Embed Size (px)
Citation preview

Spinocerebelläre Ataxien (SCA)
Update
Carola Seifried

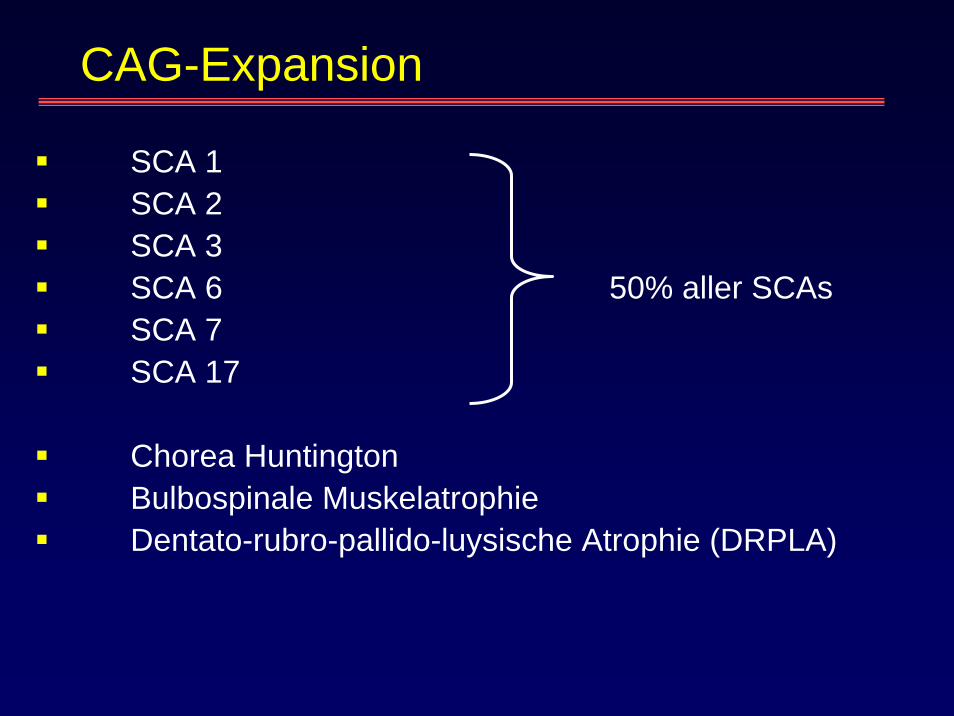
CAG-Expansion
SCA 1SCA 2SCA 3SCA 6 50% aller SCAsSCA 7SCA 17
Chorea HuntingtonBulbospinale MuskelatrophieDentato-rubro-pallido-luysische Atrophie (DRPLA)

Oligonukleotid-Expansion in nicht kodierenden Regionen
SCA 8SCA 10SCA 12

„Klassische Mutationen“
SCA 5SCA 13SCA 14SCA 27

Klassifikation der autosomal-dominantencerebellären Ataxien (ADCA) nach A. Harding
ADCA Typ 1 (SCA 1,2,3,4,8,12,13,16,17)Ataxie mit Zusatzsymptomen wie Optikusatrophie, Ophthalmoplegie, Demenz oder extrapyramidaler Symptomatik
ADCA Typ 2 (SCA 7)Ataxie mit pigmentöser Retinadegeneration
ADCA Typ 3 (SCA 5,6,10,11,14)rein cerebelläre Ataxien

Gemeinsamkeiten der SCAs
KlinikCerebelläre Symptomatik • Ataxie• Dysarthrie
Rollstuhlpflicht nach ca. 15 Jahren, Tod nach 25 Jahren
GenetikAutosomal-dominanter ErbgangPhänomen der Antizipation

SCA 1 Klinisches Bild
Kernsymptome :Gang-, Stand- sowie ExtremitätenataxieDysarthrie, Dysphagie
Additiv :PyramidenbahnzeichenPeriphere NeuropathieSpäte Sakkadenverlangsamung, OphthalmoplegieIn 20%: extrapyramidale Symptome, Inkontinenzund Demenz

