Embed Size (px)
Citation preview

1
Universidad de Chile
Facultad de Ciencias Químicas y Farmacéuticas
Departamento de Química Orgánica y Fisico Química
Caracterización Físico Química de Micelas de
Monoésteres de Sacarosa
Profesor Patrocinante: Directores:
Dr. Germán Günther S. Dr. Germán Günther S.
Dr. Antonio Zanocco L.
Memoria para optar al Título de Químico
NATALIA MÓNICA BECERRA PACHECO
Santiago de Chile 2005

2
1. INTRODUCCIÓN
En las últimas décadas la industria de los surfactantes ha reemplazado, en forma gradual
y sistemática, las materias primas derivadas de la actividad petroquímica por otros
compuestos naturales cuya producción, uso y degradación resultan menos
contaminantes. Entre estas especies se encuentran los ésteres de sacarosa, compuestos
provenientes de materias primas renovables de muy baja toxicidad, no sensibilizantes,
no alergénicos y muy adecuados como emulsificantes, emolientes, humectantes, además
de ser biodegradables.1-4 En la actualidad, a nivel tecnológico, este tipo de surfactantes
son utilizados en diversas aplicaciones de la industria farmacéutica y de los alimentos.
Entre ellas destacan la extracción y solubilización de lípidos, además del transporte de
drogas cuando se encuentran formando vesículas.5,6 Sin embargo, a pesar del amplio uso
que se ha dado a este tipo de surfactantes, los estudios realizados sobre compuestos
puros son escasos, como consecuencia de las dificultades que presenta su purificación a
partir de la mezcla compleja que suele obtenerse de la síntesis.7,8
Con el objetivo de contribuir al conocimiento del comportamiento básico de este tipo de
compuestos y sus potenciales aplicaciones, en esta memoria se estudiaron las
propiedades fisicoquímicas de soluciones acuosas de cuatro monoésteres de sacarosa
puros, con diferentes largos de cadena alquílica: 6-O-laurilsacarosa (β-D-Fructofuranosil
6-O-lauril-α-D-glucopiranósido, MLS), 6-O-miristilsacarosa (β-D-Fructofuranosil 6-O-
miristil-α-D-glucopiranósido, MMS), 6-O-palmitilsacarosa (β-D-Fructofuranosil 6-O-
palmitil-α-D-glucopiranósido, MPS), y 6-O-estearilsacarosa (β-D-Fructofuranosil 6-O-
estearil-α-D-glucopiranósido, MES). Los ésteres utilizados se obtuvieron empleando
metodologías sintéticas de trans-esterificación, para luego ser purificados por
cromatografía en columna de sílice. Ambos procesos fueron realizados en un trabajo

3
previo a esta tesis.9 La estructura general de los cuatro surfactantes incluidos en este
estudio se muestra en la Figura 1.
OOH
OHOH O
O
OH
OH
OH
OHRCOO
R = C11H23 Derivado del ácido láurico
R = C13H27 Derivado del ácido mirístico
R = C15H31 Derivado del ácido palmítico
R = C17H35 Derivado del ácido esteárico
Figura 1. Estructura general de los monoésteres de sacarosa estudiados.
La actividad superficial que muestran los tensoactivos o surfactantes tiene su origen en
la naturaleza dual de su estructura (carácter anfipático), producto de la presencia de dos
tipos de grupos con propiedades opuestas. Uno de ellos es de naturaleza hidrofóbica
(formado por una o más cadenas hidrocarbonadas), mientras que el otro corresponde a
un grupo polar, iónico o neutro. Los ésteres de sacarosa son surfactantes no iónicos cuyo
grupo polar corresponde a una molécula de azúcar.

4
La presencia de este tipo de moléculas en solución implica la existencia de interacciones
con el solvente, atractivas para un segmento y repulsivas para el otro, lo que se traduce
en la formación espontánea de agregados de tamaño coloidal. En medio acuoso, las
moléculas se orientan de modo tal que las cadenas hidrocarbonadas tengan el menor
contacto posible con el solvente, quedando las cabezas polares en contacto directo con
éste. En la Figura 2 se muestran algunas de las estructuras que comúnmente adoptan
estos microagregados en solución acuosa. Si la morfología de la molécula es apropiada,
se formarán espontáneamente microagregados denominados micelas, con 50 a 200
unidades monoméricas organizadas de acuerdo a la polaridad del entorno.
Figura 2. Microestructuras comúnmente observadas para surfactantes en solución
acuosa.
La existencia y naturaleza de este tipo de estructuras proporciona al sistema un carácter
bifásico, estableciéndose en el caso de las micelas un equilibrio dinámico entre la
formación del microagregado y su monómero en solución. El fenómeno de micelización
corresponde a un proceso reversible, espontáneo y de naturaleza principalmente
entrópica.10
Micela
esférica
Micela elongada
Vesícula
Bicapa

5
Una de las propiedades fisicoquímicas más importantes que caracteriza a los sistemas
micelares es su concentración micelar crítica (CMC), parámetro que corresponde a la
concentración de surfactante a la cual comienza la asociación de moléculas para formar
los microagregados. El fenómeno se inicia con la formación de microagregados
pequeños (o premicelas), que crecen rápidamente en un rango de concentraciones
específico para cada surfactante. La posterior adición de tensoactivo provoca un
aumento en el número de microagregados presentes en solución, mientras el tamaño y
forma micelar permanecen relativamente inalterados en un rango dado. La CMC no es
un valor preciso, corresponde a un rango de concentración y su magnitud experimental
depende normalmente de la metodología empleada para evaluarlo. Si el surfactante es
una mezcla de especies significativamente diferentes entre si, dicho rango puede llegar a
ser relativamente amplio.
Los valores de la CMC dependen tanto de las características del grupo hidrofílico (tipo,
tamaño y contraión), como de las del segmento hidrofóbico (longitud y ramificación) del
compuesto.11 Como consecuencia, es posible observar diferencias de hasta 4 órdenes de
magnitud entre los valores reportados en la literatura para este parámetro. En general,
para detergentes de tipo iónico, los valores de CMC son mayores que los observados
para moléculas no iónicas de largo de cadena similar.
El proceso de micelización es el resultado de un balance entre fuerzas repulsivas y
atractivas, cuyo inicio trae consigo cambios significativos en el valor de algunas
propiedades macroscópicas del sistema. Entre ellas se pueden mencionar: tensión
superficial, conductividad, densidad, presión osmótica y dispersión de la luz. Al analizar
el comportamiento de estas propiedades en función de la concentración de surfactante se

6
observa un claro quiebre, a partir del cual es posible la determinación directa y
relativamente sencilla de la CMC.
Otra característica importante de los sistemas micelares corresponde al número de
moléculas de surfactante que conforman cada microagregado, magnitud definida como
número de agregación, nag. Este parámetro no sólo contiene información acerca del
tamaño y forma micelar, sino que además es importante en la determinación de la
estabilidad y posibles aplicaciones prácticas del sistema en estudio.12 Algunos valores de
CMC y números de agregación para surfactantes de uso común se presentan en la Tabla
1.
Tabla 1. Parámetros micelares para algunos surfactantes comunes en solución acuosa a
temperatura ambiente. 13
Surfactante CMC /M Número de Agregación
Iónicos
Cloruro de Dodecilamonio 1,5 x 10-2 52
Dodecilsulfato de Sodio 8,0 x 10-3 62
Bromuro de Cetiltrimetilamonio 9,2 x 10-4 60
No iónicos
Polioxietileno (6) octanol 9,9 x 10-3 -
Polioxietileno (E9-10) t-octilfenol 2,6 x 10-4 143
Polioxietileno (E23) laurileter 6,0 x 10-5 40
N-dodecil-N,N’-dimetilbetaína 2,0 x 10-3 40

7
Entre los factores externos que afectan la CMC y el número de agregación de un sistema
micelar se encuentran la presencia de aditivos orgánicos, tipo y concentración de
electrolitos adicionados, la temperatura, etc.12 La presencia de alcoholes, por ejemplo,
produce generalmente una disminución en la CMC del sistema al cual son agregados, y
su influencia depende no sólo de su peso molecular y ramificación sino también de su
concentración.14 En este trabajo se determinará la dependencia de la CMC, la
microviscosidad y la micropolaridad de los microagregados con la presencia de
alcoholes alifáticos lineales y ramificados de largo de cadena intermedio.
Estudios previos muestran que la relación entre la CMC y la temperatura no es directa ni
simple.15 Cuando ésta aumenta, disminuye la hidratación del grupo hidrofílico,
favoreciéndose el proceso de micelización. Un mayor incremento de la temperatura
conlleva la desorganización creciente de las moléculas de agua que rodean al grupo
apolar, reduciéndose las interacciones repulsivas con el consecuente aumento de la
CMC. Se ha reportado que, para surfactantes iónicos el tamaño micelar es relativamente
insensible a cambios en la temperatura, mientras que, para micelas no iónicas el número
de agregación depende en forma significativa de la temperatura.11 El proceso de
micelización se describe termodinámicamente en términos de la variación de energía
libre de Gibbs (∆GMIC), la variación de entalpía (∆HMIC) y la variación de entropía
(∆SMIC). Los cambios de las funciones termodinámicas asociadas al proceso de
micelización, se pueden obtener a partir de la dependencia de la CMC con la
temperatura.
Para este estudio se emplearon medidas físicas (Tensiómetro de Dü Nouy) y fotofísicas.
Estas últimas involucran el uso de pruebas o sondas fluorescentes, cuyo comportamiento
en sistemas compartimentalizados permite la determinación de propiedades
fisicoquímicas de los microagregados en que se encuentran incorporados. Este método

8
consiste en introducir una determinada sonda en el microagregado micelar y, a partir de
los cambios observados en la emisión y del comportamiento descrito para la sonda en
medio homogéneo, trazar un cuadro estático y/o dinámico del microagregado

9
2. MATERIALES
2.1 Reactivos
El 6-dodecanoil-2-dimetilaminonaftaleno (Molecular Probes), la benzofenona, el cloruro
de hexadecilpiridinio monohidrato y la dibutilanilina (Sigma) fueron utilizados tal como
se recibieron, el cloruro de dodecilpiridinio monohidrato (Sigma) fue sometido a
ebullición con carbón activo en acetona para luego ser recristalizado tres veces desde
etanol.16 Del mismo modo, el Pireno (Sigma) fue recristalizado tres veces desde etanol.
Los solventes utilizados: n-hexanol, n-propanol e isopropanol, fueron de grado
cromatográfico o espectroscópico (Merck). El agua empleada, fue purificada con un
sistema Milli-Q Plus, Millipore.
Los surfactantes estudiados, 6-O laurilsacarosa (MLS), 6-O miristilsacarosa (MMS), 6-
O palmitilsacarosa (MPS) y 6-O estearilsacarosa (MES), fueron obtenidos previamente
en el Laboratorio de Cinética y Fotoquímica de la Facultad de Ciencias Químicas y
Farmacéuticas de la Universidad de Chile mediante el procedimiento sintético que se
detalla más adelante.9

10
2.2 Instrumentación
Las medidas de fluorescencia en condiciones de irradiación estacionaria, se realizaron en
un espectrofluorímetro Fluorolog-Tau 2 (SPEX, Jobin Yvon) controlado a través de un
computador con el software DMF 300 que permite la obtención de espectros de
fluorescencia corregidos.
Tensiómetro de Du Noüy K8 KRÜSS, rango de medida 5 – 90 mN/m, precisión ± 0,1
mN/m, anillo Pt-Ir de diámetro igual a 20 mm y cubeta de vidrio de 50 mm de diámetro.
Las experiencias resueltas en el tiempo involucraron el uso de un láser de Nitrógeno PTI
modelo PL2300 con una potencia promedio de 3 mJ y un ancho medio de pulso de 1ns a
337 nm. Luego de seleccionar la longitud de onda con un monocromador 01-001 PTI, la
emisión de la muestra se detectó con un fotomultiplicador R928 (Hamamatsu). La
adquisición de la señal se realizó mediante un osciloscopio Hewlett Packard modelo
54540A de 1 GSa/s y 500 MHz, acoplado a un computador DTK 486. El análisis de los
datos obtenidos y el control de los experimentos se realizó con un programa desarrollado
en el Laboratorio de Cinética y Fotoquímica con el software LabView en lenguaje G.
Todas las experiencias se realizaron a temperatura controlada, empleando un baño
termorregulado Fisons, HAAKE F3.

11
3. METODOLOGÍA
3.1 Síntesis de los monoésteres de sacarosa
La síntesis de los compuestos estudiados se realizó mediante el método de trans-
esterificación de la sacarosa con el éster metílico del ácido graso correspondiente (11,
13, 15 y 17 unidades metilénicas).
La reacción fue llevada a cabo en un solvente polar aprótico (dimetilformamida) en
presencia de un catalizador básico (K2CO3), con agitación y calentamiento a presión
reducida durante varias horas. La mezcla de mono, di y triésteres recuperados del crudo
de reacción, se recristalizó en acetona varias veces para eliminar la sacarosa remanente.
Los monoésteres puros se aislaron empleando cromatografía en columna de sílice con
metanol:clorformo 3:1 como mezcla de elución. Los compuestos fueron caracterizados
por los métodos tradicionales (Resonancia Magnética Nuclear 1H y 13C, Cromatografía
Gaseosa, Cromatografía Líquida, Espectrometría de Masa y Análisis Elemental).9
3.2 Determinación de la concentración micelar crítica
Como es sabido, el proceso de micelización va acompañado de cambios significativos en
muchas de las propiedades fisicoquímicas del sistema. Este hecho ha permitido

12
O
N
(CH2)10CH3
desarrollar un gran número de métodos para la determinación experimental de los
valores de CMC, entre los que destacan las metodologías de fluorescencia y las
mediciones de tensión superficial.
En todas las metodologías de fluorescencia es necesario incorporar al sistema un
compuesto capaz de emitir luz, conocido como sonda. Las más adecuadas son aquellas
cuya fotofísica se ve fuertemente influenciada por la naturaleza del medio o
microambiente en el que se encuentran. Esta dependencia posibilita la caracterización
del entorno de la sonda, haciendo de este tipo de técnicas una herramienta ampliamente
utilizada en el estudio de sistemas que presentan más de una fase o microfase. La
selección de la sonda dependerá de la propiedad que se desea evaluar.
Las sondas seleccionadas para realizar los estudios fueron Laurdan (6-dodecanoil-2-
dimetilaminonaftaleno) y Pireno (Fig. 3).
Laurdan
Pireno
Figura 3. Estructura de las pruebas fluorescentes utilizadas.

