized modelselection, which is done automatically by LASSO28 (seeSupporting Information for more details). Using the LASSO
technique (least absolute shrinkage and selection operator), weobtained the equation of best fit to be
= + + +
−
Y X X
X X
7.418083 19.144761 0.001978 0.304841
0.1766128 9
11 14
The adjusted R2 of the fit is 0.8813. Writing in terms ofvariable names, we have
= + ×
+ × + ×
− ×
Kp 7.418083 19.144461 charge on guanidyl moiety
0.001978 polarizability 0.304841 relative EA
0.176612 relative substituent binding energy
i
Therefore, we find that among the 14 parameters, the chargeon the guanidyl moiety, polarizability, relative electron affinity,and relative substituent binding energy are the best variables inthe linear model. After knowing this, we tested the level ofsignificance of each of these three variables. The variable X11, orrelative EA, is the most significant variable at level 0.01. Thismeans there is a 1% chance that the coefficient of X11 in theregression equation is zero. The binding affinity increases withan increase in the charge on the guanidyl moiety, polarizability,and relative EA and decreases as the relative substituent bindingenergy increases.We can now explain the apparent discrepancy of why the
−CF3-substituted arylguanidines have lower binding affinitythan −Cl-substituted arylguanidines, in spite of being more
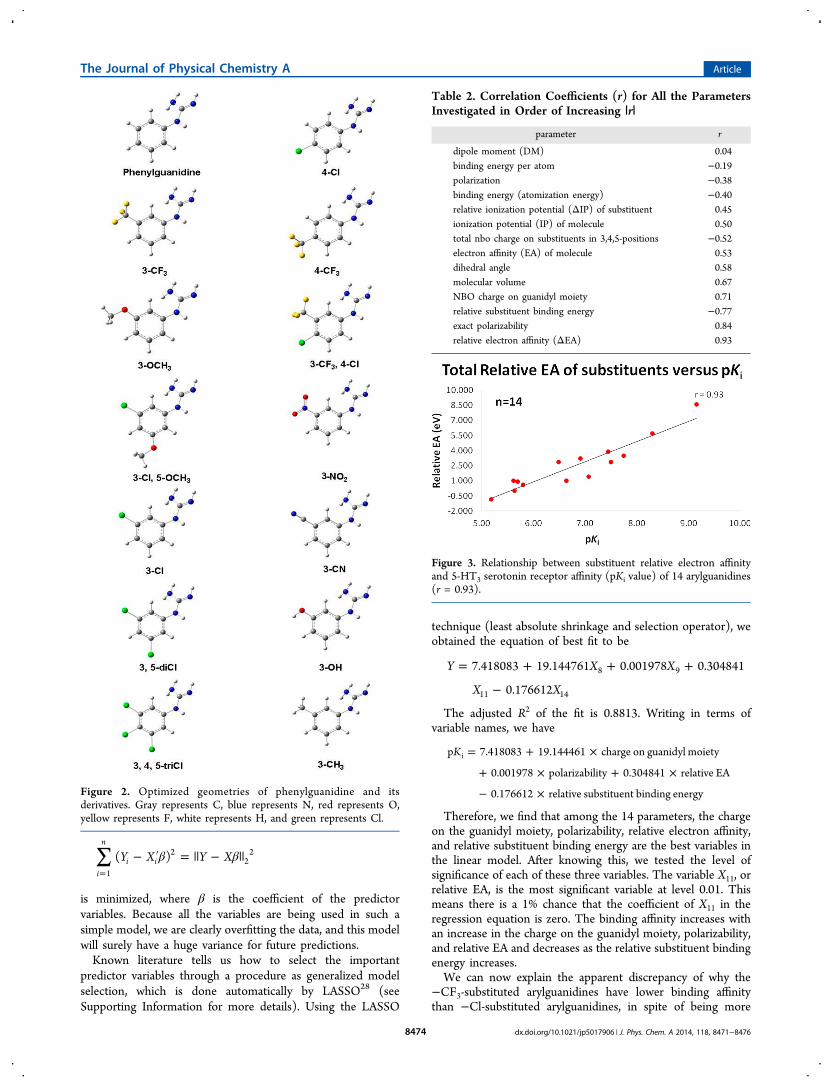
Figure 2. Optimized geometries of phenylguanidine and itsderivatives. Gray represents C, blue represents N, red represents O,yellow represents F, white represents H, and green represents Cl.
