
FDA Arthritis Advisory Committee (AAC)FDA Introductory Remarks
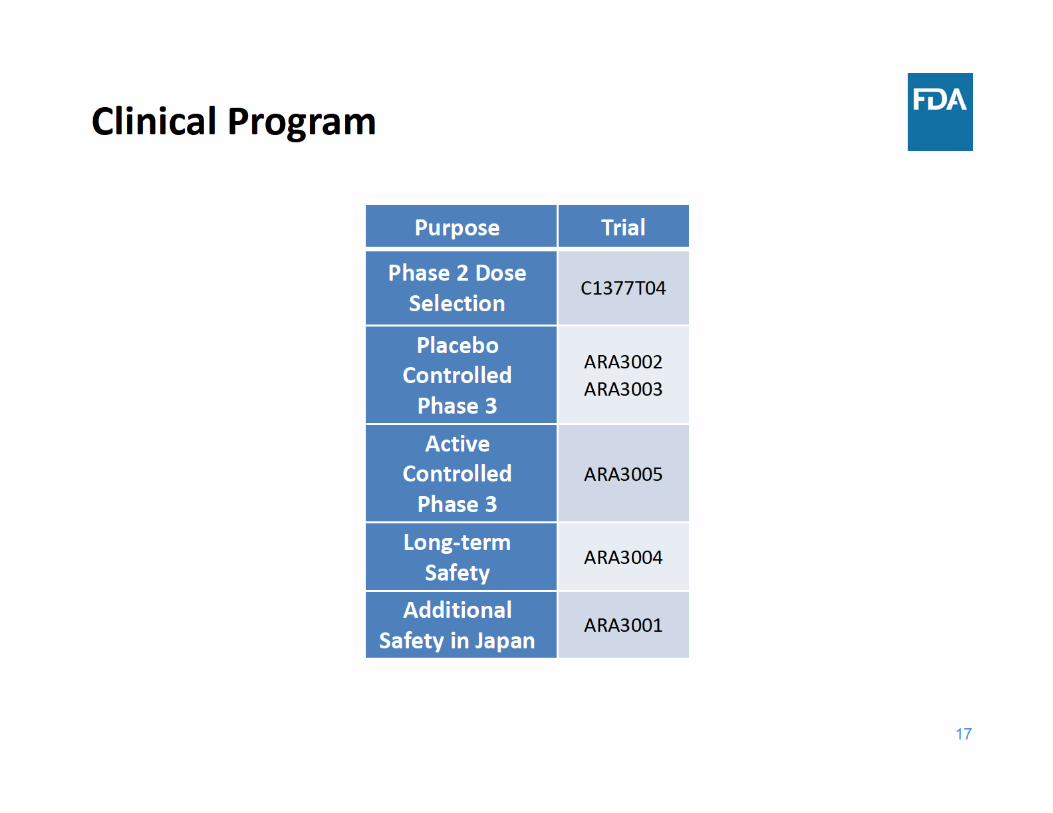
BLA 761057: Sirukumab for the treatment of adult patients with moderately to severely active rheumatoid
arthritis (RA) who have had an inadequate response or are intolerant to one or more disease modifying anti‐
rheumatic drugs (DMARDs)
Janet Maynard, MD, MHSClinical Team Leader
Division of Pulmonary, Allergy, and Rheumatology Products (DPARP)Center for Drug Evaluation and Research (CDER)
August 2, 2017

2
Rheumatoid Arthritis (RA): Overview
• Chronic, systemic, inflammatory disorder that primarily involves synovial joints
• Can result in permanent joint damage and disability• Multiple therapeutic options approved over the last 20 years

3
Overview
• Product: • Sirukumab (Plivensia®)
• Mechanism of action:• Human monoclonal antibody against interleukin (IL)‐6
• Proposed indication:• Treatment of adult patients with moderately to severely active
rheumatoid arthritis (RA) who have had an inadequate response or are intolerant to one or more disease modifying anti‐rheumatic drugs (DMARDs)
• Proposed dosage:– 50 mg subcutaneously every 4 weeks (q4w)
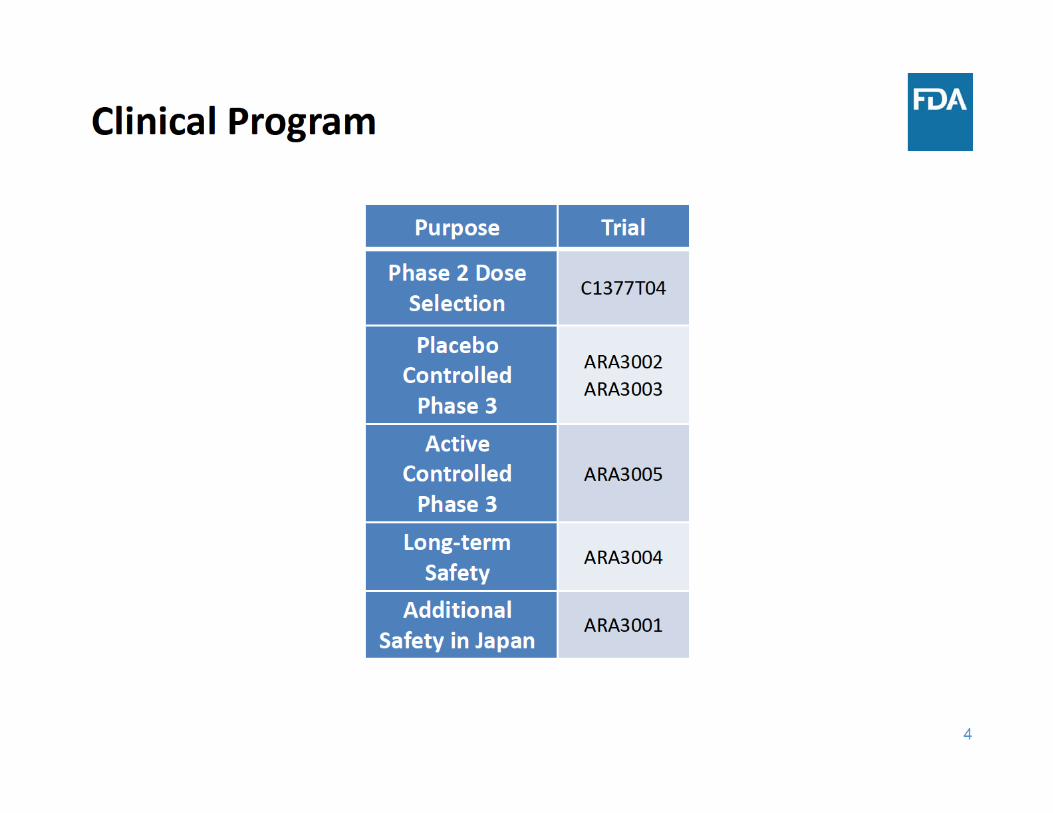
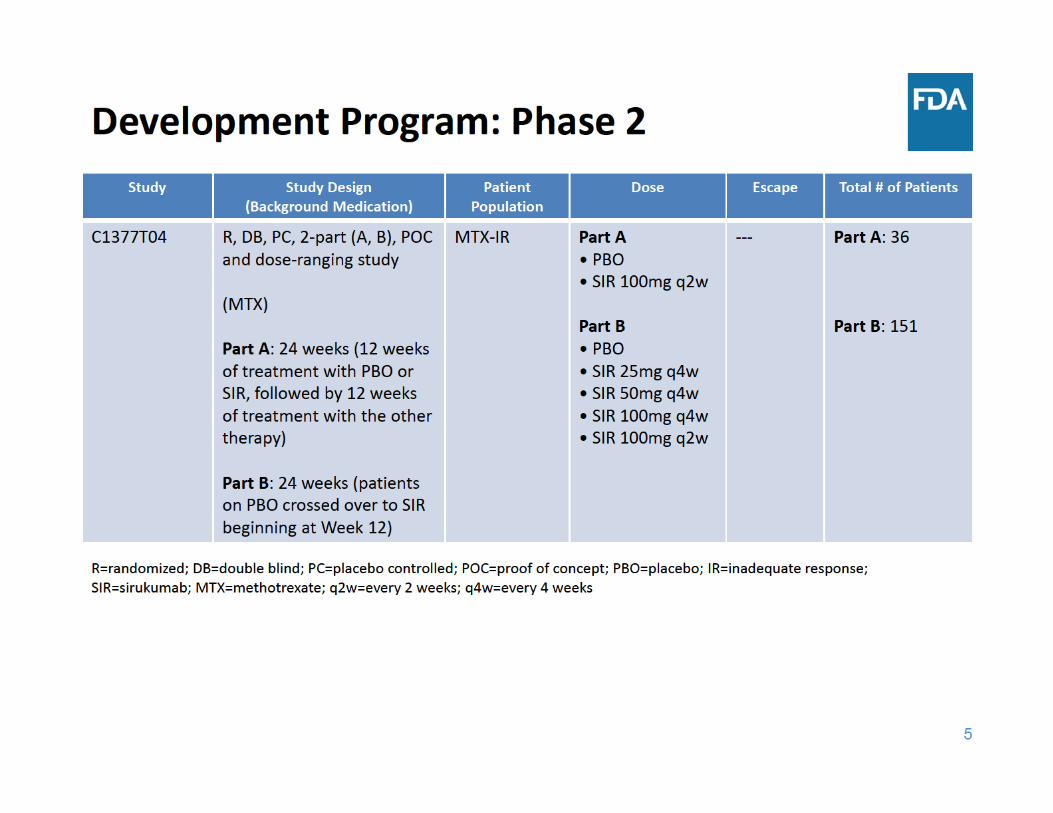
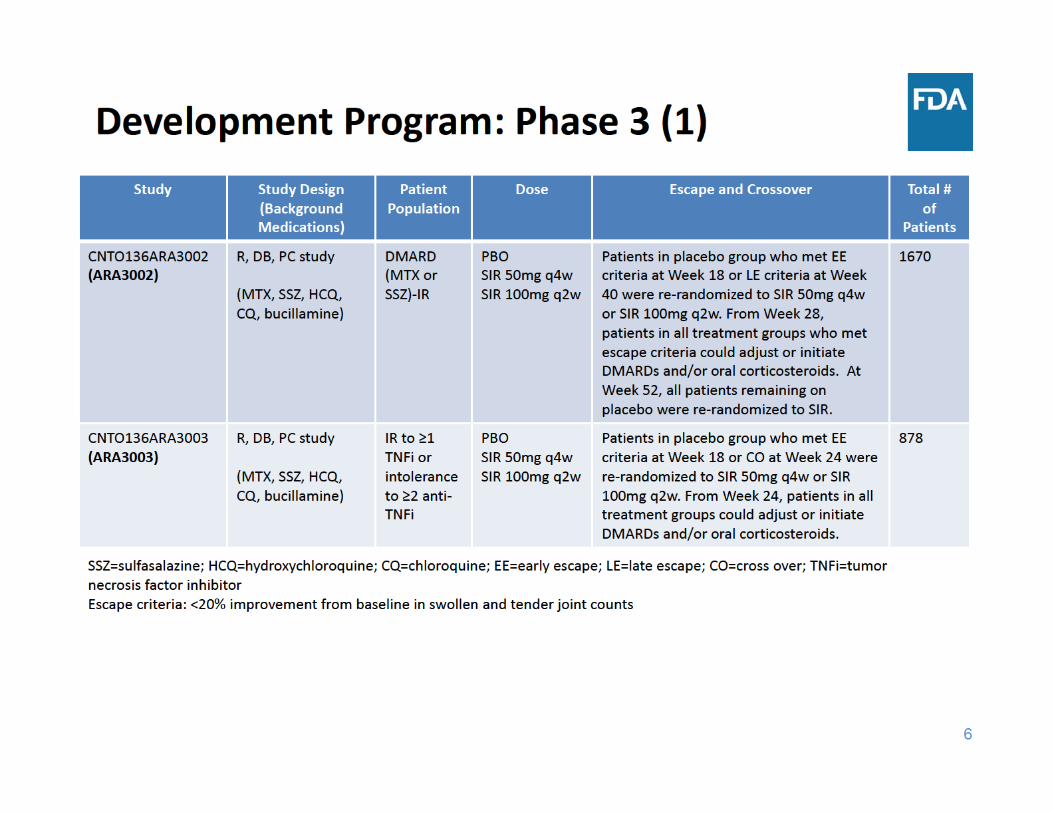
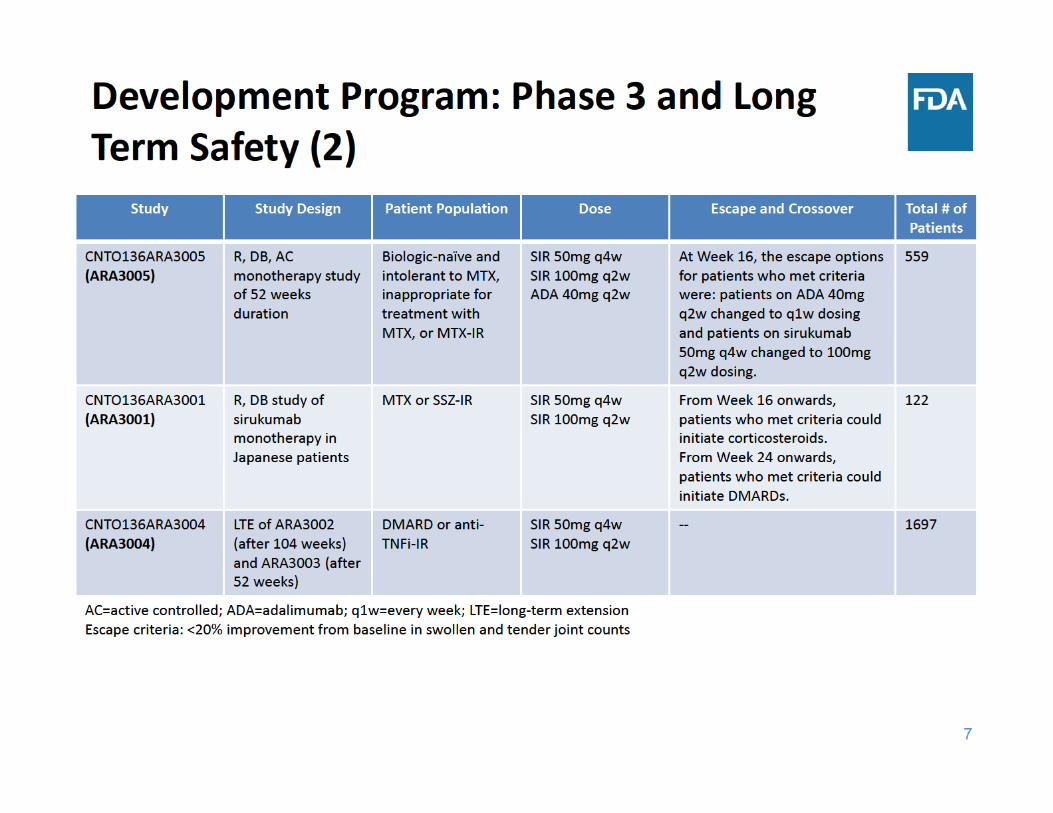

8
Efficacy Considerations
• Efficacy for signs and symptoms (ACR Responses, DAS28), physical function (HAQ‐DI), and radiographic outcomes
• Efficacy of 50 mg q4w and 100 mg q2w was similar• Sirukumab was not superior to adalimumab
ACR=American College of Rheumatology; DAS28=disease activity score 28; HAQ‐DI=Health Assessment Questionnaire‐Disability Index

9
Safety Considerations
• All‐cause death– Imbalance in all‐cause death with sirukumab over placebo– Rate of all‐cause death was similar with both sirukumab doses– Major causes of death include cardiovascular events, malignancy,
and infections
• Sirukumab associated with imbalances in serious adverse events and gastrointestinal (GI) perforation
• Laboratory abnormalities– Decreases in neutrophil and platelet counts – Elevations in lipid parameters and liver function tests

10
Issues for Consideration
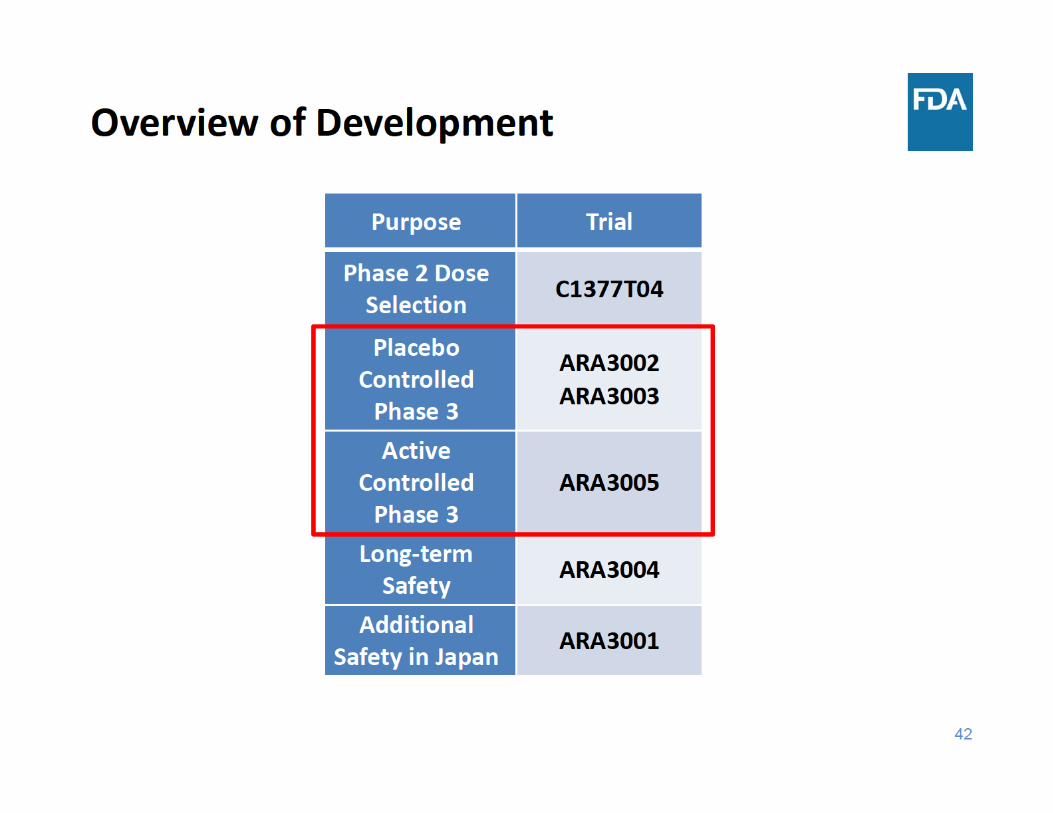
• Efficacy of sirukumab for adults with RA • Design of 52‐week placebo‐controlled radiographic study, ARA3002
• Safety findings, with particular consideration of the imbalance in all‐cause death between sirukumab and placebo
• Dose selection for phase 3 • Overall risk/benefit of sirukumab for adults with moderately to severely active RA

11
Purpose of Proceedings Before an Advisory Committee (21 CFR 14.5)
a. An advisory committee is utilized to conduct public hearing on matters of importance that come before FDA, to review the issues involved, and to provide advice and recommendations to the Commissioner
b. The Commissioner has sole discretion concerning action to be taken and policy to be expressed on any matter considered by an advisory committee
CFR=Code of Federal Regulations


FDA Arthritis Advisory Committee (AAC)Introduction and Clinical Overview
BLA 761057: Sirukumab for the treatment of adult patients with moderately to severely active rheumatoid
arthritis (RA) who have had an inadequate response or are intolerant to one or more disease modifying anti‐
rheumatic drugs (DMARDs)
Mark Borigini, MDMedical Officer
Division of Pulmonary, Allergy, and Rheumatology ProductsCenter for Drug Evaluation and Research
August 2, 2017

14
Overview of FDA Presentations
• Introduction and Clinical Overview– Mark Borigini, MD
• Dose Selection Considerations: Phase 2 Study Results– Dipak Pisal, PhD
• Review of Efficacy– William Koh, PhD
• Review of Safety and Risk/Benefit Considerations – Mark Borigini, MD

15
Overview of FDA Presentations
• Introduction and Clinical Overview– Mark Borigini, MD
• Dose Selection Considerations: Phase 2 Study Results– Dipak Pisal, PhD
• Review of Efficacy– William Koh, PhD
• Review of Safety and Risk/Benefit Considerations – Mark Borigini, MD

16
Overview
• Product: • Sirukumab (Plivensia®)
• Mechanism of action:• Human monoclonal antibody against interleukin (IL)‐6
• Proposed indication:• Treatment of adult patients with moderately to severely active
rheumatoid arthritis (RA) who have had an inadequate response or are intolerant to one or more disease modifying anti‐rheumatic drugs (DMARDs)
• Proposed dosage:– 50 mg subcutaneously every 4 weeks

18
Issues for Consideration
• Efficacy of sirukumab for adults with RA • Design of 52‐week placebo‐controlled radiographic study, ARA3002
• Safety findings, with particular consideration of the imbalance in all‐cause death between sirukumab and placebo
• Dose selection for phase 3 • Overall risk/benefit of sirukumab for adults with moderately to severely active RA

19
Evaluation of Radiographic Progression: Study Design Considerations
• Duration • Comparator • Escape

20
Escape Options in ARA3002 and ARA3003
• Escape allowed at multiple time points – Week 18 (ARA3002 and ARA3003)
• Re‐randomized to sirukumab (50 mg q4w or 100 mg q2w)
– Week 40 (ARA3002)• Re‐randomized to sirukumab (50 mg q4w or 100 mg q2w)
– From Week 28 (ARA3002) or Week 24 (ARA3003)• Initiation or up‐titration of DMARDs/oral corticosteroids
– Escape criteria: <20% improvement from baseline in swollen and tender joint count
Q2W=every two weeks; Q4W=every four weeks

21
Relevant Regulatory History (1)
• Pre‐IND (investigational new drug) meeting (March 27, 2008) – During drug development, need to develop evidence to support the choice of dose
• IND submitted (June 2008)
• End of Phase 2 (EOP2) meeting (April 5, 2011)– FDA noted concerns about the lack of efficacy data in support of the 50 mg q12w dose of sirukumab that Janssen proposed in their pivotal phase 3 trials
• Recommended either additional dose‐ranging to evaluate lower doses and/or alternative dosing intervals or choose another dose level for phase 3 utilizing a dose evaluated in phase 2 trials

22
Relevant Regulatory History (2)
• Advice/Information Request (October 12, 2012) – In proposed protocol for CNTO136ARA3002, patients randomized to
placebo could remain on placebo for up to 52 weeks• FDA noted ethical concern with respect to withholding treatment from
patients with uncontrolled disease activity for extended periods of time– Selected doses for phase 3 studies (100 mg q2w and 50mg q4w) were
acceptable and at the sponsor’s discretion, but concerns raised at EOP2 meeting were noted
• Advice/Information Request (November 21, 2012) – In follow‐up to the sponsor’s questions regarding whether the rescue
mechanisms were adequate for a 52‐week placebo control period in study CNTO136ARA3002, FDA responded that the protocol was generally acceptable
• Pre‐BLA meeting (May 18, 2016)– General agreement on content and format of BLA submission

23
Issues for Consideration
• Efficacy of sirukumab for adults with RA • Design of 52‐week placebo‐controlled radiographic study, ARA3002
• Dose selection for phase 3 • Safety findings, with particular consideration of the imbalance in all‐cause death between sirukumab and placebo
• Overall risk/benefit of sirukumab for adults with moderately to severely active RA

24
Overview of FDA Presentations
• Introduction and Clinical Overview– Mark Borigini, MD
• Dose Selection Considerations: Phase 2 Study Results– Dipak Pisal, PhD
• Review of Efficacy– William Koh, PhD
• Review of Safety and Risk/Benefit Considerations – Mark Borigini, MD

Dose Selection Considerations: Phase 2 Study Results
Dipak S. Pisal, PhDClinical Pharmacology Reviewer
Division of Clinical Pharmacology II Office of Clinical PharmacologyFood and Drug Administration
August 2, 2017

26
Outline• Dose Selection
• Phase 2 Study
• Dose Selection for Phase 3
• End of Phase 2 (EOP2) Meeting Communication
• Summary

27
Dose Selection
• The Agency’s expectation is that there will be adequate dose‐ranging in the clinical development program for Rheumatoid Arthritis (RA)
• Phase 3 Dose Selection:– PK, PD, Efficacy and Safety from dose‐ranging studies – Wide range of doses and dosing regimens– Efficacy endpoints that will be used in phase 3 studies. (e.g. ACR, DAS28)
– Dose related safety considerations (e.g. lipids, AST/ALT, hematology, etc. )
PK: pharmacokinetics, PD: pharmacodynamics; ACR 20:American College of Rheumatology score 20, DAS28: Disease Activity Score‐28, AST: aspartate aminotransferase; ALT: alanine aminotransferasehttps://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm354468.pdf

