Embed Size (px)
DESCRIPTION
fisiquicoquimica
Citation preview

Ing. Juan Montesano

Funciones Potenciales
∑≥
⎟⎟⎠
⎞∑≥
=⎟⎠⎞
∑−
QdST
TQdS
STQdS
mc
mc
gmc
mc
δ
δ
δδ
dU +PdV
pdVTdSdU −≤
pVUyT
SUdV
VUdS
SUdU
SVSV
−=⎟⎠⎞
⎜⎝⎛∂∂
=⎟⎠⎞
⎜⎝⎛
∂∂
⇒⎟⎠⎞
⎜⎝⎛∂∂
+⎟⎠⎞
⎜⎝⎛
∂∂
=

3
La trasformación de LegendreSi una función potencial Z=Z(X1, X2 …Xn) el diferencial exacta es:
dZ= C1 dX1+ C2 dX2+… Cn dXn
Las transformaciones de Legendre definen funciones T
relacionadas con C y X. Para una expresión diferencial total que contenga n variables hay 2n – 1transformaciones de Legendre posibles son:
T1 = Z-C1 X1 T2 = Z-C2 X2 … Tn = Z-Cn Xn
∑−=−i
iin X.CZT1

4
Potenciales termodinámicos
dV.PdS.TdU −= VXSXPCTC
UZ
==
−===
21
21
S.THS.TV).P(UTV).P(UT
S.TUT
−=−−−=−−=
−=
−21
2
1
Se definen como las transformadas de Legendre de la energía interna.U=U(S,V); 22-1=·3
Función de Helmhotlz A
Entalpía H
Función Gibbs GEl diferencial de las funciones T
112221
22112
11221
111122111
2211
11111
dC.XdC.XdTdC.XdX.CdTdC.XdX.CdT
dC.XdX.CdX.CdX.CdT
dX.CdX.CdZdoreemplazandC.XdX.CdZdT
−−=
−=
−=
−−+=
+=
−−=
−

5
Potenciales termodinámicos
dV.PdT.SdA −−=
dP.VdS.TdH +=
La ecuación fundamental puede expresarse como sigue:
dP.VdT.SdG +−=112221
22112
11221
dC.XdC.XdTdC.XdX.CdTdC.XdX.CdT
−−=−=−=
−
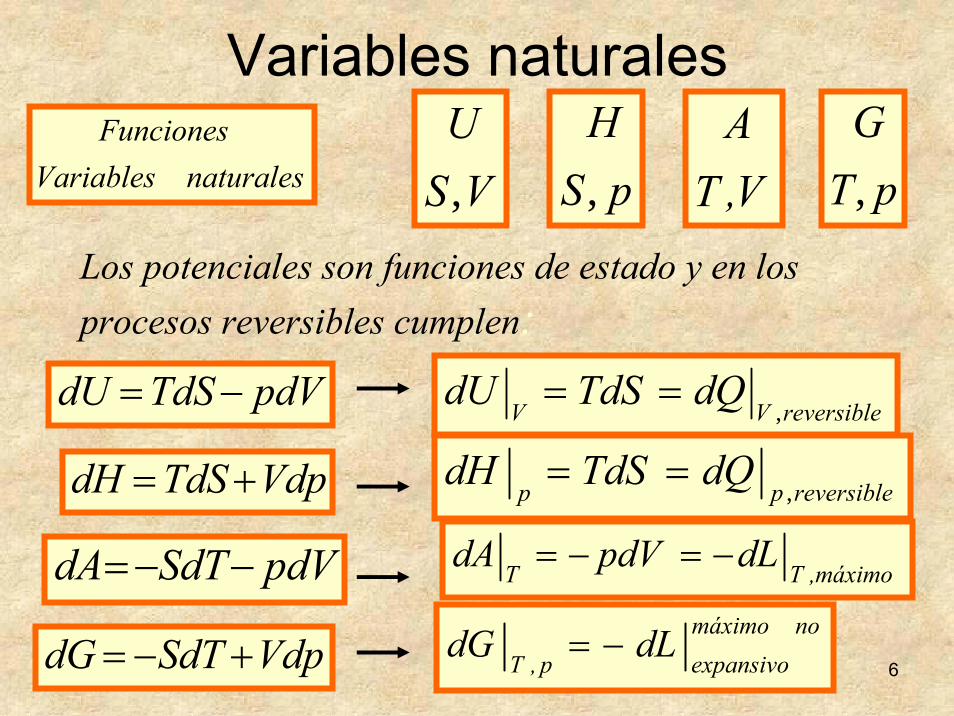
6
naturalesVariablesFunciones
VSU, pS
H, V,T
ApT
G,
Variables naturales
Los potenciales son funciones de estado y en los procesos reversibles cumplen:
pdVTdSdU −= reversibleVVdQTdSdU
,==
VdpTdSdH += reversibleppdQTdSdH
,==
pdVSdTdA −−= máximo,TT dLpdVdA −=−=
VdpSdTdG +−=nomáximo
expansivop,T dLdG −=

Ecuaciones de Maxwell
Tp
TV
Sp
SV
pGV
TGS
VAp
TAS
pHV
SHT
VUp
SUT
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎠⎞
⎜⎝⎛
∂∂
−=
⎟⎠⎞
⎜⎝⎛
∂∂
−=⎟⎠⎞
⎜⎝⎛
∂∂
−=
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎠⎞
⎜⎝⎛
∂∂
=
⎟⎠⎞
⎜⎝⎛
∂∂
−=⎟⎠⎞
⎜⎝⎛
∂∂
=
pT
VT
pS
VS
TV
pS
Tp
VS
SV
pT
Sp
VT
⎟⎠⎞
⎜⎝⎛
∂∂
−=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
⎟⎠⎞
⎜⎝⎛
∂∂
=⎟⎠⎞
⎜⎝⎛
∂∂
⎟⎠⎞
⎜⎝⎛
∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
⎟⎠⎞
⎜⎝⎛
∂∂
−=⎟⎠⎞
⎜⎝⎛
∂∂
En las funciones potenciales las
derivadas parciales cruzadas
son iguales
Una regla para obtener las ecuaciones de Maxwell es la siguiente. Se construye el esquema:
Para calcular el valor de la derivada:se opera así:
VSp
⎟⎠⎞
⎜⎝⎛
∂∂
Si las variables intensivas están en dos derivadas, el signo es negativo:
SV VT
Sp
⎟⎠⎞
⎜⎝⎛
∂∂
−=⎟⎠⎞
⎜⎝⎛
∂∂

8
Ing. Juan Montesano

9
Hipótesis de homogeneidad• En 1875, J.W.Gibbs estableció:• La energía interna de cualquier parte de un
sistema en equilibrio es función de la entropía, del volumen y de la masa de las diversas sustancias o “componentes” que la forman.
• La energía interna de una parte homogénea de un sistema en equilibrio es función homogénea de primer orden de sus variables.

10
Ecuación de Gibbs
• La hipótesis de Gibbs hace que un sistema en equilibrio homogéneo cumpla la ecuación de Gibbs:
• donde las ni representan las masas en moles de los componentes del sistema y los μi se conocen como sus “potenciales químicos:
∑ =+−=
n
i ii dndVpdSTdU1
... μ
ijnVSii n
U
≠
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=,,
μ

11
• Como U es una función homogénea:
• reconociendo las derivadas:
• se llega a:
∑≠
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎠⎞
⎜⎝⎛
∂∂
+⎟⎠⎞
⎜⎝⎛
∂∂
=i
inVSinSnV
nnUV
VUS
SUU
ij,,,,
TSU
nV
=⎟⎠⎞
⎜⎝⎛
∂∂
,p
VU
nS
−=⎟⎠⎞
⎜⎝⎛
∂∂
,i
nVSiij
nU μ=⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
≠,,
Ecuación de Gibbs-Duhem I
∑+−=i iinpVTSU μ

12
Ecuación de Gibbs-Duhem II
• Diferenciando:
• comparando con la ecuación fundamental:
• se llega a la ecuación de Gibbs-Duhem:
• de donde:
∑+−=i ii npVTSU μ
∑∑ ++−−+=i iii ii dndnpdVVdpTdSSdTdU .. μμ
0. =+− ∑ i ii dnVdpSdT μ
∑+−=i ii dnpdVTdSdU .μ
( )Tpii ,μμ =

Sistemas de multicomponentesEcuaciones gibbssianas para un solo componente
Ecuaciones gibbssianas para multicomponentes
j
j
j
j
T,p,ni
iii
T,V,ni
iii
S,p,ni
iii
S,V,ni
iii
nGμ.dnμdpVdTSdG
nAμ.dnμdVpdTSdA
nHμ.dnμdpVdSTdH
nUμ.dnμdVpdSTdU
⎟⎠⎞
⎜⎝⎛
∂∂
=⇒∑++−=
⎟⎠⎞
⎜⎝⎛
∂∂
=⇒∑+−−=
⎟⎠⎞
⎜⎝⎛
∂∂
=⇒∑++=
⎟⎠⎞
⎜⎝⎛
∂∂
=⇒∑+−=
pT
VT
pS
VS
nGdndpVdTSdG
nAdndVpdTSdA
nHdndpVdSTdH
nUdndVpdSTdU
,
,
,
,
.
.
.
.
⎟⎠⎞
⎜⎝⎛
∂∂
=⇒++−=
⎟⎠⎞
⎜⎝⎛
∂∂
=⇒+−−=
⎟⎠⎞
⎜⎝⎛
∂∂
=⇒++=
⎟⎠⎞
⎜⎝⎛
∂∂
=⇒+−=
μμ
μμ
μμ
μμ
Potenciales químicos Potenciales químicos

14
Descripción de un sistema( )inVSUU ,,=
∑= inn
nUu =(
La hipótesis de Gibbs hace: donde nison los números de moles de los componentes del sistema: .
Los valores molares de las funciones son:
nSs =(
nVv =(
nny i
i =
La relación fundamental de un sistema homogéneo es:
( )iy,v,suu ((ω( =

15
Descripción de un cuerpo puro
( ) nVpSTnVSUU ...,, μ+−==
nSTVpUH ... μ+=+=
nVpSTUA ... μ+−=−=
La hipótesis de Gibbs asegura que:
nVpSTUG ... μ=+−=“El potencial químico de un cuerpo puro es su función de Gibbs molar”:
μv.Pas.Thv.Ps.Tug =+=−=+−= ((((((((
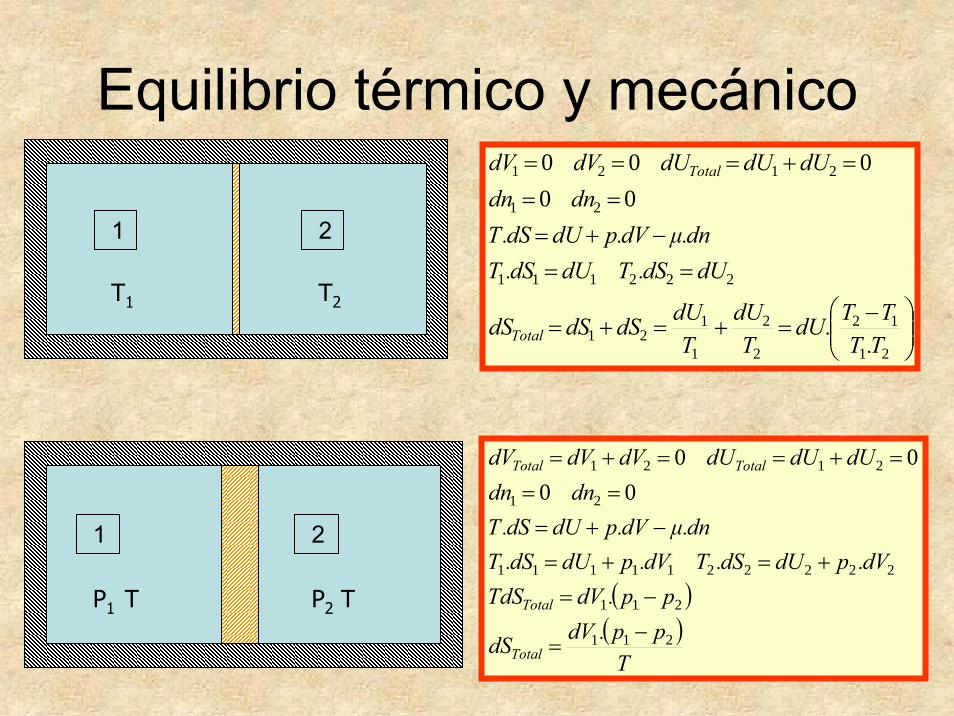
Equilibrio térmico y mecánico
⎟⎟⎠
⎞⎜⎜⎝
⎛ −=+=+=
==−+=
===+===
21
12
2
2
1
121
222111
21
2121
00000
T.TTT.dU
TdU
TdUdSdSdS
dUdS.TdUdS.Tdn.μdV.pdUdS.T
dndndUdUdUdVdV
Total
Total
1 2
T1 T2
1 2
P1 T P2 T ( )( )
Tpp.dVdS
pp.dVTdSdV.pdUdS.TdV.pdUdS.T
dn.μdV.pdUdS.Tdndn
dUdUdUdVdVdV
Total
Total
TotalTotal
211
211
2222211111
21
2121
0000
−=
−=+=+=
−+===
=+==+=
17
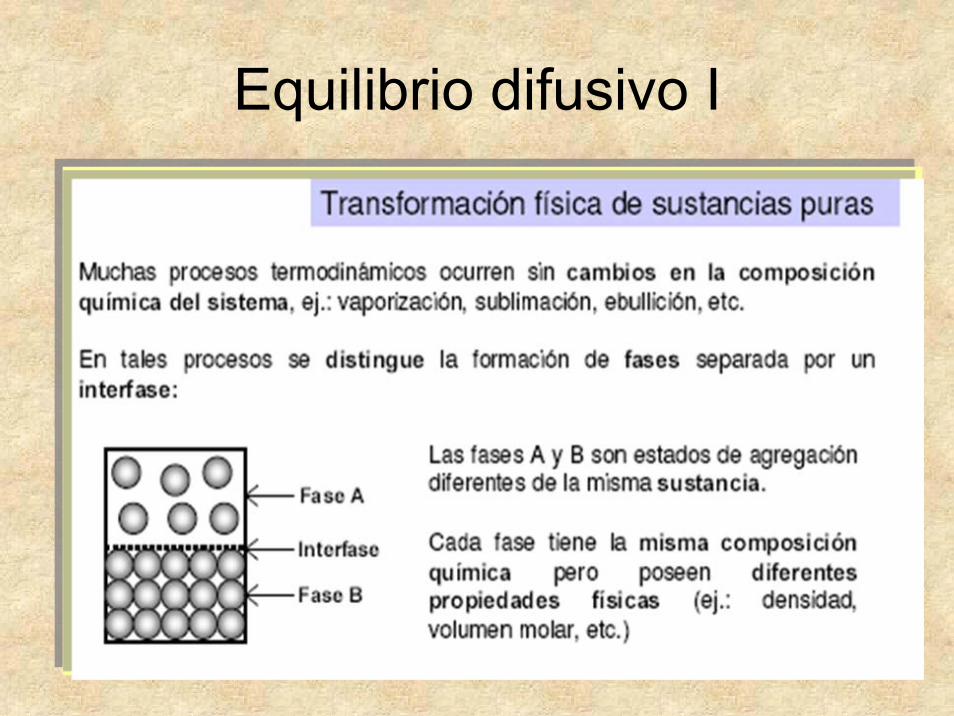
Equilibrio difusivo I
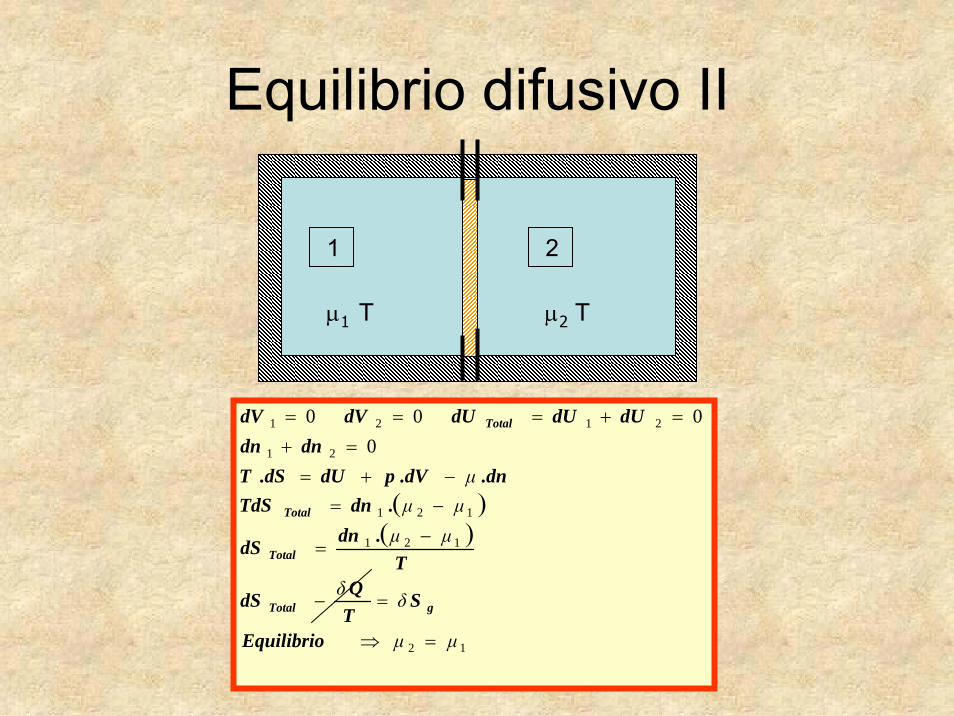
Equilibrio difusivo II
( )121
21
2121
0000
μμμ
−=−+=
=+=+===
.dnTdSdn.dV.pdUdS.T
dndndUdUdUdVdV
Total
Total1 2
μ1 T μ2 T
( )( )
12
121
121
21
2121
0000
μμ
δδ
μμμμμ
=⇒
=−
−=
−=−+=
=+
=+===
Equilibrio
STQdS
T.dndS
.dnTdSdn.dV.pdUdS.T
dndndUdUdUdVdV
gTotal
Total
Total
Total