13
La molécula de Laurdan experimenta interacciones de tipo Van der Waals entre su
cadena hidrocarbonada y la porción hidrofóbica de bicapas o micelas, lo que permite un
anclaje lo suficientemente fuerte para limitar su intercambio con el medio acuoso.
Debido a ésto, la sonda se encontrará en muy baja concentración en el agua, y
prácticamente la totalidad de su emisión provendrá de zonas hidrofóbicas o cercanas a la
interfase de los microagregados. El comportamiento espectroscópico del Laurdan
incorporado a microagregados está estrechamente relacionado no sólo con la polaridad
sino también con la fluidez de su entorno inmediato.17 Además, su rendimiento cuántico
de fluorescencia es mucho mayor cuando se localiza en un medio apolar que cuando se
encuentra en medio acuoso, los máximos de emisión en dodecano, dimetilsulfóxido y
metanol, se localizan alrededor de los 380, 460 y 490 nm respectivamente. El origen de
este desplazamiento ha sido explicado en base al efecto de relajación dipolar del
solvente,18 fenómeno que implica la reorientación de los dipolos que solvatan a la
prueba excitada cuando los tiempos de relajación y de vida de la prueba son del mismo
orden. La pérdida de energía asociada a esta difusión rotacional se ve reflejada en un
corrimiento de la emisión del Laurdan hacia el rojo. En el caso de bicapas, cuando la
estructura de la fase en la que se encuentra es de tipo gel, su emisión se ubica en la
región del azul, mientras que en fases líquidas cristalinas ésta se desplaza hacia la zona
del verde.18 El comportamiento experimental descrito para bicapas es equivalente al
observado en sistemas micelares, una variación en la viscosidad del entorno de la prueba
se ve reflejada en su espectro de emisión.
La separación de los componentes espectrales del Laurdan y la posibilidad de excitar
selectivamente moléculas de sonda en diferentes entornos, han sido utilizados para
desarrollar el concepto de polarización generalizada GP,17 definido como:

14
A B
A B
I IGPI I
−=
+ (1)
donde IA y IB corresponden a las intensidades de la emisión a 440 (A) y 490 nm (B),
longitudes de onda de máxima emisión del Laurdan en fases gel y líquido-cristalina de
bicapas, respectivamente. De esta forma, el valor de GP permite determinar
cuantitativamente la proporción de moléculas de Laurdan que se encuentran rodeadas
principalmente por fases liquido-cristalina o gel en sistemas fosfolipídicos. 17
Otro compuesto ampliamente utilizado como sonda fluorescente es el Pireno. Este
hidrocarburo aromático policíclico ha sido extensamente empleado para sensar polaridad
local o micropolaridad. Al encontrarse como monómero en solución, esta molécula
presenta cinco bandas vibrónicas principales en su espectro de emisión de fluorescencia.
La primera banda (I), localizada alrededor de los 373 nm es intensa (asociada a una
transición permitida) y su magnitud muestra escasa dependencia con la polaridad del
solvente. La intensidad de la tercera (III), ubicada a alrededor de los 384 nm, muestra un
significativo aumento en solventes polares. Las intensidades relativas de estas bandas
están determinadas por la magnitud del acoplamiento vibrónico entre el primer y
segundo estado singulete excitado.19 El comportamiento descrito para estas dos
transiciones, permitió desarrollar por primera vez hace más de 20 años una escala de
polaridades en función de la razón de ambas intensidades (I/III), denominada escala
Py.20

15
El carácter hidrofóbico del Pireno ha permitido el uso de la razón I/III para la
determinación de la CMC. La naturaleza apolar de este compuesto se traduce en su
solubilización preferente en el interior micelar, dando como resultado una fuerte
variación de la polaridad sensada por la prueba luego de la formación de los primeros
microagregados. El valor experimentalmente determinado para esta razón dependerá de
una serie de características propias del surfactante, tales como la longitud de la cadena
hidrocarbonada, la naturaleza del contraión en el caso de detergentes iónicos y
principalmente de la naturaleza del grupo polar. De acuerdo con lo anterior, ambas
sondas se emplearon para evaluar la CMC.
Para el Laurdan la concentración de trabajo en cubeta fue de 2 µM y la longitud de onda
de excitación 364 nm, valor que corresponde aproximadamente al máximo de emisión
de la sonda en el medio. Se registraron espectros de emisión desde 380 nm a 600 nm con
un tiempo de integración de 0,5 s y un incremento de 1 nm. A partir de estos espectros
se evaluó el parámetro GP de acuerdo a la ecuación (1), en función de la concentración
creciente de cada monoéster de sacarosa.
El procedimiento utilizado para el Pireno fue análogo, empleando una concentración de
0,1 µM para evitar la formación de excímero. Excitando a 337 nm, el barrido se realizó
entre 350 nm y 500 nm. A partir de las intensidades de emisión de las bandas I y III, se
obtuvo el valor de la razón I/III en función de la concentración de surfactante.
Paralelamente, se realizó la determinación de la CMC de los monoésteres utilizando un
método físico. Se evaluó la dependencia de la tensión superficial del sistema con la
concentración de surfactante mediante un tensiómetro de Du Noüy, hasta observar un
valor independiente de posteriores adiciones de monoéster. La temperatura fue
16
controlada a la décima de grado en todas las experiencias realizadas, tanto en el estudio
fotofísico como en las medidas de tensión superficial.
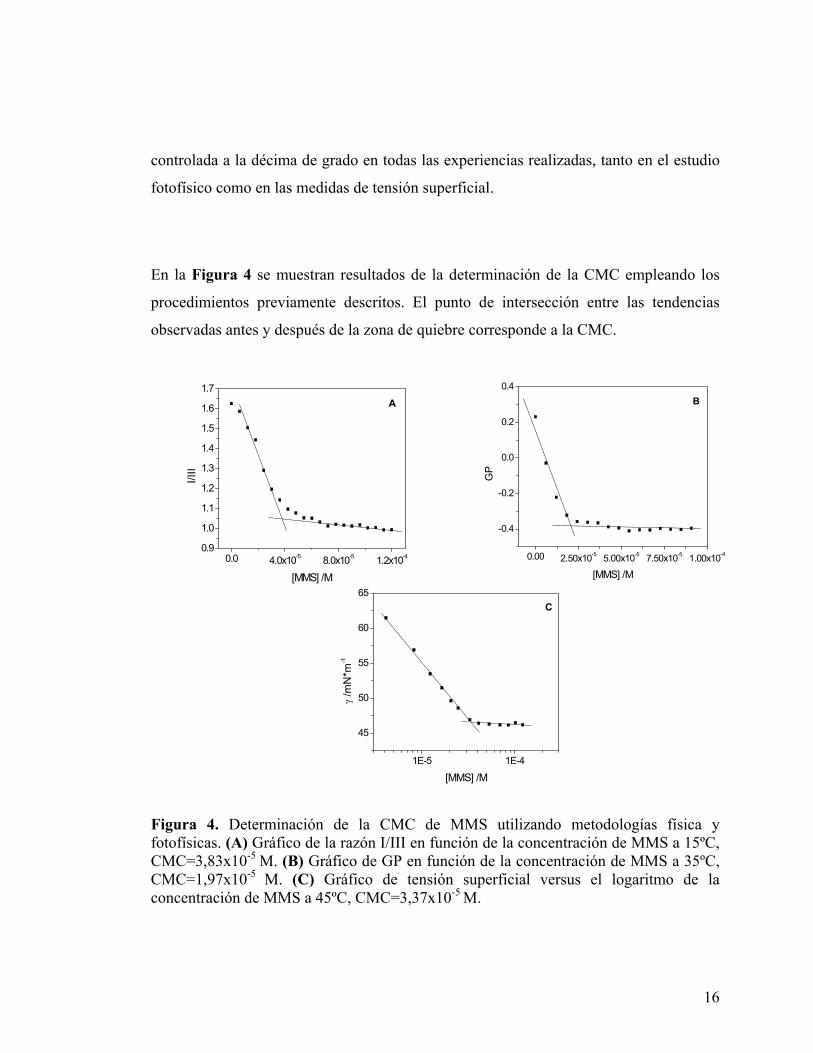
En la Figura 4 se muestran resultados de la determinación de la CMC empleando los
procedimientos previamente descritos. El punto de intersección entre las tendencias
observadas antes y después de la zona de quiebre corresponde a la CMC.
Figura 4. Determinación de la CMC de MMS utilizando metodologías física y fotofísicas. (A) Gráfico de la razón I/III en función de la concentración de MMS a 15ºC, CMC=3,83x10-5 M. (B) Gráfico de GP en función de la concentración de MMS a 35ºC, CMC=1,97x10-5 M. (C) Gráfico de tensión superficial versus el logaritmo de la concentración de MMS a 45ºC, CMC=3,37x10-5 M.
1E-5 1E-4
45
50
55
60
65C
γ /m
N*m
-1
[MMS] /M
0.00 2.50x10-5 5.00x10-5 7.50x10-5 1.00x10-4
-0.4
-0.2
0.0
0.2
0.4
GP
[MMS] /M
B
0.0 4.0x10-5 8.0x10-5 1.2x10-40.9
1.0
1.1
1.2
1.3
1.4
1.5
1.6
1.7A
I/III
[MMS] /M

17
3.3 Números de agregación
3.3.1 Determinación por desactivación de fluorescencia en estado
estacionario (SSFQ)
Este método se basa en la iluminación continua del sistema y el posterior registro de la
intensidad de emisión de una prueba fluorescente totalmente incorporada a las micelas,
mediante un espectrofluorímetro. Las medidas se realizan en ausencia y en presencia de
concentraciones crecientes de un desactivador o apagador hidrofóbico. La utilización de
esta metodología requiere satisfacer ciertas condiciones que restringen los números de
agregación que pueden ser evaluados con ella. Entre estas condiciones se pueden
mencionar: (i) la razón cinética kq / k0 (donde kq es la constante de velocidad de
desactivación intramicelar y k0 es la constante de velocidad de decaimiento de la sonda)
debe ser suficientemente grande de modo que cualquier micela que contenga una
molécula de sonda fluorescente y una o más moléculas de desactivador no emita luz, (ii)
la desactivación debe ser de naturaleza estrictamente dinámica y (iii) no debe existir
migración intermicelar de moléculas de sonda o desactivador durante la escala de tiempo
del experimento.12 Cumpliéndose estas condiciones y, suponiendo que la distribución de
moléculas de sonda y desactivador obedece la estadística de Poisson, se encuentra que:
[ ][ ]
QMicelao
Q
Ie
I
=
(2)

18
donde I0/IQ corresponde a la razón de intensidades de emisión en ausencia (I0) y en
presencia de apagador (IQ), [Q] es la concentración de desactivador y [Micela] es la
concentración micelar.
Graficando el logaritmo natural de la razón de intensidades I0/IQ evaluadas a
concentración constante de surfactante, versus la concentración de desactivador [Q], es
posible obtener el número de agregación promedio nag, según:
[ ]agC CMCn
Micela−
= (3)
donde C es la concentración total de surfactante y CMC la concentración micelar crítica.
Para estas experiencias se utilizó Pireno como sonda fluorescente en concentración igual
a 0,1 µM. Los desactivadores seleccionados fueron: benzofenona, cloruro de
hexadecilpiridinio, cloruro de dodecilpiridinio y dibutilanilina. Las concentraciones
crecientes de desactivador se obtuvieron por adición directa de solución concentrada del
compuesto. La razón entre las concentraciones de desactivador y micela se mantuvo
entre 0 y 2. El Pireno se excitó con luz de longitud de onda igual a 337 nm y su emisión
se observó a 373 nm.
3.3.2 Determinación por desactivación de fluorescencia resuelta en el
tiempo (TRFQ)

19
Este método se basa en la excitación de la sonda incorporada a las micelas con un pulso
de luz muy corto. La intensidad de emisión es registrada en función del tiempo. En
ausencia de desactivador, la prueba excitada decae en forma monoexponencial con una
constante de velocidad k0:
00
1=k
τ (4)
donde τ0 corresponde al tiempo de vida de la sonda en el entorno micelar. La presencia
de un desactivador afecta las curvas de decaimiento de modo que éstas dejan de ser
monoexponenciales y se ajustan a la ecuación:
( )1
0
− − − − =Q
o
k tt R eI I e τ
(5)
donde I(t) e I(0) son las intensidades de fluorescencia a los tiempos t=t y t=0 medidas
luego del pulso de luz, y R corresponde a la relación:
[ ][ ]
QR
Micela= (6)

20
El valor del número de agregación puede obtenerse a través de la expresión:
( )[ ]ag
C CMCn R
Q−
= (7)
donde C es la concentración total de surfactante, CMC corresponde a la concentración
micelar crítica y [Q] a la concentración de desactivador. Cuando se usa Pireno como
sonda, una de las restricciones de este método es que la razón molar entre las
concentraciones de sonda y micelas debe ser menor que 0,05 para prevenir la formación
de excímero. Además, el valor de R debe mantenerse cercano a 1, pues la presencia de
concentraciones mayores de desactivador puede afectar la estructura micelar.
A tiempos largos, 1Q
t k , la ecuación (5) se reduce a la expresión:
( )( ) 00
→∞
= − −
t
I t tLn RI τ (9)
Como consecuencia, a tiempos largos en una representación semilogarítmica las curvas
de decaimiento exhiben un comportamiento lineal con pendiente –1/τ0 e intercepto R.

21
La sonda y desactivantes empleados fueron los mismos que para las experiencias en
estado estacionario.
3.4 Efecto de la adición de alcoholes sobre algunas
propiedades de los sistemas micelares
3.4.1 Concentración micelar crítica
El procedimiento para evaluar la CMC en presencia de alcohol es el mismo que se
utilizó para su determinación en agua pura. Se registró la variación de la razón I/III de
Pireno 0,1 µM en agua con diferentes concentraciones de n-hexanol, en función de la
concentración de monoéster. Los experimentos se realizaron utilizando tres
concentraciones de alcohol: 13,10 mM, 26,16 mM y 39,18 mM. Las concentraciones
crecientes de surfactante se obtuvieron adicionando alícuotas de solución concentrada de
detergente directamente a la cubeta, siendo despreciables los cambios en la
concentración de alcohol. La temperatura de los experimentos se mantuvo constante en
25±1ºC mediante un baño termostatizado.
3.4.2 Microviscosidad y micropolaridad

22
Para determinar los cambios que sufren los microagregados con la presencia de alcohol,
se evaluó la razón I/III del Pireno y la polarización generalizada (GP) del Laurdan en
presencia de concentraciones crecientes de los mismos. Como se mencionó previamente,
estos valores dan cuenta de la polaridad y fluidez del entorno de la sonda,
respectivamente. Ambas propiedades se evaluaron utilizando razones alcohol/surfactante
micelizado entre 0 y 60 para n-hexanol y entre 0 y 150 para n-propanol e isopropanol.
Todos los experimentos se realizaron a 25±1ºC.