28
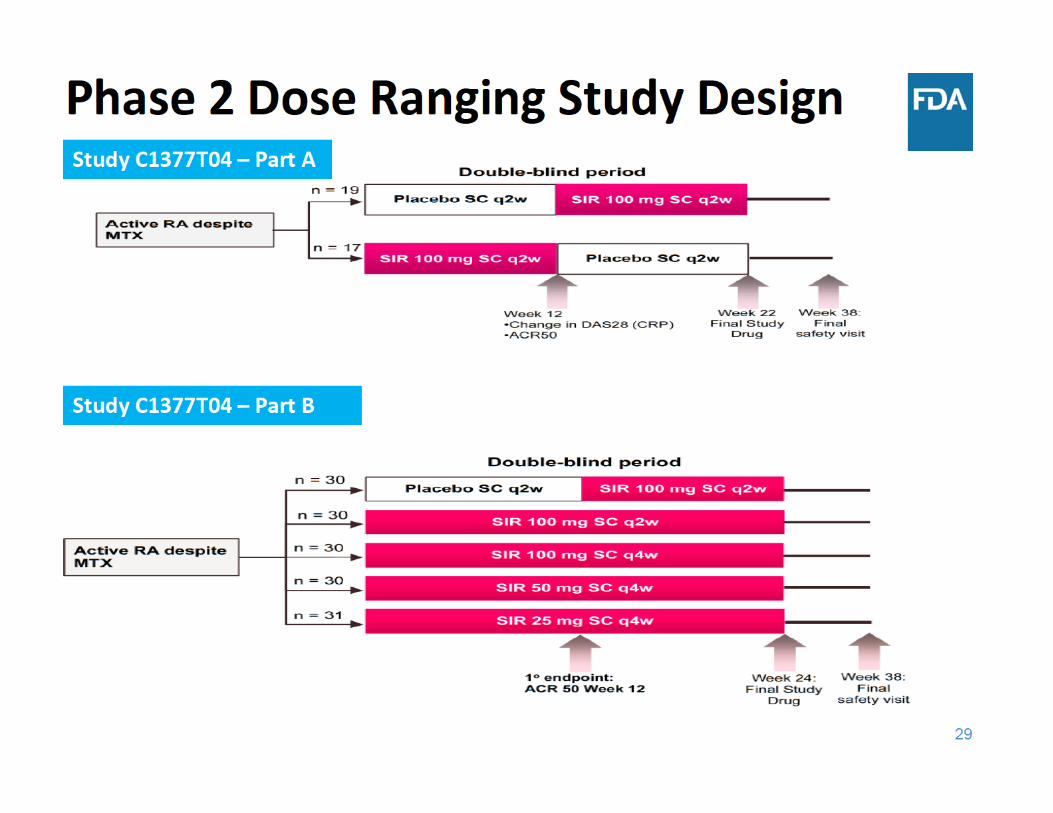
Dose Selection in Current Application
• The proposed dose is 50 mg q4w
• In the phase 3 program, doses of 50 mg q4w and 100 mg q2w were evaluated
31
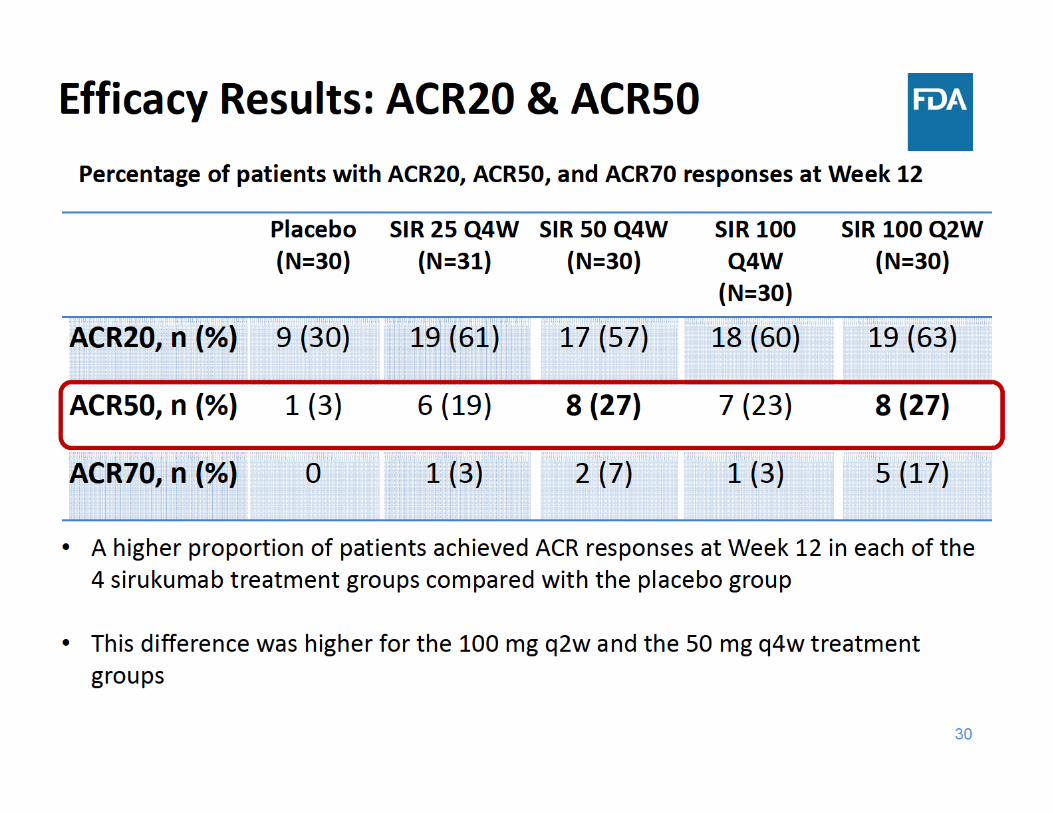
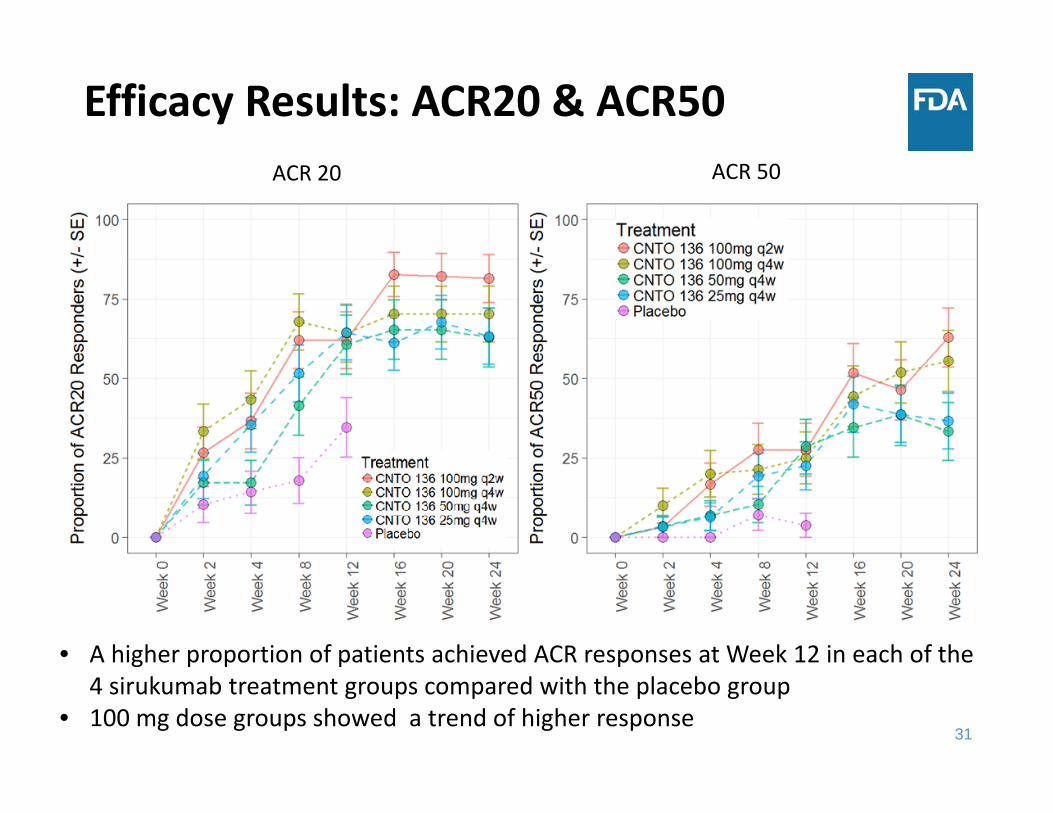
Efficacy Results: ACR20 & ACR50
• A higher proportion of patients achieved ACR responses at Week 12 in each of the 4 sirukumab treatment groups compared with the placebo group
• 100 mg dose groups showed a trend of higher response
ACR 20 ACR 50

34
Efficacy Summary• All sirukumab dose groups showed better response than placebo
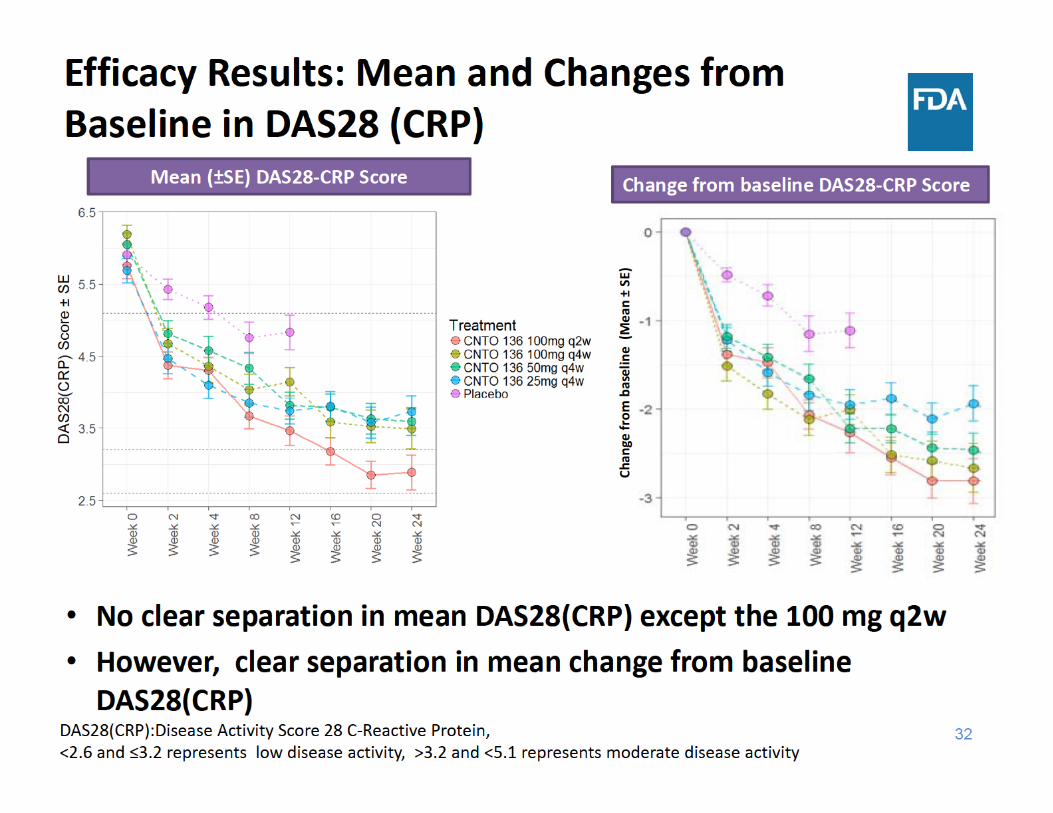
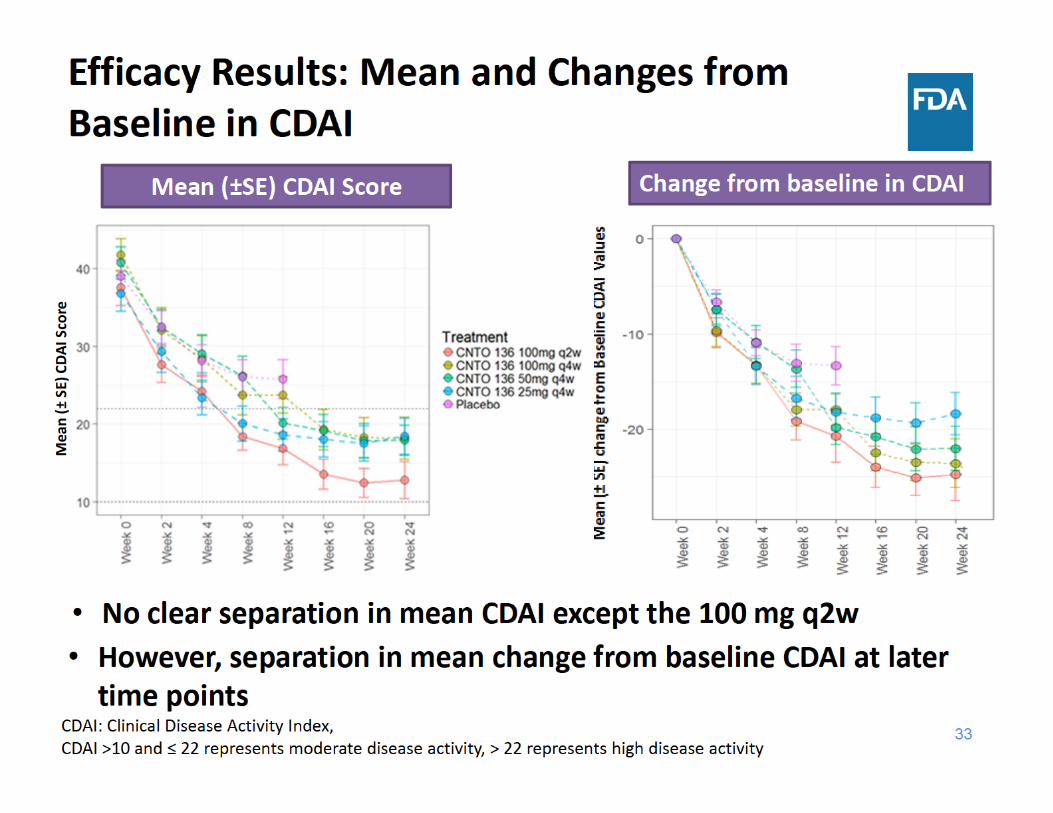
• There was a trend with higher efficacy with higher dose towards the later time points, although no clear dose response was observed up to week 12• ACR: 100 mg dose groups showed a trend of higher response • Separation in mean change from baseline DAS28(CRP) & CDAI • No clear separation in mean DAS28 (CRP) & CDAI except the 100 mg q2w
• Selection of 100 mg every two weeks & 50 mg every 4 weeks was reasonable
• It is a study with sample size (N= ~ 30) in each treatment group

36
Proposed Doses for Phase 3
• Janssen initially proposed following dosing regimens at the end of the phase 2 meeting:– Based on these efficacy, safety and exposure response modeling (serum level efficacy analysis, & PK/ACR20 E‐R model)
• 100 mg every 2 weeks • 50 mg every 4 weeks • 50 mg every 12 weeks
― It was not studied dose group in phase 2 study & lacked safety and efficacy data

37
EOP2 Meeting & Discussions
IND 101073 EOP2, April 5, 2011, IND 101073, October, 12, 2012
• FDA expressed concerns about the lack of efficacy data in support of the 50 mg Q12 week dose of sirukumab and recommended additional dose ranging study to evaluate lower doses and/or alternative dosing intervals or choose another dose level for the Phase 3 trials that utilizes a Q2 or Q4 week dosing regimen
Advice/Information Request (October 12, 2012)
• Selected doses for phase 3 studies (100 mg q2w and 50mg q4w) were acceptable and at the Janssen’s discretion, but concerns raised at EOP2 meeting were noted

38
Summary
• Efficacy:– All sirukumab dose groups showed better response than placebo
– There was a trend with higher efficacy with higher dose at later time points, No dose response was observed up to week 12
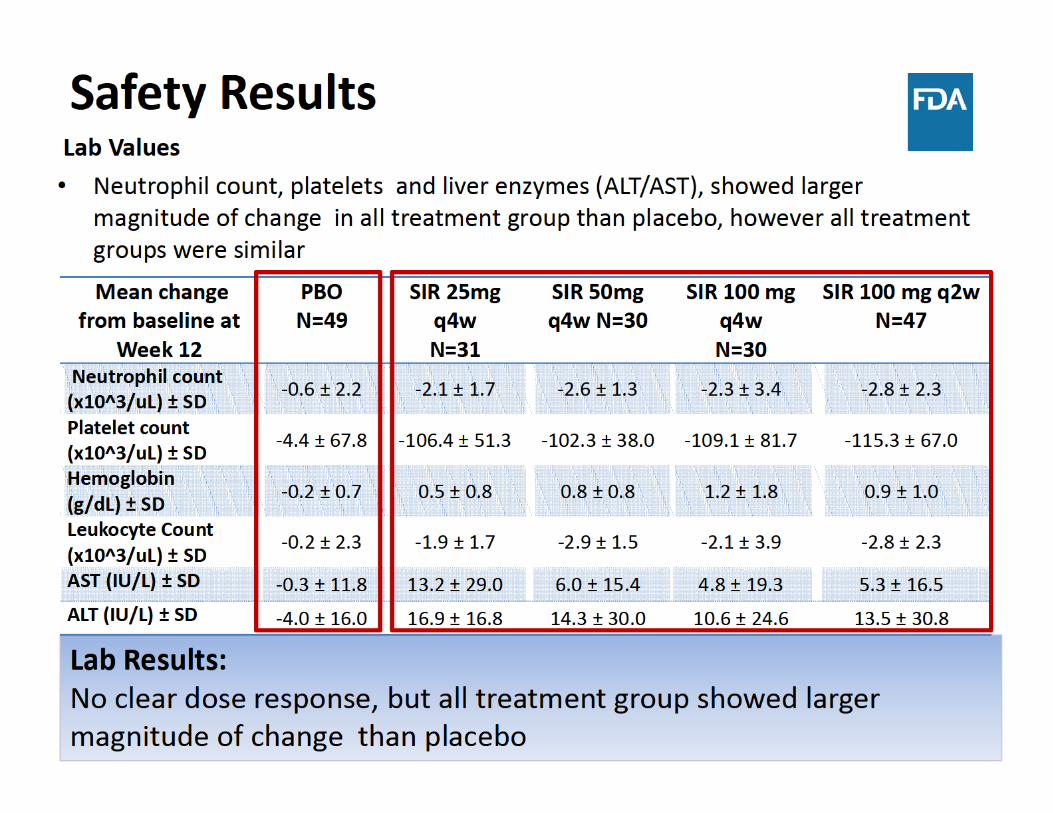
• Safety:– No dose response was observed for safety lab values, however all sirukumab treatment group showed larger magnitude of change
• Janssen selected 100 mg q2w and 50 mg q4w as their final doses for phase 3 studies

39
Overview of FDA Presentations
• Introduction and Clinical Overview– Mark Borigini, MD
• Dose Selection Considerations: Phase 2 Study Results– Dipak Pisal, PhD
• Review of Efficacy– William Koh, PhD
• Review of Safety and Risk/Benefit Considerations – Mark Borigini, MD

Arthritis Advisory Committee MeetingBLA761057: Sirukumab for RA
Review of Efficacy
William Koh, PhD Statistical Reviewer
Division of Biometrics II, Office of Biostatistics,Office of Translational Sciences, CDER
August 2, 2017

41
Outline
• Overview of phase 3 efficacy studies
– Study design
• Key efficacy results in placebo‐controlled studies
– Baseline demographics, disposition
– Signs and symptoms endpoints
– Radiographic progression results
• Key efficacy results in active‐controlled study
• Conclusions of efficacy findings
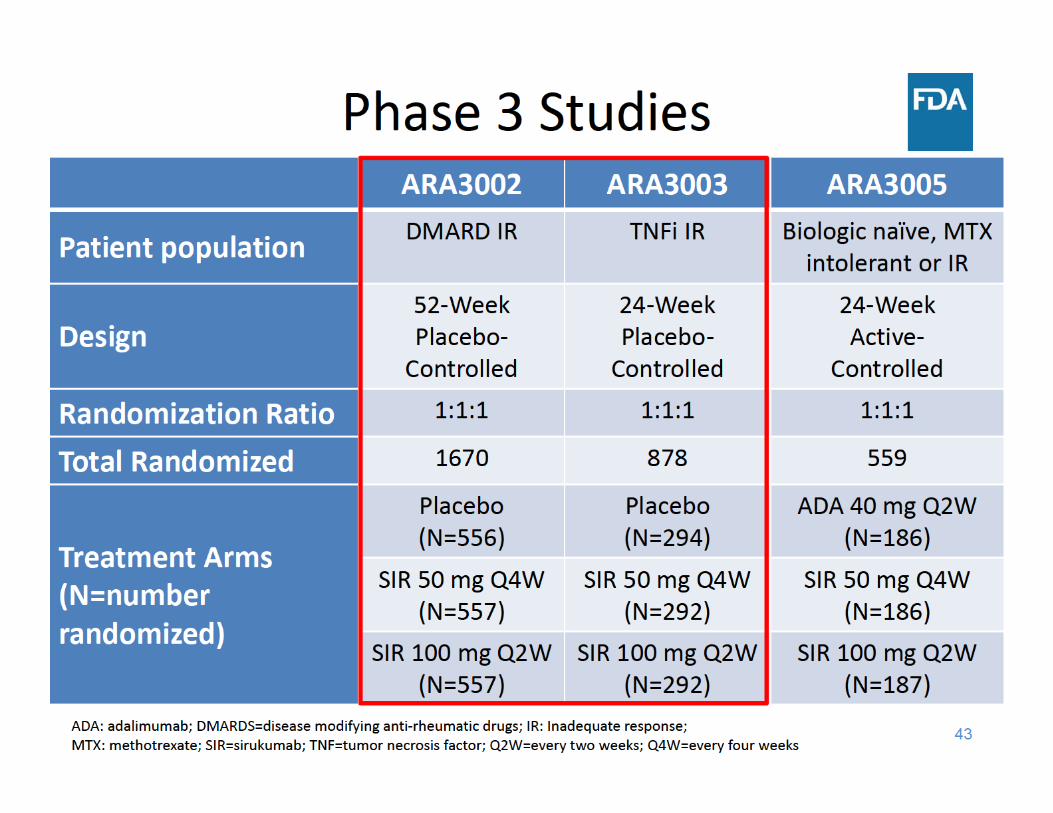

44
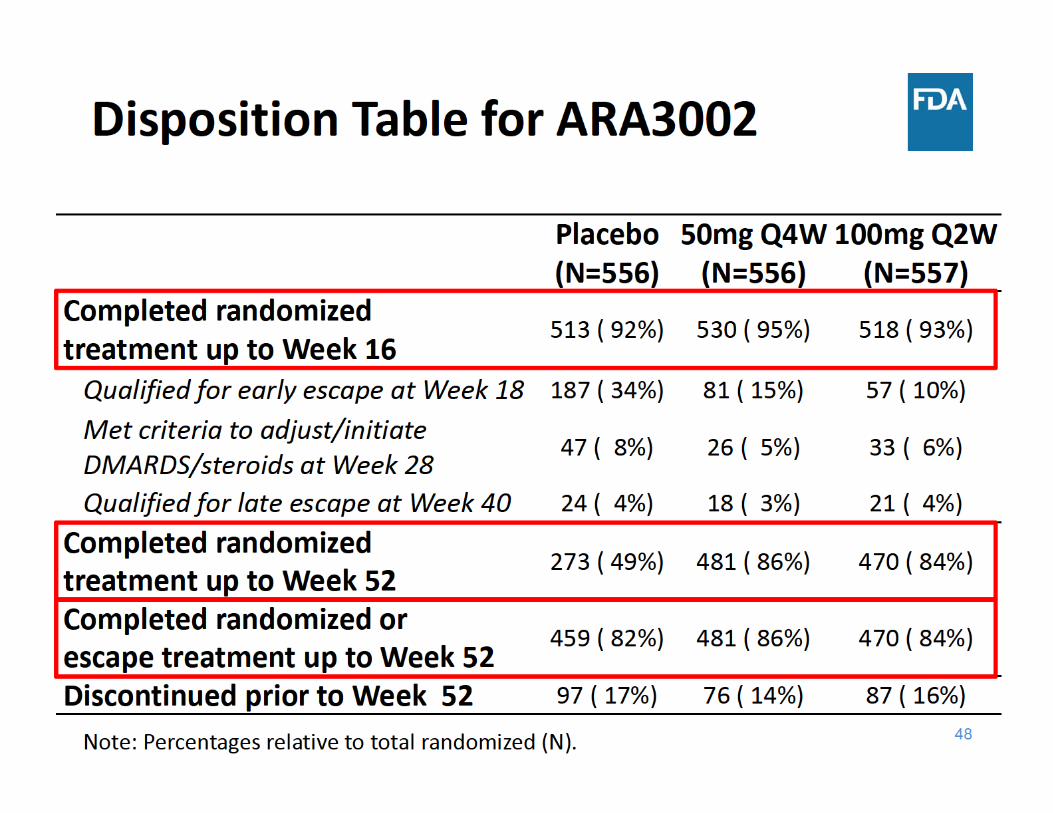
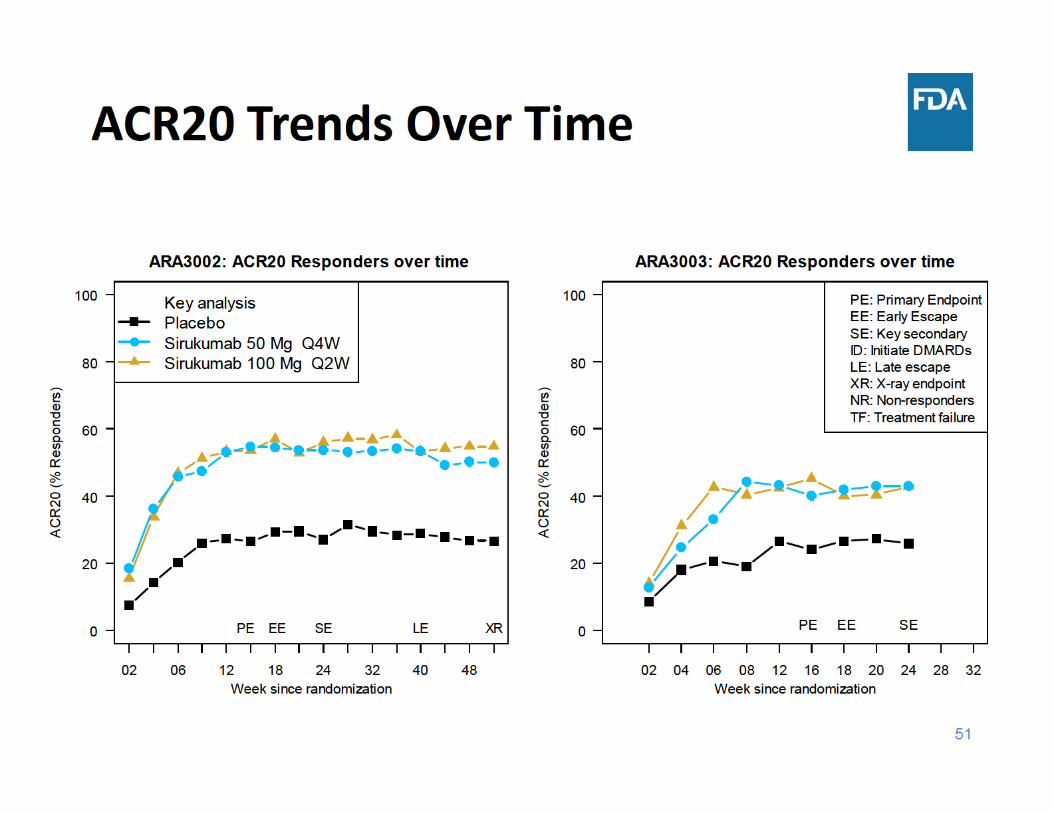
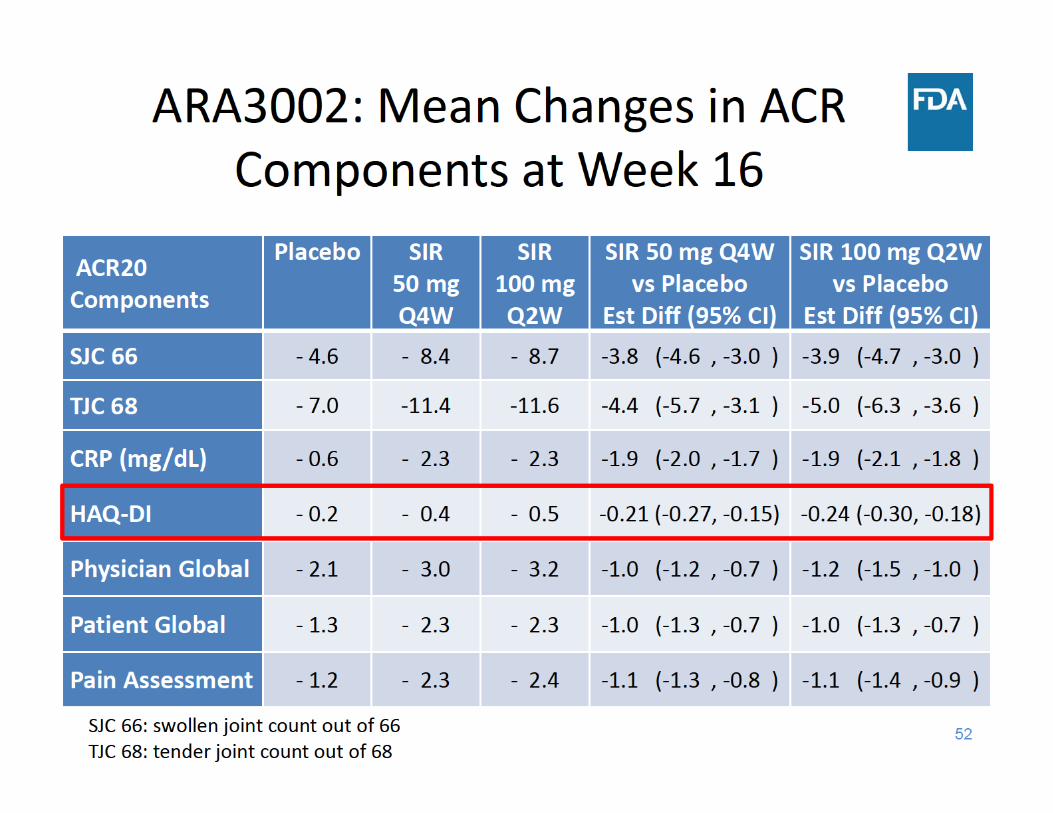
Study ARA3002 Design• 52‐week, randomized, parallel‐group, double‐blind study in
1670 patients with active rheumatoid arthritis (RA) and inadequate response to DMARDs
• Escape for placebo subjects (to sirukumab)– Early Escape *(EE) (at Week 18)– Late Escape * (LE) (at Week 40)
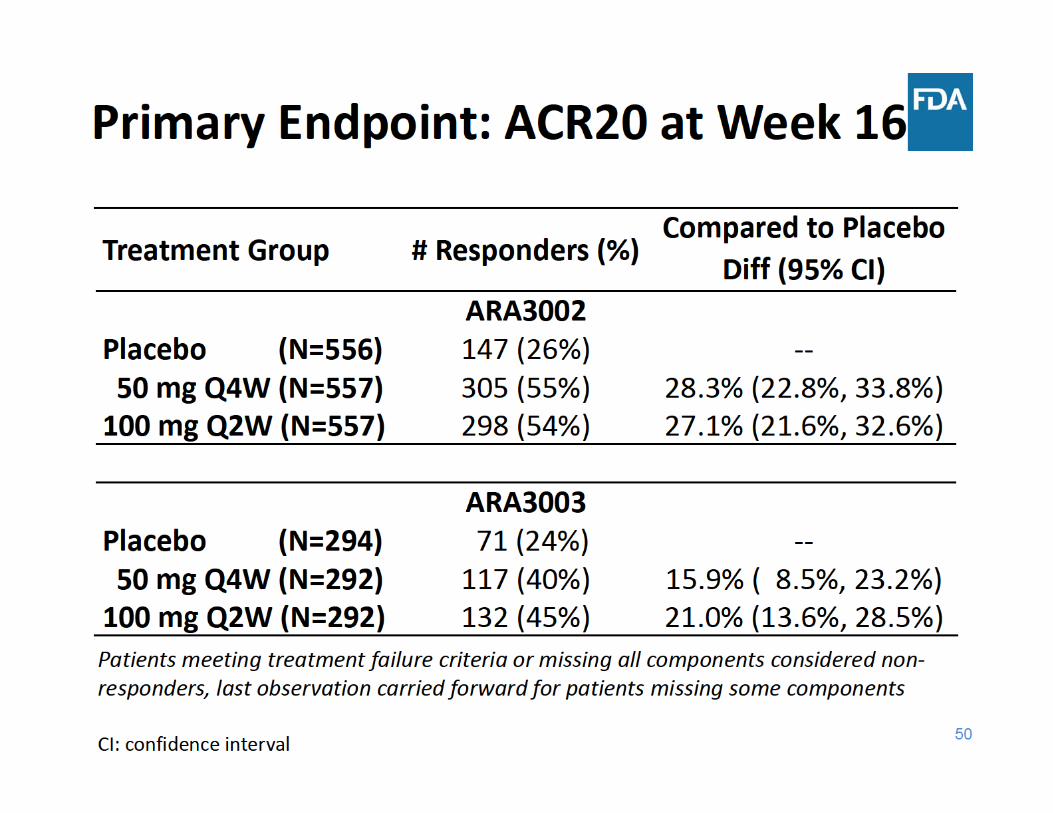
• Co‐Primary endpoints– ACR20 (at Week 16)– Change from baseline in vdH‐S score (at Week 52)
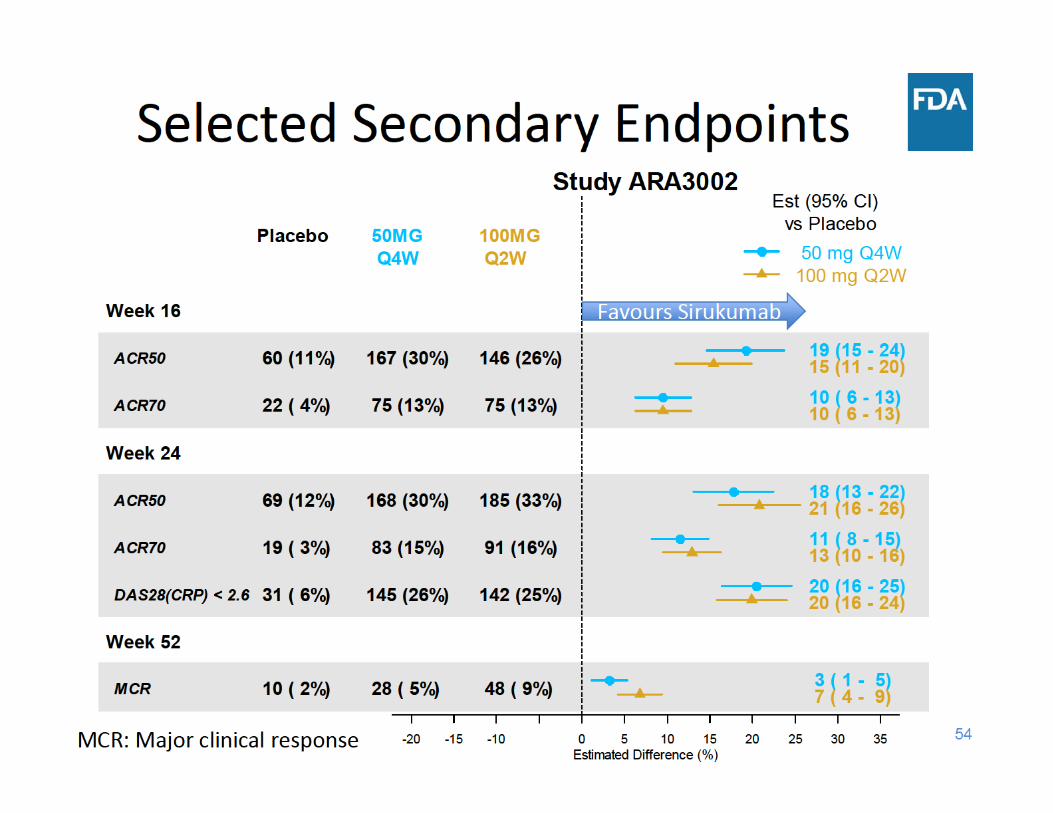
• Secondary endpoints – Change from baseline in HAQ‐DI (at Week 24) – ACR50 (at Week 24) – DAS28(CRP) < 2.6 (at Week 24)– Major Clinical Response or MCR (at Week 52)
* EE/LE Criteria: <20% improvement from baseline in both swollen and tender joint countsvdH‐S : van der Heijde modified Sharp score