19
Equilibrio difusivo III

20
Potencial químico• El potencial químico de la especie “i “en una fase es
una medida de la tendencia al escape en la fase que posee la especie” i”
• El potencial químico es una medida de la presión química ejercida por “i”en la fase considerada
• Si el potencial químico de “i “es diferente en las fases del sistema, que se halla a P y T constantes, esta sustancia i se moverá hasta que su potencial químico sea igual en todas las fases.
• Este movimiento se efectuará desde la fase de mayor potencial químico.
• El potencial químico es la fuerza impulsora que provoca el fenómeno de difusión
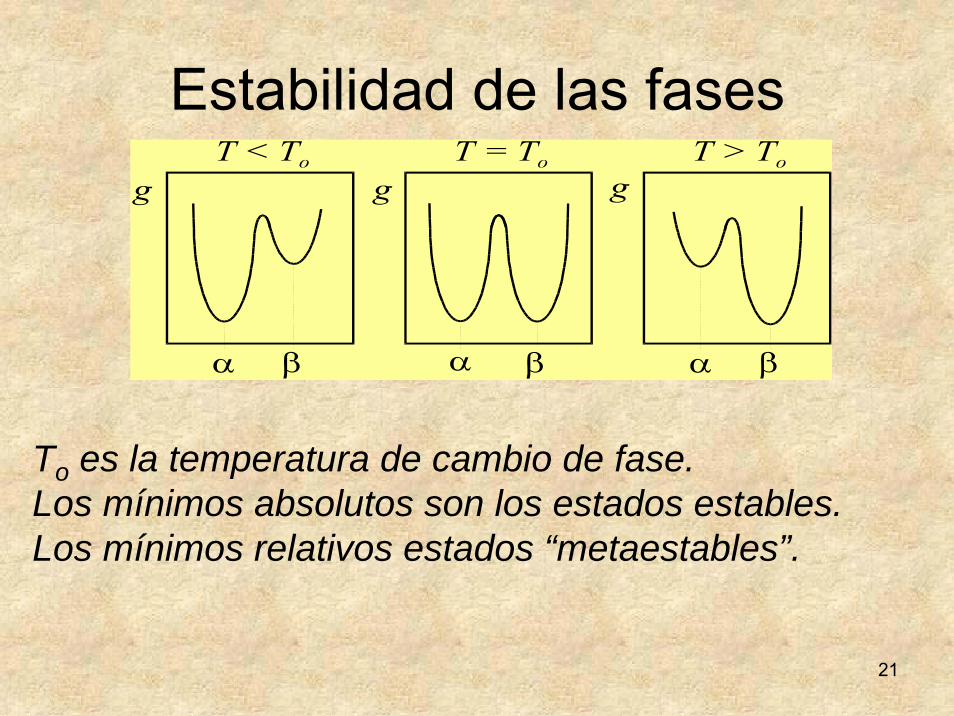
21
Estabilidad de las fases
To es la temperatura de cambio de fase.Los mínimos absolutos son los estados estables.Los mínimos relativos estados “metaestables”.
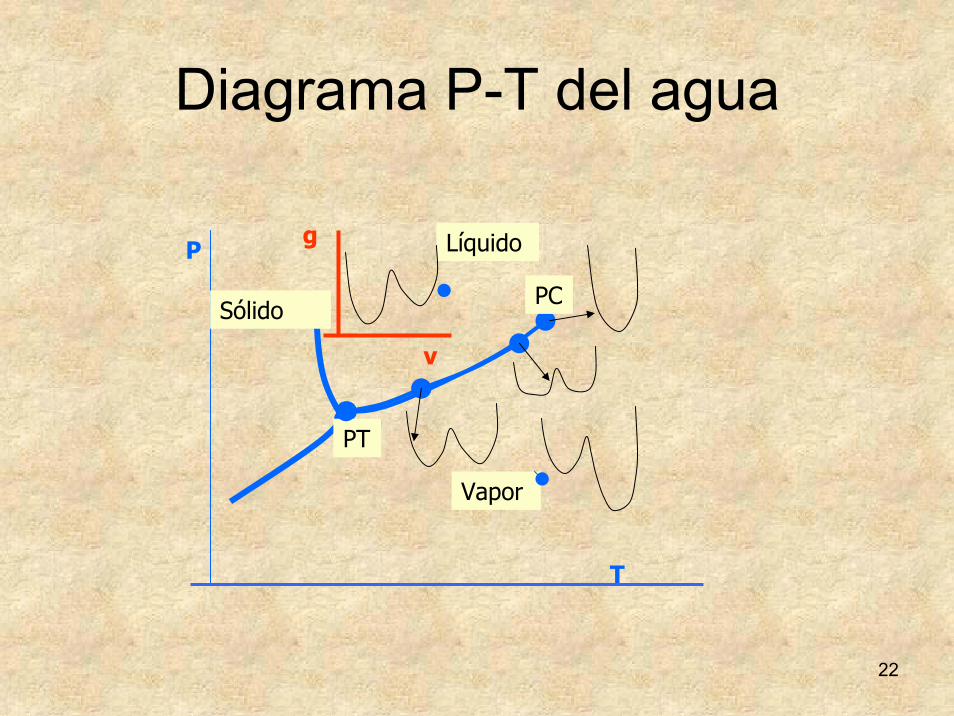
22
PC
PT
Líquido
Sólido
Vapor
T
P
v
g
Diagrama P-T del agua

23
• Presentan tres fases generales: sólida, líquida y gaseosa.
• Los sólidos pueden tener fases alotrópicas.
• Todas ellas están gobernadas por G mínimo.
vdpsdTddg +−== μ
sT p
−=⎟⎠⎞
⎜⎝⎛
∂∂μ
gaslíquidosólido sss <<
gaslíquidosólido vvv <<<≈
vp T
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂μ
Cuerpos puros
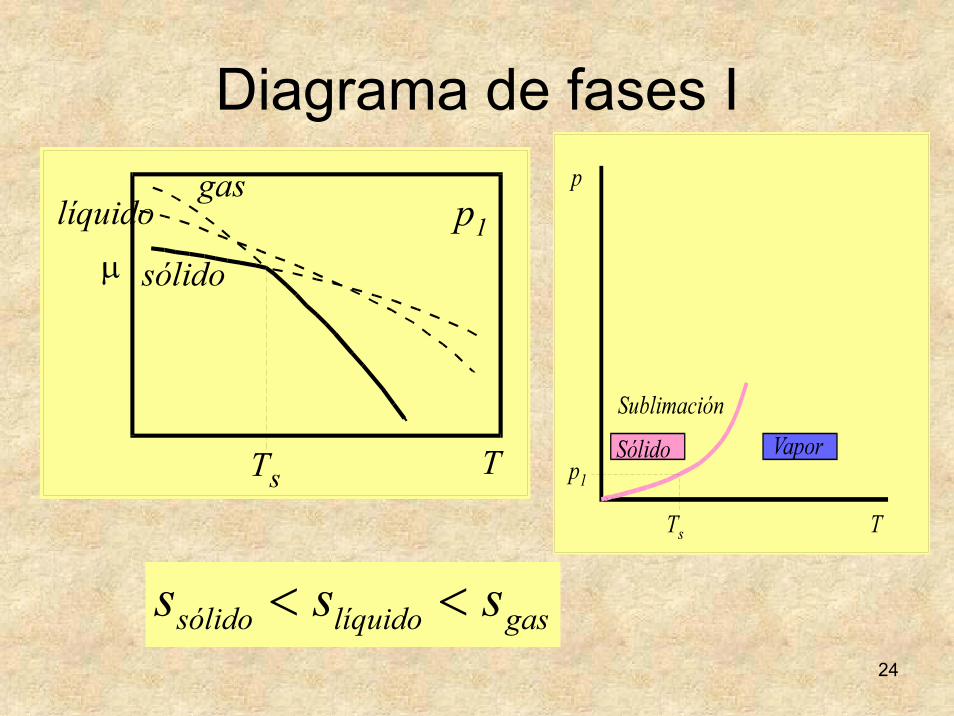
24
Diagrama de fases I
gaslíquidosólido sss <<
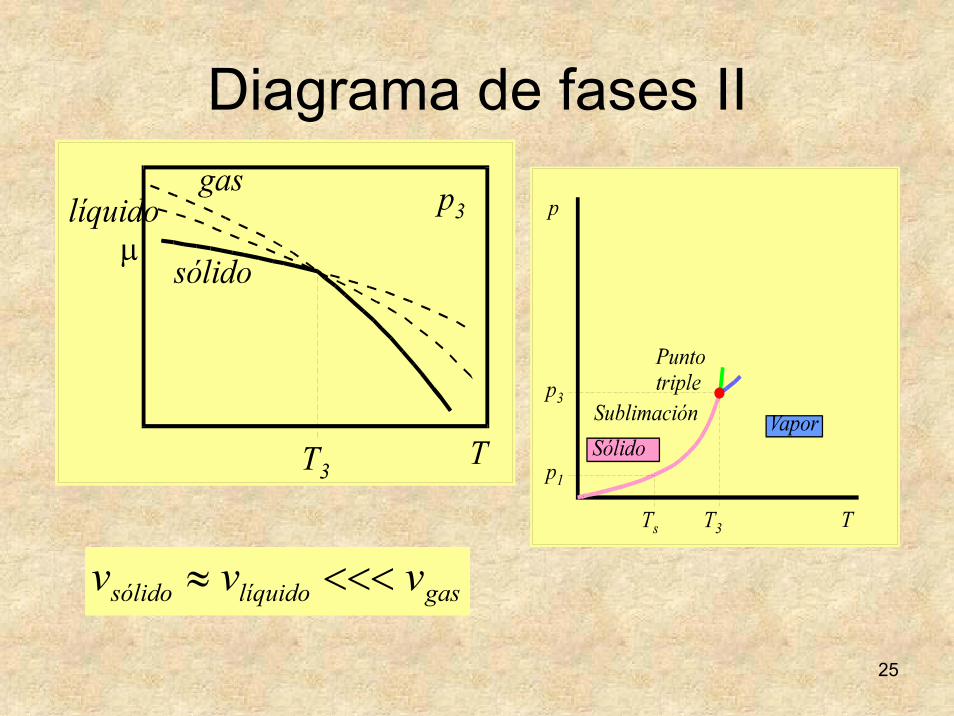
25
Diagrama de fases II
gaslíquidosólido vvv <<<≈
26
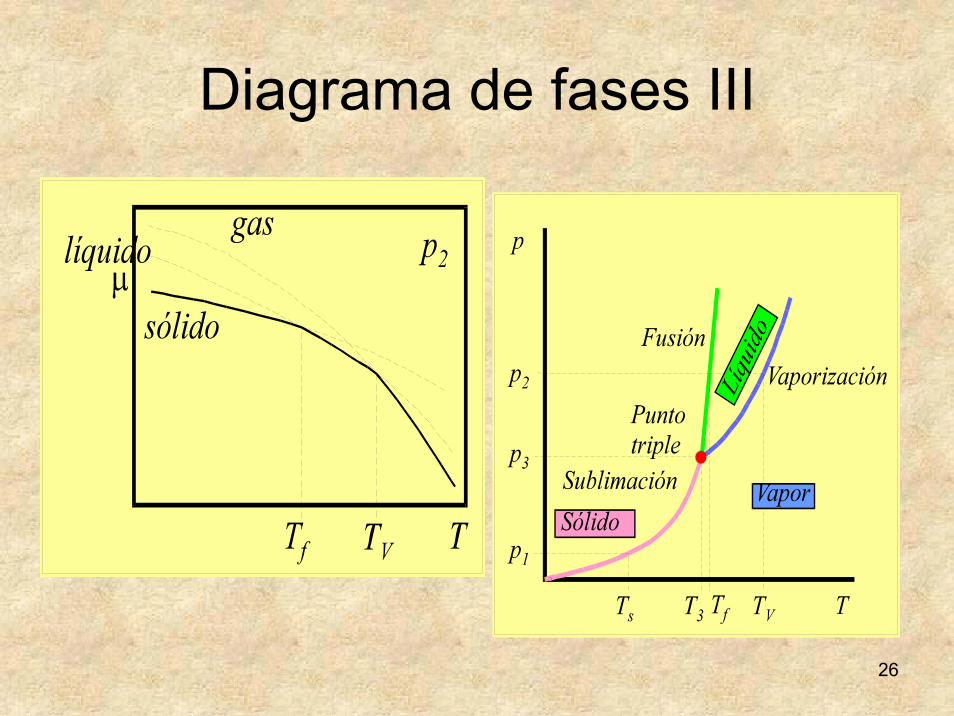
Diagrama de fases III
27
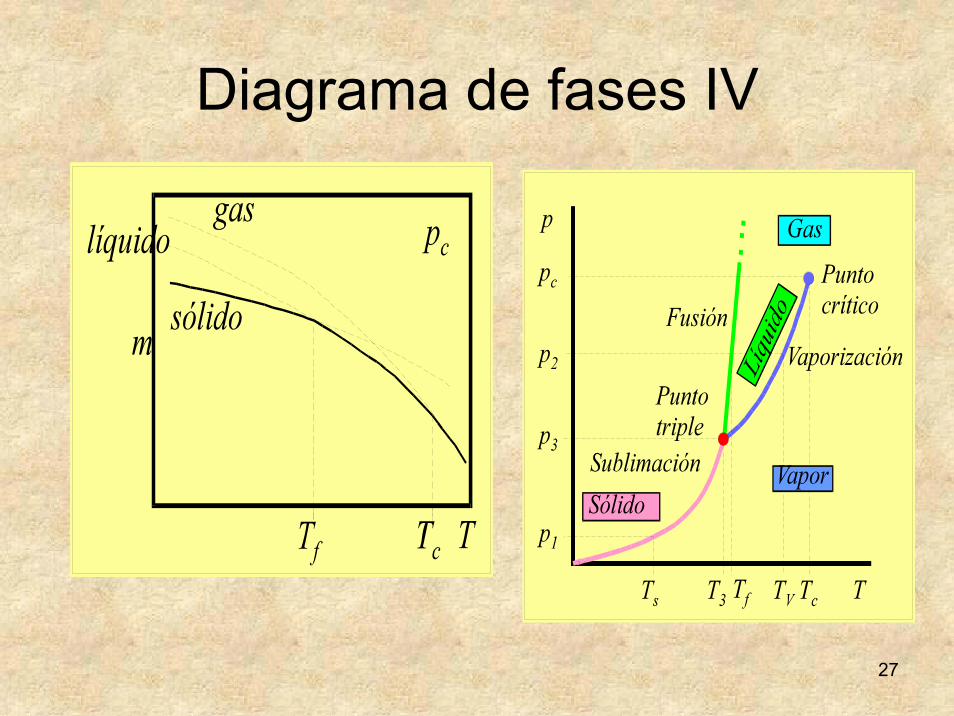
Diagrama de fases IV

