
UMIVERDIDADE DE SAO PAULOFACULDADE DE CIENCIAS FARMACEUTICAS
DEPARTAMENTO DE ANALISES CLÍNICAS E TOXICOLÓGICASLABORATORIO DE BIOLOGAI MOLECULAR ALPLICADA AO DIAGNÓSTICO
Apostila de noções de Biologia molecular
Prof. Dr. Mario H. HirataProfa. Dra Rosário D. C., Hirata
1999

ÍNDICE
Tópico Página
Aula número 1: Introdução................................................................................................................................... 2Estrutura do DNA e RNA.................................................................................................................. 2Replicação, transcrição e tradução................................................................................................... 4Organização do genoma de eucariotos, procariotos e viral............................................................... 7Enzimas de restrição e suas aplicações.......................................................................................... 10
Aula número 2: Extração do DNA genômico ....................................................................................................... 13
Aula número 3: Princípios de eletroforese.......................................................................................................... 19
Fatores que influenciam na eletroforese.................................................................................... 20Tipos de suporte....................................................................................................................... 22Tipos de eletroforese................................................................................................................ 22Aplicações da eletroforese nas técnicas moleculares................................................................. 23
Aula número 4: Reação de polimerização em cadeia (PCR)................................................................................ 28Princípios da reação de PCR........................................................................................................... 28Protocolo geral para PCR................................................................................................................ 31Fatores que influenciam na PCR..................................................................................................... 31Escolha de iniciadores para PCR..................................................................................................... 35
Aula número 5: PCR no Diagnóstico das Doenças genéticas.............................................................................. 39Polimorfismo e mutações................................................................................................................ 39Diagnóstico de alguns polimorfismos por PCR................................................................................. 41Polimorfismo de apolipoproteína...................................................................................................... 42Polimorfismo de receptores da LDL................................................................................................. 44
Aula número 6: PCR no diagnóstico de Doenças infecto-contagiosas-------------------------------------------------------- 46Meningites bacterianas-------------------------------------------------------------------------------------------------- 47 Tuberculose------------------------------------------------------------------------------------------------------------------ 50Infecção pelo virus da Hepatite C--------------------------------------------------------------------------------------- 59Infecção pelo virus HIV---------------------------------------------------------------------------------------------------- 64
Aula número 7: PCR e RFLP na identificação de indivíduos................................................................................ 68
2

APLICAÇÃO DA PCR EM LABORATÓRIO CLÍNICO E MEDICINA FORENSE
Aula número 1
Profa. Dra. Rosario Dominguez Crespo HirataProf. Assoc. Mario Hiroyuki Hirata
INTRODUÇÃO
Os avanços nos estudos da biologia molecular em eucariotos tem contribuido significantemente para
o entendimento da etiologia das doenças humanas. As técnicas de recombinação dos ácidos nucleicos têm
permitido aos pesquisadores identificar os vários genes responsáveis por doenças hereditárias, avaliar os
mecanismos de infecção dos microrganismos, conhecer os processos que regulam a diferenciação e
proliferação celular (principalmente nos processos neoplásicos), entre outros. A biologia molecular vem
também apresentando aplicação bastante expressiva na pesquisa de marcadores genéticos úteis no
diagnóstico laboratorial. Por outro lado, as diferenças nas sequências do DNA, observadas entre as
espécies ou entre os indivíduos, são extremamente úteis para as análises forenses, os testes de
paternidade, a identificação de antígenos de histo-compatibilidade, a identificação de allelos mutantes, a
identificação de gêneros e espécies de bactérias, fungos, leveduras, vírus e outros.
Nesta apostila serão abordados os princípios básicos de biologia molecular, bem como serão
descritas as aplicações da técnica de PCR no diagnóstico clínico e nos testes de identificação.
ESTRUTURA DOS ÁCIDOS NUCLEICOS (DNA E RNA)
As informações genéticas são armazenadas nos ácidos nucleicos. Há dois tipos de ácidos
nucleicos: ácido desoxiribonucleico (DNA) e ácido ribonucleico (RNA). O DNA é encontrado principalmente
nos cromossomos no núcleo das células, enquanto RNA está presente no citoplasma, havendo muito pouco
nos cromossomos.
Os ácidos nucleicos consistem de cadeias de nucleotídeos. Cada nucleotídeo é constituido por
uma base nitrogenada, uma molécula de açúcar e um grupamento de fosfato (PO4=). Há dois tipos de
açúcar nos ácidos nucleicos: 2-deoxiribose no DNA e ribose no RNA (Figura 1A). As bases nitrogenadas
são as pirimidinas: citosina (C), timina (T) e uracila (U) e as purinas: adenina (A) e guanina (G). O DNA
contém A, C, G e T, enquanto que o RNA contém U em vez de T. Em ambos DNA e RNA, os nucleotídeos
estão ligados formando uma longa cadeia cadeia polinucleotídica. Essa cadeia é formada por ligações entre
o grupo fosfato do carbono 5 de um nucleotídeo e o carbono 3 do açúcar do nucleotídeo adjacente (Figura
1B).
Em geral, o RNA consiste de uma cadeia polinucleotídica simples, enquanto que o DNA é composto
de duas cadeias polinucleotídicas pareadas, dispostas em direção contraria, sendo denominadas
antiparalelas (Figura 1B). As ligações covalentes do esqueleto de açúcar-fosfato localizam-se na parte
3

externa da estrutura e as bases nitrogenadas estão projetadas para a parte interna. O arranjo das bases na
molécula de DNA segue uma sequência em que cada base de uma cadeia é pareada com outra base na
segunda cadeia, de modo que guanina em uma cadeia é pareada com a citosina na outra cadeia e a
adenina sempre pareia com a timina (Figura 1C). Dessa forma o DNA apresenta a estrutura tridimensional
de dupla hélice, onde o ângulo e a distância entre um nucleotídeo e outro é sempre constante.
(A) (B)
O
OH
OH
4
5
CH2OH
3 2
1
2-deoxiribose
1
23
CH2OH
5
4
OH
OH
O
OH
ribose
(C)
N
N
H
N
N
H
HH
CH3
O
O
N
N
H
(para a cadeia)
(para a cadeia)
Timina Adenina
(para a cadeia)
(para a cadeia)
H
H
O
N
N
NH
H
H
H
N
N
N
N
O
H
Citosina Guanina
5’ fosfato 3’ hidroxila
OCH2
P
P
CH2 O
OCH2
P
P
CH2 O
A
G
T
C
T
C
A
G
O CH2
P
P
CH2O
O CH2
P
P
CH2O
OH
OH
3’ hidroxila 5’ fosfato
Figura 1. Características estruturais das moléculas dos ácidos nucleicos. (A) Molécula do açúcar. (B) Representação diagramática do arranjo antiparalelo das duas cadeias polinucleotídicas do DNA. P representa o grupo fosfato. (C) Pareamento da bases na molécula de DNA mostrando as pontes de hidrogênio.
E. CHARGAFF, em 1951, verificou que na molécula de DNA a relação entre as purinas e pirimidinas
era constante na maioria das espécies animais, ou seja, a quantidade da purina, adenina, é igual a da
pirimidina, timidina; da mesma forma, a quantidade da purina guanina é igual à da pirimidina citosina.
Essa característica importante é definida pela propriedade fisico-química das moléculas com formas
semelhantes se atrairem mutuamente com significante afinidade (força de atração de Van Der Waals),
somada à força de atração entre os átomos de cargas opostas (ligações iônicas ou pontes de hidrogênio).
Essas propriedades são bastante específicas e no caso do DNA elas operam de forma conjunta e
simultânea com significante energia de interação, como se fosse um molde perfeito. Em resumo, pode-se
dizer que a lei de CHARGAFF se baseia na especificidade da interação das bases puricas e pirimidicas,
onde a citosina pareia apenas com a guanina e a adenina pareia apenas com a timidina ou uracila. É
4

importante salientar que a ligação entre a A e T ou U ocorre por interação de 2 pontes de hidrogênio,
enquanto que entre a C e a G a ligação se dá por 3 pontes de hidrogênio (ligação mais forte). Devido a essa
especificidade do pareamento entre as bases; a sequência de uma das cadeias polinucleotídicas do DNA é
complementar a outra (cadeia complementar).
A estrutura secundária de dupla hélice (duplex) do DNA, descrita por Watson & Crick em 1953, está
disposta linearmente. Entretanto, pode ser também circular como nas bactérias, plasmídeos e certos vírus,
e pode ser também de fita simples como em alguns tipos de fagos. Entretanto, a disposição da molécula de
DNA nos organismos superiores é de particular importância, onde a informação genética está amplamente
concentrada nos cromossomas dentro do núcleo. O comprimento total dos 46 cromossomas humanos tem
menos que 0,5 mm, entretanto se extendido pode atingir muitos metros. Isto se deve a que o DNA é
compactado na forma super-enrolada em vários níveis: primário, duplex; secundário, ao redor das proteinas
ligadas ao DNA (histonas) formando os nucleossomos; terciário, enrolado com os nucleossomas formando
um cilindro ou estrutura solenoide; e finalmente quaternária, formando voltas ou “loops”. Esta disposição
super-enrolada é a forma natural do DNA.
Propriedades dos ácidos nucleicos
Os ácidos nucleicos podem ser dissociados e associados por processos físicos e químicos. Em
condições fisiológicas, a estrutura de dupla hélice do DNA é bastante estável. No entanto, se essa estrutura
for submetida a alta temperatura (90C a 100C) ou a pH extremos (menor que 3,0 ou maior que 10,0), ela
se separa em duas fitas simples, sofrendo o processo de dissociação, desnaturação ou “melting”. Da
mesma forma, ao submeter-se essas fitas simples a condições de associação (temperatura a 65C, ou pH
próximo de 7,0) por algumas horas, elas podem se reconstituir formando a dupla fita novamente,
obedecendo a lei da complementariedade; ou seja a dupla fita somente se reassocia se as bases forem
exatamente complementares. Esse fenômeno de associação é também denominado de renaturação,
hibridização ou “annealing”. Esses processos de desnaturação e renaturação são essenciais para
manipulação dos ácidos nucleicos in vitro, principalmente do DNA, contribuindo enormemente para o
isolamento, comparação e identificação desses compostos. Outra propriedade física muito importante dos
ácidos nucleicos é a sua capacidade de absorver energia na região do ultravioleta, apresentando o pico de
absorbância máxima em 260 nm. Esta propriedade é útil tanto para a quantificação quanto para avaliar a
pureza dessas substâncias.
REPLICAÇÃO, TRANSCRIÇÃO E TRADUÇÃO
Replicação
Toda vez que uma célula se divide, todo o seu conteúdo de DNA é duplicado na íntegra,
transmitindo em absoluto as informações genéticas contidas na célula precursora, esse processo denomina-
se replicação. A replicação ocorrre de forma semi-conservativa, com as duas cadeias precursoras (DNA
5

de dupla fita, dsDNA) servindo de molde para a síntese da nova cadeia (Figura 2). Dessa forma, para a
replicação ocorrer é necessário que dupla fita precursora se abra formando 2 cadeias independentes (fita
simples, ssDNA). Esse é um processo energeticamente desfavorável e ocorre com a ação conjunta de uma
proteina DNA-específica e várias enzimas; sendo as fitas simples precursoras replicadas, então, de forma
independente e separada. A replicação pode ter início em vários sítios, mas cada origem de replicação é
utilizada uma vez, durante um simples ciclo celular. A fita filha é sintetizada pela ação catalizadora da DNA
polimerase III, enzima que utiliza como molde a cadeia precursora e incorpora os nucleotídeos de forma
sequencial na nova cadeia, produzindo-a de acordo com a lei de pareamento das bases. Como essa
polimerase sintetiza DNA na direção de 5' para 3', a síntese de uma das cadeias complementares é
realizada de forma contínua enquanto que a outra é sintetizada discontinuamente. Os fragmentos da cadeia
discontínua são ligados pela enzima DNA ligase. Muitas outras proteínas estão envolvidas na estabilização
e manutenção da integridade das cadeias simples que são precursoras para a síntese da dupla fita, assim
como no reconhecimento dos sítios de iniciação. As DNA polimerases possuem também, além da função
de sintetizar polinucleotídeos, atividade exonucleásica ou "a correção de leitura" na qual os nucleotídeos
incorretamente incorporados são removidos. Essa propriedade é fundamental para a manutenção a
integridade da sequência original.
Figura 2. Representação da molécula de DNA durante a replicação: as duas cadeias precursoras são separadas e as cadeias complementares que são sintetizadas.
Transcrição
A síntese protéica não é resultante da leitura direta da sequência nucleotídica do DNA. Sabemos
que o DNA está localizado no cromossoma do núcleo da célula e a síntese protéica ocorre quase que na
sua totalidade nos ribossomas, localizados no citoplasma. As informações genéticas contidas na sequência
nucleotídica do DNA têm que ser transferidas para uma molécula intermediária que pode mover-se para o
citoplasma, o RNA mensageiro (mRNA), e que ordena, então, a síntese da proteina. O processo de síntese
de mRNA é conhecido por transcrição e o tamanho da molécula de mRNA é definido pela sequência de
aminoácidos que o ácido nucleico codifica.
Apenas uma das fitas do DNA é transcrita dando origem a uma única sequência de mRNA para
cada gene. A indicação de qual fita do DNA deve ser transcrita é orientada por uma sequência específica
denominada promotor, localizada antes (“upstream”) da sequência a ser transcrita, que é reconhecida pela
6

RNA polimerase (enzima sintetisadora de mRNA). Como a RNA polimerase adiciona sequencialmente
monofosfatos de ribonucleosídeo à extremidade 3’ da cadeia de RNA em crescimento, a polimerização
ocorre na direção 5’ - 3’. Isto significa que cada molécula de mRNA será identica à sequência nucleotídica
da fita de DNA que não é transcrita. O mRNA transcrito é transportado para os ribossomos (RNA
ribossômico, rRNA) no citoplasma, onde ocorre a síntese protéica.
O material genético de certos virus (retrovirus) é o RNA e a informação genética segue, portanto, a
direção reversa (de RNA para DNA). Este processo é conhecido por transcrição reversa, na qual a
sintese de DNA é dirigida pelo RNA e a enzima responsável é denominada transcriptase reversa. Essa
enzima tem importante função no ciclo vital dos retrovírus.
Tradução
A relação entre a sequência de nucleotídeos do DNA e a sequência de aminoácidos da proteina
correspondente é denominada código genético. Esse código é universal e é encontrado em todos os
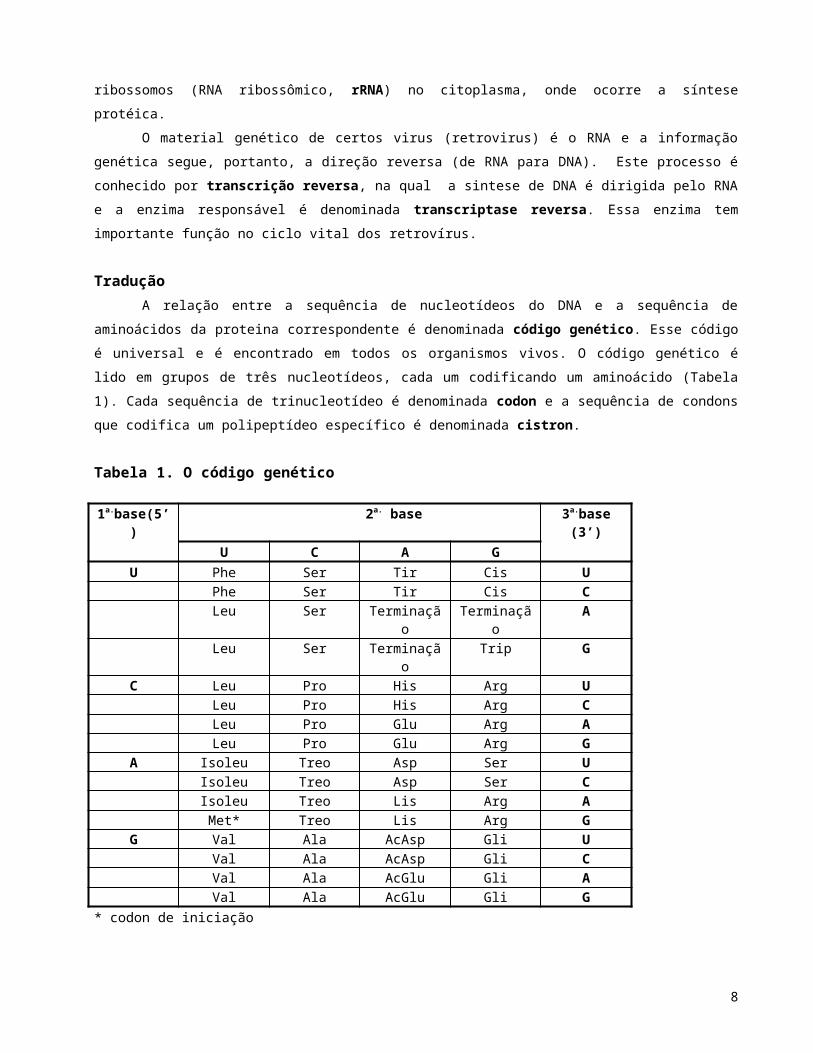
organismos vivos. O código genético é lido em grupos de três nucleotídeos, cada um codificando um
aminoácido (Tabela 1). Cada sequência de trinucleotídeo é denominada codon e a sequência de condons
que codifica um polipeptídeo específico é denominada cistron.
Tabela 1. O código genético
1a.base(5’) 2a. base 3a.base (3’)U C A G
U Phe Ser Tir Cis UPhe Ser Tir Cis CLeu Ser Terminação Terminação ALeu Ser Terminação Trip G
C Leu Pro His Arg ULeu Pro His Arg CLeu Pro Glu Arg ALeu Pro Glu Arg G
A Isoleu Treo Asp Ser UIsoleu Treo Asp Ser CIsoleu Treo Lis Arg AMet* Treo Lis Arg G
G Val Ala AcAsp Gli UVal Ala AcAsp Gli CVal Ala AcGlu Gli AVal Ala AcGlu Gli G
* codon de iniciação
O processo de decodificação pelo qual a informação genética presente na molécula de mRNA dirige
a síntese proteica é denominado tradução. Esse processo envolve o mRNA, o rRNA e o RNA de
transferência ou tRNA, encontrado no citoplasma. Cada tRNA tem 70-90 nucleotídeos em comprimento e é
de fita simples, mas devido ao pareamento de bases dentro da molécula, adquire a forma de uma folha de
trevo. Dentro dessa estrutura, um trinucleotídeo de sequência variável forma o anti-codon que pode parear
7
com um codon da molécula de mRNA. Na outra extremidade da molécula de tRNA está uma sequência para
ligação a um aminoácido específico. Portanto, um codon particular no mRNA é relacionado através de um
tRNA a um dado aminoácido. Na tradução, o ribossoma liga-se primeiramente a um sítio específico na
molécula de mRNA (codon de iniciação, ATG) que ajusta a fase de leitura ("reading frame"). O
ribossoma, então, se movimenta ao longo da molécula de mRNA, traduzindo um codon de cada vez usando
tRNAs para adicionar aminoácios ao final da cadeia polipeptídica em alongamento. A tradução é finalizada
quando o ribossoma reconhece na fita de mRNA o codon de terminação ("stop codon"). A direção da
leitura é de 5’-3’, sendo que a sequência de nucleotídeos da fita de DNA corresponde à sequência de
aminoácidos da proteina traduzida na direção do N-terminal para o C-terminal.
Em princípio as sequências de bases nos ácidos nucleicos devem ser traduzidas em qualquer fase
de leitura diferente, dependendo onde o processo de decodificação se inicia. Para uma dada sequência
UUAGCAAAGCUGCGAAUG, as três fases de leitura possíveis são :
UUA GCA AAG CUG CGA A
UAG CAA AGC UGC GAA U
AGC AAA GCU GCG AAU G
Entretanto, o código genético não se sobrepõe, pois a fase de leitura é ditada pelas sequências de iniciação
e de terminação da tradução presentes na molécula de mRNA. A fase de leitura pode ser alterada por
mutações que removem ou inserem bases na sequência nucleotídica. Esse tipo de alteração é conhecido
como descolamento de fase ("frameshift") e pode causar a perda total da função da proteina produzida.
DNA RNA ProteinasReplicação
Transcrição
Transcrição reversa
Replicação e Transcrciçãodo RNA
Tradução
Fluxo da informação genética intracelular
ORGANIZAÇÃO DO GENOMA DE EUCARIOTOS, PROCARIOTOS E VIRAL
Estrutura Gênica
Os cromossomas constituem-se de uma longa e ininterrupta sequência de nucleotídeos na qual
estão dispostos muitos genes. O gene é uma unidade de herança constituida por uma sequência
nucleotídeos que carrega as informações necessárias para a síntese de um dado polipeptídeo. O gene é
uma entidade estável mas pode sofrer mutações, sendo que a nova forma do gene é herdada de modo
estável, exatamente como a forma que lhe deu origem. Um gene pode existir em formas alternativas,
denominadas alelos, que determinam a expressão de alguma característica particular.
8

Embora todos genes funcionais sejam transcritos, a maioria do DNA genômico não é totalmente
transcrito. A quantidade total de DNA nuclear no genoma haplóide (gamético) humano compreende 3 x 106
kb. Como a maioria dos genes tem 1 a 20 kb em comprimento, deveria haver 1 milhão de genes, mas
somente 3000 locos de doença foram identificados. Mesmo considerando todos os outros genes que
conferem as características normais como altura e inteligência, uma grande porção do DNA genômico não
tem função definida. Entretanto, mais e mais tem se observado que este DNA tem funções importantes,
incluindo o controle de ação gênica.
Genes que são responsáveis pela síntese de enzimas específicas ou peptídeos são denominados
de genes estruturais, enquanto que os genes reguladores modificam os efeitos dos genes estruturais.
Controle da expressão gênica
Nas células existem mecanismos moleculares que controlam o número e a quantidade das proteinas
produzidas. As diferenças na síntese proteica resultam de sinais moleculares que controlam as taxas em
que as moléculas de mRNA são transcritas a partir do DNA (controle transcricional) ou traduzidas
(controle traducional)
Processamento pós-transcricional do mRNA
A transcrição e a tradução nos procariotos são processos concomitantes mas nos eucariotos são
processos temporal e espacialmente desconectados. Como vimos, nos eucariotos a transcrição ocorre no
núcleo e a tradução no citoplasma. Durante a passagem do núcleo para o citoplasma, o mRNA transcrito
(denominado pre-mRNA) deve ser processado para que seja obtido o mRNA maduro.
Nos procariotos, a sequência de nucleotídeos do gene é colinear com a sequência de aminoácidos
de uma dada proteína, entretanto, nos eucariotos a sequência codificante é geralmente interrompida por
sequências adicionais.
A maioria dos genes dos eucariotos são compostos por regiões codificadoras denominadas exons e
regiões não-codificadoras denominadas introns. Estes introns são sequências posteriormente removidas
durante o processamento do transcrito primário. Esse processo é denominado processamento do RNA ou
“RNA splicing”. Após a remoção dos introns, a RNA polimerase II adiciona uma sequência AAUAAA
extremidade 3’ do mRNA, conhecida como poli-A. A maioria dos mRNAs eucarióticos possuem 100 a 200
resíduos de adeninas ligadas à extremidade 3', denominada cauda de poli-A, adicionada pela enzima poli-A
polimerase. Antes que ocorra a remoção dos introns, o mRNA sofre a adição de resíduos de 7-metil-
guanosina (Caps) na extremidade 5’, denominada metilação Cap. Esse processamento sofrido pelo mRNA
torna a molécula mais estável e facilita o seu deslocamento para o citoplasma. Entretanto, mutações que
ocorrem nas regiões de junção dos exons podem modificar o mRNA processado, produzindo um mRNA
maduro que codifica para uma proteína diferente da original.
Alguns genes complexos tem moléculas de mRNA muito grandes, que são processadas de
diferentes formas para produzir diferentes mRNA maduros que são traduzidos em polipeptídeos diferentes.
Esse processo é denominado processamento alternativo (“alternative splicing”) e ocorre no mesmo tecido
9

mas em diferentes estágios de desenvolvimento e diferenciação e pode também ocorrer em diferentes
tecidos.
Estrutura gênica dos procariotos
Existem regiões específicas no DNA, denominadas promotores que identificam o sítio de início da
transcrição do RNA (região na qual a RNA polimerase se liga para iniciar a transcrição).
Nos procariotos, os genes que codificam as enzimas pertencentes a uma mesma via metabólica são
adjacentes uns aos outros e agrupados ("cluster") ao longo do cromossomos junto com sequências
regulatórias. Este conjunto é denominado operon, sendo que a sequência que regula a expressão desses
genes é denominada operador. O operador encontra-se antes (“upstream”) da sequência do promotor, nas
posições de -35 a -10 antes do início da sequencia codificante. Nos procariotos, a regulação destes operons
obedece a condições nutricionais ou ambientais. O estímulo da expressão de uma enzima em resposta à
presença de um substrato particular é denominada indução. A lactose, por exemplo, pode servir como fonte
de carbono e energia para a Escherichia coli. O metabolismo deste substrato depende da hidrólise de seus
componentes, glicose e galactose, pela enzima -galactosidase. A presença de lactose (indutor) induz a
síntese de -galactosidase por ativação do operon da lactose (operon lac), aumentando a taxa de ligação
da RNA polimerase ao DNA e a taxa de síntese do mRNA da -galactosidase. Na ausência do indutor o
operon é reprimido pela ação do repressor. O repressor liga-se a uma sequência especifica no operador,
bloqueando a ligação da mRNA polimerase ao promotor, inibindo assim a síntese de mRNA que codifica
para a galactosidase.
Estrutura gênica dos eucariotos
Os promotores dos eucariotos são elementos que sinalizam o sítio onde a transcrição deve iniciar-se
e, possivelmente, controlam o nível de expressão do gene associado. Geralmente estão localizados cerca
de 30 bases “upstream” do codon de iniciação (ATG) e ompreendemsêquencias denominadas “boxes” ou
“motifs”. As mais frequentes são as TATA, CAT e CG “boxes.
Além dos promotores, os genes dos eucariotos possuem sequências regulatórias adicionais
conhecidas como amplificadores ("enhancers"), que podem estar muito distantes do promotor (antes ou
depois, “upstream” ou “downstream”) e que potenciam a taxa de transcrição. É importante salientar que
estes amplificadores não são específicos e potenciam a transcrição de qualquer promotor que estiver na sua
vizinhança. A eficiência da expressão de um gene em um tecido específico depende da adequada
combinação e integração dos amplificadores, dos promotores e das sequencias adjacentes.
O genoma dos eucariotos pode apresentam os pseudogenes que são replicas de genes ativos mas
não produzem um produto reconhecível, geralmente ocorrem devido a mutações acumuladas no gene.
Transposons são elementos genéticos móveis que se deslocam de uma região para outra do
genoma e, enzimaticamente, se integram a estes locais. Desta forma, a localização destes segmentos de
DNA no genoma é instável. Estes transposons contém os genes que codificam para as enzimas necessárias
10

