Embed Size (px)
Citation preview

Instructions for use
Title TLR3アジュバントによるCTL依存性抗腫瘍免疫応答の解析
Author(s) 武田, 洋平
Issue Date 2017-03-23
DOI 10.14943/doctoral.k12560
Doc URL http://hdl.handle.net/2115/65967
Type theses (doctoral)
Note 配架番号:2301
File Information Yohei_Takeda.pdf
Hokkaido University Collection of Scholarly and Academic Papers : HUSCAP

学位論文
TLR3 アジュバントによる CTL 依存性抗腫瘍免疫応答の解析
(Analysis of CTL-dependent anti-tumor immune response induced by TLR3
adjuvant)
2017 年 3 月
北海道大学
武田 洋平


学位論文
TLR3 アジュバントによる CTL 依存性抗腫瘍免疫応答の解析
(Analysis of CTL-dependent anti-tumor immune response induced by TLR3
adjuvant)
2017 年 3 月
北海道大学
武田 洋平

目次
発表論文目録および学会発表目録・・・・・・・・・・・・・・・・・・1
序章・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・2
略語表・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・7
第一章: Poly(I:C)療法における DC-CTL 依存性抗腫瘍免疫誘導メカニズム
の解析
緒言・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・9
実験材料および方法・・・・・・・・・・・・・・・・・・・・・・・・9
実験結果・・・・・・・・・・・・・・・・・・・・・・・・・・・・17
考察・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・47
第二章: 抗腫瘍免疫療法における治療奏功性評価の指標となり得る CD11c
陽性 CD8+
T cell の解析
緒言・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・50
実験材料および方法・・・・・・・・・・・・・・・・・・・・・・・51
実験結果・・・・・・・・・・・・・・・・・・・・・・・・・・・・54
考察・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・74
総括および結論・・・・・・・・・・・・・・・・・・・・・・・・・77
謝辞・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・79
引用文献・・・・・・・・・・・・・・・・・・・・・・・・・・・・80

1
発表論文目録および学会発表目録
本研究は以下の論文に発表した。
1. Masahiro Azuma, Yohei Takeda, Hiroko Nakajima, Haruo Sugiyama, Takashi
Ebihara, Hiroyuki Oshiumi, Misako Matsumoto, Tsukasa Seya.
Biphasic function of TLR3 adjuvant on tumor and spleen dendritic cells promotes
tumor T cell infiltration and regression in a vaccine therapy
Oncoimmunology 5(8):e1188244. (2016)
2. Yohei Takeda, Masahiro Azuma, Misako Matsumoto, Tsukasa Seya.
Tumoricidal efficacy coincides with CD11c up-regulation in antigen-specific
CD8+
T cells during vaccine immunotherapy
Journal of Experimental and Clinical Cancer Research 35(1):143. (2016)
本研究の一部は以下の学会に発表した。
1. Yohei Takeda, Tsukasa Seya, Misako Matsumoto, Haruo Sugiyama, Hiroko
Nakajima.
BATF3 supports TLR3-derived IL-12 induction in CD8+ DC, which promotes
antitumor T cell responses by Poly(I:C)
第 74 回日本癌学会学術総会 2015 年 10 月 8 日、名古屋
2. 武田洋平、松本美佐子、瀬谷司
抗腫瘍 Poly(I:C)療法における細胞傷害性 CD11c+CD8+T 細胞の増加は、治
療著効性を評価する指標となり得る
第 20 回日本がん免疫学会総会 2016 年 7 月 28 日、大阪

2
序章
生物には下等動物から高等動物に至るまで、自己と非自己を認識し病原
体や癌細胞などの異物を排除する免疫機構が備わっている。脊椎動物の様
な高等動物の体内には様々な免疫担当細胞が存在しており、それらが協調
的に働くことで複雑な生体防御機構が構築されている。ヒトを含む脊椎動
物の免疫機構は大きく自然免疫、獲得免疫に大別されている。自然免疫は
異物の侵入に際して迅速に起動し、速やかな異物排除に寄与すると同時に、
後に続く獲得免疫を誘導する引き金となる 1,2。獲得免疫においては、自然
免疫に属する樹状細胞 (Dendritic cell; DC) の活性化を介して増殖した T細
胞や B 細胞が中心となり、より異物に対する特異性が高い応答や、異物が
排除された後の免疫記憶の確立が誘導される。即ち、自然免疫は獲得免疫
を含めた全ての免疫応答の起点となっており、如何に効果的に自然免疫
(DC の活性化) を誘導できるかが、生体防御の観点において非常に重要で
ある。
外来性異物が体内に侵入した際の自然免疫の起動を担っているのが、パ
ターン認識受容体 (Pattern-Recognition Receptor; PRR) と呼ばれる分子群
である。PRR は、病原体などの多くの微生物に特有の分子パターン
(Pathogen associated molecular patterns; PAMPs) や、傷害を受けた癌細胞な
どの自己由来細胞から放出されるダメージ関連分子パターン (Damage
associated molecular patterns; DAMPs) を認識し、異物排除に関わる様々な
免疫応答を誘導する 3,4。PRR には細胞膜上およびエンドソーム内に局在す
る Toll-like receptor (TLR) や細胞質内に局在する RIG-I like receptor (RLR)、
Nod-like receptor (NLR) など多数の分子群が報告されており、多彩な病原
体の侵入に対応することが可能となっている 5,6。
TLR ファミリーに属する TLR3 はエンドソーム内に局在しており、ウイ
ルス由来の 2 重鎖 RNA (dsRNA) を認識する。TLR3 刺激により、アダプタ
ー分子である TIR domain-containing adapter molecule-1 (TICAM-1; 別名
TRIF) を介して、下流の Interferon regulatory factor-3 (IRF3)、Nuclear
factor-kappa B (NF-B)、Activator protein-1 (AP-1) といった転写因子が活性
化する 7,8。また dsRNA は TLR3 以外にも、細胞質内に局在する Retionic acid
inducible gene-I (RIG-I) および Melanoma differentiation associated protein-5
(MDA5) といった RLR にも認識される 9。RIG-I / MDA5 は、アダプター分
子である Mitochondrial antiviral-signaling protein (MAVS; 別名 IPS-I) を介
して IRF3、NF-B、AP-1 を活性化する 10。即ち、dsRNA はエンドソーム
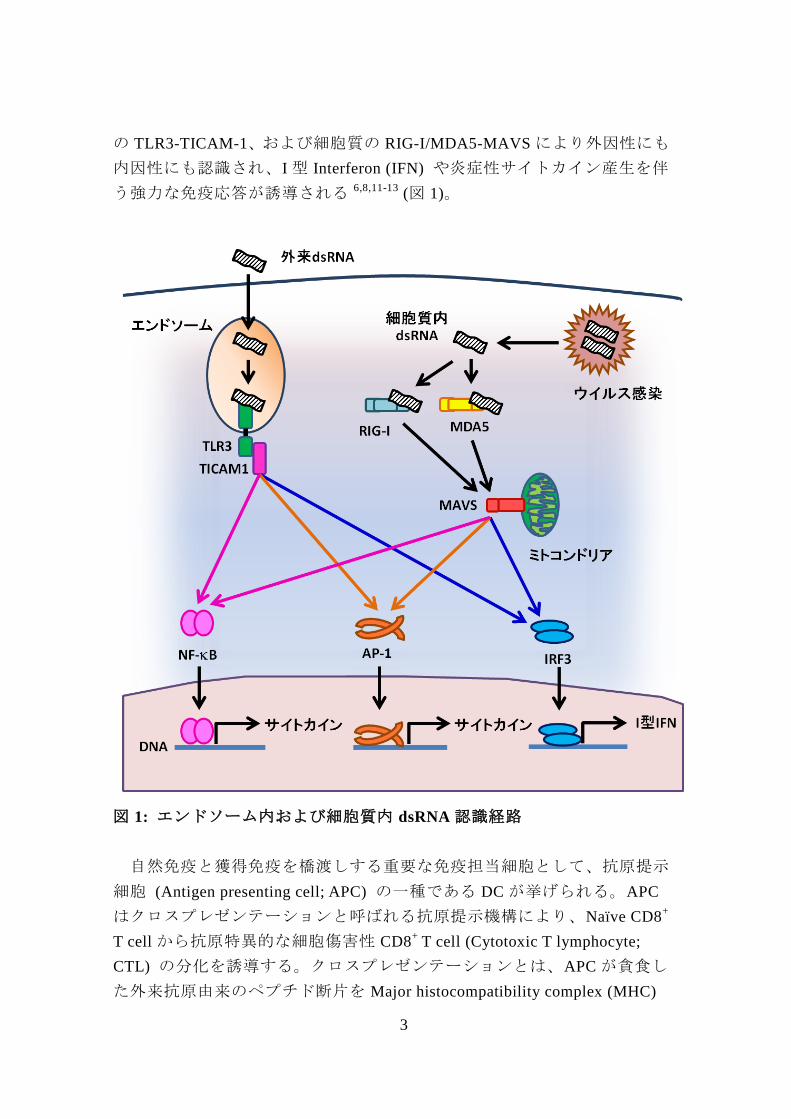
3
の TLR3-TICAM-1、および細胞質の RIG-I/MDA5-MAVS により外因性にも
内因性にも認識され、I 型 Interferon (IFN) や炎症性サイトカイン産生を伴
う強力な免疫応答が誘導される 6,8,11-13 (図 1)。
図 1: エンドソーム内および細胞質内 dsRNA 認識経路
自然免疫と獲得免疫を橋渡しする重要な免疫担当細胞として、抗原提示
細胞 (Antigen presenting cell; APC) の一種である DC が挙げられる。APC
はクロスプレゼンテーションと呼ばれる抗原提示機構により、Naïve CD8+
T cell から抗原特異的な細胞傷害性 CD8+
T cell (Cytotoxic T lymphocyte;
CTL) の分化を誘導する。クロスプレゼンテーションとは、APC が貪食し
た外来抗原由来のペプチド断片を Major histocompatibility complex (MHC)
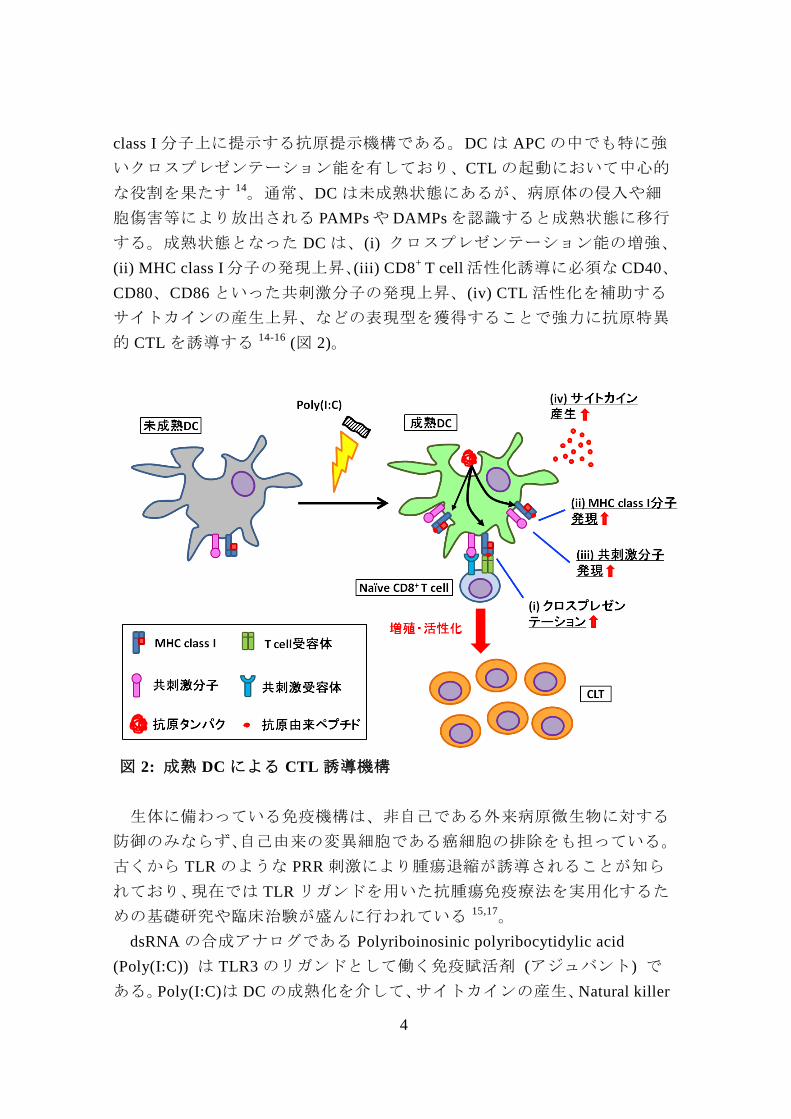
4
class I 分子上に提示する抗原提示機構である。DC は APC の中でも特に強
いクロスプレゼンテーション能を有しており、CTL の起動において中心的
な役割を果たす 14。通常、DC は未成熟状態にあるが、病原体の侵入や細
胞傷害等により放出される PAMPs や DAMPs を認識すると成熟状態に移行
する。成熟状態となった DC は、(i) クロスプレゼンテーション能の増強、
(ii) MHC class I 分子の発現上昇、(iii) CD8+
T cell 活性化誘導に必須な CD40、
CD80、CD86 といった共刺激分子の発現上昇、(iv) CTL 活性化を補助する
サイトカインの産生上昇、などの表現型を獲得することで強力に抗原特異
的 CTL を誘導する 14-16 (図 2)。
図 2: 成熟 DC による CTL 誘導機構
生体に備わっている免疫機構は、非自己である外来病原微生物に対する
防御のみならず、自己由来の変異細胞である癌細胞の排除をも担っている。
古くから TLR のような PRR 刺激により腫瘍退縮が誘導されることが知ら
れており、現在では TLR リガンドを用いた抗腫瘍免疫療法を実用化するた
めの基礎研究や臨床治験が盛んに行われている 15,17。
dsRNA の合成アナログである Polyriboinosinic polyribocytidylic acid
(Poly(I:C)) は TLR3 のリガンドとして働く免疫賦活剤 (アジュバント) で
ある。Poly(I:C)は DC の成熟化を介して、サイトカインの産生、Natural killer
5
(NK) 細胞や CTL の活性化を誘導し、腫瘍を強力に退縮させる 18-20。しか
しアジュバントとしての有効性が認められる半面、Poly(I:C)は TLR3 のみ
ならず細胞内の RIG-I や MDA-5 を活性化して、過剰な炎症性サイトカイ
ン産生に起因する有害応答をも惹起してしまう 21,22。このような問題を回
避するため、瀬谷研究室では TLRl3 に特異的に認識されるアジュバント
を開発し、その実用化を目指して研究を行っている 23。
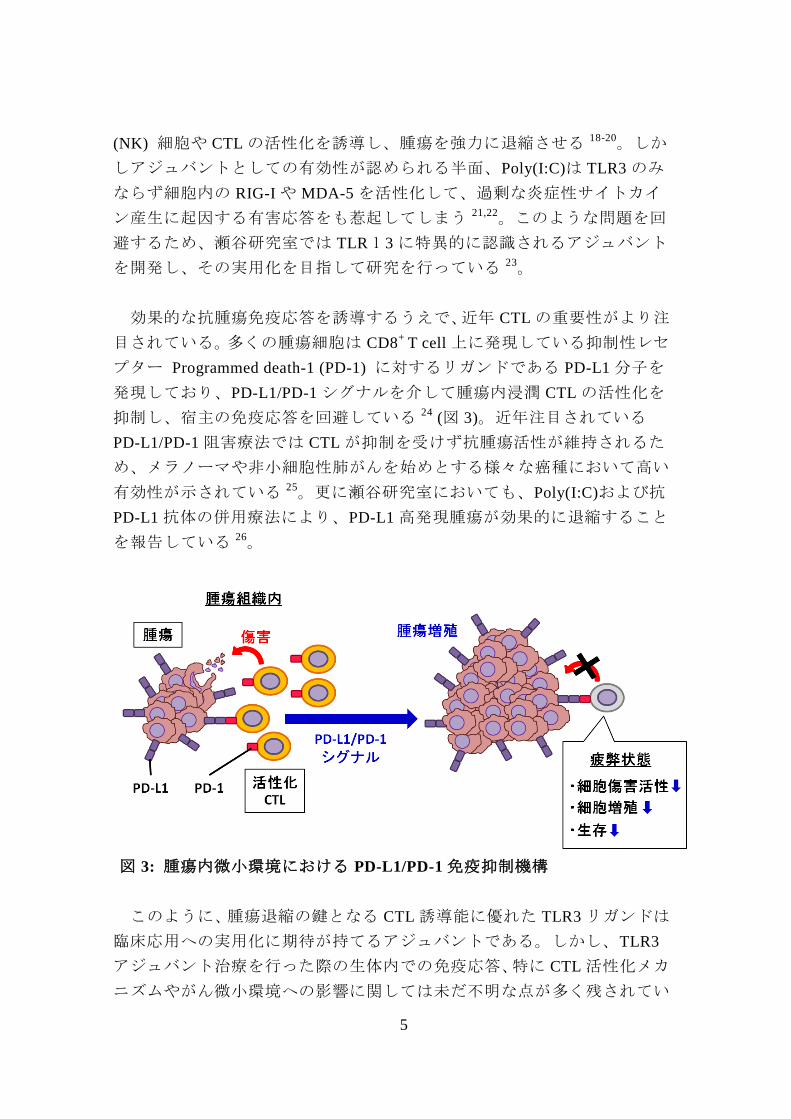
効果的な抗腫瘍免疫応答を誘導するうえで、近年 CTL の重要性がより注
目されている。多くの腫瘍細胞は CD8+
T cell 上に発現している抑制性レセ
プター Programmed death-1 (PD-1) に対するリガンドである PD-L1 分子を
発現しており、PD-L1/PD-1 シグナルを介して腫瘍内浸潤 CTL の活性化を
抑制し、宿主の免疫応答を回避している 24 (図 3)。近年注目されている
PD-L1/PD-1 阻害療法では CTL が抑制を受けず抗腫瘍活性が維持されるた
め、メラノーマや非小細胞性肺がんを始めとする様々な癌種において高い
有効性が示されている 25。更に瀬谷研究室においても、Poly(I:C)および抗
PD-L1 抗体の併用療法により、PD-L1 高発現腫瘍が効果的に退縮すること
を報告している 26。
図 3: 腫瘍内微小環境における PD-L1/PD-1 免疫抑制機構
このように、腫瘍退縮の鍵となる CTL 誘導能に優れた TLR3 リガンドは
臨床応用への実用化に期待が持てるアジュバントである。しかし、TLR3
アジュバント治療を行った際の生体内での免疫応答、特に CTL 活性化メカ
ニズムやがん微小環境への影響に関しては未だ不明な点が多く残されてい

6
る。このような抗腫瘍応答制御機構を詳細に明らかにすることで、より有
効性・安全性の高い新規アジュバントや新規治療戦略の開発、およびその
臨床応用が可能になると考えられる。そのため本研究第一章においては、
Poly(I:C)による抗腫瘍 CTL 活性化について、特に中心的役割を担うと考え
られる DC を介したメカニズムに着目し、その詳細な制御機構の解明を目
的とし解析を行った。また第二章においては、実際に臨床で利用する際に
重要な指針となり得る、Poly(I:C)療法の奏功性評価のための新規バイオマ
ーカー候補についても解析を行った。
第一章では、マウス移植がんモデルにおいて、転写因子 Basic leucine
zipper transcription factor ATF-like 3 (BATF3) 陽性の DC サブセットが TLR3
シグナル依存的腫瘍退縮を担っていることを明らかにした。TLR3 シグナ
ルにより成熟化した BATF3 陽性 DC は、クロスプレゼンテーションの促進
や IL-12、CXCL10 といった液性因子の産生により、腫瘍抗原特異的 CTL
の活性化や腫瘍内浸潤を惹起した。本章により TLR3 アジュバントによる
抗腫瘍免疫応答を担う責任 DC サブセットの同定、およびその CTL 依存的
腫瘍退縮誘導メカニズムの一端を明らかにした。
また第二章においては、担がんマウスモデルにおいて、Poly(I:C)療法に
より CD11c 分子陽性の CD8+
T cell 分画が増加することを見出した。この
CD11c 陽性分画は強い細胞傷害活性を有し、また生体内の CD11c+
CD8+
T
cell 増加レベルと、腫瘍抗原特異的 CTL 増加レベルおよび腫瘍退縮程度が
非常に良く相関することを解明した。以上より、Poly(I:C)治療時の CD11c+
CD8+
T cell の増加レベルは、治療奏功性評価の簡便かつ有用な指標となり
得る可能性が示された。
以上、本研究はより有効かつ安全性の高い TLR3 アジュバント療法の実
用化のための知見の蓄積に寄与するものである。

7
略語表
本文中および図中で使用した略語は以下のとおりである。
APC; Antigen presenting cell
AP-1; Activator protein-1
BATF3; Basic leucine zipper transcription factor ATF-like 3
CFSE; Carboxy fluorescent diacetate succinimidy ester
CTL; Cytotoxic T lymphocyte
DAMPs; Damage associated molecular patterns
DC; Dendritic cell
DLN; Draining lymph nodes
dsRNA; Double-stranded RNA
FACS; Fluorescence activated cell sorter
FBS; Fetal bovine serum
HE; Hematoxylin-Eosin
IDO; Indoleamine 2,3-dioxygenase
IFN; Interferon
IPS-1; Interferon-beta promoter stimulator-1
IRF3; Interferon regulatory factor-3
MALP2s; Macrophage activating lipopeptide-2-short
MAVS; Mitochondrial antiviral-signaling protein
MDA5; Melanoma differentiation associated protein-5
MDSC; Myeloid-derived suppressor cell
MHC; Major histocompatibility complex
NK; Natural killer
NLR; Nod-like receptor
NF-B; Nuclear factor-kappa B
OVA; Ovalbumin
PAMPs; Pathogen associated molecular patterns
PD-1; Programmed death-1
PI; Propidium iodide
Poly(I:C); Polyriboinosinic polyribocytidylic acid
PRR; Pattern-recognition receptor
RIG-I; Retionic acid inducible gene-I
RLR; RIG-I like receptor

8
STING; Stimulator of interferon genes
TAA; Tumor-associated antigen
TCR; T cell receptor
TICAM1; TIR domain-containing adapter molecule-1
TLR; Toll-like receptor
WT; Wild-type
WT1; Wilms tumor 1

