Embed Size (px)
Citation preview

2198 CANADIAN JOURNAL OF CHEMISTRY. VOL. 43, 1965
Reaction of Syrupy 2,3,4,6-Tetra-0-acetyl-a-D-glucopyranose with Iodoniu7n Di-sym-collidine Perchlorate and D-Glz~cal Triacetate
The reaction of the mixture of the a- and fi-anomers of 2,3,4,6-tetra-0-acetyl-D-glucose, obtained by equi- libration in pyridine, was performed under the same conditions as described above starting with the pure 2,3,4,6-tetra-0-acetyl-P-~-glucose.
On deacetylation, a 39% yield of virtually pure 2-deoxy-2-iodo-a-D-glucopyranosyl fi-D-glucopyranoside was obtained as evidenced by the n.m.r. spectrum and optical rotation, and its catalytic reduction and acetylation to VI.
ACICNOWLEDGMENTS
The research was supported by grants (N.R.C.-T 172) to R. U. Lemieux and three North Atlantic Treaty Organization Studentships to A. R. M. (1961-1964). The n.nl.r. spectra were determined by Mrs. Gail Conway.
REFERENCES R. U. LEZIIEUX and S. LEVINE. Can. J . Chem. 40, 1926 (1962). J . STANEK and V. SCHWARZ. Collection Czech. Chern. Commun. 20, 42 (1955). R. U. LE~I IEUX and S. LEVINE. Can. I. Chern. 42. 1473 (1964). . ,
H. CARLSOHN. Angew. Chem. 46, 747<(1933). '
H. CARLSOHN. Ber. 68, 2209 (1935). M. J. OUCHAKOW and W. 0. T,SCHITOW. Bull. Soc. Chi~n. France, Ser. 5, 3, 2142 (1935). R. U. LEarr~ux and A. R. MORGAN. Can. J . Chem. In press. 1965. P. Z. ALLEN. Methods in carbohydrate chemistry. Vol. I. R. L. Whistler and M. L. Wolfrom (Editors).
Academic Press. New York. 1962. D. 372. W. 1. WHELAN. Private communication. R. fJ. L ~ ~ r r s u x and B. FRASER-REID. Can. J . Che~n. 42, 532 (1964). J. 13. BREWSTER. J. Am. Chem. Soc. 81, 5475, 5453 (1959). R. U. L ~ h r r ~ u s , C. T. BISHOP, and G. E. PELLETIER. Can. J . Chem. 34, 1365 (1956). H. H. SCFILUBACH and I. WOLF. Ber. 62, 1507 (1929). A. GEORG. Helv. Chim. Acta, 15, 924 (1932). R. U. L ~ h r r ~ u s and C. BRICE. Can. J . Chem. 33, 109 (1955). R. U. LELIIEUX, R. I<. I<ULLNIG, H. J . BERNSTEIN, and W. G. SCHNEIDER. J . Am. Chem. Soc. 80, 6098
(19.58). ,----,- M. KAI~I>LUS. J . Chem. Phys. 30, 11 (1959). R. U. LELIIEUX and G. HUBER. Can. 1. Chern. 31, 1040 (1953). I<. M~TSUDA. Nature, 180, 985 (1957s B. HELFERICH and I. ZIRNER. Chem. Ber. 95. 2604 (1962). R. U. L E ~ I I E U X a n d 1. W. LOWN. Can. 1. ~ h k m . 42.'893 (19641 R. U. LEMIEUS. an. Carbohydrate chern. 9, 1 (1954): ' A. S. PERLIX. Can. J. Chem. 41, 399 (1963). R. I<. NESS and H. G. F ~ ~ ~ ~ ~ ~ ~ , JR. J . Am. Chem. Soc. 78, 4710 (1956). N. J . ANTIA. J. Am. Chem. Soc. 80, 6138 (1958). E. L. HIRST and J . I<. N. JONES. Discussions Faraday Soc. 7, 271 (1949). W. E. TREVELYAN. D. P. PROCTOR. and I. S. HARRISON. Nature. 166. 444 (19501. . . . , R. U. LEMIEUX and H. F. 1 3 . 4 ~ ~ ~ . ' ~ n a i Chem. 26, 920 (1954). L. HOUGH, J. I<. N. JONES, and W. H. \ .~'ADA~AN. J. Chem. Soc. 1702 (1950). S. M. PARTRIDGE. Nature, 164, 443 (1949). J . C. SCHUMACHER (Editor). Perchlorates, their properties, manufacture and uses. Reinhold Publishing
Corp., N.Y. 1960. p. 187. R. U. LEMIEUX and B. FRASER-REID. Can. J. Chem. 43, 1460 (1965). E. FISCHER and M. BERG~IANN. Ber. 52, 829 (1919).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
HE
NR
Y M
AD
DE
N L
IBR
AR
Y T
EC
H S
ER
V o
n 05
/03/
13Fo
r pe
rson
al u
se o
nly.

