
C U R S O S E M B I O L O G I A , B I O Q U Í M I C A , B I O T E C N O L O G I A , E E N G E N H A R I A
B I O L Ó G I C A
Bioinformática
João [email protected]
Aulas T9-T10

Esquema de anotação Annothaton
1. ORFs? Coordenadas? Quadro de leitura? Cadeia + ou -? (SMS ORF Finder)
2. Existem proteínas homólogas? (BLASTp, BLASTx)3. Existem domínios funcionais? Onde? (InterProScan)4. Qual o tamanho / massa molecular da proteína codificada
pela ORF em aa / kDa?5. A que organismo ou táxon pertencerá a sequência
metagenómica? (BLAST Taxonomy Report)6. Relações filogenéticas (MSA; Prof. Rita Castilho)7. Conclusões e Análise de Resultados (PONTO PRINCIPAL DA
AVALIAÇÃO)

Massa Molecular (Molecular Weight)
� 1 Da = 1 unidade de massa atómica = 1/12 massa de um átomo de C ≈ massa de um átomo de H
� 1 mole de H tem uma massa de 1 g� Logo: 1 Da => 1 g mol-1
� A massa molecular de uma macromolécula depende do nº de unidades que a forma
� Mw Proteína = somatório da Mw dos resíduos de aa� Mw DNA = somatório da Mw dos resíduos de nucleótidos� Annothaton: anotar Mw - apenas se a ORF estiver
completa!

A ORF está completa quando:
� Identificámos o seu codão STOP (a 3’)� Identificámos o seu codão START (a 5’)� O alinhamento múltiplo (MSA = multisequence
alignment) com sequências homólogas não revela a falta de qualquer sequência

Determinação do codão START por MSA
GOS_12345 MSAHNTMALAGHAHHHAIKLYVVFABD_P0001 MSAHYTMALVGHAHKHAIKLYVIFABP_P0002 MSAHYTMALVGHAHHHAIKLYVVFA
Exemplo 1 Conclusão
A ORF está completana sua extremidade 5’
GOS_12345 MALAGHAHHHAIKLYVVFABD_P0001 MSAHYTMALVGHAHKHAIKLYVIFABP_P0002 MSAHYTMALVGHAHHHAIKLYVVFA
Exemplo 2 Conclusão
A ORF está incompletana sua extremidade 5’
GOS_12345 MSAHNTMALAGHAHHHAIKLYVVFABD_P0001 MALVGHAHKHAIKLYVIFABP_P0002 MALVGHAHHHAIKLYVVFA
Exemplo 3 Conclusão
A ORF começa no2º codão START

Alinhamentos de sequências múltiplas (MSA)Para que servem?
� Determinar qual o verdadeiro codão START por comparação com sequências homólogas e corrigir a extremidade 5’ dada pela “greedyapproach”
� Procurar sequências adicionais� Montar sequências genómicas� Montar ESTs� Pontos de partida para análises filogenéticas

Alinhamentos de sequências múltiplas (MSA)Pesquisa de sequências adicionais
5’-ACTGATTAGCAACTAAGGACATAAAACTGCTTAGCCAT-3’5’-ACTGATTACCATACATTGACTTAACTGACTAATCTTAT-3’5’-ACTGATTAGCATCCAAGGACATAAAACTGCTATGTTAT-3’
ACTGATTASCAWMYAWKGACWTAAMWSWSYWWWSYYATConsensoprimer, sonda
(sequência comum usada para detectarfamílias de genes [homólogos] por hibridação)

Alinhamentos de sequências múltiplas (MSA)Montagem de sequências genómicas
5’-AGCTATTACAGGAACTTGCACATGGGCTTAGCTAGCAAATTTAGC-3’ seq123_67seq123_24 5’-CTTGCACATGGGCTTAGCTAGCAAATTTAGCTAGCT-3’
seq123_89 5’-CAAATTTAGCTAGCTTGCCATTA-3’
seq123_67 + seq123_24 + seq123_89 = contig
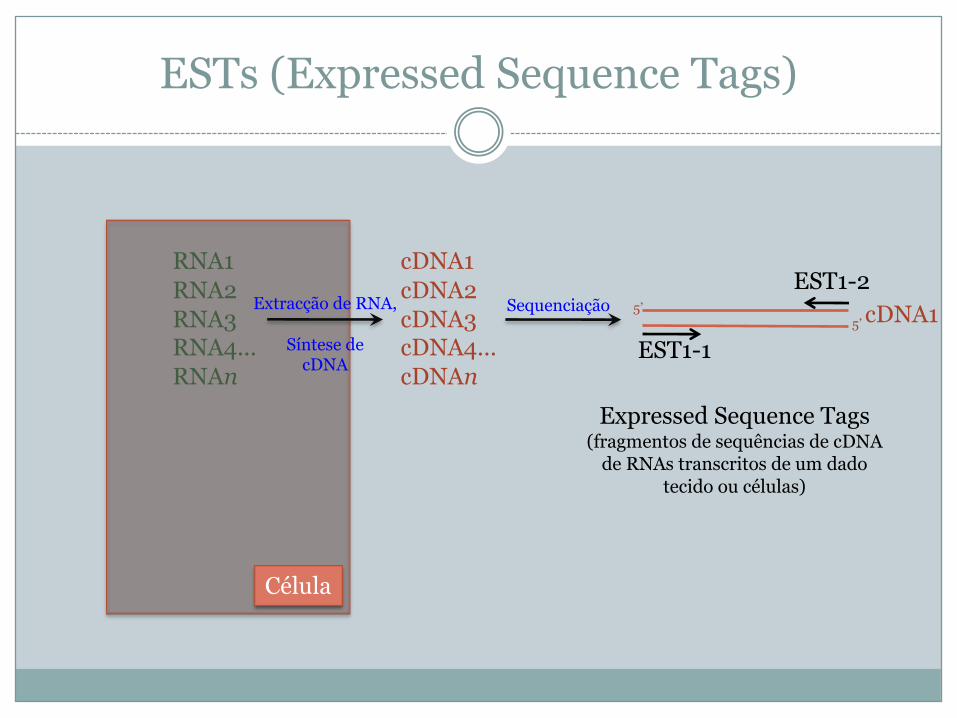
ESTs (Expressed Sequence Tags)
Expressed Sequence Tags(fragmentos de sequências de cDNA
de RNAs transcritos de um dado tecido ou células)
RNA1RNA2RNA3RNA4...RNAn
cDNA1cDNA2cDNA3cDNA4...cDNAn
Célula
Extracção de RNA,
Síntese decDNA
Sequenciação
EST1-1
EST1-2cDNA15’
5’
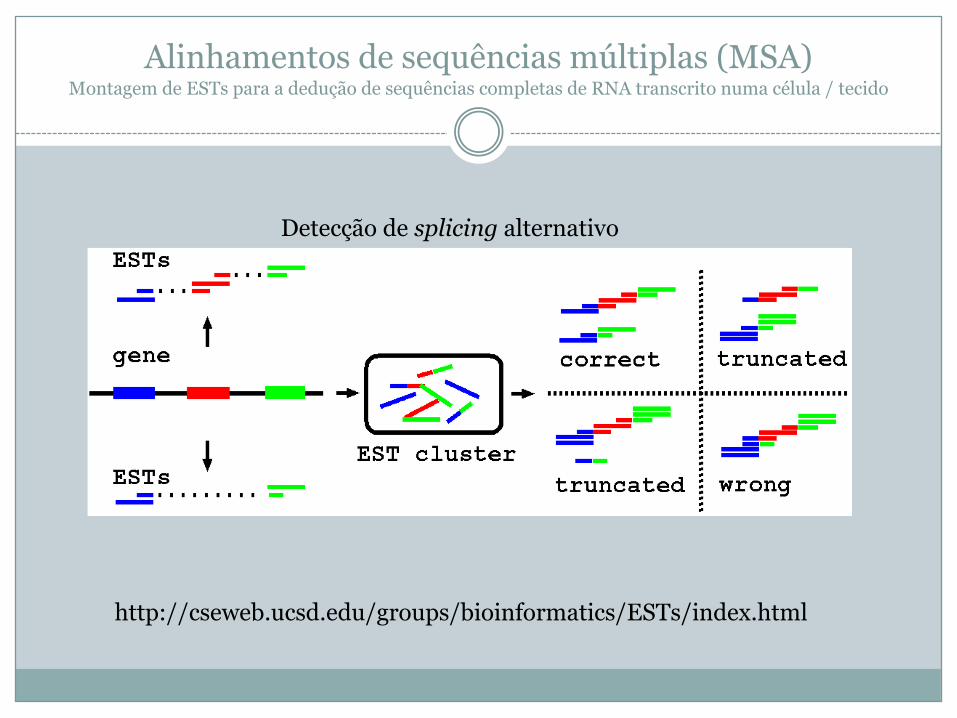
Alinhamentos de sequências múltiplas (MSA)Montagem de ESTs para a dedução de sequências completas de RNA transcrito numa célula / tecido
http://cseweb.ucsd.edu/groups/bioinformatics/ESTs/index.html
Detecção de splicing alternativo