à sua inserção e para remobilização para outro sítio. Geralmente, a inserção de um transposon produz
mutação ou rearranjo cromossômico variável que abole a função do gene que recebe esse elemento móvel.
Polimorfismo de DNA
A análise do DNA humano progrediu na medida em que se conheceu a variabilidade de sequências
entre os indivíduos. Mini e microsatélites são sequências curtas, repetidas consecutivamente, que estão
distribuidas ao longo dos cromossomas humanos, representando cerca de 40% do DNA. Sua natureza
repetitiva faz com que sejam instáveis durante a replicação do DNA de geração para geração. A
consequência é que o número de repetições dentro de uma sequência varia largamente entre os indivíduos.
Em função dessa variabilidade (polimorfismo de DNA), cada indivíduo, exceto gêmeos idênticos, terá um
padrão ou perfil de DNA pessoal (“Fingerprinting”). Esses perfis são usados para estabelecer a identidade
dos indivíduos em diversas situações.
As sequências de microsatélites mais comuns no DNA humano são CA ou GT, dependendo de qual
das fitas de DNA está sendo lida, pois há milhares dessas sequências espalhadas no DNA humano. O
tamanho da repetição pode ser muito variável e pode fornecer excelentes marcadores genéticos para
estudar o comportamento fenotípido das famílias. A maioria dessas sequências repetitivas não produz
efeito deletério, entretanto foi demonstrado que uma forma de retardo mental em homens, a síndrome do X
frágil, é causada pela expansão de um triplete (CGG) contendo 6 a 54 repetições. Verificou-se também que
disturbios neurológicos na distrofia miotônica e na doença de Huntington, estão associados a alterações no
tamanho dos microsatétiles. Esta descoberta forneceu a base científica para o fenômeno de antecipação
no qual os disturbios genéticos tornam-se mais severos nas gerações subsequentes.
IMPORTÂNCIA DAS ENZIMAS NA BIOLOGIA MOLECULAR
Muitas enzimas que atuam na replicação e transcrição tem sido purificadas e a sua atividade
biológica usada in vitro para reações moleculares específicas diretas. A maioria dessas enzimas é isolada
de bactérias, fungos, leveduras ou virus. Essas enzimas são utilizadas das mais diversas formas, a saber:
clonagem e amplificação de genes de interesse, preparo de sondas de ácidos nucleicos, ensaios de
hibridização, entre outras aplicações.
As nucleases representam uma categoria de enzimas específicas de ácidos nucleicos que
hidrolizam as ligações fosfo-diester dos polímeros de ácidos nucleicos, uma de cada vez. As nucleases
podem requerer um grupamento hidroxil terminal livre (exonucleases), com especificidade de 3' para 5', ou
podem atuar somente em ligações internas (endonucleases). As nucleases podem ser específicas para
DNA ou RNA. As nucleases DNA-específicas podem catalizar apenas cadeias únicas (por exemplo, a S1
nuclease) ou cadeias duplas (DNAse I).
As endonucleases de restrição são nucleases encontradas naturalmente em bactérias e são
denominadas enzimas de restrição porque restringem sua atividade a DNA estranhos. O DNA do
11

hospedeiro não é atacado porque os sítios vulneráveis a suas próprias enzimas são protegidos por um
processo denominado metilação do DNA. Cada enzima é designada de acordo com o organismo do qual é
derivada. A EcoRI, por exemplo, é uma enzima obtida de Escherichia coli cepa R e foi a primeira enzima
desse tipo isolada. A maioria das enzimas de restrição reconhecem sequências de 4, 5 ou 6 nucletídeos.
Muito poucas reconhecem sequências maiores. Algumas são denominadas isosquisomeros, por clivar em
diferentes sítios dentro de uma mesma sequência (Ex. XmaI e SmaI) e por clivar o mesmo sítio mesmo
sendo obtidas de diferentes microorganismos (Ex. HindIII e HsuI). Outras enzimas reconhecem diferentes
sequências com o mesmo sítio interno (Ex. BamHI e Sau3A). Para cada região da molécula de DNA onde a
sequência específica ocorre, a enzima de restrição corta ambas as cadeias de forma reprodutível, formando
extremidades assimétricas ou coesivas (“stick-end”) ou simétricas ou cegas (“blunted-end”). Estas enzimas
são utilissímas para cortar duplas fitas de grande tamanho em fragmentos reprodutíveis e para preparar
DNA de diferentes fontes utilizado em procedimentos de clonagem.
As Ligases, como a DNA-ligase usada durante a replicação, catalizam a formação de ligações
fosfo-diester entre dois segmentos de ácidos nucleicos. As DNA ligases requerem a presença de um molde
complementar, no entanto, as RNA ligases não tem essa exigência para o processamento do mRNA.
As Polimerases catalizam a síntese do polímero de ácido nucleico complementar usando uma
cadeia precursora como molde. Essas enzimas são fundamentais para a clonagem e ampificação de genes.
A Taq DNA polimerase, por exemplo, é uma enzima extremamente útil para a amplificação do DNA in vitro
pela reação de polimerização em cadeia (PCR).
A transcriptase reversa, presente apenas nos retrovírus, cataliza a síntese de DNA a partir de
moldes de RNA ou DNA. Essa enzima tem importante função no ciclo vital dos vírus, mas pode ser usada in
vitro para procedimentos de clonagem e para o preparo de sondas de DNA a partir de mRNA. A transcrição
reversa constitui-se um método útil para produzir uma copia complementar de um gene (denominado cDNA)
a partir do mRNA. Dessa forma, o genoma dos retrovirus pode ser identificado em amostras biológicas
utilizando esse método..
Enzimas de restrição e suas aplicações
Tecnologia de DNA recombinante
Como vimos, as endonucleases de restrição são enzimas que clivam a molécula de DNA em sítios
sequência-específicos. A especificidade de cada enzima proporcionou o desenvolvimento da tecnologia de
DNA recombinante. Isto significa que fragmentos de DNA de tamanho reprodutível podem ser produzidos e
esses fragmentos contendo um gene particular, por exemplo, podem ser removidos do restante da molécula
de DNA e ser inseridos em um DNA carreador, produzindo moléculas de DNA recombinado biologicamente
funcionais. Nos primeiros experimentos de recombinação foram utilizados os vetores plasmidiais para
carrear fragmentos de DNA estranhos. Plasmideos são pequenos pedaços de DNA circular extra-
cromossômicos que ocorrem naturalmente no citoplasma das bactérias e se replicam independentemente do
DNA bacteriano hospedeiro. Utilizando uma endonuclease particular, o plasmídeo pode ser aberto e um
fragmento de DNA estranho pode ser inserido, por ação de uma DNA ligase, produzindo um plasmídeo
12

recombinate. Os plasmídeos fornecem veículos úteis para carregar fragmentos de DNA ou genes e, quando
transfectados em bactérias (usualmente Escherichia coli), podem produzir clones que ao serem replicados in
vivo, produzirão cópias múltiplas do fragmento de DNA incorporado.
Identificação de genes
O DNA extraído de um fragmento de tecido, de leucócitos ou de células do líquido amniótico,
contém milhares de genes, embora em algumas células muitos destes genes estejam desligados ou
reprimidos e somente relativamente poucos estejam ativos. A identificação de um gene ou sequência
nucleotídica especifica em todo esse DNA pode ser realizada através do método de Southern blot (E.M.
Southern, 1975). Nesse método, as enzimas de restrição são utilizadas para fragmenttar o DNA e os
fragmentos resultantes são separados por eletroforese e identificados por hibridização de ácidos nucleicos,
utilizando uma sequência de nucleotídeos específica (sonda genética) que é capaz de reconhecer o gene
de interesse.
Polimorfismo de restrição
Em muitas doenças, o defeito genético é desconhecido e a produção de sondas gene-específicas é
muito difícil ou não é possível. Entretanto, o gene produtor da doença pode estar muito proximo (ligado) a
um sítio de reconhecimento de uma enzima de restrição particular. Sabe-se que variações nas sequências
de DNA ocorrem aleatoriamente através do genoma inteiro e não estão associados a uma doença
específica. Estas variações (alterações de bases) resultam na perda de um sítio de restrição existente ou de
aquisição de um novo sitio. Tais alterações nas sequências de DNA significam que os fragmentos,
produzidos por uma enzima de restrição particular, terão diferentes comprimentos entre diferentes
indivíduos e podem ser reconhecidos por suas mobilidades em eletroforese. Eles são denominados
polimorfismos de tamanho de fragmento de restrição (RFLP) e são herdados como características
genéticas simples obedecendo as leis mendelianas de herança. Se for demonstrado, em estudos de
famílias, que um gene produtor de doença está ligado a um RFLP, este pode, então, fornecer um meio de
detectar o gene defeituoso sem efetivamente conhece-lo.
Literatura recomendada
Emery, A.E. An Introduction to Recombinant DNA in Medicine, 2a. Ed., 1995, A.E.H Emery & S. Malcon, eds., Johon Wiley & Sons Ltda, Chichester, England, 206 pág.
Lewin, B. Genes V., B. Lewin, ed., 1994, Oxford University Press, New York, USA, 1272 pág.Strachan, T. Human molecular Genetics. T. Strachan & A.P.Read, eds., 1996, Bios Scientific Publications
Ltd., Oxford, UK, 596 pág.Watson, J. Recombinant DNA, 2a. Ed., J. Watson, M. Gilman, J. Witkowrk, M. Zoller, 1992, Scientific
American Inc., New York, USA, 626 pág.
13

Aula número 2
EXTRAÇÃO DE DNA GENÔMICO
Profa.Dra. Rosario Dominguez Crespo HirataMarcos de Oliveira Machado
INTRODUÇÃO
O potencial de aplicações das técnicas de DNA recombinante em laboratórios de Bioquímica Clínica
estão se tornando altamente evidentes, especialmente para a análise do diagnóstico das várias doenças
genéticas e de outras mais. Correntemente, as análises de DNA para o diagnóstico estão disponíveis
somente para algumas desordens genéticas, mas o campo de estudo está rapidamente se expandindo e
novas análises estão sendo desenvolvidas. A maioria dos testes de DNA para o diagnóstico envolve a
extração de DNA dos leucócitos humanos. Tradicionalmente essas técnicas de extração consomem muito
tempo e possuem procedimentos pouco eficientes. A maioria dos métodos populares para esse fim, envolve
digestão com proteinase K, que leva aproximadamente dois dias, e requer um grande volume de sangue
(10-15 ml), ou utilizam solventes orgânicos como o fenol, clorofórmio e álcool isoamílico, os quais são
altamente tóxicos. Embora esses métodos consigam recuperar o DNA de alto peso molecular de uma
grande variedade de amostras, eles requerem muitos passos e podem incluir a transferência dos extratos de
DNA para recipientes adicionais ou procedimentos de lavagem do material utilizando vários filtros comerciais
ou colunas. Estes passos adicionais permitem o aumento das oportunidades de transferência cruzadas de
amostras ou a introdução de contaminantes. Tais técnicas não são bem apropriadas para o uso na rotina
dos laboratórios clínicos. Portanto, um método simples, rápido e econômico é necessário para a introdução
das análises de DNA nos laboratórios de Bioquímica Clínica.
Os estudos de ligação genética utilizando a técnica de polimorfismo de tamanho de fragmentos de
restrição (RFLP), a reação em cadeia da polimerase (PCR), a técnica de transferência de DNA (“”Southern
blot””), e outras, necessitam que o DNA extraído esteja em boas condições de uso; ou esteja livre de
contaminantes e interferentes que possam prejudicar a reação.
O uso da técnica de precipitação salina para a extração de DNA utilizando como detergente o Triton
X-100, evita o uso dos perigosos solventes orgânicos e o alto custo e demorada digestão da proteinase K.
Esta técnica envolve a desidratação e precipitação das proteínas celulares com solução de cloreto de sódio
concentrada. O DNA extraído fica livre de RNA, de proteínas e da degradação de enzimas. A quantidade e a
qualidade do DNA extraído é muito boa comparada com as outras técnicas de extração. O DNA é apropiado
para as técnicas de biologia molecular, como as que utilizam o PCR e a digestão com enzimas de restrição.
14

PREPARAÇÃO E ANÁLISE DE ÁCIDOS NUCLEICOS
Os métodos de isolamento e purificação dos ácidos nucleicos consistem de três etapas: (1) lise das
células e solubilização do DNA ou RNA; (2) médodo enzimático ou químico para remover as proteínas
contaminantes e outras macromoléculas; e (3) precipitação alcoólica para isolamento do DNA. Os protocolos
básicos geralmente são aplicáveis a um ampla variedade de materiais.
1. Isolamento de DNA genômico de tecidos e células:
O método básico compreende a ruptura do tecido com um homogeinizador, a lise celular com o
detergente iônico SDS, extração fenólica das proteinas e precipitação etanólica do DNA. Este método é útil
para preparação de DNA genômico de muitas amostras de tecido ou de linhagens celulares, com um mínimo
de consumo de tempo e trabalho. O DNA genômico produzido é adequado para amplificação por PCR ou
para a disgestão com enzimas de restrição e análise de transferência de DNA. Uma quantidade substancial
de RNA acompanha o DNA genômico, o que dificulta a quantificação por espectroscopia no UV. Algumas
amostras de DNA não amplificam bem por PCR ou não são completamente digeridas com enzimas de
restricão e devem, portanto, ser reextraidas.
Outro método compreende a solubilização dos tecidos ou células com SDS, que inativa as DNAses
endógenas. A proteinase K é usada para digerir as proteinas celulares, seguida de digestão com Rnase para
remover a maioria do RNA celular. O isolamento do DNA segue basicamente o princípio anterior. Este
método é adequado para extrair DNA genômico para análise de DNA por Southern blot ou construção de
bibliotecas genômicas. Porém não é adequado para extrair DNA de tecidos pois não há etapa de ruptura do
tecido conectivo..
O protocolo mais adequado à rotina laboratorial para avaliação de polimorfismos e mutações
genéticas compreende a lise celular com SDS, remoção das proteinas por precipitação com cloreto de sódio
concentrado (“salting-out”) e isolamento do DNA genômico por precipitação etanólica.
2. Isolamento de RNA de células ou de amostras biológicas
A maioria dos procedimentos de purificação e concentração de RNA são semelhantes aos utilizados
na purificação de DNA, exceto que 2,5 volumes de etanol devem ser usados rotineiramente para
precipitação do RNA. É essencial que toda água usada diretamente ou nos tampões seja tratada com
dietilpirocarbonato (DEPC) para inativar as RNases.
O método mais adequado para preparar RNA de boa qualidade envolve a solubilização simultanea
do tecido ou células e inativação de Rnases endógenas na presença de isotiocianato de guanidina, com
subsequente separação do RNA do DNA e das proteínas por ultracentrifugação em gradiente de cloreto de
césio. Dois outros métodos que não requerem ultracentrifugação podem se utilizados: (1) lise com
detergente não-iônico para proporcionar um método rápido para preparação de amostras de RNA
citoplasmático e (2) lise celular com isotiocianato de guanidina, remoção das proteinas pela extração fenólica
em pH ácido, seguida de precipitação do RNA. O segundo método é frequentemente utilizado para
isolamento de genoma dos virus RNA a partir de amostras biológicas
15

Outros métodos de isolamento e identificação do RNA podem ser : (1) isolamento do mRNA por
cromatografia com oligo-dT celulose; (2) separação eletroforética do RNA em gel de agarose contendo
formaldeido para detecção e quantificação de espécies específicas de RNA por Northern blotting e
hibridização; entre outros.
3. Isolamento de DNA genômico de bactérias
As bactérias de uma cultura líquida saturada são lisadas e as proteinas removidas por digestão com
proteinase K. Os debris de parede celular, polissacarídeos e as proteinas remanescentes são removidas
pela extração fenólica e o DNA genômico é recuperado do sobrenadante resultante pela precipitaçao com
etanol.
PURIFICAÇÃO E CONCENTRAÇÃO DE DNA DE SOLUÇÕES AQUOSAS
Os procedimentos de isolamento de dsDNA e ssDNA a partir soluções aquosas são úteis quando
proteinas ou solutos necessitam ser removidos, ou quando soluções de DNA necessitam ser concentradas.
O protocolo básico é apropriado para purificação de DNA a 1 mg/mL a partir de volumes menores que 0,4
ml.
1. Extração fenólica e precipitação etanólica
Um dos métodos mais úteis e usados para isolamento e concentração de ácidos nucleicos de
soluções aquosas é a extração com fenol/clorofórmio seguida de precipitação com etanol. Durante a
extração orgânica, as proteinas contaminantes são denaturadas e mantidas na fase orgânica ou na interface
entre as fases orgânica e aquosa, enquanto que os ácidos nucleicos permanecem na fase aquosa. O fenol
usado nesse protocolo deve ser tamponado para prevenir que os produtos oxidados presentes no fenol
danifiquem os ácidos nucleicos. Alcool amílico frequentemente é adicionado à mistura fenol/clorofórmio
quando ocorre a formação excessiva de espuma durante a extração.
Durante a precipitação com etanol, os sais e outros solutos como os resíduos da extração
fenol/clorofórmio permanecem em solução enquanto os ácidos nucleicos formam um precipitado que pode
ser facilmente separado por centrifugação. Na presença de altas concentrações de cátions monovalentes
(0,1 a 0,5 M), o etanol induz uma transição estrutural nas moléculas de ácido nucleico promovendo a
agregação e precipitação das mesmas. Entretanto, como vários sais e pequenas moléculas orgânicas são
solúveis em etanol a 70%, a precipitação etanólica e lavagem do sedimento elimina os sais no DNA.
Embora cloreto de sódio, acetado de sódio, e acetato de amonio sejam capazes de induzir a precipitação, é
mais difícil remover cloreto de sódio devido a sua baixa solubilidade em etanol a 70%. O sal recomentado
para a maioria das aplicações de rotina deste método é o acetato de sódio a 0,3 M (concentração final), que
é mais solúvel em etanol que cloreto de sódio a 0,3 M e, portanto, menos facilmente precipitável com a
amostra de ácido nucleico. Para amostras contendo dodecil sulfato de sódio (SDS), o sal recomendado é
cloreto de sódio 0.2 M, pois o SDS é solúvel em etanol nessas condições.
16

Soluções de DNA diluídas: quando soluções de DNA estão diluidas (< 10 g/mL) ou quando
menos que 1 g de DNA está presente, deve-se aumentar a proporção de etanol para o volume aquoso
(3:1) e aumentar o tempo de precipitação (4 a 16 horas), incubando-se a -20oC (ou em gelo seco). A
centrifugação para separação do DNA precipitado deve ser feita a baixa temperatura para assegurar o
máximo de recuperação do DNA dessas soluções.
Pequenas quantidades de DNA (nanogramas): pode-se utilizar um ácido nucleico carreador, como
o tRNA de E. coli, levedura ou fígado bovino, em concentrações de 10 g/mL. Nessa condição o DNA é
coprecipitado com o tRNA. O tRNA não interfere com a maioria das reações enzimáticas, mas será
eficientemente fosforilado pela T4 polinucleotídeo quinase e não poderá ser usado se esta enzima for usada
em marcações subsequentes. Recuperação de pequenas quantidades de fragmentos curtos de DNA e
oligonucleotídeos pode ser aumentada pela adição de cloreto de magnésio a uma concentração inferior a 10
mM antes da adição do etanol. Entretanto, o DNA precipitado de soluções mais concentradas que 10 mM
de íons magnésio ou fosfato são dificeis de dissolver e devem ser dulidas antes da precipitação com etanol.
Se a amostra de DNA está em baixa concentração em um grande volume, o volume pode ser reduzido pela
extração repetida com sec-butanol (2-butanol). Cabe lembrar que apenas a água é removida com essa
extração, todos os solutos e sais são concentrados com os ácidos nucleicos. Após essa etapa, o ácido
nucleico deve ser extraido com fenol/clorofórmio e precipitado com etanol.
DNA em grandes volumes aquosos: a extração pode ser simplesmente ampliada usando o
mesmo procedimento. (tubos de poliestireno não resistem a mistura fenol/clorofórmio). As etapas de
centrifugação à temperatura ambiente não podem exceder velocidades de 2500 rpm (1200 x g), pro 5
minutos. O precipitado etanólico deve ser centrifugado em um tubo Corning de parede reforçada por 15
minutos em fotor de angulo fixo a 10.000 rpm (8000 x g), a 4oC. Tubos de vidro devem ser siliconizados para
facilitar a recuperação de pequenas quantidades de DNA (< 10g).
Remoção de oligonucleotídeos e trifosfatos: Pequenos oligonucleotídeos (< 30 bp) e
nucleotídeos não incorporados usados na marcação ou outras reações de modificação do DNA podem ser
efetivamente removidos das soluções contendo DNA por duas etapas de precipitação com etanol na
presença de acetato de amonia. Este procedimento não é suficiente para remover completamente grandes
quantidades de ligadores (“linkers”) utilizados nos procedimentos de clonagem. Acetato de amonia não é
recomendável se a amostra de ácido nucleico for fosforilada na extremidade 5’ pela T4 quinase ou na
extremidade 3’ com a transferase terminal, pois os íons de amonio tesiduais inibem essas duas enzimas.
Variações do procedimento de precipitação: O isopropanol pode substituir o etanol, quando se
deseja manter mínimo o volume dos ácidos nucleicos precipitados. O isopropanol é menos volátil que o
etanol e é, portanto, mais difícil de remover por evaporação. Alguns sais são menos solúveis em
isopropanol, e podem ser precipitados com os ácidos nucleicos. É recomendado que a precipitação com
isopropanol seja seguida imediatamente por uma precipitação onvencional com etanol para eliminar
isopopropanol residual e o sal. Alternativamente, cloreto de lítio pode ser usado como o sal de precipitação,
pois o cloreto de lítio é muito mais solúvel no etanol e o precipitado resultante é relativamente isento de sal.
Cloreto de lítio deve ser evitado, entretanto, se o precipitado de RNA for usado como molde para a
transcrição reversa após precipitação.
17

Grandes mRNA e rRNA podem ser purificados de dsDNS e pequenos RNAs (tRNA e 5S RNA) por
duas etapas de precipitação seletiva com cloreto de lítio na ausencia de alcool, seguida de uma precipitação
convencional com etanol.
Soluções concentradas de ácidos nucleicos: se a concentração de ácidos nucleicos é muito alta
( > 300 g/mL), um precipitado pode-se formar sem resfriamento durante a precipitação etanólica. Se um
precipitado visível é formado, não é necessário esfriar a amostra ou aguardar algum tempo antes de separar
o precipitado por centrifugação.
2. Fracionamento dos ácidos nucleicos
Geralmente todos os protocolo em biologia molecular requerem, em alguma etapa, o fracionamento
dos ácidos nculeicos. Técnicas cromatográficas são apropriadas para algumas aplicações e podem ser
usados para separação dos plasmídeos do DNA genômico, bem como separação de DNA genômico dos
debris no lisado celular. A eletroforese, entretanto, tem muito mais resolução que métodos alternativos e é
geralmente o método de fracionamento de escolha. As separações eletroforéticas podem ser analíticas ou
preparativas e podem envolver fragmentos com peso moleuclar variando de menos que 100 Da a mais que
108 Da. Uma variedade de sistemas eletroforéticos foram desenvolvidos para acomodar essa ampla faixa
de variação. Além disso, um fragmento individual pode ser identificado por tecnica de hibridização após a
separaração eletroforética de uma mistura complexa (Southern blot). Este método tem contribuido
grandemente para ao mapeamento e identificação de sequencias de cópia única ou múltipla em genomas
complexos, e facilitado os experimentos iniciais de clonagem de DNA eucarioto.
3. Purificação e isolamento de DNA
Os protocolos utilizados para purificação de DNA são geralmente divididos em três categorias: (1)
eluição do DNA de um gel de agarose por diálise, (2) dissolução do gel de agarose e recuperação do DNA
por ligação a partículas de vidro, e (3) eluição pelo aquecimento da agarose de baixo ponto de fusão a 70oC,
e recuperação do DNA pela extração fenólica e precipitação etanólica ou usando uma matrix que liga
especificamente o fragmento de DNA. Atualmente, há vários sistemas de purificação de fragmentos de DNA
no mercado. Esses sistemas diferem no tamanho e o tipo de ácido nucleico que pode ser eficientemente
isolado; nos efeitos dos sais, dos solventes orgânicos e de outras substâncias na eficiência de ligação da
matriz; e no tipo de solvente que é usado para eluir o oligonucletídeo ou o polinucleotídeo da matriz.
Matriz de sílica (vidro) - na presença de iodeto de sódio, DNA e RNA ligam-se seletivamente à
matriz de sílica. Outras substâncias presentes (sais, solventes, nucleotídeos, proteinas, nucleotídeos etc)
são lavados com uma solução de etanol tamponada. O polinucleotídeo ligado é eluido em um reduzido
volume de tampão Tris-EDTA (TE) ou água.
Resina de purificação - é uma resina de purificação com propriedades similares aos sistemas de
matriz de sílica (Magic1, Promega). Esta matriz liga polinucleotídeos de dupla fita mais eficientemente que os
de fita simples, e pouco eficientemente os RNAs pequenos (tRNA), DNA de dupla fita de menos que 220 bp
ou oligonucleotídeos.
18

