Embed Size (px)
Citation preview

Khảo sát một số dẫn xuất halogen, ancol,
phenol và axit cacboxylic bằng phương pháp
hóa học lượng tử
Nguyễn Hà My
Trường Đại học Khoa học Tự nhiên
Luận văn ThS. ngành: Hóa lý và hóa lý thuyết; Mã số: 60 44 31
Người hướng dẫn: PGS.TS. Phạm Văn Nhiêu ; TS. Nguyễn Họa Mi
Năm bảo vệ: 2012
Abstract. Nghiên cứu cơ sở lý thuyết hóa học lượng tử, phương trình Schrodinger,
sự gần đúng Born – Oppenheirmer (Bon-Openhemơ, phương pháp biến phân, thuyết
trường tự hợp Hartree-Fork, phương trình Roothaan. Nghiên cứu cơ sở của các
phương pháp tính gần đúng lượng tử, cơ sở lý thuyết hóa học hữu cơ. Dẫn xuất
halogen, Anco, Phenol, Axit cacboxylic.
Keywords. Hóa lý; Hóa học lượng tử
Content
MỞ ĐẦU
Hóa học lượng tử bắt đầu phát triển từ khoảng những năm 30 của thế kỷ XX và ngày
càng chứng tỏ là một lý thuyết không thể thiếu trong mọi lĩnh vực hóa học. Hóa học lượng tử
là ngành khoa học nghiên cứu các hệ lượng tử dựa vào phương trình chính tắc của cơ học
lượng tử do Schorodinger đưa ra năm 1926, và nhanh chóng trở thành công cụ hữu ích của
hóa lý thuyết để đi sâu tìm hiểu, nghiên cứu vấn đề cốt lõi nhất của hóa học là cấu trúc và các
tính chất hóa lý của các chất.
Các quy luật phản ứng thế vào một số hợp chất hữu cơ, đặc biệt là phản ứng thế vào
vòng benzen là những quy luật thực nghiệm được hình thành rất lâu, và được sử dụng nhiều
trong giảng dạy hóa học hữu cơ. Các nghiên cứu khoa học đã chỉ ra được hướng thế vào liên
kết C – H trong vòng benzen. Tuy nhiên cho đến nay chưa có tài liệu nào công bố số liệu giải
thích và làm rõ thêm những quy luật trên. GAUSSIAN là phần mềm phát triển vượt trội về
các phương pháp ab initio (DFT) khá hiệu quả, được nhiều nhà nghiên cứu chuyên nghiệp sử
dụng. Với các thuật toán được viết tốt hơn, các bước tối ưu hóa của Gaussian cần 4 chuẩn hội
tụ trong khi Hyperchem chỉ có 1. Tuy chạy hơi chậm nhưng có độ chính xác khá cao, vì thế
đây là một công cụ hữu hiệu trợ giúp các nhà hóa học thực nghiệm trong nghiên cứu của
mình.
Từ những lý do trên, chúng tôi chọn đề tài nghiên cứu :”Khảo sát một số dẫn xuất
halogen, ancol, phenol và axit cacboxylic bằng phƣơng pháp hóa học lƣợng tử”.
Luận văn gồm các phần mở đầu, nội dung, kết luận, tài liệu tham khảo và phụ lục.
Phần nội dung chính gồm 3 chương.
Chƣơng 1. Tổng quan

Chƣơng 2. Đối tƣợng và phƣơng pháp nghiên cứu
Chƣơng 3. Kết quả và thảo luận
Chúng tôi hy vọng các kết quả của luận văn có thể góp phần làm rõ hơn hướng một số phản
ứng và là tài liệu tham khảo cho việc giảng dạy hóa học ở trường phổ thông.
CHƢƠNG 1: TỔNG QUAN
1.1. CƠ SỞ LÝ THUYẾT HÓA HỌC LƢỢNG TỬ
1.1.1. Phƣơng trình Schrodinger Sự biến đổi trạng thái vi mô theo thời gian của hệ lượng tử được mô tả bởi phương trình
Schrodinger (Srodingơ, 1926) có dạng tổng quát:
it
Ĥѱ (1.1)
( , )q t – Hàm sóng mô tả trạng thái của hệ lượng tử. Đó là hàm của tọa độ (q) và thời gian t. 2
2
2H T U U
m
Với 2 2 2
2
2 2 2x y z
: toán tử Laplace
U: thế năng (năng lượng tương tác giữa các hạt lượng tử trong hệ)
Trong trường hợp thế năng của hệ không phụ thuộc vào thời gian:
U = U(q) (q: tọa độ)
Hệ kín, hoặc hệ chuyển động trong một trường ngoài không đổi, thì toán tử Hamilton
H
không phụ thuộc vào thời gian và trùng với toán tử năng lượng toàn phần H
(q), còn trạng
thái của hệ được gọi là trạng thái dừng: ( , ) ( )q t q . Khi đó phương trình Schrodinger
được viết dưới dạng:
( ) ( ) ( )H q q E q
(1.2)
Ở đây E là trị riêng năng lượng, ( )q là hàm sóng (q: tọa độ tổng quát (x, y, z)
hay (r, , ). Khi đó nghiệm của phương trình (1.1) có thể viết dưới dạng:
( , ) ( ).iET
q t q e
(1.3)
Những trạng thái (1.3), ở đó hệ lượng tử có giá trị xác định gọi là những trạng thái dừng và
phương trình Schrodinger (1.2) là phương trình Schrodinger cho những trạng thái dừng, được
dùng trong hóa lượng tử.