45
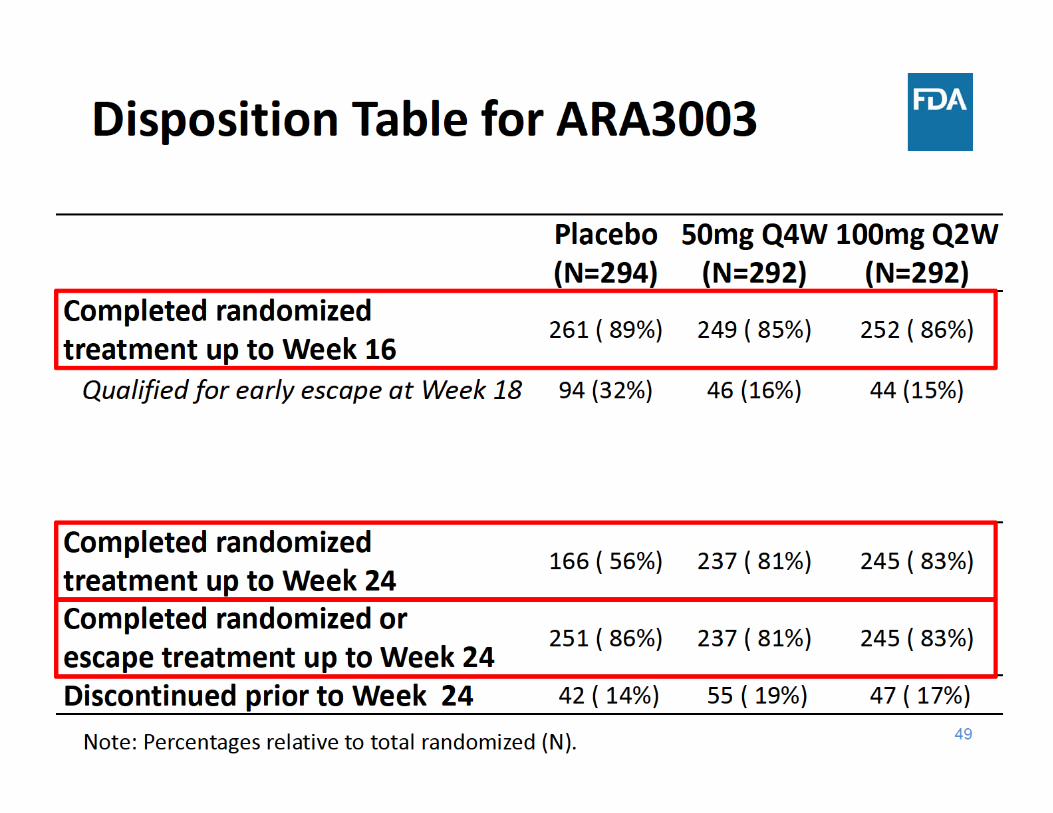
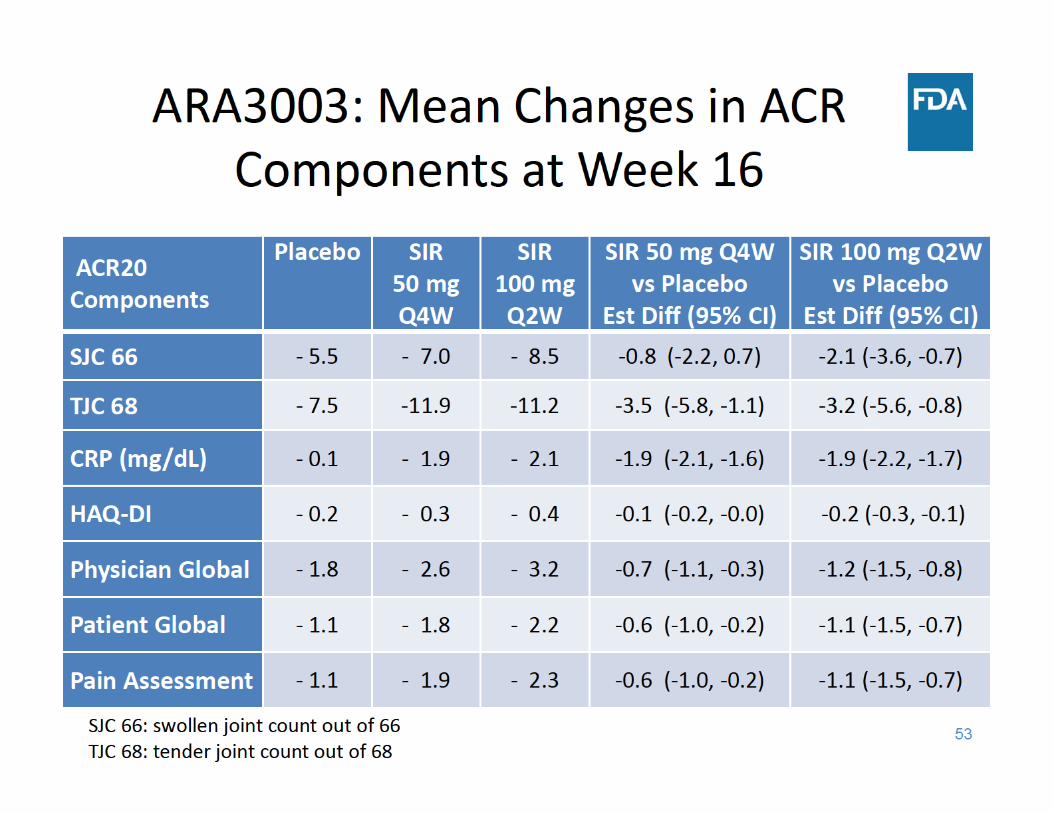
Study ARA3003 Design• 24‐week, randomized, parallel‐group, double‐blind study in
878 patients with active rheumatoid arthritis (RA) and inadequate response to TNF inhibitors
• Escape for placebo subjects– Early Escape* (EE) (at Week 18)
• Primary endpoint– ACR20 (at Week 16)
• Secondary endpoints – Change from baseline in HAQ‐DI (at Week 24)– ACR50 (at Week 24) – DAS28(CRP) < 2.6 (at Week 24)
* EE Criteria: <20% improvement from baseline in both swollen and tender joint counts

46
American College of Rheumatology (ACR) Response Criteria
• Multi‐component endpoint• Proportion of patients achieving a target % improvement from
baseline (20, 50, 70)• For ACR20
– At least 20% improvement in swollen and tender joint counts AND– At least 20% improvement in at least 3 out of the 5 components
• Patient global assessment (0 – 10 unit)• Physician global assessment (0 – 10 unit)• Patient pain (0 – 10 unit)• Health Assessment Questionnaire‐Disability Index (0 – 3 unit)• Laboratory test for inflammation (CRP or ESR)
• Treatment failure criteria: patients with certain background medication changes or who discontinued treatment
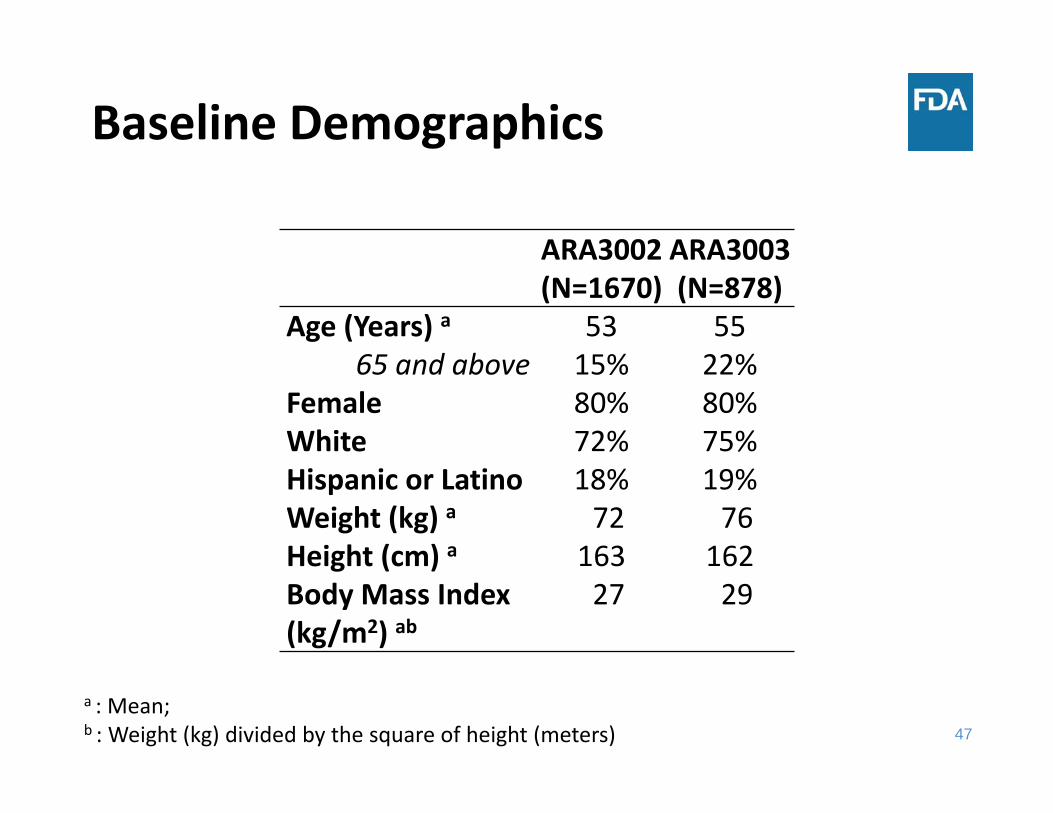
47
Baseline Demographics
ARA3002(N=1670)
ARA3003(N=878)
Age (Years) a 53 55 65 and above 15% 22%
Female 80% 80%White 72% 75%Hispanic or Latino 18% 19%Weight (kg) a 72 76 Height (cm) a 163 162 Body Mass Index (kg/m2) ab
27 29
a : Mean; b : Weight (kg) divided by the square of height (meters)

56
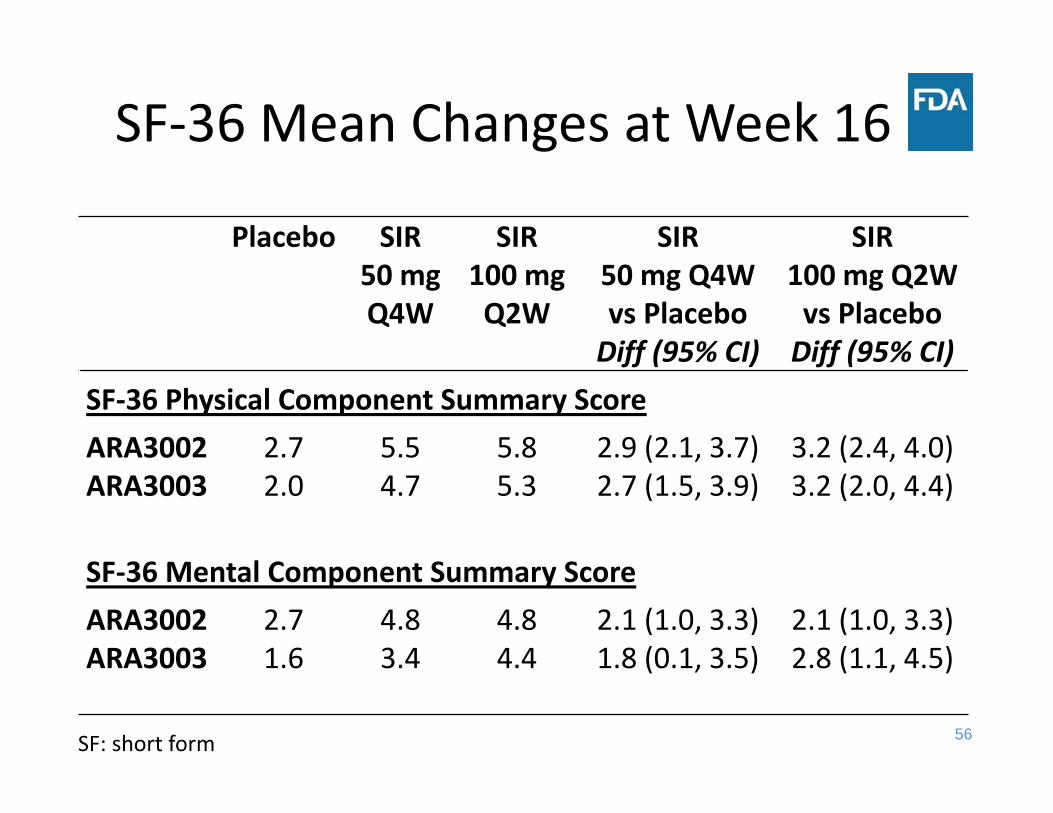
SF‐36 Mean Changes at Week 16
Placebo SIR50 mg Q4W
SIR100 mg Q2W
SIR50 mg Q4W vs Placebo Diff (95% CI)
SIR100 mg Q2W vs PlaceboDiff (95% CI)
SF‐36 Physical Component Summary ScoreARA3002 2.7 5.5 5.8 2.9 (2.1, 3.7) 3.2 (2.4, 4.0) ARA3003 2.0 4.7 5.3 2.7 (1.5, 3.9) 3.2 (2.0, 4.4)
SF‐36 Mental Component Summary ScoreARA3002 2.7 4.8 4.8 2.1 (1.0, 3.3) 2.1 (1.0, 3.3) ARA3003 1.6 3.4 4.4 1.8 (0.1, 3.5) 2.8 (1.1, 4.5)
SF: short form

57
• van der Heijde modified Sharp or vdH‐S (range: 0 – 448) – Erosion Score (0 – 280) – Joint Space Narrowing Score (0 – 168)
• Prespecified analysis
• Important supportive analyses
Radiographic Progression Evaluation in ARA3002

58
• van der Heijde modified Sharp or vdH‐S (range: 0 – 448) – Erosion Score (0 – 280) – Joint Space Narrowing Score (0 – 168)
• Prespecified analysis– Linear extrapolation (LE)
• Important supportive analyses– Observed data analysis– Mixed effects analysis
Radiographic Progression Evaluation in ARA3002
59
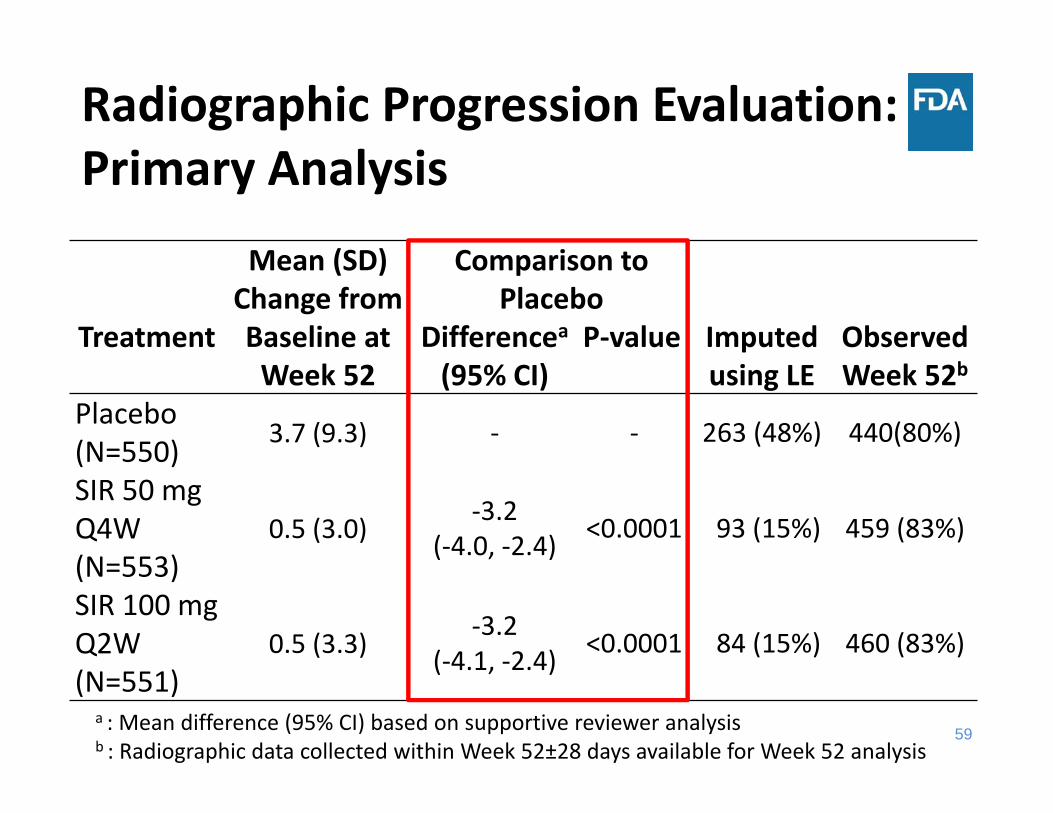
Radiographic Progression Evaluation: Primary Analysis
Mean (SD) Change from Baseline at Week 52
Comparison to Placebo
Imputed using LE
Observed Week 52b
Treatment Differencea(95% CI)
P‐value
Placebo (N=550) 3.7 (9.3) ‐ ‐ 263 (48%) 440(80%)
SIR 50 mg Q4W (N=553)
0.5 (3.0) ‐3.2 (‐4.0, ‐2.4) <0.0001 93 (15%) 459 (83%)
SIR 100 mg Q2W(N=551)
0.5 (3.3) ‐3.2(‐4.1, ‐2.4) <0.0001 84 (15%) 460 (83%)
a : Mean difference (95% CI) based on supportive reviewer analysisb : Radiographic data collected within Week 52±28 days available for Week 52 analysis
60
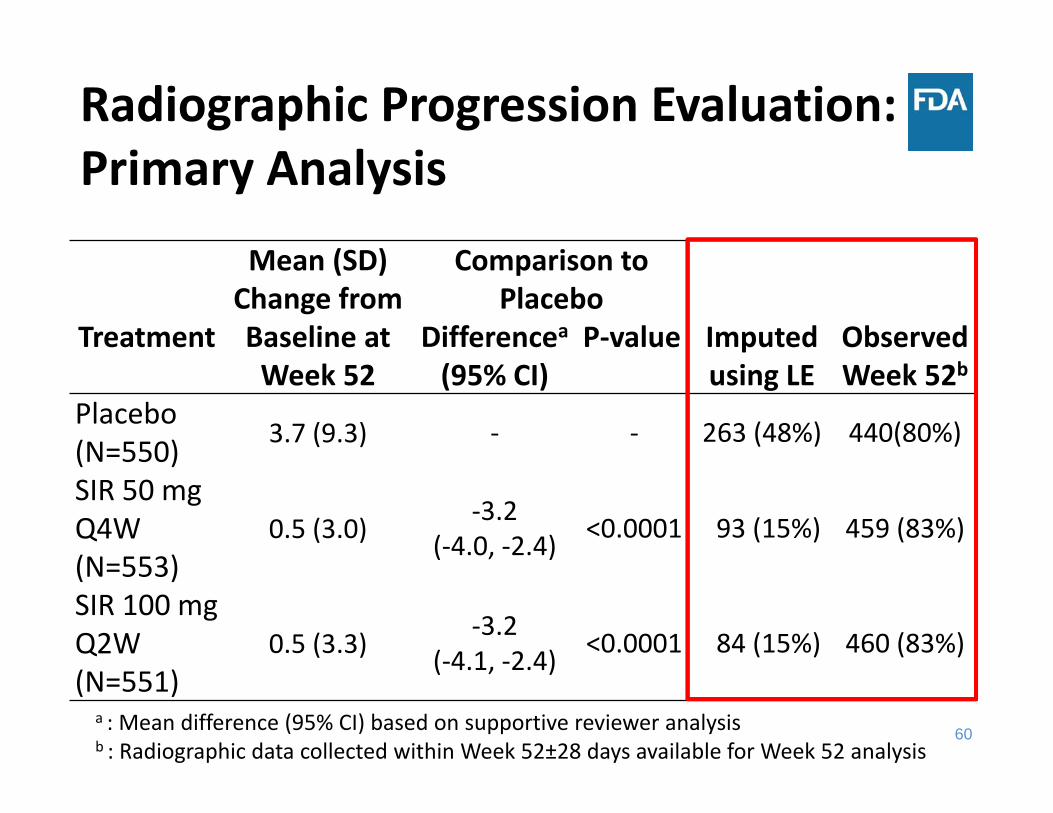
Radiographic Progression Evaluation: Primary Analysis
Mean (SD) Change from Baseline at Week 52
Comparison to Placebo
Imputed using LE
Observed Week 52b
Treatment Differencea(95% CI)
P‐value
Placebo (N=550) 3.7 (9.3) ‐ ‐ 263 (48%) 440(80%)
SIR 50 mg Q4W (N=553)
0.5 (3.0) ‐3.2 (‐4.0, ‐2.4) <0.0001 93 (15%) 459 (83%)
SIR 100 mg Q2W(N=551)
0.5 (3.3) ‐3.2(‐4.1, ‐2.4) <0.0001 84 (15%) 460 (83%)
a : Mean difference (95% CI) based on supportive reviewer analysisb : Radiographic data collected within Week 52±28 days available for Week 52 analysis
61
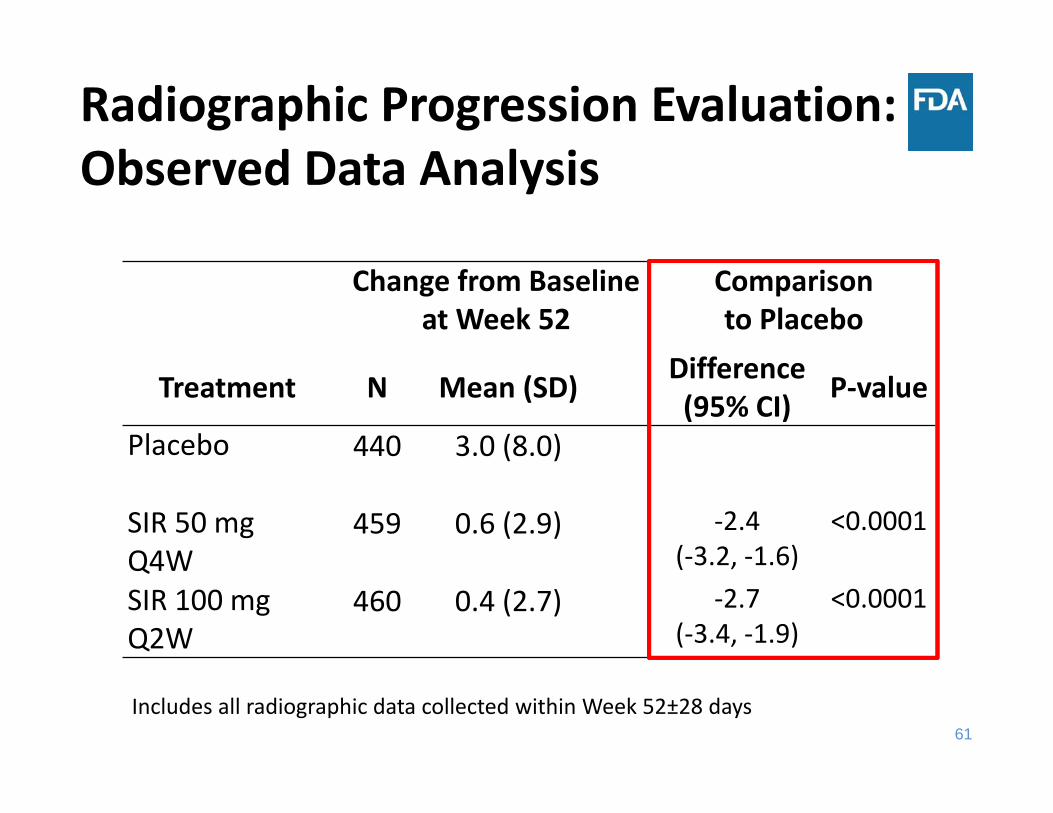
Change from Baseline at Week 52
Comparison to Placebo
Treatment N Mean (SD) Difference(95% CI) P‐value
Placebo 440 3.0 (8.0)
SIR 50 mg Q4W
459 0.6 (2.9) ‐2.4(‐3.2, ‐1.6)
<0.0001
SIR 100 mg Q2W
460 0.4 (2.7) ‐2.7(‐3.4, ‐1.9)
<0.0001
Radiographic Progression Evaluation: Observed Data Analysis
Includes all radiographic data collected within Week 52±28 days

