Embed Size (px)
Citation preview

CASE REPORT
Clin Exp Nephrol (2005) 9:69–73 © Japanese Society of Nephrology 2005DOI 10.1007/s10157-004-0334-7
Wataru Kitagawa · Naoto Miura · Harutaka YamadaKazuhiro Nishikawa · Arao Futenma · Hirokazu Imai
The increase of antiglomerular basement membrane antibody followingpauci-immune-type crescentic glomerulonephritis
Received: July 20, 2004 / Accepted: November 15, 2004
AbstractA 50-year-old woman was admitted because of high feverand fatigue. Proteinuria, hematuria, and elevated BUN(47.8mg/dl) and creatinine (3.4mg/dl) suggested rapidlyprogressive glomerulonephritis. The serological studyrevealed all negative results for rheumatoid factor,antinuclear antibody, serum cryoglobulins, MPO-ANCA,PR3-ANCA, and anti-streptolysin O. Antiglomerular base-ment membrane (GBM) antibody, as assessed by ELISA,was 11EU (normal, �10). Kidney biopsy on the eighthhospital day demonstrated pauci-immune-type crescenticglomerulonephritis without ANCA. Methylprednisolonepulse therapy (500mg/day, 3 days) and 45mg/day predniso-lone orally were started. At 3 weeks after kidney biopsy, theanti-GBM antibody value increased from 11EU/ml to116EU/ml, and MPO and PR3-ANCA were still negative.HLA type was DR8 and DR 15(2), with a genotype ofHLA-DRB1*08021 and HLA-DRB1*15011. The presentcase suggests that HLA-DR15 plays an important role onantibody production against alpha 3(IV) NC1 autoantigenafter severe nephritis or tissue damage.
Key words Anti-GBM antibody · HLA-DR 15 · Pauci-immune-type crescentic glomerulonephritis
Introduction
Crescentic glomerulonephritis is divided into four typesaccording to their causative factors: antiglomerular base-ment membrane (GBM) antibody, immunecomplex,pauci-immune with antineutrophil cytoplasmic antibody(ANCA), and pauci-immune without ANCA.1 For the
W. Kitagawa · N. Miura · H. Yamada (*) · K. Nishikawa ·A. Futenma · H. ImaiDepartment of Internal Medicine, Division of Nephrology andRheumatology, Aichi Medical University School of Medicine,Nagakute, Aichi 480-1195, JapanTel. �81-561-62-3311; Fax �81-561-63-1583e-mail: [email protected]
anti-GBM antibody type, there are two clinical phenotypes,one limited to the glomeruli, and the other, known asGoodpasture syndrome, involving both glomerulonephritisand pulmonary hemorrhage. Recently, a simple enzyme-linked immunosorbent assay (ELISA)-based system hasbeen developed to detect circulating anti-GBM antibodyrecognizing alpha 3 type IV collagen NC1-domain antigenepitope [alpha 3(IV) NC1 domain antigen]. As a clinicalproblem, there are some patients who have both anti-GBMantibody and ANCA.2
We report herein an adult patient with pauci-immunewithout ANCA-type crescentic glomerulonephritis whodeveloped anti-GBM antibodies while receiving predniso-lone, and discuss the possible mechanism underlying anti-GBM antibody production.
Case report
A 50-year-old woman was admitted to Aichi MedicalUniversity Hospital on April 3, 2003, because of high feverand fatigue. She had been treated with antihypertensivedrugs (�-blocker and diuretics) since 1999, and a recentlaboratory examination revealed a blood urea nitrogen(BUN) level of 8.9mg/dl and a serum creatinine level of0.8mg/dl. Two weeks before admission to the hospital,she had high fever and diarrhea. She developed anorexiaand easy fatigue even while receiving fosfomycin andlevofloxacin.
On admission, her vital statutes were as follows: bodytemperature, 38.1°C; pulse rate, 84 beats/min; respiratoryrate, 16 breaths/min; and blood pressure, 124/72mmHg.A physical examination showed anemic conjunctiva andbilateral knock pain on the costovertebral region of herback. Cardiolpulmonary, abdominal, and neurological ex-aminations were normal.
