
EVOLUZIONE MOLECOLARE
L’evoluzione molecolare studia la velocità e i tipi di
cambiamenti che hanno luogo nel materiale genetico o nei
suoi prodotti.
Gli studi di evoluzione molecolare si basano essenzialmente
su analisi comparative e quindi presuppongono la
conoscenza delle macromolecole biologiche almeno a livello
della struttura primaria.

DIMENSIONE
TEMPO
CINEMATICA DINAMICA
OBIETTIVI
individuazione delle costrizioni funzionali
comprensione dei processi evolutivi
studi filogenetici, relazioni tassonomiche
pura descrizione meccanismi evolutivi
EVOLUZIONE MOLECOLARE
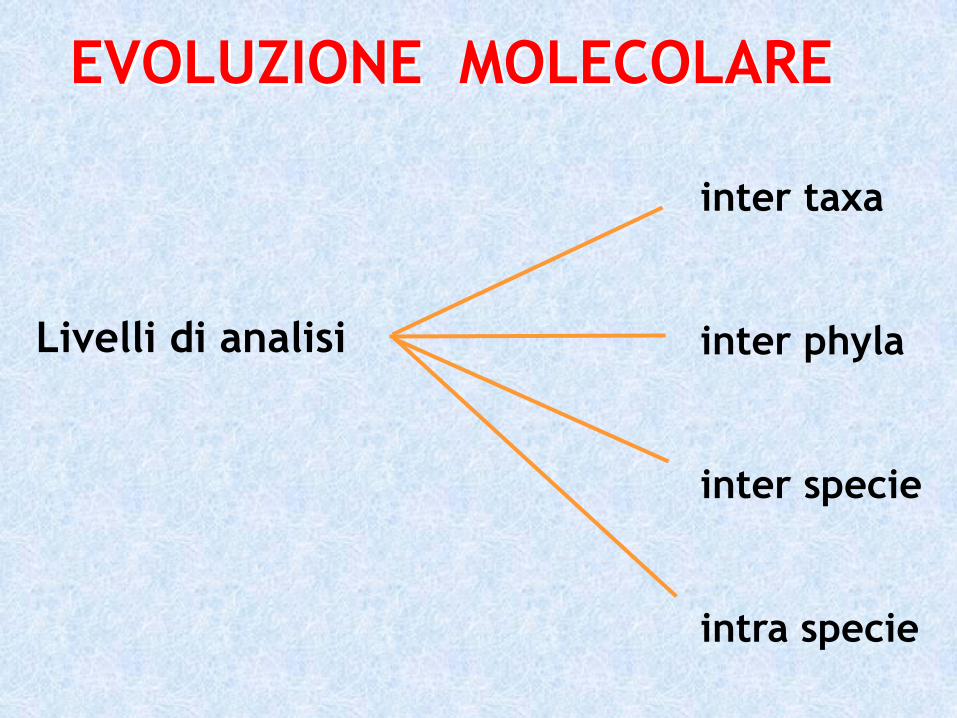
Livelli di analisi
inter taxa
inter phyla
inter specie
intra specie
EVOLUZIONE MOLECOLARE

Scelta delle
sequenze
geni ortologhi
geni paraloghi
altre regioni
(non codificanti, regolatorie)
genomi completi
EVOLUZIONE MOLECOLARE

speciazione
duplicazione genica ba
specie 1
specie 2
gene β 1gene α 1 gene α 2 gene β 2
speciazione
ort
olo
gia
e p
ara
logia

Evoluzione della famiglia genica della
beta-globina umana
5’ 3’
e g1 g2 yh d b
Embrionale Fetale Pseudogene Adulta
0
100
200
Myaduplicazione genica

Mutazioni Puntiformi: Sostituzioni nucleotidiche
•Transizioni
•Transversioni
Per capire le dinamiche della sostituzione nucletidica
dobbiamo capire la probabilità di sostituzione di un
nucleotide con un altro (diversi modelli matematici).
A
G
C
T

Sulla base delle distanze genetiche è possibile ricostruire la storia
evolutiva delle molecole, e quindi anche degli organismi.
Solitamente il numero di sostituzioni tra due sequenze è espresso in
termini di sostituzioni per sito (k) e non di sostituzioni totali. Questo
permette di confrontare sequenze di lunghezza diverse.
Generalmente il numero di sequenze che viene osservato tra coppie di
sequenze è sempre inferiore rispetto al numero di sostituzioni che
hanno effettivamente avuto luogo (sostituzioni multiple, retromutazioni
e sostituzioni convergenti). Per cui sono necessarie delle correzioni
matematiche.

A
C
T
G
A
A
C
G
T
A
A
C
G
C
A
C
T
G
A
A
C
G
T
A
A
C
G
C
A
C
T
G
A
A
C
G
T
A
A
C
G
C
T AT
G G
T A
Confronto delle
sequenze attuali
sostituzione singolasostituzione multipla
sostituzione convergente
sostituzioni coincidenti
A a T G g A C G T A a C G C
t C T G g A C G T A A C G C
Sequenza ancestrale
tempo
Sequenze discendenti

Tipi di sostituzioni nucleotidiche
sostituzione singola1 cambio, 1 differenza
A C
A C
A
T A
A C
A
C T
G C
A C
A
A G
C C
A C
A
A C
T T
A T
A
C T
A C
A A
A C
A
C A
sostituzione multipla2 cambi, 1 differenza
sostituzione coincidente2 cambi, 1 differenza
sostituzione parallela2 cambi, 0 differenze
sostituzione convergente3 cambi, 0 differenze
retro-sostituzione2 cambi, 0 differenze

Modelli
deterministici: basati sulla
osservazione dei caratteri
EVOLUZIONE MOLECOLARE
stocastici (probabilistici
dipendenti dal tempo):
basati sulla misura delle
diversità

Modelli Stocastici
Markov
Jukes Cantor
Kimura

Modello di Jukes e Cantor a un parametro
Il modello più semplice, proposto nel 1969: tutte le sostituzioni
nucleotidiche avvengono con la stessa probabilità.
Ad es., A potrà mutare in T, C o G con la stessa probabilità.
Il tasso di sostituzione in ognuna delle tre possibili direzioni è lo stesso (a)
e il tasso di sostituzione complessivo per ogni nucleotide sarà 3a
L’unico parametro da stimare è a, per cui modello a un parametro

Probabilità che un dato nucleotide i rimanga al tempo t:
Pii(t)= ¼ + ¾ e-4at
Probabilità che un dato nucleotide i diventi j al tempo t:
Pij(t)= ¼ - ¼ e-4at

Modello di Kimura a due parametri
E’ stato osservato che le transizioni sono più frequernti delle
transversioni. Per Kimura nel 1980 propone diverse velocità
per transizioni e transversioni, adottando due parametri (=
due diversi tassi di sostituzione: a e b)

Probabilità che un dato nucleotide non cambi nel tempo t:
X(t)= ¼ + ¾ e-4bt + ½ e-2(a+b)t
Indipendentemente da quale sia il nucleotide, poiché anche per
Kimura i 4 nucleotidi sono equivalenti.
Probabilità che un dato nucleotide cambi al tempo t:
Y(t)= ¼ + ¼ e-4bt - ½ e-2(a+b)t