62
Addressing Missing Data• Tipping point sensitivity analysis including all observed data regardless of treatment discontinuation or early escape were conducted for – ACR20 at Week 16 – HAQ‐DI at Week 16– vdH‐S at Week 52
• Sensitivity analyses conducted were supportive of the efficacy of sirukumab for both signs and symptoms and radiographic endpoints

64
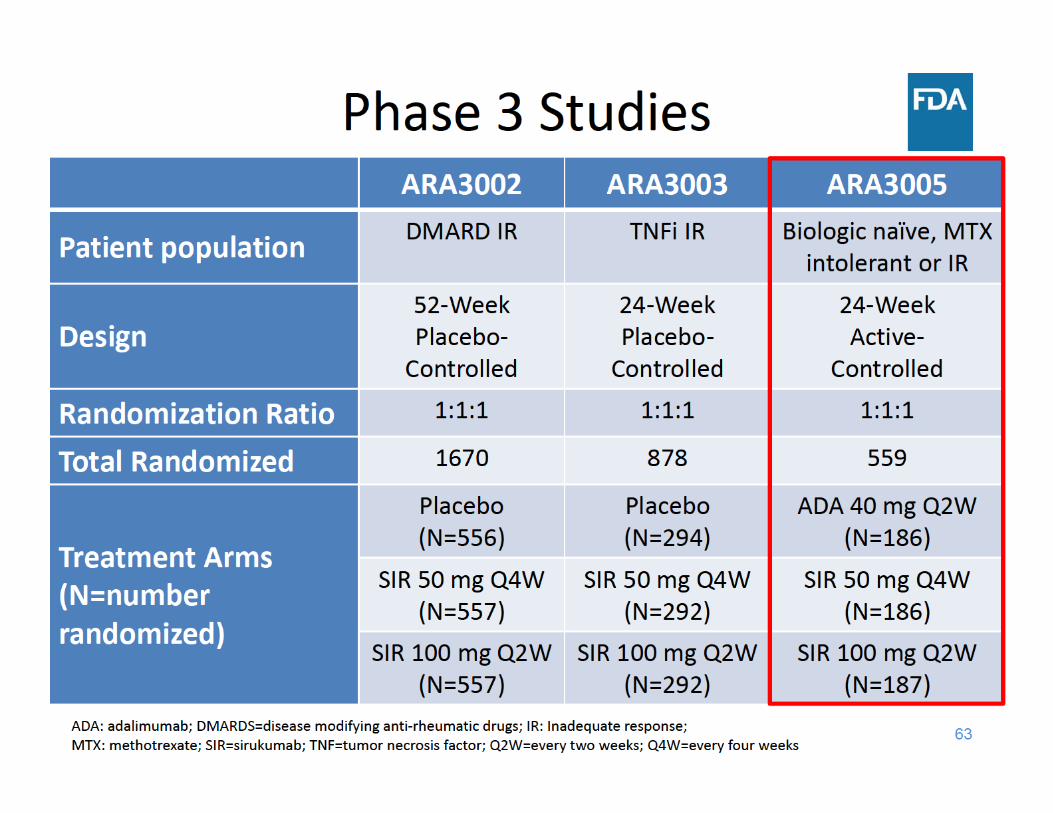
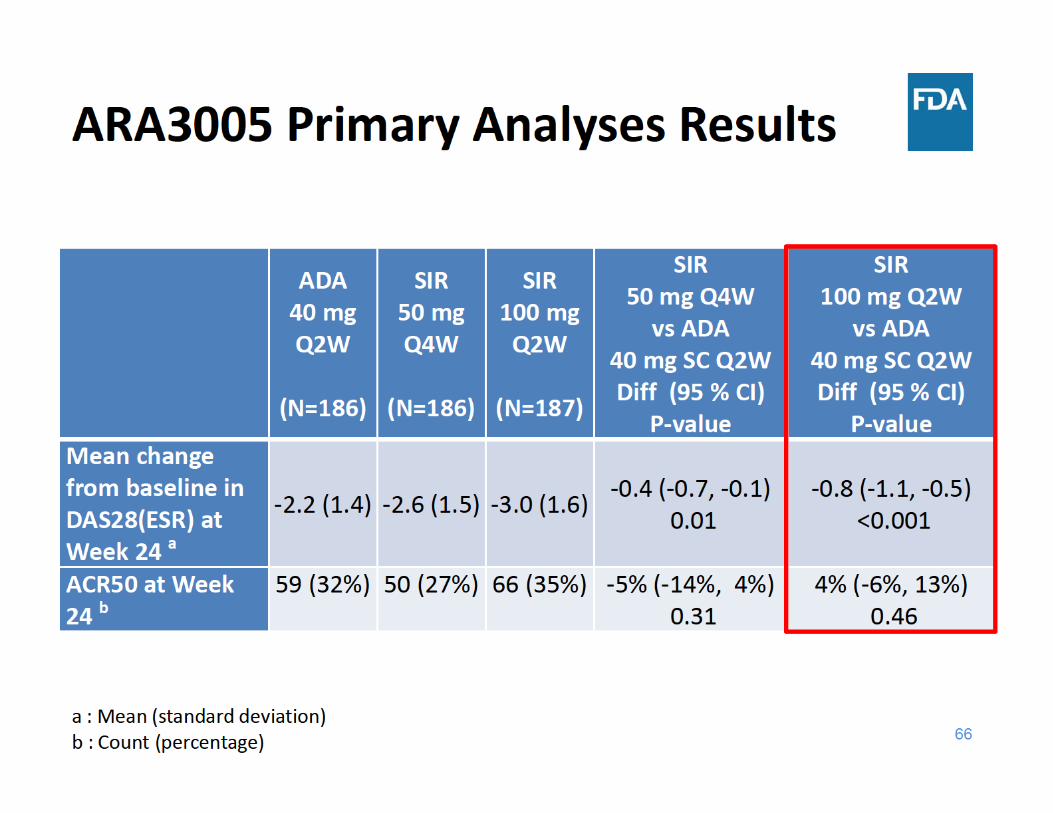
Study ARA3005 Design• 24‐week, randomized, active‐controlled, parallel‐group, double‐
blind study in 559 patients with active RA who failed methotrexate
• Study design– Monotherapy– Escape (EE) at Week 16
• ADA 40mg Q2W ADA 40mg QW• SIR 50mg Q4W SIR 100mg Q2W• Patients on SIR 100mg Q2W remained on their dosing
• Primary analyses based on SIR 100mg Q2W– Change from baseline in DAS28(ESR) score at Week 24– ACR50 at Week 24
• Secondary analyses – DAS28 (ESR) < 2.6 100 mg Q2W– Change from baseline in DAS28(ESR) score, ACR50 50 mg Q4W– ACR20 for 100mg Q2W, ACR20 for 50mg Q4W
DAS28(ESR)=disease activity score 28 joint count using erythrocyte sedimentation rate (ESR)
65
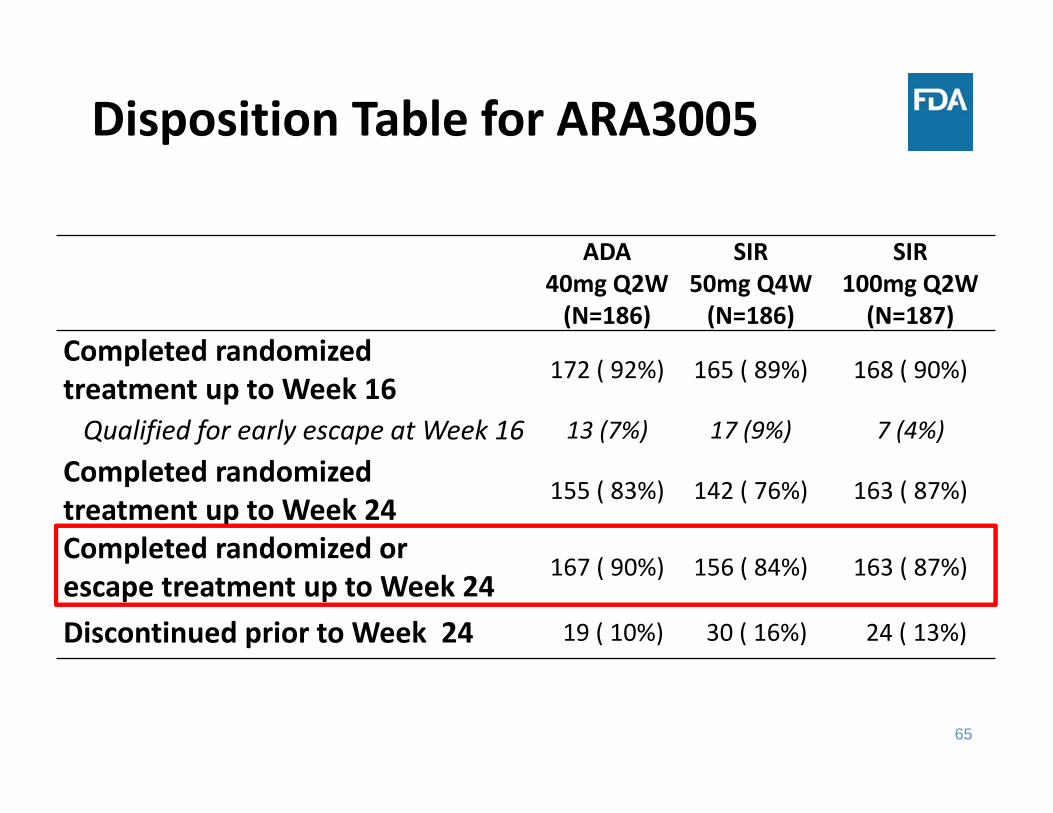
Disposition Table for ARA3005
ADA40mg Q2W(N=186)
SIR 50mg Q4W(N=186)
SIR100mg Q2W(N=187)
Completed randomized treatment up to Week 16
172 ( 92%) 165 ( 89%) 168 ( 90%)
Qualified for early escape at Week 16 13 (7%) 17 (9%) 7 (4%)
Completed randomized treatment up to Week 24
155 ( 83%) 142 ( 76%) 163 ( 87%)
Completed randomized or escape treatment up to Week 24
167 ( 90%) 156 ( 84%) 163 ( 87%)
Discontinued prior to Week 24 19 ( 10%) 30 ( 16%) 24 ( 13%)
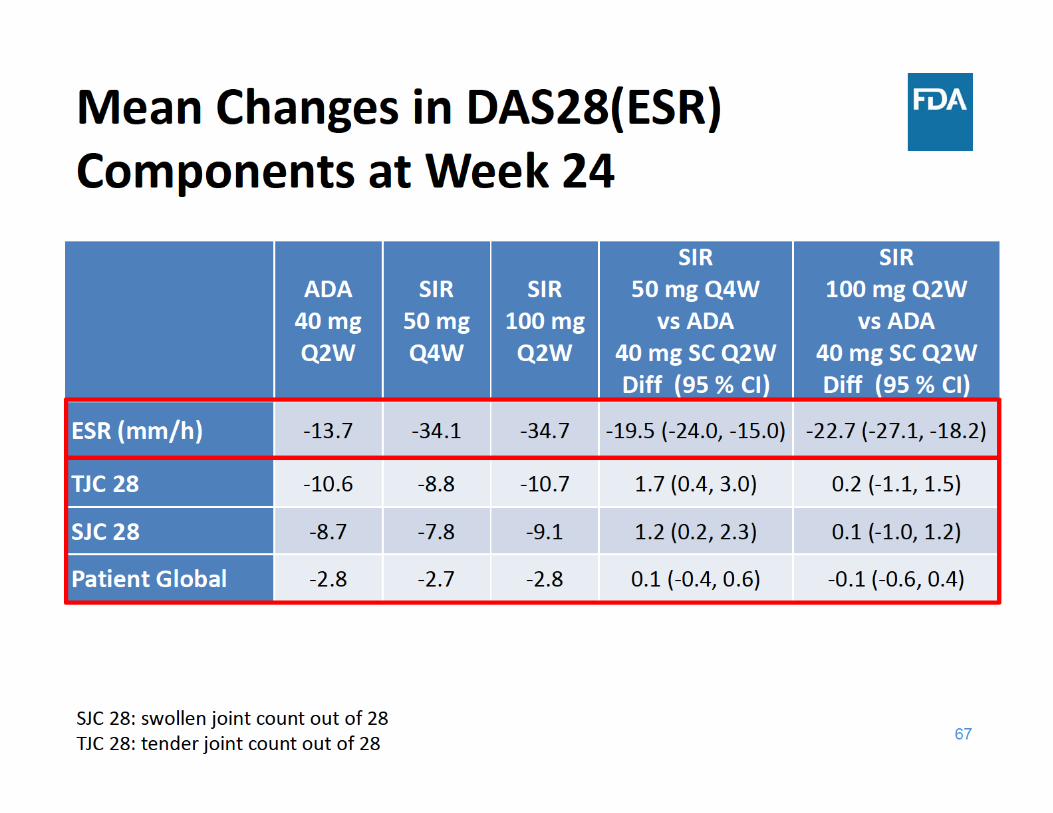
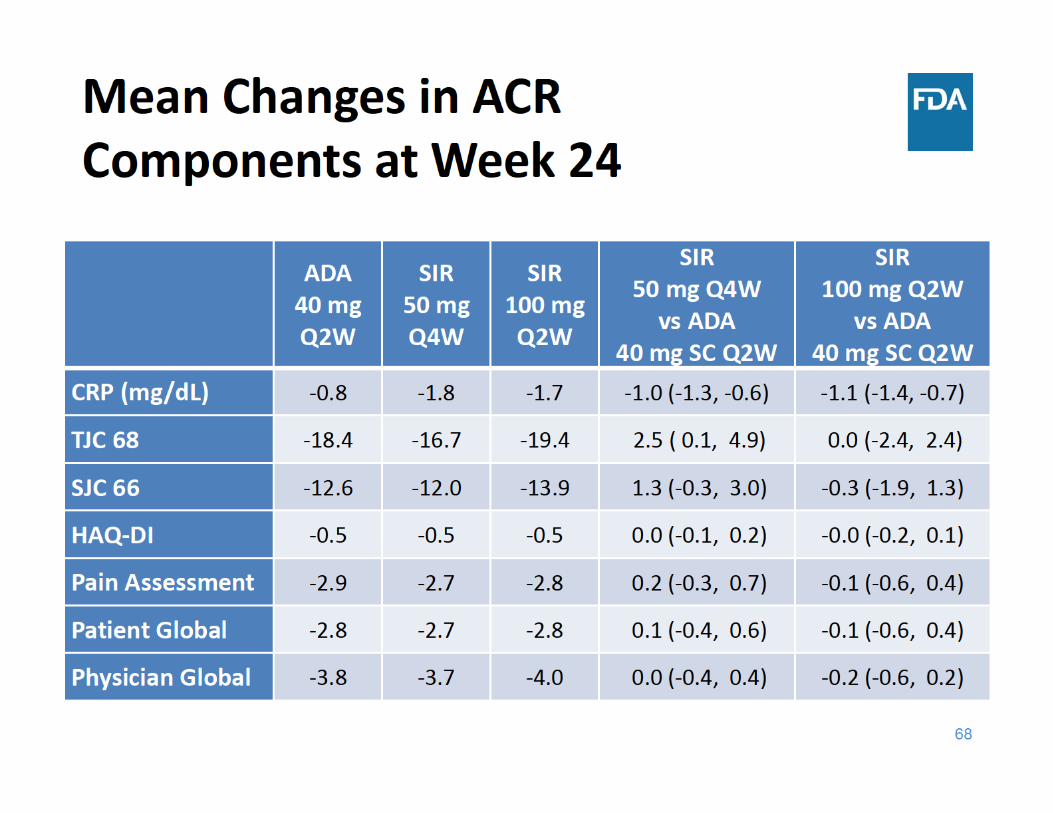

69
Overall Efficacy Findings
• Efficacy on symptoms + function in ARA3002 and ARA3003 for both sirukumab doses– Primary Week 16 ACR20 results– Results for HAQ‐DI, secondary endpoints supportive
• Evidence of inhibition of radiographic progression in ARA3002
• Results convincing despite missing data• No consistent differences between doses• No superiority but similar improvements to adalimumab in ARA3005

70
Overview of FDA Presentations
• Introduction and Clinical Overview– Mark Borigini, MD
• Dose Selection Considerations: Phase 2 Study Results– Dipak Pisal, PhD
• Review of Efficacy– William Koh, PhD
• Review of Safety and Risk/Benefit Considerations – Mark Borigini, MD
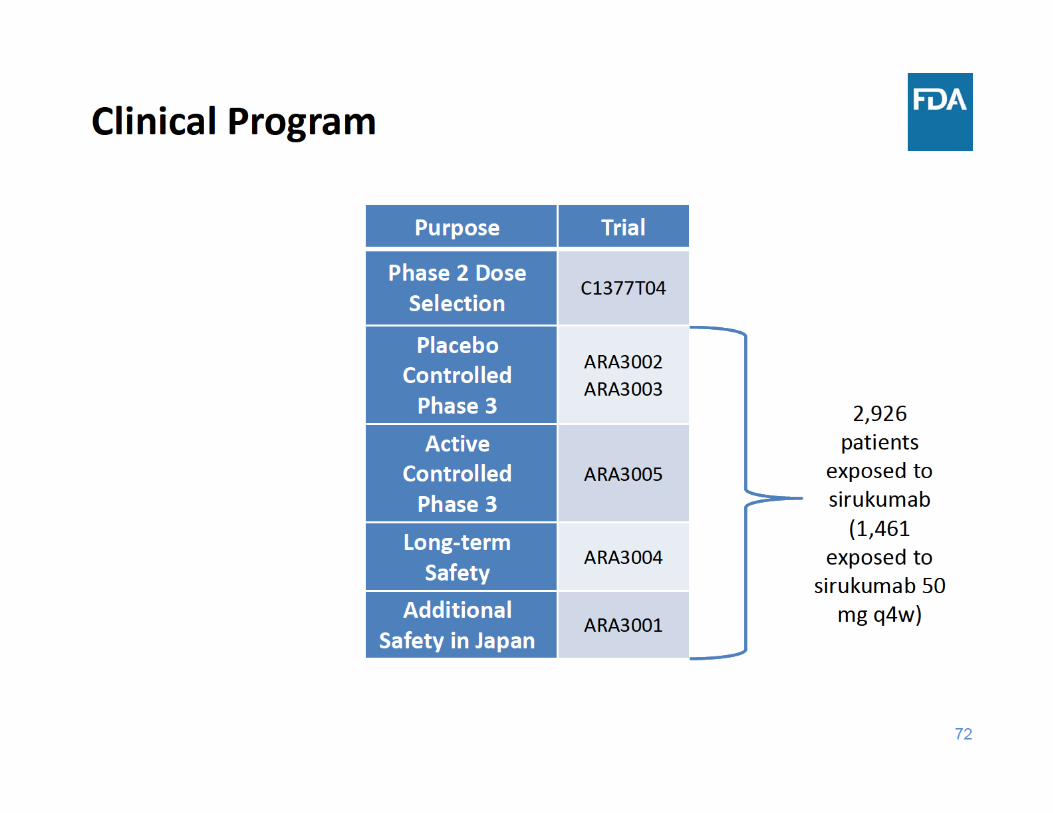
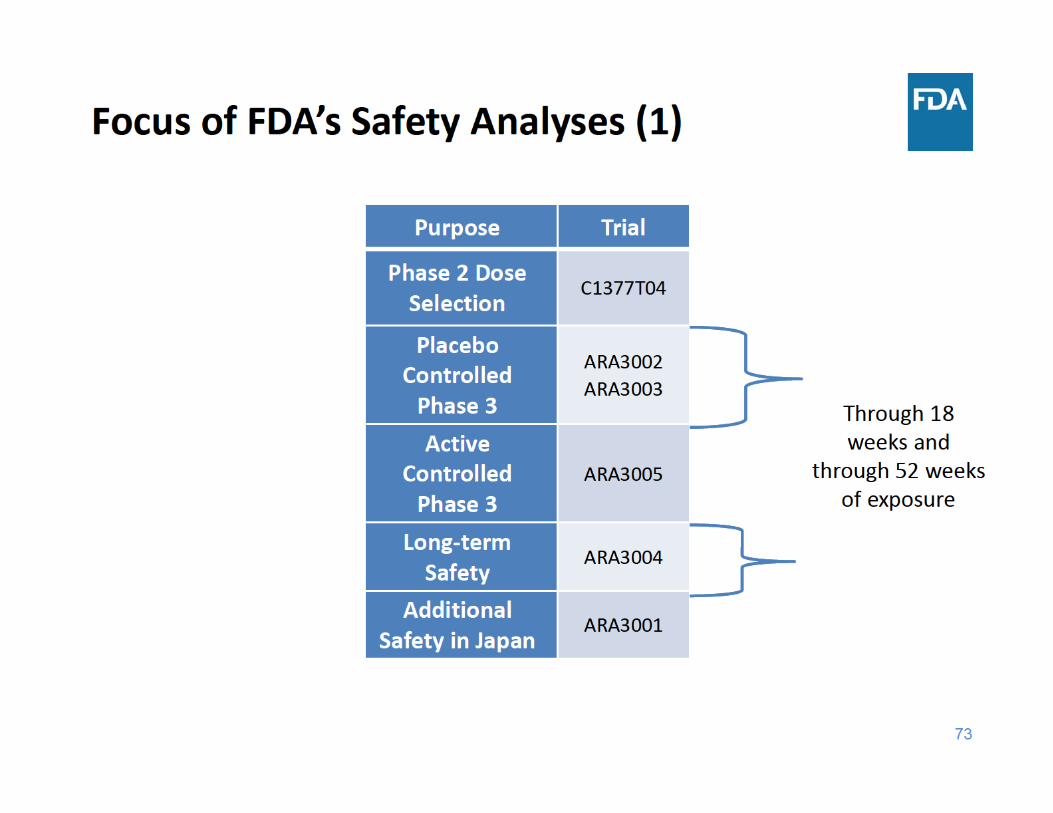
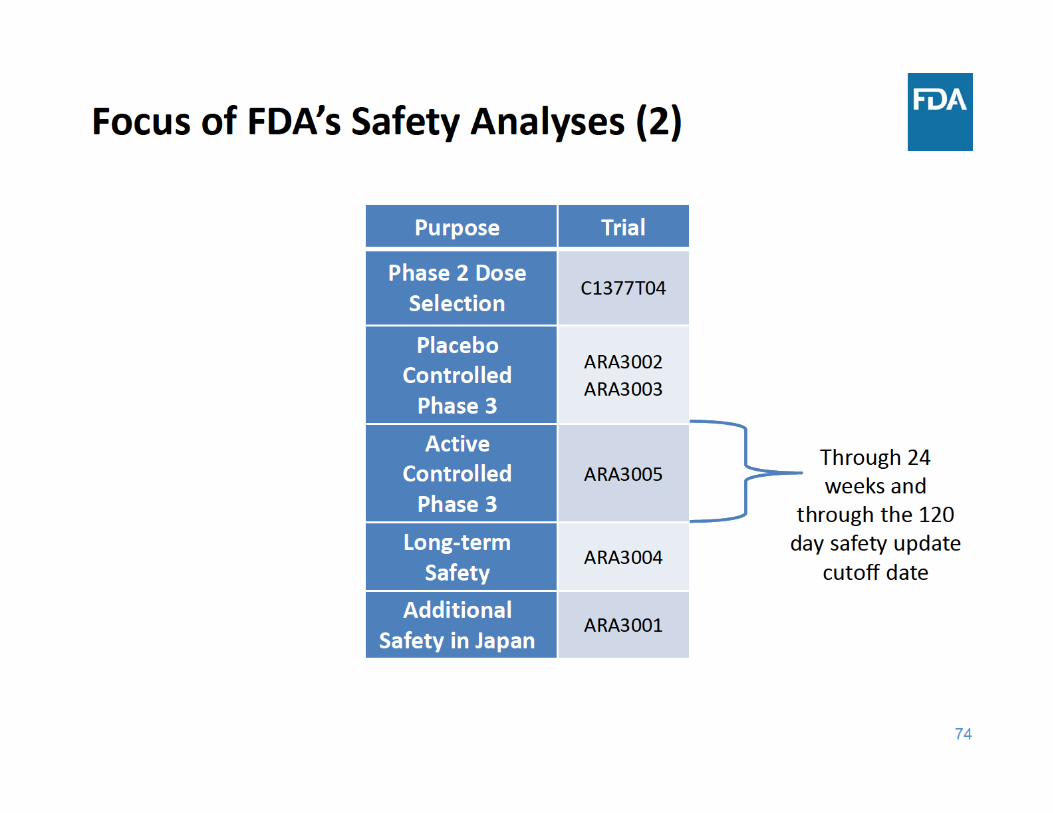
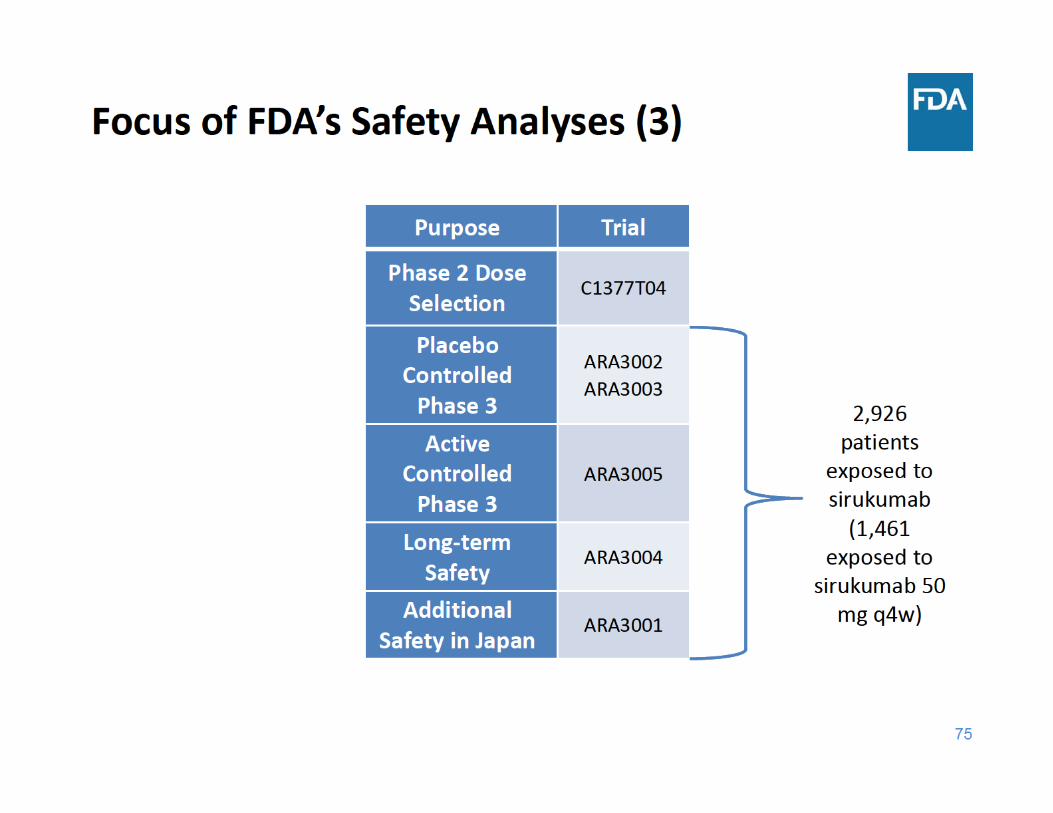

