Embed Size (px)
Citation preview

1
Termická analýza
Tyto studijní materiály vznikly v rámci projektu grantového projektu
FRVŠ 737/2012 typ A/a
„Zřízení laboratoře pro praktickou výuku termické analýzy se zaměřením na anorganické nekovové
materiály“
autoři: A. Kloužková, P. Zemenová, J. Kloužek, W. Pabst
VŠCHT PRAHA 2012

2
V rámci projektu FRVŠ 737/2012 „Zřízení laboratoře pro praktickou výuku
termické analýzy se zaměřením na anorganické nekovové materiály“ byl vypracován
tento stručný text, který je určen především pro posluchače laboratoří termické analýzy se
zaměřením na anorganické nekovové materiály. Poskytuje stručný přehled termických metod
především metod TG (Termogravimetrické Analýzy), DTA (Diferenční Termické Analýzy)
a DSC (Diferenční skenovací kalorimetrie). V závěrečné části je uveden popis obsluhy
zařízení LINSEIS STA PT 1600/1750oC HiRes, které bylo rámci tohoto projektu pořízeno, a
způsob zpracování naměření dat touto simultánní analýzou pomocí příslušného softwaru.
autoři
Obsah str.
1. ÚVOD, HISTORIE, ZÁKLADNÍ DEFINICE METOD TERMICKÉ ANALÝZY…………………3
2. TERMODYNAMIKA A TERMICKÁ ANALÝZA…………………………………………………4
3. ZÁKLADNÍ INSTRUMENTACE, ZÁKLADNÍ POJMY, TERMOANALYTICKÉ KŘIVKY........5
4. TERMOGRAVIMETRIE (TG)............................................................................................................9
4. 1 PŘÍSTROJOVÉ VYBAVENÍ TG.............................................................................................9
4. 2 PARAMETRY OVLIVŇUJÍCÍ TERMOGRAVIMETRII........... .........................................10
4. 3 VYHODNOCENÍ TERMOGRAVIMETRICKÝCH KŘIVEK..............................................12
5. DIFERENČNÍ TERMICKÁ ANALÝZA (DTA)…...........................................................................13
5. 1 PŘÍSTROJOVÉ VYBAVENÍ DTA, KALIBRACE................................................................14
5. 2 PARAMETRY OVLIVŇUJÍCÍ DIFERENČNÍ TERMICKOU ANALÝZU .........................15
5. 3 VYHODNOCENÍ KŘIVEK DTA, POUŽITÍ…...……………...............................................16
6. DIFERENČNÍ SKENOVACÍ KALORIMETRIE............................................................................. 18
6. 1 PŘÍSTROJOVÉ VYBAVENÍ...................................................................................................19
6. 2 PARAMETRY OVLIVŇUJÍCÍ DIFERENČNÍ SKENOVACÍ KALORIMETRII.................19
6. 3 VYHODNOCENÍ KŘIVEK DSC, POUŽITÍ…………………................................................20
7. KOMBINACE METOD TERMICKÉ ANALÝZY...........................................................................21
8. SIMULTÁNNÍ STA-LINSEIS...........................................................................................................21
9. VYHODNOCENÍ STA KŘIVEK PŘÍSLUŠNÝM PROGRAMEM.................................................29
9. 1. ZÁKLADNÍ PRÁCE S KŘIVKAMI...................................................................................... 30
9. 2 ZÁKLADNÍ ZPRACOVÁNÍ DTA/DSC KŘIVKY............................................................... 38
9. 3 ZÁKLADNÍ ZPRACOVÁNÍ TG KŘIVKY.............................................................................39
9. 4 EXPORT VÝSLEDKŮ INTERPRETACE……………………………………..……………40
10. UKÁZKOVÉ PŘÍKLADY…………………………………………….…………………………..43
11. LITERATURA………………..…………….……………………….………………………….…44
3
1. ÚVOD, HISTORIE, ZÁKLADNÍ DEFINICE METOD TERMICKÉ ANALÝZY
Termín termická analýza zahrnuje obecně experimentální metody, při nichž jsou
analyzovány změny složení a vlastností studovaného systému při tepelném zatížení.
Studovanými systémemy jsou různé látky popř. jejich směsi (nejčastěji pevné, např.
minerály, horniny apod.). Metodami termické analýzy jsou sledovány (přesněji analyzovány)
změny jejich složení popř. změny jejich vlastností. V průběhu tepelného zatížení vzorku
dochází k vyvolání nebo změně intenzity procesu např. chemické reakce, rozkladu,
dehydratace, fázové přeměně, které mohou být doprovázeny změnou hmotnosti, objemu,
uvolňováním nebo spotřebováním energie, změnou vodivosti atd. Podle vlastnosti, jejíž
změna je sledována jako funkce teploty se nazývá i příslušná analýza, viz tab. I [1-4].
Tepelné zatížení vzorku probíhá podle určitého programu a může být dynamické (zahřívání
nebo ochlazování) nebo statické (při konstantní teplotě v závislosti na čase). V současné
době existuje řada termoanalytických metod, mezi nejpoužívanější patří TG, DTA a DSC
(popis viz tabulka). Tyto metody lze označit jako tzv. primární, na ně navazují další,
sekundární metody (např. při TG dojde rozkladem ke změně hmotnosti s uvolněním plynných
produktů, které jsou následně detekovány další metodou EGA, Evolved Gas Analysis) [2, 4].
Současné měření více vlastností v průběhu jednoho experimentu provádí tzv. simultánní
termická analýza (STA) a popisuje se zkratkou použitých metod např. TG/DTA, TG/DTA
EGA-MS, TG/EGA-IR (MS-hmotnostní spektrometrie, IR- infračervená spektroskopie).
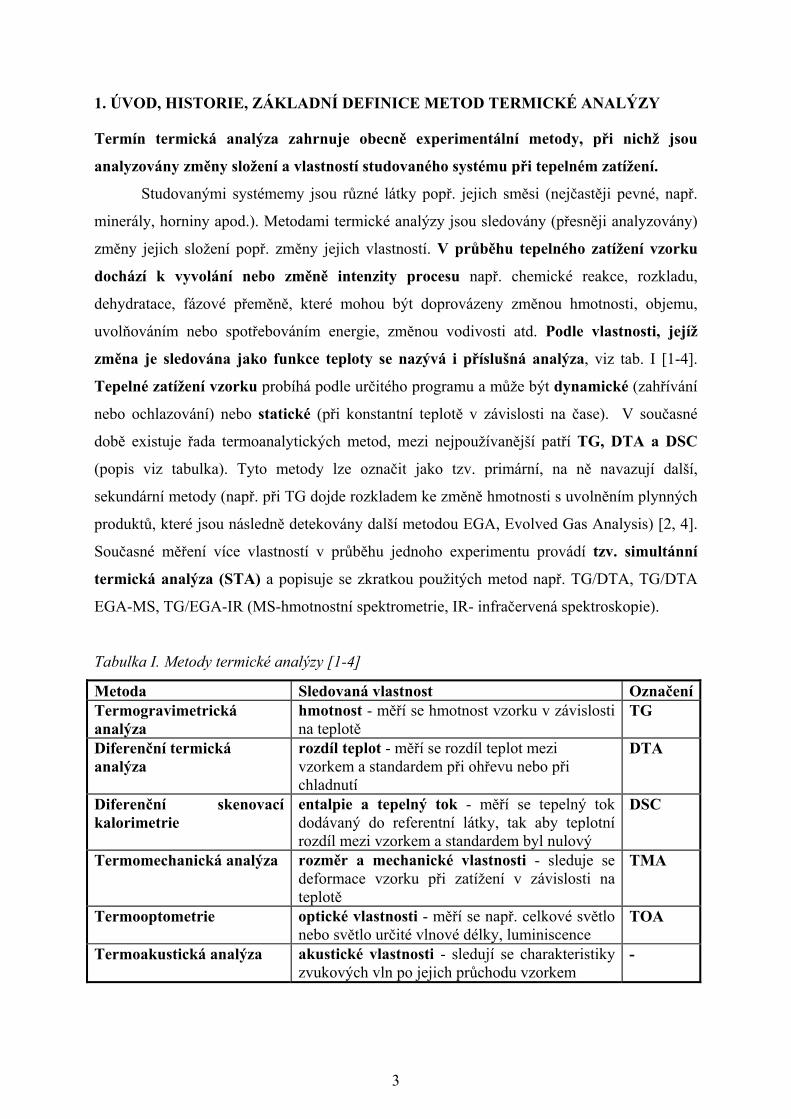
Tabulka I. Metody termické analýzy [1-4]
Metoda Sledovaná vlastnost OznačeníTermogravimetrická analýza
hmotnost - měří se hmotnost vzorku v závislosti na teplotě
TG
Diferenční termická analýza
rozdíl teplot - měří se rozdíl teplot mezi vzorkem a standardem při ohřevu nebo při chladnutí
DTA
Diferenční skenovací kalorimetrie
entalpie a tepelný tok - měří se tepelný tok dodávaný do referentní látky, tak aby teplotní rozdíl mezi vzorkem a standardem byl nulový
DSC
Termomechanická analýza rozměr a mechanické vlastnosti - sleduje se deformace vzorku při zatížení v závislosti na teplotě
TMA
Termooptometrie optické vlastnosti - měří se např. celkové světlo nebo světlo určité vlnové délky, luminiscence
TOA
Termoakustická analýza akustické vlastnosti - sledují se charakteristiky zvukových vln po jejich průchodu vzorkem
-

4
Prapočátky termických analýz lze zařadit do období, kdy naši předci poprvé
zaznamenali změny hornin při jejich zahřívání (např. kamenů v okolí ohniště). Jejich
identifikace na základě působení tepla byla poprvé popsána v Theofrastově spise " O
nerostech" [2]. Jako vědní disciplína se termická analýza začala rozvíjet až daleko později na
konci 19. století, protože do té doby nebylo k dispozici přístrojové vybavení nutné k
zaznamenání teploty a hodnoty sledované vlastnosti a k definování samotného teplotního
programu. Za zakladatele vědecké termické analýzy je považován Le Chatelier, který ji v r.
1886 poprvé použil ke studiu kalcitu. O rok později publikoval výsledky svého výzkumu
termického chování jílovitých hornin ve formě termoanalytických křivek pěti jílových
minerálů: halloyzitu, alofánu, kaolinitu, pyrofilitu a montmorillonitu. Jednalo se přímé měření
změny rychlosti teploty zkoumaného vzorku při jeho rovnoměrném ohřevu. Sledované reakce
se projevovaly s prodlevami v záznamech a určení tohoto zpoždění bylo velmi nepřesné.
Použitá metoda v podstatě odpovídala diferenční termické analýze (DTA) bez referenčního
vzorku. Dalším vývojovým stupněm byl rok 1899, kdy Roberts-Austen použil k porovnání
standardní vzorek (platinu), který nepodléhal žádným změnám při tepelném zatížení.
Principem současné DTA je měření rozdílu teplot mezi referenčním a studovaným vzorkem.
Rozvoj další nejrozšířenější termické analýzy termogravimetrie (TG) byl spojen s vynálezem
termováh Hondou (r. 1915). Významnou osobností termické analýzy byl Kurnakov, který v
r. 1904 vyvinul registrační válec umožňující současnou registraci několika křivek. Třetí
nejpoužívanější metodou od r. 1962 je diferenční skenovací kalorimetrie (DSC). [1- 4]
U nás se o rozvoj termických analýz zasloužil především Škramovský, později
Bárta, Šatava a Blažek. Díky této skupině se na Katedře silikátů Vysoké školy chemicko-
technologické konalo několik konferencí o termické analýze, které přispěly k jejímu rozšíření
a byla publikována řada prací z oblasti konstrukčních řešení a aplikací DTA a TG.
V uplynulých letech došlo k výraznému rozvoji automatizace kontroly měření a
registrace dat a samotných metod termické analýzy. Byly zlepšeny metody už známé a
vytvořeny nové. V současné době je na trhu řada zařízení pro termickou analýzu (Linseis,
Netzch, Perkins -Elmer, Setaram atd.) a tak už není hlavní pozornost zaměřena na jejich
konstrukci, ale je nyní směřována k analytickému a fyzikálně-chemickému využití těchto
metod v různých odvětvích.
2. TERMODYNAMIKA A TERMICKÁ ANALÝZA
Většina metod termické analýzy sleduje příslušné vlastnosti systému (hmotnost,
energii, rozměr, vodivost apod.) jako dynamickou funkci teploty. Základním jevem

5
důležitým pro tyto metody je změna entalpie (ΔH). Každou fyzikální a chemickou změnu
lze charakterizovat změnou Gibbsovy volné energie (ΔG), která je dána vzorcem:
ΔG = ΔH - TΔS, (1)
kde ΔH je změna entalpie, T je absolutní teplota a ΔS je změna entropie během děje.
Každý systém se snaží dosáhnout takového stavu, kterému odpovídá nižší
hodnota Gibbsovy volné energie. Jako příklad lze uvést modifikační přeměnu látky z jedné
krystalické formy do druhé, která má za dané teploty nižší hodnotu Gibbsovy volné energie a
je tedy stabilnější. K vytvoření krystalické struktury nebo jiného stavu s nižší hodnotou volné
entalpie může dojít při ohřevu i přes jednotlivé mezistupně. Příklady přeměn studované látky
vlivem rostoucí teploty v jejím okolí jsou uvedeny v tab. II [1,4,6].
Tabulka II. Fyzikální a chemické procesy identifikovatelné při ohřevu pomocí DTA, DSC
křivek
Fyzikální procesy Endotermické Exotermické Změna hmotnosti krystalizace + beze změny tání + beze změny vypařování + úbytek sublimace + úbytek fázové přeměny + + beze změny Chemické procesy Endotermické Exotermické chemisorpce + nárůst dehydroxylace + úbytek rozklad + + úbytek oxidace + nárůst
3. ZÁKLADNÍ INSTRUMENTACE, ZÁKLADNÍ POJMY, TERMOANALYTICKÉ
KŘIVKY
Termické analyzátory mají některé obecné vlastnosti pro jednotlivé termické metody
společné a skládají se z následujících částí (viz Obr. 1):
zdroje tepla - pece (trubková pec z tepelně odolného materiálu, např. křemen,
korund, MoSi nebo grafit, materiál je dán teplotou použití, elektrický ohřev je
zabezpečen vinutím okolo pece),
příslušného měřícího zařízení (termočlánky, termováhy), které registruje změny
studované vlastnosti nebo vlastností - apod. ve formě elektrického signálu, součástí
měřícího zařízení je držák a nosič - kelímek pro umístění vzorku, příslušný systém je
volen podle prováděné analýzy tj. TG/DTA, TG/DSC, TG, DSC CP,

6
vstupu pro nosný plyn a výstupu pro plynné produkty tepelného rozkladu s nosným
plynem (v případě dynamické pecní atmosféry plyn prochází pecí definovanou
rychlostí např. v ml/min, podle typu nosného plynu se rozlišuje pecní atmosféra tj.
atmosféra uvnitř pece v okolí vzorku na oxidační - vzduch, redukční - vodík, inertní
- dusík nebo argon; pokud není použit nosný plyn a atmosféra je neměnná tj. na
začátku měření je tvořena vzduchem, následně během analýzy se zvyšuje obsah
plynných produktů, jedná se o atmosféru statickou).
Obr. 1: Simultánní termický analyzátor LINSEIS STA PT 1600/1750oC HiRes, v horní části jsou uvedeny měřící systémy [8]
pec
vzorek, popř. srovnávací vzorek
nosný plyn s produkty rozkladu
měřící zařízení: termočlánky (DTA), termováhy (TG)
nosný plyn
registrace naměřených hodnot
definovaný teplotní program
7
Vlastní měření se obecně skládá z:
1. vložení kelímku s přesně naváženým vzorkem na měřící zařízení do pece zaplněné
zvolenou pecní atmosférou,
2. nastavení požadovaného teplotního režimu (změny teploty v čase určené obsluhou,
nejčastěji jde o lineární nárůst teploty, popř. prodlevu na určité teplotě s izotermním
měřením nebo chlazení apod.) pomocí příslušného programu a spuštění komunikace
měřícího zařízení s počítačem, pokud je připojen i sekundárního měřící systém (např.
hmotového spektrometru) musí rovněž komunikovat s počítačem, který zaznamenává
naměřené hodnoty,
3. interpretace naměřených dat příslušnými softwary.
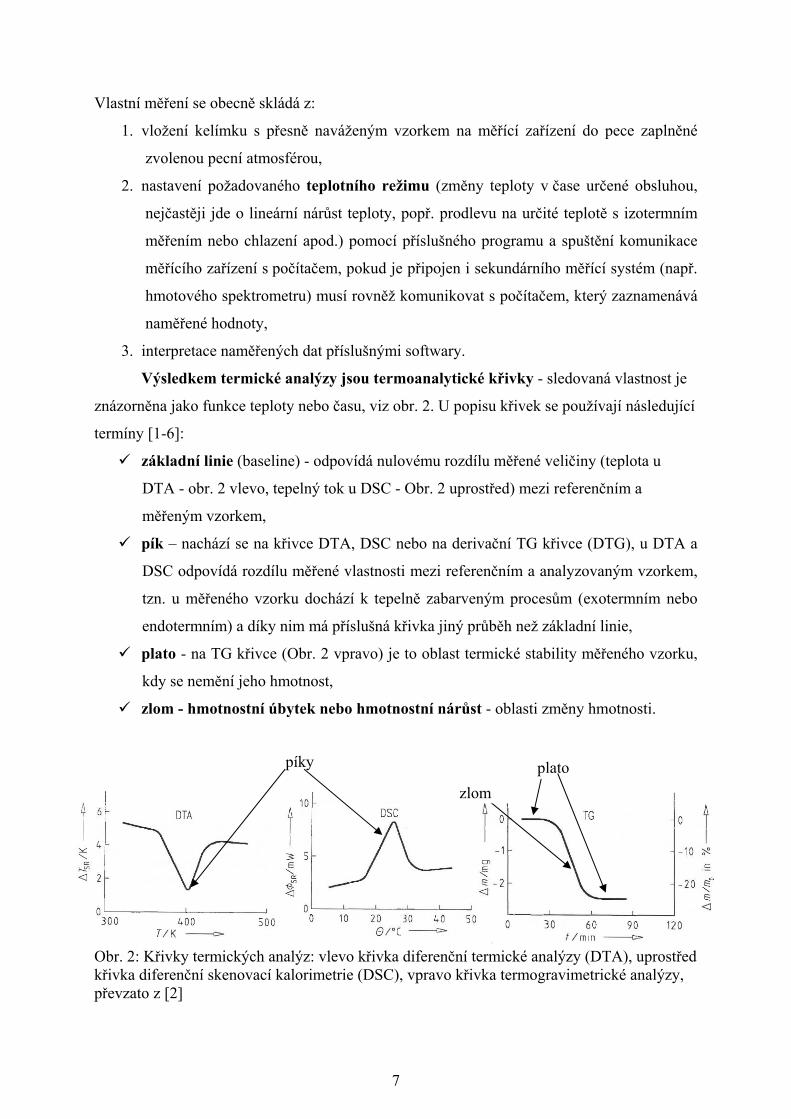
Výsledkem termické analýzy jsou termoanalytické křivky - sledovaná vlastnost je
znázorněna jako funkce teploty nebo času, viz obr. 2. U popisu křivek se používají následující
termíny [1-6]:
základní linie (baseline) - odpovídá nulovému rozdílu měřené veličiny (teplota u
DTA - obr. 2 vlevo, tepelný tok u DSC - Obr. 2 uprostřed) mezi referenčním a
měřeným vzorkem,
pík – nachází se na křivce DTA, DSC nebo na derivační TG křivce (DTG), u DTA a
DSC odpovídá rozdílu měřené vlastnosti mezi referenčním a analyzovaným vzorkem,
tzn. u měřeného vzorku dochází k tepelně zabarveným procesům (exotermním nebo
endotermním) a díky nim má příslušná křivka jiný průběh než základní linie,
plato - na TG křivce (Obr. 2 vpravo) je to oblast termické stability měřeného vzorku,
kdy se nemění jeho hmotnost,
zlom - hmotnostní úbytek nebo hmotnostní nárůst - oblasti změny hmotnosti.
Obr. 2: Křivky termických analýz: vlevo křivka diferenční termické analýzy (DTA), uprostřed křivka diferenční skenovací kalorimetrie (DSC), vpravo křivka termogravimetrické analýzy, převzato z [2]
píky plato
zlom
8
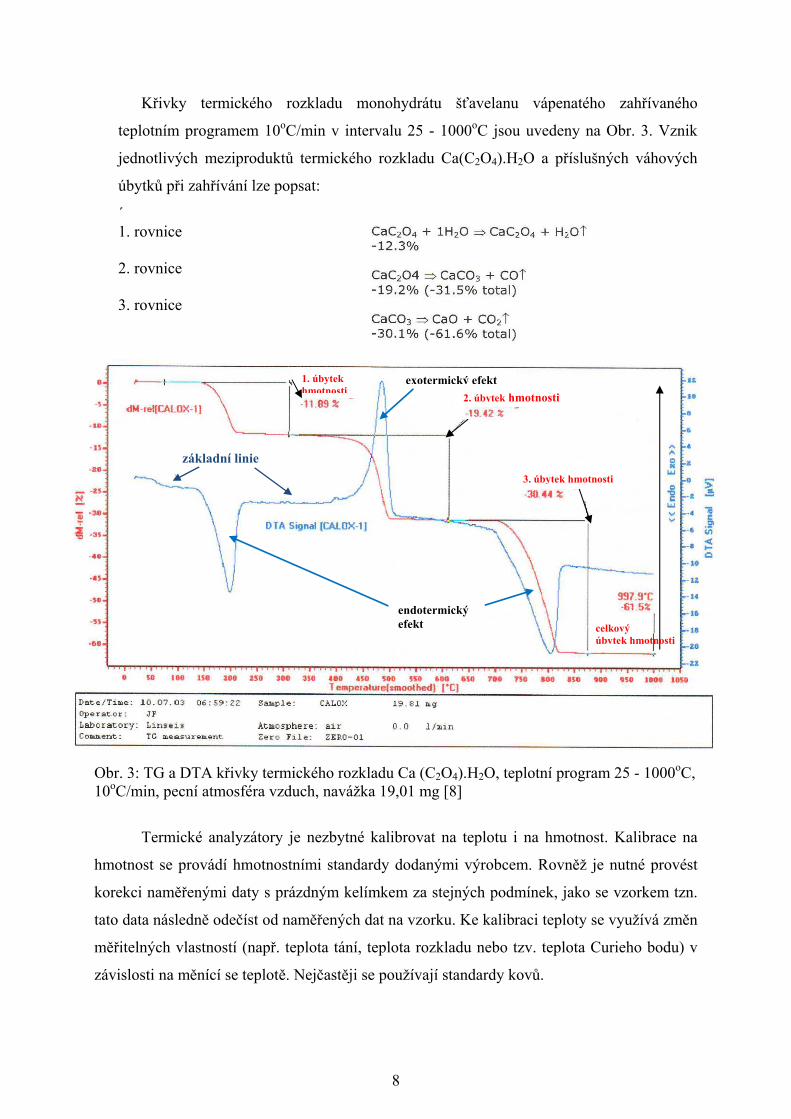
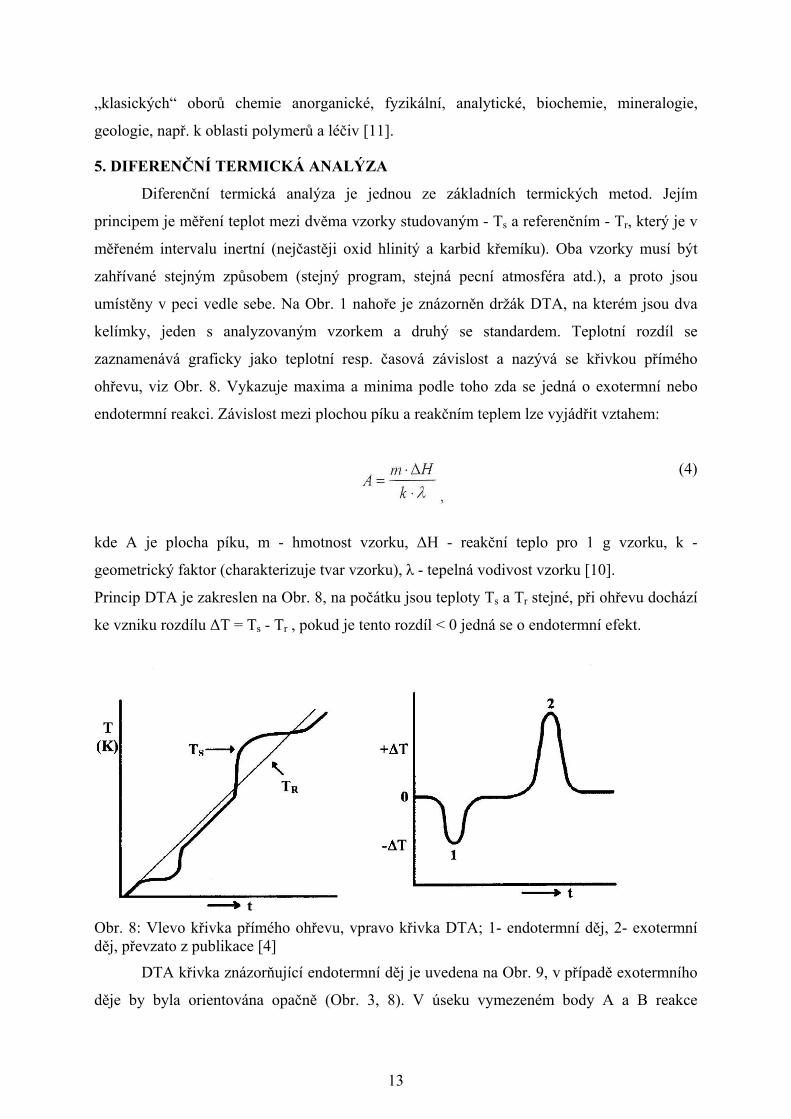
Křivky termického rozkladu monohydrátu šťavelanu vápenatého zahřívaného
teplotním programem 10oC/min v intervalu 25 - 1000oC jsou uvedeny na Obr. 3. Vznik
jednotlivých meziproduktů termického rozkladu Ca(C2O4).H2O a příslušných váhových
úbytků při zahřívání lze popsat:
´ 1. rovnice
2. rovnice 3. rovnice
Obr. 3: TG a DTA křivky termického rozkladu Ca (C2O4).H2O, teplotní program 25 - 1000oC, 10oC/min, pecní atmosféra vzduch, navážka 19,01 mg [8]
Termické analyzátory je nezbytné kalibrovat na teplotu i na hmotnost. Kalibrace na
hmotnost se provádí hmotnostními standardy dodanými výrobcem. Rovněž je nutné provést
korekci naměřenými daty s prázdným kelímkem za stejných podmínek, jako se vzorkem tzn.
tato data následně odečíst od naměřených dat na vzorku. Ke kalibraci teploty se využívá změn
měřitelných vlastností (např. teplota tání, teplota rozkladu nebo tzv. teplota Curieho bodu) v
závislosti na měnící se teplotě. Nejčastěji se používají standardy kovů.
2. úbytek hmotnosti
3. úbytek hmotnosti
1. úbytek hmotnosti
celkový úbytek hmotnosti
endotermický efekt
exotermický efekt
základní linie