Modello Kimura più realistico di Jurk e Cantor, ma presenta notevole
restrizioni:
•Non è detto che i 4 tipi di transizioni avvengano con la stessa velocità;
•I tassi di sostituzione da i verso j può non essere lo stesso che da j verso
i;
•Il processo di sostituzione possono essere influenzati dalla composizione
nucleotidica;
Modello di Tamura a 3 parametri
Modello di Felsenstein a 5 parametri

Modello reversibile (REV) o modello generale di reversibilità
(GTR) a 9 parametri
Tiene conto di:
•Composizione delle basi
•Reversibilità delle sostituzioni
•Diverse probabilità per i 6 tipi di sostituzioni nucleotidiche

Proprietà
Nessuna assunzione “a priori” sulla matrice
delle distanze ma semplice verifica della
condizione di stazionarietà
Misura accurata delle fluttuazioni statistiche
(Simulazione con il metodo del Bootstrap)
C. Saccone, C. Lanave, G. Pesole and G. Preparata
Methods in Enzymology (1990). Vol. 183, pp. 570 - 583
Modello Stazionario di Markov

Modelli di mutazione per i microsatelliti
Il numero di ripetizioni possono aumentare o diminuire con uguale
probabilità.
La probabilità di retromutazioni è molto maggiore rispetto agli SNPs,
quindi il concetto di “modello degli alleli infiniti” (ogni mutazione
produce nuovi alleli) risulta non realistico.
Stepwise Mutation Model (SMM): Le mutazioni che aumentano o
diminuiscono la lunghezza avvengono con la stessa probabilità. E’
stato adattato anche per cambiamenti multipli.
Correlazione positiva tra lunghezza dei microsatelliti e tasso di
mutazione.
Negli alleli lunghi maggiore tendenza alla delezione piuttosto che
all’inserzione.

Zuckerkandl and Pauling (1962)
IPOTESI DELL’ OROLOGIO MOLECOLARE

Orologi molecolari 1
22
L‟idea di datare gli eventi evolutivi attraverso le differenzecalibrate fra le proteine fu espressa per la prima volta nel1965 da E. Zuckerkandl e L. Pauling, che rilevarono come levelocità di evoluzione molecolare per loci con vincolifunzionali simili siano pressoché costanti su lunghi periodi ditempoDa osservazioni fatte su differenti globine, Zuckerkandl ePauling postularono che la differenza genetica tra due speciediverse, espressa dalla sequenza aminoacidica, è funzionedel tempo di divergenza dall‟antenato comuneLa verifica di tale affermazione fu ottenuta confrontando lesequenze proteiche e, quindi, i tassi di sostituzione amino-acidica, di diverse specie, con i tempi di divergenza stimatisulla base di ritrovamenti fossili

Orologi molecolari 2
23
di battiti tra due proteineomologhe appare linearmentecorrelato con la quantità ditempo trascorso dal momentoin cui la speciazione le ha fattedivergere nel loro percorsoevolutivo
Le frequenze di sostituzione in proteine omologhe erano cosìcostanti su molte decine di milioni di anni, da suggerire unparagone diretto fra l‟accumulo di cambiamenti aminoacidicied il continuo ticchettio di un orologio molecolareL‟orologio molecolare può “battere” a diverse velocità perproteine distinte, ma il numero

tempo
sostituzioni/sito
OROLOGIO MOLECOLARE

Orologi molecolari 3
25
Secondo l‟ipotesi dell‟orologio molecolare, quindi, i geni e iprodotti genici evolvono con tassi che sono approssimati-vamente costanti nel tempo e lungo le differenti lineeevolutivePerciò, se la divergenza genetica si accumula in modoregolare con il passare del tempo, allora è possibile dedurrei tempi di divergenza anche in assenza di evidenze fossiliIn pratica, una frequenza costante di variazione faciliterebbenon solo la determinazione delle relazioni filogenetiche fraspecie, ma anche dei tempi di divergenza.

Orologi molecolari 4
26
La validità dell‟ipotesi di un orologio molecolare universale èstata subito molto discussa
E. Mayr, nel 1965, affermò: “Evolution is too complex and toovariable a process, connected too many factors, for the timedependence of the evolutionary process at the molecular levelto be a simple function”……mentre gli evoluzionisti classici argomentavano che l‟anda-mento irregolare dell‟evoluzione morfologica era incompatibilecon una velocità costante di cambiamento molecolare
Inizialmente ci si riferì ad un orologio molecolare proteico,dal momento che negli anni „60, i dati sul DNA erano ancoratroppo scarsi, ed intenso fu il dibattito fino agli anni „80, cheportò fino a mettere in discussione l‟essenza stessa dell‟ideadi Zuckerkandl e Pauling, ovvero la costanza delle velocitàevolutive

Orologi molecolari 5
27
Fin dal 1971 è stato chiaro che proteine diverse evolvonocon tassi ampiamente variabiliDi conseguenza venne esclusa, da subito, la possibilità diosservare un orologio proteico universaleTest statistici condotti da Ohta e Kimura (1971), da Fitch(1976) da Gillespie e Langley (1979) hanno restituito risul-tati contrastanti, suggerendo che l‟ipotesi dell‟orologiomolecolare proteico deve essere rifiutata per la maggiorparte delle proteine sia nei confronti fra vertebrati che frainvertebrati

Orologi molecolari 6
28
La quantità di dati disponibili per testare l‟ipotesi dell‟oro-logio molecolare sta crescendo in modo esponenzialeLe frequenze di sostituzione nei ratti e nei topi sono risultatemolto similiViceversa, l‟evoluzione molecolare dell‟uomo e della scimmiaantropomorfa (es. gorilla) ha una velocità pari alla metà diquella delle scimmie del vecchio mondo (es. babbuini) dalmomento della loro speciazioneInfatti, i test di frequenza relativa eseguiti su geni omologhinei topo e nell‟uomo indicano che i roditori hanno accumu-lato un numero di sostituzioni doppio rispetto ai primati,dall‟ultimo antenato comune (speciazione dei mammiferi) da80 a 100 milioni di anni fa
Orologio molecolare non costante: l‟uso della divergenzamolecolare per datare i tempi di esistenza di due specie hasenso solo se le specie “condividono l‟orologio”

Orologi molecolari 9
29
Cause di variazione di frequenza nelle discendenzeDiversità dei tempi di generazione (durata del periodoriproduttivo)Efficienza media di riparazione, tasso metabolicoNecessità di adattamento a nuove nicchie ecologiche
Difficili da quantificare:Conosciamo le differenze attualiSappiamo che nel momento della divergenza gli organi-smi avevano attributi simili ……ma abbiamo poche informazioni sulle differenze relati-ve durante tutto il corso dell‟evoluzione

A partire dai primi anni 80, lo sviluppo delle tecniche molecolari, ha
permesso il sequenziamento di molti geni di cui erano già state
analizzate le sequenze proteiche.
L’idea dell’orologio molecolare fu quindi applicata anche al DNA.
Nel 1980 Miyata et al. hanno ottenuto un tasso di mutazione molto
simile fra i diversi gruppi di mammiferi. Mentre Bonner et al. hanno
riscontrato un tasso significativamente più basso nei primati malgasci
rispetto a tutti gli altri primati. Wu e Li hanno dimostrato differenze
significative fra l’uomo e i roditori. Tali differenze furono attribuite al
tempo di generazione, alla diversa efficienza nei meccanismi di
riparazione del DNA, al differente tasso metabolico.

