Embed Size (px)
DESCRIPTION
第四章 中药制剂的含量测定. 【 目的要求 】 掌握中药制剂含量测定的样品前处理方法及测定方法的效能指标。 掌握可见 - 紫外分光光度法、 TLCS 、 HPLC 、 GC 在中药制剂分析中的应用。 熟悉荧光分光光度法及原子吸收光度法在中药制剂分析中的应用。 了解双波长、三波长、差示、导数光谱法在中药制剂分析中的应用。. 药物分析学科. §1 样品的处理. 样品处理的主要作用 释放被测组分,制成稳定试样 除去杂质,纯化 浓缩 试样形式符合测定的要求 一、样品的粉碎 目的 —— 取样均匀,提取完全 粉碎设备 —— 粉碎机、研钵、匀浆机 - PowerPoint PPT Presentation
Citation preview

第四章
中药制剂的含量测定

【目的要求】 掌握中药制剂含量测定的样品前处理方法及测定方法
的效能指标。
掌握可见 -紫外分光光度法、 TLCS、HPLC、 GC
在中药制剂分析中的应用。
熟悉荧光分光光度法及原子吸收光度法在中药制剂分
析中的应用。
了解双波长、三波长、差示、导数光谱法在中药制剂
分析中的应用。
药物分析学科

§1 样品的处理 样品处理的主要作用 释放被测组分,制成稳定试样 除去杂质,纯化 浓缩 试样形式符合测定的要求
一、样品的粉碎目的——取样均匀,提取完全粉碎设备——粉碎机、研钵、匀浆机※ 粒度大小合适,全部过筛※ 防止污染或损失
药物分析学科

§1 样品的处理 样品处理的主要作用 释放被测组分,制成稳定试样 除去杂质,纯化 浓缩 试样形式符合测定的要求
一、样品的粉碎目的——取样均匀,提取完全粉碎设备——粉碎机、研钵、匀浆机※ 粒度大小合适,全部过筛※ 防止污染或损失
药物分析学科

§1 样品的处理二、样品的提取(一)冷浸法
操作:样品→精密称定→置容器内→精密加入溶剂→称定重量
→ 浸泡( 12~24h )→称重→补足重量→测定
测定方法:
全部测定法:过滤→滤液→蒸干→定容→测定(低浓)
部分测定法:过滤→滤液→取若干体积进行测定(高浓)
优缺点及适用范围:
操作简便,适用于热不稳定的样品,提取杂质较少,但是费
时,费溶剂药物分析学
科
掌握每种提取方法的操作、优缺点及应用范围

§1 样品的处理二、样品的提取(二)回流提取法
采用回流装置,用单一溶剂或混合溶剂于水浴上加热提取
优缺点及适用范围:
提取效率高,提取时间短,为 0.5 ~ 2 小时;提取杂质较多,
不适用于热不稳定或具有挥发性的成分
(三)连续回流提取
用索氏提取装置,少用混合溶剂,其余操作同回流提取法。
优缺点及适用范围:提取效率高,所需溶剂少,提取杂质较少
操作简便;不适用于受热易分解的成分。 药物分析学科

§1 样品的处理二、样品的提取(四)超声提取法
样品置容器中,加入提取溶剂,放入超声振荡器中进行提取
优缺点及适用范围:
提取效率高,速度快 30min ;超声波可引起化学成分的改变
(五)超临界流体提取法( SFE )
优点:速度快、萃取效率高、方法准确度高、选择性高、节省
溶剂、易于自动化,而且可避免使用易燃,有毒的有机溶剂,
能与色谱和光谱等分析仪器直接联用
药物分析学科

§1 样品的处理三、样品的分离纯化( 1 )沉淀法
样品 + 沉淀剂 →沉淀→过滤→滤液→测定
→ 沉淀→重量法或溶解后测定
药物分析学科
处理样品的核心
复方益母草口服液中水苏碱的含量测定利用雷氏盐(硫氰酸铬铵)作沉淀剂,在酸性介质中可与大部分有机碱生成难溶于水的配合物与其它杂质分离。

§1 样品的处理三、样品的分离纯化( 2)蒸馏法
适用于具挥发性的待测组分,如挥发油、小分子的生物碱、
丹皮酚等
最常用的水蒸气蒸馏法:
共水蒸馏法
通水蒸气蒸馏法
水上蒸馏法
利用盐析作用,提高蒸馏效果药物分析学
科

§1 样品的处理三、样品的分离纯化( 3)液 - 液萃取法
直接萃取法
萃取剂的选择:亲脂性有机溶剂,如苯、氯仿或乙醚;
弱亲脂性的溶剂,如乙酸乙酯、正丁醇等
多次萃取( 3-4 次)
调整水相酸度,提取有机酸、有机碱
利用盐析作用,提高提取率
药物分析学科