9
4. TERMOGRAVIMETRIE
Termogravimetrie (TG) je základní metoda termické analýzy, studuje změny
hmotnosti, které probíhají v měřeném systému v závislosti na teplotě. Výsledkem je
termogravimetrická křivka, která znázorňuje závislost hmotnosti na teplotě nebo na čase (Obr.
2 vpravo) a lze z ní odečíst teploty hmotnostních změn. Odvozenou metodou je derivační
termogravimetrie (DTG), kde výsledná křivka uvádí rychlost hmotnostní změny na teplotě,
viz Obr. 4 [9]. Ta se používá, pokud změny hmotnosti neposkytují zřetelnou změnu, je
vhodná k odlišení těsně po sobě jdoucích efektů.
Obr. 4: Porovnání křivek DTG a TG, na TG křivce je popsán zlom, BCD - schod, B-počátek schodu, C- inflexní bod, D- konec schodu, Bi je počáteční teplota, Di je konečná teplota, BiDi reakční interval, FG - výška schodu, AB - přední základní čára, DE- zadní základní čára, podle literatury [3, 9]
4. 1. PŘÍSTROJOVÉ VYBAVENÍ
Přístroje pro TG se skládají z elektrické pece, analytických vah, nosičů vzorků,
zařízení pro měření a řízení teploty a registračního zařízení - počítače. Vlastní
termogravimetrická analýza probíhá na termovahách. Používají se tři upořádání resp.
umístění termovah vůči peci:
vertikální se vzorkem položeným na mechanismu termovah - plnění shora,
vertikální se vzorkem zavěšeným na mechanismu termovah,
horizontální.
V současné době se používá u všech třech typů kompenzační metody měření hmotnosti -
vzorek je po celou dobu měření umístěn na stejném místě a případná změna hmotnosti je
kompenzována pohybem na opačnou stranu a vzorek se hned vrátí do původní polohy před
změnou. Ta je snímána nejčastěji optickým senzorem.
A
D
DTG
TG
m
dm/dt
zlom
B
Bi
C
Di
E
F
G
A
píky
10
Měřený vzorek je v kelímku umístěném na termočlánku, který snímá aktuální teplotu.
Materiálem nosiče je nejčastěji platina, korund popř. oxidová keramika (ZrO2 apod.). Vždy je
potřeba zvážit oblast stability materiálu nosiče.
4. 2 PARAMETRY OVLIVŇUJÍCÍ TERMOGRAVIMETRII
Termogravimetrickou analýzu ovlivňuje řada faktorů, které je nutné před spuštěním
experimentu zvážit. Volba těchto parametrů má zásadní vliv na výsledek analýzy resp. na tvar
TG- křivky. Jde o:
přípravu vzorku (navážka, velikost částic, homogenita, napěchování vzorku),
tvar a velikost nosiče (kelímku),
pecní atmosféra, pecní tlak a vlhkost,
teplotní režim.
Při přípravě vzorků je nutné, aby byl vzorek homogenní a reprezentoval analyzovaný
materiál, aby měl stejnou velikost částic a nebyl např. kontaminován. Obecně lze uvést, že
jemně rozetřená pevná látka je reaktivnější oproti hrubozrnnější. Dochází u ní k snížení teplot
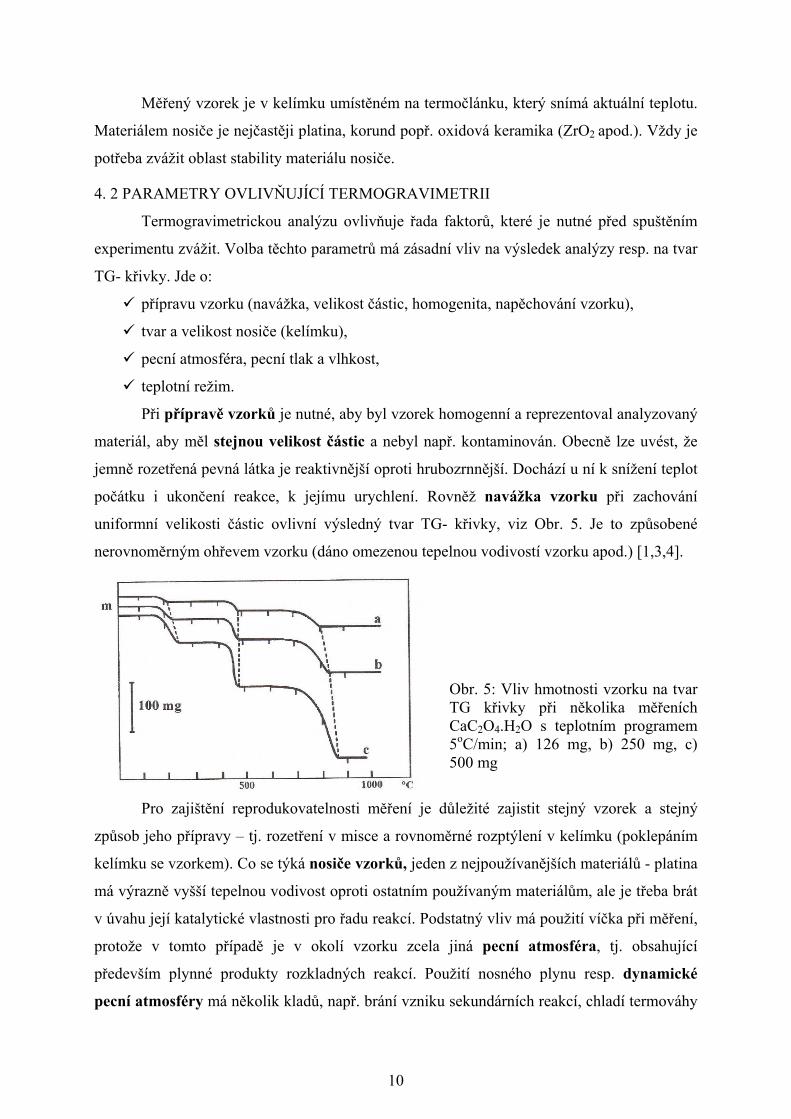
počátku i ukončení reakce, k jejímu urychlení. Rovněž navážka vzorku při zachování
uniformní velikosti částic ovlivní výsledný tvar TG- křivky, viz Obr. 5. Je to způsobené
nerovnoměrným ohřevem vzorku (dáno omezenou tepelnou vodivostí vzorku apod.) [1,3,4].
Obr. 5: Vliv hmotnosti vzorku na tvar TG křivky při několika měřeních CaC2O4.H2O s teplotním programem 5oC/min; a) 126 mg, b) 250 mg, c) 500 mg
Pro zajištění reprodukovatelnosti měření je důležité zajistit stejný vzorek a stejný
způsob jeho přípravy – tj. rozetření v misce a rovnoměrné rozptýlení v kelímku (poklepáním
kelímku se vzorkem). Co se týká nosiče vzorků, jeden z nejpoužívanějších materiálů - platina
má výrazně vyšší tepelnou vodivost oproti ostatním používaným materiálům, ale je třeba brát
v úvahu její katalytické vlastnosti pro řadu reakcí. Podstatný vliv má použití víčka při měření,
protože v tomto případě je v okolí vzorku zcela jiná pecní atmosféra, tj. obsahující
především plynné produkty rozkladných reakcí. Použití nosného plynu resp. dynamické
pecní atmosféry má několik kladů, např. brání vzniku sekundárních reakcí, chladí termováhy
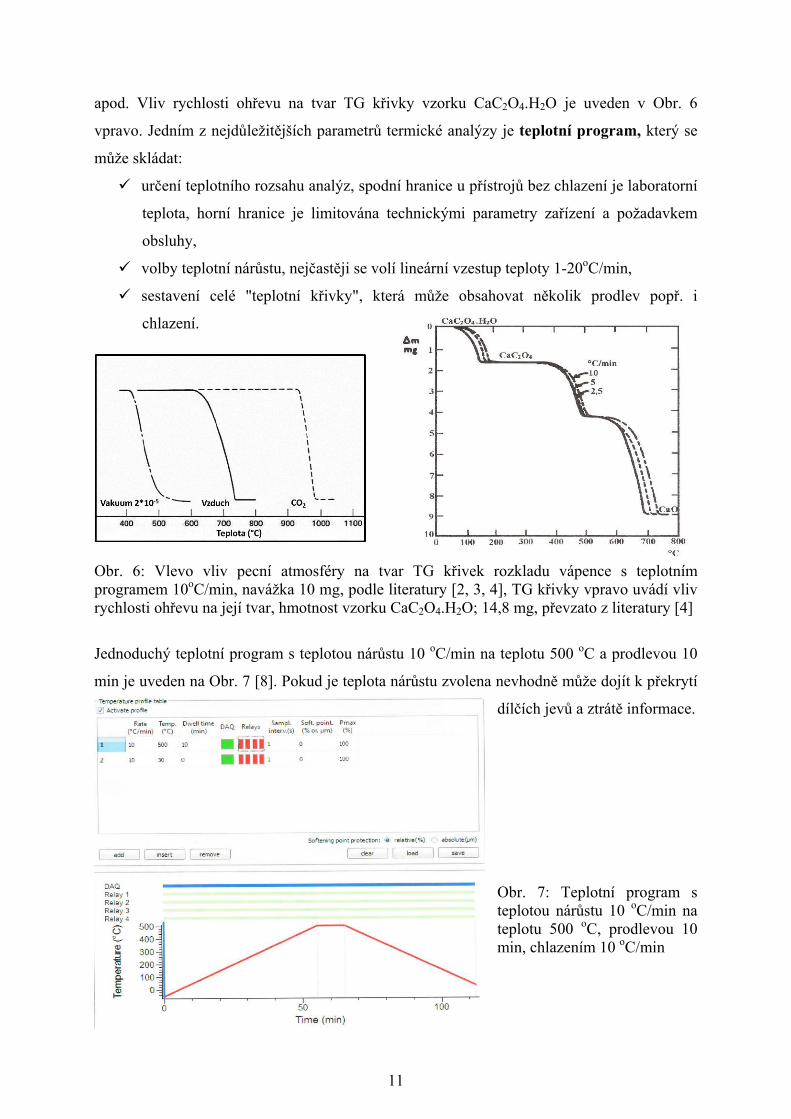
11
apod. Vliv rychlosti ohřevu na tvar TG křivky vzorku CaC2O4.H2O je uveden v Obr. 6
vpravo. Jedním z nejdůležitějších parametrů termické analýzy je teplotní program, který se
může skládat:
určení teplotního rozsahu analýz, spodní hranice u přístrojů bez chlazení je laboratorní
teplota, horní hranice je limitována technickými parametry zařízení a požadavkem
obsluhy,
volby teplotní nárůstu, nejčastěji se volí lineární vzestup teploty 1-20oC/min,
sestavení celé "teplotní křivky", která může obsahovat několik prodlev popř. i
chlazení.
Obr. 6: Vlevo vliv pecní atmosféry na tvar TG křivek rozkladu vápence s teplotním programem 10oC/min, navážka 10 mg, podle literatury [2, 3, 4], TG křivky vpravo uvádí vliv rychlosti ohřevu na její tvar, hmotnost vzorku CaC2O4.H2O; 14,8 mg, převzato z literatury [4]
Jednoduchý teplotní program s teplotou nárůstu 10 oC/min na teplotu 500 oC a prodlevou 10
min je uveden na Obr. 7 [8]. Pokud je teplota nárůstu zvolena nevhodně může dojít k překrytí
dílčích jevů a ztrátě informace.
Obr. 7: Teplotní program s teplotou nárůstu 10 oC/min na teplotu 500 oC, prodlevou 10 min, chlazením 10 oC/min

12
4. 3 VYHODNOCENÍ TERMOGRAVIMETRICKÝCH KŘIVEK
Výsledkem termografické analýzy je gravimetrická křivka. Z Obr. 2 vpravo je patrné,
že na ose y mohou být jednotky hmotnosti (mg) nebo v procentech původní hmotnosti (%),
přičemž na ose x je teplota nebo čas. Nejčastěji se z důvodu porovnávání výsledků používá
kombinace y = původní % hmotnosti, x = teplota. Z TG křivek, pro které jsou typickými
útvary zlom a plato (Obr. 4, 5) lze odečíst:
oblasti beze změn - plato (oblast termické stability),
oblasti se změnami hmotnosti (úbytek nebo nárůst),
dílčí hmotnostní úbytek,
celkový hmotnostní úbytek.
Jednotlivé termíny jsou uvedeny v Obr. 3. Na TG křivce se mohou vyskytovat
"úseky", které nesouvisí s měřeným vzorkem např. vliv klesající hustoty pecní atmosféry se
stoupající teplotou nebo vliv rychlosti průtoku plynu atd. Výrazná deformace křivek je
způsobena rozdílem mezi teplotou vzorku a teplotním programem, křivka pak ztrácí lineární
charakter a vede k deformaci křivek jednotlivých metod. Jedná se o velmi častý jev, který se
musí počítačově upravit linearizací teploty [4].
Tvary křivek TG:
křivka bez zlomu – probíhá děj bez změny hmotnosti, např. tání,
křivka na počátku s úbytkem hmotnosti, typické pro sušení a desorpci,
na křivce je jeden zlom mezi dvěmi platy, viz Obr. 6 vlevo, typické pro rozklad,
získané hodnoty lze využít k studiu kinetiky,
termický rozklad probíhá v několika krocích, které jsou odděleny zřetelnými platy, viz
Obr. 6 vpravo, jednotlivé kroky lze kvantifikovat, viz Obr. 2,
obdobné křivky jako v předchozím případě bez přesného oddělení jednotlivými platy,
(někdy lze lépe rozlišit jednotlivé děje při nižších rychlostech nárůstu teploty).
na křivce mohou být patrné i nárůsty hmotnosti, případ reakcí měřené látky a plynů
pecní atmosféry (např. oxidace železa v pecní atmosféře). [2,4]
TG se používá pro kvalitativní (identifikace děje) i kvantitativní hodnocení (odečtení
velikosti příslušných změn), např. ke sledování sušení, dehydroxylace, tepelného rozkladu,
tepelné oxidace, reakce v pevné fázi, reakce pevné a plynné fáze (oxidace, redukce, koroze),
katalýzy, studiu reakční kinetiky a reakčních mechanismů, identifikaci sloučenin popř. studiu
nových sloučenin, stanovení čistoty chemických sloučenin [4]. Její použití se rozšířilo z
13
„klasických“ oborů chemie anorganické, fyzikální, analytické, biochemie, mineralogie,
geologie, např. k oblasti polymerů a léčiv [11].
5. DIFERENČNÍ TERMICKÁ ANALÝZA
Diferenční termická analýza je jednou ze základních termických metod. Jejím
principem je měření teplot mezi dvěma vzorky studovaným - Ts a referenčním - Tr, který je v
měřeném intervalu inertní (nejčastěji oxid hlinitý a karbid křemíku). Oba vzorky musí být
zahřívané stejným způsobem (stejný program, stejná pecní atmosféra atd.), a proto jsou
umístěny v peci vedle sebe. Na Obr. 1 nahoře je znázorněn držák DTA, na kterém jsou dva
kelímky, jeden s analyzovaným vzorkem a druhý se standardem. Teplotní rozdíl se
zaznamenává graficky jako teplotní resp. časová závislost a nazývá se křivkou přímého
ohřevu, viz Obr. 8. Vykazuje maxima a minima podle toho zda se jedná o exotermní nebo
endotermní reakci. Závislost mezi plochou píku a reakčním teplem lze vyjádřit vztahem:
(4)
kde A je plocha píku, m - hmotnost vzorku, ΔH - reakční teplo pro 1 g vzorku, k -
geometrický faktor (charakterizuje tvar vzorku), λ - tepelná vodivost vzorku [10].
Princip DTA je zakreslen na Obr. 8, na počátku jsou teploty Ts a Tr stejné, při ohřevu dochází
ke vzniku rozdílu ΔT = Ts - Tr , pokud je tento rozdíl ˂ 0 jedná se o endotermní efekt.
Obr. 8: Vlevo křivka přímého ohřevu, vpravo křivka DTA; 1- endotermní děj, 2- exotermní děj, převzato z publikace [4]
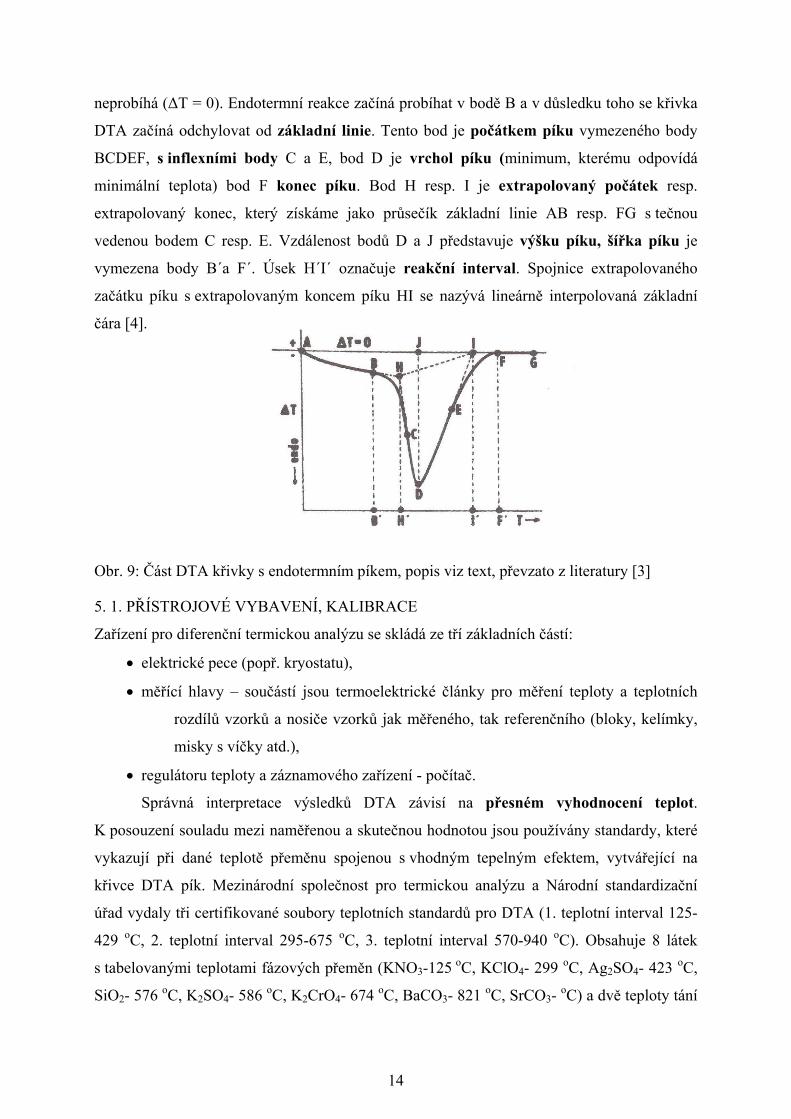
DTA křivka znázorňující endotermní děj je uvedena na Obr. 9, v případě exotermního
děje by byla orientována opačně (Obr. 3, 8). V úseku vymezeném body A a B reakce
14
neprobíhá (ΔT = 0). Endotermní reakce začíná probíhat v bodě B a v důsledku toho se křivka
DTA začíná odchylovat od základní linie. Tento bod je počátkem píku vymezeného body
BCDEF, s inflexními body C a E, bod D je vrchol píku (minimum, kterému odpovídá
minimální teplota) bod F konec píku. Bod H resp. I je extrapolovaný počátek resp.
extrapolovaný konec, který získáme jako průsečík základní linie AB resp. FG s tečnou
vedenou bodem C resp. E. Vzdálenost bodů D a J představuje výšku píku, šířka píku je
vymezena body B´a F´. Úsek H´I´ označuje reakční interval. Spojnice extrapolovaného
začátku píku s extrapolovaným koncem píku HI se nazývá lineárně interpolovaná základní
čára [4].
Obr. 9: Část DTA křivky s endotermním píkem, popis viz text, převzato z literatury [3]
5. 1. PŘÍSTROJOVÉ VYBAVENÍ, KALIBRACE
Zařízení pro diferenční termickou analýzu se skládá ze tří základních částí:
elektrické pece (popř. kryostatu),
měřící hlavy – součástí jsou termoelektrické články pro měření teploty a teplotních
rozdílů vzorků a nosiče vzorků jak měřeného, tak referenčního (bloky, kelímky,
misky s víčky atd.),
regulátoru teploty a záznamového zařízení - počítač.
Správná interpretace výsledků DTA závisí na přesném vyhodnocení teplot.
K posouzení souladu mezi naměřenou a skutečnou hodnotou jsou používány standardy, které
vykazují při dané teplotě přeměnu spojenou s vhodným tepelným efektem, vytvářející na
křivce DTA pík. Mezinárodní společnost pro termickou analýzu a Národní standardizační
úřad vydaly tři certifikované soubory teplotních standardů pro DTA (1. teplotní interval 125-
429 oC, 2. teplotní interval 295-675 oC, 3. teplotní interval 570-940 oC). Obsahuje 8 látek
s tabelovanými teplotami fázových přeměn (KNO3-125 oC, KClO4- 299 oC, Ag2SO4- 423 oC,
SiO2- 576 oC, K2SO4- 586 oC, K2CrO4- 674 oC, BaCO3- 821 oC, SrCO3- oC) a dvě teploty tání