THE PREPARATION AND CONFIGURATIONS OF TRI-0-ACETYL-a-D-GLUCOPYRANOSE 1,2-(ORTHOESTERS)'
R. U. LEMIEUX AND A. R. MORGAN^ Depart?~zent of Clzemistry, University of Alberta, Edmonton, Alberta
Received January 25, 1965
ABSTRACT
Reaction of tri-0-acetyl-2-0-acyl (acetyl, benzoyl, pivalyl, and methoxycarbony1)-a-D- glucopyranosyl halides with a variety of alcohols in sym-collidine containing a tetraalkyl- ammonium halide gave the 1,2-orthoesters in near quantitative yields. In each case one of the two diastereoisomeric orthoesters was obtained in high yield and was shown by application of mn1.r. spectroscopy to be that isomer which has the alkoxy group on the dioxolan ring trans t o the pyranose ring (exo-configuration). Participation of a methoxycarbonyloxy group allowed the preparation of a 1,2-(dimethyl orthocarbonate). 1,2-(Phenyl orthoacetates) were prepared.
INTRODUCTION
Helferich and co-workers (1, 2) observed that reaction of t e t r a - O - a ~ e t ~ l - a - ~ - ~ l u c o - pyranosyl bromide (I) with alcohols in the presence of sym-collidine (2,4,6-trimethyl- pyridine) gave tri-0-acetyl-a-D-glucopyranose 1,2-(alkyl orthoacetates). Our study of the
x CH, OR CH3
reaction of I with pyridine (3,4) led to the conclusion that the orthoesters must form from the 0-anoiner of I which arises in the reaction mixture as a result of the liberation of broinide ion. A recent study of the equilibration of the tetra-0-acetyl-D-glucopyranosyl chlorides has in fact shown that the anomerization reaction is strongly catalyzed by halide ions ( 5 ) . I t has long been established that the reaction of tetra-0-acetyl-0-D- glucopyranosyl chloride with alcol~ols, preferably in the presence of pyridine, yields the alkyl1,2-orthoacetates (6,7). Tetra-0-acetyl-0-D-glucopyranosyl bromide (11) has recently been prepared and this highly labile coinpound was shown to give the 1,2-orthoacetate when reacted with n~ethanol (8). Particularly in view of the promise that the 1,2-ortho- esters of sugars will play an iinportant role in the chemical syilthesis of a-glycopyranosides (9), it seemed iillportailt to examine their preparation from the readily available O- acylated glycopyranosyl halides which possess a 1,2-cis-configuration. 1,2-Orthoesters are readily prepared from those sugars which provide the 1,2-trans-acylated glycopyranosyl
'Presented by R. U. Lentienx at 148tl~ iMeeting of the Anzerican Clze?~~ical Society, Cl~icago, Illi?zois, August SO -September 4 , 1964.
2 T I ~ i s research was part of a thesis subrr~itted by A. R. iV1. i n partial fz~ljillment of the requiren~ents for the degree of Doctor of Plzilosophy, 1964.
Canadian Journal of Chemistry. Volume 13 (1965)
2199
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
HE
NR
Y M
AD
DE
N L
IBR
AR
Y T
EC
H S
ER
V o
n 05
/03/
13Fo
r pe
rson
al u
se o
nly.

2200 CANADIAN JOURNAL O F CHEMISTRY. VOL. 43. 1965
halide as the more stable anomer (10). This communication is concerned with the prepara- tion of a variety of 1,2-orthoesters and a 1,2-orthocarbonate from derivatives of D-glucopyranose in near quantitative yield. I t is apparent that the method should be generally applicable to the preparation of 1,2-orthoesters from 0-acylated 1,2-cis-glyco- pyranosyl halides.