Sono stati proposti diversi orologi molecolari “universali”,
come l’orologio mtDNA, secondo cui il DNA mitocondriale
animale evolve con un tasso di divergenza pari al 2% per
sequenza per milione di anni.
Nel 2002 Kumar e Subramanian hanno potuto caratterizzare
le differenze nel tasso di mutazione all’interno e fra i principali
gruppi di mammiferi.
E’ chiaro, quindi che il tasso di mutazione
in differenti gruppi di mammiferi è piuttosto
diverso

In questo scenario di continui dibattiti, fu quindi
abbandonata l’idea di un orologio universale e si cominciò
a pensare agli orologi molecolari tassonomicamente
“locali”, ovvero che possono essere trovati in particolari
geni e in taxa strettamente affini.
Gli orologi molecolari locali sono più probabili degli
universali in quanto le principali fonti di eterogeneità fra
linee sono rappresentate da differenze
• nella dimensione di popolazione
• nel tasso metabolico
• nel tempo di generazione
• nell’efficienza di riparazione del DNA
Ci si aspetta che questi parametri siano più simili fra taxa
affini che quindi potranno presentare tassi di evoluzione
molecolare piuttosto simili

Stima del MRCA (most recent common ancestry)
Se si applica il concetto di orologio molecolare alle ricostruzioni
filogenetiche, si possono datare gli eventi che sono rappresentati negli
alberi filogenetici
In altre parole è possibile ottenere una cronologia dei cambiamenti e
quindi determinare dei tempi reali in cui quei cambiamenti sono
avvenuti.

E’ necessario conoscere il ritmo a cui l’orologio, cioè
bisogna calibrare il tasso di mutazione della regione
genomica oggetto di studio.
Ci baseremo solo su marcatori uniparentali

I metodi basati su modelli popolazionistici sono complessi, perché
richiedono l’elaborazione di molte simulazioni, es. simulatori di
coalescenza.
Teoria della coalescenza: l’evoluzione degli aplotipi viene simulata
dal presente verso il passato (dagli aplotipi osservati fino ad
arrivare a quello ancestrale)

La genetica studia la trasmissione ereditaria dal passato al presente
forward

Ma quando si lavora su popolazioni si raccolgono dati sul presente e si cerca di risalire al passato
? ?
backward