15
(In- 154 oC, Sn- 230 oC ), teploty jsou vyjádřené teplotami extrapolovaného začátku píku,
v prvém případě exotermního, v druhém endotermního.
5. 2 PARAMETRY OVLIVŇUJÍCÍ DIFERENČNÍ TERMICKOU ANALÝZU
V případě DTA výsledky analýzy ovlivňuje řada faktorů, které je nezbytné předem
zvážit, obdobně jako u TG (viz kap. 4. 2). Zahrnují vliv experimentálních podmínek i
samotného vzorku a souvisí s:
konstrukcí a s materiály zařízení (včetně kelímků - jejich geometrie,
umístění čidel),
tepelnými zdroji, rychlostí ohřevu, pecní atmosféra,
přípravou vzorků (navážka, homogenita, velikost částic, plnění kelímku,
ředění inertním matriálem),
měřením teploty, teplotním gradientem, tepelnou vodivostí vzorku, referencí,
záznamem termoanalytických křivek, apod.
Parametr spojený s přípravou vzorku souvisí nejenom se způsobem jeho přípravy,
ale s jeho umístěním na nosiči (nutno dodržet i stejnou geometrii tzn. tvar a velikost kelímku).
Zahrnuje vliv velikosti částic, upěchování vzorku a ředění vzorku inertním matriálem.
Vliv velikosti částic je obdobný jako u TG (Obr. 5), viz dále 5.3. Při přípravě vzorku
je opět důležité upěchování vzorku, které ovlivňuje přenos tepla a difúzi plynů. Přílišné
stlačení práškového vzorku usnadňuje přenos tepla, ale znesnadňuje difúzi plynných
produktů. Požadované napěchování se provádí poklepem kelímku se vzorkem o desku stolu.
Výše uvedený parametr souvisí i s podmínkami pecní atmosféry. V případě statické
atmosféry jsou uvolňované plyny v kontaktu s analyzovaným vzorkem, pokud jsou
považovány za nežádoucí, musí být odsávány. V dynamické atmosféře je vzorek ve styku s
atmosférou, která je do pece zaváděna (např. proud vzduchu, dusíku, argonu apod.). K
zabránění spékání nebo smršťování měřeného vzorku se používá jeho ředění inertní látkou
(např. korundem). Ta musí obsahovat stejně velké částice jako vzorek.
Rovněž referenční vzorek musí mít určité vlastnosti:
nesmí podléhat termickým změnám v daném teplotním rozmezí,
nesmí reagovat s nádobkou, popř. držákem, ve kterém je umístěn,
jeho tepelná vodivost a kapacita by měla být co nejbližší analyzovanému
vzorku.
16
křivka A - vzduch
křivka B – CO2
křivka C – s víčkem
křivka D – ředěno inertní látkou
Příkladem srovnávací látky pro anorganické vzorky může být Al2O3 nebo SiC. Při
ředění vzorku je vhodné použít stejnou látku jako inertní i referenční. Vliv pecní atmosféry,
použití víčka a ředění inertem při DTA sideritu (FeCO3) je uvedeno na Obr. 10.
Obr. 10: Vliv pecní atmosféry, použití víčka a ředění inertem na křivku DTA sideritu [12]
Zásadním problémem všech termických analýz je korelace mezi naměřenými daty a
teplotními procesy probíhajícími ve vzorku.
5. 3 VYHODNOCENÍ DTA KŘIVEK
Výsledkem DTA analýzy je křivka znázorňující závislost rozdílu teplot mezi měřeným
a referenčním vzorkem v jednotkách elektrického napětí tj. v μV na teplotě popř. na čase.
Křivky se skládají z oblastí nulových hodnot a píků, které jsou v kladných hodnotách
nazývány exoefekty a v záporných endoefekty (Obr. 3 a 7). Charakteristickou hodnotou je
počátek píku, viz Obr. 9, někdy se uvádí maximální rozdíl ΔT = Ts - Tr (reakční interval),
který závisí na mnoha výše uvedených parametrech. Plocha píku odpovídá hodnotě změny
entalpie, Δ H (rovnice 1, kap. 2). Tvar křivky DTA je primárně dán především měřeným
vzorkem, ale je také ovlivněn výše uvedenými parametry zadávanými obsluhou (tj. navážkou,

17
úpravou vzorků, velikostí částic, režimem, pecní atmosférou). Na tvar křivky mají výrazný
vliv:
Rychlost ohřevu – zvýšením rychlosti se zvýší výška píku (zvětší se jeho plocha) a
vrchol píku se posune k vyšší teplotě, velká rychlost zahřívání může způsobit splynutí
dvou dějů probíhající těsně po sobě.
Hmotnost vzorku – je přímo úměrná ploše píku, při vzniku plynných produktů se se
zvětšující hmotností vzorku posouvá vrchol píku k vyšší teplotě. Děje, které probíhají
v úzkém teplotním intervalu lze snížením hmotnosti vzorku od sebe oddělit.
Geometrie vzorku – kelímek nebo miska musí obsahovat tolik vzorku, aby i po jeho
upěchování zasahovalo teplotní čidlo do středu vzorku. Geometrické uspořádání vzorku
ovlivňuje výšku píku, při správném umístění čidla je pík nejvyšší. K výraznému poklesu
výšky píku dochází při umístění čidla při povrchu vzorku.
Velikost částic – malé částice urychlují průběh reakce a posunují její začátek k nižším
hodnotám teplot.
Význam DTA spočívá v identifikaci dějů spojených s výměnou tepla mezi vzorkem a
okolím. Používá se pro kvalitativní (identifikace děje) i kvantitativní hodnocení
(odečtení velikosti příslušných změn). Z hlediska kvalitativní analýzy lze z tvaru a poloh
píků určit přítomnost a charakter děje (exotermní, endotermní) a příslušné teplotní intervaly,
počáteční a konečnou teplotu, vratnost dějů. Díky katalogům termoanalytických křivek lze
metodami DTA a TG identifikovat řadu minerálů, rud, hornin a jejich případné příměsi. Pro
získání přesnějších údajů je vhodné DTA doplnit další analýzou (např. TG). Kvantitativní
analýza je založena na určování velikosti ploch píků, nezbytnou součástí je sestavení
kalibračních křivek proměřením několika vzorků se známým obsahem stanovované složky.
Obr. 11: Křivky DTA některých minerálů: 1) kalcit CaCO3, 2) magnezit MgCO3, 3) dolomit CaCO3. MgCO3, 4) ankerit (Mg, Fe)CO3.CaCO3, 5) siderit FeCO3, 6) breinerit (Mg, Fe)CO3, 7) rodochrozit MnCO3, 8) aragonit CaCO3, 9) hydromagnezit 4MgCO3.Mg(OH)2.4H2O

18
Podle tvaru píků lze usuzovat na určitý druh fázové přeměny:
1. pík je tvořen ostrým vrcholem – tání čisté látky, eutektika, přeměna 1. druhu,
2. píky s oblým vrcholem – přeměny probíhající v určitém teplotním intervalu, určuje se
bod nástupu, vrchol píku atd.,
3. křivky se dvěma píky blízko za sebou – odpovídají průchodu dvoufázovou oblastí.
DTA je využívána v mnoha oblastech, především při výzkumu nových systémů, sledování
jejich čistoty, stability, při studiu reakční kinetiky, při konstrukci fázových diagramů atd.
Derivační diferenční termická analýza (DDTA)
Princip derivační diferenční termické analýzy je shodný s předchozí metodou. Registruje se
časová derivace křivky DTA, a to d(ΔT)/dt = f (T) resp. d(ΔT)/dt = f (t). Tato metoda pomáhá
přesněji zjistit teploty začínajících změn a rozlišit překrývající se jevy.
6. DIFERENČNÍ SKENOVACÍ KALORIMETRIE
Princip metody je v udržení stejné teploty studovaného a referenčního vzorku, které jsou
zahřívány současně vedle sebe. Udržení nulového teplotního rozdílu se dosahuje buď
dodáním energie do vzorku (pokud v něm probíhá endotermní děj) nebo do referenční látky
(ve vzorku probíhá exotermní děj). Přesnost měření je oproti DTA vyšší.
Používají se dva základní typy DSC analyzátorů:
• DSC s kompenzací příkonu - DSC s kompenzací příkonu se nazývá též „obrácená“ DTA.
Podstatou DSC s kompenzací příkonu je zachování nulového teplotního rozdílu mezi
měřeným a srovnávacím vzorkem. Základem jsou dvě oddělené měřící cely a dva tepelné
zdroje, oba vzorky jsou zahřívány stejnou rychlostí. Pokud v měřeném vzorku začne probíhat
endotermní reakce, je teplotní rozdíl mezi měřeným a referenčním vzorkem vynulován
dodáním energie do měřeného vzorku. A naopak, pokud probíhá exotermní děj, je teplota
měřeného vzorku vyšší oproti referenčnímu. K vyrovnání teplot dochází dodáním méně
energie do měřeného vzorku oproti referenčnímu. V obou vzorcích zůstává stejná teplota díky
kompenzaci příkonu. Měřenou veličinou je elektrický příkon, který je potřebný k udržení
konstantní teploty obou vzorků. Křivka DSC dQ / dt = f (t), kde Q značí tepelnou energii.
Tento typ DSC zařízení umožňuje zaznamenat velmi citlivé změny teploty a je vhodný
ke sledování izotermních dějů. DSC křivka, stejně jako křivka DTA, obsahuje píky, které jsou
opačně orientované vzhledem k ose x, viz obr. 2.

19
• DSC s tepelným tokem - oba vzorky, referenční i měřený, jsou umístěny na samostatných
teplotních čidlech ve společné kalorimetrické cele. Měření rozdílu příkonu je nahrazeno
měřením rozdílu teplot analyzovaného a referenčního vzorku, které jsou spojeny tepelným
mostem. Při změnách teploty v měřeném vzorku, které jsou způsobené endotermními nebo
exotermními ději, je rozdíl teplot zaznamenán jako tepelný tok od vzorku nebo do vzorku a je
považován za úměrný rozdílu teplot.
6. 1 PŘÍSTROJOVÉ VYBAVENÍ
DSC s kompenzací příkonu je složeno ze dvou oddělených obvodů:
kontrolního - měří průměrnou teplotu vzorků a automaticky vyrovnává tepelný výkon
tak, že se průměrná teplota vzorků zvyšuje lineárně,
řídícího – zaznamenává rozdíly teplot mezi měřeným a srovnávacím vzorkem, určuje,
který ze vzorků má vyšší teplotu a automaticky kompenzuje tyto teplotní rozdíly.
Studovaný i referenční vzorek mají své nosiče, které obsahují teplotní čidla a topná tělíska.
Vzorky jsou od sebe dokonale izolovány, aby se zabránilo tepelnému toku mezi nimi.
DSC s tepelným tokem se skládá z:
měřící hlavy s držákem pro vzorek a srovnávací látku,
pece,
termostatu,
zdroje plynů a zdroje napětí.
regulátoru teploty a záznamového zařízení - počítače.
Pro DSC lze použít vzorky velmi malých hmotností (1 až 100 mg), vkládají do keramických
nebo kovových misek popř. do folií aby se docílilo dokonalého kontaktu s topným tělískem a
teplotními čidly.
6. 2 PARAMETRY OVLIVŇUJÍCÍ DIFERENČNÍ SKENOVACÍ KALORIMETRII
DSC ovlivňují obdobné parametry jako u předchozích metod (např. pecní atmosféra
v okolí vzorku je výrazně ovlivněna geometrií vzorku). Před vlastní analýzou je třeba zvážit,
jaké systémy jsou studovány, jaké děje v něm mohou probíhat a jak se projeví na DSC křivce,
v jakých teplotních režimech budou probíhat a jakými dalšími metodami termické analýzy je
lze studovat. Jak již bylo uvedeno u všech metod termické analýzy je základním problémem
korelace mezi naměřenými daty a teplotními ději, které probíhají v měřeném vzorku.
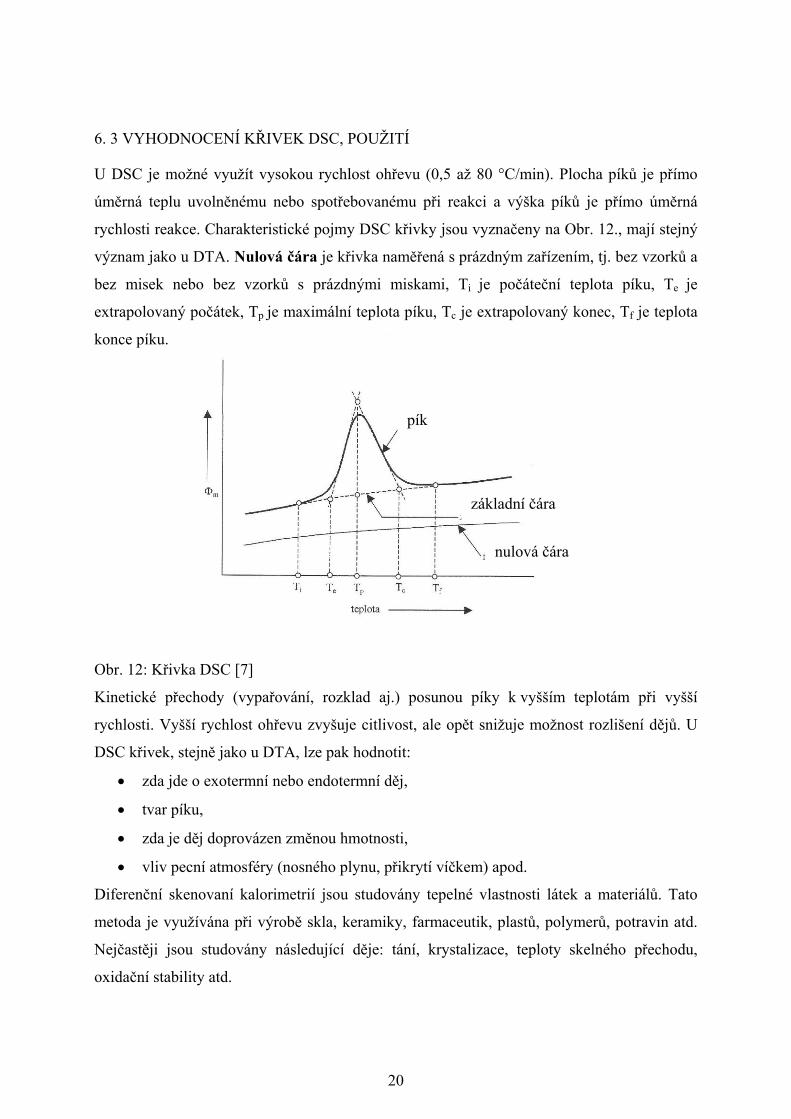
20
pík
základní čára
nulová čára
6. 3 VYHODNOCENÍ KŘIVEK DSC, POUŽITÍ
U DSC je možné využít vysokou rychlost ohřevu (0,5 až 80 °C/min). Plocha píků je přímo
úměrná teplu uvolněnému nebo spotřebovanému při reakci a výška píků je přímo úměrná
rychlosti reakce. Charakteristické pojmy DSC křivky jsou vyznačeny na Obr. 12., mají stejný
význam jako u DTA. Nulová čára je křivka naměřená s prázdným zařízením, tj. bez vzorků a
bez misek nebo bez vzorků s prázdnými miskami, Ti je počáteční teplota píku, Te je
extrapolovaný počátek, Tp je maximální teplota píku, Tc je extrapolovaný konec, Tf je teplota
konce píku.
Obr. 12: Křivka DSC [7]
Kinetické přechody (vypařování, rozklad aj.) posunou píky k vyšším teplotám při vyšší
rychlosti. Vyšší rychlost ohřevu zvyšuje citlivost, ale opět snižuje možnost rozlišení dějů. U
DSC křivek, stejně jako u DTA, lze pak hodnotit:
zda jde o exotermní nebo endotermní děj,
tvar píku,
zda je děj doprovázen změnou hmotnosti,
vliv pecní atmosféry (nosného plynu, přikrytí víčkem) apod.
Diferenční skenovaní kalorimetrií jsou studovány tepelné vlastnosti látek a materiálů. Tato
metoda je využívána při výrobě skla, keramiky, farmaceutik, plastů, polymerů, potravin atd.
Nejčastěji jsou studovány následující děje: tání, krystalizace, teploty skelného přechodu,
oxidační stability atd.

21
7. KOMBINACE METOD TERMICKÉ ANALÝZY
K přesnějšímu popisu dějů, které probíhají v analyzovaném vzorku během zahřívání
(ochlazování) je vhodné použít více metod.
Kombinované metody (dvě a více metod) se používají při analýze vzorku postupně. V tomto
případě je nutné pro každou analýzu mít nový vzorek zkoumaného materiálu, a proto jsou
kombinované metody náročnější na množství vzorku. Výhodou je, že lze kombinovat nejen
metody termické analýzy, ale také i jiné „netermické“ metody např. spektroskopii, atomovou
absorpci, rentgenostrukturní analýzu apod.
Simultánní metody (STA) umožňují během jednoho měření zkoumat více fyzikálních
vlastností a to za použití pouze jednoho vzorku. Mezi další výhody patří dodržení stejných
experimentálních podmínek, které ale musí vyhovovat všem použitým metodám. V současné
době patří mezi nejběžnější simultánní metody TG-DTA a TG-DSC. Dalšími variantami jsou
TG-EGD, TG-EGA, DTA-EGD, DTA-EGA, TG-DSC-EGA. Tyto metody slouží k upřesnění
popisu dějů probíhajících mezi pevnou a plynnou fází.
8. SIMULTÁNNÍ STA LISEIS, POPIS ZAŘÍZENÍ
8. 1 POPIS ZAŘÍZENÍ SIMULTÁNNÍ STA LINSEIS STA PT1600/1750 °C HiRes a MS
PFEIFFER VACUUM

22
Obr. 13: Zařízení LINSEIS STA PT 1600/1750oC HiRes MS Pfeiffer Vacuum s příslušenstvím v laboratoři termické analýzy AS89
8. 2 ZPROVOZNĚNÍ PŘÍSTROJŮ A POČÍTAČE S ŘÍDÍCÍMI PROGRAMY
Přístroje jsou zapínány v následujícím pořadí:
a) zapnout počítač, b) zapnout chladící okruh, c) zapnout přístroj Linseis STA PT 1600/1750 °C HiRes tlačítkem On-Off,
Obr. 14: Zařízení LINSEIS STA PT 1600/1750oC HiRes
d) zapnout hmotový spektrometr ThermoStarTM GSD320 (Pfeiffer Vacuum).
Obr. 15: Hmotový spektrometr Pfeiffer Vacuum
Jednotlivé kroky:
Nejprve je nutné se přesvědčit, zda je spuštěný chladící okruh!
Spustit přístroj na regulaci teploty (od firmy Linseis, který je přednastaven na
teplotu 200 °C).
Spustit hmotový spektrometr spínačem na zadní straně přístroje.
Na přední straně panelu zmáčknout tlačítko OK, kterým se dostaneme do
hlavního panelu pro nastavení přístroje.
Spustit vyhřívání pomocí šipek - Function → Turn heaters on.
Spustit čerpadlo pomocí šipek - Function → pump down.
Na přístroji musí být po správném spuštění zelená kontrolka pro power a normal,
oranžová kontrolka pro capillary a inlet. Na displeji se objeví hodnota tlaku ≈ 10-6
mbar, teplota kapiláry 200 °C, vstupu 110 °C a oznámení System OK.

