Embed Size (px)
DESCRIPTION
temas diversos relacionados a la biologia molecular
Citation preview

UNIVERSIDAD AUTONOMA DE COAHUILA
FACULTAD DE CIENCIAS QUIMICAS
BIOLOGIA MOLECULAR
ALUMNA: KRLA JOSEFINA SANTAMARIA LOPEZ
SEXTO SEMESTRE
QFB
ENERO-JUNIO 2014

Resumen
La pirosecuenciación es un método de secuenciación de ADN basado en la monitorización a
tiempo real de la síntesis de ADN. Esta técnica se conoce desde hace algunos años, pero gracias al
impulso dado al desarrollo de nuevas tecnologías de secuenciación más eficientes y baratas se han
conseguido una serie de mejoras que han hecho posible la aplicación de esta técnica para la
secuenciación a una escala genómica y con bajo coste. Existen varias tecnologías basadas en los
principios de la pirosecuenciación, uno de ellas es el sistema GS FLX de Roche que a continuación
describimos como ejemplo.
Concepto
Un aspecto importante de esta técnica es que todos los pasos se realizan “in vitro”, a
diferencia de la tecnología Sanger. Esta técnica comienza con el procesado y adaptación
del ADN para obtener una librería de pequeños fragmentos monocatenarios. Lo primero es
fragmentar el ADN a secuenciar mediante un proceso físico conocido como “nebulización”
donde el ADN se rompe en fragmentos de 200 a 800 pares de bases aproximadamente.
Posteriormente, mediante protocolos estandarizados de biología molecular, se añaden dos
pequeñas secuencias adaptadoras a cada fragmento de ADN obtenido en la nebulización.
El adaptador A y el B. Las secuencias adaptadores están diseñadas para cumplir funciones
en los pasos de selección, amplificación y secuenciación.
El siguiente paso es seleccionar los fragmentos de ADN a los que se han unido
correctamente los adaptadores. Para esto, los fragmentos de ADN se unen a unas esferas
especiales por la parte 3´ del adaptador B. Esta unión no es por complementariedad de
bases. Los fragmentos que solo contengan adaptador A no se unirán a las esferas y son
eliminados y los que contengan dos adaptadores B se unirán por dos puntos. Los
fragmentos de doble cadena unidos a estas esferas son sometidos a alta concentración de
NaOH que provoca la separación de las cadenas simples. Si el fragmento tiene dos
adaptadores B se separarán las cadenas pero ambas permanecerán unidas a las esferas.
Como resultado de este sencillo proceso se liberan de las esferas los fragmentos de
cadena simple que tienen un adaptador de cada clase con el B situado en el extremo 5´
del fragmento, ya que los fragmentos complementarios a estos permanecerán unidos por
el extremo 3´ del adaptador B.
Para el proceso de amplificación, los fragmentos libres de cadena simple se unen a otras
esferas que tienen un gran número de secuencias complementarias del adaptador A. Esta
unión está optimizada para que solo se una un fragmento a una esfera. Una vez unidos los
fragmentos a las esferas se introducen en una emulsión de agua y aceite de forma que
cada esfera quede dentro de una gota con todos los reactivos y encimas necesarios para la
PCR. Tras una serie de termociclos de PCR se habrán generado en cada esfera un gran
número de secuencias de doble cadena idénticas y unidas por el adaptador A. De nuevo,
las esferas son sometidas a alta concentración de NaOH para separar las cadenas
complementarias. El resultado es que cada esfera tiene adherido a su superficie un gran
número de cadenas simples idénticas unidas por el extremo 3´.

Antes de empezar la secuenciación en sí misma se añaden cebadores complementarios al
adaptador B en posición 5´, ADN polimerasas y los cofactores necesarios para la síntesis
de la cadena complementaria a los fragmentos unidos a las esferas.
El siguiente paso es cargar las esferas en un dispositivo conocido como “PicoTiterPlate”
que es el que se introduce en el secuenciador. Este dispositivo consta de más de un millón
de pocillos de 44 micras de diámetro de forma que solo quepa una esfera con fragmentos
de ADN por pocillo. Además de las esferas con ADN, se introducen otro tipo de esferas que
contienen las enzimas necesarias para detectar la incorporación de nucleótidos durante la
síntesis de la cadena complementaria.
Estas enzimas son la sulfurilasa y la luciferasa. La sulfurilasa genera ATP a partir de
pirofosfato y la luciferasa se transforma en oxiluciferina en presencia de ATP con emisión
de luz. De esta forma se construye una cascada enzimática que empieza cuando la ADN
polimerasa incorpora un nucleótido a la cadena creciente complementaria del ADN a
secuenciar liberando un pirofosfato y termina cuando la luciferasa emite luz.
Una vez cargadas las esferas con ADN y las esferas con enzimas, el “PicoTiterPlate” se
introduce en el secuenciador. El secuenciador automatiza el proceso de secuenciación, la
captura de imágenes y su interpretación. El secuenciador vierte automáticamente sobre
los pocillos los reactivos necesarios y un tipo de nucleótido cada vez. Así de forma cíclica
se irán vertiendo As, Cs, Gs y Ts. En cada pocillo, la ADN polimerasa añadirá uno o más
nucleótidos dependiendo de la secuencia que actúa como molde y se emitirá luz con una
intensidad proporcional al númerro de nucleótidos incorporados a la nueva cadena que se
va sintetizando durante el proceso de secuenciación. El secuenciador consta de un sistema
óptico especial que recoge el patrón de destellos luminosos que se emiten en el
“PicoTiterPlate”. Mediante programas informáticos se interpretan estos patrones de luz y
se generan unas gráficas que indican si ha habido incorporación o no de nucleótidos y su
número.
Después de esto se lava el exceso de nucleótidos y reactivos y se repite el proceso con
otro tipo de nucleótido de forma cíclica hasta que finalice la síntesis de una cadena
complementaria a la cadena que actúa como molde. El resultado es la secuenciación de los
fragmentos que hay en cada pocillo.
En los adaptadores hay unas secuencias conocidas que sirven para calibrar los aparatos de
recepción e interpretación de imágenes. Uno de los defectos de este sistema es la pérdida
de precisión en las regiones homopoliméricas.
La posibilidad de automatizar y de paralelizar el proceso de secuenciación que ofrece esta
técnica es una de las principales ventajas frente a otros métodos de secuenciación. La
pirosecuenciación permite la secuenciación de grandes cantidades de ADN en menos
tiempo que otras técnicas y con un coste menor.

4.1 Introducción
La evaluación de la inocuidad de los alimentos derivados de animales MG se debe fundamentar en la comprensión de los métodos de transgénesis y en las aplicaciones previstas y los resultados posibles de la integración y expresión de los transgenes. Sobre esta base, la Consulta examinó los posibles peligros de la expresión de los transgenes para los animales y para los aspectos ecológicos relacionados con la salud humana. La Consulta también examinó las perspectivas futuras de la evolución, utilización y supervisión de la transgénesis con fines de producción animal para el consumo humano.
La Consulta observó asimismo que se estaban obteniendo insectos modificados genéticamente, aunque por el momento no para la producción de alimentos. Aunque las cuestiones planteadas por los insectos modificados genéticamente requerían un debate, quedaban fuera del ámbito de esta Consulta de expertos.
4.2 Técnicas y aplicaciones
4.2.1 Técnicas
Para la transferencia de genes a animales se pueden utilizar varias técnicas (Houdebine, 2003). Difieren en su idoneidad para distintas clases de animales, su eficacia de transformación y sus repercusiones para la evaluación del riesgo.
La utilización del método de transferencia de genes depende del conocimiento de un gen codificador de un producto que confiera un rasgo de interés. El gen que se debe transferir se incorpora a un vector de expresión que también contiene elementos genéticos para controlar su expresión. El uso de distintos tipos de vectores de expresión ofrece ventajas metodológicas para distintas clases de animales y también afecta a la probabilidad de que se materialicen peligros genéticos o inmunológicos posteriores. Los biotecnólogos pueden transferir deliberadamente a un huésped un:
Gen de fusión: Un gen que codifica un producto de interés con un elemento que regulará su expresión en el huésped.
Transposón: Un elemento de ADN capaz de escindirse de un lugar del genoma e insertarse en otro lugar, que se ha modificado para contener el gen de fusión.
Retrovirus: Un virus que se puede integrar en el genoma y expresarse por medio de los procesos de replicación de la célula huésped y que se ha modificado para contener el gen de fusión.
Muchos vectores de expresión contienen genes marcadores. Algunos genes marcadores son simplemente informadores para la transferencia eficaz de un gen, mientras que otros codifican productos génicos, de manera que se

pueden seleccionar individuos transgénicos, por ejemplo mediante la aplicación de antibióticos.
Los métodos habituales para la introducción de un vector de expresión en el huésped son los siguientes:
Microinyección: Inyección directa del vector de expresión en óvulos fecundados o células huésped utilizando una aguja fina de vidrio.
Electroporación: Introducción del vector de expresión en óvulos fecundados o células huésped mediante la aplicación de impulsos eléctricos para inducir poros transitorios en la membrana de las células huésped.
Bombardeo de partículas: Fijación del vector de expresión sobre partículas de oro e introducción en las células huésped mediante el bombardeo con las partículas.
Transformación celular, seguida de clonación: Dado que es más sencillo añadir genes a células cultivadas o inactivarlos en ellas que con los óvulos fecundados, los núcleos de las células transformadas con éxito se pueden transferir a óvulos enucleados e implantarlos en hembras receptoras para generar animales clonados de células somáticas, que también son transgénicos.
Transformación de gametos: Se pueden introducir genes en oocitos o espermatocitos y utilizar los gametos transformados para la fecundación, generando un animal completo.
La aplicación de cualquiera de estos métodos dará lugar a la transformación eficaz de un pequeño porcentaje de los animales así producidos. Luego se pueden identificar los individuos transgénicos y reproducirlos para obtener una línea transgénica.

Principales Criterios
Tamaño
Tamaño ideal: 20-25 nucleótidos de
longitud
generalmente: 18-30 nucleótidos de
longitud
Base en el extremo 3' Debe ser una G o una C
Temperaturas de
fusión (Tm) 50-65 ºC
contenido GC 40-60%
auto-
complementariedad
Debe ser evitada
Para minimizar la formación de
estructuras secundarias y los dimeros
de primer
Similaridad Debe tener un 100% de
apareamiento con el molde
Cebadores para PCR o para Secuenciación
PCR:
Cuando el objetivo a amplificar es un locus, la posición de los
iniciadores debe ser relativa a los codones de inicio y
terminación. Es recomendable que el cebador "forward" se
encuentre mas o menos a 35 pb del inicio de la secuencia que
codifica, de la misma forma el cebador "reverse" debe
localizarse 35 pb despues de la región que codifica.

SECUENCIACIÓN
Cuando el objetivo es obtener la secuencia de ADN es
recomendable que los cebadores se localicen por lo menos 50
pb antes de la región que se desea secuenciar, esto para el
cebador "forward" cuando hay uno, y 50 pb despues de la
región a secuencias para el cebador "reverse", cuando hay uno.
Es importante que el fragmento a secuenciar no sea muy
grande, debido a que los errores de lectura de secuencia
aumentan cuando los fragmentos son grandes, si es necesario
secuenciar un fragmento muy largo, es conveniente diseñar
cebadores internos, para obtener la secuencia en dos partes y
luego ensamblarla.
Temperatura de Asociación
La temperatura de asociación de los cebadores es uno de los
factores mas determinantes de la reacción. Se recomienda que
se emplee como temperatura de asociación la temperatura de
fusión -5ºC, aproximadamente. Existen muchos métodos para
calcular la temperatura de fusión, pero siempre, despues de
efectuado el cálculo es necesario ir al laboratorio y ensayar con
diferentes temperaturas de asociación cercanas a la
temperatura de fusión para determinar la temperatura óptima
para cada reacción.
Cálculo de la temperatura de fusión (Tm) (2)
Existen muchos métodos para estimar el valor de la
temperatura de fusión.
Para secuencias de 20 bp o menos, existe la siguiente
aproximación:
Tm = 2ºC (A + T) + 4ºC (G + C)
donde se asume una concentración de sal de 0.9M, tipica de "dot blot" y otros ensayos de hibridización A continuación tenemos otra expresión para el calculo de Tm

Tm = 81.5 + 0.41(%GC) - 500/L + 16.6 log[M],
donde L se refiere a la longitud del oligonucleótido, y [M] es la concentración de cationes monovalentes. Esta fórmulas es unicamente aplicable a asecuencias polinucleotidicas largas.
El método mas preciso para estimar la Tm de oligonucleótidos está basado en el análisis termodinámico del proceso de fusión del cual se puede mostrar que,
Los cambios en la entalpia( ) y la entropia( ) de la formación del duplex se calculan a partir de parametros termodinámicos de vecindades. R es la constante molar de los gases (1.987 cal.K-1mol-1), y C es la concentración molar del oligonucleotido. Se puede adicionar un secundo término a la ecuación anterior para tener encuenta el efecto estabilizante de la sal sobre el duplex:
Los efectos de Na+ y K+ son equivalentes dentro de los margenes del error experimental.

BIO
QU
IMIA
VO
L. 2
5 N
O. 1
- 98
- 20
00
14
HISTORIA
El desplazamiento de sustancias bajo la acción de un campoeléctrico fue citado por Reuss en 1809 en las memorias de laSociedad Imperial Natural (Moscow) (1809) 1.
Allí se describe la experiencia de la fig. 1, donde se observa elcomportamiento migratorio de pequeñas partículas de arena enun ámbito de agua transparente contenido en un recipiente devidrio con un lecho de arena fina en su fondo y dos tubosconteniendo electrodos de una batería.
El pasaje de la corriente produce un enturbiamiento en lasproximidades del polo positivo producido por la migración departículas de arena muy pequeñas que se movilizan por su cargaeléctrica negativa.
Este experimento puede ser considerado como el primeraporte bibliográfico que revela la polarización de la sílice, pues laarena es dióxido de silicio y fundida permite obtener los capilaresque se emplean en la electroforesis capilar.
Varios años más tarde, en 1816, fue observado el transportedel agua por acción de la corriente galvánica generada por lapolarización negativa del capilar que une los dos recipienteselectródicos.
La pared del capilar, sabemos hoy, adquiere carga negativa (alpasaje de la corriente eléctrica) produciéndose la polarización delagua, la que por consiguiente tiende a desplazarse hacia el polonegativo. Esta corriente líquida que se desplaza en sentido opuestoa la dirección de la corriente eléctrica, se designa con el nombre deFuerza electroendosmótica (FEO) (fig. 2)
Definición de electroforesis
Ha sido definida como el movimiento o desplazamientodiferencial de especies cargadas (iones), sustancias neutras omigración pasiva, por atracción o repulsión en un campo eléctrico.La electroforesis en solución en medios libres sin elementossoportes (agar, almidón, geles de poliacrilamida), fue desarrolladapor Tiselius 2, 3, el que resolvió mezclas de proteínas en un tuboaplicando un campo eléctrico de corriente continua, no pulsante,estudios que lo hicieron acreedor al Premio Nobel en Química.
Los problemas experimentales que se presentaron, estuvieronvinculados a la difusión térmica y a la convección, lo que determinóel empleo de medios anticonvectantes como agar, agarosa ypoliacrilamidas en forma de geles.
Las técnicas que más se han desarrollado están en la actualidadaplicadas al campo de las proteínas y productos de la biologíamolecular, como son los geles de agarosa y proliacrilamida enplacas o films de distinto espesor verticales u horizontales 4 (fig3).
Electroforesis capilar
El empleo de capilares para la separación de sustancias neutras oiones cargados eléctricamente, apareció en 1967 en una experienciadesarrollada por Hjerten empleando capilares milimétricos, losque eran rotados a través de su sección longitudinal para evitarlos efectos de la convección.
Virtanen y Mikkers en 1979 desarrollaron las separacionesempleando electroforesis en capilares de 200 µm de diámetrointerno en vidrio y teflón respectivamente.
La separación de cloruro, aspartato y glutamato porisotacoforesis, hecha por Martin en 1942, los trabajos deKonstantinov, Oshukova en 1963 y los de Evereast en su tesisde graduación en 1964, fueron los precursores en el empleo decapilares y la medición de la absorción UV a través del mismo,permitió obtener los espectros característicos de la separación 5.
Más adelante, en 1980, Jorgenson y Lukacs empleando técnicasavanzadas en la obtención de capilares de sílica fundida empleandiámetros de 75µm y Jorgenson clarifica teóricamente lasrelaciones entre los parámetros operacionales y las cualidades dela separación revelando el elevado potencial analítico de esta técnica.
Esta separación de péptidos realizada sobre un capilar de sílicafundida a potenciales elevados 20 a 30 Kv en un campo de 400 a500 v/cm refrigerados por aire, fue el lanzamiento de la EC 6.
Estos capilares de 75 a 100 cm de largo y con diámetrosinternos de 50-70-100 µm y de 300 a 400 de diámetro externoson los que permiten una capacidad elevada de resoluciónN>200.000 platos/m.Figura 1. Migración de las partículas de sílice
Figura 2. Fuerza electroendosmotica
Figura 3. Distintos tipos de electroforesis

BIO
QU
IMIA
VO
L. 2
5 N
O. 1
- 98
- 20
00
15
La corriente electroendosmótica (FEO) generada por los grupossilanol de la superficie interna del capilar da como resultado unacorriente plana del frente del líquido que contrasta con el frenteparabólico de la cromatografía líquida de alta resolución (fig. 4).
La ventaja de esta técnica es que el capilar de sílica fundida quegeneralmente se cubre con una capa de polimina para darle mayorrigidez y resistencia, tiene una ventana a través de ella que permiteel pasaje de la luz UV de tal manera que la visualización es on-line.
Con esta técnica descrita es posible separar cationes, aniones,proteínas, macromoléculas y sustancias no cargadas en formasimultánea.
Breves nociones teóricas
La separación por electroforesis está basada en la diferencia develocidad de los solutos en un campo eléctrico. La velocidad deun ion está dada por
v = µe E (1)
donde:v : velocidad iónicaµ e : movilidad electroforéticaE : campo eléctrico
Existe una relación entre el voltaje aplicado y la longitud delcapilar en volts/cm, la movilidad de un ión es una constante, laque puede ser determinada por el coeficiente friccional a través deun medio elegido.
fuerza eléctrica (FE) µe x (2)
fuerza friccional (FF)
La fuerza eléctrica está representada en la siguiente ecuación:
FE = q E (3)
Y para un ión esférico la fuerza friccional está determinada porla ecuación:
FF = - 6 π η r v (4)donde:
q: carga iónicaη: viscosidad de la soluciónr: radio iónicov: velocidad del ión
µe= q E 6 πηrE
µe= q
6 πηr
En el estado estacionario, las fuerzas son iguales y opuestas:
q E = 6 π η r v (5)q E = 6 π η r v µe E
Con lo expuesto queda en evidencia que especies altamentecargadas tienen movilidades elevadas.
La movilidad electroforética se le encuentra usualmente en lastablas de constantes físicas.
Corriente electroendosmótica
El componente efector más importante en la EC es la electroen-dosmosis o corriente electroendosmótica, o fuerzaelectroendosmótica.
Surge como consecuencia de aplicar un campo eléctrico que gen-era una doble capa eléctrica entre la solución y la pared del capilar.
En condiciones acuosas, la fase sólida posee un exceso decargas negativas, como resultado de dos procesos físico-químicos,la ionización de la superficie (que es un equilibrio ácido-base) ypor otra parte la adsorción de especies iónicas sobre la superficiedel capilar.
Figura 4. Comparación entre EC y HPLC
Figura 5. Desarrollo de flujo electroendosmóticoa) Superficie de sílica fundida cargada negativamente (Si-O). b) Acumu-lación de cationes hidratados cerca de la superficie. c) Flujo de cargashacia el cátodo luego de la aplicación de un campo eléctrico.

BIO
QU
IMIA
VO
L. 2
5 N
O. 1
- 98
- 20
00
16
Estos procesos ocurren en el capilar al paso de la corrienteeléctrica y la FEO se encuentra altamente controlada por losnumerosos grupos silanoles (SiOH) que también pueden existircomo SiO (fig. 5a).
Los círculos en grisado representan las moléculas de sílicatransformadas por hidratación en silanoles en medio acuosocuando pasa la corriente eléctrica (fig. 5b).
Los contraiones (cationes en la mayoría de los casos) mantienenel balance de cargas; ese potencial se llama potencial zeta. Cuandose aplica el voltaje a través del capilar, los cationes que forman ladoble capa eléctrica migran hacia el polvo positivo (fig. 5 c).
Por consiguiente, la doble capa eléctrica tiene un balance queestá a su vez en equilibrio con la pared que corresponde alpotencial zeta.
La magnitud de FEO puede ser expresada en términos de lamovilidad y velocidad por la fórmula siguiente:
VFEO = (εξ / η) E oµFEO = (εξ / η)
donde:
VFEO: velocidadµFEO: movilidad FEOε : constante dieléctricaξ : potencial Z
A pH elevados los grupos silanol son desprotonados y laFEO es significativamente mayor que a pH bajo donde sonprotonados.
De la electroforesis en papel de filtro a la electroforesiscapilar 7
El papel de filtro es un conglomerado de fibras de celulosa que seembeben en una solución buffer: éstas por su composiciónquímica, al paso de una corriente eléctrica se polarizannegativamente mientras que el agua se carga positivamente y migraen sentido opuesto a la dirección de migración de la corriente.
A esta corriente líquida, conocida como corrienteelectroendosmótica, se suman o se oponen las corrientes de líquidoproducidas por la evaporación e (->) y e (< -), la migración de un
compuesto cargado positiva o negativamente será hacia el cátodoo el ánodo según actúen las fuerzas (fig. 6).
Si consideramos un capilar de sílice, el fenómeno es muy simi-lar, pero la evaporación es nula. En la pared del capilar, por pasajede la corriente eléctrica se produce una capa eléctrica negativa(silanos) que condiciona la polarización del agua yconsecuentemente tiene lugar una corriente líquida en sentidoopuesto al campo eléctrico aplicado.
La corriente líquida se transforma progresivamente en unafuerza llamada fuerza electroendosmótica (FEO), que esequivalente a la obtenida por acción de la bomba en CromatografíaLíquida de Alta Presión (HPLC).
Esta corriente de líquido actúa transportando a los compuestoscontenidos en el capilar, acorde a la carga eléctrica que adquieren enel campo eléctrico (voltajes desde 20 a 50 V/cm).
El aparato para electroforesis capilar (EC)
Está constituido por una fuente de poder (FP) de alto voltaje (20a 30 Kv) y de 0 a 200µA, y un capilar de sílica de 25 a 75 µm dediámetro interno y de 100 a 200 µm de diámetro externo, el quepuede estar refrigerado por aire o líquido por efecto Peltier.
Los electrodos de platino se encuentran ubicados en elrecipiente que contiene el buffer, que además de servir para sucontacto recibe los extremos del capilar.
El carrousel puede estar termostizado y contiene los buffers ylas muestras. Con un movimiento de ascenso y descenso puedeenviar cada una de las soluciones empleadas en la corrida al capilaro a los recipientes electródicos.
Los capilares de sílica dejan pasar la luz ultravioleta visible adistintas longitudes de onda, generando por arreglos de diodos,los espectros para la identificación de los compuestos separadospor electroforesis capilar (fig. 7).
Figura 6. Polarización de las fibres de celulosa ante el paso de unacorriente eléctrica Figura 7. Esquema de un aparato de electroforesis capilar

BIO
QU
IMIA
VO
L. 2
5 N
O. 1
- 98
- 20
00
17
Aplicaciones
El explosivo avance de la electroforesis capilar se ha extendidoal área biomédica, en el campo de las proteínas, péptidos, ADN,análisis de líquidos de perfusión, monitoreo de drogas,marcadores genéticos tumorales y neurobioquímicos, drogasxenobióticas, de abuso, y pericias forenses.
En el área biofarmacéutica, para el control de calidad deproductos farmacéuticos y biotecnológicos, quimioterápicos y deestructura quiral.
En el área de alimentos, se le aplica al fraccionamiento ycuantificación de aminoácidos, hidratos de carbono, ácidosorgánicos, aditivos y contaminantes.
En el área de control ambiental, permite la identificación decontaminantes y sus metabolitos, pesticidas, metales pesados ehidrocarburos.
Como todo desarrollo de un área analítica nueva, laincorporación de técnicas acopladas como la espectroscopia demasa, fluorescencia inducida por laser y otras variantes permitenaugurar un promisorio futuro.
Desarrollos tecnológicos que contribuyeron a la EC
Seis desarrollos tecnológicos han concurrido para el desarrollocomercial de la instrumentación, que constituye en la actualidad laelectroforesis capilar.
1. Los capilares de sílica fundido revolucionaron lacromatografía gaseosa (CG) y han producido la cámara deseparación de la electroforesis capilar.
2. La disponibilidad de fuentes de potencia de alto voltaje 20 a30 KV, altamente estabilizadas y automatizadas para elmantenimiento estable de la tensión y corriente.
3. Los detectores desarrollados para la cromatografía líquidade alta performance (HPLC), de absorbancia de altasensibilidad pudieron ser aplicados con modificacionesópticas al tubo capilar.
4. Los desarrollos obtenidos por la electroforesis en gelesde tamizado molecular como los de poliacrilamida, agarosay otros, en las técnicas de la separación de proteínas.
5. El desarrollo de los anfolitos, obtenidos por copulacióndel ácido acrílico y las polietilen poliaminas que han permitidogenerar ambientes de pH continuos y estables, donde se lograseparar proteínas de puntos isoeléctricos muy próximos.
6. El análisis computacional aplicado a la resolución de productosobtenidos por separación por HPLC o CE que pueden serestudiados, espectralmente a través de software para tal fin.
ELECTROFORESIS CAPILAR: VENTAJAS
Las separaciones se realizan empleando mecanismos tradicionales,en un ámbito capilar, que además ofrece más facilidad y velocidadque la cromatografía líquida de alta performance (HPLC).
Mientras elimina el problema de los solventes de la HPLC, latoxicidad de los mismos y su costo, pues emplea solucionesacuosas en su gran mayoría con muy baja concentración iónica,incorpora los principios de la automatización a través de un hard-ware creado especialmente con un software altamente optimizado.
Las separaciones se obtienen en pocos minutos, on line con la
corrida, obteniéndose simultáneamente resultados cuantitativos, enoposición a los procedimientos tradicionales que utilizan horas o días.
VOLUMENES EMPLEADOS
El empleo de microvolúmenes y la rapidez de la EC en oposicióna la HPLC, consumiendo microlitros de muestra y volúmenesdel orden de los nanolitros para las soluciones electrolíticas, lahacen una técnica altamente versátil y económica.
INSTRUMENTACION
Los aparatos desarrollados por la industria instrumental reciénaparecieron en 1988, y el primer simposio internacional del añosiguiente, permitió mostrar los avances extraordinarios de estelogro analítico.
Los aparatos comerciales de EC comprenden doscompartimentos electroforéticos unidos por un compartimentoque contiene un capilar, a una fuente de alta tensión de 20 a 30 kv,el que es sometido a la luz UV con un detector unido a unregistrador, que grafica las diferencias de absorbancia; a un sistemade administración de datos. Las terminales de salida permiten,por su conexión, el registro del voltaje, tiempo de corrida, yregistro de la temperatura del capilar, a través de un software queestá incluido en una workstation computarizada (fig. 8)
PRINCIPIO FISICOQUIMICO
Las moléculas son separadas por las fuerzas del campo eléctricoaplicado. Recordemos que la electroforesis ha sido definida comoel movimiento diferencial de especies cargadas (iones) o no, poratracción o repulsión en un campo eléctrico.
Las muestras separadas en el capilar son monitoreadas por eldetector (fig. 9)
Los picos del electrograma son similares a los de la HPLC, y segeneran como consecuencia de la concentración de los analitos yde su velocidad de migración (fig. 9 bis).
Las muestras son introducidas en el capilar por electroforesiso por electrocinética, o por desplazamiento por inyección.
Durante la inyección electroforética, el capilar está sumergidoen la muestra y se aplica el alto voltaje.
Los iones de la muestra migran dentro del capilar en relacióna sus movilidades electroforéticas.
Si la fuerza iónica de la solución muestra es más baja que elbuffer electroforético, los cambios en el campo eléctrico de la interfase
Figura 8. Carrusel para muestras en una EC

I1I SIMPOSIO CIENTÍFICO EN BIOLOGÍA CELULAR Y MOLECULAR
pueden expresarse o no (DNA repetitivo, pseudogenes, etc.), por lo que representan mejor al conjunto del genoma.
Otro aspecto importante es la naturaleza selectiva o neutra de los marcadores. ¿Por qué se insiste tanto en la ventaja de ser neutros respecto a la selección natural?. La razón es tanto técnica como metodológica. Es un hecho que es prácticamente imposible estimar la intensidad de la selección natural sobre caracteres concretos en la propia naturaleza, fuera de los experimentos controlados en el laboratorio, y mucho menos deducir cuál fue en el pasado la intensidad de ésta (Endler, 1986). Todo ello lleva frecuentemente a un razonamiento tautológico: de acuerdo con los principios de la selección natural aquellas alternativas favorecidas aumentarán su frecuencia, por ello todo gen frecuente tiende a suponerse como selectivamente ventajoso; la tautología proviene de olvidar que los cambios en las frecuencias génicas pueden producirse por causas distintas a la selección. La evolución puede producirse por causas diferentes a la selección natural. Por tanto si es difícil saber en algunos casos si un gen es adaptativo o no y siempre es extremadamente difícil estimar la selección natural, es obvio que cualquier modelo que incluya este parámetro es difícilmente comprobable experimentalmente. Sin embargo, para alternativas selectivamente neutras (total o prácticamente neutras) los modelos estadísticos que predicen su cambio no incluyen este parámetro, son de más fácil aplicación y, fundamentalmente, permiten probar la hipótesis de su neutralidad. De hecho, aunque a mediados de los 60 se habían desarrollado ya muchos modelos teóricos sobre la dinámica de los genes en las poblaciones, estas teorías eran usadas raramente para interpretar los datos experimentales excepto en contadas ocasiones. La situación cambió abruptamente cuando se dispuso de datos moleculares sobre el cambio evolutivo de los genes. Desde entonces las teorías se usan profusamente para probar hipótesis alternativas sobre los mecanismos de evolución. La interacción entre la teoría y los datos ha estimulado el trabajo en nuevas teorías matemáticas sobre la evolución molecular y la genética de poblaciones, que a su vez pueden usarse en la comprobación de hipótesis. De particular importancia ha sido el desarrollo de teorías para probar la hipótesis nula sobre las mutaciones neutras (Nei, 1987). Los marcadores moleculares incluyen todo tipo de secuencias: aquellas que son claramente adaptativas, alternativas alélicas neutras que no afectan la secuencia proteica codificada, secuencias sin función (DNA repetitivo, pseudogenes, por ejemplo) que pueden cambiar libremente, etc.
3. MARCADORES MOLECULARES: CARACTERÍSTICAS Y LIMITACIONES
Los marcadores moleculares de uso generalizado pueden clasificarse en función de la técnica empleada para la obtención de los segmentos discretos de DNA: aquellos que se obtienen tras la fragmentación del genoma correspondiente con endonucleasas de restricción (restrictasas), y aquellos que se obtienen por amplificación selectiva de secuencias mediante la reacción en cadena de la polimerasa (PCR, polymerase chain reaction). En algunas técnicas
250

Marcelino Pérez de la Vega
se combinan estos dos procedimientos básicos. Los polimorfismos obtenidos por el primer procedimiento son conocidos como polimorfismos de longitud de fragmentos de restricción (RFLP, restriction fragment length polymorphisms). En este caso los fragmentos discretos de DNA generados por una restrictasa s~m separados por electroforesis en gel, visualizándose selectivamente determinados fragmentos mediante la hibridación con sondas marcadas. En el segundo procedimiento se utilizan cebadores particulares en cada caso para amplificar por replicación selectiva segmentos discretos de DNA. En ambos casos podemos visualizar polimorfismos de segmentos anónimos, cuya secuencia, función y naturaleza nos son desconocidas, o segmentos conocidos, genes conocidos o parte de ellos, secuencias repetidas, etc. Ello depende de la utilización de sondas o cebadores anónimos, elegidos al azar entre miles posibles simplemente porque generan un buen polimorfismo, o que corresponden a secuencias cuya naturaleza conocemos.
Tanto en estudios teóricos de tipo evolutivo como en la utilización práctica de los marcadores moleculares es importante conocer dos de sus características: el carácter dominante o codominante de cada tipo de marcador, y el nivel de polimorfismo que genera. El carácter domínate o codominante determina el nivel de facilidad con que pueden llevarse a cabo estimaciones de parámetros genéticos y evolutivos tan básicos y de uso generalizado como, por ejemplo, frecuencias de recombinación, distancia de mapa, heterocigosidad, distancia evolutiva, etc. El nivel de polimorfismo, y también la naturaleza del propio marcador, determina su utilidad en función del conjunto de organismos a estudiar. Así, por ejemplo, un marcador que genere un nivel bajo de polimorfismo puede ser útil para estudiar especies o géneros dentro de una familia, pero ineficaz para estudiar diferencias entre individuos de una misma población o entre descendientes de un cruzamiento. Esto último es fácil de comprender, si un marcador es poco polimórfico la mayoría o todos los individuos de una población o especie tendrán el mismo fenotipo, lo que lo hace poco útil para diferenciar poblaciones o individuos.
Según Lewontin (1974), inferir la historia de las poblaciones o razas y obtener información sobre los procesos genéticos de la especiación requiere la estimación de frecuencia génicas. La teoría de la Genética de poblaciones es un ejercicio abstracto a no ser que se puedan determinar las frecuencias de alelos alternativos en varios loci, en diferentes poblaciones y en momentos diferentes de la historia de una población. Hoy día disponemos de las técnicas adecuadas para poder hacerlo, pero es obvio que la estimación de las frecuencias génicas es más precisa en genes codominantes que dominantes. Con los primeros los genotipos se observan directamente y la conversión de las frecuencias genotípicas en génicas es inmediata. Con los segundos sólo podemos calcular las frecuencias génicas si se cumplen otros requisitos en la población, por ejemplo que se ajuste a panmixia, lo que no siempre ocurre en particular en especies vegetales, o que los individuos hayan alcanzado una homocigosidad total, lo que por ejemplo es frecuente es especies vegetales autógamas. Pero en cualquier caso, para una muestra del mismo número de individuos, el error estadístico de la estima es mayor con los dominantes que con los codominantes.
251
III SIMPOSIO CIENTÍFICO EN BIOLOGÍA CELULAR Y MOLECULAR
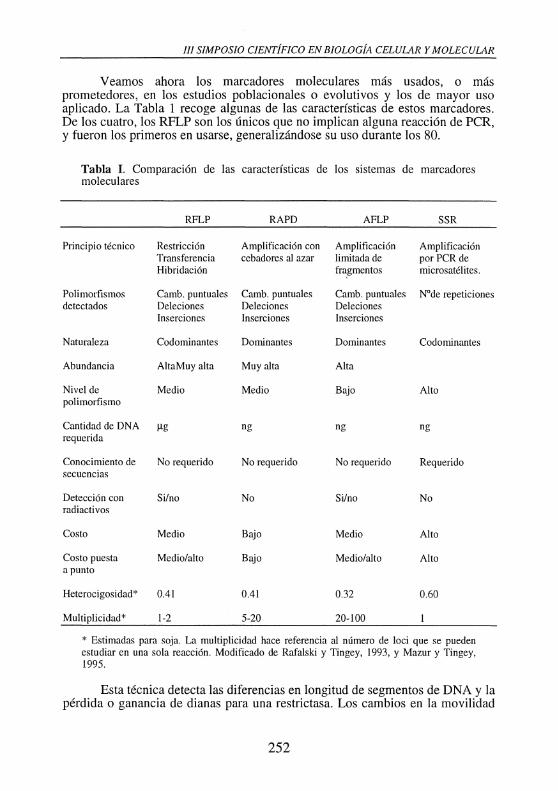
Veamos ahora los marcadores moleculares más usados, o más prometedores, en los estudios poblacionales o evolutivos y los de mayor uso aplicado. La Tabla 1 recoge algunas de las características de estos marcadores. De los cuatro, los RFLP son los únicos que no implican alguna reacción de PCR, y fueron los primeros en usarse, generalizándose su uso durante los 80.
Tabla I. Comparación de las características de los sistemas de marcadores moleculares
RFLP RAPD AFLP SSR
Principio técnico Restricción Amplificación con Amplificación Amplificación Transferencia cebadores al azar limitada de por PCR de Hibridación fra_gmentos microsatélites.
Polimorfismos Camb. puntuales Camb. puntuales Camb. puntuales Node repeticiones detectados Deleciones Deleciones Deleciones
Inserciones Inserciones Inserciones
Naturaleza Codominantes Dominantes Dominantes Codominantes
Abundancia AltaMuy alta Muy alta Alta
Nivel de Medio Medio Bajo Alto polimorfismo
Cantidad de DNA J.lg ng ng ng requerida
Conocimiento de No requerido No requerido No requerido Requerido secuencias
Detección con Si/no No Si/no No radiactivos
Costo Medio Bajo Medio Alto
Costo puesta Medio/alto Bajo Medio/alto Alto a punto
Heterocigosidad* 0.41 0.41 0.32 0.60
Multiplicidad* 1-2 5-20 20-100
* Estimadas para soja. La multiplicidad hace referencia al número de loci que se pueden estudiar en una sola reacción. Modificado de Rafalski y Tingey, 1993, y Mazur y Tingey, 1995.
Esta técnica detecta las diferencias en longitud de segmentos de DNA y la pérdida o ganancia de dianas para una restrictasa. Los cambios en la movilidad
252

Marcelino Pérez de la Vega
electroforética de un fragmento de DNA generados por una restrictasa indican diferencias en la longitud del fragmento debidas a inserciones o deleciones. Los cambios en el número de fragmentos junto con la aparición de otros indican la pérdida o ganancia de dianas de restricción para tal enzima causadas generalmente por la sustitución de bases en la diana (Figura 1). En líneas generales la técnica consiste en cortar DNA genómico extraído de una muestra de tejido con una endonucleasa de restricción. Muestras del DNA de cada individuo pueden ser cortadas con diferentes enzimas y analizadas separadamente. Los fragmentos obtenidos se separan por electroforesis en gel, normalmente de a8arosa. Puesto que el genoma de un eucarionte superior puede oscilar de 109 a 1 O 1 pares de bases, una restrictasa genera un enorme número de fragmentos discretos de muy diferentes tamaños. El resultado de la electroforesis es por tanto un rastro continuo de fragmentos, lo que implica la necesidad de usar un método que muestre sólo un conjunto de fragmentos. Para ello se requiere de sondas específicas. Después de la electroforesis los fragmentos son desnaturalizados y transferidos y fijados a una membrana (blotting) manteniendo el orden y posición relativa del gel. Los fragmentos así fijados se visualizan mediante la hibridación con una sonda marcada. Las sondas pueden haberse generado a partir de cDNA, que representa DNA codificante o de DNA genómico nuclear, codificante o no codificante. En ambos casos las sondas pueden ser de un gen conocido o de genes o segmentos anónimos. Los filtros pueden ser lavados para desprender la sonda y utilizados de nuevo con otra sonda diferente. El resultado es que cada restrictasa y cada sonda genera un patrón discreto de bandas en cada individuo de la población.
La información obtenida con esta técnica depende del número de sondas y del número de restrictasas usadas. Cada sonda híbrida con un conjunto diferente de fragmentos de DNA genómico y cada enzima corta el DNA genómico en puntos diferentes. Los fragmentos de restricción pueden asignarse a loci genéticos, y los datos interpretados en términos genéticos fácilmente, estimando directamente los niveles de variación genética en poblaciones y especies. Muchos de los fragmentos no codificantes pueden ser selectivamente neutros y representar secuencias que divergen más rápidamente que el cDNA. Algunos son muy polimórficos y por tanto útiles para distinguir entre individuos, razas, variedades, etc. Por otro lado las sondas cDNA representan secuencias más conservadas, lo que permite usarlas como marcadores a través de grupos de especies relacionadas. De hecho, restringiendo las condiciones para la hibridación se puede asegurar que una sonda obtenida en una especie hibride con fragmentos homólogos u homeólogos del genoma de otra especie, como ocurre entre maíz, arroz, trigo y otras gramíneas. Para la estimación de datos poblacionales y evolutivos existen numerosos modelos matemáticos (Nei y Miller, 1990; Weir, 1990).
253

Un plasmido, es una pieza de ADN, pequeña y circular que se encuentra frecuentemente en bacterias. Esta molécula, debido a los genes que porta, puede por ejemplo ayudar a la bacteria a sobrevivir en presencia de un antibiótico.
Los plásmidos son importantes porque se pueden (1) aislar en grandescantidades, (2) cortar, dividir e insertarles cualquier pieza de ADN, (3)devolverlo nuevamente a la bacteria donde se replicarán junto con el ADNnativo y (4) ser aislados nuevamente, obteniéndose billones de copias delADN que se les insertó. Su tamaño varía entre los 2.5 y las 20 Kb
Definiciones (2)
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
BAC es el acrónimo de “Bacterial Artificial Chromosome” y en principio seusa como los plásmidos, pudiendo construir BAC que porten ADNhumano, de ratón, etc., e insertarlos en una bacteria que hace dehospedaje. Al igual que con los plásmidos, al proliferar la bacteria tambiénse replican los BACs. En este caso se trata de entre 100 a 400 kb quepueden ser replicadas fácilmente usando BACs y ésta ha sido una de lasformas en que se ha clonado grandes porciones del genoma humano

Las técnicas de recombinación del ADN permiten transferir parte de ADN de un organismo (normalmente el que se está estudiando) a otro más simple de manipular y reproducir, como una bacteria. Al reproducirse la bacteria se reproduce el trozo de ADN en estudio que luego se puede volver a separar (con lo que se tienen grandes cantidades de ADN) y estudiar en detalle.
[ 0 ] Las enzimas de restricción permiten la separación (corte) del ADN en posiciones específicas que reconoce (no necesariamente alineadas). La línea roja representa el punto de corte de una enzima sobre la insulina –a la izquierda- y el plásmido bacterial de E. Coli.
Para secuenciar necesitamos una buena cantidad de ADN, por lo que es necesariohacer muchas copias del mismo. Para ellos se usan bacterias que crecen y sedividen rápidamente, pero antes necesitamos incorporar el ADN a estudiar en ellas.
Recombinación del ADN
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
sobre la insulina –a la izquierda- y el plásmido bacterial de E. Coli.
[ 3 ] El vector de inserta en la célula e incorpora los genes que porta en el ADN de la célula[ 4 ] Si la célula acepta los genes extraños, los pasará a sus células hijas en el proceso de división celular
[ 1 ] El ADN queda separado en los puntos de corte exponiendo sus bases nitrogenadas
[ 2 ] Se usa ADN ligasa para unir el trozo de ADN de la insulina y del plásmido de E.Coli

Históricamente hay dos métodos de secuenciación del ADN• Maxam & Gilbert, o secuenciación química• Sanger, que usa dideoxynucleotidos.
Hoy en dia el Método Sanger es el más usado en los laboratorios (aparte de las técnicas de secuenciación masiva)
Sanger y Gilbert compartieron el Nobel de Química en 1980
Las reacciones para secuenciar el ADN son similares acualquier reacción PCR (Polimerasa Chain Reaction). Lamezcla incluye una muestra de ADN, nucleótidos libres,
¿Cómo se secuencia el ADN?
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
mezcla incluye una muestra de ADN, nucleótidos libres,una enzima (generalmente una variante de la Taqpolimerasa) y un “primer” (una pieza pequeña –de 20 a 30nt- de ADN de una sola hebra) que se pueda es capaz dehibridar con una de las hebras de la muestra de ADN.
Se calienta la mezcla para separar las dos hebras de ADN,lo que permite que el “primer” se ligue a la zona deseada yla ADN-polimerasa inicie la elongación del primer.
Si el trabajo se realizara sobre una muestra de un billón decopias idénticas de ADN se obtendría un billón de copiasde una de sus hebras.

En el método Sanger sin embargo, las reacciones se realizan en presencia de un dideoxyribonucleotido. Éste es como cualquier ADN regular, salvo que no tiene el grupo hidroxil 3', por lo que, una vez que se añade al final de una cadena de ADN, no tiene forma de continuar su crecimiento
Los dideoxynucleotidos son moléculas similares a los nuclétidos normales pero les falta un grupo –OH lo que impide que otros nucleótidos se unan a él deteniendo la replicación del ADN.
El método Sanger (1)
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
Haciendo un símil con las piezas de un puzzle (4 tipos de piezas que serían los nucleótidos normales que se unen para formar el ADN), los dinucléotidos de los cuales también hay cuatro tipos (ACGT) les falta un borde y por lo tanto no permiten que una nueva pieza se enganche a él, deteniendo la replicación del ADN.A la izquierda se muestra un conjunto de piezas normales, cuyo perfil se dibuja al lado. A la derecha la representación de lo que sería su correspondiente dinucleótido

La clave del método está en que la mayor parte de losnucleótidos son regulares y que solo una pequeñafracción de ellos son dideoxy nucleotides.
Así al replicar hebras de ADN en presencia de dideoxy-T,la mayor parte de las veces cuando se necesite una 'T'para la nueva hebra, la enzima encontrará una Tcorrecta, y la replicación continuará añadiendo másnucleótidos.
Sin embargo, un porcentaje de las veces (proporcional ala cantidad de dideoxy-T que se haya incluido) la enzima
El método Sanger (2)
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
la cantidad de dideoxy-T que se haya incluido) la enzimacolocará un ddT y el crecimiento de la hebra se detendrá.
La Electroforesis en Geles se usa para separar fragmentos por su tamaño.
Los productos de una determinada reacción (hebras de diferente tamaño) secolocan en el gel y se induce su movimiento por carga eléctrica.
Los fragmentos pequeños se mueven poco (poca carga) mientras que los mayoresaparecen en la parte superior.
Ahora con un dispositivo capaz de leer imágenes (o geles) como un escáner yestimando la carga de los fragmentos es posible deducir las posiciones de lasTiminas (T) en la secuencia original.