SCA 1
ZusatzdiagnostikElektrophysiologie
Verlängerung der zentralen, aber auch der peripheren motorischen Überleitungszeiten im MEP (→ Abgrenzung gegenüber anderen SCA-Formen)Axonale Polyneuropathie
Bildgebung Olivo-ponto-cerebelläre Atrophie (OPCA)
GenetikMutation auf Chromosom 6p (Nähe des HLA-Komplexes)
In Deutschland 25% aller SCAs

SCA 2 Prävalenz
in Deutschland/Europa: 1/100.000(ca. 15% aller dominanten Ataxien)
in Holguin, Kuba : 500/100.000insgesamt >1500 Erkrankte

SCA 2 Klinisches Bild (1)
Kernsymptome :cerebelläres Syndrom
Gang -, Stand - sowie ExtremitätenataxieDysarthrie, Dysphagie
Aktions- und Halte-Tremorfrüh sensible, axonale Neuropathie früh verlangsamte BlicksakkadenHyporeflexie
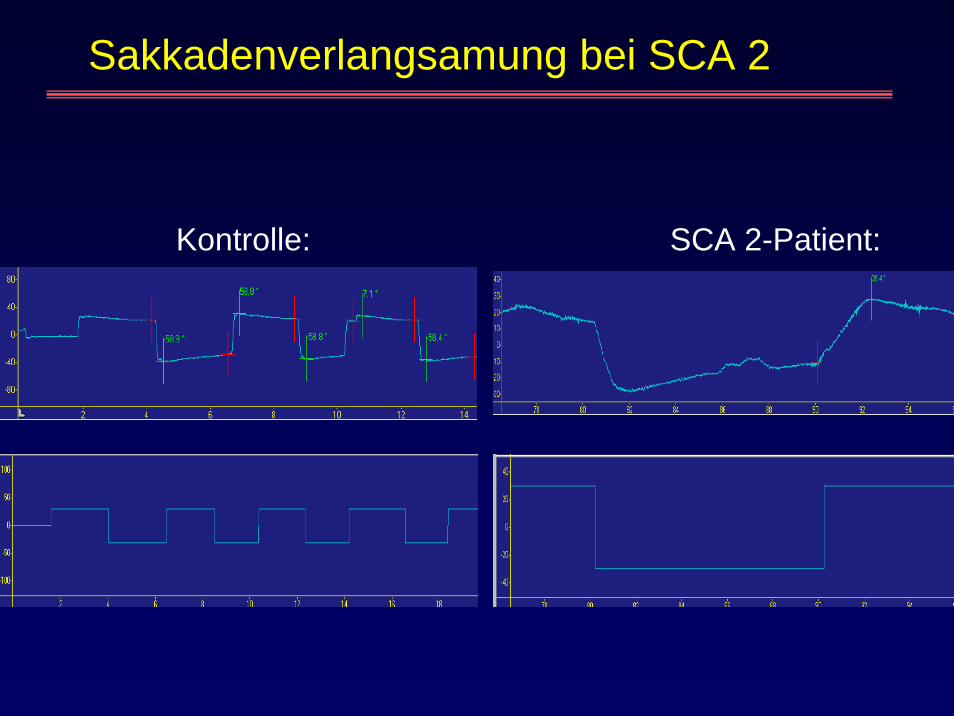
Sakkadenverlangsamung bei SCA 2
Kontrolle: SCA 2-Patient:

SCA 2 Klinisches Bild (2)
additive / spätere Symptome:
Anfangs HyperreflexieMyoklonien(L-Dopa-responsives) Parkinson-Syndrom InkontinenzSelten: dementielles Syndrom
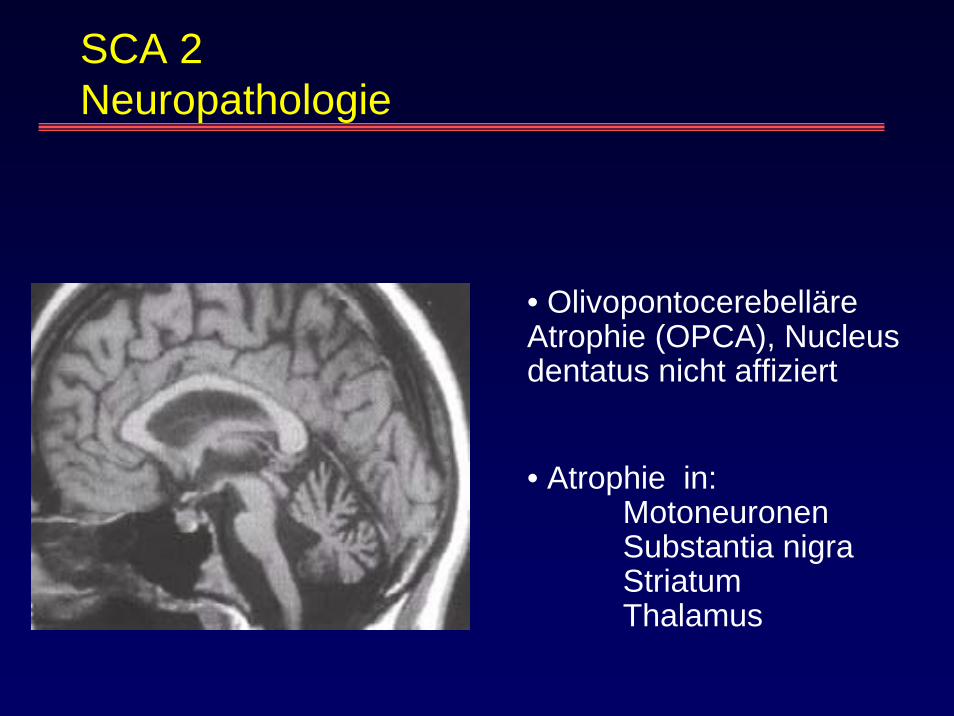
SCA 2 Neuropathologie
• OlivopontocerebelläreAtrophie (OPCA), Nucleusdentatus nicht affiziert
• Atrophie in: MotoneuronenSubstantia nigraStriatumThalamus

SCA 3/ MJD Klinisches Bild (1)
Augenbewegungsstörungen:BlickrichtungsnystagmusDoppelbilder
NeuropathieIm Verlauf DysphagieDystonie, Pyramidenbahnzeichen, spastische MuskeltonuserhöhungSchlafstörungen mit Restless-legs-Syndrom (ca. 40% - L-Dopa-responsiv)

SCA 3/ MJD Klinisches Bild (2)
Patienten mit Repeatlängen im intermediären Bereich → Symptome des peripheren Nerven-systemsFaszikulationenHyporeflexieSensomotorische axonale PolyneuropathieStörung der Tiefensensibilität

SCA 3/ MJD Zusatzdiagnostik
Elektrophysiologieaxonale Schädigung
BildgebungGeringe Atrophie im Vergleich zur ausgeprägten SymptomatikErweiterung des IV. VentrikelsAtrophie des cervikalen Myelons(Hirnstamm nur wenig betroffen)
ca. 35% aller SCAs in Deutschland

SCA 6 Klinisches Bild
„rein“ cerebelläre Symptomatik
AtaxieDysarthrieHorizontaler und/oder vertikaler BlickrichtungsnystagmusSakkadierte BlickfolgeMangelnde Suppression des VOR
ca. 25% aller SCAs in Deutschland

SCA 6 Genetik
Später Erkrankungsbeginn (60% nach dem 50. Lebensjahr), oft keine FamilienanamneseNormale LebenserwartungCAG-Repeatexpansion auf Chromosom 19q13:α 1A – Untereinheit des spannungsabhängigen neuronalen Kalziumkanals, CACNA1A-GenExpression in Purkinje-Zellen
Therapieversuch mit Acetazolamid (250-500 mg/d)

SCA 7
Klinisches Bild :einzige SCA, die einen spezifischen Phänotyp aufweist und klinisch diagnostiziert werden kanncerebelläre Ataxie in Kombination mitPigmentdegeneration der Makula
Progredienter Visusverlust

SCA 17 Klinisches Bild
Kernsymptome :Gang-, Stand- sowie ExtremitätenataxieDysphagie, Dysarthrie
Additiv :Psychiatrische SymptomeDemenzAbsence-EpilepsieExtrapyramidale Zeichen
Neuropathologie : cerebello-(olivär)Gen: TATA-binding protein (CAG45-54)

DifferentialdiagnosePolyneuropathie (axonal, sensibel oder sensomotorisch)
SCA 1 (42%, v.a. bei vielen CAGs)SCA 2 (80%)SCA 3 (54%, v.a. bei wenigen CAGs)SCA 4SCA 8
PyramidenbahnzeichenHäufig: SCA 1,3,7,12Selten: SCA 2,6,8
Akinese/Rigor/DystonieSCA 3SCA 12SCA 17
EpilepsieSCA 10SCA 17