23
Nyní lze spustit v počítači program Quadera.
e) Nastavit průtok argonu zařízením na 100 ml/min.
8.3 PŘÍPRAVA VZORKU A JEHO UMÍSTĚNÍ DO NOSIČE ZAŘÍZENÍ STA
a) Navážit vzorek na analytických vahách s přesností navážky na 0,1 mg s maximálním
zaplněním kelímku cca 2/3.
b) Zaznamenat navážku vzorku!
c) Vložit kelímek se vzorkem do pecního otvoru.
Otevřít pec tlačítkem LIFT.
Vložit pomocí pinzety kelímek se vzorkem do platinového držáku na určené místo
- na pravé straně (na levé straně je prázdný referenční kelímek!).
Zavřít pec tlačítkem LIFT.
8.4 NASTAVENÍ MĚŘENÍ V PROGRAMU LINSEIS Data Acquisition
Spustit Linseis Data Acquisition.
a) Nastavit měření v General setting, Temperature profile a Adjust TG.
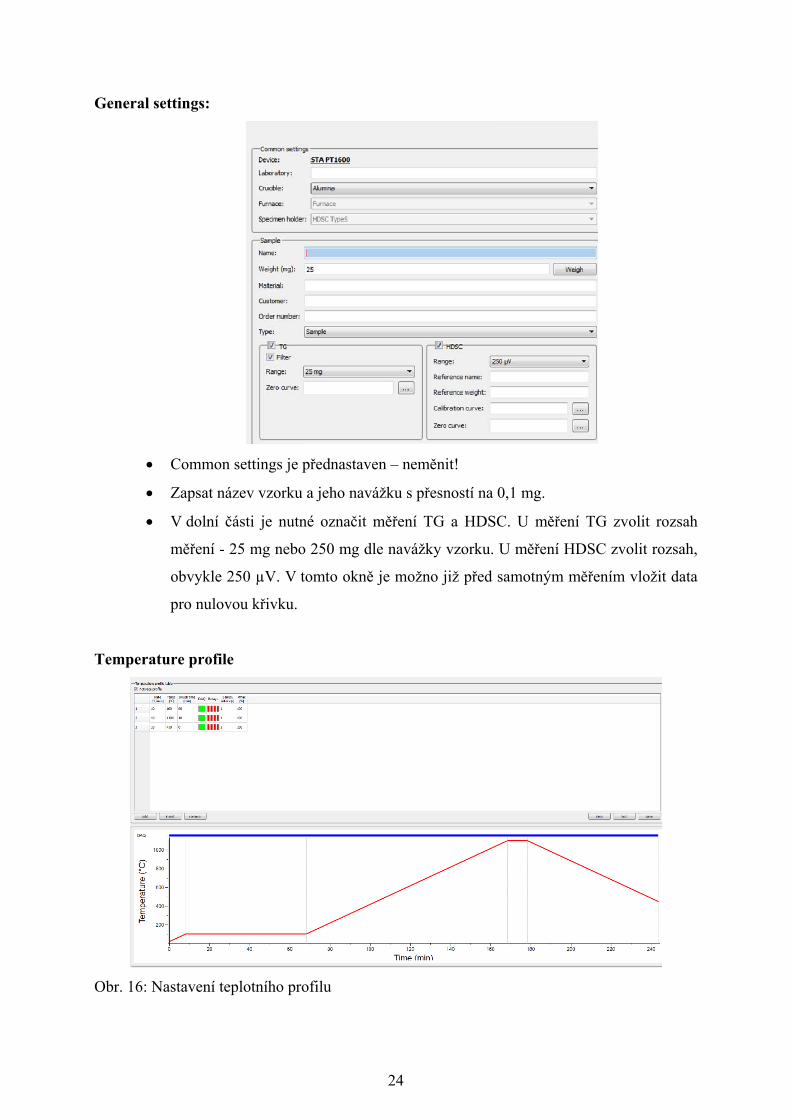
24
General settings:
Common settings je přednastaven – neměnit!
Zapsat název vzorku a jeho navážku s přesností na 0,1 mg.
V dolní části je nutné označit měření TG a HDSC. U měření TG zvolit rozsah
měření - 25 mg nebo 250 mg dle navážky vzorku. U měření HDSC zvolit rozsah,
obvykle 250 µV. V tomto okně je možno již před samotným měřením vložit data
pro nulovou křivku.
Temperature profile
Obr. 16: Nastavení teplotního profilu

25
Nastavit jednotlivé části měření, tj. předehřev, ohřev a chlazení. U každé části je
nutno nastavit rychlost ohřevu (°C/min), koncovou teplotu dané části (°C)
a výdrž na koncové teplotě (min).
Vybrat úsek teplotního programu, během kterého se budou ukládat data – zelené
políčko ve sloupci DAQ.
Nastavený teplotní profil lze také uložit jako šablonu, která může být použita pro
další měření.
Důležitou informací je, že teplotní režim lze měnit v průběhu měření!
Adjust TG
→
Adjust TG slouží k tárování (vynulování) váhy před samotným měřením.
Po označení Adjust je nutno vyčkat, až se ručička ustálí kolem 0 a bude svítit
zeleně kontrolka v pravé horní části.
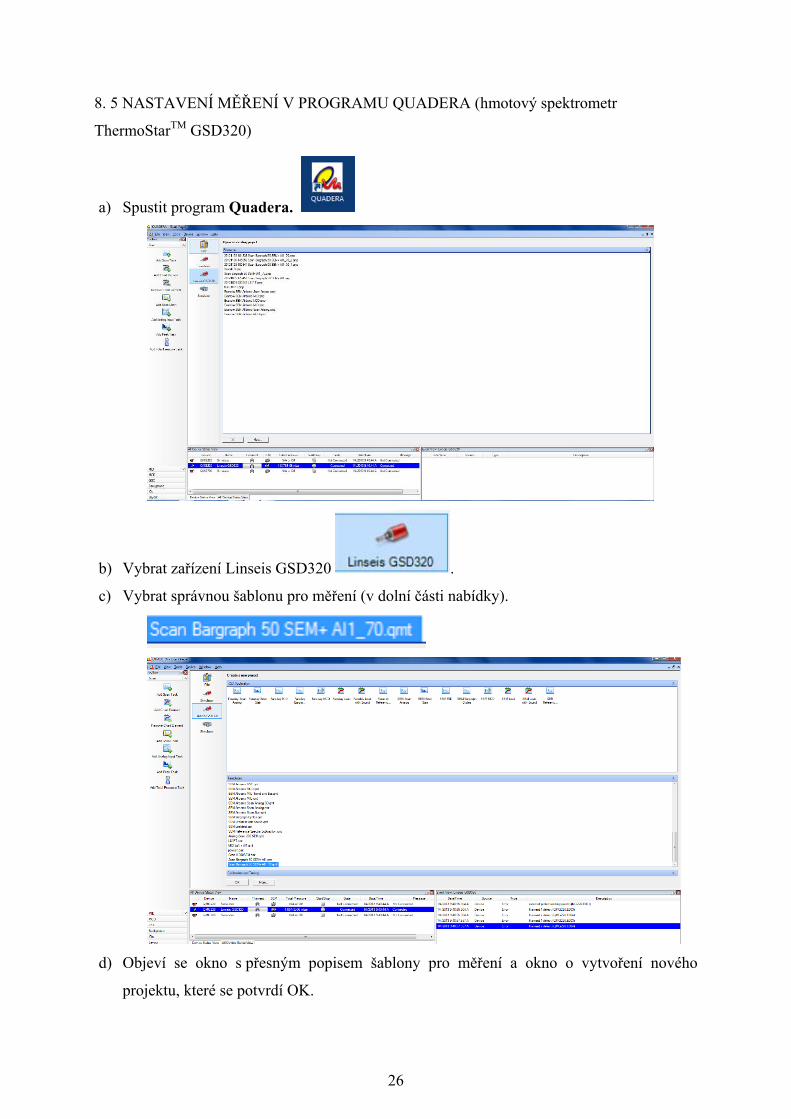
26
8. 5 NASTAVENÍ MĚŘENÍ V PROGRAMU QUADERA (hmotový spektrometr
ThermoStarTM GSD320)
a) Spustit program Quadera.
b) Vybrat zařízení Linseis GSD320 .
c) Vybrat správnou šablonu pro měření (v dolní části nabídky).
d) Objeví se okno s přesným popisem šablony pro měření a okno o vytvoření nového
projektu, které se potvrdí OK.
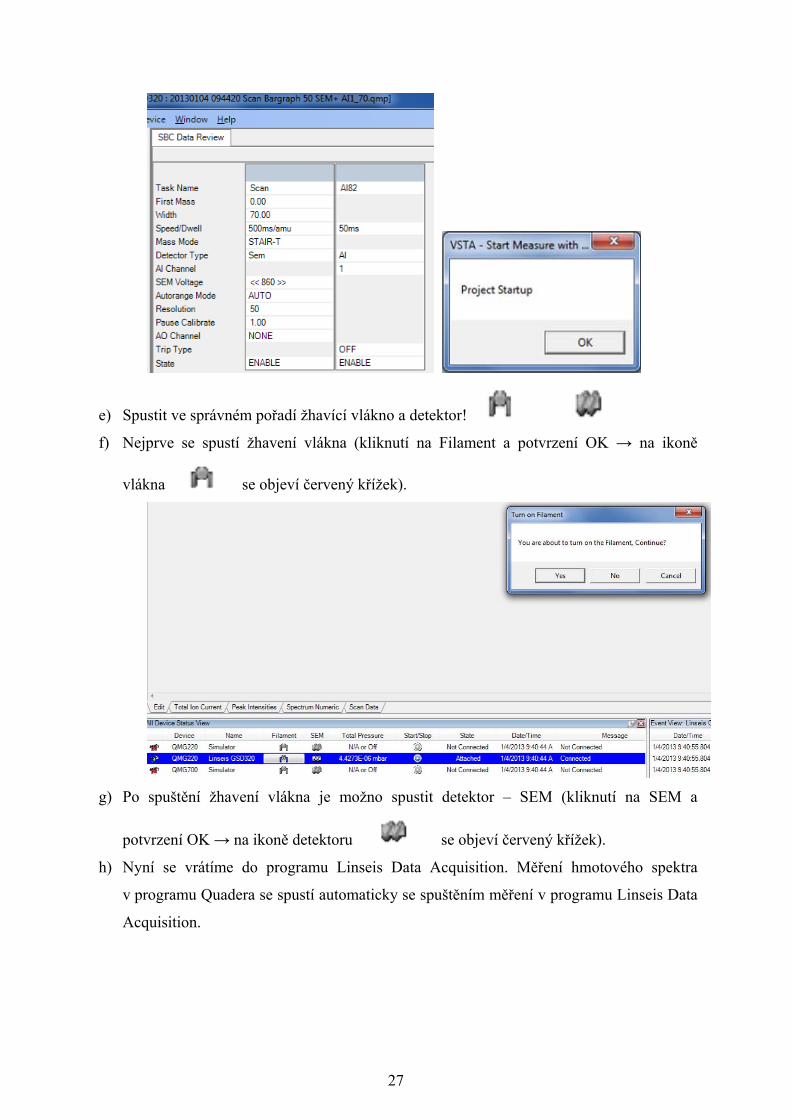
27
e) Spustit ve správném pořadí žhavící vlákno a detektor!
f) Nejprve se spustí žhavení vlákna (kliknutí na Filament a potvrzení OK → na ikoně
vlákna se objeví červený křížek).
g) Po spuštění žhavení vlákna je možno spustit detektor – SEM (kliknutí na SEM a
potvrzení OK → na ikoně detektoru se objeví červený křížek).
h) Nyní se vrátíme do programu Linseis Data Acquisition. Měření hmotového spektra
v programu Quadera se spustí automaticky se spuštěním měření v programu Linseis Data
Acquisition.
28
8.6 SPUSTIT MĚŘENÍ V PROGRAMU Linseis Data Acquisition
→
a) Po správném nastavení měření je možno jej spustit pomocí ikony .
8.7 ÚKONY PO UKONČENÍ MĚŘENÍ
a) Program měření se automaticky ukončí dle nastaveného teplotního profilu.
b) V programu Quadera je nutno vypnout detektor a žhavení vlákna ve správném pořadí,
nejprve detektor a následně žhavení vlákna!!!
c) V programu Quadera je nutno uložit naměřená hmotová spektra!
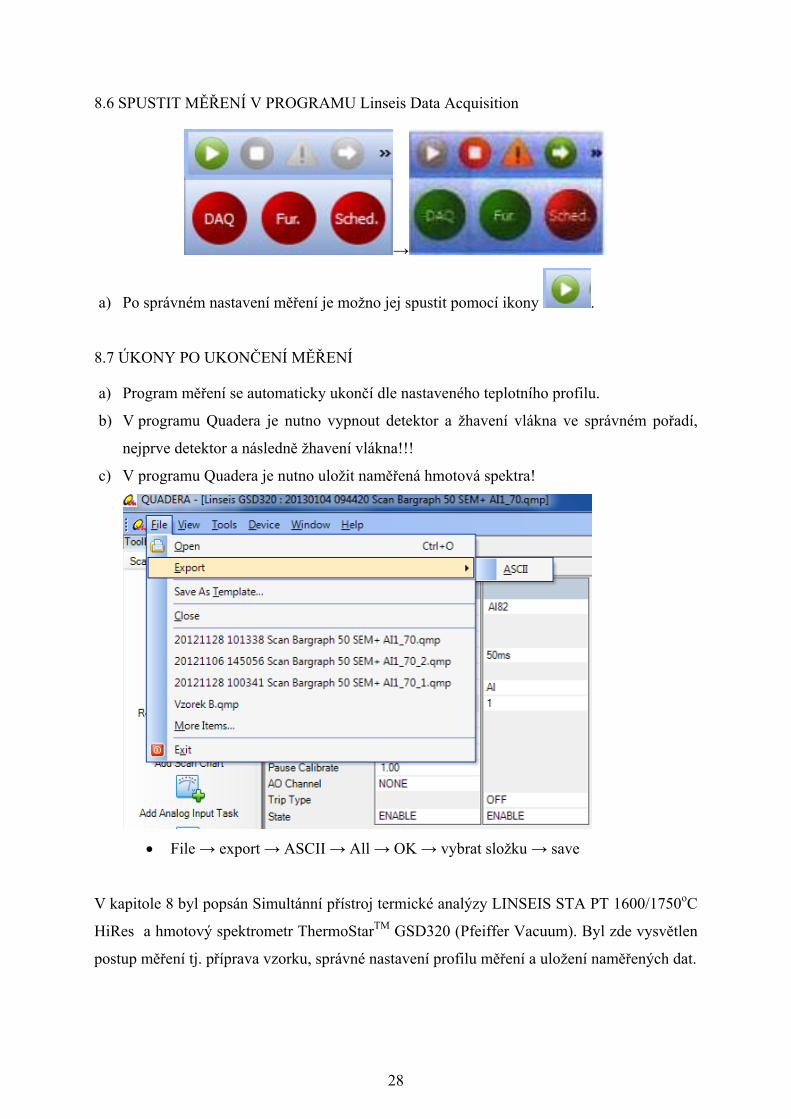
File → export → ASCII → All → OK → vybrat složku → save
V kapitole 8 byl popsán Simultánní přístroj termické analýzy LINSEIS STA PT 1600/1750oC
HiRes a hmotový spektrometr ThermoStarTM GSD320 (Pfeiffer Vacuum). Byl zde vysvětlen
postup měření tj. příprava vzorku, správné nastavení profilu měření a uložení naměřených dat.

29
9. VYHODNOCENÍ STA KŘIVEK PŘÍSLUŠNÝM PROGRAMEM
Program Linseis Evaluation slouží ke zpracování naměřených dat získaných pomocí
zařízení Linseis STA PT 1600/1750 °C HiRes a hmotového spektrometru ThermoStarTM
GSD320.
Program obsahuje hlavní lištu s menu, pod kterou jsou ikony pro vlastní práci
s naměřenými křivkami. V levé části okna je plocha (Element tree), kde je možno po
rozbalení názvu new project zjistit informace a označení jednotlivých načtených křivek. Níže
jsou detailně ukázány jednotlivé části horní ovládací lišty menu, které jsou podrobněji
vysvětleny v kapitolách 9.1-9.4.
Obr. 17: Základní pracovní plocha v programu Linseis Evaluation

30
9. 1 ZÁKLADNÍ PRÁCE S KŘIVKAMI
Otevření nového okna:
Pro zpracování nových dat je nutno vytvořit nové pracovní okno.
a) File → new project
Načtení naměřených dat vybraného měření:
V tomto programu není načtení všech křivek daného měření automatické, je nutno všechny
požadované křivky vybrat ze souboru naměřených dat.
a) File → load curve → vybrat požadovanou křivku (křivky TG a DCS/DTA jsou
zvlášť) → OK
b) ikona
Práce s křivkou:
Pracovat s křivkou lze pouze za předpokladu, že je označena!
Tj. kliknutím na vybranou křivku se na jejich koncích vytvoří malé čtverečky.
Informace o křivce:
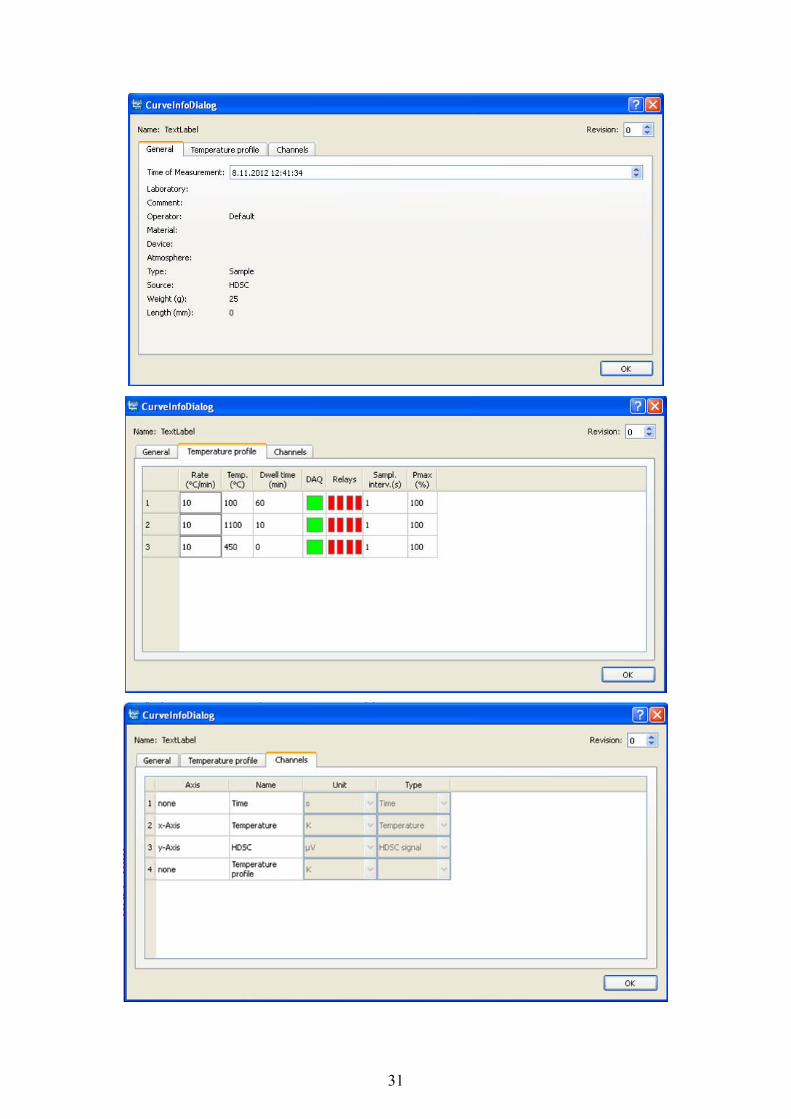
Mezi základní informace o křivce patří datum a čas měření, měřící zařízení, navážka vzorku
(v mg), teplotní profil (tj. temperance, ohřev, chlazení – rychlost ohřevu, zvolená teplota
jednotlivých částí měření, výdrž na zvolené teplotě) a označení os.
a) pravé tlačítko myši → infos
b) ikona
31
32
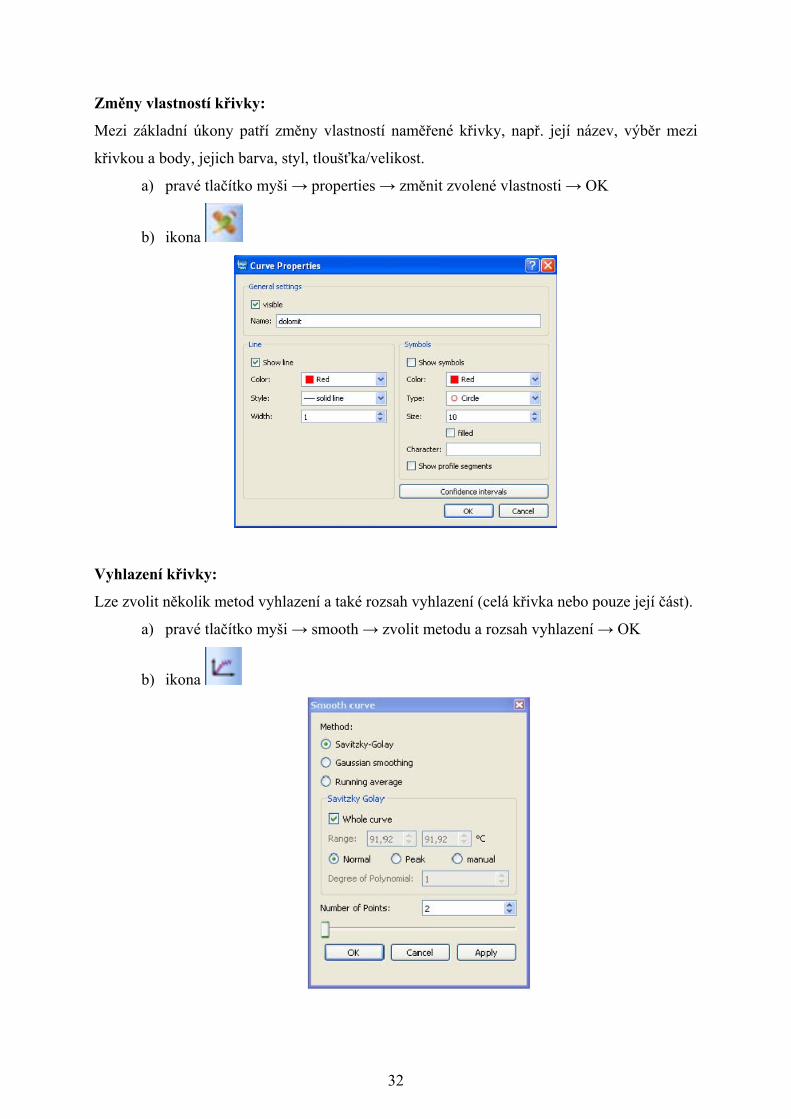
Změny vlastností křivky:
Mezi základní úkony patří změny vlastností naměřené křivky, např. její název, výběr mezi
křivkou a body, jejich barva, styl, tloušťka/velikost.
a) pravé tlačítko myši → properties → změnit zvolené vlastnosti → OK
b) ikona
Vyhlazení křivky:
Lze zvolit několik metod vyhlazení a také rozsah vyhlazení (celá křivka nebo pouze její část).
a) pravé tlačítko myši → smooth → zvolit metodu a rozsah vyhlazení → OK
b) ikona
33
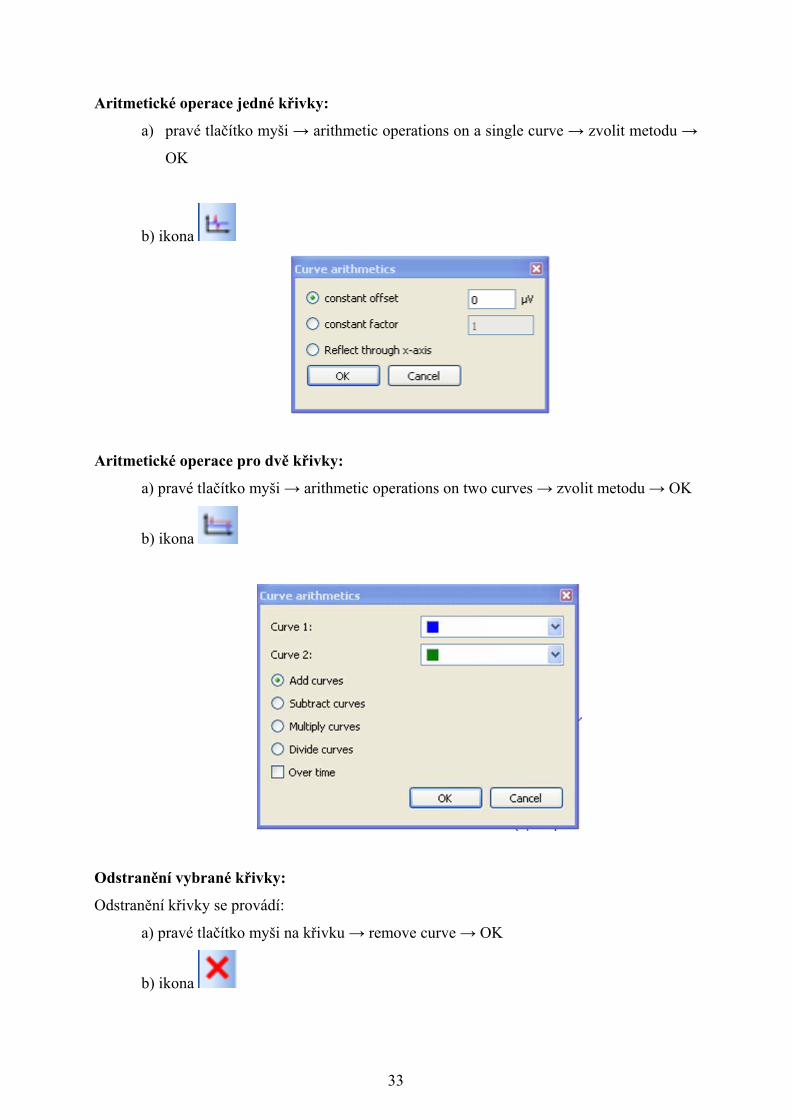
Aritmetické operace jedné křivky:
a) pravé tlačítko myši → arithmetic operations on a single curve → zvolit metodu →
OK
b) ikona
Aritmetické operace pro dvě křivky:
a) pravé tlačítko myši → arithmetic operations on two curves → zvolit metodu → OK
b) ikona
Odstranění vybrané křivky:
Odstranění křivky se provádí:
a) pravé tlačítko myši na křivku → remove curve → OK
b) ikona