Al colocarse los trozos replicados en el gel se observauna figura como la de la izquierda (en la que se hacoloreado cada nucleótido).
Para secuenciar ADN, se hace la reacción enpresencia de pequeñas cantidades de los 4terminadores dideoxi. Luego se usa un gel paraseparar los resultados y a partir de él se lee lasecuencia usando el código de colores (usualmenterojo, verde, azul y amarillo) con que se han marcadolos dd. Pueden haber hasta 96 pistas de muestrascorriendo en un gel , que podría llegar a tener entre 3y 4 metros de largo por unos 30 a 40 cms. de ancho.
El método Sanger (3)
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
y 4 metros de largo por unos 30 a 40 cms. de ancho.
El espacio entre bandas no es tan claro como seríadeseable, sino que aparece más como en la figura
El ordenador interpreta la imagen de cada pista del gelobteniendo la intensidad media de cada fila/columnacolor dominante que permite deducir de quenucleótido se trata.De esta forma se reconstruye la secuencia de ADN enlecturas de fragmentos alrededor de 700 nucleótidos.
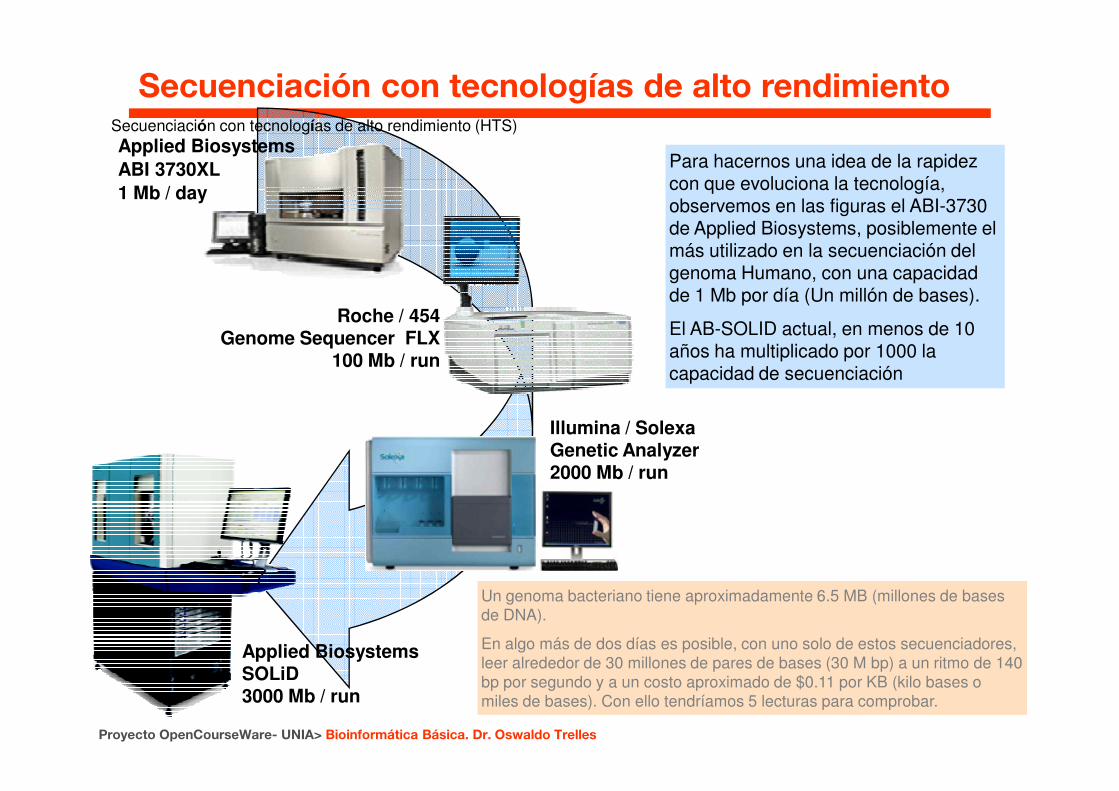
Secuenciación con tecnologías de alto rendimiento (HTS)Applied Biosystems ABI 3730XL1 Mb / day
Roche / 454 Genome Sequencer FLX
100 Mb / run
Para hacernos una idea de la rapidez con que evoluciona la tecnología, observemos en las figuras el ABI-3730 de Applied Biosystems, posiblemente el más utilizado en la secuenciación del genoma Humano, con una capacidad de 1 Mb por día (Un millón de bases).
El AB-SOLID actual, en menos de 10 años ha multiplicado por 1000 la capacidad de secuenciación
Secuenciación con tecnologías de alto rendimiento
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
Applied BiosystemsSOLiD3000 Mb / run
Illumina / Solexa Genetic Analyzer2000 Mb / run
Un genoma bacteriano tiene aproximadamente 6.5 MB (millones de bases de DNA).
En algo más de dos días es posible, con uno solo de estos secuenciadores, leer alrededor de 30 millones de pares de bases (30 M bp) a un ritmo de 140 bp por segundo y a un costo aproximado de $0.11 por KB (kilo bases o miles de bases). Con ello tendríamos 5 lecturas para comprobar.
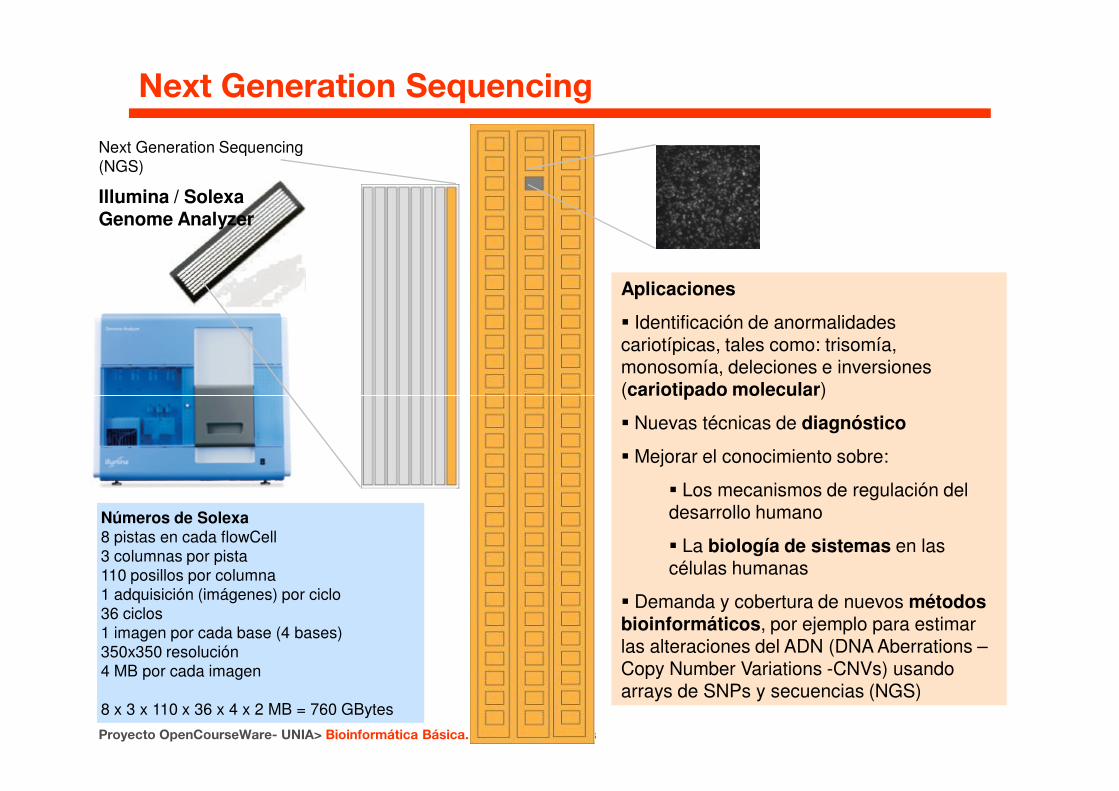
Next Generation Sequencing(NGS)
Illumina / Solexa Genome Analyzer
Aplicaciones
� Identificación de anormalidades cariotípicas, tales como: trisomía, monosomía, deleciones e inversiones (cariotipado molecular)
Next Generation Sequencing
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
Números de Solexa8 pistas en cada flowCell3 columnas por pista110 posillos por columna1 adquisición (imágenes) por ciclo36 ciclos1 imagen por cada base (4 bases)350x350 resolución4 MB por cada imagen
8 x 3 x 110 x 36 x 4 x 2 MB = 760 GBytes
(cariotipado molecular)
� Nuevas técnicas de diagnóstico
� Mejorar el conocimiento sobre:
� Los mecanismos de regulación del desarrollo humano
� La biología de sistemas en las células humanas
� Demanda y cobertura de nuevos métodos bioinformáticos, por ejemplo para estimar las alteraciones del ADN (DNA Aberrations –Copy Number Variations -CNVs) usando arrays de SNPs y secuencias (NGS)

La secuencia de nuestro genoma es 99.9% idéntico al de cualquier otro ser humano. La diferencia del 0.1% (3 millones de bases) está representada por:
... AAACGTCTA ...
... AAAC-TCTA ...
... AAACGTCTA ...
... AAAGCTCTA ...
... AAACGTCTA ...
... AAACATCTA ...
Inserciones / deleciones, Inversiones y Polimorfismos de una sola
Aplicaciones (1)
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
Inserciones / deleciones, Inversiones y Polimorfismos de una sola base “Single Nucleotide Polymorphisms o SNPs”
Cómo se detectan?: Por comparación de AND genómico proveniente
de distintos individuos (proyectos genoma)

Identificación de genes relacionados con enfermedades genéticas:– Mayor rapidez– Enfermedades multigénicas (SNPs)• Diabetes• Esquizofrenia• Identificación y/o localización de genes de interés agronómico o veterinario.• Desarrollo de vacunas Farmacogenómica• Uso de estrategias derivadas de la genómica para descubrir nuevos blancos terapéuticos• Identificar los genes que determinan la eficacia y toxicidad de medicamentos específicos
Aplicaciones (2)
Proyecto OpenCourseWare- UNIA> Bioinformática Básica. Dr. Oswaldo Trelles
Test de Paternidad:Comparando la secuencia de ADN de madre e hijo es posible identificar fragmentos en el ADN del hijo que no aparecen en la madre y por tanto deben haber sido heredados del padre. Se comparan estos fragmentos adquiridos por via paterna con el ADN del sujeto del test.
específicosFarmacogenómica (II)• Medicina personalizada– Determinar el perfil genético de cada individuo en cuanto a la sensibilidad a una determinada droga– Genes polimórficos involucrados en: metabolismo, transporte, blanco específicos, receptores, enzimas, etc.Bases de datos útileshttp://www.ncbi.nlm.nih.gov/http://www.ncbi.nlm.nih.gov/Genomes/index.html

Transferencia de ADN a células animales Las células animales pueden incorporar ADN por distintos métodos como microinyección directa, electroporación, encapsulación de ADN en membranas artificiales (liposomas) seguido de su fusión con membranas celulares, vectores basados en virus, YAC o mediada por células germinales. En general, el ADN introducido por cualquiera de estos métodos se integra en el genoma del hospedador.
La transferencia de genes depende de la introducción de secuencias de ADN en el núcleo de una célula somática, germinal, célula huevo fecundada o una célula madre embrionaria, seguido de la integración del ADN en un sitio del cromosoma.
Vectores de transferencia génica Un vector de transferencia génica es el vehículo empleado para introducir ADN en una célula u organismo.
Existen dos grandes grupos de vectores, virales y no virales. Los primeros incluyen todos aquellos que se han obtenido a partir de un virus tratando de eliminar sus características patológicas y de adaptarlos a su nueva función como transportadores de material genético heterólogo. Pretenden aprovechar la ventaja que aportan los virus como vectores naturales que han sufrido procesos de evolución complejos a lo largo de millones de años para optimizar la función de introductores de material genético en las células que invaden. Los vectores no virales siguen una estrategia de síntesis en lugar de modificación. Parten de estructuras sencillas conocidas e intentan reconstruir desde la base un sistema completamente artificial que posibilite el transporte efectivo de genes en el interior de la célula. Existen también vectores biológicos no virales (bacterianos) como portadores de genes terapéuticos, pero son los menos estudiados.
Fig1. Tipo de vectores y proceso esquematizado de obtención.
5

Las características de un vector ideal para poder adaptarlo luego a situaciones concretas serían las siguientes:
- Que sea reproducible - Que sea estable - Que permita la inserción de material genético sin restricción de tamaño - Que alcance concentraciones elevadas (> 108 partículas/ml) - Que permita la transducción tanto de células en división como
quiescentes - Que posibilite la integración específica de gen - Que reconozca y actúe sobre células específicas - Que la expresión del gen pueda ser regulada - Que carezca de elementos que induzcan una respuesta inmune - Que pueda ser caracterizado completamente - Que sea inocuo y minimice sus posibles efectos secundarios - Que sea fácil de producir y almacenar a coste razonable
El vector es una parte importante en el sistema de transferencia génica, pero el sistema consta de dos componentes: el vector (vehículo de transporte) y el cargo (material a ser transportado). El cargo a su vez se subdivide en: el efector (gen a introducir) y el soporte (los elementos que condicionan su expresión).
El cargo suele ser una estructura de tipo plasmídico (ADN bicatenario y circular), aunque puede ser de diversa naturaleza y complejidad, desde un simple oligonucleótido hasta un cromosoma artificial, que permite incorporar una cantidad elevada de material genético con capacidad de perpetuación en el tiempo.
El efector que se utilice dependerá del efecto que se persiga. Si se pretende compensar, sustituir o reparar un gen dañado, el efector debe ser una copia del gen intacto o un elemento que permita su reparación. Si se pretende inducir un efecto biológico concreto (eliminar específicamente un tipo de células, bloquear la expresión de un virus, inducir una respuesta inmune…), se pueden introducir genes naturales que potencien dicho efecto o genes que originen nuevas actividades que den lugar al efecto perseguido.
El soporte es la base para el control de la expresión del transgen e incluye distintos tipos de elementos reguladores. El primero y fundamental es el promotor, la zona de ADN anterior al gen donde se recluta la maquinaria de transcripción. La naturaleza del promotor condiciona el tipo de regulación de la expresión génica. Se pueden utilizar promotores constitutivamente activos o promotores específicos, que sólo son activos en un tipo celular concreto. De la misma manera se pueden emplear promotores inducibles, que requieren la presencia de un elemento concreto para ejercer su función. Éste puede ser un agente de adicción exógena (control externo) o un agente determinado por características fisiológicas especiales (como por ejemplo promotores activos ante situaciones de hipoxia). Otros elementos del soporte que pueden ser importantes dependiendo de la funcionalidad perseguida son los potenciadotes y represores de los promotores, secuencias de aislamiento (insulators) que impiden las influencias de secuencias reguladoras próximas, secuencias de
6

integración (para permitir la inclusión del material exógeno dentro del genoma de la célula hospedadora), secuencias de recombinación homóloga (para permitir el intercambio de material genético con el genoma hospedador con una región específica), secuencias de empaquetamiento (para introducir el material genético en el interior de un vector viral), secuencias inmunoestimulantes (secuencias bacterianas del tipo islas CpG no metiladas que sirven como coadyuvantes en las vacunas de ADN)
Los vectores son los encargados de asegurar la estabilidad del material genético transportado y de vencer todas las barreras biológicas hasta alcanzar su destino final, el núcleo de la célula diana, donde tiene lugar la regulación génica. También de ellos va a depender la vía de administración a utilizar.
En los casos de transferencia génica en individuos postnatales, el vector ha de solventar barreras en su camino hacia su destino funcional. Entre estas barreras se incluyen: la estabilidad en el medio extracelular (para evitar su degradación y eliminación), el reconocimiento de la célula diana (generalmente mediante un receptor específico), su unión a la membrana, su internalización en la célula (normalmente utilizando un sistema de transporte vesicular como la endocitosis), el escape de los sistemas de degradación intracelular (lisosomas) y su entrada final al núcleo, donde debe desmantelarse para permitir que el material genético que transporta gane acceso a su ubicación final y a la maquinaria de transcripción.
Fig2. Puntos clave para regular la eficacia de un vector de transferencia génica que se introduce en el individuo
7
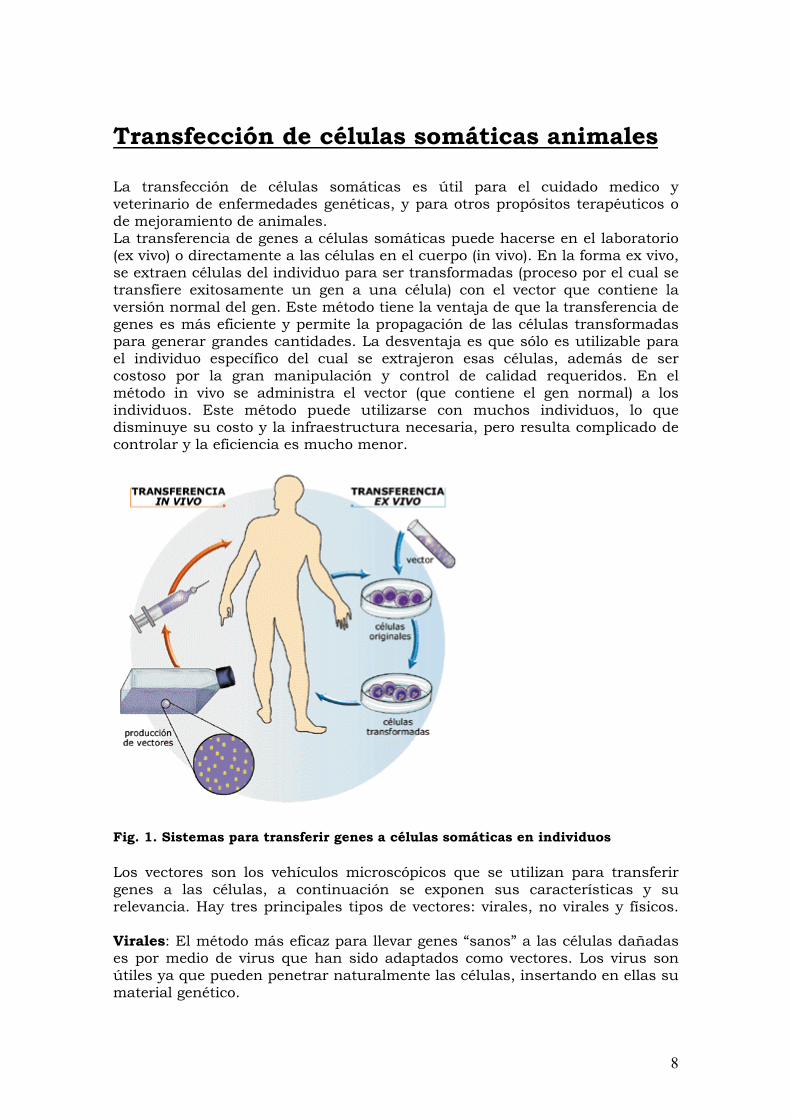
Transfección de células somáticas animales La transfección de células somáticas es útil para el cuidado medico y veterinario de enfermedades genéticas, y para otros propósitos terapéuticos o de mejoramiento de animales. La transferencia de genes a células somáticas puede hacerse en el laboratorio (ex vivo) o directamente a las células en el cuerpo (in vivo). En la forma ex vivo, se extraen células del individuo para ser transformadas (proceso por el cual se transfiere exitosamente un gen a una célula) con el vector que contiene la versión normal del gen. Este método tiene la ventaja de que la transferencia de genes es más eficiente y permite la propagación de las células transformadas para generar grandes cantidades. La desventaja es que sólo es utilizable para el individuo específico del cual se extrajeron esas células, además de ser costoso por la gran manipulación y control de calidad requeridos. En el método in vivo se administra el vector (que contiene el gen normal) a los individuos. Este método puede utilizarse con muchos individuos, lo que disminuye su costo y la infraestructura necesaria, pero resulta complicado de controlar y la eficiencia es mucho menor.
Fig. 1. Sistemas para transferir genes a células somáticas en individuos
Los vectores son los vehículos microscópicos que se utilizan para transferir genes a las células, a continuación se exponen sus características y su relevancia. Hay tres principales tipos de vectores: virales, no virales y físicos. Virales: El método más eficaz para llevar genes “sanos” a las células dañadas es por medio de virus que han sido adaptados como vectores. Los virus son útiles ya que pueden penetrar naturalmente las células, insertando en ellas su material genético.
8

Sin embargo, antes de poder usarlos como vectores deben modificarse para eliminar los genes virales que les permiten replicarse y causar enfermedad. A estos virus se les llama virus modificados o atenuados, y entre los más utilizados como vectores se encuentran los retrovirus, adenovirus y virus adeno-asociados. También se han desarrollado poxvirus (especialmente el virus de la vaccinia) para vacunas y terapias génicas. Actualmente, los vectores virales son los más eficientes para transformar células, aunque no carecen de desventajas: son de manufactura costosa, hay límites a la cantidad de genes (es decir, la longitud de los fragmentos de ADN) que pueden ser insertados en los vectores y que pueden llegar a desencadenar una respuesta inmune (inmunogenicidad).
No virales: Básicamente se trata de inyectar el fragmento de ADN que contiene el gen de interés directamente a las células o empacado dentro de otras moléculas como son los liposomas (pequeñas vesículas de grasa que pueden transportar sustancias al interior de las células). Los vectores no virales son menos eficientes para transformar células, pero no tienen limites para el tamaño del inserto (el tamaño del ADN que se va a inyectar), son menos inmunogénicos y más fáciles de elaborar.
Físicos: Involucran sobre todo inyectores sin aguja y electroporación. Los inyectores sin aguja utilizan alta presión para insertar el ADN en células de la piel o en células en cultivo, mientras que la electroporación utiliza pulsos eléctricos que abren temporalmente “agujeros” en las membranas de las células, permitiendo insertar el ADN. Los métodos físicos aún son ineficientes en la transformación y tienen un rango limitado de aplicación. Los riesgos de la terapia génica continúan siendo difíciles de cuantificar. Por ello cada ensayo debe comenzar lentamente, con un control muy cuidadoso de la dosis de vectores que contengan los genes que se pretende insertar. Estos riesgos tomaron una nueva dimensión en el otoño de 1999, cuando un joven de 18 años murió cuatro días después de haber iniciado un tratamiento con terapia génica. Los mismos riesgos podemos asumir para el resto de especies animales manipuladas genéticamente.
Transgénesis La transgénesis se puede definir como la introducción de ADN extraño en un genoma, de modo que se mantenga estable de forma hereditaria y afecte a todas las células en los organismos multicelulares. Generalmente, en animales, el ADN extraño, llamado transgen, se introduce en zigotos, y los embriones que hayan integrado el ADN extraño en su genoma, previamente a la primera división, producirán un organismo transgénico; de modo que el transgén pasará a las siguientes generaciones a través de la línea germinal (gametos).
Entre las aplicaciones de los animales transgénicos se pueden destacar:
• La posibilidad de estudiar a nivel molecular el desarrollo embrionario y su regulación.
• Manipular de forma específica la expresión génica in vivo. • Estudiar la función de genes específicos.
9

• Poder utilizar a mamíferos como biorreactores para la producción de proteínas humanas.
• La corrección de errores innatos de metabolismo mediante terapia génica.
La transgénesis se puede efectuar siguiendo dos estrategias comunes, microinyección de zigotos y la manipulación de células embrionarias, las cuales se describen brevemente a continuación:
1. Transgénesis por microinyección de zigotos
Desde que en 1982 se obtuviera un ratón transgénico, la producción de animales transgénicas es cada vez más cotidiana, existiendo ya animales transgénicos de las siguientes especies: ratón, rata, conejo, cerdo, vaca, cabra y oveja. La técnica se realiza, fundamentalmente por microinyección y se realiza de la siguiente forma:
o En la primera fase, se aíslan un número grande de óvulos fertilizados. Se consigue sometiendo a las hembras a un tratamiento hormonal para provocar una superovulación. La fertilización puede hacerse in vitro o in vivo.
o En la segunda fase, los zigotos obtenidos se manipulan uno a uno y con una micropipeta a modo de aguja, se introduce una solución que contiene ADN.
o En la tercera fase, estos óvulos son reimplantados en hembras que actuarán como nodrizas permitiendo la gestación hasta término.
Por último, tras el destete de los recién nacidos, éstos se chequean, para ver si ha ocurrido la incorporación del transgén.
2. Transgénesis por manipulación de células embrionarias.
Una estrategia más poderosa para la transgénesis implica la introducción de ADN extraño en células embrionarias totipotentes (células ES) o células embrionarias madres (células EM). Estas células se toman del interior de la blástula en desarrollo y se pasan a un medio donde se tratan con distintos productos con lo que se conseguirá que las células no se diferencien, y se mantiene su estado embrionario.
El ADN extraño se introduce en las células ES mediante diversas técnicas, posteriormente las células transfectadas son reintroducidas en una blástula y ésta reimplantada en una hembra.
Con esta técnica los neonatos son quimeras; pero mediante el cruce de éstas se consiguen animales transgénicos con aquellas quimeras que hayan incorporado el transgén en su línea germinal
10

El ganado transgénico que se emplea para producir proteínas terapéuticas, debe contener en el ADN extraño de sus células, además del gen codificante de la proteína, una secuencia o promotor que haga que se exprese dicho gen en unas determinadas células solamente. Por ejemplo, si se quiere que la proteína se produzca junto con la leche, el transgén se fusiona con una secuencia reguladora de una proteína de la leche, con lo que la proteína sólo se formará en las células de glándulas mamarias.
Esto es lo que se hizo con la oveja Tracy en 1992, la primera oveja que produjo una proteína humana, la alfa-antitripsina bajo la dirección del promotor ovino de la beta-lactoglobulina. Dicha proteína se produce en una cantidad de 35 g/l, la cual se emplea para curar el edema pulmonar.
Lo mismo se ha hecho para la famosa cerda Genie, que fabrica en su leche la proteína C humana que controla la coagulación sanguínea y es necesaria para los hemofílicos. Además de estas técnicas existen otras alternativas que se describirán más adelante, y algunas de las cuales son una combinación.
Transferencia génica por microinyección • Microinyección pronuclear: pequeñas cantidades del ADN de interés
(transgen) eran inyectadas en el pronúcleo de un embrión al estado de dos células. Aunque ampliamente aceptada y utilizada en forma rutinaria en muchos laboratorios, ha habido muy poco progreso para mejorar su eficiencia, la que se mantiene en el orden 0.1-5%, dependiendo de la especie considerada.
En la década del 80 ocurrió un importante avance en la tecnología de animales transgénicos que marcó el curso de la investigación en este campo por al menos dos décadas. Gordon y colaboradores describieron una técnica donde el ADN desnudo fue inyectado en el pronúcleo de un ovocito de ratón recientemente fertilizado, el que posteriormente se transfirió a hembras receptoras sincronizadas. Este experimento demostró que era posible usar un plásmido recombinante como vector para transferir genes foráneos directamente hacia el embrión. El ADN inyectado de esta forma se integró en el genoma y pudo ser heredado por la descendencia de los animales transgénicos fundadores. La inyección de embriones al estado de una célula fue clave para obtener una integración temprana del transgen, permitiendo al ADN foráneo contribuir en el genoma de todas las células somáticas y la línea germinal. Generación de los ratones transgénicos: En una primera fase se aíslan un número grande de ovocitos fertilizados, los que se consiguen sometiendo a la hembras a un tratamiento hormonal para provocar una mayor ovulación. En una segunda fase los ovocitos recién fertilizados se manipulan uno a uno, y con una micropipeta a modo de aguja se introduce una solución que contiene ADN. El ADN purificado es inyectado directamente en el pronucleo
11
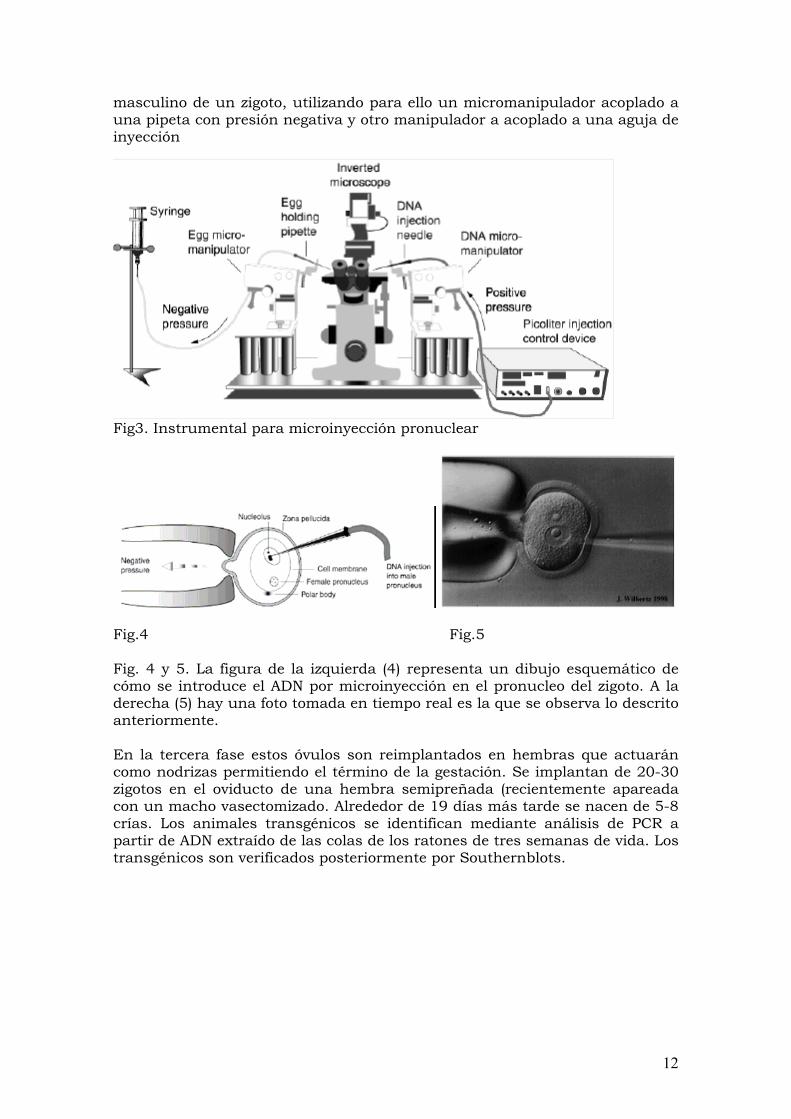
masculino de un zigoto, utilizando para ello un micromanipulador acoplado a una pipeta con presión negativa y otro manipulador a acoplado a una aguja de inyección
Fig3. Instrumental para microinyección pronuclear
Fig.4 Fig.5
Fig. 4 y 5. La figura de la izquierda (4) representa un dibujo esquemático de cómo se introduce el ADN por microinyección en el pronucleo del zigoto. A la derecha (5) hay una foto tomada en tiempo real es la que se observa lo descrito anteriormente. En la tercera fase estos óvulos son reimplantados en hembras que actuarán como nodrizas permitiendo el término de la gestación. Se implantan de 20-30 zigotos en el oviducto de una hembra semipreñada (recientemente apareada con un macho vasectomizado. Alrededor de 19 días más tarde se nacen de 5-8 crías. Los animales transgénicos se identifican mediante análisis de PCR a partir de ADN extraído de las colas de los ratones de tres semanas de vida. Los transgénicos son verificados posteriormente por Southernblots.
12

Fig.6. Implantación de óvulos transfectados e Identificación de ratones transgénicos nacidos mediante la técnica de PCR Los ratones positivos para la prueba PCR son cruzados con animales controles para identificar a los fundadores, es decir, aquellos que tienen el transgen insertado en su línea germinal. La eficiencia de transgénesis con este método es menor al 5%. La integración del transgen en el genoma del ratón es al azar y los niveles de expresión son variables. En ciertos casos la integración del transgen también ocurrió después de la primera división del zigoto, lo que resultó en animales fundadores mosaicos. Estos animales mosaicos aún transmiten el transgen a la descendencia pero lo hacen a una frecuencia menor al 50%. Desgraciadamente, la eficiencia para generar animales transgénicos utilizando esta tecnología es baja, particularmente en animales de granja. La eficiencia de la inyección pronuclear está controlada por una serie de factores como quedara demostrado por los trabajos de Brinster y colaboradores en 1985, quienes entregaron valiosa información sobre la integración de los transgenes y permitieron establecer que la concentración y la forma (circular o linear) del ADN eran los factores más críticos para una eficiente integración. No se encontraron diferencias significativas cuando el pronúcleo femenino o masculino era utilizado para la inyección, aunque este último es preferido por ser más grande. Por otro lado, el cruzamiento de ratones híbridos (por ejemplo C57BL x SJL) fue más exitoso en la producción de animales transgénicos que las líneas consanguíneas (C57Bl x C57Bl). El descubrimiento de la inyección de pronúcleos como un nuevo método para modificar el genoma de los animales revolucionó la forma en que los investigadores pudieron analizar la expresión de los transgenes y pavimentó el camino para la generación de los primeros animales transgénicos de granja en 1985. Desde entonces, la tecnología ha sido implementada con éxito en la mayoría de los animales domésticos como en conejos, por Buhler y colaboradores en 1990, ovejas, por Wright y colaboradores en 1991, cabras por Ebert y colaboradores en 1991, vacas por Krimpenfort y colaboradores en 1991 y cerdos por Wall y colaboradores en 1990. Sin embargo, además de los problemas asociados con la integración de los transgenes hay ineficiencias asociadas con la recolección, cultivo de los huevos fertilizados y transferencia
13
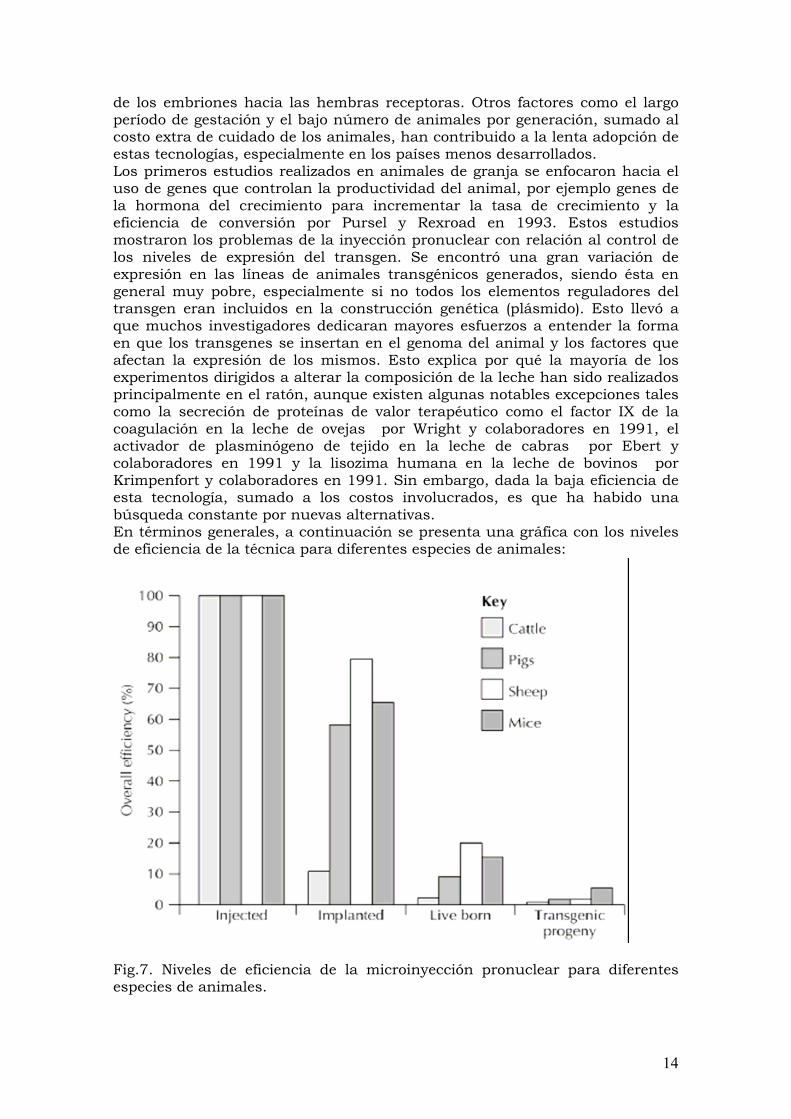
de los embriones hacia las hembras receptoras. Otros factores como el largo período de gestación y el bajo número de animales por generación, sumado al costo extra de cuidado de los animales, han contribuido a la lenta adopción de estas tecnologías, especialmente en los países menos desarrollados. Los primeros estudios realizados en animales de granja se enfocaron hacia el uso de genes que controlan la productividad del animal, por ejemplo genes de la hormona del crecimiento para incrementar la tasa de crecimiento y la eficiencia de conversión por Pursel y Rexroad en 1993. Estos estudios mostraron los problemas de la inyección pronuclear con relación al control de los niveles de expresión del transgen. Se encontró una gran variación de expresión en las líneas de animales transgénicos generados, siendo ésta en general muy pobre, especialmente si no todos los elementos reguladores del transgen eran incluidos en la construcción genética (plásmido). Esto llevó a que muchos investigadores dedicaran mayores esfuerzos a entender la forma en que los transgenes se insertan en el genoma del animal y los factores que afectan la expresión de los mismos. Esto explica por qué la mayoría de los experimentos dirigidos a alterar la composición de la leche han sido realizados principalmente en el ratón, aunque existen algunas notables excepciones tales como la secreción de proteínas de valor terapéutico como el factor IX de la coagulación en la leche de ovejas por Wright y colaboradores en 1991, el activador de plasminógeno de tejido en la leche de cabras por Ebert y colaboradores en 1991 y la lisozima humana en la leche de bovinos por Krimpenfort y colaboradores en 1991. Sin embargo, dada la baja eficiencia de esta tecnología, sumado a los costos involucrados, es que ha habido una búsqueda constante por nuevas alternativas. En términos generales, a continuación se presenta una gráfica con los niveles de eficiencia de la técnica para diferentes especies de animales:
Fig.7. Niveles de eficiencia de la microinyección pronuclear para diferentes especies de animales.
14

- Microinyección de ADN en células embrionarias: manipulación de las células madre embrionarias de ratón (ES cells) apareció como una potencial solución para muchos de los problemas encontrados con la técnica de microinyección pronuclear. Transformación genética mediante recombinación homóloga en células madre embrionarias (ES cells). El aislamiento de células madre embrionarias de ratón, en 1989 por Thompson y colaboradores, abrió nuevas posibilidades para estudiar la función génica en animales transgénicos. Las células madre embrionarias se obtienen desde el macizo celular interno de blastocistos y se pueden mantener en cultivos sin perder su estado indiferenciado gracias a la presencia en el medio de cultivo de factores inhibitorios de la diferenciación. Estas células pueden ser manipuladas in vitro vía recombinación homóloga, permitiendo así alterar la función de genes endógenos. Las células así modificadas pueden ser entonces reintroducidas en blastocistos receptores contribuyendo eficientemente a la formación de todos los tejidos en un animal quimérico incluyendo la línea germinal. Esta tecnología posibilita modificaciones genéticas muy finas en el genoma del animal, tal como la introducción de copias únicas de un gen. La incorporación de una copia única de un gen en un sitio predeterminado del cromosoma tiene las ventajas de permitir controlar el número de copias del transgen y se puede controlar la inserción de este en un sitio favorable (transcripcionalmente activo) para su expresión tejido-específica. Utilizando esta tecnología ha sido posible anular la función de genes endógenos del ratón mediante la integración de un marcador de selección, lo que ha permitido la generación de varios cientos de ratones llamados knock out que han servido como modelos de enfermedades genéticas en humanos y como modelos para analizar la función de genes endógenos (Melton, 1994; Shastry, 1998; Kolb y col., 1999; Wallace y col., 2000).
Fig.8. Representación de las técnicas de microinyección pronuclear y microinyección de células embrionarias
15

Transferencia génica con transposones Los transposones se definen como elementos transponibles que contienen genes adicionales a parte de los necesarios para la transposición y están dentro de secuencias repetidas. Dentro de esta categoría se encuentran los trasposones compuestos de procariotas, familia Tn3, elemento P de Drosophila, secuencias Alu de mamíferos, retrotrasposones…
CLASE I: MEDIANTE DNA
I.I PROCARIOTAS
Elementos IS (Is1, IS2, IS3, IS30, IS200, etc)
Transposones compuestos (Tn5, Tn10, etc)
Transposones de la familia Tn3
Bacteriófagos (Mu, lambda, P22, etc)
Sistemas de inversión
I.II. EUCARIOTAS
Elementos controladores del Zea mays (Ac, Mu, Sm, etc)
Elementos P de Drosophila CLASE II: MEDIANTE RNA
II.II EUCARIOTAS
Superfamilia vírica
Retrovirus, LINE, Ty, ...
Superfamilia no vírica
SINE Alu y B1 de mamíferos, pseudogenes procesados derivados de la polimerasa II
Fig.9. Clasificación de los elementos transponibles
16

Fig.10. Estructura básica de los trasposones
Los trasposones son secuencias de material genómico que tienen la capacidad del saltar fuera y dentro del genoma. Son capaces de autorreplicarse e integrarse aleatoriamente en nuevos sitios dentro del genoma (trasponerse o “saltar”). Pueden entrar en plásmidos y ser propagados por ellos. Estas propiedades les confieren la capacidad de ser transferidos exitosamente en células y genomas extraños.
La ingeniería genética puede manipular estos trasposones para llevar a cabo transferencia horizontal de genes. La transferencia horizontal de genes es la transferencia de material genético entre células y genomas que pertenecen a especies no relacionadas, por procesos distintos a la reproducción. Un ejemplo de ello es el uso de los trasposones de salmónidos para la transferencia génica en vertebrados. Los trasposones de vertebrados de la familia Tc1/mariner contienen como mínimo un gen único que codifica la enzima trasposasa flanqueada por dos repeticiones terminales invertidas (tir). Cada una de las tir posee uno o varios sitios de 20-30 p.b. de interacción con la trasposasa. La trasposasa interacciona con estos sitios para cortar el trasposón, luego interacciona con secuencias presentes en el genoma en otro locus y pega el trasposón en el nuevo locus. En vertebrados todos los trasposones que se han detectado están inactivados por mutaciones. Sin embargo recientemente se ha obtenido un trasposón activo de salmónidos mediante múltiple mutagénesis dirigida. Dicho transposón denominado “Bella durmiente” o “Sleeping beauty” (SB) se puede usar en todos los vertebrados analizados hasta el momento como vehículo de incorporación de nuevos genes y para inactivar genes en el genoma por inserción.
17
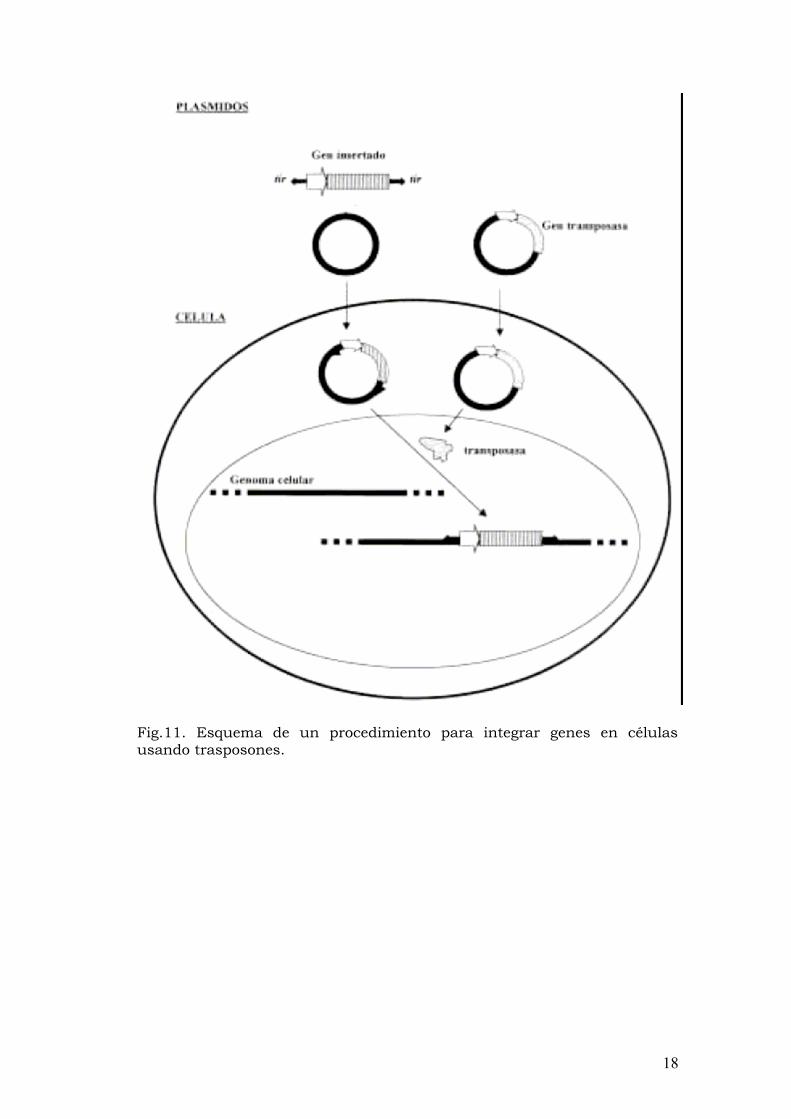
Fig.11. Esquema de un procedimiento para integrar genes en células usando trasposones.
18

Transferencia génica con vectores virales Los vectores virales son todos aquellos que han sido obtenidos a partir de un virus, tratando de eliminar sus características patológicas y adaptarlos a una nueva función como transportadores de material genético heterólogo. Pretenden aprovechar las ventajas que aportan los virus como vectores naturales que han sufrido procesos de evolución complejos a lo largo de millones de años para optimizar su función de introductores de material genético en las células que invaden. Para modificar un virus y transformarlo en un vector de transferencia génica en animales, el primer paso es bloquear su capacidad de propagación eliminando los genes responsables de su replicación. El segundo paso es la eliminación de parte del genoma viral para crear espacio para introducir el material genético de interés (gen o genes, más los elementos de control). Se han de eliminar genes que no sean esenciales, y especialmente aquellos que puedan resultar tóxicos o perjudiciales para el individuo a manipular.
La ventaja de los vectores virales en la gran eficacia de transferencia y la posibilidad, en algunos casos, de integrar el transgen en el genoma de la célula hospedadora. Por otro lado presentan desventajas como su falta de especificidad celular en la mayoría de los casos, la limitación del tamaño (debido a que han de empaquetarlo en la cápsida), su elevada inmunogenidad y el riesgo biológico que conllevan (reconstitución de partículas replicativas por recombinación, y oncogenidad inducida por integración inespecífica.
Se han usado retrovirus y lentivirus como vectores para integrar transgenes en genomas animales. Esta técnica se ha usado en ratones con éxito y se ha extendido a gallinas, vacunos y suinos.
- Vectores retrovirales
Los retrovirus son virus de ARN con envuelta. Poseen una elevada eficacia de transducción en un gran número de tipos celulares, son capaces de integrarse de forma estable en el genoma de las células que infectan sin expresar ninguna proteína viral inmunogénica, y son relativamente poco patogénicos, con excepción de algunos como el VIH. La mayoría de ellos derivan del virus de la leucemia murina (MuLV).
En general, su principal limitación es que sólo pueden transducir eficazmente células en división, ya que el acceso al núcleo del complejo de preintegración requiere la ruptura de la membrana nuclear. Hay un tipo de retrovirus, los lentivirus, que si que pueden infectar e integrarse en células quiescentes, como el VIH y SIV, pero el riesgo inherente al uso de los mismos limita su aplicación.
Otro inconveniente que tiene el uso de retrovirus como vectores es la poca estabilidad de las partículas y su baja tasa relativa de producción. Esto ha sido parcialmente resuelto con la sustitución de la proteína de la envuelta por la glicoproteína G del virus de la estomatitis vesicular que posibilita la obtención de títulos de hasta 109 p.i. /ml y estabiliza las partículas.
19