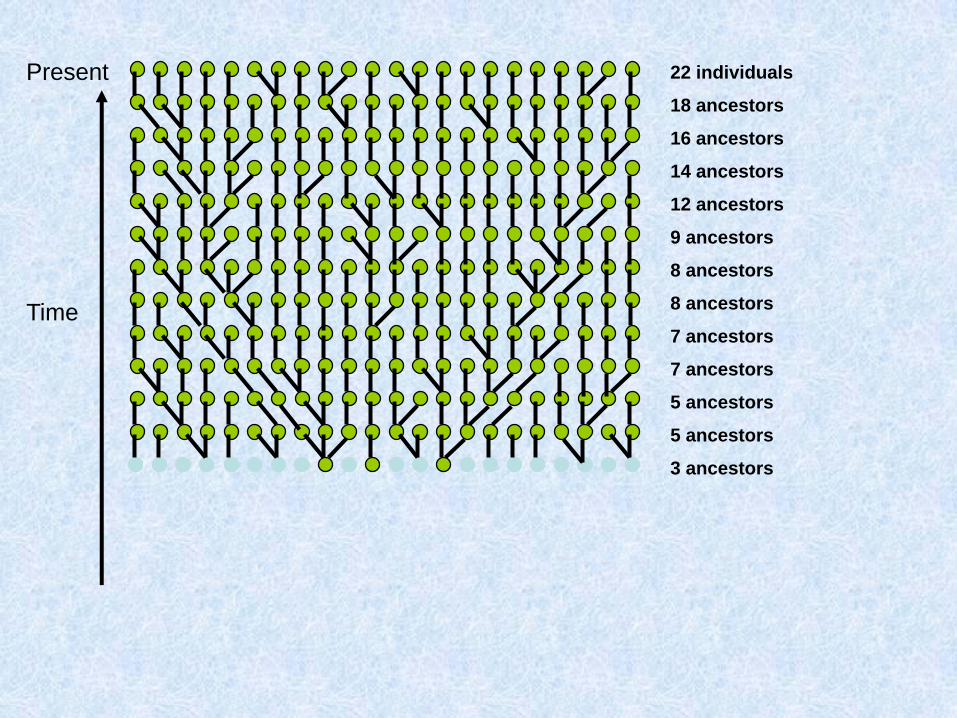
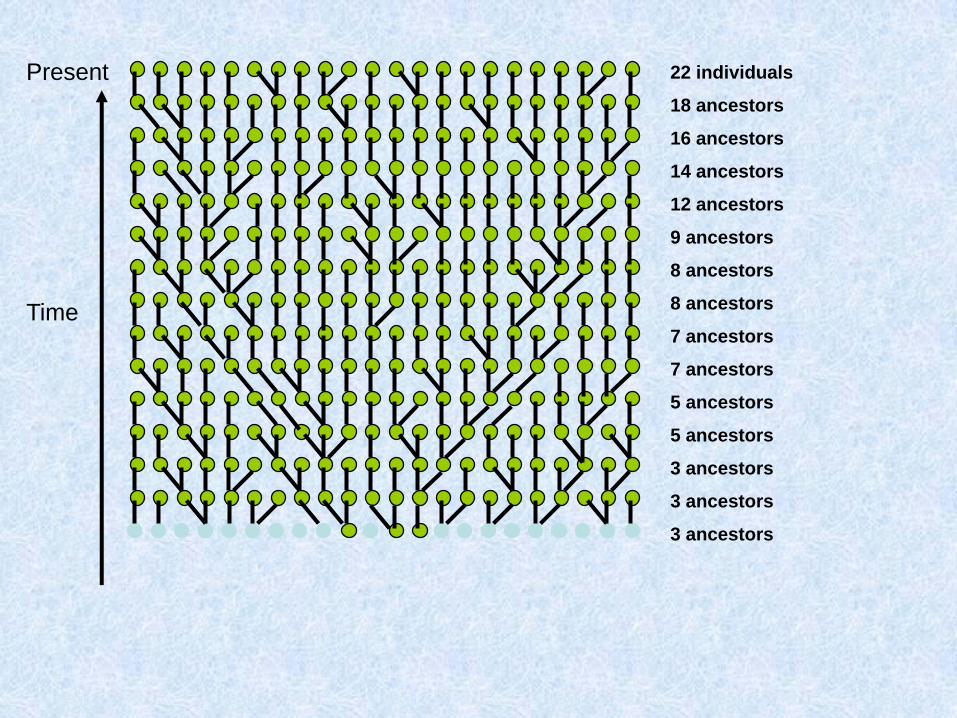
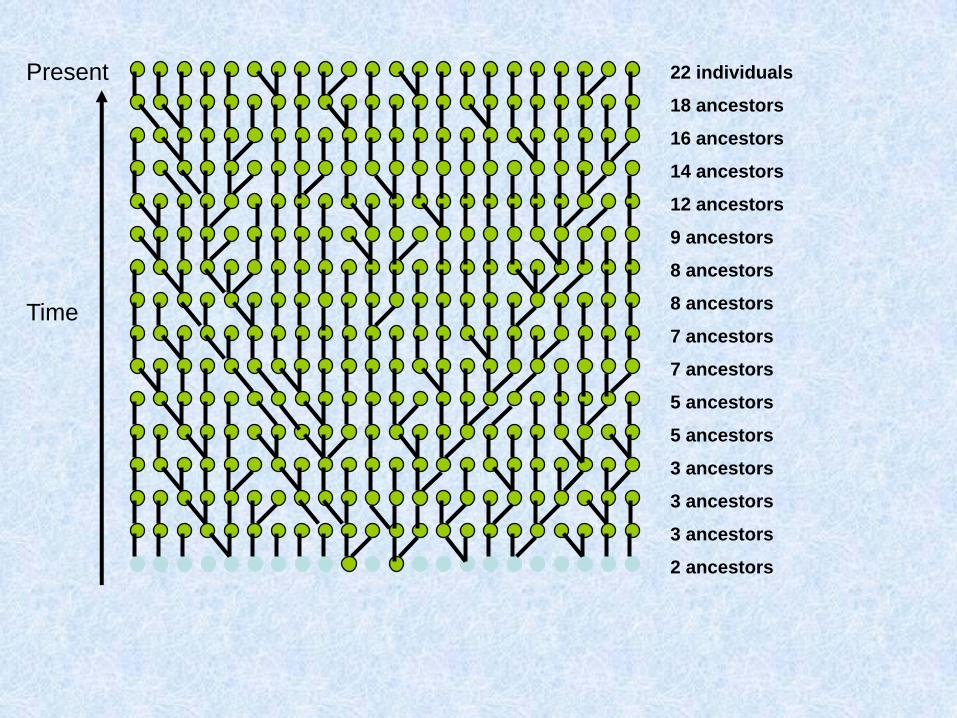
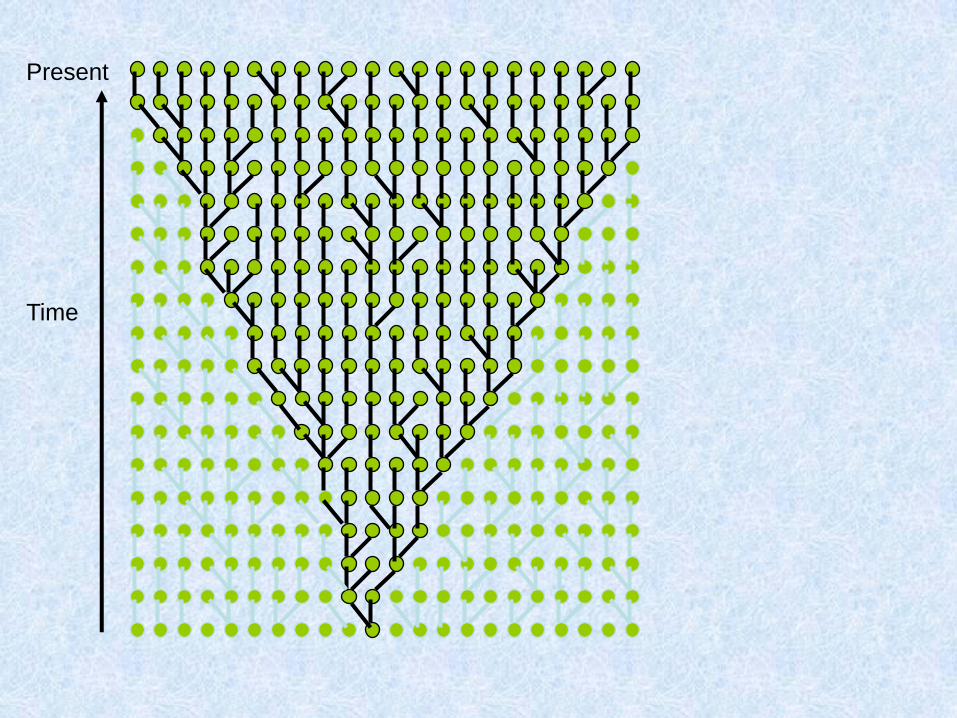
Costruiamo (procedendo verso il passato) la genealogia materna di un gruppo di individui
Due possibilità: o ogni individuo ha una madre diversa:
O due individui hanno la stessa madre:
Chiamo questo fenomeno coalescenza

Assunzioni del coalescente classico (Kingman 1982)
1. Neutralità
2. Siti infiniti
3. Se gli individui sono diploidi e le dimensioni della popolazione sono N, il modello vale per 2N copie aploidi e indipendenti
4. Unione casuale entro la popolazione
5. Dimensioni della popolazione costanti
Parliamo di caratteri a trasmissione uniparentale

Present
Time
22 individuals

Present
Time
22 individuals
18 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors
7 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors
7 ancestors
5 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors
7 ancestors
5 ancestors
5 ancestors
Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors
7 ancestors
5 ancestors
5 ancestors
3 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors
7 ancestors
5 ancestors
5 ancestors
3 ancestors
3 ancestors
Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors
7 ancestors
5 ancestors
5 ancestors
3 ancestors
3 ancestors
3 ancestors
Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors
7 ancestors
5 ancestors
5 ancestors
3 ancestors
3 ancestors
3 ancestors
2 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors
7 ancestors
5 ancestors
5 ancestors
3 ancestors
3 ancestors
3 ancestors
2 ancestors
2 ancestors

Present
Time
22 individuals
18 ancestors
16 ancestors
14 ancestors
12 ancestors
9 ancestors
8 ancestors
8 ancestors
7 ancestors
7 ancestors
5 ancestors
5 ancestors
3 ancestors
3 ancestors
3 ancestors
2 ancestors
2 ancestors
1 ancestor
Present
Time

Present
Time
Most recent common ancestor
(MRCA)