1.1.2. Phƣơng pháp biến phân
Mục đích của phương pháp dựa trên MO – LCAO là để tìm ra cij gần đúng nhất với hàm
sóng thực tế ứng với năng lượng cực tiểu theo tập hàm cơ sở đã chọn. Biến đổi từ phương
trình Schrodinger ta có: *
*
H dE
d
(1.8)
Ở đây d là thể tích vô cùng nhỏ của không gian và spin.
Nếu hàm đã chuẩn hóa thì tích phân ở mẫu bằng đơn vị và phương trình có dạng: *E H d (1.9)
Khi áp dụng phương pháp biến phân, hàm sóng gần đúng thường được biểu diễn
dưới dạng MO – LCAO ở trên, tức là:

1 1 2 2 3 3 .... n nc c c c (1.10)
1.1.3. Thuyết trƣờng tự hợp Hartree-Fock
Phương pháp gần đúng đầu tiên được sử dụng để giải phương trình (1.2) là phương
pháp gần đúng Hartree – Fock (HF). Từ quan điểm vật lý về trường thế hiệu dụng trung bình
hóa đối với mỗi electron hợp bởi thế hút của hạt nhân và thế đẩy trung bình hóa do tất cả các
electron khác sinh ra.
1.1.4. Phƣơng trình Roothaan Phương pháp Hartree – Fock đề cập ở trên thuận tiện đối với việc sử dụng trong những
trường hợp trường Coulomb đối xứng cầu, tức là đối với các nguyên tử. Tuy nhiên, nó khó
tính được đối với những phân tử, không có trường Coulomb đối xứng cầu. Roothaan (1951)
sử dụng các tập hàm cơ sở để mở rộng phần không gian (bán kính) của các hàm spin –
orbitan. Việc này giúp chuyển các phương trình HF thành một bài toán ma trận có thể giải
được.
1.2. CƠ SỞ CỦA CÁC PHƢƠNG PHÁP TÍNH GẦN ĐÚNG LƢỢNG TỬ
Các phương pháp tính gần đúng được xây dựng trên cơ sở phương trình Roothaan.
Hầu hết các phương pháp này đều tập trung giải quyết vấn đề thế năng tương tác giữa các
electron với nhau dựa vào việc giải gần đúng các phương trình chưa tích phân Coulomb và
các tích phân xen phủ giữa các electron.
Với một lượng lớn các electron các tích phân đa tâm xuất hiện trong các số hạng J, K
(trong phương trình 1.19) hầu như không thể giải được. Để khắc phục những trở ngại đó,
người ta sử dụng một số phương pháp bán kinh nghiệm khác nhau dựa vào một số giả thiết
gần đúng sau:
- Giảm bộ hàm cơ sở.
- Bỏ qua một số tích phân.
- Thay thế một số tích phân bằng các hàm đơn giản có chứa tham số rút ra từ thực
nghiệm. Những tham số đó có được bằng cách đo hay tính toán như thế ion hóa, ái lực
electron, phổ,...
- Xem xét các hệ thống các electron và các electron riêng rẽ.
Các phương pháp tính gần đúng hiện nay bao gồm các phương pháp tính không kinh nghiệm
Ab initio (tính toán các tham số trên mô hình ước lượng) và các phương pháp bán kinh
nghiệm sử dụng các tham số thực nghiệm: CNDO, NDDO, AM1, PM3, MINDO, ZINDO...
1.3. CƠ SỞ LÝ THUYẾT HÓA HỌC HỮU CƠ
Electron là tiểu phân linh động nhất trong phân tử, dù chưa tham gia liên kết hoặc đã
tham gia liên kết nó đều có thể bị dịch chuyển bởi ảnh hưởng tương hỗ của các nguyên tử
trong phân tử.
Thuyết dịch chuyển electron xuất hiện từ năm 1920, trước thời kỳ phát triển của
thuyết obitan phân tử. Đơn giản và dễ hiểu, nó đã giúp giải thích đa số các dữ liệu thực
nghiệm có liên quan đến cấu trúc, tính chất và khả năng phản ứng của các hợp chất hữu cơ, vì
vậy nó đã được áp dụng rộng rãi cho đến nay.
1.3.1. Hiệu ứng cảm ứng.
- Bản chất: hiệu ứng cảm ứng là sự phân cực các liên kết, lan truyền theo mạch các liên kết
do sự khác nhau về độ âm điện.
- Phân loại: +I I=0 -I
Y C C H C X
Y là nhóm đẩy electron gây nên hiệu ứng cảm ứng dương (+I) như các nhóm mang điện tích
âm: -S-, O
-, các gốc ankyl CnH2n+1
-....