9
第一章
Poly(I:C)療法における DC-CTL 依存性抗腫瘍免疫誘導メカニズムの解析
緒言
Naïve CD8+
T cell が腫瘍細胞に対して傷害性を発揮するためには、腫瘍
関連抗原 (Tumor-associated antigen; TAA) を貪食した DC によるクロスプ
レゼンテーションが必須である。DC には複数のサブセットが存在してお
り、各サブセットごとに生体内での局在やクロスプレゼンテーション能、
PRR 分子の発現パターン等が異なっている 15,27。マウス生体内に存在する
CD8+
DC は非常に高いクロスプレゼンテーション能を有するサブセット
であり、ヒトでもそのカウンターパートとして CD141+
(BDCA3+) DC が存
在することが知られている 28-30。Mouse CD8+
DC および human CD141+
DC
は選択的に高い TLR3 発現を示すことから 29,31、これらサブセットが生体
内での Poly(I:C)依存的免疫応答の誘導に強く関与する可能性が考えられる。
前駆細胞からの CD8+
DC の分化においては、転写因子である BATF3 が
他の転写因子と協調的に作用し分化誘導を正に制御することが知られてお
り、Batf3 欠損 (Batf3-/-
) マウスでは CD8+
DC の数が減少する 32-34。この
ような Batf3-/-マウスでは、病源体感染に対する抵抗性や抗原特異的 CD8
+ T
cell 誘導能が低下していることが示されている 32,35,36。
以上の知見から、BATF3 依存的に誘導される mouse CD8+
DC や human
CD141+
DC といったサブセットは、Poly(I:C)依存性腫瘍退縮においても重
要な役割を担っていると考えられる。そのため本章においては、Batf3-/-マ
ウスを用い、Poly(I:C)に対する抗腫瘍応答を野生型マウス (Wild-type; WT)
と比較することで、Poly(I:C)治療による CD8+
DC を介した CTL 依存性腫
瘍退縮メカニズムを詳細に解明することとした。
実験材料および方法
1. マウス
WT の C57BL/6 マウスは日本クレア社より購入した。Batf3-/-マウスは
Jackson Laboratory 社より購入した。OT-I Tg マウスは東北大学 石井直人
教授より供与された。OT-II Tg マウスは北里大学 岩淵和也教授より供与
された。Tlr3-/-マウスは大阪大学 審良静男教授より供与された。全ての動
物実験は、北海道大学動物実験に関する規程に従い、承認を受けて行った。
2. 試薬および抗体
10
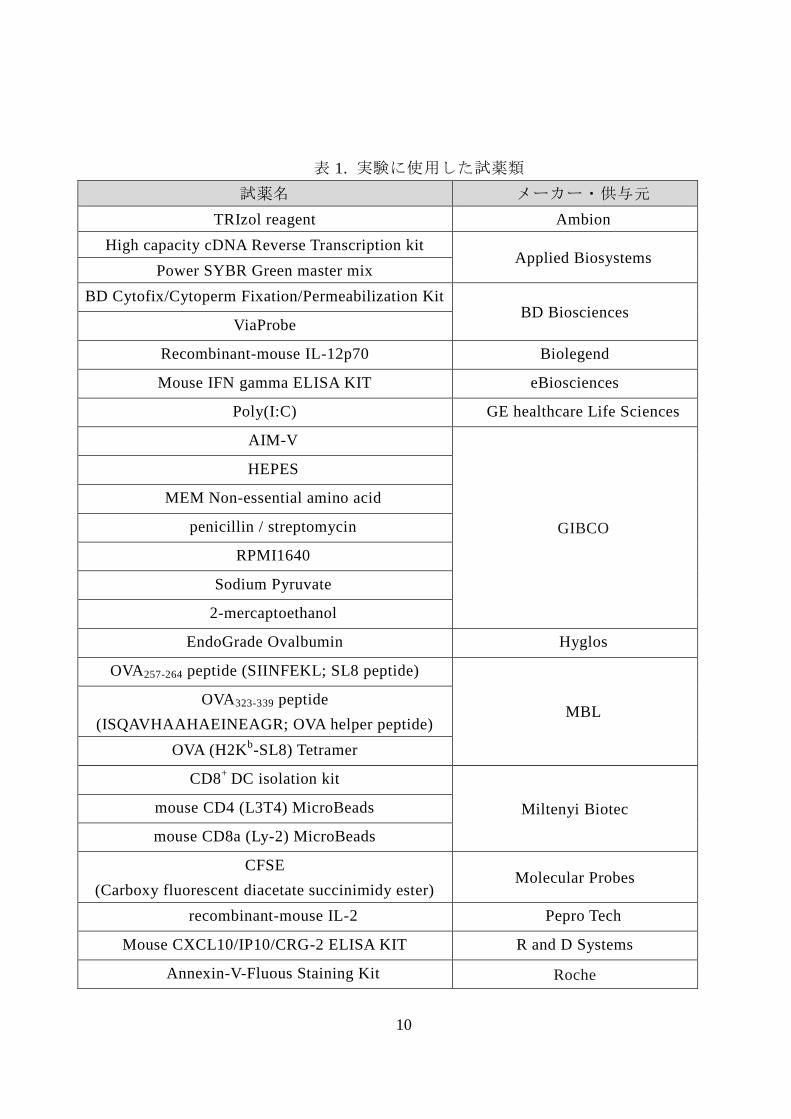
表 1. 実験に使用した試薬類
試薬名 メーカー・供与元
TRIzol reagent Ambion
High capacity cDNA Reverse Transcription kit Applied Biosystems
Power SYBR Green master mix
BD Cytofix/Cytoperm Fixation/Permeabilization Kit BD Biosciences
ViaProbe
Recombinant-mouse IL-12p70 Biolegend
Mouse IFN gamma ELISA KIT eBiosciences
Poly(I:C) GE healthcare Life Sciences
AIM-V
GIBCO
HEPES
MEM Non-essential amino acid
penicillin / streptomycin
RPMI1640
Sodium Pyruvate
2-mercaptoethanol
EndoGrade Ovalbumin Hyglos
OVA257-264 peptide (SIINFEKL; SL8 peptide)
MBL OVA323-339 peptide
(ISQAVHAAHAEINEAGR; OVA helper peptide)
OVA (H2Kb-SL8) Tetramer
CD8+
DC isolation kit
Miltenyi Biotec mouse CD4 (L3T4) MicroBeads
mouse CD8a (Ly-2) MicroBeads
CFSE
(Carboxy fluorescent diacetate succinimidy ester) Molecular Probes
recombinant-mouse IL-2 Pepro Tech
Mouse CXCL10/IP10/CRG-2 ELISA KIT R and D Systems
Annexin-V-Fluous Staining Kit Roche

11
Collagenase D
DNase I
DOTAP Liposomal Transfection Reagent
G418
Tissue-Tek O.C.T. compound Sakura Finetek Japan
Brefeldin A
Sigma-Aldrich
Collagenase I
Collagenase IV
Hank’s balanced salt solution
Hyaluronidase
DNase I Takara Bio
FBS (Fetal bovine serum) Thermo Fisher Scientific
Prolong Gold
WT1126-134 peptide (RMFPNAPYL; Db126 peptide) 大阪大学
杉山治夫教授より供与された。 WT1 (H2Db-Db126) Tetramer
表 2. 実験に使用した抗体
抗体名 クローン番号 メーカー
APC-anti-mouse CD3 17A2
Biolegend
Alexa Fluor 700-anti-mouse CD8
APC-anti-mouse CD8
FITC-anti-mouse CD8
PE-anti-mouse CD8
53-6.7
FITC-anti-mouse CD11b
PE/Cy7-anti-mouse CD11b M1/70
PE/Cy7-anti-mouse CD11c N418
Purified anti-mouse CD16/32 93
PE-anti-mouse CD103 2E7
PE-anti-mouse CLEC9A 7H11
PE-anti-mouse F4/80 BM8
PE-anti-mouse Gr1 RB6-8C5
FITC-anti-mouse I-Ab KH74
12
FITC-anti-mouse IFN- XMG1.2
PE-anti-mouse IL-12/IL-23p40 C15.6
APC-anti-mouse TLR3 11F8
APC-anti-mouse XCR1 ZET
PE-anti-mouse CD4 GK1.5
eBioscience APC-anti-mouse IL-12p35 4D10p35
PE-anti-mouse T cell receptor (TCR) V2 B20.1
3. 細胞および培養メディウム組成
マウス胸腺腫細胞 (EL4) 由来の Ovalbumin (OVA) 抗原強制発現株であ
る EG7 は ATCC 社より購入した。細胞培養には、あらかじめ 56°C で 30
分間加熱し非動化した 10% ウシ胎児血清 (Fetal bovine serum; FBS)、1 mM
Sodium Pyruvate、10 mM HEPES、55 M 2-mercaptoethanol、50 IU penicillin
/ 50 g/ml streptomycin、0.5 mg/ml G418 を含む RPMI1640 メディウムを用
いた。
マウス骨髄性白血病細胞 (C1498) 由来の Wilms Tumor 1 (WT1) 抗原強
制発現株である WT1-C1498 は大阪大学 杉山治夫教授より供与された 37。
細胞培養には、10%非動化 FBS、55 M 2-mercaptoethanol、50 IU penicillin /
50 g/ml streptomycin、0.5 mg/ml G418 を含む RPMI1640 メディウムを用い
た。
マウス脾臓細胞を採取後 5 日間培養した際には、10%非動化 FBS、55 M
2-mercaptoethanol 、 50 IU penicillin / 50 g/ml streptomycin 、 1x MEM
Non-essential amino acid を含む、45% RPMI 1640 と 45% AIM-V の混合メデ
ィウムを用いた。培養 3 日目には新たに等量のメディウムを追加し、更に
最終濃度が 20 IU/ml となるように recombinant-mouse IL-2 を添加した。
マウス脾臓細胞を採取し 5 日未満の期間培養を行った場合は、10%非動
化 FBS、55 M 2-mercaptoethanol、10 mM HEPES、 50 IU penicillin / 50 g/ml
streptomycin を含む RPMI1640 メディウムを用いた。全ての細胞は 37°C、
5% CO2 存在下で培養を行った。
4. 腫瘍移植および Poly(I:C)治療
マウスの腰背部に 1 x 106 の細胞数の WT1-C1498、あるいは 2 x 10
6 の細
胞数の EG7 を皮下投与した。その後、腫瘍体積を数日おきにノギスを用い
て測定した。腫瘍体積は以下の式を用いて算出した。

13
[腫瘍体積 (mm3) = 短径 (mm) x 短径 (mm) x 長径 (mm) x 0.52]
WT1-C1498 担がんマウスへの Poly(I:C)投与実験においては、腫瘍移植後
5 日および 12 日目に PBS または 50 g Poly(I:C)を腫瘍組織周辺の皮下に投
与した。また別の実験では、100 ng IL-12p70 の単独投与または Poly(I:C) +
IL-12p70 の併用投与を行った。更に別の実験では、100 g WT1126-134 peptide
を 20 g DOTAP で封入した後、その単独投与または Poly(I:C) + WT1126-134
peptide の併用投与を行った。
EG7 担がんマウスにおいては、腫瘍径が 200-600 mm3 程度に達した頃に
PBS、100 g OVA 単独、50 g Poly(I:C)単独、あるいは OVA + Poly(I:C)併
用投与の何れかの処置を行った。また、CD8+
T cell、NK cell 除去実験にお
いては、Poly(I:C)投与の一日前に、ハイブリドーマを移植したヌードマウ
スから採取した-CD8抗体または-NK1.1 抗体を含む腹水を、標的となる
マウスに腹腔内投与した。CD8+
DC 養子移植実験においては、野生型お
よび Batf3-/-マウス由来の脾臓細胞を Collagenase D処理 (実験材料および方
法の第 6 項参照) した後、CD8+
DC isolation kit を用いて CD8+
DC を分離
した。その後 5 x 105 の細胞数の CD8
+ DC を、PBS あるいは Poly(I:C)投与
を行う 1 日前の EG7-担がん Batf3-/-マウスへ尾静脈投与した。
5. 腫瘍内微小環境の解析
担がんマウスを安楽殺した際、WT1-C1498 および EG7 腫瘍組織塊を採
取した。腫瘍組織からの mRNA 採取、フローサイトメーター (Fluorescence
activated cell sorter; FACS) 解析、腫瘍内浸潤 IL-12 および CXCL10 の ELISA
解析を行う場合は、一定質量の組織片をカミソリを用いて細断し、0.05
mg/ml Collagenase I、0.05 mg/ml Collagenase IV、0.025 mg/ml Hyaluronidase、
0.01 mg/ml DNase I を含んだ一定液量の Hank’s balanced salt solution に懸濁
し 33°C、15 分間処理した。その後 1500 rpm、5 分間の遠心を行い、上清液
を腫瘍内浸潤 IL-12 および CXCL10 の ELISA 解析用サンプルとし、沈殿し
た細胞成分を mRNA および FACS 解析用サンプルとした。mRNA 解析用サ
ンプルは TRIzol reagent に溶解した。FACS 解析用サンプルは、さらに ACK
lysis buffer (組成: 150mM NH4Cl、10mM KHCO3、0.1 mM Na2EDTA) 内で 1
分間反応させ赤血球を溶解した後、始めに 100 m、続いて 45 m メンブ
レンフィルターに通し Filtration を行った。
免疫組織染色用の凍結組織切片サンプルを採取する場合は、腫瘍組織塊
を 4% paraformaldehyde / PBS 溶液に浸し 4°C で 30 分間固定した。その後
15% スクロース / PBS 溶液に移し、組織塊が底に沈むまで 4°C でスクロー

14
ス置換を行った。その後 30% スクロース / PBS 溶液に移し、再び組織塊
が底に沈むまで 4°C で置換操作を行った。スクロース置換が完了した後、
組織塊を Tissue-Tek O.C.T. compound に移し、液体窒素に浸し凍結させるこ
とで包埋した。包埋後の凍結組織ブロックは、組織染色を行うまで-80°C
で保存した (その後の組織染色法は実験材料および方法の第 8 項参照)。
6. 脾臓細胞、鼠径リンパ節細胞、腫瘍内浸潤細胞の FACS 解析
マウスから脾臓、鼠径リンパ節および腫瘍細胞を採取した。脾臓、ある
いは鼠径リンパ節内の DC の解析を行う場合は、採取した組織を 400
Mandle unit/ml Collagenase D を含んだ Hank’s balanced salt solution 内にてす
り潰し、37°C、25 分間コラゲナーゼ反応させた後、EDTA を最終濃度が 10
mM になるように加えて 37°C、5 分間静置することにより反応を止めた。
その後 ACK lysis buffer にて溶血を行い、FACS buffer (組成: 0.5% ウシ血清
アルブミン、 0.1% NaN3 / PBS) に再懸濁した。DC 以外の細胞ポピュレー
ションの解析を行う場合は、コラゲナーゼ処理を行わず ACK lysis buffer
反応後に FACS buffer に再懸濁した。約 1 x 106
/ml の細胞懸濁液に対し
anti-mouse CD16/32 抗体を希釈倍率が 1/500 となるように加え、4°C、5 分
間のブロッキングを行った。
その後、通常の細胞表面分子の染色を行う場合は、蛍光標識抗体を希釈
倍率が 1/200 になるように、また ViaProbe を希釈倍率が 1/50 になるように
加え、4°C、20 分間静置した後 FACS buffer で洗浄した。
WT1-tetramer および OVA-tetramer 陽性細胞の染色を行う場合は、ブロッ
キング後に蛍光標識された WT1-tetramer、または OVA-tetramer を希釈倍率
が 1/50 になるように加え、4°C、30 分間反応させた。その後 FACS buffer
で洗浄した後、細胞表面の CD3 および CD8 分子の染色を行った。
CD8+
DC における IL-12p35、IL-12p40 の細胞内染色を行う場合は、脾
臓細胞を 4 x 106 /ml の濃度で平底 96 well プレートに播種し、10 g/ml
Brefeldin A を加え、37°C で 4 時間培養した。その後、ブロッキングおよび
細 胞 表 面 分 子 の 染 色 を 行 い 、 続 い て BD Cytofix/Cytoperm
Fixation/Permeabilization Kit を用いて付属マニュアルに従い固定、透化処理
を行った。透過処理後に 2% ラット血清を加え 4°C、15 分間の細胞内ブロ
ッキングを行った。ブロッキング後、蛍光標識された anti-mouse IL-12p35
および anti-mouse IL-12/IL-23p40抗体を希釈倍率が 1/200 となるように加え、
4°C、30 分間反応させた。
CD8+
T cell、および CD4+
T cell における IFN-の細胞内染色を行う場合

15
は、脾臓細胞を 4 x 106 /ml の濃度で平底 96 well プレートに播種し、100 nM
SL8 peptide (IFN-+
CD8+
T cell を解析する場合) または 1M OVA helper
peptide (IFN-+
CD4+
T cell を解析する場合) を加えた後 37°C で 2 時間培養
した。2 時間後、10 g/ml Brefeldin A を加え、さらに 37°C で 4 時間培養し
た。培養後は既述の細胞内染色と同様の方法で細胞内 IFN-染色を行った。
また、細胞内 TLR3 分子を蛍光標識する場合も同様に細胞内染色法に従
った。
死細胞の検出を行う場合は、Annexin-V-Fluous Staining Kit を用いて
Annexin V 染色を行うと同時に、Propidium iodide (PI) を 10 g/ml の濃度に
なるように添加し、4°C で 15 分間反応させた。
作製したサンプルは FACS Aria II または FACS Calibur (BD biosciences 社)
を用いて解析した。
7. 定量 PCR
各種細胞を TRIzol reagent に溶解させたサンプルから、付属プロトコー
ルに従い RNA 精製を行った。精製した RNA は、400-1000 ng の RNA に対
し DNase I を 5 U 加え、室温で 15 分間反応させた。その後 EDTA を最終濃
度が 2.5 mM になるように加え、70°C で 10 分間インキュベーションし
DNase を不活化した。DNase 処理した RNA を用いて High capacity cDNA
Reverse Transcription kit の付属プロトコールに従い逆転写反応を行った。逆
転写反応により得られた cDNA を鋳型とし、Power SYBR Green master mix
を用いて PCR を行い、増幅産物を Step One Real-time PCR system (Applied
Biosystems 社) により検出した。得られた増幅曲線より Ct 値を算出し、鋳
型に含まれる mRNA 量をCt 法により算出した。なお、内部標準として
は Gapdh を使用した。
表 3. 実験に使用した primer の配列
Gene Primer sequences
Forward Reverse
Gapdh 5’-GCCTGGAGAAACCTGCCA-3’ 5’-CCCTCAGATGCCTGCTTCA-3’
Batf3 5’-CAGACCCAGAAGGCTGACAAG-3’ 5’-CTGCGCAGCACAGAGTTCTC-3’
Ccl5 5’-TGCCCACGTCAAGGAGTATTT-3’ 5’-TCGAGTGACAAACACGACTGC-3’
Cd40 5’-GACTCAGGCGAATTCTCAGC-3’ 5’-GGTGCAGTGTTGTCCTTCCT-3’
Cxcl9 5’-GATAAGGAATGCACGATGCTC-3’ 5’-TCTCCGTTCTTCAGTGTAGCAA-3’

16
Cxcl10 5’-GTGTTGAGATCATTGCCACGA-3’ 5’-GCGTGGCTTCACTCCAGTTAA-3’
Cxcl11 5’-GGCTGCGACAAAGTTGAAGTGA-3’ 5’-TCCTGGCACAGAGTTCTTATTGGAG-3’
Ddx58 5’-GCCCTGTACCATGCAGGTTAC-3’ 5’-AGTCCCAACTTTCGATGGCTT-3’
Gzmb 5’-TCCTGCTACTGCTGACCTTGTC-3’ 5’-ATGATCTCCCCTGCCTTTGTC-3’
Ifih1 5’-GCTGCCCAGAAGACAACACAG-3’ 5’-CGACAGCAGGCAGAAGACACT-3’
Ifna2 5’-TACTCAGCAGACCTTGAACC-3’ 5’-GGTACACAGTGATCCTGTGG-3’
Ifnb 5’-CCAGCTCCAAGAAAGGACGA-3’ 5’-CGCCCTGTAGGTGAGGTTGAT-3’
Ifng 5’-GATATCTGGAGGAACTGGCAAAAG-3’ 5’-AGAGATAATCTGGCTCTGCAGGAT-3’
Il12b 5’-AATGTCTGCGTGCAAGCTCA-3’ 5’-ATGCCCACTTGCTGCATGA-3’
Il12rb1 5’-AGTTGCGAATGGACTGGAATG-3’ 5’-GTCACCCAAGGTCCAATTCGT-3’
Il12rb2 5’-AAGTCCCCAAGGAAATGAAAGG-3’ 5’-GTGATGATAGCGATGCAAATGC-3’
Il15 5’-TTAACTGAGGCTGGCATTCATG-3’ 5’-ACCTACACTGACACAGCCCAAA-3’
Il28 5’-TCCCAGTGGAAGCAAAGGATTG-3’ 5’-TCAAGCAGCCTCTTCTCGATGG-3’
Mavs 5’-AGCCCTCCAGAGAGCATCAA-3’ 5’-GAGGCAACATTTGCTGCGT-3’
Prf1 5’-CAAGGTAGCCAATTTTGCAGC-3’ 5’-GGCGAAAACTGTACATGCGAC-3’
Ticam1 5’-TGTTGGAAAGCAGTGGCCTAT-3’ 5’-GATGACGTGGTGTTCTGCAGA-3’
Tlr3 5’-TTGCGTTGCGAAGTGAAGAA-3’ 5’-ACTTGCCAATTGTCTGGAAACA-3’
Tnsfs9 5’-CTGCCCCAACACTACACAACA-3’ 5’-TTGGCTGTGCCAGTTCAGAGT-3’
Tnsfs10 5’-CTTCACCAACGAGATGAAGCAG-3’ 5’-TCCGTCTTTGAGAAGCAAGCTA-3’
8. 腫瘍組織切片作製
腫瘍組織塊を O.C.T. compound に包埋した凍結ブロックサンプルについ
て、クライオスタット (LEICA BIOSYSTEMS 社、型番: LEICA CM1850) を
用いて 10 m の厚さの組織切片を切り出し、剥離防止処理がなされたスラ
イドグラスに張り付けた。スライドグラスを充分に乾燥させた後、-30°C
のアセトン溶液に浸し氷上で 30 分間固定した。30 分後、スライドグラス
を風乾させ、PBS で 3 回洗浄を行った。組織切片をブロッキング液 (10
g/ml アイソタイプ抗体、5% ウシ血清アルブミン / PBS) に浸し、室温で
1 時間静置した。1 時間後、蛍光標識抗体を希釈倍率が 1/100 になるように
加え、4°C で一晩反応させた。蛍光標識後、PBS で 3 回洗浄を行い、Prolong
Gold を組織上に添加し、カバーグラスをかぶせて封入を行った。

17
作製した組織切片は共焦点レーザー顕微鏡 (Zeiss 社、型番: LSM 510
META) を用いて観察した。
9. in vitro OT-I proliferation assay
マウス脾臓から CD8+
DC isolation kit を用いて CD8+
DC を単離し、5 x
104 /well の細胞数で平底 96 well プレートに播種した。その後、各種濃度の
OVA を加え、50 g/ml Poly(I:C)の存在下、および非存在下で 3 時間培養し
た。ある実験においては OVA + Poly(I:C)に加え、100 ng/ml IL-12p70 の併
用添加も行った。3 時間後、OT-I Tg マウスの脾臓から mouse CD8a (Ly-2)
MicroBeads を用いて CD8 陽性細胞 (その大部分が CD8+
T cell) を単離した。
単離した OT-I cell は 1 M CFSE / PBS に懸濁し 37°C、10 分間インキュベ
ーションし蛍光標識した後、2.5 x 104
/well の細胞数に調整し 96 well プレ
ート内で CD8+
DC と共培養した。またある実験においては、OT-II Tg マ
ウスの脾臓から mouse CD4 (L3T4) MicroBeads を用いて CD4 陽性細胞 (そ
の大部分が CD4+
T cell) を単離し、2.5 x 104
/well の細胞数に調整した後、
CD8+
DC と CFSE 標識 OT-I cell を共培養している 96 well プレートに更に
加えることで 3 種細胞培養を行った。
60 時間の共培養の後、蛍光標識抗体により CD8、TCR V2 分子を染色
した。その後 OT-I cell (CFSE+
CD8+
TCR V2+
cells) における CFSE の蛍光
強度の減弱度を FACS を用い解析し、それを基に OT-I cell の OVA 抗原特
異的分裂レベルを評価した。また FACS 解析の際に、細胞培養上清を回収
し、上清中の IFN-濃度を ELISA kit を用いて解析した。
10. 統計処理
2 群間の有意差検定では、Student’s t-検定を用い p 値を求めた。また多重
比較検定は、統計解析ソフト Prism4 を用い行った。パラメトリック解析が
可能な場合には、one-way analysis of variance (ANOVA)の後に Bonferroni’s
test を行い p 値を求めた。パラメトリック解析が適応できない場合には、
Kluskal-Wallis test の後に Dunn’s multiple comparison test を行い p 値を求め
た。(*p < 0.05)
実験結果
1. BATF3 は Poly(I:C)による CTL 依存性腫瘍退縮を担う
(i). WT1 陽性腫瘍治療モデル:

18
まず始めに CD8+
DC の分化に関わる転写因子 BATF3 の有無が Poly(I:C)
誘導性腫瘍退縮に影響するか否かを、WT1-C1498 移植マウスモデルにおい
て評価した。WT1-C1498 担がん WT マウスでは、無治療 (PBS 投与群) の
場合は腫瘍が増大してゆくのに対し、Poly(I:C)投与を行うことで腫瘍退縮
が誘導された (Fig 1-1A)。この Poly(I:C)による腫瘍退縮効果は、CD8陽性
細胞を除去することで完全にキャンセルされることから、CD8+
T cell が中
心的役割を担っていることが示された (Fig 1-1B)。しかし、WT1-C1498 担
がん Batf3-/-マウスでは、Poly(I:C)による腫瘍退縮が全く誘導されなかった
(Fig 1-1C)。なお、NK1.1 陽性細胞を除去しても WT マウスでは Poly(I:C)
により腫瘍退縮が誘導されることから、NK cell 及び NKT cell は腫瘍退縮
には関与しないことが示された (Figure 1-1D)。
WT マウスでは Poly(I:C)治療後に腫瘍内浸潤 CD8+
T cell の割合が増加
しているのに対し、Batf3-/-マウスでは浸潤増加は認められなかった (Figure
1-1E)。また、WT1 抗原中に含まれるエピトープ領域のひとつであり、MHC
class I 分子に結合する Db126 ペプチドを Poly(I:C)と併用投与した後、脾細
胞を採取し Db126 ペプチドで再刺激を行うと、WT マウスでは WT1 特異
的 CD8+
T cell の増加が認められたのに対し、Batf3-/-マウスではそのような
増加は認められなかった (Fig 1-1F, G)。
以上の結果より、Poly(I:C)により誘導される抗原特異的 CD8+
T cell が
WT1-C1498 腫瘍退縮を担っており、その CD8+
T cell 誘導には BATF3 が不
可欠であることが示された。
(ii) OVA 陽性腫瘍治療モデル:
次に、別種の腫瘍細胞株である EG7 においても Poly(I:C)誘導性腫瘍退
縮に対する BATF3 の関与を評価することとした。EG7-担がん WT マウス
に腫瘍抗原である OVA を単独投与しても、腫瘍退縮および腫瘍内におけ
る OVA 特異的 CD8+
T cell 増加が誘導されないのに対し、OVA に加え
Poly(I:C)を併用投与すると、強力な腫瘍退縮および OVA 特異的 CD8+
T cell
の腫瘍内浸潤が誘導された (Fig 1-2A)。
次に、EG7-担がん WT および Batf3-/-マウスに対し Poly(I:C)治療を行っ
た際の腫瘍退縮および抗原特異的 CD8+
T cell 増殖強度を評価した。WT お
よび Batf3-/-マウスともに無治療の場合 (PBS 群) では腫瘍は増大してゆく
が、Batf3-/-マウスでは、より強い腫瘍増殖が認められた (Fig 1-2B 左図)。
Poly(I:C)単独投与および OVA との併用投与では、WT、Batf3-/-マウスとも
に無治療群と比較して腫瘍増殖抑制効果が認められたが、Day 16 時点での

