Embed Size (px)
Citation preview

Dr. MARCELO A. MARTÍ Director y Prof. Adjunto Dto. de Química Biológica FCEN-UBA, Investigador
Independiente IQUIBICEN-CONICETAsesor Científico bitgenia (www.bitgenia.com)
Contacto: [email protected]
Mapeo Alineamiento y llamado
de variantes

Análisis de datos 1.0
Anotación

Mapeo y
Alineamiento

Mapeo y Alineamiento
Tools (runtime):● BWA: runtime 3hs per lane
(~30 hs per Whole Genome sample)
● PICARD: runtime 2hs per lane (~40 hs per WG sample)
● GATK: runtime 6hs per lane (~75 hs per WG sample)
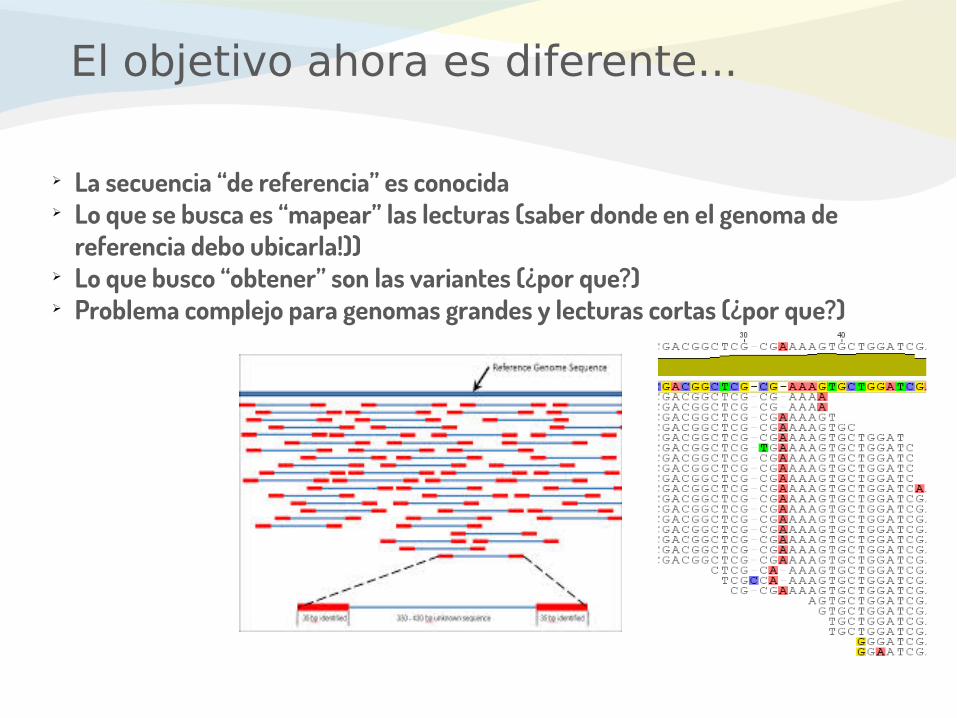
El objetivo ahora es diferente...
➢ La secuencia “de referencia” es conocida➢ Lo que se busca es “mapear” las lecturas (saber donde en el genoma de
referencia debo ubicarla!))➢ Lo que busco “obtener” son las variantes (¿por que?)➢ Problema complejo para genomas grandes y lecturas cortas (¿por que?)