28
Análisis de diagrama de fases I • Diagrama similar en
todas las sustancias.• Sólo varía la disposición.• Línea de sublimación:
desde el cero absoluto hasta el punto triple.
• Punto triple: Coexisten las tres fases: sólida, líquida y gaseosa

29
Análisis de diagrama de fases II
• Línea de vaporización: desde el punto triple al crítico.
• Punto crítico: confusión de las fases líquida y gaseosa.
• La presión en cada punto de las líneas de sublimación y vaporización es la presión de vapor del sólido y del líquido a esa temperatura.

30
Análisis de diagrama de fases III
• El punto crítico delimita la zona de gas de la zona de vapor.
• Vapor: licúa al enfriarlo.• Gas: no licúa al enfriarlo.• Línea de fusión: desde el punto triple sin
límite superior conocido.
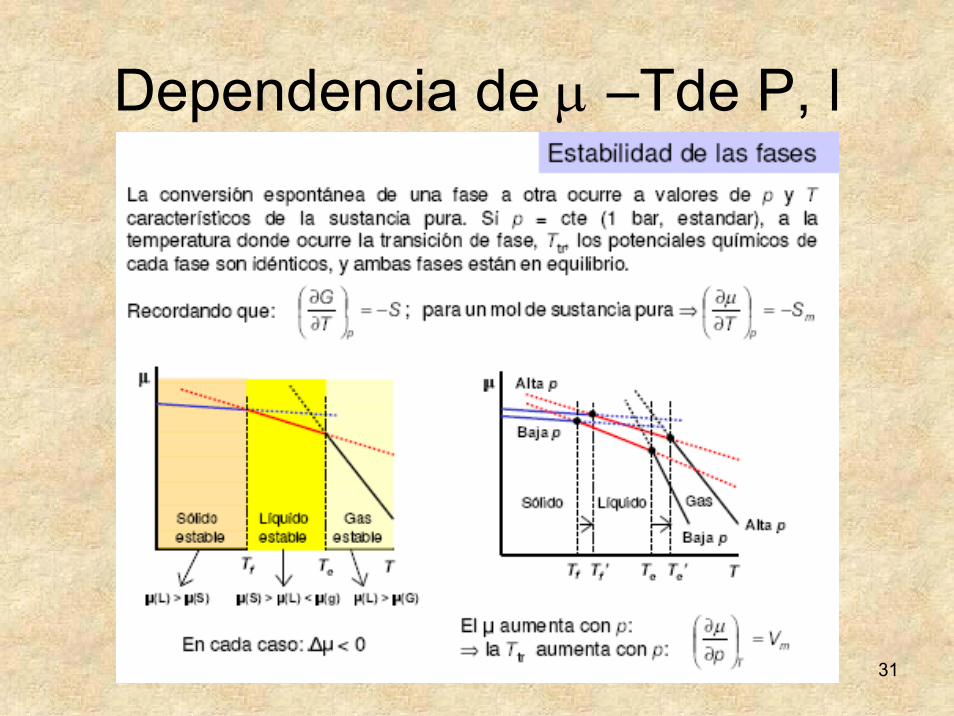
31
Dependencia de μ –Tde P, l
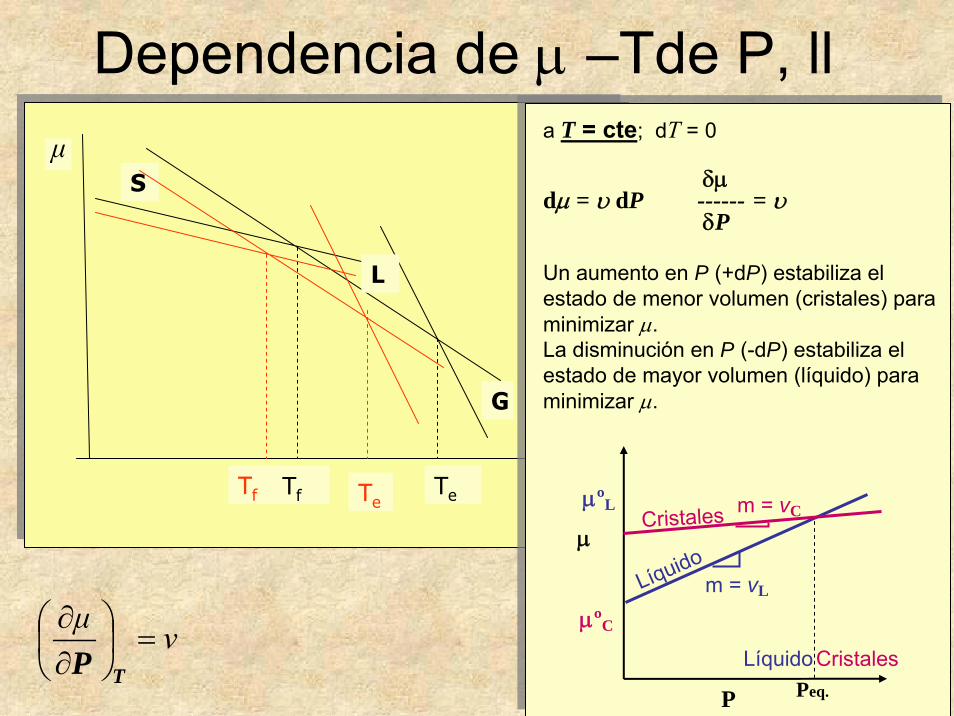
32
Dependencia de μ –Tde P, ll
νμ=⎟
⎠⎞
⎜⎝⎛
∂∂
TP
μ
T
S
L
G
Tf TeTeTf
a T = cte; dT = 0
δμdμ = υ dP ------ = υ
δP
Un aumento en P (+dP) estabiliza el estado de menor volumen (cristales) para minimizar μ.La disminución en P (-dP) estabiliza el estado de mayor volumen (líquido) para minimizar μ.
P
μ
Líquido
CristalesμºL
μºC
m = vC
m = vL
Peq.
CristalesLíquido

33
Principio de Le Chatelier:
Si un cambio ocurre en un sistema, el sistema responde de manera que la fuerza que causa el cambio sea minimizada o moderada.
Si aumenta P, el sistema responde disminuyendo V.
A alta P serán más estables las fases que ocupenmenor volumen molar (cristales). Al bajar P, el líquido, que generalmente ocupa mayor volumen molar, será más estable (ej. fusión por decompresión).
Si aumenta T el sistema responde aumentando S.A alta T serán más estables los estados más desordenados (líquido tiene mayor entropía).A baja T serán más estables los estados más ordenados (cristales tienen menor entropía)
La curva en este caso debe tener pendiente positiva (al aumentar P, la temperatura de fusión se desplaza a valores más altos).
Principio de Le Chatelier:
Si un cambio ocurre en un sistema, el sistema responde de manera que la fuerza que causa el cambio sea minimizada o moderada.
Si aumenta P, el sistema responde disminuyendo V.
A alta P serán más estables las fases que ocupenmenor volumen molar (cristales). Al bajar P, el líquido, que generalmente ocupa mayor volumen molar, será más estable (ej. fusión por decompresión).
Si aumenta T el sistema responde aumentando S.A alta T serán más estables los estados más desordenados (líquido tiene mayor entropía).A baja T serán más estables los estados más ordenados (cristales tienen menor entropía)
La curva en este caso debe tener pendiente positiva (al aumentar P, la temperatura de fusión se desplaza a valores más altos).
P
T
CristalesGC < GL
LíquidoGL < GC
Cur
va d
efu
sión
+P
-P
+T-T
En la curva de fusión coexisten cristales y líquido, GL = GC (ΔG = 0)SL > SC (ΔS > 0)VL > VC (ΔV > 0)
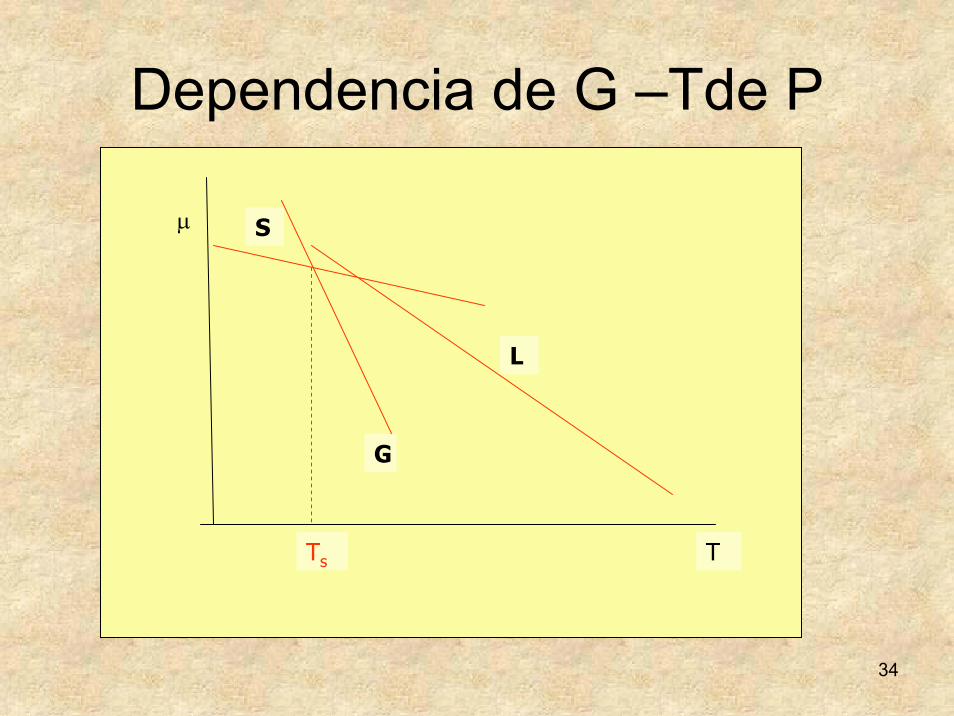
34
Dependencia de G –Tde P
T
S
L
G
Ts
μ
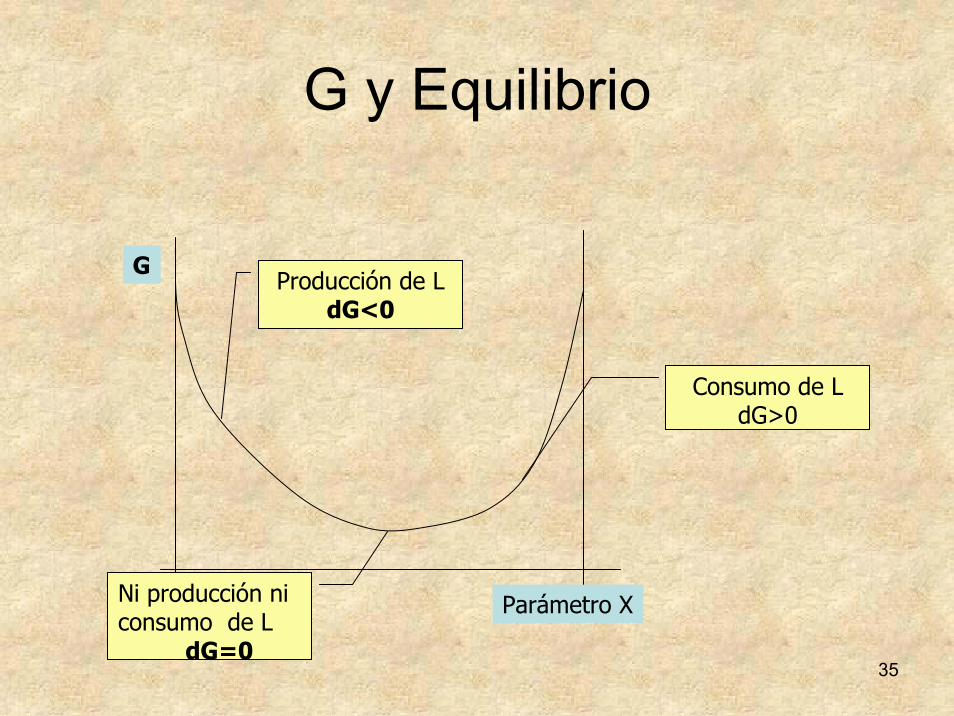
35
G y Equilibrio
Parámetro X
GProducción de L
dG<0
Ni producción ni consumo de L
dG=0
Consumo de LdG>0