DifferentialdiagnoseDemenz
SCA 2SCA 12SCA 17
Geistige RetardierungSCA 13
MyoklonusSCA 2SCA 14
Kopf/Hand-TremorSCA 12SCA 16

Genetische Testung
SCA 1SCA 2SCA 3SCA 6SCA 7SCA 8SCA 10SCA 12SCA 14SCA 17

Therapie
Keine kausale Therapie Symptomatische Therapie bei Spastik, extrapyramidalmotor. Symptomen, Restless-legs-Syndrom etc.Physiotherapie, Logopädie
Cerebelläre Ataxie
nicht-erblich erblich
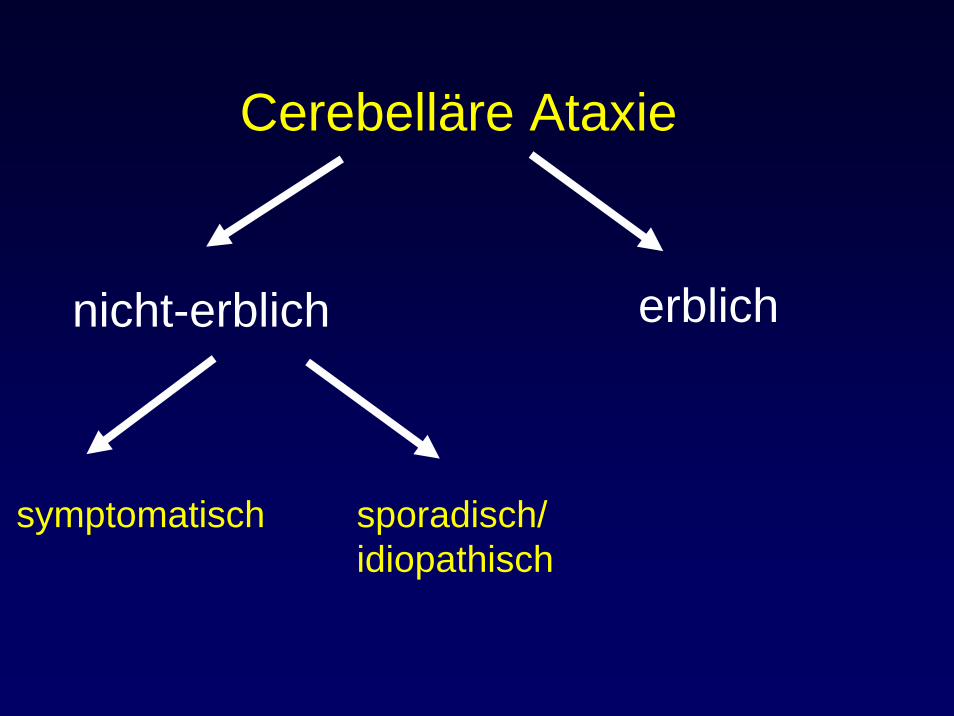
Cerebelläre Ataxie
nicht-erblich erblich
symptomatisch sporadisch/idiopathisch

