Embed Size (px)
DESCRIPTION
Elektronicky excitované stavy. Molekula přechází ze stavu o energii E i do stavu o energie E k jestliže absorbuje elektromagnetické záření o frekvenci ν a je splněna podmínka E i – E k = h ν k elektronickým přechodům dochází v UV/VIS oblasti (180 - 700 nm). - PowerPoint PPT Presentation
Citation preview

Molekula přechází ze stavu o energii Ei do stavu o energie Ek jestliže absorbuje elektromagnetické záření o frekvenci ν a je splněna podmínka
Ei – Ek = hν
k elektronickým přechodům dochází v UV/VIS oblasti (180 - 700 nm)
Elektronicky excitované stavy
Fotofyzikální procesy - dojde k relaxaci molekuly beze změny její identity
Fotochemické procesy - dojde k chemické přeměně molekuly
Kvantová chemie – popis fotofyzikálních a fotochemických procesů interpretace spekter

Charakteristika spekter
Poloha pásu – dána energií přechodu
ΔE = Ei – Ek
Intensita pásu

Lambert-Beerův zákon:
A = log (I0/I) = ε(λ)cd A - absorbancec – koncentraced – tloušťka absorbující vrstvyε(λ) – molární absorpční koeficient
εmax míra intensity elektronického přechodu
Celková plocha absorpčního pásu
f ~ ∫ ε(ν) dν f – síla oscilátoru
f ~ |Mi→k|2
Mi→k – transitní dipólový moment

Intenzita přechodu
Franck-Condon faktor
*
0 0ˆ f fM
* * * *
0 ' ' ' ' ' 'ˆ ˆ d d d des n n esf e s es v v v v e s esn eM
* * *
0 ' ' 'ˆ d d dn e sf v v e e s seM
| | | |j A Aj A
e er Z R ˆ ˆ ˆe n =
Orbitální výběrová pravidla
Spinová výběrová pravidla
Náboj a poloha elektronů a jader
Separace elektronické orbitální, vibrační a elektronické spinové funkce:

Výběrová pravidla
Formálně zakázané a povolené přechody = transitní moment povoleného přechodu může být velmi malý => velmi malá intensita
- zakázaný přechod může být pozorován ve spektru = výběrové pravidlo je relaxované nějakou poruchou
Spinová výběrová pravidla –
přechod je povolen, když oba stavy mají stejný spin(ale spin-orbitální interakce)
Orbitální výběrová pravidla –
symetrická - integrál je celkově symetrický nebo obsahuje symetrickou komponentu (porušen v důsledku ´vibronové interakce´)
malý překryv vlnových funkcí a lokální symetrie - n →π* and charge transfer přechody
*
' d ss s
*
'ˆ d ee ee
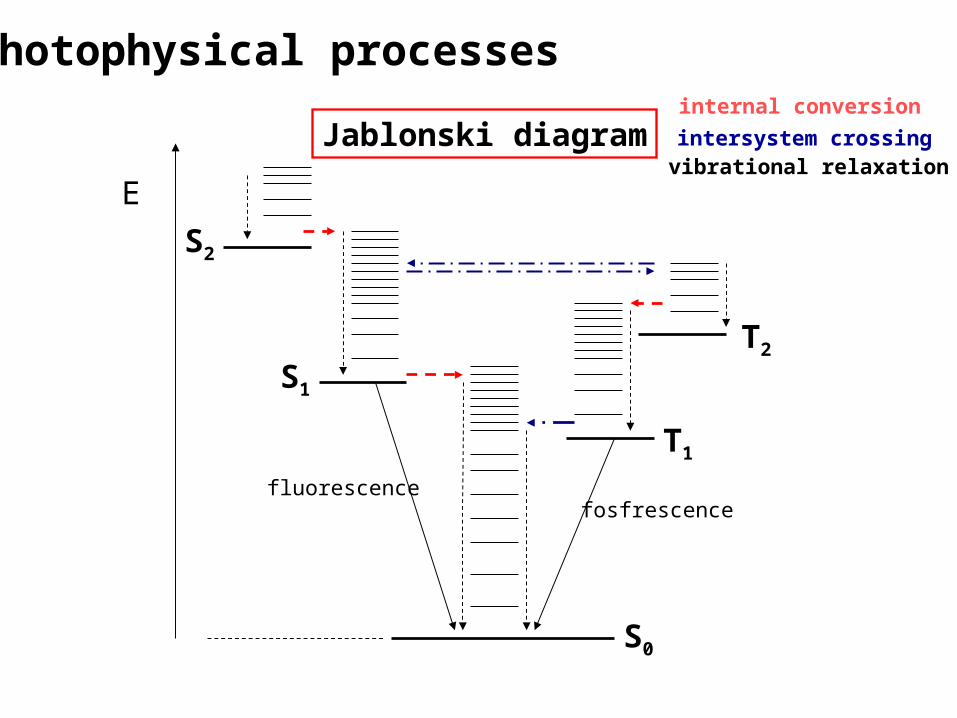
Jablonski diagram
E
S0
S1
S2
T1
T2
fluorescencefosfrescence
internal conversionintersystem crossingvibrational relaxation
Photophysical processes