“El” algoritmo: La transformación de Burrow-Wheelers (BWT)
Los programas más utilizados BWA y Bowtie(2) ambos utilizan BWT. Son 10-20 más rápidos que los alineadores de otro tipo..
La BWT es una transformación matemática general.. muy interesante (y compleja)... veamos un poco de que se trata
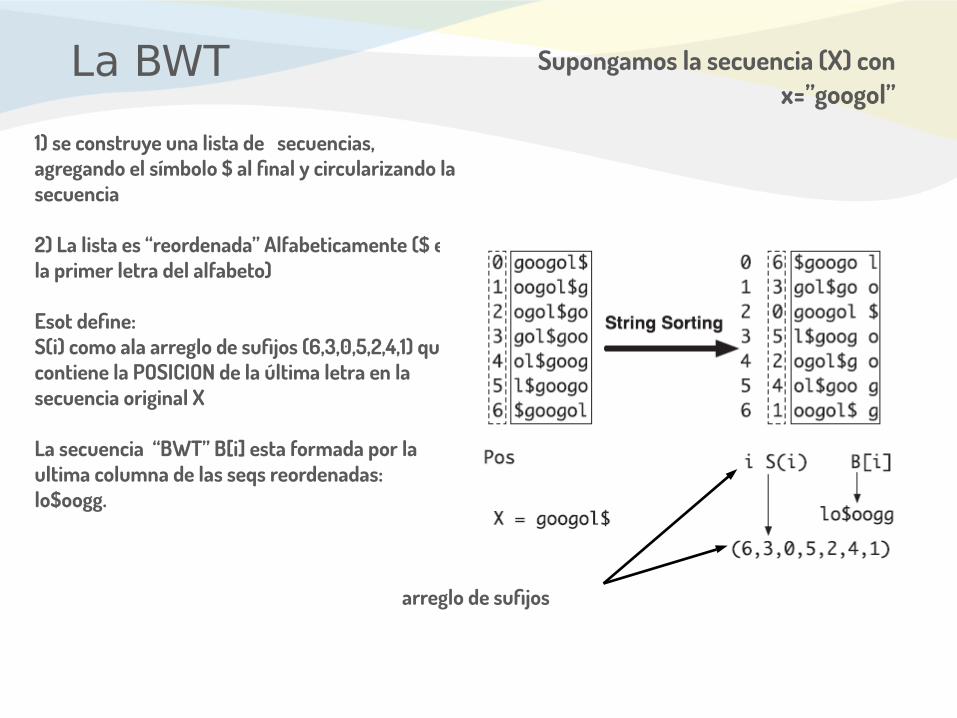
La BWT
1) se construye una lista de secuencias, agregando el símbolo $ al final y circularizando la secuencia
2) La lista es “reordenada” Alfabeticamente ($ es la primer letra del alfabeto)
Esot define:S(i) como ala arreglo de sufijos (6,3,0,5,2,4,1) que contiene la POSICION de la última letra en la secuencia original X
La secuencia “BWT” B[i] esta formada por la ultima columna de las seqs reordenadas: lo$oogg.
Supongamos la secuencia (X) con x=”googol”
arreglo de sufijos

La BWT....
Sea W una subsecuencia de X defino ahora:Rmin(W) como al “minimo entero” que corresponde al orden (en la lista re-ordenada) donde W es prefijo de XRmax(W) como al “maximo entero” que corresponde al orden donde W es prefijo de X
Ejemplos W=GO
Filas dónde GO es prefijo
El Intervalo Rmin:Rmax [1,2] es denominado SA
Rmin=1Rmax=2

Otro ejemplo.. W2=GOL
Filas dónde GOL es prefijo
El Intervalo Rmin:Rmax [1,1] es denominado SA
Rmin=1 Rmax=1
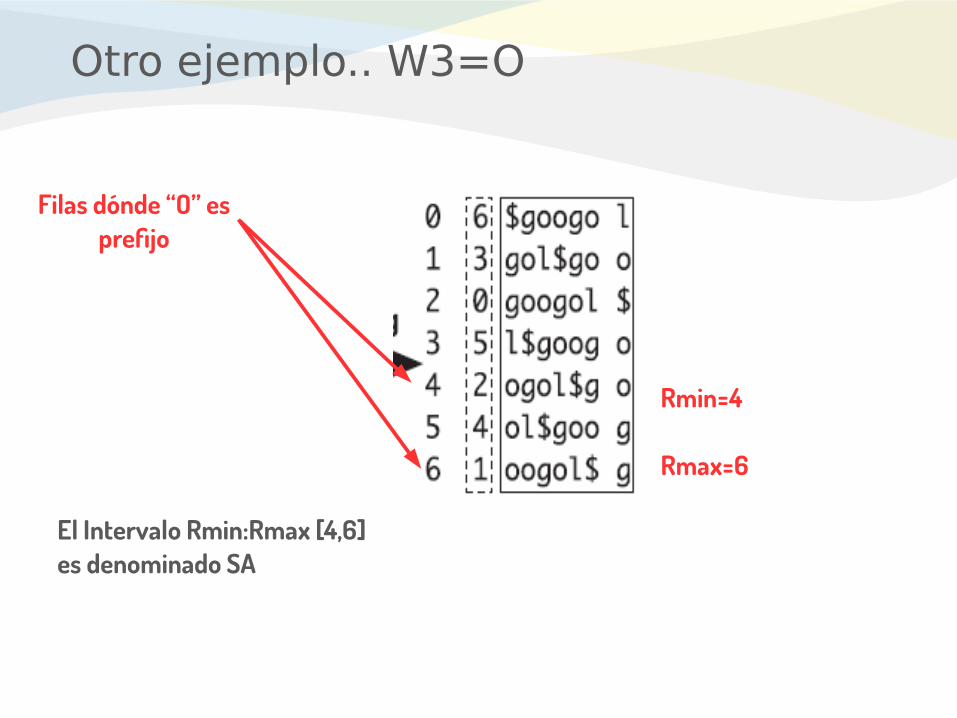
Otro ejemplo.. W3=O
Filas dónde “O” es prefijo
El Intervalo Rmin:Rmax [4,6] es denominado SA
Rmin=4
Rmax=6