34
Zobrazení křivek TG a DTA/DSC:
V tomto programu je možno měnit pozice křivek v pracovní ploše. Křivky (TG a DTA/DSC)
je možno umístit přes sebe, nad sebe nebo vedle sebe.
a) ikona (křivky jsou přes sebe), (křivky jsou nad sebou),
(křivky jsou vedle sebe)
Výřez křivky:
Výřez křivky je možno použít v případě, kdy např. při dalším zpracování křivky chceme
pracovat pouze s částí patřící ohřevu, ale při daném měření bylo zaznamenáno např. také
chlazení. To pak ve výsledné naměřené křivce může tvořit jakousi smyčku.
a) pravé tlačítko myši na křivku → cut → vybrat počáteční a koncový bod křivky →
OK
b) ikona
Tečny ke křivce:
Možnost práce s tečnami umožňuje získat informace o bodě, získaném protnutím dvou tečen
(viz následující obrázek).
a) pravé tlačítko myši na křivku → tangnets → vybrat dva body na křivce → OK
b) ikona
35
Vyrovnání píku do roviny
V některých případech, např. pro porovnání píků z několika měření, je vhodné počátek
a konec píku vyrovnat do roviny.
a) pravé tlačítko myši na křivku → rotate → označit dva body → uchopit myší
křivku a rotací křivky vyrovnat počáteční a koncový bod píku do roviny
b) pravé tlačítko myši na křivku → slope correction → označit dva body → uchopit
myší křivku a rotací křivky vyrovnat počáteční a koncový bod píku do roviny
c) ikona nebo
Extrapolace:
a) pravé tlačítko myši na křivku → extrapolation → zvolit teplotu pro extrapolaci →
OK
b) ikona
Změna parametrů vybrané osy:
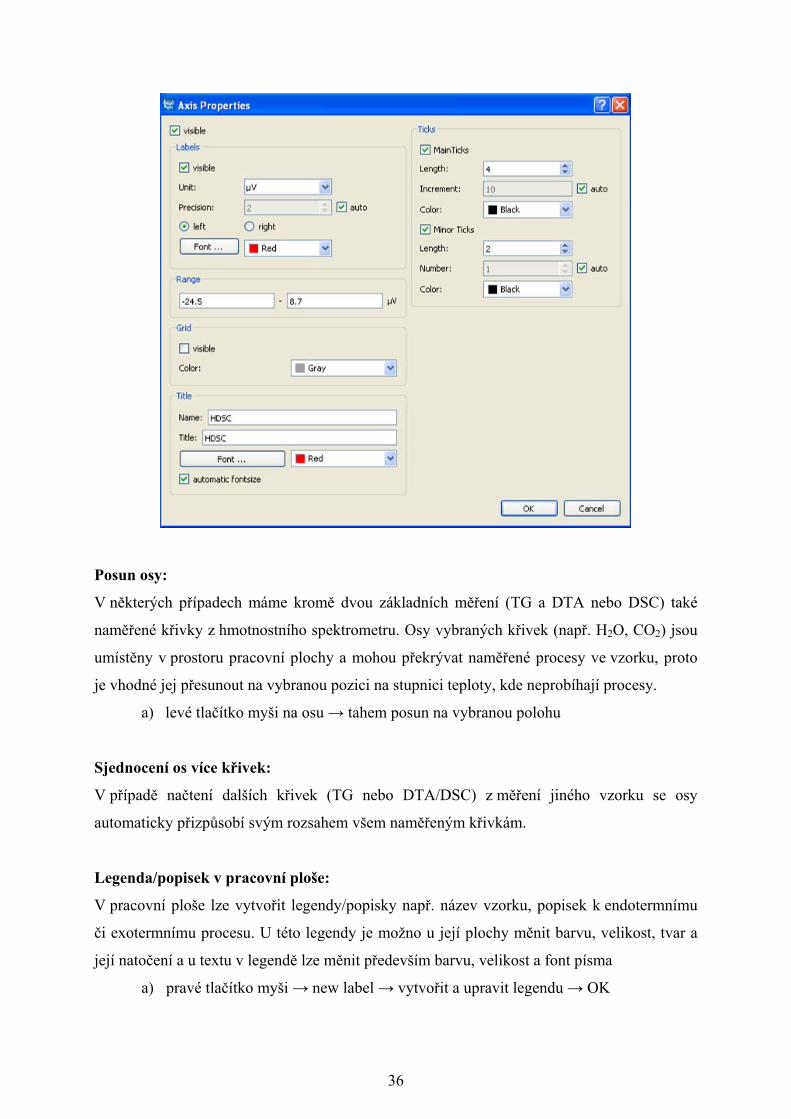
Stejně jako v případě křivek, také u os lze měnit jejich parametry. U os se mění zejména
jednotky dané veličiny V (např. na µV, kV, mV, nV), font písma, barva písma a osy, rozsah
stupnice osy, název osy. Do zpracování křivky lze přidat pomocnou mřížku a měnit její barvu.
Dále lze měnit velikost postranních bodů, hlavní i vedlejší jednotky osy.
a) pravé tlačítko myši na osu → properties → změnit zvolené vlastnosti → OK
36
Posun osy:
V některých případech máme kromě dvou základních měření (TG a DTA nebo DSC) také
naměřené křivky z hmotnostního spektrometru. Osy vybraných křivek (např. H2O, CO2) jsou
umístěny v prostoru pracovní plochy a mohou překrývat naměřené procesy ve vzorku, proto
je vhodné jej přesunout na vybranou pozici na stupnici teploty, kde neprobíhají procesy.
a) levé tlačítko myši na osu → tahem posun na vybranou polohu
Sjednocení os více křivek:
V případě načtení dalších křivek (TG nebo DTA/DSC) z měření jiného vzorku se osy
automaticky přizpůsobí svým rozsahem všem naměřeným křivkám.
Legenda/popisek v pracovní ploše:
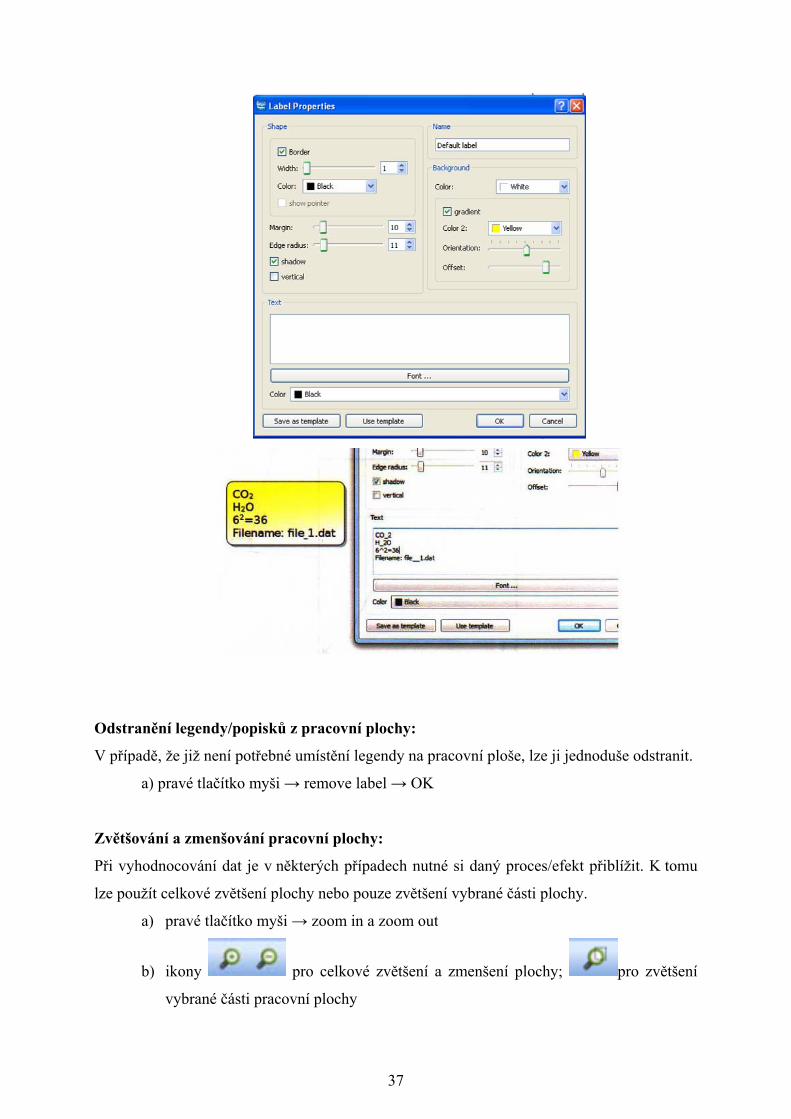
V pracovní ploše lze vytvořit legendy/popisky např. název vzorku, popisek k endotermnímu
či exotermnímu procesu. U této legendy je možno u její plochy měnit barvu, velikost, tvar a
její natočení a u textu v legendě lze měnit především barvu, velikost a font písma
a) pravé tlačítko myši → new label → vytvořit a upravit legendu → OK
37
Odstranění legendy/popisků z pracovní plochy:
V případě, že již není potřebné umístění legendy na pracovní ploše, lze ji jednoduše odstranit.
a) pravé tlačítko myši → remove label → OK
Zvětšování a zmenšování pracovní plochy:
Při vyhodnocování dat je v některých případech nutné si daný proces/efekt přiblížit. K tomu
lze použít celkové zvětšení plochy nebo pouze zvětšení vybrané části plochy.
a) pravé tlačítko myši → zoom in a zoom out
b) ikony pro celkové zvětšení a zmenšení plochy; pro zvětšení
vybrané části pracovní plochy

38
9. 2 ZÁKLADNÍ ZPRACOVÁNÍ DTA/DSC KŘIVKY
Vyhodnocení píku HDSC křivky:
Při vyhodnocení DTA/DSC křivky jsou nejdůležitější informacemi plocha píku a teploty -
maxima píku, bodu reakce, onsetu a offsetu.
a) pravé tlačítko myši na křivku → ikona píku a označení obou bodů na křivce →
OK
b) ikona

39
Derivace HDSC křivky:
K rozpoznání efektů se v některých případech používá také derivace křivky.
a) pravé tlačítko myši → derivate → lze vybrat x-osu (teplota, čas) → OK
b) ikona
9. 3 ZÁKLADNÍ ZPRACOVÁNÍ TG KŘIVKY
Relativní hmotnostní změna:
Relativní hmotnostní změna se použije, pokud chceme osu TG převést na procenta.
a) pravé tlačítko myší označit křivku → relative mass change → OK
b) ikona
Zbytková hmotnost:
Zbytková hmotnost uvádí zbylou hmotnost efektu v mg.
a) pravé tlačítko myší označit křivku → residual mass → OK
b) ikona

40
Hmotnostní změny:
Hmotnostní změna uvádí rozdíl hmotnosti označeného efektu v mg.
a) pravé tlačítko myši označit křivku → mass change → vybrat body na křivce →
OK
b) ikona
Derivace TG křivky:
K rozpoznání efektů se v některých případech používá také derivace křivky.
a) pravé tlačítko myši → derivate → lze vybrat x-osu (teplota, čas, teplotní profil)
b) ikona
9.4 EXPORT VÝSLEDKŮ INTERPRETACE
Export datových souborů křivek:
Export dat je důležitý, za předpokladu, že s naměřenou křivkou budeme dále pracovat v jiném
programu. Po otevření export dat je nutno vybrat, které křivky, s jakými jednotkami chceme
exportovat. Dále je vhodné vybrat správný formát souboru pro následné zpracování (.csv nebo
.xls).
a) pravé tlačítko myši na křivku → export curve data → vybrat křivky a typ formátu
→ OK
41
Export vyhodnocení:
Zpracování křivek lze exportovat ve formě obrázku nebo pdf.
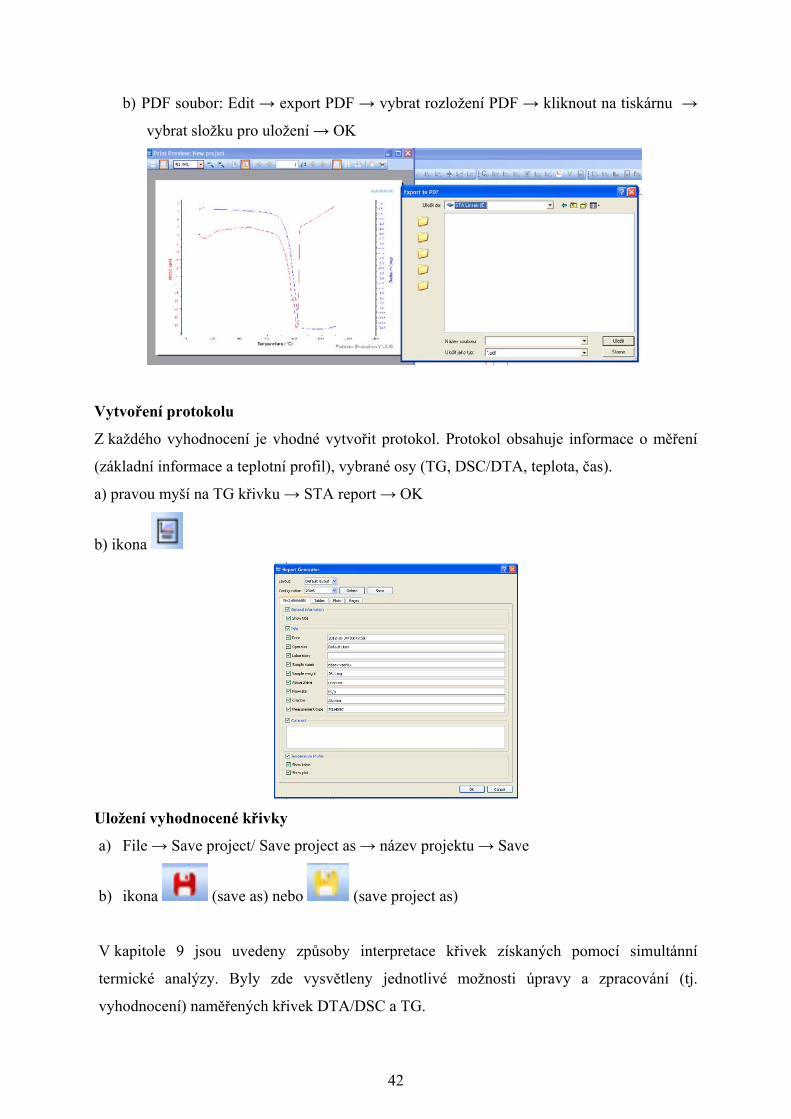
a) Obrázek: Edit → export image → vybrat typ souboru a složku umístění → OK
42
b) PDF soubor: Edit → export PDF → vybrat rozložení PDF → kliknout na tiskárnu →
vybrat složku pro uložení → OK
Vytvoření protokolu
Z každého vyhodnocení je vhodné vytvořit protokol. Protokol obsahuje informace o měření
(základní informace a teplotní profil), vybrané osy (TG, DSC/DTA, teplota, čas).
a) pravou myší na TG křivku → STA report → OK
b) ikona
Uložení vyhodnocené křivky
a) File → Save project/ Save project as → název projektu → Save
b) ikona (save as) nebo (save project as)
V kapitole 9 jsou uvedeny způsoby interpretace křivek získaných pomocí simultánní
termické analýzy. Byly zde vysvětleny jednotlivé možnosti úpravy a zpracování (tj.
vyhodnocení) naměřených křivek DTA/DSC a TG.

43
10. UKÁZKOVÉ PŘÍKLADY
10.1 Studium rozkladu kalcitu
Pomocí přístrojů Linseis STA PT 1600/1750 °C HiRes a hmotového spektrometru
ThermoStarTM GSD320 sledovat rozklad kalcitu v teplotním rozsahu 100-1000 °C
s rychlostí ohřevu 5 a 10 °C/min a navážkou 25 mg v atmosféře argonu. Cílem práce je
vyhodnocení a porovnání křivek DSC a TG pro dvě různé rychlosti ohřevu.
10.2 Studium dehydroxylace kaolinu
Pomocí přístrojů Linseis STA PT 1600/1750 °C HiRes a hmotového spektrometru
ThermoStarTM GSD320 sledovat dehydroxylaci kaolinu v teplotním rozsahu 100-1200 °C
s rychlostí ohřevu 10 °C/min a navážkou 10, 25 a 50 mg v atmosféře argonu. Cílem práce je
vyhodnocení a porovnání křivek DSC a TG (popř. určení počátku dehydroxylace) pro tři
různé navážky vzorku.
10.3 Studium teploty skelného přechodu
Pomocí přístroje Linseis STA PT 1600/1750 °C HiRes sledovat změnu Tg (teplota skelného
přechodu) a pík krystalizace pro sklo (velikost úlomků 100 µm nebo 200 µm) pro 3 rychlosti
ohřevu (10, 20 a 30 °C/min) v rozsahu teplot 100-900 °C a navážkou 50 mg.
10.4 Studium teploty tání glazur
Pomocí přístrojů Linseis STA PT 1600/1750 °C HiRes a hmotového spektrometru
ThermoStarTM GSD320 (Pfeiffer Vacuum) sledovat děje při ohřevu a identifikovat teplotu
tání tří různých typů glazur, rychlost ohřevu 10 °C/min v rozsahu teplot 100-1100 °C a
navážkou 25 mg.
10. 5 Krystalizace tetragonálního leucitu
Přístroj Linseis STA PT 1600/1750 °C HiRes použít k identifikaci teploty krystalizace
z amorfního gelu (prekurzoru) a z amorfního očkovaného prekurzoru pro rychlostí ohřevu 10
°C/min v rozsahu teplot 100-1100 °C a navážkou 25 mg.

44
11. LITERATURA
1. BLAŽEK A. Moderní metody v analytické chemii. Praha: SNTL Praha, 1972, str. 11
2. BROWN M. E. Handbook of Thermal Analysis and Calorimetry. [online], Elsevier
B.V.,1998
3. ŠULCOVÁ P., BENEŠ L. Experimentální metody v anorganické chemii. Pardubice:
Univerzita Pardubice FCHT, 2008, str. 135-198
4. ŠTARHA P., TRÁVNÍČEK Z. Termická analýza. Olomouc: Univerzita Palackého v
Olomouci, 2011
5. PINKAS J., LOSOS, Z. Úloha 8: Termická analýza
http://sci.muni.cz/chemsekce/c8870/pdf/Uloha8_Termanal.pdf (4. 1. 2013)
6. ŠESTÁK J. Měření termofyzikálních vlastností pevných látek. Praha: Academia Praha,
1982, str. 49
7. Charakterizácia DSC kriviek: Študijné materiály. v 1. letná škola termickej analýzy a
kalorimetrie, Bratislava 20. - 22. 9. 2001. Edited by P. Šimon, Š. Svestík, E. Smrčková,
M. Palou
8. Linseis Messgerate GmbH Linseis STA, Termogravimetric Analyzer Instruction Manual.
9. ROSICKÝ J. Termická analýza. Praha, Univerzita Karlova, 1989
10. ZÝKA J. Analytická příručka. Praha: SNTL Praha, 1979
11. SEILEROVÁ L, BRUSOVÁ H., KRATOCHVÍL B., KREJČÍK L. Využití termické
analýzy ve výzkumu a vývoji léčiv, Chem. Listy, 2012, 106, str. 890-895
12. ROWLAND R. A. Differential thermal analysis of clays and carbonates, Clays and clay
technology, 1955, 169, str. 151-163
13. KLOUDA, P. Moderní analytické metody. Ostrava: Pavel Klouda, 2003
45
Laboratorní návody Termická analýza – Bakalářské studium
pro obor Technologie konzervování a restaurování (N150010)
Termická analýza a mikrostruktura nízkopálené
keramiky ÚVOD Laboratorní práce je zaměřena na hodnocení mikrostruktury nízkopálené keramiky, identifikaci její teploty výpalu pomocí simultánní termické analýzy a sledování změn složek keramického střepu se vzrůstající teplotou.
K identifikaci typu mikrostruktury střepové hmoty společně s určením přibližné teploty výpalu nízkopálené keramiky se používá kombinace několika analytických metod, především rentgenové fluorescenční analýzy (XRF), rentgenové difrakční analýzy (XRD), termické analýzy (TA) a optické mikroskopie (OM). Přibližná teplota výpalu historické keramiky (je jeden z častých dotazů archeologů) se určuje na základě přítomnosti minerálů, které se mohou v archeologické keramice vyskytovat.
Nízkopálená střepová hmota je obvykle vypálena pod 1000 °C. Podle použitých vstupních surovin může obsahovat křemen, živce, slídy, uhličitany a jílové minerály (např. kaolinit, illit, montmorillonit) nebo jejich zbytky ve formě nekrystalických reaktivních produktů.
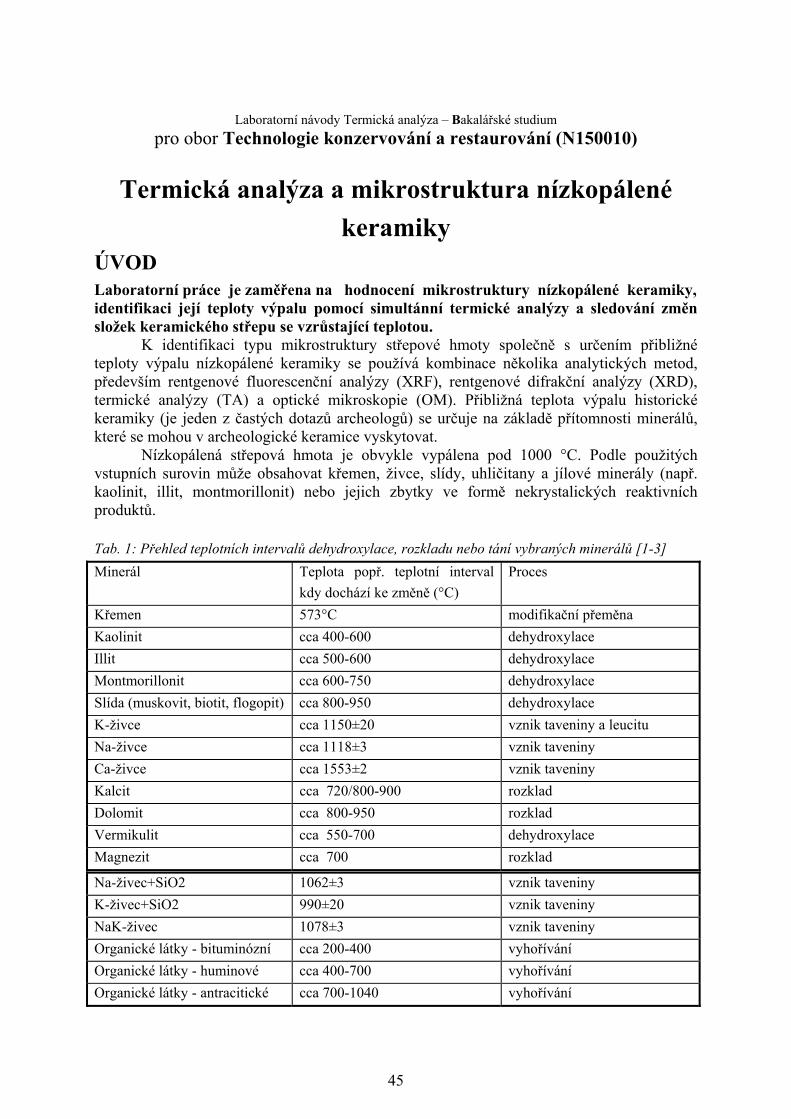
Tab. 1: Přehled teplotních intervalů dehydroxylace, rozkladu nebo tání vybraných minerálů [1-3]
Minerál Teplota popř. teplotní interval
kdy dochází ke změně (°C)
Proces
Křemen 573°C modifikační přeměna
Kaolinit cca 400-600 dehydroxylace
Illit cca 500-600 dehydroxylace
Montmorillonit cca 600-750 dehydroxylace
Slída (muskovit, biotit, flogopit) cca 800-950 dehydroxylace
K-živce cca 1150±20 vznik taveniny a leucitu
Na-živce cca 1118±3 vznik taveniny
Ca-živce cca 1553±2 vznik taveniny
Kalcit cca 720/800-900 rozklad
Dolomit cca 800-950 rozklad
Vermikulit cca 550-700 dehydroxylace
Magnezit cca 700 rozklad
Na-živec+SiO2 1062±3 vznik taveniny
K-živec+SiO2 990±20 vznik taveniny
NaK-živec 1078±3 vznik taveniny
Organické látky - bituminózní cca 200-400 vyhořívání
Organické látky - huminové cca 400-700 vyhořívání
Organické látky - antracitické cca 700-1040 vyhořívání