Nezářivé přechody
Systém nelze studovat v rámci Born-Oppenheimerovy aproximacepoužití poruchové teorie:
Pravděpodobnost nezářivého přechodu mezi dvěma stavy:PES se přiblíží
' 22 | |ˆi f f ihk H
Vnitřní konverse: '
| | |ˆ Cf if N iH
Mezisystémový přechod:'
| | |ˆ SCf if SO iH
Operátor poruchy
Operátor kinetické energie jader
Spin-orbitální interakce

Klasifikace excitovaných stavů
Valenční stavy – singletní a tripletní stavy charakterizované přechody n → π* , π → π*, n → σ*, π → σ*, σ → π*
Rydbergovy stavy - elektron excitován do ´atomic-like´ orbitalu s n > nvalence
• Excitační energie exc = i - R/(n-)2
• Systémy s uzavřenou slupkou - positivně nabité jádro a elektron v orbitalu s nval+1,2,… difúzní charakter• Spektra v plynné fázi - ostré a relativně slabé pásy

Systémy v základním stavu – v současné době lze studovat rutinními postupy pomocí ab initio metod
Tato situace neplatí pro výpočet systémů v excitovaných stavech.
Electronicky Excitované Stavy
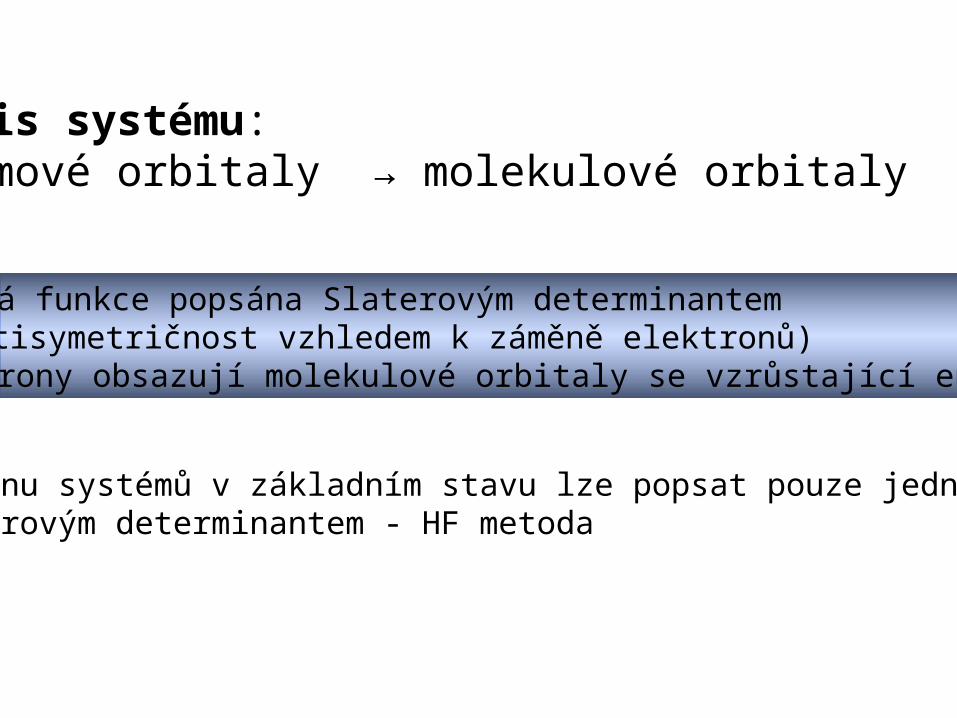
Popis systému:atomové orbitaly → molekulové orbitaly
vlnová funkce popsána Slaterovým determinantem (antisymetričnost vzhledem k záměně elektronů) Elektrony obsazují molekulové orbitaly se vzrůstající energií
Většinu systémů v základním stavu lze popsat pouze jedním Slaterovým determinantem - HF metoda