36
Equilibrio químico
i
io
i
OCO
CO
ii
αnnX
α;α
;αO.COCO
A.αA.αA.αA.αA.α
−=
==
−=+⇔
∑⇔+⇔+
211
121
0
2
222
44332211
dX.αdn
X.nX.n
X.nmolesnmolesnmolesn
i
oooo
OCO
CO
COCO
=
+=+=
−=
===
21010
12002
2
2
22
Definición
grado de avance ≠ 0
( )121
21
2121
0000
μμμ
−=−+=
=+=+===
.dnTdSdn.dV.pdUdS.T
dndndUdUdUdVdV
Total
Totalα1 A1 α2 A2
P , T
+
α3 A3 +α4 A4

37
Equilibrio químico
( ) dX.α.μTdSdn.μdV.pdUdS.T
dX.αdn
dUdV
ii
ii
ii
∑−=∑−+=
=
== 00
( )
( )
[ ]T
dX.μ.μ.μ.dSdS
TdX.α.μdS
TdX.α.μ
dS
COOCORp
RRR
ppp
2212
11 −+−=+
∑−=
∑−=
En el equilibrio:
0=∑ ii μ.α

38
Equilibrio heterogéneo Equilibrio Mecánico Térmico Químico Condición p1 = p2 T1 = T2 μi
(1) = μi(2)
“La condición de equilibrio másico en un sistema multicomponente consiste en que el potencial químico de cada componente sea el mismo en todos sus puntos”. El conjunto de las tres condiciones forman el“equilibrio termodinámico”.
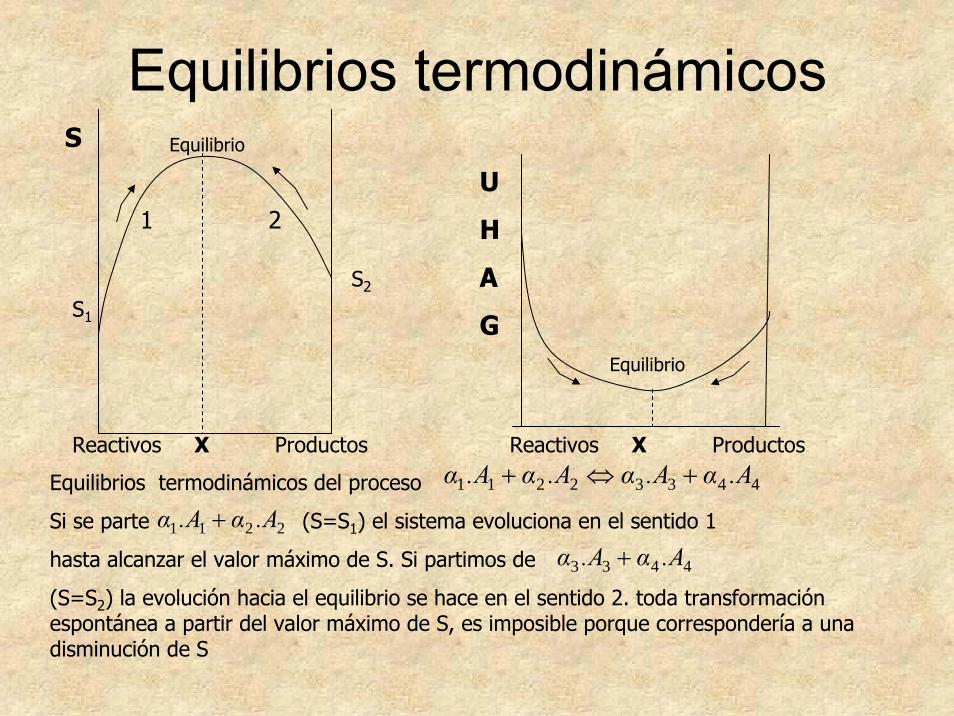
Equilibrios termodinámicos
2211 A.αA.α +44332211 A.αA.αA.αA.α +⇔+
Reactivos X Productos
S
S1
S2
Equilibrio
Reactivos X Productos
Equilibrio
U
H
A
G
Equilibrios termodinámicos del proceso
Si se parte (S=S1) el sistema evoluciona en el sentido 1
hasta alcanzar el valor máximo de S. Si partimos de
(S=S2) la evolución hacia el equilibrio se hace en el sentido 2. toda transformación espontánea a partir del valor máximo de S, es imposible porque correspondería a una disminución de S
4433 A.αA.α +
1 2

Influencia de ∆H y ∆S en una reacción química
)inicialentropíafinalentropía(aumenteSΔexotérmicareacciónuyamindisHΔ
SΔ.THΔGΔ
>
<−= 0
∆H ∆S ∆G La reacción puede verificarse...
< 0 > 0 < 0 Siempre
< 0 < 0 ? Cuando T es suficientemente baja (T∆S<I∆HI)
> 0 > 0 ? Cuando T es suficientemente alta (T∆S> ∆H)
> 0 < 0 > 0 En ningún caso
∆H ∆H/T 0 En equilibrio

41
Termodinámica y proceso de disolución
∆Gd = ∆Hd - T ∆Sd
Espontaneidad:
Si ∆Gd < 0
Proceso espontáneo (hay disolución)
Calor del procesoBalance de fuerzas intermolecularesentre los componentes purosy la disolución
Variación de entropía
Balance de desorden entre los componentes puros y la disolución

42
Balance de fuerzas atractivas intermoleculares I
1. Fuerzas soluto-soluto ≈ Fuerzas soluto – disolvente
∆Hd ≈ 0 (fuerzas similares) Se da disolución
y es ideal∆Sd > 0 (aumenta desorden)
2. Fuerzas soluto-soluto < Fuerzas soluto – disolvente
∆Hd < 0 (fuerzas más intensas en disolución) Se da disolución
no ideal∆Sd > 0 (aumenta desorden)

43
Balance de fuerzas atractivas intermoleculares II
3. Fuerzas soluto-soluto > Fuerzas soluto-disolvente
∆Hd > 0 (fuerzas menos intensas en disolución)Puede haber disolución(no ideal) si el aumento de entalpía no compensa el aumento de entropía
∆Sd > 0 (aumenta desorden)
4. Fuerzas soluto-soluto >> Fuerzas soluto-disolvente
∆Hd >> 0 (fuerzas mucho menos intensas en disolución)
No hay disolución∆Sd > 0 (aumenta desorden)
44
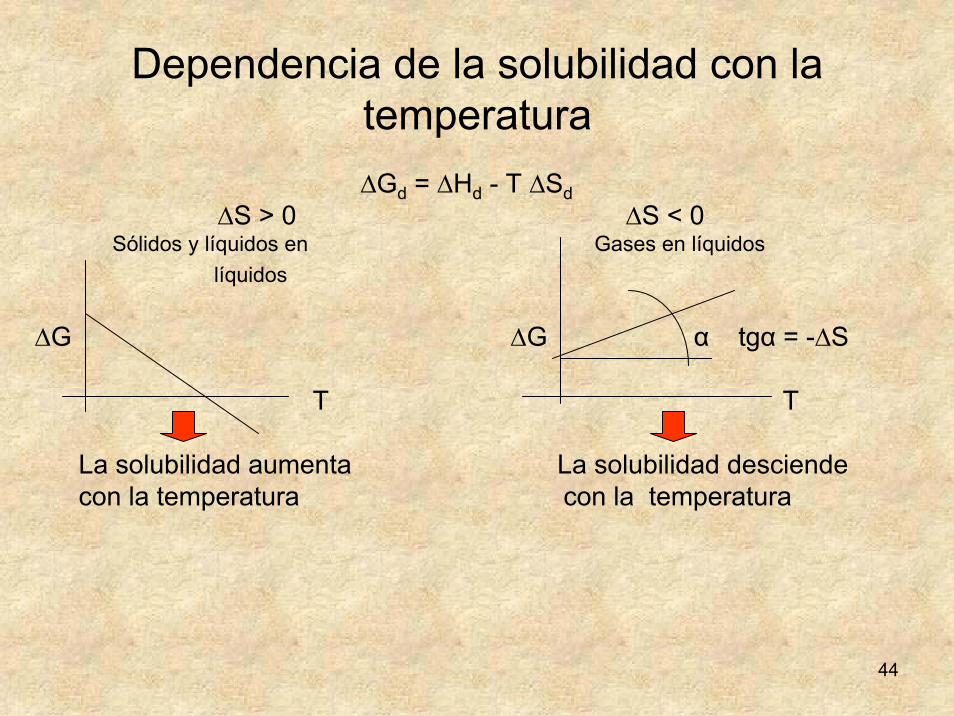
Dependencia de la solubilidad con la temperatura∆Gd = ∆Hd - T ∆Sd
∆S > 0 ∆S < 0Sólidos y líquidos en Gases en líquidos
líquidos
∆G ∆G α tgα = -∆S
T T
La solubilidad aumenta La solubilidad desciende con la temperatura con la temperatura
45
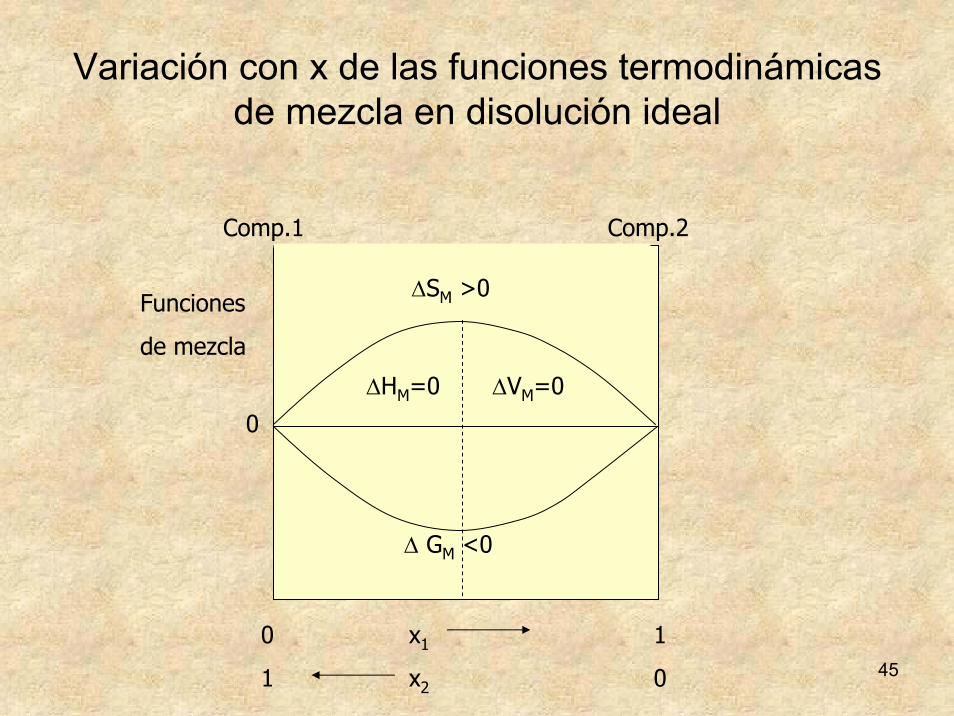
Variación con x de las funciones termodinámicas de mezcla en disolución ideal
ΔHM=0 ΔVM=0
Comp.2
0 x1 1
1 x2 0
Comp.1
Funciones
de mezcla
ΔSM >0
Δ GM <0
0

