Embed Size (px)
Citation preview

A CITOKRÓM P450 ENZIMRENDSZER FARMAKOGENETIKAILAG
JELENTŐS POLIMORFIZMUSAINAK VIZSGÁLATA
MAGYAR ÉS ROMA POPULÁCIÓBAN
PhD értekezés
Szalai Renáta
Doktori Iskola vezetője: Prof. Dr. Sümegi Balázs
Témavezető: Prof. Dr. Melegh Béla
PTE KK Orvosi Genetikai Intézet
Pécs, 2015

2
Tartalomjegyzék
RÖVIDÍTÉSEK JEGYZÉKE .................................................................................................... 3
1. BEVEZETÉS ..................................................................................................................... 4 1.1. Gyógyszermetabolizmus farmakogenetikája ............................................................... 5
1.2. Citokróm P450 enzimrendszer .................................................................................... 8 1.2.1. CYP1A2 genetikája ............................................................................................ 10 1.2.2. CYP1B1 genetikája ............................................................................................ 13 1.2.3. CYP2C8 genetikája ............................................................................................ 15 1.2.4. CYP3A5 genetikája ............................................................................................ 17 1.2.5. CYP4F2 genetikája ............................................................................................ 19
1.3. Taxán vegyületek ....................................................................................................... 20 1.4. Antikoaguláns gyógyszerek ....................................................................................... 24 1.5. Roma populáció bemutatása ...................................................................................... 26
2. CÉLKITŰZÉSEK ............................................................................................................ 28
3. ANYAGOK ÉS MÓDSZEREK ....................................................................................... 29 3.1. Vizsgált populációk ................................................................................................... 29 3.2. Molekuláris biológiai módszerek .............................................................................. 30
3.2.1. DNS izolálás ....................................................................................................... 30 3.2.2. PCR reakció ........................................................................................................ 30 3.2.3. RFLP .................................................................................................................. 32 3.2.4. Real time-PCR .................................................................................................... 34 3.2.5. Direkt szekvenálás .............................................................................................. 35
3.3. Statisztikai módszerek ............................................................................................... 35
4. EREDMÉNYEK .............................................................................................................. 36 4.1. CYP1A2 gén .............................................................................................................. 36 4.2. Taxán terápiát befolyásoló citokróm P450 gének ..................................................... 39
4.2.1. CYP1B1 gén ....................................................................................................... 39 4.2.2. CYP2C8 gén ....................................................................................................... 40 4.2.3. CYP3A5 gén ...................................................................................................... 41 4.2.4. CYP4F2 gén ....................................................................................................... 42
5. EREDMÉNYEK MEGBESZÉLÉSE ÉS KÖVETKEZTETÉSEK ................................. 43
6. EREDMÉNYEK ÖSSZEFOGLALÁSA ......................................................................... 52
7. KÖZLEMÉNYEK JEGYZÉKE ....................................................................................... 53
8. IRODALOMJEGYZÉK ................................................................................................... 58
9. KÖSZÖNETNYILVÁNÍTÁS .......................................................................................... 72

3
RÖVIDÍTÉSEK JEGYZÉKE
ABC ATP-binding casette (ATP-kötő kazetta)
BBMRI Biobanking and Biomolecular Resources Research Infrastructure
COPD chronic obstructive pulmonary disease
(krónikus obstruktív tüdőbetegség)
CYP citokróm P450
EM extenzív metabolizáló
GWAS genome-wide association study (genomszintű asszociációs tanulmány)
ht haplotípus
IM intermedier metabolizáló
NSAID nonsteroidal anti-inflammatory drug
(nem szteroid gyulladáscsökkentő gyógyszer)
OATP organic anion-transporting polypeptide
PAH policiklusos aromás szénhidrogén
PCG primer congenitalis glaucoma
PCR polymerase chain reaction (polimeráz láncreakció)
PM poor metabolizer (gyengén metabolizáló)
RBC red blood cell (vörösvértest)
RFLP restriction fragment length polymorphism
(restrikciós fragmenthossz polimorfizmus)
SLC solute carrier transporter
SNP single nucleotide polymorphism (egypontos nukleotid polimorfizmus)
UM ultragyors metabolizáló
wt wild type (vad típus)
5’-UTR 5’- untranslated region (5’- nem transzlálódó régió)

4
1. BEVEZETÉS
Az első megfigyelések a farmakogenetika történetében az 1930-as években kezdődtek
és csupán néhány ritka véletlennek köszönhetően kialakult extrém reakció tanulmányozására
vezethetők vissza. Ezek a fenotípusok a szokásos gyógyszeres kezelésre adott szokatlan
válaszon, vagy egyéb környezeti faktorok által kiváltott abnormális reakciókon alapultak. A
személyre szabott orvoslásra való igényt Sir William Osler (1849-1919) kanadai orvos már
az 1800-as években megfogalmazta, aki szerint „Ha nem lenne az egyének közötti hatalmas
variabilitás, akkor az orvoslást pusztán tudománynak tekinthetnénk és nem művészetnek”.
Mégis a farmakogenetika fogalmának bevezetése csak az 1950-es évek végére tehető (1959),
Friedrich Vogel (1925-2006) német humán genetikus nevéhez fűződik.
A gyógyszerválaszban jelentkező interindividuális variabilitás egy gyakori, komplex
probléma az orvosi klinikai gyakorlatban. Ezeket az egyedi különbségeket okozhatják a
gyógyszer adszorpciójában, eloszlásában, metabolizmusában és exkréciójában mutatkozó
eltérések, melyeknek hátterében genetikailag determinált faktorok állhatnak. A
gyógyszerhatásban megmutatkozó egyének közötti különbségek oka 20-95%-ban a
különböző genetikai háttérrel magyarázható. Az enzimek szubsztrát specificitásában
jelentkező átfedések, a genetikai polimorfizmusok nagy száma és a különböző etnikai
csoportok között észlelt variációk megnehezítik a kezelés hatékonyságának predikcióját. A
farmakogenetikai vizsgálatok középpontjában elsősorban olyan receptorok, transzport
fehérjék, target proteinek és gyógyszer metabolizáló enzimek állnak, melyek
heterogenitásáért olyan genetikai variánsok felelősek, melyek hatással vannak az egyéni
gyógyszerválaszra mind a terápiás hatás, mind pedig a mellékhatások tekintetében.
A napi szinten alkalmazott gyógyszerek a betegek 50-60%-ánál hatástalannak
bizonyulnak, a súlyos és fatális mellékhatások évente százezrek haláláért és milliók kórházi
gondozásáért tehetők felelőssé. Európai adatok alapján a súlyos gyógyszerhatás az ötödik
leggyakoribb halálok, évente csaknem 200 ezer haláleset hozható vele összefüggésbe. Az
emiatt keletkező többletköltség évente nyolcvanmilliárd euróra becsülhető. Ezeket a tényeket
szem előtt tartva az egyénre szabott orvoslás kertein belül egy korszerűbb, prevenciós,
genetikai információn alapuló szemlélet kialakítása lenne a cél farmakogenetikai vizsgálatok
segítségével. A genetikai és környezeti tényezők együttes figyelembevételével lehetőség
nyílhat az optimális terápia kiválasztására és a gyógyszer dózisának pontos meghatározására,
elkerülve a nem kívánt mellékhatásokat és a terápiás kudarcot.

5
1.1. Gyógyszermetabolizmus farmakogenetikája
Farmakokinetikailag fontos lépés a szervezetbe került gyógyszerek és más idegen
anyagok (xenobiotikumok) eliminációjában a metabolizmus folyamata, mely eredményeként
vízoldékonyabb, könnyebben kiválasztható termék keletkezik [1]. Az enzimek által katalizált
biotranszformációs folyamatok során inaktív, aktív vagy toxikus metabolitok keletkeznek [2,
3]. A metabolizmus elsődleges színtere a máj, de más szövetek, szervek is rendelkeznek
metabolikus aktivitással, mint például a tüdő, vese, vékonybél, bőr és a placenta [4]. Ezek
alapján beszélhetünk hepatikus-mikroszomális enzimekkel történő, hepatikus nem
mikroszomális enzimek segítségével végbemenő és extrahepatikus metabolizmusról (1. ábra)
[4].
1. ábra A szervezeten belüli gyógyszereliminációs utak százalékos megoszlása
(Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Zanger UM. Anal Bioanal Chem. 2008)
Farmakológiai klasszifikáció alapján a gyógyszerek metabolizmusában két fő fázist
különítenek el, a fázis I és fázis II reakciókat [5]. A fontosabb fázis I és fázis II reakciókat az
1. táblázat foglalja össze. A fázis I reakciókat alkotják a májsejtek endoplazmatikus
retikulumában lejátszódó (mikroszomális) ún. előkészítő vagy funkcionalizációs reakciók
(oxidáció, redukció, hidrolízis). Ezen folyamatok 90%-át monooxigenáz enzimek katalizálják,

6
melyek közül legfontosabb a citokróm P450 enzimrendszer [6]. A fázis II konjugációs
reakciókban a metabolit valamilyen endogén szubsztráttal konjugálódva tovább fokozza a
vegyület hidrofil tulajdonságát. Ezek az acetilációs, glükuronizációs, szulfatációs, metilációs
reakciók a citoszolban zajlanak le. Farmakogenetikai szempontból a legfontosabb fázis II
enzimeket kódoló polimorf gének a NAT2 (N-acetil-transzferáz), UGT1A1 (UDP-glükuronil-
transzferáz) és TPMT (thiopurin-S-metiltranszferáz) [2, 5, 7].
A metabolizmust követően transzportfehérjék segítségével a már poláros,
konjugálódott molekula szekretálódik a sejtből. A transzportfehérjéket kódoló génekben
(pl.: MDR1, SLCO1B1) előforduló mutációk megváltoztathatják a hatóanyag biológiai
elérhetőségét, multidrog rezisztenciához és más betegségek kialakulásához (cisztás fibrózis,
Dubin-Johnson szindróma, Tangier kór) vezethetnek [8, 9].
1. táblázat Fázis I és fázis II reakciók, enzimek és fontosabb szubsztrátjaik
Fázis I
Reakció típusa Enzim Gén Szubsztrát
Oxidáció
Citokróm P450
CYP1A2 koffein, warfarin
CYP2C9 warfarin, fenitoin
CYP2C19 omeprazol, mefenitoin
CYP2D6 debrizokvin, kodein
CYP3A4 statinok, clopidogrel
CYP2E1 etanol, halotán
Dihidropirimidin-dehidrogenáz DPYD fluorouracil
Aldehid-dehidrogenáz ALDH ciklofoszfamid, heroin
Alkohol-dehidrogenáz ADH etanol, retinol
Hidrolízis Butiril-kolinészteráz BCHE szukcinilkolin
Fázis II
Acetiláció N-acetil-transzferáz NAT2 izoniazid, koffein
Glükuronizáció UDP-glükuronil-transzferáz UGT1A1 irinotecan, bilirubin
Metiláció Tiopurin-S-metiltranszferáz TPMT azatioprin, merkaptopurin
Katekol-O-metiltranszferáz COMT katekolaminok

7
Genetikailag polimorf enzim által metabolizált, standard dózisban alkalmazott
gyógyszer klinikai hatása más-más módon és mértékben változhat meg, toxicitást, elnyújtott
terápiás hatást vagy hatástalanságot okozva. Míg egyes enzimek tekintetében a monogénes
polimorfizmusok már önmagukban is magyarázzák a variabilitás legnagyobb részét
(pl.: CYP2D6), a legtöbb esetben további polimorfizmus jelenléte, epigenetikai és környezeti
tényezők egyaránt hatással lehetnek a génexpresszióra [2, 10]. Funkcióvesztéses mutáció
csökkent clearence értéket és a gyógyszer emelkedett plazmakoncentrációját, míg
funkciónyeréses polimorfizmus emelkedett clearence-t és alacsonyabb gyógyszer
koncentrációt eredményez [10].
A nem DNS szekvencia variáción alapuló ún. epigenetikai regulátorok (DNS
metiláció, hiszton modifikáció, mikro RNS-ek) szintén fontos szerepet játszanak a
gyógyszerek metabolizmusában és a gyógyszerhatás kialakításában. Az epigenetikai mintázat
reverzibilis, lehet szövet-specifikus és környezeti tényezők által befolyásolt [10, 11]. A nem
genetikai tényezők közül a nem, kor, különböző társuló betegségek jelentősen
befolyásolhatják a szervezet gyógyszer metabolizáló képességét. Zhang és munkatársai
jelentős eltérést észleltek az mRNS expresszió mértékében nők és férfiak között különböző
citokróm P450 génekben [12]. Míg a CYP1A2, CYP3A4 és a CYP7A1 női túlsúlyt mutatott,
addig férfiakban a CYP3A5 és CYP27B1 gének mutattak fokozottabb expressziót [12]. Az
életkor szintén fontos befolyásoló tényező a gyógyszerek biotranszformációjában, különösen
a még éretlen enzimrendszerrel rendelkező újszülöttek és az idősebb populáció tekintetében
[13]. Idős betegek körében számolni kell az együttesen alkalmazott gyógyszerek potenciális
interakcióiból adódó vagy a csökkent vérkeringés és vesefunkció miatt kialakuló gyengébb
metabolizáló képességgel [14]. Ezt különösen fontos szem előtt tartani azon gyógyszerek
esetében, melyek szűk terápiás tartományúak (antidepresszánsok, antipszichotikumok,
antikoagulánsok). Továbbá az infekciók, gyulladások és daganatos megbetegedések során
felszabaduló proinflammatorikus citokinek (IL-1ß, TNF-α, IL-6), mint szignál molekulák
jelentősen befolyásolhatják a máj génexpressziós profilját, ami a gyógyszer metabolizáló
enzimek down-regulációjához vezethet [15, 16].
Funkcionális sajátosságuk, vagyis az enzimaktivitás alapján a gyógyszer metabolizáló
enzimek négy fenotípusos csoportját különböztethetjük meg [17]. Az extenzív metabolizáló
(EM) csoport normál enzimaktivitással jellemezhető a két funkcionális (vad típusú) allél
jelenlétének köszönhetően, amik az eredeti szekvencia szerinti teljes, funkcionális fehérjét
kódolják [11]. Ilyen betegek esetében a gyógyszer standard dózisban adva inaktív formává
transzformálódik, vagy a prodrug hatásos vegyületté alakul minimális mellékhatással vagy

8
mellékhatás nélkül. Az intermedier metabolizáló (IM) csoportban a génpár alléljai közül
legalább az egyik csökkent működésű enzimet kódol, így a metabolizmus kismértékű
csökkenése emelkedett hatóanyagszinthez és mérsékelt toxicitáshoz vezethet. Az ilyen
genotípussal rendelkezők körében a szokásosnál alacsonyabb dózisban kezdve a
gyógyszerelést a mellékhatások kivédhetőek [10, 11]. A gyengén metabolizáló (PM)
fenotípus esetén hiányzik az aktív allél, gyakrabban kell számolnunk a csökkent
metabolizmus miatt emelkedett gyógyszer koncentrációval és toxicitás kialakulásával.
Ultragyors metabolizálók (UM) körében nagyobb mennyiségű enzim expresszálódik, mert az
érintett gén kettő vagy több példányban van jelen génduplikáció eredményeképpen [11, 18].
Előfordulhat az is, hogy az egyik allél normál mennyiségű fehérjét, a másik pedig egy
polimorfizmusnak vagy mutációnak köszönhetően nagyobb mennyiségű, illetve fokozott
aktivitású funkcionális fehérjét produkál. Ilyen esetekben a gyors elimináció miatt a klinikai
gyakorlatban fennáll az aluldozírozás veszélye.
Gyógyszerhatás szempontjából jelentős genetikai különbségek léteznek nem csupán
egyének között, de populációs szinten (rassz, etnikum) is [17, 19-22]. Az interetnikai
polimorfizmusok lehetnek kozmopoliták és populáció specifikusak. Kozmopolitának
nevezzük őket akkor, ha ugyan eltérő frekvenciával, de minden etnikai csoportban jelen
vannak, míg a populáció specifikusak gyakoribbak a geográfiailag elszigetelt csoportokban.
1.2. Citokróm P450 enzimrendszer
A citokróm P450 (CYP) rendszer az emberi és más alacsonyabb rendű szervezetekben
univerzálisan előforduló enzim szupercsalád. A citokróm P450 enzimek legfontosabb feladata
a különböző endogén és exogén vegyületek biotranszformációja [5]. Legfontosabb fiziológiai
szubsztrátjai közé tartoznak a szteroidok, zsírsavak, prosztaglandinok, leukotriének és biogén
aminok [7]. Ezen felül számos xenobiotikum, úgy, mint gyógyszerek, növényi toxinok,
környezetből származó mérgező vegyületek átalakítását végzik [7, 23]. A CYP enzimek a
fázis I reakciókban túlnyomórészt oxidatív reakciókat katalizáló monooxigenázok, oxidázok
és peroxidázok. Fontos szerepet játszanak inaktív „prodrug”-ok aktív metabolittá
alakulásában, mint például a CYP2D6-mediálta kodein-morfin átalakulás [24]. Általában a
CYP enzimek nem specifikusak, egy enzim több különböző gyógyszert is képes átalakítani és
egy gyógyszer gyakran több enzim révén is metabolizálódhat. A CYP1, CYP2 és CYP3
enzimcsalád felelős a klinikumban alkalmazott gyógyszerek 70-80%-ának fázis I-függő

9
metabolizmusáért (2. ábra) [6, 25]. A legtöbb CYP enzim jelentős interindividuális
variabilitást mutat, mely részben a genetikai polimorfizmusoknak, részben pedig a különböző
mértékű génexpressziónak tulajdonítható. A humán citokróm P450 enzimrendszert 57
funkcionálisan aktív gén és 58 pszeudogén alkotja [10]. A Humán Genom Projekt a
szekvencia szimilaritás alapján 18 családot és 44 alcsaládot különböztet meg [26].
A citokróm P450 enzimek klasszifikációjához egy egységes nomenklatúra került
kidolgozásra (http://www.cypalleles.ki.se/). E nevezéktanban a CYP1A2*1 példáján a jelölés a
következő: a „CYP” szó a standard rövidítés a humán citkoróm P450-re. Az ezt követő első
szám (pl.: 1) jelöli a családot, melybe olyan izoenzimek tartoznak, amelyek fehérje
szekvenciájában egymással legalább 40%-os homológiát mutatnak. Az enzimek ugyanazon
alcsaládba sorolása 55%-nál nagyobb homológiára utal, megkülönböztetésükre betűt
használunk, ami az enzimcsalád rövidítést követi (pl.: A). Az alcsaládok egyes gén tagjait
rövidítjük az alcsaládot rövidítő betűt követő számmal (pl.: 2). Az ezt követő csillagos szám
jelenti az allélt. A *1 jelölést alkalmazzuk általában a normál enzimaktivitással jellemezhető,
vad típusú allélra [24].
2. ábra Citokróm P450 enzimek által metabolizált gyógyszerek százalékos eloszlása
(http://aiocm.org/uncategorized/review-on-interaction-between-herb-metabolism-and-cytochrome-p450/)

10
1.2.1. CYP1A2 genetikája
A citokróm P450 1A2 (CYP1A2) gén a 15-ös humán kromoszóma hosszú karján
helyezkedik el 15q24.1 pozícióban, 7 exon és 6 intron alkotja, fehérje terméke egy 58294 Da
molekulatömegű, fázis I típusú enzim [19, 27]. Konstitutívan expresszálódik a májban,
emellett expressziója indukálható a tüdőben, hasnyálmirigyben, gasztrointesztinális traktusban
és az agyban [28]. A CYP1A2 enzim a klinikumban alkalmazott gyógyszerek körülbelül
11%-át metabolizálja (2. ábra). A CYP1A2 mikroszómális enzim szerepet játszik számos
klinikailag fontos gyógyszer (klozapin, paracetamol, verapamil, teofillin, koffein)
metabolizmusa mellett olyan endogén szubsztrátok biotranszformációjában, mint a melatonin,
bilirubin, ösztradiol [20, 27, 29]. A CYP1A2 gén által kódolt enzim felelős bizonyos
prokarcinogén vegyületek, úgy, mint aromás- és heterociklusos aminok, policiklusos aromás
szénhidrogének (PAH) metabolikus aktivációjáért is. Az enzim aktivitását egyes környezeti
tényezők - vegyszerek, gyógyszerek (protonpumpa-gátlók, orális kontraceptivumok),
táplálkozási faktorok, dohányzás – befolyásolhatják [27, 30, 31]. A génexpresszióban
bekövetkező változások daganatos megbetegedések, infarktus, krónikus obstruktív
tüdőbetegség (COPD) és más kórkép kialakulásához vezethetnek [32-37].
A CYP1A2 gén rendkívül variábilis, a leírt polimorfizmusokat a Human Cytochrome
P450 Allele Nomenclature Committee adatbázis (http://www.cypalleles.ki.se/cyp1a2.htm)
összegzi. A fontosabb CYP1A2 alléleket, polimorfizmusokat és azok funkcióit a 2. táblázat
foglalja össze. Az egy nukleotidot érintő polimorfizmusok (SNP-k) egy része funkcionális
jelentőséggel bír, gyakoriságuk figyelemre méltó egyének közötti és interetnikai különbséget
mutat [27, 29, 38, 39]. Az interindividuális különbségek 10-200-szorosra tehetők a CYP1A2
expresszióban és aktivitásban [40].
Az általunk vizsgált négy polimorfizmus a CYP1A2 gén nem-kódoló régióiban
helyezkednek el, közülük kettő (rs2069514, rs35694136) a szabályozó régióban, kettő pedig
az első intronban (rs762551, rs12720461). A vizsgálatainkban szereplő nem-kódoló
polimorfimusok elhelyezkedését a 3. ábra szemlélteti. A legalaposabban tanulmányozott
CYP1A2 nem-kódoló régióiban elhelyezkedő polimorfizmusok a -3860G>A, -2467delT és
-163C>A SNP-k.