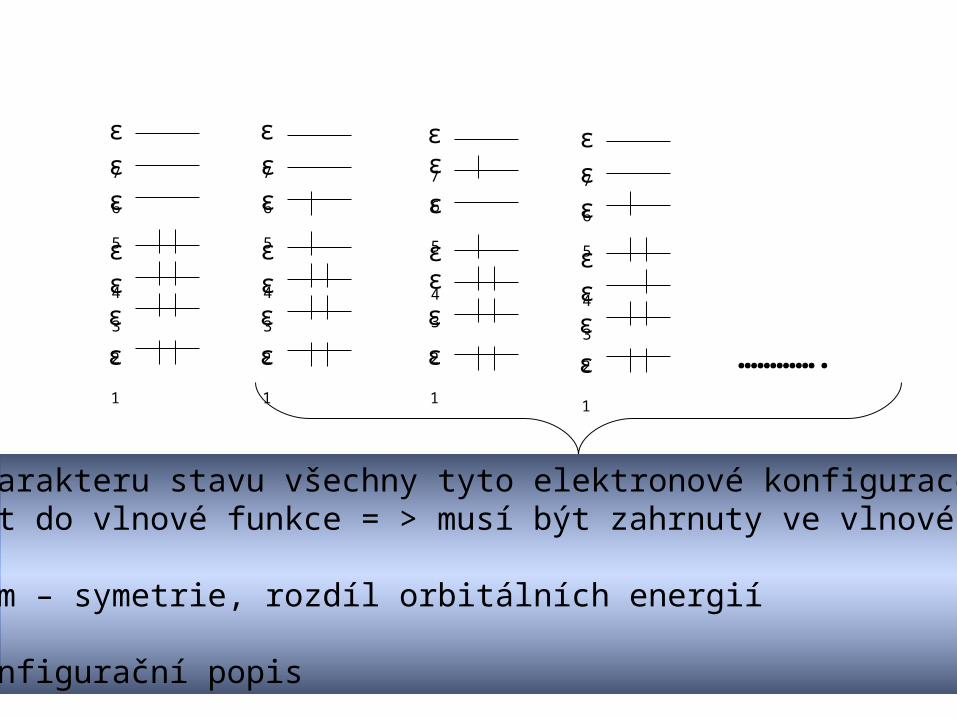
ε1
ε2
ε3
ε4
ε7
ε6
ε5
ε7
ε1
ε2
ε3
ε4
ε6
ε5
ε1
ε2
ε3
ε4
ε7ε6
ε5
ε1
ε2
ε3
ε4
ε7
ε6
ε5
………….
Podle charakteru stavu všechny tyto elektronové konfigurace mohou přispívat do vlnové funkce = > musí být zahrnuty ve vlnové funkci
Kritérium – symetrie, rozdíl orbitálních energií
Multi-konfigurační popis
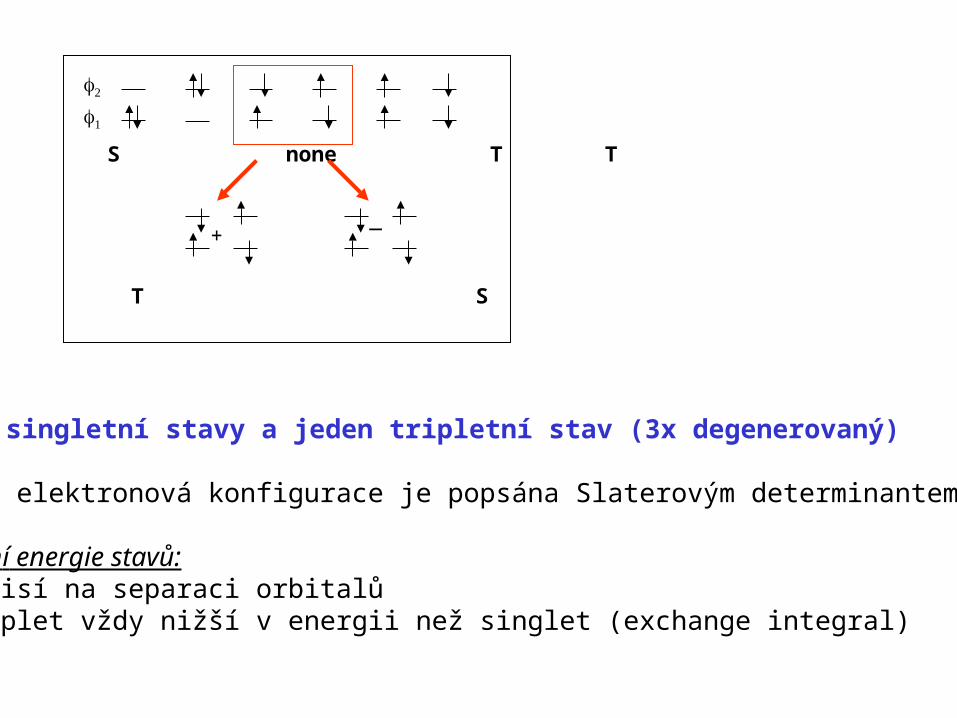
S S none T T
+_
T S
tři singletní stavy a jeden tripletní stav (3x degenerovaný)
Každá elektronová konfigurace je popsána Slaterovým determinantem
Relatiní energie stavů:- závisí na separaci orbitalů- triplet vždy nižší v energii než singlet (exchange integral)
1. Korelační energie – zásadní pro popis systémů v excitovaných stavech
Statická korelace elektronů - celková vlnová funkce obsahuje ty elektronické konfigurace, které spolu interagují
Dynamická korelace elektronů - podle své povahy mají jednotlivé stavy různou míru korelační energie – nutné zahrnout pro
získání ´přesné´ hodnoty excitační energie a správného pořadí stavů
2. Použití dostatečně flexibilní báze, flexibilněnjší než pro popis základního stavu, zvláště pro výpočet Rydbergových a iontových stavů
HF metoda - vlnová funkce popsána jedním Slaterovým determinantem - elektron se pohybuje ve zprůměrovaném poli ostatních elektronů