Urinary protein was 2�, occult blood reaction was 3�for dipsticks, and the urine sediment showed 10–15 redblood cells per high power field and 31–50 white blood cells(WBC) per high power field with hyaline cast, granular cast,

70
and WBC cast. Blood counts were as follows: erythrocytes,324 � 104/µl, hemoglobin, 10.2g/dl; hematocrit, 30.4%; leu-kocyte count, 10100/µl (neutrophils 82%, lymphocytes12%, monocytes 5%); and platelets, 54.3 � 104/µl.
Blood chemistry values were as follows: serum total pro-tein, 8.4g/dl; albumin, 3.8g/dl; blood urea nitrogen, 47.8mg/dl; creatinine, 3.4mg/dl; uric acid, 7.5mg/dl; total bilirubin,0.5mg/dl; aspirate aminotransferase, 37IU/l; alanine ami-notransferase 39IU/l; lactic dehydrogenase, 267IU/l; alka-line phosphatase, 676IU/l; and total cholesterol, 138mg/dl.Immunological data were as follows: C-reactive protein,23.4mg/dl; IgG, 1174mg/dl (normal range, 880–1800); IgA,282mg/dl (normal range, 126–517); IgM, 73mg/dl (normalrange, 52–270); C3, 220mg/dl (normal range, 84–151); C4,42mg/dl (normal range, 17–40); CH50 63U/ml (normalrange, 30–40). The patient was negative for rheumatoidfactor, antinuclear antibody, serum cryoglobulins, MPO-ANCA, PR3-ANCA, and p- and c-ANCA by immunofluo-rescent study, and antistreptolysin O. Anti-GBM antibody,as assessed by ELISA, was 11EU (normal, �10).
Cultures from arterial and venous blood were negative inall three trials, and urinary bacterial cultures were negative.Results of chest radiography and electrocardiogram werewithin normal limits.
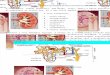
Gallium scintigram demonstrated bilateral kidney up-take (Fig. 1). A kidney biopsy was performed on the eighthhospital day. Light microscopic examination showed that 2
of 20 glomeruli were globally sclerosed, and the remaining18 glomeruli showed circumferential cellular crescents withnecrotizing lesion. Mononuclear cells had infiltrated intotubules and interstitial tissue with mild degree of interstitialfibrosis (Fig. 2). Immunofluorescence studies revealed noglomerular deposition of linear and granular IgG, IgA, IgM,kappa, lambda, C3, or C1q, but fibrinogen 2� was found inthe crescentic areas and interstitial tissue (Fig. 3). Unfortu-nately, we could not evaluate the glomerular changes be-cause the specimen mainly contained tubular damage.
Clinical course (Fig. 4): kidney biopsy on the 8th hospitalday revealed pauci-immune-type crescentic glomerulone-phritis without ANCA. We started methylprednisolonepulse therapy (500mg/day, 3 days) and 45mg/day predniso-lone orally. We reexamined the circulating IgG-class anti-GBM antibody level at 3 weeks after kidney biopsy becausethe initial value was slightly over the limit and the patientshowed a worsening serum creatinine level even while onprednisolone therapy. At this point, the anti-GBM antibodyvalue increased from 11 to 116EU/ml (no elevation ofIgA or IgM-class anti-GBM antibodies) and MPO andPR3-ANCA were still negative, leading us to perform twocourses of plasma exchange in addition to hemodialysis, andto continue administration of 30mg/day prednisolone.At discharge (the 78th hospital day), proteinuria andhematuria were 2�, the titer of anti-GBM antibody haddecreased to 13EU/ml, BUN was 60.6mg/dl, and creatininewas 2.8mg/dl (see Fig. 4). Three months after discharge, thetiter of anti-GBM antibody dropped below 10EU/ml, BUNwas 46.5mg/dl, and creatinine was 2.3mg/dl. The patient’sHLA type was DR8 and DR 15(2), with a genotype ofHLA-DRB1*08021 and HLA-DRB1*15011.