11
2. táblázat Fontosabb CYP1A2 allélek és polimorfizmusok elhelyezkedése, funkciója
Allél SNP Aminosav csere Elhelyezkedés CYP1A2 aktivitás
CYP1A2*1A -
vad típus - 15q24.1 normál
CYP1A1*1B C5347T (N516N) 7. exon ismeretlen
CYP1A2*1C G-3860A - enhancer ↓ indukció
CYP1A2*1D T-2467delT - enhancer ismeretlen
CYP1A2*1E T-739G - 1. intron ismeretlen
CYP1A2*1F C-163A - 1. intron ↑ aktivitás
CYP1A2*1J -739G; -163A - 1. intron ismeretlen
CYP1A2*1K -729T; -739G; -163A - 1. intron ↓transzkripció
CYP1A2*2 C2866G (F21L) 2. exon ismeretlen
CYP1A2*3 T5347T (D348N) 4. exon ↓ génexpresszió
CYP1A2*4 A2499T (I383F) 5. exon ↓ génexpresszió
CYP1A2*5 G3497A (C406Y) 6. exon ismeretlen
CYP1A2*6 G5090T (R431W) 7. exon ↓ génexpresszió
CYP1A2*7 G3533A - 6. intron ↓ splicing defektus
CYP1A2*8 G5166A (R456H) 7. exon ↓ génexpresszió
CYP1A2*9 C248T (T38M) 2. exon ismeretlen
CYP1A2*10 G502C (E168Q) 2. exon ismeretlen
CYP1A2*11 C558A (F186L) 2. exon ↓ génexpresszió
CYP1A2*12 A634T (S212C) 2. exon ismeretlen
CYP1A2*13 G1514A (G299S) 3. exon ismeretlen
CYP1A2*14 C5112T (T438I) 7. exon normál
CYP1A2*15 C125G (P42R) 2. exon ↓ génexpresszió
CYP1A2*16 G2473A (R377Q) 5. exon ↓ génexpresszió
(http://www.pharmacologyweekly.com/table-cytochrome-p450-cyp1a2-genetic-polymorphisms-pharmacogenetics)

12
3. ábra Általunk vizsgált CYP1A2 nem-kódoló SNP-k lokalizációja
(Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J 11, 481-494. Zhou 2009.)
A -3860G>A (CYP1A2*1C, rs2069514) variáns, mely a CYP1A2 gén enhancer
régiójában foglal helyet, csökkent enzimaktivitást eredményez megváltoztatva egy nem
meghatározott transzkripciós faktor kötőhelyét [31, 41]. Korábbi vizsgálatok bizonyították,
hogy a CYP1A2*1C variánst hordozó betegek esetében a prodrugként alkalmazott
gyógyszerek biotranszformációja alacsonyabb, ami a gyógyszer csökkent biológiai
elérhetősége és hatékonysága miatt a betegség súlyosbodásához vezet [39, 42, 43].
A CYP1A2 enzim hatását a legnagyobb mértékben a -2467delT (CYP1A2*1D,
rs35694136) polimorfizmus befolyásolja. A CYP1A2 gén promoter régiójában, -2467
pozícióban található timin deléció egy megváltozott transzkripciós faktor kötőhelyet hoz létre
egy rövid DNS szakaszon, az ún. xenobiotikum reszponzív elemben, ezáltal stimulálja a
CYP1A2 gén transzkripcióját a dohányfüstben megtalálható komponensek jelenlétében [44].
A CYP1A2*1D variáns hordozása egyes népcsoportokban fokozott kockázatot jelent COPD-
re és tüdőrák kialakulására főként dohányzó férfiakban [44, 45].

13
A CYP1A2 gén első intronjában elhelyezkedő -163C>A (CYP1A2*1F, rs762551)
polimorfizmus az enzim fokozott indukciójával hozható összefüggésbe, ami a gyakorlatban
konvencionális dózisban alkalmazott CYP1A2 szubsztrát gyógyszerre nézve csökkent
terápiás hatást eredményez [35, 46, 47]. Tian és munkatársainak munkája, valamint Bu és
kutatócsoportjának metaanalízise rávilágított, hogy kaukázusi populációban a CYP1A2*1F
SNP hordozása kapcsolatba hozható különböző daganatos megbetegedések kialakulásával
[36].
A CYP1A2 -729C>T (rs12720461) SNP fontos hatással lehet a karcinogén vegyületek
bioaktivációjára, a -729T allélnek tulajdonítható csökkent génexpresszió révén, ami hatással
lehet egyes gyógyszerek metabolizmusára [29, 41]. A*1K haplotípus (-163C>A, -739T>G,
-729C>T) fontos eleme a -729C>T polimorfizmus, ami 40%-kal csökkent enzimindukcióval
hozható összefüggésbe, szintén az első intronban foglal helyet [29].
Számos, a CYP1A2 gén kódoló régióit érintő, többnyire aminosav szubsztitúciót
eredményező, ritka SNP (CYP1A2*2 - *16) került leírásra. Ezeknek a variánsoknak a
funkciója vagy ismeretlen, vagy csökkent génexpressziót, esetleg splicing defektust okoznak
(*7), jellemzően nagyon alacsony előfordulási gyakorisággal (0,5-1%) (2. táblázat) [27].
1.2.2. CYP1B1 genetikája
A citokróm P450 1B1 (CYP1B1) gén egy hem-tiolát NADPH-függő monooxigenáz
enzimet kódol a 2p21-22 humán kromoszóma pozícióban [48, 49]. A gént 3 exon és 2 intron
alkotja 12 kbp hosszú régión keresztül [49]. A CYP1B1 elsődleges feladata szteroid
hormonok, terápiás gyógyszerek metabolizmusa és a környezetből származó toxinok
detoxifikációja [50-52]. A CYP1B1 az egyik legfontosabb fázis I enzim a prokarcinogén
vegyületek reaktív metabolittá alakításában és klinikailag fontos daganat ellenes gyógyszerek
metabolizmusában, mint a tamoxifen és a flutamid [52, 53]. A CYP1B1 konstitutívan
expresszálódik a tüdőben és szteroid célszervek szöveteiben, mint a méh, mell, petefészek,
here, prosztata és mellékvese [48, 54]. A környezeti szennyezőanyagok (pl.:PAH)
metabolikus aktivációja mellett a 17ß-ösztradiol metabolizmusban is szerepet játszik, így
jelentős mértékben járul hozzá a hormon mediált karcinogenezishez [48, 49, 55, 56].
Napjainkig a polimorf CYP1B1 génben *1-*26-ig számos allél került leírásra
(http://www.cypalleles.ki.se/cyp1b1.htm). Ezek közül jónéhány SNP-t hoztak összefüggésbe
daganatos megbetegedésekre való fokozott hajlammal. A leginkább CYP1B1 variáns

14
jelenlétével kapcsolatos rákos megbetegedések közé sorolják a petefészek-, endometrium-,
vese-, prosztata-, tüdő-, fej-nyaki daganatokat kaukázusi és ázsiai populációkban [57, 58]. A
legrészletesebben tanulmányozott variáns allélok a CYP1B1*2, *3, *4 és *7, melyek jelenléte
funkcionális vizsgálatokban fokozott CYP1B1 mRNS expressziót és erősebb hidroxilációs
aktivitást eredményezett, összehasonlítva a CYP1B1*1 vad típusú alléllal [59, 60].
A CYP1B1*2 allélen, két kapcsolt polimorfizmus, nevezetesen a c.142C>G
(p.Arg48Gly) és c.355G>T (p.Ala119Ser) együttes előfordulását értjük
(http://www.cypalleles.ki.se/cyp1b1.htm). A CYP1B1*2 hordozása összefüggésben áll fej-
nyaki daganatok kialakulásával és koraszülésre való emelkedett kockázattal [61, 62].
Egyes vizsgálatokból kiderült, hogy a CYP1B1*3 (c. 4326C>G; p.Leu432Val) variáns
hordozása és az idiopátiás férfi infertilitás, endometrium daganat, nyitott zugú glaukoma,
valamint tüdő daganat előfordulása között kapcsolat mutatható ki [57, 58]. A CYP1B1*3
polimorfizmus a leggyakoribb SNP-nek számít észak-indiai populációban 48,8%-os
frekvenciával, ezt követi a CYP1B1*4 47,3%-kal, majd a CYP1B1*2 39%-os gyakorisággal és
a CYP1B1*7 16,5%-kal [54]. A CYP1B1 *3/*3 homozigóta variáns genotípus kaukázusi
populációban mérsékelt kockázatot jelent endometrium daganatra [63]. Továbbá egyes
elképzelések szerint a CYP1B1*3 genotípus modulálhatja a docetaxel kezelésre adott választ a
fokozott ösztrogén metabolizmusnak köszönhetően [63]. A CYP1B1 nem metabolizálja a
taxán vegyület docetaxelt, mégis a CYP1B1 gén túlexpressziója docetaxel rezisztenciával
társul, mivel csökken a hatásos gyógyszer koncentráció a rákos sejtekben [64]. A CYP1B1*3
allélt hordozók körében szignifikánsan alacsonyabb gyakorisággal fordul elő hiperszenzitív
reakció taxán kezelést követően [65].
Mivel a CYP1B1 génben bekövetkező mutációk állnak elsődlegesen az autoszomális
recesszív módon öröklődő primer congenitalis glaucoma (PCG) hátterében, úgy vélik, hogy a
CYP1B1 enzim szerepet játszik egy valószínűleg szteroid természetű, a szem fejlődéséhez
nélkülözhetetlen szignál molekula metabolizmusában [66-68]. Li és munkatársai PCG-s
betegek CYP1B1 mutációit összegző összefoglaló közleményükben beszámoltak arról, hogy
roma populációban a misszensz c.7996G>A (E387K) founder mutációnak bizonyult a
CYP1B1 génben 79,63%-os prevalenciával. Kaukázusi populációban a legközönségesebben
előforduló mutációnak a c.7940G>A (R368H) bizonyult [55].

15
1.2.3. CYP2C8 genetikája
A fázis I metabolizmusban szerepet játszó citokróm P450 2C8 (CYP2C8) polipeptid
sokféle különböző xenobiotikum és endogén vegyület biotranszformációjáért felelős [69]. A
máj összes CYP enzimeinek körülbelül 7%-át adja, továbbá kisebb mértékben expresszálódik
a vesében, mellékvesében, agyban, petefészekben, uterusban és a duodenumban [70, 71]. A
CYP2C8 enzim végzi a klinikumban alkalmazott gyógyszerek 5%-ának oxidatív
metabolizmusát, köztük struktúrálisan meglehetősen heterogén vegyületek átalakítását, ami az
enzim szubsztrát kötő aktív centrumának felépítésével áll összefüggésben [72]. Az enzim
aktív centrumának mérete hasonló a CYP3A4 enzim aktív centrumának méretéhez, de
alakjában különbözik, ez magyarázhatja a két gyógyszer metabolizáló enzim nagy mértékben
átfedő szubsztrát specificitását, de eltérő metabolikus profilját [70, 73]. A CYP2C8 enzim fő
szubsztrátjai közé tartoznak az aminokinolin, antiarrythmiás, leukotrién antagonista és taxán
gyógyszerek, de kisebb mértékben részt vesz számtalan más vegyület csoport átalakításában
is, úgy, mint nem szteroid gyulladáscsökkentők (NSAID), statinok, kálcium csatorna
blokkolók, ópioid és tirozin-kináz gátló szerek [74-76]. Több korábbi tanulmány számol be
arról, hogy in vivo és in vitro vizsgálatok során a CYP2C8 enzim legfontosabb inhibitora a
koleszterin szint csökkentésére alkalmazott gemfibrozil hatóanyag [77-80].
A CYP2C alcsalád legjelentősebb mértékben indukálható tagja a CYP2C8,
transzkripciós aktivációja a nukleáris pregnán X (NR1I2) -, konstitutív androsztán (NR1I3) -
és glükokortikoid (NR3C1) receptor által közvetített. Indukciójában részt vevő in vitro
upregulátor a rifampicin, dexametazon és a fenobarbitál [69]. A CYP2C8 gén lokalizációja a
10q24.1 pozícióra tehető, a CYP2C klaszter legkisebb tagjának számít. A 31 kb hosszú gént 9
exon alkotja [71, 81]. A CYP2C8 interindividuális variábilitása, mind a fehérje
expressziójában, mind pedig az enzim katalitikus aktivitásában a nagy számú genetikai
polimorfizmus jelenlétének köszönhető. A fontosabb polimorfizmusokat a CYP2C8 génben a
4. ábra szemlélteti.
Mára több mint 450 SNP-t azonosítottak a CYP2C8 génben, melyek jelentősen
befolyásolhatják a CYP2C8 mediált metabolizmust és hatással lehetnek a gyógyszerválaszra,
különös tekintettel azokra a variánsokra, melyek a kódoló régióban foglalnak helyet [82, 83].
A CYP2C8 promoter régiójában azonosított SNP-k, a CYP2C8*1B (g.-271C>A, rs7909236)
és CYP2C8*1C (g.-370G>T, rs17110453) eltérő gyakorisággal fordulnak elő különböző
populációkban. Variáns alléljuk kaukázusi populációban gyakori frekvenciájú (23% és 12%),
azonban afrikai populációban ez a két polimorfizmus ritka, vagy egyáltalán nem fordul elő

16
[84]. A CYP2C8*1B variánst összefüggésbe hozták a promoter régió fokozott aktivitásával
egy transzkripciós faktor erősebb kötése révén, míg a CYP2C8*1C allél jelenlétének
funkcionális konzekvenciája még nem teljesen tisztázott. A CYP2C8 gén ötödik exonjában
helyet foglaló missszensz (p.I269F) c.805A>T (rs11572103) SNP-t jelölő CYP2C8*2 allél
csökkent enzimaktivitást és alacsonyabb gyógyszer clearence értéket ereményez, mint a vad
típusú gén [85, 86]. A c.805A>T variáns afrikai populációban gyakori (19%), míg kaukázusi
és ázsiai népcsoportokban ritkán fordul elő [85].
A CYP2C8 génben a *3 allél két kapcsolt variáns együttes jelenlétét feltételezi. Az
egyik ezek közül a c.416G>A (rs11572080) SNP, ami egy arginin-lizin aminosav cserét
eredményez a 139-es pozícióban (p.R139K), a gén 3. exonjában, a c.1196A>G (rs10509681)
SNP pedig egy lizin-arginin kicserélődéshez vezet a 399-es aminosav pozícióban (p.K399R),
a gén 8. exonjában [85]. A CYP2C8*3 allél frekvenciája kaukázusi fehér populációban 11-
14% [84]. Egyes kutatók vizsgálatai alapján a CYP2C8*3 allél csökkent enzimaktivitást
eredményez, ami gyengébb paclitaxel metabolizmussal társul, míg más kutatók nem találtak
összefüggést a *3 allél jelenléte és a paclitaxel clearance között [84, 87]. Néhány CYP2C8
szubsztrát esetében, úgy, mint a repaglinid és a cerivastatin a CYP2C8*3 allél megléte
fokozott metabolizmussal társult [88, 89]. A CYP2C8*3 allél eltérő gyógyszer metabolizáló
hatása hátterében álló egyik valószínűsíthető ok a nagy, több szubsztrátot felismerő CYP2C8
szubsztrátkötőhely, a másik lehetséges magyarázat a CYP2C8*3 és a CYP2C9*2 közötti erős,
részleges linkage disequilibrium lehet [88, 90]. A két variáns között fennálló kapcsoltság
egyes elképzelések szerint felelős lehet a NSAID-indukálta gasztrointesztinális vérzésekért
[91]. A CYP nomenklatúrában a CYP2C8 gén 5. exonjában a c.792C>G, rs1058930 variáns
(p.I264M) egyezményes jelölésére használt CYP2C8*4 allél kaukázusi populációban 7%
gyakorisággal mutatható ki [69]. A *4 variáns hordozása a vad típushoz mérten csökkent
enzimaktivitással és gyengébb szubsztrát metabolizmussal hozható összefüggésbe [84, 88,
92]. További ritka variánsokat írtak le CYP2C8*5-től *14-ig, melyek jellemzően ázsiai
populációban fordulnak elő <1%-os frekvenciával [93].
A paclitaxel, statinok, antidiabetikus-, antimaláriás- és nem szteroid
gyulladáscsökkentő gyógyszerek metabolizmusán túl a CYP2C8 más klinikai asszociációban
is a kutatások középpontjába került. A bifoszfonátok (pl.: zoledronsav), melyek széles körben
alkalmazott gyógyszerek a benignus és malignus csontbetegségek kezelésében ritka, de súlyos
mellékhatása lehet az állkapocscsont nekrózis. Egy GWA tanulmány számolt be a CYP2C8
rs1934951 intronikus SNP-ről, mely szignifikáns kapcsolatot mutatott az állcsont szövet
elhalás kialakulásában myeloma multiplex-es betegekben bifoszfonát kezelést követően [94].

17
Bár a bifoszfonátok nem a CYP450 enzimrendszer segítségével metabolizálódnak, a jelenség
hátterében feltételezhetően a CYP2C8-nak az angiogenezisben és/vagy gyulladás
kialakulásában játszott szerepe állhat. Smith és munkatársai párhuzamot vontak a renális
diszfunkció kialakulásának kockázata és a CYP2C8 polimorfizmusok jelenléte között, olyan
szervtranszplantált betegekben, akik a calcineurin inhibitorok csoportjába tartozó, allograft
kilökődés kezelésére használt ciklosporin és takrolimusz terápiában részesültek [95].
4. ábra A CYP2C8 génben előforduló fontosabb polimorfizmusok
(Genetic polymorphisms of CYP2C8 in Japanese population.Nakajima M, 2003)
1.2.4. CYP3A5 genetikája
A CYP3A5 gén a sokoldalú CYP3A alcsalád tagja a CYP3A4, CYP3A7 és a CYP3A43
génnel együtt, ami a 200 leggyakrabban alkalmazott gyógyszer 37%-ának eliminációjáért
felelős [4]. A CYP3A4 és CYP3A5 együttesen a máj citokróm enzimeinek körülbelül 30%-át
teszik ki, a P450 rendszeren keresztül metabolizálódó gyógyszerek fele CYP3A szubsztrát
[96]. A májban és a bélben expresszálódó CYP3A5 a 7q22.1 kromoszomális pozícióban
helyezkedik el. Kilenc kódoló exonjának fehérje terméke egy 502 aminosavból felépülő
protein [96]. A funkcionális CYP3A5*1A vad típusú allélon kívül polimorf tulajdonságának
köszönhetően több variánsa (CYP3A5*9-ig) létezik [97].
A CYP3A5 gén részben funkcionálisnak tekinthető, aminosav cserével járó CYP3A5*2
(c.1193C>A; T398N; rs28365083) variánsa is ismert, mely kaukázusi fehér populációban
0,3-1,3%-os gyakorisággal van jelen [98, 99].

18
A leggyakoribb és legrészletesebben tanulmányozott, mRNS splicingot megváltoztató
variáns a CYP3A5 génben a CYP3A5*3 (c.6986A>G, rs776746) [96, 97]. Az alternatív splice
forma egy a 3. intronból származó inszerciót tartalmaz, ami a leolvasási keret eltolódásán
keresztül a kódoló régiójában egy korai stop kodon beépülést és funkcióképtelen fehérje
létrejöttét eredményezi [97]. A CYP3A5*3/*3 homozigóta genotípusú egyéneket „nem-
expresszáló” fenotípusúnak tekintjük. Ezzel szemben a CYP3A5*1/*1 és CYP3A5*1/*3
genotípusú személyek az „expresszáló”-k, akik gyorsabban metabolizálják a CYP3A5
szubsztrát gyógyszereket, például a tacrolimust, midazolamot, az antiretrovirális szakinavirt
vagy az akut limfoblasztos leukémiában szenvedők kezelésére használt vinkrisztint [97, 100-
103]. A CYP3A5*3 allél gyakoriságában jelentős interetnikai különbségek mutatkoznak.
Előfordulási gyakorisága kaukázusi populációkban 82-95%-os, afroamerikaiakban 33%, japán
populációban 85%, kínaiban 65%, mexikói népcsoportban 75%-ra tehető [97, 104, 105]. A
változó gyógyszerválaszon kívül a különböző CYP3A5 genotípusok hajlamosító, illetve védő
szerepet tölthetnek be egyes betegségek kialakulásában. Shimada és kutatócsoportja japán
populációban végzett vizsgálatainak eredményei azt mutatják, hogy azok a nőbetegek, akik
CYP3A5 expresszálónak tekinthetők, fokozott kockázattal rendelkeznek emlődaganat
kialakulására szemben a nem-expresszálókkal [106]. Hasonló eredményhez jutottak Borst és
munkatársai, mely szerint a CYP3A5 expresszálók körében gyakoribb a gyermekkori akut
limfoblasztos leukémia kialakulása, mint a CYP3A5*3/*3 genotípus esetén [107].
További fontos CYP3A5 variánsnak tekintjük a CYP3A5*6 és CYP3A5*7 allélokat,
amelyek csonka, funkcióképtelen fehérjét eredményeznek különböző mechanizmusok révén.
A CYP3A5*6 (c.624G>A; rs10264272) variáns esetén a hetedik exonban egy G>A tranzíció
eredményeként, a CYP3A5*7 (27131–27132insT; rs76293380) tekintetében pedig egy timin
inszerció útján jön létre egy korai stop kodon a 348-as pozícióban [97, 108]. A CYP3A5*6 és
CYP3A5*7 allélok afroamerikai populációkban viszonylag gyakoriak (7-17% és 8%),
kaukázusi és ázsiai populációkban nagyon ritkán vagy egyáltalán nem fordulnak elő [97, 104,
108, 109].