Esquema de anotação Annothaton
1. ORFs? Coordenadas? Quadro de leitura? Cadeia + ou -? (SMS ORF Finder)
2. Existem proteínas homólogas? (BLASTp, BLASTx)3. Existem domínios funcionais? Onde? (InterProScan)4. Qual o tamanho / massa molecular da proteína codificada
pela ORF em aa / kDa?5. A que organismo ou táxon pertencerá a sequência
metagenómica? (BLAST Taxonomy Report)6. Relações filogenéticas 7. Conclusões e Análise de Resultados (PONTO PRINCIPAL DA
AVALIAÇÃO)

BLAST Taxonomy Reports

Lineage Report

A importância do Organism Report
Valor EScore

Critérios de decisão de taxonomia da fontebiológica de sequências metagenómicas
� Escolher o táxon que tenha valores E e scores com diferenças significativas com os restantes taxa
� Caso haja apenas taxa com valores E e scores muitopróximos (não significativos) essa sequência não édiagnosticante para esse táxon; por isso escolher um táxon mais abrangente

Esquema de anotação Annothaton
1. ORFs? Coordenadas? Quadro de leitura? Cadeia + ou -? (SMS ORF Finder)
2. Existem proteínas homólogas? (BLASTp, BLASTx)3. Existem domínios funcionais? Onde? (InterProScan)4. Qual o tamanho / massa molecular da proteína codificada
pela ORF em aa / kDa?5. A que organismo ou táxon pertencerá a sequência
metagenómica? (BLAST Taxonomy Report)6. Relações filogenéticas 7. Conclusões e Análise de Resultados (PONTO PRINCIPAL DA
AVALIAÇÃO)

Taxonomia e Ontologia Molecular
� Taxonomia (Annotathon)� Ontologia Génica (GO)� Conclusões (Annotathon)

Taxonomia
� Uma das funções das anotações (meta)genómicas é a definição do táxon da fonte de material genético

Tipos de Classificações
� Classificação hierárquica (de Lineu)� Classificação cladística

Classificação Hierárquica de Sistemas Biológicos
� Domínio� Superreino, Reino, Subreino� Superfilo, Filo (≈ divisão), Subfilo� (Superclasse,) Classe, Subclasse� Superordem, Ordem, Subordem (, Infraordem)� (Superfamília, Epifamília,) Família, Subfamília (, Tribo,
Subtribo, Infratribo)� Género, Subgénero� Espécie, Subespécie� Estirpe (≈ variedade)
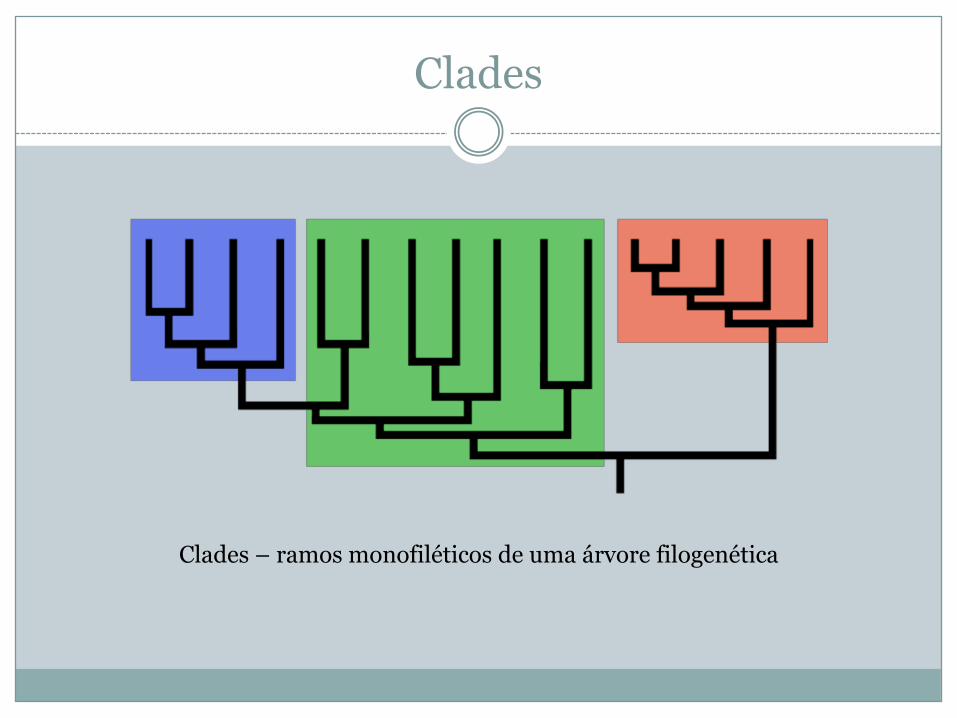
Clades
Clades – ramos monofiléticos de uma árvore filogenética

Monofilia, Parafilia e Polifilia
� Grupo monofilético – grupo taxonómico que provém de um ancestral comum cujos descendentes se encontram incluídos neste grupo (≈ grupo holofilético)
� Grupo parafilético – grupo taxonómico que provém de um ascendente comum cujos descendentes se encontram parcialmente incluídos neste grupo
� Grupo polifilético – grupo taxonómico que provém de mais que um ascendente