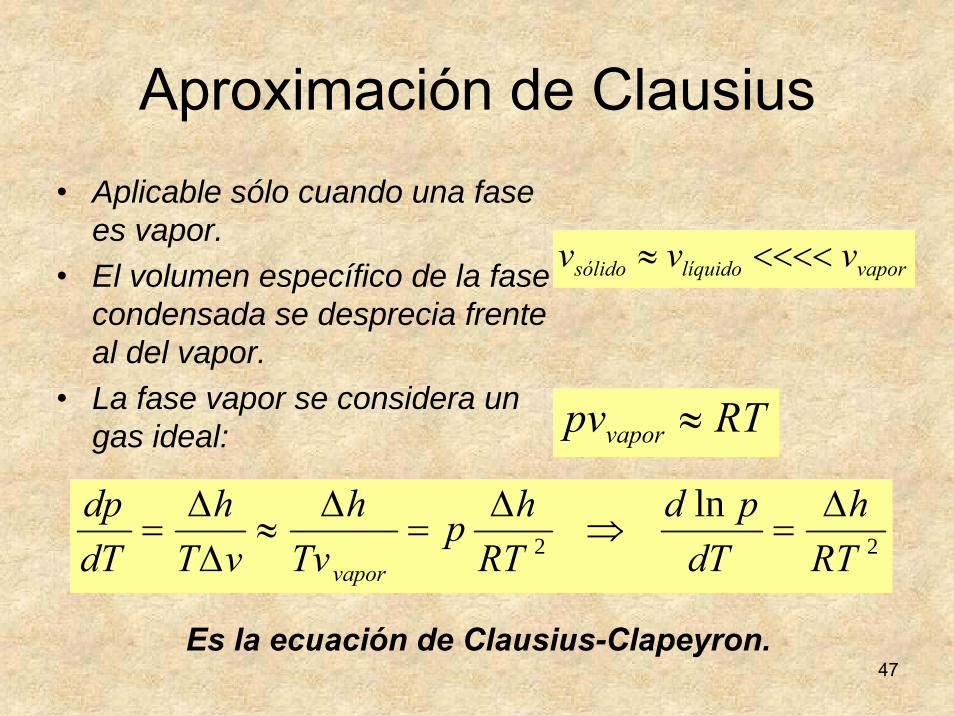
Ecuación de Clapeyron
21 μμ =
21 μμ dd =
dpvdTsdpvdTs 2211 +−=+−
En el equilibrio de las fases 1 y 2 de un cuerpo puro se cumple y a lo largo de su línea de coexistencia: .Sustituyendo los potenciales químicos:
dondey ThThhsss //)( 1212 Δ=−=−=Δ
12 vvv −=Δ
ρρρ
ΔΔ
−=Δ
=Δ
Δ=
ΔΔ
=T
hvT
LvT
hvs
dTdp
21
47
Aproximación de Clausius• Aplicable sólo cuando una fase
es vapor.• El volumen específico de la fase
condensada se desprecia frente al del vapor.
• La fase vapor se considera un gas ideal:
vaporlíquidosólido vvv <<<<≈
RTpvvapor ≈
22
lnRT
hdT
pdRT
hpTv
hvT
hdTdp
vapor
Δ=⇒
Δ=
Δ≈
ΔΔ
=
Es la ecuación de Clausius-Clapeyron.
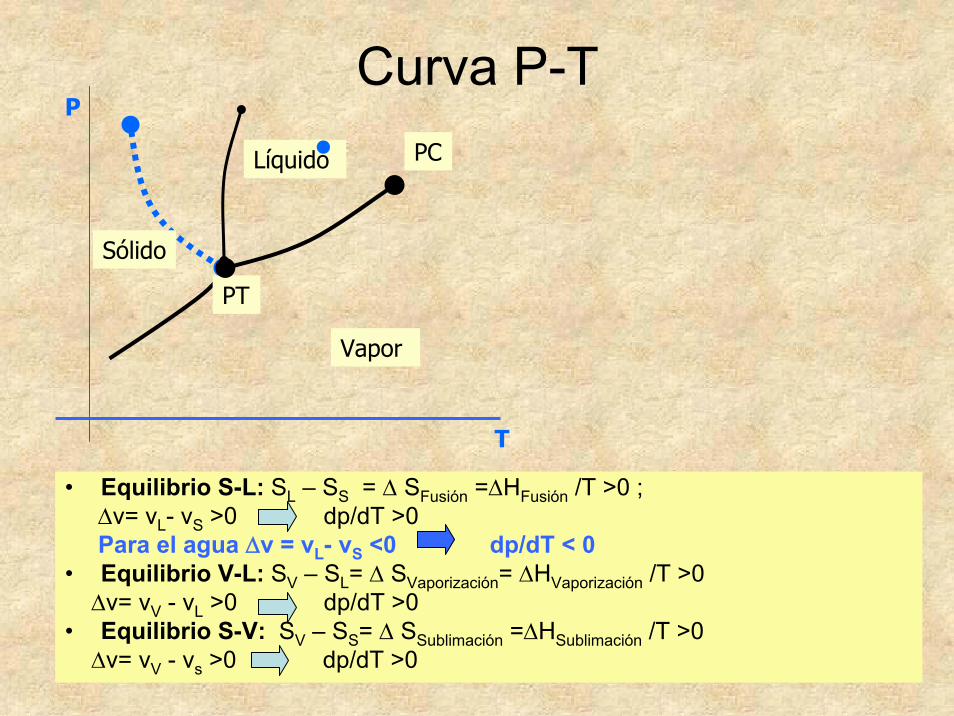
48
Curva P-T
• Equilibrio S-L: SL – SS = Δ SFusión =ΔHFusión /T >0 ;Δv= vL- vS >0 dp/dT >0Para el agua Δv = vL- vS <0 dp/dT < 0
• Equilibrio V-L: SV – SL= Δ SVaporización= ΔHVaporización /T >0 Δv= vV - vL >0 dp/dT >0
• Equilibrio S-V: SV – SS= Δ SSublimación =ΔHSublimación /T >0 Δv= vV - vs >0 dp/dT >0
PC
PT
Líquido
Sólido
Vapor
T
P

49
El punto triple• Coexiste en equilibrio las tres fases. • Si existen fases alotrópicas, pueden ser dos sólidas y una
líquida o vapor.• Según la regla de las fases, es un punto fijo.• En un ciclo, lo suficientemente cerrado a su alrededor, se
cumple:
0=−+= aciónlimsubónvaporizacifusión hhhh ΔΔΔΔ

50
El punto crítico I• Es la cúspide de la curva de
vaporización. • Sus fluctuaciones producen
la difusión de la luz que le ilumina y la opalescencia crítica.
• Según la regla de las fases, es un punto fijo.
• En el diagrama p-V, la isoterma crítica tiene un punto de inflexión, por lo que:
( ) 0,, =ccc TVpF
0=⎟⎠⎞
⎜⎝⎛
∂∂
críticopuntoV
p
02
2
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
críticopuntoV
p
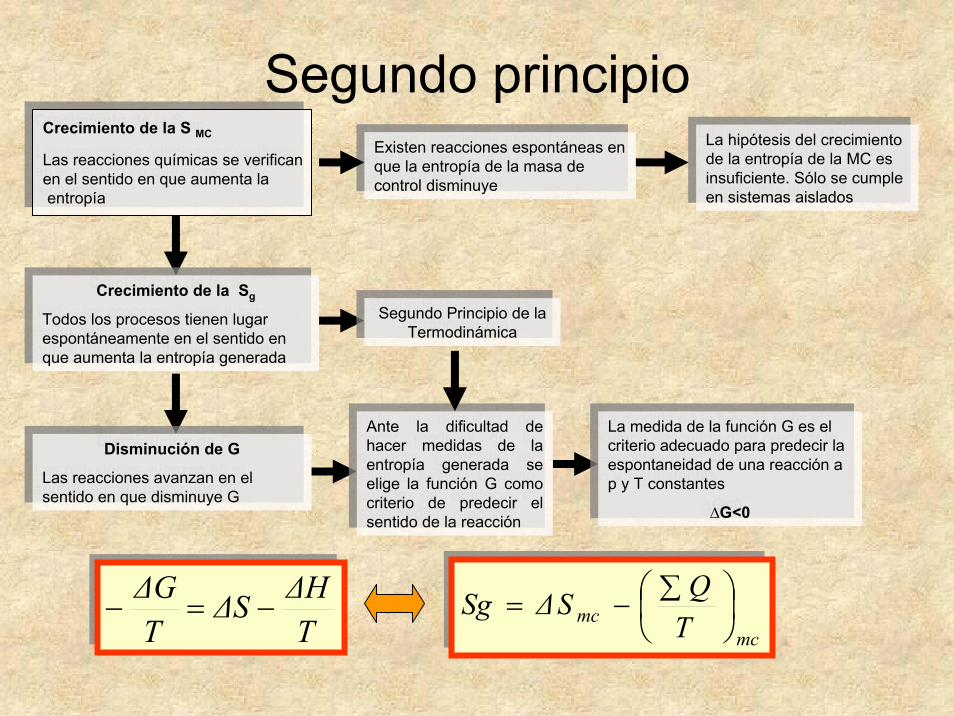
Segundo principio
THΔSΔ
TGΔ
−=−
Crecimiento de la S MC
Las reacciones químicas se verifican en el sentido en que aumenta laentropía
Crecimiento de la S MC
Las reacciones químicas se verifican en el sentido en que aumenta laentropía
Existen reacciones espontáneas en que la entropía de la masa de control disminuye
Existen reacciones espontáneas en que la entropía de la masa de control disminuye
La hipótesis del crecimiento de la entropía de la MC es insuficiente. Sólo se cumple en sistemas aislados
La hipótesis del crecimiento de la entropía de la MC es insuficiente. Sólo se cumple en sistemas aislados
Crecimiento de la Sg
Todos los procesos tienen lugar espontáneamente en el sentido en que aumenta la entropía generada
Crecimiento de la Sg
Todos los procesos tienen lugar espontáneamente en el sentido en que aumenta la entropía generada
Segundo Principio de la Termodinámica
Segundo Principio de la Termodinámica
Disminución de G
Las reacciones avanzan en el sentido en que disminuye G
Disminución de G
Las reacciones avanzan en el sentido en que disminuye G
Ante la dificultad de hacer medidas de la entropía generada se elige la función G como criterio de predecir el sentido de la reacción
Ante la dificultad de hacer medidas de la entropía generada se elige la función G como criterio de predecir el sentido de la reacción
La medida de la función G es el criterio adecuado para predecir la espontaneidad de una reacción a p y T constantes
∆G<0
La medida de la función G es el criterio adecuado para predecir la espontaneidad de una reacción a p y T constantes
∆G<0
mcmc T
QSΔSg ⎟⎠⎞
⎜⎝⎛ ∑−=

Ing. Juan Montesano

Disoluciones ideales
Presión de vapor de los componentes puros, ecuaciones de Antoine
CtBApln sat +
−=
Llamando xA y xb a las fracciones molares de A y B en la fase líquida. Según la ley de Raoult, si pA y pB son las presiones parciales de A y B en la fase vapor:
Llamando pAo la presión de vapor del líquido puro A (xA =1) a la misma
temperatura de la disolución y pBo a la del líquido puro B (xB =1)
pA = xA PAopB = xB PBo
Ley de Raoult

54
G-A y su aplicación a una pila electroquímica
PP
Pp
ansivoexpnomáximo
p
Tε.F.υSΔε
Tε.T.F.υHΔ
Tευ.F
TGΔ
υ.F.εa.femargcLGΔ
TGΔ.THΔGΔ
TSHG
⎟⎠⎞
⎜⎝⎛
∂∂
=⎥⎦
⎤⎢⎣
⎡−⎟
⎠⎞
⎜⎝⎛
∂∂
=
⎟⎠⎞
⎜⎝⎛
∂∂
−=⎟⎠⎞
⎜⎝⎛
∂∂
=−=−=
⎟⎠⎞
⎜⎝⎛
∂∂
+=
−=
-
-

Presión parcial- x
Componente de B puro
Componente de A puro
pAo
1 XA 0
0 XB 1
pBo
p A + p B = P
T = constante

56
Presión parcial- x • pA = pA
o - xB.pAo
BoA
AoA xp
pp=
−
Si A es el disolvente y B el soluto, la disolución relativa de presión de vapor del disolvente es igual a la fracción molar del soluto
A
B
AB
BoA n
nnn
np
pΔ≅
+=−
La disminución relativa de la presión de vapor del disolvente depende exclusivamente de la relación entre los números de moles del soluto y del disolvente sin que para nada influya la naturaleza de ambos
donde Δp = pA – pAo
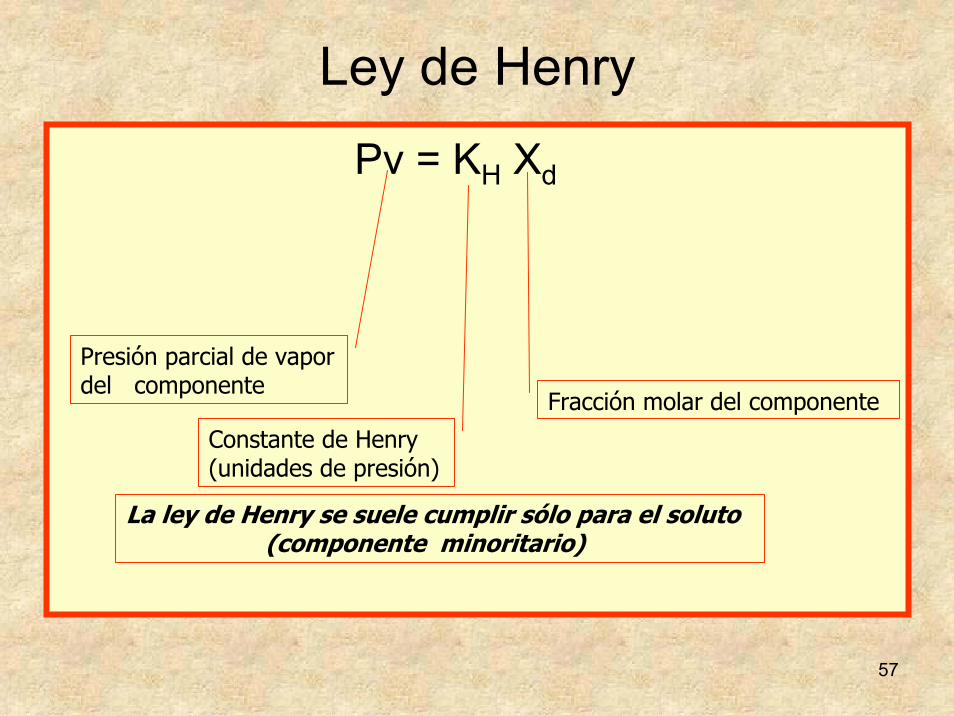
57
Ley de Henry
Pv = KH Xd
Presión parcial de vapor del componente
Constante de Henry (unidades de presión)
Fracción molar del componente
La ley de Henry se suele cumplir sólo para el soluto (componente minoritario)
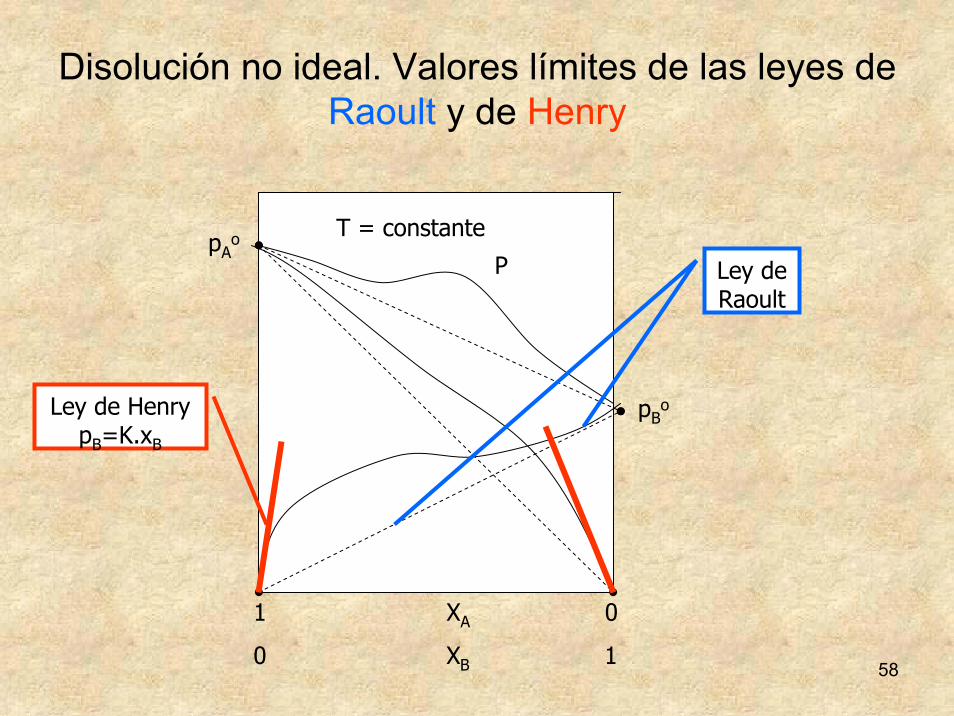
58
Disolución no ideal. Valores límites de las leyes de Raoult y de Henry
pAo
1 XA 0
0 XB 1
pBo
P
T = constante
Ley de Raoult
Ley de HenrypB=K.xB