X là nhóm hút electron gây nên hiệu ứng cảm ứng âm (-I) như các hạt mang điện tích dương:
-N+R3; các nguyên tử có độ âm điện lớn như: - F, -Cl, -OR, -SR, -NR2...; các gốc hiđrocacbon
không no: CH2=CH-; C6H5-....

Nguyên tử H coi như không có sự hút hay đẩy electron.
- Đặc điểm: hiệu ứng cảm ứng giảm rất nhanh khi kéo dài mạch truyền ảnh hưởng. Hiệu ứng
cảm ứng không bị cản trở bởi các yếu tố không gian.
1.3.2. Hiệu ứng liên hợp
- Bản chất: Hiệu ứng liên hợp là sự phân cực các liên kết π lan truyền trong hệ liên hợp (biểu
thị bằng mũi tên cong).
- Phân loại:
Hiệu ứng +C C = 0 -C
1.3.3. Hiệu ứng siêu liên hợp:
- Hiệu ứng siêu liên hợp dương là hiệu ứng đẩy electron do tương tác giữa các eletron các liên
kết C H và electron p của nối đôi hay vòng benzen. Kí hiệu +H ( từ tiếng anh
Hyperconjugation). Hiệu ứng +H tăng khi số liên kết C H tăng.
-CH3 > - CH2CH3 > - CH(CH3)2 > - C(CH3)3 (Ngược với hiệu ứng cảm ứng)
- Hiệu ứng siêu liên hợp âm
Nhóm C H liên kết với hệ thống liên kết p có thể gây nên hiệu ứng siêu liên hợp hút
electron gọi là hiệu ứng siêu liên hợp âm (-H).
1.3.4. Hiệu ứng không gian
Loại hiệu ứng do kích thước của nhóm nguyên tử gây nên được gọi là hiệu ứng không gian,
kí hiệu là S ( từ tiếng anh Steric Effect).
1.3.5. Hiệu ứng ortho
Các nhóm thế ở vị trí ortho trong vòng benzen thường gây những ảnh hưởng rất bất
ngờ đến tính vật lí cũng như tính chất hóa học của phân tử.
Chẳng hạn các axit đồng phân ortho RC6H4NH2 có hằng số pKb lớn hơn các đồng phân khác
bất kể bản chất của nhóm thế là gì. Loại ảnh hưởng đặc biệt của nhóm thế ở vị trí ortho như
vậy gọi là hiệu ứng ortho.
Hiệu ứng ortho không đơn thuần là hiệu ứng không gian mà là một hiệu ứng hỗn hợp của
nhiều yếu tố.
- Hiệu ứng không gian loại 1: Nhóm X ở vị trí ortho cản trở sự tấn công của tác nhân Y vào
nhóm chức Z.
- Hiệu ứng không gian loại 2: Nhóm X làm mất tính đồng phẳng của hệ.
- Hiệu ứng cảm ứng: Nhóm X có vị trí ortho ở gần nhóm chức hơn nên thể hiện mạnh hơn ở
các vị trí khác, ngoài ra còn có tác dụng trực tiếp nhờ hiệu ứng trường.
- Tạo liên kết hidro nội phân tử.
1.3.6. Phản ứng thế ở nhân thơm
Vòng thơm kiểu benzen là vòng liên hợp kín rất bền vững và có năng lượng thơm hóa khá
lớn (Benzen: 36kcal/mol, Naphtalen: 61kcal/mol). Vì vậy đối với một hợp chất thơm phản
ứng thế xảy ra dễ dàng hơn phản ứng cộng, do trong phản ứng cộng hệ thơm bị phá vỡ.
Những phản ứng thế ở vòng benzen có thể là thế electrophin SEAr, thế nucleophin SNAr hoặc
thế gốc SRAr. Tuy vậy, do vòng thơm có mật độ điện tích cao và dễ tương tác với tác nhân
electronphin, cho nên phản ứng thế đặc trưng và phổ biến hơn cả ở các hợp chất thơm là thế
electrophin SEAr.
CHƢƠNG II. ĐỐI TƢỢNG VÀ PHƢƠNG PHÁP NGHIÊN CỨU
2.1. ĐỐI TƢỢNG NGHIÊN CỨU
Trong một phạm vi hẹp, đối tượng nghiên cứu của luận văn này là nghiên cứu cấu trúc và

tính chất của khoảng 70 chất thuộc dãy dẫn xuất của halogen, ancol, phenol, axit cacboxylic:
2.1.1. Dẫn xuất halogen
,
CH3X, C2H5X, CH2=CHX, CH3CH=CHX. Trong đó X: H, F, Cl, Br, I
2.1.2. Ancol
CH3OH, C2H5OH, XCH2CH2OH
Trong đó X: H, F, Cl, Br, I
2.1.3. Phenol
Trong đó X: H, F, Cl, Br, I
2.1.4. Axit cacboxylic
CH3COOH, XCH2COOH, C2H5COOH, XCH2CH2COOH
Trong đó X: H, F, Cl, Br, I
2.2. PHƯƠNG PHÁP NGHIÊN CỨU
Phương pháp sử dụng nghiên cứu trong luận văn này là các phương pháp tính toán lý
thuyết hóa học lượng tử. Khi sử dụng phương pháp tính toán hóa học lượng tử với mức độ
gần đúng tốt thì các kết quả về cấu trúc hình học, phân bố điện tích và phân bố electron là rất
có giá trị. Ngoài ra, tính toán lý thuyết còn cung cấp giá trị khá chính xác về năng lượng, các
thông số nhiệt động như nhiệt tạo thành, ái lực electron, năng lượng ion hóa…tương đối phù
hợp với kết quả thực nghiệm.