19
1.2.5. CYP4F2 genetikája
A sejtek endoplazmatikus retikulumaiban lokalizálódó citokróm P450 4F2 (CYP4F2)
enzim katalizálja a hosszú szénláncú zsírsavak, K vitamin (K1, K2), E vitamin, arachidonsav
és leukotrién B4 NADPH-függő oxidációját [110]. A hepatikus CYP4F2 regulálja továbbá az
antimaláriás furamidin prodrugjának, a pafuramidinnek bioaktivációját is [111]. A CYP4F2
humán gén kromoszomális elhelyezkedése a 19p13 pozicióra tehető. A gén 13 kódoló
exonból áll, aminosav szekvenciája a CYP4F3 génnel 81-93%-os homológiát mutat [112].
Legfontosabb, funkcionális jelentőséggel bíró genetikai variáns a CYP4F2 génben a
c.1297G>A (rs2108622) polimorfizmus (CYP4F2*3), mely egy valin-metionin aminosav
cserét eredményez a fehérjében (V433M). Bár a CYP4F2*3 variáns jelenléte nem
befolyásolja sem az mRNS transzkripciót, sem az enzimaktivitást, mégis csökkent fehérje
szintet eredményez, feltehetőleg a csökkent transzláció vagy a fokozott protein degradációnak
köszönhetően [113]. A CYP4F2 1297A allél hordozása módosult warfarin metabolizmussal
hozható összefüggésbe, ami 4-12%-kal magasabb kívánt antikoaguláns dózist határoz meg
különösen ázsiai és európai populációkban [114, 115]. A CYP4F2 rs2108622 polimorfizmus
nem csupán a warfarin dózist, de az Európában szélesebb körben alkalmazott K vitamin
antagonista acenokumarol hatékony dózisát is hasonló módon befolyásolja [116]. Korábbi
vizsgálatok eredményei számolnak be arról, hogy CYP4F2*3 variáns hordozása kapcsolatban
állhat magas vérnyomás kialakulásával és csökkent E vitamin metabolizmussal [117, 118].

20
1.3. Taxán vegyületek
A széles körben alkalmazott paclitaxel (Taxol®) és docetaxel (Taxotere®) a
mikrotubulus stabilizáló vagy mikrotubulus inhibitor természetű taxán vegyületek csoportjába
tartozó, különösen aktív kemoterápiás ágensek, melyeket emlő-, petefészek- és tüdődaganatos
betegek kezelése során alkalmaznak [119-121]. A mikrotubulusok olyan α- és ß-tubulin
alegységekből álló komplex struktúrák, melyek számos sejtfunkcióban játszanak szerepet,
úgy, mint intracelluláris transzport, szekréció, a sejt alakjának fenntartása és
neurotranszmisszió [122]. A taxán gyógyszerek a tubulin ß alegységéhez kötődve a tubulin
polimerizációt stabilizálják, ezáltal különböző fázisaiban tartóztatják fel a sejtciklust, így
gátolják a mitózist a metafázis-anafázis határán [120, 122-124]. Az 1970-es évek elején a
mellrák kezelésének gyógyszeres terápiája az alkiláló ágensek (pl.: ciklofoszfamid)
methotrexáttal és 5-fluorouracillal való kombinációjára korlátozódott, ami átlagosan 14 hónap
átlagos túlélést eredményezett [125]. Az 1980-as években terjedt el az antraciklinek
(doxorubicin és epirubicin) használata, mint elsővonalbeli daganatellenes szerek, majd az
1990-es években bevezetésre került paclitaxel és docetaxel kombinációban alkalmazva
antraciklinekkel és alkiláló szerekkel jelentősen emelték a metasztatikus emlődaganatos
betegek teljes- és betegségmentes túlélését [126-128]. A taxánok elsővonalbeli használata
során a gyógyszeres válasz 25-69%-ra tehető, a betegek gyakran mutatnak rezisztenciát a
kezeléssel szemben [129-132].
A paclitaxelt 1971-ben izolálták először az amerikai tiszafa (Taxus brevifolia)
kérgéből [122, 133]. A vegyület kémiai szerkezetét az 5. ábra szemlélteti. Használatát 1992-
ben engedélyezte az Amerikai Élelmiszer- és Gyógyszerbiztonsági Felügyelet (FDA),
előrehaladott petefészekrák kezelésére használt kemoterápiás gyógyszerként került
forgalomba [134]. A paclitaxel erősen hidrofób természetű vegyület, mely megkívánja, hogy
vivőanyagként oldatban alkohollal és Cremophor® EL-lel (polyoxyetilezett ricinusolaj) együtt
alkalmazzák [135]. Ez az oldószer súlyos hiperszenzitív reakciókat válthat ki, így a terápia
részeként dexametazon egyidejű alkalmazása javasolt [135]. A paclitaxel nem lineáris
farmakokinetikát mutat, mely megnehezíti a gyógyszer helyes adagolását [122, 136, 137]. A
gyógyszer felezési ideje 15-50 óra, a májban metabolizálódik és az epével kerül kiválasztásra
[122]. Gyakori mellékhatásai a paclitaxel gyógyszernek a hajhullás, myeloszupresszió,
gasztrointesztinális tünetek, febrilis neutropenia, myalgia és perifériás neuropathia [122, 130].

21
A második generációs taxán, a docetaxel, az inaktív taxán prekurzor -
10-deacetylbaccatin III - oldalláncának észteresítésével nyert szemiszintetikus vegyület [130,
138]. A docetaxel a közönséges tiszafából (Taxus baccata) az 1980-as években került
azonosításra [134]. Kémiai struktúráját az 5. ábra szemlélteti. A docetaxel nagyobb
affinitással kötődik a ß-tubulinhoz és kétszer hatékonyabban gátolja a mikrotubulus
depolimerizációt szemben a paclitaxellel [139]. A docetaxel a sejtciklust 3 fázisban képes
blokkolni a centroszómát célozva, az S, G2 és M fázisokban, míg paclitaxel esetén a
mikrotubulus target a mitotikus orsó. Sejtciklusbeli specifikussága csak a G2 és M fázisra
terjed ki [139]. Ellentétben a paclitaxellel, a docetaxelhez használt vivőanyag, a Poliszorbát
80 jelentősen kevesebb mellékhatást eredményez a szervezetbe jutva, mint a Cremophor® EL
[122]. További különbség a két taxán vegyület között, hogy a docetaxel lineáris
farmakokinetikájú, eliminációs felezési ideje 1 óra [122, 140, 141]. A gyógyszer szedése
során hasonló mellékhatások jelentkezhetnek, mint paclitaxel terápia esetén, további tünetként
előfordulhat pleuralis ascites, oedema, neutropenia, bőr- és körömelváltozás (Palmar-plantar
erythrodysesthesia), hyperlacrimatio, melyek limitálhatják a dózist [122, 130]. Bár
hatásmechanizmusukban hasonlóság, farmakológiai és farmakokinetikai jellegükben
különbségek figyelhetők meg, a rutin klinikai gyakorlatban elérhető két taxán vegyület
jelentősen javítja a mellrákos betegek teljes- és betegségmentes túlélési rátáját, különösen
antraciklin alapú terápiában [128, 142, 143]. Ennek ellenére a taxán rezisztencia gyakori
jelenség, azonban előfordulhat szolid tumorokban szenvedő betegeknél, hogy paclitaxellel
szemben rezisztenciát mutatnak, docetaxelre mégis szenzitívnek bizonyulnak [130-132, 144,
145].
5. ábra Paclitaxel és docetaxel kémiai struktúrája
(http://pubs.rsc.org/en/content/articlehtml/2012/md/c2md00315e)

22
A taxán vegyületekre adott egyéni gyógyszerválaszt befolyásolhatják azok a genetikai
variánsok, melyek a taxán útvonalban szerepet játszó metabolizáló enzimek és transzporter
fehérjék működését érintik. A taxánok metabolikus útvonalát a 6. ábra szemlélteti. A
paclitaxel és a docetaxel egyaránt a máj mikroszómális citokróm P450 enzimrendszerén
keresztül metabolizálódik. Mindkét vegyület elsődleges metabolizálója a CYP3A4 enzim, a
paclitaxel a CYP2C8, a docetaxel pedig a CYP3A5 enzim további hozzájárulásával
biotranszformálódik. Mindkét taxán egyaránt szubsztrátja több membránhoz asszociált, aktív
transzportot végző ABC (ATP-binding casette) transzporternek, mint az ABCB1, ABCG2,
ABCC1 és ABCC2. A hepatikus influxban fontos szerepet játszó szerves anion transzportáló
polipeptid (OATP) család SLCO1B3 tagja kulcs regulátornak bizonyult mind a paclitaxel,
mind pedig a docetaxel transzportban.
6. ábra Taxán vegyületek metabolikus útvonala
(https://www.pharmgkb.org/pathway/PA154426155?previousQuery=taxane%20pathway)

23
Számos tanulmány született, mely próbálta feltárni a taxán útvonalban szerepet játszó
gének polimorfizmusai és a taxán kezelés várható kimenetele közötti kapcsolatot, az
eredmények mégis ellentmondásosak. Marsh és kutatócsoportja emlődaganatos betegek
gyógyszerválaszt befolyásoló farmakogenetikai vizsgálata során kapcsolatot találtak a
CYP1B1 gén *3 (c.4326C>G; L432V; rs1056836), leucin-valin aminosav cserét eredményező
misszensz variánsának hordozása és a progresszió mentes túlélés között, függetlenül a
paclitaxel clearance-től [146]. Sissung és munkatársai hasonlóan rosszabb prognózisról
számoltak be docetaxel kezelés kapcsán a CYP1B1 *3/*3 homozigóta genotípus esetén
összehasonlítva a CYP1B1*1 allélt hordozókkal. Ezen eredmények bizonyítékul
szolgálhatnak, hogy a CYP1B1*3 polimorfizmus egy fontos biomarkernek tekinthető a
kemoterápia kimenetelének predikciójában [64]. A CYP2C8*3 allél, mely a c.416G>A
(Arg139Lys) és c.1196A>G (Lys399Arg) aminosav cserékkel járó nukleotid szubsztitúciók
együttes jelenlétével definiálható, csökkent enzimaktivitást eredményez [147]. Gréen és
munkatársai kimutatták, hogy a CYP2C8*3 allél felelős a csökkent paclitaxel eliminációért,
illetve, hogy a paclitaxel indukálta neurotoxicitás korrelál a gyógyszer expozícióval [148].
Egy másik tanulmány számolt be docetaxel metabolizmus farmakogenetikai vizsgálata
kapcsán arról, hogy a CYP3A4*1B (-392A>G) nem-kódoló polimorfizmusa és a CYP3A5 gén
vad típusú *1A allélja jobb docetaxel clearance értékkel hozható összefüggésbe [149].

24
1.4. Antikoaguláns gyógyszerek
A K vitamin antagonista orális antikoagulánsok hatóanyagai, mint a warfarin
(Marfarin®) és az acenokumarol (Syncumar®) széles körben alkalmazott gyógyszerek a
thromboemboliás kórképek (mélyvénás trombózis, tüdőembólia) hosszú távú kezelésében
[150]. A kumarin származékok szűk terápiás tartománya miatt fontos a dózis pontos
meghatározása, ami nagy kihívást jelenthet a klinikus számára, mivel a gyógyszerválasz
nagyfokú individuális és etnikai különbségeket mutat [22, 151]. Az egyéni
gyógyszerválaszban megmutatkozó különbségek hátterében demográfiai, környezeti, klinikai
és genetikai faktorok állnak [152, 153]. Napjainkig számos nemzetközi tanulmány született
azzal a céllal, hogy megvizsgálja és azonosítsa azokat a genetikai variánsokat,
valószínűsíthető SNP-ket, melyek kapcsolatba hozhatók a véralvadásgátlók megváltozott
farmakokinetikájával, farmakodinamikájával [150, 154, 155].
Az antikoaguláns gyógyszerválasz fő genetikai determinánsai a CYP2C9, VKORC1 és
a CYP4F2, de számos további gén is részt vesz a warfarin biotranszformációjában és
hatásmechanizmusának kialakításában, mint a CYP2C19, CYP1A2, CYP1A1, CYP3A4,
GGCX, EPHX1 és CALU [156-158]. A módosult warfarin válaszért felelős kulcsfontosságú
enzimeket kódoló gének: a K vitamin ciklust, mint farmakológiai célpontot érintő VKORC1
(K vitamin epoxid-reduktáz komplex 1), az S-warfarint metabolizáló CYP2C9 (citokróm P450
2C9), a K vitamin katabolizmusban szerepet játszó CYP4F2 (citokróm P450 4F2), valamint a
K vitamin-függő véralvadási faktorokat (II., VII., IX. és X. faktor) aktiváló γ-karboxiláz
enzimet kódoló GGCX (gamma-glutamil karboxiláz) [113, 150, 159, 160]. Az FDA 2007-ben
kiadott közleményében hangsúlyozza a genetikai teszt fontosságát a warfarin kezdő dózisának
meghatározásához [161]. A CYP2C9 és VKORC1 génben található polimorfizmusok együtt
30-40%-ban felelősek a warfarin dózis variabilitásért, míg a VKORC1, CYP2C9 és CYP4F2
genotípusok a társuló klinikai faktorokkal együttesen 60%-ban magyarázzák az antikoaguláns
gyógyszerválasz változékonyságát [162-164]. A CYP2C9*2 (c.430C>T) és CYP2C9*3
(c.1075A>C) misszensz polimorfizmusok jelenléte jelentős csökkenést eredményez az enzim
aktivitásában [160]. A helyes dózis megállapításához a VKORC1 genotípus meghatározásában
a három legfontosabb funkcionális polimorfizmus a -1639G>A (VKORC1*2), c.9041G>A
(VKORC1*3) és a c.6009C>T (VKORC1*4) SNP-k [165]. A CYP4F2 gén által kódolt enzim
a K vitamin biotranszformációban szerepet játszó oxidáz, melynek variánsai csökkentik az
enzim K vitamin metabolizáló kapacitását [115]. Újabb kutatási eredmények számolnak be

25
arról, hogy a magas warfarin dózist meghatározó CYP4F2*3 (rs2108622; V433M) SNP
frekvenciája szintén szignifikáns különbségeket mutat különböző populációkban. Ez az érték
kaukázusi népcsoportokban 30%-ra, ázsiai populációkban 17%-ra tehető. A CYP4F2*3
variáns hordozása további 2-5%-ban magyarázhatja az egyéni, hatékony dózisok közötti
különbségeket [115, 166].

26
1.5. Roma populáció bemutatása
A vizsgálataink tárgyát képező magyar és roma populáció közös tulajdonsága, hogy
eredetüket tekintve mindkettő különbözik más európai kaukázusi népektől. A romák az indiai
szubkontinens északi részéről származnak. Indiából észak-nyugat felé történő migrációjukat
körülbelül 1000 évvel ezelőtt kezdték ismeretlen okból [167, 168]. Társadalomtudományi és
nyelvészeti tanulmányok a romák ázsiai eredetére utalnak. A genetikai bizonyítékok szintén a
romák indiai eredete mellett szólnak, úgy, mint az Y kromoszóma H-M82 haplocsoportjának
és a mitokondriális DNS-ben az mtDNS M haplocsoport hordozása, mely nagyon ritkán
fordul elő az indiai szubkontinensen kívül [169, 170]. Morar és munkatársai balkáni, oláh és
nyugat-európai roma csoportokban 5 betegség okozó génmutáció vizsgálata során kimutatták
a congenitalis myasthenia szindróma kialakulásáért felelős 1267delG mutációt roma és indiai
(pakisztáni) mintákban, ami a legerősebb bizonyítékul szolgál a romák indiai eredetére [171].
Világszerte számos országban él roma populáció, melynek becsült mérete körülbelül
12 millió főre tehető, ebből 8-10 millióan élnek Európában, legnagyobb számban
Romániában, Szlovákiában, Bulgáriában és Magyarországon [170, 171]. Magyarországon
több minor etnikum is előfordul, közülük is a romák képviseltetik magukat a legnagyobb
számban. A Központi Statisztikai Hivatal 2008-as felmérése szerint 6-800000 roma él
Magyarországon, ezzel hazánk a negyedik romák által legsűrűbben lakott országának számít
európai viszonylatban (www.ksh.hu). A magyarországi romák megyénkénti eloszlását az
állandó populációhoz viszonyítva a 7. ábra mutatja be. A magyarországi roma populáció 3 fő
alpopulációra osztható nyelvi és történeti szempontból; a romungrókra, az oláh és a beás
romák csoportjára. A magyar nyelvű romungrók közé tartozik a magyarországi romák
mintegy kétharmada. A magyar és lovári nyelvet beszélő, 19. században Havasalföld
irányából érkező oláh roma szubpopuláció a roma populáció 21%-át teszi ki [172]. A
harmadik csoportot a főként Dél-Dunántúl falvaiban élő beás roma emberek alkotják, akik a
magyar nyelv mellett az archaikus román nyelvet használják. Nagyjából a magyarországi
romák 8%-a a beás roma alpopuláció tagja.
A roma populáció egy kitaszított, hátrányos helyzetben lévő etnikai csoport
Európában, rossz szocioökonómiai státusszal [173]. A roma berendezkedésben élő emberek
gyakran jelentősen alacsonyabb iskolázottságúak, magasabb köztük a munkanélküliek aránya,
várható élettartamuk messze elmarad az átlag európai populációkban mért adatoktól. A romák
körében a csecsemőhalandóság szintén emelkedett értéket mutat, továbbá gyakrabban

27
szenvednek krónikus légzőszervrendszeri- (asztma, bronchitis), kardiovaszkuláris- és
daganatos betegségektől [174, 175]. A herediter motoros és szenzoros neuropátia, congenitalis
myasthenia szindróma, galaktokináz deficiencia és a policisztás vesebetegség, mint ritka,
öröklődő betegségek is azonosításra kerültek a roma populáció tagjaiban [176-178]. Kovács
és munkatársai egy kis magyarországi roma csoportban a IV-es típusú rövid borda-
polydactylia szindróma eseteit írták le [179]. Szintén magyarországi anhidrotikus
ektodermális diszpláziás roma beteg kapcsán került bemutatásra a GJA1 génben egy különös,
sporadikus, a GJB2 génben pedig egy heterozigóta mutáció [180].
7. ábra A romák megyénkénti aránya Magyarországon 2003-ban
(Janky, Kemény, Lengyel: A magyarországi cigányság 1971-2003, 2005)

28
2. CÉLKIT ŰZÉSEK
Kutatásunk célja a citokróm P450 enzimrendszer különböző, farmakogenetikai
szempontból releváns génjeiben leírt polimorfizmusok genetikai vizsgálata, mely magába
foglalja ezen polimorfizmusok gyakoriságának és eloszlásának meghatározását roma és
magyar populációban. Ezáltal rávilágíthatunk azokra a jelentős genetikai különbségekre,
melyek a gyógyszermetabolizmus fázis I enzimeit érintik és hatással lehetnek a
gyógyszerválaszra.
Kitűzött céljaink pontokban:
1. A CYP1A2 kapcsán a gén szabályozó régiójában elhelyezkedő rs2069514 és
rs35694136 és az intronikus rs762551 és rs12720461 polimorfizmusok
gyakoriságának meghatározása roma és magyar populációban.
2. A vizsgált CYP1A2 variánsok együttállásaiból a különböző haplocsoportok
frekvenciájának meghatározása a két vizsgált populációban.
3. A taxán kezelés kimenetelét befolyásoló CYP1B1, CYP2C8 és CYP3A5
rs1056836, rs1058930, rs776746 variánsok allélfrekvenciáinak vizsgálata.
4. Az antikoaguláns terápiát érintő CYP4F2 rs2108622 polimorfizmust vizsgálva
átfogó képet kapni arról, hogy az általunk tanulmányozott két népcsoport között
az említett SNP tekintetében mutatkozik-e olyan jelentős genetikai különbség,
mely eltérő módon befolyásolhatja a két populáció véralvadásgátló
gyógyszerekkel szembeni érzékenységét.
5. A CYP1A2, CYP1B1, CYP2C8, CYP3A5 és CYP4F2 gének vizsgált
polimorfizmusaiban kapott allélfrekvencia értékek összehasonlítása más kutatók
szakirodalomban leírt populációs adataival, különös tekintettel a kaukázusi és
indiai populációkra.