§1 样品的处理三、样品的分离纯化( 3)液 - 液萃取法
离子对萃取法
BH++ In- → BH+·In- (水相)→ BH+·In- (有机相)
适合于高度电离的有机酸、碱化合物的萃
水相酸度的控制
离子对试剂的选择
有机溶剂的选择
药物分析学科

§1 样品的处理三、样品的分离纯化( 4)色谱法(层析法) 分离原理:吸附色谱、分配色谱、离子交换色谱、凝胶色谱 操作方式:柱色谱、薄层色谱、纸色谱 装柱方法:湿法装柱、干法装柱 操作程序:①柱的活化;②上样;③清洗;④洗脱。 微柱色谱(固相萃取) 柱填料:硅胶、氧化铝、氧化镁、活性炭、聚酰胺、大孔硅
藻土、离子交换树脂、键合相硅胶C8、 C18等 药物分析学
科

§1 样品的处理三、样品的分离纯化( 4)色谱法(层析法)
硅胶:酸性吸附剂,适用于中性或酸性成分的层析,强烈保留碱性化合物
洗脱顺序:极性小→大
氧化铝:弱碱性,分离一些碱性中草药成分,不宜用于醛、酮、酸、内
酯等类型的化合物分离
中性氧化铝、酸性氧化铝:适用于中性、酸性成分的分离
键合相硅胶( C18或 ODS):分开脂溶性和水溶性成分
洗脱顺序:极性大→小
药物分析学科

§1 样品的处理三、样品的分离纯化
( 4)色谱法(层析法)
大孔树脂:分为极性(丙烯酰胺聚合物 )和非极性(苯乙烯和二乙烯苯
的共聚物)型
非极性型( XAD-2等):分开脂溶性和水溶性成分
※ 使用前需要用有机溶剂除去杂质,有时还需用酸、碱清洗
聚酰胺:与溶质形成氢键而产生吸附作用,常用于含酚、酸、醌类药物样
品液的净化分离
硅藻土、纤维素:以水基质液作为固定相,与水不混溶的有机溶剂为流动
相的分配色谱 药物分析学科

§1 样品的处理三、样品的分离纯化
( 4)色谱法(层析法)
离子交换树脂:用于除去样品中的离子及萃取可离解化合物 ( 弱酸、
弱碱药物 )
( 5)固相微萃取( SPME)
集萃取、浓缩、进样于一
体的样品前处理新方法
药物分析学科

§1 样品的处理三、样品的分离纯化( 6)消化法 -- 测定中药制剂中的无机元素
目的:破坏有机物,使无机物游离 湿法消化:
硝酸 -高氯酸法:适用于血,尿、组织等生物样品和含动植物药制
剂的破坏,无机金属离子均为高价态。
※ 对含氮杂环类有机物破坏不够完全。
硝酸 -硫酸法:适用于大多数有机物的破坏。
※ 与硫酸形成不溶性硫酸盐的金属离子的测定,不宜采有此法。
硫酸 -硫酸盐法:常用于含砷或锑的有机样品的破坏,破坏后得到
三价砷或锑。 药物分析学科

§1 样品的处理三、样品的分离纯化( 6)消化法 -- 测定中药制剂中的无机元素
目的:破坏有机物,使无机物游离 干法消化:
供试品 灰分
※ 控制温度在 420℃以下
※不适用于含易挥发性金属(如汞、砷等,)有机样品的破坏。
※如果灰化不完全,可以加入硝酸 - 水( 1 : 3 )或稀盐酸 - 水( 1 :
3 )
水浴上蒸干后,继续小火灰化药物分析学
科
灼烧灰化
(+NaCO3/MgO)

§2 含量测定方法的验证需验证的分析项目
1. 鉴别试验;2. 杂质定量或限度检查;3. 原料或制剂中有效成分含量测定;4. 制剂中其它成分(降解产物、防腐剂等)的测定;5. 溶出度、释放度等功能检查中的溶出量等的测试方
法。
药物分析学科

§2 含量测定方法的验证一、准确度
是指以测得结果与真实值之间的接近程度。由于真实值是未知
的,故常用回收率试验来表示。
二、精密度
是指一种均匀样品经多次取样重复分析后,各次结果之间的符
合程度。常用标准偏差或相对标准偏差表示。重复性、中间精
密度及重现性
药物分析学科