Discussion
The patient shows two unique points, that severe nephritisinduces costovertebral spontaneous and knock pain, and
Fig. 1. Gallium scintigram showing high uptake in both kidneysFig. 2. Light microscopic examination revealing cellular crescents andtubulointerstitial nephritis. Periodic acid-Schiff (PAS). �200

71
that anti-GBM antibody increases after kidney damagesuch as crescentic glomerulonephritis and tubulointerstitialnephritis, while receiving prednisolone.
Costovertebral spontaneous and knock pain has alreadybeen reported when kidney volume increased rapidly, notonly in acute pyelonephritis, but also in crescentic glomeru-lonephritis and tubulointerstitial nephritis. At this time, wecould differentiate severe nephritis from acute pyelonephri-tis, based on the positive result of the gallium scintigramand negative urinary bacterial cultures.
Regarding the production of anti-GBM antibody, threemajor factors have been proposed. One is the exposure ofalpha 3 type IV collagen NC1-domain antigen epitope[alpha 3(IV) NC1 domain antigen] that is usually hidden inthe normal three-dimensional structures. Historically, anti-GBM antibody-related kidney disease was first described inyoung men following influenza infection, as reported by
Goodpasture in 1918.3 In a study of a situation involving aninfluenza outbreak in 1958, Santon and Tange first usedthe term Goodpasture syndrome to describe severe glom-erulonephritis, mainly crescentic glomerulonephritis, to-gether with pulmonary hemorrhage.4 An analysis of linearamino acid sequence revealed a homologous sequence,SEGTGQA, between the alpha 3(IV) NC1 domain antigenand the influenza A hemagglutinin;5 however, evidence ofconcomitant influenza A2 viral infection is rare. Other trig-gers than influenza A2 virus to exposure of the antigen havebeen reported such as environmental exposure, particularlyto hydrocarbons,6 smoking,7 occupational solvents,8 anddrugs such as methicilline9 and d-penicillamine.10 Also, kid-ney tissue damage following lithotripsy and urinary obstruc-tion has been proposed as a trigger of Goodpasturesyndrome.11-13 However, a second factor, other than the an-tigen exposure, is necessary to produce anti-GBM antibody,
Fig. 3. Immunofluorescencestudy demonstrating nosignificant deposition of IgG,IgA, IgM, and C3. Fibrinogenstaining was positive in thecrescentic area and interstitialtissue

72
because the data showing the low incidence of anti-GBMantibody-related glomerulonephritis, even though there aremany causative factors in our circumstance, suggest the im-portance of the patient’s genetic background such as HLA.HLA-DR typing, HLA-DRB1*1501 and DQB1*0201,14–16
are thought to play a major role in the antigen-presentingmechanism from macrophage to lymphocytes. The presentpatient’s HLA type was DR8 and DR 15(2), with a geno-type of HLA-DRB1*08021 and HLA-DRB1*15011, indi-cating that she is susceptible to production of anti-GBMantibodies.
The last factor in the production of anti-GBM antibodyis the abnormal immune tolerance in the thymus. The find-ing that the alpha 3(IV) NC1 domain antigen is expressedin the thymus epithelial cells of normal individuals meansthat thymus instructs lymphocytes not to respond to alpha3(IV) NC1 autoantigen.17 However, the evidence thatcirculating T cells from patients with anti-GBM antibody(Goodpasture syndrome) could react with the alpha 3(IV)NC1 autoantigen suggests that this central immune toler-ance to educated T lymphocytes is disturbed in Good-pasture syndrome.18,19 At this time, unfortunately, we couldnot determine the character of the T cells.
The present case presents the possibility that severe ne-phritis such as pauci-immune-type crescentic glomerulone-phritis with tubulointerstitial nephritis may induce theexposure of alpha 3(IV) NC1 autoantigen and produce anti-
GBM antibody under the condition of HLA-DRB1*15011.However, it is still unclear whether the increased anti-GBMantibody aggravated the kidney damage.
Acknowledgments The author thanks Dr. Yasuhiko Tomino, Divisionof Nephrology, Department of Medicine, Juntendo University Schoolof Medicine, and Dr. Masayuki Endoh, Department of Medicine,Tokai University School of Medicine, for their support.