19
腫瘍体積は WT マウスと比較して Batf3-/-マウスで有意に大きかった (Fig
1-2B 中央図、右図)。また、Day 16 時点での脾臓内の OVA 特異的 CD8+
T cell
の存在割合を解析したところ、OVA + Poly(I:C)投与によるOVA特異的CD8+
T cell 誘導レベルが Batf3-/-マウスでは WT マウスよりも低下していること
が示された (Fig 1-2C)。
以上の結果より、EG7 担がんマウスでは BATF3 が欠損することで無治
療時の基礎的な抗腫瘍能が低下し、加えて Poly(I:C)による抗原特異的 CD8+
T cell 誘導能も低下するため、十分な腫瘍退縮が誘導されないことが示さ
れた。
(iii) リンパ組織における CD8+ DC サブセットの存在割合:
Batf3-/マウス体内では CD8
+ DC の数が減少することが知られている
32-34。そのため、Batf3-/マウスにおける Poly(I:C)誘導性抗腫瘍応答の低下は、
クロスプレゼンテーションを担う CD8+
DC の数が減少していることに起
因している可能性が考えられた。よって無刺激時および Poly(I:C)刺激時の
CD8+
DC の存在割合を EG7-担がん WT マウスと Batf3-/-マウスにおいて比
較することとした。脾臓および所属リンパ節 (Draining lymph nodes; DLN)
である鼠径リンパ節に局在する CD8+
DC の割合を FACS により解析した
結果 (Fig 1-3A)、無刺激時の Batf3-/-マウスでは、過去の報告と一致して
CD8+
DC の減少が認められた。また、Poly(I:C)刺激時においてもその存在
割合は WT マウスと比較して低かった (Fig 1-3B, C)。
CD8+
DC 中には発現マーカーおよび表現型により更に複数のサブセ
ットが含まれていることが知られている。その中で特にクロスプレゼンテ
ーション能が高いサブセットは XCR1 や CLEC9A といった分子を発現して
おり、これら分子は human CD141+
DC にも同様に発現していることが知ら
れている 28-30,38,39。そのため CD8+
DC におけるこれら分子の発現レベル
について解析を行った。その結果、Batf3-/-マウスにおいては XCR1
hi
CLEC9A+
CD8+
DC サブセットが著しく減少していることが明らかとなっ
た (Fig 1-3D)。
以上より、Batf3-/-マウスにおける Poly(I:C)誘導性抗腫瘍応答の低下は、
機能的な CD8+
DC サブセットの消失に起因している可能性が考えられた。
20

21
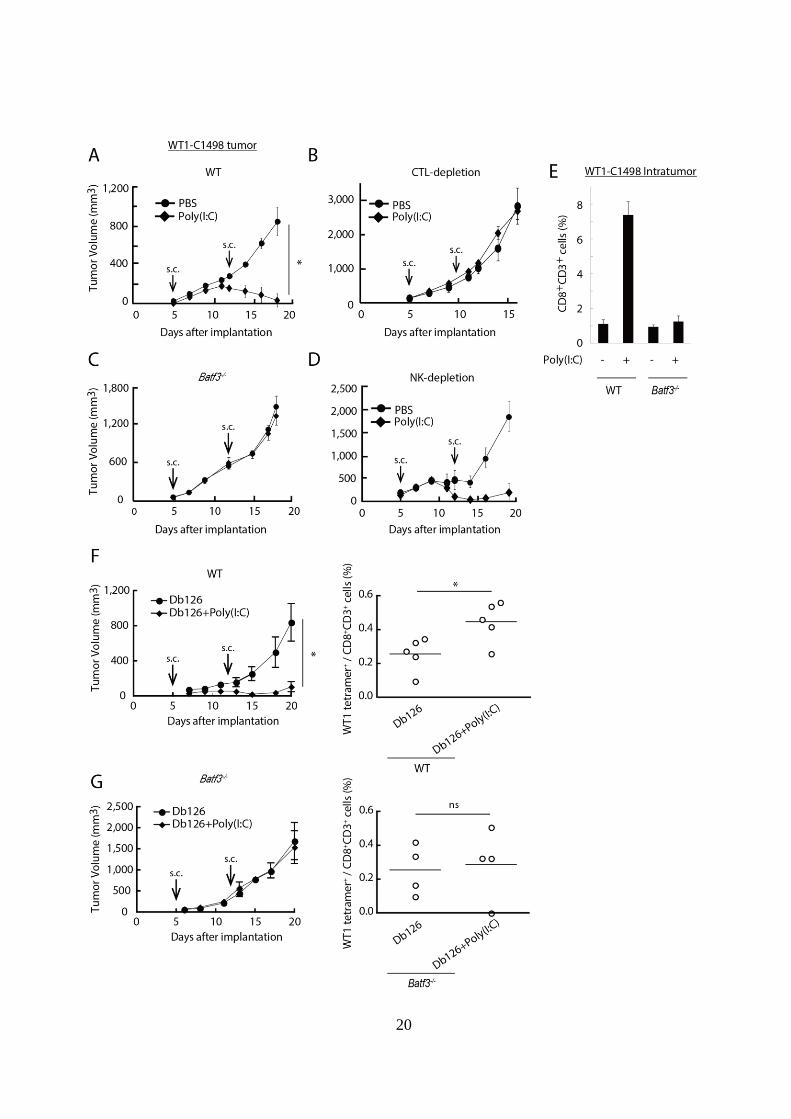
Figure 1-1. Poly(I:C)は BATF3 依存性に WT1-C1498 腫瘍退縮を誘導する。
(A, C): WT および Batf3-/-マウスに WT1-C1498 を移植し、腫瘍移植後 Day 5、
12 に PBS または Poly(I:C)投与を行い、腫瘍体積の推移を測定した。
(B, D): WT マウスに WT1-C1498 を移植した後、Day 5、12 に PBS または
Poly(I:C)投与を行った。(B)においては Day 4、11 に CD8陽性細胞の除去
操作を、(D)においては NK1.1 陽性細胞の除去操作を行い、腫瘍体積の推
移を測定した。
(E): (A)、(C)と同様の処置をしたマウスから、腫瘍移植後 Day 19 に腫瘍組
織を採取し、FACS により全腫瘍組織細胞中の CD8+
T cell の割合を解析し
た。
(F, G): WT1-C1498 担がん WT および Batf3-/-マウスにおいて、瘍移植後 Day
5、12 に Db126 ペプチド単独、または Db126 + Poly(I:C)併用投与を行い、
腫瘍体積の推移を測定した (左図)。WT マウスでは Day 20 に、Batf3-/-マウ
スでは Day 21 に脾臓を採取し、Db126 ペプチド存在下で 5 日間培養した。
その 5 日後に Tetramer assay を行い CD8+
T cell 中の WT1 tetramer+
cell の割
合を解析した (右図)。
実験には各群 3-4 匹のマウスを用いた。*p < 0.05、 ns; 統計学的有意差無
し。
22
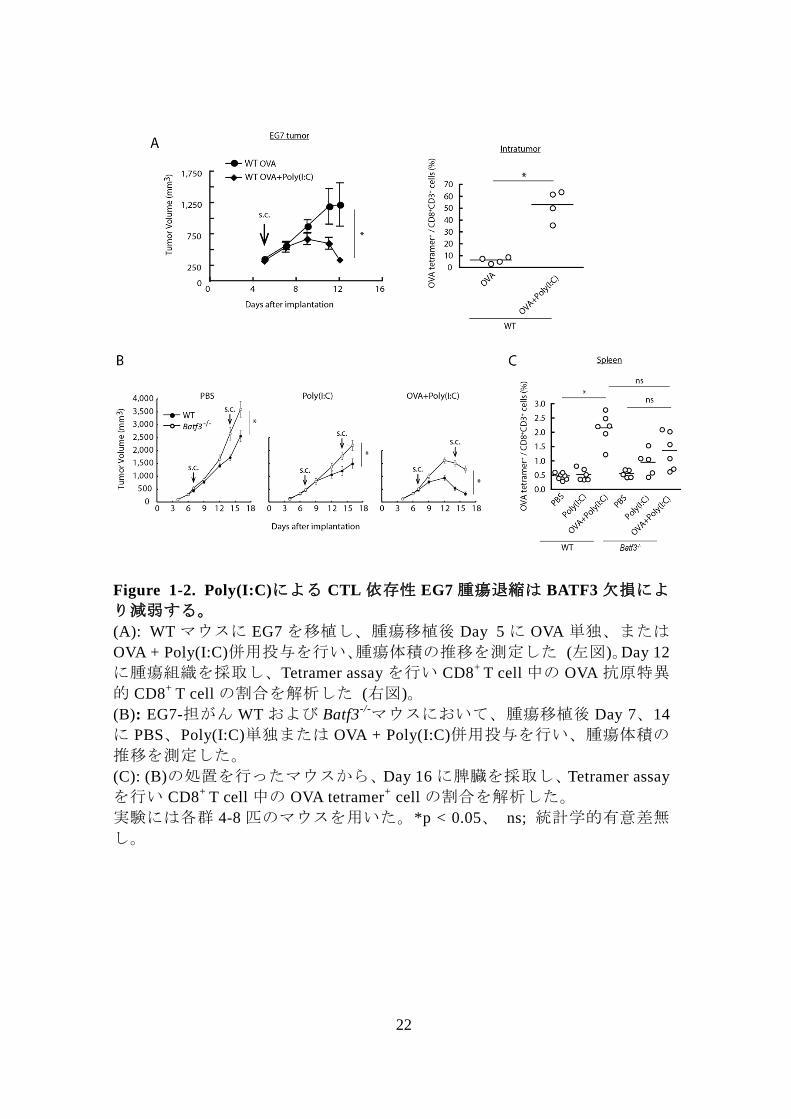
Figure 1-2. Poly(I:C)による CTL 依存性 EG7 腫瘍退縮は BATF3 欠損によ
り減弱する。
(A): WT マウスに EG7 を移植し、腫瘍移植後 Day 5 に OVA 単独、または
OVA + Poly(I:C)併用投与を行い、腫瘍体積の推移を測定した (左図)。Day 12
に腫瘍組織を採取し、Tetramer assay を行い CD8+
T cell 中の OVA 抗原特異
的 CD8+
T cell の割合を解析した (右図)。
(B): EG7-担がん WT および Batf3-/-マウスにおいて、腫瘍移植後 Day 7、14
に PBS、Poly(I:C)単独または OVA + Poly(I:C)併用投与を行い、腫瘍体積の
推移を測定した。
(C): (B)の処置を行ったマウスから、Day 16 に脾臓を採取し、Tetramer assay
を行い CD8+
T cell 中の OVA tetramer+ cell の割合を解析した。
実験には各群 4-8 匹のマウスを用いた。*p < 0.05、 ns; 統計学的有意差無
し。
23
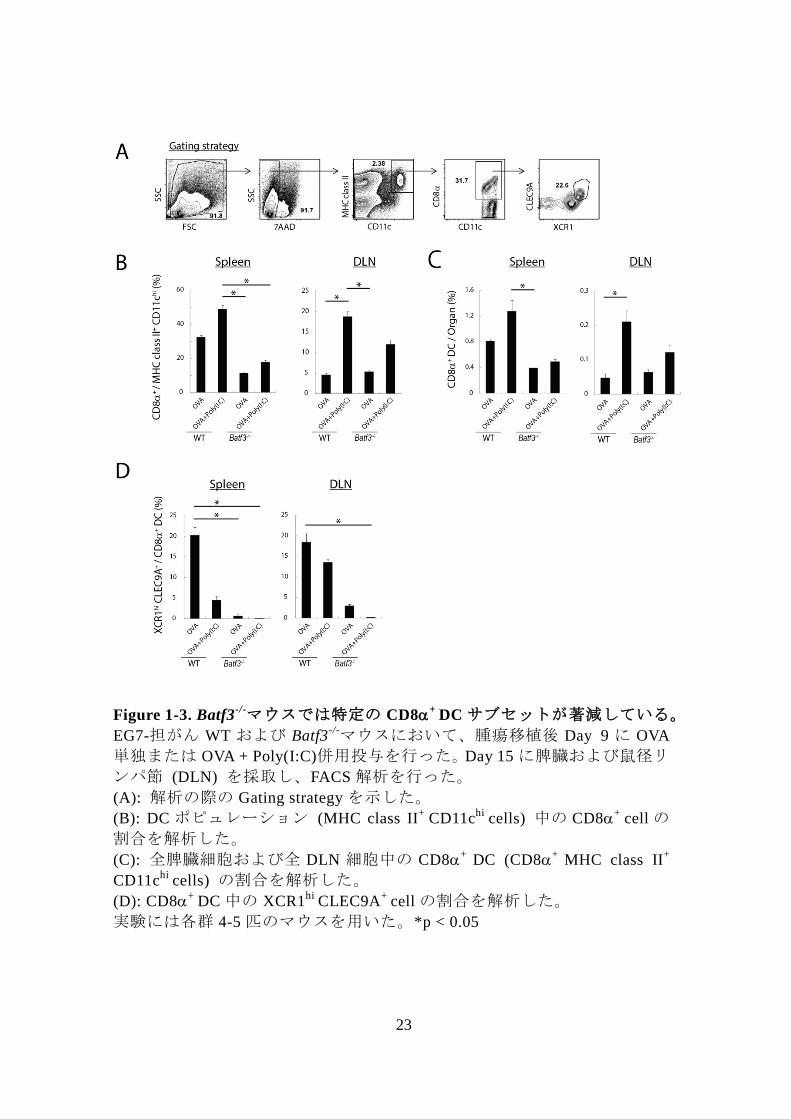
Figure 1-3. Batf3-/-マウスでは特定の CD8
+ DC サブセットが著減している。
EG7-担がん WT および Batf3-/-マウスにおいて、腫瘍移植後 Day 9 に OVA
単独または OVA + Poly(I:C)併用投与を行った。Day 15 に脾臓および鼠径リ
ンパ節 (DLN) を採取し、FACS 解析を行った。
(A): 解析の際の Gating strategy を示した。
(B): DC ポピュレーション (MHC class II+
CD11chi
cells) 中の CD8+
cell の
割合を解析した。
(C): 全脾臓細胞および全 DLN 細胞中の CD8+
DC (CD8+
MHC class II+
CD11chi
cells) の割合を解析した。
(D): CD8+
DC 中の XCR1hi
CLEC9A+
cell の割合を解析した。
実験には各群 4-5 匹のマウスを用いた。*p < 0.05

24
2. BATF3 陽性 CD8+
DC は TLR3 シグナル依存的に細胞性免疫を誘導する
前項の結果より、BATF3 依存的に分化誘導する CD8+
CD サブセットが
Poly(I:C)誘導性 CTL 活性化に重要である可能性が示唆された。よって本項
では、BATF3 による制御が、CD8+
DC の CTL 誘導に関わる表現型の獲得
に影響を与えるか否かを評価した。
(i) BATF3 陽性 CD8+ DC の poly(I:C)誘導性クロスプレゼンテーション能
の解析:
まず、非担がん WT および Batf3-/-マウスに OVA と Poly(I:C)を併用投与
した際の、in vivo における OVA 特異的 CD8+
T cell 誘導能を評価した。そ
の結果、Fig 1-2C の EG7-担がんマウスでの結果と異なり、非担がんマウス
では、BATF3 の欠損により Poly(I:C)による OVA 特異的 CD8+
T cell 誘導が
完全にキャンセルされた (Fig 2-1A)。次に、より直接的に CD8+
DC の
Poly(I:C) 誘導性クロスプレゼンテーション能を評価するため OT-I
proliferation assay を行った。OT-I cell は OVA 抗原を認識する TCR を強制
発現させた CD8+
T cell であるため、OVA 存在下で CD8+
DC と共培養した
際の OT-I cell の分裂レベルにより、CD8+
DC のクロスプレゼンテーショ
ン能を評価することが出来る。実験の結果、OVA 抗原が存在しない場合
(OVA 0 g/ml) は、たとえ Poly(I:C)刺激を行ったとしても CD8+
DC は OT-I
cell 分裂を誘導しなかった (Fig 2-1B 左図)。一方、添加した OVA 量が多い
場合 (OVA 100 g/ml) は、WT 由来 CD8+
DC は Poly(I:C)刺激により十分
なクロスプレゼンテーション能を発揮し、また、Batf3-/-由来 CD8
+ DC も
WT と同程度のクロスプレゼンテーション能を発揮した。なお、Tlr3-/-由来
CD8+
DC では、Poly(I:C)誘導性クロスプレゼンテーションが全く認められ
なかったことから、この機構は細胞質内 MAVS 経路ではなく、
TLR3-TICAM1 経路に依存していることが分かる (Fig 2-1B 右図)。更に
OVA を 10 g/ml の濃度で添加した場合も検証すると、 Batf3-/-
CD8+
DC
の Poly(I:C)誘導性クロスプレゼンテーション能は WT 由来のものと比較し
て低かった (Fig 2-1B 中央図)。また、この OVA 濃度下で共培養した後の、
培養上清液中の IFN-濃度を測定することで、クロスプレゼンテーション
を受けた OT-I cell の IFN-産生能を評価した。その結果、Batf3-/-
CD8+
DC
と共培養した際の OT-I cell からの Poly(I:C)誘導性 IFN-産生量は、WT の
場合と比較して著しく低いことが示された (Fig 2-1C)。以上の結果は、抗
原量が少ない状況で CD8+
T cell が増殖・活性化するためには、BATF3 陽性
CD8+
DC サブセットによるクロスプレゼンテーションが必要であること

25
を示唆している。
Batf3-/-
CD8+
DC でクロスプレゼンテーション能が低かった理由として、
WT と比較して細胞生存期間が短縮している可能性を考えた。そのため、
WT および Batf3-/-マウスから CD8
+ DC を単離し、PBS および Poly(I:C)刺
激後の細胞生存率を経時的に解析した。その結果、WT、Batf3-/-
CD8+
DC
共に、Poly(I:C)刺激により生存期間が延長することが、また、Batf3-/-
CD8+
DC では WT と比較して生存期間が延長する傾向が認められた (Fig 2-2)。
以上の結果より、Batf3-/-
CD8+
DC の低クロスプレゼンテーション能は細
胞生存期間に起因するものではないことが示された。
(ii) BATF3 陽性 CD8+ DC の表現型解析:
次に、WT および Batf3-/-
CD8+
DC において、生存期間以外にも Poly(I:C)
による CTL 誘導に関連する表現型に差異があるかを解析した。まず、
Poly(I:C)認識に関わる PRR 群の CD8+
DC における遺伝子発現について評
価した。具体的には Tlr3、Ticam1、Ifih1 (MDA5 遺伝子)、Ddx58 (RIG-I 遺
伝子)、Mavs について解析した。その結果、Batf3-/ -
CD8+
DC では WT と比
較して Tlr3 発現が低下しており、対照的に Ddx58 発現が上昇していた (Fig
2-3A)。また TLR3 についてタンパクレベルでの発現量を FACS により解析
したところ、遺伝子発現と一致し、Batf3-/-
CD8+
DC では TLR3 発現量が
低いことが明らかとなった(Fig 2-3B)。
更に、CTL 誘導に関わる液性因子の遺伝子群について、Poly(I:C)刺激後
の CD8+
DC における発現量を評価した。その結果、Batf3-/-
CD8+
DC で
は WT と比較して Poly(I:C)誘導性 Ifnb、Il 28 (IFN-) 発現が上昇している
傾向が認められた。対照的に、Poly(I:C)依存性 Il12b 発現は著しく低下して
いた (Fig 2-3C)。そのため、In vivo においても Poly(I:C)投与時の IL-12 産
生能を評価することとした。WT および Batf3-/-マウスに Poly(I:C)を投与し
た後の血中 IL-12p40 濃度を経時的に測定したところ、WT では血中濃度は
上昇したのに対し、Batf3-/-マウスでの血中濃度は非常に低かった (Fig
2-3D)。また、IL-12 を構成する 2 つのサブユニットの IL-12p35、IL-12p40
の細胞内染色を行うことで、脾臓内 CD8+
DC における Poly(I:C)誘導性
IL-12 産生能を評価した。その結果、WT マウスの脾臓内 CD8+
DC におい
ては Poly(I:C)に応答して IL-12p35+
IL-12p40+細胞の割合が増加するのに対
し、Batf3-/-マウスではそのような増加は認められなかった (Fig 2-3E)。