Monofilia, Parafilia e Polifilia
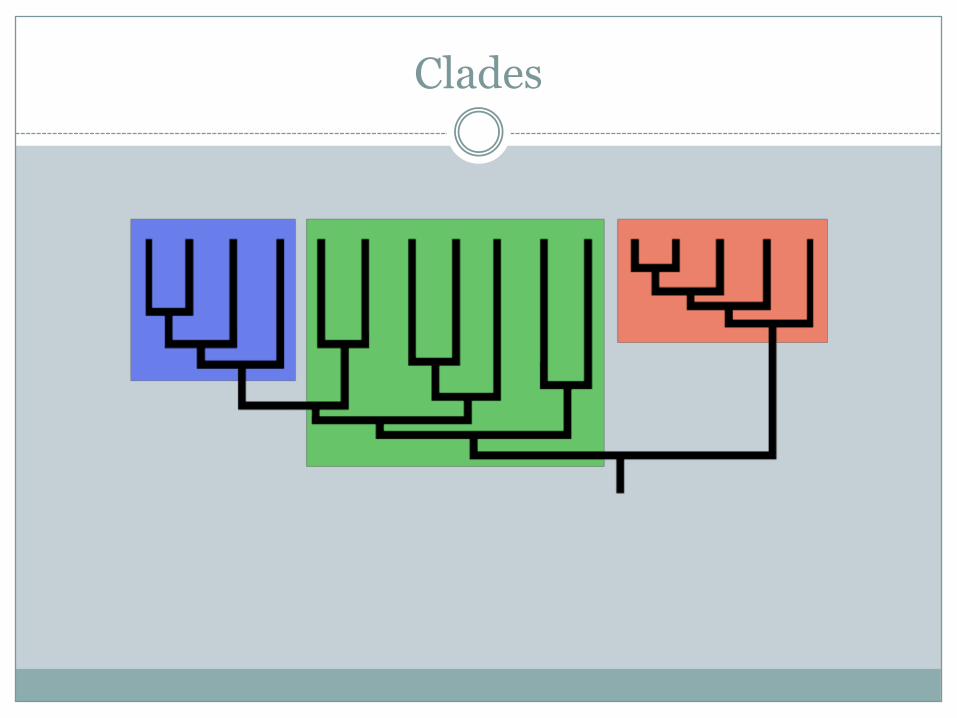
Clades

NCBI Taxonomy Database
� Cada táxon tem um nº de identificação - NCBI numerical identifier ou Taxonomy ID

Filogenia
� http://www.ncbi.nlm.nih.gov/Taxonomy/
� Pesquisa por táxon ou identificador numérico
� Colocar o identificador numérico no campo Taxonomy para o táxon menos abrangente ao qual a sequênciadeverá pertencer com uma elevada probabilidade (analisarvalores E do BLAST e o nó imediatamente superior do ramo à qualpertence a sequência em estudo das árvores filogenéticas obtidas)
� Raramente se consegue identificar até à espécie a fontebiológica de sequências metagenómicas

Determinação da fonte biológica de uma sequência metagenómica
� Os resultados do Taxonomy Report do BLAST não são indicadores suficientes para a fonte biológica de uma sequência metagenómica
� Nem sempre os melhores resultados do BLAST correspondem ao táxon indicado pela análise filogenética
� Os resultados do BLAST e da análise filogenética devem ser congruentes e significativos para a atribuição da fonte biológica ser credível

Determinação da fonte biológica de uma sequência metagenómica
� Os resultados do Taxonomy Report do BLAST não são indicadores suficientes para a fonte biológica de uma sequência metagenómica
� Nem sempre os melhores resultados do BLAST correspondem ao táxon indicado pela análise filogenética
� Os resultados do BLAST e da análise filogenética devem ser congruentes e significativos para a atribuição da fonte biológica ser credível

Fonte biológica de uma sequência metagenómica
� A fonte biológica de uma sequência metagenómica corresponde ao táxon que terá maior probabilidade de ser o dador da sequência nucleotídica em estudo
� A classificação taxonómica da fonte biológica nem sempre pode ser realizado até à espécie
� A classificação taxonómica da fonte biológica deve corresponder ao táxon menos abrangente do qual se tenha fortes indicações através da análise filogenética e da análise dos resultados do BLAST

Critérios de atribuição taxonómica
� Se o melhor resultado ter uma identidade > 98% e valores E claramente diferente do 2º melhor resultado que aponte para um táxon diferente do 1º, muito provavelmente esse será o táxon da fonte biológica
Function [species] Identity E valueGlycerol 3-P dehydrogenase [species x] 99 % 0.0Glycerol 3-P dehydrogenase [species y] 74% 1o-74
Glycerol 3-P dehydrogenase [species z] 65% 1o-46
Function [species] Identity E valueGlycerol 3-P dehydrogenase [species x] 99 % 1o-106
Glycerol 3-P dehydrogenase [species y] 98% 1o-105
Glycerol 3-P dehydrogenase [species z] 94% 1o-103
✔
?

Critérios de atribuição taxonómica
� Se o melhor resultado ter uma identidade << 98% e valor E claramente diferente do 2º melhor resultado que aponte para um táxon diferente do 1º, a espécie x poderá ser aparentada com a da fonte biológica ou poderemos a estar lidar com sequências parálogas à nossa sequência (query)
Function [species] Identity E valueGlycerol 3-P dehydrogenase [species x] 75 % 10-86
Glycerol 3-P dehydrogenase [species y] 55% 1o-74
Glycerol 3-P dehydrogenase [species z] 25% 1o-46?

Filogenia e taxonomia molecular
� A análise filogenética serve para apoiar ou não conclusões tiradas com resultados do BLAST em relação à atribuição da fonte biológica mais provável da sequência query
� A análise filogenética dá-nos também informação sobre as relações evolutivas entre sequências
� A análise filogenética é uma ferramenta essencial em taxonomia molecular (atribuição de classificação taxonómica de um ou mais organismos dadores de DNA por métodos moleculares)