Debido a que estos virus tienen un rango de infectividad muy amplio, cuando se quiere transducir selectivamente una población celular, se le incorporan ligandos heterólogos o anticuerpos monocatenarios que generan nuevas especificidades. Son vectores que poseen proteínas quiméricas en la envuelta.
Contrario a la percepción de muchos, los primeros animales transgénicos fueron producidos hace ya casi 30 años mediante la microinyección de ADN viral (SV40) en la cavidad del blastocele de embriones de ratón (Jaenish y Mintz, 1974). Los próximos intentos involucraron embriones de ratón infectados con el retrovirus Moloney de la leucemia murina (MoMuLV), lo que resultó en la transmisión estable hacia la línea germinal (Jaenish, 1976). Esto se logró reemplazando genes que no son esenciales para el virus por genes heterólogos, aprovechando así la capacidad de los virus de infectar un amplio espectro de células y con una gran eficiencia. Una de las grandes desventajas de este método radica en que la integración del ADN se produce en diferentes etapas del embrión en desarrollo, lo que implica que el ADN no se integra en todas las células somáticas o en la línea germinal y por lo tanto no hay transmisión del transgen a la descendencia. Además, los animales generados por este método tienen a menudo más de un sitio de integración, lo cual ocurre cuando más de una célula del embrión es infectada por el virus (Palmiter y Brinster, 1985). Esto implica que las líneas de ratones transgénicos deben ser cruzadas para segregar los diferentes loci conteniendo el transgen y poder así aislar líneas con un sitio de inserción único. Finalmente, los vectores retrovirales poseen una limitada capacidad de ADN foráneo que puede ser acomodado, alrededor de 8 kb, lo cual imposibilita muchos experimentos especialmente con secuencias genómicas humanas que pueden superar ampliamente este tamaño.
- Vectores Adenovirales Los adenovirus son virus de DNA sin envuelta con un genoma bicatenario hasta 36 kb. Se han seleccionado los serotipos 2 y 5 para la obtención de vectores por no estar asociados con ninguna enfermedad severa ni originar tumores en animales. Pueden transducir un número bastante amplio de tipos celulares distintos, tanto células en división como post-mitóticas, con una eficacia muy elevada. El genoma viral se mantiene episómico, lo que permite una expresión genética transitoria. Originalmente la capacidad del vector para incorporar ADN era de hasta 8 kb. Recientemente se ha conseguido eliminar casi todo el genoma viral, manteniendo las secuencias de empaquetamiento y las secuencias terminales, rindiendo vectores que pueden incorporar hasta 35 kb a los que se les ha denominado “vectores sin tripas” (gutless). La eliminación de material genómico viral además de aumentar la capacidad del vector de incorporar ADN, disminuye el riesgo de inducción de respuesta inmune. La mayoría de la población ha sido infectada en algún momento por adenovirus y presenta una respuesta inmune inicial. Una de las mayores ventajas de estos virus es su alta reproductividad con títulos de hasta 1010 p.i./ml, y su gran estabilidad, que les permiten ser concentrados adicionalmente para alcanzar valores de 1012 p.i./ml. Su falta de especificidad celular se compensa con el desarrollo de estrategias de redireccionalidad. Quimeras que introducen ligandos específicos en proteínas de la cápside y moléculas biespecíficas que reconocen tanto la cápside viral como el receptor seleccionado.
20
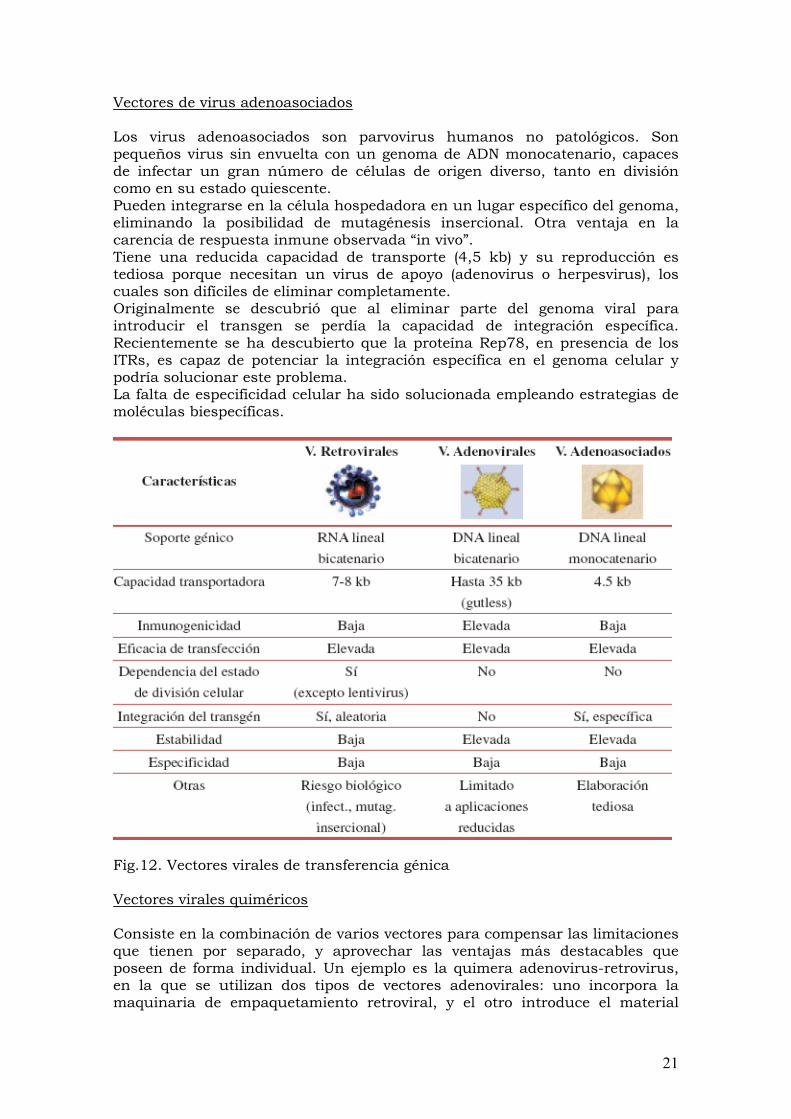
Vectores de virus adenoasociados Los virus adenoasociados son parvovirus humanos no patológicos. Son pequeños virus sin envuelta con un genoma de ADN monocatenario, capaces de infectar un gran número de células de origen diverso, tanto en división como en su estado quiescente. Pueden integrarse en la célula hospedadora en un lugar específico del genoma, eliminando la posibilidad de mutagénesis insercional. Otra ventaja en la carencia de respuesta inmune observada “in vivo”. Tiene una reducida capacidad de transporte (4,5 kb) y su reproducción es tediosa porque necesitan un virus de apoyo (adenovirus o herpesvirus), los cuales son difíciles de eliminar completamente. Originalmente se descubrió que al eliminar parte del genoma viral para introducir el transgen se perdía la capacidad de integración específica. Recientemente se ha descubierto que la proteína Rep78, en presencia de los ITRs, es capaz de potenciar la integración específica en el genoma celular y podría solucionar este problema. La falta de especificidad celular ha sido solucionada empleando estrategias de moléculas biespecíficas.
Fig.12. Vectores virales de transferencia génica Vectores virales quiméricos Consiste en la combinación de varios vectores para compensar las limitaciones que tienen por separado, y aprovechar las ventajas más destacables que poseen de forma individual. Un ejemplo es la quimera adenovirus-retrovirus, en la que se utilizan dos tipos de vectores adenovirales: uno incorpora la maquinaria de empaquetamiento retroviral, y el otro introduce el material
21

genético equivalente a un vector retroviral. Se aprovecha la eficacia de transducción de los vectores adenovirales para cotransfectar con ambos vectores, transformando las células diana en productoras de vectores retrovirales. Los vectores así generados son capaces de infectar células vecinas e integrarse en su genoma.
Transferencia génica mediada por semen Consiste en la introducción de ADN foráneo en gametos masculinos antes del proceso de fertilización.
Las células espermáticas están consideradas por algunos autores como células inertes dado que no cuentan con la mayoría de la maquinaria molecular y bioquímica que existe en las células somáticas para permitir la replicación de ADN, la transcripción de los genes y la síntesis de proteínas. Su morfología es particular, y se caracteriza por un compartimento extremadamente reducido y un núcleo que contiene en ADN compactado a modo de cromatina condensada, conectados a un largo flagelo. Estas observaciones de la morfología llevan a la conclusión de que las células espermáticas tienen como única misión actuar de vectores de su propio genoma durante la fertilización. La primera evidencia de que las células espermáticas de mamífero podían incorporar ADN foráneo cuando eran incubadas en solución con estas macromoléculas fue descrita por Brackett et al. en 1971.
En 1989 Lavitrano y col. describieron la producción de ratones transgénicos mediante inseminación artificial, utilizando semen que había sido incubado con ADN exógeno. Las células espermáticas habían incorporado ADN plasmídico. La generación F1 contenía el ADN exógeno.
La transferencia génica mediada por semen en vertebrados se ha desarrollado y sufrido muchos cambios en los últimos años. La incubación de las células espermáticas con ADN foráneo seguida de fertilización in vitro o in vivo ha generado conejos, cerdos, ratones, ovejas, vacas, peces, pollos transgénicos. La definición y el establecimiento de los protocolos según la especie a transformar es y será un tema valioso en biotecnología.
Una ventaja de la transferencia génica mediada con semen frente a la microinyección, es que la segunda requiere manipulación individual de los embriones, mientras que con la segunda se pueden transformar genéticamente un alto número de embriones en un solo paso. Esto es particularmente interesante cuando se quieren obtener especies acuáticas transgénicas.
Existen investigaciones que sugieren que el control y captación del ADN exógeno por parte de las células espermáticas de mamíferos está altamente regulada y es muy específica. De hecho, el fluido seminal antagoniza fuertemente la unión de ADN foráneo y bajo condiciones normales es una fuerte protección de las células espermáticas contra el ADN foráneo.
22
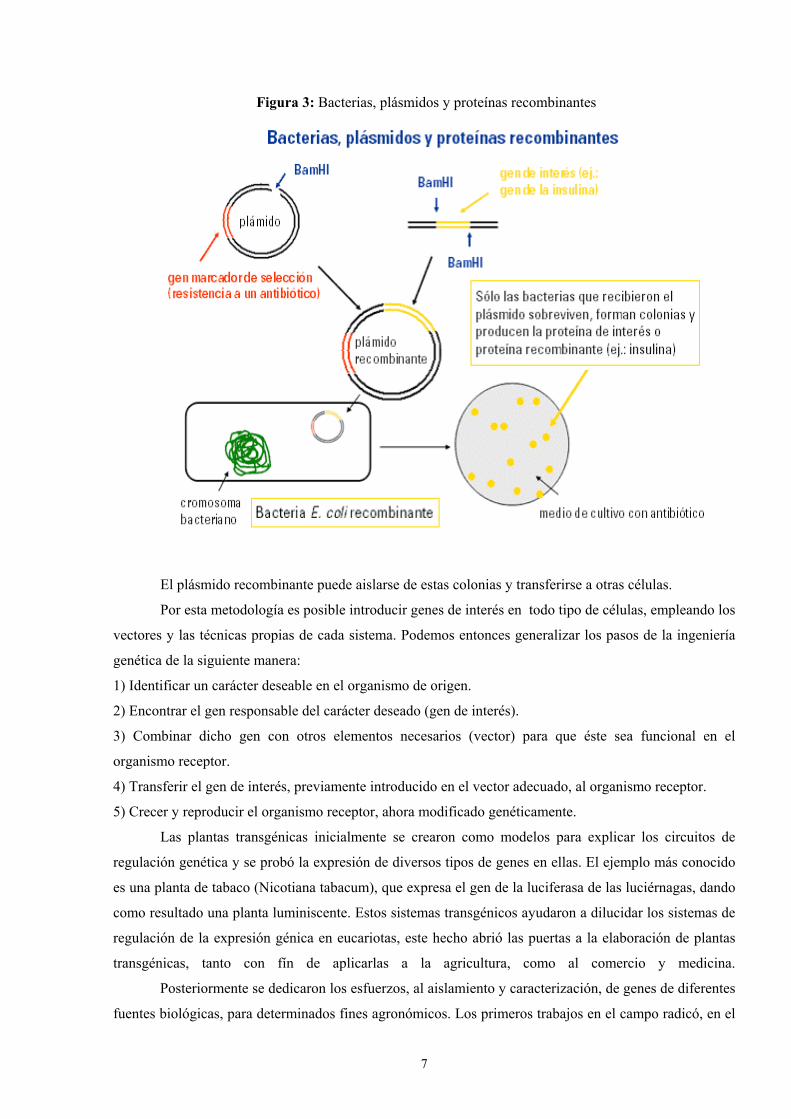
Figura 3: Bacterias, plásmidos y proteínas recombinantes
El plásmido recombinante puede aislarse de estas colonias y transferirse a otras células.
Por esta metodología es posible introducir genes de interés en todo tipo de células, empleando los
vectores y las técnicas propias de cada sistema. Podemos entonces generalizar los pasos de la ingeniería
genética de la siguiente manera:
1) Identificar un carácter deseable en el organismo de origen.
2) Encontrar el gen responsable del carácter deseado (gen de interés).
3) Combinar dicho gen con otros elementos necesarios (vector) para que éste sea funcional en el
organismo receptor.
4) Transferir el gen de interés, previamente introducido en el vector adecuado, al organismo receptor.
5) Crecer y reproducir el organismo receptor, ahora modificado genéticamente.
Las plantas transgénicas inicialmente se crearon como modelos para explicar los circuitos de
regulación genética y se probó la expresión de diversos tipos de genes en ellas. El ejemplo más conocido
es una planta de tabaco (Nicotiana tabacum), que expresa el gen de la luciferasa de las luciérnagas, dando
como resultado una planta luminiscente. Estos sistemas transgénicos ayudaron a dilucidar los sistemas de
regulación de la expresión génica en eucariotas, este hecho abrió las puertas a la elaboración de plantas
transgénicas, tanto con fín de aplicarlas a la agricultura, como al comercio y medicina.
Posteriormente se dedicaron los esfuerzos, al aislamiento y caracterización, de genes de diferentes
fuentes biológicas, para determinados fines agronómicos. Los primeros trabajos en el campo radicó, en el
7

aislamiento de los genes de las proteínas Cry de Bacillus thuringensis, una bacteria entomopatógena, y
son usadas en las plantas transgénicas como un bioinsecticida, llamado convencionalmente Bt; también se
trabajó en la construcción de genes que confieran a las plantas resistencia a herbicidas. El principal
trabajo, en lo que a resistencia a herbicidas se refiere, se hizo con el glifosfato, aunque también se
trabajaron otros herbicidas como el glufosinato. Las plantas con la tecnología RoundupReady (RR) de
Monsanto son resistentes al glifosfato de la misma empresa, denominado Roundup y sus variedades
mejoradas.
La resistencia a Roundup está dada por la expresión de una proteína bacteriana necesaria para la
síntesis de enzimas fotosintéticas, la proteína de origen celular es inhibida por el herbicida, pero no la de
origen bacteriano.
En la actualidad, las nuevas ofertas de Monsanto muestran plantas de maíz, algodón y soya que
poseen ambas características: la resistencia a insectos y la tolerancia a herbicidas. Otras empresas han
generado plantas similares y con otras características, como el tomate "Flavr–Savr de Calgene, el cual no
se ablanda y puede ser almacenado por mucho tiempo, se logro mediante la tecnología de RNA
antisentido, la cual inhibe la proteína responsable de la senescencia del fruto maduro.
Las plantas transgénicas no sólo se utiliza para cultivos, sino que también se han utilizado para la
producción de sustancias, como metabolitos y productos secundarios importantes. Uno de los avances
más impresionantes de la biotecnología vegetal ha sido la posibilidad de expresar vacunas contra una
amplia variedad de enfermedades en las plantas, incluso se han logrado expresar anticuerpos de
reconocimiento y prevención del cáncer.
Hoy, la ingeniería genética se suma a las prácticas convencionales como una herramienta más
para mejorar o modificar los cultivos vegetales.
Figura 4: Comparación entre cruzamiento tradicional y biotecnología
8

En este sentido, esta metodología ofrece tres ventajas fundamentales respecto a las técnicas
convencionales de mejora genética basadas en la hibridación: +Los genes que se van a incorporar pueden provenir de cualquier especie, emparentada o no (por
ejemplo, un gen de una bacteria puede incorporarse al genoma de la soja).
+En la planta mejorada genéticamente se puede introducir un único gen nuevo preservando en su
descendencia el resto de los genes de la planta original.
+Este proceso de modificación demora mucho menos tiempo que el necesario para el mejoramiento
por cruzamiento.
Podemos así modificar propiedades de las plantas de manera más amplia, más precisa y más rápida.
3. PROCEDIMIENTOS PARA LA OBTENCIÓN DE PLANTAS TRANSGÉNICAS
3.1. GENERALIDADES
Principalmente se emplean tres métodos para introducir genes ajenos en una planta. Todos estos
métodos obtuvieron por primera vez, con más o menos éxito, plantas transgénicas en la década de los
ochenta y muchas de ellas se comercializaron en los noventa.
a) El método se basa en el empleo de un vector vivo que lleve el material genético a la célula
blanco. Existen dos formas de introducir material genético por esta vía:
1) Mediante virus genéticamente modificados (que llevan los genes de interés en lugar de los genes
estructurales), los cuales insertan su genoma en el DNA celular para la replicación y de esta manera se
consigue la expresión de los genes foráneos.
2) el mecanismo natural de infección de la bacteria del suelo Agrobacterium tumefaciens que introduce
un gen de su plásmido en las células de la planta infectada. Recordemos que un plásmido es un fragmento
de ADN circular y extracromosómico que suele contener información no vital para la bacteria y cuyo
tamaño es del orden del 1 al 3% del cromosoma bacteriano. Este gen se integra en el genoma de la planta
provocándole un tumor o agalla. Lo que se hace con A. tumefasciens, es crear una cepa recombinante de
ésta (con los genes de interés) y se induce la formación de tumores, en los cuales se encuentran células
modificadas por la interacción, se aíslan estas células y a partir de ellas se genera el individuo transgénico
(Fig 5). Se aplicó con éxito por primera vez en 1984 en el tabaco y el girasol. Las gramíneas y en general
todas las monocotiledóneas presentan gran resistencia a Agrobacterium por lo cual este método es
portancia económica. bastante inviable en un extenso grupo de plantas de gran im
9

Figura 5: Resumen del proceso de DNA recombinante para crear plantas transgénicas
b) Otro método empleado para transformar genéticamente plantas es el uso de protoplastos, que
son células vegetales a las que se les ha liberado de la pared celular. De esta manera queda eliminada la
barrera principal para la introducción de genes foráneos. Mediante esta técnica se consiguió por primera
vez cereales transgénicos en 1988. Puede realizarse una transferencia directa de genes mediante la fusión
de protoplastos (la célula vegetal sin la pared) mediante químicos como el PEG (polietilenglicol), de
donde se obtienen híbridos nucleares y luego células transgénicas por recombinación; para este in
también puede emplearse liposomas.
c) La biolística es otro método difundido, consiste en bombardear las células con partículas
metálicas microscópicas recubiertas del DNA que se desea introducir. Si bien esta técnica ha dado buenos
resultados, tiene un componente aleatorio de efecto muy fuerte que da un amplio margen a resultados
impredecibles y un incremento significativo en la tasa de mutación celular. Igualmente costosos, pero con
menos problemas de efecto aleatorio, están los métodos de inyección (micro y macroinyección), estos
métodos consisten en inyectar el material genético foráneo al núcleo de la célula mediante equipo
sofisticado. Los métodos de microinyección tienen mayor eficacia que los de macroinyección por la
focalización dirigida de la inserción. Adicionalmente se emplean otros métodos directos como la
10

transformación del polen y la electroporación, pero no son ampliamente utilizados. Microcañón o cañón
de partículas que consiste en bombardear tejidos de la planta con micropartículas metálicas cubiertas del
fragmento de ADN que interesa se integre en el ADN de la planta. Es el procedimiento que más éxitos ha
conseguido y el que promete más avances.
3.2. DESCRIPCIÓN DE LAS TECNICAS
3.2.1. Transferencia genética con Agrobacterium tumefaciens
En 1970 se planteó la hipótesis de que la enfermedad de las plantas denominada agalla del cuello
podría ser producida por la transferencia de material genético entre una bacteria, Agrobacterium
tumefaciens, y las células vegetales. La agalla del cuello se caracteriza por la formación de voluminosas
agallas, sobretodo en el cuello del tallo (zona de contacto entre el tallo y la raíz), también en las raíces y el
tallo de numerosas plantas de interés agronómico. La enfermedad es de naturaleza tumoral y ya se había
demostrado, a finales de los años sesenta, que las células afectadas contienen unas sustancias, las opinas
(sustancias nitrocarbonadas), que no se encuentran en las células normales. También se demostró que
existen varias clases de tumores en función de la concentración de opinas y que es el material genético de
la bacteria el que determina este carácter ya que estas observaciones se realizaron en tejidos cultivados in
vitro, es decir, en ausencia de bacterias. Se concluyó que las células tumorales habían adquirido la
propiedad de sintetizar opinas durante la interacción con la bacteria. También se concluyó que la
naturaleza de las opinas depende de la cepa bacteriana y también que cada cepa degrada específicamente
sus propias opinas. Quedaba demostrada la hipótesis de la transferencia de información entre la bacteria y
la célula vegetal.
Schell (1973) anunció el descubrimiento en cepas de Agrobacterium tumefasciens de un plásmido
de un tamaño jamás observado hasta entonces y que el plásmido llamado Ti (del inglés Tumour inducing)
es portador del carácter patógeno. Más adelante se observó que todas las células de las agallas eran
portadoras de un fragmento del plásmido Ti que se denominó ADN-T (ADN transferido). Se demostró
después que el plásmido tenía varias funciones: la función de virulencia (Vir), responsable de la
transferencia del ADN-T, la oncógena (Onc), responsable del tumor (consecuencia de la síntesis de
auxina y citoquinina), la función que especifica la síntesis de opinas (Ops), moléculas que sirven de
alimento a la propia bacteria, y la función catabólica (Opc, opina catabolismo). En realidad se
encontraron varios segmentos Opc1, Opc2, que permiten la degradación de las opinas producidas por el
tumor. Se ha de distinguir dos tipos de funciones: las funciones situadas fuera del segmento ADN-T (Vir,
Opc1,Opc2) que se expresan en la bacteria y las funciones controladas por el segmento ADN-T (Onc,
Ops) que se expresan en la célula vegetal después de la transferencia de este segmento (Fig. 6).
11

Figura 6: Plásmido Ti de Agrobacterium tumefaciens
En resumen la bacteria no es patógena per se porque no segrega ninguna toxina que disuelva las
paredes celulares como hacen otras bacterias patógenas. Sus efectos se deben a la transferencia de un
segmento de ADN, el ADN-T, cuya expresión en las células vegetales es la causa de la enfermedad. La
supresión en el plásmido del segmento transferido hace que la bacteria sea inofensiva sin que ello se la
prive de la capacidad de transferir ADN a una célula vegetal. Por tanto se puede plantear su sustitución
por un fragmento de ADN extraño.
El segmento ADN-T está delimitado en ambos extremos por unas secuencias determinadas de
nucleótidos que actúan a modo de señales. La señal "promotor" al principio y la "terminador" al final. La
región transferida y que se integra en el genoma de la planta es la comprendida entre estas dos señales. En
teoría era posible transferir cualquier gen extraño colocado entre estas dos secuencias. En 1983 se
introdujo un gen bacteriano que confería resistencia al antibiótico cloramfenicol. Se escogió este gen sólo
porque es fácil poner de manifiesto su expresión: las células que han integrado el gen sintetizan el enzima
cloramfenicol transacetilasa que gobierna la síntesis del antibiótico. El gen empleado se expresa en la
bacteria Escherichia coli. Para que un gen pueda expresarse el enzima ARN polimerasa debe reconocer el
"promotor" y el "terminador". La ARN polimerasa del tabaco (una planta muy empleada en estos
experimentos de transferencia de genes) no reconoce los promotores y terminadores de E. coli y por
consiguiente no transcribe este gen. Para solucionar el problema se fabricó un gen compuesto o quimérico
a partir del gen de la resistencia al cloramfenicol de E. coli, un promotor y terminador procedentes del
segmento ADN-T de Agrobacterium tumefaciens. El gen quimérico se reincorporó en un plásmido Ti
(Fig. 7).
12

Figura 7: Gen quimérico en el plásmido Ti de Agrobacterium tumefaciens.
De esta manera el gen quimérico funcionó al poder ser detectada la actividad de la cloramfenicol
transcetilasa en tejidos tumorales. Aún quedaba una dificultad a salvar: la regeneración de una planta
entera a partir de células transformadas. Como las células transformadas eran tumorales eran incapaces de
esta regeneración y el siguiente paso consistió en eliminar los genes tumorales del segmento ADN-T. De
esta manera se pudo regenerar plantas enteras transgénicas que eran fértiles y con las que se pudo estudiar
la transmisión de caracteres a su descendencia. Además si se escogen los promotores adecuados, es
posible expresar genes en órganos específicos, como raíces, semillas y tubérculos.
El gen de la resistencia a antibióticos no tiene interés agronómico por lo que había que identificar,
aislar y clonar los genes que pudiesen mejorar las plantas cultivadas. En el caso de caracteres con base
genética compleja (donde intervienen numerosos genes), como la resistencia de una planta al frío, es
mucho más difícil la manipulación genética que con los caracteres que se expresan como consecuencia de
la actividad de un enzima.
El sueño de obtener plantas resistentes a los insectos fitófagos se ha hecho realidad con la
obtención de plantas transgénicas portadoras de un gen bioinsecticida. Bacillus thruringiensis es una
bacteria grampositiva del suelo que en los estadios de esporulación produce unos cristales de proteínas de
propiedades insecticidas. Berliner en 1909 aisló la bacteria de los cadáveres del gusano de la harina
(Ephestia kuehniella) procedente de Turingia. Al creerse que la bacteria era el causante de la muerte del
insecto, sugirió la idea de recurrir a B. thuringiensis para luchar contra la plaga de insectos. Los primeros
preparados comerciales aparecieron en 1938. Era práctica habitual en los agricultores tirar a voleo esporas
de B. thuringiensis sobre los cultivos pero se presentaba el inconveniente de tener que realizar la práctica
con una frecuencia mucho mayor que con los insecticidas químicos. A estas proteínas se las denominó cry
(del inglés crystal) por su capacidad de formar cristales o ð-endotoxinas por su acumulación en el interior
de las bacterias y su carácter tóxico. Las proteínas cry provocan la lisis de las células intestinales de los
13

insectos. Estos bioinsecticidas se caracterizan por su especificidad, pues sólo son tóxicos en escarabajos,
moscas y mariposas (grupos de insectos causantes de la mayoría de las plagas), y porque son
prácticamente inocuas en humanos. E. Schnepf y H. Whiteley aislaron en 1981 el primer gen que
codifica una proteína insecticida. Se acababa de sentar las bases para que M.D. Chilton en 1983
obtuviera las primeras plantas transgénicas de tabaco utilizando Agrobacterium tumefaciens. Le siguieron
otros experimentos en diversos laboratorios de Europa y América con el tomate y la patata. Estos
experimentos sirvieron para demostrar que la expresión de proteínas insecticidas en plantas era posible y
proporcionaba un método eficaz de lucha contra los insectos (Figura 8). Figura 8: Obtención de plantas transgénicas resistentes a los insectos mediante Agrobacterium
tumefaciens.
Todas estas investigaciones culminaron en 1996 con la entrada en el mercado de plantas
transgénicas (algodón, patata y maíz) resistentes a insectos. A todas estas plantas transformadas se las
denomina Plantas Bt (de Bacillus thuringiensis).
En 1997 el 25% de los cultivos transgénicos comercializados portaban genes cry. El problema de
la aparición de insectos resistentes a estas plantas se prevé solucionarlo con la implantación de distintas
proteínas insecticidas en una misma planta transgénica o en plantas transgénicas plantadas en años
alternativos.
3.2.2. Transferencia genética con protoplastos Como la formación de agallas no se producía en prácticamente ninguna monocotiledónea, se
investigaron al mismo tiempo otros métodos que permitiesen generar plantas transgénicas en este grupo
que abarca a las gramíneas, tan importantes en la nutrición humana. Los protoplastos son células de
14

cualquier tejido vegetal a las que se les ha liberado de la pared celular que es la barrera que impide el paso
de grandes moléculas como el ADN. La pared celular se elimina digiriéndola con un enzima. El gen que
se ha de transferir se adiciona al medio de cultivo del protoplasto. Si se somete un protoplasto a descargas
eléctricas creamos diminutos poros en la membrana por los cuales puede penetrar el ADN. A este método
se le denomina electroporación. También podemos ayudar a introducir ADN en un protoplasto empleando
sustancias como el polietilenglicol que desestabiliza la membrana celular. Otro método consiste en
emplear liposomas que contengan el ADN a transferir. La dificultad principal que plantea este método
estriba en el escaso desarrollo de las plántulas generadas a partir de protoplastos.
En 1988 se obtuvo por primera vez cereales transgénicos a partir de la regeneración de
protoplastos con genes exógenos en medio de cultivo para células vegetales.
3.2.3. Transferencia genética con el "cañón de partículas" (Biobalística)
Sanford, biólogo molecular de la Universidad de Cornell (EEUU) a principios de los años
ochenta estaba buscando el método definitivo para transformar cualquier tipo de plantas. En 1984
estableció contacto con E. Wolf director del centro de fabricación de micropartículas de su misma
universidad. Entre ambos surgió la idea de bombardear células vegetales con ADN y como éste es una
molécula flexible y frágil decidieron enganchar ADN a micropartículas metálicas. En presencia de
cloruro de calcio y espermidina el ADN queda adherido a las micropartículas metálicas por interacciones
no covalentes. En las primeras pruebas se empleó micropartículas de tungsteno de cuatro micrómetros de
di metro. Las partículas se proyectan sobre el tejido vegetal por el impulso de un chorro de aire
comprimido o la explosión de una carga de pólvora (Fig. 9). Una vez dentro del tejido vegetal el ADN se
desprende de las micropartículas debido a las modificaciones del entorno iónico. Cuando la cantidad de
partículas en una célula era superior a once había pocas probabilidades de que la célula sobreviviera. Se
pensó que el tungsteno podría ser ligeramente tóxico y por este motivo se emplearon posteriormente
micropartículas de oro. Una vez probada la penetración de ADN quedaba por demostrar la transferencia
genética. Adhirieron a las micropartículas metálicas ARN del genoma del virus del mosaico del tabaco.
Tres días después del bombardeo de células de cebolla se observaron partículas víricas, lo que demostraba
que el material genético introducido seguía siendo funcional.
15

Figura 9: Microcañón con partículas metálicas rodeadas de ADN
A partir de numerosos experimentos se cambiaron muchos factores para mejorar el rendimiento:
el tamaño de las microbolas, su velocidad, la inmovilización de las células vegetales y la cantidad de
ADN transportado. Un problema que plantea esta técnica es que se generan dos tipos de células: las
transformadas y las no transformadas dentro de un mismo órgano. Aparecen entonces competiciones
entre los dos tipos celulares disminuyendo la eficacia del método.
Pero por otra parte se evita el problema mayor que supone la regeneración de plantas a partir de
protoplastos.
3.2.4. Otras técnicas de transferencia genética
- Se ha intentado la transformación directa depositando una solución de ADN a transferir y de
polen sobre los estigmas. De esta manera se supone que el ADN penetraría a través del tubo polínico
durante su desarrollo en el estigma. Los raros éxitos conseguidos no han superado, hasta ahora, las
pruebas de la expresión de los genes en la descendencia.
- También se ha intentado inyectar en una célula vegetal una solución de ADN. La
microinyección se realiza bajo control microscópico y con microcapilares. La microinyección resulta
poco efectiva porque las puntas de los microcapilares se rompen y se obstruyen con facilidad además se
necesitan inyectar al menos 10000 células, una a una, para tener la seguridad de que al menos una de ellas
ha incorporado el material genético.
Después de los expuesto podemos decir que el objetivo de todos estas técnicas es la creación de
una planta transgénica. El proceso completo lo podemos esquematizar en la siguiente figura.
16
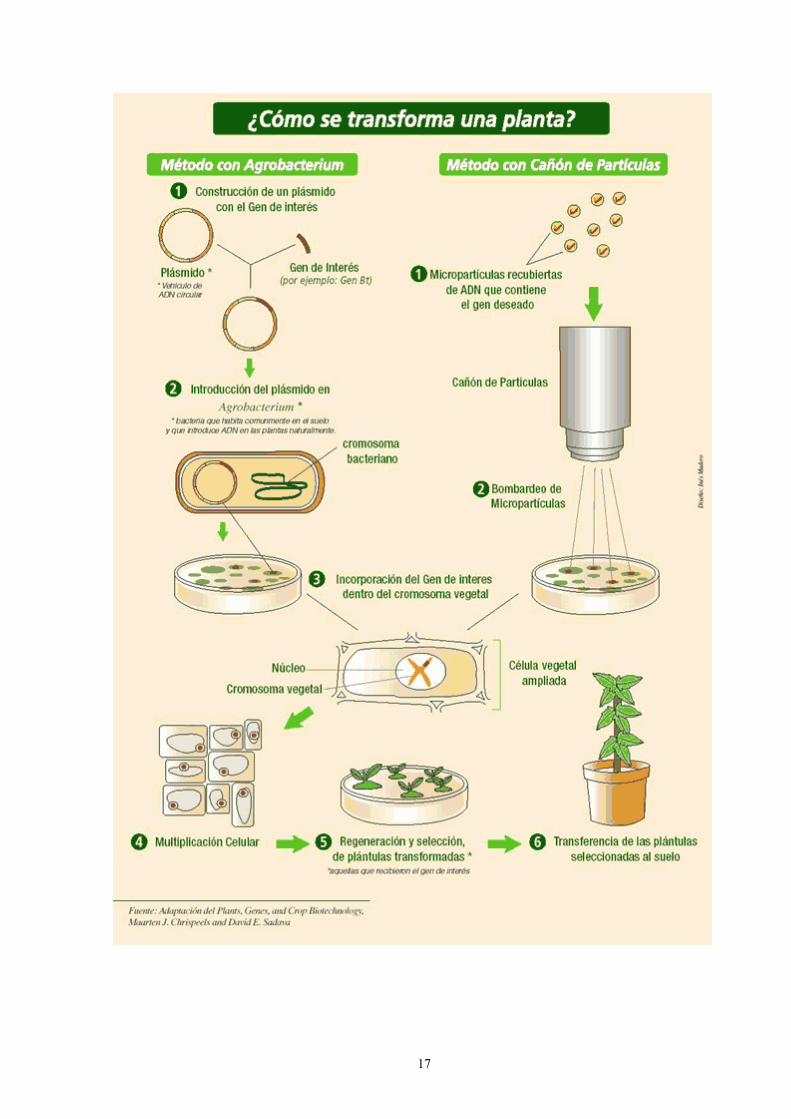
17

Pirosecuenciación
S Una alternativa más moderna (1988) a la secuenciación Sanger, más rápida y barata, aunque menos precisa S Aprovecha el trifosfato que se libera cuando el dNTP se une a
la cadena, en forma de pirofosfato (PPi) S Requiere de tres enzimas adicionales: ATP-sulfurilasa,
luciferasa y apirasa S Requiere de dos sustratos adicionales: adenosin-fosfosulfato
(APS) y luciferina S Con estas enzimas vamos a tener reacciones que de manera
natural generan señales luminosas, ahorrándonos las marcas fluorescentes, y haciendo el proceso más rápido y barato

a) Partimos, como en Sanger, de una secuencia plantilla y un primer, pero con más enzimas y sustratos, y sin etiquetar los dNTPs
b) La polimerasa une un dNTP, liberando un PPi en el proceso
c) La ATP sulfurilasa convierte el PPi en ATP con ayuda del APS
d) La luciferasa convierte el ATP en luz, con ayuda de la luciferina
e) Medimos un pulso de luz
f) La apirasa se libra de los reactivos sobrantes para que el sistema esté limpio para el siguiente dNTP
g) El resultado final es, como en Sanger, picos de intensidad

Pirosecuenciación
S Ventajas S Muy rápido (hasta 700Mbps por ejecución)
S Más barato que Sanger
S Precisión bastante alta
S Desventajas S No puede generar secuencias muy largas (<1Kbs)
S Problemas para medir homopolímeros (secuencias de varios nucleótidos iguales)

Yeast ArtificialChromosomesCarlo V Bruschi, International Centre for Genetic Engineering and Biotechnology, Trieste, Italy
Kresimir Gjuracic, International Centre for Genetic Engineering and Biotechnology, Trieste, Italy
Yeast artificial chromosomes are shuttle-vectors that can be amplified and modified in
bacteria and employed for the cloning of very large DNA inserts (up to 1–2 megabase pairs)
in the yeast Saccharomyces cerevisiae.
Introduction
Yeast artificial chromosomes (YACs) are plasmid shuttle-vectors capable of replicating and being selected in commonbacterial hosts such as Escherichia coli, as well as in thebudding yeast Saccharomyces cerevisiae. They are ofrelatively small size (approximately 12 kb) and of circularformwhen they are amplified or manipulated in E. coli, butare rendered linear and of very large size, i.e. severalhundreds of kilobases (kb), when introduced as cloningvectors in yeast. The latter process involves cleavage atstrategically located sites by two restriction enzymes, whichbreak themin two linearDNAarms.Theseare subsequentlyligatedwith the appropriateDNA insert before transforma-tion into recipient yeast cells converted to spheroplasts,where telomere sequences are added by the cell’s telomeraseenzymes. In this linear form, these specialized vectorscontain all three cis-acting structural elements essential forbehaving likenatural yeast chromosomes: anautonomouslyreplicating sequence (ARS) necessary for replication; acentromere (CEN) for segregation at cell division; and twotelomeres (TEL) for maintenance. In addition, theircapacity to accept large DNA inserts enables them to reachthe minimum size (150 kb) required for chromosome-likestability and for fidelity of transmission in yeast cells. YACshave several advantages over other large capacity vectors:these include accommodation of DNA segments thousandsof kilobases in size and stable maintenance of clonedeukaryotic DNA due to the compatibility with the yeastreplication machinery. Moreover, they are amenable tolarge-scaleplasmidamplification inE. coliand tocreationofspecific genetic changes within the exogenous DNAsequences by using the faithful and efficient yeast mechan-ism of homologous recombination.
Overview of Yeast ArtificialChromosomes (pYACs) Plasmids
Many different yeast artificial chromosomes exist as on-going refinements of the initial pYAC3 and pYAC4
plasmids (Figure 1) constructed by Burke et al. (1987). Thebasic structural features of YACs were developed from theyeast centromere shuttle-plasmids YCp series. These arecomposed of double-stranded circular DNA sequencescarrying the b-lactamase gene bla and the bacterial pMB1origin of replication, thus conferring resistance to ampi-cillin and the ability to replicate in bacteria, respectively.They also include yeast ARS1 with its associated CEN4DNA sequence, as well as theURA3 selectable marker. Onthis basic scaffoldplasmid the yeastHIS3 is cloned, flankedby a telomere-likeDNA sequence derived from the terminiof the Tetrahymena pyriformis macronuclear ribosomalDNA, which allows the formation of functional telomeresin yeast, that are adjacent to two recognition sites for theBamHI restriction enzyme. Most of these YACs alsocontain the cloning site in the middle of the SUP4suppressor of an ochre allele of a tyrosine transfer RNA(tRNA) gene; this enables restoration of the normal whitecolour phenotype in otherwise red ade1 and/or ade2nonsense mutants (Fischer, 1969). Accordingly, in the
Article Contents
Secondary article
. Introduction
. Overview of Yeast Artificial Chromosomes (pYACs)
Plasmids
. Construction of Yeast Artificial Chromosomes
. Biological Features of YACs
. Modifying YACs by Homologous Recombination
. Use of Yeast Artificial Chromosomes
. YAC Genomic Libraries
. Production of Transgenic Mice with YACs
. YACs in the Construction of Mammalian Artificial
Chromosomes
bla
ARS1CEN4
HIS3 URA3TEL
TEL
HindIII 8500
XhoI 8500
BamHI 8000
BamHI 6100
XhoI 5500
HindIII 5500
Sal I 3300
pYAC311400 bp
SUP4 ochrepMB1 ORI
Figure 1 Circular map of plasmid vector pYAC3. All the selectablemarkers, the origins of replication for bacteria and yeast, and the yeastchromosomal structural elements are indicated, together with the mapposition of the major restriction sites.
1ENCYCLOPEDIA OF LIFE SCIENCES / & 2002 Macmillan Publishers Ltd, Nature Publishing Group / www.els.net

insertional inactivation cloning process, the SUP4 gene isdisrupted by the DNA insert, thus removing the suppres-sion of the ade mutations and allowing their phenotypicexpression as red colour.
Construction of Yeast ArtificialChromosomes
After plasmid DNA purification, two distinct digestionsare performed: the first with BamHI that cuts twiceadjacent to the two telomeric DNA sequences flankingtheHIS3 gene, which therefore is excised from the plasmidand lost (Figure 2a). This first digestion generates a longlinear fragment carrying telomeric sequences at each end.The excision of theHIS3 gene is used as negative selectivemarker for uncut pYAC molecules. The second digestionconsists of the opening of the cloning site within the SUP4gene (Figure 2a). As a result of this second digestion, twolinear fragments are produced as left and right arms of thefuture linear YAC (Figure 2b). The selective markers arethus separated: TRP1 adjacent to ARS1 and CEN4 on the
left arm and URA3 on the right arm. Large DNAfragments with ends compatible to the cloning site,obtained from the desired genome source by digestionwith an appropriate restriction endonuclease, are ligatedwith phosphatase-treated YAC arms, to create a singleyeast-transforming DNA molecule (Figure 2c). Primarytransformants can be selected for complementation of theura3 mutation in the host, and successively for comple-mentation of the host trp1mutation, thereby ensuring thepresence of both chromosomal arms. Transformantcolonies containing the exogenous DNA insert within theSUP4 gene are detected by their red colour, due to theinactivation of the suppressor activity and the consequentaccumulation of a redmetabolic precursor in adehost cells.
Biological Features of YACs
The stability ofYACvectors in yeast per se is similar to thatof natural chromosomes (102 5/102 6) provided that allthree structural elements (ARS,CENandTEL) are presentand functional and, in addition, that the minimal requiredsize is reached by the insertion of enough exogenousDNA.
DNA fragments
Insert DNA URA3 TELTRP1/ARS1/CEN4TEL
(c)
URA3 TEL
Right YAC arm
Ligation andyeast transformation
TRP1/ARS1/CEN4TEL
(b)
Treatment withrestriction nucleases
TEL HIS3 TELBamHI
Circular YACBLA/ORI
TRP1/ARS1/CEN4 SUP4
Cloning site
URA3
(a)
Left YAC arm
Figure 2 Construction of the yeast artificial chromosome. (a) A circular YAC vector able to replicate in Escherichia coli due to the presence of bacterialori and bla gene (blue cylinder) and propagated in yeast cells as a linear molecule containing all necessary chromosomal elements: yeast centromereCEN4 (orange circle), autonomous replication sequence ARS1 (dark green cylinder) and two Tetrahymena telomeric sequences TEL (orange ellipse)functional in yeast after linearization with the BamHI restriction endonuclease. The yeast SUP4 gene (red cylinder) contains a cloning site used as acolour marker for selection of YACs containing exogenous insert DNA. (b) DNA fragments with ends compatible to the YAC cloning site are preparedfrom source DNA. After double digestion of the YAC vector, the markers used to select for transformants are separated on two chromosomal arms: TRP1 onthe left and URA3 on the right arm (light green cylinders). (c) Chromosomal arms ligated with exogenous DNA are selected after transformation ofappropriate yeast strain (ura3, trp1, ade2). Adapted from Burke et al., (1987).
Yeast Artificial Chromosomes
2 ENCYCLOPEDIA OF LIFE SCIENCES / & 2002 Macmillan Publishers Ltd, Nature Publishing Group / www.els.net

However, the genetic and biochemical background of thehost cell also plays an important role in determining thestability of YACs. Indeed, several mutations are known toaffect YAC stability and segregation together with naturalchromosomes. For example, alterations in the expressionsof genes such as BUB1, BUB2 and BUB3,MAD1,MAD2and MAD3, CDC6, CDC20, PDS1 and others lead tochromosome losses, including YACs. Another importantconsideration is that faithful duplication of YACs isguaranteed only if other DNA sequences incompatiblewith ARS do not exist on the construct. This point isparticularly relevant when unknown DNA inserts arecloned in the YAC vector, as is the case for genomiclibraries, in which there could be cryptic or otherwiseunknown ARS-like sequences able to interfere with theARS function.
Modifying YACs by HomologousRecombination
Depending on the experimental systems and the yeaststrains, different selectablemarkers and restriction sites areappropriate on the YAC vectors. These can be constructedin vitro by standard techniques and then used forsubcloning DNA fragments. Additionally, existing YACclones can be modified by homologous recombination inyeast, a process called ‘retrofitting’ (Figure 3). Accordingly,genetic markers can be modified by simply transformingYAC-containing yeast cells with a disruption cassettecarrying the desired genetic marker flanked by short DNAsequences homologous to one of the markers present onthe artificial chromosome. The same result can be obtainedby using one arm of the artificial chromosome carrying thenew genetic marker as the transforming molecule. Asshown in Figure3a, the introductionof the yeastLYS2 gene,together with the mammalian selectable marker Neo, intothe URA3 gene on the right arm of a YAC is achieved byusing recombination techniques. Neo is the usual markergene for subsequent selection of mammalian cells afterYAC transfection (see below). Moreover, disruption ofthis marker provides a selectable replacement marker forintroducing specific genetic changes within the exogenousDNA insert (Figure 3).A yeast integrative plasmid (YIp) carrying the mutated
copy of an exogenous DNA segment and the functionalcopy of a previously disrupted marker can be used for thispurpose (Barton et al., 1990). After plasmid integration,the target DNA is duplicated in tandem via homologousrecombination, generating a YAC with one wild-type andone mutant copy of insert DNA (‘pop-in’). At this stage,YACs with only the mutated copy of the marker can beobtained after spontaneous recombination within dupli-cated DNA regions (‘pop-out’) by growing the trans-formed cells in the absence of selection pressure for several
generations (Figure 3b). However, the frequency is not veryhigh (from 102 4 to 102 5 per generation) and there isapproximately the same probability for both the mutatedand wild-type copy of the marker to remain within the cellon the YAC vector. In the latter case it is necessary toperform additional molecular tests of the obtained ‘pop-outs’.
Use of Yeast Artificial Chromosomes
YAC vectors were initially created for the cloning of largeexogenousDNAsegments inS. cerevisiaebut soonbecamechromosomal-like platforms for a variety of in vivoexperiments. Applications of YACs range from generatingwhole DNA libraries of the genomes of higher organismsto identifying essential mammalian chromosomal se-quences necessary for the future constructionof specializedmammalian artificial chromosomes (MACs). The avail-ability of YAC libraries has greatly advanced the analysisof genomes previously cloned in cosmid vectors. Forexample, YAC clones have been used as hybridizationprobes for the screening of cDNA libraries, thus simplify-ing the characterization of unidentified genes. Anothermajor application of YACs is in the study of regulation ofgene expression by cis-acting, controlling DNA elements,that are present either upstream or downstream of largeeukaryotic genes, after the transfer of these YACs fromyeast to mammalian cells. Recent technological develop-ments allow the transfer of YACs into mouse embryonalstem (ES) cells and the subsequent generation of transgenicmice. Investigators have begun to employ these artificialchromosomes for the in vivo study of multigenic loci inmammalian cells.
YAC Genomic Libraries
Bacterial artificial chromosomes (BACs) and the P1-derived artificial chromosomes (PACs) can accommodateinserts of exogenous DNA up to 300 kb. Although thisrepresents an average insert size close to the one obtainedafter YAC preparation in vitro, it is possible to constructYACs with megabase-long inserts using the precisehomologous recombination process of yeast. The pre-requisite for this technique is the isolation of YAC clonescontaining neighbouring genomic DNA inserts. Initialscreening for specific sequences usually results in severalclones, which most probably contain overlapping inserts.Following insert fingerprinting by conventional Southernhybridization or polymerase chain reaction (PCR)-basedmethods, using tandemly repeated sequences asmarkers, itis possible to obtain detailed physical maps allowing anaccurate alignment of two inserts by comparing theirindividual restriction patterns. Further neighbouring
Yeast Artificial Chromosomes
3ENCYCLOPEDIA OF LIFE SCIENCES / & 2002 Macmillan Publishers Ltd, Nature Publishing Group / www.els.net
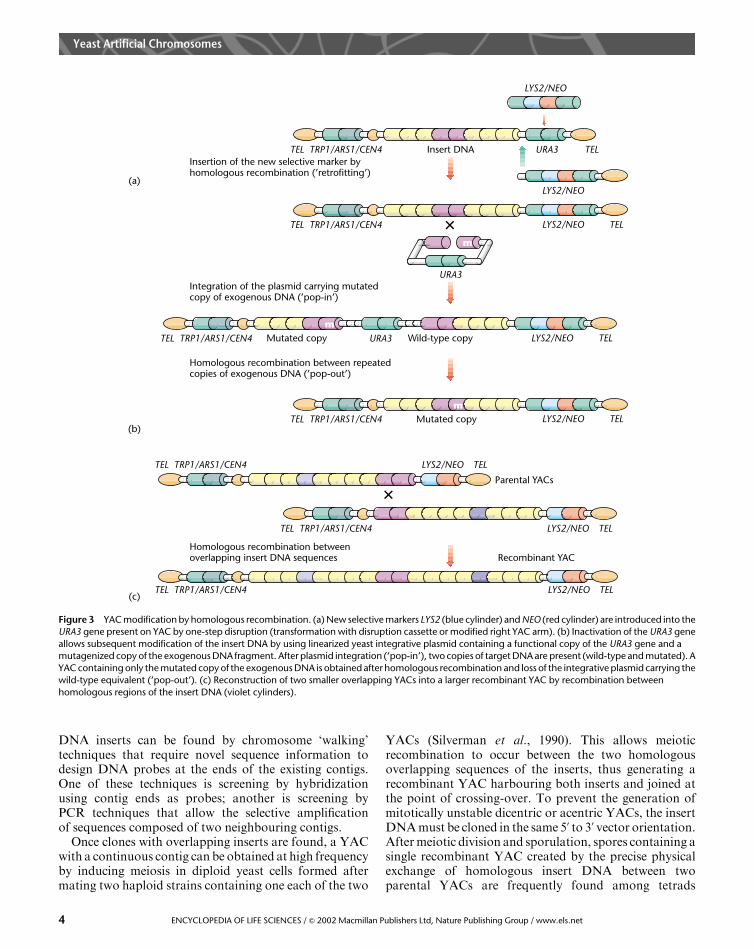
DNA inserts can be found by chromosome ‘walking’techniques that require novel sequence information todesign DNA probes at the ends of the existing contigs.One of these techniques is screening by hybridizationusing contig ends as probes; another is screening byPCR techniques that allow the selective amplificationof sequences composed of two neighbouring contigs.Once clones with overlapping inserts are found, a YAC
with a continuous contig can be obtained at high frequencyby inducing meiosis in diploid yeast cells formed aftermating two haploid strains containing one each of the two
YACs (Silverman et al., 1990). This allows meioticrecombination to occur between the two homologousoverlapping sequences of the inserts, thus generating arecombinant YAC harbouring both inserts and joined atthe point of crossing-over. To prevent the generation ofmitotically unstable dicentric or acentric YACs, the insertDNAmust be cloned in the same 5’ to 3’ vector orientation.Aftermeiotic division and sporulation, spores containing asingle recombinant YAC created by the precise physicalexchange of homologous insert DNA between twoparental YACs are frequently found among tetrads
TRP1/ARS1/CEN4TEL
m
Insert DNA URA3 TELTRP1/ARS1/CEN4TEL
(a)
LYS2/NEO
LYS2/NEO
Insertion of the new selective marker byhomologous recombination (’retrofitting’)
TRP1/ARS1/CEN4TEL × LYS2/NEO TEL
URA3
(b)
Integration of the plasmid carrying mutatedcopy of exogenous DNA (’pop-in’)
LYS2/NEO TELWild-type copyURA3Mutated copym
(c)
Homologous recombination between repeatedcopies of exogenous DNA (’pop-out’)
TRP1/ARS1/CEN4TEL LYS2/NEO TELm
Mutated copy
TRP1/ARS1/CEN4TEL LYS2/NEO TEL
×TRP1/ARS1/CEN4TEL LYS2/NEO TEL
Homologous recombination betweenoverlapping insert DNA sequences
TRP1/ARS1/CEN4TEL LYS2/NEO TEL
Parental YACs
Recombinant YAC
Figure 3 YAC modification by homologous recombination. (a) New selective markers LYS2 (blue cylinder) and NEO (red cylinder) are introduced into theURA3 gene present on YAC by one-step disruption (transformation with disruption cassette or modified right YAC arm). (b) Inactivation of the URA3 geneallows subsequent modification of the insert DNA by using linearized yeast integrative plasmid containing a functional copy of the URA3 gene and amutagenized copy of the exogenous DNA fragment. After plasmid integration (‘pop-in’), two copies of target DNA are present (wild-type and mutated). AYAC containing only the mutated copy of the exogenous DNA is obtained after homologous recombination and loss of the integrative plasmid carrying thewild-type equivalent (‘pop-out’). (c) Reconstruction of two smaller overlapping YACs into a larger recombinant YAC by recombination betweenhomologous regions of the insert DNA (violet cylinders).
Yeast Artificial Chromosomes
4 ENCYCLOPEDIA OF LIFE SCIENCES / & 2002 Macmillan Publishers Ltd, Nature Publishing Group / www.els.net