MCSCF - “Multi-configurational self-consistent field”
Definice:
• odpovídající electronické konfigurace (Slaterovy determinanty)
• MCSCF vlnová funkce |MCSCF> = I cI |I>, I - Slaterův determinant det(i ),
cI a ci, MO LCAO rozvoje jsou optimalizovány
Jak zadefinovat MCSCF vlnovou funkci ????
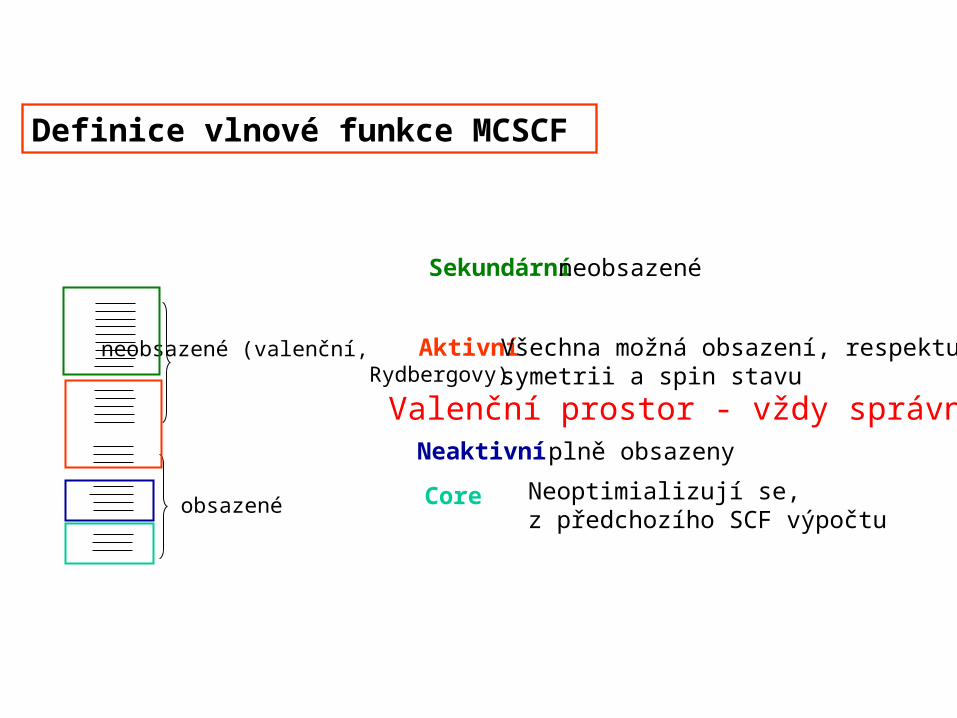
Definice vlnové funkce MCSCF
obsazené
neobsazené (valenční, Rydbergovy)
Neaktivní plně obsazeny
Aktivní Všechna možná obsazení, respektujícísymetrii a spin stavu
Sekundární neobsazené
Neoptimializují se, z předchozího SCF výpočtu
Core
Valenční prostor - vždy správně