59
Ley de Henry y Raoult
K.xp,xsi,HenrydeLeyp.xp,xsi,RaoultdeLey
BBB
oAAAA
=→=→
01
La ley de Raoult está normalmente asociada al disolvente y la ley de Henry al soluto a baja concentración

60
• Ing. Juan Montesano
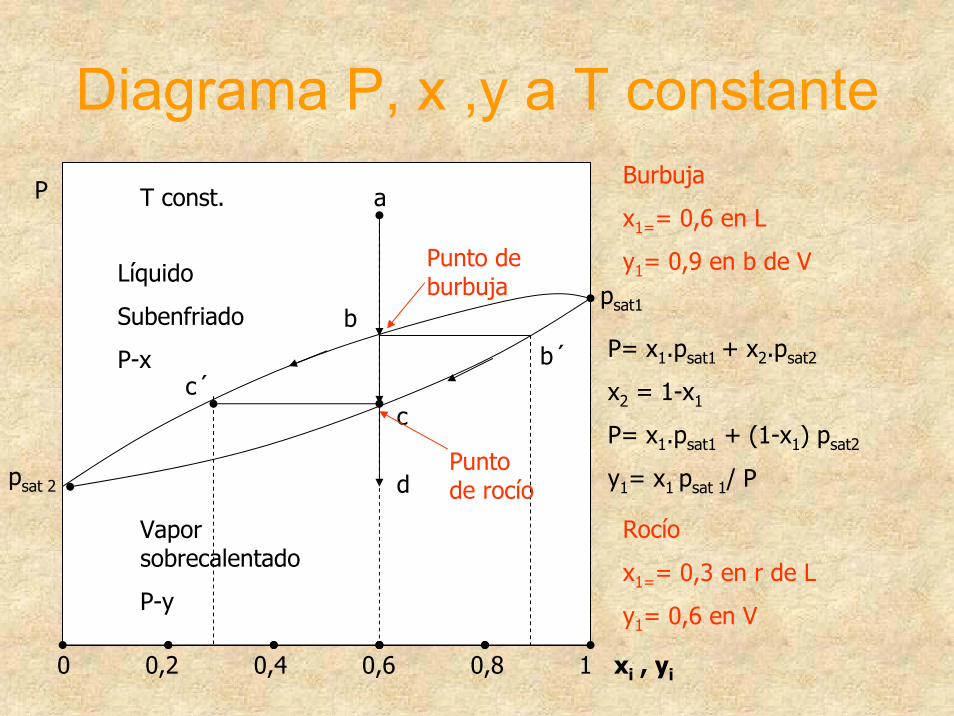
Diagrama P, x ,y a T constante
0 0,2 0,4 0,6 0,8 1 xi , yi
Líquido
Subenfriado
P-x
Vapor sobrecalentado
P-y
a
b
c
d
c´b´
P
P= x1.psat1 + x2.psat2
x2 = 1-x1
P= x1.psat1 + (1-x1) psat2
y1= x1 psat 1/ P
T const.
psat1
Punto de burbuja
psat 2Punto de rocío
Burbuja
x1== 0,6 en L
y1= 0,9 en b de V
Rocío
x1== 0,3 en r de L
y1= 0,6 en V
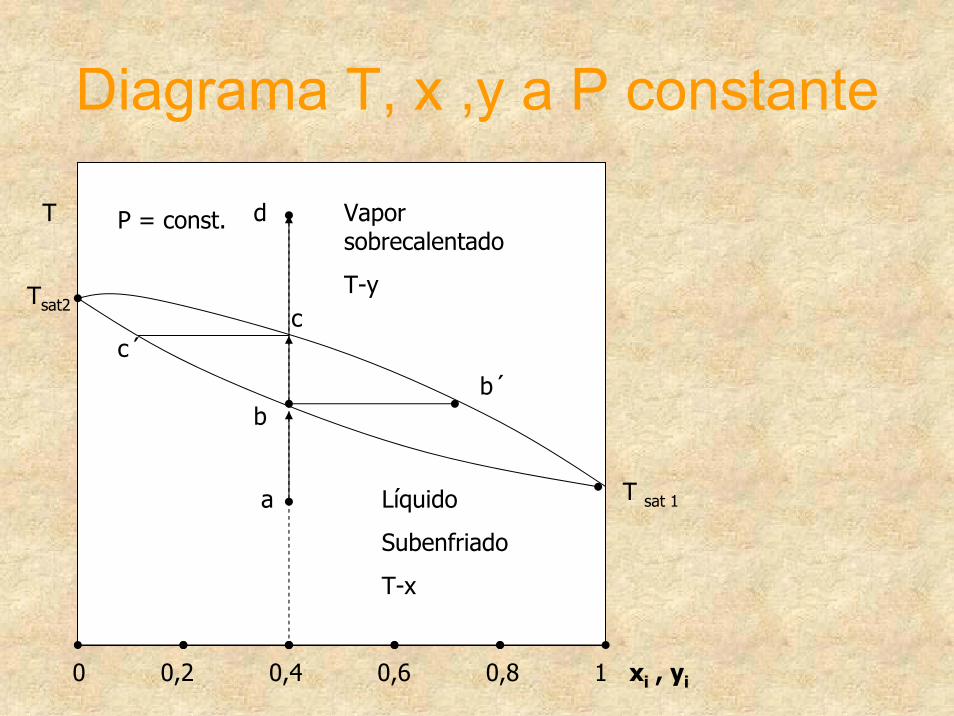
Diagrama T, x ,y a P constante
0 0,2 0,4 0,6 0,8 1 xi , yi
Líquido
Subenfriado
T-x
Vapor sobrecalentado
T-y
a
b
c
d
c´
b´
T P = const.
T sat 1
Tsat2
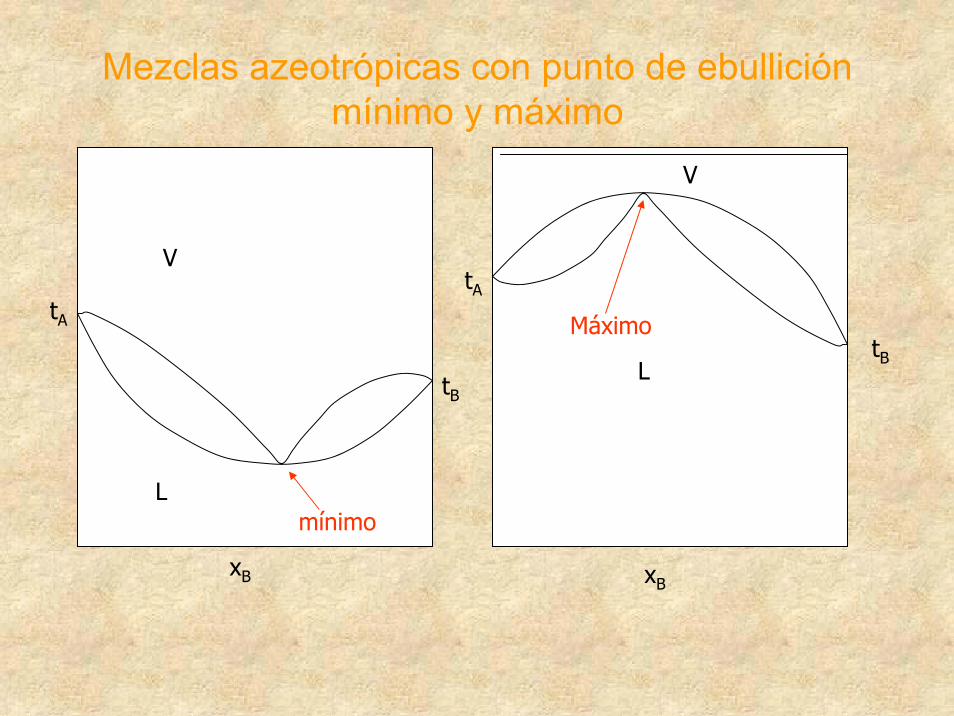
Mezclas azeotrópicas con punto de ebullición mínimo y máximo
tA
V
LtB
xB
L
V
mínimo
tA
xB
MáximotB
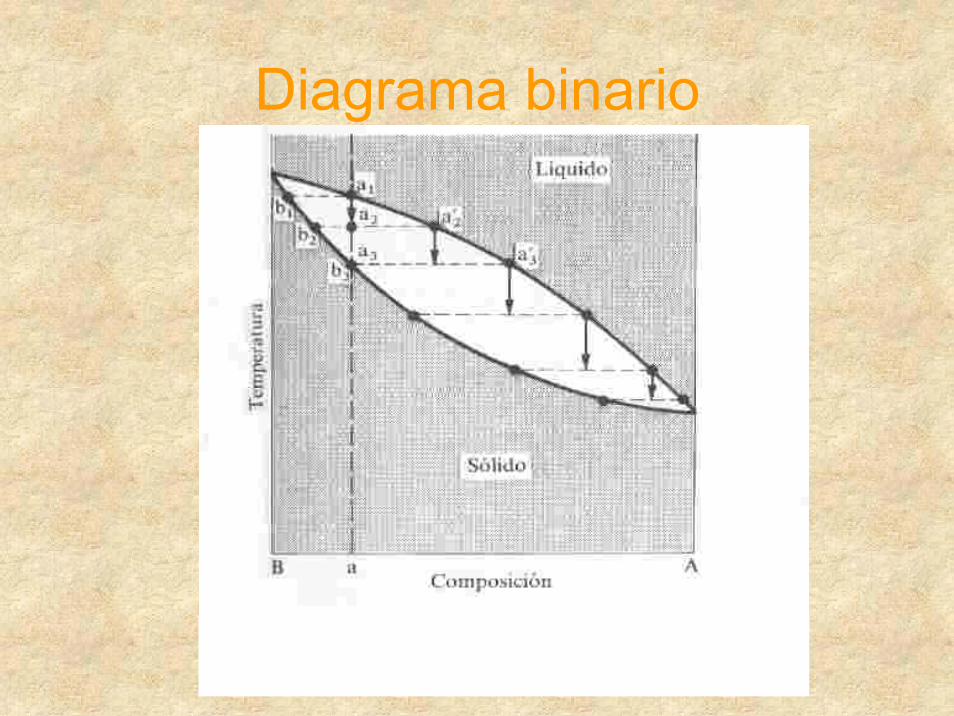
Diagrama binario
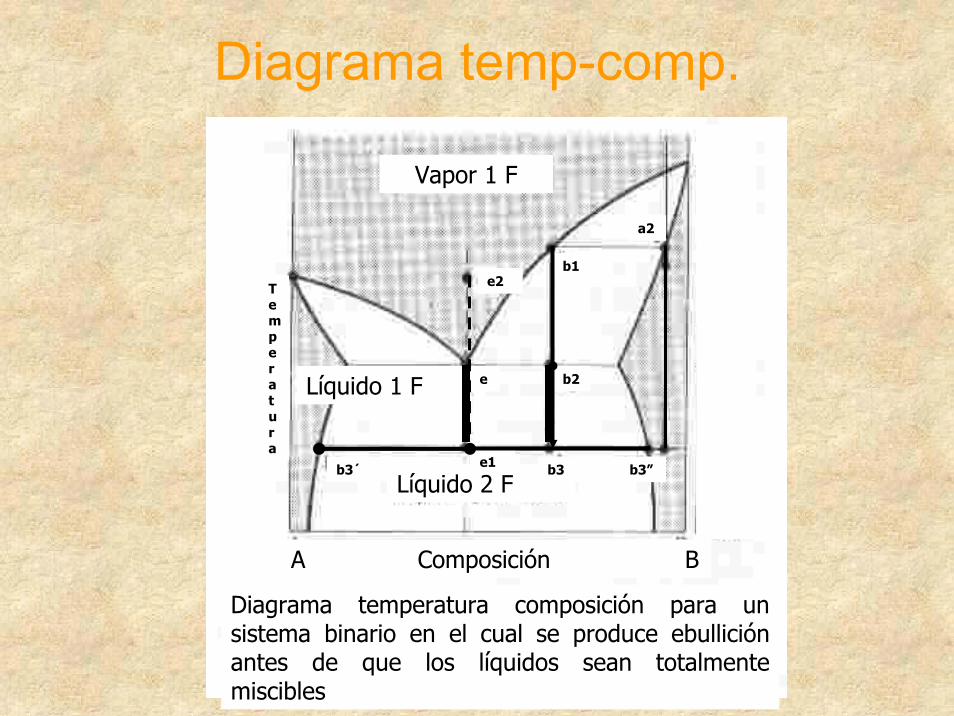
Diagrama temp-comp.
Vapor 1 F
Líquido 1 F
Líquido 2 F
A Composición B
Diagrama temperatura composición para un sistema binario en el cual se produce ebullición antes de que los líquidos sean totalmente miscibles
e1
e b2
b1
b3 b3”
e2
a2
b3´
Temperatura
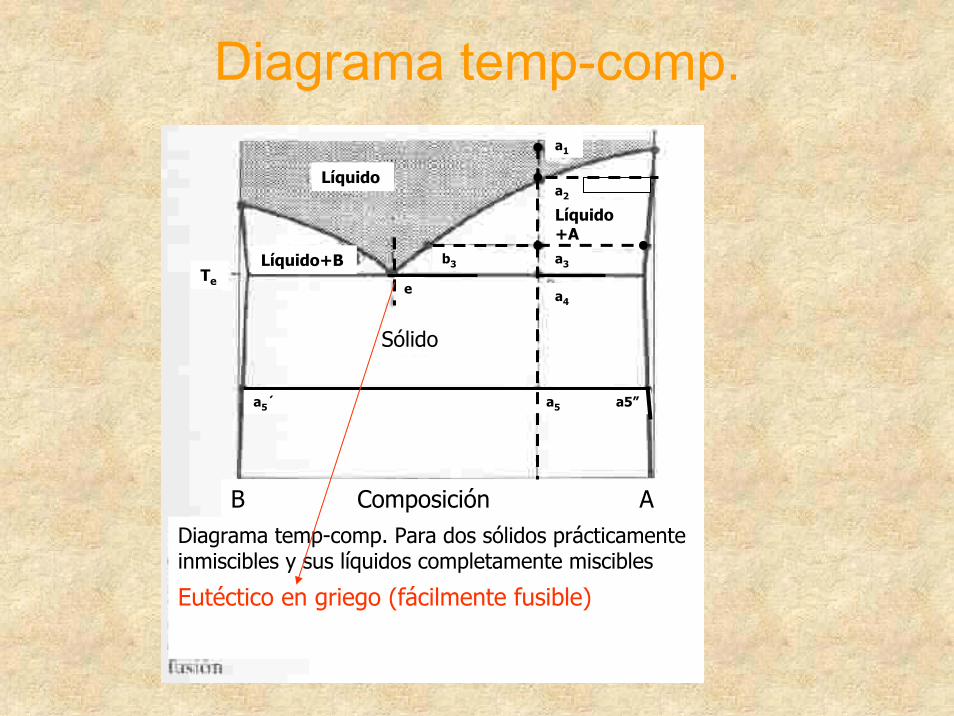
Diagrama temp-comp.
Diagrama temp-comp. Para dos sólidos prácticamente inmiscibles y sus líquidos completamente miscibles
Eutéctico en griego (fácilmente fusible)
Sólido
Líquido+B
Líquido
Líquido +A
a5´ a5 a5”
a4
a3
eTe
B Composición A
b3
a2
a1
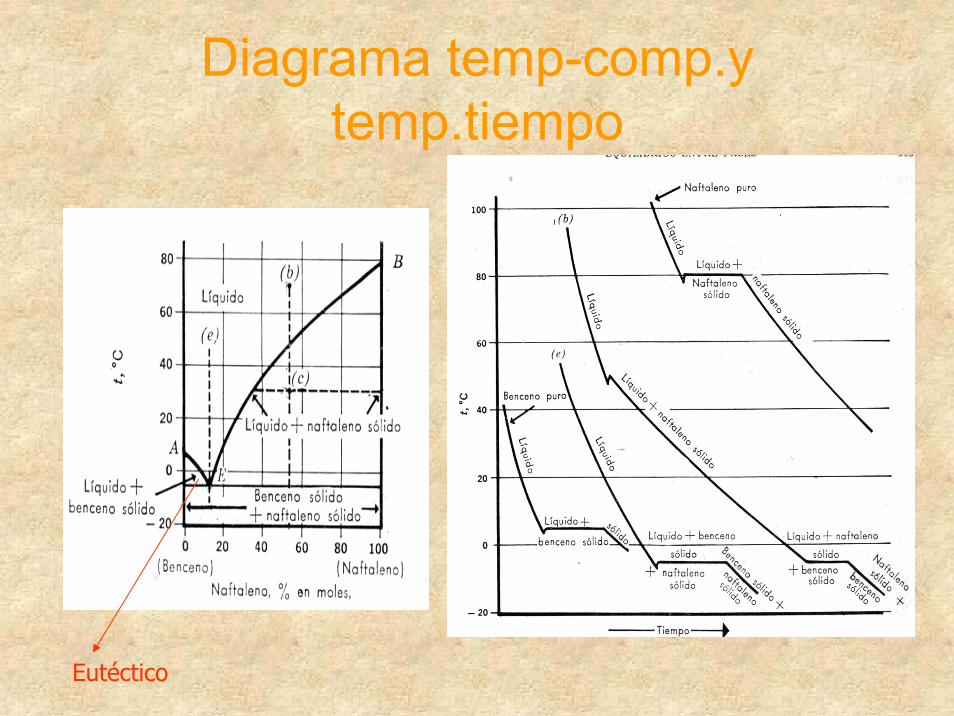
Diagrama temp-comp.ytemp.tiempo
Eutéctico
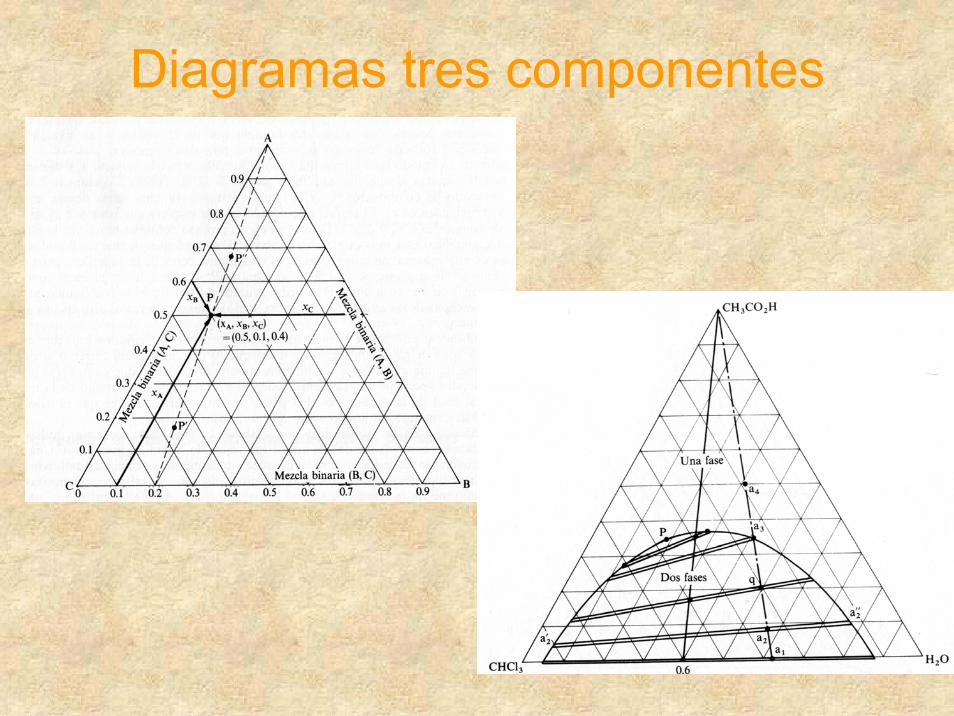
Diagramas tres componentes
69
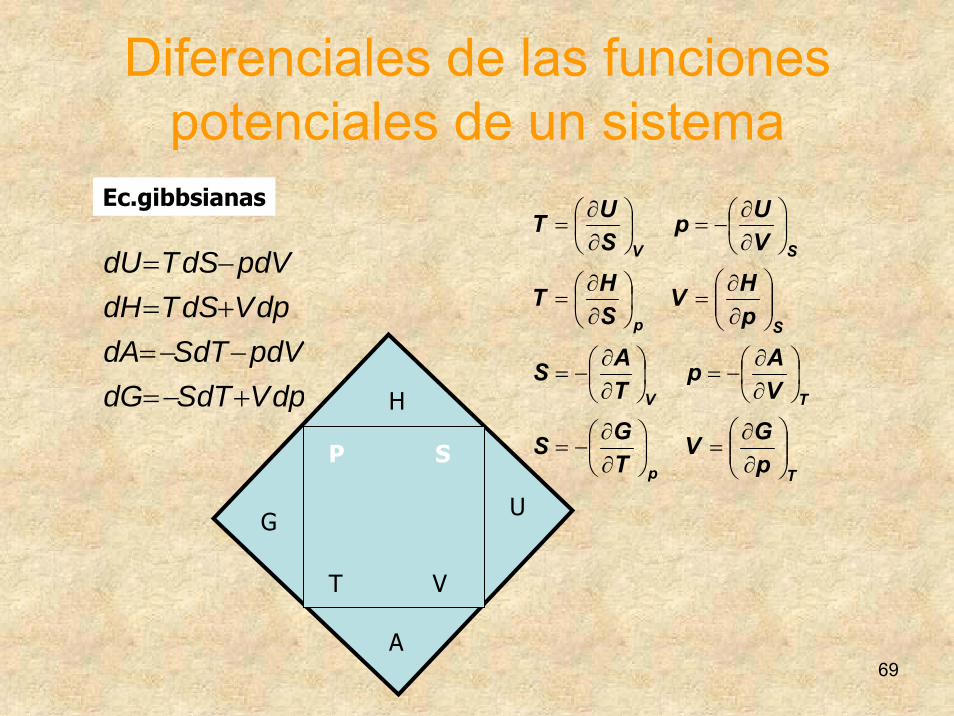
Diferenciales de las funciones potenciales de un sistema
dpVdTSdGdVpdTSdA
dpVdSTdHdVpdSTdU
+−=−−=
+=−=
Ec.gibbsianas
G
P S
T V
H
U
A
Tp
TV
Sp
SV
pGV
TGS
VAp
TAS
pHV
SHT
VUp
SUT
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎠⎞
⎜⎝⎛
∂∂
−=
⎟⎠⎞
⎜⎝⎛
∂∂
−=⎟⎠⎞
⎜⎝⎛
∂∂
−=
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
=⎟⎠⎞
⎜⎝⎛
∂∂
=
⎟⎠⎞
⎜⎝⎛
∂∂
−=⎟⎠⎞
⎜⎝⎛
∂∂
=