Filogenia
� A análise filogenética serve para apoiar ou não conclusões tiradas com resultados do BLAST em relação à atribuição da fonte biológica mais provável da sequência query
� A análise filogenética dá-nos também informação sobre as relações evolutivas entre sequências
� A análise filogenética é uma ferramenta essencial em taxonomia molecular (atribuição de classificação taxonómica de um ou mais organismos dadores de DNA por métodos moleculares)
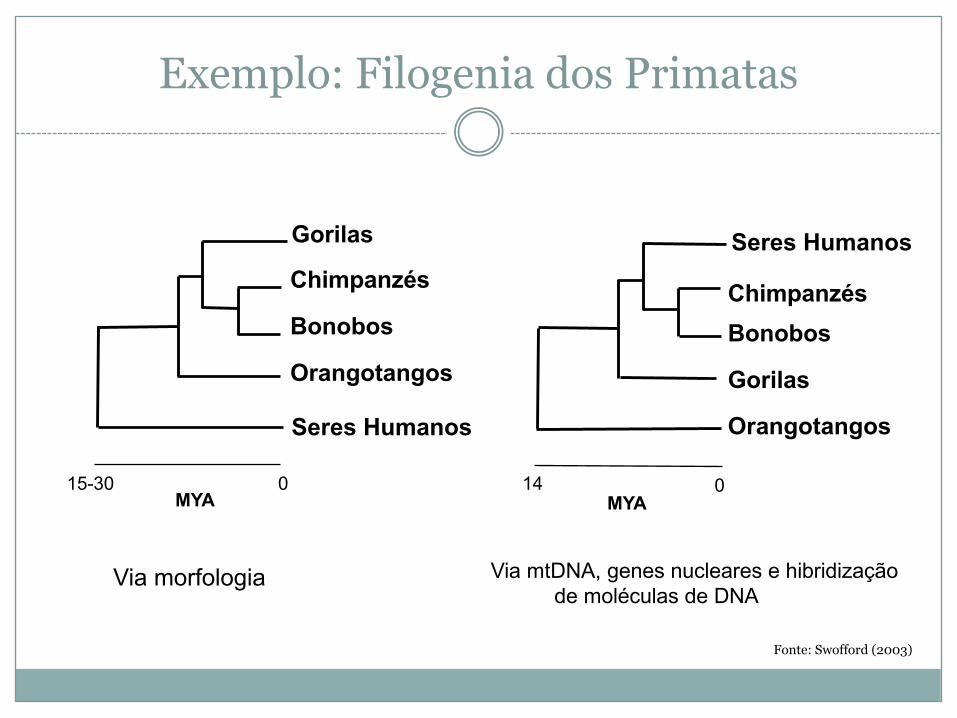
Exemplo: Filogenia dos Primatas
15-30
Seres Humanos
Gorilas
Chimpanzés
Bonobos
Orangotangos
MYA0
Via morfologia
MYA
Seres Humanos
ChimpanzésBonobos
Gorilas
Orangotangos
014
Via mtDNA, genes nucleares e hibridizaçãode moléculas de DNA
Fonte: Swofford (2003)
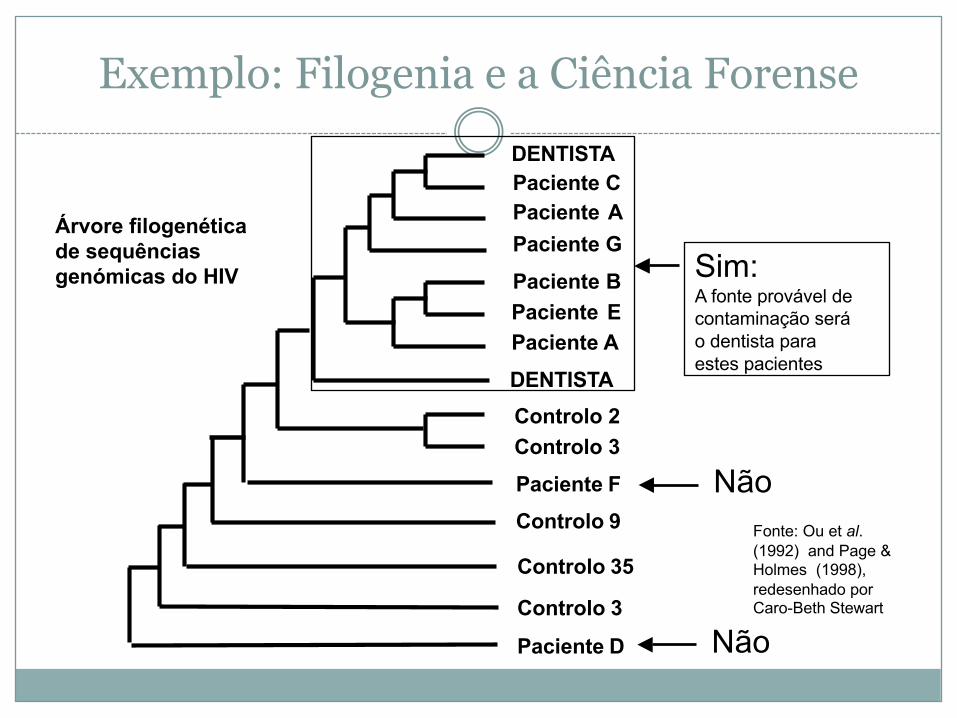
Exemplo: Filogenia e a Ciência Forense
Controlo 2Controlo 3
Controlo 9
Controlo 35
Controlo 3
Não
NãoFonte: Ou et al.(1992) and Page & Holmes (1998), redesenhado porCaro-Beth Stewart
Sim:A fonte provável de contaminação será o dentista para estes pacientes
Paciente D
Paciente F
DENTISTAPaciente CPaciente APaciente G
Paciente BPaciente EPaciente A
DENTISTA
Árvore filogenética de sequências genómicas do HIV

Filogenia melhora a atribuição de função
Eisen et al. (1998)
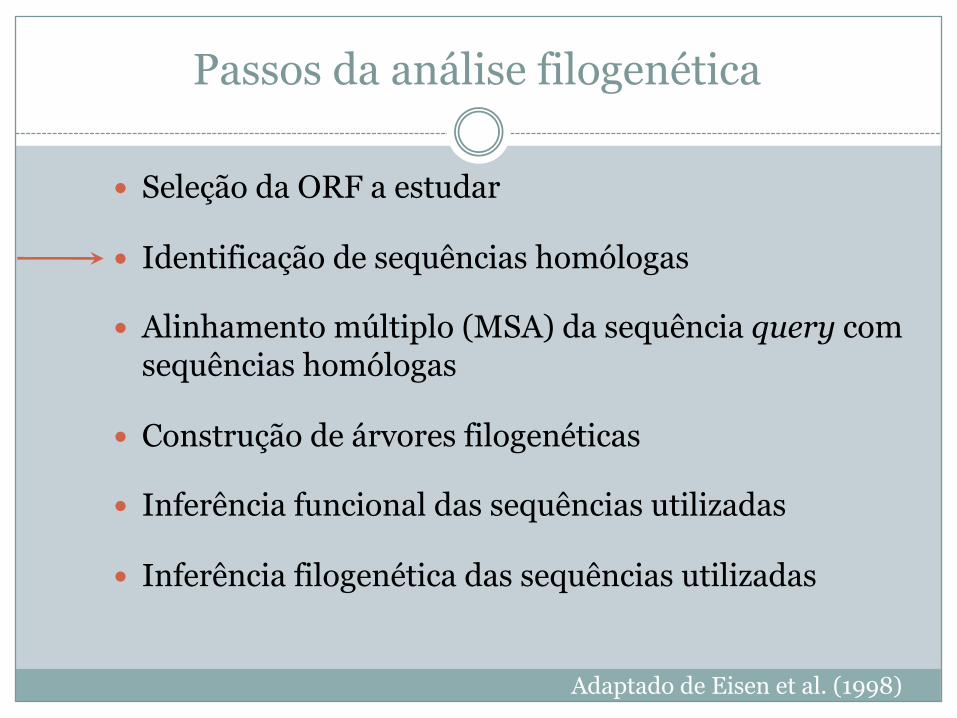
Passos da análise filogenética
� Seleção da ORF a estudar
� Identificação de sequências homólogas
� Alinhamento múltiplo (MSA) da sequência query com sequências homólogas
� Construção de árvores filogenéticas
� Inferência funcional das sequências utilizadas
� Inferência filogenética das sequências utilizadas
Adaptado de Eisen et al. (1998)
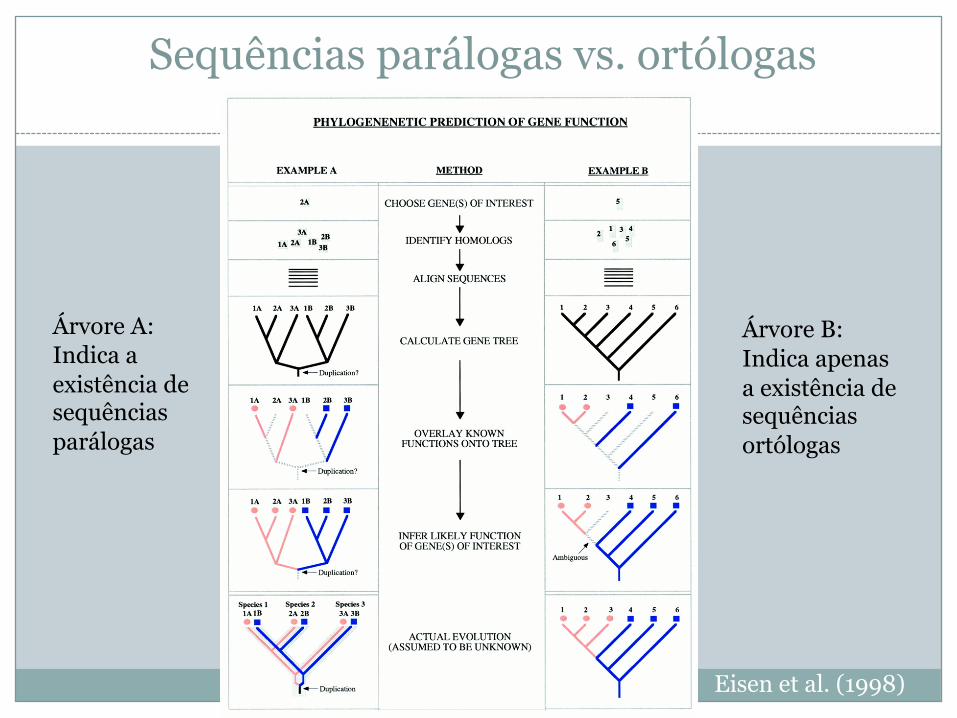
Sequências parálogas vs. ortólogas
Eisen et al. (1998)
Árvore A:Indica a existência de sequências parálogas
Árvore B:Indica apenas a existência de sequências ortólogas