46
1. Teoretická část 1. 1 Charakterizace keramického střepu [1-2] Keramika je nejstarším kompozitním materiálem, který člověk záměrně vytvořil. V určitých obdobích je pravěká keramika nejčastějším a někdy i jediným dokladem existence lidské aktivity v dané lokalitě. Často bývá používána k dataci archeologických nálezů z jiných materiálů. Obecně je definována jako anorganický, nekovový, ve vodě prakticky nerozpustný pevný materiál obsahující vedle amorfní fáze nejméně 30 % krystalické složky. Keramika získává své konečné vlastnosti až tepelným zpracováním většinou při teplotách nad 800 oC. Výpalu však předcházejí operace jako je příprava surovinové směsi, její vytváření (u historické keramiky při pokojové teplotě), sušení, povrchová úprava atd., které konečné vlastnosti keramiky rovněž ovlivňují. Vlastnosti keramického materiálu jsou většinou složitou funkcí jeho surovinového, chemického a mineralogického složení a procesů, které probíhají při jednotlivých technologických operacích. Obr. 1: Technologické schéma výroby keramiky U keramiky jsou vlastnosti závislé na její konečné struktuře. Základní schéma výroby klasické keramiky, tj. zhotovené z přírodních surovin, je od počátku objevení keramické výroby až po současnost v podstatě stále stejné (viz Obr. 1). K základním přírodním surovinám pro přípravu keramiky patří jíly a jejich směsi doplněné neplastickými surovinami tzv. ostřivy a tavivy. Typ použitých jílů, ostřiv a taviv a jejich obsah v surovinové směsi je volen podle druhu keramiky. Primárním měřítkem je teplota výpalu resp. zda je cílem připravit pórovitý keramický materiál nebo slinutou keramiku (keramika bez pórů nebo s velmi nízkou pórovitostí). Rozdíly mezi plastickými a neplastickými surovinami se výrazně projeví jak při sušení, tak zejména při výpalu. Při výpalu keramického materiálu ovlivňuje neplastická složka směsi mechanické vlastnosti, smrštění výpalem, pórovitost, rychlost slinování a optimální teplotu výpalu střepu. Chování některých neplastických příměsí se změní při výpalu při teplotě, kdy se objeví ve střepu např. eutektická tavenina nebo se začnou tavit živce. Potom působí jako taviva. Při výpalu směsí z přírodních plastických a neplastických surovin do teplot cca 1000 oC jsou relativně nejstabilnějšími krystalickými fázemi křemen a živce. U křemene probíhá pouze reverzibilní modifikační přeměna křemen při teplotě 573 oC, která způsobuje nad touto teplotou zvětšení jeho zrn. Modifikační přeměny SiO2 lze znázornit následujícím schématem:
573 oC 1050 oC 1726 oC β-křemen -křemen → -cristobalit křemenné sklo . (2) 180 oC 270 oC
β-cristobalit Pokud SiO2 obsahuje příměsi, může vznikat z - křemene i - tridymit, a to při cca 870 °C (podle Ostwaldovova pravidla popř. i přes - cristobalit), který při ochlazení přechází na β- a pak na γ- tridymit. Zrna živců jsou stabilní až do počátku svého tavení. Ve směsích se často taví při eutektických teplotách, tedy při teplotách podstatně nižších než by odpovídalo jejich teplotám tání, což je princip slinování pórovité keramiky s teplotou výpalu do 1000 oC.

47
Jílové minerály však stabilní nejsou. V intervalu do cca 300 oC nejdříve uvolňují fyzikálně vázanou vodu a potom v relativně širokém teplotním intervalu 450 až 800 oC chemicky vázanou vodu a zásadně mění svoji krystalickou strukturu. Uvolňování chemicky vázané vody tzv. dehydroxylace dvojvrstvého jílového minerálu kaolinitu začíná od teploty cca 450 oC. Je to endotermní reakce a probíhá podle následující rovnice: 450- 600 oC Al2O3 2SiO2 2H2O Al2O3 2SiO2 + 2H2O . (3) Intenzivně probíhá až do teploty cca 600 oC. Ze stabilního krystalického minerálu kaolinitu vzniká nestabilní dehydroxylovaná forma prakticky nekrystalického metakaolinitu. Při této reakci se také uvolňuje z mřížky kaolinitu cca 14 hmotn. % vody, která byla původně vázána v jeho struktuře chemickou vazbou ve formě OH- skupin a dochází ke smrštění vypalovaného tělesa. Teprve nad teplotou cca 1000 oC se metastabilní metakaolinit postupně přeměňuje na stabilní krystalické fáze. Vzniká mullit a při teplotách nad 1000 oC z uvolněného nekrystalického oxidu křemičitého ještě cristobalit (vysokoteplotní modifikace SiO2): 950-1050 oC 2 ( Al2O3. 2SiO2) → 2Al2O3. 3SiO2 defektní spinel + SiO2 amorfní, (4) cca 1100 oC 3 ( 2Al2O3.3SiO2) → 2 (3Al2O3. 2SiO2) mullit + 5SiO2 amorfní, (5) nad 1200 oC amorfní SiO2 → SiO2 cristoballit. (6) Podobně probíhá dehydroxylace trojvrstvých jílových minerálů illitu a montmorillonitu. Uvolnění chemicky vázané vody v illitu probíhá v teplotním intervalu 350 - 600 oC. Vznik mullitu může výrazně ovlivnit vyšší obsah některých reaktivních oxidů v illitu jako např. CaO, SiO2 či MgO nebo FeO. Vzniká také určitý podíl taveniny, která potom tvoří skelnou fázi. V teplotním intervalu cca 780 až 820 oC dochází k dehydroxylaci slíd a k rozkladu vápence: cca 800 oC CaCO3→ CaO+CO2 . (7) Pokud je uvolněný oxid vápenatý CaO obklopen produkty rozkladu – metakaolinitem, reaguje s ním na novou nestabilní krystalickou sloučeninu gehlenit (760-900 oC) a pak (850-950 oC) na stabilní krystalickou fázi anortit (CaO.Al2O3.2SiO2). Po výpalu keramiky z přírodních surovin do teplot cca 1000 oC tedy střep obsahuje značný podíl nekrystalické fáze, krystalickou fázi a póry. Krystalickou fázi tvoří zrna křemene případně živce, pseudomorfózy po destičkách slídy a za příznivého chemického složení výchozí směsi např. také krystaly anortitu (CaO.Al2O3.2SiO2) a případně další. Teprve po výpalu střepu na teploty vyšší než je 1000 oC vznikají ve střepu vysoce stabilní minerály jako je mullit (3Al2O3.2SiO2), cristobalit (SiO2) atd. Na Obr. 2 je uveden snímek výbrusu nízkopálené keramiky z doby bronzové s ostřivem ve formě křemičitého písku, na Obr. 3 je ostřivo ve formě keramických střepů. Základními keramickými tavivy jsou živce, alkalické hlinitokřemičitany, které se do teploty tání chovají jako ostřivo, viz Obr. 4-5.
Obr. 2: Písčité zrno, snímek výbrusu keramiky doby Obr. 3: Keramické ostřivo, snímek výbrusu keramiky

48
Obr. 4: Uprostřed snímku výbrusu z úlomku středověké dlaždice z Rokytné je zrno K- živce ortoklasu, zkřížené nikoly
Obr. 5: Uprostřed snímku výbrusu z keramiky doby bronzové je zrno plagioklasu s typickými lamelami, zkřížené nikoly
1. 2 Analytické metody Rentgenová fluorescenční analýza (XRF)
Rentgenová fluorescenční analýza se využívá k určení chemického složení analyzovaného vzorku. Tato metoda je určena pro měření práškových či kompaktních vzorků s cílem kvalitativního a kvantitativního stanovení prvků. Metoda XRF je založena na měření a vyhodnocování sekundárního záření, které je vybuzeno dopadem primárního rentgenového záření z povrchové vrstvy vzorku. Intenzita vznikajícího fluorescenčního záření je analyzována vhodným detektorem a zpracována do RTG emisního spektra, kde jsou uvedeny charakteristické čáry každého prvku obsaženého ve vzorku. Na základě poloh (energií) jednotlivých emisních čar je možné určit o jaký prvek se jedná a podle intenzit také stanovit kvantitativní zastoupení jednotlivých prvků. Přesnost detekce této metody se pohybuje v rozmezí 0,0X - 0,X %. [4-8]
Rentgenová difrakční analýza (XRD)
XRD je jednou z možných metod používaných pro určení mineralogického složení střepové hmoty. Tato metoda je univerzální, nedestruktivní pro kvalitativní a kvantitativní analýzu různých krystalických fází přítomných v práškových a pevných vzorcích. Identifikace je založena na porovnání difraktogramů neznámých vzorků s databází standardů. Zdrojem rentgenového záření bývá rentgenová anoda. Po dopadu monochromatického záření o vlnové délce λ na vzorek dojde k rozptylu (difrakci) záření na atomech krystalu. Paprsky rentgenového záření „odrážené“ na (pomyšlených) mřížkových rovinách interferují v místě detektoru. Pokud je splněna Braggova podmínka (8), vlny rentgenového záření mají shodnou fázi a dojde tedy ke konstruktivní interferenci, tj. k zesílení intenzity rozptýleného záření v určitých směrech a tím ke vzniku jednotlivých píků („reflexů“). Závislost intenzity rozptýleného záření v závislosti na dvojnásobku difrakčního úhlu (ten je dán polohou detektoru) se nazývá difraktogram.[5, 9-12] 2d.sinΘ = nλ (8) kde: d je mezirovinná vzdálenost (v Å), Θ je difrakční úhel (theta), n je řád difrakce (tj. celé číslo vyjadřující difrakci na mřížkových rovinách odpovídajícími vícenásobkům nejmenších mezirovninných vzdáleností), λ je vlnová délka rentgenového záření (v Å). Na Obr. 6 je ukázka difraktogramů pravěké keramiky. Obr. 6: Posunuté difraktogramy střepových hmot zásobnic 2240-
bronzové z Roztok u Prahy, zkřížené nikoly doby bronzové z Roztok u Prahy

49
1, 1201, urny 2516-1 a misky 2240-2 a misky 2516-2 (resp. urnového dna), vpravo nahoře detail v úseku v polohách 26-29 °2Ө, kde se nacházejí hlavní píky křemene a živců Termické analýzy (TA)
Termín termická analýza zahrnuje obecně experimentální metody, při nichž jsou analyzovány změny složení a vlastností studovaného systému při tepelném zatížení.
Termickými analýzami se sledují fyzikální a chemické procesy v látkách a systémech v závislosti na změně teploty. Při zahřívání vzorku může ve vzorku docházet k fyzikálním změnám (např. modifikační přeměně, sublimaci, absorpci, adsorpci, desorpci, krystalizaci) či chemickým změnám (např. chemisorpci, dehydroxylaci, reakci v pevných látkách, reakciv tavenině, reakci v plynné fázi). Mezi termické analýzy patří několik metod, mezi nejpoužívanější patří DTA nebo DSC a TG. DTA je označení pro Diferenční termickou analýzu, kterou se sledují tepelně zabarvené procesy. Metoda je založena na teplotním rozdílu mezi měřeným vzorkem a srovnávacím vzorkem. Na výsledné DTA křivce se zobrazuje v místech exotermického děje maximum a v místech endotermického minimum. DSC je označení pro Diferenční kompenzační kalorimetrii, která může být s kompenzací příkonu nebo s tepelným tokem. V případě DSC s kompenzací příkonu se měří elektrický příkon a u DSC s tepelným tokem se měří rozdíl teplot mezi vzorkem a srovnávacím materiálem. V druhém případě mají píky efektů stejnou orientaci jako u DTA, v prvém případě opačnou. TG je označení pro Termogravimetrickou analýzu. Analýzou se měří změna hmotnosti v závislosti na teplotě. Ze získané křivky lze určit interval teplot, kdy dochází ke změně hmotnosti důsledkem různých procesů ve vzorku (dehydroxylace, tepelný rozklad, oxidace aj.).[4, 13-18]
Současné měření více vlastností v průběhu jednoho experimentu provádí tzv. simultánní termická analýza (STA) a popisuje se zkratkou použitých metod např. TG/DTA, TG/DTA EGA-MS, TG/EGA-IR (MS-hmotnostní spektrometrie, IR- infračervená spektroskopie).
Tabulka II. Základní metody termické analýzy [4, 13-18] Metoda Sledovaná vlastnost Označení Termogravimetrická analýza hmotnost - měří se hmotnost vzorku v závislosti na teplotě TG Diferenční termická analýza rozdíl teplot - měří se rozdíl teplot mezi vzorkem a
standardem při ohřevu nebo při chladnutí DTA
Diferenční skenovací kalorimetrie
entalpie a tepelný tok - měří se tepelný tok dodávaný do referentní látky, tak aby teplotní rozdíl mezi vzorkem a standardem byl nulový
DSC
Termomechanická analýza rozměr a mechanické vlastnosti - sleduje se deformace vzorku při zatížení v závislosti na teplotě
TMA
Termooptometrie optické vlastnosti - měří se např. celkové světlo nebo světlo určité vlnové délky, luminiscence
TOA
Termoakustická analýza akustické vlastnosti - sledují se charakteristiky zvukových vln po jejich průchodu vzorkem
-
Optická mikroskopie (OM)
Optická mikroskopie je metoda používaná k získání informací o mikrostruktuře střepové hmoty. Z keramického materiálu se připraví velmi tenký výbrus (o tloušťce 20 – 30

50
μm) pro pozorování v optickém mikroskopu v procházejícím polarizovaném světle v paralelních nikolech (sleduje se štěpnost, reliéf, tvar, barva a pleochroismus minerálu) nebo ve zkřížených nikolech (sleduje se zhášení a interferenční barvy). Hodnocením výbrusů lze získat informace o orientaci textury nebo mikrostruktury ve střepu, tj. upořádání zrn ostřiva, destiček slídy a pórů. Na základě charakteru mikrostruktury lze posoudit způsob zpracování surovin a tvarování. Dále lze zjistit mineralogické složení keramického střepu a stanovit velikost jednotlivých minerálů. Lze sledovat homogenitu střepu na základě granulometrického složení krystalických fází. [19-24]
1. 3 Přístroje [25] Rentgenová fluorescenční analýza:
Pro tuto analýzu se na VŠCHT používá sekvenční vlnově disperzní spektrometr ARL 9400 XP+. Tímto přístrojem lze analyzovat kompaktní materiály, práškové vzorky (měření probíhá ve vakuu) a kapalné vzorky (měření probíhá v heliu). Přístroj je vybaven rhodiovou rentgenovou anodou, plně automatickým goniometrem a dvěmi detektory (průtokového proporcionálního a scintilačního). Toto vybavení spektrometru dovoluje analyzovat prvky od B po U, přičemž ve vzorku nelze stanovit obsah C a N.
Rentgenová difrakční analýza: Pro tuto analýzu se na VŠCHT používá práškový difraktometr X'Pert PRO v Bragg-Brentanově parafokusující geometrii s použitím vlnové délky CuKα záření (λ = 1.5418 Å,
U = 40 kV, I = 30 mA). Data jsou neskenována pomocí ultrarychlého detektoru X'Celerator (nebo scintilačního detektoru vybaveného sekundárním zakřiveným monochromátorem) v úhlovém rozsahu 5-60° (2θ) s krokem měření 0.02° (2θ) a časem čítaní 0.3 s krok-1. Termická analýza:
K hodnocení fyzikálních a chemických procesů ve vzorcích se používá přístroj simultánní termické analýzy Linseis STA PT 1750-MS s modulem DTA-TG nebo DSC-TG od firmy Linseis (Německo) s připojeným hmotnostním spektrometrem ThermoStar od firmy Pfeiffer Vacuum. K naprogramování měření se používá program Linseis Platinum Acquisition a následně k vyhodnocení program Linseis Platinum Evaluation. K dispozici bude v laboratoři podrobná příručka pro obsluhu a následné vyhodnocení naměřených dat.
Optická mikroskopie: K hodnocení výbrusů střepové hmoty se používá optický mikroskop Olympus BX 60
se zvětšením objektivu 4, 10, 20 a 40x a zvětšením okuláru 10x. Fotografie výbrusů při různých zvětšeních se pořizují fotoaparátem Olympus E-520. Fotografie se zpracovávají počítačovým programem QuickPHOTO Industrial.
1. 4 Charakteristika křivek: Výsledky termických analýz znázorňují příslušné křivky, viz obr. 7.
Obr. 7: Křivky termických analýz: vlevo křivka diferenční termické analýzy (DTA), uprostřed křivka diferenční skenovací kalorimetrie (DSC), vpravo křivka termogravimetrické analýzy, převzato z [18]
51
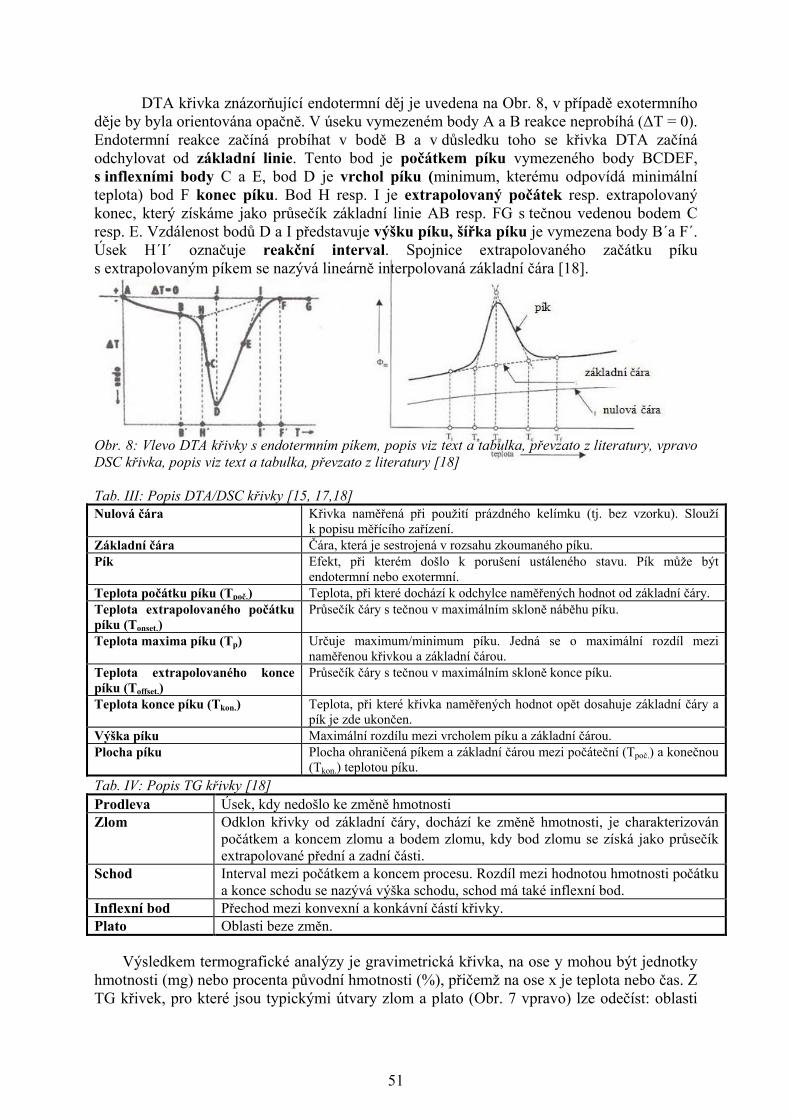
DTA křivka znázorňující endotermní děj je uvedena na Obr. 8, v případě exotermního děje by byla orientována opačně. V úseku vymezeném body A a B reakce neprobíhá (ΔT = 0). Endotermní reakce začíná probíhat v bodě B a v důsledku toho se křivka DTA začíná odchylovat od základní linie. Tento bod je počátkem píku vymezeného body BCDEF, s inflexními body C a E, bod D je vrchol píku (minimum, kterému odpovídá minimální teplota) bod F konec píku. Bod H resp. I je extrapolovaný počátek resp. extrapolovaný konec, který získáme jako průsečík základní linie AB resp. FG s tečnou vedenou bodem C resp. E. Vzdálenost bodů D a I představuje výšku píku, šířka píku je vymezena body B´a F´. Úsek H´I´ označuje reakční interval. Spojnice extrapolovaného začátku píku s extrapolovaným píkem se nazývá lineárně interpolovaná základní čára [18]. Obr. 8: Vlevo DTA křivky s endotermním píkem, popis viz text a tabulka, převzato z literatury, vpravo DSC křivka, popis viz text a tabulka, převzato z literatury [18] Tab. III: Popis DTA/DSC křivky [15, 17,18] Nulová čára Křivka naměřená při použití prázdného kelímku (tj. bez vzorku). Slouží
k popisu měřícího zařízení. Základní čára Čára, která je sestrojená v rozsahu zkoumaného píku. Pík Efekt, při kterém došlo k porušení ustáleného stavu. Pík může být
endotermní nebo exotermní. Teplota počátku píku (Tpoč.) Teplota, při které dochází k odchylce naměřených hodnot od základní čáry. Teplota extrapolovaného počátku píku (Tonset.)
Průsečík čáry s tečnou v maximálním skloně náběhu píku.
Teplota maxima píku (Tp) Určuje maximum/minimum píku. Jedná se o maximální rozdíl mezi naměřenou křivkou a základní čárou.
Teplota extrapolovaného konce píku (Toffset.)
Průsečík čáry s tečnou v maximálním skloně konce píku.
Teplota konce píku (Tkon.) Teplota, při které křivka naměřených hodnot opět dosahuje základní čáry a pík je zde ukončen.
Výška píku Maximální rozdílu mezi vrcholem píku a základní čárou. Plocha píku Plocha ohraničená píkem a základní čárou mezi počáteční (Tpoč.) a konečnou
(Tkon.) teplotou píku.
Tab. IV: Popis TG křivky [18] Prodleva Úsek, kdy nedošlo ke změně hmotnosti Zlom Odklon křivky od základní čáry, dochází ke změně hmotnosti, je charakterizován
počátkem a koncem zlomu a bodem zlomu, kdy bod zlomu se získá jako průsečík extrapolované přední a zadní části.
Schod Interval mezi počátkem a koncem procesu. Rozdíl mezi hodnotou hmotnosti počátku a konce schodu se nazývá výška schodu, schod má také inflexní bod.
Inflexní bod Přechod mezi konvexní a konkávní částí křivky. Plato Oblasti beze změn.
Výsledkem termografické analýzy je gravimetrická křivka, na ose y mohou být jednotky hmotnosti (mg) nebo procenta původní hmotnosti (%), přičemž na ose x je teplota nebo čas. Z TG křivek, pro které jsou typickými útvary zlom a plato (Obr. 7 vpravo) lze odečíst: oblasti

52
beze změn - plato (oblast termické stability), oblasti se změnami hmotnosti (úbytek nebo nárůst), dílčí hmotnostní úbytek, celkový hmotnostní úbytek. Jednotlivé termíny jsou uvedeny v Obr. 9. Na TG křivce se mohou vyskytovat "úseky", které nesouvisí s měřeným vzorkem např. vliv klesající hustoty pecní atmosféry se stoupající teplotou nebo vliv rychlosti průtoku plynu atd. Výrazná deformace křivek je způsobena rozdílem teploty vzorku od teplotního
programu, křivka pak ztrácí lineární charakter a vede k deformaci křivek jednotlivých metod. Obr. 9: Porovnání křivek DTG a TG, na TG křivce je popsán zlom, BCD - schod, B-počátek schodu, C- inflexní bod, D- konec schodu, Bi je počáteční teplota, Di je konečná teplota, BiDi reakční interval, FG - výška schodu, AB - přední základní čára, DE- zadní základní čára, podle literatury [5]
2. Praktická část Cílem práce je seznámit se s jednotlivými metodami pro hodnocení nízkopálené
keramiky, tj. s jejich analyzováním, zpracováním a interpretací. Poté získané poznatky aplikovat při identifikaci teploty výpalu nízkopálené střepové hmoty a popsání procesů , které probíhají při ohřevu střepové hmoty.
Cílem praktické práce bude identifikace teploty výpalu dvou neznámých vzorků na základě zpracování dat získaných výše uvedenými analýzami.
2. 1 Postup práce: 1) Zpracovat data chemického složení na základě protokolu z XRF analýzy pro příslušný
vzorek.
2) Vyhodnotit data z XRD analýzy (tj. difraktogramy) dvou neznámých vzorků
programem X´PertHighScore Plus a příslušné databáze.
3) Vyhodnotit pomocí programu Linseis Platinum Evaluation a Příručky naměřené
křivky DTA/DSC a TG pro dva neznámé vzorky.
4) Zhodnotit pomocí optického mikroskopu mikrostrukturu výbrusů dvou vzorků.
Literatura: Tab. 1:
1. HANYKÝŘ, V., KUTZENDÖRFER, J. Technologie keramiky, Praha: Silikátový svaz, Praha, 2008. ISBN 978-80-86821-48-1.