Estrategia de clonado en plásmido
Pasos:
1. Construcción de vector recombinante.
2. Transformación de células competentes.
3. Plaqueo en medio selectivo con antibiótico (para seleccionar las bacterias que adquirieron el plásmido) + IPTG y X-Gal (para identificar las bacterias que tienen el plásmido con inserto)
4. Validación de la presencia de inserto mediante:a. digestión de plásmidos de colonias + con ER y análisis en gel de agarosa.b. amplificación del inserto a partir de las colonias (“colony PCR”) y gel.
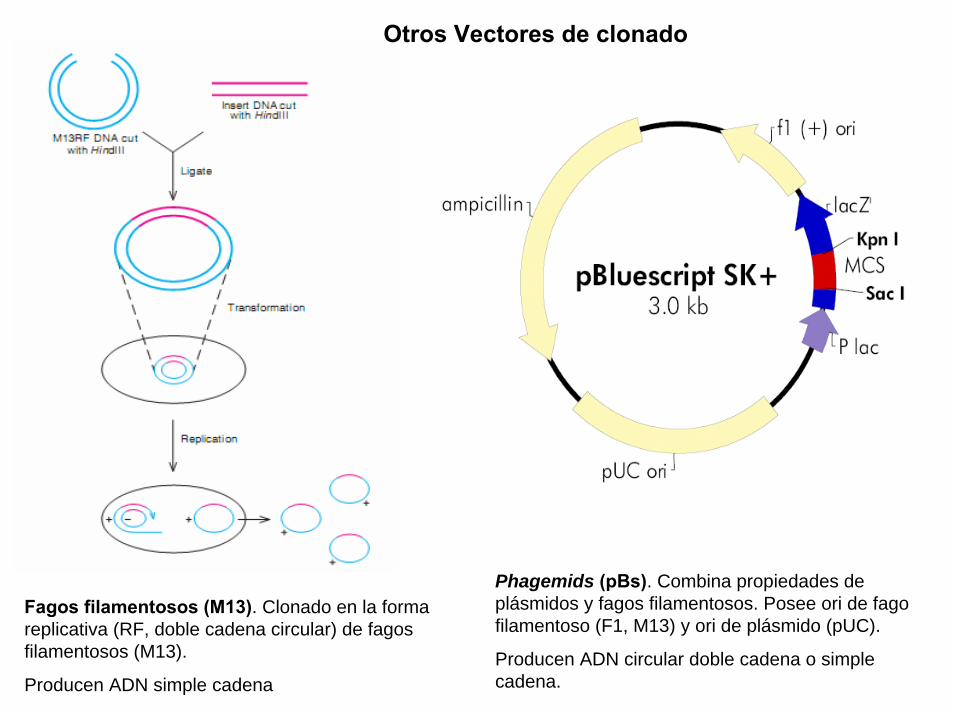
Phagemids (pBs). Combina propiedades de plásmidos y fagos filamentosos. Posee ori de fago filamentoso (F1, M13) y ori de plásmido (pUC).
Producen ADN circular doble cadena o simple cadena.
Otros Vectores de clonado
Fagos filamentosos (M13). Clonado en la forma replicativa (RF, doble cadena circular) de fagos filamentosos (M13).
Producen ADN simple cadena
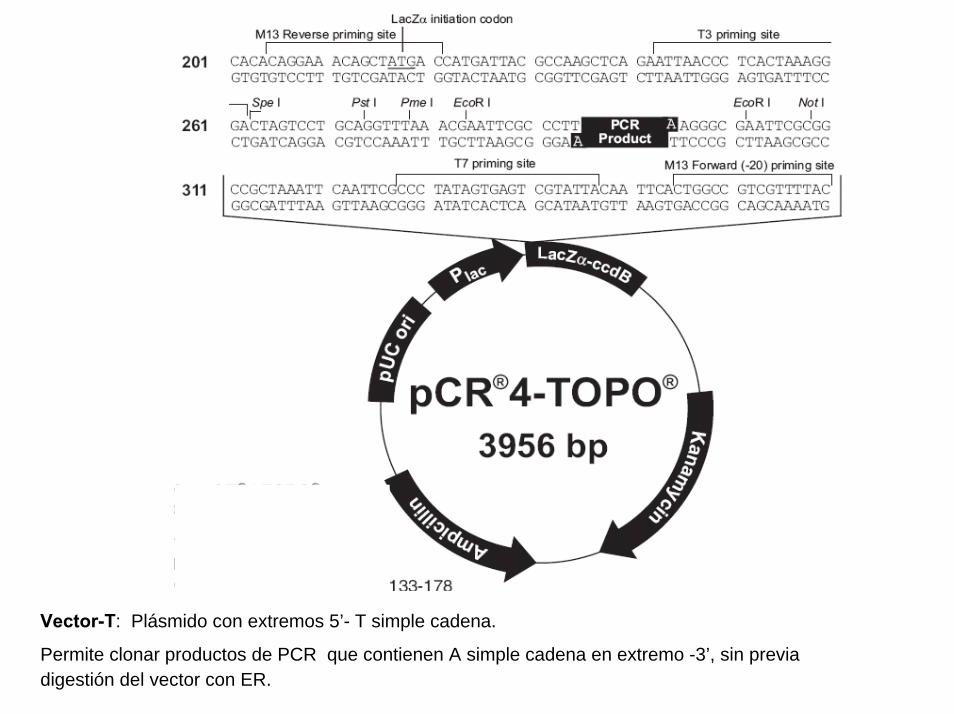
Vector-T: Plásmido con extremos 5’- T simple cadena.
Permite clonar productos de PCR que contienen A simple cadena en extremo -3’, sin previadigestión del vector con ER.
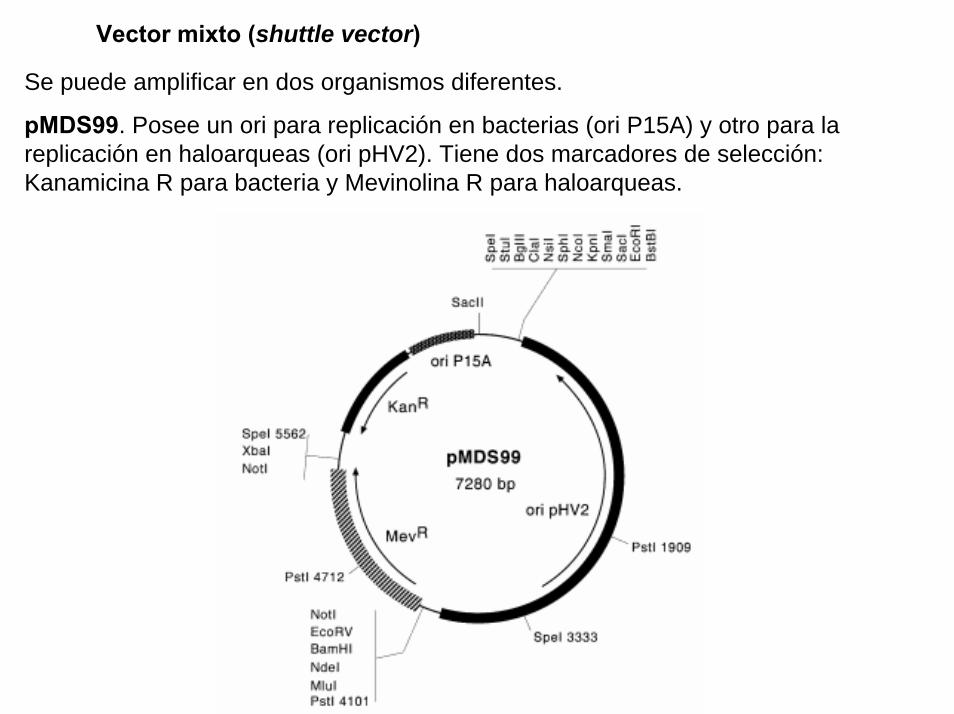
Vector mixto (shuttle vector)
Se puede amplificar en dos organismos diferentes.
pMDS99. Posee un ori para replicación en bacterias (ori P15A) y otro para la replicación en haloarqueas (ori pHV2). Tiene dos marcadores de selección: Kanamicina R para bacteria y Mevinolina R para haloarqueas.
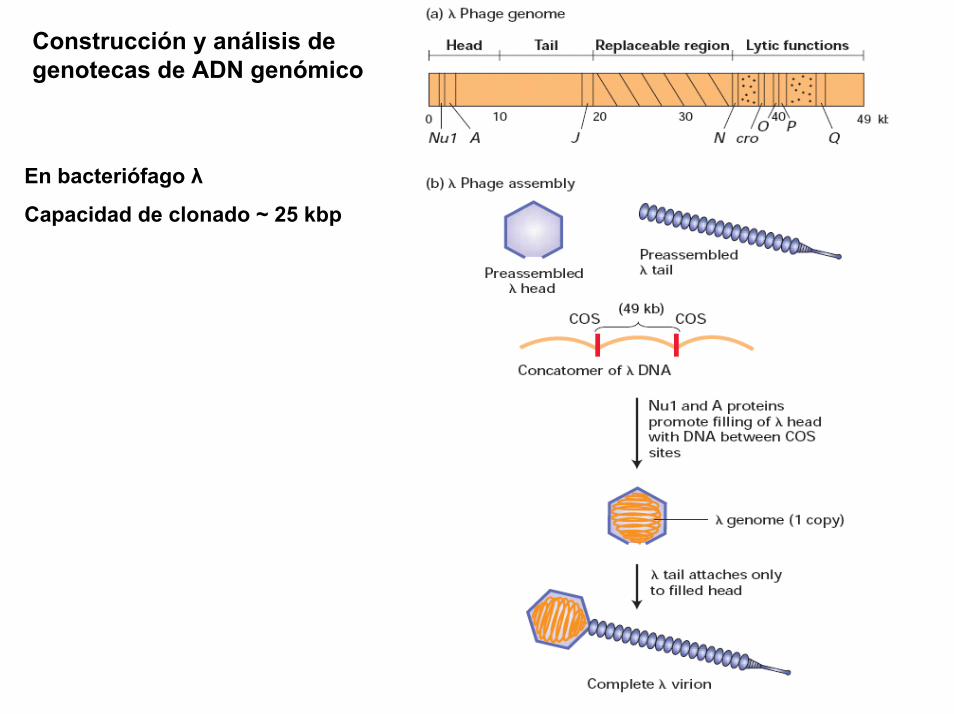
Construcción y análisis de genotecas de ADN genómico
En bacteriófago λ
Capacidad de clonado ~ 25 kbp
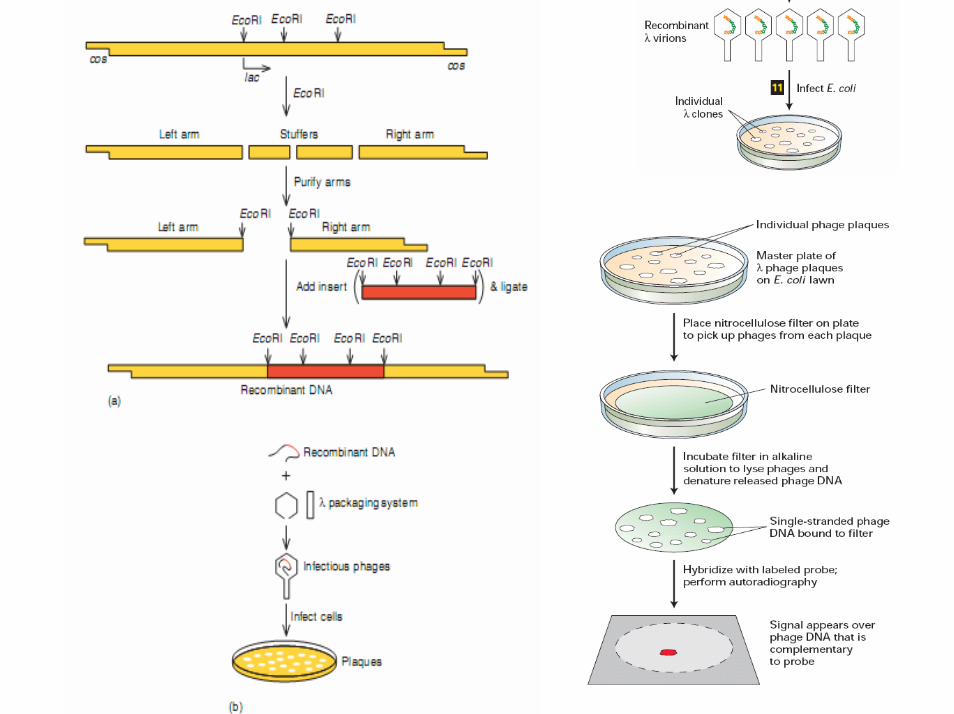
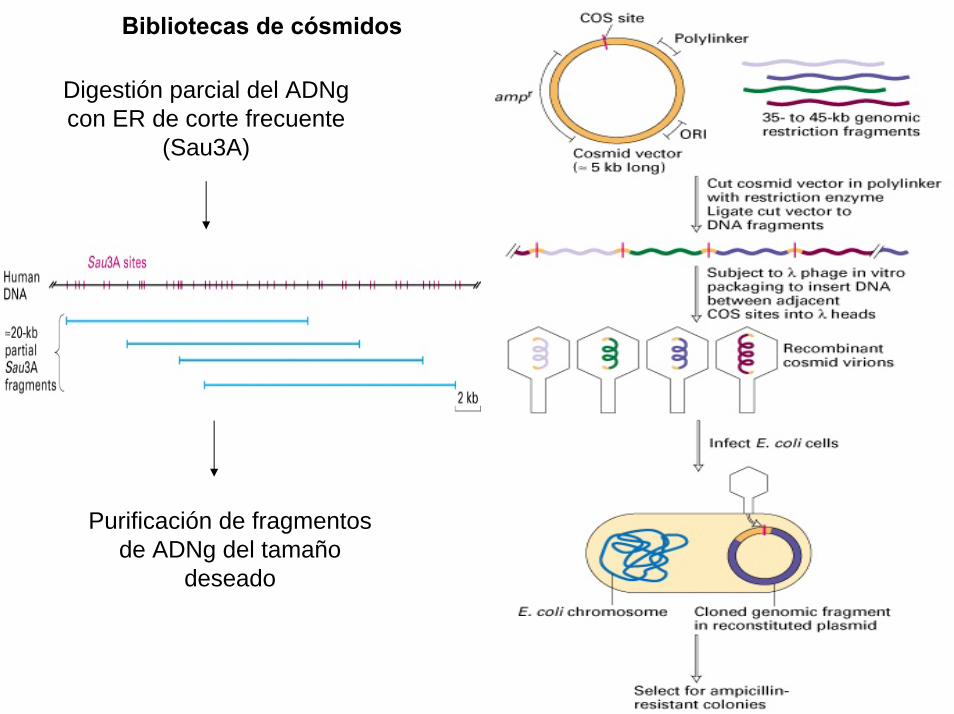
Bibliotecas de cósmidos
Digestión parcial del ADNgcon ER de corte frecuente
(Sau3A)
Purificación de fragmentosde ADNg del tamaño
deseado
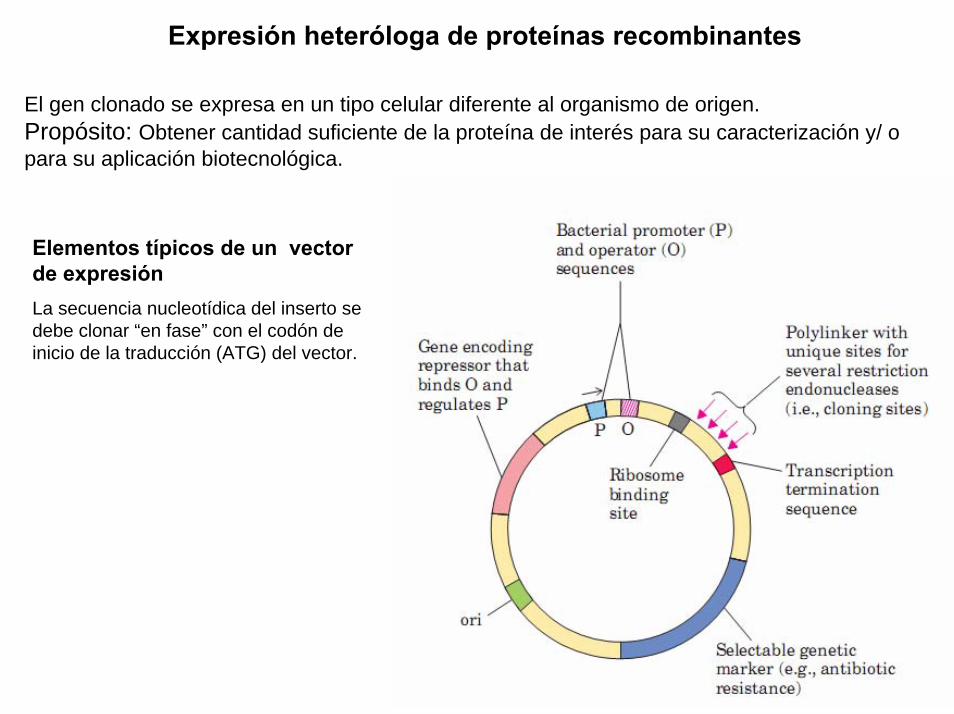
Expresión heteróloga de proteínas recombinantes
El gen clonado se expresa en un tipo celular diferente al organismo de origen.Propósito: Obtener cantidad suficiente de la proteína de interés para su caracterización y/ o para su aplicación biotecnológica.
Elementos típicos de un vector de expresiónLa secuencia nucleotídica del inserto se debe clonar “en fase” con el codón de inicio de la traducción (ATG) del vector.
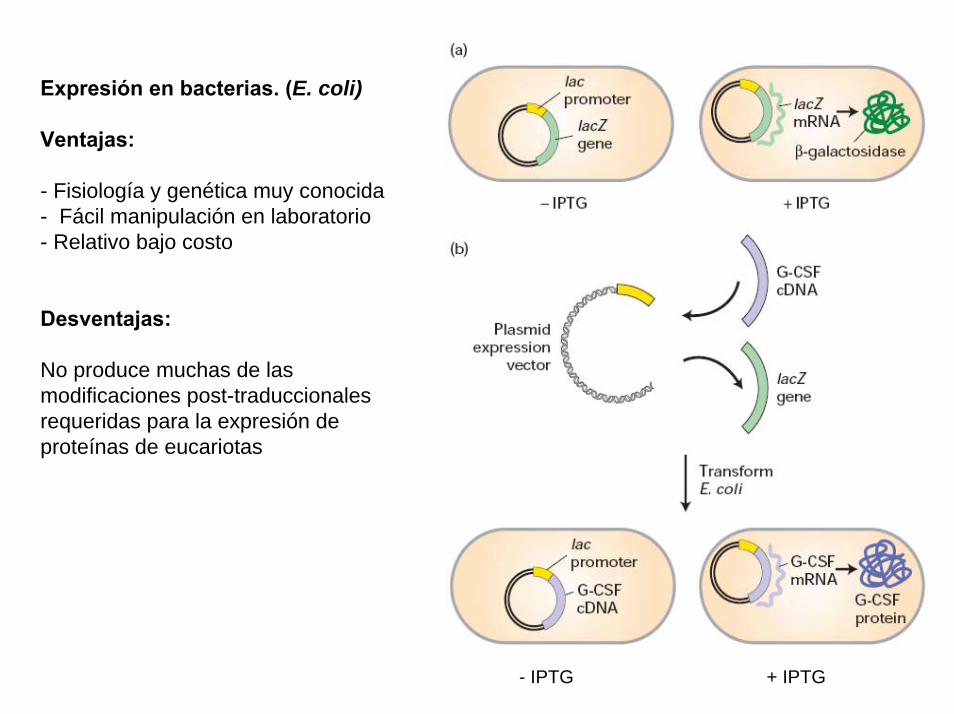
Expresión en bacterias. (E. coli)
Ventajas:
- Fisiología y genética muy conocida- Fácil manipulación en laboratorio- Relativo bajo costo
Desventajas:
No produce muchas de las modificaciones post-traduccionalesrequeridas para la expresión de proteínas de eucariotas
- IPTG + IPTG
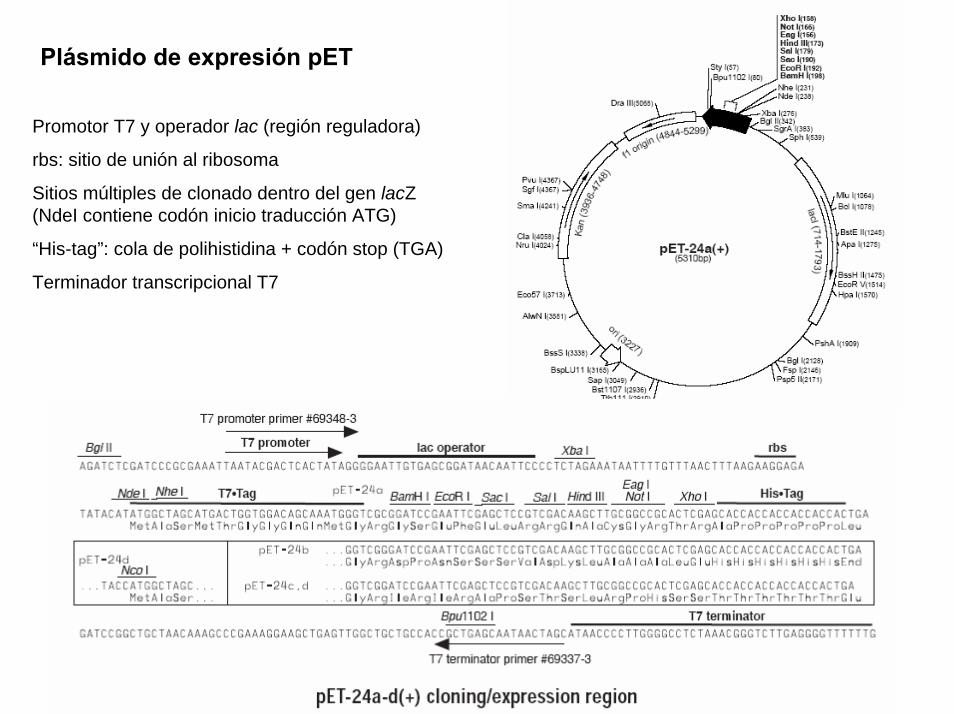
Plásmido de expresión pET
Promotor T7 y operador lac (región reguladora)
rbs: sitio de unión al ribosoma
Sitios múltiples de clonado dentro del gen lacZ(NdeI contiene codón inicio traducción ATG)
“His-tag”: cola de polihistidina + codón stop (TGA)
Terminador transcripcional T7
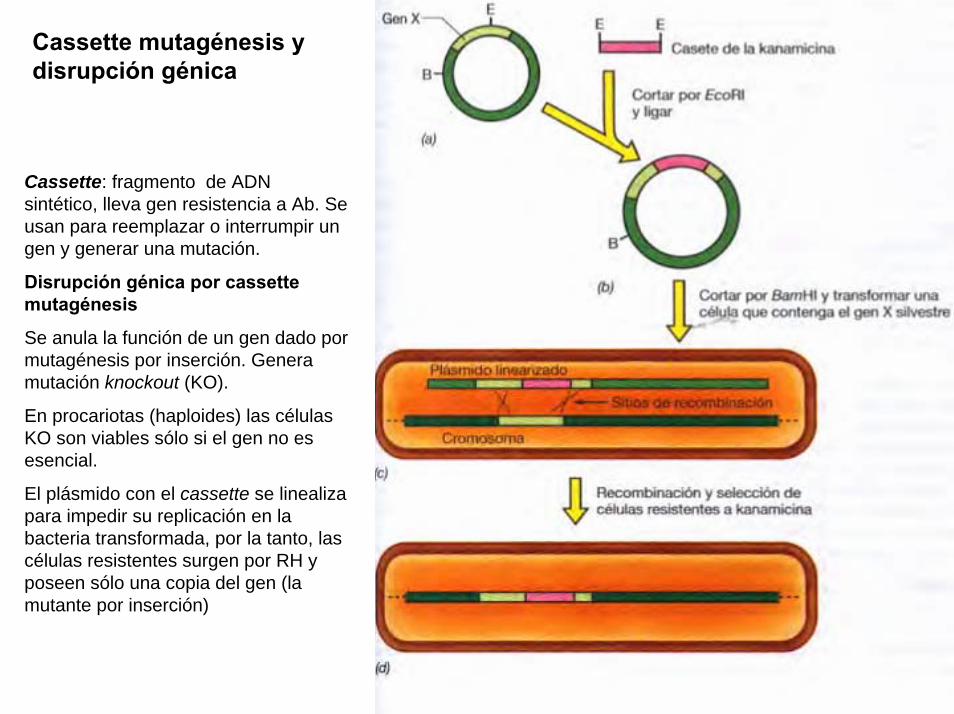
Cassette mutagénesis y disrupción génica
Cassette: fragmento de ADN sintético, lleva gen resistencia a Ab. Se usan para reemplazar o interrumpir un gen y generar una mutación.
Disrupción génica por cassettemutagénesis
Se anula la función de un gen dado por mutagénesis por inserción. Genera mutación knockout (KO).
En procariotas (haploides) las células KO son viables sólo si el gen no es esencial.
El plásmido con el cassette se linealizapara impedir su replicación en la bacteria transformada, por la tanto, las células resistentes surgen por RH y poseen sólo una copia del gen (la mutante por inserción)

cada etapa de la reacción. La automatización del proceso se debe al descubrimiento de la enzima Taq polimerasa termoestable, extraida del Thermus aquaticus2, que eliminó el inconveniente de agregar enzima fresca en cada paso de la reacción.
Fig 1.- Equipo (maquina de PCR), utilizado para la amplificación del ADN. La maquina se programa con las temperaturas necesarias para cada paso de la reacción y todo el proceso de amplificación se realiza automáticamente.
En la actualidad, la PCR es una técnica de rutina indispensable en laboratorios de investigación médica y biológica con una gran variedad de aplicaciones.
La clonación del ADN para la secuenciación, la filogenia basada en el ADN, el análisis funcional de genes, el diagnostico de trastornos hereditarios, la identificación de huellas genéticas (usada en técnicas forenses y prueba de paternidad), taxonomía, la detección y diagnostico de enfermedades infecciosas, son algunas de las aplicaciones donde la PCR ha sido implementada como una técnica de gran valor.
Componentes de la PCR
Desoxinucleótidos trifosfato (dNTPs) . Es una mezcla de deoxi - nucleótidos que sirve de sustrato para la síntesis de nuevo ADN.

Iniciadores ( primers). Dos oligonucleótidos que son, cada uno, complementarios a una de las dos hebras del ADN. Son secuencias cortas, de entre 10-30 nucleótidos, que son reconocidos por la polimerasa permitiendo iniciar la reacción. Deben estar enfrentados (uno en cada hebra del ADN), para delimitar el fragmento a amplificar).
ADN polimerasa. El descubrimiento en 1976 de la Taq polimerasa, una ADN polimerasa extraída de la bacteria termófila Thermus aquaticus que habita medios de muy alta temperatura (50-80 °C), eliminó los grandes inconvenientes del método de la PCR. Esta ADN polimerasa es estable a altas temperaturas, permaneciendo activa hasta después de la desnaturalización del ADN
ADN molde . Molécula que contiene la región de ADN que se va a amplificar.
Etapas de la PCR
La PCR consiste en una serie de cambios repetidos de temperatura llamados ciclos. Por lo general el número de ciclos es de 20 a 30, cada ciclo consiste en tres cambios de temperatura. Las temperaturas usadas y el tiempo aplicado en cada ciclo, dependen de algunos parámetros en los que se incluye, la enzima usada para la síntesis de ADN, la concentración de iones divalentes y dNTPs en la reacción así como la temperatura de unión de los iniciadores 3.
Desnaturalización del ADN
En la primera etapa, la reacción es llevada a una temperatura de 94-96ºC y se mantiene entre 30 segundos y un minuto. A estas temperaturas el ADN se desnaturaliza (se separan las dos hebras que lo constituyen). La temperatura a la cual se decide llevar a cabo esta etapa va a depender de la proporción de Guanina-Citocina (G-C) que tenga la hebra, así como también del largo de la misma.
Alineamiento/Unión del iniciador a la secuencia complementaria

En la segunda etapa también conocida como hibridación, los iniciadores se unen a las regiones 3`complementarias que flanquean el fragmento que se quiere amplificar. Para ello, es necesario bajar la temperatura a 50-65ºC durante 20-40 segundos, permitiendo así el alineamiento. La polimerasa se une entonces al hibrido formado por la cadena molde y el iniciador y empieza la síntesis del ADN. Los iniciadores actúan como límite de la región de la molécula que va a ser amplificada.
Extensión/Elongación de la cadena
En la tercera etapa, la polimerasa sintetiza una nueva hebra de ADN complementario a la hebra molde, añadiendo los dNTPs complementarios en dirección 5`� 3` uniendo el grupo 5`-fosfato de los dNTPs con el grupo 3`-hidroxilo del final de la hebra de ADN creciente. La temperatura en esta etapa depende de la ADN polimerasa que se utilice. Para la Taq polimerasa, la temperatura de máxima actividad está entre 74-80ºC, aunque por lo general se usa 74ºC. El tiempo de elongación va a depender del tipo de polimerasa empleado, así como también del tamaño del fragmento de ADN que se requiere amplificar.
Elongación final
En la cuarta etapa, la temperatura es regulada de 70-74ºC durante 5-15 minutos tras el último ciclo de PCR, con el objeto de asegurar que cualquier ADN de cadena simple restante sea totalmente amplificado.
Por último, el producto amplificado es mantenido a una temperatura de 4ºC por un periodo indefinido lo cual permite conservar el producto de la reacción
Para comprobar que la PCR ha generado el fragmento de ADN esperado, se emplean técnicas como la electroforesis, que permite separar los fragmentos de ADN generados de acuerdo a su tamaño. Por lo general se emplea la electroforesis en gel de agarosa para fragmentos grandes, en acrilamida para fragmentos más pequeños y asociada a marcaje fluorescente así como la electroforesis capilar3, 4

Fig 2.- Equipo de electroforesis utilizado para la separación de los productos amplificados por la PCR.
La detección del producto de la Reacción en Cadena de la Polimerasa se realiza normalmente mediante una corrida electroforética. Dependiendo del tamaño del producto de la amplificación y la resolución que se desea, se utilizan diferentes matrices de separación (agarosa, poliacrilamida) a distintas concentraciones. La posterior visualización se puede realizar con bromuro de etidio con una lámpara de luz UV, tinción de plata, fluorescencia o radioactividad.
De igual forma, la identidad del producto puede ser confirmada mediante hibridación (unión complementaria) con una sonda marcada radiactivamente, cuya secuencia de bases esté contenida en el fragmento de interés.
Los tamaños de los productos de PCR se determinan comparando los mismos, con marcadores que contienen fragmentos de ADN de tamaño conocido los cuales se corren en el gel junto a los productos de PCR.

Fig 4:_ Representa de manera esquemática el proceso de amplificación a partir del ADN extraído de la muestra. En cada ciclo se duplica el número de copias del ADN que se amplifica
Contaminación en la PCR
La reacción en cadena de la polimerasa PCR es una técnica altamente sensible y especifica, por lo que es muy importante evitar contaminaciones que puedan dar origen a la amplificación de ADN no deseado.
La contaminación con ADN extraño puede solucionarse con protocolos y procedimientos que separen las reacciones pre-PCR (mezcla de todos los componentes de la reacción), del ADN molde. Esto normalmente implica la separación espacial de las áreas de realización de la PCR de los de análisis o purificación de los productos, así como la limpieza exhaustiva de la superficie de trabajo entre la realización de un PCR y el siguiente.
Tipos de PCR
PCR anidada
En esta variante de PCR, el producto de una amplificación es utilizado como molde para realizar una segunda amplificación con iniciadores que se ubican dentro de la primera secuencia amplificada. Este tipo de PCR es altamente específica 5
PCR in situ
Es una PCR que se realiza en preparaciones fijadas sobre un portaobjeto. Consiste en una reacción de PCR en secciones histológicas o células, donde los productos generados pueden visualizarse en el sitio de amplificación. Se realiza para ello una primera amplificación del ADN blanco y luego detección mediante hibridación in situ convencional con sondas de ADN/ARN. De esta manera puede detectarse cantidades pequeñísimas del material genético 6

PCR multiplex
Es una PCR en la cual se amplifica más de una secuencia en una misma reacción. Emplea dos o más pares de iniciadores en un único tubo de reacción con el fin de amplificar simultáneamente múltiples segmentos de ADN. Consiste en combinar en una única reacción todos los pares de iniciadores de los sistemas que queremos amplificar simultáneamente, junto con el resto de los reactivos de la reacción en cantidades suficientes. La ventaja de esta PCR es que se obtiene la información de varios loci en una sola reacción, utilizando menor cantidad de molde para el análisis y menor cantidad de reactivos. Se puede generar rápidamente la construcción de una base de datos 7.
RT-PCR
Este tipo de PCR utiliza como molde inicial el ARN y se requiere de una transcriptasa inversa, para realizar la conversión del ARN a un tipo de ADN llamado ADNc (ADN complementario) 8.
PCR tiempo real
Es una reacción de PRC cuya principal característica es que permite cuantificar la cantidad de ADN o ARN presente en la muestra original 9. Se puede dividir en:
a- Técnica basada en fluorocromos no específicos.
b- Técnica basada en sondas específicas.
En las técnicas basadas en fluorocromos no específicos, el ADN se une al fluorocromo (por lo general SYBR Green) produciendo fluorescencia que es medida por un termociclador apto para PCR en tiempo real. Permite cuantificar sólo una secuencia por reacción, pero tiene la ventaja de utilizar iniciadores normales para su realización. Es mucho más económica que la PCR que utiliza sondas específicas.

Las técnicas basadas en sondas específicas utilizan una sonda unida a dos fluorocromos que hibrida en la zona intermedia entre el iniciador directo (forward) y el inverso (reverse). Cuando la sonda esta intacta, presentan una transferencia energética de fluorescencia por resonancia (FRET). Dicha fluorescencia no se produce cuando la sonda esta dañada y los dos fluorocromos están distantes, producto de la actividad 5`�3`exonucleasa de la ADN polimerasa. Esto permite monitorizar el cambio de patrón de fluorescencia y deducir el nivel de amplificación del gen.
Random Primer PCR
Esta variante de la técnica es muy sensible y especifica, es muy útil en el estudio y diagnóstico de organismos de una misma especie con variaciones genéticas especificas. Su aplicación permite la amplificación de moléculas de ADN y genomas que varían entre 400 pares de bases hasta 40 Mega bases. La técnica utiliza primes que consisten en un pool de de secuencias de los extremos 3’ del ADN y una región 5’ constante para la detección del primer incorporado 10.
Aplicaciones
El diagnóstico molecular está revolucionando la práctica clínica de enfermedades infecciosas. Sus efectos serán importantes en la configuración de la atención de casos agudos donde oportunas y precisas herramientas de diagnóstico son esenciales para las decisiones de tratamiento del paciente y los resultados. PCR es la técnica molecular más desarrollada hasta ahora y tiene una amplia gama de aplicaciones clínicas, incluyendo la detección de patógenos específicos o de amplio espectro, evaluación de las infecciones emergentes, la vigilancia, la detección temprana de agentes biológicos y la creación de perfiles de resistencia a los antimicrobianos. Los métodos basados en PCR también pueden asociarse a los procedimientos tradicionales de diagnóstico. Otro avance de la tecnología es necesario para mejorar la

automatización, optimizar la sensibilidad y especificidad y expandir la capacidad para detectar varios organismos simultáneamente. 11
La PCR convencional es utilizada como base para un gran número de técnicas en el laboratorio debido a su especificidad y rapidez.
Permite identificar especies de bacterias que producen determinados cuadros infecciosos. Esto se consigue, amplificando cierta zona del genoma bacteriano cuyo producto de PCR posea características particulares que permitan identificar de una manera inequívoca la bacteria o agente causal.
En el caso de infecciones virales, que implican la integración del genoma del patógeno en el ADN del hospedador, como es el caso de la infección por VIH, la PCR cuantitativa posibilita la determinación de la carga viral existente y por tanto, del estadio de la enfermedad 12, 13
De igual manera, la PCR es usada en servicios de donantes de sangre para test de rutina. Mediante la implementación de la PCR, se pueden detectar infecciones peligrosas en el donante (HIV, Hepatitis B) mientras aún están en el período de incubación.
A su vez, en el diagnostico de enfermedades hereditarias presentes en el genoma, en las cuales los procedimientos se presentan largos y engorrosos, la PCR ha venido a facilitar este problema, amplificando mediante iniciadores correspondientes los diferentes genes para la identificación de mutaciones existentes14.
En dermatología, la aplicación de la PCR en las enfermedades de la piel ha permitido conocer los mecanismos íntimos de muchas entidades, facilitando su diagnóstico e incluso su tratamiento. Esta técnica es ampliamente utilizada en el diagnóstico de enfermedades de la piel producida por parásitos, virus y bacterias; tales como leishmaniasis 15, lepra 16, oncocercosis 17 y virus de papiloma 18, entre otras, ampliamente reportadas en la literatura.
En la fig 5 se muestra un ejemplo de una reacción de polimerasa utilizada para el diagnóstico de leishmaniasis cutánea producida por Leishmania braziliensis. La reacción se lleva a cabo un par de oligonucleotidos obtenidos a partir de una secuencia repetida de ADN nuclear de Leishmania









Guía práctica sobre la técnica de PCR 517
Capítulo 17
Guía práctica sobrela técnica de PCR
Laura Espinosa Asuar
517
PCR son las siglas en inglés de Polymerase Chain Reaction o Reacción en Cadena de la Polimerasa. La idea básica de la técnica es sintetizar muchas veces un pedazo o fragmento de ADN utilizando una polimerasa que puede trabajar a temperaturas muy elevadas, ya que proviene de la bacteria Ther-mus aquaticus que vive a altas temperaturas (79ºC a 85ºC), de ahí su nombre comercial más conocido: taq polimerasa. Cuando hacemos una reacción de PCR simulamos lo que sucede en una célula cuando se sintetiza el ADN y en el tubo se mezclan todos los ingredientes necesarios para hacerlo: la polime-rasa, el ADN del organismo que queremos estudiar –donde se encuentra el fragmento que queremos sintetizar– , los oligonucleótidos (llamados también primers, iniciadores, cebadores, “oligos”, etc.) necesarios para que se inicie la transcripción, dinucleótidos (dNTPs), y las condiciones para que la enzima trabaje adecuadamente (cierto pH, determinadas cantidades de magnesio en forma de MgCl2, KCl, y pueden necesitarse otras sales o reactivos, dependiendo de cada polimerasa). Esta técnica tan ingeniosa tiene muchísimas aplicaciones distintas y se ha convertido en una herramienta muy importante en la biología molecular; sus aplicaciones van desde la genética de poblaciones, evolución molecular y genómica hasta la medicina forense.
¿Pero cómo funciona el PCR? Supongamos que ya tenemos los tubos listos con todo lo necesario para que la síntesis del fragmento que nos interesa que se lleve a cabo (taq polimerasa, dinucleótidos, ADN, agua, buffer con mag-

Las herramientas moleculares518
nesio y otras sales, y oligonucleótidos). El siguiente paso es colocar los tubos en una máquina conocida como termociclador, que básicamente sirve para calentarlos o enfriarlos a temperaturas muy precisas. ¿Cómo es que se ampli-fica el (o los) fragmento(s) que queremos? Primero, para hacer más sencilla la explicación, vamos a suponer que esperamos un solo fragmento de un tamaño determinado, y lo que sucede es lo siguiente (ver el primer ciclo de la figura 1): el termociclador calienta o enfría los tubos a tres temperaturas distintas, que se repiten una y otra vez (lo que se llama los ciclos de reacción), la primera es a 95ºC (y a este paso se le llama desnaturalización) durante la cual las dobles cadenas del ADN se abren o desnaturalizan, quedando en forma de cadenas sencillas; después el termociclador ajusta la temperatura en un intervalo en-tre 40º y 60ºC (llamada de alineamiento), a esta temperatura se forman y se rompen constantemente los puentes de hidrógeno entre los oligonucleótidos y el ADN, y aquellas uniones más estables (las que son complementarias) durarán mayor tiempo, quedando los oligonucleótidos “alineados” formando una pequeña región de doble cadena. La polimerasa se une a este pequeño pedazo de ADN de doble cadena y comienza a copiar en sentido 5’ a 3’; al agregar unas bases más, los puentes de hidrógeno que se forman entre las bases estabilizan más la unión y el oligonucleótido permanece en este sitio para el siguiente paso. Después la temperatura sube a 72ºC (paso que se conoce como extensión), ya que 72ºC es la temperatura en la cual la polimerasa alcanza su máxima actividad, y continúa la síntesis de los fragmentos de ADN a partir de los oligonucleótidos que ya se habían alineado.
En el primer ciclo, con estas tres temperaturas, se sintetizarán los prime-ros fragmentos a partir del ADN genómico. Estos primeros fragmentos no tendrán el tamaño esperado, serán un poco más grandes ya que la taq copiará hasta donde le sea posible, pero como veremos más adelante, se obtendrán en cantidades tan pequeñas que al final no podremos detectarlos. Después se repiten una vez más las tres temperaturas, pero en este segundo ciclo, los oligonucleótidos, además de unirse al ADN que pusimos al inicio, también se unirán a los fragmentos recién sintetizados del primer ciclo (ver segundo ciclo de la figura 1), por lo tanto en este segundo paso la polimerasa sintetizará 2 fragmentos largos copiados directamente del ADN y 2 fragmentos del tamaño esperado, que es el tamaño que hay entre los dos oligonucleótidos que hemos usado. De esta forma con cada ciclo aumentará el número de fragmentos del tamaño que queremos. Cabe mencionar que antes y después de estos ciclos se programan dos pasos, uno de 95ºC durante varios minutos para iniciar con desnaturalización, y al final de los ciclos, un paso último de extensión a

Guía práctica sobre la técnica de PCR 519
Figura 1. Descripción del proceso de un PCR en los primeros ciclos de reacción. Comúnmente se utilizan 35 ciclos para amplificar el fragmento que se requiere
(modificada de Parkes, 2003)
Fuente: Parkes H. 2003. Food for Thought http://www.chemsoc.org/chembytes/ezine/1999/parkes_may99.htm, accesado 08/03.
Primer cicloDesnaturalización del ADN
Alineamiento de los oligos
Extensión
Se sintetizan dos fragmentos un poco más largos de lo esperado
Segundo ciclo(Se muestra sólo la alineación y la extensión)
Se amplifica otra vez la cadena de ADN Y se amplifican los fragmentos generados en el primer ciclo. Así se obtienen los 2 primeros fragmentos del tamaño esperado
Tercer ciclo(Se muestra sólo la extensión)
Se amplifican también los fragmentos del tamaño esperado obtenidos en el ciclo 2
Y por último, se amplifican 2 fragmentos largos sobre la cadena de ADN
Se sintetizan más fragmentos del tamaño experado, amplificándose los fragmentos largos de los ciclos 1 y 2
72ºC para permitir que la taq termine de sintetizar todos los fragmentos que pueden haber quedado incompletos.

Las herramientas moleculares520
Para este tipo de PCR es necesario que uno de los oligonucleótidos tenga la misma secuencia que se encuentra en una de las cadenas del ADN, y el otro deberá llevar la secuencia complementaria que estará al final del fragmento que se quiere amplificar (por lo cual se les llama forward y reverse) para que uno sea complementario a la cadena que forma el otro; si no es así no podría amplificarse el sitio que se necesita. Como cada pedazo sintetizado sirve como base para sintetizar otros en el siguiente ciclo, el número de copias aumenta-rá en forma exponencial (ver tercer ciclo de las figuras 1 y 2). Con una sola molécula de ADN, en el ciclo 1 se producen 21=2 nuevos fragmentos, en el ciclo 2 serán 22, esto es, 4 fragmentos recién sintetizados, y así, con 35 ciclos de PCR se producirán 21+22…+ 234+235= 236 nuevos fragmentos, de los cuales sólo 70 serán fragmentos de un tamaño mayor al esperado (2 por cada ciclo) obtenidos al sintetizarlos directamente del ADN genómico; esta pequeña cantidad es casi imposible de detectar al analizar nuestros productos.
Tipos de técnicas
El PCR tiene diferentes métodos o aplicaciones en función de lo que nos intere-se investigar (como son los RAPDs, AFLPs, ISSRs, SSCP…. véase los capítulos 18 y 19 de este libro) y el primer paso es tener claro el tipo de información que necesitamos para elegir o diseñar la estrategia más apropiada para nuestro trabajo. Brevemente podemos dividir la técnica en dos categorías:
Figura 2. En una reacción de PCR los fragmentos se amplñifican en forma exponencial (tomado de Vierstraete, 2001)
Gen buscado
Plantilla de ADN1er ciclo
2do ciclo
3er ciclo
4to ciclo
Ciclo 35
Ampliación exponencial
16 copias8 copias22 =
8 copias2º =
2 copias

Guía práctica sobre la técnica de PCR 521
1) PCRs para la amplificación de un solo sitio conocido del genoma (locus). Estos PCRs requieren conocer la secuencia que se trabaja (por ejemplo cuando amplificamos un gen específico como el 16S), en cuyo caso se utilizan oligonucleótidos diseñados a partir de la secuencia de ese gen y se obtiene un solo fragmento de un tamaño ya conocido. Con este tipo de PCRs es posible hacer filogenias, y para obtener los datos hay distintos ca-minos: desde hacer geles especiales que detectan cambios hasta de una sola base entre las secuencias (SSCP), hasta utilizar enzimas de restricción para generar patrones de cada individuo, aunque lo ideal es obtener la secuencia completa del gen que amplificamos, sobre todo cuando se desea responder a preguntas relacionadas con las fuerzas evolutivas que han actuado sobre él. El gen secuenciado puede ser analizado desde varias perspectivas y muchas de ellas se encuentran en los diversos capítulos de este libro.
2) PCRs en los que no es necesario conocer la región que se está amplifican-do (se amplifican regiones no conocidas, como zonas hipervariables del genoma), por lo cual no se sabe el tamaño del fragmento (o fragmentos) que se esperan. Éstos se utilizan para determinar polimorfismo genómico y son los más comunes para fingerprint, ya que es sencillo obtener los datos (un gel de agarosa después del PCR es suficiente), se observan varios loci simultáneamente y la información de las zonas variables permite inferir los datos necesarios para análisis de genética de poblaciones. En general este tipo de PCRs utiliza un solo oligonucleótido con 2 caracte-rísticas importantes: que sea de pequeño a mediano (de 6 a 18 bases) y sobre todo que su secuencia esté presente muchas veces en el ADN del organismo que estudiamos. Existen zonas repetidas hipervariables del ADN que pueden amplificarse de esta manera, por ejemplo los sitios que sirven para iniciar la síntesis de ADN en los cromosomas, conocidas como microsatélites. En el primer ciclo de reacción lo que sucederá es que el oligonucleótido utilizado hibridará en distintas zonas del ADN, y primero comenzarán a sintetizarse fragmentos de tamaños variables e indefinidos (hasta donde la polimerasa logre copiarlos). En el segundo ciclo las cadenas sintetizadas a partir de las primeras copias formadas serán del tamaño que existe entre dos oligonucleótidos que no estén muy alejados entre sí. Estos fragmentos se copiarán una y otra vez, y de esta manera al final obtendremos muchos fragmentos de tamaños diferentes, de los que conoceremos la secuencia con que inician y terminan, pero no la secuencia completa de cada uno. En la figura 3 se explica con más detalle un PCR de este tipo.

Las herramientas moleculares522
Figura 3. Los PCR que amplifican zonas no conocidas utilizan oligonucléotidos pequeños y amplifican zonas repetidas en el genoma (tiomado de Harlocker, 2003)
Supongamos que hacemos una reacción de PCR con un individuo hipotético utilizando un oligo para microsatélites. En este primer ejemplo el oligo ha hibridado en el ADN en 6 sitios diferentes, y al ampli-ficar se generan 2 fragmentos:1) Fragmento A es sintetizado a partir de la secuencia de ADN que se encuentra entre los oligos que se han unido en las posiciones 2 y 52) El fragmento B es sintetizado a partir de los oligos unidos en las posiciones 3 y 6.No hay productos de PCR entre las posiciones 1 y 4 ya que están muy lejos entre sí como para permitir que ocurra la reacción de PCR. Los oligos unidos a las posiciones 4 y 2 ó 5 y 3 tampoco amplifican ya que no están orientados uno hacia el otro.
En el segundo ejemplo hay que suponer que es un individuo que tiene un cambio en el segundo sitio, y el oligo no se ha unido en la segunda posición, por lo tanto sólo se genera el fragmento B.
Primer ejemplo
ADN
Reacción de PCR
Fragmento A Fragmento B
1 2 3
4 5 6
Segundo ejemplo
ADN
Reacción de PCR
Fragmento B
1 3
4 5 6

Las herramientas moleculares532
de las bolsas (de marcas certificadas, libres de ADNasas, ARNasas y ADN), con lo cual logramos eliminar la contaminación.
Además del control negativo que nos permite visualizar posibles casos de contaminación, una vez que se ha ajustado el protocolo de nuestro PCR es siempre recomendable incluir un control positivo. Éste es un tubo que contiene ADN de una muestra que haya amplificado adecuadamente y cuyo patrón de bandas es conocido. De esta manera, puede esperarse que si la reacción se llevó a cabo correctamente, esta muestra siempre salga. En caso contrario, si nuestro PCR no generó bandas en ninguna de las muestras, ni siquiera en el control positivo, puede suponerse que el error radica en que se nos olvidó agregar algún ingrediente o bien que los ciclos de temperatura no se llevaron a cabo adecuadamente, ya sea por problemas de la máquina o bien por error al elegir el programa de amplificación.
Métodos para visualizar el PCR
Principios básicos de la electroforesis
La idea ahora es poder analizar el o los fragmentos obtenidos en el PCR, y la electroforesis, ya sea en geles de agarosa o de acrilamida, permite separar estos fragmentos de acuerdo al tamaño de cada uno. Tanto la agarosa como la acrilamida forman una especie de red con agujeros de tamaños diferentes, por la cual obligamos a pasar los fragmentos de ADN, “jalándolos” a través de corriente eléctrica, hacia el polo positivo, ya que la carga de una molécula de ADN es negativa por la presencia de grupos fosfato (P-). Los fragmentos más pequeños pasarán primero a través de la red de agujeros, mientras que los más grandes se irán retrasando y atorando en los hoyos; de esta manera los fragmentos de tamaños similares migrarán a ritmos similares. Si hay mu-chos fragmentos de un mismo tamaño se agruparán todos juntos, por lo que podremos verlos formando lo que llamamos una banda en el gel.
¿Hacemos nuestro gel con agarosa o acrilamida? Las moléculas de acrilami-da forman redes con tamaños de poros más uniformes y más pequeños que la agarosa, por lo que este tipo de gel es útil si los tamaños de los fragmentos que manejamos son pequeños; la separación puede ser tan fina que con algunas técnicas es posible separar moléculas de ADN que tienen una sola base de diferencia. El problema es que las técnicas de acrilamida son muy laboriosas y la tinción con plata (si no quiere usarse radioactividad) tiene muchos tru-cos, por lo que en este apéndice sólo anotaremos algunas recetas para lo más

Guía práctica sobre la técnica de PCR 533
sencillo, que son los geles de agarosa. Para más información sobre geles de acrilamida pueden consultarse los sitios de internet que sugerimos al final del capítulo. La agarosa no forma redes tan uniformes, pero permite separar las moléculas de ADN en un intervalo muy grande. Utilizarla es muy sencillo y teñir los geles también lo es.
a) Métodos para geles de agarosaSerá necesario tener en el laboratorio equipo que nos permita trabajar con
los geles: una cámara de electroforesis, una fuente de poder, un transilumina-dor de luz UV y equipo de fotografía (lo más sencillo: una cámara polaroid, un filtro para luz UV y un cono adaptado a la cámara, o también existen cámaras especiales y equipo de cómputo específico para ello) para guardar la imagen del gel. Para empezar hay que preparar el buffer de corrida, el cual tendrá el pH requerido y los iones necesarios para que fluya la corriente y pueda migrar el ADN. Si en lugar de buffer utilizáramos agua, el ADN se quedaría inmóvil y no veríamos migración en los geles, y si por el contrario utilizáramos un exceso de sales, el gel se calentaría tanto que al final acabaría por derretirse. El buffer más común es el TBE (Tris Boratos EDTA), que por ser muy estable puede reutilizarse varias veces. También se utiliza con frecuencia el buffer TAE (Tris Acetatos EDTA), que es menos estable que el TBE y tiende a ionizarse más rápido, pero permite obtener mejor separación de bandas, sobre todo si son de gran tamaño (1 kb o más). Las recetas para preparar éstas y otras soluciones están al final de este apartado. Dependiendo del tamaño de los fragmentos que esperamos se utilizará una concentración de agarosa mayor o menor para obtener agujeros menos o más grandes y una mejor resolución de nuestras bandas. Si no conocemos el tamaño, se puede empezar con agarosa al 1%, pero si se conoce, se puede usar esta tabla como guía:
Rango efectivo de separación (kb) Agarosa (%)
30 a 1 0.512 a 0.8 0.710 a 0.5 1.07 a 0.4 1.23 a 0.2 1.5
Fuente: Tomado de Sosnick, 2003.
La agarosa se disuelve en el mismo buffer que utilizaremos para la corrida, y se calienta hasta ebullición para disolver bien el polvo. Hay que tener cuidado

Las herramientas moleculares534
de retirarla del calor o del microondas en cuanto comienza a hervir, pues podría derramarse. Si es un gel muy importante, en el laboratorio pesamos el matraz donde se prepara el gel antes y después de calentarlo, y le agregamos el agua que haya perdido durante el calentamiento, para asegurar que la concentra-ción de agarosa sea la correcta. La solución se agitará suavemente para evitar la formación de burbujas, y cuando se enfríe un poco (aprox. 60ºC) se vierte de una sola vez en el contenedor de geles al que ya le colocamos el peine para que se formen los pozos en donde cargaremos las muestras. Si quedan algunas burbujas, rápidamente con la punta de una pipeta podemos picarlas y quitarlas; si se dejan pueden hacer que la electroforesis no migre en forma homogénea. Cuando se enfríe y solidifique agregamos el buffer necesario hasta cubrir BIEN el gel (hemos visto que de esta forma el peine se puede quitar más fácilmente), y después se retira el peine con cuidado para no romper el fondo de los pozos.
b) Cargando el gelCon una pipeta, cada muestra se vierte en un pozo, mezclada previamente
con 1 ó 2 microlitros de colorante de corrida (receta al final). Generalmente los colorantes de corrida llevan alguna sustancia espesa, como glicerol o sacarosa, que permite que la muestra caiga hacia el fondo del pozo, y los colorantes (como el xilen-cianol o azul de bromofenol) nos dan una idea de cómo van migrando los fragmentos (en un gel de agarosa al 1%, el azul de bromofenol migra junto con los fragmentos de 300 pb, y el xilen-cianol migra igual que los fragmentos de 4 kb). No es necesario utilizar toda la muestra de PCR en una corrida, puede utilizarse del 10% al 20% de la cantidad total del PCR que hicimos (i.e. si en total son 50 µl, se cargarán 5 µl de muestra).
Nosotros en un parafilm depositamos unas gotitas de colorante (las ne-cesarias para el número de muestras que usemos) y después agregamos la gotita de la muestra a cargar sobre la gota de colorante. Con cuidado de no hacer burbujas las mezclamos subiendo y bajando con la pipeta. La punta de la pipeta se mete un poco en el pozo (¡sin romperlo!) y lentamente se vacía la pipeta para cargar el gel. Para que las muestras no se derramen y no se mezclen unas con otras hay que evitar llenar el pozo hasta arriba. Por lo menos un carril del gel siempre deberá tener un marcador de peso molecular (muchas casas comerciales los venden) como control para saber el tamaño de las bandas que tendremos. Tampoco debe olvidarse poner en el gel los con-troles negativo y positivo. El PCR sobrante lo guardamos congelado a -20ºC por si lo necesitamos después. Cuando el gel está listo se conectan los cables, lo más común es un cable rojo para conectarlo al polo positivo y uno negro

Guía práctica sobre la técnica de PCR 535
en el negativo. El ADN migrará hacia el polo positivo ya que los fosfatos de la molécula le confieren carga negativa, por lo que hay que asegurarse que la corrida del gel sea HACIA el cable rojo, o polo positivo. Para el voltaje, se recomienda utilizar 5 volts por cada centímetro que exista entre los dos elec-trodos de nuestra cámara (i.e. si la cámara mide 30 cm se correrá a 150V), esta medida se aplica cuando tenemos fragmentos grandes, de más de 2 kb; para un PCR con fragmentos pequeños, de 100 pb hasta 1 kb, nosotros utilizamos casi siempre 90 a 100 volts para geles chicos y/o grandes, y los dejamos 1 ó 2 horas, dependiendo del largo del gel.
c) Tinción del gelLo más común es utilizar bromuro de etidio, que es una molécula con dos
propiedades importantes: se intercala en las bases del ADN y brilla con luz UV a una longitud de onda determinada (264-366 nm) con lo cual podemos observar las bandas de ADN en el gel. El bromuro de etidio es un mutágeno y es altamente tóxico, por lo cual es necesario utilizar guantes y bata para su manejo. Es recomendable apartar un área del laboratorio y material exclusivo para su uso. Los geles con bromuro deberán juntarse y desecharse con alguna compañía de desechos tóxicos, así como las puntas y guantes contaminados. Las soluciones con bromuro pueden inactivarse, al final hay una lista de 3 sitios en los que dan indicaciones de cómo hacerlo. Si de derrama una pe-queña cantidad, hay que absorber muy bien con toallas de papel y después con alcohol (las toallas se juntan en una bolsa para inactivar el bromuro que quede en ellas). Cuando no estamos seguros si algo está o no contaminado con bromuro lo exponemos a la luz UV y si no hay fluorescencia es que no hay bromuro. Hay dos maneras de teñir el gel: cuando la agarosa está a unos 60ºC, antes de vertirla, se añade el bromuro de etidio directamente para que quede a una concentración de 0.5 µg/ml en el gel. De esta forma al terminar la corrida puede verse el gel. La desventaja es que el bromuro retarda la migración de las moléculas de ADN, y además la cámara de electroforesis queda contaminada con el bromuro, es por esto que hay quien prefiere teñirlo después de la corrida: el gel se sumerge en una solución de 0.5 µg/ml de bromuro de etidio por 30 a 40 mn y así no se contaminan las cámaras y el ADN migra más rápido, pero es un método más lento. Para los dos métodos será necesario preparar una reserva de bromuro de etidio como se indica al final del capítulo.Al terminar se pone el gel en el transiluminador para verlo. La luz UV puede dañar la piel y los ojos, por lo que será necesario protegerse a través de un acrílico especial y protectores de la cara y/o los ojos.