¿Cómo sigo?
El intervalo “SA(W)” aplicado sobre el arreglo “S(i)” me da directamente todas las posiciones de W en X
Recordemos:X=googol$S(i)=(6,3,0,5,2,4,1) (con i=0,1.....)
Para W=GO, SA[1,2] … S(1)=3 y S(2)=0
¿Es cierto que esas son las posiciones de go en googol???

¿Cómo sigo?
El intervalo “SA(W)” aplicado sobre el arreglo “S(i)” me da directamente todas las posiciones de W en X
Recordemos:X=googol$S(i)=(6,3,0,5,2,4,1) (con i=0,1.....)
Para W=GO, SA[1,2] … S(1)=3 y S(2)=0
¿Es cierto que esas son las posiciones de go en googol???
googol012345googol012345

¿otros ejemplos?
El intervalo “SA(W)” aplicado sobre el arreglo “S(i)” me da directamente todas las posiciones de W en X
Recordemos:X=googol$S(i)=(6,3,0,5,2,4,1) (con i=0,1.....)
Para W2=gol, SA[1,1] … S(1)=3 Para W3=o, SA[4,6] … S(4)=2, S(5)=4 y S(6)=1 (posic 1,2y4!)
googol012345

Entonces...
Dada una secuencia “X” (un genoma) una vez calculado el S(i), basta determinar para cada lectura “W” el SA(Rmin:Rmax) para saber el intervalo en el arreglo S(i) que me dara todas las posiciones dónde esa “lectura” mapea sobre la secuencia X
El algoritmos es “perfecto” para lecturas que coinciden perfectamente (sin errores variaciones) y es adaptable para lecturas que contienen hasta “Z” variantes... esto es lo que hace BWA y BowTie2!

Llamado de
Variantes

Volver a “alinear” los “pedacitos” sobre el genoma de Referencia y...
DepthProfundidad!
Read Length

¿Dónde hay una variante?
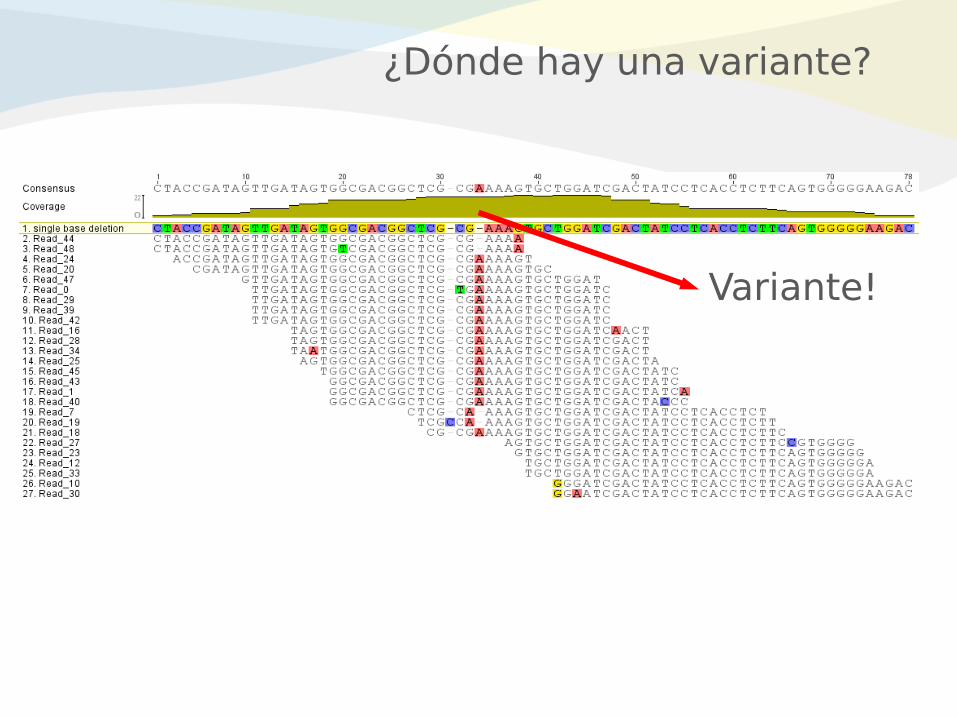
Variante!
¿Dónde hay una variante?