Definice vlnové funkce – zásadní a ne vždy triviálníkrok
Aktivní prostor - zahrnutí všech valenčních elektronů - jen pro malé systémy, pro větší je třeba aktivní prostor zredukovat
povaha stavu a jeho interakce s jinými stavy výpočty na menších systémech symetrie systému
Počet stavů zahrnutých ve výpočtu
MCSCF metoda neobsahuje dynamickou korelační energii - pořadí stavů není vždy správné, např. Rydbergovy stavy pod valenčními stavy
Systémy s vysokou symetrií – specifikace podle symetrienejnižší excitované stavy pro každou bodovou grupu dané symetrie
Nesymetrické systémy – n-tý excitovaný stav je n-tým řešením CI rozvoje - problém s přesností výpočtu a konvergencí,přehození stavů stavů o stejné symetrii, jsou-li energeticky téměř degenerované výpočet několika stavů s danou vahou pro každý stav
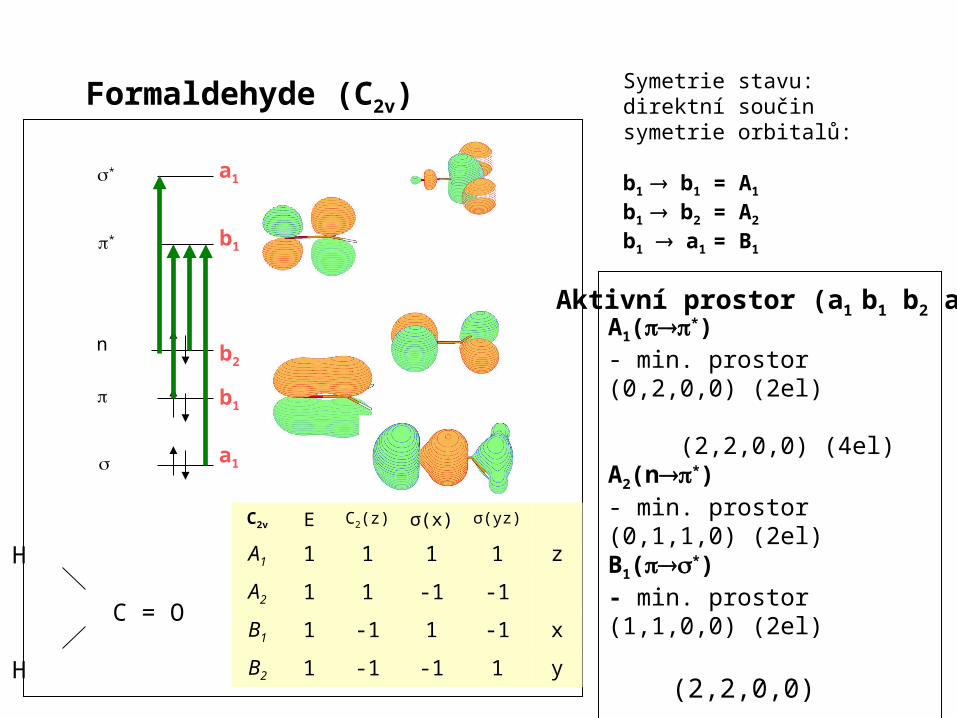
Formaldehyde (C2v) Symetrie stavu:direktní součin symetrie orbitalů:
b1 b1 = A1
b1 b2 = A2 b1 a1 = B1
*
*
n
a1
a1
b1
b1
b2
y1-1-11B2
x-11-11B1
-1-111A2
z1111A1
σ(yz)σ(x)C2(z)EC2v
H C = O
H
A1(*)- min. prostor (0,2,0,0) (2el) (2,2,0,0) (4el)A2(n*)- min. prostor (0,1,1,0) (2el)B1(*)- min. prostor (1,1,0,0) (2el) (2,2,0,0)
- libovolný prostor - n elektronů v m orbitalech
Aktivní prostor (a1 b1 b2 a2)
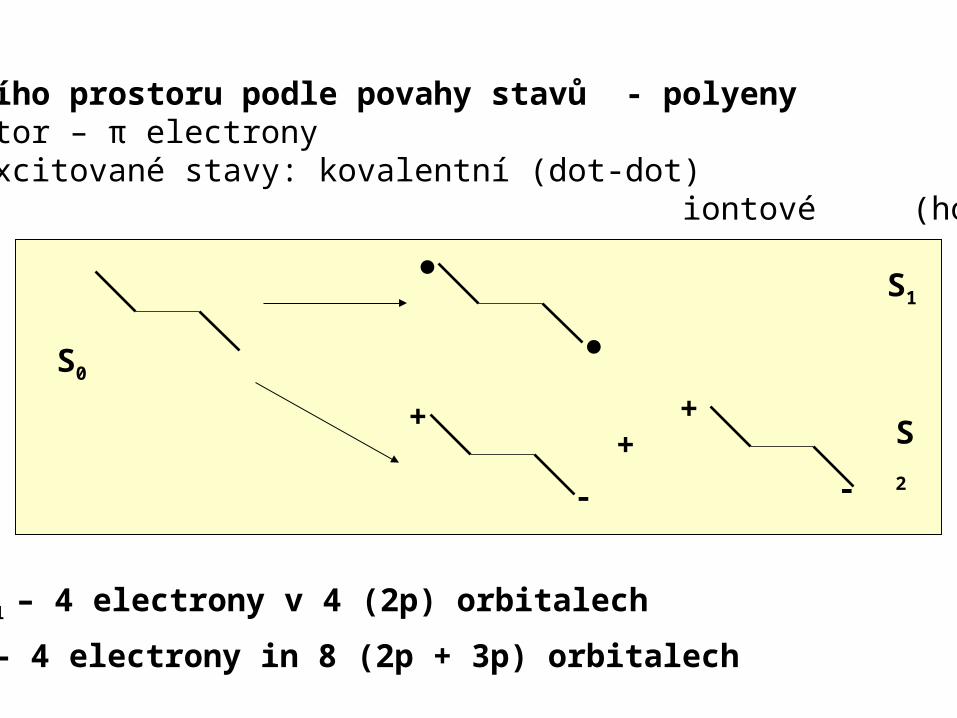
Výběr aktivního prostoru podle povahy stavů - polyenyAktivní prostor – π electronyValenční π-excitované stavy: kovalentní (dot-dot) iontové (hole-pair)
+
●
●
+
--
+
S1
S0
S2
S1 – 4 electrony v 4 (2p) orbitalech
S2 – 4 electrony in 8 (2p + 3p) orbitalech