76
Overview of Safety of Approved Monoclonal Antibodies to IL‐6 Receptor
Boxed Warning: • Serious infections that may lead to hospitalization or death. Reported infections
include: tuberculosis, invasive fungal infections, and bacterial, viral, and other infections due to opportunistic pathogens
Warnings/Precautions • Serious Infections• Gastrointestinal perforation• Laboratory abnormalities (neutropenia, thrombocytopenia, elevated liver enzymes, lipid
abnormalities)• Immunosuppression• Hypersensitivity reactions• Demyelinating disorders• Active hepatic disease and hepatic impairment• Live vaccines
*Not all Warnings/Precautions are included in the labeling of all approved IL‐6 inhibitors and there are differences in the safety information discussedIL=interleukin

77
Focus of Safety Presentation
• All‐cause death• Serious adverse events (SAEs)• Major adverse cardiovascular events (MACE)• Infections• Malignancy• Gastrointestinal (GI) Perforation• Laboratory Abnormalities (lipids, neutrophil and platelet counts, and liver function tests)
• Adalimumab comparator study (ARA3005)
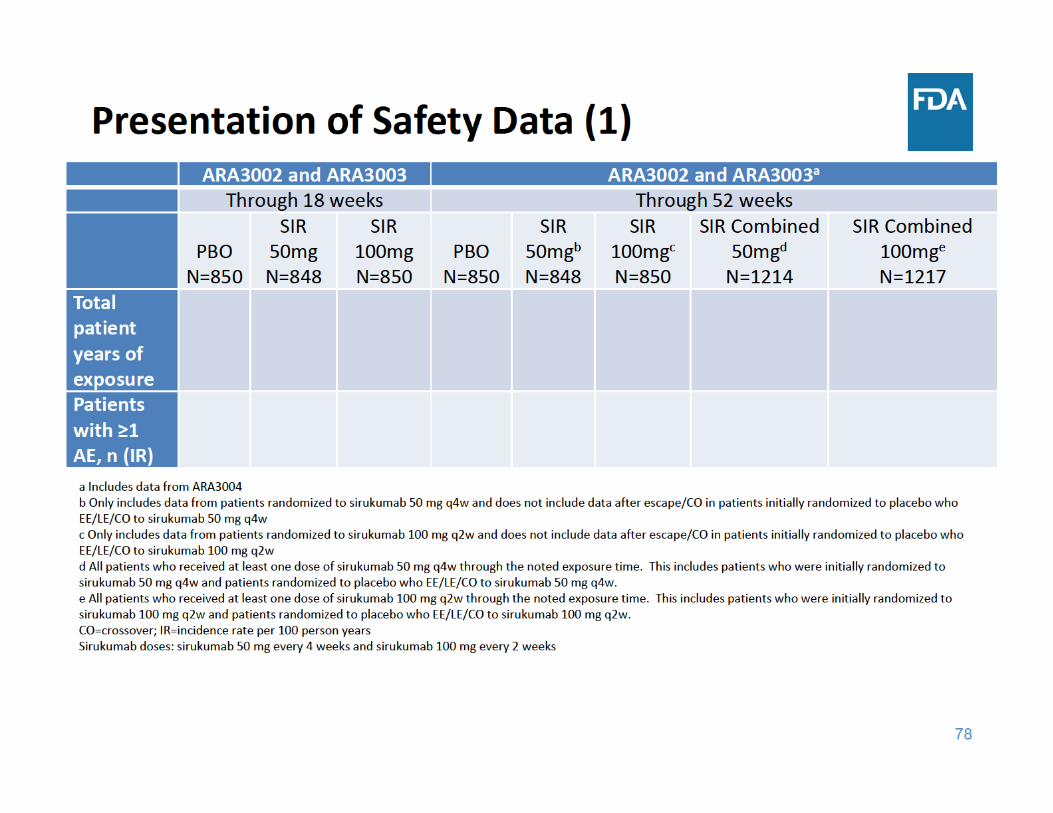

83
Focus of Safety Presentation
• All‐cause death• Serious adverse events (SAEs)• Major adverse cardiovascular events (MACE)• Infections• Malignancy• Gastrointestinal (GI) Perforation• Laboratory Abnormalities (lipids, neutrophil and platelet counts, and liver function tests)
• Adalimumab comparator study (ARA3005)

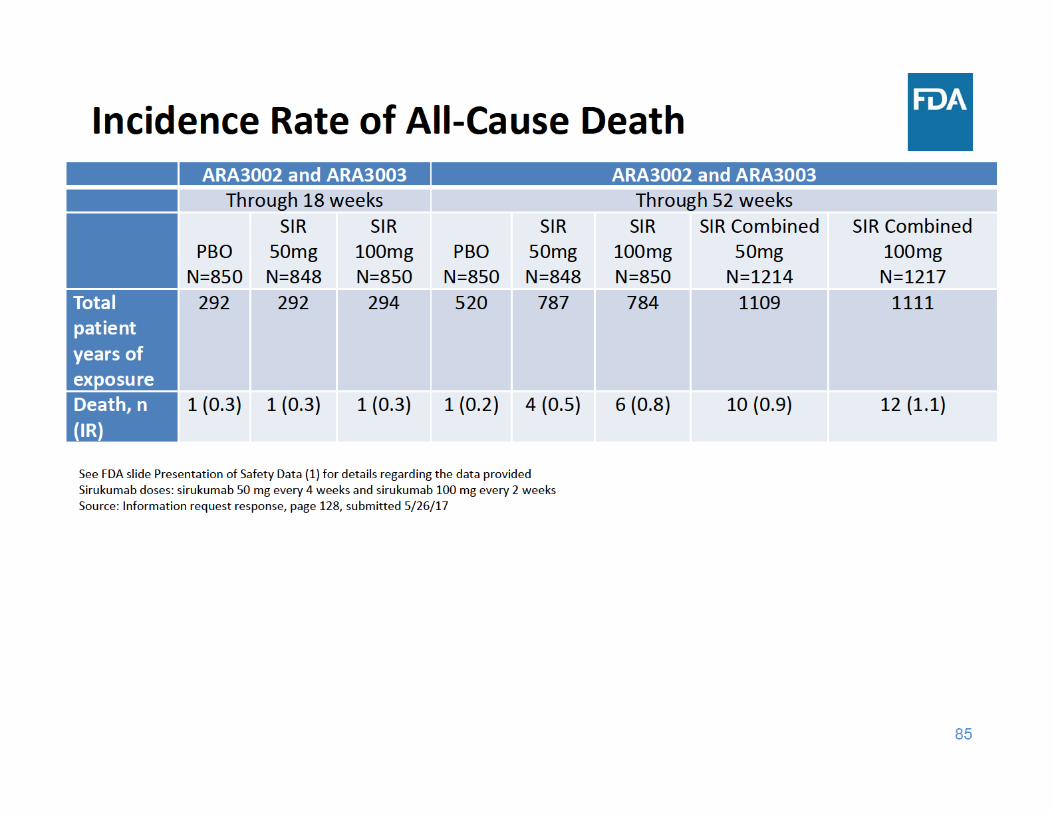
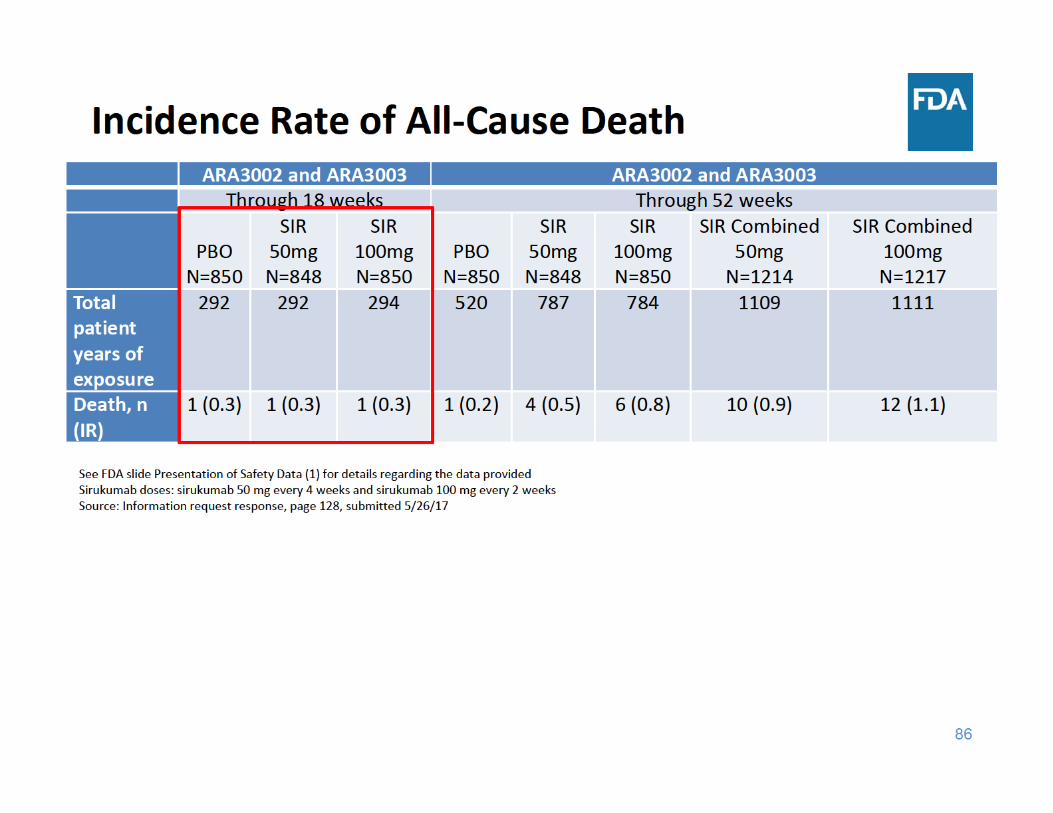
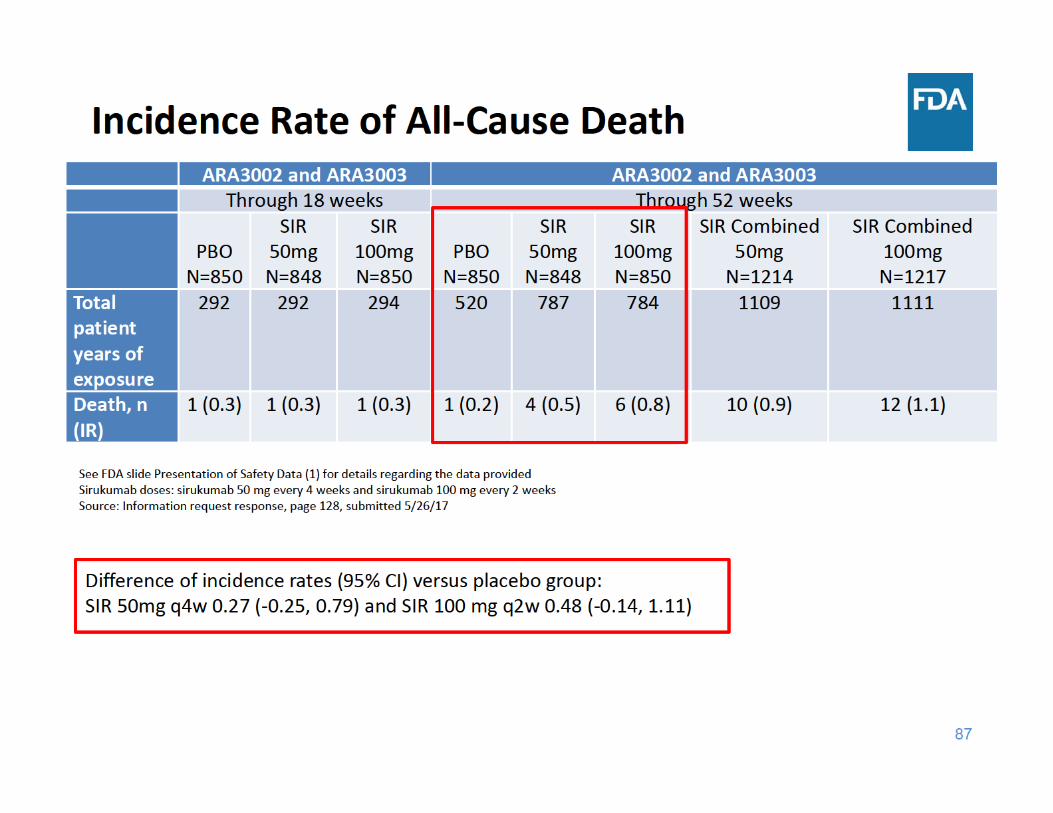
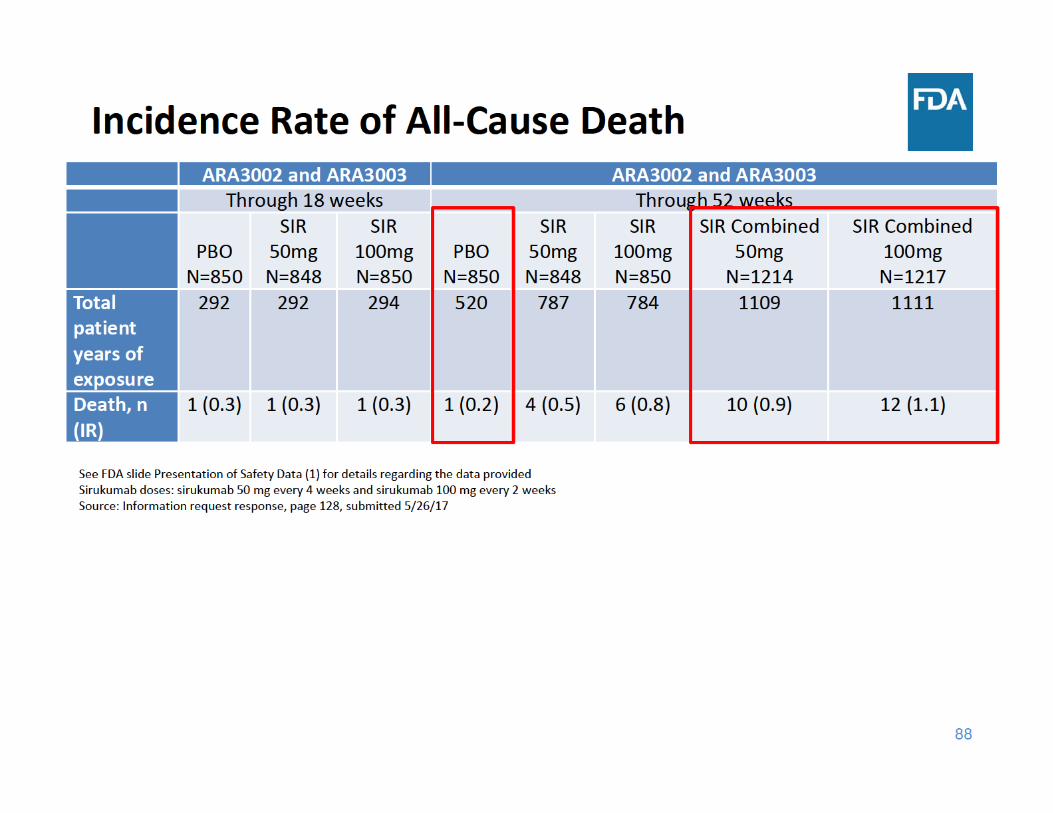
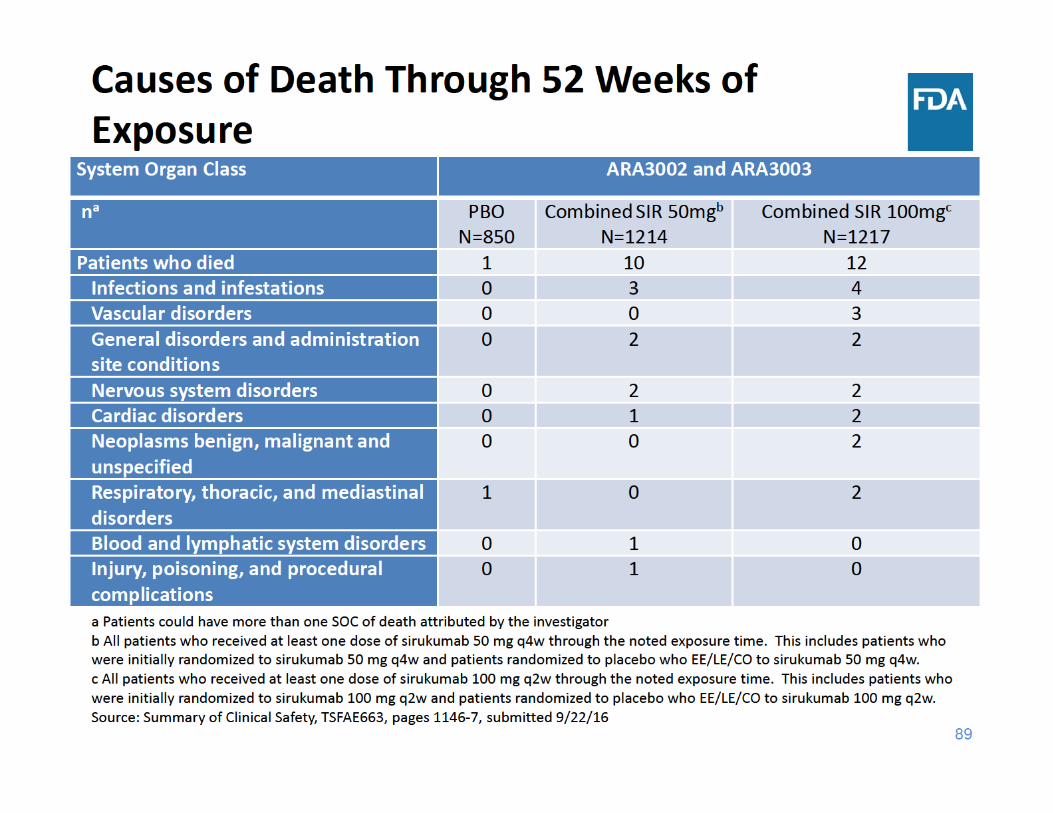

91
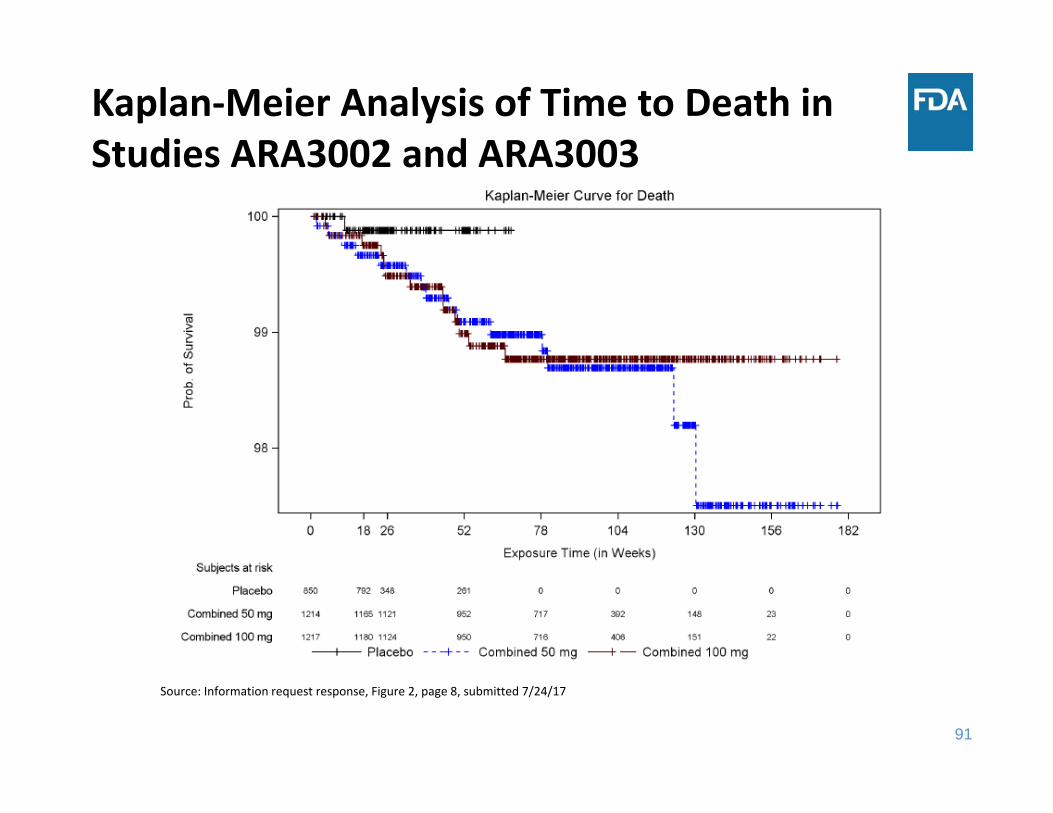
Kaplan‐Meier Analysis of Time to Death in Studies ARA3002 and ARA3003
Source: Information request response, Figure 2, page 8, submitted 7/24/17

92
All‐Cause Death Summary
• Through 18 weeks of exposure, the incidence rate of all‐cause death was the same in the placebo group and the two sirukumab groups
• Through 52 weeks of exposure, the incidence rate of all‐cause death was higher in each sirukumab group compared to placebo– The three main categories of causes of death were cardiovascular events, malignancy, and infections