CHƢƠNG 3. KẾT QUẢ VÀ THẢO LUẬN
3.1. LỰA CHỌN BỘ HÀM VÀ PHƢƠNG PHÁP TÍNH
Hầu hết các thông số lượng tử phụ thuộc vào cấu trúc hình học của phân tử. Người ta
chấp nhận cấu trúc với trạng thái năng lượng toàn phần nhỏ nhất như mô hình tồn tại trong
thực tế. Từ cấu hình tối ưu hóa hình học, chúng tôi tính được các thông số lượng tử quan tâm:
năng lượng phân tử, mật độ điện tích, độ dài liên kết,… để rút ra một số quy luật và tính chất
của các dẫn xuất halogen, ancol, phenol, axit cacboxylic.
Sử dụng chương trình Gaussian 03 (cùng sự hỗ trợ của Gauview 5.0) để tìm cấu trúc
tối ưu và năng lượng của phân tử C6H5COOH với các phương pháp tính và bộ hàm khác
nhau. Kết quả thu được được trình bày ở bảng 3.1.
Trong bảng 3.1, các phương pháp tính có độ chính xác tăng dần từ trên xuống dưới,
các bộ hàm có độ chính xác tăng dần từ trái qua phải. Năng lượng của phân tử được tính theo
đơn vị kcal/mol.
Qua kết quả tính toán thể hiện trong bảng 3.1, chúng tôi rút ra được phương pháp tính
từ cột trên xuống cột dưới, độ chính xác tăng dần, bộ hàm và bộ hàm vừa cho kết quả chính
xác, vừa chấp nhận được về thời gian tính là phương pháp DFT (B3LYP) với bộ hàm cơ sở là
6-31G.
Do đó, từ kết quả khảo sát các phương pháp tính và bộ hàm cơ sở, chúng tôi chọn
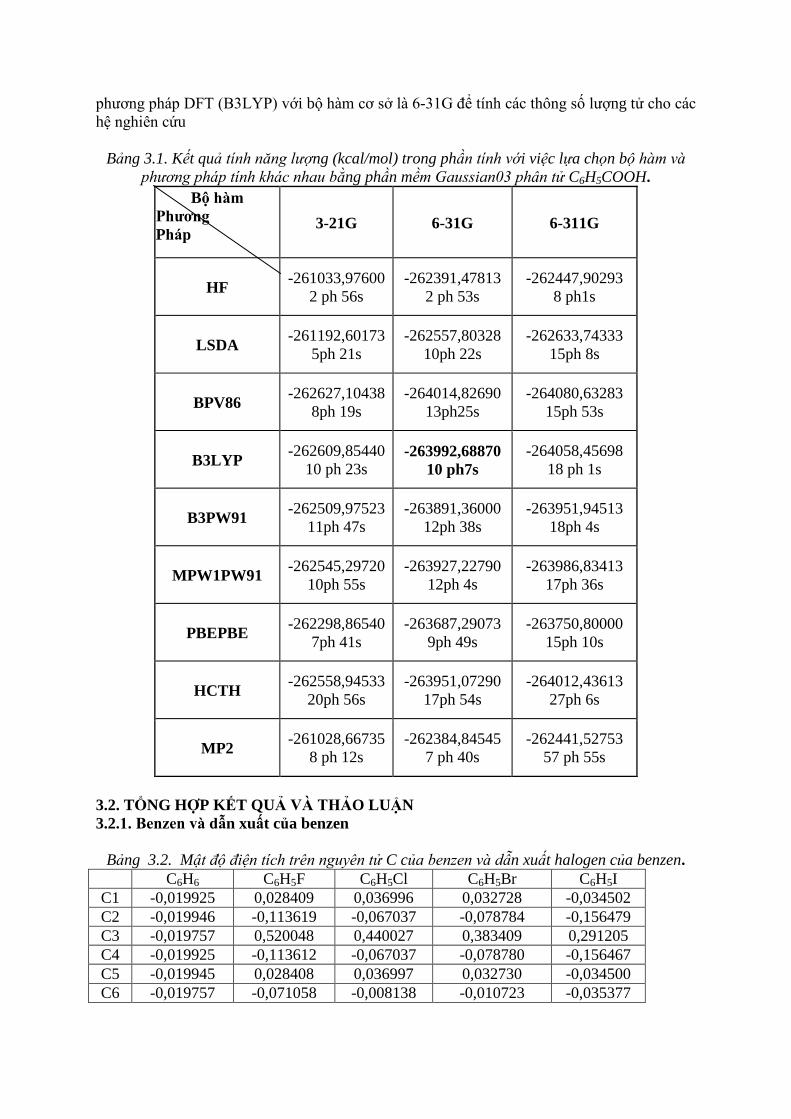
phương pháp DFT (B3LYP) với bộ hàm cơ sở là 6-31G để tính các thông số lượng tử cho các
hệ nghiên cứu
Bảng 3.1. Kết quả tính năng lượng (kcal/mol) trong phần tính với việc lựa chọn bộ hàm và
phương pháp tính khác nhau bằng phần mềm Gaussian03 phân tử C6H5COOH.