The preparation of orthoesters requires that the reaction medium be strongly buffered against acid. Although pyridine can be used for this purpose, its use requires the reaction to yield orthoesters to be substantially more rapid than the formation of the pyridinium glycosides (3). Since this need not generally be the case, it seemed desirable to employ a more hindered base. Since tetra-0-acetyl-a-D-glucopyranosyl bromide (I) did not react with sym-collidine after 5 d a t room temperature, sym-collidine was chosen as a suitable base. Of course, other 2,G-disubstituted pyridines should also be useful in this regard. In fact, Mazurek and Perlin (11) have found 2,6-dimethylpyridine to be a solvent of choice in the preparation of 1,2-orthoacetates from tetra-0-acetyl-a-D-mannopyranosyl bromide.
Since the formation of a 1,2-orthoester from a 1,2-cis-glycopyranosyl halide is dependent on the epimerization of the latter compound to the 1,2-trans-anomer, i t was evident that the anomerization reaction should be promoted by the addition of halide ion. Quaternary ammonium halide salts were used for this purpose in view of their solubilities in non-polar solvents such as sym-collidine and their effectiveness in promoting anomerization (5).
Reaction of I with a wide variety of alcohols using sym-collidine as solvent and tetra- n-butylammonium bromide as catalyst has provided near quantitative yields of the 1,2- orthoacetate (111). The preparations wherein R = ethyl, isopropyl, t-butyl, and phenyl are presented in the experimental section in order t o illustrate the generality of the method. In all cases one of the diastereoisoillers was fornled in large excess (over 85%) except for the reaction with phenol (about 70%). Lemieux and Cipera (7) have suggested that this high degree of partial asymmetric synthesis probably arose because of a more facile approach by the alcohol to the side of the 1,2-acetoxonium ion inter- mediate (IV) which is trans to the glucopyranose ring to yield the exo-orthoester (the isomer with the alkoxy group in the exo-orientation a t the 2-position of the dioxolane ring). Evidence that this is in fact the case was obtained by a comparison of the n.1n.r. spectra of the 1,2- (methyl orthoacetate), 1 ,%(methyl orthopivalate), and 1,2- (dimethyl orthocarbonate) derivatives of 3,4,6-tri-0-acetyl-a-D-glucopyranose. The orthopivalate was originally prepared in the anticipation that the bulk of the t-butyl group \\-ould prevent the formation of the exo-isomer. However, that this was probably not the case was apparent from the chemical shifts of the methoxy-group protons in the above nlentioned compounds, the chemical shifts of the pyranose-ring protons in a series of 1,2-(alkyl orthoacetates), and the difference in the conformations of the pyranose rings of the ortho- acetates and the orthopivalate as indicated by their n.m.r. spectra. As is seen in Table I, the major isomers in the reaction products obtained in preparing the orthoacetate and the orthopivalate both had the methoxy group signal a t T 6.71. This undoubtedly means that these isomers have the same configuration. The chelllical shifts of the methoxy groups in the orthocarbonate agree well with those which would be anticipated from the chemical shifts of the methoxy groups of the diastereoisolneric orthoacetates. This is to be expected since the change in the configuration of the orthoacetates places the methoxy group in one of the environnlents of the methoxy groups in the orthocarbonate. The chemical shift data reported in Table I1 show that the change in the alkyl group from methyl to ethyl, t o isopropyl, to t-butyl, and to phenyl had an appreciable effect on the cheillical shifts of the pyranose-ring protons only a t the anolneric center (H-1). Therefore, it is evident that
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
HE
NR
Y M
AD
DE
N L
IBR
AR
Y T
EC
H S
ER
V o
n 05
/03/
13Fo
r pe
rson
al u
se o
nly.