93
Focus of Safety Presentation
• All‐cause death• Serious adverse events (SAEs)• Major adverse cardiovascular events (MACE)• Infections• Malignancy• Gastrointestinal (GI) Perforation• Laboratory Abnormalities (lipids, neutrophil and platelet counts, and liver function tests)
• Adalimumab comparator study (ARA3005)
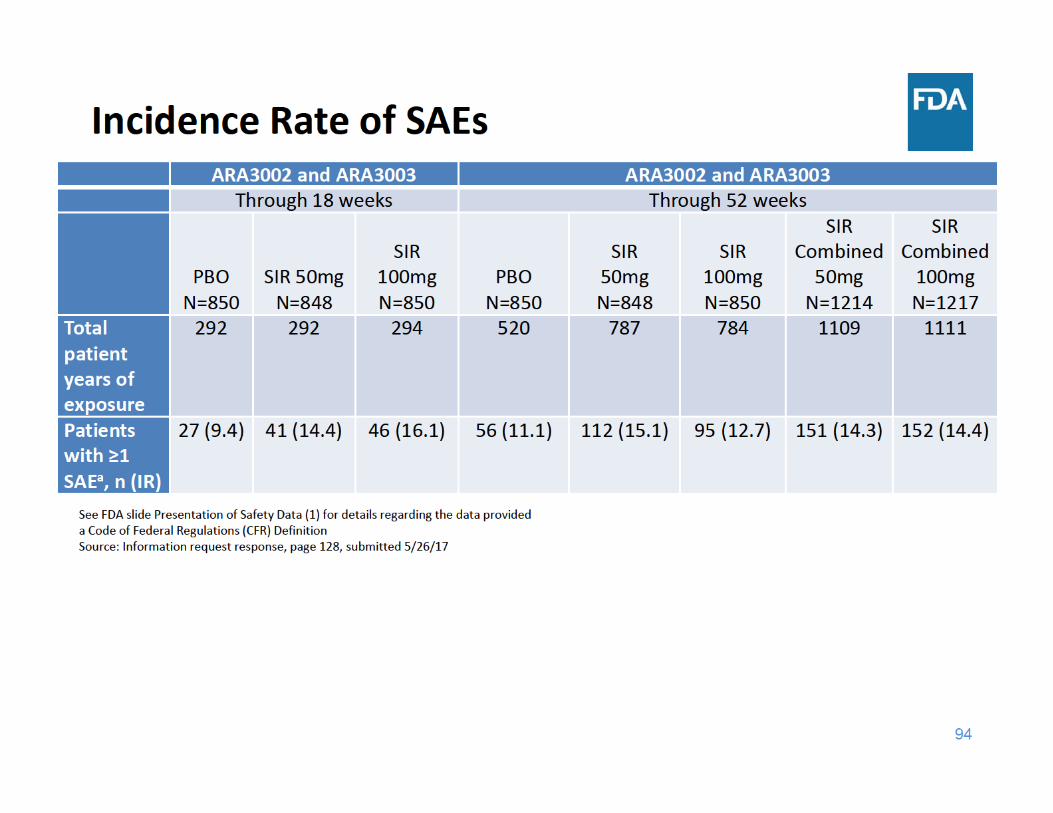
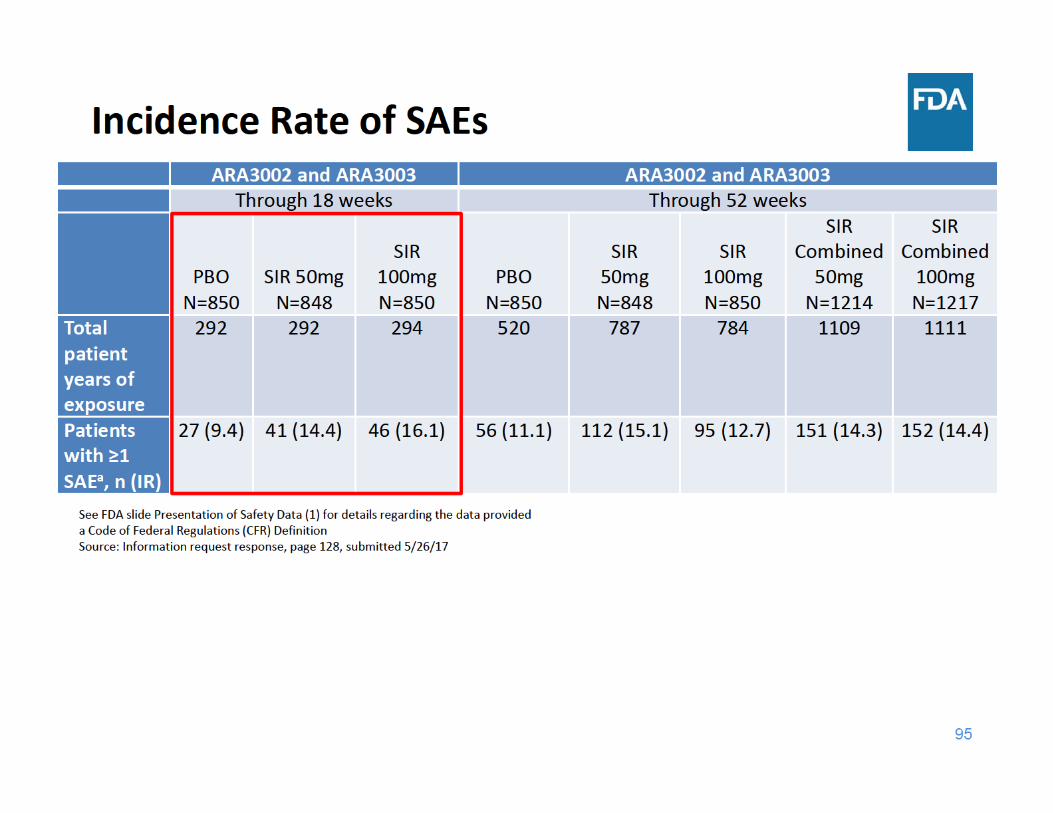
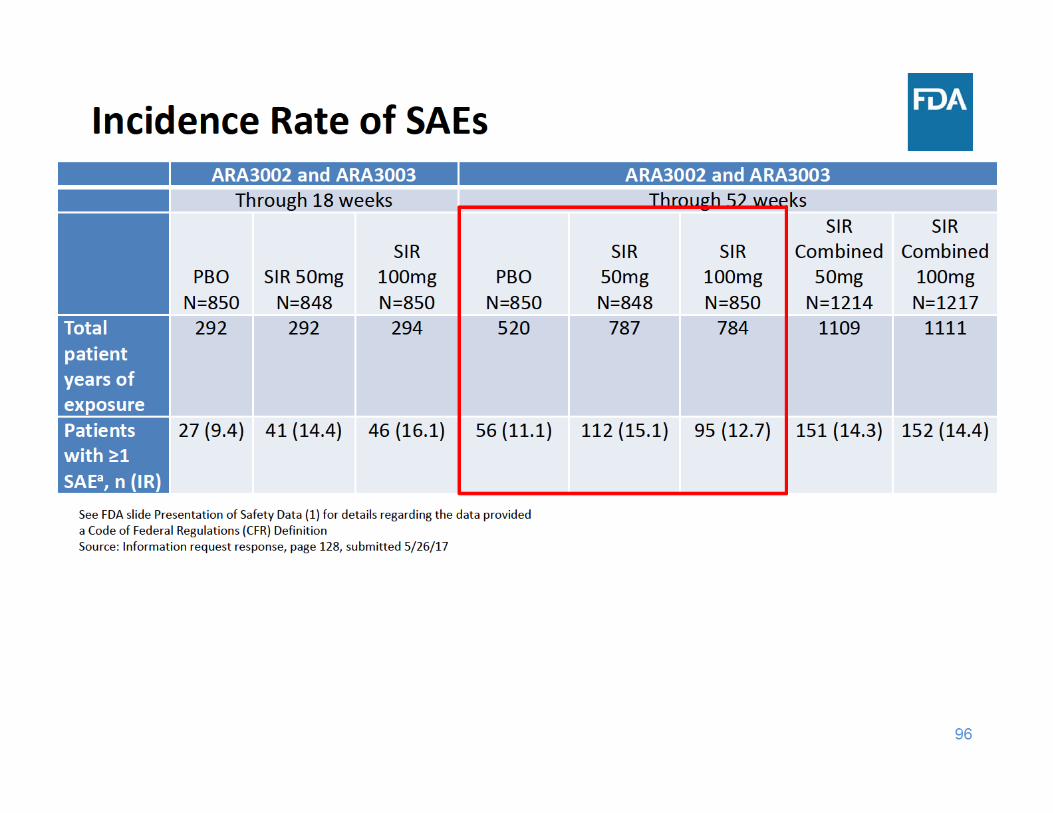
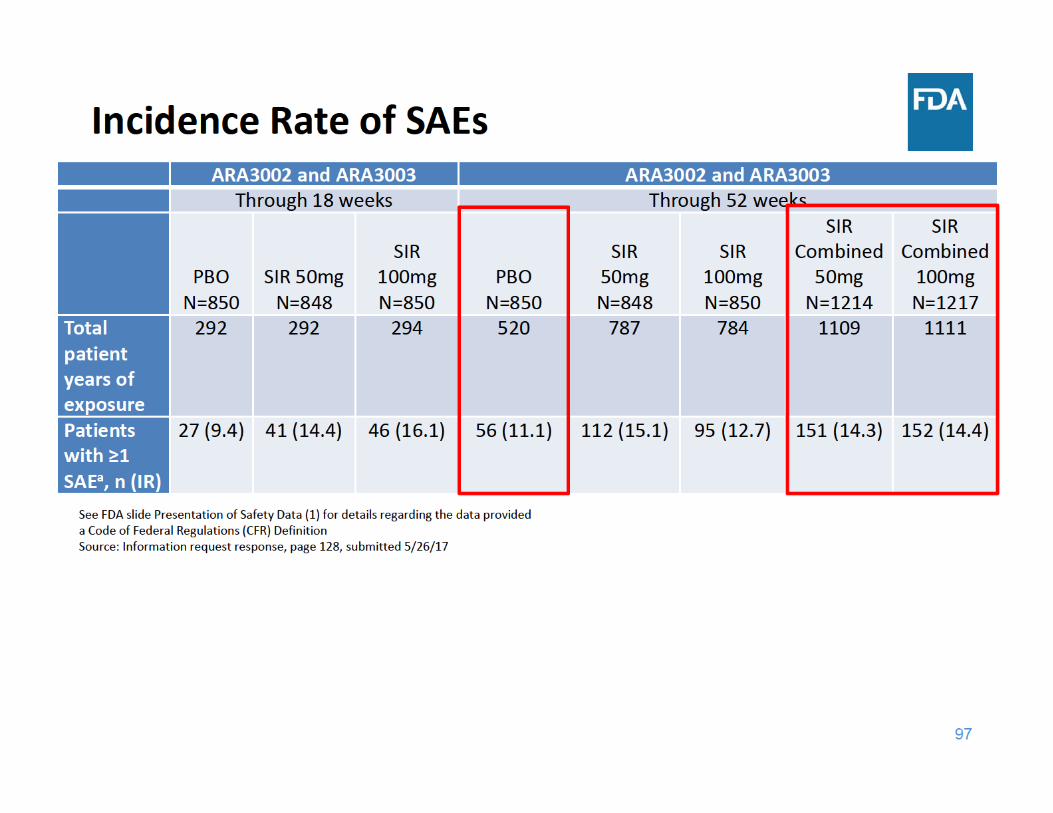

98
SAE Summary
• Through 18 and 52 weeks of exposure, the incidence rate of SAEs was higher in each sirukumab group compared to placebo– Adverse events related to infections were the most frequently reported

99
Focus of Safety Presentation
• All‐cause death• Serious adverse events• Major adverse cardiovascular events (MACE)• Infections• Malignancy• Gastrointestinal (GI) Perforation• Laboratory Abnormalities (lipids, neutrophil and platelet counts, and liver function tests)
• Adalimumab comparator study (ARA3005)
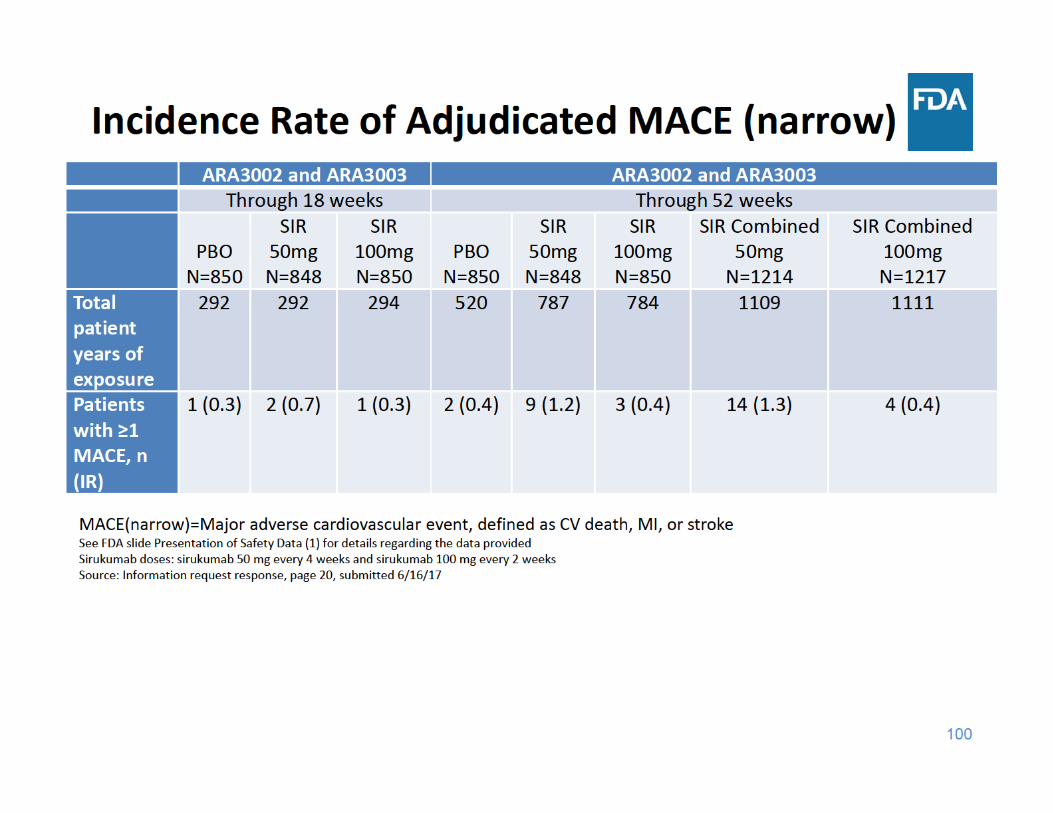
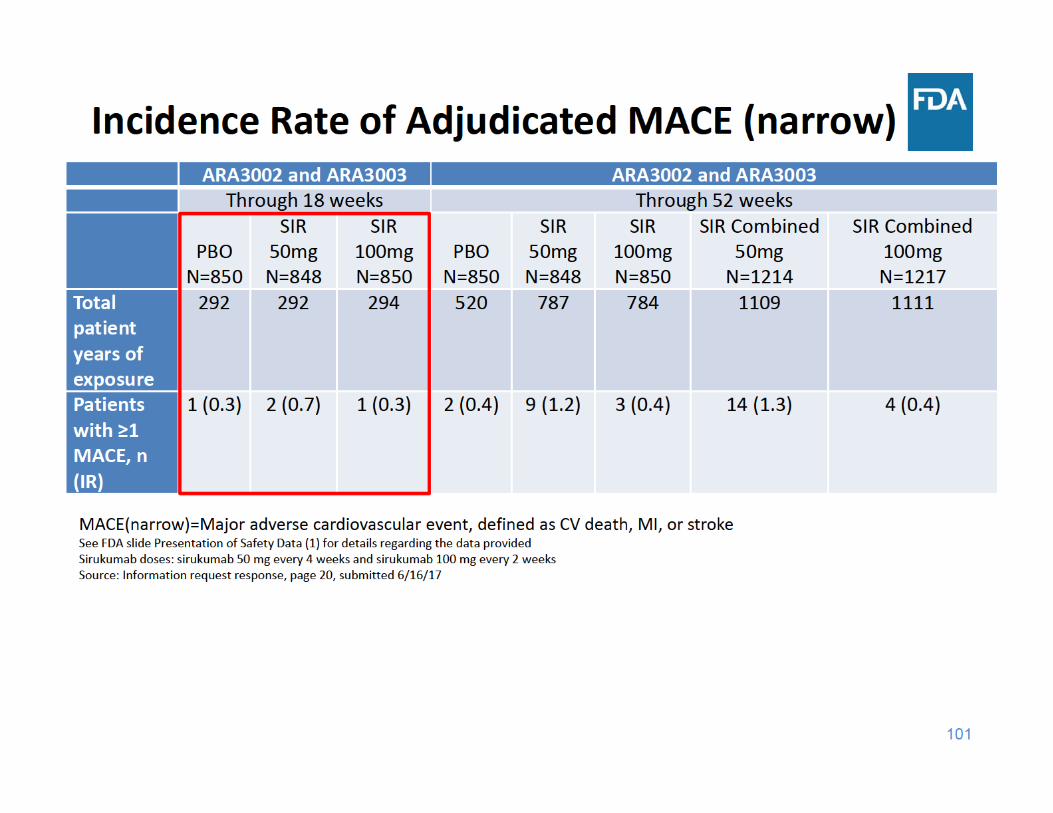
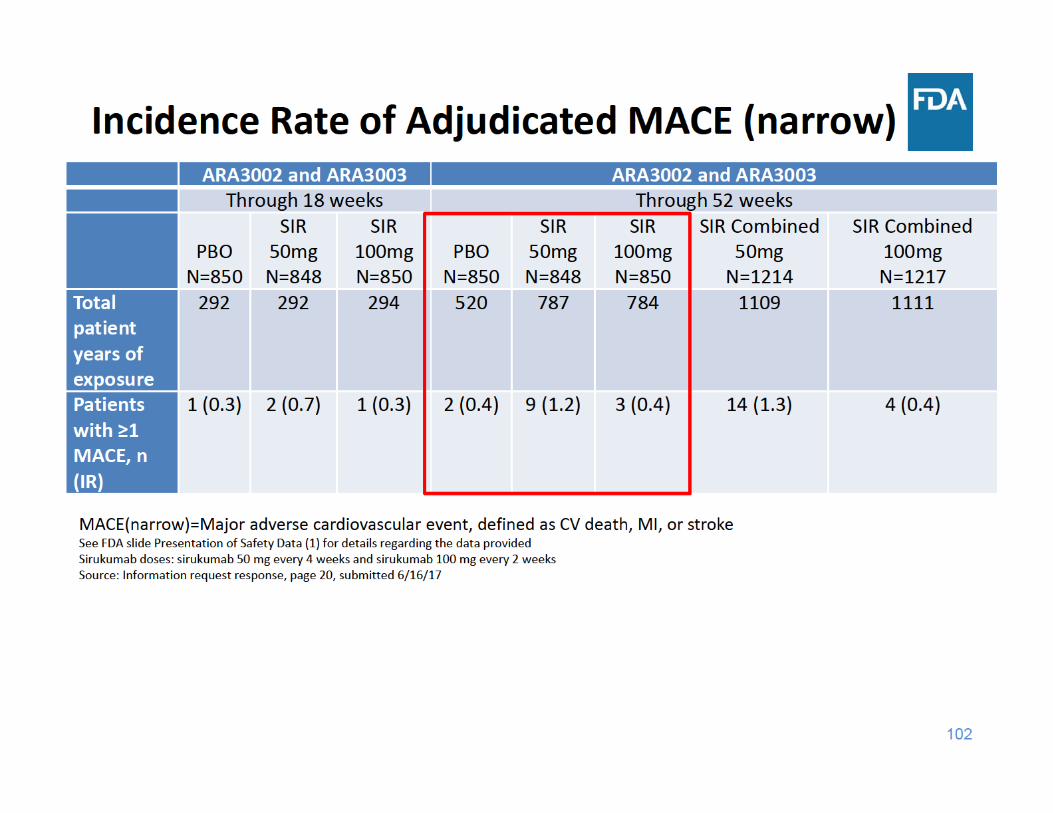
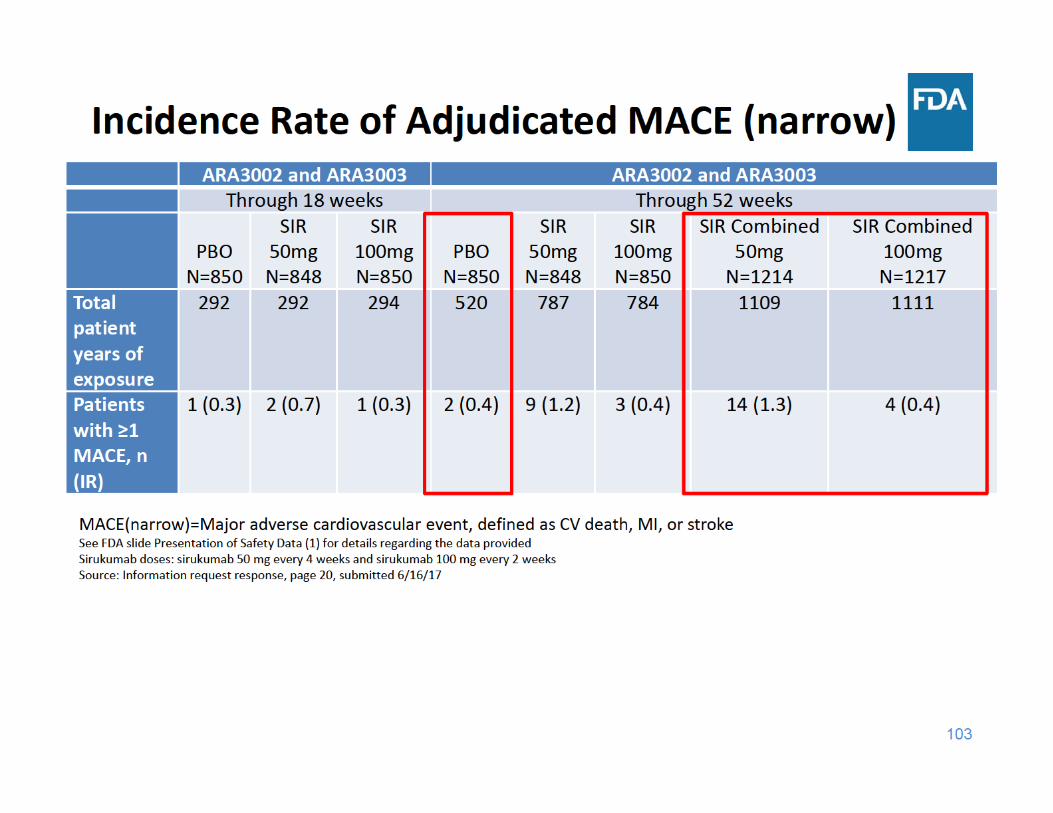

104
MACE Summary
• Through 18 and 52 weeks of exposure, the incidence rate of MACE was higher in the sirukumab 50 mg group compared to the placebo and sirukumab 100 mg groups
• Through 18 and 52 weeks of exposure, the incidence rate of MACE was similar in the sirukumab 100 mg group and the placebo group

105
Focus of Safety Presentation
• Deaths• Serious adverse events• Major adverse cardiovascular events (MACE)• Infections• Malignancy• Gastrointestinal (GI) Perforation• Laboratory Abnormalities (lipids, neutrophil and platelet counts, and liver function tests)
• Adalimumab comparator study (ARA3005)
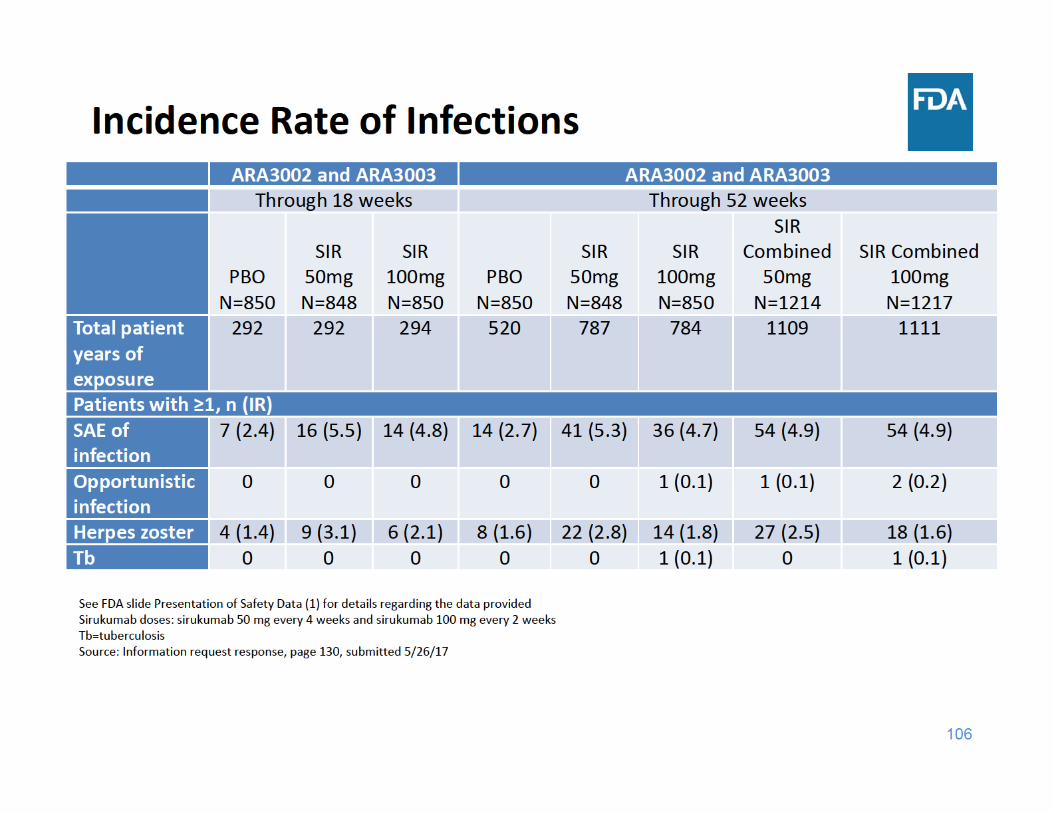
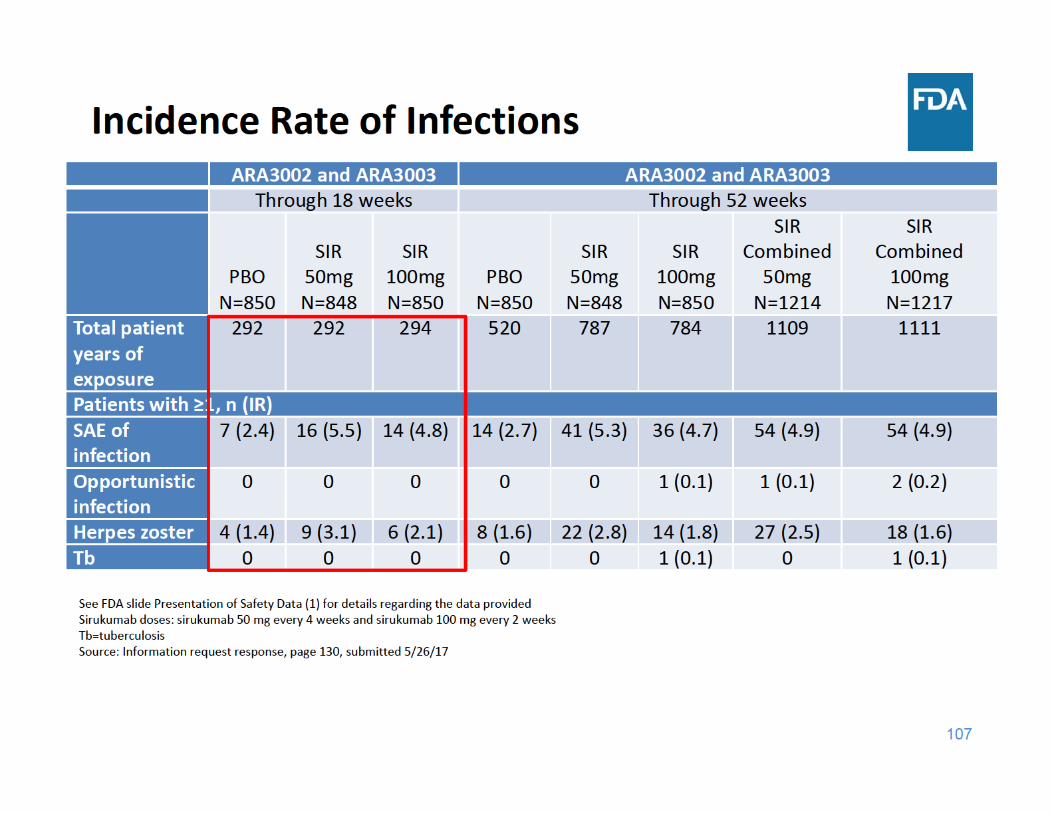