Adsorção - é um sistema de purificação de ácidos nucleicos, que contém um adsorvente estável
(NENSORB 20, DuPont-NEN) que liga quantitativamente proteinas, RNA e DNA, incluindo oligonucleotídeos
contendo menos que 12 resíduos. A ligação não é afetada por altas concentações de sais, ureia, ou outras
substâncias, mas inibida por alguns solventes orgânicos. O material não ligado é removido com água e o
DNA ou RNA (mas não proteinas) é quantitativamente eluido com um solvente alcoólico.
Gel filtração - os ácidos nucleicos podem ser purificados, separados e fracionados usando colunas
de gel filtração ou colunas de centrifugação (spin columns).
QUANTIFICAÇÃO DOS ÁCIDOS NUCLEICOS
A concentração de DNA ou RNA pode ser estimada por espectrofotometria de absorção no UV ou a
concentração de DNA pode ser realizada por espectroscopia de fluorescencia. Cubetas de quartzo devem
ser utilizadas para a espectrofotometria no UV, enquanto que as de plástico ou vidro podem ser usadas para
a região do visível. Um método alternativo é o método de placa de brometo de etídio agarose. .
Literatura recomendada
Davis, L.G. Basic Methods in Molecular Biology. 2a. Ed., L.G. Davis, W. M. Kuehl, J.F. Battey, Eds. 1994, Appleton & Lange, Norwalk, CN, USA, 777 p.
Ausubel, F.M. Short Protocols in Molecular Biology, 2nd ed., F.M. Ausubel, Ed.; Green Publising Associates and John Wiley & Sons, 1992, New York, NY.
Sambrook, J. Molecular Cloning: a laboratorial manual. J. Sambrook, E.F. Fritsch & T. Maniats, 3 volumes, 2nd ed., 1989. Cold Spring Harbor Laboratory Press, New York, NY.
19

Aula número 3
PRINCÍPIOS DE ELETROFORESEProf. Assoc. Mario Hiroyuki Hirata
Profa. Dra. Rosario Dominguez Crespo Hirata
INTRODUÇÃO
Eletroforese é uma técnica de separação que envolve o movimento de partículas com cargas em
solução, sob ação de um campo elétrico. Portanto, qualquer partícula ou molécula carregada pode se mover
nessas condições, e consequentemente podem ser separadas, desde que tenham diferentes cargas.
A separação eletroforética das partículas pode ser realizada em meio líquido(eletroforese livre de Tisellius)
ou em um suporte inerte.
A carga eletrica aplicada sob a solução é realizada através de eletrodos que são colocadas na
solução. Um ion migra através da solução na direção ao eletrodo de carga oposta. Portanto, uma partícula
carregada positivamente(catiosn) migra para o polo negativo (catodo), enquanto as partículas
negativas(anions) migram para o polo positivo(anodo).
PRINCÍPIOS BÁSICOS DA ELETROFORESE
Terminologia:
ânion: partícula carregada negativamente ou íon
cátion: partícula carregada positivamente
tampão: Uma mistura de substâncias que doam e aceitam proton com a função de manter a concentração
do proton (pH) constante ou dentro de um valor próximo. Um tampão pode ser feito com uma mistura de um
ácido fraco com o respectivo sal. Ex: ácido acético e acetato de sódio.
Condutividade: a propriedade de uma substância conduzir a corrente elétrica. Numa solução iônica, a
soma do produto da concentração da carga e a mobilidade da mesma.
eletrodos: substâncias em contato com um condutor. A substância estão conectadas a fonte elétrica.
Fonte elétrica: Equipamento que transforma corrente alternada em corrente contínua, que possa ser
controlada por um potenciometro, que permite variar a tensão, a corrente, a carga elétrica que se quer
aplicar no sistema. Esse equipamento, pode medir a tensão elétrica (em Volts), a corrente
elétrica(Amperagem), e a potência elétrica (Watts).
eletro-osmose; tendência da solução se mover em relação a uma substâncias estacionária adjacente,
quando uma diferença de potencial é aplicada.
força iônica: É a soma da concentração de todos ions na solução, medido pelo quadrado de suas cargas.
mobilidade: a velocidade de uma partícula ou ion que é dado pela voltagem aplicada. Uma medida relativa
de quanto rápido um ions se movimenta em um campo elétrico.
20

Mobilidade efetiva: A real mobilidade de uma substância sob certas condições. Geralmente é menor que a
mobilidade devido a menor carga, ou resistência do suporte ou meio.
poder de resolução: E’ a habilidade de separar substâncias que migram muito próximas.
gel: É uma malha de fibras, ou de polímeros que é sólida, mas retém uma grande quantidade de solventes
nos poros ou nos canais internos das malhas.
FATORES QUE INFLUENCIAM NA SEPARAÇÃO E MOBILIDADE
ELETROFORÉTICA
A aplicação de um campo elétrico a uma solução que contém partículas carregadas pode separá-
los. A separação depende de vários fatores que influenciam e que podem ser resumidas em: Força aplicada
nas partículas (voltagem), carga da partícula, pH do meio, condutividade do meio, resistência da matrix,
conformação da partícula, força iônica do meio, temperatura, eletroendosmose, etc
1. Força na partícula
A força exercida em uma partícula carregada depende do campo elétrico, voltagem ou volts por
centímetro, e a carga da partícula, Q. A força sob a partícula carregada é o produto:
Feletrica=QV
A força elétrica, Feletrica, quando exercida na partícula, causará o seu movimento. No entanto, o
movimento da partícula no solvente será também influenciado pela resistência, devido a viscosidade, a
resistência, a qual exerce uma força é proporcional a velocidade:
Fresistência= fv
A constante de proporcionalidade, f é chamada de coeficiente de fricção (medida de resistência de
uma partícula oferece quando em movimento em um solvente).
2. Coeficiente de fricção depende da viscosidade do solvente e do tamanho, e forma da partícula
Quanto maior a viscosidade, menor o movimento. Quanto maior ou mais assimétrica a partícula,
menor seu movimento através do solvente. O coeficiente de fricção de uma grande partícula tais como
proteinas é uma propriedade característica de cada elemento.
3. Mobilidade da partícula
Quando um campo elétrico é aplicado em uma partícula carregada, ela inicia a migração. A força
eletroforética e a fricçional se opõe uma a outra, e a velocidade dessa aumenta até as forças se igualarem
(Feletrico = Fresistencia ). A velocidade, v, que uma particula alcança em um campo elétrico, V, é determinado por
duas propriedades das partículas - sua carga e seu coeficiente de fricção. Consequentemente, o valor de
v/V é também uma propriedade característica das partículas e é importante o suficiente para receber o seu
próprio nome: mobilidade.
4. Efeito do pH do meio
21

Cada íon possui sua carga e mobilidade muito particular. No entanto, quando a solução contém uma
substância a qual o pK é próximo do pH do meio, esta substância ocorre em ambas as formas: a carregada
e a não carregada.
A fração com uma carga será dependente do pK da substância e o pH da solução. Quando o pH é
igual ao pK de um ácido fraco, somente 50% das partículas estarão carregadas. Uma unidade de pH
abaixo da pKa, 90% das partículas estarão sob a forma carregada. Desde que a carga efetiva de uma
substância varia com o pH, a sua mobilidade efetiva também varia com o pH.
O pH da solução é escolhido de tal forma, que a partícula a ser separada pela eletroforese, se
mantenha em um estado máximo de carga, para sua máxima separação. Os tampões são substâncias
iônicas por si, portanto, exercem a função de manterem o pH o mais próximo possível da ideal e fazem
parte do processo.
5. Condutividade e movimento dos íons
Em qualquer sistema elétrico, a corrente produzida é proporcional a voltagem aplicada;
V = resistência X Corrente ou V = corrente___condutividade
Na eletroforese, a corrente é um fluxo de íons(em ambas direções). A condutividade é a soma das
concentrações multiplicado pela mobilidade efetiva de todos os ions presentes. Um íon com maior
mobilidade efetiva carreia uma maior fração da corrente
A voltagem, condutividade, e a corrente portanto estão todos relacionados. Se a condutividade está
aumentada pelo aumento da concentração salina a corrente se mantém constante, a voltagem deve
decrescer. Se decresce a voltagem reduz a força elétrica, Feletrica, nas partículas carregadas, diminuindo o
movimento das macromoléculas. O aumento do tempo seria necessário para uma separação, e a resolução
diminui devido ao aumento da difusão. Se a condutividade está aumentada numa voltagem fixa, a corrente
deve aumentar, portanto aumentando o calor elétrico gerado pelo sistema, desde que o aquecimento é
proporcional ao quadrado da corrente. O excesso do aquecimento produz distúrbios convectivo nas
soluções, a qual distorce o padrão eletroforético e pode também desnaturar as macromoléculas. Desde que
o aumento da condutividade mesmo em voltagem fixa ou corrente tenha efeito deletério, resultados ótimos
podem ocorrer quando se conserva a concentração dos ions e portanto a condutividade em valores
moderados.
6. Carga e conformação
Como já se discutiu, o que determina a migração eletroforética é a carga da partícula que se quer
separar. Muitas vezes, a distribuição das cargas não estão desvinculadas a sua conformação estérica,
portanto, pode-se alterar a carga de uma partícula dependendo de sua conformação, principalmente em se
tratando de macromoléculas. As macromoléculas, em seu estado relaxado, tendem a apresentar alguns
eletrólitos ligados fortemente, portanto levando a uma alteração na sua mobilidade.
7. Atmosfera iônica e o potencial zeta
Contra-íons, ou íons de carga oposta, naturalmente tendem a circundar próximos a grupos
carregados das macromoléculas. No entanto, eles não chegam a neutralizar as cargas das macromolécuals
mas, por sua vez estão localizados próximos dos grupos carregados das macromoléculas, formando uma
dupla camada de carga em torno dessas, denominadas de atmosfera iônica.
22

As macromoléculas movem-se com a camada de hidratação e com os contra ions. Esses reduzem a carga
efetiva das macromoléculas, a um nível dado pelo potencial zeta. Esse potencial significa a energia
produzida pela carga efetiva das macromoléculas na superfície de contato, que é a região limite do
complexo macromolecular na solução e o material que o está envolvendo.
8. Suportes
A grande meta da eletroforese é a separação de uma mistura heterogênea de substâncias iônicas
seja ela macromoléculas ou íons simples, em zonas completamente identificáveis. Os suporte devem
permitir a penetração do material a ser separado o máximo possível e ainda delimitar o fluxo de volume
(eletroforese convencional). A maioria dos suportes oferecem esse princípio determinando o tamanho do
poro para o movimento eletroforético das macromoléculas. O tubo capilar também exerce esse efeito.
9. Eletroendosmose
O suporte não deve interagir com as moléculas a ser separadas, isso poderia impedir ou inibir a
separação. Uma interação comum que pode ocorrer, não é a adsorção usual do material. Mais comumente
é o efeito dos grupos carregados que estão ligados ao suporte, que resulta em eletro-endosmose. Como
exemplo típico temos o suporte ágar, que é muito utilizado na eletroforese. O ágar é uma mistura de
agarose e agaropectina. A agaropectina tem um número relativamente grande de grupos carboxil, que em
pH neutro tem contra-íons. Se o campo elétrico for aplicado, os contra-íons irão se mover, mas o grupo
carboxil ligados a matrix (agar) não migrarão. Os contra-íons carreia solvente o suficiente para que produza
um fluxo significativo nesta direção, esse fenomeno denomina-se de eletro-osmose, que as vezes é
denominado de endosmose.
TIPOS DE SUPORTE
Os tipo de suporte podem variar desde uma solução de sacarose até em substância pouco solúveis como
fibras de celulose. Os suportes de meio contínuo mais comuns são: agar, agarose, poliacrilamida e amido.
A porosidade ou a média do tamanho do poro em alguns suportes é fixo. Entretanto, em alguns suportes
podem ser controlados. Como exemplo, a mudança da concentração do gel de agarose, agar e amido, pode
mudar o tamanho do poro. O tamanho dos poros do gel de poliacrilamida mais utilizados para
macromoléculas está em torno de 5% a 10%, que separam proteínas de 15.000 a 250.000 Daltons. Esse
tipo de suporte separa tanto pelo tamanho do poro como por carga elétrica.
TIPOS DE ELETROFORESE.
a. Dependendo da matrix
Eletroforese livre de Tiselius
Eletroforese em acetato de celulose
Eletroforese em gel de agar
Eletroforese em gel de agarose
Eletroforese capilar
23

b. pH de separação
pH ácido
pH neutro
pH alcalino
APLICAÇÕES DA ELETROFORESE NAS TÉCNICAS MOLECULARES
1. Gel de Agarose
Um dos métodos eletroforéticos mais comuns utilizados na prática diária em biologia molecular é a
de gel em agarose. Essa técnica muito simples, rápida, prática e apresenta boa resolução na separação dos
fragmentos de DNA, se comparados com outras bem mais demoradas como a separação por
ultracentrifugação em gradiente de densidade de sacarose.
Uma boa separação eletroforética do DNA utilizando gel de agarose depende de vários fatores:
1.1. Tamanho molecular do DNA.
As moléculas lineares, dupla fita de DNA migram inversamente proporcional ao log10 do peso
molecular através do gel de agarose.
1.2. concentração da agarose.
O fragmento do DNA migra em diferentes proporção através do gel contendo diferentes
concentraçào da agarose. Existe uma relação linear entre a mobilidade logaritimica do DNA e a
concentração do gel que é baseada numa equação matemática(Helling e cols , 1974) figura 1.
Log=logo- Kr
Onde o é a mobilidade eletroforética livre e Kr é o coeficiente de retardamento, = a constante que
é relacionada as propriedades do gel e o tamanho e a forma da molécula em migração.
Baseada nessa correlação podemos separar várias moléculas de DNA (de variado tamanho) nas
diferentes concentrações de agarose.
Porcentagem de agarose intervalo da efficiência da separação da molécula de
DNA em Kb
0,3% 5 a 60
0,6% 1 a 20
0,7% 0,8 a 10
0,9% 0,5 a 7
1,2% 0,4 a 6
1,5% 0,2 a 4
2,0% 0,1 a 3
24

1.3. Conformação do DNA
A forma circular (forma I), forma circular contorcida(forma II) e forma linear (forma III) do DNA com
a mesmo peso molecular migra através do gel de agarose em diferentes posições (Thorne, 1966, 1967). A
mobilidade relativa das 3 formas são dependentes primariamente da concentração do gel, mas são também
influenciados pela corrente aplicada, força iônica do tampão e da densidade superhelicoidal da torção na
forma I do DNA (Johnsom and Grossman , 1977). Em algumas condiçoes, a forma I do DNA migra mais
rápido que a forma III, em outras condições pode ser de ordem inversa. Um método geral para identificar as
diferentes conformações do DNA é utilizar na corrida eletroforética em agarose diferentes concentrações de
brometo de etídio(concentração crescente). Quanto mais brometo de etidio mais se liga ao DNA. As torções
superhelicoidal negativas da a forma I do DNA vão sendo progressivamente removida e seu índice de
migração diminui. Numa concentraçao crítica do corante livre, onde não permanece a forma superhelicoidal,
o índice de migração da forma I do DNA esta no seu mínimo valor. Se ainda mais brometo de etídio for
adicionado, torções superhelicoidais positivas são gerados e a mobilidade da forma I do DNA aumenta
rapidamente. Simultaneamente, as mobilidades das formas II e formas III do DNA são diferentemente
diminuidas como consequência da neutralização da carga e da maior tenacidade é dada ao DNA pelo
brometo de etídio. Para a maioria das formas I de DNA, a concentração critica do brometo de etídio livre é
de 0,1 a 0,5 g/ml
A corrente aplicada: A baixa voltagem, a razão da migração dos fragmentos de DNA linear é proporcional
a da voltagem aplicada. Entretanto, a medida que a carga elétrica aumenta, a mobilidade dos fragmentos de
DNA de alto peso molecular aumenta diferentemente. Dessa maneira o intervalo efetivo da separação do gel
de agarose decresce a medida que a voltagem aumenta. Para obter a máxima resolução dos fragmentos de
DNA, o gel deve ser submetido a uma corrente de no máximo 5V/cm
Composição das bases e temperatura: O comportamento eletroforético do DNA em gel de agarose não é
significantemente afetado pela composição das bases do DNA ou pela temperatura do gel na corrida.
Dessa forma, os geis de agarose e a mobilidade eletroforética dos fragmentos de DNA de diferentes
tamanhos não muda entre 40C a 300C. Em geral, os geis de agarose são submetidos a corrida em
temperatura ambiente. No entanto, em concentrações muito baixas (0,5%) os géis são pouco consistentes,
sendo convenientes trabalhar em baixas temperaturas, pois esses géis ganham mais firmeza, da mesma
forma os géis de agarose de baixo ponto de fusão(“low melting” agarose).
1.4. Tampões utilizados
Existem uma variedade de tampões que podem ser utilizados para separação eletroforética de DNA.
25

TAMPÕES CONCENTRAÇÃO FORMULAÇÃO
Tris-acetato (TAE) 0,04M Tris acetato
0,01M EDTA
50X 242g tris base
57,1 ml ácido acético
100ml EDTA 0,5M(pH8,0)
Tris-fosfato (TPE) 0,08M Tris-fosfato
0,002M EDTA
10X 108 g Tris Base
15,5ml de ac. fosfórico 85%
40ml EDTA 0,5M (pH8,0)
Tris-borato (TBE) 0,089M Tris borato
0,089M ac. bórico
0,002M EDTA
5X 54g Tris-base
27,5g ác. bórico
20 ml EDTA 0,5M (pH8,0)
1.5. Tipos de Agarose
Muitos tipos de agarose estão disponíveis, o que se recomenda para uso diário é o tipo II, baixa
eletroendosmose. O inconveniente desta agarose é que possue contaminantes de polissacárides sulfatados
que inibem enzimas, como ligases, polimerases, e endonucleases. No entanto, a sua utilização é muito
difundida, pois o DNA pode ser purificado e ser utilizado com sucesso nas clonagens e como substratos de
reações com essas enzimas
1.6. Coloração do DNA em gel agarose
O método mais conveniente para visualizar o DNA no gel de agarose é a coloração por brometo de
etidio, que fluoresce sob luz ultra violeta. Esse composto contém um grupo planar, que intercala entre as
bases do DNA. A posição fixa do grupo e sua proximidade às bases causa uma ligação do corante com o
DNA, que se destaca com um aumento da fluorescência comparada com o corante em solução sob a forma
livre. A radiação UV absorvida pelo DNA a 260 nm é transmitida para o corante, ou a irradiação absorvida a
300 nm e 360 nm pelo corante ligado propriamente dito, é emitido a 590nm na região da cor vermelho
alaranjada, do espectro visível.
O brometo de etídio pode detectar tanto os ácidos nucleicos de simples ou de dupla fita. No
entanto, a afinidade do corante pela fita simples é relativamente baixa, e a fluorescência emitida é fraca.
Geralmente o brometo de etídio é incorporado no tampão de corrida e no gel a 0,5ug/ml. Nesse caso
devemos considerar que a mobilidade linear das fitas duplas está reduzida na presença do corante em
aproximadamente 15%. Uma vantagem deste procedimento é a possibilidade de visualizar o gel durante a
separação eletroforética, sem a necessidade de remover o gel do suporte e da cuba, simplesmente incidindo
a luz UV sob o gel. Mas, se houver necessidade da coloração posterior basta imergir o gel numa solução de
brometo de etídio, no próprio tampão de corrida, po r 45 minutos a temperatura ambiente. A descoloração
normalmente é dispensada, exceto se houver excesso de corante. Quando a quantidade de DNA é muito
reduzida, a pouca fluorescência do brometo de etídio pode interferir na visuallização da banda de DNA.
Neste caso recomenda-se imergir o gel em uma solução de sulfato de magnésio a 1mM por uma hora, a
temperatura ambiente
26

1.7. Documentação:
O gel de agarose é facilmente documentado, utilizando um sistema de fotografia instantânea ou
sistema automatizado de captura de imagem. O filme mais sensível para essa finalidade é fornecida pela
Polaroid, tipo 57 ou 667, asa 3000. Sob a luz UV de no mínimo 2500uW/cm 2 e uma boa máquina ,
utilizando filtro laranja, expõe-se por alguns segundos. Recomenda-se para a máquina Polaroid modelo DS
34 uma abertura de 5,6 ou 8 e uma velocidade de 4 ou 8 segundos, mas pode variar com a intensidade das
bandas e do tamanho do gel. Para padronização tente vários tempos de exposição com várias aberturas.
2. Gel de poliacrilamida
O gel de poliacrilamida é um suporte bastante utilizado em biologia molecular para separação e
caracterização de fragmentos de tamanho menores de DNA, que se tem dificudade por geis de agarose.
Descrito por Raymond & Weintrab em 1959. O produto de polimerização da acrilamida, que é o monômero,
com a N.N'metilenobisacrilamida, que realiza a reação de "cross linking" ou reação cruzada, gera o suporte
de poliacrilamida. A polimerização ocorre por ligação vinílica, sob ação de catalisadores, o perssulfato de
amônia, cuja fonte de radicais livres é o N,N,N'N' tetrametilenodiamina(TEMED). Portanto, o oxigênio é um
inibidor da reação. O polímero formado são cadeias lineares de poliacrilamida, cuja reação cruzada entre
elas é dada pela bisacrilamida. O comprimento médio das cadeias é determinado pela concentração relativa
da acrilamida e a quantidade de bisacrilamida. Portanto a relação entre a quantidade de acrilamida e
bisacrilamida determina as propriedades físicas do gel, que darão subsídios para separação nesse tipo de
gel.
O gel de poliacrilamida é utilizado para os fragmentos menores que 1kb. Eles podem ser preparados
em uma variada concentração (3,5% a 20%), dependendo do tamanho do fragmento de interesse para a
separação e identificação.
ACRILAMIDA (%) INTERVALO DE SEPARAÇÃO (NUCLEOTÍDEOS)
3,5 10-1000
5,0 80-500
8,0 60-400
12,0 40-200
20,0 10-100
Os géis de poliacrilamida devem ser preparados entre duas placas de vidro, pois necessita de um
ambiente isento de oxigêncio para se polimerizar.
O tamanho do gel pode variar de 10cm de altura por 10cm de largura até 20 a 30cm de largura por
40 a 100 cm de altura dependendo da necessidade do sistema; de espessura normalmente para DNA
utiliza-se 0,75 a 1,5 mm.
27

Literatura recomentada
Kaplan, L.A. & Pesce, A.J. Clinical Chemistry: Theory, Analysis, Correlation, 3a. Ed., J.F. Shanahar & J. Roche, eds., 1996, Mosby-Year Book, Inc., St Louis, USA.
Sambrook, J. Molecular Cloning: a laboratorial manual. J. Sambrook, E.F. Fritsch & T. Maniats, 3 volumes, 2nd ed., 1989. Cold Spring Harbor Laboratory Press, New York, NY.
28

Aula número 4
REAÇÃO DE POLIMERIZAÇÃO EM CADEIA (PCR)
Prof. Assoc.Mario Hiroyuki HirataProfa. Dra. Rosario Dominguez Crespo Hirata
PRINCÍPIOS DA REAÇÃO DE PCR
A reação de polimerização em cadeia (“Polimerase chain reaction”, PCR) é a técnica que permite a
amplificação do DNA ou RNA in vitro, utilizando-se basicamente de uma reação enzimática catalizada pela
polimerase (enzima termoestável) cuja atividade depende de ions Mg++, e ocorre em 3 etapas: “Melting”(ou
desnaturação que consiste na separação da dupla fita do DNA a ser amplificado), “annealing”( anelamento
ou hibridização- ligação do iniciador ou “primer” ao DNA a ser amplificado) e “extension”(extensão ou seja a
polimerização propriamente dita) Metodologicamente a técnica de PCR requer três passos (SAIKI et al.,
1.988):
1. Desnaturação - Inicialmente é necessário que as duas fitas de DNA a ser amplificado sejam separadas.
A elevação da temperatura entre 90 a 95 ºC promove a separação da fita dupla de DNA em duas fitas
simples. Este procedimento é chamado “Desnaturação” ou “melting”, e muitas vezes a temperatura de
desnaturação é determinada empiricamente e depende de alguns fatores como a quantidade de guanina
e citosina (GC) do fragmento DNA alvo ou de interesse.
2. Anelamento - A polimerase para assumir suas funções, isto é, anexar as bases complementares,
transformando as fitas simples em fitas duplas, necessita de um fragmento de DNA já ligado na região
previamente escolhida. A solução, então, é demarcar as extremidades do DNA-procurado ou de
interesse nas duas longas fitas simples, tornando-as duplas, apenas nesse intervalo. Para isso
adicionam-se, à solução, os “primers” ou iniciadores que são pequenos fragmentos sintéticos de DNA de
fita simples, oligonucleotídeos de 20 a 30 bases nitrogenadas de comprimento, que são sintetizados in
vitro baseado na sequência do DNA a ser amplificado sendo eles complementares à seqüência do
segmento de DNA de interesse. O conhecimento da seqüência procurada é naturalmente pré-requisito
para a síntese dos “primers”. Os “primers” perseguem na reação as regiões, as quais foram escolhidas,
para hibridizar-se às seqüências complementares nas duas fitas simples (usualmente 5’). Nas duas fitas
surgem, assim, um pequeno fragmento de DNA de dupla fita intacta. A hibridização dos “primers”
descrito como “Annealing” (do inglês), deve ser feito a uma tempertura inferior à da desnaturação (45 a
60 C).
3. Extensão - Quando os “primers” encontram e ligam-se aos segmentos complementares, a DNA-
Polimerase pode assumir sua função. Inicia-se a partir dos pequenos fragmentos de DNA de fita dupla
(resultado do anelamento do “primer”), incorporando um a um os nucleotídeos correspondentes, isto é,
as bases juntamente com as moléculas de açúcar e fosfato. A DNA-Polimerase liga os nucleotídeos
entre si, completando assim as fitas simples e tornando-as duplas (extensão). A DNA-Polimerase não 29