29
3. ANYAGOK ÉS MÓDSZEREK 3.1. Vizsgált populációk
Vizsgálataink során magyar és roma populációból származó DNS mintákkal
dolgoztunk. A DNS mintákat egészséges, magyarországi roma és magyar személyektől
gyűjtöttük. A vizsgálatainkban résztvevő személyek valamennyien előzetes tájékoztatást
követően beleegyezésüket adták a vizsgálatok elvégzéséhez és nyilatkoztak etnikai
hovatartozásukról.
A roma és magyar DNS minták a Pécsi Tudományegyetem központi biobankjából
származtak, mely része a Páneurópai Nemzetközi Biobankhálózatnak (BBMRI; Biobanking
and Biomolecular Resources Research Infrastructure). A minták gyűjtésében és tárolásában az
1975-ben az Orvos-világszövetség által megalkotott Helsinki deklarációban megfogalmazott
etikai alapelvek voltak irányadók, a biobank vezetésében és fenntartásában az Egészségügyi
Tudományos Tanács Tudományos és Kutatásetikai Bizottság (ETT-TUKEB) által jóváhagyott
elveket követtük.
Kutatásaink összesen 5 citokróm P450 gén 8 funkcionálisan jelentős
polimorfizmusának vizsgálatára terjedtek ki.
A CYP1A2 gén nem-kódoló (5’-UTR és intronikus) régióiban elhelyezkedő négy
polimorfizmus (-163C>A, -729C>T, -2467T>delT és -3860G>A) vizsgálatához 404 roma
(148 férfi, 256 nő; átlag életkor 35±11 év) és 396 magyar (236 férfi, 163 nő; átlag életkor
42±14 év) mintát analizáltunk.
A taxán terápiát befolyásoló három citokróm P450 gén (CYP1B1, CYP2C8 és
CYP3A5) polimorfizmusainak (rs1056836, rs1058930, rs776746) vizsgálata során 397 roma
(172 férfi, 225 nő; átlag életkor 36±14 év) és 412 magyar (204 férfi, 208 nő; átlag életkor
41±22 év) minta került genotipizására.
A CYP4F2*3 (rs2108622) allél gyakoriságának meghatározásához 484 roma
(230 férfi, 254 nő; átlag életkor 41±20 év) és 493 magyar
(278 férfi, 215 nő; átlag életkor 37±13 év) DNS mintáját vizsgáltuk.

30
3.2. Molekuláris biológiai módszerek
3.2.1. DNS izolálás
A DNS-izolálást EDTA-val alvadásgátolt perifériás vérminták leukocitáiból végeztük az
alább részletezett rutin kisózásos technika segítségével. A vérmintákat centrifugacsőben 4°C-
os RBC (red blood cell) lízispufferrel egészítettük ki 50 ml-es térfogatra, melyet 30 perces
jeges inkubáció követett, közben 4-5-ször átforgattuk. Ezután 30 percig tartó centrifugálás
(5000 rpm-en és 4°C-on), majd a felülúszó gondos eltávolítása következett. A térfogatot ismét
kiegészítettük lízispufferrel és a fenti folyamatot 4 alkalommal ismételtük meg. Az utolsó
lépésnél 5 ml SE puffert (pH=8; 4,39g (75mmol) nátrium-klorid + 8,41g (25 mmol) Na-
EDTA), 25 µl proteináz-K-t (10mg/ml) és 500 µl 10%-os SDS-t adtunk az üledékhez, majd
vortexelést követően 37°C hőmérsékleten egy éjszakán át 200 rpm-en rázógépen inkubáltuk a
mintákat. Másnap kiegészítettük az oldatot 3 ml telített nátrium-klorid oldattal (6 M), majd 15
másodpercig tartó vortexelést követően 15 percig 3000 rpm-en centrifugáltuk. A DNS-t
tartalmazó felülúszót egy másik 50 ml térfogatú csőbe óvatosan átöntöttük és 40 ml térfogatra
egészítettük ki 96%-os etanollal, 5-10 percig szobahőmérsékleten inkubáltuk, amíg a DNS ki
nem csapódott. A kivált DNS-t eppendorf csőbe helyeztük, 200 µl 70%-os etanolt adtunk
hozzá és 20 percig inkubáltuk, később az etanolt pipettával eltávolítottuk. Ezt követően a
DNS-t 30 percig száradni hagytuk szobahőmérsékleten, majd hozzáadtunk 500 µl TE puffer
oldatot (pH=8, 0,78 g Tris-HCl + 0,14g EDTA) a DNS-hez és egy éjszakán át 37°C-os
hőmérsékleten inkubáltuk, így lehetővé tettük a DNS teljes beoldódását.
3.2.2. PCR reakció
A DNS vizsgálatához munkánk során az első lépés minden esetben egy polimeráz
láncreakció (Polymerase Chain Reaction; PCR) útján történő amplifikáció volt. A PCR
amplifikáció 50 µl-es végtérfogatban történt, mely 200 µM dNTP-t (dATP, dTTP, dGTP,
dCTP), 1 U Taq polimerázt, 5 µl reakciópuffert (10 mM Tris-HCl, pH 9,0, 500 mM KCl, 14
mM MgCl2), 0,2 mM forward és reverse primert, valamint templátként 1 µg extrahált
genomiális DNS-t tartalmazott. A PCR-t minden esetben saját tervezésű, szekvencia-
specifikus oligonukleotidokkal - forward és reverse primerrel - végeztük. Az egyes
vizsgálatokhoz használt, általunk tervezett primerek szekvenciáit a 3. táblázat foglalja össze.
Az amplifikáció MJ Research PTC 200 thermal cycler PCR készülékkel valósult meg.

31
Az RFLP módszerekhez kapcsolódó PCR reakciók kondíciói a következők voltak a
CYP1A2 polimorfizmusok vizsgálatánál: elődenaturáció 2 min 95ºC-on, ezt követte 35
cikluson keresztül: denaturáció 30 s 95ºC-on, az annealing hőmérséklete a -163C>A és a
-3860G>A SNP-k esetén 59ºC volt 30 s-ig, -729C>T SNP-nél 60ºC volt 30 s-ig, ez után a
primer extenzió 30 s 72ºC-on és a végső lánchosszabbítás 72ºC-on 5 percig tartott.
A CYP1B1, CYP2C8, CYP4F2 és CYP3A5 SNP-k PCR amplifikációja a következő
feltételek mellett zajlott: elődenaturáció 2 min 95ºC-on, ezt követte 35 cikluson keresztül 30 s
denaturálás 95ºC-on, primerkötődés 30 s 57ºC-on (CYP1B1; rs1056836), 55ºC-on (CYP2C8;
rs1058930), 60ºC-on (CYP4F2; rs2108622), 62ºC-on (CYP3A5; rs776746). Ez után a primer
extenzió 45 s 72ºC-on, melyet 5 perc végső extenzió követett 72ºC-on. A keletkezett PCR
termékek detektálása gélelektroforézissel (2%-os agaróz gélben), etídium-bromidos festéssel
és UV megvilágítással történt.
3. táblázat Primer szekvenciák
Gén SNP Primerek (5’-3’)
CYP1A2
rs2069514 Forward: TCATCAAGCTACACATGATCGAG
Reverse: TGCGTGTCAGGTCTCTTCAC
rs762551 Forward: GGTATCAGCAGAAAGCCAGC
Reverse: CCCAGCTGGATACCAGAAAG
rs12720461 Forward: ACTCACCTAGAGCCAGAAGCTC
Reverse: AATGGCTTAGTCCAAACTGCC
CYP1B1 rs1056836 Forward: TCATCACTCTGCTGGTCAGG
Reverse: AGAATTGGATCAGGTCGTGG
CYP2C8 rs1058930 Forward: GGTCTGCAATAATTTCCCTC
Reverse: TGATATTCATCTTCAGTTTGTGG
CYP3A5 rs776746 Forward: GTGGTCCAAACAGGGAAGAGGT
Reverse: GCCCATACAGGCAACATGACTTAG
CYP4F2 rs2108622 Forward: ATCAACCCGTTCCCACCT
Reverse: ACATTGTGCTCCCAGACG

32
3.2.3. RFLP
A CYP1A2 gén kapcsán restrikciós fragmenthossz polimorfizmus (RFLP) módszert
használtunk az rs2069514, rs762551 és rs12720461 polimorfizmusokhoz kapcsolódó
genotípusok elkülönítésére. A CYP1B1, CYP2C8, CYP4F2 és CYP3A5 SNP-k genotipizálása
minden esetben szintén ezzel a módszerrel valósult meg. A módszer tervezésekor és a
restrikciós endonukleáz kiválasztásakor (a CYP4F2 gén variánsának vizsgálatától eltekintve)
minden esetben fontos szempont volt, hogy az amplifikált target szekvencia tartalmazzon egy
obligát hasító helyet is a keresett SNP-n kívül a módszer hatékonyságának ellenőrzése
szempontjából. A vizsgálatok során alkalmazott restrikciós endonukleázok, hasítási
mintázatuk és a hozzá tartozó genotípusok a 4. táblázatban kerültek feltüntetésre. A
restrikciós enzimmel történő hasítás után az emésztett PCR termékeket agaróz
gélelektroforézissel választottuk szét. A genotípusok elkülönítése a 3%-os agaróz gélben
etídium-bromid festéssel, UV megvilágítással történt standard DNS létra mellett (8. ábra).
8. ábra Az alkalmazott PCR-RFLP módszerek agaróz gélelektroforézist követő fotója az
(A) CYP2C8*4, (B) CYP3A5*3 és (C) CYP1B1*3 polimorfizmusok esetén
A CYP2C8*4 allél esetén sorban a következő genotípusok láthatók: A (1) wt/wt; (2)*4/*4 ,
(3) wt/*4. A CYP3A5*3 SNP-re: B (1) *3/*3 , (2) wt/wt, (3) wt/*3 és a CYP1B1*3 variánst
illetően: C (1) *3/*3 , (2) wt/*3, (3) wt/wt. P; PCR termék, L ; 50 bp DNS létra.

33
4. táblázat A vizsgált polimorfizmusok genotipizálásához használt restrikciós
endonukleázok és hasítási mintázatuk
Gén Variáns PCR termék (bp) Endonukleáz Fragmentek (bp) Genotípus
CYP1A2
rs2069514 611 BseLI (BslI)
123, 139, 349 GG
123, 139, 349, 488 GA
123, 488 AA
rs762551 298 Bme1390I
(ScrFI)
46, 66, 186 CC
46, 66, 186, 232 CA
66, 232 AA
rs12720461 250 Bme1390I
(ScrFI)
28, 33, 67, 122 CC
28, 33, 67, 95 122 CT
33, 95, 122 TT
CYP1B1 rs1056836 311 BseNI (BsrI)
22, 95, 194 CC
22, 95, 117, 194 CG
117, 194 GG
CYP2C8 rs1058930 500 TaqI
83, 150, 267 CC
83, 150, 233, 267 CG
233, 267 GG
CYP3A5 rs776746 309 RsaI
96, 213 AA
22, 74, 96, 213 AG
22, 74, 213 GG
CYP4F2 rs2108622 492 PvuII
176, 316 GG
176, 316, 492 GA
492 AA

34
3.2.4. Real time-PCR
A CYP1A2 -2467delT polimorfizmus vizsgálatához real-time PCR módszert
használtuk. A CYP1A2 -2467T>delT (CYP1A2*1D, rs35694136) SNP-t TaqMan Drug
Metabolism Genotyping Real-Time PCR Assay (C_60142977_10) (Applied Biosystems, CA,
USA) segítségével határoztuk meg a használati utasítás szerint. A PCR amplifikáció Chromo4
Real-Time PCR Detectorral (Bio-Rad, Hercules, CA, USA) valósult meg a következő
kondíciók szerint: kezdő inkubáció 2 min 95ºC-on, majd denaturáció 20 s 95ºC-on, annealing
és primer extenzió 40 s 60ºC-on, 40 cikluson keresztül történt. A genotípusok analizálására
MJ Opticon Monitor Analysis Software v3.1-et használtunk.

35
3.2.5. Direkt szekvenálás
Valamennyi általunk tervezett PCR-RFLP módszer specificitását és eredményeink
konfirmálását Sanger-féle bidirekcionális szekvenálással végeztünk random módon BigDye
Terminator v.1.1 cycle sequencing kit alkalmazásával, ABI 3500 Genetic Analyser (Applied
Biosystems CA, USA) szekvenátor segítségével.
9. ábra A Sanger-szekvenálás elektroferogramjai heterozigóta genotípusok esetén
A 9. ábrán látható szekvenciarészleteken a nyilak a következő polimorfizmusokat
mutatják sorban: (A) CYP2C8*4 (c.792C>G), (B) CYP3A5*3 (c.6986A>G) és (C) CYP1B1*3
(c.4326C>G).
3.3. Statisztikai módszerek
A vizsgált genetikai variánsok allélfrekvenciái között fennálló összefüggések
feltárására χ2-tesztet alkalmaztunk SPSS 20.0 programcsalád felhasználásával, a
szignifikancia szintet p<0,05-nél húztuk meg. A haplotípus analízis vizsgálatához Phase 2.1.
programot használtunk.

36
4. EREDMÉNYEK 4.1. CYP1A2 gén
A roma és magyar populációban kapott CYP1A2 genotípus- és allélfrekvencia értékek
eloszlását az 5. táblázat foglalja össze. A CYP1A2 gén nem-kódoló régióiban elhelyezkedő
polimorfizmusok közül roma és magyar populációban a -163A SNP volt a leggyakoribb
(56,9% vs. 68,6%), ezt követően a -2467delT variáns volt jelen 6,81% és 5,81%-os
gyakorisággal. A -3860A és -729T variánsok jelentősen ritkábban fordultak elő vagy nem
voltak kimutathatók a két csoportban.
A CYP1A2*1C allél (-3860G>A) vizsgálata során a -3860A variáns allél
gyakoriságában különbséget észleltünk magyar és roma populációban (2,02% vs. 0%). A
CYP1A2 -3860AA homozigóta genotípus nem volt kimutatható sem a roma, sem a magyar
mintákban.
A CYP1A2*1F allélt (-163C>A) illetően jelentős különbséget észleltünk a
-163AA genotípus frekvenciájában. Ez az érték a roma populációban 31,9%, magyar
populációban 49,5% volt (p<0,001). További szignifikáns eltérést mutattunk ki a variáns
-163A allél frekvenciájában a roma és magyar populáció között (56,9% vs. 68,6%, p=0,025).
Míg a magyar populációban a -163AA homozigóta genotípus fordult elő leggyakrabban
(49,5%), addig a roma minták fele -163CA heterozigóta genotípusúnak mutatkozott (50,0%).
A CYP1A2 gén -2467delT polimorfizmusát (CYP1A2*1D allél) vizsgálva a variáns
delT allélfrekvencia értékekben nem találtunk szignifikáns különbséget a roma csoportban a
magyarokhoz viszonyítva (6,81% vs. 5,81%). Magyar mintákban a homozigóta
-2467delT/delT genotípus nem volt kimutatható, roma populációban ez az érték 0,74% volt.
A CYP1A2 -729T variáns gyakorisága 0,25% volt magyar populációban, a roma
minták erre a polimorfizmusra nézve 100%-ban vad típusúnak bizonyultak.

37
5. táblázat Genotípus- és allélfrekvencia értékek eloszlása roma és magyar populációban a
CYP1A2 gén polimorfizmusai esetén
Polimorfizmus Genotípus Genotípus frekvencia
Allél Allélfrekvencia
Roma n=404 (%)
Magyar n=396 (%)
Roma (%)
Magyar (%)
-163C>A
C/C 73 (18,1) 49 (12,4) C 43,1 31,4
C/A 202 (50,0) 151 (38,1) A 56,9 68,6*
A/A 129 (31,9) 196 (49,5)#
-3860G>A
G/G 404 (100,0) 380 (95,9) G 100,0 98,0
G/A 0 (0,0) 16 (4,04) A 0,0 2,0
A/A 0 (0,0) 0 (0,0)
-2467delT
T/T 352 (87,1) 350 (88,4) T 93,2 94,2
T/delT 49 (12,1) 46 (11,6) delT 6,8 5,8
delT/delT 3 (0,74) 0 (0,0)
-729C>T
C/C 404 (100,0) 394 (99,5) C 100,0 99,7
C/T 0 (0,0) 2 (0,5) T 0,0 0,3
T/T 0 (0,0) 0 (0,0)
*p=0,025 #p<0,001 Az általunk vizsgált -3860G>A, -2467T>delT, -163C>A és -729C>T
polimorfizmusok a 6. táblázatban bemutatott öt különböző haplotípust (ht) határozták meg. A
különböző haplotípusok (ht1-ht5) frekvenciái sorban a következők voltak magyar minták
vizsálatát követően: 31,4%, 2,02%, 62,8%, 0,25% és 3,54%. Roma populációs mintákban a
ht2 és a ht4 haplotípus nem volt kimutatható, a ht1, ht3 és ht5 haplotípusok gyakorisági
értékei a következők voltak roma mintákban: 43,1%, 50,1% és 6,81%
(7. táblázat).

38
6. táblázat A vizsgált CYP1A2 polimorfizmusok együttállása által meghatározott 5 fő
haplotípus
Haplotípus
Allélkonstelláció
-3860G>A -2467T>delT -729C>T -163C>A
ht1 G T C C
ht2 A delT C A
ht3 G T C A
ht4 G delT T A
ht5 G delT C A
7. táblázat CYP1A2 SNP-k által alkotott haplotípusok gyakorisága roma és magyar
populációban
Haplotípus Roma n (%) Magyar n (%)
ht1 348 (43,1) 249 (31,4)
ht2 - 16 (2,02)
ht3 405 (50,1) 497 (62,8)
ht4 - 2 (0,25)
ht5 55 (6,81) 28 (3,54)

39
4.2. Taxán terápiát befolyásoló citokróm P450 gének
4.2.1. CYP1B1 gén
A CYP1B1 gén c.4326C>G polimorfizmusát tekintve a CYP1B1 4326GG homozigóta
variáns genotípus előfordulása szignifikánsan magasabb volt roma populációban, mint
magyarban (50,12% vs. 39,32%, p=0,002). A CYP1B1 4326G allél frekvenciája ezzel
szemben csak kis mértékben bizonyult emelkedettnek a roma mintákban, összehasonlítva a
magyar populációs mintákkal (69,4% vs. 64,2%). A különbség nem bizonyult szignifikánsnak
(p=0,852). A különböző CYP1B1 genotípusokhoz tartozó értékeket a 8. táblázat szemlélteti.
8. táblázat CYP1B1 c.4326C>G polimorfizmus genotípusainak és variáns
allélfrekvenciájának eloszlása magyar és roma populációban
Gén Polimorfizmus Genotípus Magyar
n=412 (%) Roma
n=397 (%)
CYP1B1 4326C>G
CC 45 (10,92) 45 (11,34)
CG 205 (49,76) 153 (38,54)
GG 162 (39,32) 199 (50,12)*
G allélfrekvencia 0,642 (64,2) 0,694 (69,4)
* p=0,002

40
4.2.2. CYP2C8 gén
A CYP2C8 c.792C>G (rs1058930) polimorfizmus vizsgálata során a CYP2C8 792GG
(*4/*4 ) homozigóta genotípus a roma csoportban nem volt kimutatható, a magyar
populációban is csak 1,46%-os gyakorisággal fordult elő. A CYP2C8 792G allél
frekvenciájában szignifikáns különbséget tapasztaltunk a két populáció között. A magyar
mintákban a 792G variáns allél jelenléte jelentősen gyakoribb volt összehasonlítva a roma
csoportban kapott értékekkel (5,83% vs. 2,14%, p=0,001). A CYP2C8 c.792C>G SNP roma
és magyar populációban kapott genotípus és allélfrekvencia értékeit a 9. táblázat foglalja
össze.
9. táblázat A CYP2C8 c.792C>G polimorfizmus gyakorisága magyar és roma mintákban
Gén Polimorfizmus Genotípus Magyar n=412 (%)
Roma n=397 (%)
CYP2C8 792C>G
CC 370 (89,80) 380 (95,72)
CG 36 (8,74) 17 (4,28)
GG 6 (1,46) 0 (0,00)
G allélfrekvencia 0,058 (5,83) 0,021 (2,14)*
* p=0,001