Las herramientas moleculares542
raramente más de cien caracteres (Sanderson y Donoghue, 1989). Los datos moleculares también tienen la ventaja de trabajar directamente con la base genética de la variación, mientras que la base genética de la mayoría de los caracteres morfológicos se asume. Asimismo, en el acercamiento molecular los caracteres se pueden seleccionar y definir de una manera relativamente objetiva (Hillis y Wiens, 2000).
En los estudios morfológicos, en cambio, los caracteres deben ser des-cubiertos y delimitados generalmente sin ningún criterio explícito para la selección o la codificación del carácter, por lo que tienen el potencial de ser arbitrarios. Por ejemplo, los morfologistas no divulgan generalmente sus criterios para incluir o excluir caracteres y cuando se dan los criterios, varían considerablemente entre estudios (Hillis y Wiens, 2000).
Sin embargo, tienen la ventaja de permitir un muestreo taxonómico mu-cho más cuidadoso que el que se realiza con análisis moleculares, lo que es importante para las revisiones sistemáticas, los estudios de la evolución del carácter y la valoración filogenética. Asimismo, los especímenes de museo ofrecen muchos caracteres morfológicos para una gran cantidad de taxa, mientras que un muestreo de este tipo para un estudio molecular puede ser difícil por el alto costo de la secuenciación, la necesidad de material relativa-mente fresco y la inaccesibilidad de las áreas donde se distribuyen algunos de ellos (Hillis y Wiens, 2000).
La morfología es también la única manera en que la mayoría de los taxa fósiles pueden analizarse filogenéticamente y las especies extintas no sólo representan una gran proporción de la biodiversidad de la tierra, sino que también pueden ser cruciales para el entendimiento de las relaciones entre los taxas vivos (Smith, 1998).
De lo anterior se puede apreciar que tanto los acercamientos moleculares como morfológicos tienen ventajas y desventajas; que ambos enfoques siguen desempeñando un papel crucial en casi todos los grupos de organismos y que hasta nuestros días las especies se describen y se identifican con base en ambas clases de datos.
Marcadores moleculares
Los marcadores moleculares son una herramienta necesaria en muchos campos de la biología como evolución, ecología, bio-medicina, ciencias forenses y estudios de diversidad. Además se utilizan para localizar y aislar genes de interés. En la actualidad existen varias técnicas moleculares que

Breve revisión de los marcadores moleculares 543
nos permiten conocer cómo se encuentran las proporciones de genes en las poblaciones naturales de manera indirecta, como con los análisis de proteínas, o de manera directa con estudios de ADN. Los diferentes tipos de marcadores se distinguen por su capacidad de detectar polimorfis-mos en loci únicos o múltiples y son de tipo dominante o co-dominante (Simpson, 1997).
ADN nuclear (nADN)
En eucariontes, la mayor parte de la información genética se encuentra conte-nida en el núcleo de la célula. El nADN se encuentra empaquetado y asociado a proteínas histonas, conformando los cromosomas.
El nADN contiene regiones únicas −de una sola copia− y no únicas −duplicadas o regiones repetitivas (Stansfield, 1992). Se considera que los organismos diploides tienen dos copias de cada región genética (locus) en los pares homólogos de los cromosomas, llamadas alelos, sin tener en cuenta si contienen regiones codificantes (exones) o no codificantes (intrones o regiones intergénicas).
ADN de cloroplasto (clADN)
En los organismos fotosintéticos con cloroplastos existe un ADN típicamente bacteriano circular, de 120 a 200 kb (Brown et al. 1979), con intrones y exo-nes que se considera muy conservado, ya que se trata fundamentalmente del mismo genoma desde las hepáticas hasta las plantas superiores. Cada cloro-plasto contiene varias regiones nucleotídicas, cada una con 8 a 10 moléculas de ADN. Un organismo unicelular como Euglena puede contener de 40 a 50 cloroplastos, por lo que la celula entera puede contener más de 500 copias del genoma del cloroplasto (Stansfield, 1992).
ADN mitocondrial (mtADN)
El genoma mitocondrial (mtADN) tiene un tamaño de 15 a 17 kb (Brown et al. 1979) y su longitud varía considerablemente entre especies: 20 micróme-tros en Neurospora; 25 micrómetros en levaduras; 30 micrómetros en plantas superiores; 5 micrómetros en algunos animales metazoarios (multicelulares). Se considera que la mayoría del mtADN de hongos y plantas no codifica, ya que el mtARN contiene intrones. El mtARN de animales carece de intrones y

Las herramientas moleculares544
se transcribe como ARN policistrónico que se parte en ARN monocistrónico antes de la traducción (Stansfield, 1992).
Las moléculas de cloroplasto y mitocondria son especialmente importan-tes para trazar historias filogeográficas y de estructura poblacional genética estrechamente relacionada al linaje, porque son de herencia uniparental y no recombinan. También nos permiten inferir cambios demográficos y de dispersión entre especies (Dirienzo y Wilson, 1991).
ADN ribosomal (RADN)
El rADN puede encontrarse en mitocondrias, cloroplasto y núcleo. Contiene la información para el ARN que conforma los ribosomas, por lo que es información que se transcribe pero no se traduce. El rADN se presenta en repeticiones tándem y está formado por tres subunidades altamente conservadas (18 rADN, 5.8 rADN y 28 rADN), separadas por dos espaciadores con elevadas tasas de sustitución (ITS1 e ITS2). Estas repeticiones en tandém se encuentran conservadas a lo largo de todo un genoma y evolucionan concertadamente, lo que se atribuye a eventos recombinatorios como entrecruzamiento desigual y conversión génica. Estas secuencias, por la baja tasa de sustitución que presentan, son extremadamente útiles en el planteamiento de hipótesis de relaciones filogenéticas de taxa con tiempos de divergencia muy antiguos (Hills et al., 1991).
Isoenzimas/aloenzimas
Las isoenzimas fueron originalmente definidas como formas moleculares múltiples de las enzimas que tienen funciones idénticas o similares y están presentes en el mismo individuo (Market y Moller, 1959).
Las isoenzimas pueden presentar varias formas alélicas, conocidas como aloenzimas (Prakash et al., 1969). En su mayoría son selectivamente neutras y se utilizan como marcadores hereditarios para cuantificar las frecuencias alélicas y genotípicas de los individuos, que son los estimadores básicos de la composición genética de una población. En estudios de genética las isoenzimas fueron usadas desde 1966, cuantificando la variación genética en poblaciones humanas y de Drosophila y un poco más tarde en estudios de plantas superiores (Harris, 1966; Johnson et al., 1966; Hubby y Lewontin, 1966; figura 1).
La aplicación de las isoenzimas está dirigida a la cuantificación de heteroci-gosis, diversidad genética, diferenciación genética y otras medidas de variación genética intra e interpoblacional. También han sido aplicadas exitosamente

Breve revisión de los marcadores moleculares 545
para evaluar y entender aspectos de biología evolutiva como los sistemas de reproducción y patrones de fecundación cruzada, relaciones entre fenotipo y ambiente, filogenias, endemismo, diversidad en plantas clonales y apomícti-cas e interacciones planta-animal (Pérez-Nasser y Piñero, 1997) (véanse los capítulos 6 y 7 de este libro).
Por ejemplo, en el estudio de los sistemas reproductivos de plantas las alo-enzimas tienen las siguientes ventajas: 1) son expresadas codominantemente, por lo que los genotipos homócigos y heterócigos pueden ser distinguidos con mucha precisión, y es poco probable que afecten el comportamiento del polinizador; 2) muchos de sus loci son polimórficos, casi cualquier especie presenta al menos uno o dos; 3) probablemente no están sujetas a fuertes fuerzas selectivas, por lo que se consideran buenos marcadores (Brown et al. 1989); 4) la técnica es barata en comparación con otras.
Por otro lado, presentan algunas limitaciones: revelan poca variación y la técnica es muy laboriosa, requiere de mucho tiempo y entrenamiento y, sobre todo, es poco reproducible entre laboratorios.
En la electroforesis de enzimas se utilizan cuatro métodos principales: a) gel de almidón (horizontal y vertical); b) gel de poliacrilamida; c) gel de agarosa y d) gel de acetato de celulosa. La electroforesis horizontal en geles de almidón sigue siendo la preferida, ya que es relativamente barata, de fácil manipulación, buena resolución, no es tóxica y se puede analizar de forma rápida la variación para uno o varios loci.
Para un locus dado, el número de bandas varía entre individuos homócigos y heterócigos (figura 1) y también varía en función de la estructura cuaternaria de la enzima; si es monomérica los individuos homócigos representan una sola banda y los heterócigos presentan dos bandas; si es una enzima dimérica, los heterócigos presentan tres bandas (dos homodímeros de los padres y un producto adicional de movilidad intermedia); en las enzimas tetraméricas se observan 5 bandas en los individuos heterócigos.
RAPDs
Los RAPDs (polimorfismos de ADN amplificados al azar) son marcadores que amplifican aleatoriamente segmentos de ADN en una gran variedad de especies. Los RAPDs se basan en la probabilidad estadística de que se presen-ten sitios complementarios al oligonucleótido de 10 pares de bases (pb) a lo largo del genoma. El polimorfismo de las bandas entre los individuos se debe a cambios en la secuencia de los nucleótidos en los sitios de acoplamiento del
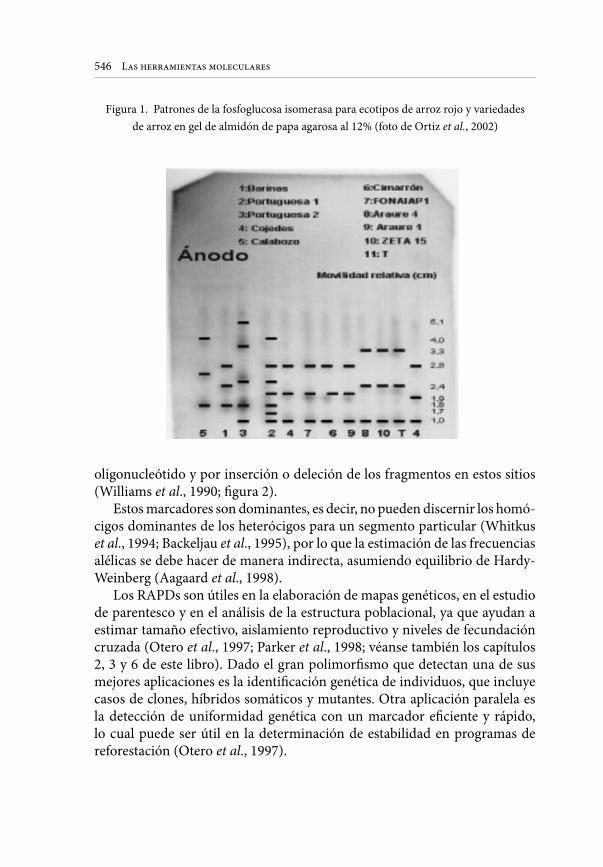
Las herramientas moleculares546
oligonucleótido y por inserción o deleción de los fragmentos en estos sitios (Williams et al., 1990; figura 2).
Estos marcadores son dominantes, es decir, no pueden discernir los homó-cigos dominantes de los heterócigos para un segmento particular (Whitkus et al., 1994; Backeljau et al., 1995), por lo que la estimación de las frecuencias alélicas se debe hacer de manera indirecta, asumiendo equilibrio de Hardy-Weinberg (Aagaard et al., 1998).
Los RAPDs son útiles en la elaboración de mapas genéticos, en el estudio de parentesco y en el análisis de la estructura poblacional, ya que ayudan a estimar tamaño efectivo, aislamiento reproductivo y niveles de fecundación cruzada (Otero et al., 1997; Parker et al., 1998; véanse también los capítulos 2, 3 y 6 de este libro). Dado el gran polimorfismo que detectan una de sus mejores aplicaciones es la identificación genética de individuos, que incluye casos de clones, híbridos somáticos y mutantes. Otra aplicación paralela es la detección de uniformidad genética con un marcador eficiente y rápido, lo cual puede ser útil en la determinación de estabilidad en programas de reforestación (Otero et al., 1997).
Figura 1. Patrones de la fosfoglucosa isomerasa para ecotipos de arroz rojo y variedades de arroz en gel de almidón de papa agarosa al 12% (foto de Ortiz et al., 2002)

Breve revisión de los marcadores moleculares 547
Entre las principales ventajas de los RAPDs esta que amplifican regiones tanto codificantes del ADN como las no codificantes y revelan niveles de variación más altos que los RFLPs e isoenzimas (Williams et al., 1990; Lynch y Milligan, 1994; Otero et al., 1997; Russell et al., 1997; Parker et al., 1998); es una técnica relativamente fácil que no necesita conocimiento previo de la secuencia de ADN, no requiere la construcción o el mantenimiento de una librería genómica, el número de loci que puede ser examinado es ilimitado y no requiere pruebas radiactivas (Reiter et al., 1992; Whitkus et al. 1994).
Sin embargo los problemas prácticos detectados con los RAPDs son la presencia de bandas “erróneas” (artefactos), la reproducibilidad de los re-sultados y la comigración de bandas. Asimismo, muchos de los alelos raros presentes en las poblaciones estudiadas con RAPDs no son detectados o pueden ser mal interpretados (Zhivotovsky, 1999). Por otro lado, como los loci son dominantes, los RAPDs dan menos información genética por locus que los marcadores codominantes.
Al analizar los datos obtenidos con RAPDs se deben tener en cuenta dos supuestos: 1) cada uno de los marcadores representa un locus mendeliano en el cual el marcador visible, el alelo dominante, está en equilibrio de Hardy-Weinberg con un alelo recesivo y 2) los alelos marcados para diferentes loci no migran a la misma posición en el gel (Lynch y Milligan, 1994).
Figura 2. Patrones de bandeo con RAPDs en Ferocactus robustus en gel de agarosa y tinción con bromuro de etidio (foto de Israel Carrillo).

Las herramientas moleculares548
Microsatélites
Los microsatélites o secuencias simples repetidas (SSRs; Litt y Luty, 1989) son secuencias de ADN formadas de 1 a 4 pares de bases, por ejemplo mononu-cleótidos (TT)n, dinucleótidos (AT)n, o tetranucleótidos (AAGG)n. Estos loci se encuentran en regiones codificantes y no codificantes del ADN y es probable que se formen por eventos de rompimiento que generan polimorfismos con valores superiores al 90% (Armour et al., 1994; Coltman et al., 1996; Gupta et al., 1996; figura 3).
Los microsatélites de ADN nuclear han sido detectados en múltiples grupos de plantas y animales, y han sido utilizados fundamentalmente para estudios de variación genética intra e interespecífica (Edwars et al., 1991; Armour et al., 1993; Devey et al., 1996; Queller et al., 1993; Weight y Bentzen, 1994), análisis de linajes (Queller et al., 1993) y de sistemas reproductivos (Awadalla y Ritland, 1997). Recientemente se han encontrado microsatélites en algunos organelos citoplasmáticos, como el cloroplasto (SSRc; Powell et al., 1995a, b; Vendramín et al., 1996; figura 3 ) y la mitocondria (SSRm; Soranzo et al., 1998) lo que ha enriquecido la fuente de estudios evolutivos, ya que estos organelos son heredados uniparentalmente y no están sujetos a recombinación, por lo que los cambios acumulados que observamos en las poblaciones se deben sólo a los procesos de mutación y demográficos (Echt et al., 1998). Esto permite contestar preguntas evolutivas muy pun-tuales relacionadas con el monitoreo del flujo genético, introgresión (Ven-dramin et al., 1998), análisis de paternidad (McCracken et al., 1999), para hacer inferencias de parámetros demográficos y para determinar patrones evolutivos de los procesos históricos del origen de especies, formas o razas (Golstein et al., 1996).
Los microsatélites han tomado ventaja sobre otros marcadores genéticos como los AFLPs, RAPDs, RFLPs, debido a que: i) tienen el más alto grado de polimorfismo; ii) segregan de manera mendeliana y son codominantes; iii) la presencia de un solo locus genético por microsatélite hace que la lec-tura de las bandas sea clara y fácil de interpretar y iv) son selectivamente neutros (Golstein y Pollock, 1994; Vendramin et al., 1996). Además, para trabajar con SRR es necesario conocer la secuencia de la región a analizar para contar con primers específicos que amplifiquen la región repetitiva (el microsatélite) responsable de la variación observada, que además es homóloga para diferentes especies o incluso géneros (Golstein et al., 1996). Esto es, los microsatélites son específicos para ciertos grupos de especies y

Breve revisión de los marcadores moleculares 549
homólogos entre sí (Vendramin et al., 1996), lo que permite hacer estudios comparativos entre especies o géneros de un mismo grupo. Por otra parte, se han realizado estimaciones de las tasas de mutación de estas regiones y se ha llegado a la conclusión de que los microsatélite del ADN nuclear tienen tasas de mutación de 1x 10 -3 a 1x 10 -6 (Wiessenbach et al., 1992; Weber y Wong, 1993), más altas que las que se presentan en el ADN de cloroplasto las cuales según Provan et al. (1999) son de 3.2-7.9 x 10-5. Conocer las tasas de mutación nos brinda una base importante para realizar análisis robustos de la genealogía de las poblaciones que nos hablen acerca de la historia evolutiva de las especies, ventaja muy importante sobre otros marcadores genéticos.
Otra ventaja que presenta el uso de microsatélites con relación al uso de secuencias, es que la mutación es homogénea: en la mayoría de los SSR las mutaciones son de un paso (una unidad repetitiva) siguiendo el modelo mutacional SSM de Ohta y Kimura (1973). Sin embargo el número de mutaciones varía si se trata de diferentes tipos de microsatélites, por ejemplo, Golstein et al. (1995b) mencionan que las mutaciones de repe-ticiones formadas por dinucleótidos o trinucleótidos son de dos pasos o incluso de múltiples pasos, lo que ha sido confirmado por estudios en trinucleótidos. Por ejemplo, en el humano el microsatélite formado por las repeticiones CAG − asociado con un desorden neurológico−, puede mutar de pocos a cientos de alelos (Ashley y Warren 1995). Sin embargo este comportamiento asimétrico del tamaño de las mutaciones de SSR ha sido raramente reportado.
Al ser los microsatélites altamente polimórficos, pueden ser ventajosos con relación al uso de secuencias, aunque la variación del ADN nuclear debe analizarse con precaución, ya que pudo haberse generado por duplicaciones o por la presencia de familias multigénicas (Rieseberg,1991), que generan un alto grado de homogeneidad dentro y entre especies, proceso conocido como evolución concertada. Estos fenómenos pueden distorsionar la historia evolutiva de los organismos en estudio y confundir las relaciones filogenéticas (Rieseberg et al., 1991b). Es decir, se pueden presentar aparentes reticulaciones o escenarios de hibridización introgresiva. Finalmente, los microsatélites han sido utilizados para hacer reconstrucciones de árboles de genes con base en la teoría de coalescencia (véase el capítulo 4 de este libro). Los fragmentos son separados en geles de acrilamida y son visualizados con nitrato de plata o radioactividad.
Las herramientas moleculares550
ISSRs
Es un marcador molecular conocido como secuencias repetidas intersimples. Esta es una técnica relativamente nueva y es similar a los RAPDs, excepto que en los ISSRs el primer es un di ó trinucleotido repetido (Culler y Wolfe, 2001 véase el capítulo 19 de este libro).
Los dos métodos que se utilizan en la electroforesis de ISSRs son: a) gel de agarosa visualizado con bromuro de etidio, y b) geles de acrilamida teñidos con nitrato de plata.
Las bandas que se obtienen con este marcador van de 100 a 2000 pb. La variación alélica en los ISSRs consiste de la presencia o ausencia de los pro-ductos amplificados.
Comparada con los primers de los RAPDs las secuencias de los ISSRs es usualmente larga lo que resulta en una mayor reproductibilidad de las bandas.
Figura 3. Patrones del microsatélite Pt63718 de cloroplasto en Pinus strobiformis en gel de acrilamida al 6% y tinción con nitrato de plata (foto de Alejandra Moreno Letelier)
Breve revisión de los marcadores moleculares 551
Los ISSRs han sido utilizado en especies cultivadas desde 1994, pero sólo recientemente se han utilizado en estudios de la variación poblacional en especies silvestres (Wolfe y Liston, 1998; Wolfe et al. 1998 a,b). Hasta la fecha sólo existen algunos estudios donde se ha comparado directamente la variación genética, incluyendo estimaciones de la tasa de fecundación cru-zada (que tradicionalmente se estima con marcadores codominantes) y se ha utilizado esta técnica en especies donde la variación poblacional con enzimas es pequeña o no existe. Dada la alta variación genética entre individuos de una misma población, sería recomendable utilizar estos marcadores para análisis de paternidad.
Entre las ventajas principales de los ISSRs es que tienen un gran número de bandas polimorficas. En un estudio realizado en Viola pubescens una planta cleistogamica se reportó una gran variación genética. A nivel de especie, el 100% de los loci fueron polimorficos aún cuando los primers (se utilizaron tres) fueron seleccionados al azar. Dentro de cada población cerca del 71% de 83 loci fueron polimorficos (Culley y Wolfe 2001). Además es una técnica relativamente fácil de montar, altamente repetible y ya existen primers uni-versales para plantas.
Figura 4. Patrones de ISSRs en Opuntia rastrera en gel de agarosa al 1.5% y tinción con bromuro de etidio (foto de Lucia Plasencia)

Las herramientas moleculares552
Por otro lado las limitaciones de los ISSRs son: que las bandas son leídas como marcadores dominantes, es decir cuando tenemos la presencia de la banda no se sabe si el individuo es homócigo dominante o herócigo, que la diversidad genética está basada considerando que cada banda representa un locus con dos alelos y que el alelo dominante esta en equilibrio de Hardy-Weinberg con un alelo recesivo y por último que es una técnica relativamente cara, ya que involucra el uso de geles de poliacrilamida y para su visualización nitrato de plata.
RFLPs
El análisis de polimorfismos de la longitud de los fragmentos de restricción (RFLPs) fue el primer marcador de ADN utilizado por biólogos poblacionales (Parker et al., 1998). Este método expresa diferencias específicas del ADN que fueron reconocidas por enzimas de restricción particulares (endonucleasas). Cada una de las endonucleasas (de origen bacteriano), reconoce y corta so-lamente una secuencia específica de bases nitrogenadas en el ADN, siempre y cuando éstas no estén protegidas (metiladas). Por consiguiente cualquier ADN que no esté metilado puede ser reconocido y cortado en fragmentos de longitud definida; y cualquier mutación dentro de esos sitios, podría cambiar el patrón del fragmento y permitir que se detecte un RFLP al comparar dos o más genomas (Valadez y Kahl, 2000). Para detectar RFLPs hay dos técnicas: southern blots e hibridización, y PCR.
Southern blots e hibridización
Es necesario en primer lugar aislar el ADN del organismo de interés (véase el capítulo 15 de este libro, purificarlo y cortarlo con una o más endonucleasas de restricción. Los fragmentos resultantes se separan en geles de agarosa por electroforesis y se transfieren a una membrana que puede ser de nylon o de nitrocelulosa (southern blotting). La subsecuente hibridación con una sonda marcada y detección con autorradiografía, luminografía o quimioluminiscencia hará visible un fragmento de ADN específico, que puede ser comparado con el fragmento resultante de otros genomas tratados de la misma forma (Parket et al., 1998). La sonda marcada se obtiene de fragmentos de ADN nuclear, de organelos o bien de ADN complementario, también denominado ADN copia (ADNc) que es una molécula de una sola hilera producida en el laboratorio usando mRNA como molde y transcripción reversa (véase el capítulo 15 de este

Breve revisión de los marcadores moleculares 553
libro). Para el caso de obtenerla a partir de ADN nuclear, se requiere construir una biblioteca genómica mediante el aislamiento, restricción y clonación del ADN en un vector apropiado (Valadez y Kahl, 2000), que se explica a continua-ción. Las bibliotecas de ADN son colecciones de vectores recombinantes, que cuentan con secuencias de ADN insertadas. La figura 5 muestra la construcción de una biblioteca de ADNc en el vector lambda gt10. En la figura se puede ob-servar cómo, mediante ADN transcriptasas, polimerasas o ligasas se construye un fragmento de ADN (el de interés) y se inserta en el fago a utilizar (parte A de la figura); una vez obtenido el fago se procede a infectar una colonia de E. coli en una caja de Petri. Cada infección producirá una placa característica en la caja de Petri (en una caja de Petri de 150 mm pueden caber 50 mil placas). Una vez que las placas se han formado se coloca un filtro de nitrocelulosa en contacto con las colonias de la caja de Petri. El ADN transferido al filtro es desnaturalizado y fijado en el filtro mediante el uso de luz ultravioleta. De esta forma el filtro puede ser hibridizado con una sonda radiactiva de ADNc de interés y sometido a autorradiografia; una mancha en la autorradiografia permitirá identificar la placa de la caja de Petri que contiene el fago con el ADN de interés. El fago puede entonces ser extraído del agar de la caja de Petri y reinfectar a E. coli. La repetición de este proceso de separación de un fago recombinante, permite clonar el ADN que contiene el fago. Este proceso ha permitido desarrollar bibliotecas de ADN de diferentes tejidos.
RFLP-PCR
El segundo método que se utiliza para la técnica de RFLPs es la PCR. Los RFLPs son causados por rearreglos del ADN, tales como pérdidas, inserciones o sustituciones de secuencias o nucleótidos únicos, lo que genera una ganancia o pérdida de sitios de restricción. Esto es detectado a través de diferencias en el peso molecular de los fragmentos homólogos de restricción del ADN genómico (figura 6).
Con el producto amplificado de PCR se realiza una digestión con diferentes enzimas de restricción. Los fragmentos resultantes de la digestión son sepa-rados por electroforesis en geles de agarosa teñidos con bromuro de etidio o con acrilamida teñidos con nitrato de plata (Karl et al., 1992); este método es más barato y rápido que el anterior.
Los RFLPs generalmente se han utilizado para construir mapas genéticos, para la clonación de genes basados en mapas y para ayudar a resolver proble-mas taxonómicos o filogenéticos.

Las herramientas moleculares554
El grado de polimorfismo detectado con esta técnica difiere ampliamente entre las especies dependiendo de la sonda utilizada. Entre las principales desventajas que presenta está el requerimiento de grandes cantidades de ADN de buena calidad para la detección de loci de copias únicas, además requiere de muchas manipulaciones y solamente se detecta una fracción de la variabilidad de secuencias existentes en el genoma, es decir, su información es limitada.
AFLPs
Los AFLPs (polimorfismos de longitud de los fragmentos amplificados) son una técnica que combina la digestión de dos enzimas de restricción, generalmente Mse I que reconoce y corta 4 pb y Eco RI que reconoce y corta 6 pb dentro de una secuencia, con la unión de secuencias específicas al nucleótido ligado a los extremos de los fragmentos de restricción y dos
Figura 5. Construcción de una biblioteca de ADN en el vector lambda gt10 (A). En (B) se muestra en forma esquematizada el cernimiento para obtener la clona pura
mARN AAA
mARNTranscriptasa de reversa dNTPs
AAA
+ oligo (dT)
mARN
cADNAAATTT
ARNasa HPolimerasade ADNdNTPsmARN
cADNAAATTT
Ligasa Eco Rl
GGAATTCCCCAATTCC
GGAATTCCCCAATTCC
Digestión por Eco Rl
GGAATTCCGG
GGCCAATTCC
Eco Rl dirigido, ? GT 10, Ligasa de ASDN
COS Inserto COS
Paquete de ADN recombinante de ? gt 10 en el fago
Biblioteca de ? gt 10
A B
Caja de E. coli infectadas con el fgo
Transferencia al filtro
ADN fijo en el filtro
Hibridización con ADN o ARN
Señal en la placa donde está el fango
Autorradiografía
Autorradiografía
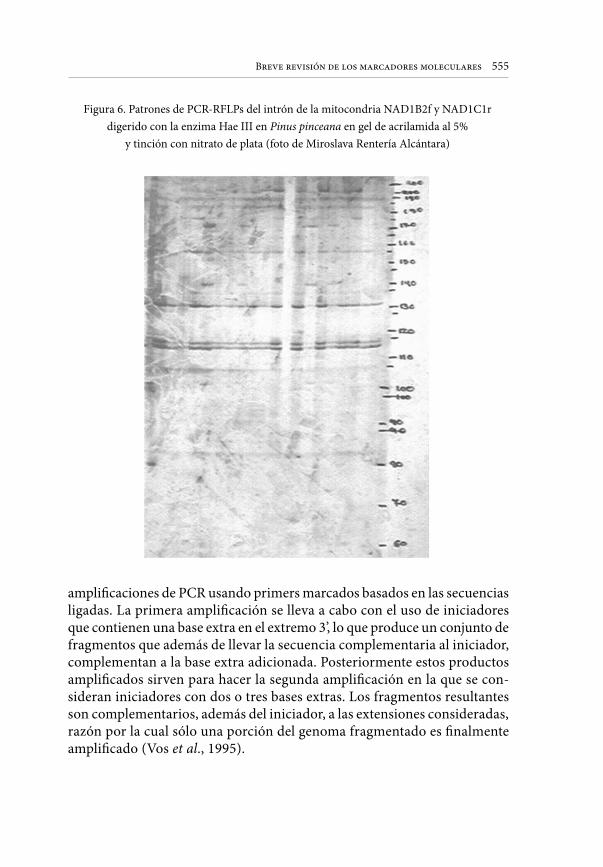
Breve revisión de los marcadores moleculares 555
amplificaciones de PCR usando primers marcados basados en las secuencias ligadas. La primera amplificación se lleva a cabo con el uso de iniciadores que contienen una base extra en el extremo 3’, lo que produce un conjunto de fragmentos que además de llevar la secuencia complementaria al iniciador, complementan a la base extra adicionada. Posteriormente estos productos amplificados sirven para hacer la segunda amplificación en la que se con-sideran iniciadores con dos o tres bases extras. Los fragmentos resultantes son complementarios, además del iniciador, a las extensiones consideradas, razón por la cual sólo una porción del genoma fragmentado es finalmente amplificado (Vos et al., 1995).
Figura 6. Patrones de PCR-RFLPs del intrón de la mitocondria NAD1B2f y NAD1C1r digerido con la enzima Hae III en Pinus pinceana en gel de acrilamida al 5%
y tinción con nitrato de plata (foto de Miroslava Rentería Alcántara)
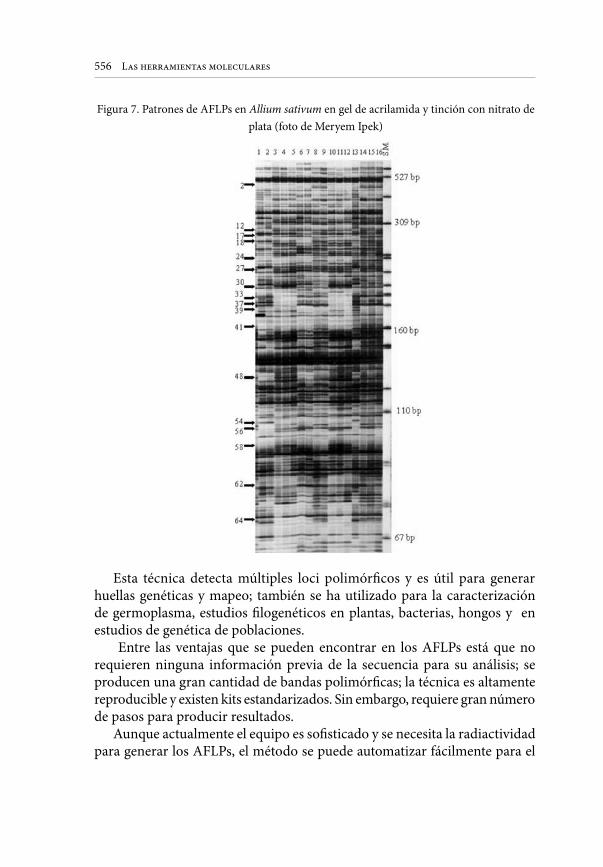
Las herramientas moleculares556
Esta técnica detecta múltiples loci polimórficos y es útil para generar huellas genéticas y mapeo; también se ha utilizado para la caracterización de germoplasma, estudios filogenéticos en plantas, bacterias, hongos y en estudios de genética de poblaciones.
Entre las ventajas que se pueden encontrar en los AFLPs está que no requieren ninguna información previa de la secuencia para su análisis; se producen una gran cantidad de bandas polimórficas; la técnica es altamente reproducible y existen kits estandarizados. Sin embargo, requiere gran número de pasos para producir resultados.
Aunque actualmente el equipo es sofisticado y se necesita la radiactividad para generar los AFLPs, el método se puede automatizar fácilmente para el
Figura 7. Patrones de AFLPs en Allium sativum en gel de acrilamida y tinción con nitrato de plata (foto de Meryem Ipek)

Breve revisión de los marcadores moleculares 557
alto rendimiento del procesamiento de muestras. Las marcas fluorescentes están substituyendo a la radiactividad y el número de datos producidos en un tiempo corto compensa los costos.
Las bandas observadas en los geles de AFLPs son clasificadas como pre-sencia o ausencia de cada individuo y el análisis se desarrolla como un sistema dominante recesivo (Simpson, 1997).
Secuenciación de ADN
Los análisis más detallados de diferenciación de ADN pueden obtenerse se-cuenciando la región de interés para diferentes individuos. Hasta hace poco tiempo, el uso extenso de secuencias de ADN para los estudios de poblaciones no habían sido prácticos porque los fragmentos de ADN para cada indivi-duo tenían que ser aislados para librerías de ADN subgenómico después de haber sido identificados por southern blotting e hibridización. Sin embargo, actualmente el uso de secuencias es muy común: se ha aplicado en análisis de genética de poblaciones y problemas taxonómicos.
Este método incluye cuatro pasos: i) identificar secuencias que tenga la variación necesaria; ii) aislar y purificar un número elevado de la secuencia (ya sea por clonación o amplificación); iii) secuenciar; iv) alinear la secuencia (con los programas MALIGN, Clustal V para Macintosh ver. 1.5, JACK ver. 4.2).
Las técnicas que existen para secuenciar son la química, la enzimática y la automática.
La química diseñada por Alan Maxan y Walter Gilbert consiste en dividir el ADN en cuatro muestras y tratar cada una con agentes químicos que rompen el ADN específicamente en una base, bajo condiciones en las que solo unos pocos nucleótidos del fragmento se afectan. Los fragmentos se marcan con radiactividad y se auto radiografían.
En el método enzimático de Fred Sanger el ADN a ser secuenciado se utiliza como patrón de referencia para producir una síntesis in vitro, mediante una polimerasa y una réplica de ADN que inicia la síntesis del ADN siempre en el mismo sitio. El punto crítico es el uso de trifosfato de dideoxirribonucleó-sido: la deoxirribosa no cuenta con el grupo 3-OH, de tal forma que cuando el nucleótido es incorporado a la cadena de ADN, se bloquea la adición del siguiente nucleótido, por lo que cada molécula de ADN sintetizada va a termi-nar aleatoriamente en dicho nucleótido. Esta síntesis se lleva a cabo en cuatro muestras; en cada muestra se incorpora un dideoxinucleósido con una base diferente (ddATP, ddCTP, ddTTP, ddGTP) y los otros tres deoxinucleótidos

Las herramientas moleculares558
normales. Los fragmentos sintetizados durante el proceso van a terminar en sitios diferentes de la cadena del ADN, generando una escalera de moléculas de ADN que pueden ser detectadas por auto radiografía. Las cuatro muestras se corren en electroforesis en líneas paralelas para determinar el orden de los nucleótidos, identificando la secuencia de ADN que será complementaria al ADN que sirvió de modelo para la síntesis (figura 8).
Figura 8. Método se secuenciación enzimática de ADN. 1) Síntesis en presencia del ADN de interés, polimerasa, un fragmento de ADN como iniciador, nucleótidos y dideoxinucleótidos atípicos; 2) Síntesis por separado para cada nucleótido, generando fragmentos de ADN con
el dideoxinucleótido incorporado; 3) separación de los fragmentos en un gel para identificar la secuencia
A
GGATTG A
A
A
A
A
C
C
C
CC
G
G
GG
G
TT
TT
A ddATP
Polimerasa
1
5’
3’ 5’CGTAACT
G•T• T •C• T• A• A •C• TC •A• A
ADN PolimerasadATPdCTPdTTPdGTP
+2
3
+ d d C T P + d d T T P + d d G T P+ dd AT P
C •A• A G C A
C •A• A C•G• A• T•T•G A
C •A• A G C C •A• A G C A T AC •A• A
C •A• A C G A T C •A• A C G A T T G
A C T G
AGTTACG
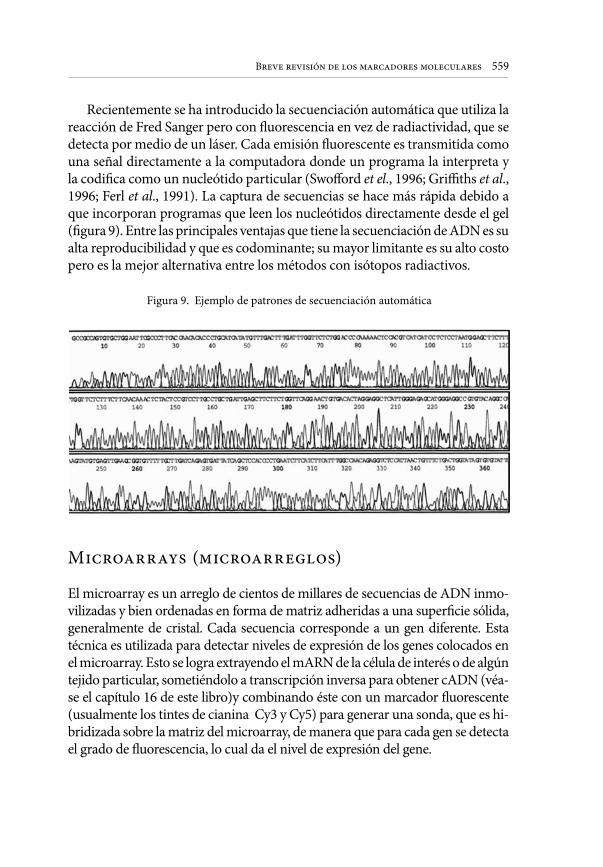
Breve revisión de los marcadores moleculares 559
Recientemente se ha introducido la secuenciación automática que utiliza la reacción de Fred Sanger pero con fluorescencia en vez de radiactividad, que se detecta por medio de un láser. Cada emisión fluorescente es transmitida como una señal directamente a la computadora donde un programa la interpreta y la codifica como un nucleótido particular (Swofford et el., 1996; Griffiths et al., 1996; Ferl et al., 1991). La captura de secuencias se hace más rápida debido a que incorporan programas que leen los nucleótidos directamente desde el gel (figura 9). Entre las principales ventajas que tiene la secuenciación de ADN es su alta reproducibilidad y que es codominante; su mayor limitante es su alto costo pero es la mejor alternativa entre los métodos con isótopos radiactivos.
Figura 9. Ejemplo de patrones de secuenciación automática
Microarrays (microarreglos)
El microarray es un arreglo de cientos de millares de secuencias de ADN inmo-vilizadas y bien ordenadas en forma de matriz adheridas a una superficie sólida, generalmente de cristal. Cada secuencia corresponde a un gen diferente. Esta técnica es utilizada para detectar niveles de expresión de los genes colocados en el microarray. Esto se logra extrayendo el mARN de la célula de interés o de algún tejido particular, sometiéndolo a transcripción inversa para obtener cADN (véa-se el capítulo 16 de este libro)y combinando éste con un marcador fluorescente (usualmente los tintes de cianina Cy3 y Cy5) para generar una sonda, que es hi-bridizada sobre la matriz del microarray, de manera que para cada gen se detecta el grado de fluorescencia, lo cual da el nivel de expresión del gene.

Las herramientas moleculares560
Los microarrreglos pueden ser divididos en dos tipos principales que di-fieren en su construcción: spotted microarrays (microarreglos punteados) y arreglos de oligonucleótidos de alta densidad. Los microarreglos punteados usan generalmente muestras de ADN de 500 a 5000 bases (Ekins et al., 1999) producidos por PCR a partir de bibliotecas o de bases de datos como Gen-Bank, UniGene, dbEST, etc. Las secuencias son depositadas sobre la superficie sólida por un robot en lugares definidos. Hay dos métodos de depositar los puntos:
Distribuidor activo: basado en la tecnología de las impresoras de inyección de tinta.
Distribuidor pasivo: aplica la solución de ADN con un alfiler que toca la superficie sólida. Se puede obtener una mayor densidad de manchas usando este método.
La superficie sólida generalmente es un portaobjetos especialmente cubierto pero también se pueden usar membranas de nylon y portaobjetos cubiertos de oro. El tamaño de los puntos de la matriz típicamente varía de 80 a 150 µm de diámetro y en un solo portaobjetos cabe un máximo de 80,000 puntos. En los microarrays punteados es común comparar las expresiones genéticas de dos muestras biológicas (tejido con tumor vs. tejido sin tumor, por ejemplo) en un mismo portaobjetos, así que el mRNA es preparado para que los dos niveles de expresión puedan ser medidos (Schena et al., 1995; figura 9).
Por su parte, los microarreglos de oligonucleótidos de alta densidad son construidos comercialmente y tienen una precisión y densidad muy alta al usar oligonucleótidos cortos de 20 a 25 bases.
Dos de las compañías que construyen estas clases de micrroarreglos son Affymetrix y Agilent Technologies. Los microarreglos de Affymetrix son los más populares y son conocidos como GeneChipsTM, producidos sintetizando decenas de miles de oligonucleótidos cortos in situ usando técnicas de fotoli-tografía. Affymetrix produce diferentes microarrays, cada uno con diferente composición de genes (Lockart et al., 1996).
Los microarreglos necesitan ser leídos por un instrumento apropiado para convertir la distribución de florescencia o radioactividad en una imagen de computadora. En el caso de las sondas fluorescentes, se pueden usar escáners de láser o cámaras CCD. En la imagen de computadora cada intensidad del punto corresponde a un nivel de expresión de la sonda. Puesto que un punto contiene múltiples pixeles, es normal promediar todos los pixeles de un mismo punto junto con los pixeles del borde exterior. Sin embargo, la cuantificación

Breve revisión de los marcadores moleculares 561
Figura 10. Técnica de microarrays para detectar niveles de expresión de genes (tomada de la página web del Instituto de Neurobiología, UNAM)
del grado de fluorescencia está sujeta a las fluctuaciones de intensidad dentro de cada punto, aunque éstas pueden ser superadas con las técnicas desarro-lladas por Brown et al. (1999).
Los microarreglos de Affymetrix presentan problemas similares, pero también se han desarrollado algoritmos para combatirlos.Esta tecnología permite a los investigadores estudiar la relación de genes individuales de enfermedades (por ejemplo el cáncer), y después aplicar el conocimiento al tratamiento de pacientes.
También se ha utilizado en el estudio de tejidos finos, en células normales durante el desarrollo y estudios de animales transgenicos.
La tecnología de los microarreglos también tiene uso potencial en el desarrollo de nuevas drogas así como en la investigación de farmacogenó-micos (cuyo fin es encontrar la correlación entre las respuestas terapéuticas a las drogas y los perfiles genéticos de los pacientes) y de toxicogenómicos (cuya meta es encontrar correlaciones entre los toxicantes y los cambios en los perfiles genéticos de los objetos expuestos a los toxicantes; Leming Shi, 1998, 2002).
Preparar la sonda de ADN complementario (ADNc)
“Normal” Tumor
RT/PCR
Mercado con compuestos
fluorescentes
Combinar en cantidades
iguales
Hibridar la sonda a la micromatriz
Tecnología de micromatrices
SCAN
Preparar la colección de ADNc en una micromatriz

Las herramientas moleculares562
Los dos grandes problemas que se presentan con los datos de microarreglos son por un lado los niveles de expresión, que varían de experimento a experi-mento, y por las diversas fuentes de errores aleatorios y sistemáticos durante el análisis. Por otro lado, hay un pequeño número de muestras comparado con el gran número de variables, lo que causa que las técnicas estadísticas tradicionales fracasen. Otra gran limitante de los microarrays proviene, iró-nicamente, de su productividad notable, y actualmente los científicos apenas pueden hacer frente al volumen enorme de información producida por los microarreglos, además de tener un alto costo.
Bibliografía
Aagaard J.E., V.K. Krutovskii y H.S. Strauss. 1998. RAPDs and allozymes exhibit similar levels of diversity and differentiation among populations and races of Douglas-fir. Heredity 81:69-78.
Armour A. L., R. Neuman, S. Gobert y A.J. Jeffreys. 1994. Isolation of human simple repeat loci by hybridization selection. Human Molecular Genetics 3(4):599-605.
Ashley C.T. y S.T. Warren. 1995. Trinucleotide repeat expansion and human disease. Annual Review Genetics 29:703-728.
Ayala F.J. 1984. Genética Moderna. Fondo Educativo Interoamericano, Barcelona.Avise, C.J. 1994. Molecular markers, natural history and evolution. Chapman & Hall,
New York.Backeljau T., L. De Brun, H. De Wolf, K. Jordaens, S. Dongen, R. Verhagen y B. Win-
nepenninckx. 1995. Random amplified polymorphic DNA (RAPD) and parsimony methods. Cladistics 11:119-130.
Berovides V. y M.A. Alfonso. 1995. Biología evolutiva. Pueblo y Educación, La Ha-bana.
Brettschneider R. 1998. Molecular tools for screening biodiversity in plants and animals. Chapman & Hall, Londres.
Brown A.H.D. 1979. Enzyme polymorphisms in plant populations. Theoretical Po-pulation Biology 15:1-42.
Brown W.M., M. George Jr. y A. C. Wilson. 1979. Rapid evolution of mitochondrial DNA. Proc. Natl. Acad. Sci. USA 76:1967-1971.
Brown, P.O. y D. Botstein. 1999. Exploring the new world of the genome with DNA microarrays. Nature Genetics 21: 33–37.
Coltman D.W., D.W. Bowen y J.M. Weith. 1996. PCR primers for harbour seal (Phoca vitulina concolour) microsatellites amplify polymorphic loci other pinniped spe-cies. Molecular Ecology 5:161-163.