23
4. RESULTADOS Y DISCUSIÓN
4.1 Evaluación de la concentración micelar crítica
De acuerdo a la metodología detallada en la sección previa, la variación de la razón I/III
del Pireno y la dependencia de la tensión superficial con la concentración de surfactante
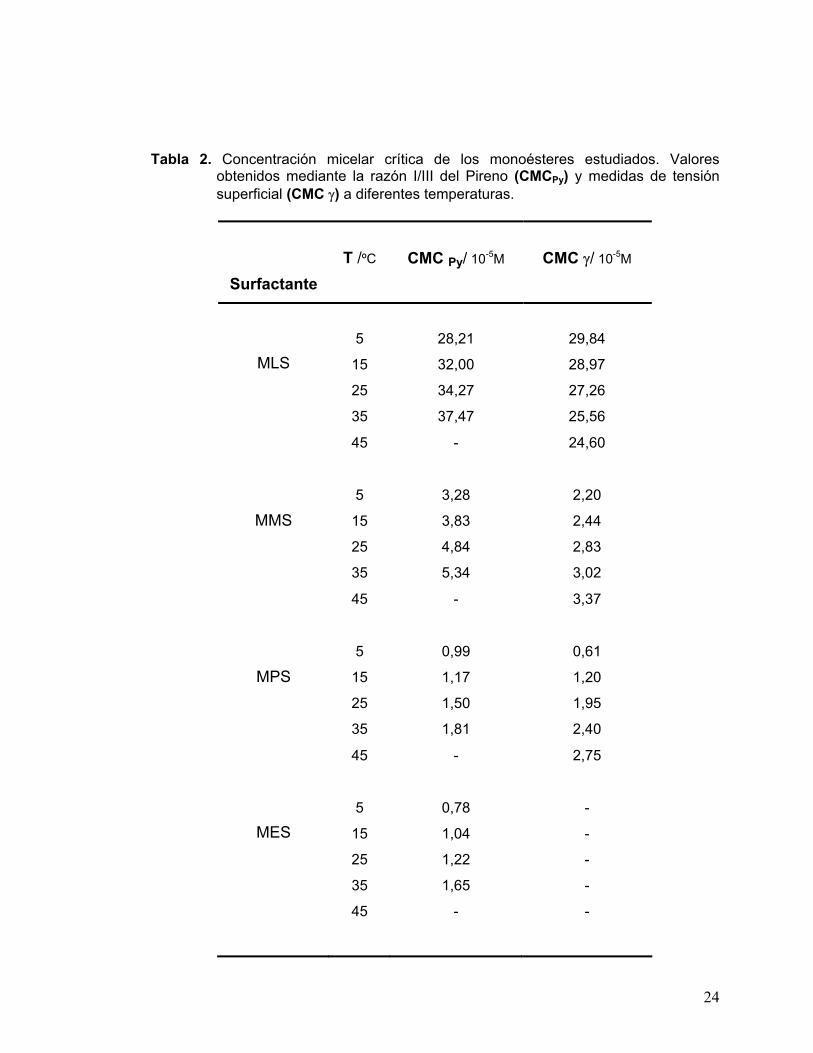
permiten la determinación gráfica de la CMC. En la Tabla 2 se resumen los valores de
CMC determinados mediante medidas de fluorescencia para los cuatro compuestos y por
medidas de tensión superficial para tres de ellos. En ambos casos las determinaciones se
realizaron a varias temperaturas.
La caracterización de MES a través del método físico no fue posible ya que los valores
de tensión superficial medidos a cada concentración de surfactante resultaron
irreproducibles, impidiendo la determinación de la CMC.
Adicionalmente, se determinó la CMC de los cuatro monoésteres utilizando la
polarización generalizada del Laurdan, en función de la temperatura. Los resultados
obtenidos en estas condiciones presentaron una tendencia en apariencia anómala; para
MLS la dependencia de la CMC con la temperatura resultó ser directa, para MPS es
inversa, mientras que para MMS y MES resultó ser prácticamente independiente. De
acuerdo con las observaciones mencionadas, los resultados obtenidos con esta
metodología no fueron empleados para determinar los cambios en las funciones
termodinámicas asociadas al proceso de micelización.
24
Tabla 2. Concentración micelar crítica de los monoésteres estudiados. Valores obtenidos mediante la razón I/III del Pireno (CMCPy) y medidas de tensión superficial (CMC γ) a diferentes temperaturas.
Surfactante T /ºC CMC Py/ 10-5M CMC γ/ 10-5M
5 28,21 29,84
15 32,00 28,97
25 34,27 27,26
MLS
35 37,47 25,56
45 - 24,60
5 3,28 2,20
15 3,83 2,44
25 4,84 2,83
MMS
35 5,34 3,02
45 - 3,37
5 0,99 0,61
15 1,17 1,20
25 1,50 1,95
MPS
35 1,81 2,40
45 - 2,75
5 0,78 -
15 1,04 -
25 1,22 -
35 1,65 -
MES
45 - -

25
Los valores obtenidos mediante medidas de tensión superficial resultaron muy similares
a los reportados en la literatura21 para 6-O-laurilsacarosa y 6-O-palmitilsacarosa
sintetizados enzimáticamente: 25 y 2,8x10-5 M, respectivamente (valores obtenidos
mediante el mismo método a 25 ºC).
Los resultados obtenidos por fluorescencia con Pireno como sonda y a partir de medidas
de tensión superficial, indican que el valor de la CMC es inversamente proporcional al
largo de cadena del surfactante; un mayor número de unidades metilénicas en la cadena
hidrocarbonada implica una menor solubilidad en solventes de carácter polar,
traduciéndose en la formación de microagregados a una menor concentración. Al
estudiar la dependencia de la CMC con el largo de la cadena hidrocarbonada, se observa
que la relación no es lineal, véase Figura 5. Si bien la CMC para el MES no pudo ser
determinada mediante el tensiómetro de Du Noüy, se aprecia que los resultados
obtenidos con ambos métodos presentan la misma tendencia. Por otra parte, los datos
experimentales no se ajustan a la ecuación empírica de Klevens 22, que predice una
dependencia lineal del logaritmo de la CMC con el número de unidades metilénicas de
la cadena hidrocarbonada, para surfactantes polioxietilénicos. Este comportamiento no
lineal puede observarse también al analizar datos de CMC informados en la literatura
para distintos ésteres de hidratos de carbono (obtenidos mediante tensión superficial). Si
visualizamos el proceso de micelización como un balance entre el efecto de la cabeza
hidrofílica y el de la cadena apolar, estos resultados indicarían que la variación de
energía libre está determinada principalmente por la alta hidratación de la sacarosa y su
efecto en el proceso de asociación.

26
11 12 13 14 15 16 17
1x10-5
1x10-4
A
C
MC
/M
n11 12 13 14 15
1x10-5
1x10-4
10-3
B
CM
C /M
n
Figura 5. Gráficos de CMC obtenida mediante fluorescencia (A) y tensión superficial (B) en función del número de átomos de carbono de la cadena hidrofóbica. (●) 5, (●) 15, (●) 25, (●) 35 y (●) 45ºC.
4.2 Evaluación de las propiedades termodinámicas del proceso
de micelización
Como se mostrará en la sección 4.5, los surfactantes estudiados forman micelas con
números de agregación menores que 100, hecho que permite utilizar el modelo de acción
de masas para determinar los parámetros termodinámicos asociados al sistema.23 Para
detergentes iónicos, graficando el logaritmo natural de la CMC en función de la
temperatura (Fig. 6), es posible evaluar los parámetros termodinámicos del proceso de
micelización haciendo uso de las ecuaciones (10) y (11):

27
2 ln1MP
m CMCH RTn T
∂ ∆ = − + ∂ (10)
1 lnMmG RT CMCn
∆ = + (11)
donde ∆HM corresponde al cambio de entalpía y ∆GM a la variación de energía libre de
Gibbs del proceso de micelización. Las variables n y m corresponden al número de
agregación y al número de contraiones respectivamente, mientras que R es la constante
de los gases y T la temperatura del proceso en grados Kelvin. La ausencia de contraiones
en el caso de micelas no iónicas, hace m igual a 0, por lo que ambas expresiones se
reducen a:
2 lnM
P
CMCH RTT
∂ ∆ = − ∂ (12)
lnMG RT CMC∆ = (13)
Una vez determinados ∆GM y ∆HM, es posible calcular la variación de entropía del
sistema según la relación:
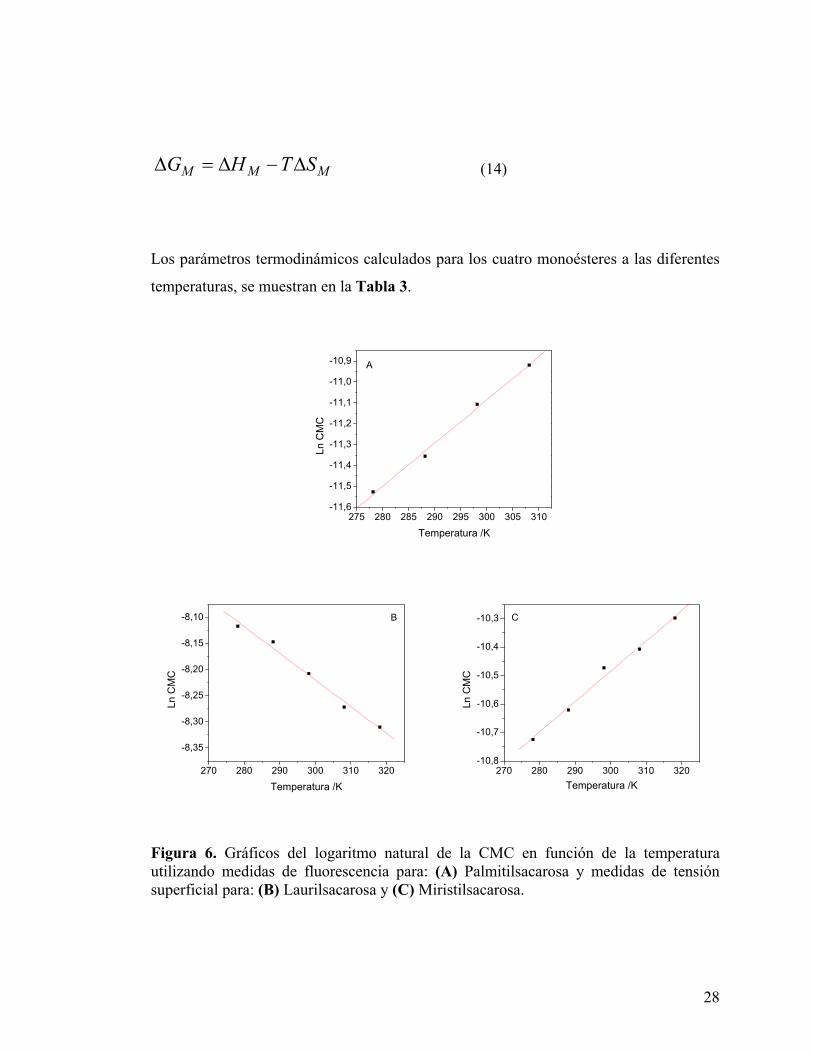
28
M M MG H T S∆ = ∆ − ∆ (14)
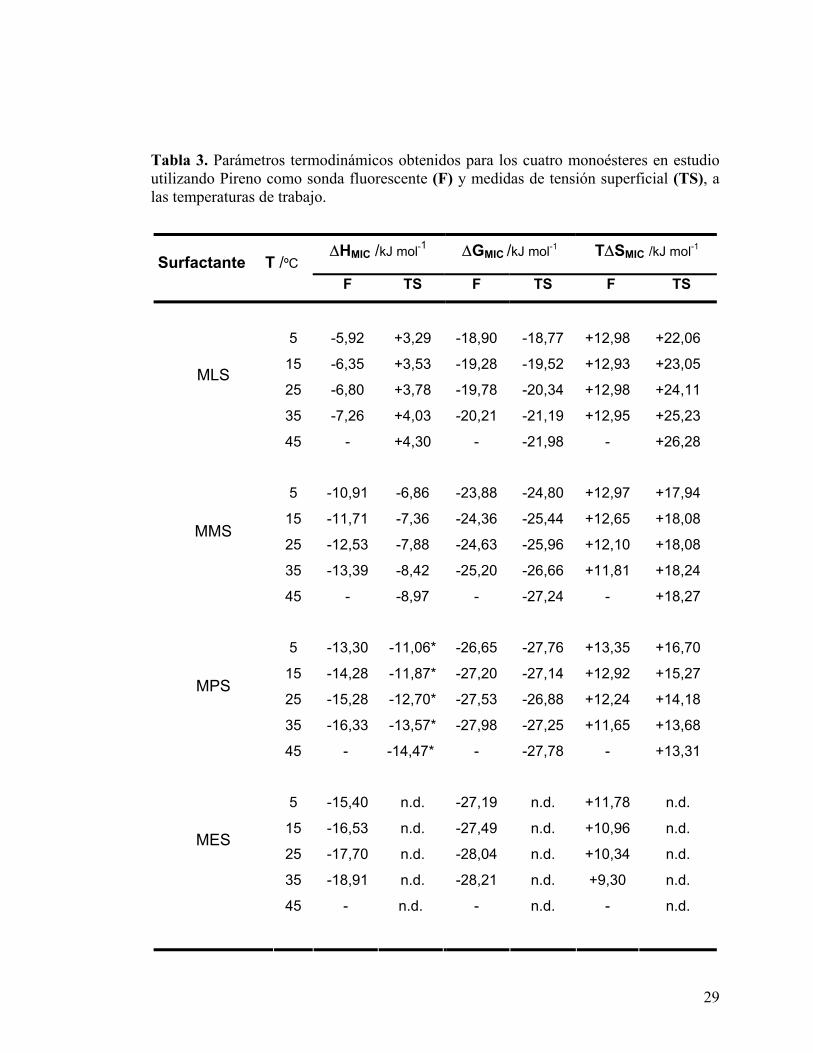
Los parámetros termodinámicos calculados para los cuatro monoésteres a las diferentes
temperaturas, se muestran en la Tabla 3.
Figura 6. Gráficos del logaritmo natural de la CMC en función de la temperatura utilizando medidas de fluorescencia para: (A) Palmitilsacarosa y medidas de tensión superficial para: (B) Laurilsacarosa y (C) Miristilsacarosa.
270 280 290 300 310 320-10,8
-10,7
-10,6
-10,5
-10,4
-10,3 C
Ln C
MC
Temperatura /K270 280 290 300 310 320
-8,35
-8,30
-8,25
-8,20
-8,15
-8,10 B
Ln C
MC
Temperatura /K
275 280 285 290 295 300 305 310-11,6
-11,5
-11,4
-11,3
-11,2
-11,1
-11,0
-10,9 A
Ln C
MC
Temperatura /K
29
Tabla 3. Parámetros termodinámicos obtenidos para los cuatro monoésteres en estudio utilizando Pireno como sonda fluorescente (F) y medidas de tensión superficial (TS), a las temperaturas de trabajo.
∆HMIC /kJ mol-1 ∆GMIC /kJ mol-1 T∆SMIC /kJ mol-1 Surfactante T /ºC
F TS F TS F TS
5 -5,92 +3,29 -18,90 -18,77 +12,98 +22,06
15 -6,35 +3,53 -19,28 -19,52 +12,93 +23,05
25 -6,80 +3,78 -19,78 -20,34 +12,98 +24,11
35 -7,26 +4,03 -20,21 -21,19 +12,95 +25,23
MLS
45 - +4,30 - -21,98 - +26,28
5 -10,91 -6,86 -23,88 -24,80 +12,97 +17,94
15 -11,71 -7,36 -24,36 -25,44 +12,65 +18,08
25 -12,53 -7,88 -24,63 -25,96 +12,10 +18,08
35 -13,39 -8,42 -25,20 -26,66 +11,81 +18,24
MMS
45 - -8,97 - -27,24 - +18,27
5 -13,30 -11,06* -26,65 -27,76 +13,35 +16,70
15 -14,28 -11,87* -27,20 -27,14 +12,92 +15,27
25 -15,28 -12,70* -27,53 -26,88 +12,24 +14,18
35 -16,33 -13,57* -27,98 -27,25 +11,65 +13,68
MPS
45 - -14,47* - -27,78 - +13,31
5 -15,40 n.d. -27,19 n.d. +11,78 n.d. 15 -16,53 n.d. -27,49 n.d. +10,96 n.d. 25 -17,70 n.d. -28,04 n.d. +10,34 n.d. 35 -18,91 n.d. -28,21 n.d. +9,30 n.d. 45 - n.d. - n.d. - n.d.
MES