reconhece apenas o bloco de início como também consegue diferenciar as duas extremidades dos
“primers” (3’ e 5’). Ela inicia a reação sempre pela extremidade 3’ do “primer”, completando a fita simples
daí em diante, portanto, sempre na direção do fragmento procurado.
Os fragmentos de DNA recém-formados fornecem mais moldes para a montagem de novas fitas
nos ciclos subseqüentes, no entanto, com região já determinada, resumindo, temos uma reação em cadeia.
Após cada ciclo, o número de fragmentos se duplica: de um formando-se 2, e então novamente 4, 8, 16, 32,
64, 128... de tal forma que pode ser definida matematicamente por:
N = número de moléculas amplificadasNo = número de moléculas iniciaisn = número de ciclos de amplificação
O modelo teórico considera que a eficiência do processo de amplificação corresponde a 100%, o
que não é observado na prática. Experimentalmente, a eficiência da reação está abaixo da ideal e gira
entorno de 78% a 97%, dependendo do gene amplificado. Teoricamente após 20 ciclos têm-se cerca de um
milhão; após 30 ciclos, cerca de um bilhão de cópias do fragmento de DNA de interesse.
Automação da PCR
O procedimento da PCR foi tão aperfeiçoada desde a sua primeira descrição, que na atualidade
transcorre de forma totalmente automática. Uma importante condição para isto foi a substituição da DNA-
Polimerase originalmente utilizada (extraída de E. coli) por uma DNA-Polimerase termoestável extraída da
bactéria Thermus aquaticus (Taq-polimerase). Desta forma, os vários ciclos transcorrem, seguidamente,
em um único tubo de ensaio, sem necessidade de interrrupção para a adição de nova DNA-Polimerase
ativa. Todos os reagentes necessários (o DNA-template, os “primers”, a DNA-Polimerase, e os dNTPs) são
misturados com uma solução tampão que ajusta o pH ótimo da reação.
Um aparelho de PCR ou um termociclo repete o correspondente programa de temperaturas. O
operador deve apenas programar e dar início para que transcorra a amplificação. Um ciclo dura em média
três minutos. Assim, em uma hora, podem ser produzidas cerca de um milhão de cópias e, em menos de
duas horas, cerca de um bilhão de cópias da seqüência de DNA de interesse.
O produto amplificado pode alcançar aproximadamente 106 cópias em 30 ciclos e pode ser realizado em 2
horas.
A PCR é uma ferramenta na biologia molecular tão poderosa quanto a descrição das enzimas de
restrição e o Southern blot. Anunciado na Conferência da Sociedade Americana de Genética Humana de
1985, a técnica de PCR tem sido utilizada de forma multidisciplinar com um número crescente de
publicações por ano(exemplo de 1985 a 1990 mais de 600). Técnicamente muitas mudanças tem sido
introduzidas melhorando cada vez mais e ampliando a sua utilização. No entanto, é preciso salientar que
N = No 2n
30

para cada situação experimental, novas modificações têm de ser introduzidas, de tal forma que é muito
difícil descrever um único procedimento para um uso generalizado. Por outro lado, é fundamental o
conhecimento básico das etapas envolvidas na técnica, para um desenho experimental com sucesso.
A utilização de PCR é tão ampla que podem ser editados compêndios sobre a metodologia
de PCR em cada especialidade diagnóstica. Na aplicação diagnóstica e, particularmente, na Microbiologia a
atuação da biologia molecular é muito ampla, apesar de estar ainda no cunho de afirmação e de uso restrito
à pesquisa e ao ensino. Porém, muitas doenças infecciosas dependem de um diagnóstico rápido e seguro,
que com certeza a única solução está na PCR. A liberação para uso rotineiro no laboratório clínico ainda não
é permitido para toda as possíveis aplicações, no entanto, a necessidade de meios mais sensíveis e
precisos farão com que a técnica de PCR se torne uma ferramenta impressindível e quase obrigatória no
diagnóstico precoce de várias doenças, apesar do custo ainda estar um pouco elevado.
A técnica de PCR é tão sensível que é capaz de amplificar uma simples molécula de DNA e
tal é a sua especificidade, que uma simples cópia de gene pode ser extraida de uma mistura complexa da
sequência genômica de forma rotineira e visualizada como uma simples banda no gel de Agarose. Como se
pode notar, a sensibilidade e a especificidade que o método pode oferecer é algo singular. A PCR utilizando
DNA polimerases termoestáveis foi descrita pela primeira vez por Kary Mullis e patenteada nos EUA sob o n
4.683.195, em julho de 1987 e n 4.683.202.
Saiki e cols, 1985, Mullis e cols,1986, Mullis e Faloona 1987 introduziram as diversas aplicações do
PCR para a comunidade científica e atualmente é inesgotável a sua aplicação prática tanto na pesquisa
básica como na aplicada.
Em termos práticos, podemos dizer que um protocolo geral pode ser seguido, mas com a ressalva
da necessidade da optimização para cada tipo de aplicação. Os principais problemas citados na literatura
para a padronização pode-se assim resumir: Não detecção do produto, baixo rendimento do produto
desejado, presença de bandas não específicas devido a “mispriming” ou “misextension” dos “primers”,
formação de dímeros dos “primers” que competem pela amplificação com o produto desejado, mutação ou
heterogeneicidade devido a erro de incorporação.
Primer: é um seguimento de DNA ou RNA de 15 a 40 bases, geralmente baseado na
sequência já conhecida do ácido nucleico, geralmente sintetizado quimicamente por instrumentos
automáticos, que podem ser adquiridos no comércio para fins já definidos ou se escolhe a região
desejada do DNA ou RNA a ser amplificada.
Sonda (probe): É um segmento de DNA ou RNA que foi marcado seja com enzimas, substratos
antigenicos, substâncias quimioluminescentes ou radioativas, que podem ligar-se com alta
especificidade a uma sequência complementar do ácido nucleico alvo (escolhido).
31

PROTOCOLO GERAL PARA PCR
1. A reação pode ser realizada no volume de 100l ou 50l. Escolha o volume a ser usado.
2. Para o volume de 100l, colocar em um tubo de 0,5 ml com tampa com fechamento hermético.
2.1. DNA a ser amplificado 105 a 106 moléculas(“target DNA” ou “Template”)
2.2. 20 pmol de cada “primer” (ou iniciadores,Tm>55C é preferido)
2.3. 20 mM Tris-HCl (pH8,3, 20C)
2.4. MgCl 2 1,5 mM
2.5. Kcl 25 mM
2.6. Tween 20 a 0,05%
2.7. 100g/ml de gelatina autoclavada ou albumina bovina livre de nuclease
2.8. 50 M de cada dNTP(dATP, dGTP, dCTP, dTTP)
2.9. 0,2 a 2 unidades de Taq polimerase
3. Pré-incubar (se for DNA genômico) 10 minutos a 94o C
4. Programar o termociclo em:
Desnaturação: 96 C (pode variar de 94 a 96C), por 15 segundos a 2 minutos
Hibridização (Primer Annealing): 55 C (pode variar de 45 a 62C), por 30 segundos a 2
minutos
Polimerização (Extension): 72C(pode variar de 55 a 72C) por 1,5 minutos
Extensão final do programa: de 25 a 30 ciclos, de 72C por 5 a 10 minutos
Estabilização e parada de reação: 4C ou adição de 10 mM de EDTA.
FATORES QUE INFLUENCIAM A PCR
1. Atividade enzimática da Taq polimerase
Se os outros componentes da reação estiverem optimizados, de 1 a 2,5 U são suficientes
para uma boa amplificação em 100l de volume total.
Quando em etapa de optimização, as quantidades da enzima a serem utilizadas
coompreendem o intervalo maior entre 0,5 a 5 U/100l. Obs: A quantidade de enzima necessária varia com
as características do “template” e do “primer.”
Se a concentração de enzima for muito alta, ocorre o aparecimento de produtos
inespecíficos que podem se acumular; e ao contrário, se baixo, a quantidade de produto desejado será
insuficiente.
32

A origem comercial da Taq polimerase é um importante fator, se a aquisição for de vários
fornecedores, pode-se obter resultados diferentes, devido as diferentes formulações, diferentes condições
de ensaio e pode diferir na definição da unidade.
2. Deoxinucleotídeo trifosfato:
Se a solução de uso for preparada a partir de solução estoque é necessário neutralizar para
pH 7,0, seguida de determinação espectrofotométrica da concentração. A solução primária deve ser
preparada a 10mM, distribuida em alíquotas e estocada a -20C. A solução de trabalho recomendada é de
1mM de cada dNTP. A estabilidade dos dNTPs após os vários ciclos de amplificação permanece em
aproximadamente 50% da concentração inicial. Na reação, a concentração recomendada varia de 20 a
200M para um resultado com boa especificidade e reprodutibilidade. Deve-se utilizar as mesmas
concentrações de dNTP ou valores equivalentes, isto minimiza os erros de incorporação. Quanto menor a
concentração de dNTPs menor o risco de “mispriming” ou erro de incorporação de nucleotídeos. A
quantidade mínima recomendada é de 20M de cada dNTP para 100l de reação, quantidade suficiente
para sintetizar 2,6 g de DNA ou 10pmol de uma sequência de 400bp. Ultimamente se estabeleceu que 2M
foi capaz de dar uma alta sensibilidade de detecção e amplificação de pontos de mutação do gene ras. De
qualquer modo, para cada experimento deve-se encontrar uma quantidade ótima.
3. Concentração de Magnésio.
A concentração de magnésio é bastante importante, pois afeta a hibridização(“annealing”), a
temperatura de separação do DNA alvo (“melting”), o produto de PCR, a especificidade do produto, a
atividade da enzima Taq, leva à formação de dimerização de “primer”, prejudicando a fidelidade do
experimento. A concentração dos ions Mg++ deve estar entre 0,5 a 2,5 mM acima da concentração total do
dNTPs. Sempre deve-se considerar a concentração de EDTA na amostra que pode interferir na
concentração dos íons Mg++.
4. Outros componentes
O tampão recomendado para o PCR é o TRIS-HCl na concentração de 10 a 50 mM com pH
entre 8,3 a 8,8 medido a 20C. Este tampão dipolar tem um pKa de 8,3 a 20C e um pKa de -0,0021/C,
portanto, o pH verdadeiro de 20mM de TRIS pH 8,3 a 20C varia de 7,8 e 6,8 durante as condições de
termociclagem.
Pode-se adicionar cerca de 50mM de KCl na mistura de reação para facilitar a hibridaçào
dos “primer”. O Cloreto de sódio a 50 mM, ou KCl abaixo de 50 mM inibe a atividade da Taq polimerase.
O DMSO (dimetilsulfoxido) é util para a reação de amplificação com Klenow fragmento de E.
coli DNA polimerase I; 10% de DMSO inibe a atividade da Taq polimerase em 50%, apesar de Chamberlain
e cols. o recomendarem para amplificação de sequências multiplas, na mesma reação.
33

A gelatina, a proteína (albumina bovina, 100(g/ml) e o detergente não iônico como o Tween
20(0,05-0,1%) podem ser utilizados para estabilizar as enzimas, mas muitos protocolos tem dispensado a
sua utilização.
5. HIbridação dos iniciadores (“Primer Annealing”)
A temperatura e tempo requerido para o hibridação de “primer ” depende da composição
em bases, do comprimento e da concentração. A temperatura básica aplicável seria de 5C abaixo da Tm
verdadeira dos primers. Devido a Taq DNA polimerase ser ativa abaixo do intervalo de temperaturas, a
extensão dos primers ocorrerá em baixas temperaturas, incluindo a etapa de hibridação. O intervalo de ação
da atividade enzimática varia na ordem de 2 vezes a magnitude entre 20 a 85C. A temperatura de
“annealing” no intervalo de 55 a 72C, geralmente dá melhor resultado. Numa concentração típica de
primers(0,2M), o “annealing” só requer poucos segundos.
O aumento da temperatura de “annealing” aumenta a discriminação contra os erros de
hibridização dos primers “mispriming”, reduzindo a extensão ou polimerização dos nucleotídeos incorretos
no final 3’ dos “primers”. Portanto, as temperaturas de “annealing” mais estringentes, especialmante nos
primeiros ciclos poderá aumentar a especificidade. Para se obter uma especificidade máxima, no início dos
ciclos aumenta-se a temperatura de “annealing” e adiciona-se a Taq polimerase após a primeira etapa de
desnaturação e “annealing” do primer.
Baixas temperaturas de etapas de “extension” com altas concentrações de dNTPs favorecem os
erros de polimerização, ou seja, os nucleotídeos são incorporados erroneamente, motivo pelo qual alguns
autores recomendam a utilização de “primers” mais longos e de 2 temperaturas para o PCR. As
temperaturas são de 55 a 75C para “annealing” e “extension” e 94 a 97C para desnaturação e separação
de cadeias.
6. Extensão (ou polimerização)
O tempo de polimerização ou “extension” também depende de alguns fatores como a concentração
e o comprimento do DNA a ser amplificado. A extensão dos “primers” tradicionalmente é realizada a 72C,
porque esta temperatura é próxima do ótimo para extensão de “primers” no modelo de DNA do M13. A
média de incorporação dos nucleotídeos a 72C varia de 35 a 100 nucleotídeos por seg-1 dependendo do
tampão, pH, concentração salina e natureza do DNA alvo.
O tempo de 1 minuto a 72C é considerado suficiente para a extensão de produtos de até
2kb. No entanto, tempos mais prolongados podem ser úteis em casos de poucos ciclos e se a concentração
do substrato for muito baixa e também em ciclos prolongados quando a concentração do produto ultrapassa
a concentração da enzima.
7. Tempo e temperatura de desnaturação
34

A maioria das causas de insucesso do PCR é a incompleta desnaturação do DNA a ser amplificado
ou do produto de PCR. Uma temperatura típica de desnaturação é 95C por 30 segundos, ou 97C por 15
segundos, porém, temperaturas mais altas podem ser apropriadas especialmente para DNA ricos em G+C.
Apenas alguns segundos são necessários para desnaturar e separar as cadeias de DNA, porém, um maior
tempo é necessário para se atingir a temperatura dentro do tubo de reação. Seria desejável a monitoração
da temperatura dentro do tubo de reação utilizando uma sonda térmica de medida exata. A desnaturação
incompleta reduz a possibilidade hibridização entre o DNA alvo e os “primers”, diminuindo o rendimento do
produto do PCR. Por outro lado, a desnaturação por longo tempo ou alta temperatura leva a perda de
atividade da Taq polimerase, que é maior que 2 horas, 40 minutos e 5 minutos nas temperaturas de 92,5;
95 e 97,5C, respectivamente.
8. Número de ciclos.
O número de ciclos dependerá da quantidade de DNA inicial, quando todos os parâmetros
estão optimizados. Um erro comum que se comete é um excesso de número de ciclos. Segundo Kary Mullis
“Se você tem que ir mais que 40 ciclos para amplificar uma simples cópia de gene, existe alguma coisa
seriamente errada com o seu PCR”. Muitos ciclos pode aumentar a quantidade e a complexidade de
produtos não específicos. Obviamente, um número muito baixo dará um baixo rendimento do produto
desejado.
9. Iniciadores (“Primers”)
A concentração dos “primers” entre 0,1 a 0,5 mM são, na maioria das vezes, satisfatórios.
Concentrações maiores podem promover “mispriming” e acúmulo de produtos não específicos e podem
aumentar a probabilidade de gerar um artefato independente do “template” denominado de “primer-
dimer”(dimerização de primer). Produtos não específicos e dimerização de “primer” são por si só substratos
para o PCR e competem com o produto desejado pela enzima, dNTPs, e os primers, resultando em baixo
rendimento do produto desejado.
Algumas das regras gerais para um bom desenho de primers eficientes seriam: de 18 a 28
nucleotídeos em comprimento, tendo cerca de 50 a 60% de G+C na sua composição. A temperatura de
“melting” (Tm) dos pares de primers deve ser adequada. Para esse propósito, pode-se usar a regra da
prática de 2C para o A ou T e 4C para o G ou C. Dependendo da aplicação, o Tm entre 55C e 80C são os
recomendados. Deve também evitar a complementaridade na porção 3’ do par de "primers", pois isso pode
promover a formação de artefatos de dimerização de primers e reduzir a produção do produto desejado.
Outra observação é evitar três ou mais C+G na porção 3’ dos primers que pode promover “mispriming” na
sequência rica em G+C. Deve-se evitar, quando possível, a presença de sequências palindrômicas dentro
dos primers. Se ainda, depois desses cuidados, continuar a falhar, é salutar então tentar outro par de
primers. Uma razão menos óbvia para alguns primers falharem é a presença de estrutura secundária no
DNA “template”. Neste caso, substituir com 7-deasa-2’deoxiGTP o dGTP e avaliar. O desenho de primers
35

para propósitos especiais também podem ser elaborados tais como a adição de sítios de enzimas de
restrição, mutação in vitro, ATG “start codon”, e outros.
10. Efeito Plateau
O termo efeito plateau é usado para descrever a atenuação exponencial da taxa de
formação de produto de PCR, que pode ocorrer durante a fase tardia do PCR concomitantemente com o
acúmulo de 0,3 a 1 pmol do produto desejado. Dependendo das condições da reação e da ciclagem térmica,
um ou mais parametros podem ser afetados:
1)utilização dos substratos (dNTP ou “primers”),
2)estabilidade dos reagentes(dNTP ou enzimas),
3)inibição por produto formado(pirofosfato,DNA duplex),
4)competição por reagentes pelos produtos inespecíficos ou dimerização de primers,
5) “reannealing” de produto específico na concentração acima de 10-8 (pode diminuir a eficiência da
extensão ou o processamento da Taq DNA polimerase ou causar subdivisão na migração das cadeias e
deslocamento de primers) e
6) incompleta desnaturação dos “templates” ou DNAs formados ou separação do produto em alta
concentração.
Um importante fator que determina o aparecimento do efeito plateau é a presença inicial de
baixas concentração de produtos desejado com presença de produtos não específicos resultantes de
eventos de “mispriming” que podem continuar a ser amplificados preferencialmente. A optimização do
número de ciclos de amplificação é a melhor maneira de evitar a amplificação de produtos indesejáveis.
ESCOLHA DOS INICADORES PARA PCR
A. Protocolo básico para desenhar um iniciador (“primer”) 5’para 3’( sense)
Regras gerais:
Determinar a prioridade da utilização(clonar, PCR, hibridizar, outros)
Se for para clonar: analisar a sequência do cDNA estudando as enzimas de restrição do DNA alvo e
seguir o protocolo a seguir:
1. Copiar a sequência e região desejada do cDNA de interesse (não mais que 6 aminoácidos
ou 18 bp)
2.Adicione no início 6 bases aleatórias
3.Adicione a sequência a enzima de restrição desejada (Hind III, EcoRI, etc)
4. Acrescentar a sequência que codifica o início da síntese de aminoácido(metionina -ATG)
5. Checar a sequência em voz alta e verificar a porcentagem de Base C+G, que não deve
ultrapassar 50%
36

Caso o primer for utilizado para outras finalidades como: hibridização, PCR diagnóstico etc, omita as
etapas 2, 3, 4. e acrescente mais pares de base para ajustar a relação C+G se necessário(não > 30).
Ex 5’ AAA AAT CTA AAA TCT CCT 3’
GAT ATG CAT ATG AAA AAT CTA AAA TCT CCT 3’
random NdeI+iniciador
B. Protocolo básico para desenhar um iniciador (“primer”) 3’para 5’ (anti-sense)
Regras gerais:
Determinar a prioridade da utilização
Se for para clonagem seguir os mesmos critérios acima para o “sense primer”
1. Copiar a sequencia da região desejada do cDNA de interesse( não mais que 6 aminoácidos ou
18 bp)
2. Adicione a sequência que codifica o “stop codon” no final da sequência(TAG TAA)
3. Adicione a sequênicia da enzima de restrição
4. Adicione 6 bases aleatoriamente(acertar a relação C+G)
5. Faça a complementar dessa sequência
6.Copie a sequencia complementar em direção oposta ou seja do 5’ para o 3’(pois o sintetizador
sempre iniciado 5’)
7. Checar a sequência em voz alta, calcular a relação C+G, não deixando ultrapassar 50%
(aceite com as base aleatórias acrescidas).
8. Se não usar para clonagem apenas copie a sequencia desejada, faça a complementariedade e
copie a sequencia inversa, checando a sequencia em voz alta.
Ex. 5’ CGC AAA GCT CAG ATT GGT 3’
5’CGC AAA GCT CAG ATT GGT TAG TAA AAG CTT AGA AGC
STOPSTOPHindIII bases aleatórias
3’ GCG TTT CGA GTC TAA CCA ATC ATT TTA GAA TCT TCG 5’
5’GCT TCT AAG ATT TTA CTA ACC AAT CTG AGC TTT GCG 3’
Recomenda-se avaliar o primer desenhado em um programa de computador, para avalia-los
adequadamente antes de requerer a síntese.
Literatura recomendada
37

Chamberlain, J.S. et al 1988. Deletion screening of the Duchenne muscular dystrophy locus via
multiplex DNA amplification. Nucleic Acids Res, 16:11141-11156.
Ehln T. & Dubeau, L. 1989. Detection of ras point mutations by polymerase chain reaction using
mutation-specific,m inosine containing oligonucleotide primers. Biochem Biophy. Res. Commun. 160:441-7.
Mullis, K.B. & Faloona, F.A. 1987. Specific synthesis of DNA in vitro via a polymerase catalyzed
reaction. Methods Enzymol.155:335-50.
Mullis K.B. et al 1986. Specific enzymatic amplification of DNA in vitro: the polymerase chain
reaction. Cold Spring Harbor Symp. Quant. Biol. 51:263-73.
Saiki, R.K.& Gelfand, D.H. 1989. Introducing amplitaq DNA polymerase . Amplifications1:4-6.
Saiki R.K. et al 1985. Enzymatic amplification of (globin genomic sequences and restriction site
analysis for diagnosis of sickle cell anemia. Science 230: 1350-1354.
38

MODELO DE PROTOCOLO DE PCR
Data: ___/ ___/
REAGENTES ORDEM DE VOLUME CONCENTRAÇÃO FINAL
ADIÇÃO
ÁGUA ESTÉRIL 1 ____ ____ ____ ____
Tampão PCR 10X 2 ____ ____ ____ ____
dATP (___mM) .... . 3 ____ ____ ____ ____
dCTP (___mM) ..... 4 ____ ____ ____ ____
dGTP (___mM) ..... . 5 ____ ____ ____ ____
dTTP (___mM) ..... 6 ____ ____ ____ ____
Primer 1 _________ 7 ____ ____ ____ ____
Primer 2 _________ 8 ____ ____ ____ ____
MgCl2 25 mM 9 ____ ____ ____ ____
Dimetilsulfoxido 10 ____ ____ ____ ____
DNA ( amostra) 11 ____ ____ ____ ____
DNA (controle +) 12 ____ ____ ____ ____
água (controle - )... .13......... ____ ____ ____ ____
Taq DNA polimerase 12 ____ ____ ____ ____
Se necessário fazer um mistura dos reagentes comuns evitando contaminação e erros na pipetagem.
39

Aula número 5
PCR NO DIAGNÓSTICO DE DOENÇAS GENÉTICAS
Marcos de Oliveira MachadoProfa. Dra. Rosario Dominguez Crespo Hirata
Prof. Assoc. Mario Hiroyuki Hirata
DOENÇAS GENÉTICAS
Atualmente já foram mapeados aproximadamente de 800 a 2800 locos gênicos. As doenças
genéticas são causadas por alterações, denominadas mutações, nesses locos ou em outros a serem ainda
mapeados.. A maioria das doenças genéticas são raras, mas a ocorrência de uma doença em particular
representa um urgente problema para uma família afetada. As doenças determinadas geneticamente não
são apenas um grupo marginal para a medicina, mas representam juntas uma considerável proporção de
todos os processos das doenças médicas.
Até o presente, em torno de 2800 genes foram mapeados nos cromossomas autossômicos
humanos, e aproximadamente 230, no cromossoma X. Mutações nestes genes levam a doenças genéticas
monogênicas. Para mais de 800 desordens clínicas a lesão intragênica é conhecida. Os métodos de
mapeamento são muitos e variados, e eles complementam um ao outro. Dos vários métodos desenvolvidos
para isolar e identificar os genes defeituosos (Ex: loco no qual as mutações causam uma doença), os
seguintes tem sido mais freqüentemente utilizados: (1) posição do clone baseado na posição conhecida do
cromossomo, (2) estratégia do gene candidato, onde um gene previamente conhecido com um papel
funcional normal sabe-se conter mutações, e (3) mapeamento de pequenas regiões para verificar deleções
ou para analisar pontos de ruptura (“breakpoints”) com translocações de cromossomas.
Numerosas doenças são clinicamente similares (fenótipo similar), mas tem diferentes causas
(princípio da heterogeneidade genética). A heterogeneidade alélica (devido a diferentes alelos mutados em
um loco genético) e heterogeneidade hetero-alélica (devido a mutações em diferentes locos genéticos)
podem ser diferenciadas. Na prática, isto é importante para o diagnóstico e aconselhamento genético, já que
o modo da herança e o curso da doença podem diferir.
POLIMORFISMOS E MUTAÇÕES
A sequência de nucleotídeos do DNA determina uma correspondente sequência de aminoácidos
através da transcrição e tradução. Se a sequência de DNA sofre mutações, a sequência de aminoácidos
pode estar alterada no sítio correspoondente (mutação à nível protéico). Assim, toda mutação tem uma
posição definida. O código do DNA e seu correspondente polipeptídeo são colineares. Uma mutação na
sequência de bases do DNA produz um códon dferente. A posição onde resultou a mudança da sequência
de aminoácidos corresponde a posição da mutação.
40

Basicamente, há três diferentes tipos de mutação envolvendo um só nucleotídeo (mutação
pontual): substituição (troca), deleção (perda), e inserção (adição). Na substituição o codon é alterado e
pode codoficar para um diferente aminoácido. Dois tipos de substituição podem ocorrer: transição (troca de
uma purina por outra purina ou uma pirimidina por outra) e transversão (mudança de uma purina por uma
pirimidina ou vise versa). Uma substituição pode alterar um códon de modo que um aminoácido diferente
estará presente neste sítio, mas isto não tem efeito na fase de leitura, caracterizando a mutação de
sequencia trocada (“missense mutation”), enquanto que uma deleção ou inserção causam uma mudança na
fase de leitura (mutação de sequencia descolada ou “frameshift mutaion”). Na mutação de sequencia
deslocada, as sequências seguintes não codificam para um produto genético funcional (mutação de
sequencia interrompida ou nonsense mutation). As mutações podem ocorrer em vários sítios do DNA, mas
existem determinados sítios em que ela ocorre com maior frequência - os chamados pontos quentes
(‘hotspots’).
A maioria dos pares de base incorretamente formados durante o processo de polimerização não se
torna permanentemente incorporados ao DNA. As DNA-polimerases possuem uma atividade exonucleásica
3’ 5’ que garante uma remoção de praticamente todo nucleotídeo erroneamente pareado. Além disso,
cerca de um em cada dez nucleotídeos corretamente pareados é removido pela atividade de exonucleásica
3’ 5’da DNA- polimerase III, que exerce função de revisar a polimerização.
Todos os seres vivos sofrem um certo número de mutações como resultado de funções celulares
normais ou interações aleatórias com o ambiente, denominadas de mutações espontâneas. Alguns agentes
podem aumentar o grau de mutações, os agentes mutagênicos, e as modificações que eles causam são
denominadas mutações induzidas. Estes agentes podem ser químicos (ex: ácido nitroso) ou físicos (ex:
raios U.V.). Os agentes químicos possuem estruturas suficientemente similares às bases normais para
serem metabolizadas e incorporadas ao DNA durante a replicação. Contudo, elas são suficientemente
diferentes para aumentar a frequência de pareamento errado e, portanto, de mutação. As radiações
aumentam a excitabilidade dos átomos presentes no DNA, e isto é a base dos efeitos mutagênicos da luz
ultravioleta e da radiação ionizante.
Na natureza, os organismos das mesmas espécies usualmente diferem em alguns aspectos quanto
a sua aparência. As diferenças são geneticamente determinadas e são denominadas de polimorfismo. Em
muitos locos gênicos, dois ou mais alelos podem ocorrer, constituindo o polimorfismo genético. O
polimorfismo genético é definido como a ocorrência em uma população de dois ou mais fenótipos
alternativos determinados geneticamente devido a diferentes alelos, pelo qual o menos frequênte alelo não
pode ser mantido somente por repetidas mutações. Um loco genético é definido como polimórfico se o (s)
alelo (s) raro (os) tem uma frequência de pelo menos 0,01 (1%), e como um resultado, heterozigotos para
tais alelos ocorrerem com uma frequência de pelo menos 2%. O polimorfismo pode ser observado à nível do
indivíduo como um todo (fenótipo), em formas variantes de proteínas e substâncias de grupos sanguíneos
(polimorfismo bioquímico), características morfológicas dos cromossomos (polimorfismo cromossomal), ou
à nível de diferenças na sequência de nucleotídeos do DNA (polimorfismo de DNA ou genético).
41