§2 含量测定方法的验证1. 重复性 在相同条件下,由同一个分析人员测定所得结果的精密度;在
规定的范围内,至少用 9次测定结果评价,如制备三个不同浓度样品各测
三次;或把被测物浓度当作 100% ,至少测 6次进行评价。
2. 中间精密度 同一实验室,不同时间由不同分析人员用不同设备所得结
果的精密度。考察随机变动因素的影响。变动因素为不同日期、不同分
析人员、不同设备。
3. 重现性 不同实验室,不同分析人员测定结果的精密度。当分析方法将
被法定标准采用时,应进行重现性试验。如建立药典分析方法时通过协
同检验得出重现性结果,协同检验的过程、重现性结果均应记载在起草
说明中。药物分析学
科

§2 含量测定方法的验证三、专属性
是指在样品中存在干扰成分的情况下,分析方法能够准确,专一
地测定分析物的能力。
四、检测限
检测限是指在规定的实验条件下,分析方法能够检测(但不需要
定量)样品中分析物的最低浓度。
1.非仪器分析目视法
用已知浓度的被测物,试验出能被可靠地检测出的最低浓度或量。
2. 信噪比法药物分析学
科

§2 含量测定方法的验证五、定量限
是指在规定的实验条件下和具有可接受的精密度和准确度前提
下,分析方法能够测定样品中分析物的最低浓度。
六、线性
七、范围
八.耐用性
是指测定条件有小的变动时,测定结果不受影响的程度为常规检
验提供依据。衡量方法不受参数微小变差影响的能力。
药物分析学科

§2 含量测定方法的验证 分析方法效能指标的要求
1 、用于鉴别试验:只对专属性、耐用性二项指标有所要求,其余
均无须要求
2 、杂质测定或制剂中降解物测定:当用于定量除检测限不必要求
外,其余七项指标均应有所要求;用于限度检查只对专属性、检
测限、和耐用性有要求。
3 、用于原料药中主成分或制剂中有效成分含量测定及溶出度测
定,除了检测限和定量限外,其余均要求。
药物分析学科

§3 常用定量分析方法一、化学分析法
适用于含量较高的成分及无机成分的测定
(一)重量分析
挥发法:如水分测定
萃取法:如冰片散中冰片的含量测定
沉淀法:如苦参片中苦参总碱的含量测定
药物分析学科

§3 常用定量分析方法一、化学分析法
(二)滴定分析
酸碱滴定法:测定生物碱、有机酸、内酯类
沉淀滴定法
银量法:生物碱的氢卤酸盐及有机卤化物
四苯硼钠法: + 生物碱→白色沉淀
配位滴定法( EDTA 法和硫氰酸铵法):测定鞣质、生物碱及
Ca2+ 、 Mg2+ 、 Fe3+ 、 Hg2+ 等矿物类制剂的含量
氧化还原滴定法:酚类、糖类及矿物药中 Fe 、 As 等 药物分析学科

§3 常用定量分析方法二、可见 - 紫外分光光度法
(一)单波长分光光度法
1 、吸收系数法( CHP最为常用)例题左金丸的含测
A=ECL (百分吸收系数,单位 g/100ml )
2 、对照品比较法
对照品溶液浓度应与供试品浓度接近( 100±10%)
3 、标准曲线法
药物分析学科

§3 常用定量分析方法二、可见 - 紫外分光光度法
(二)计算分光光度法
1 、双波长分光光度法法
系数倍率法
等吸收点法
2 、三波长分光光度法
3 、导数光谱法
讨论:计算分光光度法在中药及其制剂中的应用前景
药物分析学科

§3 常用定量分析方法三、薄层扫描法 TLCS
(一)薄层吸收扫描法
光源:氘灯 (190-340nm) 和 W灯 (340-900nm
※ 薄层板引起的散射现象(散射系数 SX )→标准曲线
( Kubella-
Munk曲线)弯曲→校正(曲线校直法和计算机回归法)
(二)薄层荧光扫描法
光源:汞灯 (250-580nm)
直线式扫描,无需曲线校直 药物分析学科

§3 常用定量分析方法三、薄层扫描法 TLCS
(三)定量分析方法
1 、方法学考察 工作曲线:①检查所选散射参数 SX 是否适宜;②考察工作曲线是否过原
点,以便确定是用外标一点法还是二点法测定;③确定点样量的线性范围。 分离度: R≥1.0
重复性考察: RSD≤3%
准确度考察: 95%~105%
2 、定量方法(讨论常用定量方法) 外标法:一点法、两点法、标准曲线法 内标法 药物分析学
科