Table 2. Correlation Coefficients (r) for All the ParametersInvestigated in Order of Increasing |r|
parameter r
dipole moment (DM) 0.04binding energy per atom −0.19polarization −0.38binding energy (atomization energy) −0.40relative ionization potential (ΔIP) of substituent 0.45ionization potential (IP) of molecule 0.50total nbo charge on substituents in 3,4,5-positions −0.52electron affinity (EA) of molecule 0.53dihedral angle 0.58molecular volume 0.67NBO charge on guanidyl moiety 0.71relative substituent binding energy −0.77exact polarizability 0.84relative electron affinity (ΔEA) 0.93
Figure 3. Relationship between substituent relative electron affinityand 5-HT3 serotonin receptor affinity (pKi value) of 14 arylguanidines(r = 0.93).
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5017906 | J. Phys. Chem. A 2014, 118, 8471−84768474

electron withdrawing in nature. This is because though CF3 ismore electron withdrawing than Cl, it has a lower relative EA(Table S2 in Supporting Information). The significance of therelative EA shows that more electron deficient substituents helparylguanidines to bind more strongly to the receptor. Althoughwe have identified four variables as being important inpredicting the variable pKi, we cannot claim that these arethe only important variables. It should be noted that the datamatrix, X, had columns that were highly correlated. Thecorrelation matrix (in absolute values) is given in Table S3(Supporting Information). We find that there is strong absolutecorrelation between X8 and X4 and between X8 and X6. So,there is a chance that variables X4 and X6, viz, dihedral angleand ionization potential, might also be important in predictingthe Y values. But because of high absolute correlation betweenthese variables, it is enough to include just one in the model.We should mention that traditionally, for developing such
regression models, the molecules are randomly divided into twogroups called the training set and the test set, respectively. Thetraining set is used to develop the regression equation. Usingthis equation, the Y values of the test set of molecules aredetermined. The goodness of the fit is determined by looking atthe errors between the predicted and actual Y values of the testset. Our approach using the LASSO method is superior to thistechnique. In our method, we assigned the 14 molecules to 5groups at random. We chose four of the groups as the trainingset and calculated the error in predicting Y of the remaininggroup, the test set. Out of the 5C4 ways of choosing the trainingset and the test set, we chose the model that minimizes the testerror. This process is repeated, starting from the initialrandomization, 200 times to develop a robust model.The parameters listed above correspond to the optimized
structures presented in Figure 2. We recognize that severalrotational isomers are possible for each of the structures, whichmay influence the dipole moment, molecular volume, polar-ization, dihedral angle, polarizability, etc. To accuratelycalculate these values, the relative abundance of the rotamersat a particular temperature needs to be known. However,because most rotational isomers are within 0.2 eV of eachother, which is within the accuracy of the DFT calculations,they can be considered to be energetically degenerate in thiscase. Therefore, correlation between the total energy and pKi orrelative electron affinity and pKi is not expected to change.In general, we can conclude that the most important
parameters that affect binding are the electronic properties. Atthis stage, we cannot model the influence of the position of thesubstituent on binding. However, we observed that when Cl, asubstituent with higher EA, is moved from the 3- to the 4-position, the binding affinity decreases whereas in the case ofCF3, a substituent with lower EA, the opposite effect isobserved. We further noted from the literature that the pKi’s of4-OCH3 phenylguanidine (EA of OCH3 is 0.68 eV) and 4-CH3phenylguanidine (EA of CH3 is −1.27 eV) are 6.00 and 6.35,respectively.21 In this case also, as the electron affinity of theligand decreases, the binding affinity increases when the ligandis moved from the 3- to the 4-position. We should emphasizethat the calculations are carried out on free-standing molecules(i.e., clusters) whereas the affinity measurements are performedin real samples where the environment may play a role. Thus, itis important to recognize trends and overall agreement ratherthan focusing solely on quantitative agreement.Clearly, we have found that both electronic factors (charge
on the guanidyl moiety, polarizability, and relative electron
affinity of substituent) as well as geometric or topologicalfactors (such as position of substituent on the phenyl ring) areresponsible for binding affinity for this set of arylguanidineligands for the serotonin 5-HT3 receptor. A method tocomputationally include the positional dependence needs tobe developed for ligand binding, such as that done by Parr andYang for chemical reactivity.29,30