Geralmente, o polimorfismo não é perceptível pelo fenótipo. Os métodos laboratoriais são o meio
pelo qual ele é detectável. Diferenças individuais nas sequências das bases dos nucleotídeos do DNA
podem ser determinadas. Se a diferença na sequência levar a uma mudança no códon, um diferente
aminoácido será incorporado no sítiocorrespondente, Isto pode ser demonstrado pela análise do produto do
gene.
O polimorfismo de uma proteína (produto do gene) pode ser demonstrado por eletroforese em gel
quando a forma variante difere de outras pela presença de um aminoácido com carga elétrica diferente.
Neste caso, o alelo forma um produto gênico que pode ser distinguido devido sua diferente velocidade de
migração em um campo elétrico (eletroforese). O polimorfismo que não leva a uma mudança na carga
elétrica não pode ser identificado dessa maneira.
À nível molecular, o polimorfismo pode ser identificado pelo uso de enzimas de restrição que
reconhecem no DNA determinadas sequências de bases nucleotídicas (sítios), específicas para a sua
atividade de clivagem. Cada enzima (obtidas de uma ampla variedade de bactérias) tem em particular um
alvo na dupla fita de DNA, geralmente uma sequência específica de 4 a 6 bp. A mudança em uma das
bases da sequência determinará a atividade ou não da enzima naquele sítio específico. Dessa forma, pode-
se identificar este tipo de polimorfismo, após a atividade dessas enzimas, por eletroforese em gel.
ANÁLISE DO POLIMORFISMO DE DNA POR PCR
O surgimento de novas técnicas em Biologia Molecular tem alterado significativamente as formas de
abordagem dos problemas básicos e aplicados nessa área. O desenvolvimento da técnica de amplificação
de segmentos de DNA utilizando a reação de polimerização em cadeia (PCR) abriu enormes perspectivas
para a análise de genes, diagnóstico de doenças genéticas, detecção de agentes infecciosos, entre outros
exemplos.
A análise do polimorfismo de DNA por PCR é realizada a partir de iniciadores adjacentes a uma
determinada região polimórfica do DNA, tornando possível amplificar-se um segmento de DNA que pode ser
então, analisado de diferentes formas, como por exemplo, por sequênciamento direto do DNA, por clivagem
com endonucleases de restrição (RFLP), ou por hibridização com sondas genéticas. A análise do produto de
PCR por determinação da sequência de nucleotídeos é a mais informativa em relação aos polimorfismos.
Quando já se tem informação sobre a presença de polimorfismo em um determinado loco, em nível
da sequência de nucleotídeos, métodos rápidos e simples podem ser utilizados para detecção dos produtos
de PCR por técnicas de hibridização com sondas genéticas específicas, usando-se condições em que a
diferença de uma única base pode ser detectada. Esse método tem sido aplicado na detecção de mutações
em doenças genéticas, como a anemia falciforme, talassemias e fibrose cística, bem como na tipagem de
HLA. O polimorfismo de DNA também pode ser detectado diretamente na reação de PCR. Nesse caso,
iniciadores contendo a região 3’correspondente à sequência polimórfica são utilizados e somente ocorre a
amplificação quando há o pareamento correto das bases na região 3’.
42

A técnica de PCR foi utilizada, pela primeira vez, na amplificação de DNA genômico para a detecção
de anemia falciforme. Desde então, várias outra doenças, como a - Talassemias, distrofia muscular de
Duchenne, síndrome de Lesch-Nyham, fenilcetonúria, fibrose cística, doença de Tay-Sachs e doença de
Gaucher, tem sido diagnosticada por PCR.
Para algumas doenças em que a predisposição genética é bastante alta, tais como doenças
cardiovasculares e doenças auto-imunes, o uso da PCR tem sido de grande utilidade. Mutações no gene do
receptor da lipoproteína de baixa densidade (LDL) e polimorfismos nos genes da apolipoproteína B e E (Apo
B e Apo E) têm sido detectados por PCR e relacionados com o risco de doenças cardiovasculares. Doenças
auto-imunes, como a diabete melito insulina-dependente, esclerose múltipla e artrite reumatóide, estão
associadas aos alelos específicos dos locos HLA classe II.
POLIMORFISMO DE APOLIPOPROTEÍNAS
Apolipoproteína B
A apo B ocorre no plasma em duas formas primárias designadas apo B-100 e apoB-48, que
possuem massa molecular relativa (Mr) de 513 Kda e 246 kDa, respectivamente. A apo B-100 é uma das
maiores proteínas já conhecidas, contendo 4536 resíduos de amino ácidos e 8-10% de carboidratos.
Defeitos genéticos da apo B têm sido descritos, como a hiperapoliproteinemia B (hiperapo-B) e a
apo B defeituosa familiar, que podem resultar em diminuição da remoção plasmática dos remanescente de
VLDL e das LDL. A hiperapo-B consiste em uma alteração do gene da apo B-100, resultado da substituição
da arginina, da posição 4019 na sequência primária da molécula de proteína, pelo triptofano. A hiperapo-B é
caracterizada pelo aumento de partículas de LDL pequenas e densas, devido a uma superprodução de LDL,
secundária ao aumento da síntese de apo B-100 e na remoção plasmática da LDL. Essas alterações
metabólicas estão fortemente associadas com a doença coronariana prematura, embora alguns pacientes
com hiperapo-B apresentem níveis normais de colesterol e triacilgliceróis no plasma.
A apo B Defeituosa Familiar ocorre por substituição do amino ácido arginina (Arg) pela glutamina
(Gln) no resíduo 3500. Essa mutação altera a corformação do domínio de ligação da apo B-100 aos
receptores da LDL, prejudicando a interação da LDL com esses receptores. Com isso há uma redução na
remoção plasmática da lipoproteínas, que se acumula no plasma, levando a hipercolesterolemia moderada
ou severa que é importante fator de risco para o desenvolvimento de doenças ateroscleróticas.
Recentemente, a clonagem e o sequênciamento do gene da apo B, localizado no braço curto do
cromossoma 2 (2p 23-24), tem permitido estudar a variação do gene à nível de DNA.
As endonucleases (enzimas de restrição) reconhecem sitios específicos na sequência de
nucleotídeos do DNA (sítios de restrição), onde se ligam e cortam, produzindo diversos fragmentos de
acordo com o número de sítios para aquela enzima. O polimorfismo ocorre quando as mutações na
sequência de DNA criam ou abolem um sítio de restrição. Analisando estes fragmentos de restrição
polimórficos, pode-se identificar alelos de um loco genético em particular que estão associados com um
43

fenótipo clínico, caracterizando o polimorfismo de tamanho de fragmentos de restrição (RFLP). A
associação do polimorfismo genético com uma dada doença ou com um fator de risco pode existir por
algumas das seguintes razões: o polimorfismo por sí só pode constituir uma mutação funcional; pode não ter
um efeito direto, mas estar acoplado a um desequilíbrio através da mutação funcional; ou a associação pode
ser devido a outros fatores.
Alguns desses RFLP podem estar associados com variações nos níveis plasmáticos de lipídeos e
aterosclerose. Diversos RFLP da apo B foram descritos. A mais consistente associação entre os RFLP e os
lipídeos plasmáticos envolve o polimorfismo XbaI, localizado na terceira base do códon 2488 do gene da apo
B. Uma única mudança na terceira base deste códon, sem alteração na sequência de aminoácidos da apo
B, é suficiente para determinar dois tipos de alelos para este tipo de polimorfismo, os quais podem ser
detectados pela enzima XbaI: o alelo X+ (presença do sítio de restrição) e o alelo X- (ausência do sítio de
restrição); formando assim três genótipos: X-X-; X-X+ e X+X+. Muitos estudos foram feitos relacionando os
alelos e/ou genótipos do polimorfismo XbaI com os níveis plasmáticos de colesterol total, colesterol da LDL,
apo B e triglicerídeos. Alguns dados revelam que a LDL de indivíduos com o genótipo X-X- possuem maior
afinidade pelo receptor da LDL quando comparada com a LDL de indivíduos com o genótipo X+X+.
Outros RFLP do gene da apo B, tais como o polimorfismo EcoRI, MspI e Ins/Del, também vem
sendo pesquisados com relação a eventuais influências no mecanismo das doenças coronarianas
ateroscleróticas e no seus fatores de risco ambientais, como a dieta.
Apolipoproteína E
A apo E é o principal constituinte protéico das VLDL e possui um papel importante no metabolismo
das lipoproteínas. Ela também está presente nos quilomícrons e seus remanescentes, e nas IDL. A sua
principal função está na contribuição para a interação das lipoproteínas com os receptores celulares
responsáveis pela captação destas lipoproteínas.
A função da apo E no desenvolvimento da aterosclerose não está totalmente esclarecida e
especialmente o mecanismo de regulação dos níveis de colesterol através desta apolipoproteína ainda é
obscuro. Por outro lado, tem se observado que o polimorfismo da apo E influência na concentração de
colesterol da LDL em diferentes popula!ões.
A estrutura e conformação da apo E em solução, e especialmente sua topografia na superfície das
partículas lipoprotêicas, provavelmente influênciam no metabolismo dessas lipoproteínas sob condições
normais e patológicas. O catabolismo de partículas ricas em triglicerídeos, através de receptores pelos quais
a apo E serve como ligante, podem desta maneira ser modulados pela conformação da apo E, a qual pode
influênciar na afinidade pelo receptor ligante. A apo E é o único ligante para o receptor da VLDL em
algumas lipoproteínas tais como os quilomícrons e seus remanescentes. Em outras lipoproteínas, incluindo
a VLDL e a LDL, a apo E divide a propriedade de ligante-receptor com a apo B-100.
Em humanos o gene da apo E está localizado no cromossoma 19 cercado nas proximidades pelos
genes da apo C-I e C-II e bastante distante do gene do receptor da LDL. Dois tipos de polimorfismos da apo
E tem sido detectados, um à nível de gene e outro à nível de pós-tradução; o primeiro polimorfismo da apo
44

E é devido a presença de três alelos comuns (2, 3 e 4) presentes em um único loco. A determinação
desses alelos pode gerar seis diferentes genótipos, sendo três heterozigóticos (E3/2, E4/2 e E4/3) e três
homozigóticos (E2/2), E3/3 e E4/4). O genótipo E3/E3 é o mais comum e está presente em 60% da
população.
No primeiro tipo de polimorfismo da apo E, o genótipo E2/2 (associado ao fenótipo E2) difere do
E3/3 (E3) pela substituição da arginina pela cisteína na posição 158 da sequência de amino ácidos da apo
E, enquanto que o genótipo E4/4 (E4) difere do E3/3 por substituição da cisteína-112 pela arginina. Essas
diferenças na estrutura primária influenciam na capacidade de ligação da apo E pelo receptor da LDL (B,E),
sendo que o fenótipo E2 possui afinidade aproximadamente 100 vezes menor por esse receptor, que o E3.
Os indivíduos que apresentam o fenótipo E2 têm níveis plasmáticos das VLDL e de seus remanescentes
aumentados, que estão associados com a disbetalipoproteinemia familiar (Hiperlipoproteinemia tipo III) e
representam riscos em potencial para o desenvolvimento da aterosclerose. No fenótipo E4 a capacidade de
ligação do receptor da LDL, é normal, porém os níveis plasmáticos de colesterol e de LDL estão
aumentados. Esse polimorfismo da apo E também pode ser analisado pela técnica da PCR seguido do uso
de enzimas de restrição, específicas para estas regiões polimórficas.
POLIMORFISMO DO RECEPTOR DA LDL
Além das apolipoproteínas os receptores celulares das lipoproteínas também estão relacionados
com as variações nos níveis séricos lipídicos e lipoprotêicos. Os receptores são estruturas glicoprotêicas
que estão agrupados em regiões definidas da membrana celular, onde a lipoproteína se liga por intermédio
da apolipoproteína. Eles fazem parte do mecanismo de remoção das lipoproteínas do plasma e fornecimento
do colesterol e triacilglicerídeos para as células. Por isso são responsáveis pela manutenção da homeostase
dos lipídeos e das lipoproteínas no plasma e nos tecidos e, ao mesmo tempo, protegem as células do
acúmulo desses lipídeos, principalmente do colesterol.
Uma variedade de células humanas contêm receptores de alta afinidade para a LDL, também
denominados receptores B,E. A LDL liga-se a estes receptores por intermédio da apo B-100 presente nas
partículas. O receptor da LDL humano é constituído de 839 resíduos de amino ácidos e apresenta cinco
domíneos.
Mutações no gene do receptor da LDL causa uma doença hereditária do metabolismo lipídico
chamada de Hipercolesterolemia Familiar (FH). A deficiência funcional no receptor da LDL na FH resulta no
acúmulo de LDL no sangue. O resultado deste alto nível de LDL induz enfim a uma aterosclerose prematura
e ao infarto do miocárdio. Alguns anos atrás um grupo de pesquisadores sugeriu que os genes normais do
loco do receptor da LDL poderiam contribuir para variação populacional nos níveis séricos de colesterol. Nos
meados de 1980, com o advento da tecnologia do DNA, surgiram novas possibilidades para examinar este
problema.
45

O gene do receptor humano para a LDL foi clonado e mapeado no cromossoma 19p 13-2. Um
amplo espectro de mutações localizadas na parte estrutural do gene do receptor-LDL tem sido analisadas.
Vários RFLP foram encontrados no gene do receptor da LDL. Estudando tais polimorfismos detectou-se
correlação com os níveis de colesterol total e LDL alterados. O alelo P- (ausência do sítio de restrição
PvuII), do polimorfismo PvuII do gene do repector da LDL, foi associado ao aumento dos níveis séricos de
colesterol total e da LDL em várias populações européias. Outros mostraram que a frequência do alelo N- do
polimorfismo NcoI) foi significantemente maior em pacientes com FH do que nos indivíduos do grupo
controle. Também, uma alta frequência do alelo A- do polimrfismo AvaII foi encontrada em pacientes
brancos Africanos com FH. A ligação entre o polimorfismo do loco do receptor da LDL e um perfil
lipoprotéico aterogênico sugere que uma mutação no gene do receptor da LDL deve ser responsável por
este fenótipo.
Diante de todos estes dados podemos concluir que o polimorfismo genético das apolipoproteínas B
e E, e do receptor da LDL contribuem de alguma forma para a elucidação dos casos em que ocorrem
respostas individual e populacional diferentes frente a determinados fatores de risco, como a dieta, para as
doenças cardiovasculares.
Literatura recomendada
HALLMAN, D.M., VISVIKINS, S., STEINMETZ, J., BOERWINKLE, E. The effect of variation in the apolipoprotein B gene on plasma lipid and apolipoprotein B levels: A likelihood-based approach to cladistic analysis. Ann. Hum. Genet., v. 58, p 35-84, 1994.
PASSAGLIA, L.M.P. Mutação, mecanismos de reparo do DNA e recombinação. In: ZAHA, A. coord. Biologia molecular básica. Porto Alegre. Mercado Aberto ed. cap. 6, p. 116-155, 1996.
PASSAGLIA, L.M.P., ZAHA, A. Técnicas de DNA recombinante. In: ZAHA, A. coord. Biologia molecular básica. Porto Alegre. Mercado Aberto ed., cap. 15, p. 307-331, 1996.
PASSARGE E. Color atlas of genetics/Eberhard Passarge, Thieme ed. p. 46,156-158. 1995WATSON, J.D., HOPKINS, N.H., ROBERTS, J.W., STEITZ, J.A. & WEINER, A.M. Molecular Biology of
the Gene. 4rd ed. Menlo Park: The Benjamin/Cummings Publishing, Inc., 1987
46

Aula número 6
APLICAÇÃO DO PCR NO DIAGNÓSTICO DE DOENÇAS INFECTO-CONTAGIOSAS
Prof.Assoc. Mario Hiroyuki Hirata
Moseley e colaboradores, em 1980, foram os pioneiros da aplicação de tecnologia de DNA
em doenças infecciosas. Eles mostraram no primeiro estudo que a bactéria E. coli poderia ser detectada por
Hibridização de DNA-DNA em amostras impregnadas em papel de filtro e incubadas uma noite em placa de
agar MacConkey, mostrando sensibilidade e especificidade quando comparados com os métodos de cultura
até então conhecidos. Na década seguinte probes de DNA capazes de identificar varias bactérias
patogênicas já eram disponíveis no mercado. A seguir surgiram probes dirigidos para DNA de virus, fungos,
parasitas e helmintos patogênicos. Ultimamente, a utilização de técnicas em biologica molecular tornou-se
importante em muitos laboratórios inclusive para diagnóstico em cortes histológicos. Com o aparecimento
dos sintetizadores automáticos de oligonucleotídeos houve um grande avanço na tecnologia de probes
sintéticos baseados nas sequências publicadas e disponíveis nos bancos dos genes.
Técnicas disponíveis de PCR de importância epidemiológica
PCR para pesquisa de bactérias patogênicasNeisseria menigitidisMycobacterium tuberculosisRichettsia rickettsiiBorrelia burgdorferiYersinia pestisTreponema pallidumChlamydia trachomatisMycoplasma pneumoniaeLegionella pneumophilaGenes de toxinas de Vibrium cholerae 01Genes de toxinas de termosensível e termolábil de “Shiga-like” em E. coliEspécies de Shigella enteroevasiva e de E. coliHelicobacter pylori
PCR para pesquisas de VirusHIV IHepatite BRNA de Hepatite CHerpes simplexEpstein-BarrCytomegalovirusVaricella-ZosterParvosvirus B19RotavírusAdenovirosesRubeolaEnterovirosesPapilomavírusPapilomavírus tipo 16 e 18mRNA E6 e E7 por 3S
PCR para detecção de locus de resistência a antibióticosRifampicina em Micobacterium tuberculosis e M. lepraeEritromicin e methicilina para Staphylococcus aureusPenicilinase em N. gonorrhoeaeAminoglycoside modifying enzymeBeta lactamases de espectros estendidos.
PCR para pesquisa de parasitas Trypanosoma cruziLeishmania spPlasmodium spEntamoeba histolyticaBabesia microtiGiardia lambliaMicrosporidium
Toxoplasma gondii
47

PCR NO DIAGNÓSTICO DAS MENINGITES BACTERIANAS
Professora Dra. Elza Masae MamizukaJane Atobe
A meningite é um conjunto de síndromes cujo denominador comum é a inflamação meníngea
(Gomes, 1991). O período de incubação varia de acordo com o agente etiológico (bactérias, vírus, fungos,
protozoários, helmintos ou espiroquetídeos), sendo as bactérias causadoras mais frequentes de meningite.
As meningites bacterianas constituem um sério problema de saúde pública em todo o mundo. Elas
representam um capítulo de importância pelos altos índices de morbi-mortalidade, principalmente em
crianças. A meningite bacteriana é a mais notável e comum infecção que ataca o Sistema Nervoso Central
(SNC). Dados da literatura relatam que 70% das ocorrências se dão em menores de 5 anos de idade e a
taxa de mortalidade chega a 28% (Kilpe et al., 1993). As sequelas variam desde os problemas de
comportamento até danos cerebrais irreversíveis.
O risco de morte é muito alto, especialmente quando não se realiza um tratamento adequado. Assim
sendo, é fundamental que o diagnóstico e tratamento apropriados realizem-se especifica e prontamente
(Radstrom et al., 1994). No Brasil, o problema se agrava devido ao baixo nível sócio-econômico da
população, e seus determinantes: pobreza, falta de higiene e baixa nutrição que contribuem para a
disseminação e implantação da meningite, além do precário sistema de Saúde Pública que dificulta o
diagnóstico precoce e o tratamento adequado. Na Grande São Paulo, o coeficiente de incidência das
meningites meningocócicas, no ano de 1995, foi de 7,95 casos/100 mil habitantes, com uma letalidade de
20,42% (Dados provisórios do Centro de Vigilância Epidemiológica - CVE).
O diagnóstico da meningite meningocócica inicia-se com a suspeita clínica, porém a confirmação é
feita pelo isolamento e identificação da Neisseria meningitidis em amostras biológicas como tecido, sangue e
líquor, ou pela detecção de diplococos Gram - negativos. A cultura de sangue apresenta-se positiva em,
aproximadamente, 50% dos pacientes.
A mortalidade dos indivíduos com doença meningocócica é reduzida quando os antibióticos são
administrados em casos suspeitos, antes da admissão em hospital. Entretanto, na vigência da
antibióticoterapia, a taxa de isolamento do meningococo no sangue ou no líquor é reduzida de 50% para
menos de 5% (NI et al., 1992).
Atualmente, o método de Reação em Cadeia da Polimerase (PCR) tem sido empregado para
amplificar e detectar DNA microbiano em amostras clínicas. Além da alta sensibilidade, a vantagem deste
método é a detecção de bactérias mortas ou inibidas pelo antibiótico. Como a meningite bacteriana deve ser
tratada de imediato, há necessidade de rápida detecção de microrganismos para o diagnóstico clínico. As
desvantagens da microscopia direta e dos testes imunológicos para detecção de antígenos, são bem
conhecidos no que diz respeito à sensibilidade e especificidade (Gray & Fedorko, 1992). O método mais
específico para diagnosticar a meningite bacteriana é através do isolamento do patógeno em cultura,
seguida de identificação (necessidade de 12 a 24 horas de incubação).
48

Para o diagnóstico da meningite meningocócica, emprega-se, atualmente, três estratégias de PCR.
A primeira emprega o PCR para detectar o DNA de um único patógeno na amostra biológica. Essa
estratégia tem sido utilizada para detectar DNA de Neisseria meningitidis, Listeria monocytogenes (Kilpi et
al., 1993) e de Haemophilus influenzae (Ketil et al., 1990). O DNA do meningococo pode ser
especificamente detectado pela amplificação do gene da hidropteroato sintetase (dhps) . Esse gene foi
clonado a partir do DNA isolado de cepas de Neisseria meningitidis sulfonamida sensível e resistente
pertencentes aos sorogrupos A, B e C. O sequenciamento desse gene permitiu construir dois iniciadores
para a amplificação do gene dhpa por PCR, denominados NM1 e NM2. O produto de amplificação tem 950
bp de tamanho.. Outro gene que pode ser utilizado para detecção de Neisseria meningitidis por PCR é o
1S1106, descrito por Ni, H. et al. (1992). Nesse caso são utilizados iniciadores adjacentes à extremidade 5’
do gene na posição 850 e na extremidade 3’ na posição 1445 do gene, produzindo um fragmento de 596 bp.
A segunda estratégia é utilizar duas etapas de amplificação que permitem a detecção de amplo
espectro de patógenos (Radstrom et al., 1994). A primeira etapa de amplificação utiliza um par de
iniciadores, denominados marcadores universais de eubactérias, correspondente a um gene de uma região
conservada no genoma como o gene da subunidade 16S do rRNA. A finalidade desta etapa é de detectar
qualquer agente bacteriano na amostra biológica. Na segunda etapa, os produtos da primeira etapa são
amplificados utilizando iniciadores específicaos para Neisseria meningitidis,, através da técnica de PCR
"semi-nested" ou são hibridizados com sondas de DNA espécie-específicas. Dados da literatura mostram
que o método de PCR como teste de triagem permite uma rápida detecção do DNA bacteriano de várias
espécies que causam meningite (Greiser et al., 1994).
A terceira estratégia é a amplificação por PCR multiplex, empregando-se iniciadores universais para
amplificar o gene de qualquer tipo de bactéria gram-negativa (ex. sonda universal COR28), e iniciadores
específicos para N. meningitidis (ex., sonda RDR 47D1). Na PCR multiplex , as duas sequências (universais
e espécie-específicas), de 370 bp (universal) e 279 bp (N. meningitidis) são amplificadas em uma única
etapa de PCR, o que reduz consideravelmente o tempo utilizado na detecção do meningococo.
Devido à sua alta sensisibilidade e especificidade, a prova da PCR em líquor é um teste rápido,
extremamente útil para a confirmação precoce do diagnóstico da meningite meningocócica e em situações
onde a cultura é dificultada devido à prévia terapia antimicrobiana muitas vezes necessária ou
imprescindível. Estudos demonstram que cerca de 23% dos casos com diagnóstico clínico de meningite
aguda não apresentam confirmação em exames microscópicos ou em culturas (Grey & Fedorko, 1992).
Pela gravidade da enfermidade, justificada tanto pela letalidade como pelo grau de sequelas
neurológicas, é de suma importância que o diagnóstico etiológico seja determinado o mais rápido possível,
orientando, desta maneira, a conduta clínica e terapêutica.
Literatura recomendada
Gomes, M. C. O. - Como diagnosticar e tratar meningite. Rev. Bras. Med. 48: 9-27, 1991.Greisen, K.; Loftelholz, M.; Purohit, A.; Leong, D. - PCR primers and probes for 16S rRNA gene of most
species of pathogenic bacteria, including bacteria found in cerebrospinal fluid. J. CLin. Microbiol. 32: 335-351, 1994.
49