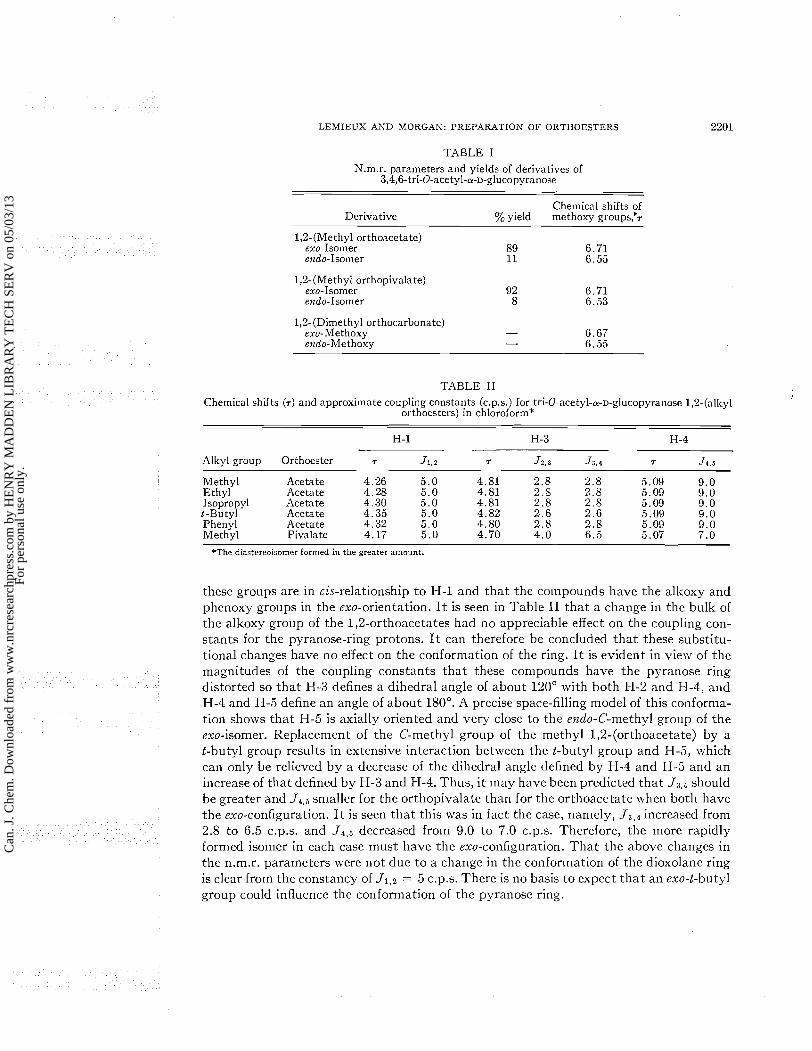
LEMIEUX AND MORGAN: PREPARATION O F ORTHOESTERS
TABLE I
N.m.r. parameters and yields of derivatives of 3,4,6-tri-0-acetyl-a-D-glucopyranose
Chemical shifts of Derivative %yield methoxy groups,'~
1,2-(Methyl orthoacetate) exo-Isomer 89 6.71 endo-Isomer 11 6.55
1,2-(Methyl orthopivalate) exo-Isomer 92 6.71 endo-Isomer 8 6.53
1,2-(Dimethyl orthocarbonate) ero-Methoxy - 6.67 endo-Methoxy - 6.55
TABLE I1
Chemical shifts (7) and approximate coupling constants (c.p.s.) for tri-0-acetyl-a-D-glucopyranose 1,2-(alkyl orthoesters) in chloroform*
H-1 H-3 H-4
Alkyl group Orthoester 7 J 1 . z T J z , ~ J 3 . 4 7 J4.5
Methyl Acetate 4.26 5 .0 4.81 2.8 2.6 5.09 9.0 Ethyl Acetate 4.28 5 .0 4.81 2.8 2. 8 5.09 9.0 Isopropyl Acetate 4.30 5 .0 4.81 2 .8 2.8 5.09 9 . 0 t-Butyl Acetate 4.35 5.0 4.82 2 .6 2.6 5.09 9.0 Phenyl Acetate 4.32 5 .0 4. 80 2 .8 2 .8 5.09 9.0 Methyl Pivalate 4.17 5 .0 4.70 4.0 6 .5 5.07 7.0
*The diastereoisomer formed in the greater amount.
these groups are in cis-relationship to H-1 and that the colnpounds have the allcoxy and phenoxy groups in the exo-orientation. I t is seen in Table I1 tha t a change in the bulk of the alkoxy group of the 1,2-orthoacetates had no appreciable effect on the coupling con- stants for the pyranose-ring protons. I t can therefore be concluded that these substitu- tional changes have no effect on the conformation of the ring. I t is evident in view of the magnitudes of the coupling constants that these conlpounds have the pyranose ring distorted so that 1-1-3 defines a dihedral angle of about 120" with both H-2 and H-4, and H-4 and H-5 define an angle of about 180". A precise space-filling model of this conforma- tion shows that H-5 is axially oriented and very close to the endo-C-methyl group of the exo-isomer. Replacement of the C-methyl group of the methyl 1,2-(orthoacetate) by a t-butyl group results in extensive interaction between the t-butyl group and H-5, ~irhich can only be relieved by a decrease of the dihedral angle defined by H-4 and H-5 and an increase of that defined by H-3 and H-4. Thus, i t may have been predicted that Jj,i should be greater and J4,5 smaller for the orthopivalate than for the orthoacetate when both have the exo-configuration. I t is seen tha t this was in fact the case, namely, J 3 , , increased from 2.8 to 6.5 c.p.s. and J4,5 decreased froin 9.0 to 7.0 c.p.s. Therefore, the inore rapidly formed isomer in each case must have the exo-configuration. Tha t the above changes in the n.m.r. parameters were not due to a change in the conforlnation of the dioxolane ring is clear from the constancy of J l S z = 5 c.p.s. There is no basis to expect tha t an exo-t-butyl group could influence the conformation of the pyranose ring.