�Plásmidos���� Moléculas de DNA circular que se replican aparte del cromosoma bacteriano
�Tamaño entre 5.000 y 400.000 pares de base�Transformación����Proceso por el cual se introduce un plásmido en la célula bacteriana.
�Incubación células y plásmidos a 0ºC en solución de Cl2Ca.�Choque térmico (37-45ºC)
�Plámido seleccionable ����Plásmido que contenga un gen que la célula huésped necesite para crecer bajo ciertas condiciones.�El gen es generalmente el de resistencia a un antibiótico� Sólo aquellas células que hayan sido transformadas por el plásmido serán resistentes al antibiótico y podrá crecer en presencia del mismo.�Características importantes de un vector de clonación plasmídico
�Necesita un origen de replicación� propagar el plásmido y mantenerlo a nivel de 10 a 20 copias por célula. �Genes que confiere resistencia a antibióticos� permite la selección�Varias secuencias únicas de reconocimientos para diferentes endonucleasas de restricció n�generar sitios por donde se puede insertar las secuencias foráneas de DNA�Tamaño global pequeño� facilita la entrada del plásmido en la célula
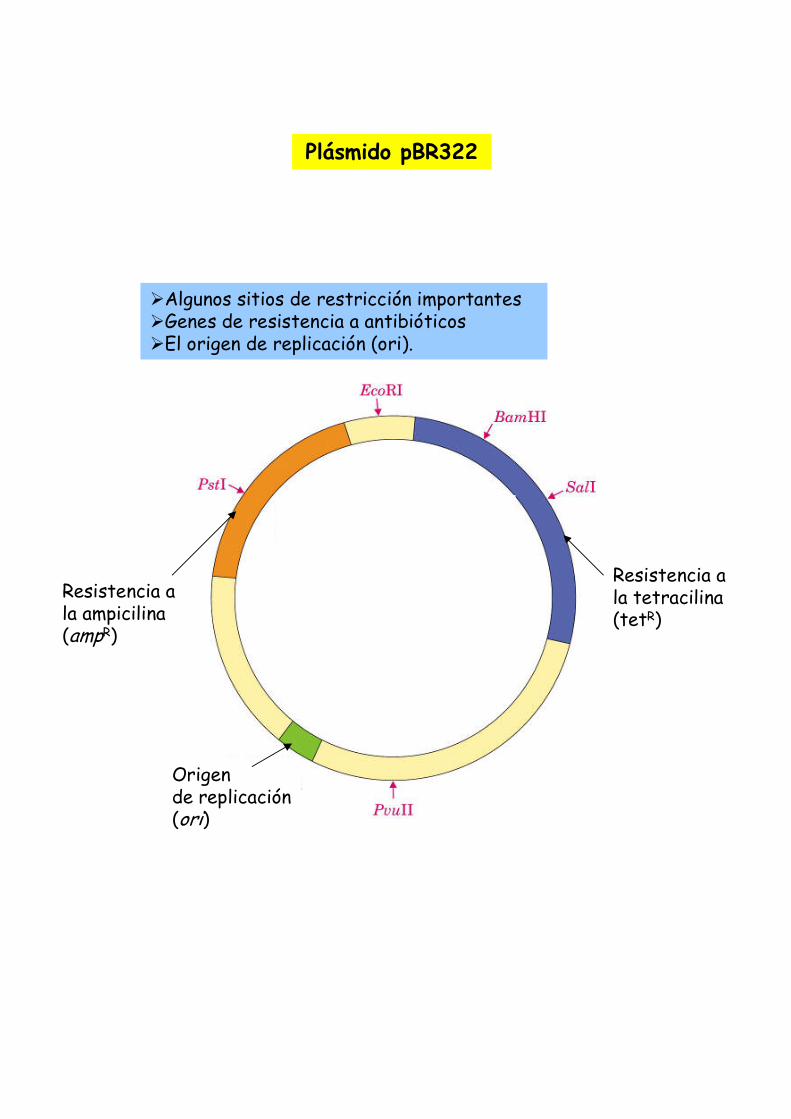
�Algunos sitios de restricción importantes�Genes de resistencia a antibióticos�El origen de replicación (ori).
Plásmido pBR322
Resistencia ala ampicilina(ampR)
Origende replicación(ori)
Resistencia ala tetracilina(tetR)
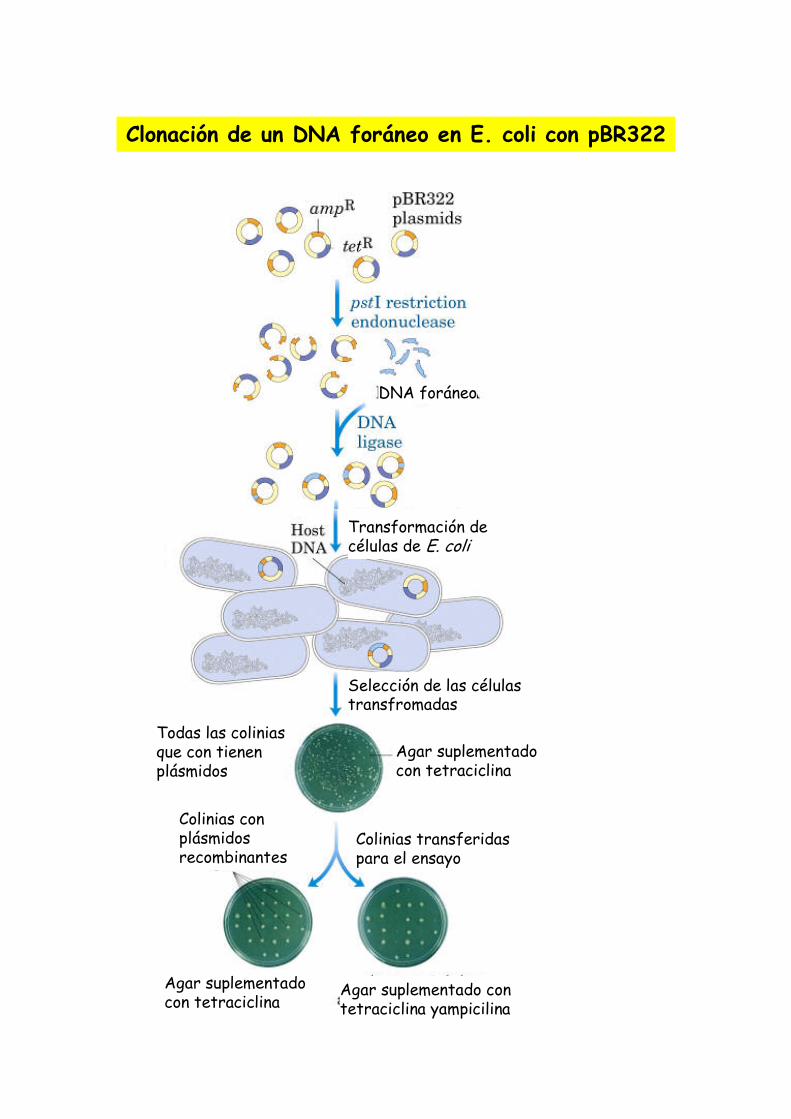
Clonación de un DNA foráneo en E. coli con pBR322
DNA foráneo
Transformación de células de E. coli
Selección de las células transfromadas
Agar suplementadocon tetraciclina
Todas las coliniasque con tienenplásmidos
Colinias con plásmidosrecombinantes
Colinias transferidas para el ensayo
Agar suplementado con tetraciclina
Agar suplementado con tetraciclina y ampicilina

�Bacteriófago λλλλ� Permite clonar segmentos de DNA más largos� Caracteríticas del genoma del fago:
� Alrededor de un tercio del genoma no es esencial� permite reemplazarlo por DNA foráneo.
� El DNA se empaqueta en partículas de fago sólo cuando tenga un tamaño entre 40.000 y 50.000 pares de base.
�Empaquetamiento in vitro���� Cuando los fragmentos de DNA foráneo de tamaño adecuadose han ligado a los fragmentos del fago� DNA recombinante se empaqueta dentro de partícula de fago al añadir extrato crudo de células bacterianas( que contienen todas las proteínas necesarias para ensamblar un fago completo)
�Cósmidos����Plásmidos recombinantes que combinan características útiles de plásmidos y del bacteriófago λ.
�Permite la clonación de fragmentos más largos�Moléculas de DNA pequeñas (5.000 a 7.000 pb)�Características:
�Origen de replicación plasmídico�Uno o más marcadores seleccionables�Un número de sitios de restricción únicos donde se puede insertar el DNA foráneo�Un sitio cos� Una secuencia de DNA del bacteriófago λ que se necesita para el empaquetamiento.
�No contienen ningún gen más del bacteriófago�Se propaga en E. coli como un plásmido

�Librería de DNA���� Una colección de los fragmentos derivados del genoma de un organismo determinado insertado, uno por uno, en unvector de clonación.
�Rotura del DNA purificado del vector con una enzima de restricción.�DNA genómico a clonar se reduce a fragmentos de tamaño apropiado por una enzima de restricción (la misma que el vector).�Separación de fragmentos (centrifugación isopícnica)�Mezcla de fragmentos con el vector linearizado y ligación.�Transformación de células bacterianas o empaquetamiento en partículas de fago.�Resultado���� gran población de bacteria o fagos, siendo cada uno de ellos portador de una molécula de DNA diferente.�Librería genómica���� Prácticamente todo el DNA genómico estará representado en la librería.
�Librería de cDNA���� Librería de DNA más especializada, sólo incluye aquellos genes que están siendo expresados en un organismo determinado e incluso ciertos tejidos o células.
�Se extrae mRNA total del organismo�Se produce DNA complementario (cDNA) mediante la transcriptasa inversa.�Los fragmentos de doble hebra se insertan en un vector adecuado y se clonan.
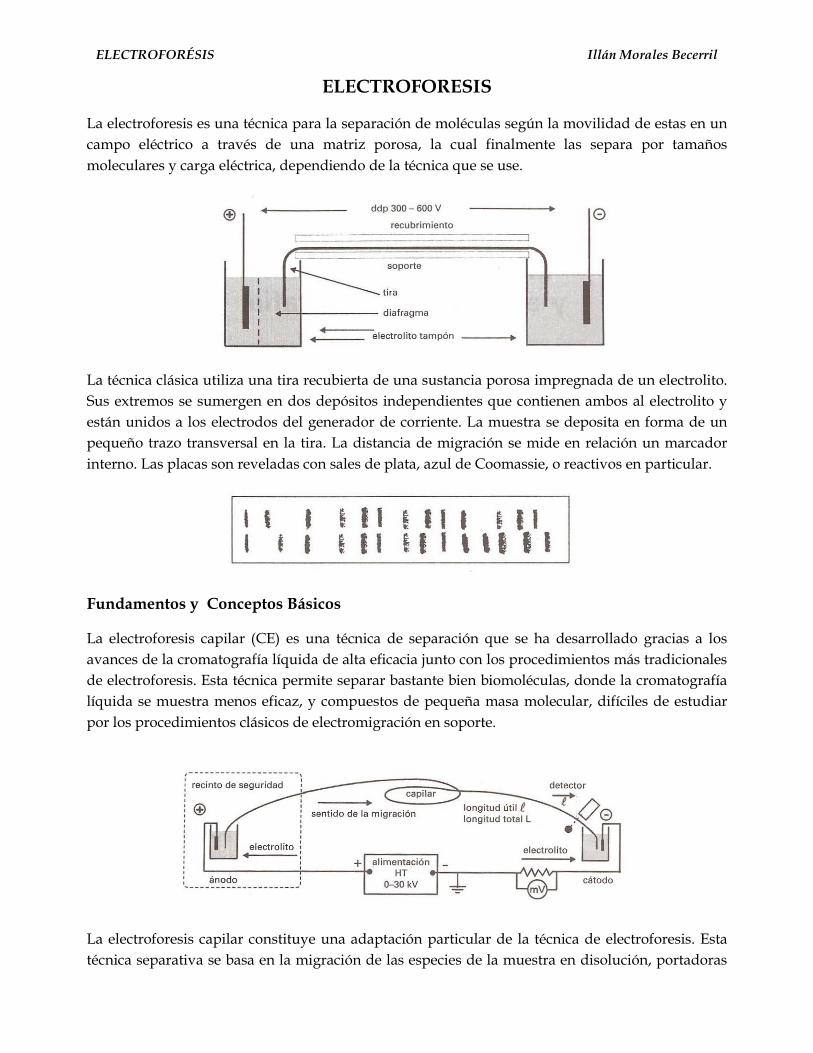
ELECTROFORÉSIS Illán Morales Becerril
ELECTROFORESIS
La electroforesis es una técnica para la separación de moléculas según la movilidad de estas en un campo eléctrico a través de una matriz porosa, la cual finalmente las separa por tamaños moleculares y carga eléctrica, dependiendo de la técnica que se use.
La técnica clásica utiliza una tira recubierta de una sustancia porosa impregnada de un electrolito. Sus extremos se sumergen en dos depósitos independientes que contienen ambos al electrolito y están unidos a los electrodos del generador de corriente. La muestra se deposita en forma de un pequeño trazo transversal en la tira. La distancia de migración se mide en relación un marcador interno. Las placas son reveladas con sales de plata, azul de Coomassie, o reactivos en particular.
Fundamentos y Conceptos Básicos
La electroforesis capilar (CE) es una técnica de separación que se ha desarrollado gracias a los avances de la cromatografía líquida de alta eficacia junto con los procedimientos más tradicionales de electroforesis. Esta técnica permite separar bastante bien biomoléculas, donde la cromatografía líquida se muestra menos eficaz, y compuestos de pequeña masa molecular, difíciles de estudiar por los procedimientos clásicos de electromigración en soporte.
La electroforesis capilar constituye una adaptación particular de la técnica de electroforesis. Esta técnica separativa se basa en la migración de las especies de la muestra en disolución, portadoras
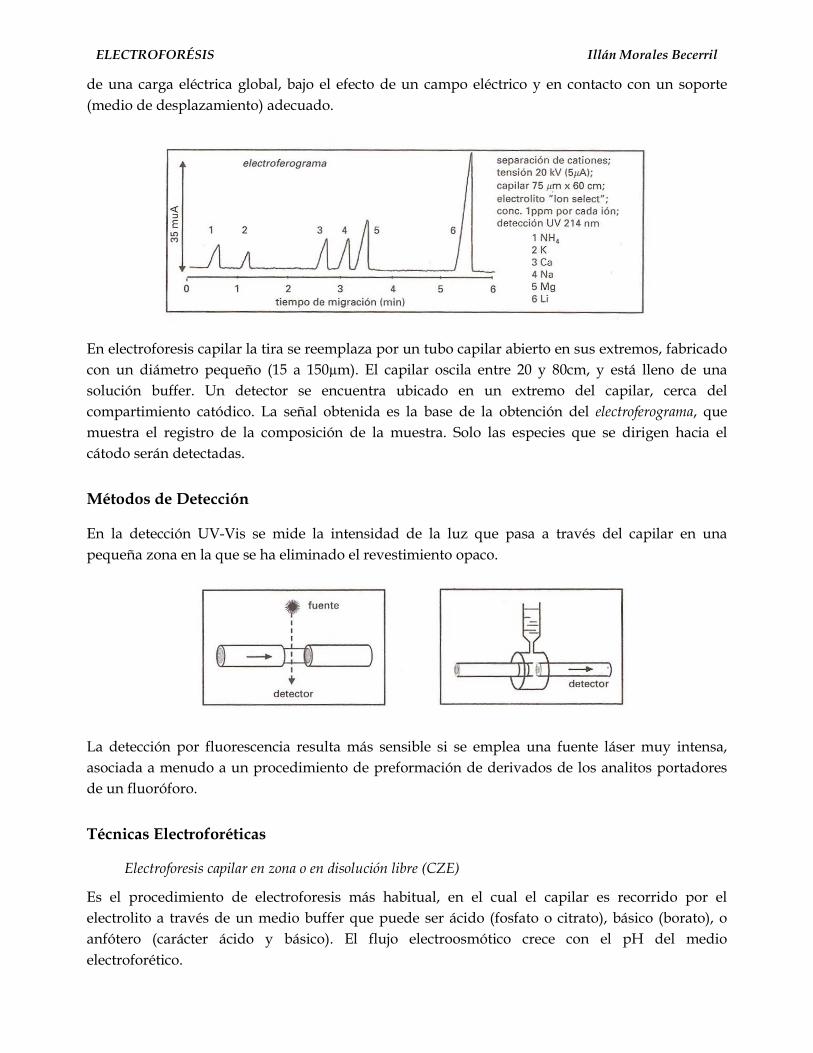
ELECTROFORÉSIS Illán Morales Becerril
de una carga eléctrica global, bajo el efecto de un campo eléctrico y en contacto con un soporte (medio de desplazamiento) adecuado.
En electroforesis capilar la tira se reemplaza por un tubo capilar abierto en sus extremos, fabricado con un diámetro pequeño (15 a 150µm). El capilar oscila entre 20 y 80cm, y está lleno de una solución buffer. Un detector se encuentra ubicado en un extremo del capilar, cerca del compartimiento catódico. La señal obtenida es la base de la obtención del electroferograma, que muestra el registro de la composición de la muestra. Solo las especies que se dirigen hacia el cátodo serán detectadas.
Métodos de Detección
En la detección UV-Vis se mide la intensidad de la luz que pasa a través del capilar en una pequeña zona en la que se ha eliminado el revestimiento opaco.
La detección por fluorescencia resulta más sensible si se emplea una fuente láser muy intensa, asociada a menudo a un procedimiento de preformación de derivados de los analitos portadores de un fluoróforo.
Técnicas Electroforéticas
Electroforesis capilar en zona o en disolución libre (CZE)
Es el procedimiento de electroforesis más habitual, en el cual el capilar es recorrido por el electrolito a través de un medio buffer que puede ser ácido (fosfato o citrato), básico (borato), o anfótero (carácter ácido y básico). El flujo electroosmótico crece con el pH del medio electroforético.

ELECTROFORÉSIS Illán Morales Becerril
Electroforesis capilar electrocinética micelar (MEKC)
En esta variante del procedimiento anterior se añade a la fase móvil un compuesto catiónico o aniónico para formar micelas cargadas. Estas pequeñísimas gotitas inmiscibles con la disolución retienen a los compuestos neutros de un modo más o menos eficaz, por afinidad hidrófila-hidrófoba. Se puede utilizar este tipo de electroforesis para moléculas que tienen tendencia a migrar sin separación, como es el caso de algunos enantiomeros.
Electroforesis capilar en gel (CGE)
Esta es la transposición de la electroforesis en egel de poliacrilamida o de agarosa. El capilar está relleno con un electrolito que contiene al gel. Se produce un efecto de filtración que ralentiza a las grandes moléculas y que minimiza los fenómenos de convección o de difusión. Los oligonucleótidos, poco frágiles, se pueden separar de este modo.
Isoelectroenfoque capilar (CIEF)
Esta técnica, también conocida como electroforesis en soporte, consiste en crear un gradiente de pH lineal en un capilar con pared tratada que contiene un anfótero. Cada compuesto migra y se enfoca al pH que tenga igual valor que su punto isoeléctrico (al pI su carga neta es nula). Seguidamente, bajo el efecto de una presión hidrostática y manteniendo el campo eléctrico, se desplazan las especies separadas hacia el detector. Las altas eficiencias obtenidas con este procedimiento permiten separar péptidos con pI que apenas difieren entre sí 0.02 unidades de pH.
Electrocromatografía Capilar
Este tipo de separación asocia la electromigración de los iones, propio de la electroforesis, y los efectos de separación entre fases presentes en la cromatografía. La técnica consiste en la utilización de un capilar relleno con una fase estacionaria, cuyo papel es doble: actúa como material selectivo que debe, por otro lado, participar en la migración del electrolito.
El factor de separación es muy elevado, pero existen un cierto número de problemas que limitan su aplicación, como el efecto de los modificadores orgánicos sobre el flujo electroosmótico y la dificultad de un control preciso del volumen de muestra introducido en el capilar.

Curso: Introducción a la Bioinformática
Lic. en Genética Ernesto Martín Giorgio 1
Introducción al diseño de primers INTRODUCCIÓN
Esta guía es una breve aproximación al diseño de primers, utilizando programas
bioinformáticos y pretende dar una orientación a aquellas personas que están incursionando
por primera vez en esta amplísima disciplina que es la Bioinformática. Si esta guía orienta y
ayuda al científico a realizar sus propios primers o cebadores, ha logrado su cometido.
En la primera parte de esta guía, revisaremos algunos conceptos generales pertinentes
al diseño de primer y a la amplificación enzimática de ADN mediante PCR, y en la segunda se
detalla un taller que le permitirá poner en práctica la mayoría de los conceptos revisados.
PARTE I
Fundamentos teóricos de la amplificación enzimática de ADN mediante la Reacción en
Cadena de la Polimerasa (PCR; polymerase chain raction)
La Reacción en Cadena de la Polimerasa es una técnica que ha revolucionado el modo
de manipular, clonar y detectar fragmentos de ADN, lo que le ha valido a su descubridor K. B.
Mullis el Premio Nobel. Después de su descubrimiento en 1983, se ha convertido en una de las
técnicas básicas de Biología Molecular, ampliamente utilizada debido a su rapidez,
especificidad, flexibilidad y aplicabilidad. Entre sus aplicaciones generales encontramos la
generación de sondas, clonación, secuenciación, diagnóstico clínico y tipificación de individuos
y microorganismos, siendo entonces muy utilizada en medicina forense y en estudios
filogenéticos.
Un ciclo de PCR comienza con un calentamiento inicial que desnaturaliza el ADN
blanco a 94-95 °C o más. Este primer paso del ciclo dura en promedio 90 s a 3 minutos. En el
proceso de desnaturalización, las dos cadenas del ADN se separan una de la otra y permiten
que la matriz (“template”) se encuentre como cadena simple, necesaria para la actividad
llevada a cabo por la polimerasa termoestable durante los pasos de amplificación.
En el siguiente paso del ciclo, la temperatura se reduce a un valor que oscila en el
rango de los 40 °C a los 60 °C. A esta temperatura, cada uno de los oligonucleótidos (primers)
hibrida con la secuencia complementaria en cada una de las cadenas simples de ADN, que se
han separado en el paso anterior, y sirven como cebadores para la síntesis, por la polimerasa
termoestable.
La síntesis de ADN se inicia cuando la temperatura de la reacción llega al valor óptimo
para la actividad de la polimerasa. Para la mayoría de las polimerasas esta temperatura se
encuentra en, aproximadamente los 72 °C. Luego se permite la síntesis durante 30 s a 2 min.
Este paso completa un ciclo y el siguiente ciclo se inicia con el retorno a los 94-95 °C para la
desnaturalización.
La cantidad de amplificado resultante va a depender de la disponibilidad de sustrato
para la reacción por eso, los oligonucleótidos y los desoxirribonuclótidos trifosforilados se

Curso: Introducción a la Bioinformática
Lic. en Genética Ernesto Martín Giorgio 2
añaden en exceso respecto al ADN a amplificar. Como es fácil inferir, la concentración de ADN,
oligonucleótidos, de y ADN polimerasa activa disminuyen después de cada ciclo debido a la
síntesis que se está produciendo por lo que la reacción lleva a un máximo de amplificación,
luego del cual el rendimiento y el número de copias obtenidas no aumentan. A esto se lo
conoce como el efecto “Platteau”.
En la reacción de PCR ideal, existen tres fragmentos de ácidos nucleicos. El fragmento
de ADN de doble cadena a ser amplificado (molécula blanco o target) y dos oligonucleótidos de
cadena simple que hibridizan en alguna región del ADN blanco (iniciadores o primers).
Además están presentes: un componente proteico (la enzima ADN polimerasa),
desoxirribonucleótidos trifosfatos (dATP, dCTP, dTTP y dGTP), buffers y sales.
Los primers se añaden en un vasto exceso comparado con el ADN blanco a ser
amplificado. Estos se hibridizan a las cadenas opuestas del blanco y se orientan enfrentándose
con sus respectivos terminales 3’, de manera tal que la síntesis llevada a cabo por la ADN
polimerasa se extiende a través de todo el segmento del ADN blanco situado entre ellos.
Recordemos que la enzima ADN polimerasa cataliza el crecimiento de cadenas de ADN nuevas
en la dirección 5’ > 3’.
La primera ronda de síntesis resulta en la generación de cadenas de ADN nuevas sin
una longitud determinada, las cuales como las cadenas parentales, se pueden hibridizar a los
primers luego de la desnaturalización e hibridización. Estos productos se acumulan con una
progresión aritmética a través de los ciclos siguientes.
Sin embargo, el segundo ciclo de desnaturalización, hibridización y síntesis, produce
dos productos de cadena simple que componen en conjunto un producto de doble cadena en
el cual posee la longitud exacta del fragmento original delimitado entre los primers. Cada
cadena de este producto discreto es complementaria a uno de los dos primers incluidos en la
reacción y puede en consecuencia participar como blanco en los ciclos subsiguientes.

Curso: Introducción a la Bioinformática
Lic. en Genética Ernesto Martín Giorgio 3
La cantidad de este producto se duplica con cada ciclo subsiguiente, acumulándose
exponencialmente. Así, treinta ciclos de desnaturalización, hibridización y síntesis resultan
teóricamente en un factor de amplificación de 236 veces (68 billones de copias) del producto
molecular original.
Una PCR ideal debería ser altamente específica, fiel y de gran rendimiento. Cada uno
de estos parámetros está influido por varios componentes de la propia reacción por lo que
muchos casos ajustando las condiciones para la máxima especificidad no se obtiene buen
rendimiento, o bien optimizando para la fidelidad no se obtiene buena eficiencia. En
consecuencia, cada vez que se optimiza una PCR hay que tener en cuenta distintos aspectos
que hacen que dicha reacción sea exitosa.
¿Cuáles son los factores de los que depende una PCR exitosa?
Aunque el concepto de la PCR es simple, el desarrollo de una reacción exitosa depende de
un número de factores. Algunos de ellos son:
- Diseño de los oligonucleótidos iniciadores o cebadores (primers):
El diseño cuidadoso de primers es uno de los aspectos más importantes de la PCR. Primers mal diseñados pueden amplificar otros fragmentos de ADN distintos a los buscados (amplificación inespecífica). En el diseño de los mismos algunas reglas se han demostrado como útiles, por ejemplo:
I – Cada primer individual debe contar con una longitud de 18-24 bases. II – Se debe mantener un contenido de G:C (Guanina:Citosina) entre 40 y 60 %. III – Los dos primers del par deben de tener temperatura de fusión “Tm” cercanos, dentro de los 5 °C. IV – La secuencia de los primers individuales debe iniciarse y terminarse con 1 o 2 bases púricas. V – Evitar regiones con potencialidad para formar estructuras secundarias internas. VI – Evitar poli X. VII – Secuencias adicionales pueden ser agregadas en el extremo 5’ del primer (no incluir cuando se estima la Tm del primer). VIII – Se pueden agregar degeneraciones en algunas posiciones del primer:
a - Se incrementa el riesgo de amplificación inespecífica. b - Se disminuye la concentración en la mezcla de cada uno de los primers posibles c - No se recomienda utilizar más de 64 primers diferentes en la mezcla. d - Código IUPAC/IUB:
A adenina R A o G C citosina W A o T G guanina S C o G T timidina en el ADN Y C o T Uracilo en el ARN K G o T N A o C o G o T H A o C o T; no G M A o C B C o G o T; no A V A o C o G; no T D A o G o T; no C

2
Un marcador es un polimorfismo, pero no siempre un polimorfismo sirve de marcador.
Hasta mediados de la década de los 60’, los marcadores usados en genética y
mejora animal estaban controlados por genes asociados a caracteres polimórficos, en
general fáciles de identificar visualmente (fenotipo), pero solo un pequeño número de esos
marcadores morfológicos permitían encontrar asociaciones significativas entre estos y los
caracteres con importancia económica.
Por sus limitaciones surgieron los marcadores isoenzimáticos o marcadores de
proteínas, que eran accesibles a un mayor número de especies y brindaban mejores
resultados. Se expresan en forma codominante, son compatibles con el normal
funcionamiento de la proteína y son relativamente abundantes, pero son técnicas muy
caras y dificulta los proyectos de mapeos, además no más del 3% del ADN genómico es
finalmente traducido a proteínas y que sólo unas pocas de estas pueden mutar sin alterar la
viabilidad de los animales. Los primeros marcadores fueron los grupos sanguíneos y luego
otros marcadores polimórficos bioquímicos o las inmunoglobulinas pero presentaban escasa
variantes polimórficas lo que limito su uso y aplicaciones Dejaron de utilizarse y aparecen así
nuevos marcadores moleculares.
MARCADORES MOLECULARES
Con el advenimiento de las técnicas de biología molecular aparecieron diversos
métodos de detección de polimorfismo genético directamente a nivel de ADN: los
marcadores genéticos moleculares basados en ADN que sirven de referencia para detectar
la transmisión de un segmento de cromosoma de una generación a otra.
El término marcador se utiliza aquí en el sentido de marcador genético, y muchas
veces se lo utiliza como sinónimo de marcador de locus; un locus polimórfico que indica el
genotipo del individuo que lo lleva (con este
propósito se utilizan marcadores en genética de
poblaciones), o el genotipo de uno o de varios
loci ligados al marcador. Inicialmente el
descubrimiento de las enzimas de restricción
permitió el análisis de los polimorfismos de longitud
de los fragmentos de restricción. Posteriormente,
con el desarrollo de la reacción en cadena de la
polimerasa (PCR), se desarrollaron nuevos
métodos de detección de marcadores
moleculares.
Los marcadores “ideales” cumplen una serie de características, entre las que se encuentran:
Elevada capacidad de detectar altos niveles de polimorfismo,
Alta heredabilidad,

3
Gran capacidad para acceder a todas las regiones del genoma,
Independencia del estado físico y de desarrollo del individuo,
Facilidad de obtención,
Detección por métodos económicos,
Independencia de las condiciones ambientales y
La posibilidad de determinación en cualquier tipo de células que contenga
núcleo.
Estas constituyen algunas de las ventajas de los marcadores moleculares sobre los
morfológicos e isoenzimas; otras radican en su capacidad de detección de mutaciones
silenciosas, que no originan cambios de aminoácidos.
Los polimorfismos detectados en el ADN se deben a diferencias al número de
determinadas regiones no codificantes dispersas en el genoma (regiones repetidas) o a
mutaciones puntuales.
En función de estas características se pueden clasificar los diferentes métodos de
detección de polimorfismos:
A- los marcadores asociados a variaciones debidas al número de repeticiones en su
secuencia (microsatélites y minisatélites) y
B- los marcadores que detectan cambios puntuales en el genoma (RFLP, AFLP, RADP,
SNP, etc.), que pueden ser detectables o no, por enzimas de restricción.
A- MARCADORES ASOCIADOS A VARIACIONES DEBIDAS AL NÚMERO DE REPETICIONES
EN SU SECUENCIA
ADN REPETITIVO DENTRO DE UN GENOMA
Se conoce que una gran proporción variable del genoma eucariota está
compuesto por secuencias repetitivas de ADN, las cuales difieren en el número y la
composición de nucleótidos.
Secuencias repetitivas en tándem (se presenta un resumen en el siguiente cuadro)
ADN satélite ADN altamente repetitivo
Minisatélites ADN telomérico Son secuencias muy conservadas.
Contribuye a la estabilidad del cromosoma.
VNTR ADN moderadamente repetitivo
Microsatélites SSLP, SSR o STR ADN moderadamente repetitivo

4
MINISATÉLITES o VNTR (número variable de repeticiones en tándem)
(Variable Number of Tandem Repeats)
En 1980 A. R. Wyman y R. White descubrieron, por azar, la primera región
hipervariable del ADN humano, y subsecuentemente se fueron hallando otras regiones de
ese tipo cerca de los genes de la globina, la insulina, la mioglobina y otros. Estas secuencias
no codificantes e hipervariables del ADN (de una longitud aproximada de entre 6 y 25 pb)
repetidos en tándem un número variable de veces (VNTR), están ubicados en regiones
centroméricas y teloméricas de los cromosomas y forman secuencias de entre 5 y 50 kb de
longitud.
Ejemplo: 7 repeticiones en tándem de la secuencia de 16 pares de bases
5’GACTGCCTGCTAAGATGACTGCCTGCTAAGATGACTGCCTGCTAAGATGACTGCCTGCTAAG
ATGACTGCCTGCTAAGATGACTGCCTGCTAAGATGACTGCCTGCTAAGAT
Figura 1. VNTR (número variable de
repeticiones en tándem).
Se genera de este modo una gran cantidad de alelos diferenciados por el número
de repeticiones que posee cada uno. Esta última característica genera un alto porcentaje
de heterocigosis (generalmente mayor al 80%) alcanzado para estos loci, lo que los
convierte en marcadores altamente informativos en estudios de análisis de ligamiento y
pruebas de identificación.
En estas técnicas, los sitios de restricción de las enzimas se encuentran flanqueando
las secuencias repetidas en tándem y se visualizan mediante transferencia de Southern
usando secuencias VNTR como sonda observando un patrón de bandas. Las sondas
moleculares desarrolladas por Jeffreys y colaboradores en el año 1985 a partir de secuencias
repetitivas de un intrón de la mioglobina humana, dieron origen a la técnica conocida
como “fingerprinting de ADN”. La misma ha revolucionado el campo de la medicina forense

5
y los estudios de paternidad en todo el mundo. El fingerprinting de ADN es el patrón de
bandas único (huella molecular) es específico de cada individuo (excepto para los gemelos
monocigóticos) obtenido por la hibridación de muestras de ADN digerido con sondas
capaces de detectar múltiples minisatélites a lo largo del genoma.
Una limitación importante del análisis de huellas moleculares de ADN con VNTR es
que precisa una muestra relativamente grande de ADN (100.000 células o 50µg) y el ADN
debe estar relativamente intacto.
Hasta el momento, se han descripto y mapeado cientos de loci de VNTR en el
genoma humano y de algunos animales, aunque se calcula que su número podría ser de
varios miles por genoma.
MICROSATELITES o SSR (secuencias simples repetidas) (simple sequence repeats)
o SSLP (polimorfismo de longitud de secuencias simples) (simple sequence length
polymorphisms) o STR (repeticiones simples en tándem) (short tandem repeat)
Los microsatélites, también conocidos como SSR, SSLP o STR, consisten en pequeñas
secuencias con 2-5 nucleótidos repetidos en tándem, altamente variables en tamaño. Se
identificaron en el año 1989 al descubrirse que existían zonas del ADN no codificante. Se
llamaron primero VNTR (variable number of tandem repeats), pero luego fue reemplazada
por la de microsatélite o STR (Short Tandem Repeat) para los segmentos mas cortos y para
los mas largos la denominación VNTR o minisatélites.
En genomas eucariotas las secuencias de los microsatélites son muy frecuentes, bien
distribuidas y mucho más polimórficas que los minisatélites, constituyendo la clase de
marcadores moleculares más polimórficos que se conocen.
Se ha señalado que los elementos más repetidos en mamíferos son extensiones de
dinucleótidos (CA)n y (GA)n y los más típicos constan de 10-30 copias de una repetición que
usualmente no son superiores a 4 pb de longitud.
Por el momento, se desconoce exactamente el origen y la función de estas
secuencias repetidas. Respecto a su origen, la aparición inicial de los microsatélites pudo
deberse al azar, o podrían haber surgido como producto de mutaciones en la secuencia
poli-A en el extremo 3’.
Esta fuente de mutación y cambio en el número de repeticiones podría proveer una
fuente de variación cuantitativa para una rápida adaptación evolutiva de las especies a
cambios ecológicos. La nutrición y el estrés podrían incrementar la velocidad de mutación
en los microsatélites debido a un descenso de la regulación de reparación de errores. De
esta forma, el estrés que acompaña al cambio evolutivo podría inducir en una población el
incremento de mutaciones requeridas para adaptarse al cambio.
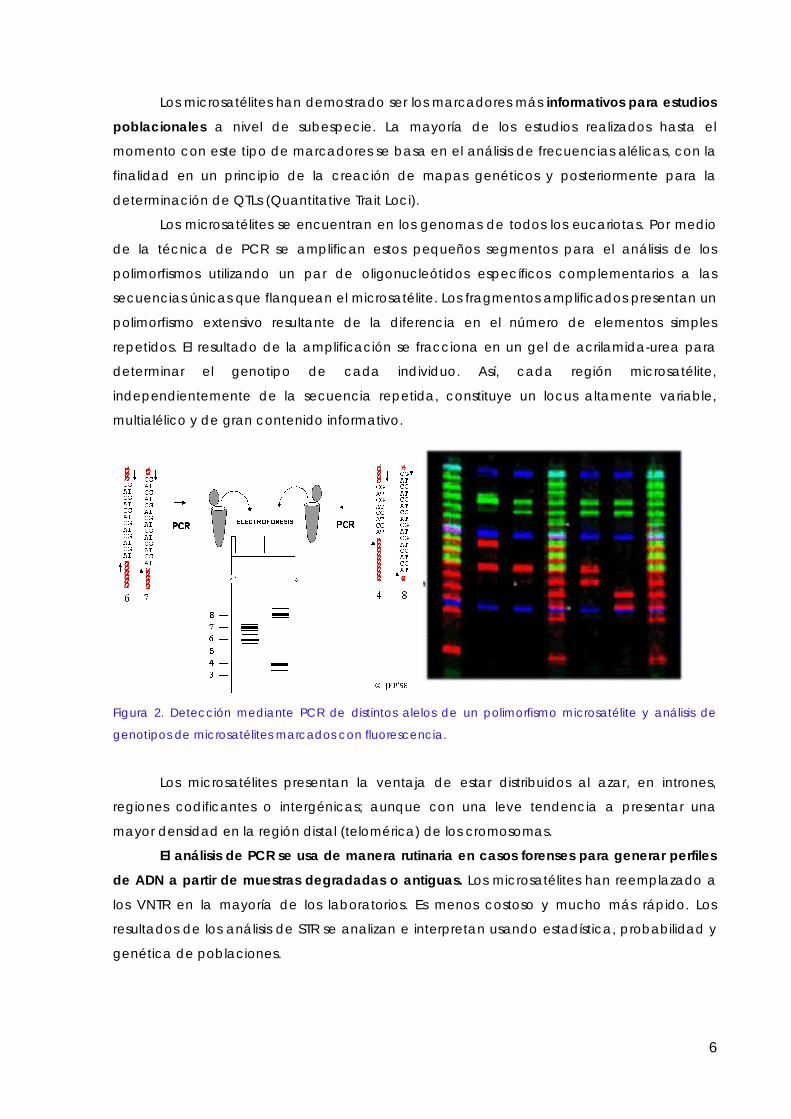
6
Los microsatélites han demostrado ser los marcadores más informativos para estudios
poblacionales a nivel de subespecie. La mayoría de los estudios realizados hasta el
momento con este tipo de marcadores se basa en el análisis de frecuencias alélicas, con la
finalidad en un principio de la creación de mapas genéticos y posteriormente para la
determinación de QTLs (Quantitative Trait Loci).
Los microsatélites se encuentran en los genomas de todos los eucariotas. Por medio
de la técnica de PCR se amplifican estos pequeños segmentos para el análisis de los
polimorfismos utilizando un par de oligonucleótidos específicos complementarios a las
secuencias únicas que flanquean el microsatélite. Los fragmentos amplificados presentan un
polimorfismo extensivo resultante de la diferencia en el número de elementos simples
repetidos. El resultado de la amplificación se fracciona en un gel de acrilamida-urea para
determinar el genotipo de cada individuo. Así, cada región microsatélite,
independientemente de la secuencia repetida, constituye un locus altamente variable,
multialélico y de gran contenido informativo.
Figura 2. Detección mediante PCR de distintos alelos de un polimorfismo microsatélite y análisis de
genotipos de microsatélites marcados con fluorescencia.
Los microsatélites presentan la ventaja de estar distribuidos al azar, en intrones,
regiones codificantes o intergénicas; aunque con una leve tendencia a presentar una
mayor densidad en la región distal (telomérica) de los cromosomas.
El análisis de PCR se usa de manera rutinaria en casos forenses para generar perfiles
de ADN a partir de muestras degradadas o antiguas. Los microsatélites han reemplazado a
los VNTR en la mayoría de los laboratorios. Es menos costoso y mucho más rápido. Los
resultados de los análisis de STR se analizan e interpretan usando estadística, probabilidad y
genética de poblaciones.

7
B- MARCADORES QUE PRESENTAN CAMBIOS PUNTUALES EN EL GENOMA
RFLP: Polimorfismo de Longitud de Fragmentos de Restricción. (Restriction Fragment Length
Polimorphism)
A finales de la década del 60, el descubrimiento de las endonucleasas de bacterias
(enzimas de restricción con muy alta especificidad), las cuales cortaban el ADN en
secuencias definidas de usualmente 4-8 pb, conllevó al desarrollo de técnicas para el
aislamiento y la manipulación de fragmentos de ADN. Una de las mayores aplicaciones de
esta técnica fue el ensamblaje de los mapas genéticos con el polimorfismo en longitud de
los fragmentos de restricción.
Esta técnica explota las variaciones en las secuencias del ADN por la creación o
abolición de sitios para el corte de las endonucleasas, dadas por los cambios de bases, las
adiciones o deleciones de fragmentos de ADN entre y en estos sitios. Además, algunas
endonucleasas son incapaces de cortar el ADN si la secuencia de reconocimiento contiene
uno o más sitios de citosina metilada, por lo tanto tales enzimas pueden detectar
polimorfismo si hay variaciones en los patrones de metilación.
El estudio de los polimorfismos de este tipo consiste en el análisis en cuanto a número
y tamaño de los fragmentos de ADN que se originan al digerir con las mencionadas enzimas
el material genómico o un fragmento amplificado del genoma, de manera que un sitio de
restricción polimórfico o una deleción o inserción entre dos sitios de corte será detectado
como un polimorfismo en la longitud de los fragmentos de restricción generados a nivel
fenotípico. La técnica consta de las siguientes etapas (figura 3): digestión del ADN con
enzimas de restricción; separación por electroforesis de los fragmentos resultantes;
transferencia de los fragmentos separados a membranas de nitrocelulosa (Southern Blotting)
e hibridación con sondas moleculares radiactivas y exposición de la membrana a un filme
de rayos X. La identificación del polimorfismo también puede hacerse por PCR. Este método
se ha empleado en la caracterización de los genes de algunas de las proteínas lácteas
bovinas.
El uso de los RFLP como marcadores para trazar la transmisión de los genes asociados
a ellos, es de una utilidad considerable en genética pues tienen como ventajas el no estar
influenciados por el ambiente, no depender del estado ontogénico del animal, permitir un
análisis de todo el genoma, poder ser detectados en estadios muy tempranos del desarrollo
independientemente de la expresión del gen, una vez heredados, y además poseen la
característica de ser codominantes en la expresión fenotípica, es decir, permiten discernir
entre el individuo homocigótico dominante y el heterocigótico; son muy comunes y
cualquier gen debe tener sitios de restricción polimórficos en una vecindad de varios cientos
de pares de bases.

8
Pero tienen como desventajas que son muy costosos, necesitan de un personal
calificado y entrenado para trabajarlo, precisan el uso de radioactividad y además,
consumen mucho tiempo. En animales domésticos se ha realizado este tipo de análisis del
ADN desde 1985, con numerosos trabajos en una amplia diversidad de especies.
Otras de las aplicaciones de esta tecnología ha sido el desarrollo de detallados
mapas de ligamiento en una amplia variedad de organismos. Aunque ésta es la más
conocida, el RFLP se ha empleado también en la caracterización del genoma de organelos,
en la identificación de líneas o individuos y en las investigaciones sobre el tema de la
diversidad genética. La identificación y el mapeo de RFLP han revolucionado la genética
humana por el desarrollo del diagnóstico asistido por marcadores y la detección de una
amplia variedad de enfermedades hereditarias. A pesar de haber sido ignorados este tipo
de marcadores en sus inicios por los genetistas veterinarios, los mismos métodos para el
mapeo en humanos son empleados para estudios de diferentes especies animales, respecto
a los sitios polimórficos, produciéndose una revolución en el mejoramiento animal por la
introducción de la selección asistida por marcadores (Marker Asisted Selection: MAS).
RAPD: Amplificación aleatoria de ADN polimórfico (Random Amplified Polymorphic DNA)
La técnica RAPD, conocida también como AP-PCR es básicamente una variación
del protocolo de la PCR con dos características distintivas: utiliza un cebador único (en lugar
de un par de primers) con una secuencia arbitraria, por lo tanto desconocida. Para que
Figura 3. A: Representación
esquemática del principio
genético del RFLP.
B: Gel de azarosa 1.5% teñido
con bromuro de etidio
conteniendo los productos
obtenidos de ADN de diferentes
individuos, amplificados por PCR

9
ocurra la amplificación, dos secuencias complementarias al cebador arbitrario deben estar
lo suficientemente adyacentes (~400pb) y en orientación opuesta que permita la
amplificación exponencial por la ADN-polimerasa (Figura 4).
En los RAPD se emplean cebadores cortos (10 pares de bases) que se utilizan de
forma individual en reacciones de PCR con un nivel de especificación muy bajos. De esta
manera se genera un patrón de bandas sencillos que dependerá de la capacidad de los
cebadores para encontrar zonas en el ADN, parcial o totalmente complementario.
Figura 4. Detección de un loci
polimórfico a través de la técnica
RAPD. (las líneas horizontales
representan los cromosomas de
las líneas consanguíneas de
ratones C3Hy B6
Durante la fase de alineamiento de la reacción de PCR-RAPD, el cebador o primer
arbitrario se unirá a los sitios de la secuencia complementaria en el ADN molde
desnaturalizado. Si dos moléculas se alinean con el molde en cadenas opuestas, con la
orientación apropiada y con una distancia amplificable (menor o igual a 2500 bases)
entonces será amplificado un fragmento discreto de ADN. Cada primer es potencialmente
capaz de amplificar un número de fragmentos (1-10 o más) de diferentes loci durante la
misma reacción de PCR resultando en varias bandas. Los segmentos amplificados pueden
ser visualizados en un gel de agarosa teñido con bromuro de etidio o en geles de
poliacrilamida de alta resolución visualizados por autorradiografía o teñidos con plata
detectándose el polimorfismo como ausencia o presencia de bandas de un fragmento
particular.
Los polimorfismos RAPD resultan de la diferencia de secuencias en uno o ambos sitios
de unión de los primers, los cuales evitan el alineamiento de la secuencia molde. Otras
fuentes de polimorfismo RAPD pueden incluir diferencias de apenas un par de bases
(mutaciones puntuales) que son suficientes para causar complementariedad del primer en
el sitio de iniciación; deleciones o inserciones adyacentes en el sitio de iniciación de modo

10
En este tipo de marcadores factores como calidad y concentración del ADN molde, presencia de contaminantes precipitables con etanol, concentración de cloruro de magnesio, contenido de G+C en los primers y su concentración y la eficiencia del termociclador, influyen en la amplificación y afectan la sensibilidad de la reacción, además de la fuente de la ADN polimerasa.
que provoquen la acción de la ADN-polimerasa, por lo que los marcadores RAPD tienen
naturaleza binaria, el segmento amplificado está presente o no; son marcadores dominantes
porque solo se amplifica una variante alélica, aquella que cumple con los requisitos de
complementariedad entre el cebador y las cadenas de ADN a una distancia menor o igual
a 2Kb.
La distribución de marcadores RAPD a lo largo del genoma es una consideración
importante cuando se evalúa la utilidad de estos marcadores, los cuales están ampliamente
ubicados más o menos al azar por todo el genoma. Existen reportes que verifican su
presencia en el genoma de numerosas especies. Los RAPDs se han utilizado también para la
caracterización racial en ganado bovino.
Una característica importante en cualquier marcador genético es que posean una
reproducibilidad consistente.
Para esta técnica no es necesario
conocer previamente la secuencia del ADN.
Esta técnica se diferencia
fundamentalmente de otras y muestra ventajas,
por ejemplo, sobre RFLP, en que no se basa en
hibridación sino en la amplificación de ADN, lo
que implica simplicidad y rapidez, lo cual se
debe a la posibilidad de detectar polimorfismo por la visualización directa de las bandas en
el gel, eliminando todas las etapas de transferencia de ADN para membranas (Southern
Blot), hibridación con sondas específicas y autorradiografía; no requiere del desarrollo de
una librería de sondas específicas, sino que un conjunto de primers arbitrarios puede utilizarse
para cualquier organismo; se elimina la necesidad de isótopos radiactivos, se requiere 10-3
veces menos concentración de ADN que para RFLP y la principal desventaja es el bajo
contenido de información genética por loci asociado a la dominancia de los marcadores
RAPD, además de que para su desarrollo requiere que se garanticen condiciones de
adecuada repetibilidad, además de ser un método muy trabajoso y costoso.
AFLP: Polimorfismos de longitud de fragmentos amplificados (Amplified fragment length
polymorphisms)
Se basa en la amplificación arbitraria de fragmentos de restricción de ADN ligados a
adaptadores: se utilizan cebadores semiespecíficos con secuencia complementaria al
adaptador en el extremo 5´. Estos marcadores combinan dos enzimas de restricción,
(generalmente la EcoR1 y la Mse1) y dos amplificaciones de PCR usando primers marcados.
Se generan patrones de bandas complejos en donde es fácil encontrar polimorfismos

11
claramente diferenciales. Consiste en la amplificación de múltiples regiones arbitrarias del
genoma.
Involucra cuatro etapas:
digestión con dos enzimas de restricción, una de corte raro (reconoce secuencias de
6pb) y otra de corte frecuente (reconoce secuencias de 4pb);
acoplamiento de adaptadores específicos de doble cadena a los extremos de los
fragmentos de restricción;
amplificación selectiva de fragmentos con cebadores específicos. Se lleva a cabo con
el uso de iniciadores que contienen una base extra en el extremo 3´ lo que produce un
conjunto de fragmentos que además de llevar la secuencia complementaria al primer o
iniciador, complementan a la base extra adicional. Hay una segunda amplificación en la
cual se conocen iniciadores con dos o tres bases extras. Los fragmentos son
complementarios a las extensiones consideradas.
separación de los fragmentos por electroforesis en geles de poliacrilamida.
Se pueden utilizar varios métodos para revelar el patrón de bandas: empleo de
isótopos radiactivos y la tinción con nitrato de plata.
Mediante la selección del número de nucleótidos selectivos es posible controlar el
número de fragmentos a amplificar, en una relación inversamente proporcional. El
polimorfismo es detectado por la presencia o ausencia de bandas debido a mutaciones en
los sitios de restricción o en secuencias adyacentes a estos, los cuales se complementan o se
diferencian de los nucleótidos selectivos añadidos a los cebadores de la PCR, y por
inserciones o deleciones dentro del fragmento amplificado.
Tiene como ventajas que se obtiene un alto grado de polimorfismo, un mayor número
de marcadores por gel analizado y no requiere del conocimiento de la secuencia de ADN.
Debido a la cantidad de marcadores que pueden ser generados, el mapeo genético
puede realizarse con mayor rapidez y más fácilmente. Es una técnica para estudios de
biodiversidad. Comparado con RFLP es más rápido, menos laborioso y provee mayor
información. Revela fundamentalmente el polimorfismo dominante, que puede ser por la
presencia o ausencia del sitio de restricción o por una simple variación de un nucleótido en
el genoma.
Los marcadores AFLP pueden ser aislados del gel y usarlo como sondas para
caracterizar nuevos clones, o secuenciarlos para diseñar cebadores y emplearlos en otras
técnicas basadas en PCR.