41
4.2.3. CYP3A5 gén
A CYP3A5 c.6986A>G polimorfizmus vizsgálatát követően a CYP3A5 6986G variáns
allél gyakorisága a magyar populációban emelkedettnek mutatkozott a roma populációval
történő összehasonlítás során, de a különbség statisztikailag nem volt szignifikáns (92,0% vs.
75,7%, p=0,091). Ezzel szemben a CYP3A5 6986GG homozigóta variáns genotípus
frekvenciája szignifikánsan magasabb volt magyarokban, szemben a roma mintákkal (84,95%
vs. 53,90%, p<0,001). A vad típusú CYP3A5 6986AA genotípus alacsony gyakorisággal volt
kimutatható mintkét vizsgált populációban. Romákban ez a genotípus több mint kétszer
gyakrabban fordult elő, mint magyarokban (2,52% vs. 0,97%) (10. táblázat).
10. táblázat CYP3A5 c.6986A>G polimorfizmus allél- és genotípus frekvenciáinak
gyakorisága a vizsgált magyar és roma populációkban
Gén Polimorfizmus Genotípus Magyar n=412 (%)
Roma n=397 (%)
CYP3A5 6986A>G
AA 4 (0,97) 10 (2,52)
AG 58 (14,08) 173 (43,58)
GG 350 (84,95) 214 (53,90)*
G allélfrekvencia 0,920 (92,0) 0,757 (75,7)
* p<0,001

42
4.2.4. CYP4F2 gén
A CYP4F2 c.1297G>A variáns genotipizálását követően eredményeink azt mutatják,
hogy a CYP4F2 rs2108622 polimorfizmushoz kapcsolódó GG, GA, AA genotípusok és az
1297A variáns allél gyakorisága roma populációban 46,5%, 42,6%, 10,9% és 32,2% volt,
magyar populációban pedig egyenként 50,1%, 42,2%, 7,7% és 22,8% (11. táblázat). A
CYP4F2 1297AA homozigóta genotípus előfordulási gyakoriságában jelentős, de
statisztikailag nem szignifikáns (p<0,08) különbséget észleltünk a két populáció között. A
roma populációs minták több mint fele CYP4F2 1297A variáns allélt hordozó (GA+AA:
53,5%), ez az érték a magyar populációban szintén magasnak bizonyult (GA+AA: 49,9%).
11. táblázat A CYP4F2 c.1297G>A polimorfizmus allél- és genotípus frekvenciáinak
gyakorisága magyar és roma populációban
Gén Polimorfizmus Genotípus Magyar n=493 (%)
Roma n=484 (%)
CYP4F2 1297G>A
GG 247 (50,1) 225 (46,5)
GA 208 (42,2) 206 (42,6)
AA 38 (7,7) 53 (10,9)
A allélfrekvencia 0,228 (22,8) 0,322 (32,2)

43
5. EREDMÉNYEK MEGBESZÉLÉSE ÉS KÖVETKEZTETÉSEK
CYP1A2
Jelen dolgozatban a CYP1A2 gén nem-kódoló - szabályozó és intronikus - régióiban
elhelyezkedő genetikai eltérések gyakoriságát vizsgáltuk, melyek hatást gyakorolhatnak a
gyógyszer metabolizmusra és különféle betegségek kialakulására.
A csökkent CYP1A2 enzimaktivitással jellemezhető CYP1A2 -3860G>A
polimorfizmus roma mintákban nem volt kimutatható. A magyar populációs mintákban mért
2%-os allélfrekvencia hasonló értéket mutat más kutatók korábbi vizsgálataiból származó
európai (svájci és francia) populációk adataival (12. táblázat). Eredményeink arra engednek
következtetni, hogy a CYP1A2 lassan metabolizáló fenotípus ritkán fordul elő az általunk
vizsgált roma és magyar populációban.
A CYP1A2 gén promoter régiójában helyet foglaló -2467delT variáns, ami a CYP1A2
gén indukciójában játszik fontos szerepet, roma populációban emelkedett gyakorisággal volt
jelen, összehasonlítva a magyar mintákkal. Ezek alapján elmondhatjuk, hogy roma
személyekben, különösen dohányzókban, a CYP1A2 enzim fokozott indukciójával és
aktivitásával kell számolnunk, valamint emelkedett rizikóval a különféle daganatos
betegségek kialakulására nézve. A magyar csoportban mért deletált timin frekvenciája a
CYP1A2 gén regulátor régiójának -2467-es pozíciójában, megközelítette más kaukázusi, úgy,
mint szerb és brit populációk frekvenciaértékeit, de jóval alacsonyabbnak bizonyult, mint
olasz és svéd populációkban (12. táblázat).
A CYP1A2*1F variáns allél frekvenciáját és a homozigóta variáns genotípus
gyakoriságát is statisztikailag szignifikánsan magasabbnak találtuk magyar populációban,
mint romában. Ez az emelkedett érték a magyar populáció tagjait fokozottabban
hajlamosíthatja daganatos megbetegedések kialakulására, mint a romákat a fokozott CYP1A2
enzim indukciója révén. A magyar minták vizsgálata során kapott *1F allélfrekvencia érték
(68,6%) összhangban van a korábban, mások által vizsgált átlag európai populációk értékeivel
(61,1-72%). A CYP1A2*1F allélfrekvenciáról roma populációban (56,9%) - hasonlóképpen a
magyarokhoz - elmondható, hogy közel azonos eredményhez jutottunk, mint amiről korábbi
kutatók számoltak be indiai népcsoportban végzett vizsgálataik alapján (58%) (12. táblázat).

44
A CYP1A2 -729T allél roma populációban nem volt kimutatható, hasonlóan, ahogy a
HapMap projekt adatai mutatják európai, japán és nigériai minták kapcsán. Ezzel szemben
magyar populációban alacsony gyakorisággal ugyan, de detektálni tudtuk (13. táblázat).
Az általunk vizsgált polimorfizmusok együttállásait vizsgálva a haplotípus analízis
során a leggyakoribb allélikus konstelláció a -3860G/-2467T/-729C/-163A (*1F) volt mind
roma, mind pedig magyar populációban, ezt követte második leggyakoribb előfordulással a
-3860G/-2467T/-729C/-163C (*1A) haplotípus. A -3860G/-2467delT/-729C/-163A kialakítva
a ht5 haplotípust, jelentősen gyakrabban fordult elő roma mintában, mint magyar mintákban.
Az allélek -3860A/-2467delT/-729C/-163A és -3860G/-2467delT/-729T/-163A együttállása
magyar populációból származó mintákban is ritkán fordult elő, roma mintákban egyáltalán
nem volt megfigyelhető.
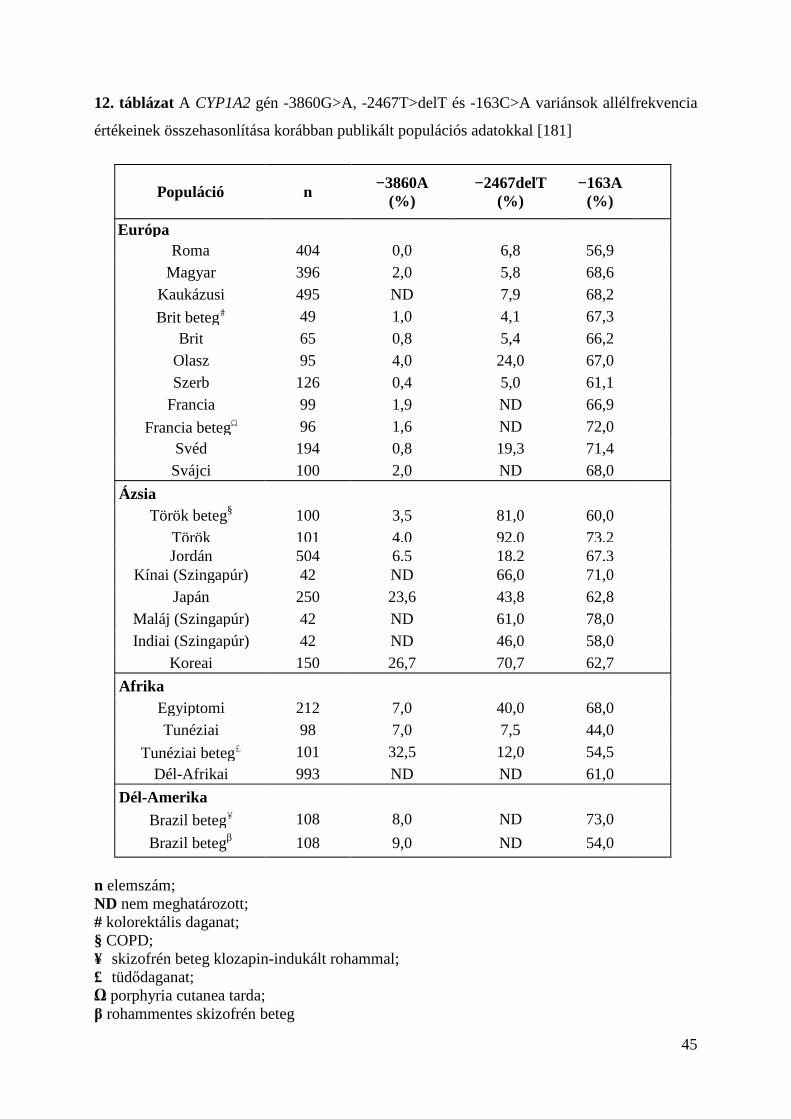
45
12. táblázat A CYP1A2 gén -3860G>A, -2467T>delT és -163C>A variánsok allélfrekvencia
értékeinek összehasonlítása korábban publikált populációs adatokkal [181]
n elemszám; ND nem meghatározott; # kolorektális daganat; § COPD; ¥ skizofrén beteg klozapin-indukált rohammal; £ tüdődaganat; Ω porphyria cutanea tarda; β rohammentes skizofrén beteg
Populáció n −3860A (%)
−2467delT (%)
−163A (%)
Európa Roma 404 0,0 6,8 56,9
Magyar 396 2,0 5,8 68,6
Kaukázusi 495 ND 7,9 68,2
Brit beteg# 49 1,0 4,1 67,3
Brit 65 0,8 5,4 66,2
Olasz 95 4,0 24,0 67,0
Szerb 126 0,4 5,0 61,1
Francia 99 1,9 ND 66,9
Francia betegΩ 96 1,6 ND 72,0
Svéd 194 0,8 19,3 71,4
Svájci 100 2,0 ND 68,0
Ázsia Török beteg§ 100 3,5 81,0 60,0
Török 101 4,0 92,0 73,2 Jordán 504 6,5 18,2 67,3
Kínai (Szingapúr) 42 ND 66,0 71,0 Japán 250 23,6 43,8 62,8
Maláj (Szingapúr) 42 ND 61,0 78,0
Indiai (Szingapúr) 42 ND 46,0 58,0
Koreai 150 26,7 70,7 62,7
Afrika
Egyiptomi 212 7,0 40,0 68,0
Tunéziai 98 7,0 7,5 44,0
Tunéziai beteg£ 101 32,5 12,0 54,5
Dél-Afri kai 993 ND ND 61,0
Dél-Amerika Brazil beteg¥ 108 8,0 ND 73,0
Brazil betegβ 108 9,0 ND 54,0

46
13. táblázat A CYP1A2 -729C>T polimorfizmus gyakorisága különböző populációkban a
HapMap Projekt adatai alapján [181]
Populáció n -729T (%)
Roma 404 0,00
Magyar 396 0,25
Európai 130 0,00
Olasz 204 1,50
Indiai (Gujarati) 202 0,50
Japán 90 0,00
Kínai 88 2,30
Afrikai (Nigéria) 124 0,00
Afrikai (Kenya) 312 0,30
A taxán vegyületek metabolizmusát és hatását befolyásoló citokróm P450 gének
polimorfizmusainak vizsgálata során roma és magyar populációban lényeges különbségeket
észleltünk, melyek befolyásolhatják a taxán terápia kimenetelét.
CYP1B1
A CYP1B1*3 variáns allél vizsgálatával kapott eredményeink alapján elmondható,
hogy a CYP1B1 4326G (*3 allél) enyhén, a GG (*3/*3 ) homozigóta variáns genotípus
szignifikánsan emelkedettnek számít roma populációban összevetve a magyarokkal. Magyar
populációban az általunk kapott CYP1B1*3 allél frekvenciája 1,5-szerese a korábban, más
kutatók kaukázusi populációkban mért adatainak. A *3 variáns gyakorisága roma
populációban is magasabbnak bizonyult, mint más észak-indiai populációs eredmények
(14. táblázat). E polimorfizmus tekintetében összességében elmondhatjuk, hogy roma
populációban gyakrabban kell számolnunk fokozott CYP1B1 mRNS expresszióval, ezáltal
erősebb enzimaktivitással, továbbá, hogy a roma népesség tagjai fokozott hajlamot

47
mutathatnak idiopátiás férfi infertilitásra, glaukoma, valamint tüdő- és endometrium daganat
kialakulására a magyarokhoz képest. Taxán terápia kapcsán a roma betegek körében a teljes
túlélési ráta alacsonyabb lehet, mint magyarok esetén, illetve paclitaxel és docetaxel terápiát
követően a romák nagyobb eséllyel mutathatnak elégtelen gyógyszerválaszt a CYP1B1*3
variáns hordozása kapcsán.
14. táblázat A CYP1B1*3 allél frekvenciája különböző populációkban
Populáció n 4326G
allélfrekvencia (%)
Referencia
Roma 397 69,4 [182]
Magyar 412 64,2 [182]
Kaukázusi 95 43,0 [65]
Német 1262 42,5 [183]
USA 406 42,2 [184]
USA 99 42,4 [185]
Brit 2695 54,7 [186]
Európai 262 41,0 [187]
Cseh 495 42,2 [188]
Indiai 200 21,3 [189]
Indiai 150 21,7 [62]
Észak-Indiai 168 48,8 [54]
Kína 227 15,9 [190]
Japán 324 15,4 [191]
Afroamerikai 126 71,8 [192]
Afroamerikai 30 81,7 [58]
Inuit (Grönland) 249 12,9 [187]

48
CYP2C8
A CYP2C8*4 variáns esetében szintén jelentős különbségeket észleltünk a két
populáció között. A vizsgált SNP-nek, mely csökkent enzimaktivitással jellemezhető, a
magyar populácóban való gyakoribb előfordulása azt sejteti, hogy a magyarok gyakrabban
tekinthetők lassú metabolizálónak a CYP2C8 szubsztrátjait illetően, a több, mint kétszer
magasabb *4 allél frekvenciájának köszönhetően, szemben a romákkal. Mivel a paclitaxel
metabolizmusért is felelős enzimről van szó, a paclitaxel-indukálta toxicitás esélye is
magasabb lehet a magyar népcsoportban, mint romák körében.
Korábbi populációs adatokkal összevetve (15. táblázat) a magyar csoportban kapott
értékeink közel azonosak voltak más kutatók kaukázusi populációban mért allélfrekvencia
értékeivel, ezzel szemben a roma CYP2C8*4 allélfrekvencia eredmények fele akkorának
mutatkoztak, mint más indiai eredetű népcsoportban mért értékek.
15. táblázat CYP2C8*4 allél frekvenciája különböző populációkban
Populáció n 792G
allélfrekvencia (%)
Referencia
Roma 397 2,14 [182]
Magyar 412 5,83 [182]
Kaukázusi 95 6,00 [65]
Brit 107 7,50 [84]
Cseh 161 5,90 [193]
Német 122 7,40 [194]
Japán 360 0,00 [195]
Kínai 288 0,00 [196]
Afrikai (Zanzibár) 165 0,60 [197]
Észak-Indiai 251 4,00 [198]
Indiai 123 0,00 [196]
Maláj 137 0,00 [196]

49
CYP3A5
Szemben a romákkal, a magyar populációban észlelt szignifikánsan magasabb
előfordulási gyakorisága a CYP3A5*3/*3 nem-expresszáló genotípusnak magyarázhatja a
CYP3A5 szubsztrát gyógyszerekkel szembeni gyakoribb, lassabb metabolizmust magyar
populációban. A csökkent metabolizmus megnövekedett docetaxel toxicitáshoz vezethet a
gyógyszer normál dózisban való alkalmazása során. Mivel az expresszáló CYP3A5
genotípusok (*1/*3 és *1/*1 ) megnövekedett kockázatot jelenthetnek akut limfoblasztos
leukémia és emlődaganat kialakulására, a magyar populációban ezen genotípusok ritkább
előfordulása a magyarokat kevésbé hajlamosítja az említett betegségek kialakulására, mint a
roma populáció tagjait.
Különböző populációkkal történő összehasonlítás után összefoglalva elmondható,
hogy a roma populációs minták vizsgálata során kapott 75,7%-os variáns allélfrekvencia
magasabb volt, mint a korábbi vizsgálatok indiai és észak-indiai populációkban mért értékei.
Ezzel szemben magyar mintáink CYP3A5 6986G alléljának 92%-os értéke jól közelít más
kaukázusi populációk (spanyol, finn és norvég) értékeihez (16. táblázat).
16. táblázat A roma és magyar populációban mért CYP3A5*3 allél frekvenciájának
összehasonítása különböző populációs adatokkal
Populáció n 6986G allélfrekvencia (%)
Referencia
Roma 397 75,7 [182]
Magyar 412 92,0 [182]
Spanyol 163 90,8 [199]
Finn 726 93,0 [200]
Norvég 199 91,1 [201]
Bolgár 160 93,1 [99]
Észak-Afrikai (Algéria) 159 81,0 [202]
Nyugat-Afrikai (Ghána) 90 19,0 [202]
Indiai 208 67,1 [203]
Indiai 241 39,0 [204]
Észak-Indiai 183 67,5 [203]

50
CYP4F2
A CYP4F2 gén rs2108622 variáns genotipizálását követően arra a következtetésre
jutottunk, hogy a genotípusok frekvenciájának eloszlását nagymértékben befolyásolja az
etnikum. A minor allél frekvenciája magyarokban (22,8%) alacsonyabbnak bizonyult, mint
roma populációban (32,2%). A kapott két érték jól tükrözi más kutatók kaukázusi
populációkban mért adatainak szélső értékeit (23,3-38,0%). Roma populációs mintákban a
tanulmányozott SNP variáns allélfrekvenciájának értékét összevetve más indiai populációs
adatokkal, sokkal alacsonyabb értéket kaptunk, mely inkább a kaukázusi értékekhez közelít
[206-208]. Hasonló a helyzet a magyar minták esetében is, ugyanis a CYP4F2 1297A variáns
allél értékei közelebb állnak korábbi vizsgálatokból származó ázsiai értékekhez, mint a
kaukázusiakhoz [166].
A CYP4F2 c.1297G>A variánshoz tartozó genotípusok gyakoriságát különböző
populációkban a 17. táblázat összegzi. Populációs összehasonlítást követően összefoglalva
elmondható, hogy a CYP4F2 rs2108622 variáns legritkábban afroamerikai népcsoportban
fordul elő (8%), ennél gyakoribb maláj (14%) és brazil populációban (15%) [166, 209]. A
vizsgált SNP legmagasabb frekvencia értéket szingapúri indiai populációban mutat (44%),
míg relatíve gyakori egyiptomi (42%) és spanyol (38%) populációkban [166, 208, 210].

51
17. táblázat CYP4F2 c.1297G>A SNP genotípus és allélfrekvencia értékeinek eloszlása roma
és magyar mintákban, összehasonlítva különböző eredetű populációkkal [205]
Populáció n c.1297G>A (%)
GG GA AA A allél
Roma 484 46,5 42,6 10,9 32,2
Magyar 493 50,1 42,2 7,70 22,8
Kaukázusi 300 42,7 46,3 11,0 34,2
Kaukázusi beteg 141 46,0 42,0 12,0 32,0
Francia 283 48,8 43,6 7,60 29,4
Hispán 300 58,7 36,0 5,30 23,3
Spanyol 100 41,0 42,0 17,0 38,0
Ázsiai 300 48,0 43,0 9,00 30,5
Japán 440 53,0 39,8 7,20 27,2
Han Kínai 115 41,7 47,8 10,4 34,4
Han Kínai 222 52,0 41,0 7,00 27,0
Kínai 88 67,0 28,4 4,50 19,0
Maláj 88 73,9 23,8 2,20 14,0
Indiai 88 32,9 45,4 21,5 44,0
Egyiptomi 195 35,4 45,3 19,3 42,0
Askenázi zsidó 500 43,4 47,6 9,00 32,8
Afroamerikai 50 84,0 16,0 0,00 8,00
Afroamerikai 300 78,0 20,7 1,30 11,7
Brazil (fehér) 184 50,0 39,0 11,0 30,0
Brazil (fekete) 74 72,0 27,0 1,00 15,0

52
6. EREDMÉNYEK ÖSSZEFOGLALÁSA
1. Szignifikánsan emelkedett variáns allélfrekvencia értéket észleltünk a vizsgált magyar
populációban a CYP1A2*1F és CYP2C8*4 allélokra nézve a roma populációs mintákhoz
viszonyítva.
2. A homozigóta variáns genotípus gyakorisága magyarokban a CYP3A5*3 és a CYP1A2*1F,
romákban pedig a CYP1B1*3 SNP-k esetén volt szignifikánsan magasabb.
3. Mindkét populációban a CYP1A2*1C variáns alacsony frekvenciájának köszönhetően a
lassan metabolizáló fenotípus, így a CYP1A2 szubsztrát-gyógyszer toxicitás esélye is
valószínűsíthetően ritka.
4. A CYP1A2 gén genetikai profiljának vizsgálatát követően megállapíthatjuk, hogy roma
populációban a CYP1A2*1D allél gyakori előfordulása növelheti a daganatos megbetegedések
kialakulásának rizikóját.
5. A magyar populációban a CYP1A2*1F funkcionális allél szignifikánsan emelkedett
gyakorisága fokozott kockázatot jelenthet emlő-, tüdő-, és petefészek daganat kialakulására,
különösen hajlamosító környezeti tényezőknek való kitettség esetén.
6. A CYP1A2 gén -3860G>A, -2467T>delT, -729C>T és -163C>A nem-kódoló
polimorfizmusai 5 fő haplotípust határoztak meg magyar populációban, míg roma mintákban
a ht2 és ht4 együttállás nem volt detektálható.
7. Roma populációs mintákban a CYP1B1*3 homozigóta genotípus gyakori előfordulása
állhat a romák taxán kezelésre adott esetleges csökkent gyógyszerválasza és a daganatos
megbetegedésekben szenvedők alacsonyabb teljes túlélési rátája hátterében.
8. A CYP2C8 és CYP3A5 gének tekintetében a CYP2C8*4 és CYP3A5*3 csökkent aktivitású
variánsok gyakorisága magyarokban nagyobb volt, mint romákban. Így mind docetaxel, mind
paclitaxel alkalmazásakor a magyarok nagyobb valószínűséggel tekinthetők taxán
gyógyszerek lassú metabolizálóinak, ezáltal feltételezhetően nagyobb az esélyük taxán-
indukált toxicitás kialakulására.
9. Roma populációban a CYP4F2*3 allél gyakoribb hordozása nagyobb dózisú kumarin
származékokkal történő kezelést tehet indokolttá a megfelelő antikoaguláns válasz elérése
érdekében.