Bộ hàm
Phƣơng
Pháp 3-21G 6-31G 6-311G
HF -261033,97600
2 ph 56s
-262391,47813
2 ph 53s
-262447,90293
8 ph1s
LSDA -261192,60173
5ph 21s
-262557,80328
10ph 22s
-262633,74333
15ph 8s
BPV86 -262627,10438
8ph 19s
-264014,82690
13ph25s
-264080,63283
15ph 53s
B3LYP -262609,85440
10 ph 23s -263992,68870
10 ph7s
-264058,45698
18 ph 1s
B3PW91 -262509,97523
11ph 47s
-263891,36000
12ph 38s
-263951,94513
18ph 4s
MPW1PW91 -262545,29720
10ph 55s
-263927,22790
12ph 4s
-263986,83413
17ph 36s
PBEPBE -262298,86540
7ph 41s
-263687,29073
9ph 49s
-263750,80000
15ph 10s
HCTH -262558,94533
20ph 56s
-263951,07290
17ph 54s
-264012,43613
27ph 6s
MP2 -261028,66735
8 ph 12s
-262384,84545
7 ph 40s
-262441,52753
57 ph 55s
3.2. TỔNG HỢP KẾT QUẢ VÀ THẢO LUẬN
3.2.1. Benzen và dẫn xuất của benzen
Bảng 3.2. Mật độ điện tích trên nguyên tử C của benzen và dẫn xuất halogen của benzen.
C6H6 C6H5F C6H5Cl C6H5Br C6H5I
C1 -0,019925 0,028409 0,036996 0,032728 -0,034502
C2 -0,019946 -0,113619 -0,067037 -0,078784 -0,156479
C3 -0,019757 0,520048 0,440027 0,383409 0,291205
C4 -0,019925 -0,113612 -0,067037 -0,078780 -0,156467
C5 -0,019945 0,028408 0,036997 0,032730 -0,034500
C6 -0,019757 -0,071058 -0,008138 -0,010723 -0,035377

Nhận xét
- Trong phân tử C6H6 mật độ điện tích phân bố đều trên 6 nguyên tử cacbon.
- Trong phân tử C6H5X, mật độ điện tích trên nguyên tử cacbon ở các vị trí liên kết với X (C3)
dương hơn mật độ điện tích trên các nguyên tử cacbon khác. Do đó, liên kết giữa C – X bị phân
cực mạnh. Độ phân cực của liên kết giảm dần theo dãy C – F > C – Cl > C – Br > C - I. KQ này
hoàn toàn phù hợp với cơ sở lý thuyết và kết quả thực nghiệm.
- Loại hợp chất này chỉ phản ứng trong điều kiện rất khắc nghiệt (nhiệt độ, áp suất cao)
Bảng 3.3. Năng lượng phân tử, mật độ điện tích trên nguyên tử C (C-X), nguyên tử X, độ dài
liên kết C – X của benzen và dẫn xuất halogen của benzen.
Chất Năng lượng
(kcal/mol)
Mật độ điện tích
trên C (C-X)
Mật độ điện tích
trên H hoặc X
Độ dài liên kết
C-X hoặc C-H
(o
A )
C6H6 -145704,67140 -0,019757 0,019887 1,09968
C6H5F -207961,22545 0,520048 -0,496813 1,35000
C6H5Cl -434088,54439 0,440027 -0,371808 1,76000
C6H5Br -1758989,76102 0,383409 -0,280580 1,91000
C6H5I -4465717,02158 0,291205 -0,140580 2,10000
Nhận xét:
-Năng lượng phân tử của C6H6 và dẫn xuất halogen của nó có năng lượng phân tử âm dần theo
thứ tự:
C6H6> C6H5F> C6H5Cl> C6H5Br> C6H5I.
Do đó độ bền của phân tử tăng dần:
C6H6> C6H5F> C6H5Cl> C6H5Br> C6H5I
- Mật độ điện tích trên nguyên tử cacbon tăng khi có nhóm thế halogen theo thứ tự:
H < I < Br < Cl < F.
Mật độ điện tích trên nguyên tử halogen âm hơn nguyên tử hiđro. Mật độ điện tích trên nguyên
tử halogen âm dần từ I đến F. Do đó độ phân cực của liên kết tăng dần từ C – I đến C – F.
Độ dài liên kết tăng dần:
C – H < C – F < C – Cl < C – Br < C – I
Liên kết càng phân cực, tính ion càng cao, liên kết ion càng bền. Độ dài liên kết càng
lớn, càng dễ bị đứt. Do đó khả năng tham gia phản ứng thế (ví dụ bằng nhóm –OH), tăng dần từ
C6H5F đến C6H5I, phù hợp với thực tế khả năng phản ứng của các hợp chất đó.