30
n.d.= no detectado
* = valores calculados considerando los datos de CMC obtenidos entre 25-45ºC
Diversos estudios reportan que, para un gran número de surfactantes iónicos y no
iónicos, la curva de CMC versus temperatura presenta un mínimo15. La disminución
inicial de la CMC es consecuencia directa de la disminución en la hidratación del grupo
hidrofílico, efecto que predomina por sobre el de la cadena hidrocarbonada en cierto
rango de temperaturas. Este fenómeno es provocado por la menor posibilidad de
formación de enlaces de hidrógeno entre el grupo polar y las moléculas de agua que lo
rodean, al aumentar la temperatura. Debido a esto, la estabilidad de los monómeros en
solución disminuye, favoreciéndose la formación de micelas a menores concentraciones.
En contraste, el aumento de la temperatura causa además la ruptura de la estructura
ordenada del agua alrededor de la cadena hidrocarbonada, disminuyendo el aumento en
la entropía del sistema al comenzar la micelización. Consecuentemente, a mayores
temperaturas, la micelización se verá desfavorecida. Así, a medida que aumenta la
temperatura, el efecto de los grupos hidrofóbicos comienza a ejercer mayor influencia y
finalmente predomina al alcanzar la temperatura de cambio de pendiente. Un posterior
incremento en la temperatura provoca un aumento en la CMC. La existencia de una
temperatura a la cual la dependencia se invierte es resultado de un balance entre estos
dos fenómenos opuestos. Estudios realizados sobre surfactantes polioxietilénicos,
muestran que existe una dependencia inversa entre la temperatura a la cual se alcanza el
valor mínimo de la CMC y el largo de la cadena hidrofóbica. Cadenas de 10, 12 y 14
unidades metilénicas presentan temperaturas de cambio de pendiente de 61, 52 y 46ºC
respectivamente.15
Al emplear metodologías fluorescentes, con Pireno como sonda, todos los monoésteres
presentan dependencia directa de la CMC con la temperatura, lo que no implica la

31
ausencia de un mínimo o cambio de signo de la pendiente fuera del rango estudiado. Sin
embargo, las determinaciones realizadas a través de medidas de tensión superficial
muestran que, la CMC de los monoésteres con largo de cadena de 13 y 15 átomos de
carbono, depende de forma directa con la temperatura, mientras que el surfactante de 12
átomos de carbono exhibe una tendencia inversa (Fig. 6). Este comportamiento puede
atribuirse a que el rango de temperatura de trabajo es inferior a la temperatura de cambio
de pendiente para este compuesto, posiblemente debido a que el efecto de la cadena
hidrofóbica no es lo suficientemente importante para que prevalezca sobre el de la
sacarosa a las temperaturas estudiadas. Aunque ambos métodos son útiles para
determinar la CMC, los fenómenos frente a los cuales son sensibles son diferentes.
Mediante las medidas de tensión superficial es posible detectar la saturación de la
interfase, fenómeno que da lugar al comienzo de la formación de microagregados,
mientras que los experimentos realizados con medidas de fluorescencia son sensibles a
los cambios que presenta el Pireno en su emisión, cuando se encuentra asociado a los
microagregados. De acuerdo con lo anterior, las medidas de tensión superficial podrían
detectar antes el comienzo de la formación de micelas, ya que al comienzo del proceso,
la emisión del Pireno podría provenir mayoritariamente del medio acuoso como
consecuencia del reducido número de agregados formados. Por otra parte, la posible
presencia de pequeñas fracciones de di o poliésteres podría traducirse en una mayor
tendencia a ocupar la interfase, lo que traería consigo valores de CMC menores. En el
caso del Pireno, una posible formación de pre-agregados, como pequeños trozos de
bicapas o incluso la presencia de una bicapa en la interfase, podrían dar lugar a la
asociación temprana de la sonda a ambientes apolares.
Si bien la dependencia de la CMC con la temperatura es ampliamente utilizada para
estimar del cambio de entalpía en procesos de micelización, se han reportado algunas
dificultades en su uso. El número de agregación podría variar con la temperatura, lo que
se traduciría en valores de entalpía que contienen un término originado a partir de

32
cambios del tamaño micelar. Además, el proceso de micelización va acompañado de
cambios importantes en la capacidad calorífica, lo que significa una importante
dependencia de la entalpía con la temperatura. Como consecuencia, no se observaría
linealidad en gráficos del logaritmo de la CMC en función del inverso de la temperatura.
Este tipo de análisis normalmente muestra dependencias lineales, cuyo origen podría
deberse a la imprecisión en la medida de los valores de CMC o a que el rango de
temperaturas cubierto no es lo suficientemente amplio.24
A temperatura ambiente, las fuerzas responsables de la micelización son principalmente
interacciones de tipo hidrofóbico. Al comenzar la micelización, éstas provocan un
aumento importante en la entropía del sistema producto de la liberación de moléculas de
agua contenidas en la esfera de solvatación de las cadenas hidrofóbicas del surfactante,
lo que se traduce en valores de ∆SMIC positivos.25 Este aumento en la entropía del
sistema contribuye de manera importante al cambio de energía libre, prevaleciendo
incluso sobre los términos entálpicos desfavorables observados en algunos sistemas.26
Los valores de ∆GMIC calculados para los sistemas estudiados en este trabajo, son
similares a los descritos en la literatura para ésteres de sacarosa comerciales. Para el
derivado del ácido palmítico (mezcla de 70% de monoéster y 30% de diéster) se he
reportado un ∆GMIC -38.2 kJ mol-1.27
Existe evidencia de que la mayor contribución al aumento de la entropía al comenzar el
proceso de micelización corresponde a la transferencia de cadenas hidrocarbonadas
desde el medio acuoso hacia el interior micelar.24 Durante el proceso, el grupo polar sólo
sufriría cambios menores y prácticamente independientes de la temperatura en su
solvatación. No obstante, existen antecedentes28 que muestran que para la sacarosa, el
alto grado de hidratación del grupo polar provocado por el número de grupos hidroxilo

33
presentes, trae como resultado un cambio menos favorable en la entalpía asociada al
proceso que el reportado para surfactantes derivados de monosacáridos.
En cuanto al efecto del segmento hidrofóbico de los compuestos estudiados, los
resultados muestran que a medida que aumenta el largo de la cadena, el cambio de
entropía del sistema disminuye, sin embargo el ∆HMIC también decrece y lo hace en
mayor medida que el término entrópico. Por ello, para los surfactantes con cadena de 15
y 17 átomos de carbono, el término predominante es el entálpico a diferencia de lo
observado en los compuestos de cadena más corta. El balance de ambos términos trae
como resultado una disminución en el ∆GMIC y por lo tanto un aumento en la
espontaneidad del proceso de micelización a medida que la hidrofobicidad del
surfactante aumenta. Un comportamiento similar ha sido reportado para haluros de 1-
metil-4-alquilpiridinio, surfactantes iónicos cuyo proceso de micelización ha resultado
ser más exotérmico a medida que aumenta el largo de cadena.29 Se ha observado
también, que en soluciones de alcoholes alifáticos con largo de cadena de cuatro
carbonos o más, donde la influencia del grupo hidroxilo es reducida, más de dos tercios
de la energía libre de disolución corresponden a factores entálpicos, a diferencia del
predominio del término entrópico observado en soluciones de alcoholes de cadena corta.
Esta tendencia ha sido descrita también para series de óxidos de dimetil-n-alquilamino
con largo de cadena entre 8 y 12 átomos de carbono los cuales presentan valores de
∆SMIC prácticamente constantes.30 La disminución en los valores de ∆GMIC con el
aumento del largo de cadena hidrofóbica está estrechamente ligada con la disminución
en el cambio de entalpía del proceso. Los antecedentes recopilados y el comportamiento
observado indican que, termodinámicamente, el cambio de entropía es lo que impulsaría
el proceso de micelización en sistemas de compuestos con largo de cadena pequeño,
mientras que el término entálpico es el que predominaría en sistemas de surfactantes de
mayor largo de cadena.

34
Los valores de ∆HMIC calculados a partir de los experimentos realizados con Pireno
resultaron de signo negativo para los cuatro monoésteres estudiados en todo el rango de
temperaturas, mientras que utilizando medidas de tensión superficial este parámetro
resultó positivo para el MLS a todas las temperaturas de trabajo. Dado que los valores de
CMC determinados por tensión superficial coinciden con los reportados en la
literatura,21 es complejo dar cuenta de la diferencia de signo para la entalpía de
micelización determinada mediante ambos métodos. A pesar de las diferencias
mencionadas, la dependencia de este parámetro con la temperatura es semejante para los
dos métodos. Un aumento en la temperatura provoca una disminución en ambos
parámetros, ∆HMIC y ∆SMIC, sin embargo la disminución en el término entálpico es más
marcada que la correspondiente al término entrópico, lo que se traduce en valores de
∆GMIC levemente menores a medida que la temperatura se incrementa. Se ha observado
además, que la formación de micelas a temperaturas altas se debe principalmente a
factores entálpicos altamente favorables, mientras que a temperatura ambiente es el
aumento de entropía la principal contribución al valor negativo del ∆GMIC.25 El rango de
temperaturas estudiado en esta tesis no es suficiente para llegar a apreciar un posible
predominio del término entálpico.
4.3 Polaridad y microviscosidad de las micelas
Como es bien sabido, la razón entre las intensidades de las bandas I y III del Pireno es
sensible a la polaridad su entorno. Esta dependencia permitió el desarrollo de la escala
Py de polaridades,20 ya descrita en la sección 3.2.

35
Por otra parte, como se indicó previamente, el parámetro GP del Laurdan da cuenta de la
microviscosidad y fluidez de su entorno. Como consecuencia de su estructura, la
molécula de Laurdan se ancla preferentemente en zonas hidrofóbicas o cercanas a la
interfase de los microagregados.17
Se evaluó la razón I/III y el parámetro GP en la CMC y en la zona del plateau. Los
valores obtenidos se resumen en la Tabla 4.
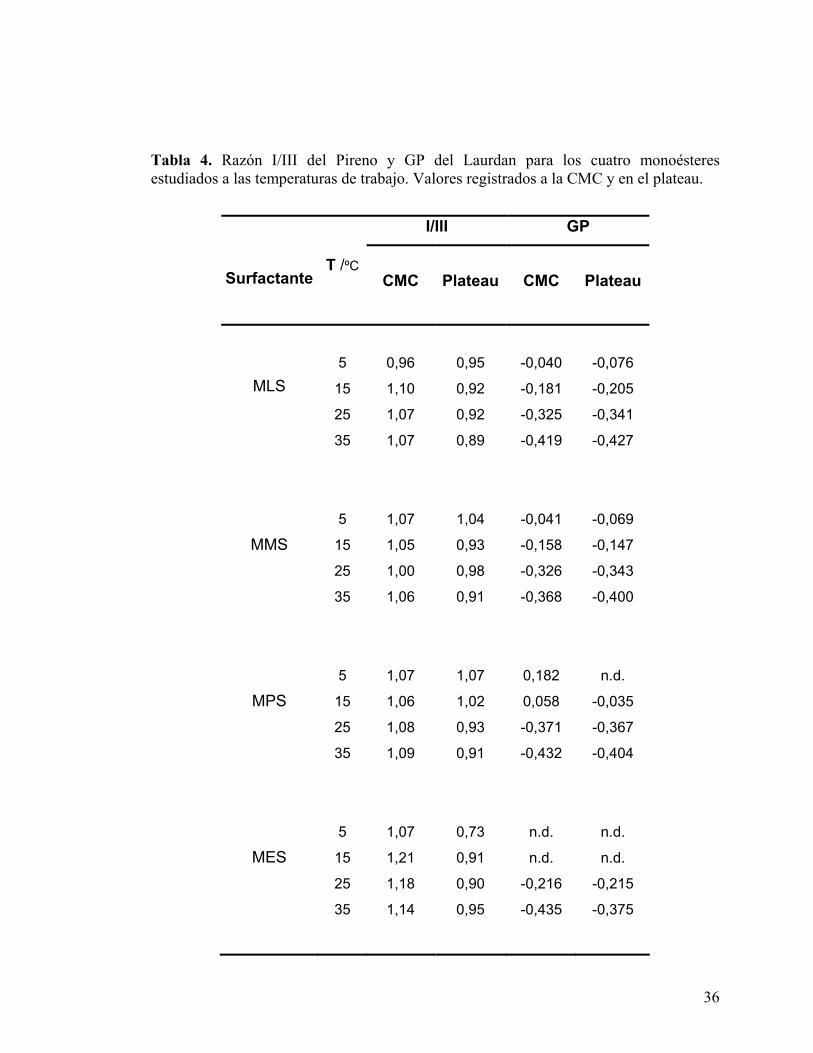
Los valores determinados para la razón I/III no muestran una dependencia clara con la
temperatura ni con el largo de cadena, sugiriendo que el Pireno sensa ambientes
parecidos en todos los casos. Este comportamiento puede deberse a que los
microagregados formados proporcionan microambientes similares o porque existe una
migración de la sonda hacia ambientes con características semejantes. Utilizando la
escala Py como referencia, se advierte que el entorno sensado por el Pireno sería similar
a aquel percibido por la prueba en n-octanol puro. En todos los casos se observa una leve
disminución de la razón I/III con el aumento en la concentración de micelas.
36
Tabla 4. Razón I/III del Pireno y GP del Laurdan para los cuatro monoésteres estudiados a las temperaturas de trabajo. Valores registrados a la CMC y en el plateau.
I/III GP
Surfactante
T /ºCCMC Plateau CMC Plateau
5 0,96 0,95 -0,040 -0,076
15 1,10 0,92 -0,181 -0,205
25 1,07 0,92 -0,325 -0,341
MLS
35 1,07 0,89 -0,419 -0,427
5 1,07 1,04 -0,041 -0,069
15 1,05 0,93 -0,158 -0,147
25 1,00 0,98 -0,326 -0,343
MMS
35 1,06 0,91 -0,368 -0,400
5 1,07 1,07 0,182 n.d.
15 1,06 1,02 0,058 -0,035
25 1,08 0,93 -0,371 -0,367
MPS
35 1,09 0,91 -0,432 -0,404
5 1,07 0,73 n.d. n.d.
15 1,21 0,91 n.d. n.d.
25 1,18 0,90 -0,216 -0,215
MES
35 1,14 0,95 -0,435 -0,375

37
n.d. = no detectado
El Laurdan resultó ser una sonda inadecuada para la determinación de los parámetros
termodinámicos de los microagregados, sin embargo es útil para dar cuenta de las
características de su entorno. Las experiencias realizadas sobre los compuestos de
cadena más corta, MLS y MMS, dieron como resultado curvas con un quiebre claro a
todas las temperaturas, sin embargo, MPS y MES no pudieron ser caracterizados a bajas
temperaturas debido a la imposibilidad de determinar la CMC: no existe quiebre ni zona
de plateau. Valores altos de GP implican una baja accesibilidad del agua al sitio de
solubilización de la sonda y por lo tanto una menor fluidez del entorno. Los valores de
GP evaluados en la CMC y el plateau, no presentan una variación significativa para un
mismo surfactante, indicando que las características del ambiente sensado por la prueba
son independientes de la concentración de micelas presentes en el sistema. A mayor
largo de cadena hidrofóbica, se observa un leve aumento en los valores de GP del
plateau, indicando una posible diferencia en la distribución de las moléculas de sacarosa
de acuerdo con el radio de la micela. La accesibilidad del agua estaría principalmente
limitada por la presencia de las cabezas voluminosas con gran número de grupos
hidroxilo. La diferencia en el largo de cadena de los microagregados no es suficiente
para modificar de manera significativa esta accesibilidad. Se observa además que al
aumentar la temperatura, el valor límite de GP decrece de forma sistemática, indicando
una mayor posibilidad de acceso del agua producido probablemente por la mayor
agitación térmica de las moléculas de surfactante.