Ketel Van, R. J.; Wever, B.; Alphen Van, L. - Detection of Haemophilus influenzae in cerebrospinal fluids by polymerase chain reaction DNA amplification. J. Med. Microbiol. 33: 271-276, 1990.
Kilpi, T.; Antiba, M.; Markku, J. T.; Pitold, M. - Length of prediagnostic history related to the course and sequele of childhood bacterial meningitis. Pediat. Infec. Dis. 12: 184-188, 1993.
Kristiansen, B. E.; Ask, E.; Jenkins, A.; Fermer, C. ; Radstrem, P.; Skold, O. - Rapid diagnosis of meningococcal meningitis by polymerase chain reaction. Lancet. 337: 1568-1569, 1991.
Gray, L. D.; Fedorko, D. P. - Laboratory diagnosis of bacterial meningitis. Clin. Microbiol. Rev. 5: 130-145, 1992.
Ni, H.; Knight, A. I.; Cartwright, K.; Palmer, W. H.; MacFadden, J. - Polymerase Chain Reaction for diagnosis of meningococcal meningitis. Lancet. 340: 1432-1434, 1991.
Rasdtrom, P.; Backman A.; Qian, N.; Kragsbjerg, P.; Pahlson, C.; Olcen, P. - Detection of bacterial DNA in cerebrospinal fluid by an assay for simultaneous detection of Neisseria meningitidis, Haemophilus influenzae and Streptococci using a seminested PCR strategy. J. Clin. Microbiol. 32: 2738-2744, 1994.
50

PCR NO DIAGNÓSTICO DA TUBERCULOSE
Prof. Assoc. Mario Hiroyuki HirataProfa. Dra. Rosario Dominguez Crespo Hirata
Profa. Dra. Elza Masae Mamizuka
INTRODUÇÃO
A infecção por micobactérias, acomete pacientes de qualquer idade, sexo ou grupo étnico e não
sendo tratada oportunamente poderá ser fatal ou trazer consigo um alto risco de seqüelas graves,
principalmente nos casos em que a terapia seja retardada ou inadequada (VIAMONT, 1.992). Atualmente,
com a Síndrome da Imunodeficiência Adquirida (AIDS), o ressurgimento de micobactérias não tuberculosas,
tem aumentado dramaticamente, sendo as de maior interesse em saúde pública as pertencentes aos
complexos M. avium e M. fortuitum que apresentam distinto significado clínico, tanto no tratamento como no
prognóstico dos pacientes acometidos, e principalmente com implicações epidemiológicas diferentes,
(KIEHN & EDWARDS, 1.987, GUTHERTZ et al., 1.989).
A tuberculose como doença continua a figurar entre as principais enfermidades que afetam a
humanidade (BOOM et al., 1.990, BRISSON-NOEL et al., 1.991). Estima-se que aproximadamente 1/3 da
população mundial esteja infectada por M. tuberculosis, com incidência anual de 8 milhões de casos novos
da doença (KOCHI et al., 1.992). Simultaneamente ao aumento da incidência da tuberculose, também
surgiram cepas de M. tuberculosis multi-resistentes às drogas como a rifampicina e isoniazida. Os dados
sobre resistência primária ou secundária no Brasil variam de região para região, sendo de 16,5% no estado
de São Paulo e de 19,1 % no Rio de Janeiro (CVE/SP, 1995). Sem dúvida, os principais problemas que se
enfrentam na tuberculose são: o problema sócio-econômico, diagnóstico clínico-laboratorial adequado e
rápido, tratamento e controle profilático da doença (WHITE & GOLD, 1.992; WILLIANS et al., 1.994;
WHELEN et al., 1.995; COOKSEY, et al 1.996).
Os recentes avanços da biotecnologia vem sendo conhecidos nos países subdesenvolvidos no
entanto, ainda são escassos os recursos econômicos e a falta de apoio aos centros de pesquisa
tecnológica, especialmente aos pesquisadores que atuam na área de saúde. Entretanto, a técnica de
amplificação do DNA pela Reação em Cadeia da Polimerase (PCR) tem sido aplicada nos vários centros de
pesquisa em diagnóstico e indica ser um procedimento simples e rápido, onde os reagentes são estáveis e
os equipamentos de baixo custo (CLARRIGDE 1994). Estudos iniciais têm demonstrado que o custo de
insumos e recursos humanos para a realização da técnica da PCR e da cultura são semelhantes.
Em razão disso, a técnica da PCR deve ser proposta também como tecnologia apropriada até para
laboratórios de saúde pública do terceiro mundo. Com o aprimoramento de sondas genéticas não radioativas
específicas para detecção de micobatérias com emprego de técnicas eletroforéticas e de hibridização, a
PCR provavelmente, representa um instrumento alternativo interessante para o controle da infecção
especialmente na detecção precoce da tuberculose e da identificação de cepas bacterianas resistentes aos
quimioterápicos (KIRSCHNER et al., 1.993; KOX et al., 1.994; 1.995).
51

Um fator bastante relevante na identificação de micobactérias é a demora no crescimento
bacteriano na cultura, além da dificuldade da realização dos testes de sensibilidade às drogas pelos
métodos tradicionais. Outro problema é que os métodos tradicionais apresentam baixa sensibilidade e
especificidade, além de ter baixa reprodutibilidade. No entanto, o abandono do tratamento propicia o
aumento de transmissão da doença e também contribui para o aparecimento de cepas resistentes por
seleção de mutantes na população das micobactérias.
Considerando as dificuldades encontradas com esses métodos e a gravidade atual da situação da
tuberculose a nível mundial, seria muito importante estabelecermos uma metodologia que gere diagnóstico
mais rápido com garantia de alta sensibilidade, especificidade e reprodutibilidade. A introdução de tal técnica
na rotina diagnóstica será de grande utilidade não somente pela abreviação do tempo mas também, pela
facilidade de treinamento técnico para a formação de recursos humanos nesta área (HERNANDEZ, 1996).
DIAGNÓSTICO LABORATORIAL DA TUBERCULOSE
Microscopia Direta
A microscopia ou baciloscopia é um método rápido para pesquisar micobactérias, mas requer
aproximadamente 104 bactérias/mL de amostra para ser detectado com segurança na maioria das espécies.
A especificidade das baciloscopias positivas é alta (84 - 100 %) (DANIEL, 1.990), mas esta porcentagem
pode ser consideravelmente reduzida em zonas endêmicas de infecções pelas bactérias do Complexo
Mycobacterium avium (MAC).
A coloração fluorescente tem melhorado a sensibilidade da microscopia direta, sem superar de forma
considerável à obtida com a coloração de Ziehl-Neelsen, que apesar de apresentar especificidade de 99 %,
tem uma sensibilidade baixa, aproximadamente de 30 % a 40 % por necessitar de uma concentração
aumentada de bacilos na amostra a ser estudada. Exames múltiplos das amostras, aumentam a
sensibilidade, melhorando o diagnóstico (KENNEDY & FALLON, 1.979)
Cultura
A cultura é um método mais sensível e específico do que a microscopia, mas requer de 3 até 8 semanas
para o crescimento. A optimização de meios seletivos acoplados a métodos radiométricos (BATEC) de
detecção de crescimento precoce de micobactérias, pode diminuir, consideravelmente, o tempo do
diagnóstico, especialmente se o inóculo inicial é alto e adequado. Entretanto, o alto custo do equipamento e
a necessidade do 14C, torna-o inacessível para a maioria dos laboratórios de médio e pequeno porte. A
sensibilidade da cultura varia entre 20 e 90 %, dependendo da quantidade da amostra submetida à cultura e
da infraestrutura do laboratório (KENNEDY & FALLON, 1.979; MOLAVI & LEFROCK, 1.985). De todas as
desvantagens apresentadas pelos métodos anteriormente mencionados, a biologia molecular pode ser
considerada uma excelente alternativa no diagnóstico deste gênero bacteriano.
52

PESQUISA DE MICOBACTÉRIAS POR TÉCNICAS MOLECULARES
Os recentes avanços da engenharia genética e da biotecnologia trazem interesse àqueles que
estão familiarizados em inovação e desenvolvimento. E, com o aprimoramento de sondas genéticas (DNA-
Probe) não radioativas e iniciadores mais específicos para detecção com eletroforese, a PCR
provavelmente, representará um instrumento alternativo para o controle da tuberculose nos países em
desenvolvimento. Dentre as principais técnicas de biologia molecular, utilizadas no diagnóstico de
micobactérias conforme segue (CLARRIDGE et al., 1.991; 1.993):
Hibridização de Ácidos Nucléicos
Esta técnica está baseada nas propriedades de complementaridade e estabilidade das fitas do DNA.
A estrutura nativa do DNA é mantida por pontes de hidrogênio entre as bases complementares das duas
fitas opostas. Entretanto, em temperaturas próximas a 100ºC, ou em pH alcalino, as pontes de hidrogênio
desestabilizam-se e as fitas separam-se. Já as ligações covalentes entre os nucleotídeos da mesma fita
permanecem estáveis. Nesta forma, o DNA é dito desnaturado. A desnaturação não é irreversível, pois
com a diminuição da temperatura da solução para 50 ºC as duas fitas pareiam-se novamente, seguindo o
princípio da complementariedade das bases (MUSIAL et al., 1.988). Durante a renaturação as fitas tanto
podem ligar-se entre si como ligadas previamente, ou podem parear-se a qualquer outra seqüência de
nucleotídeos, mesmo que seja RNA, desde que haja complementariedade de bases (TENOVER, 1.991).
Uma aplicação comum da hibridização de ácidos nucléicos é a identificação de um determinado
segmento gênico em uma amostra. Procede-se à desnaturação do DNA da amostra seguida da incubação
com uma sonda genética complementar ao segmento gênico de interesse. A sonda genética pode consistir
de oligonucleotídeos sintéticos ou fragmentos de cDNA (DNA complementar). Em ambos os casos, alguns
dos nucleotídeos da sonda serão marcados com elementos radioativos como 32P. Caso exista na amostra o
segmento gênico procurado, a sonda irá hibridizar especificamente e será detectada em auto-radiografia.
Há uma tendência recente ao emprego de nucleotídeos marcados com enzimas ou compostos
luminescentes, como a digoxigenina marcada, que não teriam os incovenientes inerentes ao uso de
radioisótopos (ROBERT et al., 1.991; SHANWAR et al., 1.993).
A hibridização com sondas marcadas é, freqüentemente, empregada na identificação dos
fragmentos de DNA e de moléculas de RNA separadas e imobilizadas pelas técnicas de Southern e
Northern blot, respectivamente. Dispondo de sondas apropriadas é possível investigar um determinado gene
ou molécula de DNA ou RNA quanto à presença do agente etiológico na amostra. Como a hibridização é
quantitativa, pode ser inferida a quantidade do mRNA em questão pela comparação da intensidade do sinal
obtido na auto-radiografia utilizando densitometria (SOUTHERN, 1.975; FRIES et al., 1.991).
Os ácidos nucléicos podem ser hibridizados com sondas genéticas, ainda que mantenha intacta a
estrutura morfológica dos tecidos, mediante a técnica denominada hibridização in situ. É um método
extremamente sensível, que permite a detecção de quantidades mínimas de ácido nucléico não acessíveis
por outros métodos. A hibridização in situ evidencia a distribuição de um determinado gene ou RNA entre
53

as várias células de um mesmo tecido, o que não é possível por Southern ou Northern blot. Há sondas
comerciais disponíveis para detecção de genoma de diversos microrganismos (COATES, 1.987).
Também os métodos de hibridização de ácidos nucleicos podem utilizar sondas genéticas de DNA
que são capazes de hibridizar com seqüências complementares de RNA (35) ribossomal 16S, podendo
estas apresentar uma alta especificidade e sensibilidade para detectar bactérias deste gênero (ROBERTS
et al., 1.987; STOCKMAN et al., 1.993). As sondas genéticas de DNA com capacidade de hibridizar com as
seqüências de RNA ribossomal 16S para as espécies mais comuns do gênero Mycobacterium (M.
tuberculosis, M. avium, e M. intracellulare), são comerciais e podem ser marcadas com elementos
radioativos ou não, diminuindo, consideravelmente, o tempo de detecção e quando associados aos métodos
de cultura radiométrica podem reduzir o tempo em até 2 semanas (ELLENER et al., 1.988; KEMP et al.,
1.990). No entanto, a técnica de hibridização de ácidos nucleicos é limitada por sua baixa sensibilidade.
Este método requer (aproximadamente 104 bactérias para a detecção) quantidade similar a que necessita a
baciloscopia, isto não é tão satifatório para a identificação de micobactérias a partir de amostras clínicas, já
que é muito difícil encontrar tal quantidade de microrganismos na maioria destas amostras. O tempo gasto
para o diagnóstico pelo método de hibridização torna-se sempre dependente da cultura bacteriana e do
inóculo inicial (WILSON et al., 1.993), exceto por esses inconvenientes, a técnica de hibridização pode
apresentar uma alta especificidade e sensibilidade (GONZALES & HANNA, 1.987) .
Endonucleases de Restrição e RFLP
A tecnologia atual da Biologia Molecular tornou-se possível graças à identificação e purificação de
enzimas nucleares. Merecem destaque, as endonucleases de restrição, pela ampla variedade de seus
membros e pelas aplicações práticas. São enzimas bacterianas com a propriedade de clivar moléculas de
DNA estranho, que porventura, penetram a célula. Representam, portanto, uma defesa primitiva de
organismos procariontes. A clivagem da dupla hélice de DNA ocorre em pontos conhecidos como sítios de
restrição, que são seqüências de 4 a 8 nucleotídeos específicos para cada enzima.
Há uma grande variedade de enzimas de restrição que reconhecem uma ampla gama de sítios de
restrição. A distribuição dos sítios de restrição num determinado gene é específica para a endonuclease,
em função da seqüência de nucleotídeos do gene. Este é o princípio em que se baseia a técnica de RFLP,
que é a sigla inglesa relativa a polimorfismo do tamanho de fragmentos gerados por endonucleases de
restrição.
Quando um segmento de DNA é monomórfico, ou seja, não exibe variação individual na sua
seqüência de nucleotídeos, o padrão de fragmentos gerados por uma determinada endonuclease de
restrição é sempre o mesmo. Caso haja variação na seqüência de nucleotídeos, poderá mudar alguns sítios
de restrição e conseqüente há variação no número de fragmentos gerados pela respectiva endonuclease de
restrição. Deleções ou inserções fora dos sítios de restrição poderão também ser identificados por
ocasionarem alterações no tamanho de alguns dos fragmentos de restrição gerados (PLIKAYTIS et al.,
1.992; VAN ECHOUTTE et al., 1.993).
54

O estudo do RFLP envolve técnicas de digestão de DNA, separação eletroforética, “Southern blot” e
hibridização de ácidos nucléicos. A amostra de DNA a ser estudada é inicialmente digerida por uma ou mais
endonucleases de restrição e separada eletroforeticamente. Após a transferência para membranas de
nitrocelulose ou nylon (Southern blot) e fixadas, sob luz UV ou calor, posteriormente esta é incubada com a
sonda genética (marcada) complementar a segmentos do gene de interesse. A hibridização da sonda
identifica os fragmentos de restrição em um gene, o tamanho e número dos fragmentos gerados obedecerá
às variações na seqüências de nucleotídeos. Esta variação pode ser normal no caso de regiões
polimórficas do genoma ou pode traduzir uma mutação específica no caso de genes monomórficos. Boa
parte dos estudos utilizando RFLP não analisa diretamente o gene de interesse, e sim regiões polimórficas
vizinhas, para as quais existem mapas de restrição definidos (TELENTI et al., 1.993).
Reação em Cadeia da Polimerase (PCR)
As limitações dos métodos de diagnóstico utilizando ácidos nucléicos, podem ser superadas com a
amplificação de fragmentos específicos destes ácidos, mediante a Reação em Cadeia da Polimerase (PCR)
(SAIKI et al., 1.988). Introduzida em 1.986 por MULLIS & FALOONA (1.987). A PCR é uma técnica que
permite a amplificação in vitro de um fragmento de DNA, milhões de vezes em poucas horas, pois com a
sua utilização é possível marcar todo e qualquer segmento de informação genética entre, bilhões de
unidades e reproduzi-lo milhões de vezes. O princípio da reação em cadeia da polimerase é simples e
baseia-se no mecanismo natural de duplicação do material genético que ocorre sempre que uma célula se
divide para formar duas novas. Portanto, na PCR, reproduz-se o processo de replicação natural do DNA
com as vantagens de extrema rapidez e de ser possível determinar, precisamente, o trecho do DNA a ser
amplificado, ao passo que na replicação normal todo o genoma é amplificado lentamente. Assim, a PCR
deve satisfazer as condições básicas do processo de replicação do DNA, realizado pelas DNA polimerases,
enzimas que agem, exclusivamente, sobre fitas pareadas de DNA.
Recentemente, vários trabalhos têm descrito esta técnica como uma boa alternativa para a
identificação de micobactérias, discutindo-se melhorias na técnica da PCR para a pesquisa dessas
bactérias em material biológico. Apesar do sucesso realizado nos laboratórios de pesquisa mediante a
utilização da PCR, esta técnica enfrenta alguns problemas, como: a contaminação do produto amplificado
(BOOM et al., 1.991), presença de inibidores da Taq - polimerase quando se preparam amostras de escarro
para a obtenção de uma boa purificação do DNA para a aplicação pela PCR (AMICOSANTE, et al., 1.995)
Detecção e Identificação de Micobactérias por Amplificação do DNA.
A detecção da presença ou ausência de micobactérias pode ser feita por hibridização de ácidos
nucléicos (DRAKE et al., 1.987), sendo a quantidade do DNA presente na amostra, muitas vezes
insuficiente para ser detectada. Têm sido feitas pesquisas de novos métodos que permitem a amplificação
de seqüências de DNA específicas. Uma vez que o processo de amplificação detecta a presença de
micobactérias não viáveis, ou seja não haja necessidade de crescimento in-vitro, a sensibilidade desta
técnica pode superar a cultura. A amplificação da seqüência do DNA micobacteriano tem permitido a
55

detecção e identificação rápida destas bactérias em amostras clínicas. O trabalho publicado por
ALTAMIRANO et al. (1.992) para o diagnóstico de tuberculose pulmonar, mostra uma sensibilidade e
especificidade aumentadas sendo próximas a 100%. SHANKAR et al. (1.991), identificou M. tuberculosis
em diferentes tipos de amostras de pacientes com diagnóstico clínico presuntivo de tuberculose. KANEKO
et al. (1.990) utilizando a PCR detectou M. tuberculosis no LCR em 84% do pacientes com diagnóstico
clínico de meningite tuberculosa.
Para a identificação de micobactérias foram elaborados alguns tipos de “primers” para detectar
gênero ou espécies de micobactérias (HANCE et al., 1989; TAKEWAKI et al., 1.993). Tudo depende do
nível ou sítio em que vão se ligar os “primers” e os genes que se procura, existindo técnicas baseadas na
amplificação de seqüências específicas de DNA ou RNA que estão presentes em todas ou em algumas
espécies de micobactérias (BÖDDINGHANS et al., 1.990 a )
Existem “primers” que são utilizados para amplificar genes que codificam proteínas conservativas
nas diferentes espécies de micobactérias, como as proteínas “heat shock”. Exemplificamos, os genes groEL
e phoS, que codificam as proteínas 65 e 38 KDa respectivamente (HANCE et al., 1.989) ou seqüências
repetitivas de inserção como as IS6110, IS6116 ou IS986, onde existem seqüências repetitivas de 1 a 20
vezes no cromossomo de membros do Complexo M. tuberculosis (EISENACH et al., 1.990; HERMANS et
al., 1.990 a,b ; THIERRY et al., 1.990; SOINI et al., 1.992).
Também estão sendo utilizadas seqüências de oligonucleotídeos da subunidade menor do
ribosomas 16S (rRNA) para amplificar fragmentos de DNA capazes de hibridizar com sondas genéticas
gênero e espécie específicas. Estas zonas são empregadas devido à capacidade de serem altamente
conservativas nos diferentes grupos bacterianos (CHEN et al., 1.989). Esta alternativa está sendo utilizada
para a identificação a partir de amostras clínicas das diferentes espécies de micobactérias (WOESE et al.,
1.987; EDWARDS et al., 1.989; BÖDDINGHANS et al., 1.990b; ROGALL et al., 1.990; BÖTTGER et al.,
1.992; KIRSCHNER et al., 1.993a,b; VUORINEN et al., 1.995).
Outros autores classificam as reações de amplificação existentes para micobactérias em gerações.
As de primeira geração incluem, principalmente, as reações diretas, como a utilização de genes que
codifican proteínas como a IS6110. As de segunda geração incluem metodologias mais complicadas que
geralmente constam de duas ou mais técnicas: NASBA (nucleic acid sequence based amplification), TMA
(transcription - mediated RNA amplification), SDA (Strand, displacent amplification) e em ambos casos
podem ser utilizadas seqüências que codificam IS6110 e 16S rRNA, OLA (oligonucleotide ligation assay)
(VAN SOOLINGEN et al., 1.991; MENDIOLA et al., 1.992; VANDER ULIET et al., 1.993; KENT et al.,
1.995; KLATSER, 1.995).
Os parâmetros de sensibilidade da técnica do PCR para identificar Mycobacterium spp. podem ser
afetados diretamente pelos métodos de extração do DNA, já que se utilizam métodos de extração em
condições extremas (exemplo: pH). Pela natureza química da bactéria muitas vezes perde-se uma
considerável quantidade de DNA, e isto é agravado quando se processam amostras com um alto índice de
contaminação, onde existe a necessidade de se descontaminá-los e concentrá-los (exemplo: escarros,
aspirado ganglionar). Teoricamente, sabe-se que para detectar micobactérias precisa-se de
56

aproximadamente um fentograma de DNA bacteriano (o que equivalente a 1/5 do microrganismo) (PATEL
et al., 1.993), embora em amostras clínicas como escarro seja necessário de 100 a 1.000 organismos
(HANCE et al., 1989; HERMANS et al., 1.990b).
A sensibilidade pode ser recuperada se utilizarmos métodos de extração com digestão enzimática,
extração com fenol clorofórmio e precipitação do DNA com etanol. Este procedimento é mais
recomendável quando se processam amostras clínicas. Já que as extrações com detergentes iônicos
resultam em uma queda da sensibilidade (SHANWAR et al., 1.993).
A sensibilidade também pode ser afetada pelo tempo de detecção do produto pós-PCR. O
procedimento mais comum é a visualização do produto em gel de agarose corado com brometo de etídio
sob luz ultravioleta, tendo sempre um marcador de peso molecular como referência para caracterizar as
bandas de DNA observadas. Outro método comumente utilizado é a hibridização com sondas genéticas
complementares as regiões internas dos produtos amplificados.
Geralmente os sistemas de hibridização são variados, mas em geral, os produtos pós-PCR podem-
se ligar a uma matriz ou membrana de nylon ou nitrocelulose (dot-blot ou slot-blot), onde se adicionam as
sondas genéticas marcadas com radioisótopos como o 32P, conjugados enzimáticos ligados a fosfatase
alcalina, digoxigenina, biotina - avidina (SHANWAR et al., 1.993; WILSON et al., 1.993) ou métodos
quimioluminescentes (BRISSON et al., 1.989, 1.991; KUSUNOKI et al., 1.991) e dependendo do sistema de
revelação utilizado pode-se melhorar a sensibilidade em relação ao método de coloração com brometo de
etídio.
Por combinação da hibridização e PCR ou digestão do produto amplificado utilizando enzimas de
restrição (RFLP) as micobactérias podem ser identificadas prontamente, seja a nível de gênero ou espécie
(PLIKAYTIS et al., 1.992; HAAS et al., 1.993; TAKEWAKI et al., 1.993).
Estas aplicações dependem das condições de cada laboratório e de acordo com o tipo de
identificação necessário. A sensibilidade pode ainda ser melhorada submetendo-se os produtos pós-PCR a
outra amplificação utilizando “primers” que sejam capazes de amplificar um fragmento interno do produto
pós-PCR. A este novo processo denomina-se “Nested-PCR” ou segunda amplificação (SHANWAR et al.,
1.993; WILSON et al., 1.993).
Para uma melhor ilustração dos métodos que estão sendo aplicados na identificação de
micobactérias a partir de amostras clínicas, utilizando PCR, seja direto ou ligado a outros sistemas. A tabela
3 apresenta alguns resultados referentes ao tipo de “primer” utilizado (PAO et al., 1.988, 1.990; BRISSON
et al., 1989, 1.991; EISENACH et al., 1.990; 1.991; COUISINS et al.,1.992; SHANWAR et al.,1.993;
WILSON et al.,1.993; VUORINEN et al., 1.995), onde a sensibilidade varia de 55 a 100 %, mas a
especificidade é alta, variando de 95 a 100 %.
57

Tabela 3: Detecção de M. tuberculosis diretamente de amostras clínicas
Referência Número de Amostras
Primers Sensibilidade Especificidade
PAO et al., 1.990 248 65 k prot 100 % 63 %
BRISSON, et al., 1.991 514 65 k prot IS6110 80 - 97 % *
EISENACH et al.,1.991 162 IS6110 100 % 95 %
COUSINS et al., 1.992 177 MPB70 prot 97 % 76 %
WILSON et al., 1.993 171 IS6110 75 - 92 % 99 - 100 %
SHANWAR et al., 1.993 389 IS6110 55 - 74 % 95 - 98 %
VUORINEN, et al., 1.995 243 rRNA 16S + Hibridização
86.2 % 100%
* Não avaliado pelo excesso de culturas negativas das amostras PCR-positivas
Como podemos verificar existe uma grande variedade de métodos diagnósticos para
micobacterioses, e muitos deles apresentam alguns problemas na correlação da sensibilidade e
especificidade (BOOM et al., 1.991, MANJUNATH et al.,1.991; BUCK et al., 1.992; WILSON et al., 1.993).
Para superar os diferentes inconvenientes apresentados pelas técnicas descritas na literatura,deve-se
procurar optimizar as técnicas tornando-as viáveis para aplicação em laboratórios de rotina.
A eleição dos “primers” baseada nos trabalhos de ANDREAS et al.(1.991), BÖTTGER (1989,
1.992); KIRSCHNER et al. (1.993)a,b; INDERLIED, et al., 1.993; CARPENTIER et al., 1.995; VUORINEN et
al. (1.995) os quais identificam, seqüências específicas de oligonucleotídeos da subunidade menor do
ribossoma 16S (rRNA). Os dois primeiros “primers” que serão utilizados (MB1 - MB2) permitem identificar o
gênero Mycobacterium. A partir deste produto pós-PCR, poderemos identificar espécies, submetendo este
produto a uma segunda amplificação (Nested-PCR) utilizando “primers” para os Complexos: M. tuberculosis
e M. avium (esquema 1).
Assim, poderemos contar com um método rápido e relevante para a identificação através da
amplificação gênica por “PCR” e “Nested-PCR”, utilizando fragmentos de nucleotídeos altamente específicos
para o gênero Mycobacterium e espécies de micobactérias de importância na medicina humana.
Literatura recomendada
BOOM, R., SOL, C.J., SALIMANS, M., JANSEN P.M., WERTHEIM-VAN DILLEN, J. VAN DER NOORDAS. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol., Washington, v.28, p.495-503, 1991.
BRASIL, Ministéro da Saúde. MS/FNS/CRPHF/PNCT. Reunião de avaliação operacional e epidemiológica do PNCT, na decáda de 80 - 90. Brasília, p.15-27, 1992.
58