Symptomatische Ataxien
AlkoholabususSonstige IntoxikationenCerebelläre EnzephalitisImmunvermittelt
Encephalitis disseminataMalabsorptionssyndromParaneoplastische Kleinhirndegeneration (PCD)
Ataxie bei erworbenem Vitaminmangel oder metabolischen StörungenTumore im Kleinhirnbereich
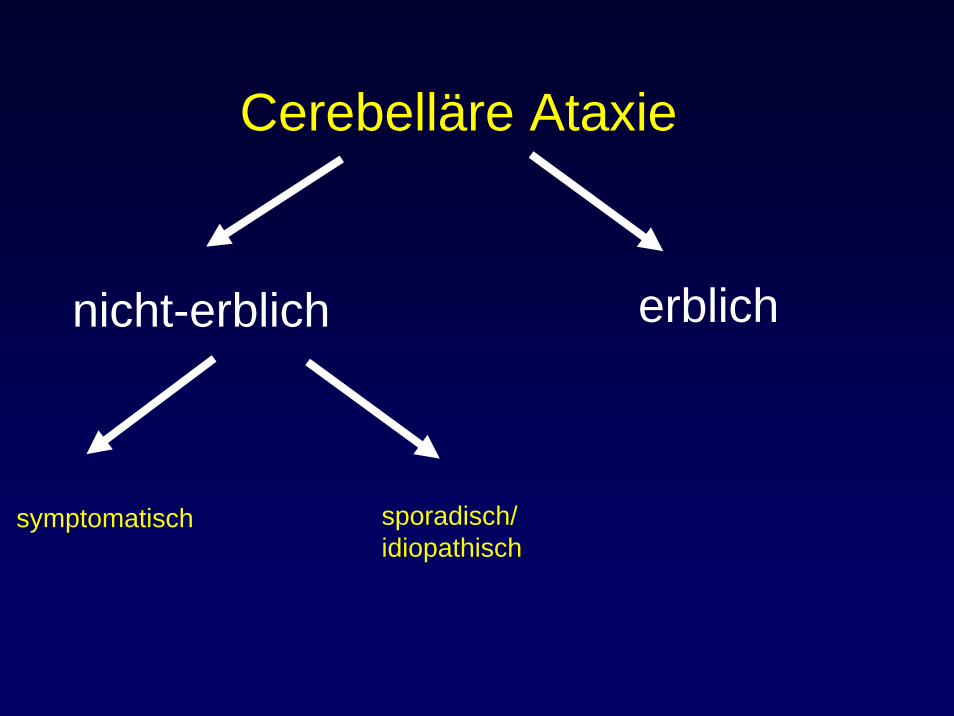
Cerebelläre Ataxie
nicht-erblich erblich
symptomatisch sporadisch/idiopathisch

Sporadische / idiopathische Ataxien
Multisystematrophie (MSA), cerebellärerTyp (MSA-C)
Sporadische, im Erwachsenenalter beginnende Ataxie (sporadic adult onsetataxie, SAOA)
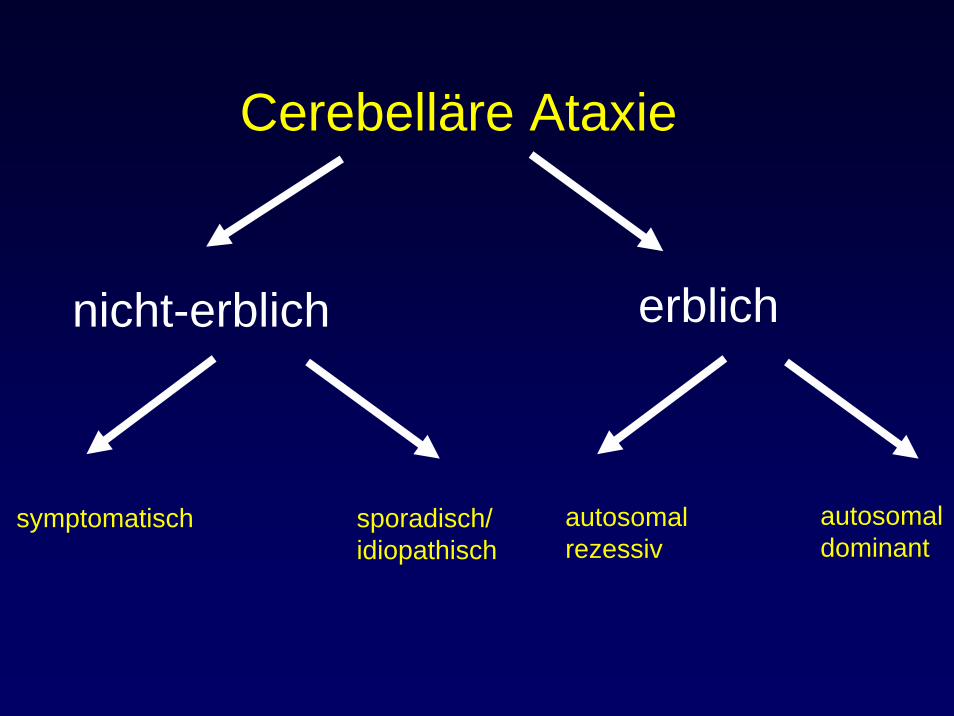
Cerebelläre Ataxie
nicht-erblich erblich
symptomatisch sporadisch/idiopathisch
autosomalrezessiv
autosomaldominant