Función Gibbs
( ) ( ) ( ) ( )20210121012021 )( .STH.STHVVPSS.TUULu −−−=−+−+−≤
21120 )( AALVVP u −≤+−
S.THG −=
21 GGL u −≤
Es decir, en un proceso a p y T conste., la disminución de la función G mide el máximo L distinto del correspondiente a las fuerzas de presión que
se puede obtener en dicha transformación
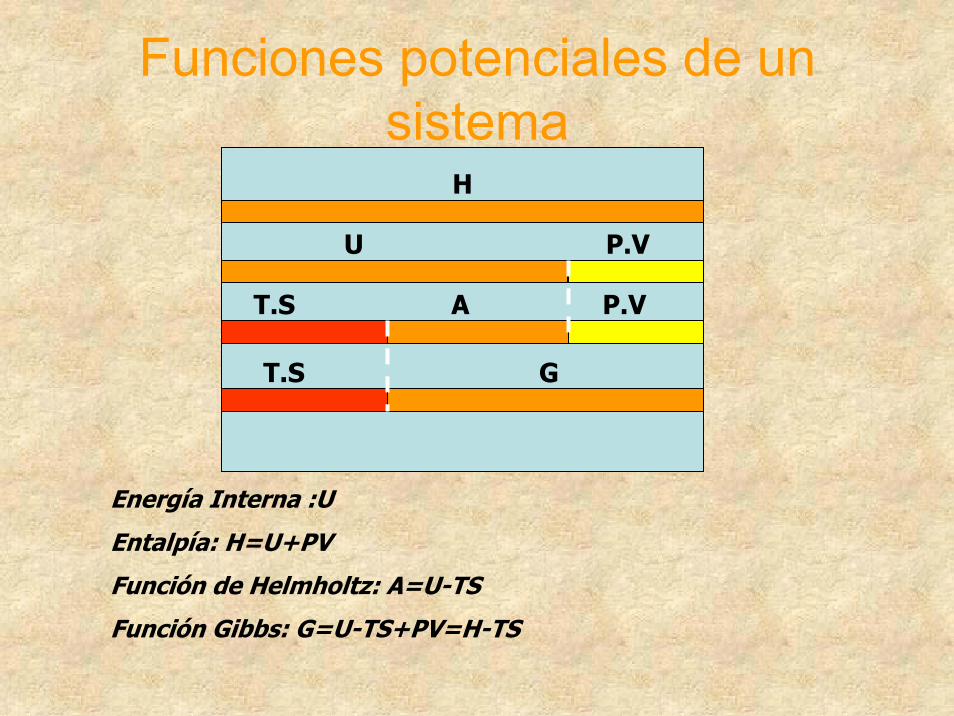
Funciones potenciales de un sistema
H
U P.V
T.S A P.V
T.S G
Energía Interna :U
Entalpía: H=U+PV
Función de Helmholtz: A=U-TS
Función Gibbs: G=U-TS+PV=H-TS
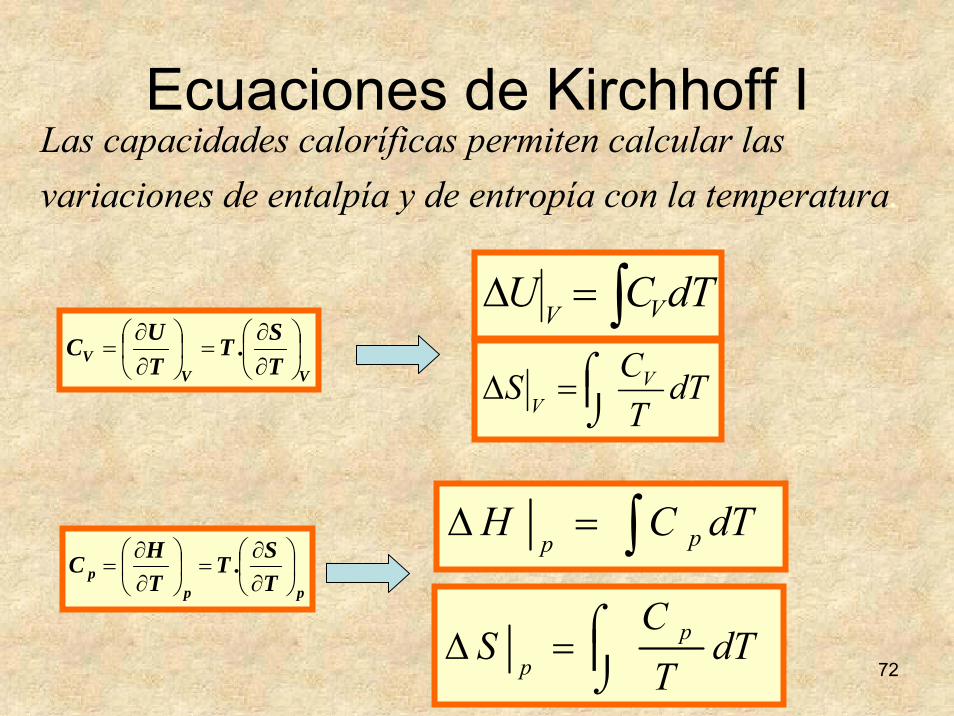
72
Ecuaciones de Kirchhoff I
VVV T
S.TTUC ⎟
⎠⎞
⎜⎝⎛
∂∂
=⎟⎠⎞
⎜⎝⎛
∂∂
=∫=Δ dTCU VV
⎮⌡⌠=Δ dT
TCS V
V
Las capacidades caloríficas permiten calcular las variaciones de entalpía y de entropía con la temperatura.
ppp T
S.TTHC ⎟
⎠⎞
⎜⎝⎛
∂∂
=⎟⎠⎞
⎜⎝⎛
∂∂
=∫=Δ dTCH pp
⎮⌡⌠=Δ dT
TC
S pp
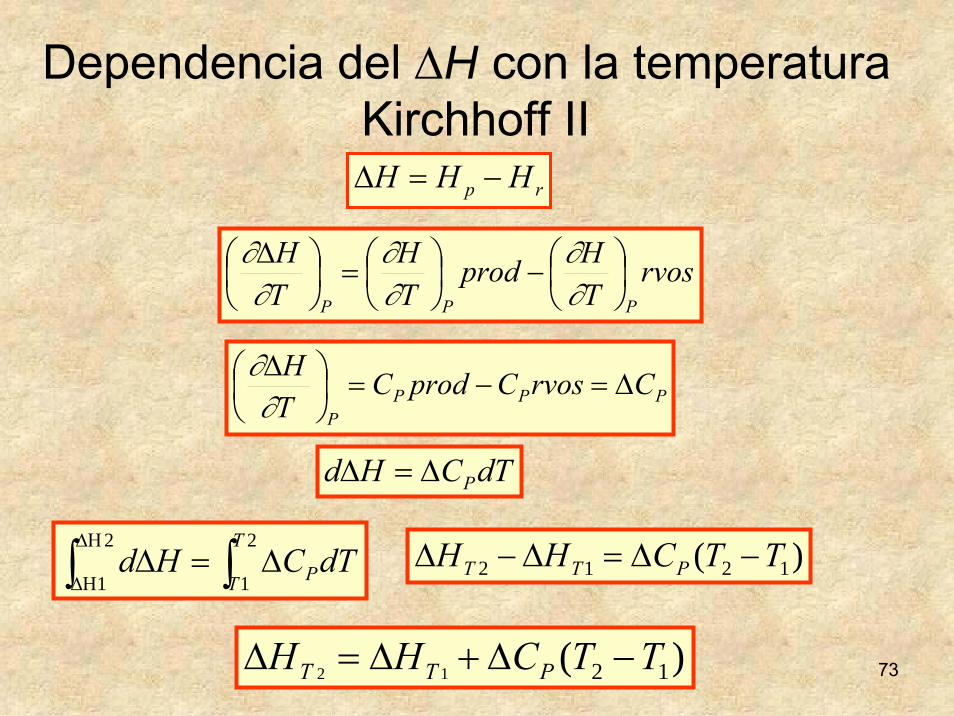
73
rp HHH −=Δ
rvosTHprod
TH
TH
PPP⎟⎠⎞
⎜⎝⎛−⎟
⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛ Δ
∂∂
∂∂
∂∂
Dependencia del ΔH con la temperaturaKirchhoff II
PPPP
CrvosCprodCTH
Δ=−=⎟⎠⎞
⎜⎝⎛ Δ
∂∂
dTCHd PΔ=Δ
∫ ∫ΔΗ
ΔΗΔ=Δ
2
1
2
1
T
T PdTCHd )( 1212 TTCHH PTT −Δ=Δ−Δ
)( 1212 TTCHH PTT −Δ+Δ=Δ