3.2. 2. Toluen và dẫn xuất halogen của toluen
Nhận xét:
- Năng lượng của các phân tử giảm dần :
C6H5CH3 > F-C6H4CH3 > Cl-C6H4CH3 > Br-C6H4CH3 > I-C6H4CH3 - Mật độ điện tích trên nguyên tử cacbon liên kết với halogen dương hơn so với nguyên tử
cacbon tương ứng cùng vị trí trong phân tử toluen. Mật độ điện tích trên nguyên tử
halogen âm dần từ I đến F. Như vậy, nhóm thế halogen làm giảm mật độ điện tích trong
vòng làm tăng độ phân cực của liên kết

C – X. Tính ion của liên kết tăng, liên kết càng bền. Do đó phản ứng thế xảy ra càng khó
khăn.
- Độ dài liên kết C-X tăng từ I đến F. Vì thế, độ bền liên kết C – X giảm dần theo dãy:
C – I < C – Br < C – Cl < C – F
Do đó, khả năng thế tăng dần theo thứ tự: F < Cl < Br < I. Ở mỗi dẫn xuất halogen của
toluen, liên kết C – X ở vị trí meta là phân cực nhất, tính ion lớn nhất nên bền nhất. Liên
kết C – X ở các vị trí octo và para ít phân cực hơn. Khả năng tham gia phản ứng thế tăng,
phù hợp với thực tế.
3.2.3. Khả năng thủy phân của dẫn xuất halogen
Bảng 3.24. Năng lượng, mật độ điện tích trên nguyên tử C (C-Cl), mật độ điện tích trên
nguyên tử Cl, độ dài liên kết C-Cl của một số dẫn xuất clo.
Chất Năng lượng
(kcal/mol)
Mật độ điện
tích trên C (C-
Cl)
Mật độ
điện tích
trên Cl
Độ dài liên
kết C-Cl
(o
A )
CH2=CH-CH2Cl -1515831,01953 0,51234 -0,43900 1,92596
CH3-CH2Cl -338467,92476 0,45192 -0,39299 1,90358
C6H5Cl -434088,54439 0,44003 -0,37181 1,76000
CH2=CHCl -337688,40491 0,36477 -0,34425 1,82368
Nhận xét:
- Mật độ điện tích trên nguyên tử cacbon gắn với nhóm thế halogen giảm dần theo thứ tự:
CH2=CH-CH2Cl > CH3-CH2Cl > C6H5Cl > CH2=CHCl. Mật độ điện tích trên nguyên tử
clo tăng dần cũng theo thứ tự đó. Vì thế, độ phân cực của liên kết C – Cl giảm dần, khả
năng tham gia phản ứng thế bởi nhóm –OH trong phản ứng thủy phân giảm dần.
- Độ dài liên kết C – Cl của CH2=CH-CH2Cl lớn hơn của CH3-CH2Cl lớn hơn của C6H5Cl
và xấp xỉ CH2=CHCl nên khả năng thủy phân giảm dần.
Kết luận: Các kết quả trên hoàn toàn phù hợp với thực nghiệm:
- Khả năng phản ứng thủy phân giảm dần theo thứ tự
CH2=CH-CH2Cl > CH3-CH2Cl > C6H5Cl CH2=CHCl
Cụ thể như sau:
CH2=CH-CH2Cl hay anlyl halogenua nói chung bị thủy phân khi đun nóng.
CH2=CH-CH2Cl + H2O
otCH2=CH-CH2OH + HCl
Etyl clorua hay ankyl halogenua nói chung không bị thủy phân khi đun nóng, chỉ phản
ứng với dung dịch NaOH khi đun nóng.
CH3-CH2Cl + NaOH otCH3- CH2OH + NaCl
Vinyl halogenua và phenol halogenua chỉ bị thủy phân bởi dung dịch NaOH đặc ở nhiệt
độ cao và áp suất cao.
CH2=CH-Cl + NaOHđặc
ot cao
p caoCH3CHO + NaCl
C6H5Cl + NaOHđặc
ot cao
p cao C6H5OH + NaCl
3.2.4. Ảnh hƣởng của gốc ankyl đến tính axit của axit cacboxylic
Bảng 3.25. Năng lượng, mật độ điện tích trên nguyên tử O (O-H), độ dài liên kết O – H
của một số axit cacboxylic.
Chất Năng lượng
(kcal/mol)
Mật độ điện tích
Trên O (O-H)
Mật độ điện
tích trên H
HCOOH -119022,46036 -0,669871 0,286062

CH3COOH -143701,03042 -0,700114 0,278652
C2H5COOH -168364,11397 -0,701616 0,278793
C3H7COOH -193021,65305 -0,712558 0,277326
Nhận xét:
- Năng lượng phân tử tăng dần từ HCOOH đến C3H7COOH.