Autosomal rezessive Ataxien (1)
Friedreich Ataxie (1-2/100 000)GAA-Repeat auf Chromosom 9 (Intron-Bereich)Leitsymptome
• Langsam fortschreitende Ataxie mit Beginn vor der Pubertät
• frühe Areflexie• Gestörter Lage- und Vibrationssinn• Sklelettdeformitäten• Kardiomyopathie

Autosomal rezessive Ataxien (2)
Ataxie mit isoliertem Vitamin E-Defizit (AVED) – therapierbar !Ataxia teleangiectatica (AT)Ataxie mit okulomotorischer Apraxie(AOA)Metabolische Störungen u.a. Lipidstoffwechselstörung
Refsum-KrankheitAbetalipoproteinämie
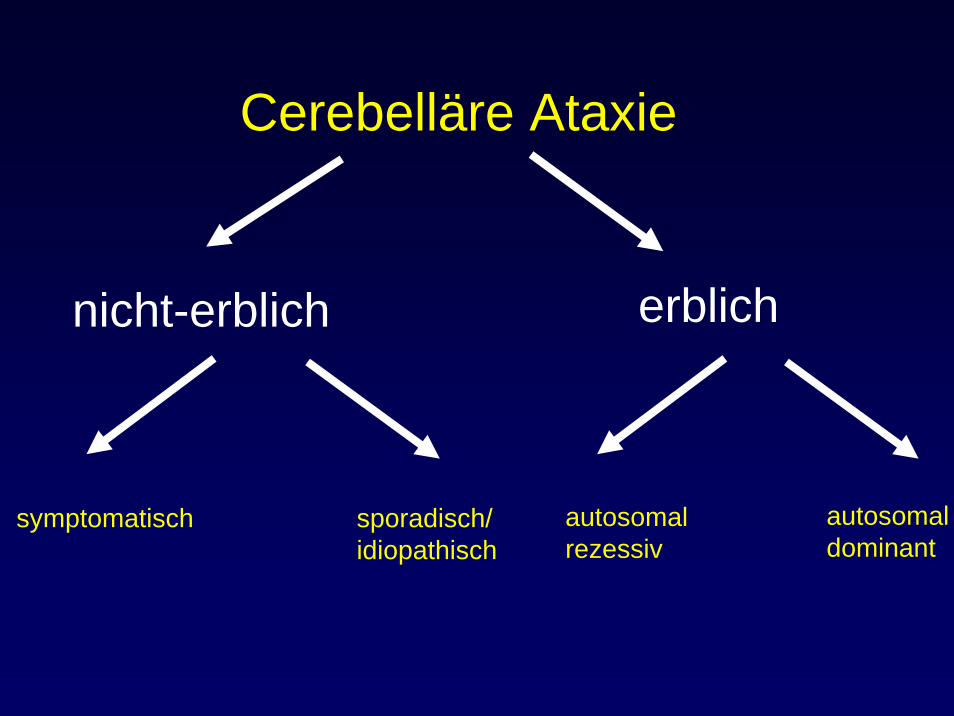
Cerebelläre Ataxie
nicht-erblich erblich