§3 常用定量分析方法三、薄层扫描法 TLCS健脾丸中熊果酸的含量测定[ 含量测定 ] 取小蜜丸或重量差异项下的大蜜丸,剪碎,混匀,取约 4g ,精密称定,加水 30ml ,放置使溶散,滤过,残渣用水 30ml洗涤,在室温干燥至呈松软粉末状或低温干燥后研细,连同滤纸一并置索氏提取器内,加乙醚适量,加热回流提取 4 小时,提取液回收乙醚至干,残渣用石油醚( 30~60℃)浸泡两次,每次 10ml (浸泡约 2 分钟),倾去石油醚,残渣加适量三氯甲烷 -无水乙醇( 2:3 )混合液使溶解,转移至2ml 量瓶内,并稀释至刻度,摇匀,作为供试品溶液。另取熊果酸对照品适量,精密称定,加无水乙醇制成每 1ml 含 0.5mg 的溶液,作为对照品溶液。照薄层色谱法(附录 VI B )试验,精密吸取供试品溶液 4ul 与 6ul ,对照品溶液 2ul 与 4ul ,分别交叉点于同一硅胶G薄层板上,以环已烷 - 三氯甲烷 -乙酸乙酯 -冰醋酸( 20:5:8:0.5 )为展开剂,展开,取出,晾干,喷以 10% 硫酸乙醇溶液,在 105℃加热至斑点显色清晰,取出,在薄层板上覆盖同样大小的玻璃板,周围用胶布固定,进行扫描,波长 λS=540nm ,λR=700nm ,测量供试品吸收度积分值与对照品吸收度积分值,计算,即得。本品含山楂以熊果酸( C30H48O3 )计,小蜜丸每 1g 不得少于 0.10mg ;大蜜丸每丸不得少于 0.90mg 。 药物分析
学科

§3 常用定量分析方法四、气相色谱法 GC
(一)应用范围 挥发油及其它挥发性组分:冰片、桉叶素、樟脑、丁香酚、龙脑等 中药制剂含水量、含醇量
(二)系统适应性试验 理论塔板数 n= 5.54(tR/W1/2)2
※最小理论塔板数,柱长,柱填充、载体性能等有影响 分离度 R=2(tR1-tR2)/(W1+W2)
※ R≥1.5 (两峰完全分离) 重复性,相对标准偏差 RSD≤2.0% (n=5)
拖尾因子 T=W0.05h/2d1 (d1峰极大至峰前沿之间的距离 )
※ 1.05≥T≥0.95 药物分析学科

样品沸点范围 柱温 固定相配比300~400 ℃ 200~250 ℃ 1~5%
200~300 ℃ 150~180 ℃ 5~10%
100~200 ℃ 各组分的平均沸点 2/3左右
10~15%
气体等低沸点样品 室温或 50℃下
15~25%
§3 常用定量分析方法四、气相色谱法 GC
(三)实验条件的选择1 、固定相的选择气固色谱-硅胶、石墨、化学键合相、高分子多孔微球等气液色谱-烃类、硅氧烷类、醇类、酯类,特殊固定液(手性)2 、柱温的选择
药物分析学科
宽沸程样品:程序升温法

§3 常用定量分析方法四、气相色谱法 GC
(三)实验条件的选择3 、载气的选择
H2 、 He :流速大,柱长,热导检测器
N2 :流速小,氢焰检测器,电子捕获检测器
流速: 20- 80ml/min
4 、其它条件的选择进样方式:直接进样、顶空进样(兼有分离分析作用)进样口(汽化室)温度:样品的沸点或稍高于沸点,
30 / 50 +T℃ ℃ 柱≤ T 汽≤ 50 +T℃ 沸点
检测室温度: 30 +T℃ 柱≤ T 检≈ T 汽
药物分析学科

§3 常用定量分析方法四、气相色谱法 GC
(三)实验条件的选择4 、其它条件的选择进样时间: 1秒以内进样量:理论允许最大进样量使下降的塔板数不超过 10%。气体: 0.1- 1ml ;液体: 0.1-1μl
分流器分流进样: 1/10~1/100
检测器: TCD, FID, NPD, ECD
药物分析学科

§3 常用定量分析方法
四、气相色谱法 GC
(四)含量测定方法(例题见 P84 )
1、内标法加校正因子
求算校正因子
待测组分含量测定
2、外标法
3、面积归一化法
4、标准溶液加入法
%/
%/
/
/
RR
SS
SS
RR
s
R
CA
CA
AW
AW
f
ff
%% Ss
XX C
A
AfC
R
XRXA
ACC
%100%
i
ii
A
AC
药物分析学科