26
次に、BATF3 陽性および陰性 CD8+
DC における TLR3 発現量と Poly(I:C)
誘導性 IL-12 産生能に相関関係があるかを評価した。まず、WT および
Batf3-/-マウスに Poly(I:C)投与を行った後、脾臓内の TLR3
+ CD8
+ DC にお
ける TLR3 発現強度を FACS により測定した。その結果、Batf3-/-マウスで
は TLR3hi のポピュレーションが著しく減少していることが明らかとなっ
た。そこで WT マウスにおいて、TLR3lo-med のポピュレーション (BATF3
陰性 CD8+
DC) と TLR3hiのポピュレーション (BATF3 陽性 CD8
+ DC) を
区別し、IL-12p40 の細胞内染色を行った。その結果、IL-12p40 産生能が高
いのは TLR3hi の BATF3 陽性 CD8
+ DC であることが明らかとなった (Fig
2-4A)。
また、BATF3 陽性および陰性 CD8+
DC における XCR1 発現量と IL-12
産生能の相関関係も評価した。FACS 解析の結果、Batf3-/-マウスでは XCR1
hi
のポピュレーションが著しく減少していたため、WT マウスにおいて
XCR1negative-medのポピュレーション (BATF3陰性 CD8
+ DC) と XCR1
hiのポ
ピュレーション (BATF3 陽性 CD8+
DC) を区別し、Poly(I:C)投与後の
IL-12p40 の細胞内染色を行った。その結果、IL-12p40 産生能が高いのは
XCR1hi の BATF3 陽性 CD8
+ DC であることが明らかとなった (Fig 2-4B)。
また、WT マウスの CD8+
DC における TLR3 と XCR1 の発現レベルを
測定したところ、XCR1 発現が高いポピュレーションほど TLR3 発現も比
例して高いことが明らかとなり (Fig 2-4C)、BATF3陽性CD8+
DCは TLR3、
XCR1 発現が高く、それに相関して高い Poly(I:C)誘導性 IL-12 産生能を有
することが示された。
以上より、TLR3 発現が高く、Poly(I:C)誘導性クロスプレゼンテーション
や IL-12 産生を担っている特定の CD8+
DC サブセットの欠損が、Batf3-/-
マウスで十分に腫瘍退縮を誘導出来ない要因となっていることが示唆され
た。
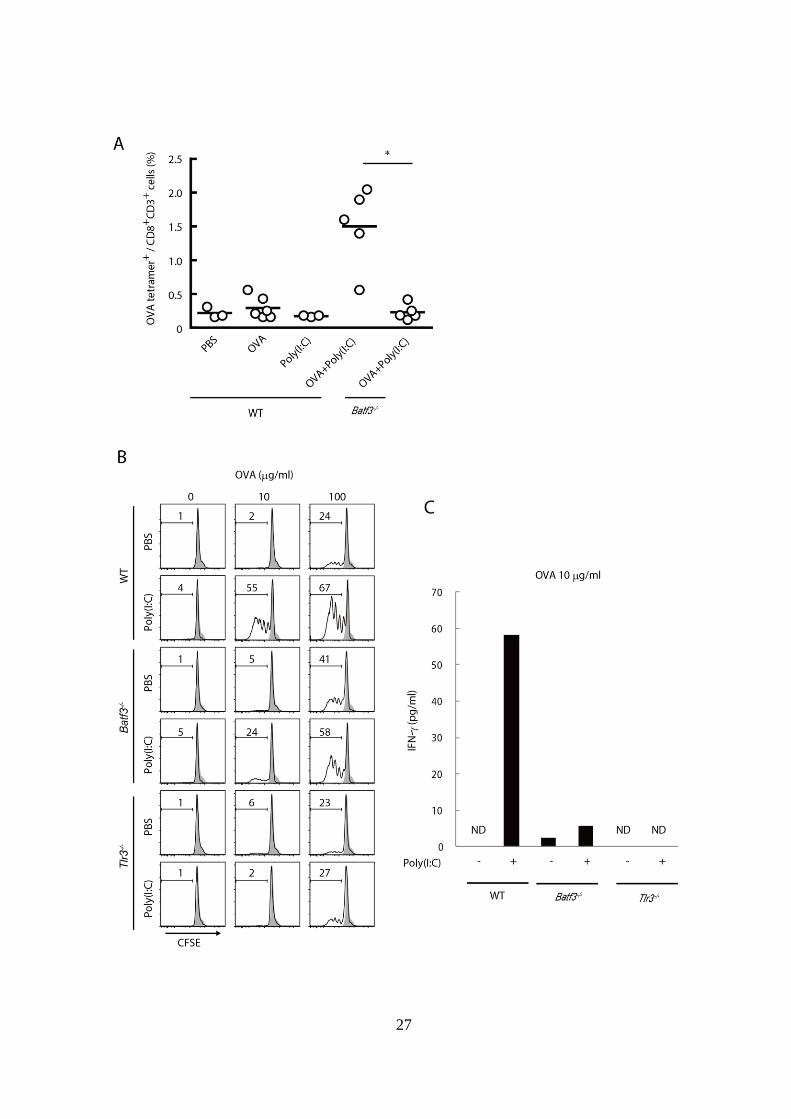
27
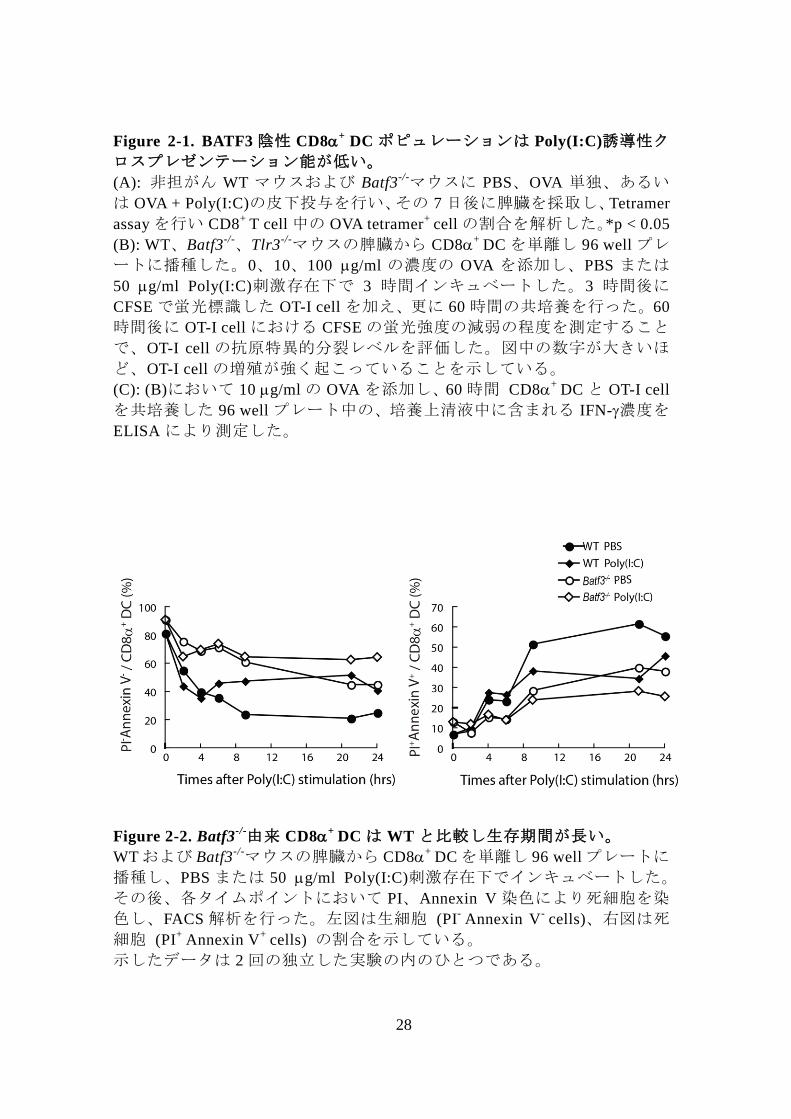
28
Figure 2-1. BATF3 陰性 CD8+
DC ポピュレーションは Poly(I:C)誘導性ク
ロスプレゼンテーション能が低い。
(A): 非担がん WT マウスおよび Batf3-/-マウスに PBS、OVA 単独、あるい
は OVA + Poly(I:C)の皮下投与を行い、その 7 日後に脾臓を採取し、Tetramer
assay を行い CD8+
T cell 中の OVA tetramer+
cell の割合を解析した。*p < 0.05
(B): WT、Batf3-/-、Tlr3
-/-マウスの脾臓から CD8+
DC を単離し 96 well プレ
ートに播種した。0、10、100 g/ml の濃度の OVA を添加し、PBS または
50 g/ml Poly(I:C)刺激存在下で 3 時間インキュベートした。3 時間後に
CFSE で蛍光標識した OT-I cell を加え、更に 60 時間の共培養を行った。60
時間後に OT-I cell における CFSE の蛍光強度の減弱の程度を測定すること
で、OT-I cell の抗原特異的分裂レベルを評価した。図中の数字が大きいほ
ど、OT-I cell の増殖が強く起こっていることを示している。
(C): (B)において 10 g/ml の OVA を添加し、60 時間 CD8+
DC と OT-I cell
を共培養した 96 well プレート中の、培養上清液中に含まれる IFN-濃度を
ELISA により測定した。
Figure 2-2. Batf3-/-由来 CD8
+ DC は WT と比較し生存期間が長い。
WT および Batf3-/-マウスの脾臓から CD8
+ DC を単離し 96 well プレートに
播種し、PBS または 50 g/ml Poly(I:C)刺激存在下でインキュベートした。
その後、各タイムポイントにおいて PI、Annexin V 染色により死細胞を染
色し、FACS 解析を行った。左図は生細胞 (PI- Annexin V
- cells)、右図は死
細胞 (PI+
Annexin V+
cells) の割合を示している。
示したデータは 2 回の独立した実験の内のひとつである。
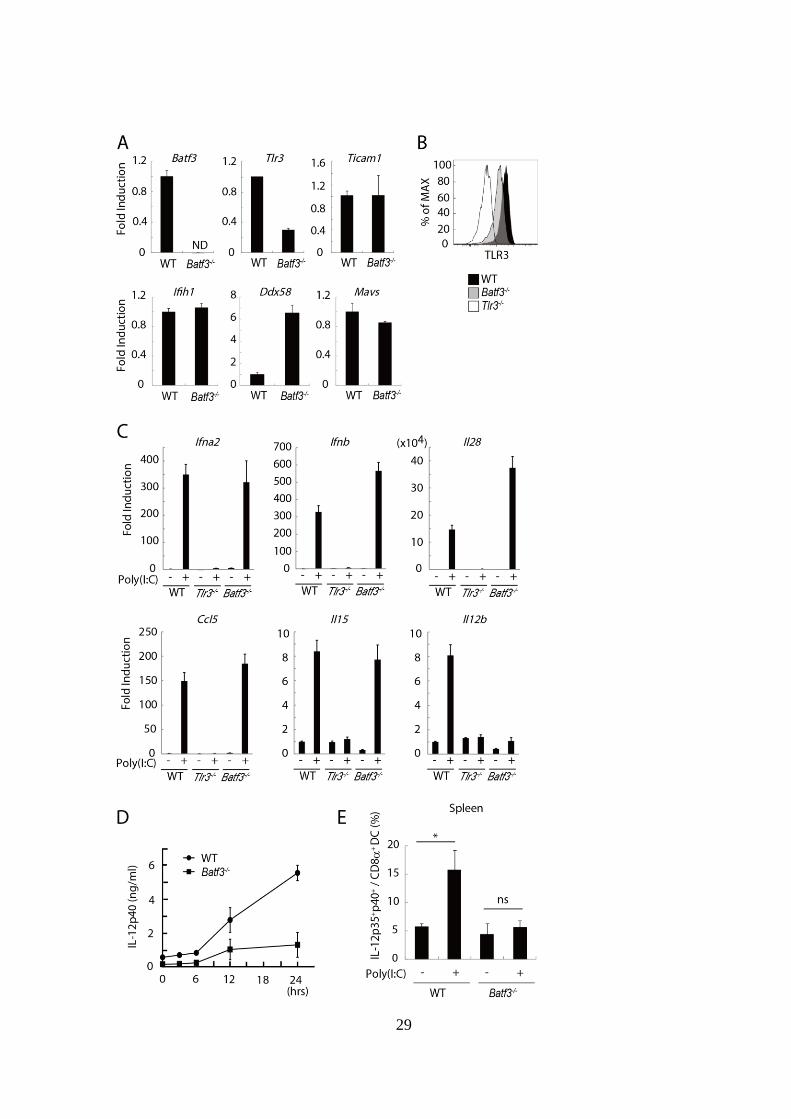
29

30
Figure 2-3. BATF3 陰性 CD8+
DC ポピュレーションは TLR3 発現が低く
Poly(I:C)誘導性 IL-12 産生能が欠如している。
(A): WT および Batf3-/-マウスの脾臓から CD8
+ DC を単離し、mRNA を採
取した。その後、定量 PCR 法により各遺伝子の発現レベルを解析した。
(B): WT、Batf3-/-、Tlr3
-/-マウスの脾臓から CD8+
DC を単離し、FASC によ
り TLR3 分子の発現レベルを解析した。
(C): WT、Batf3-/-、Tlr3
-/-マウスの脾臓から CD8+
DC を単離し 96 well プレ
ートに播種し、PBS または 50 g/ml Poly(I:C)刺激存在下で 4 時間インキュ
ベートした。その後 mRNA を採取し、各遺伝子の発現レベルを解析した。
(D): WT 及び Batf3-/-マウスに 50 g Poly(I:C)を皮下投与した後、各タイムポ
イントにおいて尾静脈より採血を行い、血清中の IL-12p40 濃度を ELISA
により測定した。
(E): WT 及び Batf3-/-マウスに PBS および 50 g Poly(I:C)を皮下投与した 12
時間後に脾臓細胞を採取した。採取した脾細胞を 96 well プレートに播種し、
Blefeldin A 存在下で 4 時間インキュベーションした。その後 FACS により
CD8+DC 中の IL-12p35+
IL-12p40+
cell の割合を解析した。
(D)、(E)の実験においては各群 3-6 匹のマウスを用いた。*p < 0.05、 ns; 統
計学的有意差無し。示したデータは 2 回の独立した実験の内のひとつであ
る。
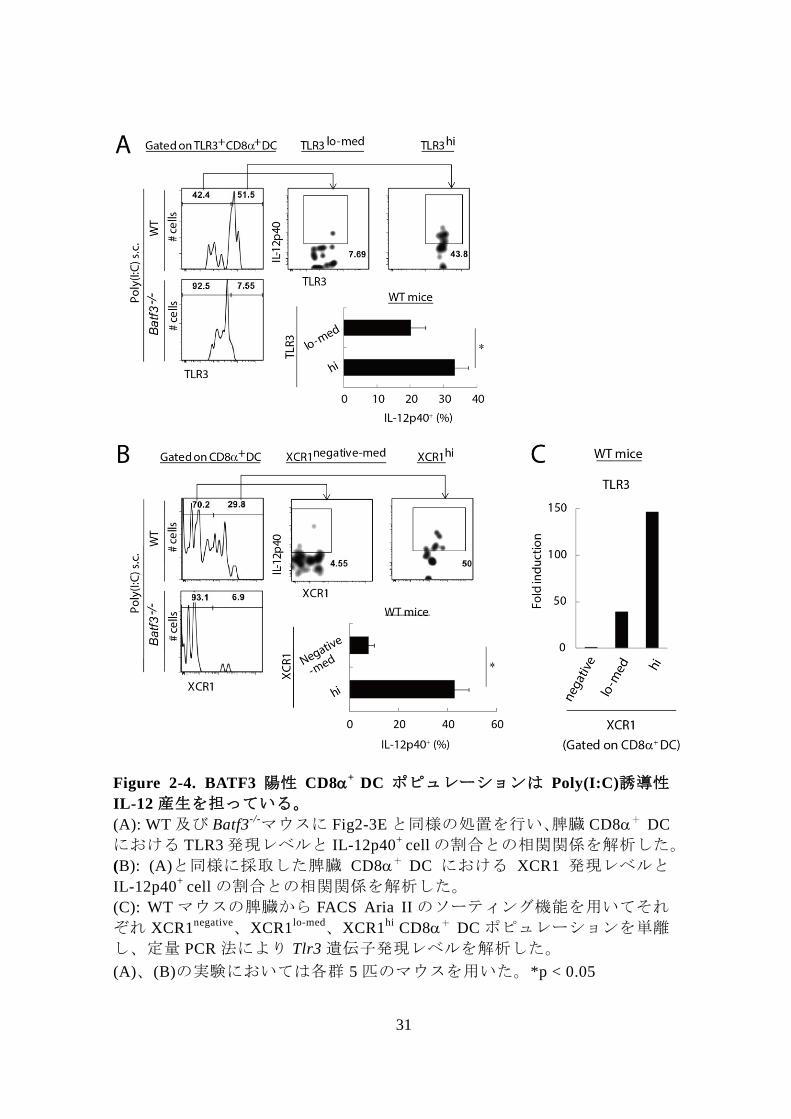
31
Figure 2-4. BATF3 陽性 CD8+
DC ポピュレーションは Poly(I:C)誘導性
IL-12 産生を担っている。
(A): WT 及び Batf3-/-マウスに Fig2-3E と同様の処置を行い、脾臓 CD8+
DC
における TLR3 発現レベルと IL-12p40+
cell の割合との相関関係を解析した。
(B): (A)と同様に採取した脾臓 CD8+ DC における XCR1 発現レベルと
IL-12p40+
cell の割合との相関関係を解析した。
(C): WT マウスの脾臓から FACS Aria II のソーティング機能を用いてそれ
ぞれ XCR1negative、XCR1
lo-med、XCR1hi
CD8+ DC ポピュレーションを単離
し、定量 PCR 法により Tlr3 遺伝子発現レベルを解析した。
(A)、(B)の実験においては各群 5 匹のマウスを用いた。*p < 0.05

32
3. BATF3 は Poly(I:C)による腫瘍内 CTL 浸潤を担う
次に、BATF3 の欠損が Poly(I:C)治療時の腫瘍内微小環境変化に影響する
か否かを評価した。まず、PBS、Poly(I:C)単独または OVA + Poly(I:C)併用
投与を行った EG7-担がん WT および Batf3-/-マウスの腫瘍組織を採取し、
組織切片の観察を行った。組織切片の Hematoxylin-Eosin (HE) 染色を行っ
たところ、腫瘍組織はそれぞれ構造の異なる辺縁部と中心部により構成さ
れていることが観察された (Fig 3-1A)。この辺縁部および中心部において
CD8分子を免疫染色したところ、WT マウスでは Poly(I:C)投与により両領
域に CD8陽性細胞が強く浸潤してくることが確認された。しかし、Batf3-/-
マウスではこのような Poly(I:C)誘導性の浸潤は非常に弱かった (Fig 3-1B,
C)。また、CD8分子と同時に TCR V2 分子の染色を行ったところ、Batf3-/-
マウスでは Poly(I:C)誘導性の TCR V2+
CD8+
T cell 腫瘍内浸潤が完全にキ
ャンセルされていることが確認された (Fig 3-1D)。同様の結果は FACS 解
析によっても確認されたことから (Fig 3-1E)、Poly(I:C)による CD8+
T cell
の腫瘍内浸潤には BATF3 が必要であることが示された。
次に腫瘍組織における Poly(I:C)誘導性サイトカイン、ケモカイン産生を
評価した。前項において、Batf3-/-マウスでは Poly(I:C)誘導性 IL-12 産生能
が著減していることが示されたため、腫瘍組織内においても IL-12 産生を
解析した。その結果、遺伝子発現、タンパク量共に Batf3-/-マウスでは OVA
+ Poly(I:C)併用投与による IL-12p40 産生が誘導されないことが示された
(Fig 3-2A, B)。また、CD8+
T cell のリクルートを担う遊走性因子である Cxcl9,
10, 1140 の遺伝子発現を、また CXCL10 についてはタンパク量も解析した。
その結果、Batf3-/-マウスでは OVA + Poly(I:C)併用投与によるこれらケモカ
インの産生誘導がキャンセルされていた (Fig 3-2A, C)。更に、CTL の細胞
傷害活性に関わる遺伝子群 (Ifng、Gzmb、Prf1、Il12rb1、Il12rb2) の腫瘍内
発現量についても解析を行ったところ、Batf3-/-マウスでは OVA + Poly(I:C)
併用投与によるこれら遺伝子群の発現誘導もキャンセルされていた (Fig
3-2D)。
また、腫瘍内浸潤 CD ポピュレーション中の CD8+
DC の割合を FACS
により解析したところ、WT マウスでは OVA + Poly(I:C)併用投与により
CD8+
DC の割合が上昇したのに対し、Batf3-/-マウスではこのような上昇
は認められなかった (Fig 3-2E)。
腫瘍組織は、腫瘍細胞とレシピエントマウス由来の細胞が混在する組織

33
であるため、Poly(I:C)刺激が直接腫瘍細胞に作用し、腫瘍内微小環境にお
けるサイトカイン産生や細胞死に影響を与えている可能性がある。そのた
め、In vitro において WT1-C1498 および EG7 腫瘍細胞株を Poly(I:C)刺激し
た際の応答性を評価した。まず WT1-C1498 および EG7 における TLR3 発
現レベルを評価したところ、両腫瘍細胞における TLR3 発現レベルは低い
ことが確認された (Fig 3-3A)。次に Ifnb、Cxcl9, 10, 11、Prf1、Gzmb、Ifng
といった遺伝子群の発現レベルを評価したところ、Poly(I:C)によるこれら
遺伝子群の発現誘導は全く認められなかった (Fig 3-3B)。また、Poly(I:C)
刺激 18 時間後の死細胞の割合を評価したところ、Poly(I:C)の有無は死細胞
の割合に影響を与えなかった (Fig 3-3C)。以上より、Poly(I:C)刺激は腫瘍
細胞に直接的には認識されないことが示された。
Fig 3-2E において BATF3 の有無により腫瘍内浸潤 CD8+
DC の割合が異
なっていたことより、他の腫瘍内浸潤ミエロイド細胞の存在割合も影響を
受けている可能性が考えられた。そのため、FACS 解析により OVA 単独お
よび OVA + Poly(I:C)併用投与を行ったマウスの腫瘍内浸潤マクロファージ、
Myeloid-derived suppressor cell (MDSC) の割合を WT と Batf3-/-マウスにお
いて比較した。その結果、OVA 単独群、OVA + Poly(I:C)併用投与群共に腫
瘍内浸潤マクロファージ、MDSC の割合は BATF3 の有無に影響を受けない
ことが示された。また、それぞれの細胞における TLR3 発現を評価したと
ころ、MDSC では発現が認められなかったのに対し、マクロファージでは
低い TLR3 発現が認められたが、この発現レベルも BATF3 の有無に影響を
受けていなかった (Fig 3-4A, B)。
以上の結果より、BATF3 は Poly(I:C)による腫瘍組織からの CD8+
T cell
遊走性因子の放出、CD8+
T cell の腫瘍内浸潤、腫瘍組織内における腫瘍細
胞の傷害といった一連の応答に必要であり、更にこれら応答を腫瘍内浸潤
CD8+
DC が正に制御している可能性が示唆された。
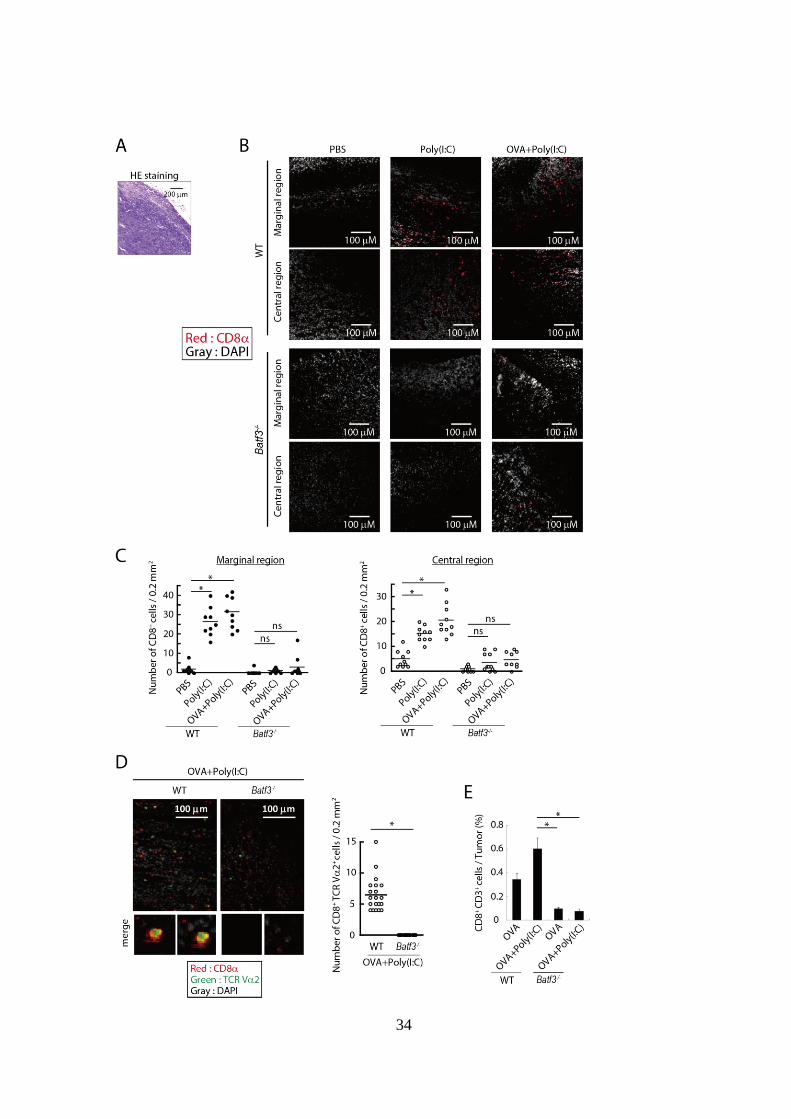
34