Semelhança vs. Homologia
� Sequências muitos semelhantes entre si são geralmente homólogas (i.e., têm um ancestral comum) e têm a mesma função
� Sequências semelhantes podem ter a mesma função, mas não ser homólogas se a semelhança resultar de evolução convergente
� Árvores filogenéticas podem indicar a presença de sequências parálogas caso haja sequências da mesma espécie com scores e valores E significativamente diferentes
Adaptado de Eisen et al. (1998)
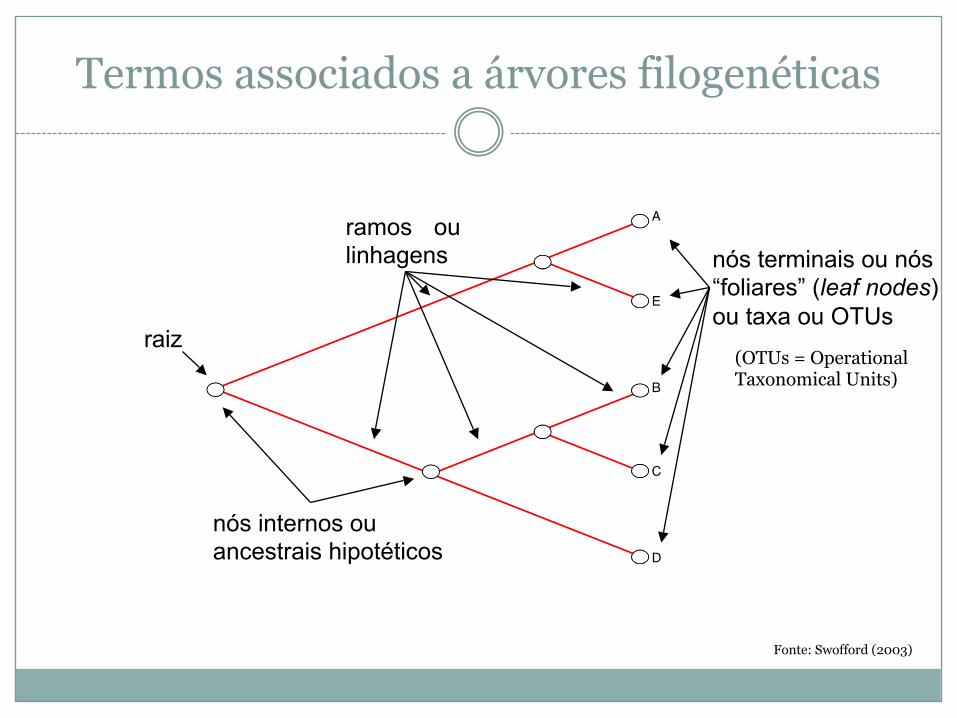
Termos associados a árvores filogenéticas
A
E
B
C
D
ramos oulinhagens
nós internos ouancestrais hipotéticos
raiz
nós terminais ou nós “foliares” (leaf nodes) ou taxa ou OTUs
Fonte: Swofford (2003)
(OTUs = OperationalTaxonomical Units)
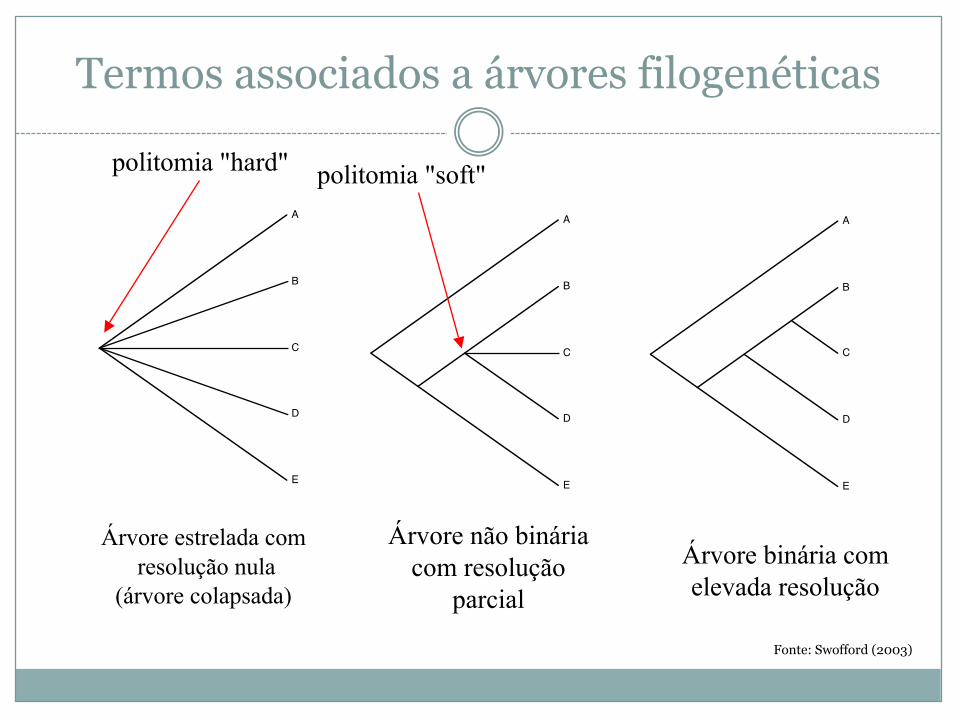
Termos associados a árvores filogenéticas
A
B
C
D
E
Árvore binária com elevada resolução
A
B
C
D
E
politomia "hard"
A
B
C
D
E
Árvore não bináriacom resolução
parcial
politomia "soft"
Árvore estrelada comresolução nula
(árvore colapsada)
Fonte: Swofford (2003)
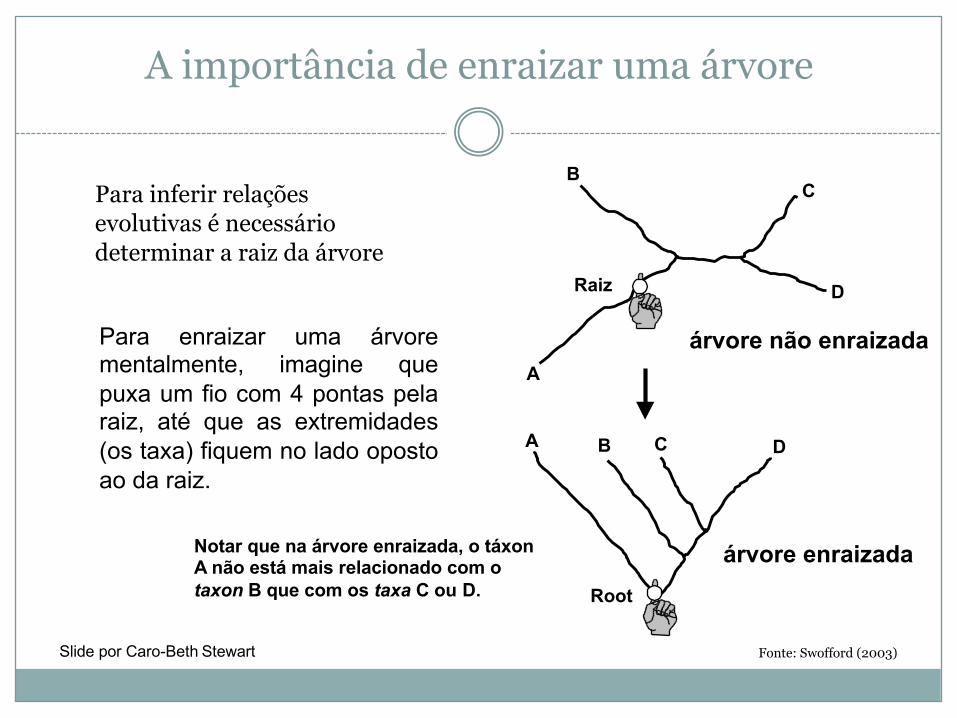
A importância de enraizar uma árvore
Fonte: Swofford (2003)
Para enraizar uma árvorementalmente, imagine quepuxa um fio com 4 pontas pelaraiz, até que as extremidades(os taxa) fiquem no lado opostoao da raiz.