symptomatisch sporadisch/idiopathisch
autosomalrezessiv
autosomaldominant
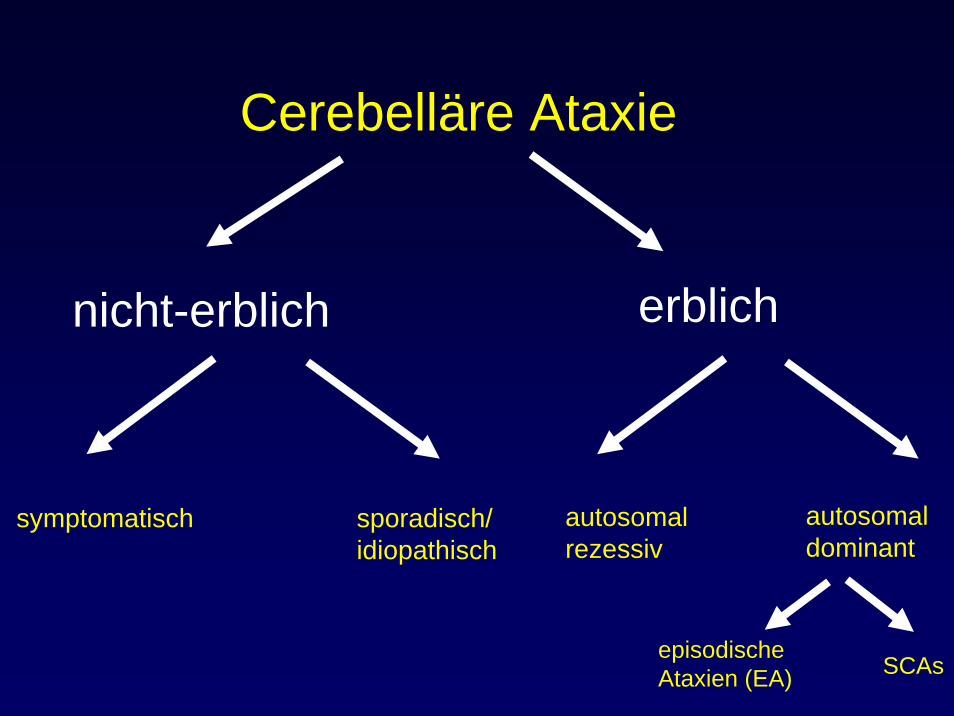
Cerebelläre Ataxie
nicht-erblich erblich
symptomatisch sporadisch/idiopathisch
autosomalrezessiv
autosomaldominant
episodische Ataxien (EA) SCAs
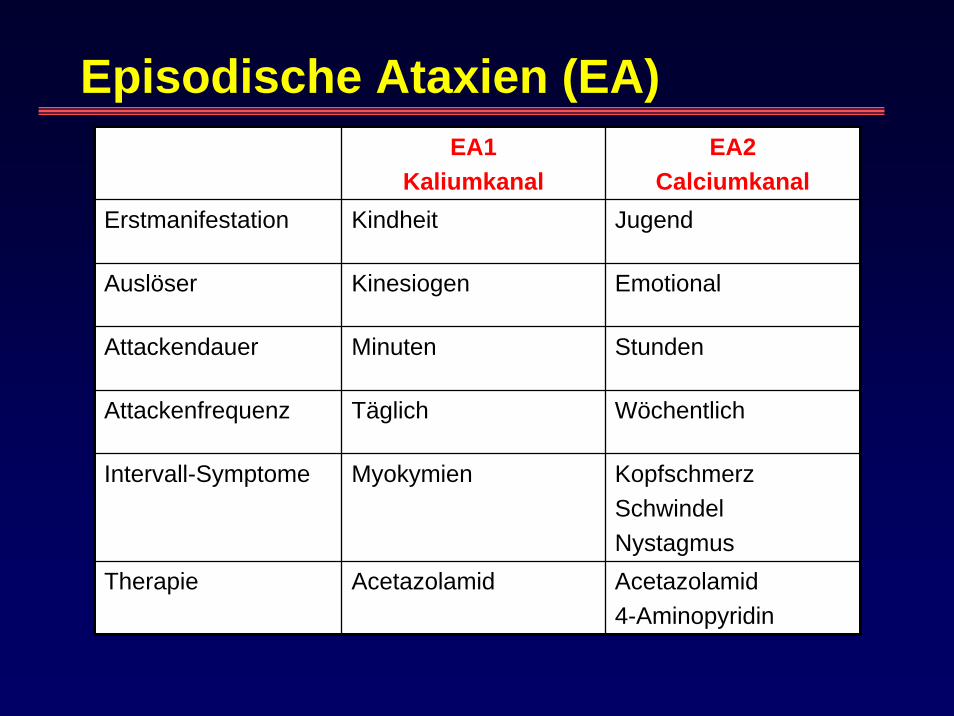
Episodische Ataxien (EA)EA1
KaliumkanalEA2
CalciumkanalErstmanifestation Kindheit Jugend
Auslöser Kinesiogen Emotional
Attackendauer Minuten Stunden
Attackenfrequenz Täglich Wöchentlich
Intervall-Symptome Myokymien KopfschmerzSchwindelNystagmus
Therapie Acetazolamid Acetazolamid4-Aminopyridin





