Datos reales
Variante Homocigota

Datos reales
Variantes Heterocigotas

VQSR
Información útil de cada Variante
Falsos Positivos

Que son los SNPs, que otras variantes genéticas existen?
Los genomas de cada individuo presentan “diferencias”, estas son de tres clases principales● Polimorfismo de Nucleotido Unico (SNPs)● Inserciones y Deleciones● Variaciones Estructurales
Mi (o TU) Genoma NO es otra cosa que una LISTA de variantes respecto de Mi (o TU) Genoma NO es otra cosa que una LISTA de variantes respecto de UNA REFERENCIA!UNA REFERENCIA!

¿Cuántas variantes puede tener un paciente?
Anotación
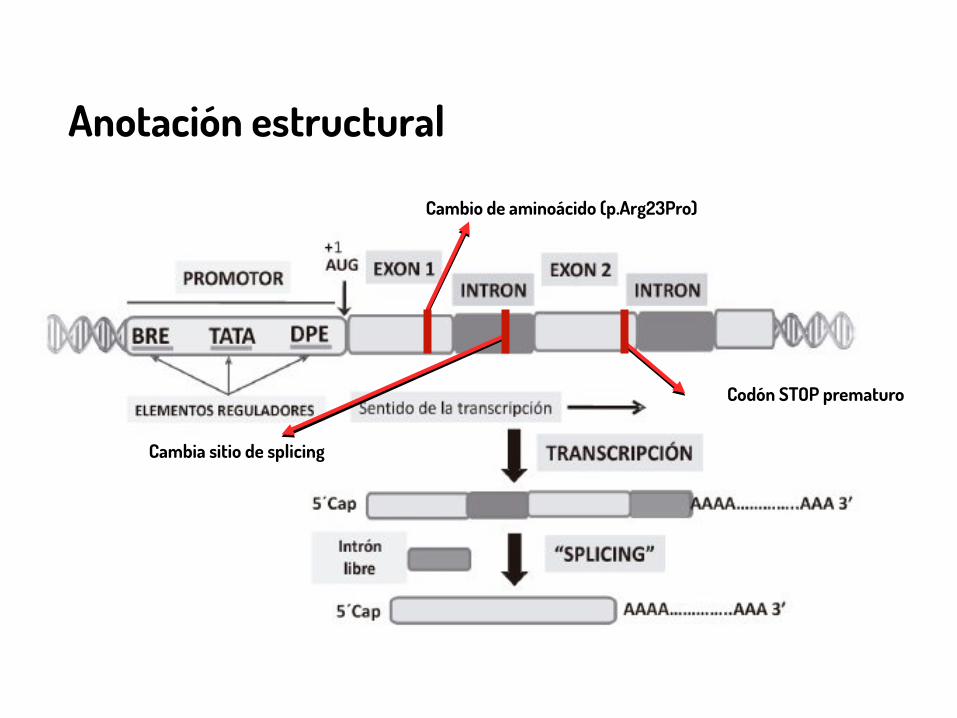
Anotación estructural
Cambio de aminoácido (p.Arg23Pro)
Codón STOP prematuro
Cambia sitio de splicing
Anotación funcional
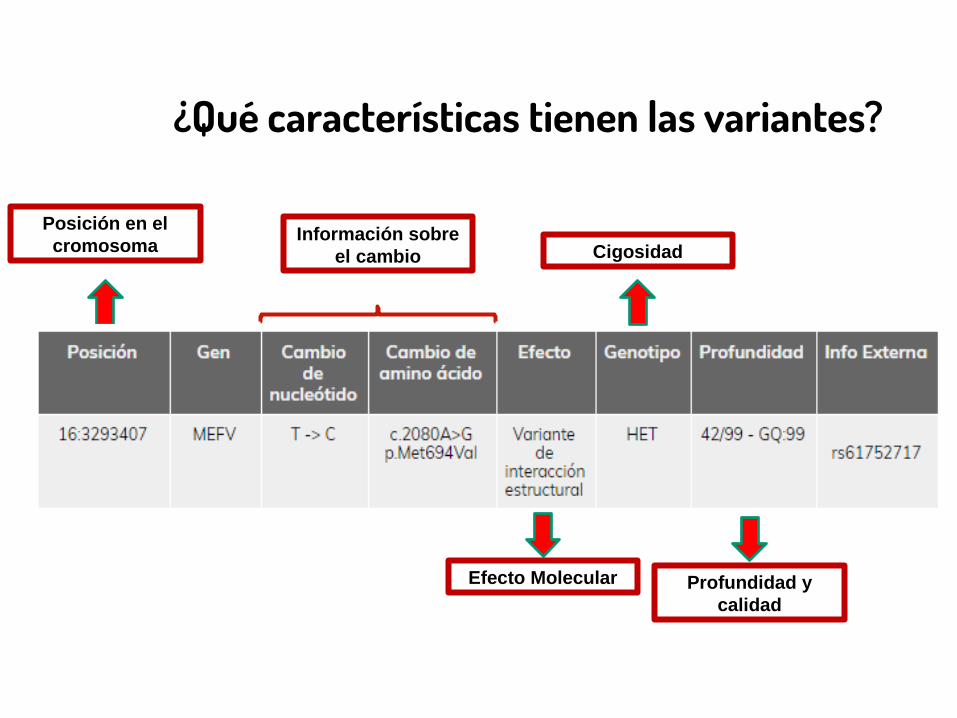
¿Qué características tienen las variantes?
Posición en el cromosoma
Efecto Molecular
CigosidadInformación sobre
el cambio
Profundidad y calidad

Criterio ACMG para la clasificación de variantes
Dos pasos:
I) Cada característica de las variantes se asocia a un código (que equivale a un puntaje) compuesto por 6 niveles (PVS, PS, PM, PP,
BA y BS).
PS3 Estudios funcionales in vitro o in vivo que apoyan un efecto dañino sobre el gen o el