A
C
Raiz D
A B C D
Root
Notar que na árvore enraizada, o táxonA não está mais relacionado com otaxon B que com os taxa C ou D.
árvore enraizada
árvore não enraizada
Slide por Caro-Beth Stewart
BPara inferir relações evolutivas é necessário determinar a raiz da árvore
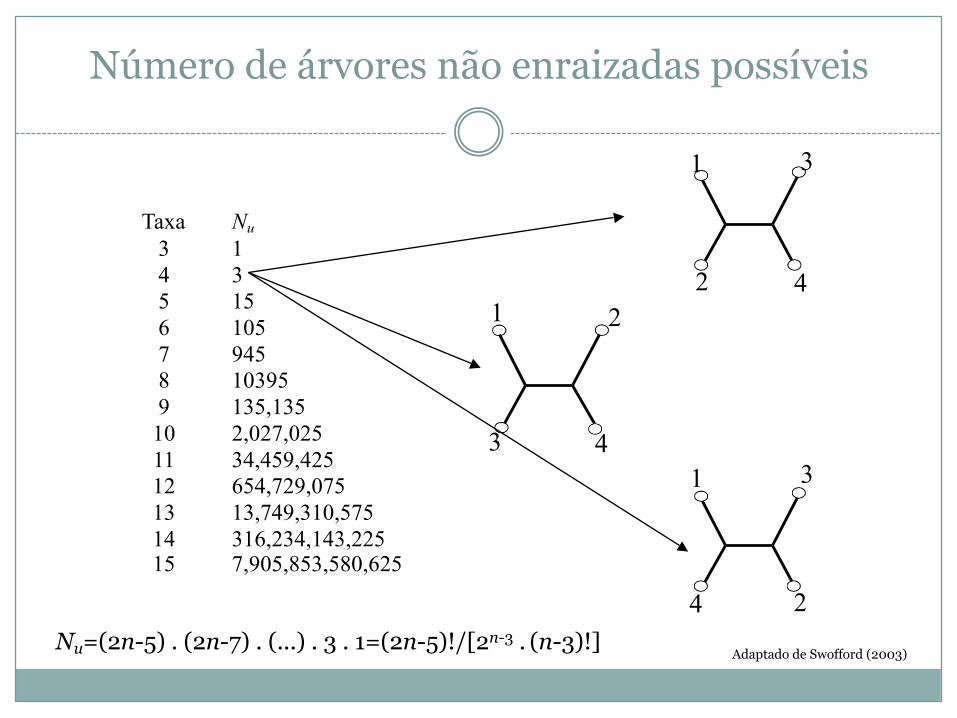
Número de árvores não enraizadas possíveis
Taxa Nu3 14 35 156 1057 9458 103959 135,13510 2,027,02511 34,459,42512 654,729,07513 13,749,310,57514 316,234,143,22515 7,905,853,580,625
1 3
421
3 4
2
1 3
4 2Nu=(2n-5) . (2n-7) . (...) . 3 . 1=(2n-5)!/[2n-3 . (n-3)!]
Adaptado de Swofford (2003)
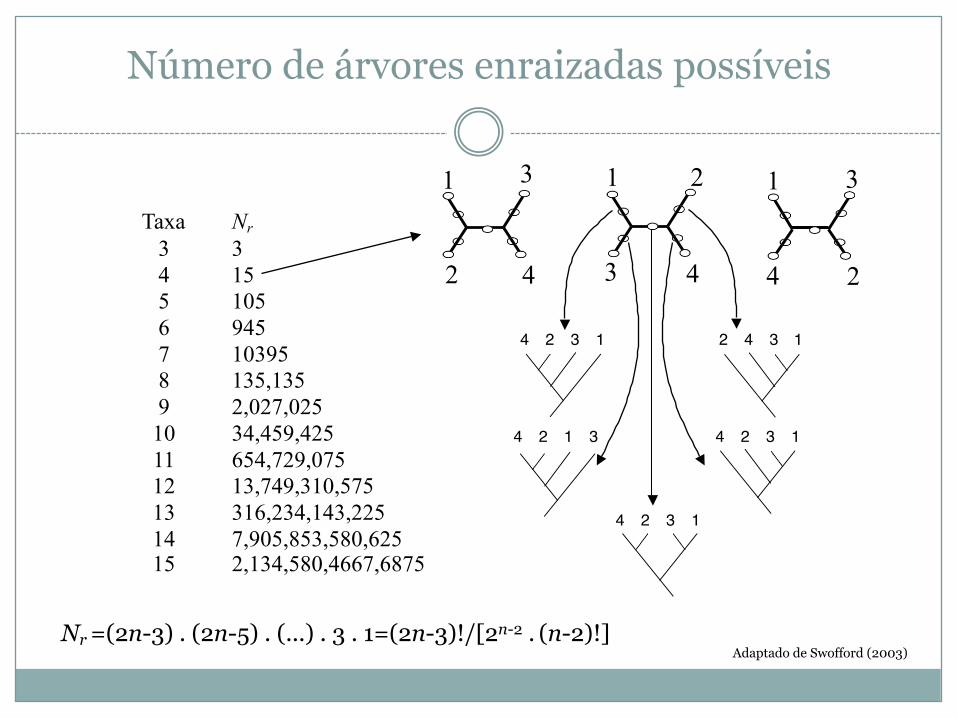
Número de árvores enraizadas possíveis
Taxa Nr3 34 155 1056 9457 103958 135,1359 2,027,02510 34,459,42511 654,729,07512 13,749,310,57513 316,234,143,22514 7,905,853,580,62515 2,134,580,4667,6875
Nr=(2n-3) . (2n-5) . (...) . 3 . 1=(2n-3)!/[2n-2 . (n-2)!]Adaptado de Swofford (2003)
1 3 1 2 1 3
4 2 3 1
2 4 3 4 4 2
4 2 3 1 2 4 3 1
4 2 1 3 4 2 3 1
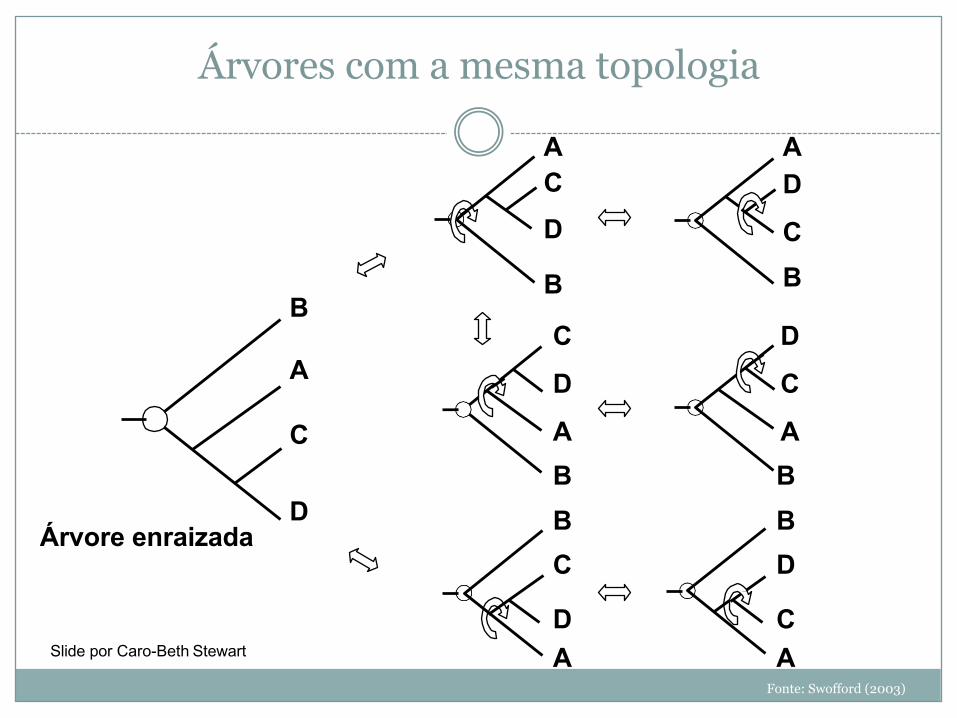
Árvores com a mesma topologia
Fonte: Swofford (2003)
B
A
C
D
C
D
ABBC
DA
B
A CD
Árvore enraizada
A D
C B
D
C
ABBD
CASlide por Caro-Beth Stewart