IV. CONCLUSIONWe have studied a total of 14 substituted arylguanidinemolecules using a cluster approach with a broad range ofbinding affinities for 5-HT3 receptors. We have calculatedstructure and electronic properties from first-principles andanalyzed the results using a multivariate-regression method toexplain the experimentally observed binding affinity. Our resultshave led to the following conclusions: (1) Both electronic andgeometric properties of the ligands guide binding. Bindingaffinity increases as the polarizability of the ligand increases.The charge on the guanidyl moiety as well as the relativebinding energy of the substituent to the arylguanidine backbonealso influences the binding. However, the most important factorguiding the binding is the relative electron af f inity of thesubstituents in the ligand. Substituents with higher electronaffinity favor stronger binding. We also note prior studies onlocal reactivity indices21 to help explain the dependence ofbinding of these compounds with the 5-HT3 receptors. (2)Multivariate regression analysis reveals that there is substantialcorrelation between the predictor variables. For example, thedihedral angle between the guanidyl moiety and the benzenering shows high correlation with the charge on the guanidylmoiety. Therefore, either of the parameters can be used todetermine strong binding. (3) Another important factor thatguides the binding is the position of the substituent. Thoughthe present study cannot fully account for change in bindingwith change in substituent position, we have observed thatwhen Cl (high EA) is moved from the 3-position to the 4-position, the binding affinity decreases. However, the bindingaffinity increases when CF3 (low EA) is moved from the 3- tothe 4-position. Due to limited data we are unable to make anygeneralization, except that in each case, the lower energystructure binds more strongly. We hope that with more data,this model can be further improved.Our results show that the electron affinity of the substituent
group does explain some apparent anomalous results in theexperimental findings for the complete set of substitutedarylguanidine compounds. This work also provides a morefundamental understanding of the basic science underlyingreceptor−ligand interactions for the 5-HT3 serotonin receptor.Both electronic and geometric parameters are important, in amanner not previously understood. The relative electronaffinity suggests the importance of the chemical potential forthese ligands in their interaction with the 5-HT3 receptor.Further, beyond this set of ligands, this may be a generalprinciple driving binding in other ligand−receptor interactions.Thus, a computationally-derived binding index developed fromfirst-principles, similar in nature to the local reactivity index ofParr and Yang,29,30 may be possible to explain receptor binding.Future work will involve additional investigation into thisconcept. In summary, we believe that the results of thesestudies provide a broader illumination on the basic scienceunderlying the ligand−receptor interactions for the serotonin 5-HT3 receptors, and potentially beyond. Further, a cluster modelmay help to explain reactivity of complex biological systems.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5017906 | J. Phys. Chem. A 2014, 118, 8471−84768475

■ ASSOCIATED CONTENT*S Supporting InformationElectron affinity and total relative electron affinity ofsubstituents, relative substituent binding energies and valuesof pKi and relative electron affinity for all the molecules, anddetails of the LASSO method are presented in the SupportingInformation. This material is available free of charge via theInternet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*P. Jena: e-mail, [email protected].
NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis was supported in part by a grant from the Department ofEnergy and used resources of the National Energy ResearchScientific Computing Center, which is supported by the Officeof Science of the U.S. Department of Energy under ContractNo. DE-AC02-05CH11231. In memory of B. K. Rao whoinitiated this study.
■ REFERENCES(1) Jena, P.; Castleman, A. W., Jr. Nanoclusters − A Bridge acrossDisciplines; Elsevier: Amsterdam, 2010.(2) Castleman, A. W., Jr.; Jena, P. Clusters: A bridge betweendisciplines. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 10552−10553.(3) Castleman, A. W., Jr.; Jena, P. Clusters: A bridge across thedisciplines of environment, materials science, and biology. Proc. Natl.Acad. Sci. U. S. A. 2006, 103, 10554−10559.(4) Jena, P.; Castleman, A. W., Jr. Clusters: A bridge across thedisciplines of Physics and Chemistry. Proc. Natl. Acad. Sci. U. S. A.2006, 103, 10560−10569.(5) Barnes, N. M.; Sharp, T. A Review of Central 5-HT Receptorsand Their Function. Neuropharmacology 1999, 38, 1083−1152.(6) Hoyer, D.; Hannon, J. P.; Martin, G. R. Molecular,Pharmacological and Functional Diversity of 5-HT Receptors.Pharmacol., Biochem. Behav. 2002, 71, 533−554.(7) Hannon, J.; Hoyer, D. Serotonin Receptors and Systems: EndlessDiversity? Acta Biologica Szegediensis 2002, 46, 1−12.(8) Hannon, J.; Hoyer, D. Molecular Biology of 5-HT Receptors.Behav. Brain Res. 2008, 195, 198−213.(9) Hoyer, D.; Clarke, D. E.; Fozard, J. R.; Hartig, P. R.; Martin, G.R.; Mylecharane, E. J.; Saxena, P. R.; Humphrey, P. P. A. InternationalUnion of Pharmacology Classification of Receptors for 5-Hydroxy-tryptamine (Serotonin). Pharmacol. Rev. 1994, 46, 157−203.(10) Glennon, R. A.; Dukat, M. Serotonin Receptors and DrugsAffecting Serotonergic Neurotransmission. In Foye’s Principles ofMedicinal Chemistry, 6th ed.; Williams, Lemke, T., Lippincott Williams& Wilkins: Baltimore, MD, 2008; pp 417−443.(11) Zifa, E.; Fillion, G. 5-Hydroxytryptamine Receptors. Pharmacol.Rev. 1992, 44, 401−458.(12) Leonard, B. E. Subtypes of Serotonin Receptors - Biochemical-Changes and Pharmacological Consequences. International ClinicalPsychopharmacology 1992, 7, 13−21.(13) Kuntz, I. D.; Blaney, J. M.; Oatley, S. J.; Langridge, R.; Ferrin, T.E. A Geometric Approach to Macromolecule-Ligand Interactions. J.Mol. Biol. 1982, 161, 269−288.(14) Halperin, I.; Ma, B. Y.; Wolfson, H.; Nussinov, R. Principles ofdocking: An Overview of Search Algorithms and a Guide to ScoringFunctions. Proteins-Struct. Funct. Genet. 2002, 47, 409−443.(15) Kitchen, D. B.; Decornez, H.; Furr, J. R.; Bajorath, J. Dockingand Scoring in Virtual Screening for Drug Discovery: Methods andApplications. Nat. Rev. Drug Discovery 2004, 3, 935−949.