38
4.4 Actividad superficial de los monoésteres
Las medidas de tensión superficial permiten calcular una serie de parámetros propios de
cada compuesto. Graficando la tensión superficial del sistema en función del logaritmo
natural de la concentración de detergente, y suponiendo que la adsorción de moléculas
de surfactante en la interfase es descrita por la isoterma de adsorción de Gibbs, es
posible calcular el exceso superficial, Γ, y el área ocupada por molécula, A, según las
siguientes expresiones:
,
1ln T PRT Cγ∂ Γ = − ∂ (15)
1
AA
N=
Γ (16)
donde R es la constante universal de los gases, T la temperatura absoluta y NA el número
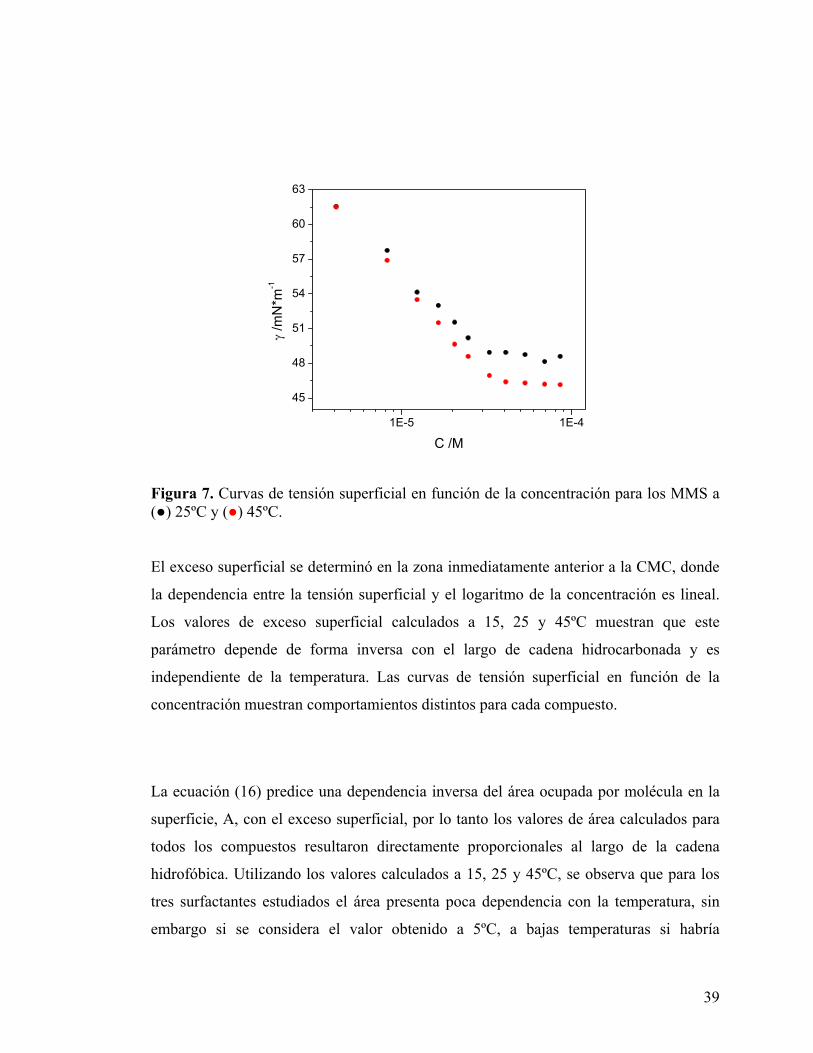
de Avogadro. Los perfiles de las curvas obtenidas para MMS se muestran en la Figura
7.
39
1E-5 1E-4
45
48
51
54
57
60
63
γ /m
N*m
-1
C /M
Figura 7. Curvas de tensión superficial en función de la concentración para los MMS a (●) 25ºC y (●) 45ºC. El exceso superficial se determinó en la zona inmediatamente anterior a la CMC, donde
la dependencia entre la tensión superficial y el logaritmo de la concentración es lineal.
Los valores de exceso superficial calculados a 15, 25 y 45ºC muestran que este
parámetro depende de forma inversa con el largo de cadena hidrocarbonada y es
independiente de la temperatura. Las curvas de tensión superficial en función de la
concentración muestran comportamientos distintos para cada compuesto.
La ecuación (16) predice una dependencia inversa del área ocupada por molécula en la
superficie, A, con el exceso superficial, por lo tanto los valores de área calculados para
todos los compuestos resultaron directamente proporcionales al largo de la cadena
hidrofóbica. Utilizando los valores calculados a 15, 25 y 45ºC, se observa que para los
tres surfactantes estudiados el área presenta poca dependencia con la temperatura, sin
embargo si se considera el valor obtenido a 5ºC, a bajas temperaturas si habría

40
dependencia para el MPS. Los valores evaluados para este parámetro se muestran en la
Tabla 5. De acuerdo con lo mencionado, el valor de A depende no sólo de las
dimensiones moleculares del grupo hidrofílico del surfactante incluyendo una posible
esfera de solvatación, sino también de la estereoquímica y el empaquetamiento de la
estructura completa.31
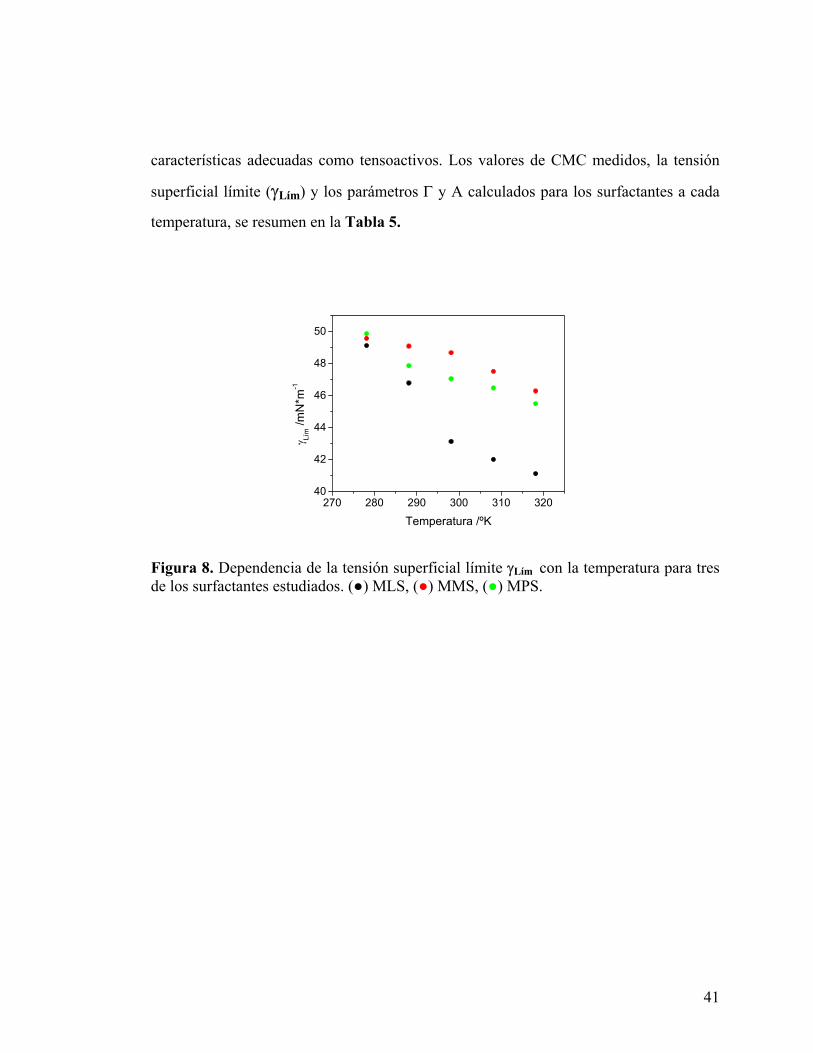
Adicionalmente, a partir de las curvas de tensión superficial, Fig. 7, es posible
determinar la tensión superficial límite (γLím) para cada monoéster. Este parámetro
constituye una medida de la capacidad de un compuesto para disminuir la tensión
superficial del sistema al cual es agregado y por lo tanto de su capacidad como
surfactante. Los resultados obtenidos muestran una disminución de la γLím con el
aumento de la temperatura para los tres monoésteres estudiados, Fig. 8. Este
comportamiento es análogo a la dependencia de la tensión superficial con la temperatura
para líquidos puros, sin embargo, los resultados observados en las soluciones micelares
muestran una leve desviación del comportamiento lineal observado en soluciones
homogéneas. Estas desviaciones de la linealidad pueden estar relacionadas con las
interacciones tipo puente de hidrógeno que tienen lugar entre las moléculas de
surfactante y las de agua, que además de estar presentes en un alto grado, son sensibles a
la temperatura. Este tipo de interacciones son las responsables de la solubilidad de los
monoésteres en agua.32 La pendiente observada resulta mucho más marcada en el caso
del MLS, disminuyendo la tensión superficial del agua en alrededor de 5 mN m-1 más
que los otros surfactantes a 45ºC. Los experimentos realizados revelan que, a
temperaturas menores que la ambiente, la γLím depende levemente del largo de la cadena
hidrocarbonada. A mayores temperaturas, la diferencia entre las γLím alcanzadas por
MMS y MPS continúa siendo leve, mientras que para el MLS esta diferencia se hace
cada vez mayor. Los valores de γLím determinados para los cuatro monoésteres,
levemente superiores a los reportados en la literatura,31 muestran que estos presentan
41
características adecuadas como tensoactivos. Los valores de CMC medidos, la tensión
superficial límite (γLím) y los parámetros Γ y A calculados para los surfactantes a cada
temperatura, se resumen en la Tabla 5.
270 280 290 300 310 32040
42
44
46
48
50
γ Lí
m /m
N*m
-1
Temperatura /ºK
Figura 8. Dependencia de la tensión superficial límite γLím con la temperatura para tres de los surfactantes estudiados. (●) MLS, (●) MMS, (●) MPS.
42
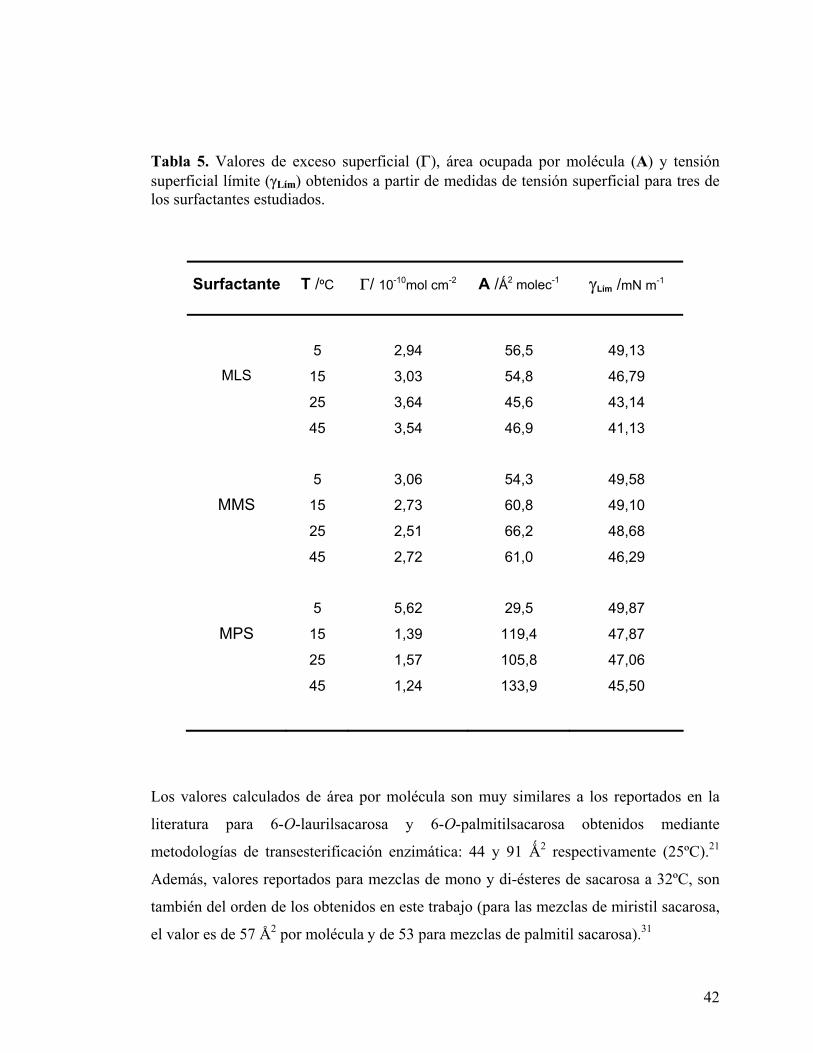
Tabla 5. Valores de exceso superficial (Γ), área ocupada por molécula (A) y tensión superficial límite (γLím) obtenidos a partir de medidas de tensión superficial para tres de los surfactantes estudiados.
Surfactante T /ºC Γ/ 10-10mol cm-2 A /Ǻ2 molec-1 γLím /mN m-1
5 2,94 56,5 49,13
15 3,03 54,8 46,79
25 3,64 45,6 43,14
MLS
45 3,54 46,9 41,13
5 3,06 54,3 49,58
15 2,73 60,8 49,10
25 2,51 66,2 48,68
MMS
45 2,72 61,0 46,29
5 5,62 29,5 49,87
15 1,39 119,4 47,87
25 1,57 105,8 47,06
MPS
45 1,24 133,9 45,50
Los valores calculados de área por molécula son muy similares a los reportados en la
literatura para 6-O-laurilsacarosa y 6-O-palmitilsacarosa obtenidos mediante
metodologías de transesterificación enzimática: 44 y 91 Ǻ2 respectivamente (25ºC).21
Además, valores reportados para mezclas de mono y di-ésteres de sacarosa a 32ºC, son
también del orden de los obtenidos en este trabajo (para las mezclas de miristil sacarosa,
el valor es de 57 Å2 por molécula y de 53 para mezclas de palmitil sacarosa).31

43
El valor evaluado para el área ocupada por molécula, A, permite realizar una estimación
teórica de la geometría de los microagregados formados, aplicando el Parámetro de
Empaquetamiento Crítico, Cpp, definido por Söderberg:33
o cCpp
a lυ
= (17)
donde ν corresponde al volumen de la cadena hidrocarbonada, ao al área del grupo polar
y lc al largo promedio de la cadena hidrofóbica.
Para el caso de cadenas hidrocarbonadas saturadas se han desarrollado las siguientes
relaciones:33
27.4 26.9nυ = + (en Ǻ3) (18)
1.5 1.265c maxl l n≤ = + (en Ǻ) (19)
donde n es el número de átomos de la cadena. Dado que las ecuaciones (18) y (19) están
definidas para hidrocarburos saturados, el largo efectivo de cadena de los ácidos grasos

44
podría calcularse de manera aproximada usando n = nc–1, donde nc es el número de
átomos de carbono del ácido graso. Según su magnitud, el Cpp predice formas esféricas,
cilíndricas, bicapas y estructuras inversas.
Si suponemos ao = A, es posible predecir el tipo de microagregado formado para cada
surfactante a las temperaturas de trabajo (Tabla 6). La forma de los microagregados
predicha por este método es sólo una estimación basada en consideraciones geométricas
simples, y no proporciona información acerca de la polidispersidad de los
microagregados presentes en el sistema.