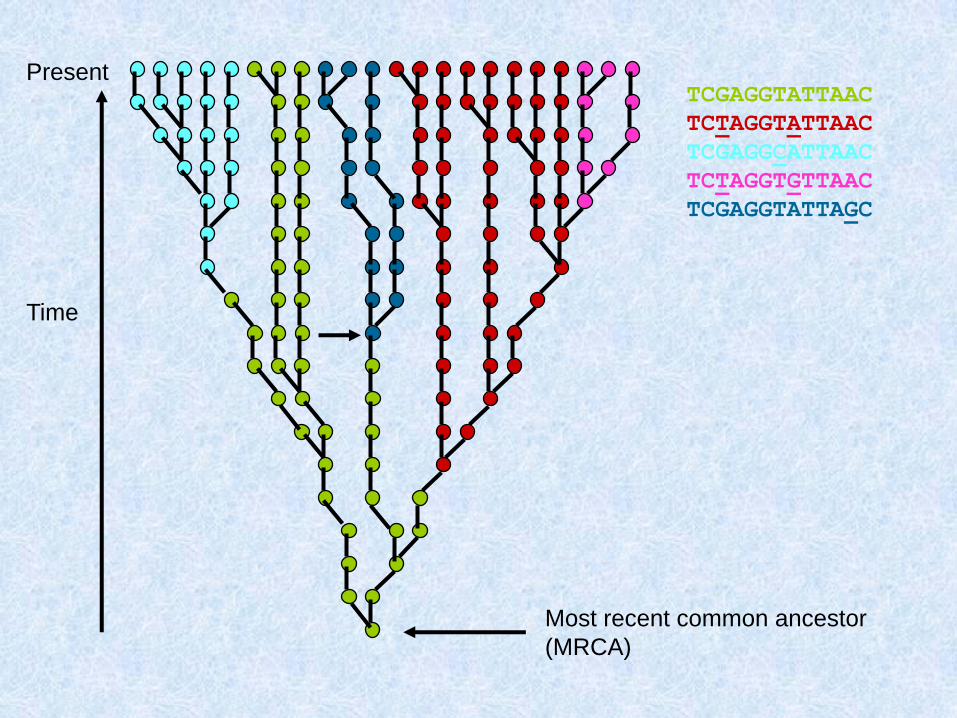
Mutational events can now be added to
the genealogical tree, resulting in
polymorphic sites. If these sites are
typed in the modern sample, they can
be used to split the sample into sub-
clades (represented by different
colours)

Present
Time
mutation
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC

Present
Time
mutation
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC

Present
Time
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC
TCGAGGCATTAAC

Present
Time
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC
TCGAGGCATTAAC

Present
Time
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC
TCGAGGCATTAAC
TCTAGGTGTTAAC

Present
Time
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC
TCGAGGCATTAAC
TCTAGGTGTTAAC

Present
Time
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC
TCGAGGCATTAAC
TCTAGGTGTTAAC
TCGAGGTATTAGC
Present
Time
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC
TCGAGGCATTAAC
TCTAGGTGTTAAC
TCGAGGTATTAGC

Present
Time
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC
TCGAGGCATTAAC
TCTAGGTGTTAAC
TCGAGGTATTAGC
TCTAGGTATCAAC

Present
Time
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC
TCGAGGCATTAAC
TCTAGGTGTTAAC
TCGAGGTATTAGC
TCTAGGTATCAAC

Present
Time
Most recent common ancestor
(MRCA)
TCGAGGTATTAAC
TCTAGGTATTAAC
TCGAGGCATTAAC
TCTAGGTGTTAAC
TCGAGGTATTAGC
TCTAGGTATCAAC
* ** * *

Per confrontare le sequenze è necessario
prima di tutto allinearle

L’evoluzione delle parole
• Tutte le parole delle lingue moderne cheindicano lo “zucchero” discendono da unaparola antenata comune
• Tutte dalla stessa (“sukkar” - parola usatadagli arabi), alcune da un’antenata piùvicina nel tempo (“zuckre” in Francia)

Sukkar
Açucar Azucar
Zuckre
Sucre
Zucker
Zucchero
Sakari
SuikerSugar
Sokker
Europa, circa 700 dC

L’evoluzione delle parole
• Immaginiamo di non conoscere le parole “antenate” dello
zucchero, e di doverci chiedere se due parole moderne in
due lingue differenti sono “simili” tra loro
SUGAR
SUCRE
SUGR
SUCR

Allineamento
• L’allineamento” è un modo di rappresentare
schematicamente i legami evolutivi tra due
o più parole (o sequenze), indicando
sostituzioni, inserzioni e delezioni
S U G A R -
S U C - R E
Sostituzione(mutazione)
Inserzioni(delezioni)

Allineamento (multiplo)
S U G - A R -S U C – - R E
Z U C K E R -
S O K K E R -
A Z U C - A R -
S A K - A R I
A ç U C - A R -
--------------------- S U C(K)A R -
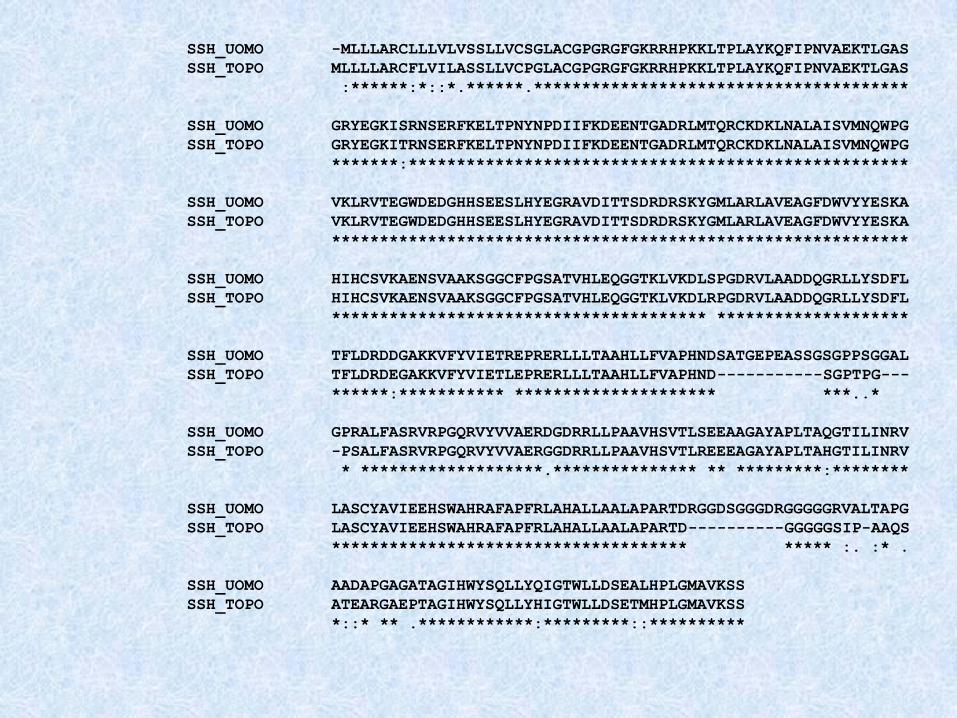
SSH_UOMO -MLLLARCLLLVLVSSLLVCSGLACGPGRGFGKRRHPKKLTPLAYKQFIPNVAEKTLGAS
SSH_TOPO MLLLLARCFLVILASSLLVCPGLACGPGRGFGKRRHPKKLTPLAYKQFIPNVAEKTLGAS
:******:*::*.******.***************************************
SSH_UOMO GRYEGKISRNSERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPG
SSH_TOPO GRYEGKITRNSERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPG
*******:****************************************************
SSH_UOMO VKLRVTEGWDEDGHHSEESLHYEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKA
SSH_TOPO VKLRVTEGWDEDGHHSEESLHYEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKA
************************************************************
SSH_UOMO HIHCSVKAENSVAAKSGGCFPGSATVHLEQGGTKLVKDLSPGDRVLAADDQGRLLYSDFL
SSH_TOPO HIHCSVKAENSVAAKSGGCFPGSATVHLEQGGTKLVKDLRPGDRVLAADDQGRLLYSDFL
*************************************** ********************
SSH_UOMO TFLDRDDGAKKVFYVIETREPRERLLLTAAHLLFVAPHNDSATGEPEASSGSGPPSGGAL
SSH_TOPO TFLDRDEGAKKVFYVIETLEPRERLLLTAAHLLFVAPHND-----------SGPTPG---
******:*********** ********************* ***..*
SSH_UOMO GPRALFASRVRPGQRVYVVAERDGDRRLLPAAVHSVTLSEEAAGAYAPLTAQGTILINRV
SSH_TOPO -PSALFASRVRPGQRVYVVAERGGDRRLLPAAVHSVTLREEEAGAYAPLTAHGTILINRV
* *******************.*************** ** *********:********
SSH_UOMO LASCYAVIEEHSWAHRAFAPFRLAHALLAALAPARTDRGGDSGGGDRGGGGGRVALTAPG
SSH_TOPO LASCYAVIEEHSWAHRAFAPFRLAHALLAALAPARTD----------GGGGGSIP-AAQS
************************************* ***** :. :* .
SSH_UOMO AADAPGAGATAGIHWYSQLLYQIGTWLLDSEALHPLGMAVKSS
SSH_TOPO ATEARGAEPTAGIHWYSQLLYHIGTWLLDSETMHPLGMAVKSS
*::* ** .************:*********::**********