producto génico resultante.
PVS1 Variante nula (sin sentido, desplazamiento de marco) en un gen donde “loss-of-function”
(LOF) es un mecanismo conocido de la enfermedad.
BA1 La frecuencia del alelo es mayor al 5%.

II) La suma del puntaje determina (derivado de las characterísticas) determina la clasificación de la variante en:
1 Muy fuerte (PVS1) Y 1 Moderada (PM1–PM6)
≥2 Fuertes (BS1–BS4)
Se detecta en heterocigosis la variante missense c.322C>T (p.Arg108Cys), descrita previamente en la base de datos Human Gene Mutation Database (HGMD) asociada a epilepsia mioclónica progresiva, y en ClinVar como variante patogénica (PS1, PP4). La variante presenta predicción in-silico como patogénica (PP3), posee muy baja frecuencia en ExAC (sin la presencia de individuos homocigotas) (PM2) y se encuentra acompañada por una segunda variante patogénica/posiblemente patogénica en trans (PM3). De acuerdo con el criterio ACMG, la variante se clasifica como Patogénica (PS1 + 2PM + 2 PP).
Patogénica
Probablemente patogénica
Benigna
Posiblemente Benigna
Significado incierto
Criterio ACMG para la clasificación de variantes

La bioinformática en La bioinformática en la Genómica Clínicala Genómica Clínica





