53
2. HANYKÝŘ, V., KLOUŽKOVÁ, A., BOUŠKA, P., VOKÁČ, M. Stárnutí pórovitého keramického střepu. In Sborník Objemové změny pórovité keramiky: Hevlín. Praha: Silikátový svaz, 2009, p. 33 – 43, ISBN 978-80-86821-54-2.
3. AKYUZ, S., AKYUZ, T., BASARAN, S., BOLCAN, C., GULEC, A. Analysis of ancient pottery using FT-IR, micro-Raman and EDXRF spektrometry. Vibrational Spectroscopy. 2008, Vol. 48, Issue 2, p. 276-280.
4. KLOUDA, P. Moderní analytické metody. Ostrava: Pavel Klouda, 2003, ISBN 80-86369-07-2.
5. NOVOTNÁ, M., KARHAN, J., PECHOVÁ, D. Metody instrumentální analýzy při průzkumu památek, 1st ed.; Praha: Společnost pro technologie ochrany památek, Praha, 2001, ISBN 80-902668-7-8.
6. ČECHÁK, T., HLOŽEK, M., MUSÍLEK, L., TROJEK, T. X-ray fluorescence as a tool for investigating archaeological finds. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 2007, Vol 580, Issue 1, p. 717-720.
7. AL-MEREY, R., KARAJOU, J., ISSA, H. X-ray fluorescence analysis of geological samples: exploring the effect of sample thickness on the accuracy of results. Applied Radiation and Isotopes. 2005, Vol 62, Issue 3, p. 501-508.
8. ČECHÁK, T., HLOŽEK, M., MUSÍLEK, L., TROJEK, T. X-ray fluorescence in investigation of archaeological finds. Nuclear Instruments and Methods in Physics Research B 2007, 263, p. 54-57.
9. Analytické metody v mineralogii. http://geologie.vsb.cz/malis/Analytické%20metody.pdf (online 10.10.2012).
10. STANJEK, H., HÄUSLER, W. Basics of X-ray Diffraction. Hyperfine Interactions, 2004, Vol. 154, 2004, p. 107-119.
11. KOCKELMANN, W., PANTOS, E., KIRFEL, A. Neutron and synchrotron radiation studies of archaeological objects. Radiation in Art and Archaeometry. Elsevier Science B.V., 2000.
12. MOROPOULOU, A., BAKOLAS, A., BISBIKOU, K. Characterization of ancient, byzantine and later historic mortars by thermal and X-ray diffraction techniques. Themochimica Acta, 1995, Vol. 269-270, p. 779-795.
13. WENDLANDT, W.: Thermal analysis techniques: Part I. Thermobalances (continued). J. Chem. Educ. 1972, Vol. 49, No.11, p A623.
14. PAPADOPOULOU, D. N., LALIA-KANTOURI, M., KANTIRANIS, N., STRATIS, J. A., Thermal and mineralogical contribution to the ancienit ceramics and natural clays characterization, Journal of Thermal Analysis and Calorimetry, 2006, Vol. 84, No. 1, 39–45.
15. ŠULCOVÁ, P. BENEŠ, L. Experimentální metody v anorganické technologii, Univerzita Pardubice, Pardubice 2008, ISBN 978-80-7395-058-3.
16. MOROPOULOU, A., BAKOLAS, A., BISBIKOU, K. Thermal analysis as a method of characterizing ancient ceramic technologies. Thermochimica Acta 2570, 1995, Vol. 269-270, p. 743-753.
17. BLAŽEK, A. Termická analýza, SNTL Praha. 1974. 18. BROWN, M. E. Handbook of Thermal Analysis and Calorimetry. [online], Elsevier
B.V., 1998. 19. GREGEROVÁ, M. Petroarcheologie keramiky v historické minulosti Moravy a
Slezska. Brno: Masarykova univerzita, 2010, ISBN 978-80-210-5168-3. 20. ERNEÉ, E., HANYKÝŘ, V., MARYŠKA, M. Výsledky přírodovědných analýz
gotických kamnových kachlů z Českého Krumlova. Památky archeologické. Praha, 2004, vol. XCV.

54
21. REEDY, CH. Thin-section Petrography of Stone and Ceramic Cultural Materials. Archetype Publications Ltd. 2008, ISBN 978-1-904982-33-3.
22. PERKINS, D., HENKE, K. R. Minerals in Thin Section, 2nd edition. Pearson Education, Inc.: New Jersey, 2004, ISBN 0–13–142015–1 .
23. MacKENZIE, W. S., GUIFORD, C. Atlas of Rock–forming Minerals in Thin Sections, Longman Scientific & Technical, 1980, ISBN 978-0470269213.
24. GREGEROVÁ, M., FOJT, B., VÁVRA, V. Mikroskopie horninotvorných a technických minerálů. Moravské zemské muzeum. Brno, 2002, ISBN 80-7028-195-2.
25. http://www.vscht.cz/clab/rtg/index.html (online 10.10.2012).

55
Laboratorní návody Termická analýza – Magisterské studium pro obor Technologie konzervování a restaurování (N148054)
Termická analýza keramických a sklářských surovin
Úvod
Laboratorní práce je zaměřena na identifikaci keramických a sklářských surovin pomocí termické analýzy a sledování změn těchto látek při vzrůstající teplotě.
1. Teoretická část 1. 1 Keramické suroviny [1-2]
Historické keramické materiály (viz Obr. 1) jsou prvním umělým kompozitním materiálem, který člověk vytvořil. Díky vynikající stabilitě vlastností můžeme dnes zkoumat keramiku z různých historických epoch. Keramické materiály jsou obecně definovány jako materiály anorganické, nekovové, ve vodě prakticky nerozpustné a nejméně z 30 % krystalické. Výrobky z těchto materiálů zpravidla vznikají tak, že se při pokojové teplotě z vhodné surovinové směsi vytvarují tělesa, která po vysušení získají své typické materiálové vlastnosti až tepelným zpracováním většinou při teplotách nad 800 oC.
Výroba keramiky probíhá podle technologického schématu, jehož počáteční část je uvedena na Obr. 2. Vlastnosti keramického materiálu jsou většinou složitou funkcí jeho surovinového, chemického a mineralogického složení a procesů, které probíhají při jednotlivých technologických operacích. Vzniklý keramický střep je obecně tvořen krystalickou a nekrystalickou ev. skelnou fází a póry. Uvedená charakteristika střepu platí pro pórovitou keramiku z přírodních surovin. Moderní keramické materiály, které se vyrábějí převážně ze surovin syntetických, často obsahují pouze fáze krystalické a prakticky žádné póry. Obr. 2: Technologické schéma výroby keramiky
Keramika s pórovitým střepem, která reprezentuje i keramiku historickou je vytvářena z přírodních surovin, které se dělí na plastické a neplastické. Základem plastických surovin jsou zeminy- jíly a hlíny. Neplastickými surovinami, které plní funkci ostřiv a taviv, jsou horniny obsahující především křemen a živce. 1. 1. 1 Plastické suroviny
Mezi plastické suroviny patří jíly a hlíny, které obsahují především jílové minerály a to dvojvrstvé (kaolinit, halloysit) nebo trojvrstvé (illit, motmorillonit). Z hlediska struktury patří mezi fylosilikáty. Vzhledem k tomu, že sem patří jednoduché trojvrstvé vrstevnaté minerály, lze k nim zařadit díky své struktuře také slídy (muskovit; biotit; flogopit), mastek a pyrofylit. Mezi plastické suroviny se přiřazuje také vermikulit.
Obr1.Věstonická venuše

56
Obecně je jíl definován jako směsný jemnozrnný přírodní materiál, který se po zvlhčení stane plastickým a po vysušení či vypálení dojde k jeho zpevnění. Nejvýznamnějšími jílovými minerály z hlediska keramické technologie jsou:
kaolinit, dvojvrstvý minerál, prvkový (strukturní) vzorec je Al2Si2O5(OH)4, oxidový vzorec je ve tvaru Al2O3.2SiO2.2H2O,
illit, trojvrstvý minerál, prvkový (strukturní) vzorec je ve tvaru K2 – x Al4(Si6 + xAl2 -
x)O20(OH)4, montmorillonit, prvkový (strukturní) vzorec trojvrstvého jílového minerálu je uváděn
ve tvaru (Si8-x Alx)(Al4-y-z Fe3+y Mgz)4O20(OH)4 Me+x
+z.
U jílů používaných pro přípravu klasické keramiky je nejčastějším jílovým minerálem kaolinit. Destičkovitý charakter částic kaolinitu je vidět na mikrosnímku kaolinu Sedlec, viz Obr. 3. Surový kaolin obvykle obsahuje 20 až 80 hmotn. % kaolinitu. Velmi často je dominantní jílový minerál v jílu doprovázen různým obsahem pískových zrn, případně úlomky hornin a slídou. Mohou být přítomny i další příměsi např. železité oxidy či hydroxidy, vápenaté a/nebo hořečnaté uhličitany apod. Tyto příměsi tvoří přirozenou neplastickou součást jílů. Obr. 3: Snímek destiček kaolinitu v kaolinu Sedlec pořízený SEM Jílové minerály při tepelném zatížení v intervalu do cca 300 oC nejdříve uvolňují fyzikálně vázanou vodu a potom v relativně širokém teplotním intervalu 450 až 800 oC chemicky vázanou vodu a zásadně mění svoji krystalickou strukturu. Uvolňování chemicky vázané vody tzv. dehydroxylace dvojvrstvého jílového minerálu kaolinitu začíná od teploty cca 450 oC, je to endotermní proces a probíhá až do teploty cca 600 oC. Ze stabilního krystalického minerálu kaolinitu (Al2Si2O5(OH)4) vzniká nestabilní forma prakticky nekrystalického metakaolinitu (Al2Si2O7), při této reakci se uvolňuje z mřížky kaolinitu cca 14 hmotn. % vody. Nad teplotou cca 950 až 1000 oC se metastabilní metakaolinit postupně přeměňuje na stabilní krystalické fáze, vzniká mullit (Al6Si2O13) a při teplotách nad 1000 oC z uvolněného nekrystalického oxidu křemičitého ještě cristobalit (vysokoteplotní modifikace SiO2). U ostatních jílových minerálů probíhá tento proces obdobně jako u kaolinitu.
Chování jílu při výpalu velmi dobře znázorňuje termická analýza. Na Obr. 4 jsou uvedeny křivky diferenční termické analýzy, termogravimetrické analýzy a délkových změn kaolinu Sedlec při ohřevu.
450- 600o
C Al2O
3. 2SiO
2 .2H
2O → Al
2O
3 .2SiO
2 + 2H
2O metakaolinit
600o
C - 950o
C nestabilní nekrystalické fáze
950-1050o
C 2 ( Al2O
3. 2SiO
2) → 2 Al
2O
3. 3SiO
2 defektní spinel + SiO
2amorfní
nad 1100o
C 3 ( Al2O
3.3SiO
2) → 2 (3Al
2O
3. 2SiO
2) mullit + SiO
2 amorfní
nad 1200o
C amorfní SiO2 → SiO
2 cristoballit

57
Obr. 4: Křivky diferenční termické analýzy (DTA), termogravimetrické analýzy (TG) a délkových změn (DZ) vzorku kaolinu Sedlec při ohřevu.
1. 1. 2 Neplastické suroviny Neplastické suroviny se dělí na: ostřiva – v průběhu tepelného zpracování keramické směsi se nemění, pozitivně
ovlivňuje reologické vlastnosti, během výpalu zpevňuje střepovou hmotu, plniva – během výpalu vyhoří, taviva- tvoří při výpalu keramické směsi taveninu, která spojuje krystalické fáze
střepu, reaguje s nimi a často se zúčastňuje vzniku nové krystalické složky. Mezi neplastické suroviny patří křemen, hlinitokřemičitá ostřiva (šamot, lupek, drcené
střepy), zvláštní ostřiva a plniva (korund; cordieritové ostřivo; spalitelná plniva – dřevěné piliny; práškové uhlí; uhelné kaly), uhličitany (vápenec – tj. kalcit; dolomit), dále pak wollastonit, magnezit a alkalická taviva – živce (draselné, sodné, vápenaté a směsné).
Chování některých neplastických příměsí se mění během výpalu při teplotě, kdy se objeví ve střepu např. eutektická tavenina nebo se začnou tavit živce. Ty jsou základními keramickými tavivy, avšak do své teploty tání se chovají jako ostřivo. Při výpalu směsí z přírodních plastických a neplastických surovin do teplot cca 1000 oC jsou relativně nejstabilnějšími krystalickými fázemi křemen a živce. U křemene probíhá pouze reverzibilní modifikační přeměna křemen při teplotě 573 oC, která je doprovázena objemovým nárůstem.Modifikační přeměny SiO2 je možné znázornit schématem:
573 oC 1050 oC 1726 oC β-křemen -křemen → -cristobalit křemenné sklo 180 oC 270 oC
β-cristobalit Zrna živců jsou stabilní až do počátku svého tavení. Ve směsích se taví při
eutektických teplotách, tedy při teplotách podstatně nižších než by odpovídalo jejich teplotám tání, viz Tab. I. Tabulka I: Teploty tání živců a leucitu
Druh živce Vzorec Teplota tání (° C) Vytvořená fáze draselný živec, ortoklas K2O. Al2O3. 6 SiO2 1150 20 tavenina + leucit leucit (podobný živcům, tzv. „foid“)
K2O. Al2O3. 4 SiO2 1686 5 tavenina
sodný živec, albit Na2O. Al2O3. 6 SiO2 1118 3 tavenina vápenatý živec, anortit CaO.Al2O3.2SiO2 1553 3 tavenina
1. 1. 3 Suroviny s vysokým obsahem CaO, popř. MgO
Velmi častým zdrojem vápenaté složky je vápenec s vysokým obsahem kalcitu CaCO3 nebo wollastonit (CaSiO3). U MgO je to magnezit MgCO3 nebo mastek Mg3Si4O10(OH)2. Společně je vápenatá a hořečnatá složka zastoupeny v dolomitu v CaMg(CO3)2.

58
I u těchto surovin dochází v průběhu tepelného zatížení k rozkladu, např. při teplotách nad cca 800 oC dochází k rozkladu kalcitu: CaCO3→ CaO+CO2 . V této první části je uvedeno stručné rozdělení keramických surovin (více viz laboratorní práce LOTR I – Příprava a vytváření keramiky). Keramický a sklářský průmysl používá některé stejné suroviny, jednou z nejdůležitějších je např. vápenec. Některé suroviny těžené v odlišné hornině obsahují stejné minerály v např. sklářské písky (surovina obsahující převážně křemenné písky) a štěrkopísky (surovinový zdroj především křemene a živců). Z tohoto důvodu zde není uvedena samostatná kapitola sklářských surovin. V rámci laboratorní práce budou studovány suroviny užívané jak pro přípravu keramické surovinové směsi, tak sklářského kmene.
1. 2. Termické analýzy Termín termická analýza zahrnuje obecně experimentální metody, při nichž jsou
analyzovány změny složení a vlastností studovaného systému při tepelném zatížení. Studovaným systémem jsou různé látky popř. jejich směsi (nejčastěji pevné, např. jíly, minerály, horniny apod.). Metodami termické analýzy jsou analyzovány změny jejich složení popř. změny jejich vlastností. V průběhu tepelného zatížení vzorku dochází k vyvolání nebo změně intenzity procesu např. chemické reakce, rozkladu, dehydratace, fázové přeměně, které mohou být doprovázeny změnou hmotnosti, objemu, uvolňováním nebo spotřebováním energie, změnou vodivosti atd. Podle vlastnosti, jejíž změna je sledována jako funkce teploty se nazývá i příslušná analýza, viz Tab. II [3-6].
V současné době existuje řada termoanalytických metod, mezi nejpoužívanější patří TG, DTA a DSC. Tyto metody lze označit jako tzv. primární, na ně navazují další, sekundární metody, např. při TG dojde rozkladem ke změně hmotnosti s uvolněním plynných produktů, které jsou následně detekovány další metodou EGA, Evolved Gas Analysis [4, 6].
Tabulka II. Základní metody termické analýzy [3-6] Metoda Sledovaná vlastnost Označení Termogravimetrická analýza hmotnost - měří se hmotnost vzorku v závislosti na
teplotě TG
Diferenční termická analýza rozdíl teplot - měří se rozdíl teplot mezi vzorkem a standardem při ohřevu nebo při chladnutí
DTA
Diferenční skenovací kalorimetrie
entalpie a tepelný tok - měří se tepelný tok dodávaný do referentní látky, tak aby teplotní rozdíl mezi vzorkem a standardem byl nulový
DSC
Termomechanická analýza rozměr a mechanické vlastnosti - sleduje se deformace vzorku při zatížení v závislosti na teplotě
TMA
Termooptometrie optické vlastnosti - měří se např. celkové světlo nebo světlo určité vlnové délky, luminiscence
TOA
Termoakustická analýza akustické vlastnosti - sledují se charakteristiky zvukových vln po jejich průchodu vzorkem
-
Současné měření více vlastností v průběhu jednoho experimentu provádí tzv. simultánní termická analýza (STA) a popisuje se zkratkou použitých metod např. TG/DTA, TG/DTA EGA-MS, TG/EGA-IR (MS-hmotová spektrometrie, IR- infračervená spektroskopie). 1. 2. 1 Termodynamika a termická analýza
Většina metod termické analýzy sleduje příslušné vlastnosti systému (hmotnost, energii, rozměr, vodivost apod.) jako dynamickou funkci teploty. Základním jevem

59
důležitým pro tyto metody je změna entalpie (ΔH). Každou fyzikální a chemickou změnu lze charakterizovat změnou Gibbsovy volné energie (ΔG), která je dána vzorcem: ΔG = ΔH - TΔS, (1) kde ΔH je změna entalpie, T je absolutní teplota a ΔS je změna entropie během děje. Každý systém se snaží dosáhnout takového stavu, kterému odpovídá nižší hodnota Gibbsovy volné energie. Jako příklad lze uvést modifikační přeměnu látky z jedné krystalické formy do druhé, která má za dané teploty nižší hodnotu Gibbsovy volné energie a je tedy stabilnější. Vytvoření krystalické struktury nebo jiného stavu s nižší hodnotou volné entalpie může dojít při ohřevu i přes jednotlivé mezistupně. 1. 2. 2 Metody
Diferenční termická analýza (DTA)
Metoda je založena na měření teplotního rozdílu mezi měřeným vzorkem a vzorkem srovnávacího materiálu (α-Al2O3, MgO). Referenční materiál musí být v celém intervalu teplot měření inertní, a proto děje spojené s výměnou tepla jsou vztaženy ke vzorku – jeho rozdíl vůči srovnávacímu materiálu. Při zahřívání vzorku může docházet k fyzikálním změnám (např. modifikační přeměna, sublimace, absorpce, adsorpce, desorpce, krystalizace) či chemickým změnám (např. chemisorpce, reakce v pevných látkách, reakce v tavenině, reakce v plynné fázi). Pokud je rozdíl teplot mezi vzorkem a srovnávacím materiálem ΔT<0 jedná se o endotermní efekt v opačném případě tj. ΔT>0 se jedná o exotermní efekt. Efekty lze charakterizovat pomocí teplotních bodů na křivce nebo plochou daného efektu. Metodu DTA lze použít ke kvalitativní a semikvantitativní analýze, sledování kinetiky, stanovení Cp a fázových přeměn. Průběh křivky DTA (stejně jako následně u křivek DSC a TG) je ovlivněn konstrukcí aparatury, velikostí a geometrií měřící nádobky, rychlostí zahřívání, množstvím a rozměrem vzorku, zhutněním vzorku a atmosférou v peci.[3-7]
Při kvalitativním vyhodnocení se na křivce sleduje poloha efektů, jejich tvar a velikost. Endotermní pík je charakteristický pro dehydrataci, změnu skupenství, redukci či chemický rozklad a exotermní pík je charakteristická pro krystalizaci, oxidaci, polymorfní přeměny a některé reakce chemického rozkladu.
Diferenční kompenzační kalorimetrie (DSC)
DSC s kompenzací příkonu: metoda, při které se měří elektrický příkon, který je potřeba k udržení konstantní teploty obou vzorků. Vzorek a srovnávací materiál jsou umístěny do oddělených cel a jsou vystaveny stejnému kontrolovanému teplotnímu programu ve specifické atmosféře. V tomto případě pravé DSC jsou křivky podobné jako u DTA, pouze poloha píků je otočena o 180 °.
DSC s tepelným tokem: Tato metoda je označována jako nepravé DSC. Zde se měří rozdíl teplot vzorku a srovnávacího materiálu. Tyto vzorky jsou umístěny ve společné peci a jsou spojeny tepelným mostem. U této metody mají píky stejnou orientaci jako u DTA [3-7].
Termogravimetrie (TG)
TG je metoda, při které se měří hmotnost zkoumané látky v závislosti na teplotě. Vzorek je vystaven kontrolovanému teplotnímu režimu, který se lineárně zvyšuje (tj. dynamické měření) nebo je funkcí času při konstantní teplotě (izometrická měření). Z naměřené křivky lze určit interval teplot, kdy dochází ke změně hmotnosti důsledkem různých procesů ve vzorku (dehydratace, tepelný rozklad, oxidace aj.). Někdy je vhodné pro lepší odlišení procesů následujících v krátkém intervalu za sebou zvolit následně derivaci TG