ALLINEAMENTO DI SEQUENZE
A COPPIE
AGTTTGAATGTTTTGTGTGAAAGGAGTATACCATGAGATGAGATGACCACCAATCATTTC
||||||||||||||||||| |||||||| ||| | |||||| |||||||||||||||||
AGTTTGAATGTTTTGTGTGTGAGGAGTATTCCAAGGGATGAGTTGACCACCAATCATTTC
MULTIPLO
KFKHHLKEHLRIHSGEKPFECPNCKKRFSHSGSYSSHMSSKKCISLILVNGRNRALLKTl
KYKHHLKEHLRIHSGEKPYECPNCKKRFSHSGSYSSHISSKKCIGLISVNGRMRNNIKT-
KFKHHLKEHVRIHSGEKPFGCDNCGKRFSHSGSFSSHMTSKKCISMGLKLNNNRALLKRl
KFKHHLKEHIRIHSGEKPFECQQCHKRFSHSGSYSSHMSSKKCV----------------
KYKHHLKEHLRIHSGEKPYECPNCKKRFSHSGSYSSHISSKKCISLIPVNGRPRTGLKTs

Allineamento GLOBALE o LOCALE
GLOBALE considera la similarita’ tra due sequenze in tutta
la loro lunghezza (da N- a C-terminale)
LOCALE considera solo specifiche REGIONI simili tra alcune parti delle sequenze in analisi (solo regioni a ↑ densità di
similarità generando più sub-allineamenti)
Global alignment
LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK
||. | | | .| .| || || | ||
TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHKAG
Local alignment
LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK
||||||||.||||
TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHK

ALLINEAMENTI DI SEQUENZA
• Per confrontare delle sequenze queste devono essere allineate
• ALLINEAMENTO: procedura per confrontare 2 o più sequenze residuo per
residuo in modo da massimizzare la similarità tra esse e ridurre il numero
di operazioni da effettuare per convertirle l’una nell’altra. E’ volto a stabilire
una relazione biunivoca tra le coppie di residui delle sequenze considerate.
• L’allineamento di sequenze è strumento indispensabile per:
- CONFRONTO tra due sequenze;
- RICERCA DI SEQ SIMILI a una in esame NELLE BANCHE DATI;
- Determinazione di PATTERN e DOMINI CONSERVATI;
- PREDIZIONE DI STRUTTURA 3D;
- Stimare L’APPARTENENZA a UN CERTO FOLD;
- COSTRUIRE UN ALBERO FILOGENETICO:
- PREDIZIONE DI STRUTTURA SECONDARIA

ALLINEAMENTO DI SEQUENZA
• PER ESEGUIRE UN ALLINEAMENTO DI SEQUENZA
SONO NECESSARI ESSENZIALMENTE 3 STRUMENTI:
- Avere a disposizione una MATRICE DI SOSTITUZIONE. La matrice
definisce la il GRADO di SIMILARITA’ tra amminoacidi;
- Avere a disposizione un ALGORITMO DI ALLINEAMENTO cercando
di massimizzare il punteggio dato dalla matrice e valutando quanti gap
(interruzioni) inserire;
- Avere a disposizione per evitare allineamenti senza senso una
PENALITA’ per l’introduzione dei GAP.
LLTTVRNN LLTTVRNN
LLVRNN LL--VRNN
I GAP riflettono
inserzioni/delezioni avvenute
durante l’evoluzione

Similarità e distanza
Esistono due modi per misurare il grado di omologia tra
due sequenze:
1. Calcolare la similarità contando i match
2. Calcolare la distanza contando mismatch e indels
Similarità elevata ↔ bassa distanza
Due sequenze identiche hanno una distanza pari a zero

Matrici di sostituzione
• Le matrici di sostituzione tengono conto dei criteri di similarità tra aminoacidi
• Comprendono 210 valori: • 20 (sulla diagonale) relativi al punteggio dell’appaiamento di
ciascun aminoacido con se stesso• 190 relativi a tutte le possibili sostituzioni aminoacidiche
• I 190 valori sono riportati anche nella loro parte speculare in modo che queste matrici hanno un formato 20 x 20 valori
• Le matrici di sostituzione più semplici considerano solo il criterio di identità e sono costituite da valori 0 o 1
• Altre matrici considerano la similarità chimica tra gli aminoacidi o il numero minimo di mutazioni per passare da un codon all’altro e attribuiscono un punteggio alle diverse sostituzioni

Similarità chimico-fisicaGli aminoacidi possono essere raggruppati in base alle caratteristiche fisico-chimiche delle loro
catene laterali. Su questa base un aminoacido può essere definito simile ad un altro
R K
basici
R K H D E
carichi
I V F L
idrofobici
R K N Q
polari
G A
poco ingombro
sterico