45
Tabla 6. Parámetro de empaquetamiento crítico y posible geometría de los microagregados en estudio.
Cpp Geometría
< 0,33 Esférica
0,33 – 0,50 Cilíndrica
0,50 - 1 Bicapas
> 1 Estructuras inversas
Surfactante T /ºC Cpp
5 0,37
15 0,38
25 0,45 MLS
45 0,44
5 0,38
15 0,34
25 0,32 MMS
45 0,34
5 0,70
15 0,18
25 0,20 MPS
45 0,16

46
De acuerdo con los valores obtenidos para este parámetro, se observa que los
monoésteres de cadena corta formarían micelas de geometría cilíndrica, mientras que los
microagregados formados por MPS tendrían forma esférica.
4.5 Números de agregación
El número de agregación es un parámetro muy importante en la caracterización de
sistemas micelares y, como se mencionó anteriormente, ha sido obtenido mediante
numerosos métodos.12 Entre las diferentes técnicas empleadas para su obtención, las más
frecuentemente utilizadas son aquellas basadas en la fluorescencia, debido a que la
propiedad medida es independiente de factores como la forma de los microagregados y
las interacciones intermicelares, además de la amplia disponibilidad de los instrumentos
requeridos. Debido a que este método no se basa en propiedades difusionales, una de las
mayores ventajas de esta técnica radica en que el número de agregación puede ser
determinado incluso cuando los microagregados interactúan con interfases o polímeros
en solución. Por otra parte, como en todos los métodos invasivos, el efecto provocado
por la presencia de la prueba y/o el desactivador sobre el sistema debe ser mantenido lo
más bajo posible.34
La determinación del número de agregación por métodos fotofísicos se basa en la
desactivación de la fluorescencia de una sonda totalmente incorporada a la micela con
un desactivador hidrofóbico. El número discreto de microagregados influye en la
distribución de las moléculas de apagador entre las micelas, de esta forma, variando la
concentración de surfactante es posible promover o inhibir totalmente las reacciones
bimoleculares de la sonda excitada (en el caso del Pireno, impidiendo o favoreciendo la
formación de excímero). Los métodos fluorescentes pueden ser clasificados en dos tipos

47
de acuerdo al modo de excitación y detección de la emisión: desactivación de
fluorescencia en estado estacionario (SSFQ) y desactivación de fluorescencia resuelta en
el tiempo (TRFQ), Fig. 9. Ambas técnicas fueron descritas en las secciones 3.3.1 y 3.3.2
respectivamente.
Las medidas en estado estacionario son mucho más fáciles de realizar y analizar que las
resueltas en el tiempo, sin embargo existen ciertas restricciones en su uso que reducen el
rango de números de agregación que es posible estudiar. Una de las principales
limitantes para el uso de SSFQ es que la constante de velocidad de desactivación kQ
debe ser mucho mayor que la constante de decaimiento de la sonda en ausencia de
desactivador, k0. Los datos experimentales12 muestran que cuando la razón kQ/k0
decrece, los valores de nag obtenidos son cada vez menores, produciéndose un error por
defecto. Por otra parte, el método de SSFQ requiere que sólo opere el mecanismo de
desactivación dinámica y no exista desactivación de tipo estática. La desactivación
estática se produce cuando la sonda en su estado basal forma un complejo con el
desactivador que no emite. Cuando ambos mecanismos operan simultáneamente, es
posible que el error por defecto (observado cuando el valor de la razón kQ/k0 no es lo
suficientemente alto), sea compensado parcialmente o incluso superado, obteniéndose
nag mayores que los reales.
Los experimentos de TRFQ no presentan las limitaciones debidas a la magnitud relativa
de las constantes y el hecho de que exista desactivación estática en conjunto con la
dinámica sólo afecta los valores de la intensidad de emisión inicial de la prueba, no los
valores del número de agregación ni de kQ. La principal dificultad de este método se
presenta cuando el valor de la razón kQ/k0 es cercano a 1. En este caso, el
comportamiento límite lineal esperado para los decaimientos de la sonda se alcanza sólo
a tiempos muy largos, lo que requiere necesariamente el uso de un contador de fotones y

48
tiempos de acumulación prolongados. Si esta condición no es satisfecha, es posible que
los ajustes de las curvas de decaimiento originen valores del tiempo de vida de la prueba
menores en presencia de desactivador que en ausencia de él, sugiriendo erróneamente
migración de la sonda y/o desactivador en la escala de tiempo de la fluorescencia.
Debido a que los nag son determinados a partir de estos parámetros de ajuste, los valores
obtenidos serán también erróneos. Las razones cinéticas kQ/k0 de los sistemas estudiados
varían entre 0,75 y 9 por lo que se espera que los números de agregación obtenidos
mediante TRFQ sean confiables.
A diferencia del método resuelto en el tiempo, los experimentos realizados en estado
estacionario requieren que ambos, sonda y desactivador, posean un tiempo de vida
menor que el tiempo de residencia en el microagregado. Empleando números de
ocupación mucho menores que uno, sondas hidrofóbicas como el Pireno exhiben un
decaimiento monoexponencial y un tiempo de vida mucho mayor que el observado en
agua, sugiriendo su total asociación a los microagregados. Dado que la constante de
desactivación kQ depende del entorno donde se encuentra el desactivador y la prueba, la
migración de ésta en la escala de tiempo del experimento puede hacer que incluso
cuando los gráficos de I0/IQ versus concentración de desactivador son lineales, los
números de agregación obtenidos a partir de ellos resulten erróneos.
En este trabajo se determinó el número de agregación de sistemas micelares de diferente
concentración de los cuatro monoésteres en estudio mediante SSFQ y TRFQ. Las
soluciones micelares de cada surfactante fueron preparadas con una concentración por lo
menos 3 veces mayor que la CMC determinada para el compuesto a la temperatura de
trabajo. El comportamiento observado indica que el número de agregación es
independiente de la concentración de surfactante, por lo que los valores informados

49
corresponden a un promedio. Los resultados obtenidos utilizando Pireno como sonda
fluorescente y diferentes desactivadores, se resumen en la Tabla 7.
Tabla 7. Números de agregación de sistemas micelares de los cuatro monoésteres estudiados. Valores obtenidos a partir de la desactivación de fluorescencia del Pireno en estado estacionario (nEE) y resuelta en el tiempo (nRT) mediante diferentes sustratos. Se incluyen además los valores de tiempo de vida del Pireno τ0 y la constante de desactivación kQ para cada compuesto.
Surfactante Desactivador nEE nRT τ0 /10-7 s kQ /106 s-1
BP 52,9 57 0,26
HDPC 72,1 70,1 24,1 DDPC 52 75 0,40 MLS DBA 50
3,20
26,0
BP 51 50 0,26 HDPC 62,1 67,1 45,4 DDPC 55 70,7 0,60 MMS DBA 55 90
3,30
9,0
BP 41,7 HDPC 54,9 72,6 37,1 DDPC MPS DBA 95 80
3,23
12,0
BP 14,3 46 0,23 HDPC 32,8 DDPC 45 0,26 MES DBA n.d. n.d.
3,26
n.d. = no detectado
BP: benzofenona, HDPC: cloruro de hexadecilpiridinio, DDPC: cloruro de
dodecilpiridinio y DBA: dibutilanilina.
50
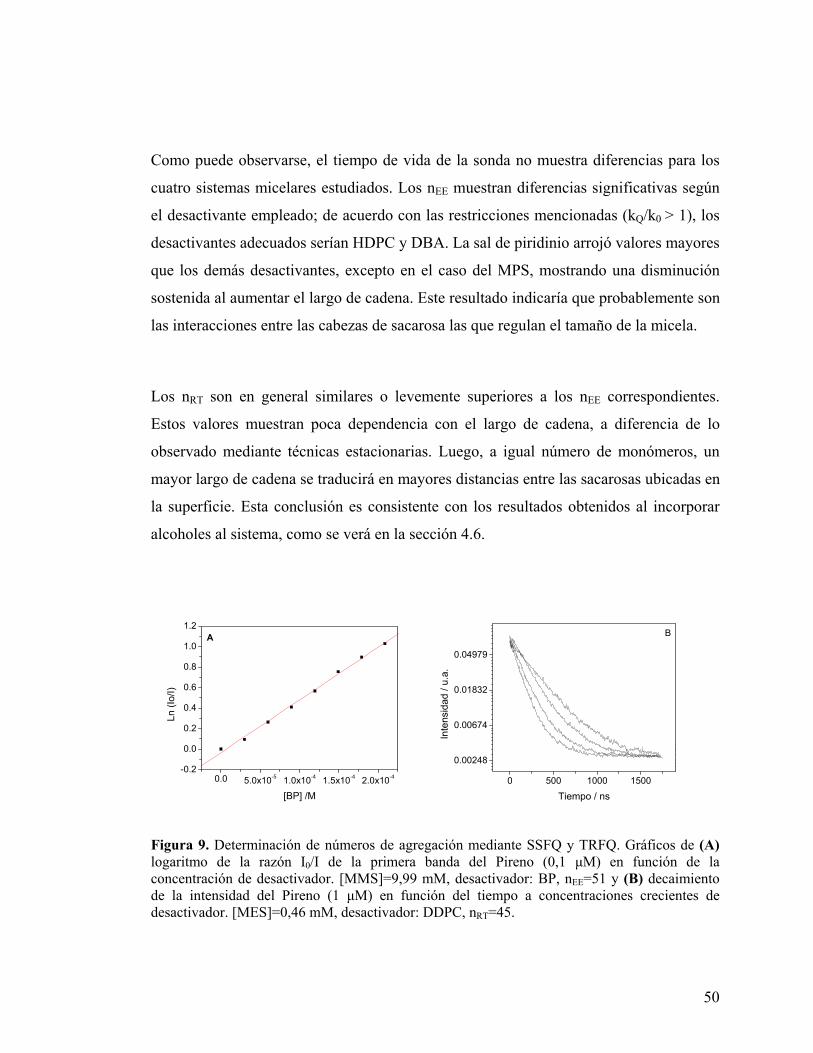
Como puede observarse, el tiempo de vida de la sonda no muestra diferencias para los
cuatro sistemas micelares estudiados. Los nEE muestran diferencias significativas según
el desactivante empleado; de acuerdo con las restricciones mencionadas (kQ/k0 > 1), los
desactivantes adecuados serían HDPC y DBA. La sal de piridinio arrojó valores mayores
que los demás desactivantes, excepto en el caso del MPS, mostrando una disminución
sostenida al aumentar el largo de cadena. Este resultado indicaría que probablemente son
las interacciones entre las cabezas de sacarosa las que regulan el tamaño de la micela.
Los nRT son en general similares o levemente superiores a los nEE correspondientes.
Estos valores muestran poca dependencia con el largo de cadena, a diferencia de lo
observado mediante técnicas estacionarias. Luego, a igual número de monómeros, un
mayor largo de cadena se traducirá en mayores distancias entre las sacarosas ubicadas en
la superficie. Esta conclusión es consistente con los resultados obtenidos al incorporar
alcoholes al sistema, como se verá en la sección 4.6.
0.0 5.0x10-5 1.0x10-4 1.5x10-4 2.0x10-4-0.2
0.0
0.2
0.4
0.6
0.8
1.0
1.2A
Ln (I
o/I)
[BP] /M0 500 1000 1500
0.00248
0.00674
0.01832
0.04979
B
Inte
nsid
ad /
u.a.
Tiempo / ns
Figura 9. Determinación de números de agregación mediante SSFQ y TRFQ. Gráficos de (A) logaritmo de la razón I0/I de la primera banda del Pireno (0,1 µM) en función de la concentración de desactivador. [MMS]=9,99 mM, desactivador: BP, nEE=51 y (B) decaimiento de la intensidad del Pireno (1 µM) en función del tiempo a concentraciones crecientes de desactivador. [MES]=0,46 mM, desactivador: DDPC, nRT=45.