110
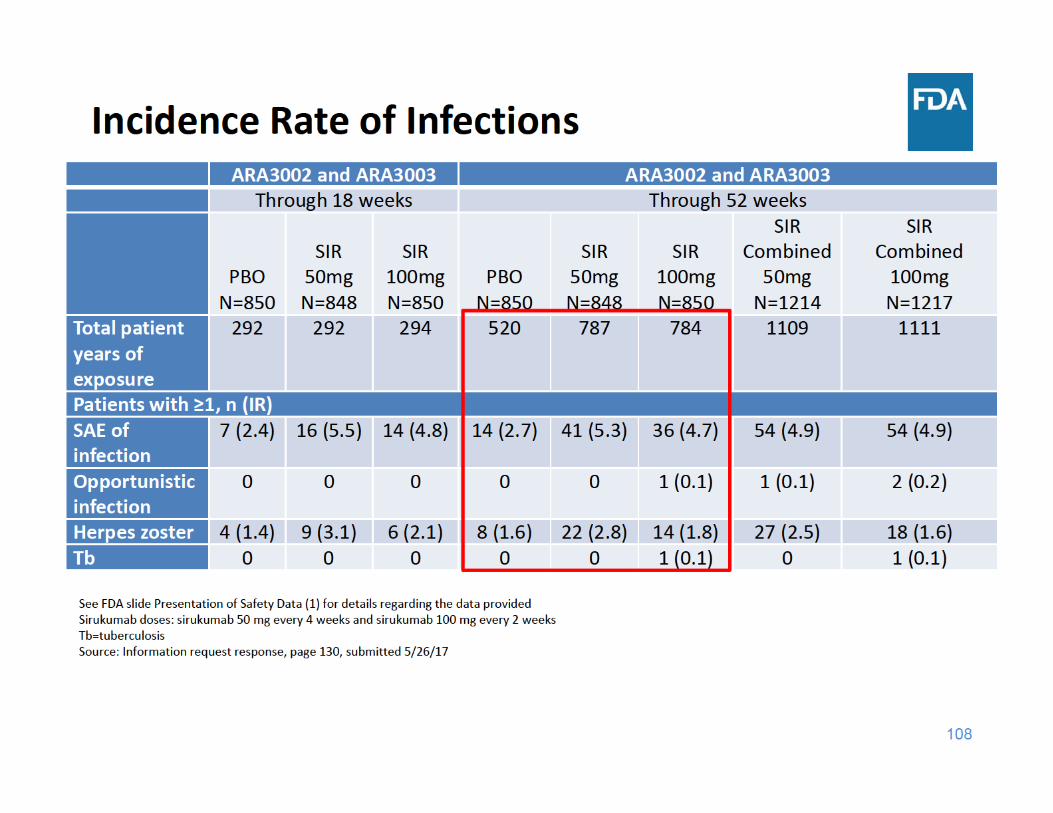
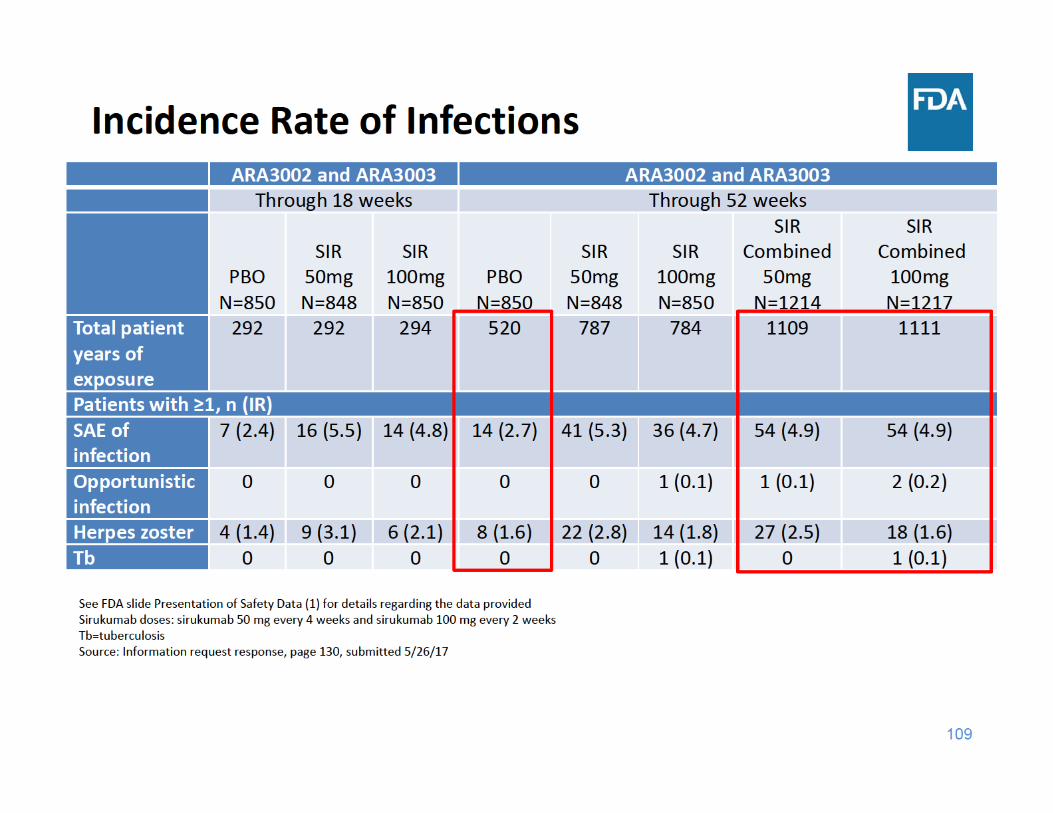
Infections Summary
• Through 18 and 52 weeks of exposure, the incidence rate of SAEs of infection and herpes zoster were higher in each sirukumab group compared to placebo– There were a limited number of cases of tuberculosis and opportunistic infections, but these cases occurred in the sirukumab arms and not in the placebo arms

111
Focus of Safety Presentation
• Deaths• Serious adverse events (SAE)• Major adverse cardiovascular events (MACE)• Infections• Malignancy• Gastrointestinal (GI) Perforation• Laboratory Abnormalities (lipids, neutrophil and platelet counts, and liver function tests)
• Adalimumab comparator study (ARA3005)
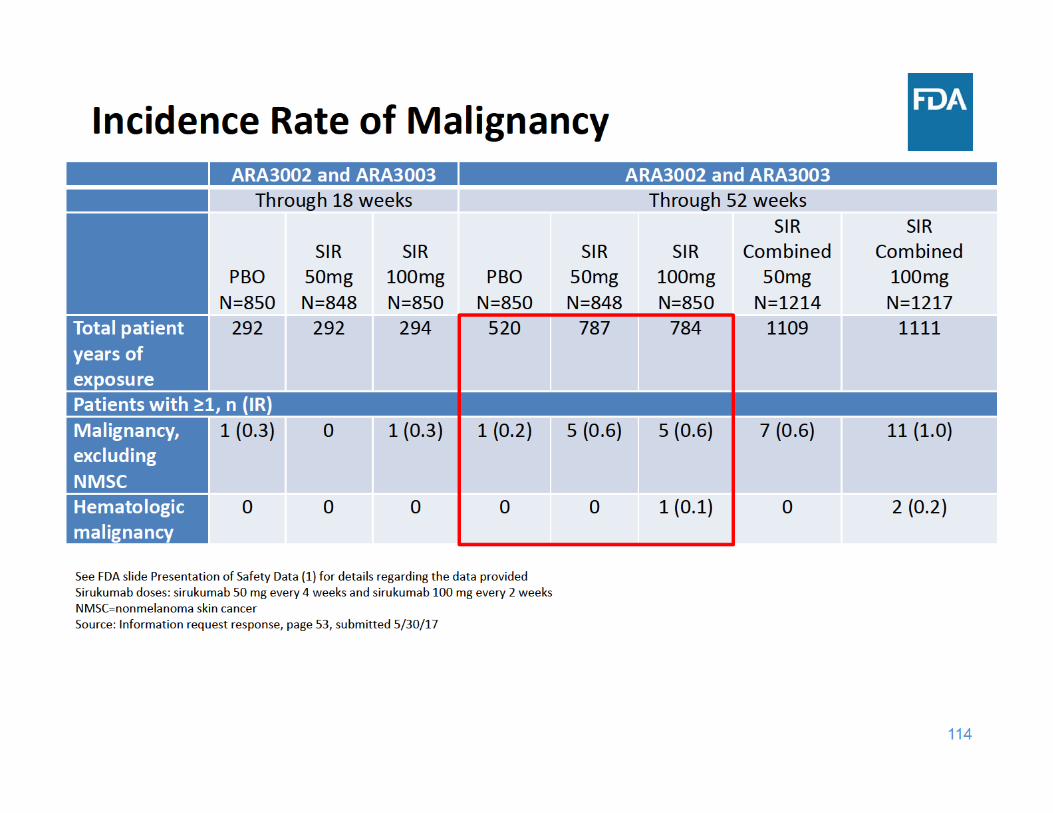
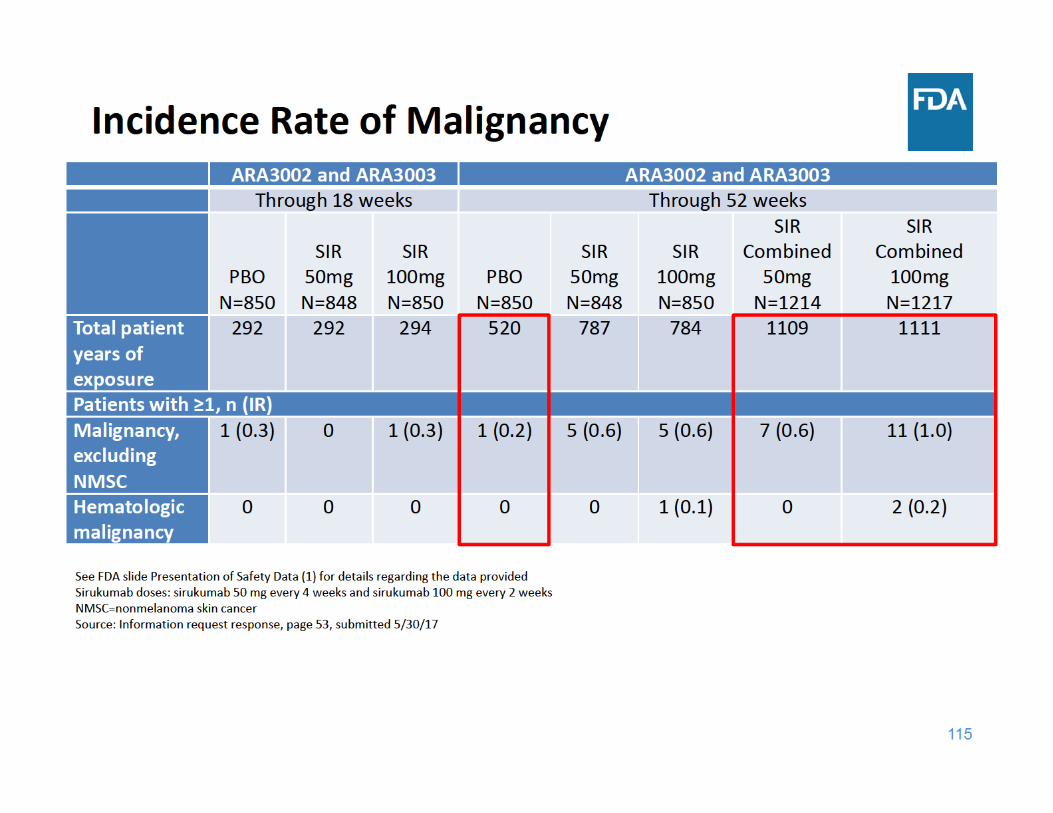

117
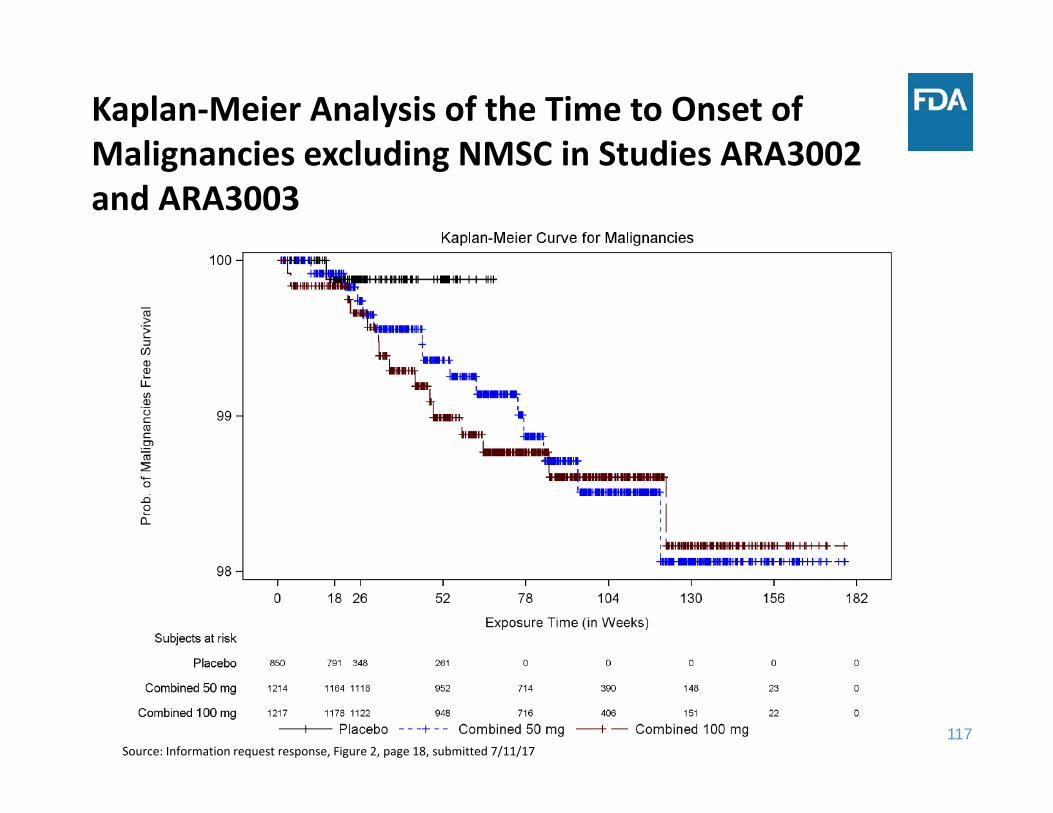
Kaplan‐Meier Analysis of the Time to Onset of Malignancies excluding NMSC in Studies ARA3002 and ARA3003
Source: Information request response, Figure 2, page 18, submitted 7/11/17

118
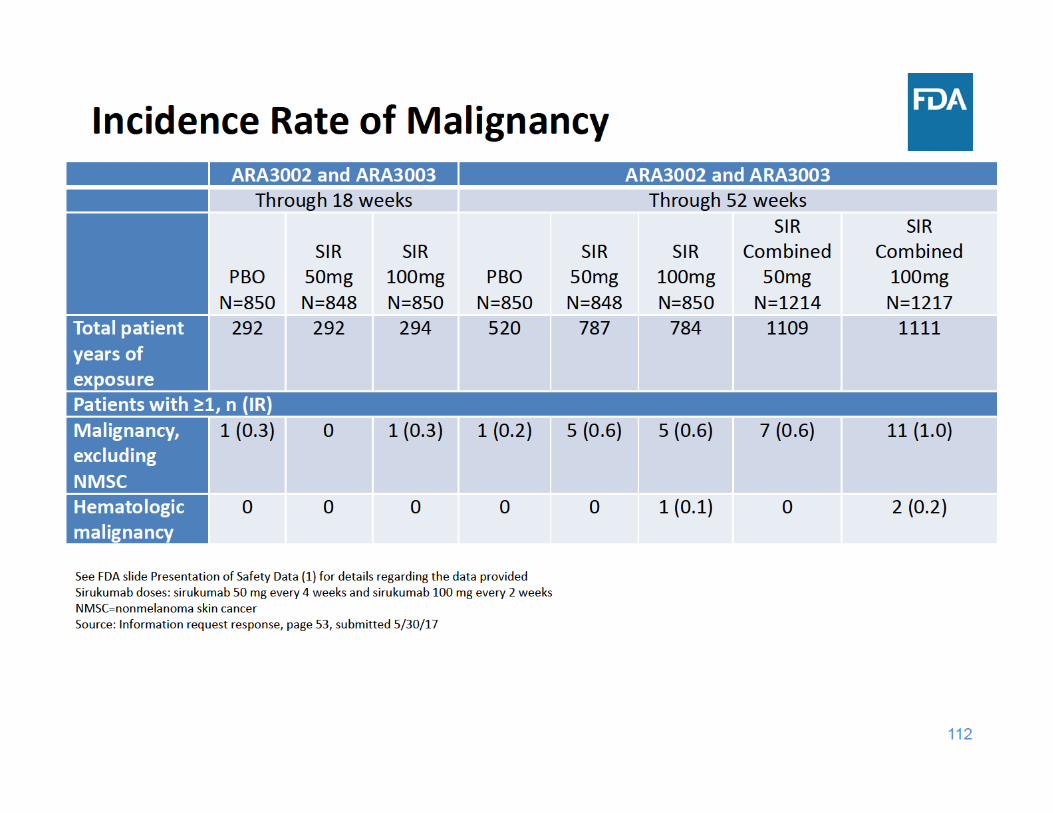
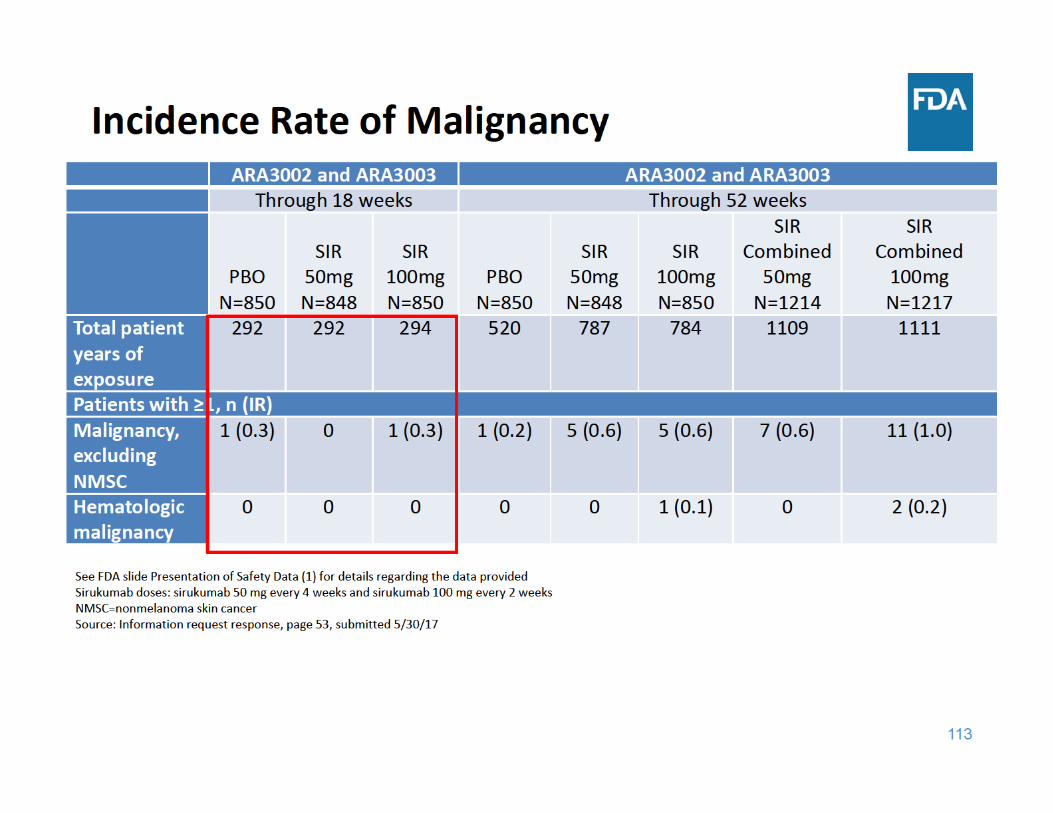
Malignancy Summary
• Through 18 weeks of exposure, the incidence rate of malignancy was the same for the placebo and sirukumab 100 mg groups and lower for the sirukumab 50 mg group
• Through 52 weeks of exposure, the incidence rate of malignancy was higher in each sirukumab group compared to placebo

119
Focus of Safety Presentation
• Deaths• Serious adverse events (SAE)• Major adverse cardiovascular events (MACE)• Infections• Malignancy• Gastrointestinal (GI) Perforation• Laboratory Abnormalities (lipids, neutrophil and platelet counts, and liver function tests)
• Adalimumab comparator study (ARA3005)
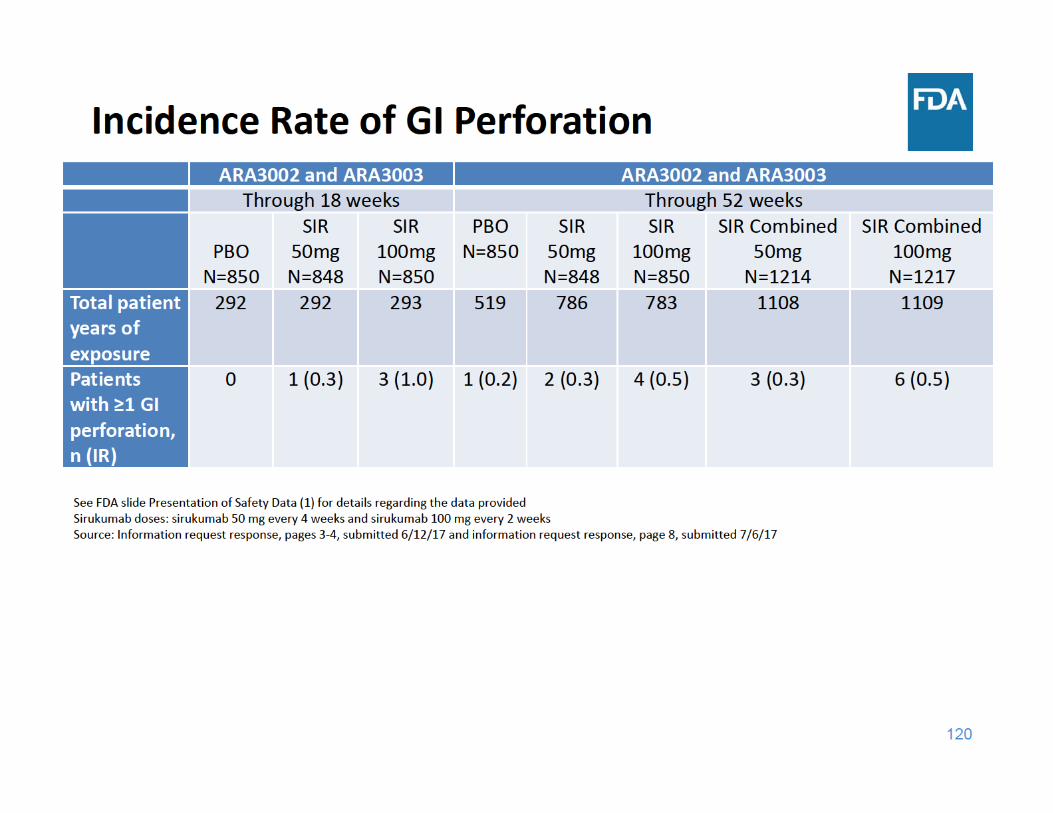
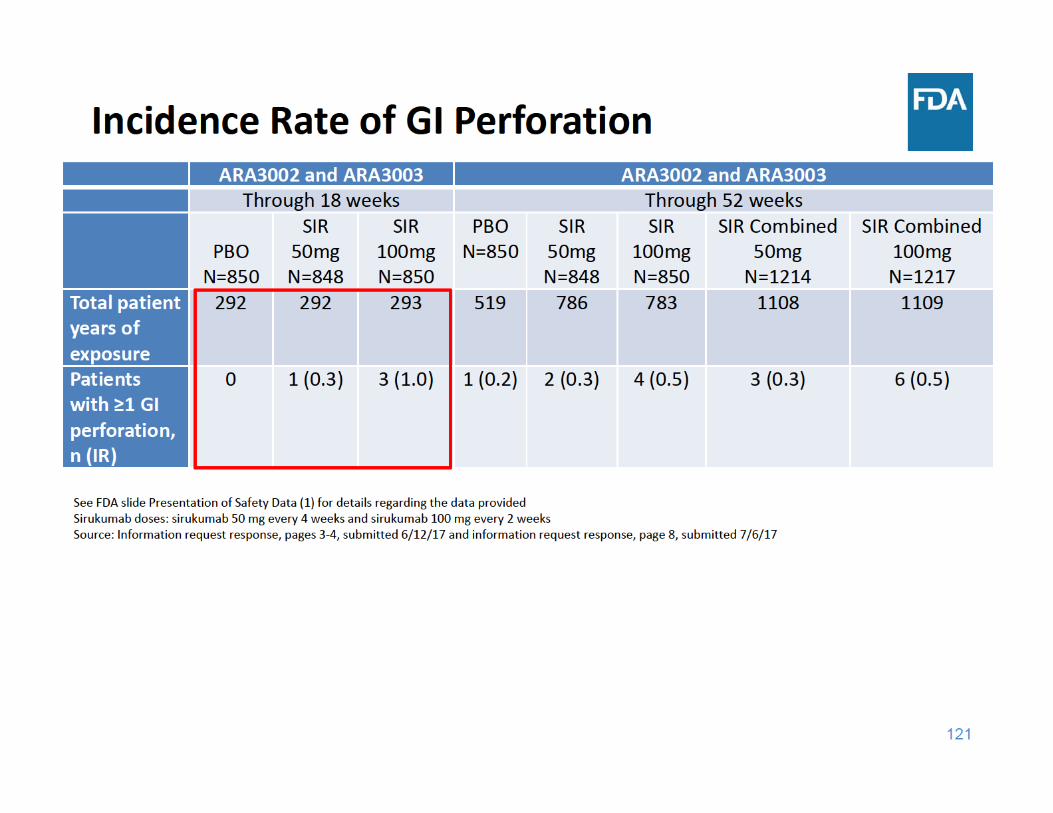
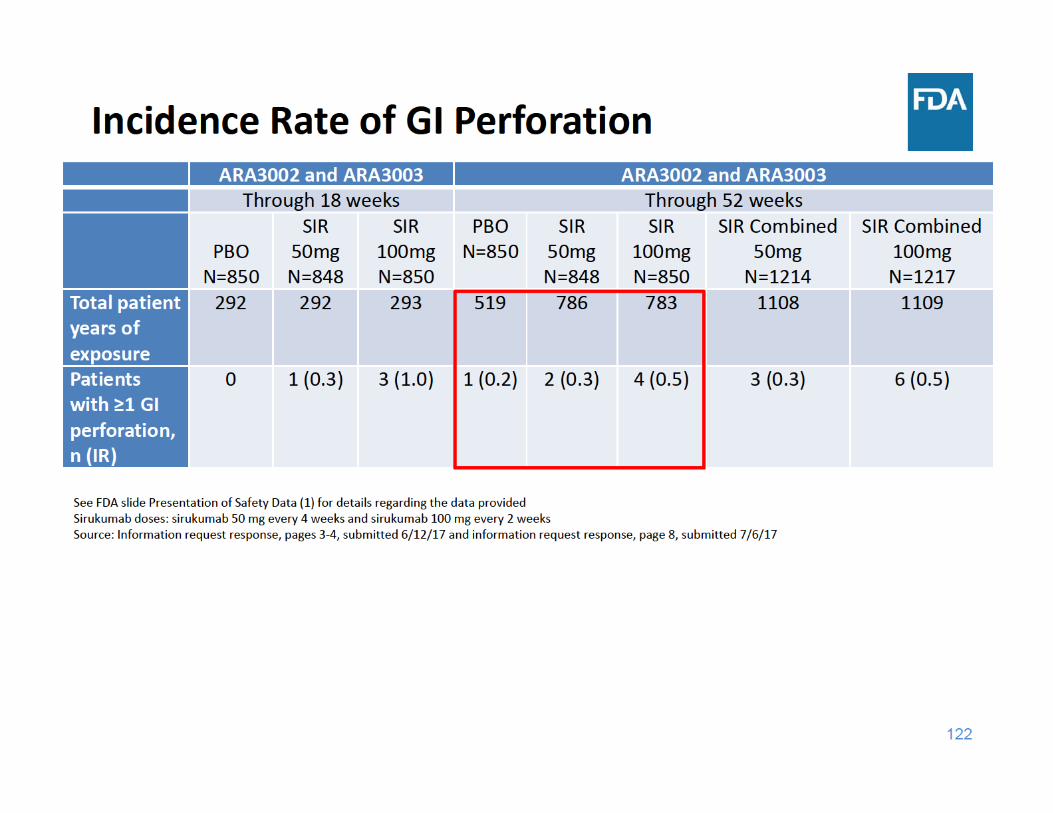
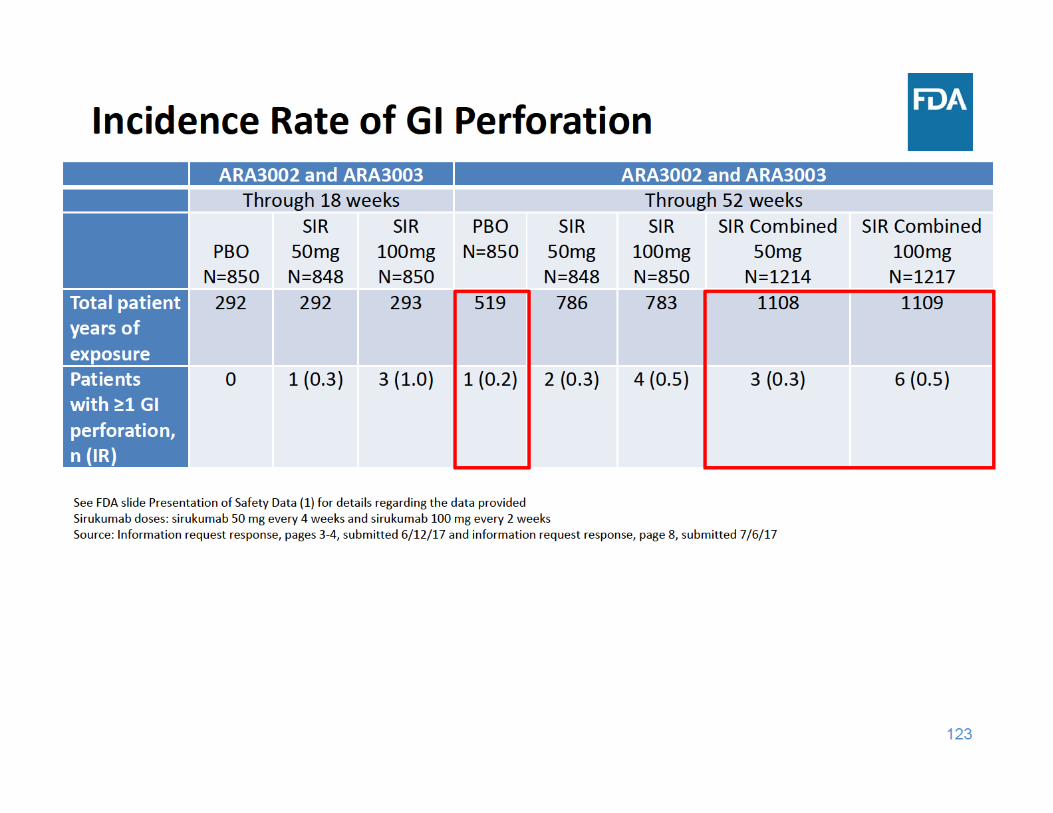