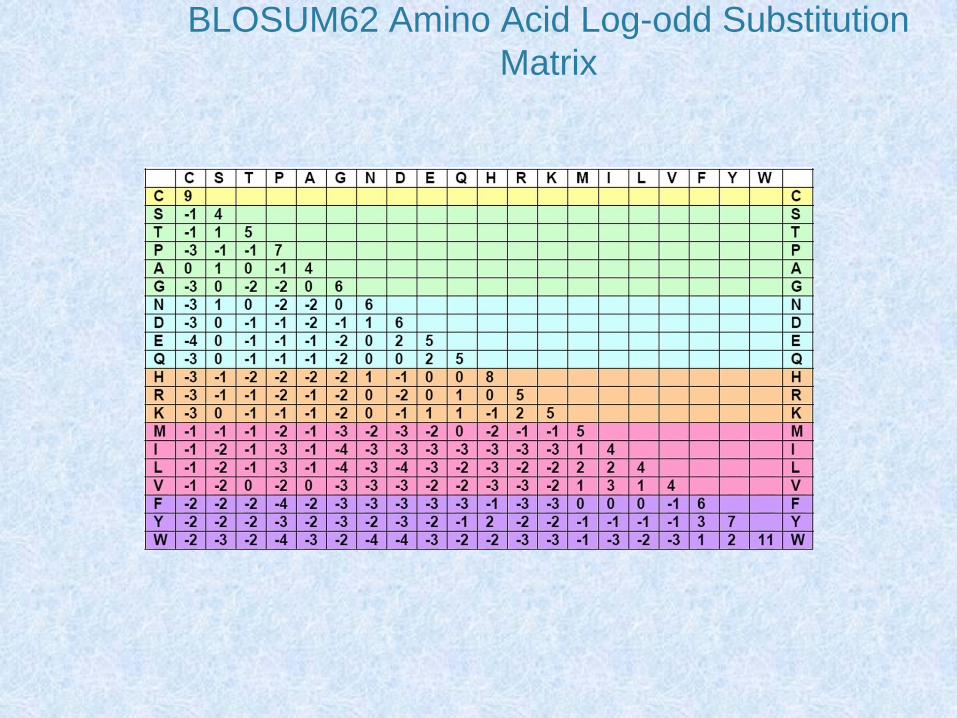
BLOSUM62 Amino Acid Log-odd Substitution
Matrix

Possiamo Valutare un
Allineamento
• Match = +2
• Mismatch = -1
• Gap = -2
G A T T C C G T
| | | | |
G A A T - C C T
+2 +2 -1 +2 -2 +2 -1 +2
=6 punti

- metodi di confronto per distanza.
-metodi filogenetici
L’albero filogenetico descrive la variabilità
riscontrata tra le popolazioni e le relazioni evolutive
tra di esse.

Terminologia
• Node/nodo: un punto di
ramificazione su un
albero filogenetico
Alberi filogenetici
E. coli
Arabidopsis
Riso
Danio
Uomo
Topo
Ratto
Ramo

• Taxon: Un livello di classificazione, una
specie, un genere, una famiglia. Usato
nella filogenesi molecolare anche per
descrivere un OTU.
• OTU (Operational Taxonomic Unit), una
“foglia” di un albero filogenetico, può
essere una specie oppure una
sequenzaE. coli
Arabidopsis
Riso
Danio
Uomo
Topo
Ratto
Ramo
Nodo
Taxon/OTU
Taxon

• Clade/Gruppo : un gruppo che contiene tutti gli OTU che sono discesi da un nodo.
E. coli
Arabidopsis
Riso
Danio
Uomo
Topo
Ratto
Nodo Ancestrale
Clade/Gruppo
monofiletico

Cladogrammi Cladogrammimostrano l‟ordine delle ramificazioni, lunghezze dei rami non significano
niente
E. coli
Arabidopsis
Riso
Danio
Uomo
Topo
Ratto
E. coli
Arabidopsis
Riso
Danio
Uomo
Topo Ratto

Filogrammile lunghezze dei rami indicano il
grado di divergenzaE. coli
Arabidopsis
Riso
Danio
Uomo
Topo Ratto
Filogrammi

Alberi senza radice o unrooted:
Alberi qualitativi
Descrivono semplicemente le
relazioni evolutive fra le OTU
Alberi filogenetici

Alberi basati sullo stato dei caratteri
- Massima Parsimonia
Alberi basati sulla misura delle distanze
- UPGMA
- Massima Verosimiglianza
- Neighbor - Joining
Alberi filogenetici

Metodi basati sulla Misura delle distanze
-UPGMA (albero con radice applicabile solo se rispettato
l’orologio molecolare)
UPGMA è un sistema di clustering basato su “Unweighted Pair Group Method
using aritmetic Average”. Raggruppa successivamente le sequenze o gli aplotipi a
partire dai più simili ed aggiungendo via via un nodo all’albero. Le distanze tra
due taxa, tra un nodo e un taxon, o tra due nodi (ovvero le lunghezze dei bracci)
sono dati dalla media aritmetica delle distanze.
- » Lunghezza dei rami proporzionale ai tempi di divergenza
- la velocità dell’evoluzione lungo tutti i rami è la stessa
Alberi filogenetici

UPGMA
A B C D E F G
A -
B 63 -
C 94 79 -
D 111 96 47 -
E 67 16 83 100 -
F 23 58 89 106 62 -
G 107 92 43 20 96 102 -

UPGMA
A B C D E F G
A -
B 63 -
C 94 79 -
D 111 96 47 -
E 67 16 83 100 -
F 23 58 89 106 62 -
G 107 92 43 20 96 102 -
GD

A B C E F DG
A -
B 63 -
C 94 79 -
E 67 16 83 -
F 23 58 89 62 -
DG 94 84 35 88 94 -
UPGMA
GD C

A B E F CDG
A -
B 63 -
E 67 16 -
F 23 58 62 -
CDG 61 64 61 74 -
UPGMA
GD C A F

UPGMA
AF B E CDG
AF -
B 98 -
E 106 16 -
CDG 112 64 61 -
B EGD C A F

UPGMA
AF BE CDG
AF -
BE 188 -
CDG 112 108 -
B E GD C A F
Root
B EGD C A F
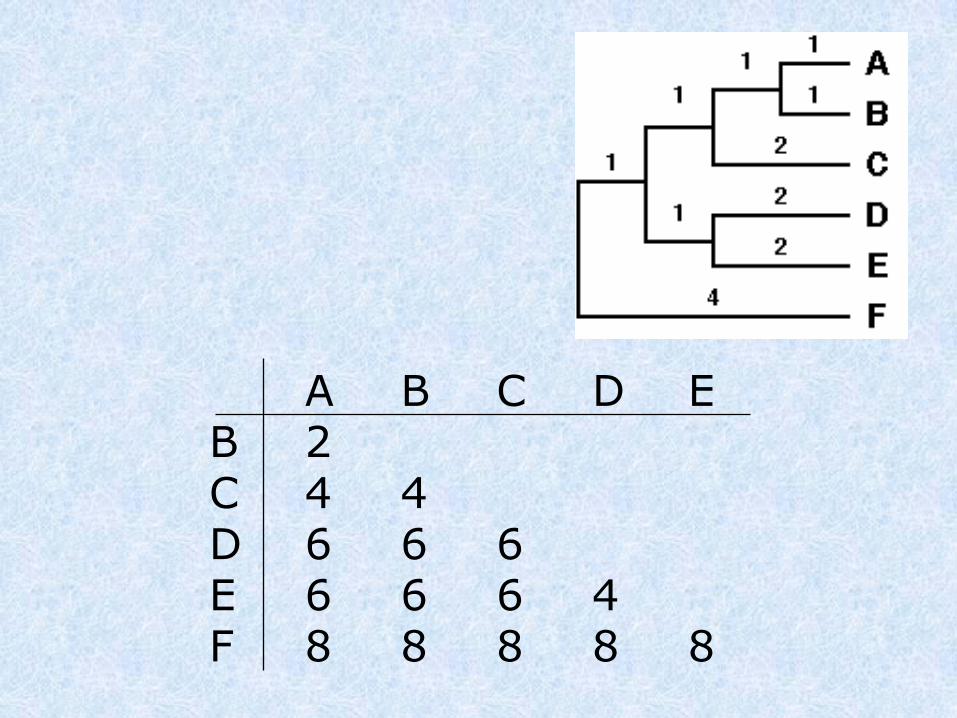
A B C D E B 2C 4 4D 6 6 6E 6 6 6 4F 8 8 8 8 8