BRISSON-NOEL, A., AZNAR, C., CHUREAU, C., NGUYEN, S., PIERRE, C., BARTOLI, M., BONETE, R., PAILOUX, G., GICQUEL, B., GARRIGUE G. Diagnosis of tuberculosis by DNA amplification in clinical practice evaluation. Lancet, London, v.338, p.364-366, 1991.
CLARRIDGE, J.E., SHAWAR, R.M., SHINNICK, T.M., PLIKAYTIS, B.B. Large scale use of polymerase chain reaction for detection of Mycobacterium tuberculosis in a routine mycobacteriology laboratory. J. Clin. Microbiol., Washington, v.31, p.2049-2056, 1994.
COOKSEY, R.C. A rapid method for method for screening antimicrobial agents for activities against, a strain of Mycobacterium tuberculosis expressing firefly luciferase. Antimicrob. Agents Chemother. Washington, v.37, p.1348-1352, 1993
COOKSEY, R.C; MORLOCK, G.P.; McQUEEN, M.; GLICK MAN, S.E. and CRAWFORD, J.T. Characterization of Streptomycin Resistance Mechanisms among Mycobacterium tuberculosis Isolates from Patiens in New York City. Antimicrob. Agents Chemother. Washington, v 40, p2820-2824, 1996
CVE/SP. Tuberculose - uma emergencia mundial. São Paulo. Secretaria de Estado de São Paulo, 1.995 (folheto).
GUTHERTZ, L. S., DANISKER, B., BOTTONE, E. J., FORD, E. G., MIDURA, F., JANDA, J. M. Mycobacterium avium and Mycobacterium intracelulare in patients with and without AIDS. J. Infect. Dis., Chicago, v.160, p.1037-1041, 1989.
HERNANDEZ, A. M. Idenficação rápida de Mycobacterium spp. a partir de amostras clínicas. São Paulo, 1996. [Tese de Doutorado Universidade de São Paulo]
KIEHN, T.E., EDWARDS, F.F. Rapid identification using a specific DNA probe of Mycobacterium avium complex from patients with acquired inmunodeficiency syndrome. J. Clin. Microbiol., Washington, v.25, p.1551-1552, 1987.
KIRSCHNER, P., SPINGER, B., VOGEL, U., MEIER, A., WREDE, A., KIEKENBECK, M., BANCE, F.B., BÖTTGER, E.C. Genotypic identification of mycobacteria by nucleic acid sequence determination: report of a 2-year experience in a clinical laboratory. J. Clin. Microbiol., Washington, v.31, p.2882-2889, 1993.
KOCHI, A. The global tuberculosis situation and the new control strategy of the World Health Organization. Tubercle Lung Dis., London, v.72, p.1-6, 1992
KOX, L.F.F., RHIENTHONG, A.D., MIRANDA, M., UNDOMSANTISUK, N., ELLIS, K. VANLEEUWEN, J., VAN HEUSDEM, S., KUIJPER, S., KOLK, H.J. A more reliable PCR for detection of M. tuberculosis in clinical samples. J. Clin. Microbiol., Washington, v.32, p.672-675, 1.994
KOX, L.F.F., VANLEEUWEN, J., KNIJPER, S., JANSEN, H.M., KOLK, H.J.A. PCR assay based on DNA coding for 16S rRNA for detection and identification of Mycobacteria in clinical samples. J. Clin. Microbiol., Washington, v.33, p.3225-3233, 1995.
VIAMONT A. Diagnóstico de meningite tuberculosa através de enzimoimunoensaio (ELISA), utilizando PPD como antígeno. São Paulo, 1992. Tese de Mestrado. Escola Paulista de Medicina.
WHITE, D.A. and GOLD, J.W.M. Clínicas médicas de América do Norte, 1992 v.1, p.121-172.WHELEN, A.C., FELMLEE, T.A., HUNT, J.M., WILLIAMS, D.L., ROBERTS, G.D., STOCKMAN, L.,
PERSING, D.H. Direct genotypic detection of Mycobacterium tuberculosis rifampin resistance in clinical specimens by using a single-tube heminested PCR assay J. Clin. Microbiol., Washington, v.33, p.1558-1561, 1995.
WILLIANMS, D.L.C., WAGUESPACK, C., GILLIS, T.P., EISENACH, K., CRAWFORD, J., PORTAELS, F., SALFINGER, M., NOLAN, C.M., ABE, C., STICH-GROH, V. Characterization of rifampin resistance in pathogenic mycobacteria. Antimicrob. Agents Chemother. Washington, v 38, p2380-2386, 1994
WILKINS, E.G.L. and IVANYI, J. Potential value of serology for diagnosis of extrapulmonary tuberculosis. Lancet, London, v.336, p.641-644, 1990.
WILSON, S.M., MCNERNEY, R., NYE, P,M., GODFREY, F., STOKER, V. Progress toward a simplified polymerase chain reaction and its application to diagnosis of tuberculosis. J. Clin. Microbiol., Washington, v.31, p.776-782, 1993.
59

PCR NO DIAGNÓSTICO DA INFECÇÃO PELO VÍRUS DA HEPATITE C (HCV)
Rozangela Verlengia, MsiProfa. Dra. Rosario Dominguez Crespo Hirata
Prof. Assoc. Mario Hiroyuki Hirata
A infecção pelo vírus da hepatite C é uma doença infecciosa, usualmente crônica e comumente
assintomática por muitos anos, estando relacionada com o desenvolvimento de cirrose e carcinoma
hepatocelular.
O conceito e a importância da hepatite não-A, não-B surgiram da observação que cerca de 2 à 20%
dos pacientes transfundidos desenvolviam hepatite, dos quais 90% não apresentavam evidência de infecção
aguda pelo vírus da hepatite A (VHA), vírus da hepatite B (VHB), citomegalovírus (CMV) e vírus Epstein-
Barr. Estas hepatites passaram a ter a denominação de hepatite não-A, não-B pós-transfusional (HNANB-
PT). Doença idêntica foi também verificada em pacientes expostos a derivados do sangue (especialmente
nos receptores de fatores de coagulação, como é o caso dos hemofílicos) e entre os usuários de droga
endovenosas. Paralelamente, observou-se que alguns pacientes sem antecedentes de transfusão, com uso
de drogas injetáveis ou de outra forma de exposição parenteral também podiam apresentar clínica,
laboratorial e histologicamente a doença identica à hepatite transfusional. Tais casos passaram a ser
denominados de "forma esporádica". Atualmente, após a identificação do agente etiológico dessas hepatites
e constatação de tratar-se de um agente único, essas hepatites passaram a receber a denominação de
hepatite C.
A descoberta do vírus da hepatite C tem sido descrita como um evento totalmente dependente das
técnicas da Biologia Molecular. No final da década de 70, estudos baseados na transmissão da hepatite C à
chimpanzés, através do plasma de pacientes portadores de hepatite C, já haviam sugerido que o agente
etiológico dessa doença era um vírus. Porém, dificuldades relacionadas principalmente com a baixa
concentração do antígeno obtida, impediam de se obter resultados conclusivos nos ensaios imunológicos
empregados. Choo et al. (1989), utilizando técnicas de Biologia Molecular, construiram uma biblioteca de
cDNA a partir do ácido nucleíco proveniente do plasma de chimpanzé infectado com hepatite C, obtida
através do isolamento por ultracentrifugação e clonagem em bacteriófago gt 11. A triagem dos fagos
recombinantes gt 11 permitiram a identificação de dois clones positivos, denominados 5-1-1 e c100-3. Os
quais, foram juntamente com outros antígenos clonados, utilizados no desenvolvimento de ensaios
imunológicos (ELISA e RIBA) para a detecção de anticorpos anti-HCV.
Análise da sequência de DNA do HCV revelou que a organização genômica e o perfil hidrofóbico
eram similares aos dos vírus pertencentes à família do Flaviviridae. Consequentemente, o HCV foi
classificado como um gênero separado dentro desta família.
O genoma do HCV é uma molécula de RNA fita simples, sense positiva de aproximadamente 9.400
nucleotídeos. O genoma do HCV contém uma única e longa fase de leitura aberta (“Open Reading frame”,
ORF), a qual codifica para uma poliproteína de 3.010 à 3.033 aminoácidos. Esta poliproteína é clivada em
proteínas funcionais através de proteases virais e do hospedeiro. A região N-terminal codifica para as
60

proteínas estruturais do núcleo (“core”) e do envelope (E1 e E2/ NS1). A região C terminal codifica para as
proteínas não estruturais (NS) as quais estão envolvidas no ciclo de replicação.
Uma das características dos vírus RNA é a alta frequência de mutações devido a perda da atividade
na correção de leitura “proof reading” da polimerase dependente de RNA. Como resultado, os vírus RNA in
vivo forma populações diversas, chamadas de “quasispecies” que contém uma ou mais sequências
principais e um largo espectro de variantes completamente relacionadas. Dentro deste contexto, estudos
recentes indicam que o genoma do HCV é heterogêneo tanto entre indíviduos quanto no mesmo indíviduo. A
diversidade na sequência de nucleotídeos do genoma viral, possibilitou classificar o HCV em 9 grandes
grupos e 28 subtipos.
A fase aguda da hepatite C é usualmente subclínica ou moderada e clinicamente indistinguível de
outras forma de hepatites virais. Pacientes apresentando sinais clínicos de doença hepática, tais como
icterícia, são raros. Sintomas semelhantes a fadiga e sensibilidade hepática são eventualmente observados,
porém a maioria dos casos são assintomáticos por um período significativamente prolongado, até que se
desenvolva disfunção hepática crônica.
Apesar da manifestação clínica discreta, as infecções com HCV apresentam tendência significativa
em cronificar-se, cujo processo é bastante lento. Além do desenvolvimento de cirrose hepática, tem-se
observado também a presença do carcinoma hepatocelular (HCC), associado a infecção crônica com o vírus
C.
A hepatite C é transmitida principalmente via produtos do sangue, destacando a transfusão de
sangue e seus derivados. O uso de drogas injetáveis, tem-se apresentado como um fator de risco
importante para a infecção com o vírus da hepatite C. A transmissão por outros fluídos biológicos, tais como,
saliva, lágrima, urina, sêmem e secreção vaginal ocorre com menor freqüência, provalvelmente devido ao
baixo título do vírus C no sangue e no fluído biológico dos portadores de HVC (10 2 a 107 cópias/ml).
Recentemente foi mostrado que a transmissão do HCV a partir de mães infectadas para às crianças, estava
relacionada ao título do RNA do HCV na mãe. Neste estudo, as mães que apresentaram anticorpos anti-
HCV e que foram RNA negativas não transmitiram HCV para seus bebês. Mães RNA-HCV positivas, cujas
crianças tornaram-se infectadas, mostraram uma tendência a ter um título 100 vezes maior do que mães
RNA-HCV positiva, cujas crianças não adquiriram o HCV. Vale a pena salientar, que a exposição a uma
quantidade relativamente pequena de sangue ou de outros fluídos biológicos pode tornar-se um fator
significante de risco para HCV nos casos em que os níveis de vírus circulantes são elevados. O fato de ser
uma doença transmissível, somado à alta taxa de cronificação com o desenvolvimento de cirrose e
carcinoma hepatocelular, torna a hepatite C uma doença de importância tanto social quanto médica.
A presença de anticorpos anti-HCV em pacientes infectados com o vírus C e a clonagem de vários
segmentos do genoma viral, por exemplo 5-1-1, c100-3, c-33c, c-22-3 e c-200, levaram ao desenvolvimento
de ensaios imunológicos, correntemente aprovados para a triagem do sangue dos doadores, possibilitando
um grande avanço na descoberta do controle da infecção por HCV, evitando uma disseminação
desenfreada da doença.
61

O primeiro teste imunológico a ser utilizado, foi um imunoensaio enzimático de primeira geração
(EIA) contendo um antígeno recombinante, C 100-3, proveniente da região não estrutural do vírus. A alta
freqüência de resultados falso-positivo e falso-negativo (ambos acima de 20%), particularmente quando
usado para triar populações de baixo risco, tais como, doadores de sangue, levou a necessidade de se
desenvolver novos ensaios.
Atualmente, os imunoensaios consistem da detecção de anticorpos contra vários antígenos
recombinantes HCV-específicos fixados sob uma fase sólida, denominados testes de ELISA de segunda e
terceira geração. Nos testes de segunda geração, os antígenos utilizados são: C100-3; NS4; C33c (NS3) e
C22-3 (core). Já os de terceira geração utilizam os antígenos: NS3; NS4; NS5/5N1S (core). Esses
antígenos tem sido também empregados em ensaios denominados de RIBA (Radio imunoblot assay - Ortho
Diagnosis, Raritan, N.J., USA) para confirmação da triagem de resultados anti-HCV. O RIBA consiste de
uma fita de nitrocelulose contendo proteínas recombinantes indíviduais associadas com a proteína
Superoxido Dismutase (SOD), com as quais os anticorpos anti-HCV presentes no soro podem reagir. Nesse
ensaio, é introduzido um controle com a proteína SOD sozinha, para detectar auto-anticorpos anti-SOD, que
poderiam resultar em teste falso-positivo.
Usando esses ensaios atuais de ELISA, a sensibilidade para a detecção dos anticorpos anti-HCV é
de cerca de 94% à 100% e a especificidade é maior que 97%. Entre as possíveis causas de resultados
falso-negativos com os ensaios correntes é a heterogeneidade viral. Os testes são produzidos a partir de
sequências de genótipos predominantes de HCV (tipo 1a), cujos antígenos não reagem com anticorpos de
pacientes infectados com genótipos menos comuns. Já condições associadas com resultados sorológicos
falso-positivo incluem hipergamaglobulenemia e desordens do tecido conjutivo.
Outras limitações constadadas com uso dos ensaios imunológicos são as relacionadas com o perfil
de soroconversão. Normalmente a soro-conversão do anti-HCV é tardia, ou seja, ocorre após o início dos
sintomas clínicos da doença ou a elevação das transaminases (ALT e AST), podendo então o anticorpo anti-
HCV ser detectado entre a oitava e décima semana após o contato com o vírus. Já no caso dos pacientes
imunossuprimidos, este período torna-se mais significativamente prolongado, uma vez que, o tempo de
soro-conversão e a evolução da resposta imune são manifestados tardiamente. Outro aspecto, é o fato de
que os ensaios de anticorpos podem promover evidências de exposição ao HCV porém, são incapazes de
diferenciar entre uma infecção presente ou passada. Pacientes na fase inicial da soro-conversão
apresentam resultados inconclusivos pelos ensaios imunológicos. Claramente, pacientes infectados
recentemente apresentam soro com reação fraca (indeterminada) que, ocasionalmente, torna-se positivo,
com o passar do tempo se obtidas outras amostras em coletas posteriores. Comumente, a soro conversão
para o anti-HCV é completamente tardia após o início da ictéricia ou elevação dos níveis das transaminases
e o anticorpo pode não ser detectado até 8 a 12 semanas após a infecção. Neste estágio, a reação EIA é
fraca e deve ser confirmada através do RIBA ou Western blot.
As falhas metodológicas observadas nos ensaios imunológicos para o diagnóstico da hepatite C,
definiu a necessidade de novos ensaios diagnósticos que proporcionem maior confiabilidade quanto: (i)
62

especificidade e sensibilidade (ii) definição de infecção passada ou recente e (iii) avaliação da atividade viral,
em relação a doença hepática, seu prognóstico e a conveniência a terapia antiviral.
Considerando as limitações observadas no uso dos ensaios imunológicos e a ausência de um
sistema de cultura in vitro para o HCV, os métodos de detecção do genoma viral devem ser mais indicados
para solucionar estes problemas, aprofundando o conhecimento dos mecanismos das infecções por HCV e,
teoricamente, oferecendo um parâmetro mais adequado para diferenciar os pacientes virêmicos dos
pacientes sem virêmia. Nesse campo de pesquisa, destaca-se a reação em cadeia da polimerase (PCR),
onde ocorre a amplificação do genoma viral, para detectar o RNA-HCV no soro.
A detecção do vírus da hepatite C processa-se através da transcrição reversa e subsequente
amplificação do genoma viral através da PCR (RT-PCR), que possibilita o diagnóstico da infecção pelo vírus
C antes de qualquer outro método tradicional. Por essa técnica, o RNA-HCV pode ser detectado no soro
uma a duas semanas após a transfusão sanguínea, antes do início clínico da infecção pós-transfusional pelo
HCV. A PCR, também, tem significativa aplicação nos casos onde o imunoensaio RIBA fornece resultados
indeterminados ou não-reativos.
Um ponto crucial na detecção do RNA-HCV é a escolha dos iniciadores para o uso diagnóstico, os
quais devem corresponder as sequências genômicas bem conservadas nas diferentes variantes do HCV, de
modo a garantir uma boa sensibilidade. No caso do HCV, pesquisa-se a região 5' não transcrita, que é a
mais conservada dentro do genoma viral. Todos os protocolos dependem da reação de transcrição reversa,
que faz a cópia de DNA complementar (cDNA), a partir do RNA viral extraído. O cDNA pode ser, então,
amplificado pela PCR cujo produto pode ser analisado por eletroforese ou por um sistema de hibridização.
Alguns pesquisadores tem utilizado, além da amplificação convencional, um ciclo adicional de amplificação
com a utilização de iniciadores internos (Nested PCR). Essa técnica aumenta significativamente a
especificiade e a sensibilidade da técnica de PCR original.
Cada vez mais, tem-se demonstrado a importância da PCR na investigação da infecção pelo vírus
da hepatite C, seja no reconhecimento do tipo de vírus infectante e posterior classificação ou na avaliação
da carga viral. Estudos recentes tem sugerido que o genótipo do HCV e o nível de viremia estão associados
com o desenvolvimento clínico da doença. Com relação ao genótipo, o HCV tipo 1b parece estar
particularmente associado com doença crônica do fígado mais severa, cirrose e carcinoma hepatocelular,
com ou sem o passo intermediário da cirrose. A quantidade de partículas virais no sangue também parece
ser dependente do genótipo viral, uma vez que, pacientes infectados com HCV tipo 2 mostra um nível
significantemente menor de RNA-HCV circulante.
Outra importante aplicação da PCR para o HCV é o acompanhamento da resposta à terapia antiviral
em pacientes com hepatite C crônica, em particular ao interferon (IFN). O IFN promove o rápido
decréscimo dos níveis séricos de alanina aminotransferase (ALT), a erradicação do genoma do HCV do soro
e a melhora do estado de inflamação e necrose do fígado. Alta viremia do HCV (superior a 107 cópias/ml de
soro) é prognóstico negativo para uma completa e adequada resposta ao tratamento com o IFN . Portanto,
a avaliação da eficácia terapêutica ao IFN é mais adequadamente realizada pela quantificação do RNA-
HCV, que somente é possível por técnicas moleculares.
63

A quantificação do RNA viral, tem sido realizada por vários métodos de amplificação do RNA-HCV, o
mais usualmente empregado é a PCR competitiva (Roche Monitor Kit). Nessa técnica, a amostra é diluida
seriadamente e cada diluição é submetida à reação de amplificação juntamente com um controle interno,
com o qual compete, revelando diferentes sinais. Outro ensaio também disponível é o “branched DNA”
(Quantiplex, Chiron Corp), um método de amplificação de sinal de fase-sólida. Neste método, o soro é
adicionado sob um suporte sólido, onde ocorre a lise do vírus, a captura e a hibridização do ácido nucleíco a
ser detectado e a amplificação do sinal. Embora essas técnicas sejam muito sensíveis, os resultados
obtidos tem mostrado variação do nível de viremia dos pacientes infectados. No caso do “branched DNA”,
este demonstrou ser significativamente menos sensível (detecção limite 350.000 cópias/ml) do que o PCR
competitivo (detecção limite aproximadamente 2000 cópias/ml). Um estudo recente de comparação entre 31
laboratórios mostrou uma substancial variação de sensibilidade e especificidade desses métodos.
Portanto, verifica-se a necessidade de aprimoramento metodológico e aplicação das técnicas
moleculares de forma mais rotineira na busca do diagnóstico precoce e do acompanhamento terapêutico
adequado.
Literatura recomendada
CHOO, et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science, 244: 359-62, 1989.
DAVIS, G.L. et al. Quantitative detection of hepatitis C virus RNA with a solid-phase signal amplification method: definition of optimal conditions for specimen collection and clinical application in interferon-treated patients. Hepatology, 19: 1337-41, 1994.
DI BISCEGLIE, A. et al. Long-term clinical and histopathological follow-up of chronic posttrasnfusion hepatitis. Hepatology, 14: 969-74, 1991.13
MATTSSON, L. et al. Outcome of acute symptomatic non-A, non-B hepatitis: a 13year follow-up study of hepatitis C virus markers. Liver, 13: 274-278, 1993.
McPHERSON, R.A. . laboratory Diagnosis of Human Hepatitis Viruses. J. Clin. Lab. Analysis. 8: 369-377, 1994.
MILLER, R.H. PURCELL, R.H. . Hepatitis C virus shares amino acid sequence similarity with Pestiviruses and Flaviviruses as well as members of two plant virus supergroups. Proceedings of the National Academy of Sciences USA. 87: 2057-2061, 1990.
OTHO, H. et. al. Transmission of hepatitis C virus from mothers to infants. N. Engl. J. Med. 300: 744-750, 1994.
REICHARD, O. Treatment of chronic hepatitis C. Sacand. J. Infec. Diseases, 22 (4): 509, 1990. RUBIN, R. A. chronic hepatitis C Advances in Diagnostic testing and Therapy. Arch. Intern. Med. 154: 387-
392, 1994.RÜSTER, B. et al. Quantification of hepatitis C virus RNA by competitive Reverse Transcription and
Polymerase chain Reaction using a modified Hepatitis C Virus RNA transcript. Analytical Biochemistry 224: 597-600, 1995.
SHINDO, M. et al. Decrease in hepatitis C viral RNA during alpha-interferon therapy for chronic hepatitis C. Ann. Intern. Med. 115: 700-4, 1990.
STEINHAUER, D.A. et al. Sequential change of the hypervariable region of the hepatitis C virus genome in acute infection. J. Med. Virol. 42: 103-108, 1987.
64

PCR NO DIAGNÓSTICO DA INFECÇÃO PELO VÍRUS HIV
Claudio Mortensen
1. Detecção e Quantificação do Vírus HIV Por PCR:
1.1. Introdução:
O vírus HIV é membro da família dos retrovírus sendo que, seus vírions (partículas individuais),
possuem uma forma aproximadamente esférica com algo em torno de 110nm de diâmetro. É um vírus
contendo um envelope com nucleocapsídeo em forma de cone. O genoma viral é composto por 2 moléculas
de RNA simples-fita, de 9,2kb, com polaridade positiva em relação a tradução (Fields).
O genoma HIV já foi totalmente seqüenciado e analisado, sendo que seu genes receberam a
seguinte nomenclatura (6): gag, pol, vif, vpr, tat, rev, vpu, env, nef. As proteínas sintetizadas a partir do
gene gag compõem o núcleo (“core”) que envolve o genoma viral. O gene gag é traduzido na forma de uma
proteína precursora Pr55gag, que é posteriormente clivada para compor as proteínas p17 (proteína de
matriz), p24 (proteína estrutural do capsídio), p9 (proteína do núcleocapsídio que se liga fortemente ao RNA
viral) e p7. O gene pol também é traduzido na forma de um precursor, Pr160gag-pol e codifica para as enzimas
virais: Protease (p10), Transcriptase Reversa e RNase-H (p51/66) e Integrase (p32). O gene vif (proteína
p23), parece atuar como um fator de infectividade, mas sua função ainda não foi totalmente elucidada. A
função do gene vpr (proteína p15) ainda não é conhecida. A proteína p14, codificada pelo gene tat, atua
como transativador transcripcional, interagindo com fatores de transcrição celulares e a seqüência TAR viral,
promovendo a iniciação e elongação dos transcritos virais. O gene rev (proteína p19), indispensável à
replicação viral, age como transativador pós-transcripcional, ligando-se à seqüência RRE viral e fatores
celulares, promovendo "splicing" e/ou transporte do mRNA viral. O gene vpu (proteína p16) influencia a
liberação das partículas virais e aumenta o turnover do antígeno CD4+. O gene env codifica a glicoproteína
precursora gp160, que posteriormente é clivada para formar a gp41, transmembranar, e a gp120,
glicoproteína de face externa, responsáveis pela ligação do vírus ao receptor celular. Por fim, o gene nef
(proteína p27) potencializa a infectividade viral (18).
LTR
gag
pol
vif vpu env
nef
LTR
vprrev
tat
Genoma do HIV
65