124
GI Perforation Summary
• Through 18 and 52 weeks of exposure, the incidence rate of GI perforations was higher in each sirukumab group compared to placebo

125
Focus of Safety Presentation
• Deaths• Serious adverse events (SAE)• Major adverse cardiovascular events (MACE)• Infections• Malignancy• Gastrointestinal (GI) Perforation• Laboratory Abnormalities (lipids, neutrophil and platelet counts, and liver function tests)
• Adalimumab comparator study (ARA3005)
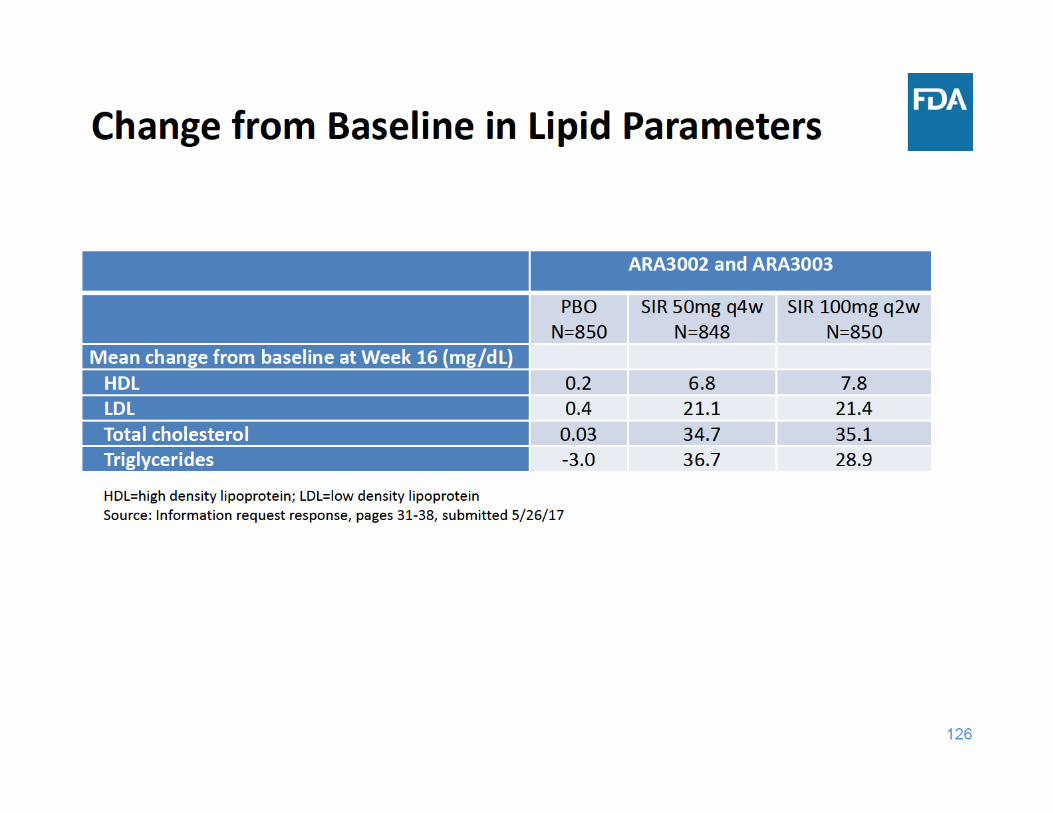
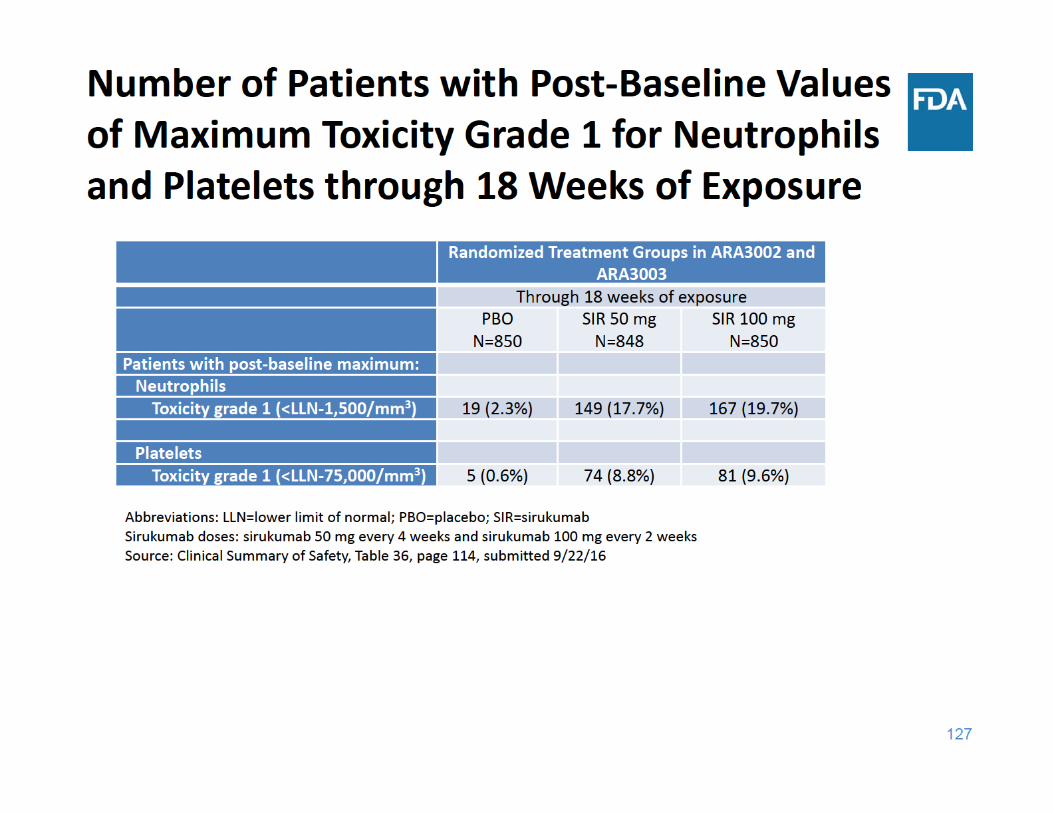
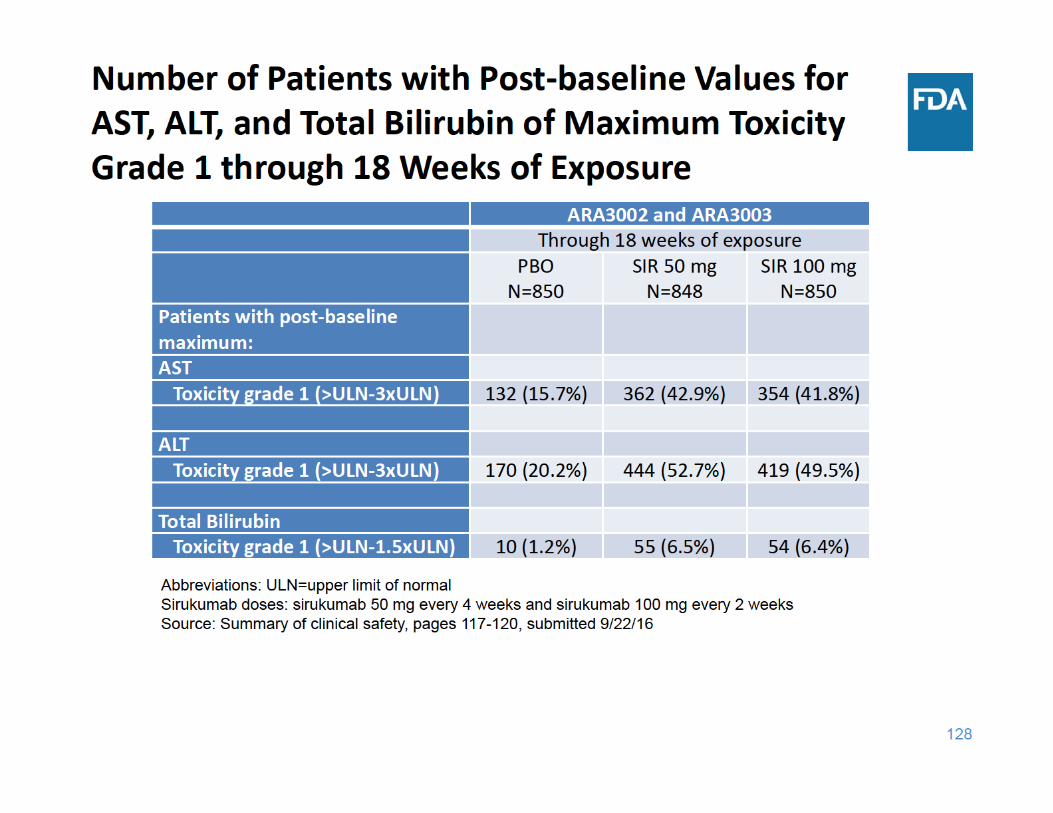

129
Laboratory Abnormalities Summary
• Sirukumab was associated with increases in lipid parameters and liver function tests and decreases in neutrophil and platelet counts – There was no dose‐response for these laboratory changes

130
Focus of Safety Presentation
• Deaths• Serious adverse events (SAE)• Major adverse cardiovascular events (MACE)• Infections• Malignancy• Gastrointestinal (GI) Perforation• Laboratory Abnormalities (lipids, neutrophil and platelet counts, and liver function tests)
• Adalimumab comparator study (ARA3005)
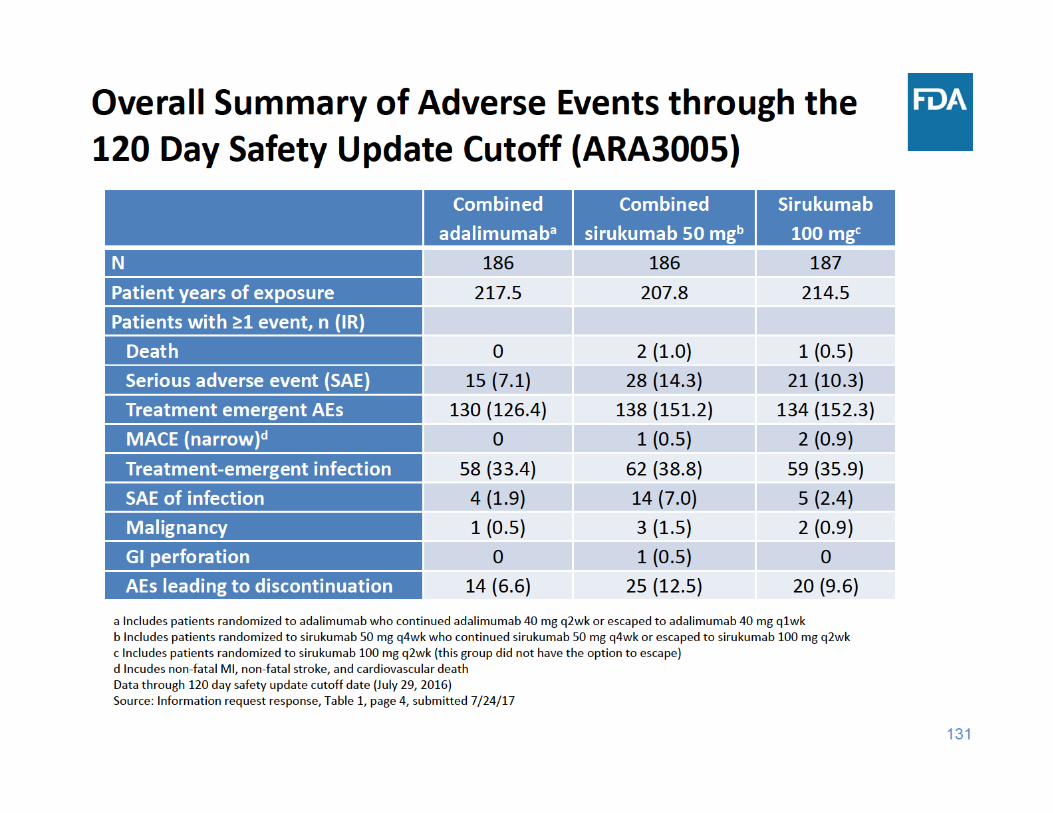
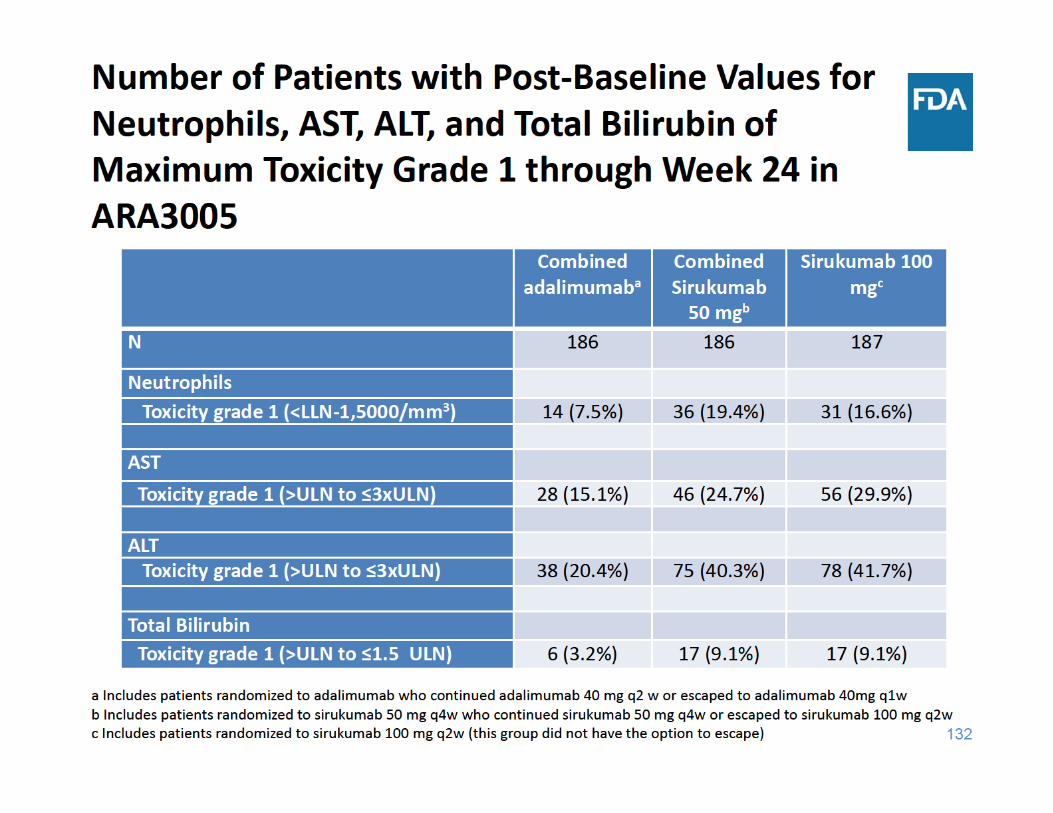
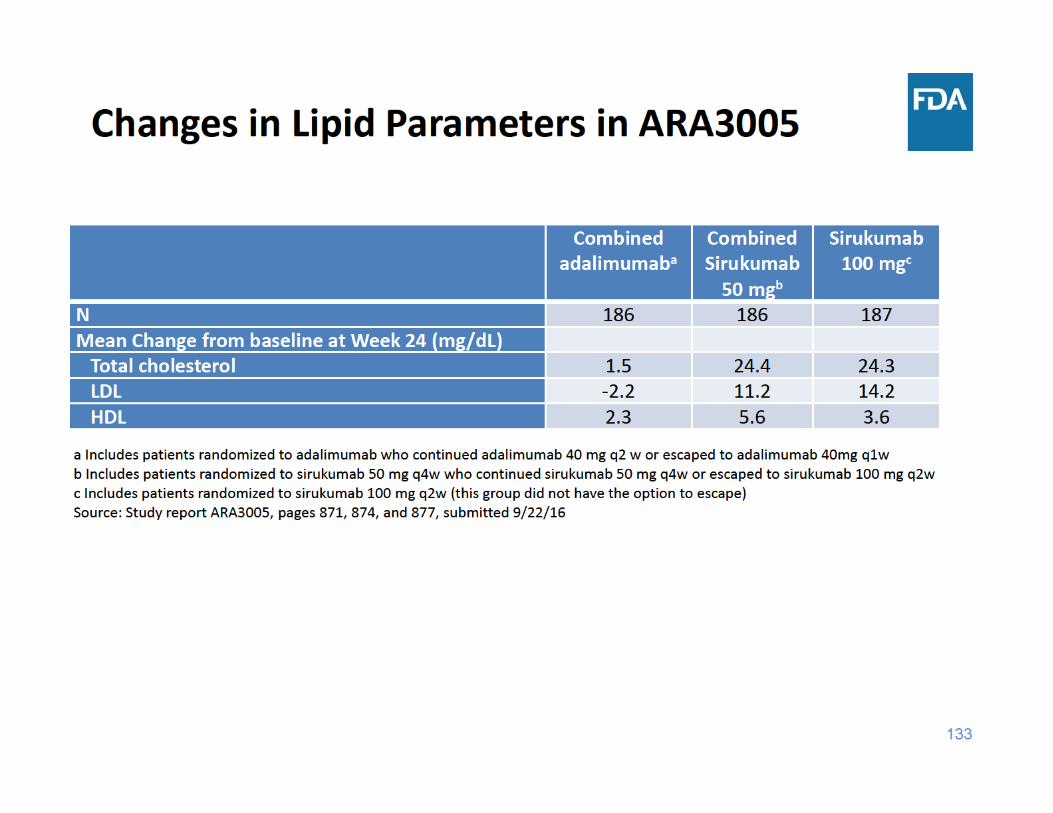

134
ARA3005 Safety Summary
• Compared to adalimumab: – More adverse events of special interest, such as death, malignancy, MACE, and serious infection, with sirukumab
– Sirukumab associated with greater decreases in neutrophil counts
– Sirukumab associated with more elevations in AST, ALT, and total bilirubin, and greater mean changes in lipid parameters

135
Safety summary
• Imbalances in death, serious adverse events, MACE, serious infection, and malignancy noted in the sirukumab program
• Laboratory abnormalities – Lipid elevations, neutropenia, thrombocytopenia, and liver function test elevations
• Additional risks include hypersensitivity and GI perforation

136
Overall Risk/Benefit
• Benefits– Sirukumab superior to placebo for:
– Signs and symptoms– Physical function – Inhibition of radiographic progression
• Risks– Imbalances noted
• Death, MACE, and malignancy
– Serious infection– GI perforation – Laboratory abnormalities
– Hypersensitivity reactions


FDA Arthritis Advisory CommitteeCharge to the Committee
BLA 761057: Sirukumab for the treatment of adult patients with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response or are intolerant to one or more disease modifying anti‐
rheumatic drugs (DMARDs)
Janet Maynard, MD, MHSClinical Team Leader
Division of Pulmonary, Allergy, and Rheumatology ProductsCenter for Drug Evaluation and Research
August 2, 2017

139
Efficacy Considerations
• Efficacy for signs and symptoms (ACR Responses, DAS28), physical function (HAQ‐DI), and radiographic outcomes
• Efficacy of 50 mg q4w and 100 mg q2w was similar• Sirukumab was not superior to adalimumab

140
Safety Considerations
• All‐cause death– Imbalance in all‐cause death with sirukumab over placebo– Rate of all‐cause death was similar with both sirukumab doses– Major causes of death include cardiovascular events, malignancy,
and infections
• Sirukumab associated with imbalances in serious adverse events and gastrointestinal (GI) perforation
• Laboratory abnormalities– Decreases in neutrophil and platelet counts – Elevations in lipid parameters and liver function tests

141
Approval of an Application 21 CFR 314.105 (c)
• “FDA will approve an application after it determines that the drug meets the statutory standards for safety and effectiveness, manufacturing and controls, and labeling”

142
Efficacy Standard21 CFR 314.125 Refusal to Approve an Application
• (b)(5) “… substantial evidence consisting of adequate and well‐controlled investigations … that the drug product will have the effect it purports or is represented to have under the conditions of use prescribed, recommended, or suggested in the proposed labeling.”

143
Safety Standard21 CFR 314.125 Refusal to Approve an Application
(b)(2) “… do not include adequate tests by all methods reasonably applicable to show whether or not the drug is safe for use under the conditions prescribed, recommended, or suggested in its proposed labeling.” (b)(3) “The results of the test show that the drug is unsafe for use under the conditions prescribed, recommended, or suggested in its proposed labeling or the results do not show that the drug product is safe for use under those conditions.” (b)(4) “There is insufficient information about the drug to determine whether the product is safe for use under the conditions prescribed, recommended, or suggested in its proposed labeling.”

144
Question 1 (Discussion)
• Discuss the efficacy of sirukumab for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or are intolerant to one or more disease‐modifying anti‐rheumatic drugs (DMARDs).

145
Question 2 (Voting)
• Overall, do the data provide substantial evidence of the efficacy of sirukumab for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or are intolerant to one or more DMARDs?
a. If not, what data are needed?

146
Question 3 (Discussion)
• Discuss the design of the 52‐week placebo‐controlled radiographic study, ARA3002.

147
Question 4 (Discussion)
• Discuss the safety findings in the phase 3 program, with particular consideration of the imbalance in all‐cause death between sirukumab and placebo.

148
Question 5 (Discussion)
• Discuss the dose selection for the phase 3 program.

149
Question 6 (Voting)
• Is the safety profile of sirukumab adequate to support approval of sirukumab for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or are intolerant to one or more DMARDs?
a. If not, what data are needed?

150
Question 7 (Voting)
• Do you recommend approval of sirukumab at the proposed dose of 50 mg subcutaneously every 4 weeks for the proposed indication of the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or are intolerant to one or more DMARDs?
a. If not, what data are needed?


152
BACK UP SLIDES
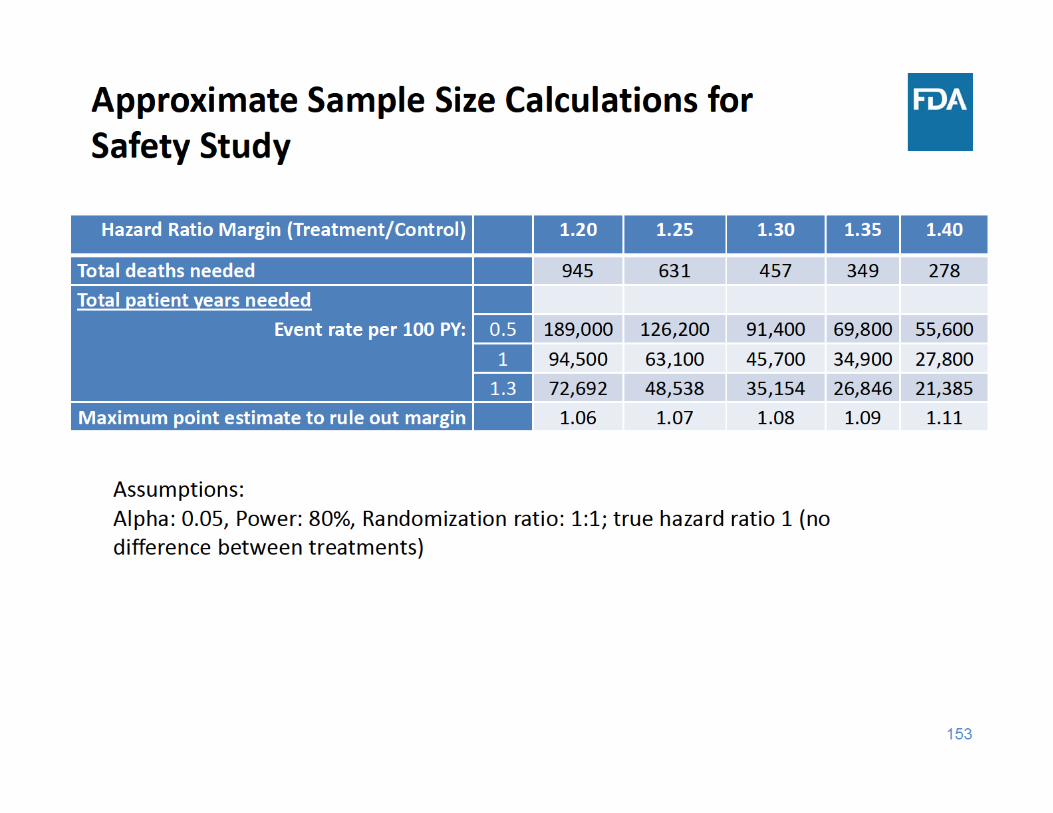
Recommended

