35
Figure 3-1. Batf3-/-マウスでは Poly(I:C)による腫瘍内 CD8
+ T cell 浸潤が
誘導されない。
(A, B): EG7-担がん WT および Batf3-/-マウスにおいて、腫瘍移植後 Day 7、
14 に PBS、OVA 単独または OVA + Poly(I:C)併用投与を行った。Day 16 に
腫瘍組織を採取し、腫瘍組織切片を作製した後、共焦点レーザー顕微鏡で
観察した。(A)では HE 染色を行った組織切片の代表的画像を示した。(B)
では各群のマウス由来の腫瘍組織切片においてCD8分子およびDAPI (核)
の蛍光標識を行った。CD8分子は赤、核は灰色で表示した。各群それぞれ
腫瘍組織周辺部および中心部の 2 箇所の代表的画像を示した。
(C): 腫瘍組織内の複数箇所において画像を撮影し、各画像中の一定面積
(0.2 mm2) 内の CD8陽性細胞の数をカウントした結果をグラフ化した。左
図は辺縁領域、右図は中心領域における結果を示している。
(D, E): EG7-担がん WT および Batf3-/-マウスにおいて、腫瘍移植後 Day 9 に
OVA 単独または OVA + Poly(I:C)併用投与を行った。Day 15 に腫瘍組織を
採取し、腫瘍組織切片の観察および FACS 解析を行った。(D)では OVA +
Poly(I:C)を投与された WT および Batf3-/-マウス由来の腫瘍組織切片におい
て CD8、TCR V2 および DAPI (核) の蛍光標識を行った。CD8分子は赤、
TCR V2 分子は緑、核は灰色で表示した。左図は各群の代表的画像を示し
ており、右図は(C)と同様の方法で 0.2 mm2 内の CD8
+ TCR V2
+ 細胞数
をカウントした結果のグラフを示している。(E)では FACS により全腫瘍組
織細胞中の CD8+
T cell の割合を評価した。
実験においては各群 5-6 匹のマウスを用いた。*p < 0.05
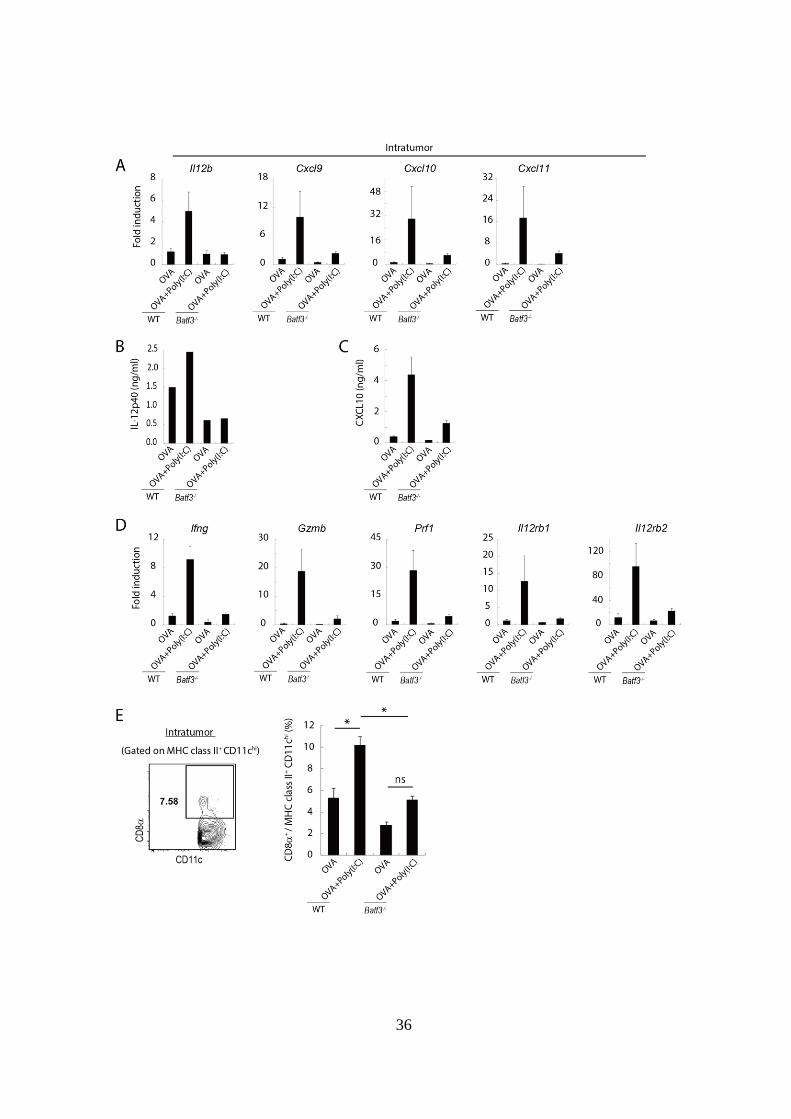
36

37
Figure 3-2. Poly(I:C)による腫瘍内微小環境改善は Batf3-/-マウスでは誘導
されない。
(A): EG7-担がん WT および Batf3-/-マウスにおいて、腫瘍移植後 Day 9、14
に OVA 単独または OVA + Poly(I:C)併用投与を行った。Day 15 に腫瘍組織
における mRNA を採取し、定量 PCR により Il12b および T cell 遊走性因子
の遺伝子発現レベルを解析した。
(B): (A)の実験で腫瘍採取を行った際に、一定体積の腫瘍組織塊を一定量の
メディウム中で細断化することで、腫瘍内浸潤 IL-12 をメディウムに溶解
させ、その存在量を ELISA により測定した。
(C): (B)と同様に腫瘍内浸潤 CXCL10 の存在量を ELISA により測定した。
(D): (A)と同様の操作を行い、腫瘍内浸潤 CD8+
T cell の細胞傷害活性に関
わる遺伝子群の発現レベルを解析した。
(E) 腫瘍組織浸潤 DC 中の CD8+
cell の割合を FASC により解析した。左
図は Gating strategy を示している。右図は各群の平均値をグラフ化して示
している。
実験においては各群 4-7 匹のマウスを用いた。*p < 0.05
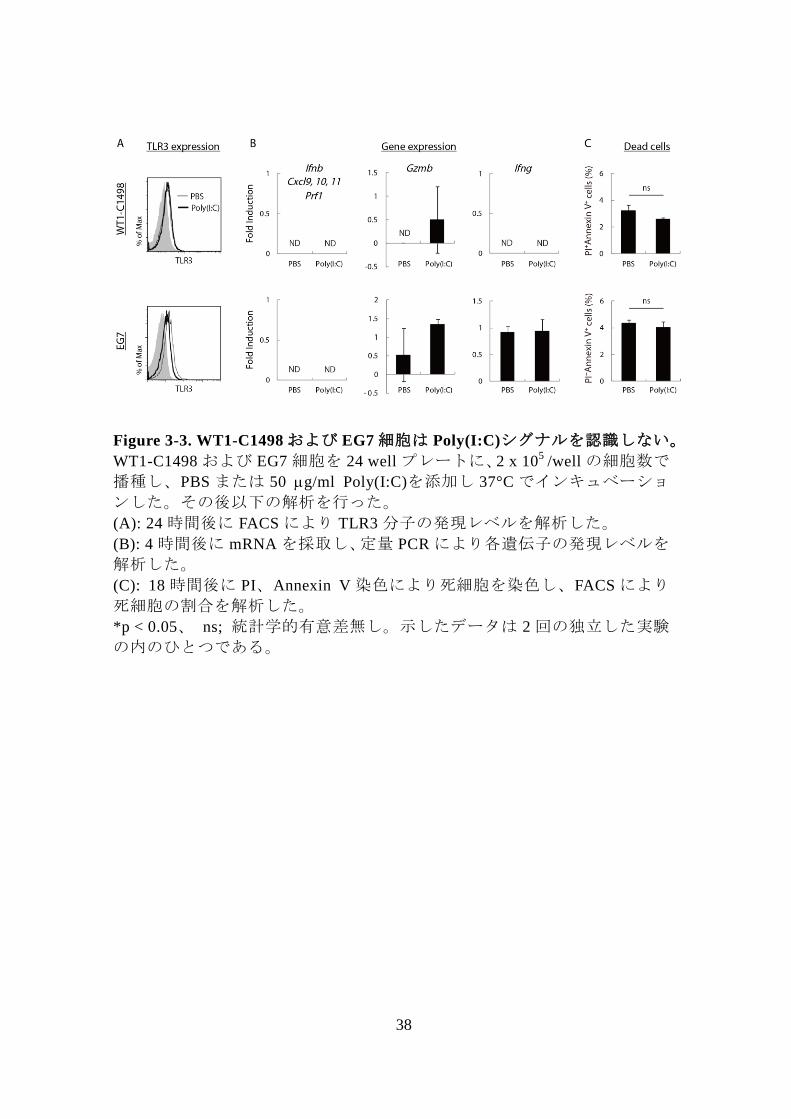
38
Figure 3-3. WT1-C1498 および EG7 細胞は Poly(I:C)シグナルを認識しない。
WT1-C1498 および EG7 細胞を 24 well プレートに、2 x 105
/well の細胞数で
播種し、PBS または 50 g/ml Poly(I:C)を添加し 37°C でインキュベーショ
ンした。その後以下の解析を行った。
(A): 24 時間後に FACS により TLR3 分子の発現レベルを解析した。
(B): 4 時間後に mRNA を採取し、定量 PCR により各遺伝子の発現レベルを
解析した。
(C): 18 時間後に PI、Annexin V 染色により死細胞を染色し、FACS により
死細胞の割合を解析した。
*p < 0.05、 ns; 統計学的有意差無し。示したデータは 2 回の独立した実験
の内のひとつである。
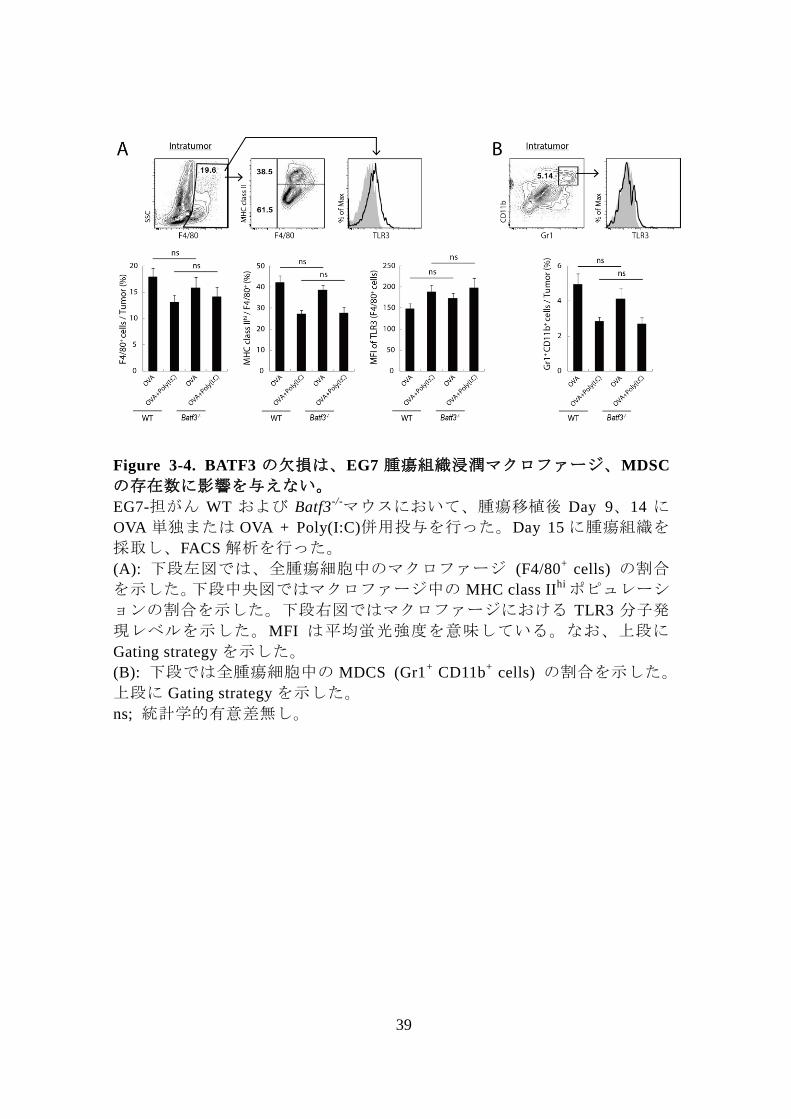
39
Figure 3-4. BATF3 の欠損は、EG7 腫瘍組織浸潤マクロファージ、MDSC
の存在数に影響を与えない。
EG7-担がん WT および Batf3-/-マウスにおいて、腫瘍移植後 Day 9、14 に
OVA 単独または OVA + Poly(I:C)併用投与を行った。Day 15 に腫瘍組織を
採取し、FACS 解析を行った。
(A): 下段左図では、全腫瘍細胞中のマクロファージ (F4/80+
cells) の割合
を示した。下段中央図ではマクロファージ中の MHC class IIhi ポピュレーシ
ョンの割合を示した。下段右図ではマクロファージにおける TLR3 分子発
現レベルを示した。MFI は平均蛍光強度を意味している。なお、上段に
Gating strategy を示した。
(B): 下段では全腫瘍細胞中の MDCS (Gr1+
CD11b+
cells) の割合を示した。
上段に Gating strategy を示した。
ns; 統計学的有意差無し。

40
4. Poly(I:C)による IL-12 産生は CTL 依存性腫瘍退縮を増強する
これまでの結果より、Poly(I:C)治療時に BATF3 が欠損すると、CD8+
DC
を起点とした CTL 活性化誘導機構が十分に機能せず腫瘍退縮が誘導出来
ない可能性が示された。そのため、Poly(I:C)誘導性腫瘍退縮における BATF3
陽性 CD8+
DC の重要性を直接的に評価するため、CD8+
DC の養子移植
実験を行った。その結果、WT1-C1498 担がん Batf3-/-マウスに WT マウス由
来 CD8+
DC を移入し Poly(I:C)治療を行うと、腫瘍退縮が誘導されたのに
対し (Fig 4-1 左図)、Batf3-/-マウス由来 CD8
+ DC を移入し Poly(I:C)治療を
行っても腫瘍退縮は誘導されなかった (Fig 4-1 右図)。この結果より、
Poly(I:C)による腫瘍退縮には BATF3 陽性 CD8+
DC の存在が必要不可欠で
あることが明らかとなった。
次に BATF3 欠損により産生が大きく減少する Poly(I:C)誘導性 IL-12 が腫
瘍退縮に関与するかを評価するため、WT1-C1498 担がん Batf3-/-マウスに
Poly(I:C)と IL-12p70 の併用投与を行い腫瘍退縮の有無を観察した。その結
果、Poly(I:C) + IL-12p70 併用投与及び IL-12p70 単独投与により腫瘍増殖の
抑制が認められた (Fig 4-2)。
上記結果より IL-12p70 の抗腫瘍効果が明らかとなったため、そのメカニ
ズムをより詳細に解析することとした。まず、非担がん Batf3-/-マウスに
OVA + Poly(I:C)を投与した際、更に IL-12p70 を併用投与し、その 7 日後に
脾臓における OVA 特異的 CD8+
T cell の存在割合を評価した。その結果、
OVA + Poly(I:C)に加え IL-12p70 を投与すると、OVA 特異的 CD8+
T cell の
増加が認められた (Fig 4-3A)。 これにより、IL-12 は Poly(I:C)による抗原
特異的 CD8+
T cell 誘導を増強することが示された。
次に、非担がん WT および Batf3-/-マウスに OVA + Poly(I:C)投与した 7 日
後に脾臓を採取し、OVA 特異的 CD8+
T cell、 CD4+
T cell における IFN-
産生能を評価した。その結果、WT マウスでは OVA + Poly(I:C)投与により
IFN-産生能を有する CD8+
T cell、CD4+
T cell が誘導されていたのに対し、
Batf3-/-マウスでは T cell からの IFN-産生誘導は認められなかった (Fig
4-3B)。このように、BATF3 欠損により、CTL 活性のみならず Poly(I:C)誘
導性 CD4+
T cell (Th1cell) 活性も抑制されていたことから、BATF3 依存的
に産生される IL-12 は Th1 cell を活性化させることで間接的に CTL 活性化
を補助している可能性を考えた。その可能性を検証するため、非担がん
Batf3-/-マウスに OVA + Poly(I:C) + IL-12p70 を投与する際、CD4 陽性細胞

41
(その大部分が CD4+
T cell) を除去した群と除去しなかった群を設け、2 週
間後に OVA 特異的 CD8+
T cell の IFN-産生能を評価した。その結果、CD4
陽性細胞除去により IFN-産生細胞の割合の低下が認められたため、IL-12
は Th1 cell を介して CTL 活性化を増強していることが示唆された (Fig
4-3C)。
上記 Figure 4-3 の結果より、in vivo における CD4+
T cell を介した IL-12
のアシスト機能が示されたため、CD8+
DC による OT-I proliferation assay
を行い、in vitro においても同様の機構が観察されるかを解析した。まず、
Figure 2-1B と同様に WT および Batf3-/-
CD8+
DC と OT-I cell を OVA 抗原
存在下で共培養し、その際 IL-12p70 単独、Poly(I:C)単独、Poly(I:C) +
IL-12p70 をそれぞれ添加し、各々のクロスプレゼンテーション誘導能を評
価した。その結果、WT、Batf3-/-
CD8+
DC 共に IL-12p70 添加による OT-I cell
の分裂増強は認められず、IL-12p70 は抗原特異的 CD8+
T cell に直接的には
作用しないことが示された (Fig 4-4A)。次に、Batf3-/-
CD8+
DC と OT-I cell
の共培養時に、更に OVA 特異的 TCR を強制発現させた CD4+
T cell である
OT-II を加えた群と加えなかった群を設定し、Figure 4-4A と同様の実験を
行った。その結果、OT-II 存在下でも IL-12p70 単独添加ではクロスプレゼ
ンテーション増強は認められなかったが、Poly(I:C) + IL-12p70 併用添加で
は OT-II 非存在下に比較してクロスプレゼンテーション能が増強すること
が示された (Fig 4-4B)。
以上の結果より、Poly(I:C)による腫瘍退縮においては BATF3 陽性 CD8+
DC サブセットが必要であり、大部分がこのサブセットから産生される
IL-12 は、CD4+
T cell を介して間接的に CTL 活性を増強することが示され
た。
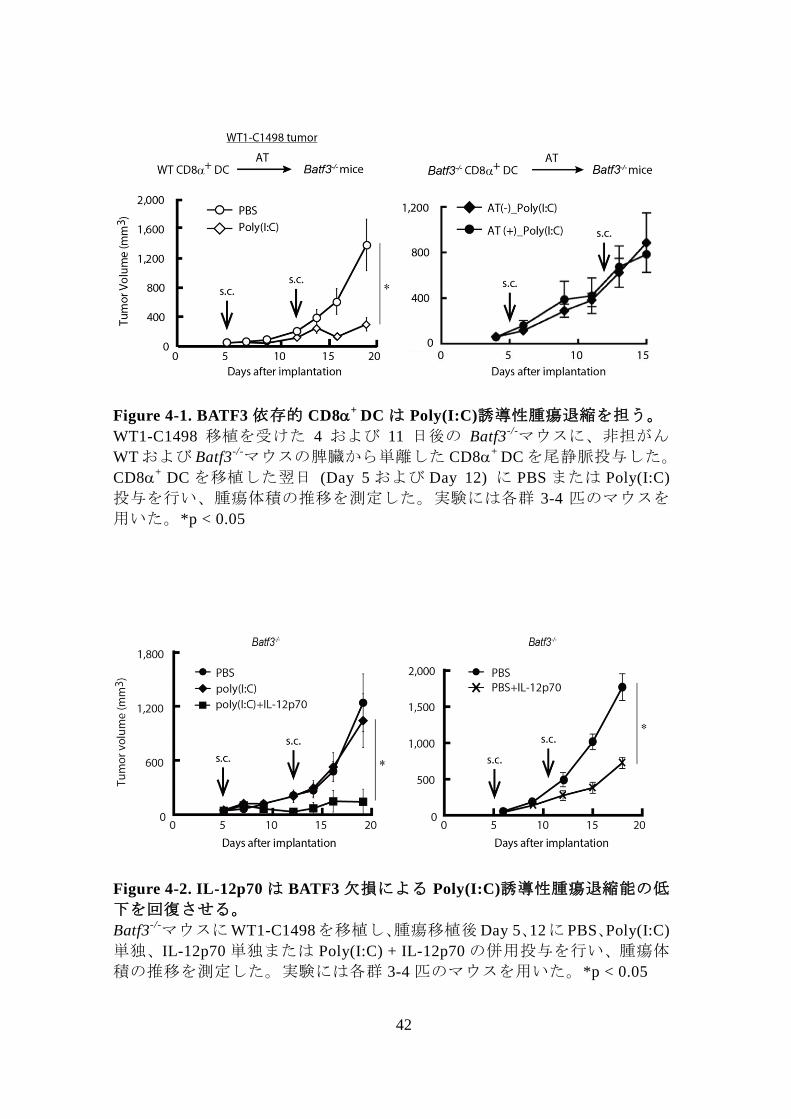
42
Figure 4-1. BATF3 依存的 CD8+
DC は Poly(I:C)誘導性腫瘍退縮を担う。
WT1-C1498 移植を受けた 4 および 11 日後の Batf3-/-マウスに、非担がん
WT および Batf3-/-マウスの脾臓から単離した CD8
+ DC を尾静脈投与した。
CD8+
DC を移植した翌日 (Day 5 および Day 12) に PBS または Poly(I:C)
投与を行い、腫瘍体積の推移を測定した。実験には各群 3-4 匹のマウスを
用いた。*p < 0.05
Figure 4-2. IL-12p70 は BATF3 欠損による Poly(I:C)誘導性腫瘍退縮能の低
下を回復させる。
Batf3-/-マウスにWT1-C1498を移植し、腫瘍移植後Day 5、12に PBS、Poly(I:C)
単独、IL-12p70 単独または Poly(I:C) + IL-12p70 の併用投与を行い、腫瘍体
積の推移を測定した。実験には各群 3-4 匹のマウスを用いた。*p < 0.05
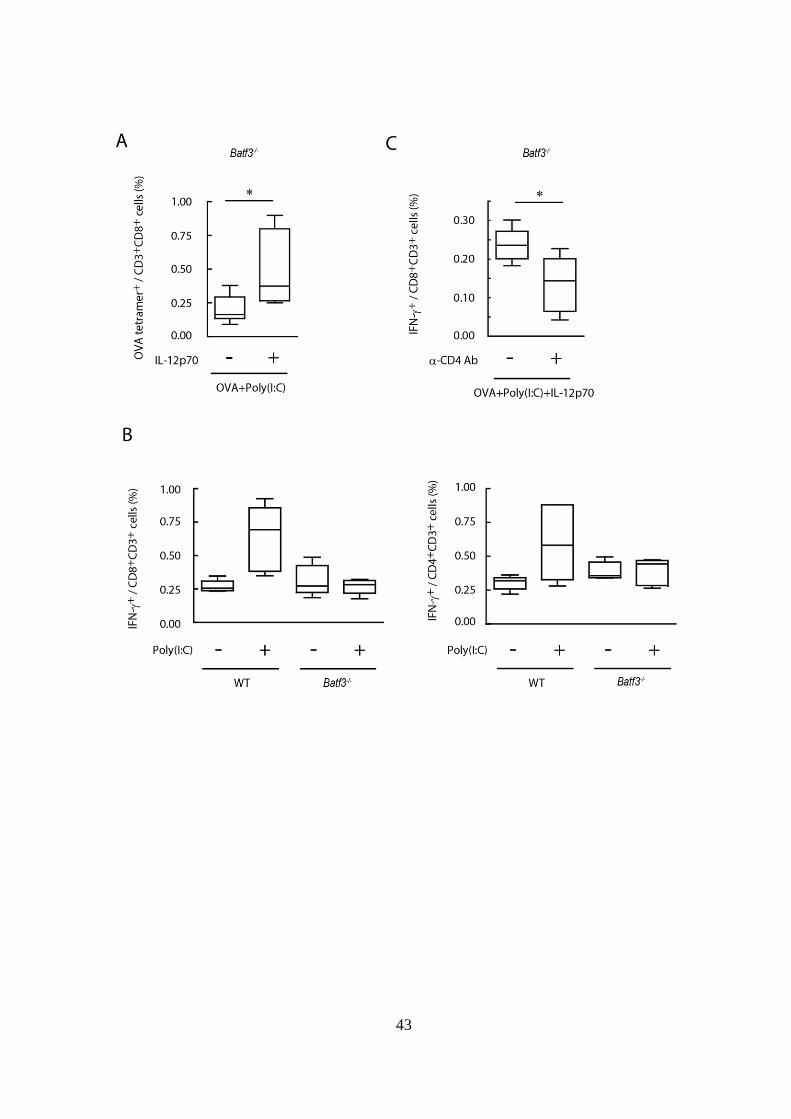
43