A B C D E B 2C 4 4D 6 6 6E 6 6 6 4F 8 8 8 8 8
dist(A,B),C = (distAC + distBC) / 2 = 4
dist(A,B),D = (distAD + distBD) / 2 = 6
dist(A,B),E = (distAE + distBE) / 2 = 6
dist(A,B),F = (distAF + distBF) / 2 = 8
Clusteriziamo le 2 seq più vicine, generiamo una nuova matrice dove queste seq. vengono considerate come un cluster unico.

A,B C D EC 4D 6 6E 6 6 4F 8 8 8 8
dist(D,E),C = (distDC + distEC) / 2 = 6
dist(D,E),F = (distDF + distEF) / 2 = 8
Dist(D,E)(A,B)= (distD(AB) + distE(AB)) / 2 = 6

AB C DEC 4DE 6 6F 8 8 8
dist(ABC),F = (dist(AB)F + distCF) / 2 = 8
dist(ABC),(DE) = (dist(AB)(DE) + distC(DE)) / 2 = 6

AB,C DEDE 6F 8 8
dist(ABC,DE)F = (dist(ABC)(F) + dist(DE)(F)) / 2 = 8

ABC,DEF 8

A B C D E B 2C 4 4D 6 6 6E 6 6 6 4F 8 8 8 8 8

-Neighbor Joining (albero senza radice quando
non è rispettato l’orologio molecolare)
Il sistema usato da neighbour-joining per trovare i
neighbour si basa sulla valutazione della distanza
tra due foglie sottraendo la distanza media di
ciascuna di queste rispetto a tutte le altre foglie. In
altre parole, neighbor-joining non considera
semplicemente la distanza tra le coppie per
costruire l’albero ma valuta la distanza rispetto a
tutti gli altri punti
- » Differente velocità nei diversi rami
si basa sul principio della minima evoluzione

Maximum Parsimony
Identifica l‟albero che richiede il minimo numero dicambiamenti evolutivi per spiegare le differenzeosservate tra le sequenze
Spesso non si può identificare un unico albero
per grandi set di dati una ricerca esaustiva non èpossibile
Metodi Deterministici

Massima Parsimonia
Sito
Sequenza1234
Sito 2
Sito 3
Sito 5
Sito7
Sito 9
Non utilizzano le distanze
Vengono costruiti più alberi, si
sceglie quello che richiede il mino
numero di sostituzioni

Maximum Parsimony (MP)
I
II
III
b
A
cA
dT
outgroupT
b
A
cA
dT
outgroupT
b
AcA
dT
outgroupT
T A/
TA/
T A/
TA/
T A/
TA/
outgroup
a
b
c
A
A
A A AA
A A
C
C
C
C C
G G G
G G G
G G G
G G G
T TT
T TT
T
outgroup
a
b
c
A AA
A
ACG G G
ACG G G
ACG G G
ACG G G
T TT
T TT
TC
- Valuta tutti I possibili alberi per ogni colonna
verticale dei caratteri delle sequenze (nucleotide
position)
- Vengono considerati solo i siti informativi
- ad ogni albero è assegnato un punteggio basato
sul numero dei cambiamenti evolutivi
- gli alberi che producono il minor numero di
cambiamenti (shortest trees) per tutte le posizioni
sono presi in considerazione

Maximum Likelihood (ML)
I
II
III
b
A
cA
dT
outgroupT
b
A
cA
dT
outgroupT
b
AcA
dT
outgroupT
T A/
TA/
T A/
TA/
T A/
TA/
outgroup
a
b
c
A
A
A A AA
A A
C
C
C
C C
G G G
G G G
G G G
G G G
T TT
T TT
T
outgroup
a
b
c
A AA
A
ACG G G
ACG G G
ACG G G
ACG G G
T TT
T TT
TC
- utilizza un calcolo delle probabilità basato su un
modello specifico di evoluzione delle sequenze
per trovare il miglior albero (minor numero di
mutazioni)
- sono considerati tutti gli alberi per ogni
nucleotide
- simile al MP, ma utilizza un modello evolutivo
- il metodo è adatto ad analizzare le relazioni tra
diversi taxa

Perche’ ?
– Per ricostruire la storia evolutiva delle
specie o delle popolazioni in esame
– Tempo di divergenza fra 2 specie = tempo
impiegato nel processo di speciazione ovvero
dell’isolamento riproduttivo
Alberi filogenetici

I dati per la costruzione degli alberi
filogenetici: un solo gene puo essere sufficiente?
Quasi mai
Soluzione:
– Analizzare piu’ di un gene
– Effettuare una stima della significatività
statistica degli alberi ottenuti
Alberi filogenetici

Alberi ottimali multipli• Parsimonia può generare piu di un
albero più parsimonioso
• Possiamo poi selezionare il
“migliore” con criteri addizionali
• Tipicamente relazioni comuni fra
tutti gli alberi ottimali vengono
riassunte in un albero consensus

Assegnare la significatività di un albero filogenetico
(robustezza dell’albero)
Si utilizza il metodo Bootstrap.
attraverso sottocampionamenti casuali dei dati
I valori in corrispondenza dei nodi rappresentano il
numero di volte che un dato raggruppamento è stato
ottenuto nei set campionati a partire dai dati originari.
Si usano di solito campionature da 100 a 1000 set.

bootstrap
+---------HUMAN
+100.0
! ! +----RHESUS
! +100.0
+-91.0 +----G MONKEY
! !
! ! +----COW
! ! +100.0
! +-40.0 +----SHEEP
+100.0 !
! ! +---------CAT
! !
! ! +----MOUSE
+-43.0 ! +100.0
! ! +------69.0 +----RAT
! ! !
+-66.0 ! +---------HAMSTER
! ! !
! ! +------------------------CHICKEN
! !
! +-----------------------------XENOPUS
!
+----------------------------------TROUT


Networks :
representation of character-state evolution amongst ancestors and descendants that have complex relationships due to non-dichotomous historical events

Network
Metodi per l’analisi di dati molecolari intraspecifici, che nonassumono uno schema evolutivo gerarchico strettamente dicotomico.
Permettono l’inclusione nell’albero e la rappresentazione grafica di processi quali la ricombinazione, il flusso
genico, le mutazioni ricorrenti
Formazione di reticolazioni
Maggior quantità di informazione

ALBERO FILOGENETICO NETWORK

ESEMPIO L1c RETICOLAZIONE
MEDIAN VECTORS
Recommended

