- Mật độ điện tích trên nguyên tử oxi của nhóm –OH âm dần từ HCOOH đến
C3H7COOH, còn điện tích dương trên nguyên tử hiđro giảm nên độ phân cực của liên kết
O – H giảm dần do gốc ankyl đẩy electron về phía nhóm cacboxyl, do đó khả năng đứt
liên kết O – H trong các phân tử axit đó giảm dần, nên tính axit yếu dần theo dãy:
HCOOH > CH3COOH > C2H5COOH > C3H7COOH
3.2.5. Tính linh động của nguyên tử hiđro trong nhóm OH
Bảng 3.26. Năng lượng, mật độ điện tích trên nguyên tử O (O-H), độ dài liên kết O –H
của một số hợp chất chứa nhóm OH
Chất Năng lượng
(kcal/mol)
Mật độ điện tích
trên O
(O-H)
Mật độ
điện tích
trên H (O-
H)
Độ dài liên
kết O-H
(o
A )
C2H5OH -97256,33708 -0,567682 0,218068 0,97811
C6H5OH -192886,09423 -0,688151 0,284833 0,97640
CH3COOH -143701,03042 -0,700114 0,278652 0,98222
Nhận xét:
- Ở C2H5OH có nhóm etyl là nhóm đẩy electron làm giảm độ phân cực của liên kết O –
H. Ở C6H5OH gốc phenyl hút electron làm tăng độ phân cực của liên kết O – H. Ở
CH3COOH, nhóm cacbonyl hút electron làm tăng khả năng phân cực của liên kết O –
H, nhưng đồng thời nhóm metyl lại đẩy electron làm giảm khả năng phân cực của liên
kết O – H.
- Những điều phân tích ở trên phù hợp với những kết quả thu được ở bảng 3.26.
Kết quả tính toán lý thuyết thu được giải thích tính linh động của nguyên tử hiđro của
nhóm – OH tăng dần theo dãy C2H5OH < C6H5OH < CH3COOH. Điều này được khẳng
định bằng các kết quả thực nghiệm thu được dưới đây:
- Phenol có tính axit yếu hơn axit axtetic, mạnh hơn ancol etylic. Cụ thể là:
C2H5OH chỉ phản ứng với kim loại kiềm, không phản ứng với dung dịch kiềm.
C2H5OH + Na C2H5Na + 1
2H2
C6H5OH không những phản ứng với kim loại kiềm mà còn phản ứng được với
dung dịch kiềm mạnh, ví dụ NaOH:
C6H5OH + NaOH C6H5ONa + H2O
Tuy vậy, C6H5OH là axit rất yếu, không làm đổi màu quỳ tím.
Axit axetic có đầy đủ các tính chất của một axit, tuy vẫn là axit yếu. Axit axetic tác
dụng với những kim loại đứng trước hiđro, dung dịch kiềm, muối cacbonat và làm đổi
màu quỳ tím thành đỏ.
3.2.6. Quy tắc thế ở vòng benzen

Bảng 3.27. Năng lượng và mật độ electron trên các nguyên tử cacbon ở vòng benzen của
phenol và axit benzoic.
Chất Mật độ electron trên nguyên tử C
Octo Meta Para
C6H5OH -0,144434 0,069020 -0,122421
C6H5COOH 0,009942 -0,067460 0,041965
Nhận xét:
- Ở phân tử phenol, mật độ electron trên các nguyên tử cacbon ở các vị trí octo và para âm
hơn mật độ electron trên nguyên tử cacbon ở vị trí meta. Do đó trong phản ứng thế, các hạt
mang điện dương (như Br+, NO2
+,….) hướng vào các vị trí octo và para, thay thế đồng thời
các nguyên tử hiđro ở các vị trí đó.
- Ở phân tử axit benzoic, mật độ electron trên nguyên tử cacbon ở vị trí meta âm hơn mật độ
electron trên nguyên tử cacbon tại các vị trí octo và para. Vì vậy, các hạt mang điện dương
hướng vào vị trí meta để thế nguyên tử hiđro ở vị trí đó.
Kết quả thu được ở trên hoàn toàn phù hợp với thực nghiệm và đúng với quy luật thế vào
vòng benzen.
Ví dụ : Phenol không những tác dụng được với Br2 nguyên chất mà còn tác dụng với nước
Br2, ba nguyên tử hiđro ở các vị trí octo và para bị thế đồng thời.
Br2 (dung dich)
trắng
+ 3HBr
Phenol 2,4,6-tribromphenol
KẾT LUẬN
Sau một thời gian nghiên cứu, chúng tôi đã thu được một số kết quả chính như sau:
1. Chọn được phương pháp bán kinh nghiệm thích hợp DFT – B3LYP của phần mềm
Gaussian 03 để tính các tham số hóa học lượng tử cho các hợp chất dẫn xuất halogen,
ancol, phenol và axit cacboxylic.
2. Tính được các tham số hóa học lượng tử của các nhất nêu trên. Kết quả thu được gồm:
- Hình học phân tử: cấu trúc hình học phân tử, độ dài liên kết,…
- Sự phân bố mật độ điện tích trên các nguyên tử
- Năng lượng toàn phần
của các chất nghiên cứu. Trên cơ sở đó xác định hướng phản ứng.