(16) Taylor, R. D.; Jewsbury, P. J.; Essex, J. W. A Review of Protein-Small Molecule Docking Methods. J. Comput.-Aided Mol. Des. 2002,16, 151−166.(17) Gozlan, H. 5-HT3 receptors. In Serotonin Receptors and TheirLigands; Olivier, B., van Wijngaarden, I., Soudin, W., Eds.; Elsevier:Amsterdam, 1997; p 221.(18) Glennon, R. A.; Dukat, M.; Westkaemper, R. B. InPsychopharmacology, a Generation of Progress; Watson, S. J., Ed.;Lippincott-Raven: New York, 1998 (CD ROM Version).(19) Dukat, M.; Abdel-Rahman, A. A.; Ismaiel, A. M.; Ingher, S.;Teitler, M.; Gyermek, L.; Glennon, R. A. Structure-Activity Relation-ships for the Binding of Arylpiperazines and Arylbiguanides at 5-HT3Serotonin Receptors. J. Med. Chem. 1996, 39, 4017−4026.(20) Dukat, M.; Choi, Y. N.; Teitler, M.; Du Pre, A.; Herrick-Davis,K.; Smith, C.; Glennon, R. A. The Binding of Arylguanidines at 5-HT3Serotonin Receptors: A Structure-Affinity Investigation. Bioorg. Med.Chem. Lett. 2001, 11, 1599−1603.(21) Glennon, R. A.; Daoud, M. K.; Dukat, M.; Teitler, M.; Herrick-Davis, K.; Purohit, A.; Syed, H. Arylguanidine and ArylbiguanideBinding at 5-HT3 Serotonin Receptors: A QSAR study. Bioorg. Med.Chem. 2003, 11, 4449−4454.(22) Becke, A. D. Density-Functional Thermochemistry. 3. The Roleof Exact Exchange. J. Chem. Phys. 1993, 98, 5648−5652.(23) Becke, A. D. Density-Functional Exchange-Energy Approx-imation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38,3098−3100.(24) Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-SalvettiCorrelation-Energy Formula into a Functional of the Electron Density.Phys. Rev. B 1988, 37, 785−789.(25) Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. Self-Consistent Orbital Methods. XX. A Basis Set for Correlated WaveFunctions. J. Chem. Phys. 1980, 72, 650−654.(26) Mclean, A. D.; Chandler, G. S. Contracted Gaussian basis setsfor molecular calculations. I. Second row atoms, Z=11−18. J. Chem.Phys. 1980, 72, 5639−5648.(27) Frisch, M. J.; Trucks, G. N.; Schlegel, H. B.; Scuseria, G. E.;Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.;Kudin, K. N.; Burant, J. C.; et al. GAUSSIAN 03, Revision B.04;Gaussian, Inc.: Pittsburgh, PA, 2003.(28) Tibshirani, R. Regression shrinkage and selection via the lasso.Journal of the Royal Statistical Society B 1996, 58, 267−288.(29) Parr, R. G.; Yang, W. T. Density-Functional Theory of Atoms andMolecules; Oxford University Press Inc.: New York, 1989; pp 92−102.(30) Parr, R. G.; Yang, W. T. Density Functional-Approach to theFrontier-Electron Theory of Chemical-Reactivity. J. Am. Chem. Soc.1984, 106, 4049−4050.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp5017906 | J. Phys. Chem. A 2014, 118, 8471−84768476


![Ph.D. Thesis Erika Lehoczkyné Birkás · 3.6. Ligand-stimulated [35 S] ... as in the case . 2 ... GABA B receptors have 2 subunits, which are encoded by 2 different genes](https://img.pdfslide.tips/doc/110x75/5ad922b87f8b9ae1768b6f21/phd-thesis-erika-lehoczkyn-ligand-stimulated-35-s-as-in-the-case-2-.jpg)


