Em seu processo de replicação (produção de novas partículas virais), o HIV necessita da formação
de uma molécula intermediária de DNA, a qual integra-se ao genoma celular, permanecendo como parte
integrante deste, durante todo o período de vida da célula em questão. A informação genética para a síntese
dos componentes do vírus, inclusive do genoma RNA, é proveniente deste DNA (denominado DNA pró-viral)
integrado. Esta molécula intermediária de DNA é sintetizada a partir do "molde" de RNA viral, pela enzima
Transcriptase Reversa, codificada pelo vírus e transportada no interior da partícula viral. A seguir, através de
mecanismos ainda não muito bem definidos, a molécula de DNA recém sintetizada, é transportada para o
núcleo celular onde será integrada ao genoma por ação de outra enzima viral, também presente no vírion, a
Integrase. Tanto a Transcriptase Reversa quanto a Integrase, são produtos do gene pol.
1.2. Detecção do Genoma Viral por PCR:
O mecanismo, bastante peculiar, de replicação dos retrovírus, possibilita dois caminhos para a
detecção da presença do vírus HIV no organismo humano: a demonstração da presença do DNA pró-viral
integrado ao genoma celular e a detecção do RNA viral presente nas partículas livres circulantes.
A este ponto, é importante esclarecer determinados aspectos da história natural da infecção pelo
vírus HIV. Até pouco tempo atrás, havia um certo consenso entre os cientistas e os clínicos de que muito
poucas células estariam infectadas ou expressando o genoma viral e que existiria muito pouco vírus
circulante, livre no plasma, durante a maior parte do curso clínico da doença. Sendo assim, foram
desenvolvidos modelos para a doença AIDS, os quais consideravam um período de latência virológica
durante a fase assintomática da mesma. Entretanto, mais recentemente, trabalhos publicados por Ho, D. e
Wei, X., demostraram que o vírus HIV replica-se muito mais rapidamente e muito mais precocemente do
que era imaginado. Estes trabalhos provaram que o período de latência é na verdade clínico, pois o vírus
replica-se numa velocidade bastante alta, já imediatamente após infectar a primeira célula e, um equilíbrio
entre esta produção de vírus e a reposição de células CD4+ acaba por estabelecer-se e, até que se esgote a
capacidade do organismo em manter este dispendioso equilíbrio, o paciente poderá não apresentar
sintomas.
Em vista disso fica claro que em qualquer momento pós-infecção, poderão ser encontradas
partículas virais circulantes, contudo, é necessário que haja uma quantidade mínima de partículas virais para
que a técnica de PCR seja capaz de detectá-las. Sob condições ideais, ou seja, tendo-se um protocolo
muito bem otimizado para a reação de PCR, seu limite de sensibilidade será de 200 cópias do genoma por
ml de plasma. É muito importante que se tenha consciência destes dados, pois haverá um período de tempo
pós-contaminação, embora muito menor do que para os testes imunológicos, em que não será possível
detectar-se a presença do vírus. Este período de tempo foi estimado em aproximadamente 9 dias pós-
contaminação. Outro ponto que deve ser observado, com relação ao PCR aplicada à AIDS, diz respeito ao
diagnóstico de crianças nascidas de mães portadoras do HIV. O risco de um recém nascido, de mãe
portadora do HIV, ter sido infectado por via placentária (transmissão vertical) foi calculado em um valor
66

aproximado de 30%, portanto é bastante importante que se possa diagnosticar o quanto antes o nível de
infeção da criança. Entretanto, os métodos imunológicos ficam sujeitos a resultados falso-positivos devido
ao fato da criança, até 3-6 meses após o nascimento, estar portanto anticorpos provenientes da mãe,
herdados durante a gestação. Sendo assim, o método mais seguro de detectar-se a presença do vírus, é
comprovar o genoma próviral integrado no genoma celular. A pesquisa de partículas livres no plasma da
criança também poderia levar a resultados falsos, pois uma grande parte dos vírions produzidos pelas
células, são inativos (não infectantes) e, sendo assim, a criança poderia portar partículas virais inativas em
sua circulação, que não teriam estabelecido uma infecção produtiva, mas que estariam sendo detectadas
pela alta resolução da técnica de PCR.
Assim sendo, apresentamos aqui um método de PCR para o diagnóstico de infecção pelo vírus HIV
que utiliza a técnica de “Nested PCR” na detecção do DNA pró-viral integrado ao genoma celular. O
protocolo descrito a seguir utiliza um conjunto de primers, desenvolvidos por este laboratório,
complementares a regiões altamente conservadas, presentes na seqüência do gene gag, gerando um
produto de amplificação de 495pb.
O DNA é isolado das células mononucleares do sangue periférico utilizando-se o mesmo protocolo
já descrito nesta apostila. Este DNA, normalmente estoca em geladeira ou freezer, é aquecido a 58OC
durante 20 min. antes de ser adicionado à reação.
Ao adicionar-se os componetes de reação aos tubos, é importante seguir uma determinada ordem.
Portanto, após descongelar todos os componentes, o primeiro a ser adicionado é a água, a seguir o Tampão
de Reação, dNTP, Primers e, só então, o DNA, que pode ser mantido no banho-maria a 58OC até o
momento de ser adicionado aos tubos. Cuidados especiais devem ser tomados a este ponto, pois o DNA
genômico humano é uma molécula bastante grande, tornando a solução em que está contida, muito viscosa.
Recomenda-se utilizar ponteiras especiais de ponta larga ou, na falta destas, cortar as pontas para torna-las
mais largas em sua abertura.
Após a adição do DNA aos tubos de reação, estes são transferidos ao aparelho de PCR para a
primeira etapa de reação, o “HOT START”, ou pré-aquecimento da amostra a 98OC durante 10min. O
objetivo desta prática é desfazer possíveis estruturas secundárias da molécula de DNA, as quais poderiam
afetar sensivelmente a assessibilidade dos primers aos seus sítios correspondentes. Quando terminados os
10min., aciona-se a função PAUSA do aparelho, retira-se os tubos colocando-os em gelo imediatamente. No
interior do fluxo laminar, adiciona-se a Taq DNA polimerase, retornando os tubos ao aparelho e liberando-se
a função pausa. A reação prosseguirá por 35 ciclos.
Finalizado o primeiro PCR, serão retirados 10l desta reação e adicionados em outra contendo o
segundo conjunto de primers (nested), sempre respeitando a ordem de adição dos componentes. A
diferença desta reação com a primeira, é a inexitência de “HOT START”, pois aqui as moléculas alvo já
possuem um tamanho milhares de vezes menor do que as da amostra inicial.
Literatura recomendada
67

Amato Neto, V.; Medeiros, E.A.S.; Kallás; E.G.; Levi, G.C.; Baldy, J.L.S.; Medeiros, R.S.S. (1996). AIDS na Prática Médica. Sarvier, São Paulo.
Cohen, P.T.; Sande, M.A.; Volberding, P.A. (1994) The AIDS Knoledge Base. Second Edition, Little, Brown and Company, Boston.
DeVita, V.T.; Hellman, S.; Rosemberg, S.A. (1991) AIDS: Etiology, Diagnosis, Treatment, and Prevention. Third Edition. J.B. Lippincott Company. Philadelphia.
Fields, B.N.; Knipe, D.M.; Howley, P.M. (1996). Fields Virology, Third Edition. Lippincott-Raven, Philadelphia.Ho, D.D., Neumann, A.U., Perelson, A.S., Chen, W., Leonard, J.M., Markowitz, M. (1995). Rapid turnover of
plasma virions and CD4+ lymphocytes in HIV-1 infection. Nature, 373: 123.Kwok, S.; Mack, D.H.; Mullis, K.B. (1987). J. Virol., 61: 1690.Phair, I.P., Wolinsky S. (1989). Diagnosis of infection with the human immunodeficiency virus. J. Infect. Dis.,
159: 320.Varmus, H.E., Swastrom, R. (1985). Replication of retrovirus. in Weiss, R., Teich, N., Varmus, H.E., et al.
(eds.): RNA Tumor Virus, ed. 2. Cold Spring Harbor, N.Y., Cold Spring Harbor Laboratory, pp 74.Wei, X., Ghosh, S.K., Taylor, M.E., Johnson, V.A., et al. (1995). Viral dynamics in human immunodeficiency
virus type 1 infection. Nature, 373: 117.White, D.O.; Fenner, F.J. (1994). Medical Virology, Fourth Edition. Academic Press, San Diego.
68

Aula número 7
PCR E RFLP NA IDENTIFICAÇÃO DE INDIVÍDUOSProf. Assoc. Mario Hiroyuki Hirata
Adriana OzakiErik Martins Ueda
Introdução
A necessidade de confirmar disputas de paternidade ou relacionamento familiar sempre existiram. O
progresso no desenvolvimento de um meio seguro para a determinação de relacionamento tem ocorrido
muito lentamente. A descoberta de Landsteiner do grupo sanguíneo ABO em 1900, estabeleceu o que se
tornou a base para os estudos de parentescos dos dias modernos, porém a aplicação da descoberta para a
determinação de paternidade, só foi demostrado em 1910 por von Dungern e Hirszfeld, conhecida como
herança Mendeliana do grupo sanguíneo. O próximo evento significante ocorreu em 1927 quando o sistema
MN foi descrito por Landsteiner e Levine. Este foi seguido pela descoberta, em 1939-1940, do sistema Rh-
Hr por Levine, Stetsun, Landsteiner e Wiener. A aplicação do conhecimento desses sistemas fornece uma
média de exclusão da paternidade de aproximadamente 55% dos homens supostamente acusados.
A combinação dos sistemas ABO, Rh e MN, tornou possível excluir 70% dos homens caucasianos
supostamente acusados da paternidade, e os testes de parentesco foram reconhecidos pelo seu valor de
exclusão, mesmo que estes tivessem alcance limitado. Apesar dessas deficiências, esses testes foram
utilizados até fins de 1960 e meados de 1970, quando a utilidade do sistema de antígenos leucocitários
humanos (HLA) foi reconhecido legalmente e científicamente como um método para avaliação nos testes de
paternidade. Os testes, usando múltiplos sistemas, incluindo o HLA, então forneceram um meio prático para
a exclusão de pelo menos 95% dos supostos pais, no entanto, ainda não totalmente adequada para esse
fim. Recentemente, técnicas baseadas no estudo de sequências polimórficas do DNA tem dado de forma
muito segura a definição adequada para a identificação de indivíduos.
A análise de sequências polimórficas no DNA é, atualmente, a ferramenta mais poderosa para
distinguir geneticamente os indivíduos ou determinar níveis de relacionamento familiar. Os testes com DNA
superam os resultados obtidos pelos métodos tradicionais, e podem solucionar de forma segura os
resultados tidos como inconclusivos pelos métodos considerados tradicionais. Por essas razões, foram
realizadas uma série de estudos de marcadores genéticos para determinar a real definição de um vínculo
familiar. A crescente demanda de testes de identificação, estimulou a comunidade científica a desenvolver
metodologias com maior grau de discriminação que permite com segurança incluir ou excluir a paternidade
ou relação familiar.
Esses marcadores genéticos são características herdadas que diferenciam os indivíduos uns dos
outros e são controlados por genes localizados em um par de cromossomos, denominados alelos. Apenas
dois alelos estão envolvidos na formação de uma característica específica, um do pai outro da mãe. Quando
o par de genes for idêntico, é chamado de homozigoto; quando o par de genes for diferente, de
69

heterozigoto. É necessário também uma escolha adequada dos alelos para um estudo detalhado da
ocorrência, a fim de aumentar a precisão e exatidão no resultado final.
Repetições Sucessivas de Número Variável (VNTR) e Repetições Sucessivas Pequenas (STR)
Vários locos de DNA são amplamente utilizados em análises forenses, os mais utilizados são
aqueles contendo as denominadas Repetições Sucessivas de Número Variável ((Variable Number “
Tandem Repeat). Estas sequências não codificam um gene específico, podendo assim serem utilizadas
para análises forenses sem nenhuma consequência ao indivíduo. Isto é uma vantagem pois as VNTR são
menos suscetíveis a seleção natural, que poderia levar a diferentes frequências de alelos em populações
diferentes. As VNTR tem sido também denominadas de minisatélites.
Uma região típica de VNTR consiste de 500 a 10,000 bp, compreendendo várias unidades de
repetições sucessivas, cada uma com 15 a 35 bp em comprimento. O número exato de repetições assim
como o comprimento da região de VNTR, varia de um alelo para outro, e diferentes alelos podem ser
identificados pelo seu comprimento. Os locos VNTR são particularmente convenientes como marcadores
para identificação, pois eles possuem um grande número de alelos diferentes, geralmente 100 ou mais,
embora apenas 15 a 25 possam ser distinguidas (A palavra alelo é tradicionalmente aplicada como forma
alternativa de um gene; entretanto, pode-se extrapolar o significado para regiões não gênicas do DNA, como
as VNTR).
As VNTR tem uma alta taxa de mutação, levando as mudanças no comprimento. Uma mutação
individual usualmente muda o comprimento por apenas uma ou poucas unidades de repetição. O resultado é
um grande número de alelos na população com diferentes sequências. O número de possíveis genótipos em
um loco é muito maior que o número de alelos, e quando diversos locos são combinados, o número total de
genótipos é muito grande.
Famílias de microsatélites de DNA compreendem arranjos de repetições consecutivas de pequeno
tamanho (“Short Tanden Repeat”, STR), que apresentam sequências simples (1-4 bp) e estão
localizados ao longo do genoma. Normalmente repetições dos mononucleotídeos A e T, são comuns e
juntos são responsáveis por cerca de 10 Mb ou 0,3% do genoma nuclear. Em contraste, sequências de G e
C são bem mais raras. No caso de repetições de dinucleotídeos, arranjos de repetições CA (repetições de
TG na fita complementar) são comuns, compreendendo 0,5% e são altamente polimórficos. Repetições
CT/TG são também comuns, ocorrendo em cada 50 kb, em média, e sendo responsável por cerca de 0,2%
do genoma. Por outro lado, sequências de CG/GC são raras. Isso se deve ao fato de que resíduos de C
ligados em sua extremidade 3’ por um resíduo G (CpG) são suscetíveis à metilação e desaminação,
resultando em TpG (ou CpA na fita complementar). Repetições consecutivas de trinucleotídeos e
tetranucleotídeos são raríssimas, porém altamente polimórficas, sendo candidatas a marcadores genéticos
do polimorfismo entre indivíduos.
70

RFLP na Identificação de Indivíduos
A maioria do DNA no genoma humano não parece codificar genes específicos, mas parece ter um
papel na integridade estrutural do cromossomo ou em sua replicação. Como vimos, o DNA não codificante
contém várias sequências repetitivas, sendo possível a análise de DNA para identificação de indivíduos. A
garantia de que o DNA apresenta repetição única para cada indivíduo, ou polimorfismo, o torna singular pelo
número característico dessas repetições. Neste contexto, essa caracteríctica muito singular pode servir
como parâmetro muito importante na caracterização de um indivíduo, porque diferentes tamanhos de
fragmentos de DNA são encontrados, após digestão do DNA com enzimas de restrição, e hibridização com
sondas alelo-específicas. Considerando a frequência desses fragmentos polimórficos na população e
possíveis variações étnicas, o teste de DNA tem a capacidade de fornecer as probabilidades mais exatas de
inclusão ou exclusão de indivíduos num contexto forense ou de vínculo familiar. Consequentemente muita
atenção tem sido dada para a padronização desses novos métodos em laboratórios forenses. Técnicas
similares tem sido usadas para teste de paternidade em casos legais.
Uma aplicação potencial é a análise da sequência dos nucleotídeos nos gens HLA para definir
identidade imunológica. Essa aplicação fornece uma oportunidade para se encontrar os melhores doadores.
Uma outra aplicação seria estabelecer suscetibilidade a patologias autoimunes em indivíduos com
alterações específicas nos genes HLA.
Os locos de VNTR consistem em numerosas cópias de sequências simples de DNA, em um
intervalo de comprimento que varia de 9 a 14 pares de base (bp), alinhados de forma contínua. Estudos
sobre populações humanas tem revelado que o número de repetições sucessivas presentes em um
determinado loco pode exibir uma grande variação de um indivíduo para outro, dessa forma se estabelecem
como marcadores genéticos que são altamente informativos de acordo com o espécime biológico. O uso de
vários locos VNTR independentes e hipervariáveis fornecem informação muito importante condiderando
uma fonte de espécime biológico a ser identificado.
A análise por RFLP de locos de VNTR envolve a purificação do DNA de fluídos corporais ou
esfregaços de biópsias, seguida da digestão do DNA com a enzima de restrição (p. ex. Hae III). Os
fragmentos de DNA resultantes são separados por eletroforese, utilizando gel de agarose; Os fragmentos
separados são então desnaturados e transferidos para uma membrana de nylon (Southern Blot). A
membrana é então hibridizada com sondas marcadas isotopicamente ou por métodos não-isotópicos (p. ex.,
enzimático) específicas para o locos de VNTR de interesse. Os métodos não-isotópicos são preferidos, pois
não necessitam dos cuidados na manipulação que a conduta de segurança exige para os materiais
radioativos. A enzima fosfatase alcalina é muito utilizada na revelação, onde são utilizados substratos
coloridos ou quimioluminescentes. O método luminescente tem seus incovenientes, pois requer um sistema
com produção contínua de luz para que o sinal possa ser coletado com o decorrer do tempo (para aumentar
a sensibilidade) e se for necessário, a exposição múltipla do filme. Os sistemas quimioluminescentes mais
sensíveis são aqueles que emitem luz continuamente e a reação enzimática envolve a clivagem de
substratos 1,2-dioxitanos estabilizados. Esses sistemas tem sido amplamente aplicados em pesquisas 71
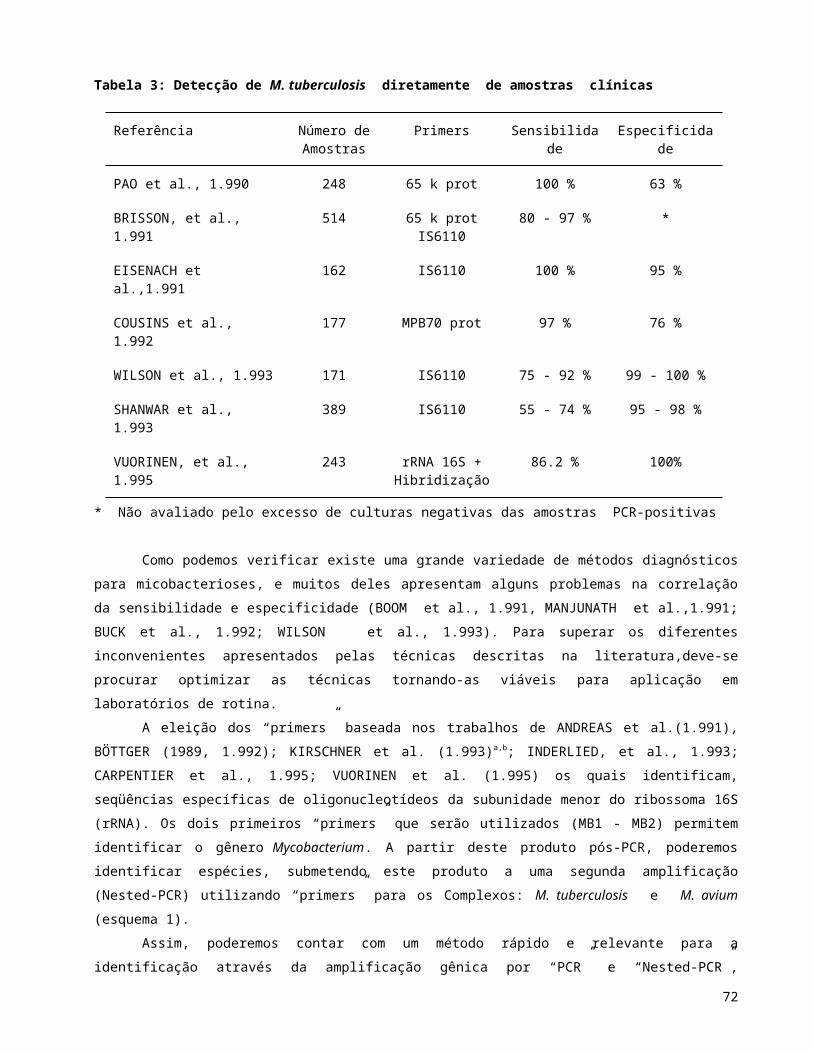
genéticas. O sistema de detecção quimioluminescente pode substituir as sondas marcadas com 32P usadas
nos sistemas de quantificações de DNA e tipagem por RFLPs.
Tipos de Sondas utilizadas no RFLP
Para análise de regiões hipervariáveis (VNTR) são utilizadas sondas moleculares complementares a
estas sequências repetitivas, o que possibilita observar as características de cada indivíduo e suas relações
com os pais. No método de RFLP, há sondas multilocais que hibridizam com vários locos ao mesmo tempo,
sendo analisadas várias bandas de DNA, que correspondem aos alelos respectivos. Os testes com sondas
unilocais utilizam a mesma metodologia, entretanto, identifica-se um loco polimórfico do DNA de cada vez,
permitindo com simplicidade observar um alelo de origem materna e outro de origem paterna. O limite de
detecção das sondas multilocais é de 500 ng de DNA, as sondas unilocais apresentam limites muito
menores onde quantidades em torno de 30 ng podem ser visualizadas, que permitem espécimes de até 1 L
de sangue ou sêmem para análise. Essa sensibilidade significa que embora as sondas multilocais
fornecerem melhor distinção individual as sondas unilocais são as mais utilizadas no contexto forense. Outra
vantagem é a possibilidade do uso das sondas unilocais em amostras misturadas.
PCR na identificação de indivíduos
Uma alternativa que vem dominando as técnicas moleculares para fins de identificação é baseada
na técnica de amplificação do gene utilizando a reação em cadeia da polimerase (PCR). Esse procedimento
baseia-se na amplificação específica de VNTR, presentes em determinados alelos humanos, através da
hibridização de oligonucleotídeos (iniciadores ou “primers”) flanqueando a região que contém a VNTR. Essa
técnica tem sido denominada Polimorfismo de Tamanho de Fragmento amplificado (AMP-FLP).
Atualmente, estes locos altamente polimórficos demonstrados por PCR são os marcadores genéticos mais
informativos para a caracterização do indivíduo.
A PCR permite um aumento da sensibilidade de um procedimento subanalítico para níveis analíticos
em um período de tempo relativamente curto. A técnica permite o aumento da quantidade de DNA de tal
forma que uma simples sequência pode ser analisada, após a PCR, em um simples procedimento sem a
utilização de material radioativo ou qualquer outro procedimento mais complexo. As vantagens em relação
aos outros métodos estão no aumento da sensibilidade, especificidade e a redução no tempo de ensaio.
Outra vantagem na utilização da PCR é que ela pode ser realizada com muito pouca quantidade de amostra
(a técnica de PCR pode detectar até 1 fentograma de DNA alvo) e em material fixado com formaldeído,
parafinados e em peças de necrópsia ou manchas de sangue secas.
Dentre os genes polimórficos utilizados para a caracterização de indivíduos, podemos citar a
amplificação de minisatélites nos locos: Apo B (apolipoproteína B), D1S80, D17S30 e microsatélites SE33,
D12S67, Y-27H39, vWF (von Willerbrand). Esses alelos podem ser amplificados de amostras contendo
pouca quantidade de DNA, o que facilita a utilização tanto para fins de identificação criminal como para
testes de identificação de paternidade. Porém, nem todos os locos de minisatélites e microsatélites 72

existentes podem ser utilizados na investigação de paternidade, sendo os mais adequados os que
apresentam maior polimorfismo e maior grau de heterozigose. A análise dos polimorfismos genéticos tem
sido utilizada como parâmetro importante na área médica e também em análises forenses.
Cálculo da Frequência dos Alelos
A frequência dos alelos é baseada na ocorrência de todos os alelos para um dalo loco estudado
dentro de mais ou menos 3% do tamanho do banco de dados, que se construiu. Em adição, pode-se
escolher alelos com frequência mínima de 2%.
Cálculo estatístico
1. Probabilidade de Exclusão
A Probabilidade de Exclusão (PE) é definida como a probabilidade de excluir ao acaso, indivíduos
da população dados os alelos da criança e da mãe. A informação genética do suposto pai não é considerada
dentro da determinação da Probabilidade de Exclusão. A Probabilidade de Exclusão é igual a frequência de
todos os homens da população que não contém os alelos compartilhado obrigatoriamente do alelo do pai e
da criança.
A equação utilizada para determinar esta probabilidade é:
PE = (1-a)2
Onde, a = frequência do alelo da criança herdado do seu pai biológico
(obrigatoriamente o alelo paterno).
2. Índice de Paternidade
O Índice de Paternidade (PI) é a razão de verossimilhança nas quais os testes apresentam duas
probabilidades condicionais. A probabilidade do teste de paternidade supor que o suspeito pai é o pai
biológico, comparado com a probabilidade de que ele não seja o pai biológico. Se ambos os pais são
heterozigotos, então:
PI = (0,5)x(0,5)/(0,5)x(fPa) ou
PI = (0,5)/(fPa)
Onde (fPa) é a frequência do alelo paterno.
Padrão de banda simples do suposto pai são tratadas como heterozigotos para o cálculo de PI.
3. Probabilidade de Paternidade
A Probabilidade de Paternidade (W) é baseada sobre o Teorema de Baye’s nas quais proporciona a
determinação de posterior probabilidade para paternidade baseados nos resultados genéticos dos testes da
mãe, da criança e do suposto pai. Em ordem de determinação da Probabilidade de Paternidade, a suposição
deve ser marcada (antes do teste) como uma prévia probabilidade de que o suposto pai seja o verdadeiro
73

pai biológico. A equação usada para o cálculo da posterior probabilidade (W) da prévia probabilidade de 0,5
é:
W = _PI_ PI + 1
PI = Índice de Paternidade.
Literatura recomendada
Alford, L; Hammond, H. A.; Coto, I.; Caskey, C. T.; (1994). Rapid and Efficient Resolution of Parentage by Amplification of Short Tandem Repeats. Am. J. Hum. Genet. 55: 190-195.
Arroyo, E.; García-Sánchez, F.; Gómez-Reino, F.; Ruiz de la Cuesta, J. M.; Vicario, J. L.; (1994). Prenatal Exclusion of Paternity by PCR-FLP Analysis. J. Forensic Sciences 39 (2): 566-572.
Boerwinkle, E.; Xiong, W.; Fourest, E.; Chan, L.; (1989). Rapid typing of tandemly repeated hypervariable loci by the polymerase chain reaction: Application to the apolipoprotein B 3' hypervariable region. Proc. Natl. Acad. Sci. USA 86: 212-216.
Budowle, B.; Chakraborty, R.; Giusti, A. M.; Eisenberg, A. J.; Allen, R. C.; (1991). Analysis of the VNTR Locus D1S80 by the PCR Followed by High-Resolution PAGE. Am. J. Hum. Genet. 48: 137-144.
Budowle, B.; Baechtel, F. S.; Comey, C. T.; Giusti, A. M.; Klevan, L.; (1995). Simple protocols for typing forensic biological evidence: Chemiluminescent detection for human DNA quantification and restriction fragment length polymorphism (RFLP) analyses and manual typing of polymerase chain reaction (PCR) amplified polymorphisms. Electrophoresis 16: 1559-1567.
Decorte, R.; Cuppens, H.; Maynen, P.; Cassiman, J. J.; (1990). Rapid Detection of Hypervariable Regions by the Polymerase Chain Reaction Technique. DNA and Cell Biology 9 (6): 461-469.
Horn, G. T.; Richards, B.; Kingler, K. W.; (1989). Amplification of the highly polymorphic VNTR segment by the polymerase chain reaction. Nucleic Acids Res. 17: 2140.
Jeffreys, A. J.; Wilson, V.; Thein, S. L.; (1985). Hypervariable ' minisatellite' regions in human DNA. Nature 314 (7): 67-73.
Silver, H.; (1989). Paternity Testing. Critical Reviews in Clinical Laboratory Sciences 25: 391-408.
74
Recommended