Rydbergovy stavy přítomny v experimentu
Rydbergovy stavy
při výpočtech při použití difusní báze na atomech - mixování valenčních a Rydbergových stavů => někdy nelze určit charakter stavu (rozsah interakce závisí na energetickém rozdílu) zvětšují velikost valenčního prostoru při nedostatečné korelaci - špatné pořadí stavů
konstrukce speciálních bázových funkcí pro popis Rydbergových stavů - s, p, d funkce lokalizované v centru náboje kationtu molekuly
výpočty excitovaných stavů v matrixích

Ab initio metody pro excitované stavy
Metody založené na jedno-referenčním Slaterově determinantu
• CIS (CI-singles)• Propagator (response metody) RPA • Coupled-clusters metody: EOM-CCSDT, SAC-CI
Metody založené na multi-referenčním popisu
• multireferenční CI (MRCI) • multireferenční poruchová (CASPT2, …)• multireferenční coupled cluster (MR-CCSD)

Výhody: variační, size-konzistentní vyvážený popis jednotlivých excitovaných stavů.
Lze použít k optimalizaci excitovaných stavů Problémy: velmi nepřesné energie, chyba 1-3 eV, špatné pořadí stavů
Configuration interaction Singles (CIS)J. B. Foresman, M. Head-Gordon, J. A. Pople, and M. J. Frisch. J Phys Chem. 96, 135 (1992).
Excitované stavy - lineární kombinace konfigurací, které jsou single-excitované vzhledem k referenčníHF konfiguraci základního stavu
ECIS = EHF + ia C2ia(a - I) - ijab Cia Cjb(ja||ib)
Neobsahuje žádnou dynamickou korelaciVíce případů selhání než “úspěchů”

Coupled cluster methods
V ýhody: velmi přesné výsledky pro jedno-referenční problémy (0.1-0.3 eV), pokud jsou zahrnuty ‘triple’ excitace
analytický gradientProblémy: výpočetně velmi náročné
nepřesné v případě multi-referenčních problémů
SAC-CI (H. Nakatsuji, Chem.Phys.,67,329,334(1979)
|SAC> = exp(ICISI)|0>
|SAC-CI> = KdKRK)| |SAC>
SI , RK - symmetry adapted excitační operatory
CI ,dK - koeficienty
Multi-referenční verse - H. Nakatsuji, J.Chem.Phys., 83, 713 (1985)
EOM-CCSD(T) (J. D. Watts, R. J. Bartlett, Chem. Phys. Letters, 258, 581 (1996)
|CC> = expT|0> , T = T(1) + T(2) + …. + T(n)
K = RK CC , RK single, double, …. excitační operatory
Multi-referenční - similarity transformed EOM-CCSD(T) - M. Nooijen, J. Phys. Chem. A, 104, 4553 (2000)

Multi-referenční metody
|MCSCF> = I cI |I>, I je Slaterův determinant det(i ), cI and ci, MO LCAO jsou optimalizovány
• MCSCF - referenční vlnová funkce pro výpočty zahrnující dynamickou korelaci
Single, Double, Triple, ….. CI excitace (MRCI(SDT), …..)
• není size-konsistentní, alternativní téměř size-konzistentní - MR-ACPF, MR-AQCC• aplikovatelné na velmi malé systémy
Coupled cluster excitations (MR-CCSD)
• aplikovatelná na velmi malé systémy
Möller-Plesset Perturbation - CASPT2
• aplikovatelná na středně velké systémy• problemy s určením aktivního prostoru

Time-dependent DFT
Time-dependent Kohn-Sham equations
TD charge density in terms of TD KS orbitals
Adiabatic approximation - using time-independent density functional
Linear response to a perturbation - changes in charge density
Obecné trendy - valenční excitované stavy - přesnost srovnatelná s CASPT2 problémy s popisem Rydbergových stavů a -systémů- asymptotické chování standartních funkcionálů - lze opravit
problémy s popisem “charge-transfer” stavů
The dynamic response of the charge density for a system, initially in ground state, that is exposed to time-dependent perturbation, is described via the time-dependent Kohn-Sham equation





