§3 常用定量分析方法
五、高效液相色谱法 HPLC
(一)特点
分离效能高、分析速度快及应用范围广
(二)适用范围
挥发性低、热稳定性差、分子量大的高分子化合物及离子型化合
物,如蛋白质、氨基酸、生物碱、甾体、类脂、核酸、维生素及无
机盐类 (三)系统适应性试验
药物分析学科

§3 常用定量分析方法五、高效液相色谱法 HPLC
(四)实验条件的选择
色谱柱的选择(固定相的选择)
流动相的选择—优级纯或色谱纯,脱气,过滤( 0.45μm)
部分含水溶剂:适用于分离中等极性、弱极性药物
非水溶剂:分离疏水性物质
缓冲溶液:适用于可溶于水并具可解离特性的化合物,如蛋白质、
肽及弱酸、弱碱类化合物。
洗脱方式:等度洗脱与梯度洗脱
检测器: UVD, FD, ELSD, ECD,RID
药物分析学
科

§3 常用定量分析方法
五、高效液相色谱法 HPLC
(五)含量测定方法
1 、外标法
2 、内标法
3 、内加法
4 、归一化法
例题 P90
药物分析学科
§3 常用定量分析方法
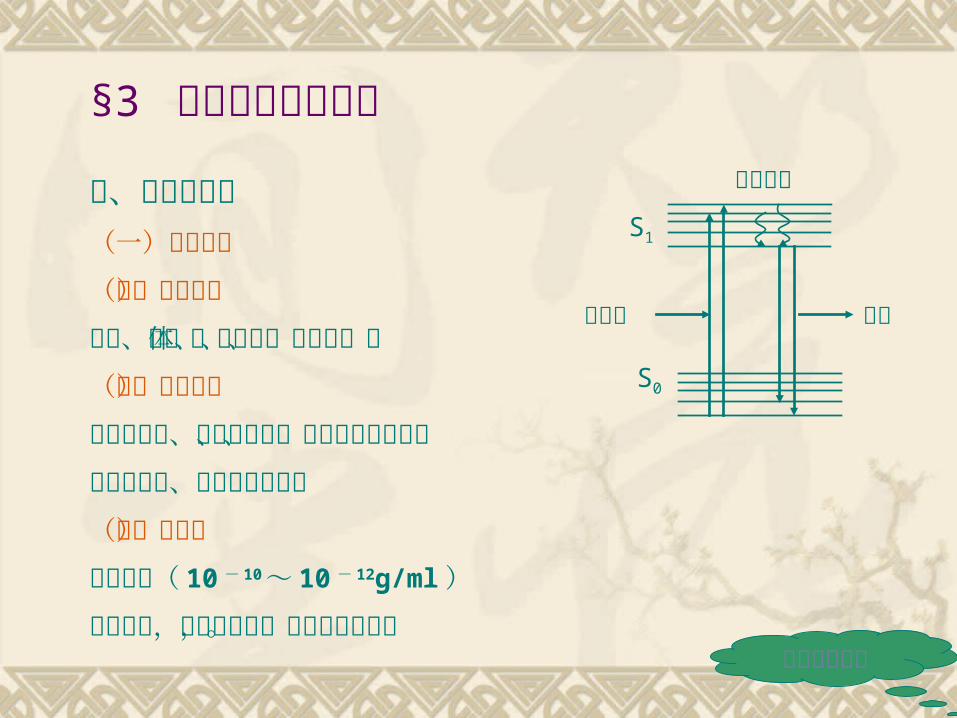
六、荧光分析法(一)基本原理(二)适用范围胺类、甾体、蛋白质、氨基酸、酶(三)测定方法直接荧光法、制备荧光法、化学引导荧光法、荧光淬灭法、胶束增敏荧光法(四)优缺点灵敏度高( 10 - 10 ~ 10 - 12g/ml )选择性好,干扰因素多,应用范围有限。
S1
S0
激发光 荧光
振动弛豫
药物分析学科

§3 常用定量分析方法
七、原子吸收分光光度法(一)基本原理A=K’ ·C
(二)测定对象呈原子状态的金属元素及部分非金属元素(三)含测方法标准曲线法标准加入法
药物分析学科

习 题
1 、薄层扫描法方法学考察时作标准曲线的目的何在?
2 、薄层扫描法测定中药制剂中有效成分含量时为何常用随行外标法?
3 、简述 GC 、 HPLC建立中药制剂有效成分含量测定方法时系统适用
性试验的各项参数及具体指标。
4 、试述 HPLC 法测定中药制剂有效成分含量时实验条件及参数的选择。
5 、定量分析方案设计:丹参为某合剂君药,请分别以水提、醇提制备
工艺,设计 HPLC 法测定制剂中丹参含量的分析方法。
药物分析学科





