51
Los valores de número de agregación reportados en la literatura para surfactantes con
azúcares como grupo hidrofílico son escasos. Medidas resueltas en el tiempo realizadas
a 25ºC sobre compuestos cuyo grupo polar corresponde a un monosacárido, han arrojado
nag de 92 y 76 para octilglucósido y Hecameg, respectivamente.35 Asimismo, estudios
resueltos en el tiempo para el n-dodecil-β-D-maltósido, (cuya área ocupada por
molécula, entre 50 y 60 Ǻ2, es similar a la determinada en este trabajo para los
monoésteres de sacarosa), han dado como resultado un nag de 82.36 Aunque un poco
menores, los valores de nag obtenidos en este trabajo son del orden de los reportados en
la literatura para surfactantes no iónicos de características similares a los estudiados.
4.6 Efecto de la adición de alcoholes sobre algunas
propiedades de los sistemas micelares
Se estudió el efecto de la presencia de concentraciones crecientes de n-hexanol, n-
propanol e isopropanol sobre el entorno de Pireno y Laurdan asociados a sistemas
micelares.
4.6.1 Concentración micelar crítica
Se evaluó el efecto de la adición de n-hexanol sobre la CMC utilizando tres
concentraciones de alcohol: 13,10 mM, 26,16 mM y 39,18 mM. Los experimentos
fueron realizados a 25ºC, utilizando Pireno como sonda fluorescente en concentración
igual a 0,1 µM. Los resultados obtenidos se muestran en la Tabla 8.
52
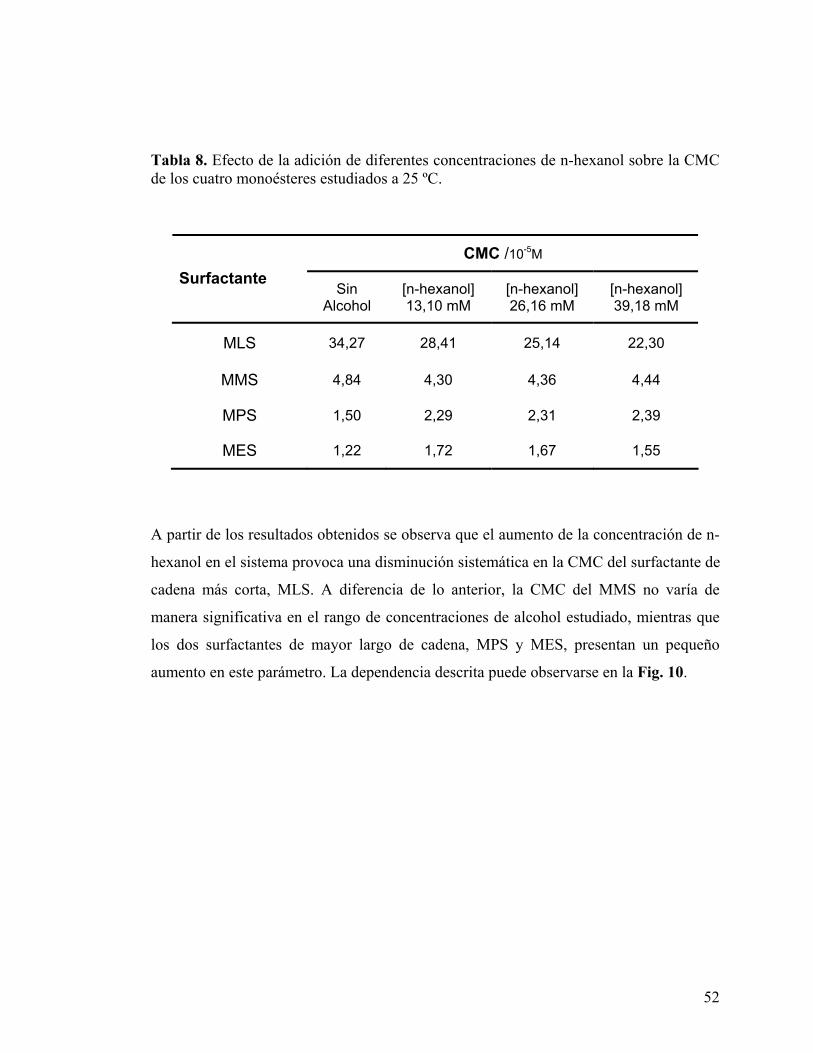
Tabla 8. Efecto de la adición de diferentes concentraciones de n-hexanol sobre la CMC de los cuatro monoésteres estudiados a 25 ºC.
A partir de los resultados obtenidos se observa que el aumento de la concentración de n-
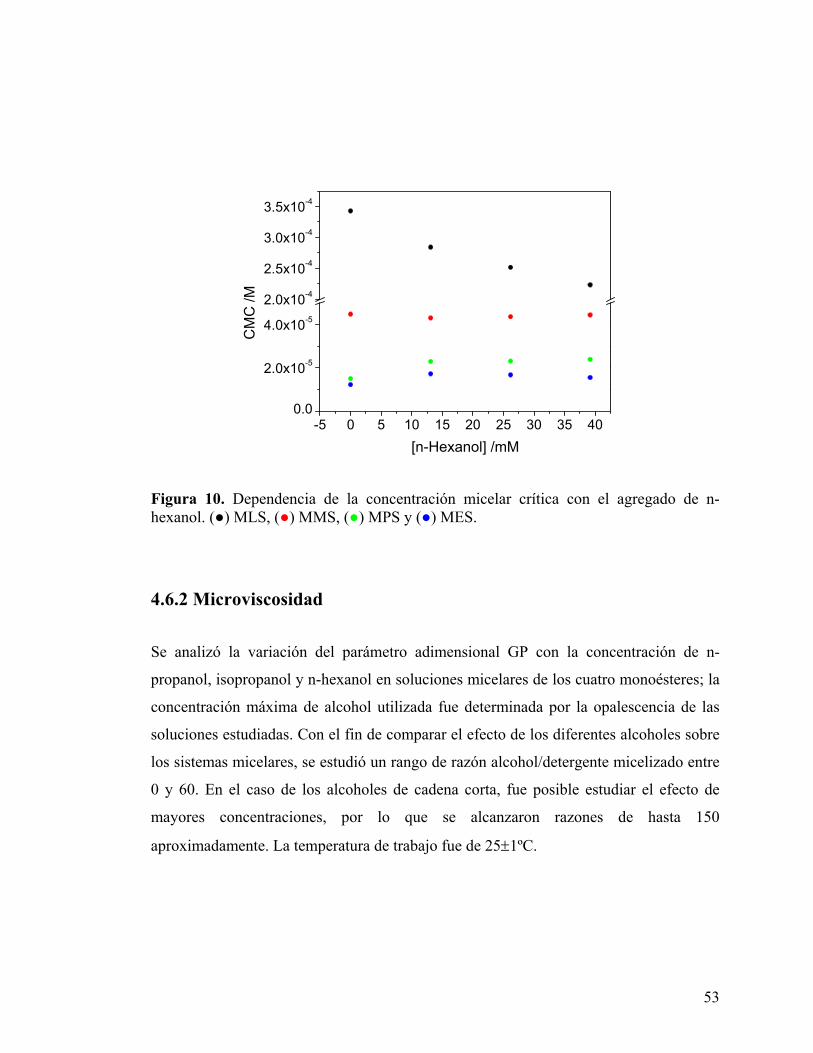
hexanol en el sistema provoca una disminución sistemática en la CMC del surfactante de
cadena más corta, MLS. A diferencia de lo anterior, la CMC del MMS no varía de
manera significativa en el rango de concentraciones de alcohol estudiado, mientras que
los dos surfactantes de mayor largo de cadena, MPS y MES, presentan un pequeño
aumento en este parámetro. La dependencia descrita puede observarse en la Fig. 10.
CMC /10-5M Surfactante
Sin Alcohol
[n-hexanol] 13,10 mM
[n-hexanol] 26,16 mM
[n-hexanol] 39,18 mM
MLS 34,27 28,41 25,14 22,30
MMS 4,84 4,30 4,36 4,44
MPS 1,50 2,29 2,31 2,39
MES 1,22 1,72 1,67 1,55
53
-5 0 5 10 15 20 25 30 35 400.0
2.0x10-5
4.0x10-5
2.0x10-4
2.5x10-4
3.0x10-4
3.5x10-4
CM
C /M
[n-Hexanol] /mM
Figura 10. Dependencia de la concentración micelar crítica con el agregado de n-hexanol. (●) MLS, (●) MMS, (●) MPS y (●) MES.
4.6.2 Microviscosidad
Se analizó la variación del parámetro adimensional GP con la concentración de n-
propanol, isopropanol y n-hexanol en soluciones micelares de los cuatro monoésteres; la
concentración máxima de alcohol utilizada fue determinada por la opalescencia de las
soluciones estudiadas. Con el fin de comparar el efecto de los diferentes alcoholes sobre
los sistemas micelares, se estudió un rango de razón alcohol/detergente micelizado entre
0 y 60. En el caso de los alcoholes de cadena corta, fue posible estudiar el efecto de
mayores concentraciones, por lo que se alcanzaron razones de hasta 150
aproximadamente. La temperatura de trabajo fue de 25±1ºC.
54
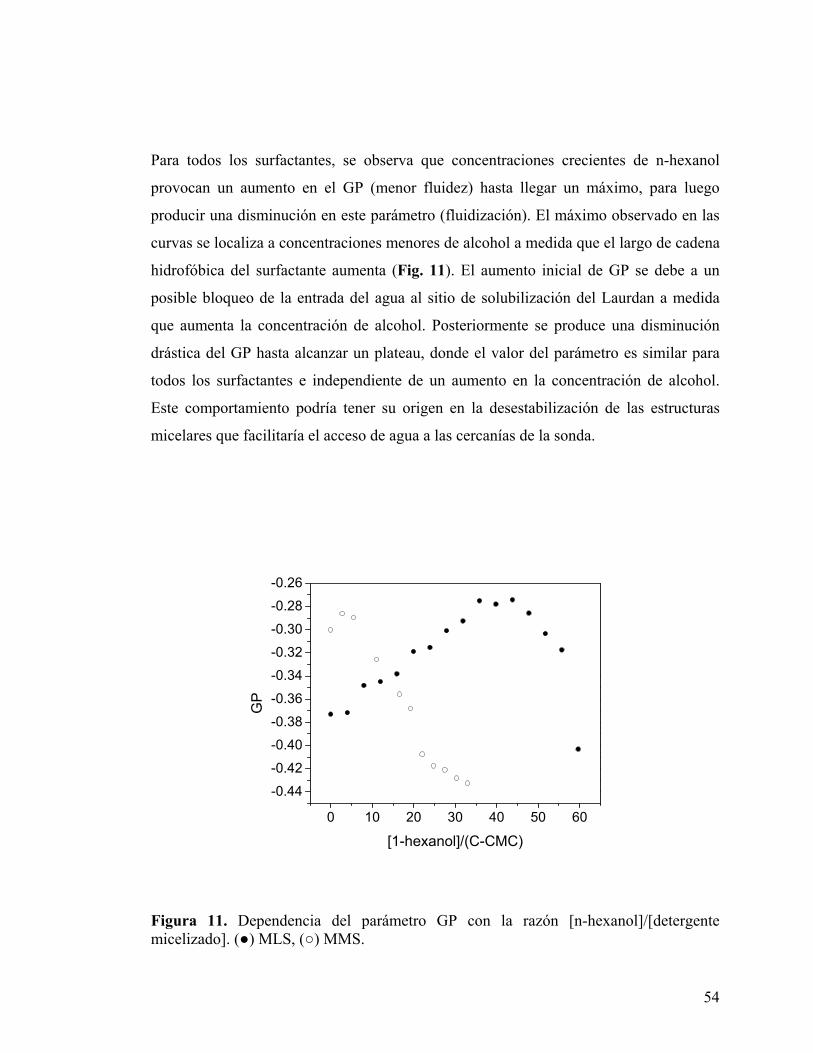
Para todos los surfactantes, se observa que concentraciones crecientes de n-hexanol
provocan un aumento en el GP (menor fluidez) hasta llegar un máximo, para luego
producir una disminución en este parámetro (fluidización). El máximo observado en las
curvas se localiza a concentraciones menores de alcohol a medida que el largo de cadena
hidrofóbica del surfactante aumenta (Fig. 11). El aumento inicial de GP se debe a un
posible bloqueo de la entrada del agua al sitio de solubilización del Laurdan a medida
que aumenta la concentración de alcohol. Posteriormente se produce una disminución
drástica del GP hasta alcanzar un plateau, donde el valor del parámetro es similar para
todos los surfactantes e independiente de un aumento en la concentración de alcohol.
Este comportamiento podría tener su origen en la desestabilización de las estructuras
micelares que facilitaría el acceso de agua a las cercanías de la sonda.
0 10 20 30 40 50 60
-0.44
-0.42
-0.40
-0.38
-0.36
-0.34
-0.32
-0.30
-0.28
-0.26
GP
[1-hexanol]/(C-CMC)
Figura 11. Dependencia del parámetro GP con la razón [n-hexanol]/[detergente micelizado]. (●) MLS, (○) MMS.