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
HE
NR
Y M
AD
DE
N L
IBR
AR
Y T
EC
H S
ER
V o
n 05
/03/
13Fo
r pe
rson
al u
se o
nly.

2202 CANADIAN JOURNAL O F CHEMISTRY. VOL. 43, 1965
The 1,2-(methyl orthopivalate) and a 1,2-(isopropyl orthobenzoate) were prepared a s syrups by reaction of the tri-0-acetyl-2-0-pivalyl-a-~-glucopyranosy bromide and tri-0- acetyl-2-0-benzoyl-a-D-glucopyranosyl bromide with the appropriate alcohol in sym- collidine containing tetra-n-butylaminoniu~n bromide. Reaction of tri-O-acetyl-2-0- methox.~,carbonyl-a-D-glucopyranosyl chloride with methanol in sym-collidine containing tetraethylammonium chloride gave the 1,2-(dimethyl carbonate). However, the syrupy product was considerably less pure than tha t obtained on reaction of tri-O-acetyl-2-0- methox?.carbonyl-0-D-glucopyranosyl chloride with methanol and silver di-(sym-collidine) perchlorate (12) using methylene chloride as solvent. Both the preparation of this 0- chloride and of the orthocarbonate show that , as expected, the methoxycarbonyloxy group can participate effectively in replacements a t a neighboring position. Although the methoxycarbonyloxy group is probably less nucleophilic than the acetoxy group, the delocalization of the positive charge in the resulting oxocarbonium ion is greater than in the acetoxoniu~n ion.
ESPERIlMENTAL
Tri-0-acetyl-a-D-glucopyranose I,$-(Ethyl Orthoacetate) Tetra-0-acetyl-a-D-glucopyranosyl bromide, 4.1 g (10 mmoles), was dissolved in 10 in1 of synz-collidine and
0.6 ml of dry ethanol (10 mmoles). Tetra-n-butyl-ammonium bromide, 1 g (3.1 mmoles), was added, and when the mixture was heated to 50' with shaking, a homogeneous solution was obtained. Crystals of sym- collidinium hydrobromide soon began to separate. After 12 h a t 50°, the almost solid reaction mixture was dissolved in the minimum chloroform and washed with just sufficient hydrochloric acid to neutralize the collidine, then with aqueous sodi~im bicarbonate, and finally water. After drying the chloroform layer by filtration through chloroform-wetted filter paper, evaporation a t reduced pressure left a semicrystalline mass. The i1.m.r. spectrum of this product indicated that the two diastereoisomers of the 1,2-orthoacetate had formed in theoretical yield, 3.8 g (10 mmoles). Recrystallization from hot ethanol with a drop of sym- collidine and water added just to turbidity resulted in a heavy crystalline precipitate. The crystals were collected by filtration, washed with water, and dried i n uacuo. The yield of the ero-isomer was 85y0, m.p. 95-96"; reported 97-97.5" (7), after two recrystallizations from ethanol. When a 10 M excess of ethanol over the tetra-0-acetyl-ol-D-glucopyranosyl bromide was used, the product still contained only 14y0 of the endo-isomer.
Tri-0-acetyl-a-D-glucopyranose I,$-(Isopropyl Orthoacetate) The standard conditions above but with isopropanol in place of ethanol were used to obtain a theoretical
yield of the exdo- and exo-diastereoison~ers. Crystallization from ethanol with a trace of sym-collidine, and water added to turbidity, gave an 85y0 yield of the ero-isomer, m.p. 116-117". On repeated recrystallization, the m.p. was raised to 120-121°, [ a ] ~ f 30" (c, 2.4 in chloroform).