44
Figure 4-3. IL-12p70 は CD4+
T cell を介して CTL 活性化を増強する。
(A): 非担がん Batf3-/-マウスに 1mg OVA + 150 g Poly(I:C)を腹腔内投与し
た。その際、さらに 100 ng IL-12p70 を併用投与した群、しなかった群の 2
群を設定した。その 7 日後に脾臓を採取し、Tetramer assay を行い CD8+
T cell
中の OVA tetramer+
cell の割合を解析した。
(B): 非担がん WT および Batf3-/-マウスに 60 g OVA 単独または OVA +
Poly(I:C)の腹腔内投与を行った。その 7 日後に脾細胞を採取し、SL8 peptide
または OVA helper peptide 存在下で 6 時間インキュベートした。その後、
SL8 peptide パルス後の CD8+
T cell および OVA helper peptide パルス後の
CD4+ T cell 中の IFN-
+ cell の割合を解析した。
(C): 非担がん Batf3-/-マウスに CD4 除去抗体を含むヌードマウス由来腹水
を Day 0、7 の 2 回腹腔内投与し、CD4 陽性細胞を除去した。またネガテ
ィブコントロールとして PBS 投与群を設けた。その 24 時間後 (Day 1、8) に
OVA + Poly(I:C) + IL-12p70 の併用投与を行った。その後 Day 15 に脾細胞を
採取し、(B)と同様に SL8 peptide パルスを行い、CD8+
T cell 中の IFN-+
cell
の割合を解析した。
実験には各群 3-4 匹のマウスを用いた。*p < 0.05
示したデータは 2 回の独立した実験の内のひとつである。
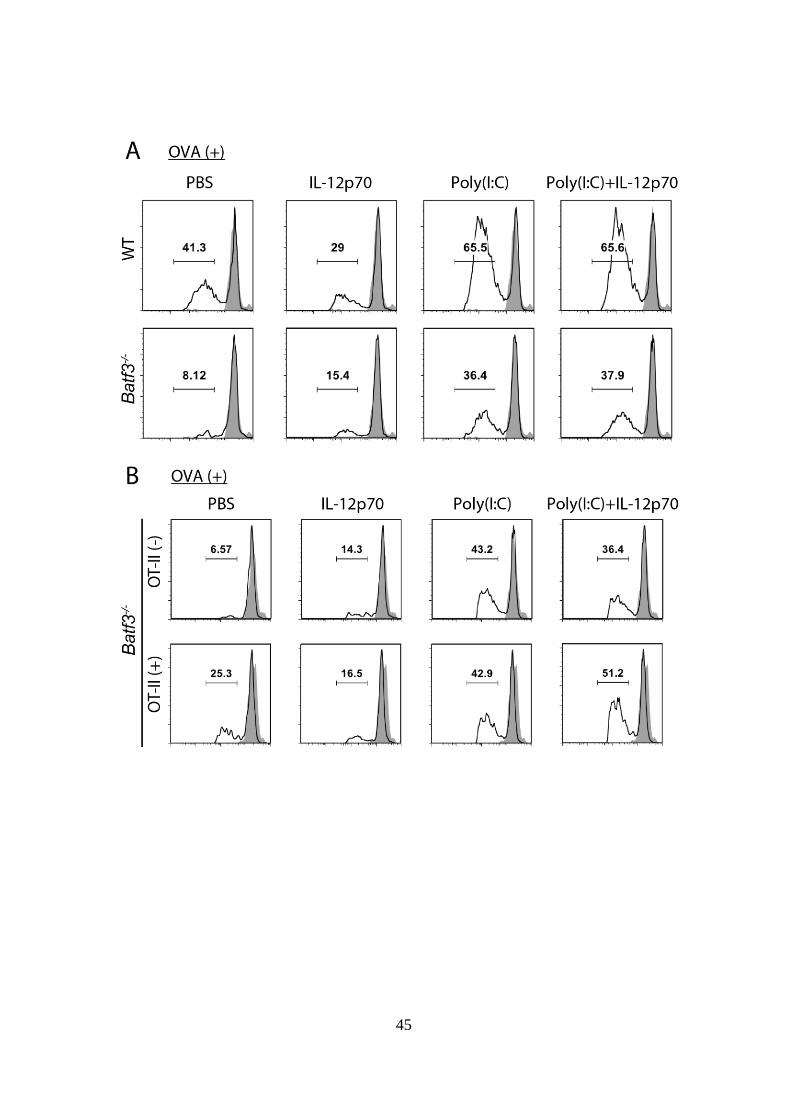
45

46
Figure 4-4. IL-12p70 は CD4+
T cell に作用し、間接的に CD8+
DC の
Poly(I:C)誘導性クロスプレゼンテーション能を増強させる。
(A): WT および Batf3-/-マウスの脾臓から CD8
+ DC を単離し 96 well プレー
トに播種した。1.5 g/ml OVA 存在下で、更に PBS、IL-12p70 単独、Poly(I:C)
単独または Poly(I:C) + IL-12p70 を添加して 3 時間インキュベートした。3
時間後に CFSE で蛍光標識した OT-I cell を加え、60 時間の共培養を行った。
60 時間後に CFSE の蛍光強度の減弱度により OT-I cell の抗原特異的分裂レ
ベルを評価した。
(B): (A)と同様に OVA および各物質を添加し、Batf3-/-マウス由来 CD8
+ DC
と CFSE 標識した OT-I cell の共培養を行った。その際、さらに OT-II cell
を共培養に加えた群、加えなかった群を設定し、60 時間の共培養後に OT-I
cell の抗原特異的分裂レベルを評価した。
示したデータは 2 回の独立した実験の内のひとつである。

47
考察
第一章では、Poly(I:C)による CD8+
DC の抗腫瘍免疫誘導メカニズムを
解析した。BATF3 陽性 CD8+
DC は TLR3 を選択的に高発現しており、こ
の TLR3 シグナル経路によりクロスプレゼンテーション促進、IL-12 産生が
惹起され、腫瘍抗原特異的 CTL が誘導される。また、BATF3 陽性 CD8+
DC
は腫瘍組織内にも浸潤しており、リンパ組織での CTL プライミングのみな
らず、腫瘍内への CTL 浸潤をも正に制御していることが示唆された。
BATF は AP-1 ファミリーに属する転写因子であり、DC や T cell の分化
制御を担う分子群である。そこに属する BATF3 は IRF8 といった他の転写
因子と共に、前駆細胞からの CD8+
DC の分化促進を担っている。しかし
ながら、C57BL/6 系統のマウスでは、BATF3 を欠損した場合でも同じ BATF
ファミリーの BATF2 が代償的に働くことで、数は少ないものの CD8+
DC
が分化誘導される 33,34。
本研究において、 C57BL/6 を遺伝的背景に持つ Batf3-/- マウスに
WT1-C1498 腫瘍を移植し Poly(I:C)治療を行ったところ、CTL 依存性腫瘍退
縮が完全にキャンセルされた (Fig 1-1)。既に述べたように、Batf3-/-マウス
であっても WT マウスと比較した場合、1/2 – 1/3 程度の CD8+
DC が残存
する。しかしながら、それにも関わらず腫瘍退縮が完全にキャンセルされ
ていたことから、BATF3 の機能について 2 つの可能性が考えられた。ひと
つは、CD8+
DC の分化誘導のみならず、Poly(I:C)誘導性クロスプライミン
グ経路の促進に BATF3 が関与している可能性、また 2 つ目として、CD8+
DC が TLR3 発現や高いクロスプレゼンテーション能を獲得するためには
BATF3 が必須である可能性が挙げられた。その後の解析により、Batf3-/-マ
ウスでは、XCR1hi
CLEC9A+の分子発現パターンを示す特定の CD8
+ DC サ
ブセットが消失していることが示され (Fig 1-3)、Poly(I:C)応答性を有する
CD8+
DC サブセットの分化誘導には BATF3 が必須であることが明らかと
なった。更にこの BATF3 陽性サブセットは、CD8+
DC の中でも選択的に
TLR3 を高発現していることが明らかとなり (Fig 2-3A, B, Fig 2-4)、これら
の分子発現パターンは human CD141+
DC と一致していた 28-30,38,39。このこ
とより、複数の分画が存在する CD8+
DC ポピュレーションの中でも、
BATF3 陽性サブセットこそが human CD141+
DC の真のカウンターパート
である可能性が示された。
Human CD141+
DC はクロスプレゼンテーション能が高いサブセットで

48
あることから 28、BATF3 陽性 CD8+
DC の Poly(I:C)誘導性クロスプレゼン
テーション能を in vivo 実験で評価した。その結果、抗原量が少ない環境下
では BATF3 陽性 CD8+
DC が必須となるのに対し、十分量の抗原量が存在
する場合は BATF3 陰性 CD8+
DC であっても中程度のクロスプライミング
を誘導出来ることが示された (Fig 2-1B)。Batf3-/-マウス由来 CD8
+ DC で
は WT と比較して TLR3 発現が著減していたのに対し、Ddx58 (RIG-I 遺伝
子) 発現はむしろ増加していたことから (Fig 2-3A)、抗原が十分量存在す
る環境では、細胞質内 MAVS 経路が TLR3 経路の代償として機能する可能
性が考えられる。また Batf3-/-マウスを用いた in vivo 実験において、非担が
ん状態では Poly(I:C)による抗原特異的 CD8+
T cell誘導は検知されなかった
が、担がん状態では中程度検知された (Fig 2-1A, Fig1-2C)。この差異とし
て、in vitro の場合と同様に、抗原量が少ない非担がん状態に対し、抗原が
多い担がん状態では BATF3 陰性 CD8+
DC の MAVS 経路が代償的に働い
た可能性がある。また、担がん状態では傷害された腫瘍細胞から放出され
る DNA が細胞質内 DNA 認識経路である Stimulator of interferon genes
(STING) - IRF3 - I 型 IFN 軸を活性化し、CTL 依存性腫瘍退縮を誘導するこ
とが知られている 41。そのため、担がんマウスで認められた BATF3 非依存
的な CTL 増加は、腫瘍由来 DNA により 2 次的に STING 経路が活性化した
結果もたらされた応答である可能性もある。
しかし、いずれの場合においても、実際の臨床症例ではマウス担がんモ
デルのように抗原性が強い腫瘍は少なく、そのような症例でも TLR3 アジ
ュバントにより強力に腫瘍退縮を誘導するためには、BATF3 陽性 DC (ヒト
では CD141+
DC) における TLR3-TICAM1 経路が重要であると考えられる。
また、BATF3 陽性 CD8+
DC における TLR3-TICAM1 経路は、細胞性免
疫を惹起する IL-12 産生を担っており、CD4+
T cell活性化誘導を介して CTL
活性を更に増強することも明らかとなった (Fig 2-3, Fig 4-3)。加えて、
Poly(I:C)による腫瘍組織からのケモカイン産生および腫瘍内への CD8+
T
cell 浸潤が BATF3 依存的に誘導されることも示された (Fig 3-1, Fig 3-2)。
本研究においては、腫瘍内の BATF3 陽性 CD8+
DC が CTL の腫瘍内浸潤
を担っているという直接の証拠を示せなかった。しかし、WT マウスでは
Poly(I:C)刺激により腫瘍組織内の CD8+
DC の割合が増加するのに対し、
Batf3-/-マウスでは増加が認められなかったことや (Fig 3-2E)、Batf3
-/-マウス
では Poly(I:C)刺激時の腫瘍内での IL-12 産生がキャンセルされていたこと
から (Fig 3-2A, B)、Poly(I:C)刺激により BATF3 陽性 CD8+
DC が腫瘍内に

49
浸潤し、腫瘍内における IL-12 産生やケモカイン産生を担っている可能性
がある。
以上、本研究により BATF3 陽性 CD8+
DC における TLR3-TICAM1 シグ
ナルは、リンパ組織での CTL 活性化、腫瘍組織への CTL 遊走、腫瘍内微
小環境における免疫状態の改善といった 3 つの段階で抗腫瘍免疫に寄与す
ることを示した。また、過去の瀬谷研究室の研究および本研究により、
Poly(I:C)誘導性クロスプレゼンテーションにおいては MAVS 経路の関与は
低く、TLR3-TICAM1 経路が重要であることを示した 18。MAVS 経路の下
流では強く I 型 IFN や炎症性サイトカイン産生が誘導されるため 9、この
過剰な炎症性応答が Poly(I:C)投与時の全身的な有害事象の引き金となるこ
とが報告されている。そのため瀬谷研究室では、TLR3 により選択的に認
識される核酸アジュバントを合成し、その低いサイトカイン産生能および
高い抗腫瘍活性を報告した 23。TLR3 は Poly(I:C)誘導性腫瘍退縮において
中心的役割を担う BATF3 陽性 DC サブセットに選択的に高発現しているこ
とから、TLR3 特異的アジュバントを用いた抗腫瘍療法は合理的な戦略で
あるといえる。本研究は、DC を起点とした TLR3 アジュバントの抗腫瘍性
応答誘導メカニズムの一端を明らかにしたものであり、より効果的で安全
性の高い TLR3 アジュバント療法の確立のための一助となると期待する。

50
第二章
抗腫瘍免疫療法における治療奏功性評価の指標となり得る
CD11c 陽性 CD8+
T cell の解析
緒言
多くの抗腫瘍免疫療法においては、如何に効果的に腫瘍抗原特異的 CTL
の増加・活性化を誘導できるかが治療奏功の鍵となる 42。成熟化した DC
により腫瘍抗原のクロスプレゼンテーションを受けた naïve CD8+
T cell は、
腫瘍抗原特異的に分裂増殖し細胞傷害活性を獲得する 14。担がんマウスを
用いた抗腫瘍実験において、腫瘍抗原および TLR3 アジュバントによる併
用療法を行うと、Tetramer+
CD8+
T cell、即ち腫瘍抗原特異的 CD8+
T cell の
増加が強く誘導される。この Tetramer+
CD8+
T cell の増加レベルは、腫瘍細
胞に対する傷害活性や腫瘍退縮程度と良く相関しているため、Tetramer
assay を行うことで直接的に腫瘍特異的 CD8+
T cell 誘導レベルを解析でき、
かつ治療奏功性を評価することが可能である 18,23。同様に実際の臨床現場
においても、治療に伴う腫瘍抗原特異的 CD8+
T cell の増加や活性化レベル
を評価することは、治療継続の可否や予後の予測に重要である。しかしな
がら、がん患者においてはマウスモデルとは異なり各人の HLA タイプや
腫瘍抗原は多種多様であり、Tetramer assay を評価の指標として用いること
は困難である。以上より、実際の臨床現場においても使用可能な、抗原特
異的 CTL 増加・活性化レベルを反映し得る、簡便かつ侵襲性の低い新たな
指標を探索することは非常に意義深いと言える。
そこで本章においては、評価の指標となり得る候補として CD11c+
CD8+
T
cell に着目した。多くの病原体感染モデルマウスを用いた過去の報告にお
いて、病原体に感染した宿主の体内では、免疫応答が誘導されるに伴い全
身的に CD11 分子を発現している CD8+
T cell 分画が増加する現象が認めら
れている 43-46。その後、この CD11c+
CD8+
T cell 分画は免疫応答の終息に伴
い再び減少するという動態を示しており、免疫応答により惹起される CTL
活性化状態を反映している可能性が示唆された。そのため、抗腫瘍免疫療
法においても同様に CD11c+
CD8+
T cell の増加が誘導される可能性が考え
られた。よって本研究において、担がんマウスに Poly(I:C)治療を行った際
の CD11c+
CD8+
T cell の動態およびその表現型を解析し、治療奏功性判定
の新たな指標となり得る可能性を評価することとした。
51
実験材料および方法
1. マウス
第一章でも使用したマウスの詳細は、第一章実験材料および方法の第 1
項を参照。Ticam1-/-マウス、Mavs
-/-マウスおよび Ticam1-/-
/ Mavs-/-マウスは
瀬谷研究室で作出されたもので、C57BL/6 マウスに戻し交配を 8 回以上行
った。全ての動物実験は、北海道大学動物実験に関する規程に従い、承認
を受けて行った。
2. 試薬および抗体
第一章でも使用した試薬および抗体の詳細は、第一章実験材料および方
法の第 2 項を参照。
表 4. 実験に使用した試薬類
試薬名 メーカー・供与元
Macrophage activating lipopeptide-2-short
(Pam2-CGNNDE; MALP2s) BioLogica
Chromium-51 Radionuclide PerkinElmer
CellAmp Whole Transcriptome Amplification Kit Takara Bio
表 5. 実験に使用した抗体
抗体名 クローン番号 メーカー
FITC-anti-mouse CD62L MEL-14
Biolegend
PE-anti-mouse CD127 SB/199
APC-anti-mouse/human KLRG1 2F1/KLRG1
PE-anti-mouse IL-2 JES6-5H4
APC-anti-mouse PD-1 RMP1-30
PE-anti-mouse TNF- MP6-XT22
3. 細胞および培養メディウム組成
EG7、WT1-C1498 および初代培養細胞の培養条件は、第一章実験材料お
よび方法の第 3 項を参照。マウス胸腺腫細胞株である EL4 は ATCC 社より
購入した。細胞培養には 10% 非動化 FBS、50 IU penicillin / 50 g/ml
streptomycin を含む RPMI1640 メディウムを用いた。
52
4. 腫瘍移植および TLR アジュバント治療
EG7 担がんマウスにおける OVA 単独、あるいは OVA + Poly(I:C)併用投
与実験は、第一章実験材料および方法の第 4 項と同様の方法で行った。ま
た WT1-C1498 担がんマウスにおける PBS、あるいは Poly(I:C)単独投与実
験も第一章実験材料および方法の第 4 項と同様の方法で行った。
EG7 担がんマウスへの MALP2s 投与実験においては、腫瘍移植後 5 日お
よび 12 日目に 100 g OVA 単独または OVA + 50 nmol MALP2s を腫瘍組織
周辺の皮下に投与した。
5. 定量 PCR
細胞を TRIzol reagent に溶解させたサンプルから RNA を精製し cDNA を
作製する場合は、第一章実験材料および方法の第 7 項と同様の方法で行っ
た。
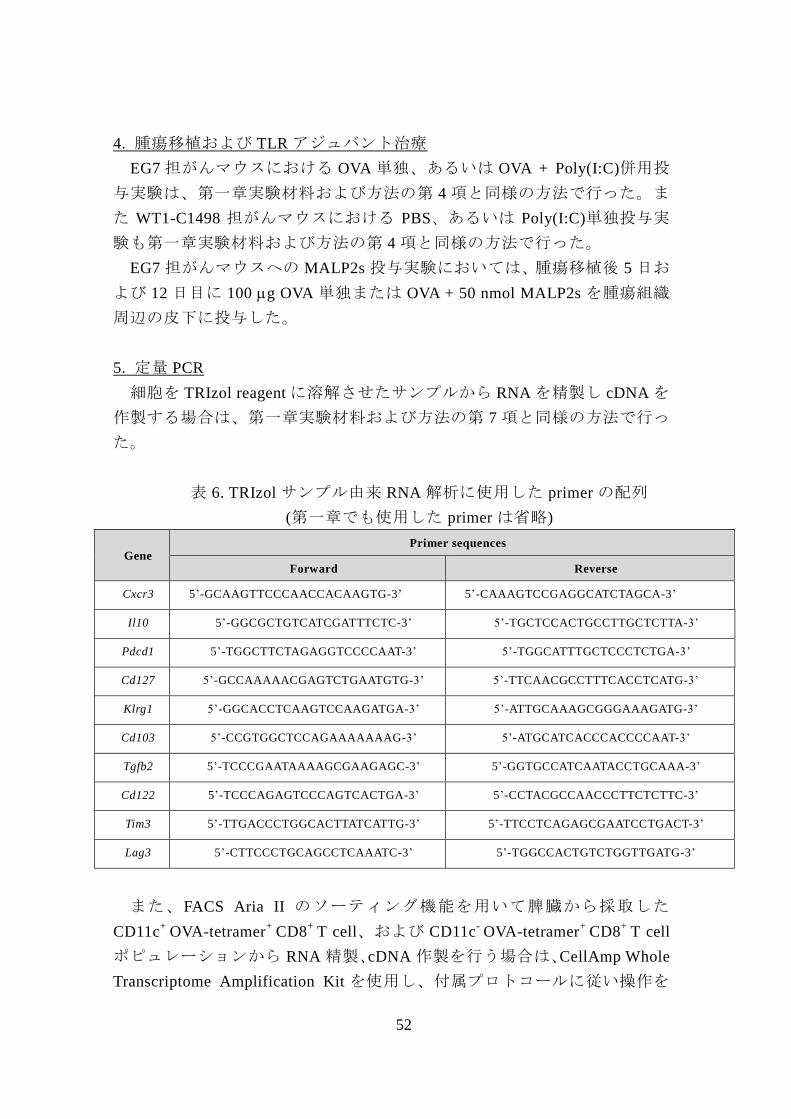
表 6. TRIzol サンプル由来 RNA 解析に使用した primer の配列
(第一章でも使用した primer は省略)
Gene Primer sequences
Forward Reverse
Cxcr3 5’-GCAAGTTCCCAACCACAAGTG-3’ 5’-CAAAGTCCGAGGCATCTAGCA-3’
Il10 5’-GGCGCTGTCATCGATTTCTC-3’ 5’-TGCTCCACTGCCTTGCTCTTA-3’
Pdcd1 5’-TGGCTTCTAGAGGTCCCCAAT-3’ 5’-TGGCATTTGCTCCCTCTGA-3’
Cd127 5’-GCCAAAAACGAGTCTGAATGTG-3’ 5’-TTCAACGCCTTTCACCTCATG-3’
Klrg1 5’-GGCACCTCAAGTCCAAGATGA-3’ 5’-ATTGCAAAGCGGGAAAGATG-3’
Cd103 5’-CCGTGGCTCCAGAAAAAAAG-3’ 5’-ATGCATCACCCACCCCAAT-3’
Tgfb2 5’-TCCCGAATAAAAGCGAAGAGC-3’ 5’-GGTGCCATCAATACCTGCAAA-3’
Cd122 5’-TCCCAGAGTCCCAGTCACTGA-3’ 5’-CCTACGCCAACCCTTCTCTTC-3’
Tim3 5’-TTGACCCTGGCACTTATCATTG-3’ 5’-TTCCTCAGAGCGAATCCTGACT-3’
Lag3 5’-CTTCCCTGCAGCCTCAAATC-3’ 5’-TGGCCACTGTCTGGTTGATG-3’
また、FACS Aria II のソーティング機能を用いて脾臓から採取した
CD11c+
OVA-tetramer+
CD8+
T cell、および CD11c- OVA-tetramer
+ CD8
+ T cell
ポピュレーションから RNA 精製、cDNA 作製を行う場合は、CellAmp Whole
Transcriptome Amplification Kit を使用し、付属プロトコールに従い操作を
53
行った。引き続く定量 PCR 解析の際には、以下のプライマーを用いた。
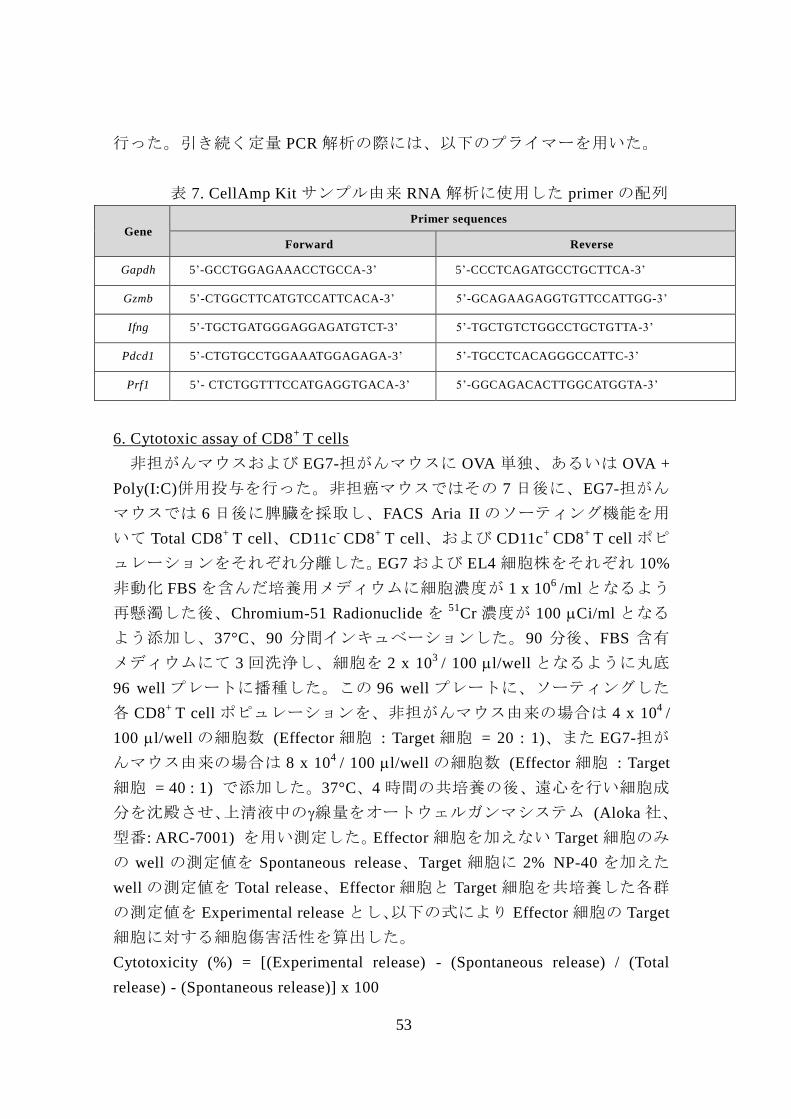
表 7. CellAmp Kit サンプル由来 RNA 解析に使用した primer の配列
Gene Primer sequences
Forward Reverse
Gapdh 5’-GCCTGGAGAAACCTGCCA-3’ 5’-CCCTCAGATGCCTGCTTCA-3’
Gzmb 5’-CTGGCTTCATGTCCATTCACA-3’ 5’-GCAGAAGAGGTGTTCCATTGG-3’
Ifng 5’-TGCTGATGGGAGGAGATGTCT-3’ 5’-TGCTGTCTGGCCTGCTGTTA-3’
Pdcd1 5’-CTGTGCCTGGAAATGGAGAGA-3’ 5’-TGCCTCACAGGGCCATTC-3’
Prf1 5’- CTCTGGTTTCCATGAGGTGACA-3’ 5’-GGCAGACACTTGGCATGGTA-3’
6. Cytotoxic assay of CD8+
T cells
非担がんマウスおよび EG7-担がんマウスに OVA 単独、あるいは OVA +
Poly(I:C)併用投与を行った。非担癌マウスではその 7 日後に、EG7-担がん
マウスでは 6 日後に脾臓を採取し、FACS Aria II のソーティング機能を用
いて Total CD8+
T cell、CD11c- CD8
+ T cell、および CD11c
+ CD8
+ T cell ポピ
ュレーションをそれぞれ分離した。EG7 および EL4 細胞株をそれぞれ 10%
非動化 FBS を含んだ培養用メディウムに細胞濃度が 1 x 106
/ml となるよう
再懸濁した後、Chromium-51 Radionuclide を 51Cr 濃度が 100 Ci/ml となる
よう添加し、37°C、90 分間インキュベーションした。90 分後、FBS 含有
メディウムにて 3 回洗浄し、細胞を 2 x 103
/ 100 l/well となるように丸底
96 well プレートに播種した。この 96 well プレートに、ソーティングした
各 CD8+
T cell ポピュレーションを、非担がんマウス由来の場合は 4 x 104
/
100 l/well の細胞数 (Effector 細胞 : Target 細胞 = 20 : 1)、また EG7-担が
んマウス由来の場合は 8 x 104
/ 100 l/well の細胞数 (Effector 細胞 : Target
細胞 = 40 : 1) で添加した。37°C、4 時間の共培養の後、遠心を行い細胞成
分を沈殿させ、上清液中の線量をオートウェルガンマシステム (Aloka 社、
型番: ARC-7001) を用い測定した。Effector 細胞を加えない Target 細胞のみ
の well の測定値を Spontaneous release、Target 細胞に 2% NP-40 を加えた
well の測定値を Total release、Effector 細胞と Target 細胞を共培養した各群
の測定値を Experimental release とし、以下の式により Effector 細胞の Target
細胞に対する細胞傷害活性を算出した。
Cytotoxicity (%) = [(Experimental release) - (Spontaneous release) / (Total
release) - (Spontaneous release)] x 100