55
Un efecto distinto se observó con la adición de n-propanol e isopropanol. A
concentraciones bajas de alcohol, las micelas de surfactantes de cadena más corta, MLS
y MMS, presentan una disminución del GP para luego, a mayores concentraciones,
llegar a un plateau. A diferencia de lo anterior, MPS y MES presentan un leve aumento
inicial del GP para luego estabilizarse a razones alcohol/surfactante micelizado similares
a las observadas en los surfactantes de cadena corta.
El efecto de n-propanol sobre MLS y MMS es muy parecido, llegándose a valores de GP
en el plateau similares para ambos monoésteres. La adición de isopropanol, en cambio,
provoca una disminución levemente mayor en el GP de las micelas de MMS. El MPS
sufre variaciones mucho más leves que los demás surfactantes, el aumento inicial de GP
provocado por la adición de alcohol es pequeño y similar para ambos compuestos, los
valores de GP finales son aproximadamente iguales. En el caso del MES, el valor de GP
inicial es mayor que el observado para los demás surfactantes. La adición de ambos
alcoholes provoca un incremento del GP, sin embargo el efecto de isopropanol es más
marcado que el del alcohol lineal, Fig. 12. Este efecto se debe posiblemente que el
alcohol ramificado produce un mayor bloqueo de la entrada de agua al sitio de anclaje
del Laurdan, disminuyendo la fluidez del entorno sensado por la prueba. El
comportamiento observado es consistente con los resultados obtenidos mediante
medidas de tensión superficial, que predicen una mayor área ocupada por molécula a
medida que el largo de cadena aumenta, lo que indicaría una mayor posibilidad de
movimiento para las cabezas de sacarosa. Este hecho debería traducirse en una mayor
facilidad de ubicación del alcohol entre las cabezas de sacarosa, con el consiguiente
aumento de GP en el rango de concentraciones estudiado.
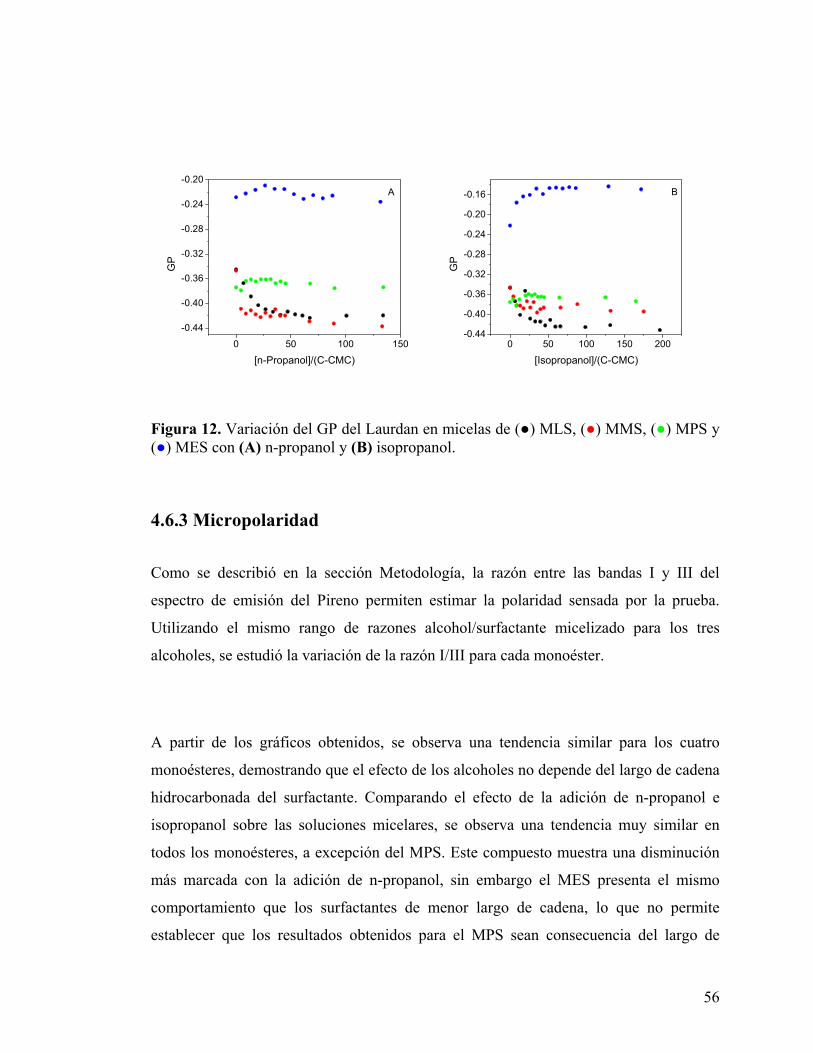
56
0 50 100 150
-0.44
-0.40
-0.36
-0.32
-0.28
-0.24
-0.20A
G
P
[n-Propanol]/(C-CMC)
0 50 100 150 200-0.44
-0.40
-0.36
-0.32
-0.28
-0.24
-0.20
-0.16 B
GP
[Isopropanol]/(C-CMC)
Figura 12. Variación del GP del Laurdan en micelas de (●) MLS, (●) MMS, (●) MPS y (●) MES con (A) n-propanol y (B) isopropanol.
4.6.3 Micropolaridad
Como se describió en la sección Metodología, la razón entre las bandas I y III del
espectro de emisión del Pireno permiten estimar la polaridad sensada por la prueba.
Utilizando el mismo rango de razones alcohol/surfactante micelizado para los tres
alcoholes, se estudió la variación de la razón I/III para cada monoéster.
A partir de los gráficos obtenidos, se observa una tendencia similar para los cuatro
monoésteres, demostrando que el efecto de los alcoholes no depende del largo de cadena
hidrocarbonada del surfactante. Comparando el efecto de la adición de n-propanol e
isopropanol sobre las soluciones micelares, se observa una tendencia muy similar en
todos los monoésteres, a excepción del MPS. Este compuesto muestra una disminución
más marcada con la adición de n-propanol, sin embargo el MES presenta el mismo
comportamiento que los surfactantes de menor largo de cadena, lo que no permite
establecer que los resultados obtenidos para el MPS sean consecuencia del largo de
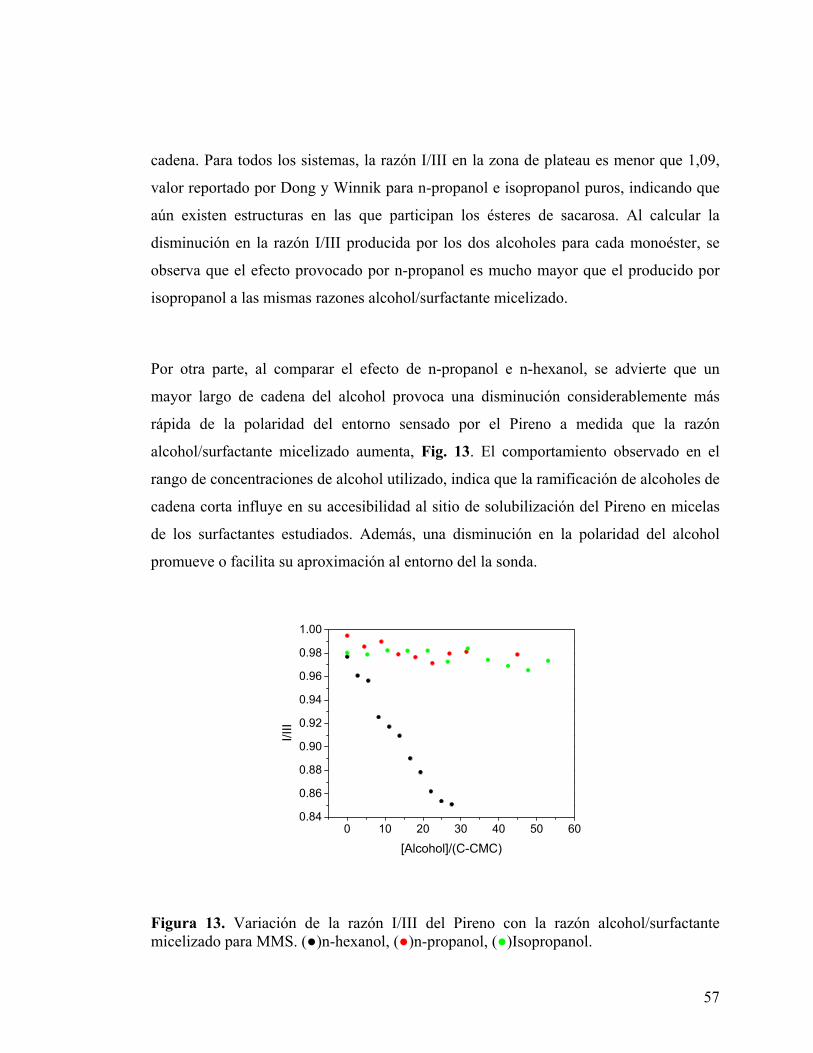
57
cadena. Para todos los sistemas, la razón I/III en la zona de plateau es menor que 1,09,
valor reportado por Dong y Winnik para n-propanol e isopropanol puros, indicando que
aún existen estructuras en las que participan los ésteres de sacarosa. Al calcular la
disminución en la razón I/III producida por los dos alcoholes para cada monoéster, se
observa que el efecto provocado por n-propanol es mucho mayor que el producido por
isopropanol a las mismas razones alcohol/surfactante micelizado.
Por otra parte, al comparar el efecto de n-propanol e n-hexanol, se advierte que un
mayor largo de cadena del alcohol provoca una disminución considerablemente más
rápida de la polaridad del entorno sensado por el Pireno a medida que la razón
alcohol/surfactante micelizado aumenta, Fig. 13. El comportamiento observado en el
rango de concentraciones de alcohol utilizado, indica que la ramificación de alcoholes de
cadena corta influye en su accesibilidad al sitio de solubilización del Pireno en micelas
de los surfactantes estudiados. Además, una disminución en la polaridad del alcohol
promueve o facilita su aproximación al entorno del la sonda.
0 10 20 30 40 50 600.84
0.86
0.88
0.90
0.92
0.94
0.96
0.98
1.00
I/III
[Alcohol]/(C-CMC)
Figura 13. Variación de la razón I/III del Pireno con la razón alcohol/surfactante micelizado para MMS. (●)n-hexanol, (●)n-propanol, (●)Isopropanol.
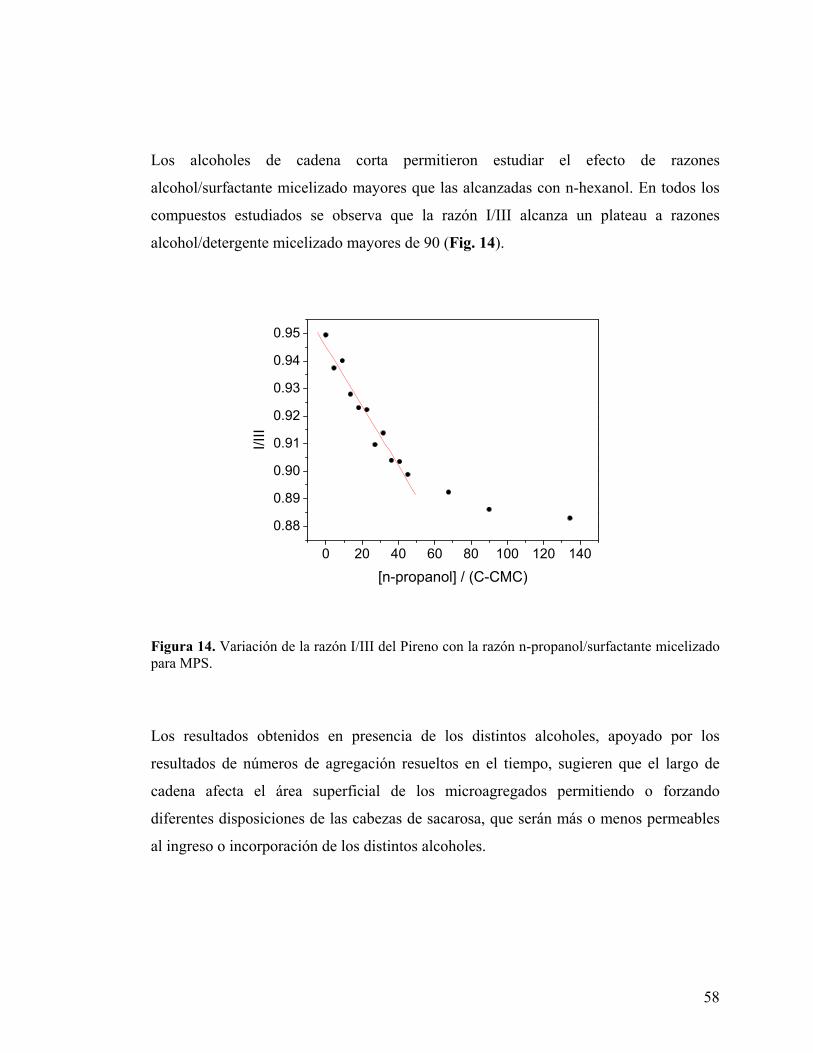
58
Los alcoholes de cadena corta permitieron estudiar el efecto de razones
alcohol/surfactante micelizado mayores que las alcanzadas con n-hexanol. En todos los
compuestos estudiados se observa que la razón I/III alcanza un plateau a razones
alcohol/detergente micelizado mayores de 90 (Fig. 14).
0 20 40 60 80 100 120 140
0.88
0.89
0.90
0.91
0.92
0.93
0.94
0.95
I/III
[n-propanol] / (C-CMC)
Figura 14. Variación de la razón I/III del Pireno con la razón n-propanol/surfactante micelizado para MPS.
Los resultados obtenidos en presencia de los distintos alcoholes, apoyado por los
resultados de números de agregación resueltos en el tiempo, sugieren que el largo de
cadena afecta el área superficial de los microagregados permitiendo o forzando
diferentes disposiciones de las cabezas de sacarosa, que serán más o menos permeables
al ingreso o incorporación de los distintos alcoholes.

59
5. CONCLUSIONES
• Los resultados obtenidos mediante medidas de tensión superficial y metodologías
fotofísicas indican que la magnitud de la concentración micelar crítica (CMC) es
inversamente proporcional al largo de la cadena hidrocarbonada de los monoésteres.
• La dependencia del logaritmo de la CMC con el número de unidades metilénicas de
la cadena hidrofóbica no es lineal (El ∆GMIC por unidad metilénica no es constante).
• A partir de la dependencia de la CMC con la temperatura fueron calculados los
cambios en las funciones termodinámicas, ∆HMIC, ∆GMIC y ∆SMIC, de los cuatro
sistemas en estudio, obteniéndose valores del orden de los reportados en la literatura
para ésteres de sacarosa comerciales. Los valores de ∆HMIC obtenidos para MLS
mediante metodologías fotofísica y física resultaron de signo opuesto.
• La espontaneidad del proceso de micelización de todos los surfactantes estudiados
aumenta con el largo de cadena.
• La micropolaridad de los microagregados formados resultó similar e independiente
de la concentración de micelas y de la temperatura en el rango estudiado.
• La microviscosidad de las micelas aumenta levemente con el largo de cadena y
depende inversamente con la temperatura.
• Los valores de exceso superficial, Γ, muestran una dependencia inversa con el largo
de cadena. No se observó dependencia de este parámetro con la temperatura en el
rango estudiado.
• El área ocupada por molécula, A, depende de forma directa con el largo de cadena.
Los valores obtenidos para este parámetro son del orden de los reportados en la
literatura para mezclas de mono y diésteres de sacarosa.

60
• La tensión superficial límite, γLÍM, depende de forma inversa con la temperatura. El
valor para MMS y MPS es similar en el rango de temperaturas estudiado, sin
embargo, para el MLS, disminuye en mayor medida.
• El parámetro de empaquetamiento crítico, Cpp, predice micelas cilíndricas para
MLS y MMS. La geometría predicha para las estructuras micelares formadas por
MPS es de tipo esférica.
• Los números de agregación evaluados muestran dependencias distintas con el largo
de cadena de acuerdo con la técnica empleada. Las medidas resueltas en el tiempo
arrojan resultados consistentes con el comportamiento general observado, mostrando
valores similares para todos los ésteres.
• La adición de n-hexanol provoca una disminución sistemática de la CMC del MLS
con el aumento de la concentración de alcohol. La CMC de MMS no se ve afectada
por la presencia de este alcohol, mientras que la de MPS y MES aumenta levemente
con la concentración de n-hexanol.

61
6. BIBLIOGRAFÍA
1 Hill, K.; Rhode, O. Fett-Lipid 1999, 101, 25-33.
2 Rybinski, W. v. Curr. Opin. Colloid Interface Sci. 2001, 6, 146-147.
3 Holmberg, K. Curr. Opin. Colloid Interface Sci. 2001, 6, 148-159.
4 Garcia, M. T.; Ribosa, I.; Campos, E.; Leal, J. S. Chemosphere 1997, 35, 545-556.
5 Stradner, A.; Mayer, B.; Sottmann, T.; Hermetter, A.; Glatter, O. J. Phys. Chem. B
1999, 103, 6680-6689.
6 Uchegbu, I. F.; Vyas, S. P. Int. J. Pharm. 1998, 172, 33–70.
7 Thevenet, S.; Wernicke, A.; Belniak, S.; Descotes, G.; Bouchu, A.; Queneau, Y.
Carbohyd. Res. 1999, 318, 52-66.
8 Liu, X.; Gong, L.; Xin, M.; Liu, J. J. Mol. Cat. A: Chem. 1999, 147, 37-40.
9 De la Nuez, L. R., Trabajo de Tesis Doctoral no publicado, 2004.
10 Wishnia, A. J. Phys. Chem.1963, 67, 2079-2082.
11 Salager, J. L., Surfactantes en solución acuosa, 1993.
12 Alargova, R. G.; Kochijashky, I. I.; Sierra, M. L.; Zana, R. Langmuir 1998, 14, 5412-
5418.
13 Kalyanasundaram, K. Photochemistry in Microheterogeneous Systems; Academic
Press Inc., Orlando, 1987.
14 Abuin, E. A.; Lissi, E. A. J. Colloid Interf. Sci. 1983, 95, 198-203.

62
15 Chen, L.-J.; Lin, S.-Y.; Huang, C.-C. J. Phys. Chem. B 1998, 102, 4350-4356.
16 Chu, D. Y.; Thomas, J. K. J. Am. Chem. Soc. 1986, 108, 6270-6276.
17 Parassasi, T.; Krasnowska, E. K.; Bagatolli, L.; Gratton, E. J. Fluoresc. 1998, 8, 365-
373.
18 Viard, M.; Gallay, J.; Vincent, M.; Paternostre, M. Biophys. J. 2001, 80, 347-359.
19 Karpovich, D. S.; Blanchard, G. J. J. Phys. Chem. 1995, 99, 3951-3958.
20 Dong, D. C.; Winnik, M. A. Can. J. Chem. 1984, 62, 2560-2565.
21Ferrer, M.; Cosmelles, F.; Plou, F. J.; Cruces, M. A.; Fuentes, G.; Parra, J. L.;
Ballesteros, A. Langmuir 2002, 18, 667-673.
22 Kratzat, K.; Finkelmann, H. Langmuir 1996, 12, 1765-1770.
23 Shinoda, K.; Hutchinson, E. J. Phys. Chem. 1962, 66, 577-582.
24 Olofsson, G. J. Phys. Chem. 1983, 87, 4000-4004.
25 Paula, S.; Süs, W.; Tuchtenhagen, J.; Blume, A. J. Phys. Chem. 1995, 99, 11742-
11751.
26 Benjamin, L. J. Phys. Chem. 1964, 68, 3575-3581.
27 Soultani, S.; Ognier, S.; Engasser, J.-M.; Ghoul, M. Colloids Surf. A: Physicochem.
Eng. Asp. 2003, 227, 35-44.
28 Pestman, J. M.; Kevelam, J.; Blandamer, M. J.; Doren, H. A. v.; Kellogg, R. M.;
Engberts, J. Langmuir 1999, 15, 2009-2014.

63
29 Zajac, J.; C.Chorro; Lindheimer, M.; Partyka, S. Langmuir 1997, 13, 1486-1495.
30 Bijma, K.; Engberts, J. Langmuir 1994, 10, 2578-2582.
31 Garofalakis, G.; Murray, B. S.; Sarney, D. B. J. Colloid Interf. Sci. 2000, 229, 391–
398.
32 Crook, E. H.; Trebbi, G. F.; Fordyce, D. B. J. Phys. Chem. 1964, 68, 3592-3599.
33 Söderberg, I.; Drummond, C. J.; Furlong, D. N.; S.Godkin; B.Matthews Colloids Surf.
A: Physicochem. Eng. Asp. 1995, 102, 91-97.
34 Ghosh, S. K.; Khatua, P. K.; Bhattacharya, S. C. J. Colloid Interf. Sci. 2004, 275, 623-
631.
35 Frindi, M.; Michels, B.; Zana, R. J. Phys. Chem. 1992, 96, 8137-8141.
36 Kjellin, M.; Reimer, J.; Hansson, P. J. Colloid Interf. Sci. 2003, 262, 506-515.





