Anal. Calcd. for C17H26010(390.4): C, 51.8; H, 6.67. Found: 384; C, 51.54; H, 6.82.
Tri-0-acetyl-ol-D-glz~copyranose I,$-(t-Butyl Orthoacetate) Again the above conditioils were employed but with t-butanol, except that the acid washing was omitted
because of the very high sensitivity of the orthoacetate. The sym-collidine was finally removed i n uacuo with warming a t 80' to give a crystalline mass in theoretical yield. The pure exo-isomer was obtained on dissolving the crude crystals in hot ethyl acetate and adding Slcellysolve B to turbidity. A 75y0 yield of the orthoester, n1.p. 152.5-154.5", [ o l ] ~ f 34.5" (c, 1.6 in chloroforn~), was obtained.
Anal. Calcd. for C ~ S H ~ B O ~ O (404.4): C, 53.46; H, 6.98. Found: 394; C, 53.1; H , 6.71.
Tri-0-acetyl-ol-D-glucopyranose I,$-(Phenyl Orthoacetate) The standard conditions above with pheilol as the alcohol were used to obtain a syrup which could not be
induced to crystallize. The n.m.r. spectrum required the formation of the diastereoisomeric orthoacetates as the only products with 68y0 of the exo- and 32% of the endo-isomers as judged from the relative intensities of the signals for the orthoacetyl group a t 7 8.22 and 7 8.42, respectively. The compounds are exceedingly sensitive to hydrolysis.
Tri-0-acetyl-ol-D-glucopyranose I,$-(Isopropyl Orthobenzoate) 1,3,4,6-Tetra-0-acetyl-ol-D-glucopyraiose (12) was benzoylated in pyridine solution using benzoyl chloride
under the usual conditions. The syrupy product resisted crystallization and was treated with 30y0 hydrogen bromide in acetic acid to produce tri-0-acetyl-2-0-bei1zoyl-a-~-glucopyranosyl bromide. This product also failed to crystallize. Nevertheless, its n.m.r. spectrum was precisely that expected for this compouild in that the presence of three 0-acetyl groups and one beilzoyl group was evident. The signal for the anomeric proton
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
HE
NR
Y M
AD
DE
N L
IBR
AR
Y T
EC
H S
ER
V o
n 05
/03/
13Fo
r pe
rson
al u
se o
nly.

LEMIEUX A N D MORGAN: PREPARATION OF ORTHOESTERS 2203
was a doublet a t T 3.23, spacing 3.8 c.p.s.; H-3 and H-4 gave triplet signals a t T 4.21 and 4.72, respectively, with spacings of 9.5 c.p.s. The signal for H-2 was a quartet centered a t 7 4.90. The signals for the protons a t the 5- and 6-positions were in the usual positions with the signal for H-5 to somewhat lower field, about 7 5.6. The syrup, 2.3 g, was dissolved in 4 n ~ l of sym-collidine and 0.25 g of tetra-n-butylammonium bromide, and 0.47 ml of isopropanol were added. When the mixture was warmed to obtain a homogeneous solution, a fine precipitate appeared, and after 20 h a t 42', the solution had set solid with crystals. The chloroform solution of the reaction mixture was washed with excess 2 N sulfuric acid, followed by aqueous sodiutn bicarbonate. The n.m.r. spectrum of the syrup obtained on evaporation of the chloroform layer had doublets each of spacing 5 c.p.s. a t 7 4.13 and 4.24 with relative intensities of about 2 : l . This evidence that both the diastereo- isomeric orthobenzoates had formed was supported by the complex structures of the signals for the acetyl groups (five signals) and the methyl groups of the isopropyl group. The pattern of signals for the protons on the pyranose ring was very similar to that observed for the tri-0-a~etyl-a-~-gl~c0pyran0Se 1,2-(alkyl ortho- esters). Crystalline material was not obtained. I t was apparent that these orthoesters were substantially more susceptible to hydrolysis than the corresponding orthoacetates.
Tri-0-acetyl-a-D-glucopyranose 1,b-(Metliyl Orthopivalate) 1,3,4,6-Tetra-0-acetyl-a-D-glucopyranose (12), 3 g (8.62 mmoles), in 9 ml of pyridine was treated with
pivalyl chloride, 3.08 g (25.6 mmoles). After 3 h, the product was isolated in the normal way, and crystals were obtained from the syrup on evaporation a t high vacuum. Two recrystallizations from methanol-water yielded crystal plates, m.p. 109.5-110°, [ a ] ~ $93.2" (c, 1.2 in chloroform). The n.nI.r. spectrum was in complete agreement with the assigned structure.