3. Khảo sát quy luật phản ứng trên cơ sở định lượng bằng các tham số lượng tử thu được
ở trên. Kết quả tính toán hoàn toàn phù hợp với kết quả thực nghiệm. Đó là:
a. Phản ứng thủy phân của các dẫn xuất halogen tăng dần theo dãy:
vinyl, phenyl, halogenua-ankyl halogenua-anlyl halogenua
b. Tính linh động của nguyên tử hiđro ở nhóm OH tăng dần theo dãy:
C2H5OH, C6H5OH, CH3COOH.
c. Tính axit giảm dần theo chiều tăng dần của gốc ankyl:
HCOOH – CH3COOH – C2H5COOH - C3H7COOH

Những kết quả thu được của luận văn được thực nghiệm khẳng định. Điều đó cho thấy
hóa học lý thuyết (cấu tạo chất, hóa học lượng tử) với sự phát triển của công nghệ thông
tin có thể đi trước một bước để định hướng cho các thí nghiệm cần thực hiện.
References
Tiếng Việt
1. Nguyễn Đình Huề, Nguyễn Đức Chuy; Thuyết lượng tử về nguyên tử và phân tử; Tập
I, Nhà xuất bản Giáo dục, 1986.
2. Nguyễn Đình Huề, Nguyễn Đức Chuy; Thuyết lượng tử về nguyên tử và phân tử; Tập
II, Nhà xuất bản Giáo dục, 1986.
3. Trần Thành Huế; Tài liệu dùng cho việc bồi dưỡng học sinh giỏi phổ thông; Hà Nội
tháng 1 năm 2002.
4. Trần Thành Huế; Hóa học đại cương, Tập 1; Nhà xuất bản Giáo dục 2000.
5. Trần Thành Huế, Bài giảng dành cho học viên cao học, Trường Đại học Sư phạm Hà
Nội, 2002.
6. Hoàng Nhâm, Hóa Hóa học vô cơ, Tập 2, NXB Giáo dục, 1994.
7. Đỗ Đình Rãng, Đặng Đình Bạch, Lê Thị Anh Đào, Nguyễn Mạnh Hà, Nguyễn Thị
Thanh Phong (2006), Hóa học hữu cơ 1, NXB Giáo dục.
8. Đỗ Đình Rãng, Đặng Đình Bạch, Lê Thị Anh Đào, Nguyễn Mạnh Hà, Nguyễn Thị
Thanh Phong (2006), Hóa học hữu cơ 2, NXB Giáo dục.
9. Đỗ Đình Rãng, Đặng Đình Bạch, Lê Thị Anh Đào, Nguyễn Mạnh Hà, Nguyễn Thị
Thanh Phong (2006), Hóa học hữu cơ 3, NXB Giáo dục.
10. Phan Tống Sơn, Trần Quốc Sơn, Đặng Như Tại, Cơ sở Hóa học hữu cơ,tập 1, NXB
Đại học và trung học chuyên nghiệp, Hà Nội, 1976.
11. Trần Quôc Sơn, Giáo trình cơ sở lý thuyết hóa học hữu cơ, NXB Giáo dục, 1989.
12. Trần Quốc Sơn, Cơ sở lý thuyết hóa hữu cơ, tập 1, NXB Giáo dục 1979.
13. Trần Quốc Sơn, Cơ sở lý thuyết hóa hữu cơ, tập 2, NXB Giáo dục 1979.
14. Lâm Ngọc Thiềm, Phạm Văn Nhiêu, Lê Kim Long (2008), Cơ sở hóa học lượng tử,
NXB Khoa học và kỹ thuật. (mục 1.2.1)
15. Đào Đình Thức (1980), Cấu tạo nguyên tử và liên kết hóa học, NXB ĐH và THCN.
16. Đặng Ứng Vận (1998), Tin học ứng dụng trong hóa học, NXB Giáo dục.
17. Nguyễn Thị Lan Anh, Luận văn thạc sĩ khoa học hóa học, trường Đại học Khoa học
tự nhiên – ĐHQGHN, 2011.
18. Nguyễn Thị Bích Loan, Luận văn thạc sĩ khoa học hóa học, trường Đại học Sư phạm
Hà Nội, 2003.
Tiếng Anh
19. Foresman J.B. Frisek E (1993), Exploring Chemistry with electronic structure
methods, second edition, Gaussian, Inc. Pitburgh, PA.
20. Ramachandran K.I., Deepa G., Namboori K. (2008). Computational chemistry and
molecular modeling: principle and application, Sringer-Verlag Berlin Heidelberg.
21. Pople J. A. Beveridge D. L. (1970). Approximate Molecular Orbital Theory, Mc
Graw Hill book company.
22. Levine I. N. (2000). Quantum chemistry (1th
Edition) Prentice Hall, Upper Saddle
River, New Jersey. 07458.
23. Jensen F. (2007) Introduction Computationnal Chemistry, Second edition, John
Willey & Sons Ltd.

24. Lewars E. G (2003) Computationnal Chemistry introduction to the theory and
applications of the molecular and quantum mechanics, second printing (2004).
Kluwer academic Publishers.
Website: http://vi.wikipedia.org





