Como enraizar uma árvore
Fonte: Swofford (2003)
Por definição de “outgroup”:Definido por um táxon ou taxa exterior aogrupo em estudo (“ingroup”), mas requerum conhecimento taxonómico prévio (usadono Annotathon). Pode ser também umafamília molecular (não usado noAnnotathon).
Por ponto de meia-distância (midpoint):A raiz é o ponto de meia-distância entre ostaxa mais distintos da árvore e édeterminado pelo tamanho dos ramos.Porém, assume que a evolução molecular éigual para todos os ramos (i.e., assume aexistência de um ”relógio molecular”).
Adaptado de um slide por Caro-Beth Stewart
Existem duas maneiras:
A
B
C
D
10
2
3
5
2
outgroup
d (A,D) = 10 + 3 + 5 = 18Midpoint = 18 / 2 = 9

Métodos de inferência filogenética: caracteres
Fonte: Swofford (2003) Adaptado de um slide por Caro-Beth Stewart
Métodos de caracteres: Alinhamento de caracteres, nucleótidos ou aminoácidos, para construir uma árvore e inferir filogenia:
Taxa CaracteresSpecies A ATGGCTATTCTTATAGTACGSpecies B ATCGCTAGTCTTATATTACASpecies C TTCACTAGACCTGTGGTCCASpecies D TTGACCAGACCTGTGGTCCGSpecies E TTGACCAGTTCTCTAGTTCG

Métodos de inferência filogenética: distância
Fonte: Swofford (2003) Adaptado de um slide por Caro-Beth Stewart
Métodos de distância: Transforma as dissemelhanças entre sequências em distâncias, sendo utilizada a respetiva matriz para calcular a árvore.
A B C D ESpecies A ---- 0.20 0.50 0.45 0.40Species B 0.23 ---- 0.40 0.55 0.50Species C 0.87 0.59 ---- 0.15 0.40Species D 0.73 1.12 0.17 ---- 0.25Species E 0.59 0.89 0.61 0.31 ----
Distância não corrigida(= % de diferença entre sequências)
Parâmetro de distância Kimura 2(estimativa corrigida da verdadeira distância de substituições de caracteres (nt. ou aa.) entre taxa.
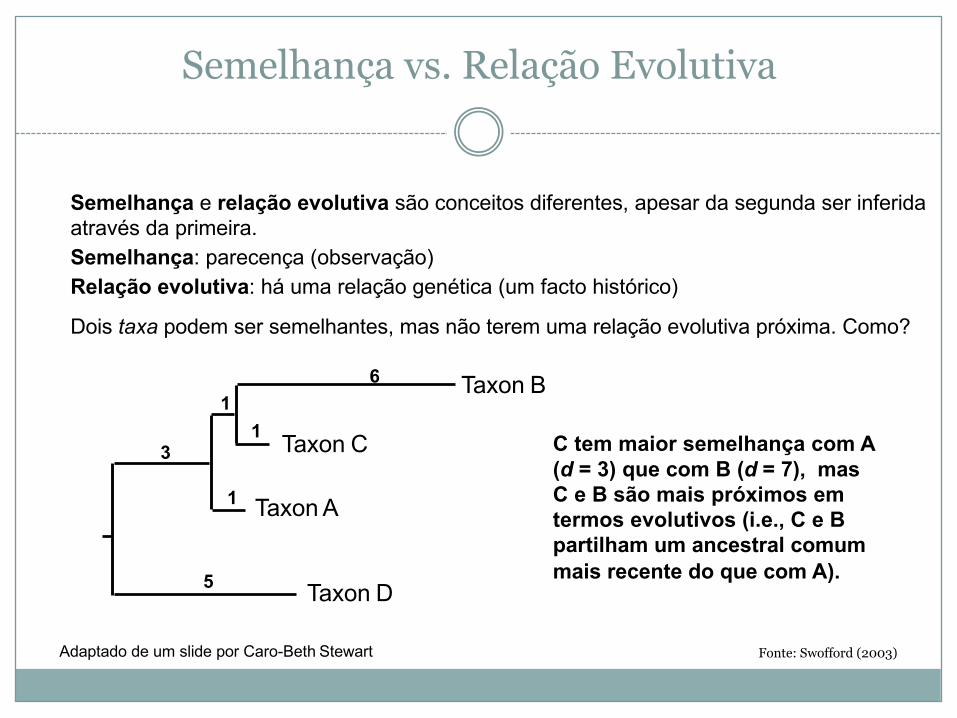
Semelhança vs. Relação Evolutiva
Fonte: Swofford (2003) Adaptado de um slide por Caro-Beth Stewart
Semelhança e relação evolutiva são conceitos diferentes, apesar da segunda ser inferida através da primeira.Semelhança: parecença (observação) Relação evolutiva: há uma relação genética (um facto histórico)
Dois taxa podem ser semelhantes, mas não terem uma relação evolutiva próxima. Como?
Taxon A
Taxon C
Taxon D
11
1
3
5
C tem maior semelhança com A (d = 3) que com B (d = 7), masC e B são mais próximos em termos evolutivos (i.e., C e Bpartilham um ancestral comum mais recente do que com A).
Taxon B6