54
なお本実験は、北海道大学放射線障害予防規程に従い、承認を受けて行
った。
7. 統計処理
2 群間の有意差検定では、Student’s t-検定を用い p 値を求めた。多重比較
検定は、統計解析ソフト Prism4 を用い、Kluskal-Wallis test の後に Dunn’s
multiple comparison test を行い p 値を求めた。(*p < 0.05)
実験結果
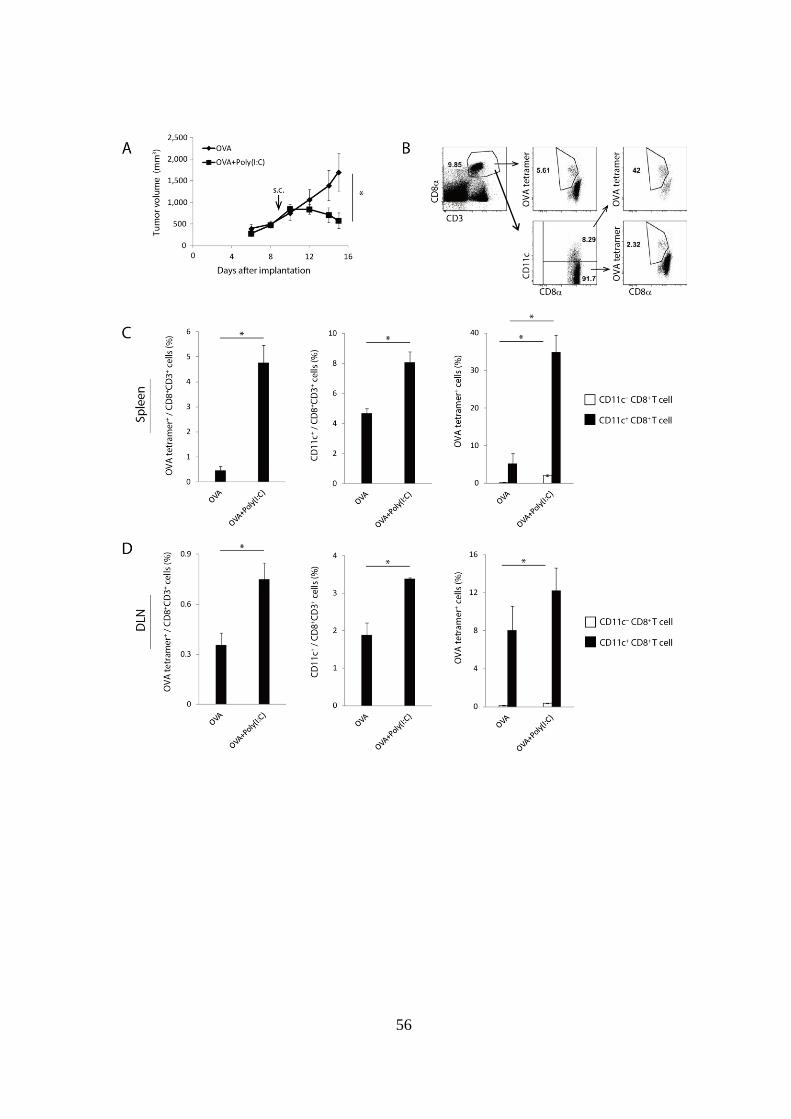
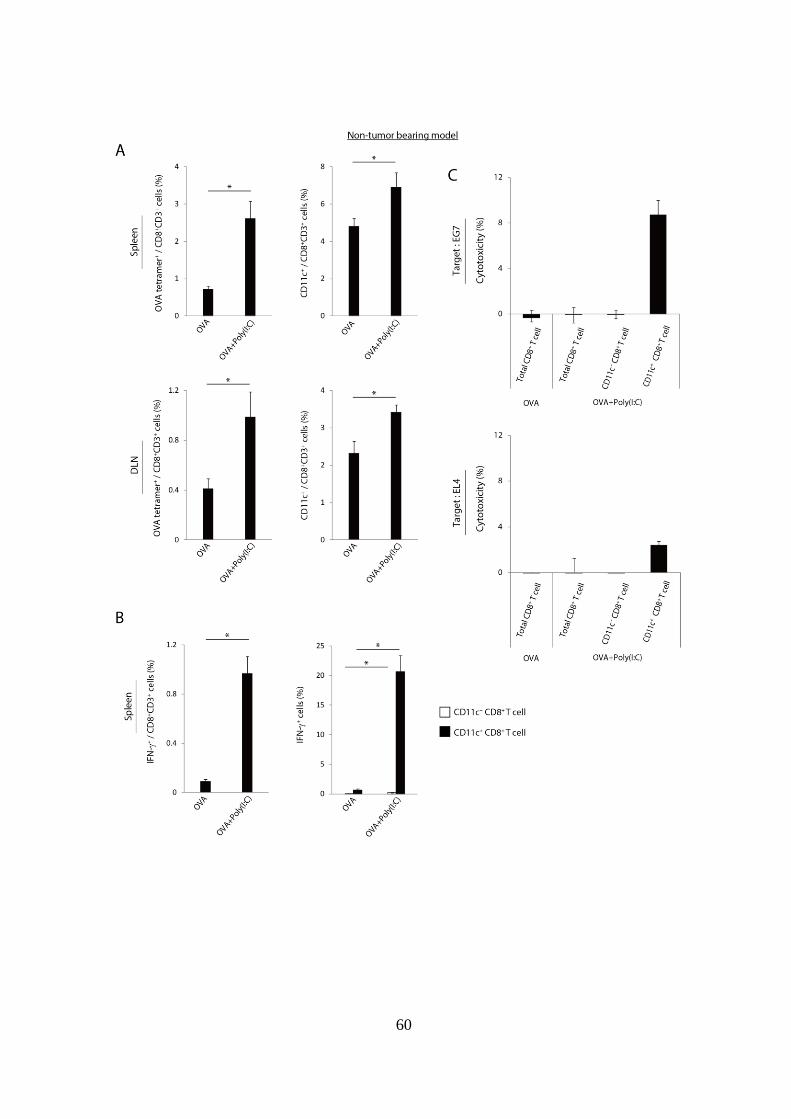
1. 抗原 + Poly(I:C)投与は CD11c+
CD8+
T cell の増加を誘導する
まず始めに、EG7 担がんマウスに腫瘍抗原 + Poly(I:C)治療を行った際に、
多くの病原体感染時と同様に CD11c+
CD8+
T cell の増加が誘導されるか否
かを評価した。第一章でも示したように、EG7-担がんマウスに腫瘍抗原で
ある OVA を単独投与しても腫瘍退縮および抗原特異的 CD8+
T cell の増加
は誘導されず、Poly(I:C)を併用投与することで強力な腫瘍退縮、抗原特異
的 CD8+
T cell の増加が誘導された (Fig 1-1A, C および D 左図)。その際、
脾臓および DLN において CD8+
T cell 中の CD11c 陽性細胞の割合を解析し
たところ、OVA 単独投与と比較して、OVA + Poly(I:C)投与で CD11c+
CD8+
T
cell 分画が有意に増加していた (Fig 1-1C, D 中央図)。このとき CD11c- 分
画および CD11c+ 分画中の OVA tetramer陽性細胞の割合を解析したところ、
CD11c- 分画中よりも CD11c
+ 分画中に高い割合で tetramer 陽性細胞が含ま
れていた (Fig 1-1C, D 右図)。以上の結果より、多くの病原体感染と同様に、
OVA + Poly(I:C)治療を受けた EG7-担がんマウスにおいても CD11c+
CD8+
T
cell 分画の増加が認められ、この分画の多くが抗原特異的 CD8+
T cell であ
ることが示された。
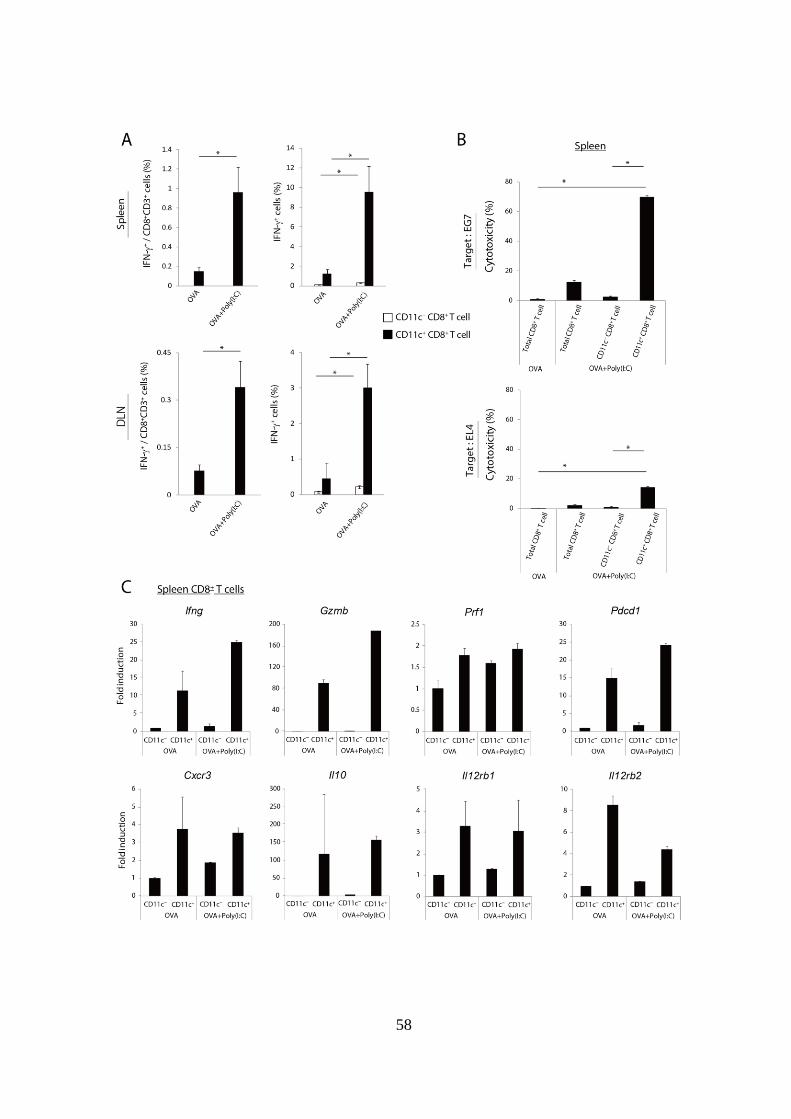
次に、OVA + Poly(I:C)投与により誘導された CD11c+ 分画の表現型を評
価することとした。まず、誘導された CD11c+
CD8+
T cell における抗原特
異的な IFN-産生能を評価した。OVA 単独または OVA + Poly(I:C)投与を受
けた EG7-担がんマウスから脾臓および DLN を採取し、SL8 peptide で再刺
激した際の CD8+
T cell 中の IFN-陽性細胞の割合を評価した。その結果、
OVA + Poly(I:C)の併用投与により IFN-産生細胞の割合が有意に増加した
(Fig 1-2A 左図)。このとき CD11c- 分画および CD11c
+ 分画中の IFN-陽性
細胞の割合を解析したところ、CD11c- 分画中よりも CD11c
+ 分画中に高い

55
割合で IFN-陽性細胞が含まれていた (Fig 1-2A 右図)。
続いて、CD11c- 分画と CD11c
+ 分画の腫瘍細胞に対する細胞傷害活性を
比較することとした。OVA 単独および OVA + Poly(I:C)投与を受けた EG7-
担がんマウスから全 CD8+
T cell、CD11c- CD8
+ T cell および CD11c
+ CD8
+ T
cell ポピュレーションを単離し、EL4 (OVA 陰性腫瘍細胞株) および EG7
(OVA 陽性-EL4 細胞株) と共培養し、細胞傷害活性を解析した。その結果、
OVA + Poly(I:C)治療を受けたマウス由来の CD11c+ 分画は EG7、EL4 の両
腫瘍細胞株に対して非常に高い細胞傷害活性を示したのに対し、CD11c- 分
画は殆ど細胞傷害活性を有していないことが明らかとなった (Fig 1-2B)。
更に OVA 単独および OVA + Poly(I:C)投与を受けた EG7-担がんマウスの
脾臓から CD11c-および CD11c
+ CD8
+ T cell 分画を単離し、その遺伝子発現
パターンを解析した。その結果、CTL の細胞傷害活性を担う Ifng、Gzmb、
Prf1 といった遺伝子や Il12r は OVA 単独、OVA + Poly(I:C)投与の両群にお
いて CD11c-に比較して CD11c
+ 分画で高い発現が認められた。また、T cell
のリンパ組織から局所への遊走を担うケモカインレセプター40 である
Cxcr3 発現や、抑制性因子である Pdcd1 (PD-1 遺伝子) や Il10 も CD11c+ 分
画で高かった。これらの遺伝子のうち、CD11c+ 分画における Ifng、Gzmb、
Pdcd1 発現は OVA 単独投与群と比較し、OVA + Poly(I:C)群では更に増加し
ていた (Fig 1-2C)。このような CD11c+ 分画の遺伝子発現パターンは、
Poly(I:C)刺激により誘導される CTL の表現型と一致していた。
また、上記 Figure 1-1C、D および Figure 1-2A、B と同様の解析を OVA
単独または OVA+Poly(I:C)投与を受けた非担がんマウスにおいても行った。
その結果、非担がんマウスにおいても EG7-担がんマウスと同様、OVA +
Poly(I:C)投与により、高い IFN-産生能、高い細胞傷害活性を有する CD11c+
CD8+
T cell 分画の増加が誘導された (Fig 1-3A-C)。
以上の結果より、OVA + Poly(I:C)投与により誘導される CD11c+
CD8+
T
cell は腫瘍抗原特異的 CTL であり、腫瘍細胞退縮を担う細胞集団であるこ
とが示された。
56

57
Figure 1-1. 腫瘍抗原および Poly(I:C)の併用投与によりリンパ組織におけ
る抗原特異的 CD11c+
CD8+
T cell の増加が誘導される。
(A): WT マウスに EG7 を移植し、腫瘍移植後 Day 9 に OVA 単独、または
OVA + Poly(I:C)併用投与を行い、腫瘍体積の推移を測定した。
(B-D): OVA 単独、OVA + Poly(I:C)投与を行った 6 日後に、脾臓および鼠径
リンパ節 (DLN) を採取し、Tetramer assay を行った。(B)は脾臓における
gating strategy を示している。(C)は脾臓の解析結果を、(D)は DLN の解析
結果を示しており、それぞれの左図では CD8+
T cell 中の OVA tetramer+
cell
の割合を示している。中央図では CD8+
T cell 中の CD11c+
cell の割合を示
している。右図では CD11c- CD8
+ T cell 中の OVA tetramer
+ cell の割合を白
い棒グラフで、また CD11c+
CD8+
T cell 中の OVA tetramer+
cell の割合を黒
い棒グラフで示している。
実験には各群 4-5 匹のマウスを用いた。*p < 0.05
示したデータは 3 回の独立した実験の内のひとつである。
58

59
Figure 1-2. 抗原 + Poly(I:C)治療により増加した脾臓の CD11c+
CD8+
T cell
は抗腫瘍性の表現型を示す。
(A): WT マウスに EG7 を移植し、腫瘍移植後 Day 9 に OVA 単独、または
OVA + Poly(I:C)併用投与を行った。Day 15 に脾臓および DLN を採取し、
SL8 peptide 存在下で 6 時間インキュベートした。左図では SL8 peptide パ
ルス後の CD8+
T cell 中の IFN-+
cell の割合を示している。右図では CD11c-
CD8+
T cell 中の IFN-+
cell の割合を白い棒グラフで、また CD11c+
CD8+
T
cell 中の IFN-+
cell の割合を黒い棒グラフで示している。
(B): 上記(A)で採取した脾細胞から全 CD8+
T cell、CD11c- CD8
+ T cell およ
び CD11c+
CD8+
T cell 分画を単離し、51Cr で標識した EG7 または EL4 細胞
と 40 : 1 の細胞比で共培養した。その 4 時間後に Effector 細胞の Target 細
胞に対する細胞傷害活性を評価した。上図は EG7 に対する傷害活性を、下
図は EL4 に対する傷害活性を示している。
(C): 上記(A)で採取した脾細胞から CD11c- CD8
+ T cell および CD11c
+ CD8
+
T cell 分画を単離し、定量 PCR により各遺伝子発現レベルを解析した。
実験には各群 4-5 匹のマウスを用いた。*p < 0.05
示したデータは 2 回の独立した実験の内のひとつである。
60

61
Figure 1-3. 抗原 + Poly(I:C)投与により、非担がんマウスにおいても細胞
傷害活性の高い CD11c+
CD8+
T cell の増加が誘導される。
(A): 非担がん WT マウスに OVA 単独、または OVA + Poly(I:C)併用投与を
行った。Day 7 に脾臓および DLN を採取し、Tetramer assay を行った。左
図では CD8+
T cell 中の OVA tetramer+
cell の割合を、右図では CD8+
T cell
中の CD11c+
cell の割合を示している。
(B): 上記(A)で採取した脾細胞を SL8 peptide 存在下で 6 時間インキュベー
トした。左図では SL8 peptide パルス後の CD8+
T cell 中の IFN-+
cell の割
合を、右図では CD11c- CD8
+ T cell 中の IFN-
+ cell の割合を白い棒グラフで、
また CD11c+
CD8+
T cell 中の IFN-+
cell の割合を黒い棒グラフで示してい
る。
(C): 上記(A)で採取した脾細胞から全 CD8+ T cell、CD11c- CD8
+ T cell およ
び CD11c+
CD8+
T cell を単離し、51Cr で標識した EG7 または EL4 細胞と 20 :
1 の細胞比で共培養した。その 4 時間後に Effector 細胞の Target 細胞に対
する細胞傷害活性を評価した。上図は EG7 に対する傷害活性を、下図は
EL4 に対する傷害活性を示している。
実験には各群 5 匹のマウスを用いた。*p < 0.05

62
2. 腫瘍内浸潤 CD11c+
CD8+
T cell は活性化 CTL 様の表現型を示す
前項において脾臓や DLN における CD11c+ 分画の解析を行ったため、次
に実際に細胞傷害活性が発揮される場である腫瘍内微小環境における
CD11c+ 分画の存在割合および表現型を解析することとした。まず、OVA
単独および OVA + Poly(I:C)投与を受けた EG-7 担がんマウスにおいて、腫
瘍内浸潤 CD8+
T cell 中の OVA tetramer 陽性細胞および CD11c 陽性細胞の
割合を解析した。その結果、既述の結果と一致し、OVA + Poly(I:C)投与に
より非常に高い割合で OVA 抗原特異的 CD8+
T cellが増加していた (Fig 2A
左図)。また、CD11c 陽性細胞の割合解析では、Poly(I:C)刺激が無い OVA
単独群においても CD8+
T cell 中の約 70%が CD11c+ 分画と非常に高い割合
を示し、更に OVA + Poly(I:C)投与によりその存在割合が上昇することが確
認された (Fig 2A 右図)。このとき、CD11c+
CD8+
T cell 分画における CD11c
分子の発現レベルを解析したところ、OVA 単独と比較して OVA + Poly(I:C)
投与群で発現レベルは有意に上昇していた (Fig 2B)。次に、CD11c- 、
CD11clo-mid および CD11c
hi CD8
+ T cell 分画中の OVA tetramer 陽性細胞の割
合を解析したところ、OVA 単独、OVA + Poly(I:C)投与の両群において、
CD11c の発現レベルと OVA tetramer 陽性細胞の割合が正に相関することが
示された (Fig 2C)。
続いて、腫瘍内浸潤 CD11c- 、CD11c
lo-mid および CD11chi
CD8+
T cell 分画
における遺伝子発現パターンを解析した。その結果、OVA 単独、OVA +
Poly(I:C)投与の両群において、Ifng、Gzmb、Prf1、Pdcd1、Il10 といった遺
伝子発現が CD11c 分子の発現レベルに比例して高くなってゆくことが示
された (Fig 2D)。また、OVA + Poly(I:C)投与群において OVA 特異的 CD8+
T
cell のみに限定し、CD11c-および CD11c
+ 分画の遺伝子発現パターンを解析
したところ、CD11c-に比較し CD11c
+ 分画で Ifng、Prf1、Pdcd1 の発現が上
昇している傾向が認められた (Fig 2E)。
以上の結果は、腫瘍内浸潤 CD11c+
CD8+
T cell は活性化状態の抗原特
異的 CTL であり、その CD11c 分子の発現レベルは細胞傷害活性程度と良
く相関することを示している。

63

64
Figure 2. 腫瘍内浸潤 CD8+
T cell における CD11c 発現レベルは抗原特異的
細胞傷害活性の強度と相関する。
(A): WT マウスに EG7 を移植し、腫瘍移植後 Day 9 に OVA 単独、または
OVA + Poly(I:C)併用投与を行い、その 6 日後に腫瘍組織を採取し、Tetramer
assay を行った。左図では CD8+
T cell 中の OVA tetramer+
cell の割合を示し、
右図では CD8+
T cell 中の CD11c+
cell の割合を示している。
(B): 腫瘍内浸潤 CD11c+
CD8+
T cell 分画における CD11c 発現レベルを
FACS により解析した。左図では Gating strategy を示し、右図では CD11c
の MFI を棒グラフ化して示した。
(C): 腫瘍内浸潤 CD11c- 、CD11c
lo-mid および CD11chi
CD8+
T cell 分画中の
OVA tetramer+
cell の割合をそれぞれ解析した。左図では Gating strategy を
示し、右図では棒グラフを示した。
(D): 腫瘍内浸潤 CD11c- 、CD11c
lo-mid および CD11chi
CD8+
T cell 分画を単離
し、定量 PCR により各遺伝子発現レベルを解析した。
(E): OVA + Poly(I:C)治療を受けた EG-7 担がんマウスから腫瘍内浸潤 OVA
tetramer+ CD11c
- および OVA tetramer+ CD11c
+ CD8
+ T cell 分画を単離し、定
量 PCR により各遺伝子発現レベルを解析した。
実験には各群 5 匹のマウスを用いた。*p < 0.05
示したデータは 3 回の独立した実験の内のひとつである。