53
7. KÖZLEMÉNYEK JEGYZÉKE
ÉRTEKEZÉS ALAPJÁUL SZOLGÁLÓ KÖZLEMÉNYEK:
1. Szalai R, Magyari L, Matyas P, Duga B, Banfai Z, Szabo A, Kovesdi E, Melegh B.
Genetic polymorphisms in promoter and intronic regions of CYP1A2 gene in Roma and
Hungarian population samples. Environ Toxicol Pharmacol. 2014;38(3):814-20.
IF: 2,084
2. Szalai R, Ganczer A, Magyari L, Matyas P, Bene J, Melegh B.
Interethnic differences of cytochrome P450 gene polymorphisms may influence outcome
of taxane therapy in Roma and Hungarian populations. Drug Metab Pharmacokinet. 2015.
IF: 2,568 (2014)
3. Sipeky C, Weber A, Melegh BI, Matyas P, Janicsek I, Szalai R, Szabo I, Varnai R, Tarlos
G, Ganczer A, Melegh B.
Interethnic variability of CYP4F2 (V433M) in admixed population of Roma and
Hungarians. Environ Toxicol Pharmacol. 2015;40(1):280-3.
IF: 2,084 (2014)

54
EGYÉB KÖZLEMÉNYEK:
Szalai R, Matyas P, Varszegi D, Melegh M, Magyari L, Jaromi L, Sumegi K, Duga B,
Kovesdi E, Hadzsiev K, Melegh B.
Admixture of beneficial and unfavourable variants of GLCCI1 and FCER2 in Roma
samples can implicate different clinical response to corticosteroids. Mol Biol Rep. 2014
Nov;41(11):7665-9.
IF: 2,024
Weber A, Szalai R, Sipeky C, Magyari L, Melegh M, Jaromi L, Matyas P, Duga B, Kovesdi
E, Hadzsiev K, Melegh B.
Increased prevalence of functional minor allele variants of drug metabolizing CYP2B6
and CYP2D6 genes in Roma population samples. Pharmacol Rep. 2015 Jun;67(3):460-4.
IF: 1,928
Sipeky C, Matyas P, Melegh M, Janicsek I, Szalai R, Szabo I, Varnai R, Tarlos G,
Ganczer A, Melegh B.
Lower carrier rate of GJB2 W24X ancestral Indian mutation in Roma samples from
Hungary: implication for public health intervention . Mol Biol Rep. 2014 Sep;41(9):6105-
10.
IF: 2,024
Magyari L, Varszegi D, Sarlos P, Jaromi L, Melegh BI, Duga B, Kisfali P, Kovesdi E, Matyas
P, Szabo A, Szalai R, Melegh B.
Marked differences of haplotype tagging SNP distribution, linkage, and haplotype
profile of IL23 receptor gene in Roma and Hungarian population samples. Cytokine.
2014 Feb;65(2):148-52.
IF: 2,664

55
Nagy A, Szalai R, Magyari L, Bene J, Toth K, Melegh B.
Extreme differences in SLCO1B3 functional polymorphisms in Roma and Hungarian
populations. Environ Toxicol Pharmacol. 2015 May;39(3):1246-51.
IF: 2,084
Sumegi K, Jaromi L, Magyari L, Kovesdi E, Duga B, Szalai R, Maasz A, Matyas P, Janicsek
I, Melegh B.
Functional Variants of Lipid Level Modifier MLXIPL, GCKR, GALNT2, CILP2,
ANGPTL3 and TRIB1 Genes in Healthy Roma and Hungarian Populations. Pathol
Oncol Res. 2015 Jan 9.
IF: 1,855
Nagy A, Sipeky C, Szalai R, Melegh B I, Matyas P, Ganczer A, Toth K, Melegh B
Marked differences in frequencies of statin therapy relevant SLCO1B1 variants and
haplotypes between Roma and Hungarian populations. BMC Genetics.
IF: 2,40
Az értekezés alapjául szolgáló közlemények összesített impakt faktora: 6,736
Egyéb közlemények összesített impakt faktora: 14,979
Összesített impakt faktor: 21,715

56
IDÉZHETŐ ABSZTRAKTOK:
R. Szalai, C. Sipeky, L. Jaromi, L. Magyari, P. Matyas, J. Bene, B. Duga, E. Kovesdi, A.
Szabo, B. Melegh.
Polymorphisms of CYP2B6 and CYP2D6 genes associated with drug metabolism in
Roma population samples
Eur J Hum Genet. 2013;21 Suppl. 2, 383.
J. Bene, K. Sumegi, L. Jaromi, L. Magyari, E. Kovesdi, B. Duga, R. Szalai, P. Matyas, A.
Szabo, Z. Banfai, B. Melegh.
Distribution of eight SNPs of lipid level modifier genes in healthy Roma and Hungarian
population samples
Eur J Hum Genet. 2013;21 Suppl. 2, 404.
B. Duga, B. I. Melegh, L. Jaromi, L. Magyari, K. Sumegi, Z. Banfai, K. Komlosi, K.
Hadzsiev, A. Szabo, R. Szalai, E. Kovesdi, J. Bene, P. Kisfali, B. Melegh
Marked differences of haplotype tagging SNP distribution, linkage and haplotype
profile of APOA5 gene in Roma population samples
Eur J Hum Genet. 2013;21 Suppl. 2, 556.
L. Magyari, D. Varszegi, P. Sarlos, L. Jaromi, J. Bene, B. Duga, K. Hadzsiev, P. Kisfali, K.
Komlosi, E. Kovesdi, P. Matyas, A. Szabo, R. Szalai, B. Melegh
Marked differences of haplotype tagging SNP distribution, linkage and haplotype
profile of IL23 receptor gene in Roma population samples
Eur J Hum Genet. 2013;21 Suppl. 2, 565.
R. Szalai, P. Matyas, L. Magyari, J. Bene, B. Duga, Z. Banfai, A. Szabo, B. Melegh
CYP1A2 gene non-coding region polymorphisms in Roma and Hungarian population
samples
Eur J Hum Genet. 2014;22 Suppl. 1, 298.

57
A. Szabó, Z. Bánfai, B. Duga, R. Szalai, L. Magyari, P. Mátyás, E. Kövesdi, K. Sümegi,
B. Melegh
Genetic relationship of European Roma people and eight ethnic groups from the
Caucasus area which suggest Romas probably admixed with during their migration
Eur J Hum Genet. 2014;22 Suppl. 1, 320.
Z. Bánfai, A. Szabó, K. Sümegi, B. Duga, L. Magyari, E. Kövesdi, P. Mátyás, R. Szalai,
B. Melegh
Admixture of Turks, Hungarians and Romas in the Carpathian Basin
Eur J Hum Genet. 2014;22 Suppl. 1, 509.
L. Magyari, R. Szalai, E. Kovesdi, Z. Banfai, B. Duga, P. Matyas, A. Szabo, B. Melegh
CYP1A1 variant in Roma population samples
Eur J Hum Genet. 2014;22 Suppl. 1, 303.
P. Matyas, R. Szalai, L. Magyari, B. Duga, E. Kovesdi, Z. Banfai, A. Szabo, B. Melegh
Asthma related FCER2 variant in Roma and Hungarian populations
Eur J Hum Genet. 2014;22 Suppl. 1, 501.

58
8. IRODALOMJEGYZÉK
1. Bertz RJ, Granneman GR. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin Pharmacokinet. 1997; 32(3):210-258.
2. Weinshilboum R. Inheritance and drug response. N Engl J Med. 2003; 348(6):529-537.
3. Preissner SC, Hoffmann MF, Preissner R, Dunkel M, Gewiess A, Preissner S. Polymorphic cytochrome P450 enzymes (CYPs) and their role in personalized therapy. PLoS One. 2013; 8(12):e82562.
4. Zanger UM, Turpeinen M, Klein K, Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 2008; 392(6):1093-1108.
5. van der Weide J, Hinrichs JW. The influence of cytochrome P450 pharmacogenetics on disposition of common antidepressant and antipsychotic medications. Clin Biochem Rev. 2006; 27(1):17-25.
6. Eichelbaum M, Ingelman-Sundberg M, Evans WE. Pharmacogenomics and individualized drug therapy. Annu Rev Med. 2006; 57:119-137.
7. Bozina N, Bradamante V, Lovric M. Genetic polymorphism of metabolic enzymes P450 (CYP) as a susceptibility factor for drug response, toxicity, and cancer risk. Arh Hig Rada Toksikol. 2009; 60(2):217-242.
8. Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst. 2000; 92(16):1295-1302.
9. Keppler D. The roles of MRP2, MRP3, OATP1B1, and OATP1B3 in conjugated hyperbilirubinemia. Drug Metab Dispos. 2014; 42(4):561-565.
10. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013; 138(1):103-141.
11. Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 2007; 116(3):496-526.
12. Zhang Y, Klein K, Sugathan A, Nassery N, Dombkowski A, Zanger UM, Waxman DJ. Transcriptional profiling of human liver identifies sex-biased genes associated with polygenic dyslipidemia and coronary artery disease. PLoS One. 2011; 6(8):e23506.
13. Kinirons MT, O'Mahony MS. Drug metabolism and ageing. Br J Clin Pharmacol. 2004; 57(5):540-544.
14. Cotreau MM, von Moltke LL, Greenblatt DJ. The influence of age and sex on the clearance of cytochrome P450 3A substrates. Clin Pharmacokinet. 2005; 44(1):33-60.
15. Slaviero KA, Clarke SJ, Rivory LP. Inflammatory response: an unrecognised source of variability in the pharmacokinetics and pharmacodynamics of cancer chemotherapy. Lancet Oncol. 2003; 4(4):224-232.
16. Aitken AE, Richardson TA, Morgan ET. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol. 2006; 46:123-149.
17. Scott SA. Personalizing medicine with clinical pharmacogenetics. Genet Med. 2011; 13(12):987-995.
18. Brockmoller J, Tzvetkov MV. Pharmacogenetics: data, concepts and tools to improve drug discovery and drug treatment. Eur J Clin Pharmacol. 2008; 64(2):133-157.

59
19. Lim JS, Singh O, Ramasamy RD, Ramasamy S, Subramanian K, Lee EJ, Chowbay B. Pharmacogenetics of CYP1A2, novel polymorphisms and haplotypes in three distinct Asian populations. Drug Metab Pharmacokinet. 2010; 25(6):616-623.
20. Ghotbi R, Christensen M, Roh HK, Ingelman-Sundberg M, Aklillu E, Bertilsson L. Comparisons of CYP1A2 genetic polymorphisms, enzyme activity and the genotype-phenotype relationship in Swedes and Koreans. Eur J Clin Pharmacol. 2007; 63(6):537-546.
21. Sipeky C, Weber A, Szabo M, Melegh BI, Janicsek I, Tarlos G, Szabo I, Sumegi K, Melegh B. High prevalence of CYP2C19*2 allele in Roma samples: study on Roma and Hungarian population samples with review of the literature. Mol Biol Rep. 2013; 40(8):4727-4735.
22. Kim KA, Song WG, Lee HM, Joo HJ, Park JY. Multiplex pyrosequencing method to determine CYP2C9*3, VKORC1*2, and CYP4F2*3 polymorphisms simultaneously: its application to a Korean population and comparisons with other ethnic groups. Mol Biol Rep. 2014; 41(11):7305-7312.
23. Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002; 360(9340):1155-1162.
24. Rogers JF, Nafziger AN, Bertino JS, Jr. Pharmacogenetics affects dosing, efficacy, and toxicity of cytochrome P450-metabolized drugs. Am J Med. 2002; 113(9):746-750.
25. Ingelman-Sundberg M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol Sci. 2004; 25(4):193-200.
26. Nelson DR. Cytochrome P450 nomenclature, 2004. Methods Mol Biol. 2006; 320:1-10.
27. Zhou SF, Yang LP, Zhou ZW, Liu YH, Chan E. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009; 11(3):481-494.
28. Jorge-Nebert LF, Jiang Z, Chakraborty R, Watson J, Jin L, McGarvey ST, Deka R, Nebert DW. Analysis of human CYP1A1 and CYP1A2 genes and their shared bidirectional promoter in eight world populations. Hum Mutat. 2010; 31(1):27-40.
29. Aklillu E, Carrillo JA, Makonnen E, Hellman K, Pitarque M, Bertilsson L, Ingelman-Sundberg M. Genetic polymorphism of CYP1A2 in Ethiopians affecting induction and expression: characterization of novel haplotypes with single-nucleotide polymorphisms in intron 1. Mol Pharmacol. 2003; 64(3):659-669.
30. Pavanello S, Pulliero A, Lupi S, Gregorio P, Clonfero E. Influence of the genetic polymorphism in the 5'-noncoding region of the CYP1A2 gene on CYP1A2 phenotype and urinary mutagenicity in smokers. Mutat Res. 2005; 587(1-2):59-66.
31. Han XM, Ouyang DS, Chen XP, Shu Y, Jiang CH, Tan ZR, Zhou HH. Inducibility of CYP1A2 by omeprazole in vivo related to the genetic polymorphism of CYP1A2. Br J Clin Pharmacol. 2002; 54(5):540-543.
32. Kobayashi M, Otani T, Iwasaki M, Natsukawa S, Shaura K, Koizumi Y, Kasuga Y, Sakamoto H, Yoshida T, Tsugane S. Association between dietary heterocyclic amine levels, genetic polymorphisms of NAT2, CYP1A1, and CYP1A2 and risk of stomach cancer: a hospital-based case-control study in Japan. Gastric Cancer. 2009; 12(4):198-205.
33. Djordjevic N, Ghotbi R, Jankovic S, Aklillu E. Induction of CYP1A2 by heavy coffee consumption is associated with the CYP1A2 -163C>A polymorphism. Eur J Clin Pharmacol. 2010; 66(7):697-703.

60
34. El-Sohemy A, Cornelis MC, Kabagambe EK, Campos H. Coffee, CYP1A2 genotype and risk of myocardial infarction. Genes Nutr. 2007; 2(1):155-156.
35. Uslu A, Ogus C, Ozdemir T, Bilgen T, Tosun O, Keser I. The effect of CYP1A2 gene polymorphisms on Theophylline metabolism and chronic obstructive pulmonary disease in Turkish patients. BMB Rep. 2010; 43(8):530-534.
36. Tian Z, Li YL, Zhao L, Zhang CL. Role of CYP1A2 1F polymorphism in cancer risk: evidence from a meta-analysis of 46 case-control studies. Gene. 2013; 524(2):168-174.
37. Park KW, Park JJ, Jeon KH, Kang SH, Oh IY, Yang HM, Cho HJ, Lee HY, Kang HJ, Koo BK et al. Enhanced clopidogrel responsiveness in smokers: smokers' paradox is dependent on cytochrome P450 CYP1A2 status. Arterioscler Thromb Vasc Biol. 2011; 31(3):665-671.
38. Aitchison KJ, Gonzalez FJ, Quattrochi LC, Sapone A, Zhao JH, Zaher H, Elizondo G, Bryant C, Munro J, Collier DA et al. Identification of novel polymorphisms in the 5' flanking region of CYP1A2, characterization of interethnic variability, and investigation of their functional significance. Pharmacogenetics. 2000; 10(8):695-704.
39. Nakajima M, Yokoi T, Mizutani M, Kinoshita M, Funayama M, Kamataki T. Genetic polymorphism in the 5'-flanking region of human CYP1A2 gene: effect on the CYP1A2 inducibility in humans. J Biochem. 1999; 125(4):803-808.
40. Gunes A, Dahl ML. Variation in CYP1A2 activity and its clinical implications: influence of environmental factors and genetic polymorphisms. Pharmacogenomics. 2008; 9(5):625-637.
41. Thorn CF, Aklillu E, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for CYP1A2. Pharmacogenet Genomics. 2012; 22(1):73-77.
42. Obase Y, Shimoda T, Kawano T, Saeki S, Tomari SY, Mitsuta-Izaki K, Matsuse H, Kinoshita M, Kohno S. Polymorphisms in the CYP1A2 gene and theophylline metabolism in patients with asthma. Clin Pharmacol Ther. 2003; 73(5):468-474.
43. Tiwari AK, Deshpande SN, Rao AR, Bhatia T, Mukit SR, Shriharsh V, Lerer B, Nimagaonkar VL, Thelma BK. Genetic susceptibility to tardive dyskinesia in chronic schizophrenia subjects: I. Association of CYP1A2 gene polymorphism. Pharmacogenomics J. 2005; 5(1):60-69.
44. Pavanello S, Fedeli U, Mastrangelo G, Rota F, Overvad K, Raaschou-Nielsen O, Tjonneland A, Vogel U. Role of CYP1A2 polymorphisms on lung cancer risk in a prospective study. Cancer Genet. 2012; 205(6):278-284.
45. Korytina GF, Akhmadishina LZ, Kochetova OV, Zagidullin Sh Z, Viktorova TV. [Association of cytochrome P450 genes polymorphisms (CYP1A1 and CYP1A2) with the development of chronic obstructive pulmonary disease in Bashkortostan]. Mol Biol (Mosk). 2008; 42(1):32-41.
46. Sachse C, Brockmoller J, Bauer S, Roots I. Functional significance of a C-->A polymorphism in intron 1 of the cytochrome P450 CYP1A2 gene tested with caffeine. Br J Clin Pharmacol. 1999; 47(4):445-449.
47. Ozdemir V, Kalow W, Okey AB, Lam MS, Albers LJ, Reist C, Fourie J, Posner P, Collins EJ, Roy R. Treatment-resistance to clozapine in association with ultrarapid CYP1A2 activity and the C-->A polymorphism in intron 1 of the CYP1A2 gene: effect of grapefruit juice and low-dose fluvoxamine. J Clin Psychopharmacol. 2001; 21(6):603-607.
48. Sutter TR, Tang YM, Hayes CL, Wo YY, Jabs EW, Li X, Yin H, Cody CW, Greenlee WF. Complete cDNA sequence of a human dioxin-inducible mRNA identifies a new

61
gene subfamily of cytochrome P450 that maps to chromosome 2. J Biol Chem. 1994; 269(18):13092-13099.
49. Tang YM, Wo YY, Stewart J, Hawkins AL, Griffin CA, Sutter TR, Greenlee WF. Isolation and characterization of the human cytochrome P450 CYP1B1 gene. J Biol Chem. 1996; 271(45):28324-28330.
50. Long JR, Shu XO, Cai Q, Cai H, Gao YT, Jin F, Zheng W. Polymorphisms of the CYP1B1 gene may be associated with the onset of natural menopause in Chinese women. Maturitas. 2006; 55(3):238-246.
51. Sowers MR, Wilson AL, Kardia SR, Chu J, McConnell DS. CYP1A1 and CYP1B1 polymorphisms and their association with estradiol and estrogen metabolites in women who are premenopausal and perimenopausal. Am J Med. 2006; 119(9 Suppl 1):S44-51.
52. Sharma M, Shubert DE, Lewis J, McGarrigle BP, Bofinger DP, Olson JR. Biotransformation of tamoxifen in a human endometrial explant culture model. Chem Biol Interact. 2003; 146(3):237-249.
53. Rochat B, Morsman JM, Murray GI, Figg WD, McLeod HL. Human CYP1B1 and anticancer agent metabolism: mechanism for tumor-specific drug inactivation? J Pharmacol Exp Ther. 2001; 296(2):537-541.
54. Kumar V, Singh S, Ahmed RS, Banerjee BD, Ahmed T, Pasha ST. Frequency of common CYP1B1 polymorphic variations in Delhi population of Northern India. Environ Toxicol Pharmacol. 2009; 28(3):392-396.
55. Li N, Zhou Y, Du L, Wei M, Chen X. Overview of Cytochrome P450 1B1 gene mutations in patients with primary congenital glaucoma. Exp Eye Res. 2011; 93(5):572-579.
56. Hayes CL, Spink DC, Spink BC, Cao JQ, Walker NJ, Sutter TR. 17 beta-estradiol hydroxylation catalyzed by human cytochrome P450 1B1. Proc Natl Acad Sci U S A. 1996; 93(18):9776-9781.
57. Roos PH, Bolt HM. Cytochrome P450 interactions in human cancers: new aspects considering CYP1B1. Expert Opin Drug Metab Toxicol. 2005; 1(2):187-202.
58. Wenzlaff AS, Cote ML, Bock CH, Land SJ, Santer SK, Schwartz DR, Schwartz AG. CYP1A1 and CYP1B1 polymorphisms and risk of lung cancer among never smokers: a population-based study. Carcinogenesis. 2005; 26(12):2207-2212.
59. Aklillu E, Oscarson M, Hidestrand M, Leidvik B, Otter C, Ingelman-Sundberg M. Functional analysis of six different polymorphic CYP1B1 enzyme variants found in an Ethiopian population. Mol Pharmacol. 2002; 61(3):586-594.
60. Sasaki M, Tanaka Y, Kaneuchi M, Sakuragi N, Dahiya R. Alleles of polymorphic sites that correspond to hyperactive variants of CYP1B1 protein are significantly less frequent in Japanese as compared to American and German populations. Hum Mutat. 2003; 21(6):652.
61. Mustafa M, Sharma T, Banerjee BD, Phil M, Ahmed RS, Tripathi AK, Guleria K. Genetic polymorphisms in Cytochrome P 4501B1 and susceptibility to idiopathic preterm labor in North Indian population. Clin Biochem. 2013; 46(18):1812-1815.
62. Singh AP, Shah PP, Mathur N, Buters JT, Pant MC, Parmar D. Genetic polymorphisms in cytochrome P4501B1 and susceptibility to head and neck cancer. Mutat Res. 2008; 639(1-2):11-19.
63. Doherty JA, Weiss NS, Freeman RJ, Dightman DA, Thornton PJ, Houck JR, Voigt LF, Rossing MA, Schwartz SM, Chen C. Genetic factors in catechol estrogen metabolism in relation to the risk of endometrial cancer. Cancer Epidemiol Biomarkers Prev. 2005; 14(2):357-366.