Tipos de semelhanças: Simplesiomorfia e Sinapomorfia
Fonte: Swofford (2003) Adaptado de um slide por Caro-Beth Stewart
A semelhança observada entre dois taxa pode ser devido a:
Relação evolutiva:com caracteres ancestrais partilhados: simplesiomorfiacom caracteres ancestrais derivados: sinapomorfia
caracterancestral
caracterderivado
CCGG
Cplesiomorfia
apomorfia
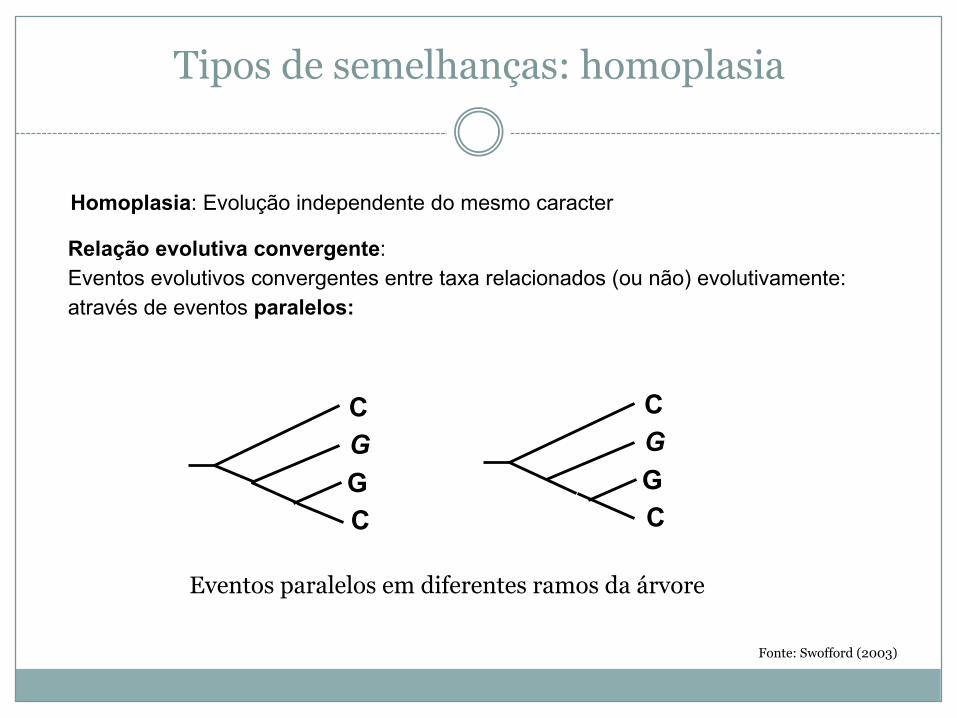
Tipos de semelhanças: homoplasia
Fonte: Swofford (2003)
Homoplasia: Evolução independente do mesmo caracter
Relação evolutiva convergente:Eventos evolutivos convergentes entre taxa relacionados (ou não) evolutivamente:através de eventos paralelos:
CGG C
CGG C
Eventos paralelos em diferentes ramos da árvore
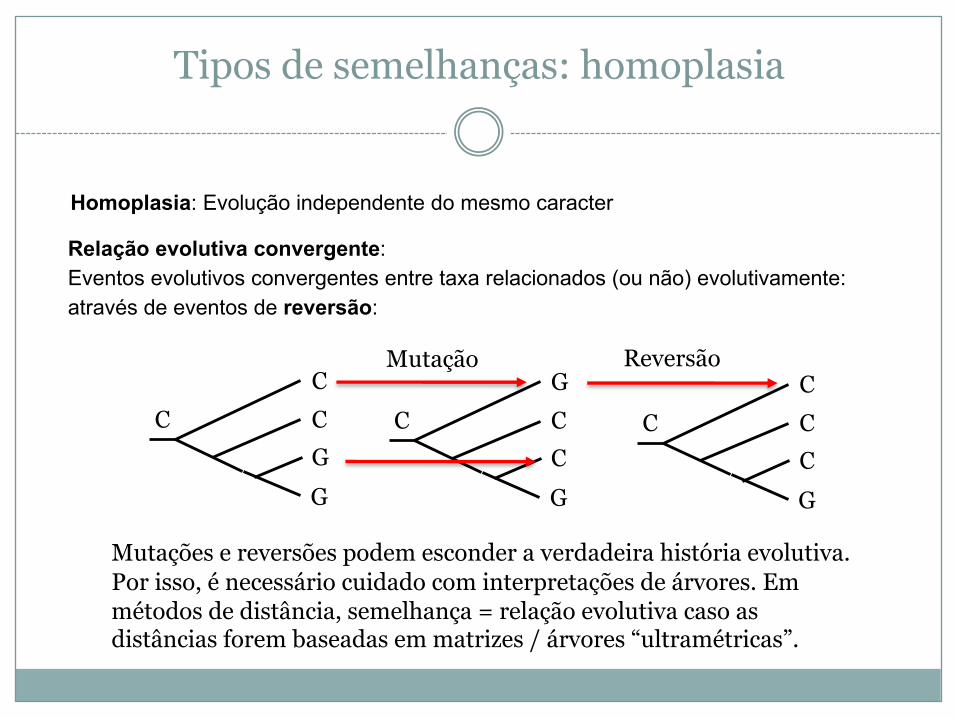
Tipos de semelhanças: homoplasia
Homoplasia: Evolução independente do mesmo caracter
Relação evolutiva convergente:Eventos evolutivos convergentes entre taxa relacionados (ou não) evolutivamente:através de eventos de reversão:
CCGG
CGCCG
CCCCG
C
ReversãoMutação
Mutações e reversões podem esconder a verdadeira história evolutiva. Por isso, é necessário cuidado com interpretações de árvores. Em métodos de distância, semelhança = relação evolutiva caso as distâncias forem baseadas em matrizes / árvores “ultramétricas”.

Distâncias / matrizes / Árvores Ultramétricas
� Satisfazem a hipótese do “relógio molecular”
� Todas as distâncias desde a raiz até aos nós foliares são iguais
� O tamanho dos ramos é proporcional ao tempo evolutivo
Isto é verdade para a maioria das árvores?
Recommended

