12
Figura 5. Representación esquemática de los AFLP
SSCPs: Polimorfismo de Conformación de las Cadenas Simples
El ADN de cadena simple tiende a doblarse y formar estructuras complejas
estabilizadas por enlaces intramoleculares débiles, especialmente por puentes de
hidrógeno. Consiste en la detección de las diferencias entre fragmentos de ADN
amplificados, con distinta secuencia. Esto permite la detección de mutaciones puntuales de
ADN sin necesidad de conocer su secuencia y son capaces de identificar entre el 30 y 70%
de todos los polimorfismos debidos a una mutación simple. Su detección se lleva a cabo en
un gel de acrilamida en condiciones no desnaturalizantes. Los primers pueden estar
radioactivamente marcados, o puede realizarse la detección de los productos amplificados
por tinción con plata y siempre deben incluirse muestras controles para identificar el tipo
salvaje del mutado.

13
El microarray es un arreglo de cientos a millares de secuencias de ADN inmovilizadas y ordenadas en forma de matriz adheridas a una superficie sólida (generalmente de cristal). Cada secuencia es un gen diferente.
SSCP es simple y adecuadamente sensible para fragmentos de hasta 200pb de
longitud, y han sido utilizados en la identificación de variantes alélicas de las proteínas
lácteas, pero para el desarrollo de esta metodología se requiere una amplia batería de
oligonucleótidos que encarece su utilización.
SNPs: Polimorfismo de nucleótido simple (Single Nucleotide Polymorphism)
Se consideran la más reciente generación de marcadores moleculares, basados en
la identificación de la sustitución de un nucleótido por otro, representando solamente dos
alelos simples. Su frecuencia oscila entre 1 cada 600 y 1 cada 1000 pares de bases. Ellos
incluyen la técnica clásica de RFLPs pero no exactamente detectando la aparición o
abolición de un sitio de restricción. Constituye una ventaja el hecho de que puedan ser
detectados sobre “arrays” (chips) de fase sólida sin necesidad del empleo de electroforesis
en geles.
El método más directo para la detección es la secuenciación de segmentos de ADN,
previamente amplificados por PCR, de varios individuos que representen la diversidad de la
población. Se diseñan cebadores para amplificar fragmentos de ADN de 400-700 pb,
derivados fundamentalmente de genes de interés o de secuencias reportadas en bases de
datos correspondientes a secuencias expresadas (expressed sequence tag, EST). Los
productos amplificados se secuencian en ambas direcciones y sus secuencias se comparan
en busca de polimorfismos.
La mayoría de los métodos de detección de
SNPs se basan en la amplificación usando “multiplex”
de una secuencia diana y la hibridación con
oligonucleótidos anclados al “microarray”, cada uno
de los cuales está terminado en un nucleótido
polimórfico.
Un paso sencillo de extensión del primer se
lleva a cabo sobre el “array”, usando una mezcla de cuatro dideoxinucleótidos marcados
con fluorescencia. Las marcas se adicionan a los cebadores que se unen perfectamente al
ADN, no siendo así con aquellos que no lo hacen en el extremo 3´. La lectura de la
presencia o ausencia de fluorescencia en el “array” permite obtener el tipo de cada SNP
sobre el “array”.
Cualquiera de los cuatro nucleótidos puede estar presente en cualquier posición en
el genoma, por lo tanto se debe suponer que cada SNP tendría cuatro alelos. Teóricamente
esto es posible, pero en la práctica la mayoría de los SNP tienen solamente dos variantes, la
secuencia original y la versión mutada. Esto se debe a la vía en que estos aparecen y se
distribuyen en una población. Un SNP se origina cuando una mutación puntual ocurre en el

14
genoma, convirtiendo un nucleótido en otro. Si la mutación ocurre en las células
reproductivas de un individuo, pudiendo ser heredada por uno o más descendientes y
después de muchas generaciones, el SNP puede establecerse en la población. Para que se
produzca un tercer alelo, una nueva mutación debe ocurrir en la misma posición en el
genoma de otro individuo, y este individuo y su descendencia deben reproducirse de forma
tal que el nuevo alelo quede establecido. Esto no es imposible, pero es poco probable, por
lo tanto la mayoría de los SNP son bialélicos.
Figura 6. Los SNP son marcadores basados en la identificación
de la sustitución de un nucleótido por otro.
Los SNP pueden aparecer tanto fuera de los
genes (que no afecten la producción o función de
alguna proteína) como en un gen específico, donde
pueden ubicarse en regiones codificantes (relacionados
con cambios en la cantidad de proteína producidas) o
no codificantes (que afectan solo la secuencia de aminoácidos).
Estos han sido empleados como marcadores para el diagnóstico de rasgos
específicos por ser abundantes en el genoma bovino, genéticamente estables y de análisis
fácilmente automatizable, lo que ha permitido además, su utilización como marcadores
para la identificación animal y pruebas de paternidad en ganado bovino.
Un haplotipo es una combinación de alelos
de diferentes loci de un cromosoma que son
trasmitidos juntos, están ligados en un mismo
cromosoma. Un haplotipo puede ser un locus, varios
loci, o un cromosoma entero dependiendo del
número de eventos de recombinación que han
ocurrido entre un conjunto dado de loci.
Figura 7. Un haplotipo es un conjunto específico de SNP y otras variantes genéticas observadas en un
cromosoma individual o en parte de un cromosoma.
Dada la alta variabilidad alélica, la probabilidad de que dos individuos no
relacionados presenten un mismo haplotipo, es prácticamente nula. Es por esto que el
estudio de haplotipos se ha convertido en una herramienta útil en la determinación de
relaciones genéticas entre individuos. Los SNPs (polimorfismos de nucleótido simple) se
heredan en grupos que se encuentran estrechamente relacionados en el ADN. A este grupo
de SNPs que se heredan en bloque, es a lo que hemos denominado haplotipos.

15
EST: Etiquetas de secuencia expresada (Expressed sequence tags)
Los EST son secuencias cortas obtenidas de clones de ADN complementario (ADNc) y
sirven como pequeños identificadores del gen. Puede usarse como un marcador, para
buscar el resto del gen o para ubicarlo en un segmento más grande de ADN.
Típicamente tienen entre 200 y 400 nucleótidos de longitud. Los datos de los EST son
capaces de proporcionar una estimación aproximada de los genes que están expresados
activamente en un genoma bajo una condición fisiológica en particular.
Esto debido a que las frecuencias para EST’s particulares reflejan abundancia en los
ARNm de una célula, que corresponde con los niveles de expresión de genes bajo esa
condición. Otro potencial beneficio del muestreo por EST es que, al secuenciar clones de
ADNc de manera aleatoria es posible encontrar nuevos genes. Sin embargo la utilización de
EST también presenta varios inconvenientes, para el análisis de perfiles de expresión. Tienen
como desventaja:
Baja calidad, debido a que se generan de forma automatizada, sin verificación
(conlleva a altos índices de error)
Errores de lectura
Contaminación común por vectores de secuencia, intrones (de ARN no
empalmado), ARN ribosomal, ARN mitrocondrial entre otros.
A pesar de estas limitaciones, la tecnología EST es todavía ampliamente utilizada,
debido a que las bibliotecas de EST pueden ser generadas fácilmente de diferentes tipos de
células (tejidos, órganos). Además facilita la identificación exclusiva de un gen de una
biblioteca de ADNc. Aunque los EST son propensos a errores, una colección completa de EST
contiene información muy útil.
La rápida acumulación de secuencias de EST, ha impulsado el establecimiento de
bases de datos públicas y privadas para almacenar los datos. Genbank tiene una base de
datos EST, dbEST (http://www.ncbi.nlm.nih.gov/projects/dbEST/).
Uno de los objetivos de las bases de datos de EST es organizar y consolidar un gran
número redundante de datos para mejorar la calidad de la información.
Este proceso incluye el preprocesamiento de los datos el cual remueve vectores
contaminantes y enmascara repeticiones. Después sigue un proceso de agrupamiento o
clustering que agrupa secuencias EST asociadas a genes únicos.
El siguiente paso es obtener el consenso mediante la fusión de secuencias
redundantes, superposición de EST y errores de lectura.
Una vez que la secuencia de codificación es identificada puede ser anotada
traduciéndola a una secuencia de proteínas para búsqueda de similitudes en base de
datos.
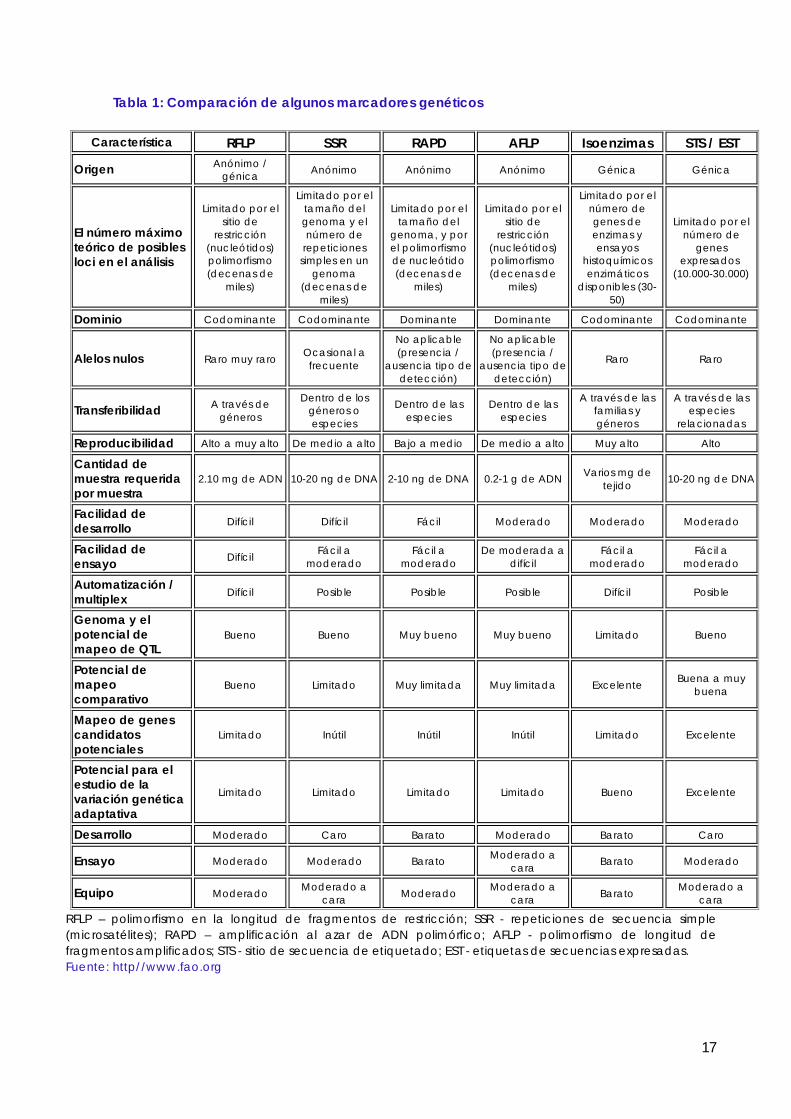
17
Tabla 1: Comparación de algunos marcadores genéticos
Característica RFLP SSR RAPD AFLP Isoenzimas STS / EST
Origen Anónimo / génica Anónimo Anónimo Anónimo Génica Génica
El número máximo teórico de posibles loci en el análisis
Limitado por el sitio de
restricción (nucleótidos) polimorfismo (decenas de
miles)
Limitado por el tamaño del genoma y el número de
repeticiones simples en un
genoma (decenas de
miles)
Limitado por el tamaño del
genoma, y por el polimorfismo de nucleótido (decenas de
miles)
Limitado por el sitio de
restricción (nucleótidos) polimorfismo (decenas de
miles)
Limitado por el número de genes de enzimas y ensayos
histoquímicos enzimáticos
disponibles (30-50)
Limitado por el número de
genes expresados
(10.000-30.000)
Dominio Codominante Codominante Dominante Dominante Codominante Codominante
Alelos nulos Raro muy raro Ocasional a frecuente
No aplicable (presencia /
ausencia tipo de detección)
No aplicable (presencia /
ausencia tipo de detección)
Raro Raro
Transferibilidad A través de géneros
Dentro de los géneros o especies
Dentro de las especies
Dentro de las especies
A través de las familias y géneros
A través de las especies
relacionadas
Reproducibilidad Alto a muy alto De medio a alto Bajo a medio De medio a alto Muy alto Alto
Cantidad de muestra requerida por muestra
2.10 mg de ADN 10-20 ng de DNA 2-10 ng de DNA 0.2-1 g de ADN Varios mg de tejido 10-20 ng de DNA
Facilidad de desarrollo Difícil Difícil Fácil Moderado Moderado Moderado
Facilidad de ensayo Difícil Fácil a
moderado Fácil a
moderado De moderada a
difícil Fácil a
moderado Fácil a
moderado
Automatización / multiplex Difícil Posible Posible Posible Difícil Posible
Genoma y el potencial de mapeo de QTL
Bueno Bueno Muy bueno Muy bueno Limitado Bueno
Potencial de mapeo comparativo
Bueno Limitado Muy limitada Muy limitada Excelente Buena a muy buena
Mapeo de genes candidatos potenciales
Limitado Inútil Inútil Inútil Limitado Excelente
Potencial para el estudio de la variación genética adaptativa
Limitado Limitado Limitado Limitado Bueno Excelente
Desarrollo Moderado Caro Barato Moderado Barato Caro
Ensayo Moderado Moderado Barato Moderado a cara Barato Moderado
Equipo Moderado Moderado a cara Moderado Moderado a
cara Barato Moderado a cara
RFLP – polimorfismo en la longitud de fragmentos de restricción; SSR - repeticiones de secuencia simple (microsatélites); RAPD – amplificación al azar de ADN polimórfico; AFLP - polimorfismo de longitud de fragmentos amplificados; STS - sitio de secuencia de etiquetado; EST - etiquetas de secuencias expresadas. Fuente: http//www.fao.org

9
3. Ventajas y desventajas de la PCR como método de clonación
3.1 Principales ventajas de la reacción I. Rapidez y sencillez de uso. La PCR permite clonar ADN en pocas horas, utilizando equipos relativamente poco sofisticados. Una reacción de PCR típica consiste en 30 ciclos de desnaturalización, síntesis y reasociación. Cada ciclo dura típicamente de 3 a 5 minutos y se utiliza un termociclador que lleva un microprocesador para programar los cambios de temperaturas y el número de ciclos deseado. Esto supera ampliamente el tiempo requerido para la clonación en células, que suele ser de semanas, o incluso meses. Por supuesto, el diseño y síntesis de los oligonucleótidos cebadores también lleva tiempo, pero este proceso ha sido simplificado gracias a la aparición de programas informáticos para el diseño de los cebadores, y a la proliferación de casas comerciales especializadas en la síntesis de oligonucleótidos por encargo. Una vez que se pone a punto, la reacción puede ser repetida de forma sencilla.
II. Sensibilidad. La PCR puede amplificar secuencias a partir de cantidades ínfimas de ADN diana, incluso a partir de ADN contenido en una sola célula. Esta elevada sensibilidad ha permitido el desarrollo de nuevos métodos para el estudio de la patogénesis molecular y la aparición de numerosas aplicaciones (ciencia forense, diagnóstico, estudios de paleontología molecular, etc) donde las muestras pueden contener muy pocas células. Sin embargo, el hecho de que el método tenga una sensibilidad tan elevada significa también que se deben extremar las precauciones para evitar la contaminación de la muestra con ADN extraño.
III. Robustez. La PCR permite la amplificación de secuencias específicas de material que contiene ADN muy degradado, o incluido en un medio que hace problemática su purificación convencional. Esto hace que el método resulte muy adecuado para estudios de antropología y paleontología molecular, por ejemplo para el análisis de ADN recuperado de individuos momificados y para intentar identificar ADN de muestras fósiles que contienen poquísimas células de criaturas extintas hace ya mucho tiempo. El método se ha empleado con éxito también para la amplificación de ADN de muestras de tejidos fijadas con formol, lo cual ha tenido importantes aplicaciones en patología molecular.

10
3.2 Principales desventajas de la reacción I. Necesidad de disponer de información sobre la secuencia del ADN diana Para poder construir oligonucleótidos específicos que actúen como cebadores para la amplificación selectiva de una secuencia particular de ADN se necesita disponer de alguna información previa sobre la propia secuencia a amplificar. Esto implica, por regla general, que la región de interés ya haya sido parcialmente caracterizada, a menudo mediante la aplicación de métodos de clonación basados en sistemas celulares. Sin embargo, y para casos concretos, se han desarrollado varias técnicas que reducen o incluso hacen desaparecer esta necesidad de disponer de información previa sobre la secuencia del ADN diana.
II. Tamaño corto de los productos de la PCR Una desventaja clara de la PCR como método de clonación de ADN ha sido el tamaño de las secuencias de ADN que permite clonar. A diferencia de la clonación de ADN en células, donde pueden clonarse secuencias de hasta 2 Mb, la información de que se dispone sobre la mayor parte de secuencias clonadas por PCR sitúa el tamaño de los fragmentos clonados entre 0 y 5 Kb, tendiendo hacia el extremo inferior. Los fragmentos pequeños se amplifican muy fácilmente, pero conforme aumenta su tamaño se hace más difícil obtener una amplificación eficiente. En la actualidad sin embargo ya es posible amplificar secuencias por PCR de tamaños entre 20 y 40 Kb.
III. Infidelidad en la replicación del ADN La clonación del ADN en células pasa por la replicación del ADN in vivo, proceso asociado a una gran fidelidad de copiado debido a la existencia, en la célula, de mecanismos de lectura y corrección de errores. Sin embargo, cuando el ADN se replica in vitro la tasa de errores cometidos durante el copiado se dispara. Ya se ha comentado anteriormente que la Taq polimerasa utilizada en la reacción no tiene actividad exonucleásica.
IV. Peligro de contaminación La facilidad con que se amplifica el ADN exige evitar el peligro de contaminación inherente al poder multiplicador de la reacción. En un tubo en el que se ha realizado una reacción de PCR hay tal cantidad de ADN, que al salir caliente del termociclador y abrir este, el vapor alcanza el ambiente del laboratorio. "Si se pasa un papel de filtro por los pomos de las puertas, o la superficie del termociclador, se puede rescatar suficiente ADN como para obtener una señal".

11
4. Aplicaciones de la PCR
Construcción de bibliotecas de ADNc Una biblioteca de ADNc es el conjunto de clones obtenidos a partir de los ARNm de un tejido determinado de un organismo completo. El ADNc se sintetiza a partir de un mensajero y un cebador por acción de la transcriptasa inversa. Los clones de ADNc tienen una utilidad especial, ya que al proceder de los mensajeros correspondientes, carecen de intrones. Los procariotas carecen de intrones en sus mensajeros y por eso no son capaces de procesar los ARNm eucarióticos correctamente. Si lo que se pretende es expresar un gen eucariota en un sistema procariótico, se deberá clonar a partir de su ADNc y no de su secuencia genómica. La mayoría de los genes eucarióticos clonados han sido aislados a partir de bibliotecas de ADNc. La elaboración de una biblioteca de ADNc implica el aislamiento de la población total de mensajeros celulares y su transcripción revertida a ADN.
Figura 3
Modo de acción de la transcriptasa inversa en el proceso de construcción de una biblioteca de ADNc.
i) La transcriptasa inversa utiliza el ARNm como molde para sintetizar una hebra de ADN
complementaria utilizando como cebador una cola de oligo-dT
ii) La misma polimerasa (gracias a su actividad ARNasa) destruye la hebra de ARNm molde y forma
una especie de "gancho" dejando un extremo 3' OH libre. El gancho servirá de cebador a la ADN
polimerasa I para sintetizar la segunda cadena del ADNc y posteriormente se eliminará por la acción
de la nucleasa S1 que corta específicamente cadenas sencillas del ADN.
iii) El ADNc así obtenido será convenientemente tratado para ser incorporado a un vector y ser clonado
in vivo para obtener una biblioteca de ADNc.

13
4.2. Clonación de secuencias desconocidas de ADN que flanquean una región conocida. Una de las limitaciones con la que se encuentra la reacción de la PCR estándar es que amplifica secuencias de ADN comprendidas entre dos cebadores convergentes, es decir, cada uno de ellos inicia la síntesis del ADN en la dirección del otro (extremos 3' OH enfrentados). Muchas veces puede interesar conocer qué tipo de secuencias de ADN no caracterizadas ocupan la región adyacente externa a una región conocida (por ejemplo, conocer la región reguladora flanqueante de un gen de secuencia ya conocida). Una variante de la técnica estándar, la PCR inversa, lo permite. El método se basa en el empleo de cebadores divergentes, es decir, cebadores cuyos extremos 3' OH se dirijan hacia el exterior de la secuencia conocida. Con cebadores de este tipo, una reacción PCR estándar no funciona. En la PCR inversa, se trata el ADN con una enzima de restricción de forma que la región deseada quede comprendida en un pequeño fragmento de restricción, al que se permite la circularización por complementariedad de los extremos cohesivos. Una vez circularizado el fragmento, se procede a una reacción de PCR estándar utilizando los cebadores divergentes, que ahora sí permiten que se desarrolle la reacción.
Figura 4 Clonación de secuencias de ADN flanqueantes a partir de un molde circularizado. Las regiones X e Y representan las secuencias desconocidas que se desea clonar. Los cebadores se diseñan de forma que se unan a la secuencia de ADN caracterizada, pero con sus extremos 3' orientados en sentidos opuestos. Tras la recuperación de la secuencia caracterizada y de sus regiones flanqueantes en forma de un fragmento de restricción, se circulariza la molécula, de manera que las secuencias flanqueantes quedan en contacto y los extremos 3' de los cebadores quedan orientados para que pueda tener lugar ahora la PCR estándar.

14
4.3 Secuenciación del ADN y obtención de sondas para hibridaciones El método de secuenciación de ADN requiere que el ADN esté en forma de cadena simple antes de su utilización. Pese a que la desnaturalización del ADN de los productos de PCR puede proporcionar los sustratos monocatenarios necesarios para la secuenciación, la calidad de la secuencia de ADN aumenta si el producto de partida es monocatenario en origen. La PCR asimétrica es una variante de la PCR estándar que permite generar un gran número de moléculas de ADN de banda simple que se pueden utilizar directamente para secuenciar o como sondas para hibridaciones. El método consiste en poner proporciones diferentes de cebadores de manera que uno de los dos esté a concentración limitante. Así, hasta los primeros 20 a 25 ciclos se amplifican las dos hebras del ADN, pero al agotarse uno de los cebadores, los 5 a 10 ciclos siguientes serán de ADN de banda simple. La concentración de uno de los cebadores suele fijarse en 100 veces la concentración del otro; utilizando, por ejemplo, una relación de 50 picomoles de un cebador y 0,5 del otro, se obtiene después de 30 ciclos, de 1 a 3 picomoles de ADN de banda simple.
Figura 5 Esquema del procedimiento empleado en la PCR asimétrica para generar ADN de banda simple

18
4.4.2 Huella del ADN Las secuencias del genoma humano que se estudian para obtener la huella genética de un individuo corresponden a regiones altamente variables del genoma, caracterizadas normalmente por ser ADN no codificante, es decir sin información para la elaboración de proteínas o ARN. Estas regiones no codificantes tienen un margen de variación más amplio que las regiones codificantes ya que las mutaciones que han sufrido a lo largo de la evolución no han sido cribadas por la selección natural y por ello, constituyen regiones hipervariables del genoma. Las regiones hipervariables del genoma están formadas por distintas repeticiones de una secuencia unidad cuyo número de pares de bases puede ser pequeño, entre 2 y 7 pares de bases, son las secuencias microsatélite, o de mayor tamaño, entre 10 y 60 pares de bases llamadas en este caso secuencias minisatélite. Las unidades de repetición de cada una de estas regiones hipervariables se disponen una a continuación de la otra en el genoma o en tándem, siendo muy alto el número de veces que se repiten las secuencias unidad de los loci minisatélites y mucho menor el número de repeticiones de las secuencias de los loci microsatélites. El número de repeticiones consecutivas en una posición genómica en particular varia de individuo a individuo. Marcadores genéticos consistentes en la unidad repetitiva pueden ser utilizados como sondas radiactivas para detectar un gran número de variantes polimórficas, exclusivas y únicas de cada individuo, pero que a la vez presentan una gran estabilidad somática entre distintos tejidos, incluyendo los germinales. El patrón de restricción observado en un individuo al ser tratado su ADN con una enzima de restricción que no corte dentro de la unidad repetida, y posteriormente transferido el ADN a nailon e hibridado con una sonda radiactiva de un minisatélite, se denomina huella genética o huella de ADN (ADN "fingerprint") (porque arroja un perfil propio de cada individuo). La probabilidad estimada experimentalmente de que una sola banda de las múltiples que se observan en un individuo esté presente en otro individuo tomado al azar es aproximadamente de 0,2; de esto se deduce que es bajísima la probabilidad (5x10-19) de encontrar otro individuo sin parentesco directo con el primero y con una huella genética idéntica. La huella genética es completamente específica del individuo. No hay diferencia en los patrones de restricción de ADN obtenida de distintos fluidos corporales, sangre o semen de un mismo individuo. Tampoco la hay entre gemelos univitelinos. Cuando se preparan y analizan de forma rigurosa, las huellas de ADN pueden proporcionar identificaciones fiables de los individuos. Una pequeña cantidad de sangre, de saliva o de semen que puedan ser encontrados en las ropas de una víctima o de un sospechoso de haber cometido un crimen dan suficiente ADN como para aplicar esta metodología. Las aplicaciones de la técnica de análisis de huellas en el ADN son muy numerosas, cabe destacar las siguientes:
- Diagnóstico de paternidad biológica y parentesco biológico - Identificación de vestigios biológicos tales como sangre, semen, saliva, raíces de
cabello, tejidos, piezas dentales, etc., en procedimientos penales (por ejemplo en la identificación de individuos sospechosos de delitos), así como identificación de individuos post-mortem.

21
Sondas unilocus Además de las sondas que reconocen muchas regiones hipervariables dispersas por el genoma (multiloci), existen otras que detectan un solo lugar en el genoma, igualmente hipervariable (unilocus). Con las sondas unilocus hay solamente dos bandas de distinto tamaño por individuo (alelo paterno y materno). Una de las ventajas que ofrece la utilización de sondas unilocus es que dan lugar a patrones de ADN con menos bandas y por tanto más fáciles de interpretar que los que proporcionan las sondas multiloci. El análisis con sondas unilocus requiere además menos ADN. Al marcar un único locus se renuncia al poder de identificación individual de la huella de ADN. El problema queda resuelto utilizando varios marcadores diferentes (de 5 a 10, por ejemplo) originando así un número no muy elevado de bandas específicas que también resulta fácil de interpretar y que dan mayor solidez al diagnóstico.
a) b)
Figura 9 a) Análisis de la movilidad de los fragmentos de restricción originados en una región unilocus para
identificar los alelos paternos (P) y maternos (M) de los cuatro hijos de un matrimonio. Una muestra del ADN de los implicados se trata con una enzima de restricción que no corte dentro de la región hipervariable, se corren las muestras en un gel de agarosa, se desnaturaliza el ADN y se transfiere a nailon. La identificación se hace por hibridación con la sonda unilocus marcada radiactivamente. Los alelos paterno y materno pueden ser identificados en todos los hijos excepto en el primero de ellos, en el que aparece un nuevo alelo.
b) Determinación de la paternidad entre dos posibles padres, P1 y P2, de un niño (N) empleando una mezcla de cinco sondas unilocus. El padre P2 muestra total identidad con la mitad de las bandas observadas en el niño.

22
Amplificación de minisatélites y microsatélites por PCR El análisis de regiones hipervariables del genoma utilizando sondas multiloci o unilocus requiere elevadas cantidades de ADN que además debe de haber estado conservado en condiciones óptimas. Alec Jeffreys, en los años noventa propuso combinar el método de la PCR con la obtención de la huella genética del individuo: amplificación de minisatélites por PCR (Minisatellite Variant Repeat-Polymerase Chain Reaction, MVR-PCR). El procedimiento a seguir en el laboratorio consiste en amplificar una secuencia de minisatélite mediante PCR y generar posteriormente un perfil de ADN. El perfil de ADN basado en la amplificación tiene la ventaja de ser más sensible que el proporcionado por las técnicas de Souther e hibridación. Permite la identificación de individuos por huella genética incluso en aquellos casos en que el material genético de que se dispone es muy escaso (por ejemplo, cuando el resto biológico es una sola raíz de cabello, o la saliva depositada en la boquilla de un solo cigarrillo) o bien cuando el resto biológico proporciona ADN muy degradado, tal como sucede en manchas de sangre sometidas a la radiación solar o a temperaturas muy altas, o se trata de restos biológicos post mortem. La amplificación de microsatélites (STR) es otro sistema utilizado por criminalistas y está adquiriendo cada vez más importancia en ciencia forense. A causa del pequeño tamaño de los framentos, los sistemas STR se hacen muy apropiados para el análisis de especímenes viejos o mal conservados.
Figura 10 Alec Jeffreys ha amplificado por PCR distintos minisatélites. Una vez amplificados, los minisatélites se caracterizan con el método de Souther. Mediante PCR se pueden amplificar restos de ADN degradado y obtener información a partir de poca cantidad de ADN. La fotografía muestra una membrana con hibridación de varios






Extracción y Purificación de ADN 4
Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos Sesión nº 4
A continuación se recogen los principios de algunos de los métodos de extracción y
purificación de ácidos nucleicos que más se emplean en la actualidad.
Métodos de extracción
Para extraer los ácidos nucleicos del material biológico es preciso provocar una lisis
celular, inactivar las nucleasas celulares y separar los ácidos nucleicos de los restos
de células. El procedimiento de lisis idóneo suele consistir en un equilibrio de
técnicas y ha de ser suficientemente fuerte para romper el material inicial complejo
(un tejido, por ejemplo), pero suficientemente suave para preservar el ácido nucleico
diana. Entre los procedimientos usuales de lisis figuran los siguientes:
• rotura mecánica (trituración, lisis hipotónica, etc.);
• tratamiento químico (detergentes, agentes caotrópicos, reducción con tioles,
etc.);
• digestión enzimática (Proteinasa K, etc.).
Es posible romper la membrana celular e inactivar las nucleasas intracelulares al
mismo tiempo. Por ejemplo, una misma solución puede contener detergentes que
solubilicen las membranas celulares y sales caotrópicas fuertes que inactiven las
enzimas intracelulares. Tras la lisis celular y la inactivación de las nucleasas, los
restos de células se eliminan fácilmente por filtración o precipitación.
Métodos de purificación
Los métodos empleados para purificar los ácidos nucleicos de extractos celulares
suelen ser combinaciones de dos o más técnicas de las siguientes:
• extracción/precipitación;
• cromatografía;
• centrifugación;
• separación por afinidad.
En los párrafos siguientes se describen brevemente estas técnicas (Zimmermann et
al., 1998).
Extracción/precipitación
A menudo se efectúa una extracción con disolventes para eliminar los
contaminantes de los ácidos nucleicos. Por ejemplo, suele emplearse una
combinación de fenol y cloroformo con objeto de eliminar las proteínas. Para
concentrar los ácidos nucleicos suele realizarse una precipitación con isopropanol o
etanol. Si la cantidad de ácido nucleico diana es escasa, puede añadirse a la mezcla

Extracción y Purificación de ADN 5
Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos Sesión nº 4
un portador inerte (como el glucógeno) para favorecer la precipitación. También
puede realizarse una precipitación selectiva con concentraciones salinas elevadas
(precipitación salina) o una precipitación de las proteínas mediante cambios del pH.
Cromatografía
Pueden emplearse diversas técnicas cromatográficas de separación: permeación
sobre gel, intercambio iónico, adsorción selectiva o unión por afinidad. La
permeación sobre gel aprovecha las propiedades de las partículas porosas del gel
para tamizar moléculas. Se emplea una matriz con poros de tamaño definido, que
dejan pasar las moléculas más pequeñas por difusión, pero no las más grandes, que
quedan eluidas en el volumen vacío. Así pues, las moléculas quedan eluidas por
orden de tamaño decreciente. La cromatográfica de intercambio iónico es otra
técnica, que se basa en la interacción electrostática de la molécula diana con un
grupo funcional de la matriz en columna. Los ácidos nucleicos (polianiones lineales
con fuerte carga negativa) pueden eluirse de las columnas de intercambio iónico con
simples Tampones salinos. En la cromatografía de adsorción, los ácidos nucleicos
se fijan selectivamente en sílice o vidrio, en presencia de determinadas sales (por
ejemplo, sales caotrópicas), mientras que otras moléculas biológicas no se fijan. A
continuación, los ácidos nucleicos pueden eluirse con agua o un tampón hiposalino y
se obtiene una muestra que puede emplearse directamente en las aplicaciones
posteriores.
Centrifugación
La centrifugación selectiva constituye un método de purificación eficaz. Así, por
ejemplo, la ultracentrifugación en gradientes autogenerados de CsCl con fuerzas g
elevadas se lleva empleando mucho tiempo para purificar plásmidos. La
centrifugación suele asociarse a otros métodos, como la cromatografía de columna
centrifugada, en cuyo caso se combina la permeación sobre gel y la centrifugación
para separar el ADN o ARN de contaminantes más pequeños (sales, nucleótidos,
etc.), intercambiar tampones o seleccionar según el tamaño. Algunos procedimientos
combinan la adsorción selectiva en matriz cromatográfica (véase el apartado anterior
«Cromatografía») y la elución por centrifugación para purificar de forma selectiva un
tipo de ácido nucleico.
Separación por afinidad
En los últimos años, cada vez son más los métodos de purificación que combinan la
inmovilización por afinidad de ácidos nucleicos y la separación magnética. Por
ejemplo, los ARNm poli A + pueden unirse a partículas magnéticas revestidas de

Extracción y Purificación de ADN 6
Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos Sesión nº 4
estreptavidina mediante oligo dT marcados con biotina, y el complejo de partículas
puede eliminarse de la solución (y de los contaminantes libres) con un imán. Esta
técnica en fase sólida simplifica la purificación de los ácidos nucleicos, pues permite
sustituir las etapas de centrifugación, extracción orgánica y separación de fases por
una operación de separación magnética, única y rápida.
Método de extracción y purificación con CTAB
El protocolo del método del bromuro de cetil-trimetil amonio (CTAB) fue elaborado
por Murray y Thompson en 1980 (Murray y Thompson, 1980) y publicado
posteriormente, en 1987, por Wagner y sus colaboradores (Wagner et al., 1987). El
método es adecuado para extraer y purificar ADN de vegetales y alimentos
derivados de vegetales y está especialmente indicado para eliminar los polisacáridos
y los compuestos polifenólicos, que, de otro modo, alterarían la pureza del ADN y,
por tanto, su calidad. Este procedimiento se ha aplicado con frecuencia en genética
molecular de los vegetales y ha sido probado en ensayos de validación para detectar
OGM (Lipp et al., 1999; 2001). Se han elaborado algunas variantes adicionales para
adaptar el método a una amplia gama de matrices de alimentos transformados o sin
transformar (Hupfer et al., 1998; Hotzel et al., 1999; Meyer et al., 1997; Poms et al.,
2001).
Principios del método del CTAB: lisis, extracción y precipitación
Las células vegetales pueden lisarse con el detergente iónico bromuro de cetil-
trimetil amonio (CTAB), que forma un complejo insoluble con los ácidos nucleicos en
medio hiposalino. De ese modo, los polisacáridos, los compuestos fenólicos y los
demás contaminantes permanecen en el sobrenadante y pueden eliminarse por
lavado. El complejo de ADN se solubiliza aumentando la concentración salina y se
precipita con etanol o isopropanol. En esta sección se describirán los principios de
las tres etapas principales: la lisis de la membrana celular, la extracción del ADN
genómico y su precipitación.
Lisis de la membrana celular: Tal como se ha mencionado anteriormente, la
primera etapa de la extracción del ADN es la rotura de la membrana celular y
nuclear, para lo cual la muestra homogeneizada se trata primero con el tampón de
extracción, que contiene EDTA, Tris-HCl y CTAB. Todas las membranas biológicas
presentan la misma estructura general, integrada por moléculas de lípidos y de
proteínas, unidas por interacciones no covalentes.

Extracción y Purificación de ADN 7
Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos Sesión nº 4
Figura 1. Representación simplificada de las membranas celulares1.
Tal como muestra la figura 1, las moléculas lipídicas están ordenadas en una doble
capa continua, en la que las moléculas proteínicas están «disueltas». Las moléculas
lipídicas están formadas por extremos hidrófilos, denominados «cabezas», y
extremos hidrófobos, denominados «colas». En este método, el detergente (CTAB)
contenido en el tampón de extracción provoca la lisis de la membrana. Dado que la
composición de los lípidos y del detergente es similar, el componente de CTAB del
tampón de extracción captura los lípidos que integran la membrana celular y nuclear.
La figura 2 ilustra el mecanismo de solubilización de los lípidos con un detergente.
Figura 2. Solubilización de los lípidos.
1 Las ilustraciones que figuran en esta página y en las siguientes proceden del Genetic Science Learning Center, Universidad de Utah, http://gslc.genetics.utah.edu.
ADN
Membrana nuclear

Extracción y Purificación de ADN 8
Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos Sesión nº 4
La figura 3 ilustra cómo se libera el ADN genómico cuando el detergente captura los
lípidos y las proteínas al entrar en contacto la membrana celular y el tampón de
extracción con CTAB. Con una concentración salina (NaCl) determinada, el
detergente forma un complejo insoluble con los ácidos nucleicos. El EDTA es un
componente quelante, que se une al magnesio, entre otros metales. El magnesio es
un cofactor de la desoxirribonucleasa. Al unir el magnesio al EDTA, la actividad de la
desoxirribonucleasa presente disminuye. El Tris-HCl confiere a la solución la
capacidad de amortiguar el pH (un pH inferior o superior daña el ADN). Es
importante señalar que, como los ácidos nucleicos se degradan fácilmente en esta
fase de la purificación, debe reducirse al mínimo el tiempo transcurrido entre la
homogeneización de la muestra y la adición de la solución tampón con CTAB. Una
vez que se han roto las membranas de la célula y de los orgánulos (como los que se
encuentran alrededor de las mitocondrias y los cloroplastos), se purifica el ADN.
Figura 3. Rotura de la membrana celular y extracción del ADN genómico.
Extracción - En esta etapa, se separan los complejos formados por los ácidos
nucleicos y el CTAB de los polisacáridos, los compuestos fenólicos, las proteínas y
los demás lisados celulares disueltos en la solución acuosa. Es especialmente
importante eliminar los polisacáridos y los compuestos fenólicos, pues pueden inhibir
numerosas reacciones enzimáticas. En concentraciones hiposalinas (NaCl < 0,5 M),
los contaminantes de los complejos de ácidos nucleicos no precipitan y pueden
eliminarse extrayendo la solución acuosa con cloroformo. El cloroformo
desnaturaliza las proteínas y facilita la separación de las fases acuosa y orgánica. La
fase acuosa suele constituir la fase superior. No obstante, si dicha fase es densa
debido a la concentración salina (> 0,5 M), formará la fase inferior. Además, si el pH
de la solución acuosa no se ha equilibrado debidamente (pH 7,8 – 8,0), los ácidos
nucleicos tenderán a repartirse en la fase orgánica. Si es preciso, la extracción con

Extracción y Purificación de ADN 9
Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos Sesión nº 4
cloroformo se realizará dos o tres veces, con objeto de eliminar por completo las
impurezas de la capa acuosa. Para perfeccionar la extracción de los ácidos
nucleicos, puede retroextraerse la fase orgánica con una solución acuosa, que se
añade a continuación al extracto anterior. Una vez purificados los complejos de
ácidos nucleicos, puede procederse a la precipitación, última etapa del
procedimiento.
Precipitación - En esta última etapa se separan los ácidos nucleicos del detergente,
para lo cual la solución acuosa se trata primero con una solución de precipitación
compuesta por una mezcla de CTAB y NaCl en concentración elevada (NaCl >
0,8 M). Una alta concentración salina es necesaria para que se forme un precipitado
de ácidos nucleicos. Puede ser preferible emplear acetato de sodio en lugar de NaCl
por su capacidad tampón. En estas condiciones, el detergente, que es más soluble
en alcohol que en agua, puede eliminarse por lavado, mientras que los ácidos
nucleicos precipitan. El posterior tratamiento con etanol al 70 % permite una mayor
purificación o elución de los ácidos nucleicos de la sal residual.
Cuantificación del ADN mediante espectrofotometría
El ADN, el ARN, los oligonucleótidos e incluso los mononucleótidos pueden
cuantificarse directamente en soluciones acuosas, en forma diluida o sin diluir,
midiendo la absorbancia A (o densidad óptica, DO) de luz ultravioleta (también
puede hacerse en el intervalo visible). Si la muestra es pura, es decir, no contiene
cantidades significativas de contaminantes como proteínas, fenol o agarosa, la
medición espectrofotométrica de la irradiación ultravioleta absorbida por las bases es
sencilla y exacta. En este método, los tampones acuosos con escasa concentración
iónica (por ejemplo, tampón TE) resultan idóneos. La concentración de ácidos
nucleicos suele determinarse midiendo a 260 nm y comparando con un blanco. La
interferencia de contaminantes puede determinarse calculando un «cociente». Dado
que las proteínas absorben a 280 nm, se emplea el cociente A260/A280 para calcular
la pureza de los ácidos nucleicos. Los cocientes respectivos del ADN y el ARN puros
son aproximadamente de 1,8 y 2,0. Una absorción a 230 nm significa que la muestra
está contaminada con hidratos de carbono, péptidos, fenoles, compuestos
aromáticos u otras sustancias. El cociente A260/A230 de las muestras puras es de 2,2
aproximadamente.
Cuando la cantidad disponible de ácidos nucleicos es escasa, puede emplearse el
método de la placa de agarosa con bromuro de etidio. Permite calcular la cantidad
de ácidos nucleicos a partir de la intensidad de la fluorescencia emitida por el

Extracción y Purificación de ADN 10
Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos Sesión nº 4
bromuro de etidio irradiado con luz UV, comparándola con unos patrones de
concentración.
Principios de la cuantificación de ADN por espectrofotometría
El espectrofotómetro se funda en la transmisión de la luz a través de una solución
para determinar la concentración de un soluto presente en la misma. El aparato
funciona conforme a un principio sencillo: se irradia una muestra con una radiación
luminosa de longitud de onda conocida y se mide la energía luminosa transmitida
con una célula fotoeléctrica situada detrás de la muestra.
Tal como muestra la figura 4, el espectrofotómetro de haz único consta de una
fuente luminosa, un prisma, un recipiente con la muestra y una célula fotoeléctrica.
Los distintos elementos están conectados a los sistemas eléctricos o mecánicos
adecuados para controlar la intensidad luminosa y la longitud de onda y para
convertir en variaciones de tensión la energía recibida en la célula fotoeléctrica. Las
variaciones de tensión se miden en un contador o se registran en un ordenador para
su posterior análisis.
Fig Figura 4. Representación esquemática de la transmisión de la luz.
Cada molécula absorbe la energía radiante a una longitud de onda específica, a
partir de la cual es posible extrapolar la concentración de un soluto en una solución.
Con arreglo a la ley de Lambert-Beer, existe una relación lineal entre la absorbancia
A (también denominada densidad óptica, DO) y la concentración de la
macromolécula, conforme a la ecuación siguiente:
A = DO = εlc (1)
donde ε es el coeficiente de extinción molar, c es la concentración y l es el paso de
luz de la cubeta. Las proteínas y los ácidos nucleicos absorben la luz en el intervalo

Extracción y Purificación de ADN 11
Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos Sesión nº 4
ultravioleta, a longitudes de onda comprendidas entre los 210 y los 300 nm. Tal
como se ha señalado anteriormente, la absorbancia máxima de las soluciones de
ADN y ARN corresponde a 260 nm y la de las soluciones de proteínas, a 280 nm.
Dado que las soluciones de ADN y ARN absorben parcialmente la luz a 280 nm y las
que contienen proteínas hacen lo propio a 260 nm, el cociente de los valores
obtenidos a 260 nm y a 280 nm (A260/A280) proporciona una estimación del grado de
pureza de los ácidos nucleicos. Los cocientes A260/A280 respectivos del ADN y el
ARN puros son aproximadamente de 1,8 y 2,0. Con un paso de luz de 10 mm y una
longitud de onda de 260 nm, una absorbancia A = 1 corresponde aproximadamente
a 50 µg/ml de ADN bicatenario, 37 µg/ml de ADN monocatenario, 40 µg/ml de ARN o
30 µg/ml de oligonucleótidos. Si la muestra también contiene proteínas, el cociente
A260/A280 será considerablemente inferior a dichos valores y no podrá determinarse
con exactitud la cantidad de ácidos nucleicos. Debe precisarse que la
espectrofotometría no permite identificar de forma fiable impurezas de ARN
presentes en las soluciones de ADN. Puede emplearse la absorbancia a 325 nm
para poner de manifiesto la presencia de restos en la solución o la suciedad de la
cubeta.
Determinación de la concentración de ácidos nucleicos
Elección de la cubeta. La cantidad de solución de ácidos nucleicos necesaria para
medir la absorbancia A depende de la capacidad de la cubeta. Debe elegirse una
cubeta apropiada atendiendo al intervalo de concentración de la muestra, el factor
de dilución y el volumen de la muestra. En la mayor parte de los procedimientos
empleados para detectar OGM, el volumen de ADN genómico disponible varía entre
50 y 100 µl. Para la cuantificación espectroscópica de pequeños volúmenes de
ácidos nucleicos se emplean diversos tipos de cubetas de microvolumen, cuya
capacidad oscila entre 5 y 70 µl.
Preparación. Para calibrar el espectrofotómetro es importante:
• establecer el paso de luz correcto;
• establecer el factor correcto (ADN bicatenario, ADN monocatenario o ARN);
• medir la solución en blanco (establecer la referencia), que será agua o solución
tampón (A260 = 0);
• cerciorarse de que la referencia establecida se renueva periódicamente;
• medir una cantidad conocida de ácidos nucleicos puros para comprobar la
fiabilidad de la referencia establecida.