62
64. Sissung TM, Danesi R, Price DK, Steinberg SM, de Wit R, Zahid M, Gaikwad N, Cavalieri E, Dahut WL, Sackett DL et al. Association of the CYP1B1*3 allele with survival in patients with prostate cancer receiving docetaxel. Mol Cancer Ther. 2008; 7(1):19-26.
65. Rizzo R, Spaggiari F, Indelli M, Lelli G, Baricordi OR, Rimessi P, Ferlini A. Association of CYP1B1 with hypersensitivity induced by taxane therapy in breast cancer patients. Breast Cancer Res Treat. 2010; 124(2):593-598.
66. Achary MS, Reddy AB, Chakrabarti S, Panicker SG, Mandal AK, Ahmed N, Balasubramanian D, Hasnain SE, Nagarajaram HA. Disease-causing mutations in proteins: structural analysis of the CYP1B1 mutations causing primary congenital glaucoma in humans. Biophys J. 2006; 91(12):4329-4339.
67. Hollander DA, Sarfarazi M, Stoilov I, Wood IS, Fredrick DR, Alvarado JA. Genotype and phenotype correlations in congenital glaucoma: CYP1B1 mutations, goniodysgenesis, and clinical characteristics. Am J Ophthalmol. 2006; 142(6):993-1004.
68. Vincent AL, Billingsley G, Buys Y, Levin AV, Priston M, Trope G, Williams-Lyn D, Heon E. Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am J Hum Genet. 2002; 70(2):448-460.
69. Aquilante CL, Niemi M, Gong L, Altman RB, Klein TE. PharmGKB summary: very important pharmacogene information for cytochrome P450, family 2, subfamily C, polypeptide 8. Pharmacogenet Genomics. 2013; 23(12):721-728.
70. Totah RA, Rettie AE. Cytochrome P450 2C8: substrates, inhibitors, pharmacogenetics, and clinical relevance. Clin Pharmacol Ther. 2005; 77(5):341-352.
71. Klose TS, Blaisdell JA, Goldstein JA. Gene structure of CYP2C8 and extrahepatic distribution of the human CYP2Cs. J Biochem Mol Toxicol. 1999; 13(6):289-295.
72. Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. Structure of human microsomal cytochrome P450 2C8. Evidence for a peripheral fatty acid binding site. J Biol Chem. 2004; 279(10):9497-9503.
73. Johnson EF, Stout CD. Structural diversity of human xenobiotic-metabolizing cytochrome P450 monooxygenases. Biochem Biophys Res Commun. 2005; 338(1):331-336.
74. Hamman MA, Thompson GA, Hall SD. Regioselective and stereoselective metabolism of ibuprofen by human cytochrome P450 2C. Biochem Pharmacol. 1997; 54(1):33-41.
75. Fischer V, Johanson L, Heitz F, Tullman R, Graham E, Baldeck JP, Robinson WT. The 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor fluvastatin: effect on human cytochrome P-450 and implications for metabolic drug interactions. Drug Metab Dispos. 1999; 27(3):410-416.
76. Projean D, Morin PE, Tu TM, Ducharme J. Identification of CYP3A4 and CYP2C8 as the major cytochrome P450 s responsible for morphine N-demethylation in human liver microsomes. Xenobiotica. 2003; 33(8):841-854.
77. Wang JS, Neuvonen M, Wen X, Backman JT, Neuvonen PJ. Gemfibrozil inhibits CYP2C8-mediated cerivastatin metabolism in human liver microsomes. Drug Metab Dispos. 2002; 30(12):1352-1356.
78. Niemi M, Backman JT, Granfors M, Laitila J, Neuvonen M, Neuvonen PJ. Gemfibrozil considerably increases the plasma concentrations of rosiglitazone. Diabetologia. 2003; 46(10):1319-1323.
79. Jaakkola T, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics of pioglitazone. Clin Pharmacol Ther. 2005; 77(5):404-414.

63
80. Deng LJ, Wang F, Li HD. Effect of gemfibrozil on the pharmacokinetics of pioglitazone. Eur J Clin Pharmacol. 2005; 61(11):831-836.
81. Inoue K, Inazawa J, Suzuki Y, Shimada T, Yamazaki H, Guengerich FP, Abe T. Fluorescence in situ hybridization analysis of chromosomal localization of three human cytochrome P450 2C genes (CYP2C8, 2C9, and 2C10) at 10q24.1. Jpn J Hum Genet. 1994; 39(3):337-343.
82. Lai XS, Yang LP, Li XT, Liu JP, Zhou ZW, Zhou SF. Human CYP2C8: structure, substrate specificity, inhibitor selectivity, inducers and polymorphisms. Curr Drug Metab. 2009; 10(9):1009-1047.
83. Naraharisetti SB, Lin YS, Rieder MJ, Marciante KD, Psaty BM, Thummel KE, Totah RA. Human liver expression of CYP2C8: gender, age, and genotype effects. Drug Metab Dispos. 2010; 38(6):889-893.
84. Bahadur N, Leathart JB, Mutch E, Steimel-Crespi D, Dunn SA, Gilissen R, Houdt JV, Hendrickx J, Mannens G, Bohets H et al. CYP2C8 polymorphisms in Caucasians and their relationship with paclitaxel 6alpha-hydroxylase activity in human liver microsomes. Biochem Pharmacol. 2002; 64(11):1579-1589.
85. Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI, Goldstein JA. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001; 11(7):597-607.
86. Parikh S, Ouedraogo JB, Goldstein JA, Rosenthal PJ, Kroetz DL. Amodiaquine metabolism is impaired by common polymorphisms in CYP2C8: implications for malaria treatment in Africa. Clin Pharmacol Ther. 2007; 82(2):197-203.
87. Rodriguez-Antona C, Niemi M, Backman JT, Kajosaari LI, Neuvonen PJ, Robledo M, Ingelman-Sundberg M. Characterization of novel CYP2C8 haplotypes and their contribution to paclitaxel and repaglinide metabolism. Pharmacogenomics J. 2008; 8(4):268-277.
88. Yu L, Shi D, Ma L, Zhou Q, Zeng S. Influence of CYP2C8 polymorphisms on the hydroxylation metabolism of paclitaxel, repaglinide and ibuprofen enantiomers in vitro. Biopharm Drug Dispos. 2013; 34(5):278-287.
89. Kaspera R, Naraharisetti SB, Tamraz B, Sahele T, Cheesman MJ, Kwok PY, Marciante K, Heckbert SR, Psaty BM, Totah RA. Cerivastatin in vitro metabolism by CYP2C8 variants found in patients experiencing rhabdomyolysis. Pharmacogenet Genomics. 2010; 20(10):619-629.
90. Yasar U, Lundgren S, Eliasson E, Bennet A, Wiman B, de Faire U, Rane A. Linkage between the CYP2C8 and CYP2C9 genetic polymorphisms. Biochem Biophys Res Commun. 2002; 299(1):25-28.
91. Blanco G, Martinez C, Ladero JM, Garcia-Martin E, Taxonera C, Gamito FG, Diaz-Rubio M, Agundez JA. Interaction of CYP2C8 and CYP2C9 genotypes modifies the risk for nonsteroidal anti-inflammatory drugs-related acute gastrointestinal bleeding. Pharmacogenet Genomics. 2008; 18(1):37-43.
92. Singh R, Ting JG, Pan Y, Teh LK, Ismail R, Ong CE. Functional role of Ile264 in CYP2C8: mutations affect haem incorporation and catalytic activity. Drug Metab Pharmacokinet. 2008; 23(3):165-174.
93. Hichiya H, Tanaka-Kagawa T, Soyama A, Jinno H, Koyano S, Katori N, Matsushima E, Uchiyama S, Tokunaga H, Kimura H et al. Functional characterization of five novel CYP2C8 variants, G171S, R186X, R186G, K247R, and K383N, found in a Japanese population. Drug Metab Dispos. 2005; 33(5):630-636.
94. Sarasquete ME, Garcia-Sanz R, Marin L, Alcoceba M, Chillon MC, Balanzategui A, Santamaria C, Rosinol L, de la Rubia J, Hernandez MT et al. Bisphosphonate-related osteonecrosis of the jaw is associated with polymorphisms of the cytochrome P450

64
CYP2C8 in multiple myeloma: a genome-wide single nucleotide polymorphism analysis. Blood. 2008; 112(7):2709-2712.
95. Smith HE, Jones JP, 3rd, Kalhorn TF, Farin FM, Stapleton PL, Davis CL, Perkins JD, Blough DK, Hebert MF, Thummel KE et al. Role of cytochrome P450 2C8 and 2J2 genotypes in calcineurin inhibitor-induced chronic kidney disease. Pharmacogenet Genomics. 2008; 18(11):943-953.
96. Lamba J, Hebert JM, Schuetz EG, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for CYP3A5. Pharmacogenet Genomics. 2012; 22(7):555-558.
97. Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001; 27(4):383-391.
98. Ekhart C, Doodeman VD, Rodenhuis S, Smits PH, Beijnen JH, Huitema AD. Polymorphisms of drug-metabolizing enzymes (GST, CYP2B6 and CYP3A) affect the pharmacokinetics of thiotepa and tepa. Br J Clin Pharmacol. 2009; 67(1):50-60.
99. Petrova DT, Yaramov N, Toshev S, Nedeva P, Maslyankov S, von Ahsen N, Oellerich M, Toncheva D. Genotyping of CYP3A5 polymorphisms among Bulgarian patients with sporadic colorectal cancer and controls. Onkologie. 2007; 30(11):559-563.
100. Passey C, Birnbaum AK, Brundage RC, Oetting WS, Israni AK, Jacobson PA. Dosing equation for tacrolimus using genetic variants and clinical factors. Br J Clin Pharmacol. 2011; 72(6):948-957.
101. Frohlich M, Hoffmann MM, Burhenne J, Mikus G, Weiss J, Haefeli WE. Association of the CYP3A5 A6986G (CYP3A5*3) polymorphism with saquinavir pharmacokinetics. Br J Clin Pharmacol. 2004; 58(4):443-444.
102. Lin YS, Dowling AL, Quigley SD, Farin FM, Zhang J, Lamba J, Schuetz EG, Thummel KE. Co-regulation of CYP3A4 and CYP3A5 and contribution to hepatic and intestinal midazolam metabolism. Mol Pharmacol. 2002; 62(1):162-172.
103. Egbelakin A, Ferguson MJ, MacGill EA, Lehmann AS, Topletz AR, Quinney SK, Li L, McCammack KC, Hall SD, Renbarger JL. Increased risk of vincristine neurotoxicity associated with low CYP3A5 expression genotype in children with acute lymphoblastic leukemia. Pediatr Blood Cancer. 2011; 56(3):361-367.
104. Lee SJ, Usmani KA, Chanas B, Ghanayem B, Xi T, Hodgson E, Mohrenweiser HW, Goldstein JA. Genetic findings and functional studies of human CYP3A5 single nucleotide polymorphisms in different ethnic groups. Pharmacogenetics. 2003; 13(8):461-472.
105. Kurose K, Sugiyama E, Saito Y. Population differences in major functional polymorphisms of pharmacokinetics/pharmacodynamics-related genes in Eastern Asians and Europeans: implications in the clinical trials for novel drug development. Drug Metab Pharmacokinet. 2012; 27(1):9-54.
106. Shimada N, Iwasaki M, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, Kusama R, Hamada GS, Nishimoto IN, Iyeyasu H et al. Genetic polymorphisms in estrogen metabolism and breast cancer risk in case-control studies in Japanese, Japanese Brazilians and non-Japanese Brazilians. J Hum Genet. 2009; 54(4):209-215.
107. Borst L, Wallerek S, Dalhoff K, Rasmussen KK, Wesenberg F, Wehner PS, Schmiegelow K. The impact of CYP3A5*3 on risk and prognosis in childhood acute lymphoblastic leukemia. Eur J Haematol. 2011; 86(6):477-483.
108. Hustert E, Haberl M, Burk O, Wolbold R, He YQ, Klein K, Nuessler AC, Neuhaus P, Klattig J, Eiselt R et al. The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics. 2001; 11(9):773-779.

65
109. Park SY, Kang YS, Jeong MS, Yoon HK, Han KO. Frequencies of CYP3A5 genotypes and haplotypes in a Korean population. J Clin Pharm Ther. 2008; 33(1):61-65.
110. Alvarellos ML, Sangkuhl K, Daneshjou R, Whirl-Carrillo M, Altman RB, Klein TE. PharmGKB summary: very important pharmacogene information for CYP4F2. Pharmacogenet Genomics. 2015; 25(1):41-47.
111. Wang MZ, Wu JQ, Bridges AS, Zeldin DC, Kornbluth S, Tidwell RR, Hall JE, Paine MF. Human enteric microsomal CYP4F enzymes O-demethylate the antiparasitic prodrug pafuramidine. Drug Metab Dispos. 2007; 35(11):2067-2075.
112. Hsu MH, Savas U, Griffin KJ, Johnson EF. Regulation of human cytochrome P450 4F2 expression by sterol regulatory element-binding protein and lovastatin. J Biol Chem. 2007; 282(8):5225-5236.
113. McDonald MG, Rieder MJ, Nakano M, Hsia CK, Rettie AE. CYP4F2 is a vitamin K1 oxidase: An explanation for altered warfarin dose in carriers of the V433M variant. Mol Pharmacol. 2009; 75(6):1337-1346.
114. Danese E, Montagnana M, Johnson JA, Rettie AE, Zambon CF, Lubitz SA, Suarez-Kurtz G, Cavallari LH, Zhao L, Huang M et al. Impact of the CYP4F2 p.V433M polymorphism on coumarin dose requirement: systematic review and meta-analysis. Clin Pharmacol Ther. 2012; 92(6):746-756.
115. Caldwell MD, Awad T, Johnson JA, Gage BF, Falkowski M, Gardina P, Hubbard J, Turpaz Y, Langaee TY, Eby C et al. CYP4F2 genetic variant alters required warfarin dose. Blood. 2008; 111(8):4106-4112.
116. Cerezo-Manchado JJ, Roldan V, Rosafalco M, Anton AI, Arroyo AB, Garcia-Barbera N, Martinez AB, Padilla J, Corral J, Vicente V et al. Effect of VKORC1, CYP2C9 and CYP4F2 genetic variants in early outcomes during acenocoumarol treatment. Pharmacogenomics. 2014; 15(7):987-996.
117. Ward NC, Croft KD, Puddey IB, Phillips M, van Bockxmeer F, Beilin LJ, Barden AE. The effect of a single nucleotide polymorphism of the CYP4F2 gene on blood pressure and 20-hydroxyeicosatetraenoic acid excretion after weight loss. J Hypertens. 2014; 32(7):1495-1502; discussion 1502.
118. Bardowell SA, Stec DE, Parker RS. Common variants of cytochrome P450 4F2 exhibit altered vitamin E-omega-hydroxylase specific activity. J Nutr. 2010; 140(11):1901-1906.
119. Bergstralh DT, Ting JP. Microtubule stabilizing agents: their molecular signaling consequences and the potential for enhancement by drug combination. Cancer Treat Rev. 2006; 32(3):166-179.
120. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004; 4(4):253-265.
121. Ring AE, Ellis PA. Taxanes in the treatment of early breast cancer. Cancer Treat Rev. 2005; 31(8):618-627.
122. Gligorov J, Lotz JP. Preclinical pharmacology of the taxanes: implications of the differences. Oncologist. 2004; 9 Suppl 2:3-8.
123. Huizing MT, Misser VH, Pieters RC, ten Bokkel Huinink WW, Veenhof CH, Vermorken JB, Pinedo HM, Beijnen JH. Taxanes: a new class of antitumor agents. Cancer Invest. 1995; 13(4):381-404.
124. Dumontet C, Sikic BI. Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J Clin Oncol. 1999; 17(3):1061-1070.
125. Tubiana-Hulin M. How to maximize the efficacy of taxanes in breast cancer. Cancer Treat Rev. 2005; 31 Suppl 4:S3-9.

66
126. Fisher B, Redmond C, Wickerham DL, Bowman D, Schipper H, Wolmark N, Sass R, Fisher ER, Jochimsen P, Legault-Poisson S et al. Doxorubicin-containing regimens for the treatment of stage II breast cancer: The National Surgical Adjuvant Breast and Bowel Project experience. J Clin Oncol. 1989; 7(5):572-582.
127. Nabholtz JM, Cantin J, Chang J, Guevin R, Patel R, Tkaczuk K, Vodvarka P, Lindsay MA, Reese D, Riva A et al. Phase III trial comparing granulocyte colony-stimulating factor to leridistim in the prevention of neutropenic complications in breast cancer patients treated with docetaxel/doxorubicin/cyclophosphamide: results of the BCIRG 004 trial. Clin Breast Cancer. 2002; 3(4):268-275.
128. Henderson IC, Berry DA, Demetri GD, Cirrincione CT, Goldstein LJ, Martino S, Ingle JN, Cooper MR, Hayes DF, Tkaczuk KH et al. Improved outcomes from adding sequential Paclitaxel but not from escalating Doxorubicin dose in an adjuvant chemotherapy regimen for patients with node-positive primary breast cancer. J Clin Oncol. 2003; 21(6):976-983.
129. Paridaens R, Biganzoli L, Bruning P, Klijn JG, Gamucci T, Houston S, Coleman R, Schachter J, Van Vreckem A, Sylvester R et al. Paclitaxel versus doxorubicin as first-line single-agent chemotherapy for metastatic breast cancer: a European Organization for Research and Treatment of Cancer Randomized Study with cross-over. J Clin Oncol. 2000; 18(4):724-733.
130. McGrogan BT, Gilmartin B, Carney DN, McCann A. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta. 2008; 1785(2):96-132.
131. Zhou J, Giannakakou P. Targeting microtubules for cancer chemotherapy. Curr Med Chem Anticancer Agents. 2005; 5(1):65-71.
132. Materna V, Surowiak P, Kaplenko I, Spaczynski M, Duan Z, Zabel M, Dietel M, Lage H. Taxol-resistance-associated gene-3 (TRAG-3/CSAG2) expression is predictive for clinical outcome in ovarian carcinoma patients. Virchows Arch. 2007; 450(2):187-194.
133. Jordan MA. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr Med Chem Anticancer Agents. 2002; 2(1):1-17.
134. Gueritte F. General and recent aspects of the chemistry and structure-activity relationships of taxoids. Curr Pharm Des. 2001; 7(13):1229-1249.
135. Roy V, Perez EA. New therapies in the treatment of breast cancer. Semin Oncol. 2006; 33(3 Suppl 9):S3-8.
136. Sparreboom A, van Tellingen O, Nooijen WJ, Beijnen JH. Nonlinear pharmacokinetics of paclitaxel in mice results from the pharmaceutical vehicle Cremophor EL. Cancer Res. 1996; 56(9):2112-2115.
137. Karlsson MO, Molnar V, Freijs A, Nygren P, Bergh J, Larsson R. Pharmacokinetic models for the saturable distribution of paclitaxel. Drug Metab Dispos. 1999; 27(10):1220-1223.
138. Ringel I, Horwitz SB. Studies with RP 56976 (taxotere): a semisynthetic analogue of taxol. J Natl Cancer Inst. 1991; 83(4):288-291.
139. Haldar S, Basu A, Croce CM. Bcl2 is the guardian of microtubule integrity. Cancer Res. 1997; 57(2):229-233.
140. Clarke SJ, Rivory LP. Clinical pharmacokinetics of docetaxel. Clin Pharmacokinet. 1999; 36(2):99-114.
141. Bruno R, Hille D, Riva A, Vivier N, ten Bokkel Huinnink WW, van Oosterom AT, Kaye SB, Verweij J, Fossella FV, Valero V et al. Population pharmacokinetics/pharmacodynamics of docetaxel in phase II studies in patients with cancer. J Clin Oncol. 1998; 16(1):187-196.