References
1. Falk RJ, Jennette JC, Nachman PH. Primary glomerular disease.In: Brenner MB, editor. Brenner and Rector’s the kidney. Phila-delphia: Sanders, 2004;1293–380.
2. Short AK, Esnault VL, Lockwood CM. Anti-neutrophil cyto-plasmic antibodies and anti-glomerular basement membraneantibodies: two coexisting distinct autoreactivities detectablein patients with rapidly progressive glomerulonephritis. Am JKidney Dis 1995;26:439–45.
3. Goodpasture E. The significance of certain pulmonary lesions inrelation to the etiology of influenza. Am J Med Sci 1919;158:863–70.
4. Santon MC, Tange JD. Goodpasture’s syndrome (pulmonaryhaemorrhage associated with glomerulonephritis). Aust Int J Med1958;7:132–44.
5. Feng L, Xia Y, Wilson CB. Alternative splicing of the NC1 domainof the human α3(IV) collagen gene. Differential expression ofmRNA transcripts that predict three protein variants with distinctcarboxyl regions. J Biol Chem 1994;269:2342–8.
Fig. 4. Clinical course. Anti-GBM, antiglomerular basement membrane; PSL, prednisolone; HD, hemodialysis; Cr, creatinine in serum

73
6. Zimmerman SW, Groehler K, Beirne GJ. Hydrocarbon exposureand chronic glomerulonephritis. Lancet 1975;2:199–201.
7. Donaghy M, Rees AJ. Cigarette smoking and lung haemorrhage inglomerulonephritis caused by autoantibodies to glomerular base-ment membrane. Lancet 1983;2:1390–3.
8. Daniell WE, Couser WG, Rosenstock L. Occupational solventexposure and glomerulonephritis. A case report and review of theliterature. JAMA 1988;259:2280–3.
9. Williams PS, Davenport A, McDicken I, Ashby D, Goldsmith HJ,Bone JM. Increased incidence of anti-glomerular basement mem-brane antibody (anti-GBM) nephritis in the Mersey Region,September 1984–October 1985. Q J Med 1988;68:727–33.
10. Peces R, Riera JR, Arboleya LR, et al. Goodpasture’s syndrome ina patient receiving penicillamine and carbimazole. Nephron 1987;45:316–20.
11. Lubec G. Anti-glomerular basement membrane disease afterlithotripsy. Lancet 1990;335:1405.
12. Guerin V, Rabian C, Droz D, et al. Anti-glomerular basementmembrane disease after lithotripsy. Lancet 1990;335:856–7.
13. Weber M, Pullig O, Boesken WH. Anti-glomerular basementmembrane disease after renal obstruction. Lancet 1990;336:512–13.
14. Dunkley H, Chapman JR, Burke J, et al. HLA-DR and –DQgenotyping in anti-GBM disease. Dis Markers 1991;9:249–56.
15. Huey B, McCormick K, Capper J, et al. Association of HLA-DRand HLA-DQ types with anti-GBM nephritis by sequence-specificologonucleotide probe hybridization. Kidney Int 1993;44:307–12.
16. Fisher M, Pusey CD, Vaughan RW, Rees AJ. Susceptibility to anti-glomerular basement membrane disease is strongly associated withHLA-DRB1 genes. Kidney Int 1997;51:222–9.
17. Wong D, Phelps RG, Turner AN. The Goodpasture antigen isexpression in the human thymus. Kidney Int 2001;60:1777–83.
18. Cairns LS, Phelps RG, Bowie L, Hall AM, Saweirs WWM, ReesAJ, et al. The fine specificity and cytokine profile of T-helper cellsresponsive to the a3 chain of type IV collagen in Goodpasture’sdisease. J Am Soc Nephrol 2003;14:2801–12.
19. Wu J, Borillo J, Glass WF II, Hicks J, Ou CN, Lou YH. T-cellepitope of a3 chain of type IV collagen induces severe glomerulo-nephritis. Kidney Int 2003;64:1292–301.