60
křivky (vznikne tzv. DTG křivka). Tato DTG křivka je srovnatelná s DTA/DSC křivkou a plocha píku souvisí se změnou hmotnosti.[2-7]
Simultánní DTA-TG/ DSC-TG
Simultánní DTA-TG nebo DSC-TG zahrnuje spojení obou metod (TG a DTA/DSC) v jedno měření za použití pouze jednoho vzorku. Při tomto měření jsou pro obě metody zajištěny stejné podmínky, tj. stejný průběh měření, navážka, atmosféra atd.[5]
Detekce (EGD) a analýza (EGA) uvolněných plynů
U obou metod dochází k programovanému ohřevu vzorku a registrace uvolněných plynu se provádí během celého teplotního režimu. Uvolněné plyny (nejčastěji H2O, CO2, SO2, v menší míře O2, H2 a CO) jsou do detektoru unášeny nosným plynem (N nebo Ar) a k detekci se používá infračervený spektrometr nebo hmotnostní spektrometr. [5]
1. 2. 3 Charakteristika křivek [4]:
Výsledky termických analýz znázorňují příslušné křivky, viz Obr. 5.
Obr. 5: Křivky termických analýz: vlevo křivka diferenční termické analýzy (DTA), uprostřed křivka diferenční skenovací kalorimetrie (DSC), vpravo křivka termogravimetrické analýzy, převzato z [4]
Tab. III: Popis DTA/DSC křivky [3-6] Nulová čára Křivka naměřená při použití prázdného kelímku (tj. bez vzorku).
Slouží k popisu měřícího zařízení. Základní čára Čára, která je sestrojená v rozsahu zkoumaného píku. Pík Efekt, při kterém došlo k porušení ustáleného stavu. Pík může být
endotermní nebo exotermní. Teplota počátku píku (Tpoč.) Teplota, při které dochází k odchylce naměřených hodnot od
základní čáry. Teplota extrapolovaného počátku píku (Tonset.)
Průsečík čáry s tečnou v maximálním skloně náběhu píku.
Teplota maxima píku (Tp) Určuje maximum/minimum píku. Jedná se o maximální rozdíl mezi naměřenou křivkou a základní čárou.
Teplota extrapolovaného konce píku (Toffset.)
Průsečík čáry s tečnou v maximálním skloně konce píku.
Teplota konce píku (Tkon.) Teplota, při které křivka naměřených hodnot opět dosahuje základní čáry a pík je zde ukončen.
Výška píku Maximální rozdíl mezi vrcholem píku a základní čárou. Plocha píku Plocha ohraničená píkem a základní čárou mezi počáteční (Tpoč.) a
konečnou (Tkon.) teplotou píku.
DTA křivka znázorňující endotermní děj je uvedena na Obr. 6, v případě exotermního děje by byla orientována opačně. V úseku vymezeném body A a B reakce neprobíhá (ΔT = 0).
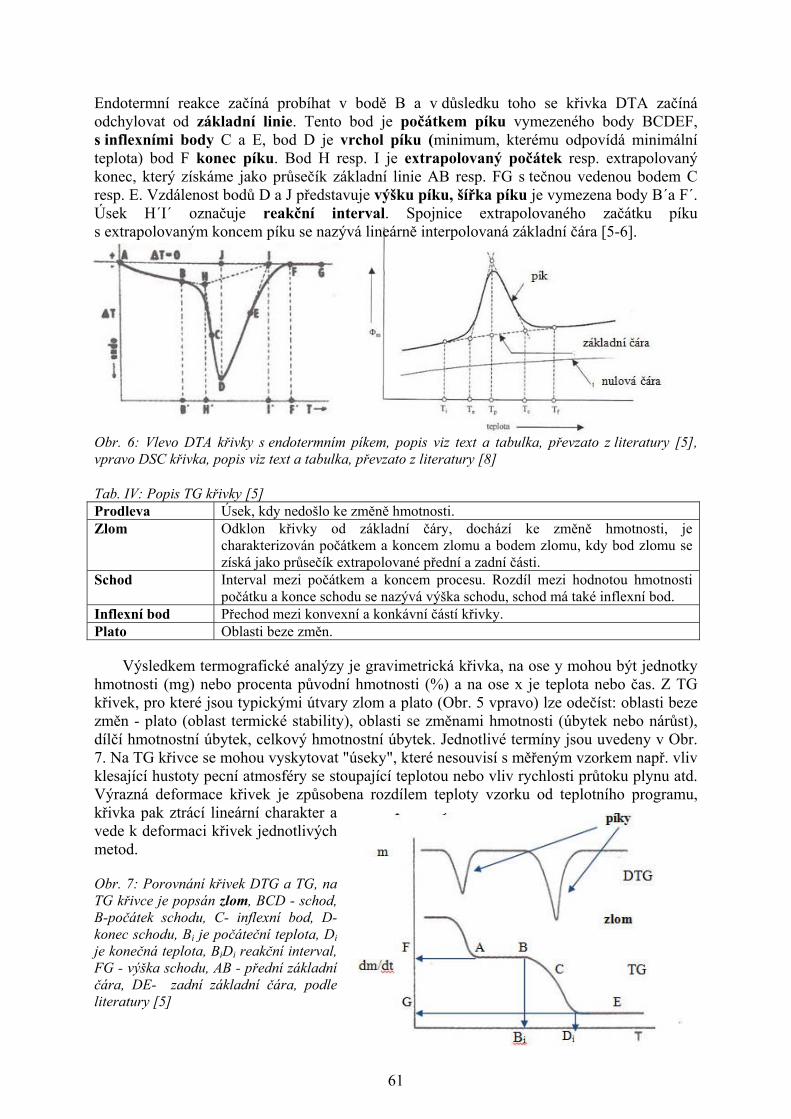
61
Endotermní reakce začíná probíhat v bodě B a v důsledku toho se křivka DTA začíná odchylovat od základní linie. Tento bod je počátkem píku vymezeného body BCDEF, s inflexními body C a E, bod D je vrchol píku (minimum, kterému odpovídá minimální teplota) bod F konec píku. Bod H resp. I je extrapolovaný počátek resp. extrapolovaný konec, který získáme jako průsečík základní linie AB resp. FG s tečnou vedenou bodem C resp. E. Vzdálenost bodů D a J představuje výšku píku, šířka píku je vymezena body B´a F´. Úsek H´I´ označuje reakční interval. Spojnice extrapolovaného začátku píku s extrapolovaným koncem píku se nazývá lineárně interpolovaná základní čára [5-6].
Obr. 6: Vlevo DTA křivky s endotermním píkem, popis viz text a tabulka, převzato z literatury [5], vpravo DSC křivka, popis viz text a tabulka, převzato z literatury [8] Tab. IV: Popis TG křivky [5] Prodleva Úsek, kdy nedošlo ke změně hmotnosti. Zlom Odklon křivky od základní čáry, dochází ke změně hmotnosti, je
charakterizován počátkem a koncem zlomu a bodem zlomu, kdy bod zlomu se získá jako průsečík extrapolované přední a zadní části.
Schod Interval mezi počátkem a koncem procesu. Rozdíl mezi hodnotou hmotnosti počátku a konce schodu se nazývá výška schodu, schod má také inflexní bod.
Inflexní bod Přechod mezi konvexní a konkávní částí křivky. Plato Oblasti beze změn.
Výsledkem termografické analýzy je gravimetrická křivka, na ose y mohou být jednotky hmotnosti (mg) nebo procenta původní hmotnosti (%) a na ose x je teplota nebo čas. Z TG křivek, pro které jsou typickými útvary zlom a plato (Obr. 5 vpravo) lze odečíst: oblasti beze změn - plato (oblast termické stability), oblasti se změnami hmotnosti (úbytek nebo nárůst), dílčí hmotnostní úbytek, celkový hmotnostní úbytek. Jednotlivé termíny jsou uvedeny v Obr. 7. Na TG křivce se mohou vyskytovat "úseky", které nesouvisí s měřeným vzorkem např. vliv klesající hustoty pecní atmosféry se stoupající teplotou nebo vliv rychlosti průtoku plynu atd. Výrazná deformace křivek je způsobena rozdílem teploty vzorku od teplotního programu, křivka pak ztrácí lineární charakter a vede k deformaci křivek jednotlivých metod. Obr. 7: Porovnání křivek DTG a TG, na TG křivce je popsán zlom, BCD - schod, B-počátek schodu, C- inflexní bod, D- konec schodu, Bi je počáteční teplota, Di je konečná teplota, BiDi reakční interval, FG - výška schodu, AB - přední základní čára, DE- zadní základní čára, podle literatury [5]
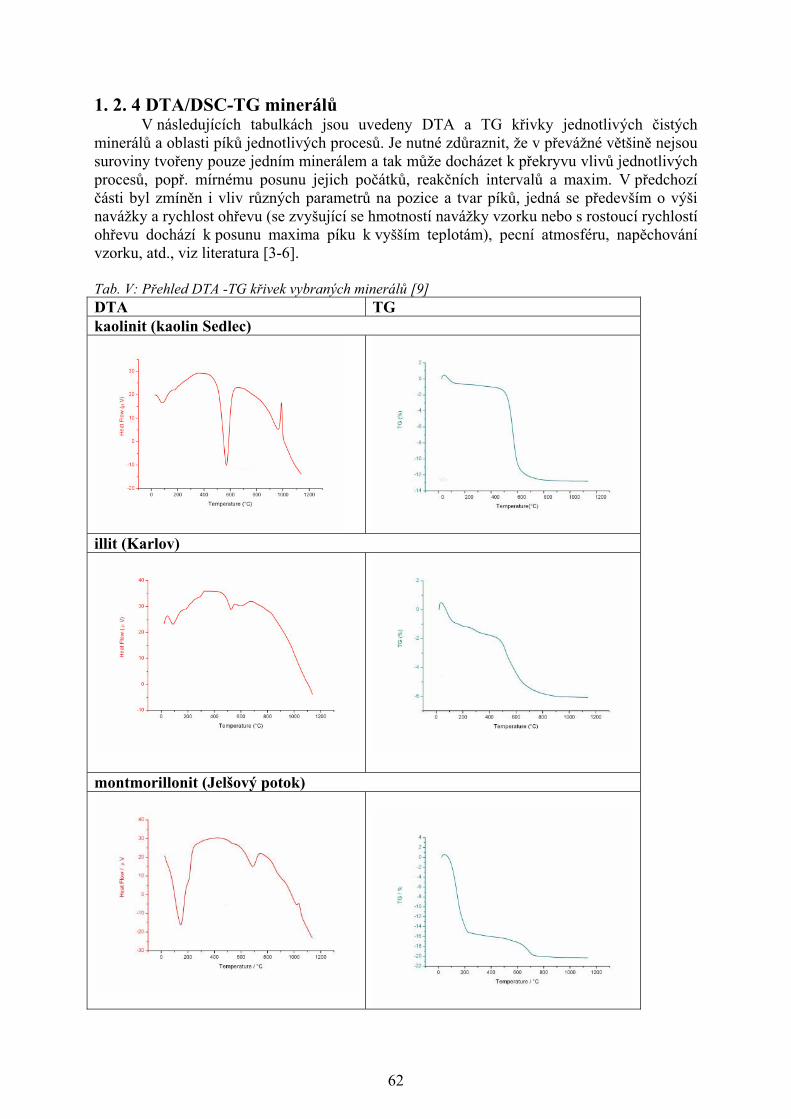
62
1. 2. 4 DTA/DSC-TG minerálů V následujících tabulkách jsou uvedeny DTA a TG křivky jednotlivých čistých
minerálů a oblasti píků jednotlivých procesů. Je nutné zdůraznit, že v převážné většině nejsou suroviny tvořeny pouze jedním minerálem a tak může docházet k překryvu vlivů jednotlivých procesů, popř. mírnému posunu jejich počátků, reakčních intervalů a maxim. V předchozí části byl zmíněn i vliv různých parametrů na pozice a tvar píků, jedná se především o výši navážky a rychlost ohřevu (se zvyšující se hmotností navážky vzorku nebo s rostoucí rychlostí ohřevu dochází k posunu maxima píku k vyšším teplotám), pecní atmosféru, napěchování vzorku, atd., viz literatura [3-6]. Tab. V: Přehled DTA -TG křivek vybraných minerálů [9] DTA TG kaolinit (kaolin Sedlec)
illit (Karlov)
montmorillonit (Jelšový potok)
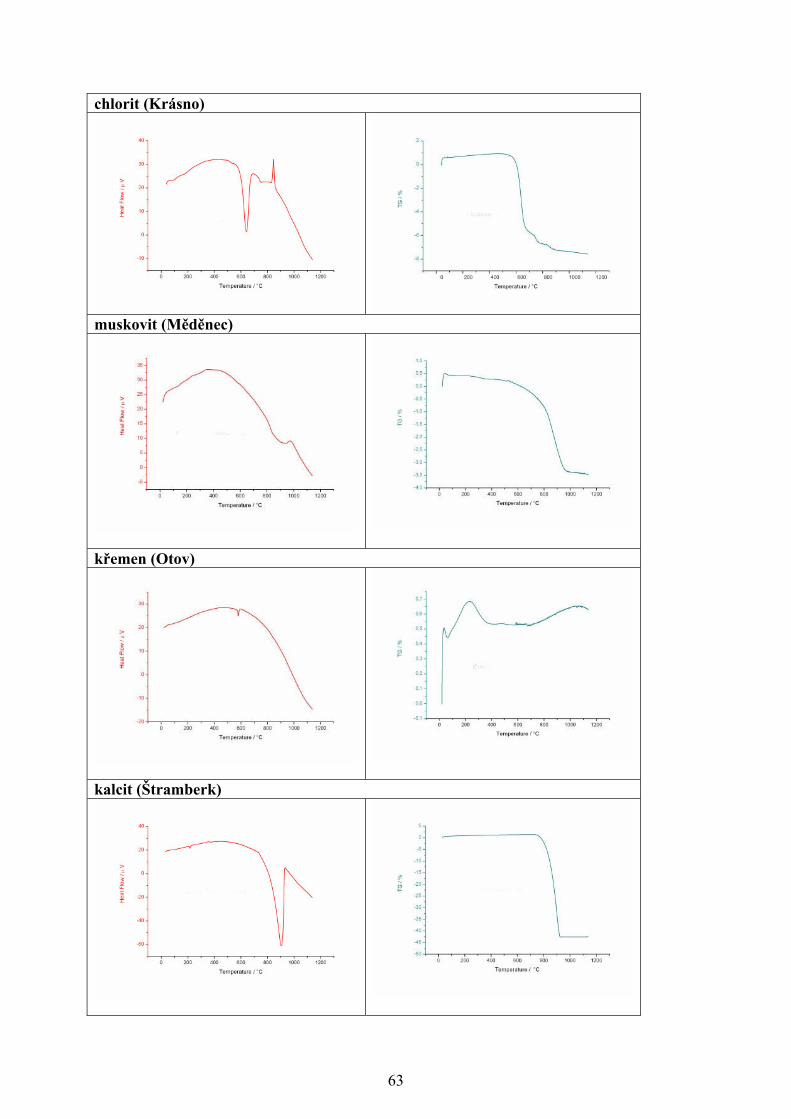
63
chlorit (Krásno)
muskovit (Měděnec)
křemen (Otov)
kalcit (Štramberk)
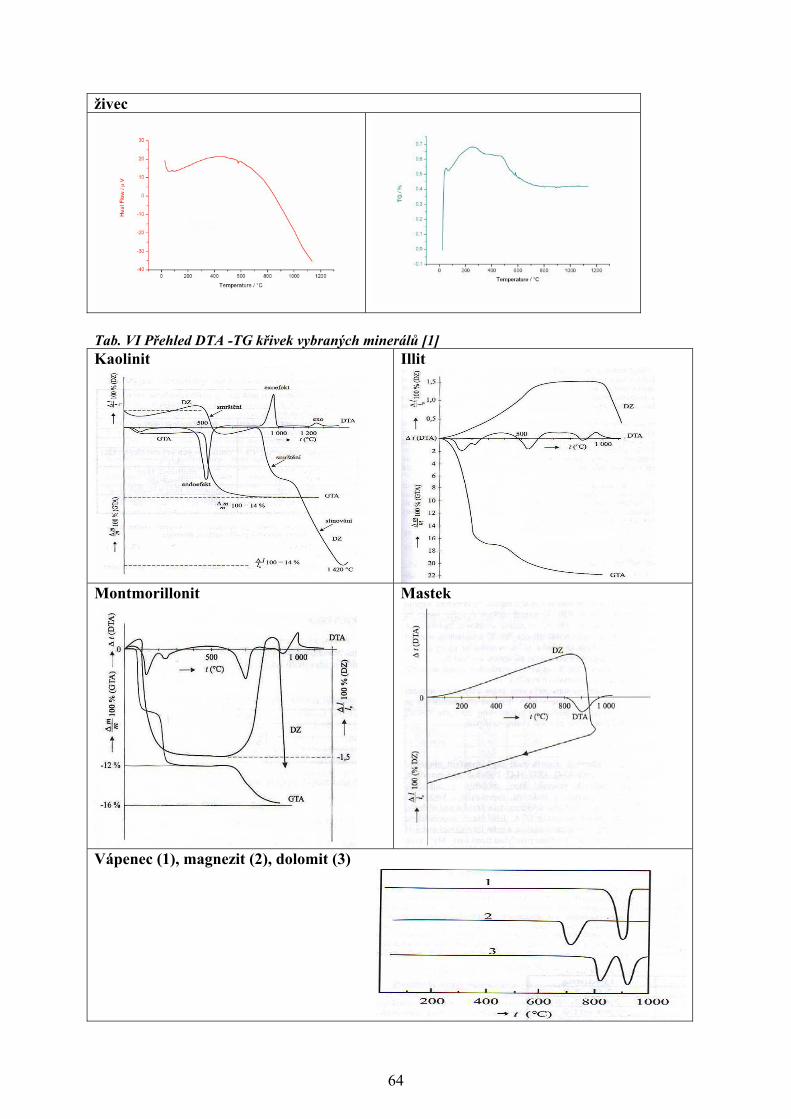
64
živec
Tab. VI Přehled DTA -TG křivek vybraných minerálů [1] Kaolinit
Illit
Montmorillonit Mastek
Vápenec (1), magnezit (2), dolomit (3)

65
2. úbytek hmotnosti
3. úbytek hmotnosti
1. úbytek hmotnosti
celkový úbytek hmotnosti
endotermický efekt
exotermický efekt
základní linie
Tab. VII. Fyzikální a chemické procesy identifikovatelné při ohřevu pomocí DTA křivek [10]
1. 2. 4 DTA/DSC-TG minerálů
Příklad výsledku simultánní STA termické analýzy měřené na zařízení Linseis STA PT 1600/1750 °C HiRes je uveden na Obr. 8 [11], jedná se o křivky termického rozkladu monohydrátu šťavelanu vápenatého ohřev teplotním programem 10oC/min v intervalu 25 - 1000oC, pecní atmosféra vzduch, navážka 19,01 mg. Vznik meziproduktů termického rozkladu Ca(C2O4).H2O a příslušných hmotnostních úbytků je uveden uvnitř obrázku.
Obr. 8: TG a DTA křivky termického rozkladu Ca (C2O4).H2O [11]

66
2. Praktická část V úvodní části byl uveden přehled keramických surovin a stručně popsány základní
termické analýzy včetně příkladů termických analýz vybraných surovin. Praktická část je zaměřena na zpracování dat získaných simultánním přístrojem
Linseis STA PT 1600/1750 °C HiRes s modulem DTA-TG nebo DSC-TG firmy Linseis s připojeným hmotnostním spektrometrem ThermoStarTM GSD320 firmy Pfeiffer Vacuum.
Cílem této laboratorní práce je: seznámit se s možnostmi aplikace STA termických analýz, seznámit se s programem Linseis Data Acquisition a Data Evaluation
(laboratorní příručka a video na webových stránkách (http://www.linseis.net/html_en/thermal/software.php,http://tresen.vscht.cz/sil/cs/skupina_keramika2) a následně vyhodnotit známý vzorek včetně jednotlivých váhových úbytků a popsat probíhající procesy probíhající při ohřevu vzorku,
analyzovat neznámý vzorek přírodních surovin, popř. keramických směsí a vyhodnotit naměřená data.
Základem při vyhodnocení všech typů vzorků je pečlivé zpracování jednotlivých křivek. U křivky TG se sledují změny hmotnosti vzorku (v mg nebo hmotn. %) U DTA/DSC křivky se nejprve určuje typ píku (tj. zda je endotermní nebo exotermní) a následně dle pozice píku reakce, která danému procesu přísluší. Důležitými informacemi u píků DTA/DSC křivek jsou maximální rychlost reakce, maximální teplota reakce (Tp), onsetová teplota (Tonset), offsetová teplota (Toffset) a plocha píku. Získané data z TG a DTA nebo DSC se dále používají např. k vyhodnocení stupně přeměny, popř. pro různé kinetické výpočty.
Literatura: 1. HANYKÝŘ, V.; KUTZENDÖRFER, J. Technologie keramiky. Praha: Silikátový svaz,
2008. ISBN 978-80-86821-48-1. 2. HANYKÝŘ, V Vliv keramických surovin na strukturu a vlastnosti pórovitého
keramického střepu, Zpravodaj STOP, 2010, 12 (4), 4-11. 3. BLAŽEK, A. Moderní metody v analytické chemii. Praha: SNTL Praha, 1972. 4. BROWN, M. E. Handbook of Thermal Analysis and Calorimetry. [online], Elsevier
B.V.,1998. 5. ŠULCOVÁ, P., BENEŠ, L. Experimentální metody v anorganické chemii. Pardubice:
Univerzita Pardubice FCHT, 2008, str. 135-198, ISBN 978-80-7395-058-3. 6. ŠTARHA, P., TRÁVNÍČEK, Z. Termická analýza. Olomouc: Univerzita Palackého v
Olomouci, 2011. 7. ŠAŠEK, L. A KOL.: Laboratorní metody v oboru silikátů. Praha: SNTL-Nakladatelství
technické literatury, Praha, 1981, 116-125. 8. Charakterizácia DSC kriviek: Študijné materiály. v 1. letná škola termickej analýzy a
kalorimetrie, Bratislava 20. - 22. 9. 2001. Edited by P. Šimon, Š. Svestík, E. Smrčková, M. Palou. 2001.
9. VACULÍKOVÁ, L., PLEVOVÁ, E: Identification of clay minerals and micas in sedimentary rocks. Acta Geodyn. Geomater. 2005, Vol. 2, No. 2, (138) 165-175.
10. JELÍNEK, E.: Moderní analytické metody v geologii, Praha, 2008. 11. Linseis Messgerate GmbH Linseis STA, Termogravimetric Analyzer Instruction Manual.



![ODDELENIE FYZIKY KOVOV · 0 20 40 60 80 100 120 140 160-2500 0 2500 5000 7500 10000 12500 I n t e n s i t y [c o u n t s] 2 Theta [deg] ODDELENIE FYZIKY KOVOV Termická analýza Štúdium](https://img.pdfslide.tips/doc/110x75/5e7972b95b12602a4737b5b8/oddelenie-fyziky-0-20-40-60-80-100-120-140-160-2500-0-2500-5000-7500-10000-12500.jpg)



![ED]t - tresen.vscht.cztresen.vscht.cz/kot/wp-content/uploads/2014/09/Cytogenetika-2016.pdfnejnovější modifikace metody mnohobarevné FISH (M-FISH zpřesnění cytogenetické diagnostiky](https://img.pdfslide.tips/doc/110x75/5e9f719e68a1e07f61215c70/edt-nejnovj-modifikace-metody-mnohobarevn-fish-m-fish-zpesnn-cytogenetick.jpg)





