67
142. Martin M, Pienkowski T, Mackey J, Pawlicki M, Guastalla JP, Weaver C, Tomiak E, Al-Tweigeri T, Chap L, Juhos E et al. Adjuvant docetaxel for node-positive breast cancer. N Engl J Med. 2005; 352(22):2302-2313.
143. Roche H, Fumoleau P, Spielmann M, Canon JL, Delozier T, Serin D, Symann M, Kerbrat P, Soulie P, Eichler F et al. Sequential adjuvant epirubicin-based and docetaxel chemotherapy for node-positive breast cancer patients: the FNCLCC PACS 01 Trial. J Clin Oncol. 2006; 24(36):5664-5671.
144. Verschraegen CF, Sittisomwong T, Kudelka AP, Guedes E, Steger M, Nelson-Taylor T, Vincent M, Rogers R, Atkinson EN, Kavanagh JJ. Docetaxel for patients with paclitaxel-resistant Mullerian carcinoma. J Clin Oncol. 2000; 18(14):2733-2739.
145. Valero V, Jones SE, Von Hoff DD, Booser DJ, Mennel RG, Ravdin PM, Holmes FA, Rahman Z, Schottstaedt MW, Erban JK et al. A phase II study of docetaxel in patients with paclitaxel-resistant metastatic breast cancer. J Clin Oncol. 1998; 16(10):3362-3368.
146. Marsh S, Somlo G, Li X, Frankel P, King CR, Shannon WD, McLeod HL, Synold TW. Pharmacogenetic analysis of paclitaxel transport and metabolism genes in breast cancer. Pharmacogenomics J. 2007; 7(5):362-365.
147. Gao Y, Liu D, Wang H, Zhu J, Chen C. Functional characterization of five CYP2C8 variants and prediction of CYP2C8 genotype-dependent effects on in vitro and in vivo drug-drug interactions. Xenobiotica. 2010; 40(7):467-475.
148. Green H, Soderkvist P, Rosenberg P, Mirghani RA, Rymark P, Lundqvist EA, Peterson C. Pharmacogenetic studies of Paclitaxel in the treatment of ovarian cancer. Basic Clin Pharmacol Toxicol. 2009; 104(2):130-137.
149. Baker SD, Verweij J, Cusatis GA, van Schaik RH, Marsh S, Orwick SJ, Franke RM, Hu S, Schuetz EG, Lamba V et al. Pharmacogenetic pathway analysis of docetaxel elimination. Clin Pharmacol Ther. 2009; 85(2):155-163.
150. Fohner AE, Robinson R, Yracheta J, Dillard DA, Schilling B, Khan B, Hopkins S, Boyer BB, Black J, Wiener H et al. Variation in genes controlling warfarin disposition and response in American Indian and Alaska Native people: CYP2C9, VKORC1, CYP4F2, CYP4F11, GGCX. Pharmacogenet Genomics. 2015; 25(7):343-353.
151. Sipeky C, Lakner L, Szabo M, Takacs I, Tamasi V, Polgar N, Falus A, Melegh B. Interethnic differences of CYP2C9 alleles in healthy Hungarian and Roma population samples: relationship to worldwide allelic frequencies. Blood Cells Mol Dis. 2009; 43(3):239-242.
152. Hirsh J, Dalen J, Anderson DR, Poller L, Bussey H, Ansell J, Deykin D. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 2001; 119(1 Suppl):8S-21S.
153. Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet. 2001; 40(8):587-603.
154. Xie HG, Prasad HC, Kim RB, Stein CM. CYP2C9 allelic variants: ethnic distribution and functional significance. Adv Drug Deliv Rev. 2002; 54(10):1257-1270.
155. Aithal GP, Day CP, Kesteven PJ, Daly AK. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet. 1999; 353(9154):717-719.
156. Chang M, Soderberg MM, Scordo MG, Tybring G, Dahl ML. CYP2C19*17 affects R-warfarin plasma clearance and warfarin INR/dose ratio in patients on stable warfarin maintenance therapy. Eur J Clin Pharmacol. 2015; 71(4):433-439.
157. Wypasek E, Branicka A, Awsiuk M, Sadowski J, Undas A. Genetic determinants of acenocoumarol and warfarin maintenance dose requirements in Slavic population: a

68
potential role of CYP4F2 and GGCX polymorphisms. Thromb Res. 2014; 134(3):604-609.
158. Scott SA, Patel M, Martis S, Lubitz SA, van der Zee S, Yoo C, Edelmann L, Halperin JL, Desnick RJ. Copy number variation and warfarin dosing: evaluation of CYP2C9, VKORC1, CYP4F2, GGCX and CALU. Pharmacogenomics. 2011; 13(3):297-307.
159. Yuan HY, Chen JJ, Lee MT, Wung JC, Chen YF, Charng MJ, Lu MJ, Hung CR, Wei CY, Chen CH et al. A novel functional VKORC1 promoter polymorphism is associated with inter-individual and inter-ethnic differences in warfarin sensitivity. Hum Mol Genet. 2005; 14(13):1745-1751.
160. Wen MS, Lee M, Chen JJ, Chuang HP, Lu LS, Chen CH, Lee TH, Kuo CT, Sun FM, Chang YJ et al. Prospective study of warfarin dosage requirements based on CYP2C9 and VKORC1 genotypes. Clin Pharmacol Ther. 2008; 84(1):83-89.
161. Thompson CA. FDA encourages genetics-aided warfarin dosing. Am J Health Syst Pharm. 2007; 64(19):1994-1996.
162. Bodin L, Verstuyft C, Tregouet DA, Robert A, Dubert L, Funck-Brentano C, Jaillon P, Beaune P, Laurent-Puig P, Becquemont L et al. Cytochrome P450 2C9 (CYP2C9) and vitamin K epoxide reductase (VKORC1) genotypes as determinants of acenocoumarol sensitivity. Blood. 2005; 106(1):135-140.
163. Sconce EA, Khan TI, Wynne HA, Avery P, Monkhouse L, King BP, Wood P, Kesteven P, Daly AK, Kamali F. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: proposal for a new dosing regimen. Blood. 2005; 106(7):2329-2333.
164. McDonagh EM, Whirl-Carrillo M, Garten Y, Altman RB, Klein TE. From pharmacogenomic knowledge acquisition to clinical applications: the PharmGKB as a clinical pharmacogenomic biomarker resource. Biomark Med. 2011; 5(6):795-806.
165. Lam MP, Cheung BM. The pharmacogenetics of the response to warfarin in Chinese. Br J Clin Pharmacol. 2012; 73(3):340-347.
166. Singh O, Sandanaraj E, Subramanian K, Lee LH, Chowbay B. Influence of CYP4F2 rs2108622 (V433M) on warfarin dose requirement in Asian patients. Drug Metab Pharmacokinet. 2011; 26(2):130-136.
167. Kalaydjieva L, Gresham D, Calafell F. Genetic studies of the Roma (Gypsies): a review. BMC Med Genet. 2001; 2:5.
168. Hajioff S, McKee M. The health of the Roma people: a review of the published literature. J Epidemiol Community Health. 2000; 54(11):864-869.
169. Kalaydjieva L, Morar B, Chaix R, Tang H. A newly discovered founder population: the Roma/Gypsies. Bioessays. 2005; 27(10):1084-1094.
170. Gresham D, Morar B, Underhill PA, Passarino G, Lin AA, Wise C, Angelicheva D, Calafell F, Oefner PJ, Shen P et al. Origins and divergence of the Roma (gypsies). Am J Hum Genet. 2001; 69(6):1314-1331.
171. Morar B, Gresham D, Angelicheva D, Tournev I, Gooding R, Guergueltcheva V, Schmidt C, Abicht A, Lochmuller H, Tordai A et al. Mutation history of the roma/gypsies. Am J Hum Genet. 2004; 75(4):596-609.
172. Zalan A, Beres J, Pamjav H. Paternal genetic history of the Vlax Roma. Forensic Sci Int Genet. 2011; 5(2):109-113.
173. Paulik E, Nagymajtenyi L, Easterling D, Rogers T. Smoking behaviour and attitudes of Hungarian Roma and non-Roma population towards tobacco control policies. Int J Public Health. 2011; 56(5):485-491.
174. Parry G, Van Cleemput P, Peters J, Walters S, Thomas K, Cooper C. Health status of Gypsies and Travellers in England. J Epidemiol Community Health. 2007; 61(3):198-204.

69
175. Petek D, Rotar Pavlic D, Svab I, Lolic D. Attitudes of Roma toward smoking: qualitative study in Slovenia. Croat Med J. 2006; 47(2):344-347.
176. Chandler D, Angelicheva D, Heather L, Gooding R, Gresham D, Yanakiev P, de Jonge R, Baas F, Dye D, Karagyozov L et al. Hereditary motor and sensory neuropathy--Lom (HMSNL): refined genetic mapping in Romani (Gypsy) families from several European countries. Neuromuscul Disord. 2000; 10(8):584-591.
177. Hunter M, Heyer E, Austerlitz F, Angelicheva D, Nedkova V, Briones P, Gata A, de Pablo R, Laszlo A, Bosshard N et al. The P28T mutation in the GALK1 gene accounts for galactokinase deficiency in Roma (Gypsy) patients across Europe. Pediatr Res. 2002; 51(5):602-606.
178. Kalaydjieva L, Lochmuller H, Tournev I, Baas F, Beres J, Colomer J, Guergueltcheva V, Herrmann R, Karcagi V, King R et al. 125th ENMC International Workshop: Neuromuscular disorders in the Roma (Gypsy) population, 23-25 April 2004, Naarden, The Netherlands. Neuromuscul Disord. 2005; 15(1):65-71.
179. Kovacs N, Sarkany I, Mohay G, Adamovich K, Ertl T, Kosztolanyi G, Kellermayer R. High incidence of short rib-polydactyly syndrome type IV in a Hungarian Roma subpopulation. Am J Med Genet A. 2006; 140(24):2816-2818.
180. Kellermayer R, Keller M, Ratajczak P, Richardson E, Harangi F, Merei E, Melegh B, Kosztolanyi G, Richard G. Bigenic connexin mutations in a patient with hidrotic ectodermal dysplasia. Eur J Dermatol. 2005; 15(2):75-79.
181. Szalai R, Magyari L, Matyas P, Duga B, Banfai Z, Szabo A, Kovesdi E, Melegh B. Genetic polymorphisms in promoter and intronic regions of CYP1A2 gene in Roma and Hungarian population samples. Environ Toxicol Pharmacol. 2014; 38(3):814-820.
182. Szalai R, Ganczer A, Magyari L, Matyas P, Bene J, Melegh B. Interethnic differences of cytochrome P450 gene polymorphisms may influence outcome of taxane therapy in Roma and Hungarian populations. Drug Metab Pharmacokinet. 2015.
183. Timofeeva MN, Kropp S, Sauter W, Beckmann L, Rosenberger A, Illig T, Jager B, Mittelstrass K, Dienemann H, Bartsch H et al. CYP450 polymorphisms as risk factors for early-onset lung cancer: gender-specific differences. Carcinogenesis. 2009; 30(7):1161-1169.
184. Cote ML, Yoo W, Wenzlaff AS, Prysak GM, Santer SK, Claeys GB, Van Dyke AL, Land SJ, Schwartz AG. Tobacco and estrogen metabolic polymorphisms and risk of non-small cell lung cancer in women. Carcinogenesis. 2009; 30(4):626-635.
185. Church TR, Haznadar M, Geisser MS, Anderson KE, Caporaso NE, Le C, Abdullah SB, Hecht SS, Oken MM, Van Ness B. Interaction of CYP1B1, cigarette-smoke carcinogen metabolism, and lung cancer risk. Int J Mol Epidemiol Genet. 2010; 1(4):295-309.
186. Bethke L, Webb E, Sellick G, Rudd M, Penegar S, Withey L, Qureshi M, Houlston R. Polymorphisms in the cytochrome P450 genes CYP1A2, CYP1B1, CYP3A4, CYP3A5, CYP11A1, CYP17A1, CYP19A1 and colorectal cancer risk. BMC Cancer. 2007; 7:123.
187. Ghisari M, Long M, Bonefeld-Jorgensen EC. Genetic polymorphisms in CYP1A1, CYP1B1 and COMT genes in Greenlandic Inuit and Europeans. Int J Circumpolar Health. 2013; 72:21113.
188. Hlavata I, Vrana D, Smerhovsky Z, Pardini B, Naccarati A, Vodicka P, Novotny J, Mohelnikova-Duchonova B, Soucek P. Association between exposure-relevant polymorphisms in CYP1B1, EPHX1, NQO1, GSTM1, GSTP1 and GSTT1 and risk of colorectal cancer in a Czech population. Oncol Rep. 2010; 24(5):1347-1353.

70
189. Shah PP, Singh AP, Singh M, Mathur N, Mishra BN, Pant MC, Parmar D. Association of functionally important polymorphisms in cytochrome P4501B1 with lung cancer. Mutat Res. 2008; 643(1-2):4-10.
190. Liang G, Pu Y, Yin L. Rapid detection of single nucleotide polymorphisms related with lung cancer susceptibility of Chinese population. Cancer Lett. 2005; 223(2):265-274.
191. Watanabe J, Shimada T, Gillam EM, Ikuta T, Suemasu K, Higashi Y, Gotoh O, Kawajiri K. Association of CYP1B1 genetic polymorphism with incidence to breast and lung cancer. Pharmacogenetics. 2000; 10(1):25-33.
192. Cote ML, Wenzlaff AS, Bock CH, Land SJ, Santer SK, Schwartz DR, Schwartz AG. Combinations of cytochrome P-450 genotypes and risk of early-onset lung cancer in Caucasians and African Americans: a population-based study. Lung Cancer. 2007; 55(3):255-262.
193. Pechandova K, Buzkova H, Matouskova O, Perlik F, Slanar O. Genetic polymorphisms of CYP2C8 in the Czech Republic. Genet Test Mol Biomarkers. 2012; 16(7):812-816.
194. Weise A, Grundler S, Zaumsegel D, Klotzek M, Grondahl B, Forst T, Pfutzner A. Development and evaluation of a rapid and reliable method for cytochrome P450 2C8 genotyping. Clin Lab. 2004; 50(3-4):141-148.
195. Nakajima M, Fujiki Y, Noda K, Ohtsuka H, Ohkuni H, Kyo S, Inoue M, Kuroiwa Y, Yokoi T. Genetic polymorphisms of CYP2C8 in Japanese population. Drug Metab Dispos. 2003; 31(6):687-690.
196. Muthiah YD, Lee WL, Teh LK, Ong CE, Ismail R. Genetic polymorphism of CYP2C8 in three Malaysian ethnics: CYP2C8*2 and CYP2C8*3 are found in Malaysian Indians. J Clin Pharm Ther. 2005; 30(5):487-490.
197. Cavaco I, Stromberg-Norklit J, Kaneko A, Msellem MI, Dahoma M, Ribeiro VL, Bjorkman A, Gil JP. CYP2C8 polymorphism frequencies among malaria patients in Zanzibar. Eur J Clin Pharmacol. 2005; 61(1):15-18.
198. Minhas S, Setia N, Pandita S, Saxena R, Verma IC, Aggarwal S. Prevalence of CYP2C8 polymorphisms in a North Indian population. Genet Mol Res. 2013; 12(3):2260-2266.
199. Gervasini G, Garcia-Martin E, Ladero JM, Pizarro R, Sastre J, Martinez C, Garcia M, Diaz-Rubio M, Agundez JA. Genetic variability in CYP3A4 and CYP3A5 in primary liver, gastric and colorectal cancer patients. BMC Cancer. 2007; 7:118.
200. Vaarala MH, Mattila H, Ohtonen P, Tammela TL, Paavonen TK, Schleutker J. The interaction of CYP3A5 polymorphisms along the androgen metabolism pathway in prostate cancer. Int J Cancer. 2008; 122(11):2511-2516.
201. Kristiansen W, Haugen TB, Witczak O, Andersen JM, Fossa SD, Aschim EL. CYP1A1, CYP3A5 and CYP3A7 polymorphisms and testicular cancer susceptibility. Int J Androl. 2011; 34(1):77-83.
202. Bains RK, Kovacevic M, Plaster CA, Tarekegn A, Bekele E, Bradman NN, Thomas MG. Molecular diversity and population structure at the Cytochrome P450 3A5 gene in Africa. BMC Genet. 2013; 14:34.
203. Bajpai P, Tripathi AK, Agrawal D. Genetic polymorphism of CYP3A5 in Indian chronic myeloid leukemia patients. Mol Cell Biochem. 2010; 336(1-2):49-54.
204. Rao DN, Manjula G, Sailaja K, Surekha D, Raghunadharao D, Rajappa S, Vishnupriya S. Association of CYP3A5*3 polymorphism with development of acute leukemia. Indian J Hum Genet. 2011; 17(3):175-178.
205. Sipeky C, Weber A, Melegh BI, Matyas P, Janicsek I, Szalai R, Szabo I, Varnai R, Tarlos G, Ganczer A et al. Interethnic variability of CYP4F2 (V433M) in admixed

71
population of Roma and Hungarians. Environ Toxicol Pharmacol. 2015; 40(1):280-283.
206. Pautas E, Moreau C, Gouin-Thibault I, Golmard JL, Mahe I, Legendre C, Taillandier-Heriche E, Durand-Gasselin B, Houllier AM, Verrier P et al. Genetic factors (VKORC1, CYP2C9, EPHX1, and CYP4F2) are predictor variables for warfarin response in very elderly, frail inpatients. Clin Pharmacol Ther. 2010; 87(1):57-64.
207. Scott SA, Khasawneh R, Peter I, Kornreich R, Desnick RJ. Combined CYP2C9, VKORC1 and CYP4F2 frequencies among racial and ethnic groups. Pharmacogenomics. 2010; 11(6):781-791.
208. Perez-Andreu V, Roldan V, Anton AI, Garcia-Barbera N, Corral J, Vicente V, Gonzalez-Conejero R. Pharmacogenetic relevance of CYP4F2 V433M polymorphism on acenocoumarol therapy. Blood. 2009; 113(20):4977-4979.
209. Perini JA, Struchiner CJ, Silva-Assuncao E, Suarez-Kurtz G. Impact of CYP4F2 rs2108622 on the stable warfarin dose in an admixed patient cohort. Clin Pharmacol Ther. 2010; 87(4):417-420.
210. Shahin MH, Khalifa SI, Gong Y, Hammad LN, Sallam MT, El Shafey M, Ali SS, Mohamed ME, Langaee T, Johnson JA. Genetic and nongenetic factors associated with warfarin dose requirements in Egyptian patients. Pharmacogenet Genomics. 2011; 21(3):130-135.

72
9. KÖSZÖNETNYILVÁNÍTÁS
Elsősorban szeretnék köszönetet mondani témavezetőmnek, Dr. Melegh Béla
Professzor Úrnak, aki lehetővé tette, hogy doktori disszertációm alapjául szolgáló
kutatómunkámat a Pécsi Tudományegyetem Általános Orvostudományi Karának Orvosi
Genetikai Intézetében a Multidiszciplináris Orvostudományok keretén belül zajló „Humán
molekuláris genetika” témában végezhessem, szakmai tevékenységemet mindvégig
figyelemmel kísérte, munkámat hasznos meglátásaival, útmutatásával irányította és segítette.
Köszönetemet fejezem ki Dr. Berenténé Dr. Bene Juditnak a szakmai, tudományos
és emberi segítségért, amit tőle mindvégig kaptam.
Köszönettel tartozom az Orvosi Genetikai Intézet minden asszisztensnőjének, akik
kezdettől fogva szakmai tapasztalatukkal és lelkiismeretes munkájukkal segítették
tanulmányaimat.
Köszönettel tartozom továbbá azoknak a kollégáknak, doktornőknek, akik
közreműködtek a minták gyűjtésével.
Hálával tartozom és köszönöm családom megértő türelmét, szeretetét és bátorítását.


















![Doktori (PhD) értekezés Szerző: Dr. Mikor András …aok.pte.hu/phd_dolgozatok/docs/phd/file/dolgozatok/2017/...és 17%-a közé tehető [2, 3]. Egy 2011-ben, 28 európai országban](https://img.pdfslide.tips/doc/110x75/5e28f464fcad8910157438c6/doktori-phd-rtekezs-szerz-dr-mikor-andrs-aokptehuphddolgozatokdocsphdfiledolgozatok2017.jpg)