Anal. Calcd. for CIBI-12~011: C, 52.77; H, 6.52. Found: C, 52.63; H, 6.20. The above crystals, 1.486 g (3.44 mmoles), were dissolved in 5 ml of 30% hydrogen bromide in acetic
acid, 1 ml of acetic acid, and 1 ml of acetic anhydride. After 9 h, the product was isolated in the normal fash- ion. The syrup obtained could not be crystallized, but had the n.m.r. spectrum required for tri-O-acetyl-2-0- pivalyl-a-D-glucopyranosyl bromide. The yield was quantitative.
The above syrup, 0.55 g (1.21 mmoles), was dissolved in 2 ml of sym-collidine and I ml of methanol (24.7 mmoles), and 0.2 g of tetra-n-butylammonium bromide were added. The reaction time required was significantly longer than that for the formation of the methyl orthoacetate. After 1 d a t 60°, the reaction mixture was shaken with carbon tetrachloride and the sym-collidinium bromide was removed by filtration. The carbon tetrachloride filtrate was washed with water and then evaporated to a syrup, 0.3 g (61% yield). The n.m.r. spectrum of the compound is described in Tables I and I1 and is discussed in detail in the introduction to this paper.
Tri-0-ucetyl-a-D-glzicopyrunose 1,2-(Dinzethyl Orthocarbonate) 3,4,6-Tri-0-acetyl-0-D-glucopyranosyl chloride (13), 20 g (61.5 mmoles), was dissolved in 20 ml of sym-
collidine and 100 ml of methyl chloroformate. On warming the solution, gases were evolved. Carbon dioxide and methyl chloride are presumably formed by nucleophilic attack of the chloride ion produced in the reaction on the methyl chloroformate. This reaction amounts to a decomposition of the methyl chloroformate which must therefore be added in a large excess. The reaction was carried out until almost all the nlethyl chloroformate had disappeared, around 4 h. The products were extracted with carbon tetrachloride, and the insoluble salts were removed by filtration. T h e carbon tetrachloride solution was washed with dilute sulfuric acid and aqueous sodium bicarbonate, and finally evaporated to a syrup. The n.m.r. spectrum of the product was in all respects precisely that expected for tri-0-acetyl-2-0-methoxycarbonyl-a-~-glucopyranosyl chloride. The yield was 90%.
The above syrup, 21 g (55 mmoles), was dissolved in 100 ml of acetic acid and 10 ml of acetic anhydride. Silver acetate, 15 g (89.8 mmoles), was added and the mixture was refluxed for 2 h. T h e silver salts were removed by filtration on Celite and the filtrate was evaporated down to a semi-crystalline mass in vacuo. . T h e ethanolic solution of the crystals was decolorized with Darco G 60 and water was added to the clear solution. Shiny needle-like crystals, m.p. 129.5-13O0, [ a ] ~ +-13.4" (c, 1.12 in chloroform) were obtained. The n.m.r. spectrum was that expected for tetra-0-acetyl-2-0-methoxycarbonyl-~-~-glucopyranose.
Anal. Calcd. for C1~H?2012: C, 47.29; H, 5.46. Found: C, 46.98; H , 5.20. The above crystals, 0.5 g (1.23 mmoles), reacted with aluminium chloride, 0.164 g (1.23 mmoles), in 6 1111
of methylene chloride over half an hour with stirring. Dry benzene, 15 ml, was added to the reaction mixture, which was passed through a bed of l / 2 g of dry silicic acid. The benzene - methylene chloride solution was washed with ice-cold water and evaporated to a crystalline mass, 0.31 g (My0 yield). Recrystallization from benzene-Skellysolve B yielded material, m.p. 55-80', [ a ] ~ $5.45" (c, 3 in chloroform). Recrystallization did not alter the properties, but crystallization from benzene alone, followed by grinding the crystals to a fine powder before drying i n vaczlo for I d , raised the m.p. t o 103.5-104°. The n.m.r. spectrum in deuterated chloroform indicated the presence of about one-third mole of benzene per mole of tri-0-acetyl-2-0-methoxy- carbonyl-0-D-glucopyranosyl chloride.