Extracción y Purificación de ADN 12
Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos Sesión nº 4
Medición en una muestra desconocida. Para medir la concentración, se utilizan
cantidades determinadas de solución de ADN en función de la capacidad de la
cubeta empleada (por ejemplo, 5 µl de ADN diluidos en 195 µl de agua si la
capacidad de la cubeta es inferior a 0,2 ml). Después de calibrar el
espectrofotómetro y de añadir la solución de ácidos nucleicos, se tapa la cubeta, se
mezcla la solución y se mide la absorbancia. Con objeto de reducir los errores de
pipeteado, debe efectuarse la medición al menos dos veces y siempre con 5 µl de
solución de ADN, como mínimo. Se desaconsejan los valores de A260 inferiores a
0,02 o comprendidos entre 1 y 1,5 (según el instrumental empleado) por el elevado
margen de error que entrañan.
La concentración c de un ácido nucleico determinado presente en una solución se
calcula conforme a las ecuaciones siguientes:
• ADN monocatenario: c(pmol/µl) = A260/0,027
• ADN bicatenario: c(pmol/µl) = A260/0,020
• ARN monocatenario: c(pmol/µl) = A260/0,025
• Oligonucleótido: c(pmol/µl) = A260100/1,5NA+0,71NC+1,20NG + 0,84NT
donde A260 es la absorbancia medida a 260 nm.
El cuadro 2 recoge un ejemplo de los valores de la absorbancia de ADN de timo de
ternera muy purificado, en suspensión en un tampón TNE 1x, considerando que el
ADN de referencia es ADN bicatenario, con una A260 = 1 a 50 µg/ml, en una cubeta
cuyo paso de luz es de 10 mm. La concentración nominal de ADN era de 25 µg/ml.
Cuadro 2. Valores de la absorbancia de ADN de timo de ternera muy purificado, en
tampón TNE 1x.
Longitud de onda
Absorbancia A260/A280 Conc. (µg/ml)
325 0,01 - - 280 0,28 - - 260 0,56 2,0 28 230 0,30 - -

Electroforesis de Proteínas Las proteínas son moléculas cuya carga neta depende del contenido de una serie de aminoácidos (fundamentalmente ácido glutámico, ácido aspártico, lisina, arginina e histidina) y del grado de ionización de éstos al pH considerado (figura 7).
Fig. 7. Principales aminoácidos responsables de la carga neta de una proteína, dependiendo del pH.
En los soportes no restrictivos (en los que el entramado no interfiere en la migración, ya que el tamaño de las proteínas es muy pequeño en comparación con el tamaño de poro del soporte), la separación depende de la densidad de carga de las moléculas y, así, cuanto mayor sea la densidad, mayor será la velocidad de migración en un campo eléctrico hacia el polo que determine su carga neta.
La primera etapa del proceso es la aplicación de la muestra. En papel o acetato de celulosa, esto se puede efectuar de forma puntual (permite el análisis de varias muestras simultáneamente en pequeñas cantidades) o longitudinalmente (permite el estudio de una sola muestra pero en mayor cantidad). La muestra se aplica disuelta en el tampón de electroforesis, del que está impregnado el soporte y que se encuentra en los reservorios de la cubeta, y se deposita en una pequeña zona, lo más estrecha posible, en el centro o en un extremo del soporte (si se conoce cuál va a ser la dirección de desplazamiento de la misma) y de forma perpendicular a la dirección del campo eléctrico. Conviene evitar la proximidad de los bordes del papel, ya que allí el campo eléctrico no es homogéneo y se distorsionan las bandas según avanzan. El volumen que se aplica suele ser inferior a 10 µL y si se necesitan mayores volúmenes porque la muestra se encuentre muy diluida, pueden hacerse aplicaciones sucesivas sobre la misma zona, dejando secar entre aplicación y aplicación. Es necesario humedecer el soporte (papel o acetato de celulosa) para que sea conductor; para ello, se impregna el soporte de tampón de electroforesis por ambos extremos y de forma simultánea para que por capilaridad se desplace hacia la zona de aplicación de la muestra.
En los geles de agarosa, se perfora el gel con ayuda de un troquel o una pipeta de diámetro adecuado y se elimina esa porción por succión. La perforación puede ser cilíndrica o rectangular, dependiendo del número y cantidad de muestra a analizar y la muestra se deposita en el orificio practicado sin que llegue a rebosar. En este caso, el tampón está embebido en el soporte (agarosa).
En todos los casos, la zona de aplicación debe ser lo más estrecha posible, para aumentar la resolución y evitar solapamientos entre bandas que migren en posiciones cercanas.
La electroforesis termina cuando se haya producido la máxima separación de los componentes de la muestra, pero sin sobrepasar los límites del soporte. Para evitar que se sobrepasen estos límites, se utilizan los marcadores electroforéticos que son moléculas coloreadas con una movilidad electroforética superior a cualquier componente de la muestra (poseen elevada carga respecto a su tamaño). Entre los marcadores más utilizados se encuentran la dinitrofenil-lisina (DNP-Lys) y el azul de bromofenol.
Una vez acabada la electroforesis, se procede a la etapa de tinción. El soporte se retira de la cubeta y se sumerge en un recipiente que contiene una disolución de un colorante que se une de manera específica a los componentes de la muestra y que precipita a los componentes separados en la posición en la que se encuentren. Las disoluciones más utilizadas en el caso de proteínas son el Negro Amido al 1% (p/v) en ácido acético y el Azul de Coomasie al 0.1% (p/v) en metanol/agua/ácido acético (9:9:2 v/v/v).
Si la electroforesis se pretende desarrollar a escala preparativa, ha de utilizarse un papel de mayor grosor (175 gr/cm2), en lugar del utilizado en la electroforesis analítica (85-100 gr/cm2; 0.15mm de espesor), lo que permite la aplicación de una mayor cantidad de muestra (50-100µL).
Inmunoelectroforesis. Es una combinación de una electroforesis en geles de agarosa seguida de una inmunodifusión (figura 8). En la primera parte, se realiza una aplicación puntual de la muestra, por lo que al final las distintas proteínas estarán distribuidas a lo largo del gel según el eje de migración. Acabada la electroforesis, y sin efectuar la tinción, se practica en el gel un canal (“trinchera”) paralelo a la dirección de migración con un bisturí de doble hoja (la separación de las hojas determina la anchura de la trinchera) en el que se deposita un antisuero que contenga anticuerpos específicos frente a una o varias de las proteínas separadas en la electroforesis.
Los geles se mantienen así a temperatura ambiente (20-22°C) durante 24-48 horas. Los anticuerpos y las proteínas (que actúan como antígenos) difunden, y si se encuentran en la proporción adecuada, producen complejos antígeno-anticuerpo insolubles que precipitan y aparecen en el gel como bandas elipsoidales.
Estos complejos solamente se producen cuando ambos compuestos se hallan en lo que se conoce como relación de equivalencia y que no son idénticas para todas las proteínas presentes en la muestra; por eso, deben determinarse en experimentos preliminares las cantidades idóneas de antisuero y de la muestra, así como las distancias entre el pocillo de aplicación de la muestra y la trinchera, ya que si las distancias son muy pequeñas, el antígeno (las proteínas) difunde rápidamente y puede no formarse el precipitado, y si la distancia es mayor de la idónea, hay que emplear mayor cantidad de antisuero y la duración del proceso se alarga.
Una vez formadas las bandas de precipitación, el gel puede lavarse para eliminar las proteínas que no hayan inmunoprecipitado y, posteriormente, teñirse como se ha descrito anteriormente. Cada proteína de la muestra produce una banda de precipitación independiente, por lo que es posible identificar el número de constituyentes de la muestra que son reconocidos por los anticuerpos.
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 1 de 17

La gran especificidad y sensibilidad de las reacciones de precipitación, permite diferenciar substancias con movilidades electroforéticas idénticas y detectar componentes que se encuentren en concentraciones mínimas (µg).
Fig. 8. Fases de una inmunoelectroforesis (IEF).
El número de bandas observado en una IEF debe entenderse como el número mínimo de componentes presentes, es decir, puede haber más componentes que no sean detectados por el antisuero empleado o que no hayan alcanzado la relación de equivalencia antígeno-anticuerpo adecuada.
La IEF es una técnica principalmente cualitativa, que se desarrolla en condiciones nativas y es especialmente apropiada para el análisis de líquidos fisiológicos y extractos celulares. Permite identificar substancias en mezclas muy complejas y en concentraciones muy bajas. Sin embargo, requiere que dichas substancias sean inmunogénicas y depende de los anticuerpos utilizados, por lo que es difícil de estandarizar.
Electroforesis en Geles de Poliacrilamida (PAGE). Se utiliza mayoritariamente para la separación de proteínas, aunque también puede ser útil para ácidos nucleicos. Los geles de poliacrilamida son soportes restrictivos del tipo II, por lo que, además de evitar la convección y minimizar la difusión, participan directamente en el proceso de separación.
Se preparan de modo que sus poros sean de un tamaño comparable al de las proteínas, de manera que produzcan un efecto de tamizado molecular; la separación electroforética depende entonces de la densidad de carga de las moléculas y de su tamaño, por lo que dos proteínas con idéntica densidad de carga, pero de tamaño diferente pueden ser separadas, ya que el soporte dificulta más el avance de la de mayor tamaño.
Son químicamente inertes frente a las moléculas biológicas, transparentes y estables en un amplio intervalo de valores de pH, temperatura y fuerza iónica; son también resistentes a agentes desnaturalizantes (urea, detergentes), no presentan efecto EEO y son mecánicamente estables, por lo que pueden ser deshidratados y reducidos a una fina película, lo que facilita su almacenamiento.
• Preparación del soporte. El soporte se prepara a partir del monómero de acrilamida (figura 9) que forma largas cadenas lineales, con abundantes grupos polares que las hace solubles en medios acuosos.
Al no estar las cadenas unidas entre sí, formarían un gel sin consistencia mecánica y totalmente inmanejable. Para evitar esto, se utiliza otro monómero, la N,N’-metilen-bisacrilamida que forma parte de dos cadenas que quedan así unidas covalentemente.
Fig. 9. Estructura de la acrilamida, bisacrilamida y poliacrilamida
En función de la concentración de acrilamida se obtienen entramados más o menos densos, mientras que la cantidad de bisacrilamida condiciona el grado de entrecruzamiento de las cadenas. Ambos componentes determinan, en conjunto, las características físicas del gel resultante: su resistencia mecánica, fragilidad, elasticidad, grado de reticulación (tamaño de poro), etc.
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 2 de 17

La polimerización de la acrilamida se obtiene por la adición de catalizadores de la polimerización que inician y aceleran el proceso de formación de un gel tridimensional. Normalmente, el proceso se inicia con la adición de persulfato amónico a una disolución acuosa (tampón) de ambos monómeros (acrilamida + bisacrilamida), seguido de la adición de TEMED (N,N,N’,N’-tetrametilendiamina) que actúa como propagador de la reacción de polimerización a pH básico (el pH ácido retarda la polimerización). Por lo tanto, ajustando las concentraciones de persulfato y TEMED puede controlarse la velocidad de polimerización. Siempre debe evitarse la presencia de oxígeno, pues inhibe la polimerización; por ello, la mezcla de polimerización debe desgasificarse a vacío (lo que impide, además, la formación de burbujas durante la polimerización que distorsionan el gel y alteran el campo eléctrico). La temperatura óptima de polimerización es de 25-30°C y el proceso ocurre en pocos minutos, aunque conviene dejarlo transcurrir más tiempo para que no queden restos de monómeros o de pequeñas cadenas libres.
Los monómeros (ya sea en polvo o en disolución) son neurotóxicos por absorción a través de la piel o por inhalación. Por ello, deben manejarse en una campana extractora con guantes y mascarilla. Una vez realizada la polimerización, la toxicidad se reduce al mínimo, pero es recomendable continuar manipulando el gel con guantes, debido a la posible presencia de radicales libres.
El tamaño de poro de los geles de poliacrilamida viene determinado por la concentración total de monómeros. Así, un gel de poliacrilamida se define por el reticulado (%T) que es la concentración total de monómeros (acrilamida + bisacrilamida; % p/v) y la dureza (%C) que viene dada por la relación de la cantidad de bisacrilamida al total de monómeros (es un valor bastante constante y, normalmente, inferior al 1%). Incrementando %T el tamaño de poro decrece (los geles con %T inferiores a 2.5-3.0% son casi líquidos y los geles con un %T del 30% presentan un reticulado tan denso que moléculas tan pequeñas como 2000-3000 Da difícilmente pueden atravesarlos). El tamaño medio de poro es 800Å, 50Å y 20Å para valores de 2.5, 7.5 y 30%T respectivamente. En la tabla I se muestran los tamaños aproximados de un conjunto de proteínas y los geles que se suelen utilizar para su análisis.
Tabla I. Tamaño aproximado de algunas proteínas y concentraciones de acrilamida (%T) utilizada en PAGE, en función de los distintos tamaños
Proteína M (kDa) Longitud (Å) Diámetro (Å)
Albúmina 69 150 38
Transferrina 90 190 37
BB1 lipoproteína 1300 185 185
Inmunoglobulina 160 235 44
Fibrinógeno 400 700 38
Concentración de acrilamida (%T) kDa
3-5 >100
5-12 20-150
10-15 10-80
>15 <15
Al polimerizar la acrilamida, adquiere la forma del recipiente, por lo que es necesario un molde para preparar el gel; los hay de dos tipos:
Tubo. Consiste en un tubo de vidrio de dimensiones variables, aunque normalmente es de 15-20 cm de largo × 0.5-0.8 cm de diámetro interno, abierto por los extremos. Se tapa el orificio inferior y se rellena con la disolución de polimerización sobre la que se deposita una pequeña cantidad de butanol (con esto se evita que al polimerizar el gel forme menisco y se excluye el oxígeno que interfiere en la polimerización). Una vez formado el gel, se elimina el butanol y se retira la tapa inferior del tubo. En la actualidad, la utilización de PAGE en tubo ha quedado restringida a la primera dimensión en electroforesis bidimensionales, PAGE preparativa y algunos casos muy específicos de análisis de proteínas radioactivas.
Placa. Es la forma más habitual en la que se desarrolla la PAGE. Puede llevarse a cabo en posición horizontal o vertical (dependiendo del tipo de cubeta empleada), aunque lo más habitual (sobre todo para la separación de proteínas) es el modo vertical (figura 10).
Fig. 10. Preparación del gel para una electroforesis en placa. Elementos para ensamblar el molde. Los espaciadores (sujetados con pinzas de presión) colocados sobre los cristales forman el molde. Adición de la solución de monómeros. Colocación del peine en la parte superior. El gel formado presenta los pocillos donde aplicar las muestras. Las dimensiones habituales de los geles son: 10-20 cm (L) × 10-15 cm (A) × 0.5-1.5 mm (E).
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 3 de 17

Si se aumenta la longitud del gel, aumenta la resolución de la electroforesis, aunque también se incrementa su duración. Asimismo, cuanto mayor sea la anchura del gel, mayor será el número de pocillos y, por lo tanto, el de muestras que se pueden analizar simultáneamente y en idénticas condiciones. El espesor del gel y las dimensiones de los pocillos viene determinado por el grosor de los espaciadores y el tamaño del peine determina la cantidad de muestra que puede aplicarse.
Una modalidad de la electroforesis vertical es la realizada con geles en gradiente en los que la concentración de acrilamida (%T) aumenta progresivamente desde la parte superior del gel hasta la inferior, es decir, el tamaño de poro decrece de forma inversa. Este gradiente de concentración de acrilamida puede ser lineal o no (cóncavo, en pasos, etc.), aunque los primeros son los más utilizados. Con estos geles el intervalo de tamaños de proteínas que pueden separarse se incrementa considerablemente frente a los geles homogéneos y presentan una mayor resolución. Además, las moléculas de la parte delantera de la banda se frenan más (encuentran antes los poros más pequeños) que las moléculas de la parte trasera, con lo que las bandas se estrechan, concentrándose sobre su parte delantera, produciendo bandas muy finas y con una gran resolución.
En la cubeta, el tampón del reservorio superior cubre el gel y los pocillos en los que se ha de cargar la muestra. Con el fin de que ésta, disuelta en el mismo tampón, no difunda al depositarla en los pocillos, se le puede añadir un compuesto que aumente su densidad (normalmente, glicerol o sacarosa). El volumen de muestra que puede aplicarse a cada pocillo depende del tamaño de la propia muestra y del pocillo, pero oscila entre 10-100µL con un máximo de 200µL. En el tampón de la muestra, también se añade el marcador electroforético (el más utilizado es el azul de bromofenol) para poder determinar el final del proceso.
• PAGE en condiciones no desnaturalizantes. La modalidad más sencilla es la descrita en apartados anteriores en las que la muestra se carga directamente en el gel en el cual se va a producir la separación, por lo tanto, la concentración del gel es uniforme y hay un único tampón. La PAGE se desarrolla en condiciones en las que no se altera la conformación nativa de las proteínas separadas, por lo que finalizada la electroforesis, pueden realizarse estudios de funcionalidad de dichas proteínas separadas (actividad enzimática, capacidad de unión de anticuerpos, unión a receptores, etc.)
Otra modalidad de electroforesis no desnaturalizante es aquella en la que el tampón presente en los reservorios es diferente al que impregna el gel, el cual tampoco es homogéneo, ya que, en realidad, hay dos geles diferentes en uno solo: una zona de resolución (donde tiene lugar la separación de las proteínas) que se polimeriza sin colocar el peine (por lo que la parte superior presenta un frente plano) y otra pequeña zona de concentración de 1-3 cm, que se polimeriza sobre la anterior (quedan fundidas), que contiene los pocillos de aplicación de la muestra y cuya finalidad es concentrar la muestra en la zona de contacto con la zona de resolución (figura 11).
Este hecho es debido a que la zona de concentración tiene un tamaño de poro muy grande (2.5-3.0%T), por lo que no es restrictiva frente al de la zona de resolución cuyo tamaño de poro sí es restrictivo.
Fig. 11. PAGE en condiciones no desnaturalizantes en sistemas de tampón discontinuo.
• PAGE en condiciones desnaturalizantes. En presencia de algunos compuestos químicos, las proteínas pierden su estructura nativa; tales compuestos, llamados agentes desnaturalizantes, producen el desplegamiento de la proteína que queda, así, sin la organización tridimensional característica de su funcionalidad biológica.
En algunas proteínas hay puentes disulfuro entre los residuos de Cys (para formar cistinas) de una misma cadena polipeptídica (puentes intracatenarios) o de diferentes cadenas (puentes intercatenarios) de algunas proteínas con estructura cuaternaria. Estos puentes se pueden romper mediante tratamiento con determinados agentes reductores, como por ejemplo, el ß-mercaptoetanol (ß-ME) o el ditiotreitol (DTT).
Otros agentes desnaturalizantes de proteínas, son la urea y determinados detergentes. La urea interfiere con las reacciones hidrofóbicas de las proteínas y debe incluirse en el tampón de la muestra y en todos los que se empleen para la preparación de los geles; las principales aplicaciones de la urea es en la separación de las histonas, así como de proteínas nucleares y ribosomales (ya que tienen mucha tendencia a agregar y son insolubles) y para el análisis conformacional de proteínas (mediante geles con gradientes transversales de urea).
Los detergentes afectan a la estructura nativa de las proteínas y a las interacciones con otras moléculas (proteínas, lípidos, etc.), ya que las interacciones hidrofóbicas de las proteínas son sustituidas por interacciones detergentes-proteína. Existen tres tipos principales de detergentes:
Detergentes no iónicos. Son débilmente desnaturalizantes y no alteran la carga de las proteínas a las que se unen; se pueden utilizar en geles con urea y en electroenfoque. Los más empleados son el Triton X-100 y el Octilglucósido, particularmente para solubilizar proteínas de membrana. Se utilizan a concentraciones de 0.1-3.0% (p/v) y deben incluirse en el tampón de la muestra y en el del gel.
Detergentes iónicos. Pueden tener carga positiva (catiónicos) que se usan para la separación de proteínas muy ácidas o muy básicas; el más utilizado es el cetiltrimetilamonio (CTAB). Otros poseen carga negativa y un fuerte carácter desnaturalizante; el más empleado es el dodecilsulfato sódico o lauril sulfato sódico (SDS)
Detergentes anfóteros. Son débilmente desnaturalizantes y no afectan a la carga de las proteínas; algunos, como el 3-(cloroamido propil)-dimetilamonio-1-propanosulfato (CHAPS) solubilizan muy bien las proteínas de membrana. Son compatibles con la utilización de urea y su uso es frecuente como alternativa a los detergentes no iónicos.
• PAGE-SDS. Permite el cálculo de parámetros moleculares (al contrario que el resto de los tipos de electroforesis), pues los complejos SDS-proteína se separan estrictamente según su tamaño molecular. El SDS interacciona con las proteínas formando complejos de características
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 4 de 17
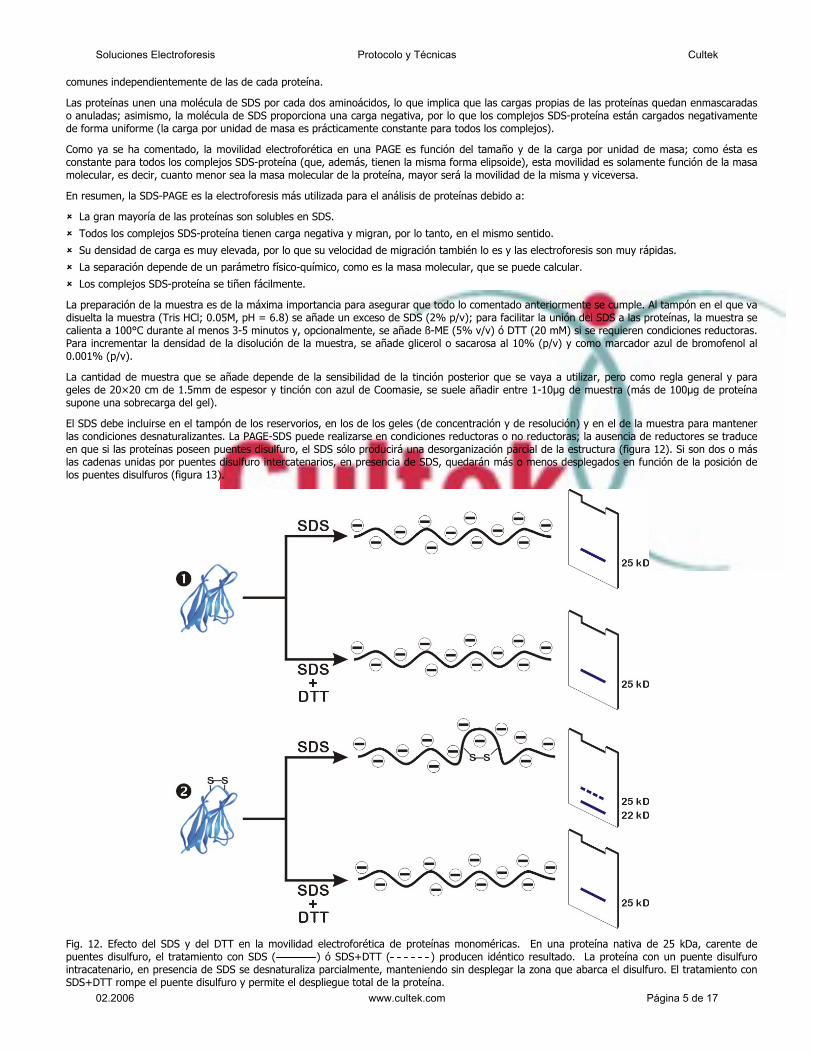
comunes independientemente de las de cada proteína.
Las proteínas unen una molécula de SDS por cada dos aminoácidos, lo que implica que las cargas propias de las proteínas quedan enmascaradas o anuladas; asimismo, la molécula de SDS proporciona una carga negativa, por lo que los complejos SDS-proteína están cargados negativamente de forma uniforme (la carga por unidad de masa es prácticamente constante para todos los complejos).
Como ya se ha comentado, la movilidad electroforética en una PAGE es función del tamaño y de la carga por unidad de masa; como ésta es constante para todos los complejos SDS-proteína (que, además, tienen la misma forma elipsoide), esta movilidad es solamente función de la masa molecular, es decir, cuanto menor sea la masa molecular de la proteína, mayor será la movilidad de la misma y viceversa.
En resumen, la SDS-PAGE es la electroforesis más utilizada para el análisis de proteínas debido a:
La gran mayoría de las proteínas son solubles en SDS.
Todos los complejos SDS-proteína tienen carga negativa y migran, por lo tanto, en el mismo sentido.
Su densidad de carga es muy elevada, por lo que su velocidad de migración también lo es y las electroforesis son muy rápidas.
La separación depende de un parámetro físico-químico, como es la masa molecular, que se puede calcular.
Los complejos SDS-proteína se tiñen fácilmente.
La preparación de la muestra es de la máxima importancia para asegurar que todo lo comentado anteriormente se cumple. Al tampón en el que va disuelta la muestra (Tris HCl; 0.05M, pH = 6.8) se añade un exceso de SDS (2% p/v); para facilitar la unión del SDS a las proteínas, la muestra se calienta a 100°C durante al menos 3-5 minutos y, opcionalmente, se añade ß-ME (5% v/v) ó DTT (20 mM) si se requieren condiciones reductoras. Para incrementar la densidad de la disolución de la muestra, se añade glicerol o sacarosa al 10% (p/v) y como marcador azul de bromofenol al 0.001% (p/v).
La cantidad de muestra que se añade depende de la sensibilidad de la tinción posterior que se vaya a utilizar, pero como regla general y para geles de 20×20 cm de 1.5mm de espesor y tinción con azul de Coomasie, se suele añadir entre 1-10µg de muestra (más de 100µg de proteína supone una sobrecarga del gel).
El SDS debe incluirse en el tampón de los reservorios, en los de los geles (de concentración y de resolución) y en el de la muestra para mantener las condiciones desnaturalizantes. La PAGE-SDS puede realizarse en condiciones reductoras o no reductoras; la ausencia de reductores se traduce en que si las proteínas poseen puentes disulfuro, el SDS sólo producirá una desorganización parcial de la estructura (figura 12). Si son dos o más las cadenas unidas por puentes disulfuro intercatenarios, en presencia de SDS, quedarán más o menos desplegados en función de la posición de los puentes disulfuros (figura 13).
Fig. 12. Efecto del SDS y del DTT en la movilidad electroforética de proteínas monoméricas. En una proteína nativa de 25 kDa, carente de puentes disulfuro, el tratamiento con SDS ( ) ó SDS+DTT ( ) producen idéntico resultado. La proteína con un puente disulfuro intracatenario, en presencia de SDS se desnaturaliza parcialmente, manteniendo sin desplegar la zona que abarca el disulfuro. El tratamiento con SDS+DTT rompe el puente disulfuro y permite el despliegue total de la proteína.
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 5 de 17
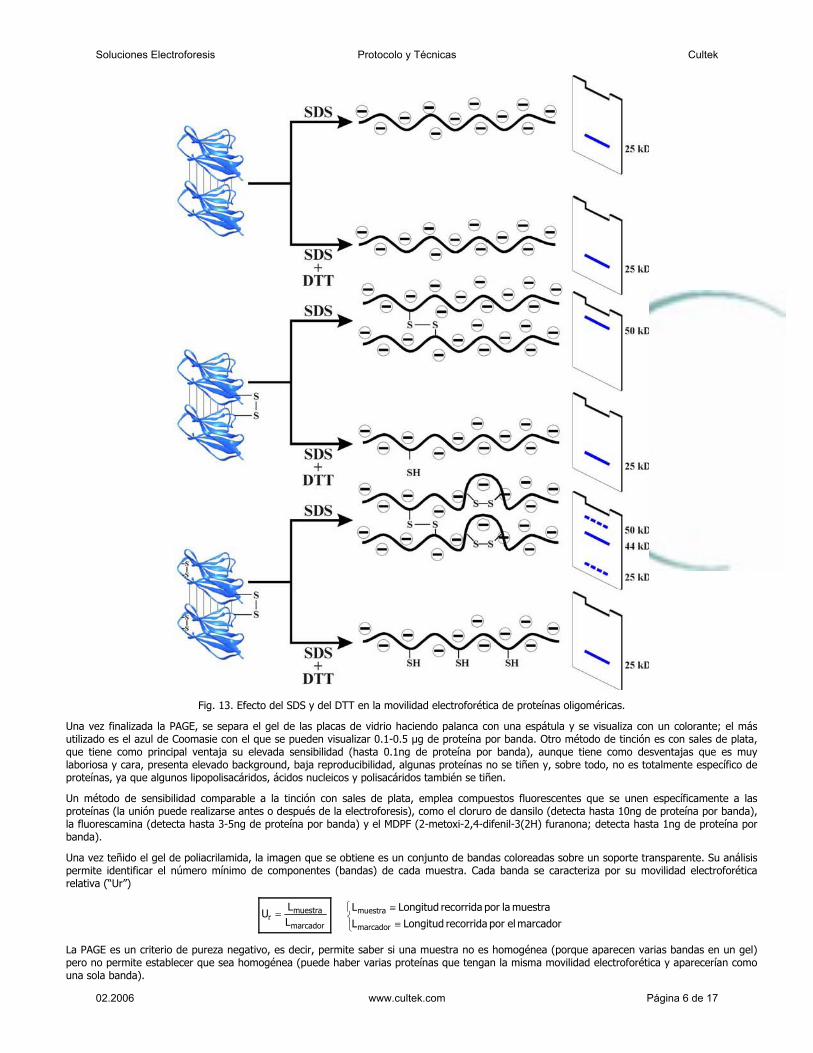
Fig. 13. Efecto del SDS y del DTT en la movilidad electroforética de proteínas oligoméricas.
Una vez finalizada la PAGE, se separa el gel de las placas de vidrio haciendo palanca con una espátula y se visualiza con un colorante; el más utilizado es el azul de Coomasie con el que se pueden visualizar 0.1-0.5 µg de proteína por banda. Otro método de tinción es con sales de plata, que tiene como principal ventaja su elevada sensibilidad (hasta 0.1ng de proteína por banda), aunque tiene como desventajas que es muy laboriosa y cara, presenta elevado background, baja reproducibilidad, algunas proteínas no se tiñen y, sobre todo, no es totalmente específico de proteínas, ya que algunos lipopolisacáridos, ácidos nucleicos y polisacáridos también se tiñen.
Un método de sensibilidad comparable a la tinción con sales de plata, emplea compuestos fluorescentes que se unen específicamente a las proteínas (la unión puede realizarse antes o después de la electroforesis), como el cloruro de dansilo (detecta hasta 10ng de proteína por banda), la fluorescamina (detecta hasta 3-5ng de proteína por banda) y el MDPF (2-metoxi-2,4-difenil-3(2H) furanona; detecta hasta 1ng de proteína por banda).
Una vez teñido el gel de poliacrilamida, la imagen que se obtiene es un conjunto de bandas coloreadas sobre un soporte transparente. Su análisis permite identificar el número mínimo de componentes (bandas) de cada muestra. Cada banda se caracteriza por su movilidad electroforética relativa (“Ur”)
marcador
muestrar L
LU =
⎩⎨⎧
≡
≡
marcador elpor recorrida LongitudL
muestra lapor recorrida LongitudL
marcador
muestra
La PAGE es un criterio de pureza negativo, es decir, permite saber si una muestra no es homogénea (porque aparecen varias bandas en un gel) pero no permite establecer que sea homogénea (puede haber varias proteínas que tengan la misma movilidad electroforética y aparecerían como una sola banda).
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 6 de 17

En las condiciones de la SDS-PAGE, si se representan los valores de logaritmo del peso molecular (logM) frente a la movilidad electroforética (Ur) de un conjunto de proteínas, se obtiene una relación lineal (figura 14). Normalmente, se utiliza un conjunto de proteínas de peso molecular conocido (marcadores de peso molecular) que se someten a SDS-PAGE en el mismo gel que se analizan las proteínas cuyos peso molecular se desea conocer.
Fig. 14. Cálculo de pesos moleculares de proteínas por SDS-PAGE.
Es importante señalar que la relación lineal entre logM y Ur se mantiene para una concentración dada de poliacrilamida (%T) y sólo en un corto intervalo de peso molecular. De forma general, en geles del 15%T la relación lineal es válida para proteínas comprendidas entre 12 y 45 kDa; en geles del 10%T, el intervalo lineal va entre 15 y 70 kDa, y en los del 5%T, entre 25 y 200 kDa.
Hay proteínas con un comportamiento anómalo en SDS-PAGE; en las glicoproteínas y las lipoproteínas, que llevan unidos azúcares o lípidos, la parte no proteica no une SDS y puede impedir el acceso del mismo a la proteína. Por ello, las glicoproteínas y lipoproteínas unen menos SDS que la mayoría de las proteínas; esto hace que la relación carga/tamaño sea menor y, por lo tanto, tengan menor movilidad electroforética y se comporten como si tuvieran mayor tamaño.
Electroforesis bidimensional. Es una sucesión de dos electroforesis distintas (o en distintas condiciones) realizadas sobre una misma muestra. En la primera de ellas (primera dimensión) se separan los componentes de la muestra según un criterio (carga, tamaño o pI), y en la segunda (segunda dimensión) según un parámetro distinto del anterior. De esta manera, se combinan dos modos de separación diferentes y se consigue el máximo de resolución posible mediante técnicas electroforéticas.
La primera dimensión puede llevarse a cabo, por ejemplo, en condiciones no desnaturalizantes y la segunda en condiciones desnaturalizantes; las proteínas ribosomales se separan en un gel discontinuo en la primera dimensión, seguido por otro continuo en la segunda, y ambos en presencia de urea; las histonas se separan en geles con urea y ácido en la primera dimensión y utilizando tritón/ácido/urea en la segunda.
Sin embargo, normalmente, la idea de electroforesis bidimensional suele referirse a la combinación de los dos tipos de electroforesis más resolutivos: electroenfoque (primera dimensión) y SDS-PAGE (segunda dimensión) (figura 15).
La electroforesis bidimensional puede considerarse como un criterio de pureza positivo debido a su gran poder de resolución, aceptándose que la aparición de una sola mancha indica una muestra homogénea.
Fig. 15. Electroforesis bidimensional. Primera dimensión (SDS-PAGE). En el pocillo I se aplica el marcador de pesos moleculares y en los pocillos II y III la muestra. El carril del pocillo III (sombreado) se corta y el resto del gel se tiñe. El carril III se coloca sobre un nuevo gel que se ha polimerizado sin pocillos, donde se desarrolla la segunda dimensión (EEF) perpendicularmente a la primera. Obsérvese que algunas bandas producen más de una mancha en la segunda dimensión.
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 7 de 17

Fig. 23. Iluminación y fotografía de geles de agarosa. Iluminación incidente. Iluminación por transmisión (transiluminación). Videocámara con la imagen del gel sobre el transiluminador. Las flechas amarillas representan la radiación emitida (fluorescencia) por los complejos DNA-BrEt.
Electroforesis en geles de agarosa en condiciones desnaturalizantes. Se lleva a cabo en modo submarino como ya se ha descrito. Se utiliza para la separación de fragmentos de DNA monocatenario grandes (1000-2000 nucleótidos) y especialmente para el análisis de mRNA, que se transfiere posteriormente a membranas para su detección (Northern Blot). La transferencia e hibridación son similares a las descritas para el DNA, pero teniendo en cuenta que el mRNA es monocatenario.
Para lograr una buena separación y para calcular con fiabilidad los tamaños es necesario que las moléculas estén completamente desnaturalizadas, ya que la estructura secundaria y terciaria de los ácidos nucleicos está mantenida esencialmente por el apareamiento (tanto inter- como intracatenario) de bases complementarias; para asegurar esta desnaturalización se utilizan agentes desnaturalizantes como la temperatura, el pH básico y diversos agentes químicos dependiendo del soporte y del ácido nucleico. No pueden utilizarse agentes desnaturalizantes que rompan los puentes de hidrógeno (urea, formamida) ya que afectan también a la agarosa (sus fibras se mantienen mediante puentes de hidrógeno).
Transferencia a membranas (Southern y Northern Blot). Las moléculas de DNA o RNA separadas en los geles de agarosa pueden ser transferidas a membranas de nitrocelulosa o de nylon. La transferencia se realiza tras la desnaturalización del DNA o RNA. La unión a la membrana ocurre a través del esqueleto azúcar-fosfato de la molécula de DNA o RNA, quedando libres y expuestas las bases nitrogenadas. Esto permite localizar e identificar secuencias específicas en los fragmentos transferidos, incubando la membrana con sondas específicas radiactivas o de otra naturaleza, que hibridan con la secuencia complementaria, revelando su posición por autorradiografía de la membrana.
De esta manera puede identificarse un fragmento específico de DNA o RNA entre toda la población de estructuras separadas por electroforesis y transferidas a la membrana. La sensibilidad del método es muy elevada y pueden detectarse <0.1pg de DNA o RNA usando una sonda marcada con 32
P (figura 24); por ejemplo, una secuencia única de 1.000pb presente en el genoma humano (1 parte en 3 millones) puede ser detectada por este método a partir de 10µg de DNA genómico.
Las membranas de nitrocelulosa dan mejor resolución que las de nylon, pero éstas últimas presentan varias ventajas por lo que su uso es más extendido: tienen una mejor consistencia (las de nitrocelulosa son bastante frágiles y pueden desmenuzarse con la manipulación) y poseen mayor capacidad de unión de DNA, pudiendo ser utilizadas para hibridaciones sucesivas; además, producen poca fluorescencia de fondo (fluorescencia inespecífica) al ser iluminadas con radiación UV, lo que es muy útil para la detección de bandas de DNA.
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 14 de 17

La desnaturalización de los fragmentos de DNA, previa a la transferencia, se realiza introduciendo el gel en una disolución de NaOH que se neutraliza posteriormente. Las cadenas de DNA desnaturalizadas no deben ser muy largas para que la transferencia sea efectiva y, así, fragmentos mayores de 15-20kb deben ser fragmentados previamente con radiación UV o tratamiento ácido.
La transferencia puede realizarse por capilaridad, por electroforesis (electrotransferencia), por centrifugación o por succión (mediante una bomba de vacío), aunque los dos últimos sistemas necesitan dispositivos específicos, por lo que su uso es más restringido. Normalmente, la electrotransferencia se utiliza cuando la separación del DNA se ha realizado en geles de poliacrilamida (dado que el reticulado es más denso que el de la agarosa). El tiempo de transferencia depende del grosor del gel, del porcentaje de agarosa y del tamaño de los fragmentos de DNA. Una vez en la membrana, el DNA se fija a ella mediante calentamiento a 80°C o iluminación con radiación UV.
Al igual que en el Western Blot, las regiones de la membrana no ocupadas por el DNA transferido deben bloquearse, para evitar que al añadir la sonda, ésta se una inespecíficamente a la membrana y produzca un fondo en el revelado. Para realizar este bloqueo, la membrana se sumerge en una disolución que contenga substancias de elevado peso molecular, como ficoll (400kDa), polivinilpirrolidona (360kDa) o seroalbúmina bovina (66kDa), además de DNA heterólogo de diversos orígenes (bacteriano, timo de ternera, esperma de salmón, etc.).
La formación de las estructuras híbridas (heterodúplex) entre la sonda y la secuencia complementaria buscada es muy dependiente de las condiciones experimentales (particularmente de la temperatura); para realizarla se utilizan dos sistemas: en el primero, la membrana se introduce en una bolsa de plástico hermética que contiene la disolución con la sonda, controlándose la temperatura por inmersión en un baño termostatizado; el segundo, utiliza hornos de hibridación, introduciendo las membranas en botellas que contienen la disolución de la sonda.
Fig. 24. Esquema de desarrollo de un Southern Blot (izquierda) y un Northern Blot (derecha).
Electroforesis en campos pulsantes (PFGE). Con las electroforesis descritas hasta ahora, se pueden separar fragmentos de hasta 20kpb, e incluso hasta 50kpb con geles de agarosa de muy baja concentración. Schwartz y Cantor desarrollaron en 1984 un sistema denominado electroforesis en gel de campo pulsante (PFGE) con el que consiguieron separar cromosomas de levadura de varios cientos de kpb. La PFGE consigue la separación de grandes fragmentos de DNA induciendo su reorientación mediante cambios periódicos en el campo eléctrico, cuya duración determina el intervalo de tamaños que se pueden separar (cuanto mayor sea el tamaño de los fragmentos a separar, mayor ha de ser la duración de los campos aplicados.
En una PFGE los fragmentos se someten a dos campos eléctricos de distinta orientación. Al aplicarse el primero (E1) durante cierto tiempo, los fragmentos se estiran y se orientan según la dirección de dicho campo. Si ahora se aplica un segundo campo (E2), perpendicular al anterior, se obliga a los fragmentos de DNA a relajarse y elongarse y alinearse de acuerdo con este segundo campo eléctrico. Cuanto menos tarde una molécula en reorientarse, antes migra en la dirección de E2; el tiempo que se consume en esta reorientación depende del tamaño de los fragmentos, tardando más los mayores. Aplicando sucesivos cambios de campo eléctrico (E1/E2/E1/E2/E1/E2 etc.) se consigue la separación de los fragmentos en función de su tamaño.
El ángulo α que forman las direcciones de los campos eléctricos E1 y E2 se denomina ángulo de reorientación, y determina lo que deben “girar” los fragmentos de DNA cada vez que cambia la orientación del campo. Normalmente se utilizan ángulos >100°; cuando se opera a un ángulo fijo, éste es de 120°, aunque si el dispositivo permite seleccionar diferentes ángulos, se trabaja entre 100° y 120°; esto es particularmente útil para la separación de fragmentos muy grandes de DNA (>2Mb).
La duración de los campos eléctricos que se alternan se denomina duración del pulso y determina el intervalo de los tamaños de DNA que se pueden separar; estos intervalos son muy variables, desde fracciones de segundo para fragmentos muy pequeños hasta 1-2 horas para fragmentos muy grandes (>5Mb) (Tabla III).
Para una duración de pulso dada, aquellos fragmentos de gran tamaño que no hayan tenido tiempo a reorientarse en la nueva dirección del campo no se separan, pero no permanecen inmóviles en el gel; se mueven muy lentamente, pero juntos, sin resolverse en bandas distintas y aparecen como una zona más o menos ancha en la parte superior del gel; dicha zona se denomina zona de compresión.
Es importante destacar que la utilidad de la PFGE es análoga a la de la electroforesis convencional, salvo que los fragmentos de DNA separados son de mayor tamaño. Esto hace que la transferencia a membranas no pueda hacerse directamente, ya que para tamaños superiores a las 20kb los fragmentos se han de dividir en otros menores mediante radiación UV o tratamiento con ácido (HCl). También puede ser utilizada para electroforesis bidimensionales, cambiando las condiciones en cada dimensión; esto permite el mapeo genético de grandes regiones de DNA o de cromosomas enteros pequeños.
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 15 de 17
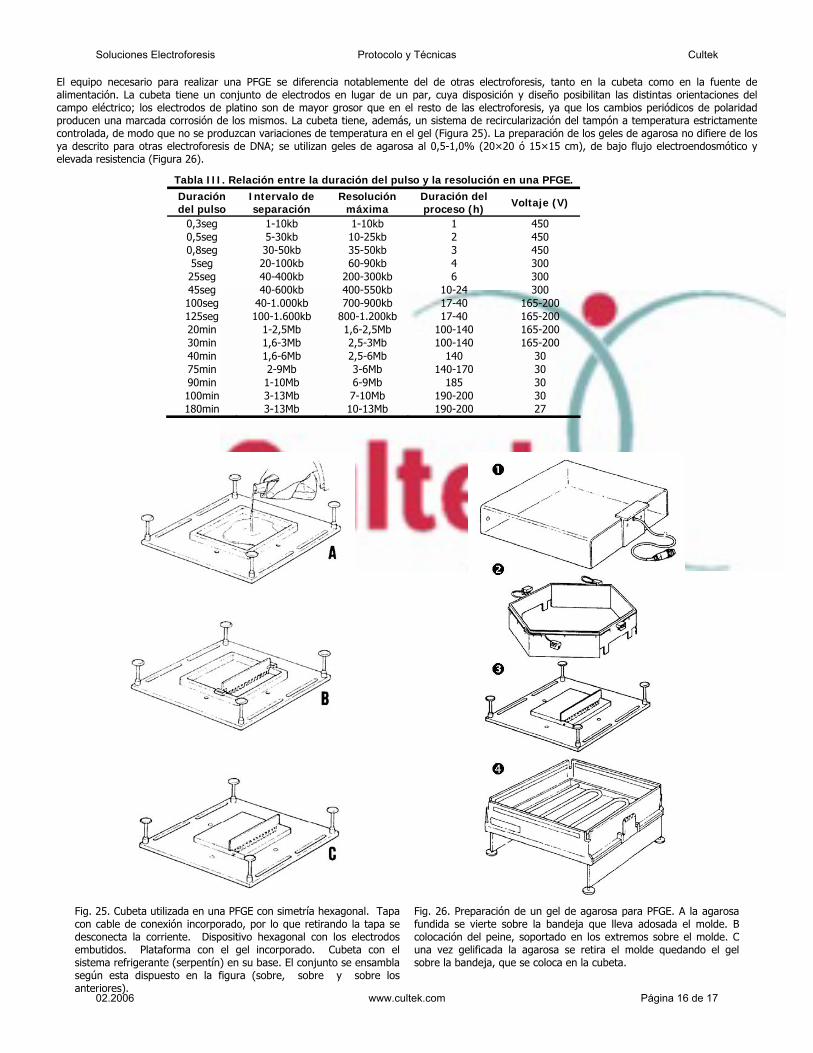
El equipo necesario para realizar una PFGE se diferencia notablemente del de otras electroforesis, tanto en la cubeta como en la fuente de alimentación. La cubeta tiene un conjunto de electrodos en lugar de un par, cuya disposición y diseño posibilitan las distintas orientaciones del campo eléctrico; los electrodos de platino son de mayor grosor que en el resto de las electroforesis, ya que los cambios periódicos de polaridad producen una marcada corrosión de los mismos. La cubeta tiene, además, un sistema de recircularización del tampón a temperatura estrictamente controlada, de modo que no se produzcan variaciones de temperatura en el gel (Figura 25). La preparación de los geles de agarosa no difiere de los ya descrito para otras electroforesis de DNA; se utilizan geles de agarosa al 0,5-1,0% (20×20 ó 15×15 cm), de bajo flujo electroendosmótico y elevada resistencia (Figura 26).
Tabla III. Relación entre la duración del pulso y la resolución en una PFGE. Duración del pulso
Intervalo de separación
Resolución máxima
Duración del proceso (h) Voltaje (V)
0,3seg 1-10kb 1-10kb 1 450 0,5seg 5-30kb 10-25kb 2 450 0,8seg 30-50kb 35-50kb 3 450 5seg 20-100kb 60-90kb 4 300 25seg 40-400kb 200-300kb 6 300 45seg 40-600kb 400-550kb 10-24 300 100seg 40-1.000kb 700-900kb 17-40 165-200 125seg 100-1.600kb 800-1.200kb 17-40 165-200 20min 1-2,5Mb 1,6-2,5Mb 100-140 165-200 30min 1,6-3Mb 2,5-3Mb 100-140 165-200 40min 1,6-6Mb 2,5-6Mb 140 30 75min 2-9Mb 3-6Mb 140-170 30 90min 1-10Mb 6-9Mb 185 30 100min 3-13Mb 7-10Mb 190-200 30 180min 3-13Mb 10-13Mb 190-200 27
Fig. 25. Cubeta utilizada en una PFGE con simetría hexagonal. Tapa con cable de conexión incorporado, por lo que retirando la tapa se desconecta la corriente. Dispositivo hexagonal con los electrodos embutidos. Plataforma con el gel incorporado. Cubeta con el sistema refrigerante (serpentín) en su base. El conjunto se ensambla según esta dispuesto en la figura (sobre, sobre y sobre los anteriores).
Fig. 26. Preparación de un gel de agarosa para PFGE. A la agarosa fundida se vierte sobre la bandeja que lleva adosada el molde. B colocación del peine, soportado en los extremos sobre el molde. C una vez gelificada la agarosa se retira el molde quedando el gel sobre la bandeja, que se coloca en la cubeta.
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 16 de 17

La preparación y aplicación de la muestra es la fase más delicada de la PFGE por el gran tamaño de las estructuras que se manejan, lo que las hace fácilmente fragmentables. El DNA suele cargarse en el gel como una disolución concentrada (de mucha viscosidad), junto con agarosa fundida o embebido en pequeños bloques o tacos de agarosa sólida (Figura 27). La utilización de una u otra forma depende del tamaño del DNA a analizar y de la naturaleza de la muestra. Los fragmentos de 200-300kb pueden cargarse como disoluciones directamente en el gel, pero para fragmentos mayores de 500kb no se suele emplear el DNA aislado como tal y sí las propias células que lo contiene.
Fig. 27. Preparación de la muestra para PFGE en bloques de agarosa. A partir de un bloque grande, que contiene las células cuyo DNA se desea analizar, se cortan pequeños tacos (parte superior) del tamaño de los pocillos practicados en el gel (parte inferior). Todos los procesos de lisis celular y digestiones enzimáticas se realizan sobre el taco de agarosa.
Soluciones Electroforesis Protocolo y Técnicas Cultek
02.2006 www.cultek.com Página 17 de 17

Biología
y
Geología
SECUENCIACIÓN DE ADN
Una técnica propia de la Ingeniería Genética es la determinación de la secuencia de nucleótidos de un ADN, conocida como secuenciación de dicho ácido nucléico.
Se ha conseguido gracias a las enzimas de restricción y a la posibilidad de obtener numerosas copias de un ácido nucléico por clonación.
Se pueden conseguir muchos fragmentos, de diversos tamaños y lo que se trata es de recomponer la molécula original como si se tratase de un puzzle.
Así se han secuenciado genes, desde virus hasta el genoma humano.
El método más usado para la secuenciación es el conocido como técnica de terminación de cadena de Sanger, la cual se ha perfeccionado conociéndose actualmente como la técnica del didesoxi.
Se denomina así por ser necesario los didesoxirribonucleótidos (figura 1), que han perdido su grupo alcohol OH del carbono 3'. En la figura 2 vemos un nucleótido normal con el grupo alcohol en el carbono 3'.
Figura 1
Como los nucleótidos se unen por este grupo OH del carbono 3', hay que pensar que si nos encontramos con un didesoxinucleótido será imposible que se una otro nucleótido y la cadena terminará aquí
Abreviadamente, éste sería el método a seguir: Como la técnica se basa en la síntesis de ADN, para hacer la reacción de secuenciación se necesita:
• Como "molde" se utiliza una de las cadenas del fragmento de ADN que se va a secuenciar.
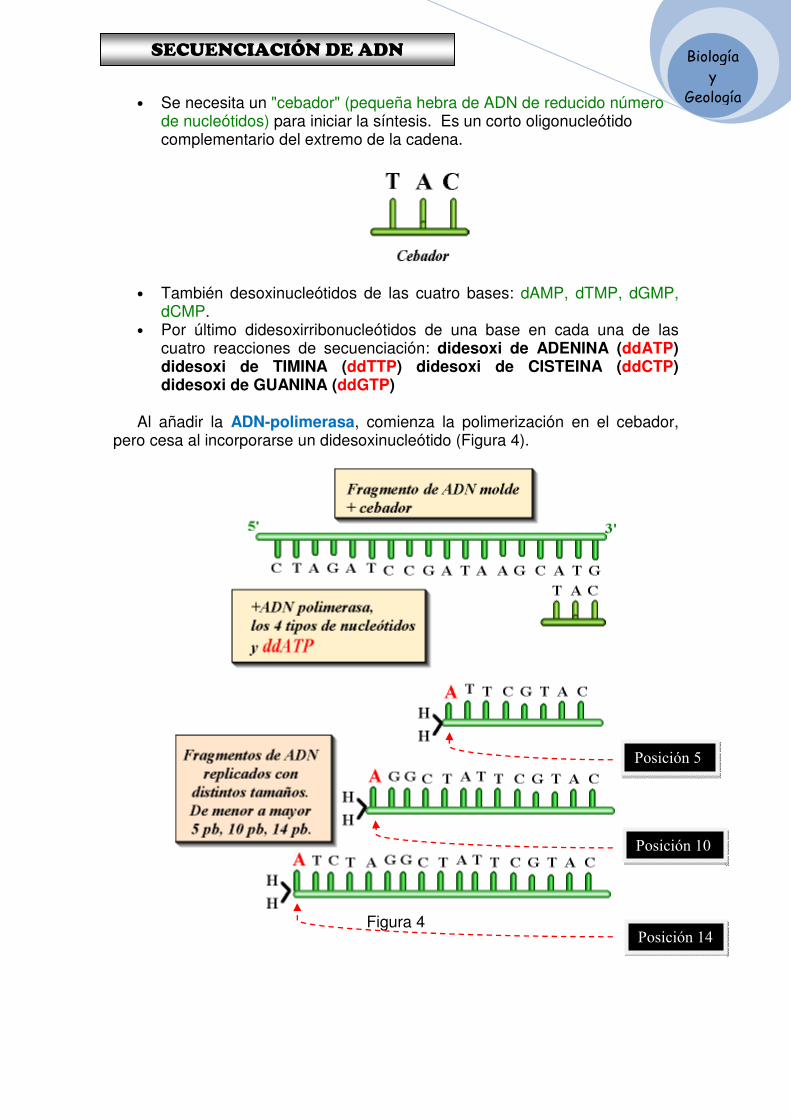
Biología
y
Geología
SECUENCIACIÓN DE ADN
• Se necesita un "cebador" (pequeña hebra de ADN de reducido número de nucleótidos) para iniciar la síntesis. Es un corto oligonucleótido complementario del extremo de la cadena.
• También desoxinucleótidos de las cuatro bases: dAMP, dTMP, dGMP, dCMP.
• Por último didesoxirribonucleótidos de una base en cada una de las cuatro reacciones de secuenciación: didesoxi de ADENINA (ddATP) didesoxi de TIMINA (ddTTP) didesoxi de CISTEINA (ddCTP) didesoxi de GUANINA (ddGTP)
Al añadir la ADN-polimerasa, comienza la polimerización en el cebador, pero cesa al incorporarse un didesoxinucleótido (Figura 4).
Figura 4
Posición 5
Posición 14
Posición 10

Biología
y
Geología
SECUENCIACIÓN DE ADN
Se produce un conjunto de cadenas dobles cuyas longitudes dependen de la situación del didesoxinucleótido incorporado. En el caso expuesto en el dibujo, esta reacción de secuenciación se hace con un didesoxi de ADENINA, lo que significa que cuando se incorpore esta Adenina no podrá continuarse la polimerización de nuevos nucleótidos, nos queda por tanto un pequeño fragmento de ADN que termina en la ADENINA, lo que nos dice que en el ADN original, en ese lugar estará el nucleótido complementario que lleva TIMINA.
En el caso expuesto deducimos tres fragmentos con 5, 10 y 14 nucleótidos (sin contar el cebador) en cuyos extremos tenemos ADENINA.
Deben prepararse cuatro reacciones de secuenciación, cada una con un didesoxirribonucleotido distinto. Los fragmentos resultantes se separan por tamaño mediante electroforesis, se autorradiografían, y la sucesión de bandas de cada una de las cuatro reacciones, comparándolas entre sí,dan la secuencia del ADN. (Figura 5)
COMENTARIO
1. ¿Qué importancia tiene el uso de didesoxirribonucleótidos? 2. ¿Qué posible aplicación se le puede dar a este método? 3. ¿Qué es la Taq ADN polimerasa? ¿Para qué se emplea?