74
Ecuaciones de Kirchhoff II (respecto a T0)
( ) ( ) ∫ Δ+Δ=ΔT
TVo
o
dTCVTUVTU ,, ,
( ) ( ) ∫ Δ+Δ=ΔT
Tpo
o
dTCpTHpTH ,, ,
( ) ( ) ∫ Δ+Δ=ΔT
TVo
oTdTCVTSVTS ,,
Cambios de las funciones con la temperatura:
( ) ( ) ∫ Δ+Δ=ΔT
Tpo
oTdTCpTSpTS ,,

75
Ecuaciones de Gibbs-Helmholtz
VTATUTSUA ⎟
⎠⎞
⎜⎝⎛
∂∂
+=−=pT
GTHTSHG ⎟⎠⎞
⎜⎝⎛
∂∂
+=−=
221
TU
TA
TTA
V=⎟
⎠⎞
⎜⎝⎛
∂∂
− 22
1TH
TG
TTG
p
=⎟⎠⎞
⎜⎝⎛
∂∂
−
( )2T
UT
TA
V−=⎟
⎠⎞
⎜⎝⎛
∂∂ ( )
2TH
TTG
p
−=⎟⎠⎞
⎜⎝⎛
∂∂
( )2T
UT
TA
V
Δ−=⎟
⎠⎞
⎜⎝⎛
∂Δ∂ ( )
2TH
TTG
p
Δ−=⎟
⎠⎞
⎜⎝⎛
∂Δ∂
En los procesos heterogéneos:

76
La masa como variable
• Hemos estudiado sistemas cerrados en los que la masa era un parámetro del sistema.
• La masa como variable necesita una relación con las otras variables.
• Además, se debe conocer la química para caracterizar las diversas formas de masa.
• Los efectos de superficie del sistema deben ser despreciables.

77
Funciones homogéneas
• Una función es homogénea si:
donde λ es un parámetro cualquiera y α su orden.• Teorema de Euler: Toda función homogénea de
primer grado es igual a la suma de los productos de sus derivadas parciales por las variables:
( )yxzz ,=
( ) ( )yxfyxfz ,.,.. αα λλλλ ==
( ) yzxzyxzyx ∂
∂+
∂=,
∂
78
1
2
.
.
.
.
.
C
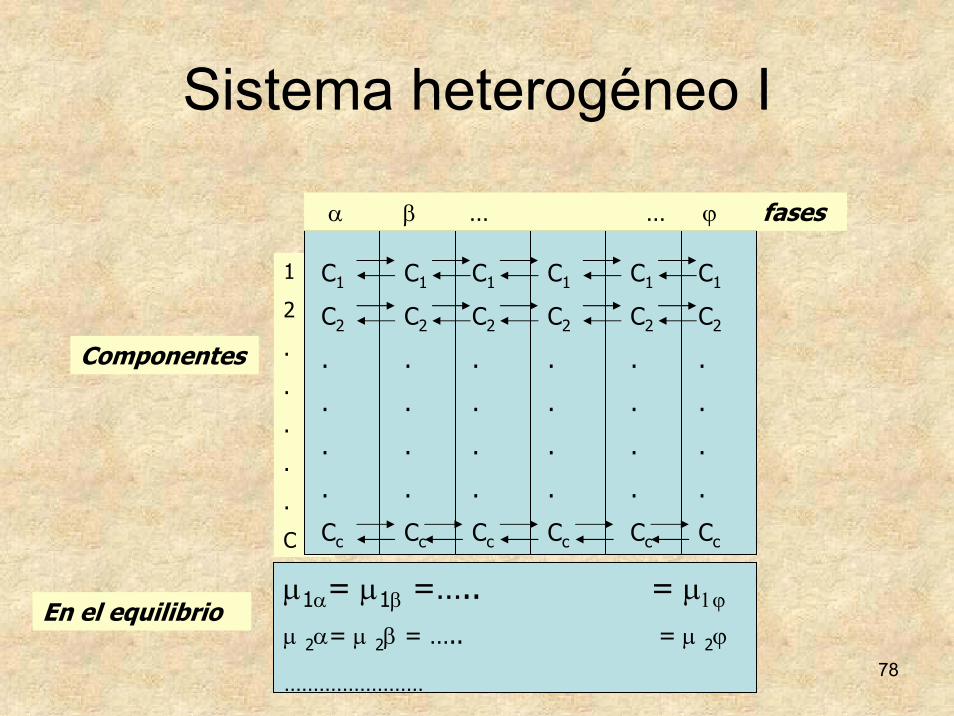
Sistema heterogéneo I
C1
C2
.
.
.
.
Cc
C1
C2
.
.
.
.
Cc
C1
C2
.
.
.
.
Cc
C1
C2
.
.
.
.
Cc
C1
C2
.
.
.
.
Cc
C1
C2
.
.
.
.
Cc
α β … … ϕ fases
Componentes
En el equilibrioμ1α= μ1β =….. = μ1ϕ
μ 2α= μ 2β = ….. = μ 2ϕ
……………………

79
Sistema heterogéneo IILas masas de los componentes en las diferentes fases se pueden expresar en función de las fracciones molares
⎪⎪⎩
⎪⎪⎨
⎧ X1,α + X2,α + … + XC,α =1
X1,β + X2,β + … + XC,β =1
.
.
X1,ϕ + X2,ϕ+ … + XC,ϕ =1
μ1α= μ1β =….. = μ1ϕ
μ2 α= μ 2 β =….. = μ 2ϕ
μ C α= μ C β =….. = μ Cϕ
Ecuaciones
ϕ
+
(ϕ -1).c

80
Sistema heterogéneo IIIVariables del sistema :
2 (la presión y la temperatura) + c.ϕ ( las concentraciones de cada componente en cada una de las fases)
La diferencia entre el nº de variables y las ecuaciones daráel nº de varianzas o grados de libertad termodinámicos del sistema Φ
Φ=(2+ c.ϕ ) – [c. (ϕ -1) + ϕ ]= 2+ c - ϕ
Esta expresión se conoce como la regla de las fases

81
Equilibrio y homogeneidadTodo sistema termodinámico en equilibrio, sea homogéneo o heterogéneo, satisface las condiciones de equilibrio termodinámico.Un sistema homogéneo (determinante de su matriz de coeficientes positivo), es también homogéneo según Gibbs. Un sistema no homogéneo se fractura en porciones homogéneas, las “fases”, con superficies que son ligaduras intrínsecas.

⎥⎥⎦
⎤
⎢⎢⎣
⎡−⎟
⎠⎞
⎜⎝⎛
∂∂
=⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
⇒
⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎠⎞
⎜⎝⎛
∂∂
−+=
⎥⎥⎦
⎤
⎢⎢⎣
⎡+⎟⎟
⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎠⎞
⎜⎝⎛
∂∂
=
+⎥⎥⎦
⎤
⎢⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎠⎞
⎜⎝⎛
∂∂
=+=⇒=
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
+⎟⎠⎞
⎜⎝⎛
∂∂
=⇒=
vTv.T.
cpTeCoeficient
dp.Tv.TvdT.cdh
dp.vT.psdT.
Ts.Tdh
dp.vdp.psdT.
Ts.Tdp.vds.Tdh)p,s(hh
dp.psdT.
Tsds)p,T(ss
pph
pp
Tp
Tp
Tp
1
AplicacionesCoeficiente de Joule-Thomson
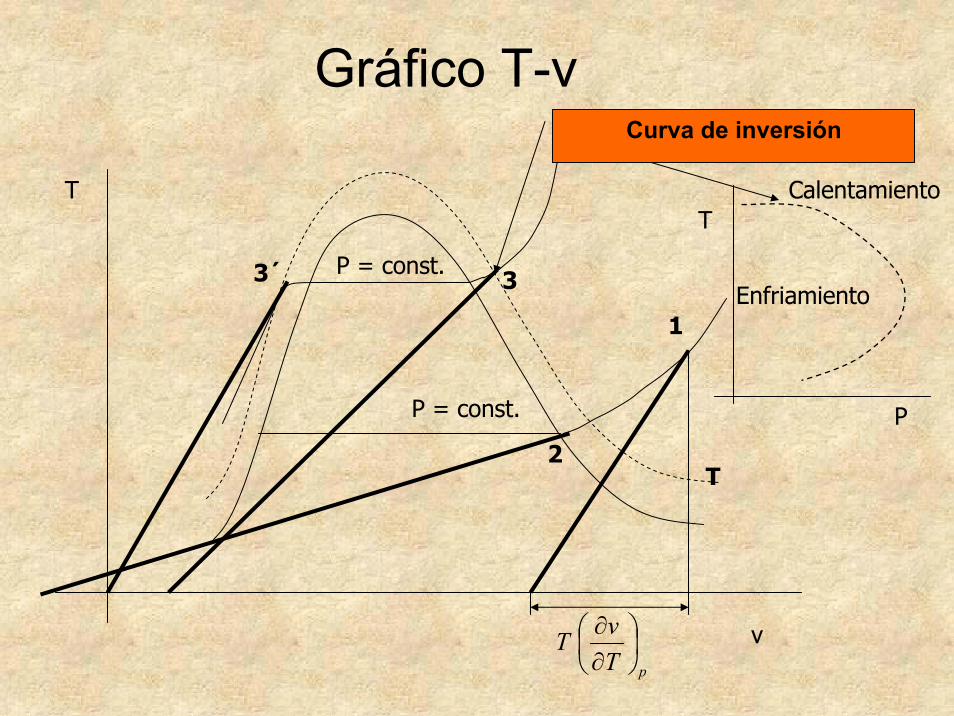
Gráfico T-v
T
pTvT ⎟
⎠⎞
⎜⎝⎛
∂∂
P = const.
P = const.
T
3
1
2
3´
v
Curva de inversión
T
Enfriamiento
Calentamiento
P

84
La presión de vapor
2
lnRT
hdT
pd Δ=
• Al aplicarse, deben cuidarse las dimensiones del segundo miembro.
Para pequeños intervalos de temperatura, se puede considerar que el calor latente es constante. Integrando la ecuación anterior:
Se tabulan las constantes a y b para cada sustancia.
Tbap
RThConstp −=⇒
Δ−= ln.ln



![[XLS]fmism.univ-guelma.dzfmism.univ-guelma.dz/sites/default/files/le fond... · Web view1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1](https://img.pdfslide.tips/doc/110x75/5b9d17e509d3f2194e8d827e/xlsfmismuniv-fond-web-view1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1-1.jpg)