Anal. Calcd. for C1~HlIO1~CI. 1/3 COHO: C, 47.18; H, 5.20. Found: C, 47.44; H, 5.30. The above &chloride, 0.765 g (2 mmoles), silver di-sym-collidine perchlorate (12), 0.9 g (2 mmoles), and
methanol, 0.5 ml (12.13 mmoles), were allowed to react in 3 ml of ~nethylene chloride with stirring overnight. After the removal of insoluble salts on Celite, the filtrate was washed with aqueous sodium bicarbonate and
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
HE
NR
Y M
AD
DE
N L
IBR
AR
Y T
EC
H S
ER
V o
n 05
/03/
13Fo
r pe
rson
al u
se o
nly.

2204 C.L\KADIAN JOURNAL OF CHEMISTR1'. VOL. 43 , 1965
water and finally evaporated to a syrup. The n.m.r, spectrum required that virtually pure tri-0-acetyl-a-D- glucopyranose l,2-(dimethyl orthocarbonate) was the product of the reaction. The same compound was formed but in a less pure condition by reacting the syrupy tri-0-acetyl-2-0-methoxycarbonyl-a-D-gluco- pyranosyl chloride with methanol in synz-collidine containing tetraethylammonium chloride. The spacing of the signal for the anomeric proton a t 7 4.32 was 5.2 c.p.s. The signal for H-3 was a quartet centered a t 7 4.69 with spacings of about 4 and 6 c.p.s. which must correspond closely to J 2 . 3 and J3,1, respectively. The quartet signal for H-4 was centered a t 7 5.11 and showed a spacing of 9 c.p.s. which must correspond to J~,L. Thus the elldo-methoxy group distorts the pyranose ring from the conformation in the ero-l,2-(alkyl orthoacetates) toward that in the exo-1,2-(methyl orthopivalate).
ACKNOWLEDGMENTS
The research was supported by grants (N.R.C.-T 172) to R. U. L. and three North Atlantic Treaty Organization Studentships to A. R. M. (1961-1964). The n.m.r. spectra were determined a t 60 Mc.p.s. by Mrs. Gail Conway.
REFERENCES 1. B. HELFERICH, A. DOPPSTADT, and G. A. GOTTSCHLICH. Naturwissenschaften, 40, 441 (1953). 2. B. HELFERICH and I<. WEIS. Chem. Ber. 89, 314 (1956). 3. R. U. LENIEUX and A. R. MORGAN. Can. J. Chem. In press. 1965. 4. R. U. LG~IIEUX and A. R. MORGAN. J . Am. Chem. Soc. 85, 1889 (1963). 5. R. U. L ~ a r ~ ~ u x and JUN-ICHI HAYAMI. Can. J. Chem. In press. 1965. 6. R. U. LEJIIEUX and C. BRICE. Can. J. Chem. 33, 109 (1955). 7. R. U. L E ~ ~ I E U X and J. D. T . CIPERA. Can. J. Chem. 34, 906 (1956). 8. F. WEYGAND, H. ZIEMANN, and H. J. RESTYANN. Chem. Ber. 91, 2534 (1958). 9. R. U. L ~ > i ~ a u x and A. R. MORGAN. Can. J. Chem. In press. 1965.
10. E. PACSU. Advan. Carbohydrate Chem. 1, 77 (1945). 11. M. MAZUREK and A. S. PERLIN. Private communication. 12. R. U. LEYIEUX and A. R. MORGAN. International Symposium on the Chemistry of Natural Products,
ICyoto, Japan. May 12-18, 1964. Abstracts of Papers, p. 151. 13. P. BRIGL. Z. Physiol. Chem. 122, 245 (1922).
Can
. J. C
hem
. Dow
nloa
ded
from
ww
w.n
rcre
sear
chpr
ess.
com
by
HE
NR
Y M
AD
DE
N L
IBR
AR
Y T
EC
H S
ER
V o
n 05
/03/
13Fo
r pe
rson
al u
se o
nly.







![Nov 23 2018.ppt [Kompatibilitätsmodus] - Limes-Institut-Bonn · Figure 11-5 The anomeric monosaccharides -D-glucopyranose and -D-glucopyranose, drawn as both Haworth projections](https://img.pdfslide.tips/doc/110x75/5f7f813523e6cd4b030ea89a/nov-23-2018ppt-kompatibilittsmodus-limes-institut-bonn-figure-11-5-the-anomeric.jpg)





