65
3. CD8+
T cell の CD11c 発現レベルは抗原特異的分裂の強度と相関する
前節の解析により、CD8+
T cell 上の CD11c 分子の発現レベルと細胞傷害
活性の強度との相関関係が明らかとなった。このことより、強い抗原特異
的シグナルを受容した CD8+
T cell ほど CD11c 発現が強く誘導される可能
性が考えられた。その可能性を検証するため、 in vitro において OT-I
proliferation assay を行い、OT-I cell の抗原特異的分裂回数と CD11c 発現レ
ベルの相関関係を解析した。その結果、Poly(I:C)刺激の有無に関わらず、
抗原特異的に分裂した回数が多い OT-I cell ほど CD11c発現レベルも高くな
ってゆき、三回以上分裂した細胞集団では、全体の 80%以上が CD11c 陽性
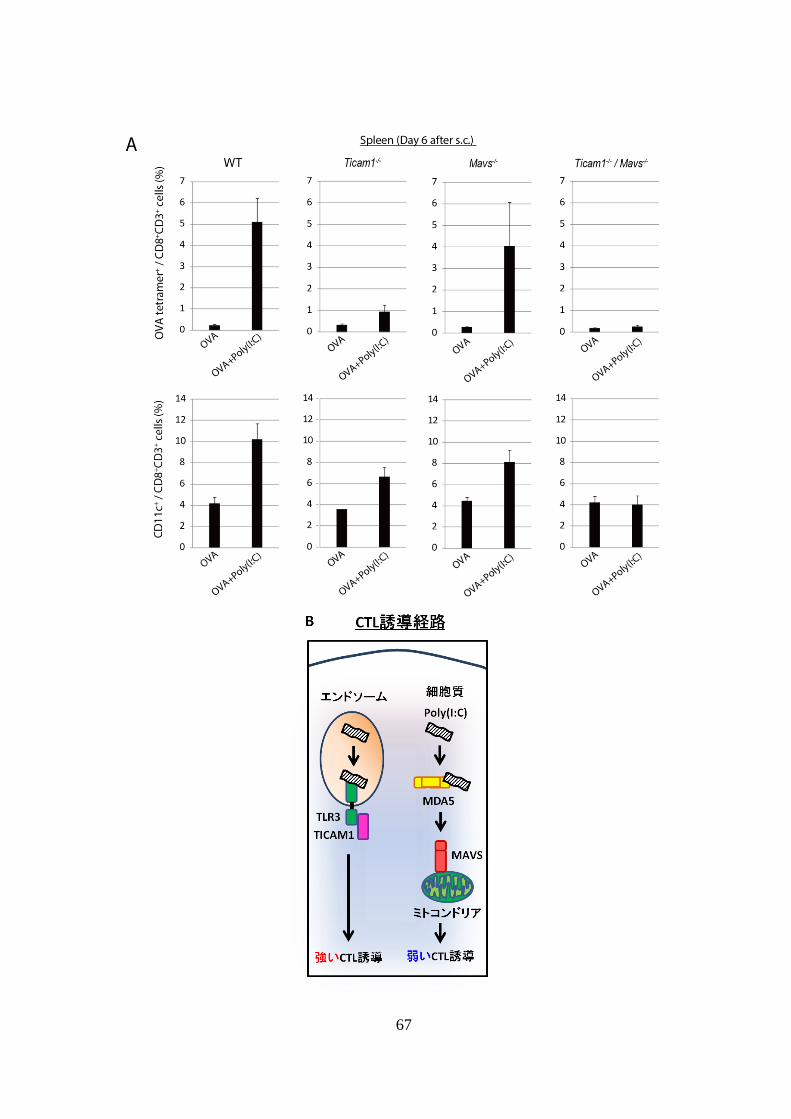
となっていた (Fig 3-1A, B)。
次に、抗原提示細胞である DC が Poly(I:C)刺激を受けた場合にはエンド
ソーム内の TLR3-TICAM1 経路と細胞質内の MAVS 経路の 2 つの経路が活
性化するため、どちらの経路が CD11c 発現誘導に貢献しているかを解析し
た。解析には WT、Ticam1-/-、Mavs
-/-、Ticam1-/-
/ Mavs-/- マウスを用い、OVA
単独および OVA + Poly(I:C)投与後の OVA 抗原特異的 CD8+
T cell および
CD11c+
CD8+
T cell 誘導レベルを比較した。その結果、OVA + Poly(I:C)投与
により誘導される Tetramer 陽性細胞の割合は WT マウスで最も高く、次い
で Mavs-/-、Ticam1
-/-マウスの順に低下してゆき、Ticam1-/-
/ Mavs-/- マウスで
は Tetramer 陽性細胞の誘導は完全にキャンセルされていた (Fig 3-2A 上図)。
これは、DC を介した CTL 誘導 (クロスプレゼンテーション ) は主に
TLR3-TICAM1 経路に依存しているという過去の報告と一致していた 18
(Fig 3-2B)。また、CD11c+
CD8+
T cell の増加レベルは、Tetramer 陽性細胞
の増加レベルと一致していた (Fig 3-2A 下図)。
以上の結果は、CD8+
T cell における Poly(I:C)誘導性 CD11c 発現増強は、
DC 側の TLR3-TICAM1 経路または MAVS 経路のどちらか一方に依存する
のではなく、CD8+ T cell に受容された抗原特異的増殖シグナルの強度に依
存することを示している。
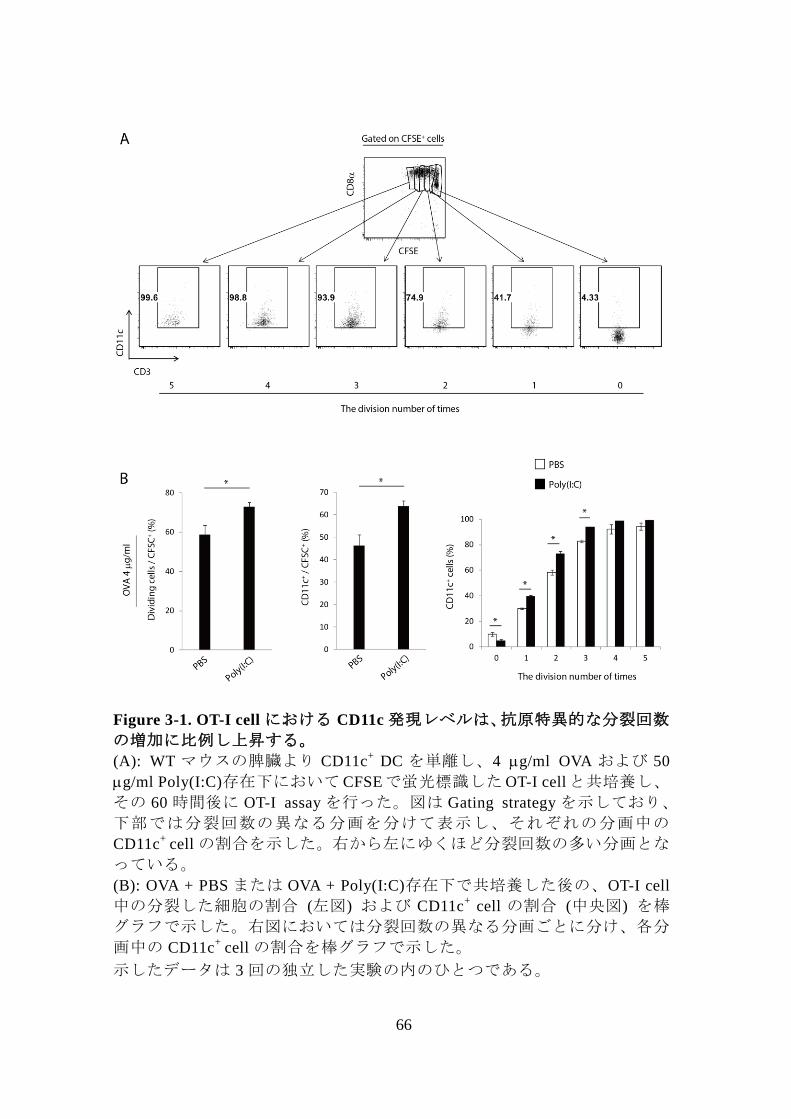
66
Figure 3-1. OT-I cell における CD11c 発現レベルは、抗原特異的な分裂回数
の増加に比例し上昇する。
(A): WT マウスの脾臓より CD11c+
DC を単離し、4 g/ml OVA および 50
g/ml Poly(I:C)存在下においてCFSEで蛍光標識したOT-I cellと共培養し、
その 60 時間後に OT-I assay を行った。図は Gating strategy を示しており、
下部では分裂回数の異なる分画を分けて表示し、それぞれの分画中の
CD11c+
cell の割合を示した。右から左にゆくほど分裂回数の多い分画とな
っている。
(B): OVA + PBS または OVA + Poly(I:C)存在下で共培養した後の、OT-I cell
中の分裂した細胞の割合 (左図) および CD11c+
cell の割合 (中央図) を棒
グラフで示した。右図においては分裂回数の異なる分画ごとに分け、各分
画中の CD11c+
cell の割合を棒グラフで示した。
示したデータは 3 回の独立した実験の内のひとつである。
67

68
Figure 3-2. Poly(I:C)による CD11c+
CD8+
T cell 誘導は特定の自然免疫シグ
ナル経路に依存しない。
(A): 様々な遺伝子欠損マウスに EG7 を移植し、OVA 単独、または OVA +
Poly(I:C)治療を行った。その 6 日後に脾臓を採取し、CD8+
T cell 中の OVA
tetramer+
cell (上図)および CD11c+
cell (下図) の割合を解析した。
実験には各群 2-3 匹のマウスを用いた。
(B): DC を Poly(I:C)刺激した際の、エンドソーム経路および細胞質内経路
の下流で誘導される CTL 活性化レベルの概念図を示した。

69
4. CD11c+
CD8+
T cell の増加は複数の腫瘍治療モデルにおいても認められ
る
前節までの OVA + Poly(I:C)治療による EG7 腫瘍退縮モデルの解析では、
CD11c+
CD8+
T cell 増加レベルは抗原特異的 CTL 誘導レベルおよび腫瘍退
縮程度と相関しており、治療奏功性を評価するひとつの指標となり得る可
能性が示された。そこで次に、上記のモデル以外の腫瘍退縮モデルにおい
ても同様の現象が認められるか解析した。まず、TLR3 と異なるシグナル
経路である TLR2-MyD88 経路依存的に DC の成熟化、CTL 活性化を誘導す
る TLR2 アジュバント 47-50 を用いて腫瘍退縮実験を行った。実験には
TLR2/6のリガンドである Macrophage activating lipopeptide-2 (MALP2) のペ
プチド配列のうち、Pam2 配列とそれに引き続く 5 つのペプチドのみを有
する MALP2s (Pam2-CGNNDE) をアジュバントとして用いた。EG7-担がん
マウスに OVA + MALP2s 併用投与を行ったところ、OVA 単独投与と比較し
有意な腫瘍退縮および DLN、腫瘍組織内での OVA 抗原特異的 CD8+
T cell
増加が誘導された (Fig 4-1A, B左図)。このとき、DLN、腫瘍組織共に CD11c+
CD8+
T cell の割合も増加しており (Fig 4-1B 右図)、TLR2 アジュバント治
療時にも Poly(I:C)治療時と同様の現象が起こることが示された。
次に、EG7 以外の腫瘍細胞株である WT1-C1498 担がんマウスを用いた
Poly(I:C)投与実験においても解析を行った。WT1-C1498 担がんマウスに
Poly(I:C)単独投与を行ったところ、PBS 投与と比較し有意に腫瘍退縮が誘
導された (Fig 4-2A)。その際、脾臓および腫瘍組織における WT1 tetramer+
CD8+
T cell の増加は検出されなかった (Fig 4-2B 左図)。しかし CD8+
T cell
の活性化の指標 51 のひとつである CD62L-
CD8+
T cell の割合が Poy(I:C)投
与で増加していること (Fig 4-2B 右図)、また第一章の Figure 1-1B, F の結
果から、tetramer assay では検出限界以下であるが Poly(I:C)投与により抗原
特異的 CD8+
T cell が誘導されていると考えられた。またこのとき、CD11c+
CD8+
T cell の割合も増加していた (Fig 4-2B 中央図)。Poly(I:C)単独治療で
は腫瘍抗原との併用時よりも腫瘍退縮誘導能は弱いため、Poly(I:C)群内に
は強力に腫瘍退縮が誘導された個体がいた一方、腫瘍退縮があまり誘導さ
れなかった個体も存在した (Fig 4-2C 左図)。そこで PBS 群内の一個体、
Poly(I:C)群内の Poly(I:C)に対する応答性が強かった一個体、および応答性
が弱かった一個体の脾臓、腫瘍中の CD11c+
CD8+
T cell の割合を比較した
ところ、Poly(I:C)応答性が弱かった個体では CD11c+ 分画の増加は小さか
ったのに対し、応答性の強かった個体では大きく増加していた (Fig 4-2C
右図)。また、腫瘍内 CD11c-および CD11c
+ CD8
+ T cell 分画を単離し、Ifng、

70
Gzmb、Prf1 の遺伝子発現を解析したところ、EG7-担がんモデルと同様に、
腫瘍内 CD11c+ 分画は活性化 CTL の表現型を示していた (Fig 4-2D)。
以上の結果より、抗原特異的 CD11c+
CD8+
T cell の増加は、複数の抗腫
瘍治療モデルにおいて共通に認められる現象であり、抗腫瘍免疫療法にお
ける治療著効性評価の有用な指標となり得ることが明らかとなった。
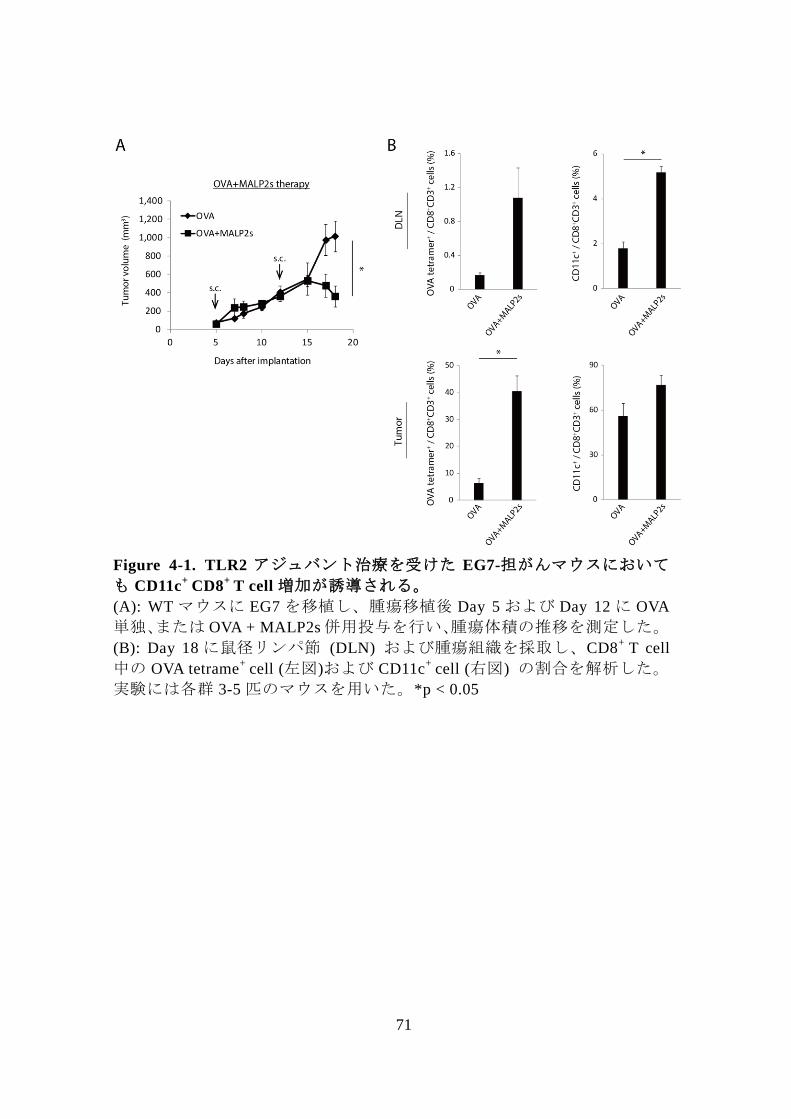
71
Figure 4-1. TLR2 アジュバント治療を受けた EG7-担がんマウスにおいて
も CD11c+
CD8+
T cell 増加が誘導される。
(A): WT マウスに EG7 を移植し、腫瘍移植後 Day 5 および Day 12 に OVA
単独、または OVA + MALP2s 併用投与を行い、腫瘍体積の推移を測定した。
(B): Day 18 に鼠径リンパ節 (DLN) および腫瘍組織を採取し、CD8+
T cell
中の OVA tetrame+
cell (左図)および CD11c+
cell (右図) の割合を解析した。
実験には各群 3-5 匹のマウスを用いた。*p < 0.05
72

73
Figure 4-2. Poly(I:C)治療を受けた WT1-C1498-担がんマウスにおいても
CD11c+
CD8+
T cell 増加が誘導される。
(A): WT マウスに WT1-C1498 を移植し、腫瘍移植後 Day 5 および Day 12
に PBS、または Poly(I:C)単独投与を行い、腫瘍体積の推移を測定した。
(B): Day 14 に脾臓および腫瘍組織を採取し、CD8+
T cell 中の WT1 tetramer+
cell (左図)、CD11c+
cell (中央図)、および CD62L- cell (右図)の割合を解析し
た。
(C): 上記 A において、PBS 群の中の代表的な一個体を、また Poly(I:C)群
の中で腫瘍退縮があまり誘導されなかった一個体および強く腫瘍退縮が誘
導された一個体を選出し、腫瘍径の推移 (左図) と脾臓、腫瘍内の CD8+
T
cell 中の CD11c+
cell の割合を示した (右図)。
(D): 腫瘍内浸潤 CD11c-および CD11c
+ CD8
+ T cell 分画を単離し、定量 PCR
により Infg、Gzmb、Prf1 遺伝子発現レベルを解析した。
実験には各群 3-4 匹のマウスを用いた。*p < 0.05

74
考察
第二章では、アジュバントを用いた抗腫瘍免疫療法において、CD8+
T cell
が DC により抗原提示を受けると、その増殖シグナルの強さに比例して
CD11c 分子の発現増強が誘導されることを示した。増殖した CD11c+
CD8+
T
cell 分画は高い細胞傷害活性を有し、EG7 と WT1-C1498 の 2 種類の腫瘍移
植マウスに対する腫瘍退縮誘導に寄与することが明らかとなった。この
CD11c+
CD8+
T cell 分画の増殖レベルは腫瘍抗原特異的 CD8+
T cell の増殖
レベルおよび腫瘍退縮程度と相関しており、抗腫瘍免疫療法の奏功性を評
価する新たな指標となり得ることが示された。
CD11c はインテグリンX 鎖であり、ミエロイド系細胞、特に DC で高発
現している分子である 52,53。この分子は、定常状態のマウスの脾臓やリン
パ節、胸腺、肝臓、骨髄といった全身の各種臓器中に局在する CD8+
T cell
のうち、一部の分画にも発現している 54,55。また CD11c+ 分画は小腸にも
存在しており、小腸における CD8+
T cell は他の臓器と比較し、非常に高い
割合で CD11c 分子を発現していることが知られている 54,56。またマウスが
病原性細菌や原虫、ウイルスに感染した場合には、血中やリンパ組織、感
染局所において CD11c+ 分画の大幅な増加が誘導される。このような感染
時に増加した CD11c+ 分画は高い IFN-産生能および細胞傷害活性を有し、
分子発現パターンから short-lived effector CD8+
T cell と同様の表現型を示
すことが報告されている 43-46。Listeria monocytogenes 感染モデルマウスに
おいては CD11c+
CD8+
T cell の細胞傷害活性は CD11c 分子を阻害すること
で減弱しており、この分子が単なる活性化マーカーではなく、機能的な作
用を有していると考えられる 43。
本研究において、非担がんマウスや EG7-担がんマウスを用いた実験によ
り、アジュバントを用いず OVA を単独投与した場合では、抗原特異的 CD8+
T cell 増加や腫瘍退縮は誘導されないことが示された。しかし、そのよう
な場合においてもリンパ組織中には少数の CD11c+
CD8+
T cell が存在して
いた (Fig 1-1C,D, Fig 1-3A)。この CD11c+ 分画は活性化 CTL 様の遺伝子発
現パターンを示していることより (Fig 1-2C)、たとえアジュバントの様な
強力な免疫応答が無くとも、内因性の抗原刺激により生体内にはごく少数
の活性型 CD11c+
CD8+
T cell が一定数含まれている可能性が示唆された。
しかしながら、Poly(I:C)の様なアジュバント刺激が無い場合では、腫瘍抗
原特異的 CD11c+ 分画は量的、質的に不十分であり腫瘍退縮を誘導するこ

75
とが出来ない。従って、DC を介して抗原特異的 CD11c+ CD8
+ T cell の増殖、
細胞傷害活性の増強を惹起するアジュバントの存在は抗腫瘍免疫療法にお
いて不可欠といえる。
CD11c+
CD8+
T cell の細胞傷害活性について複数報告がなされている半
面、対照的に免疫抑制性作用を有するという報告もなされている 57。Listeria
感染モデルマウスにおいては、CD11chi 分画が活性化した CD4
+ T cell を殺
傷し、過剰な組織傷害を防いでいることが示されている 43。また、4-1BB
により誘導されるリウマチ性関節炎モデルマウスや自己免疫性ぶどう膜網
膜炎モデルマウスにおいては、CD11c+ 分画より産生される IFN-が、マク
ロファージや DC における免疫抑制性分子 Indoleamine 2,3-dioxygenase
(IDO) の発現を増加させることで病態を改善させることが示されている58,59。以上のように、報告されている CD11c
+ 分画の免疫抑制能の一部はそ
の細胞傷害能や IFN-産生能に起因するものであるため、高い細胞傷害活
性を有するがゆえに引き起こされ得る過剰な傷害を防ぐための機構である
と推察できる。
本研究の Fig 1-3C の実験においては、OVA + Poly(I:C)により誘導された
非担がんマウス由来 CD11c+ 分画が、標的になる抗原を全く発現していな
い EL4 細胞に対しても僅かに細胞傷害活性を発揮していた。これは CD11c+
分画が非特異的な細胞傷害能をも有していることを示しており、長期的な
存続は標的細胞以外の傷害を引き起こす可能性を示唆している。この観点
からみれば、病源体感染時に増加した CD11c+ 分画が感染の終息とともに
再び減少するという動態を示すのも、上記のような過剰な組織傷害を防ぐ
ための機構の一つであると考えられる。
リンパ組織においてクロスプライミングを受け活性化した抗原特異的
CTL は、遊走能を獲得し腫瘍組織に移動した後、腫瘍細胞を傷害する。し
かし、腫瘍内では PD-L1 等をはじめとする抑制性因子により CTL が疲弊
状態に陥る場合もある。そのため、腫瘍内浸潤 CD8+
T cell の存在数や活性
化の状態は、抗腫瘍免疫療法の成否に大きく影響するといえる。このよう
な腫瘍内浸潤 CD8+
T cell の解析を行ったところ、Poly(I:C)刺激の有無に関
わらず、腫瘍内 CD8+
T cell 中にはリンパ組織と比較して非常に高い割合で
CD11c+ 分画が含まれていることが示された (Fig 2A)。またその表現型は
活性化 CTL 様の表現型を示していた (Fig 2D, E)。このことより、アジュバ
ント刺激が無い場合でも、腫瘍内に浸潤している CD8+
T cell の大部分は活

76
性型 CTL であることが分かる。Poly(I:C)はリンパ組織でのクロスプライミ
ングおよび CTL の遊走を促進することで、腫瘍への活性化 CD11c+
CD8+
T
cell の浸潤を増加させ、腫瘍退縮を導くと考えられる。このように誘導さ
れた腫瘍内浸潤 CD11c+ 分画は Pdcd 遺伝子を高発現していることから、
PD-L1 による抑制機構のターゲットになり易いと推察される。そのため、
Poly(I:C)と PD-1/PD-L1 阻害の併用療法は理にかなった効果的な戦略であ
ると言え、事実、Poly(I:C)単独に比較して抗 PD-L1 抗体の併用投与により、
より強力に腫瘍退縮が誘導されることが示されている 26,60。
本研究では主に、OVA 抗原特異的 CTL 解析法が確立されている、EG-7
担がんマウスに腫瘍抗原 OVA + Poly(I:C)を併用投与するという実験系を用
いた。しかし実際の人の臨床においては、腫瘍抗原エピトープを含む短鎖
ペプチドを用いた抗原ペプチド療法では、上記マウス実験系ほど強力な抗
腫瘍免疫応答を誘導することは難しい 61,62。その理由としては、単一エピ
トープしか含まれていないことによる免疫原生の低さや HLA 拘束性が挙
げられる 63。また腫瘍抗原タンパクや長鎖ペプチドを投与する場合は、こ
れらの問題を回避し得る可能性があるが、質の高い長鎖ペプチド抗原を充
分量合成することは現時点では困難である 64。そのため、EG7 実験モデル
よりもより実際の臨床に近いモデルとして、WT1-C1498 担がんマウスに対
する Poly(I:C)単独投与実験を行った (Fig 4-2)。その結果、この実験系にお
いても CD8+
T cell の活性化、腫瘍退縮に相関して CD11c+ 分画の増加が認
められた。更に TLR2 アジュバントを用いた治療モデルにおいても同様の
現象が認められた (Fig 4-1)。これらの知見は、実際の臨床において様々な
癌種に対する抗腫瘍免疫療法を行う際、CD11c+ CD8
+ T cell の増加レベルが、
治療奏功性評価の有用かつ簡便な指標となり得る可能性を示している。
77
総括および結論
本研究により、以下の知見が得られた。
第一章
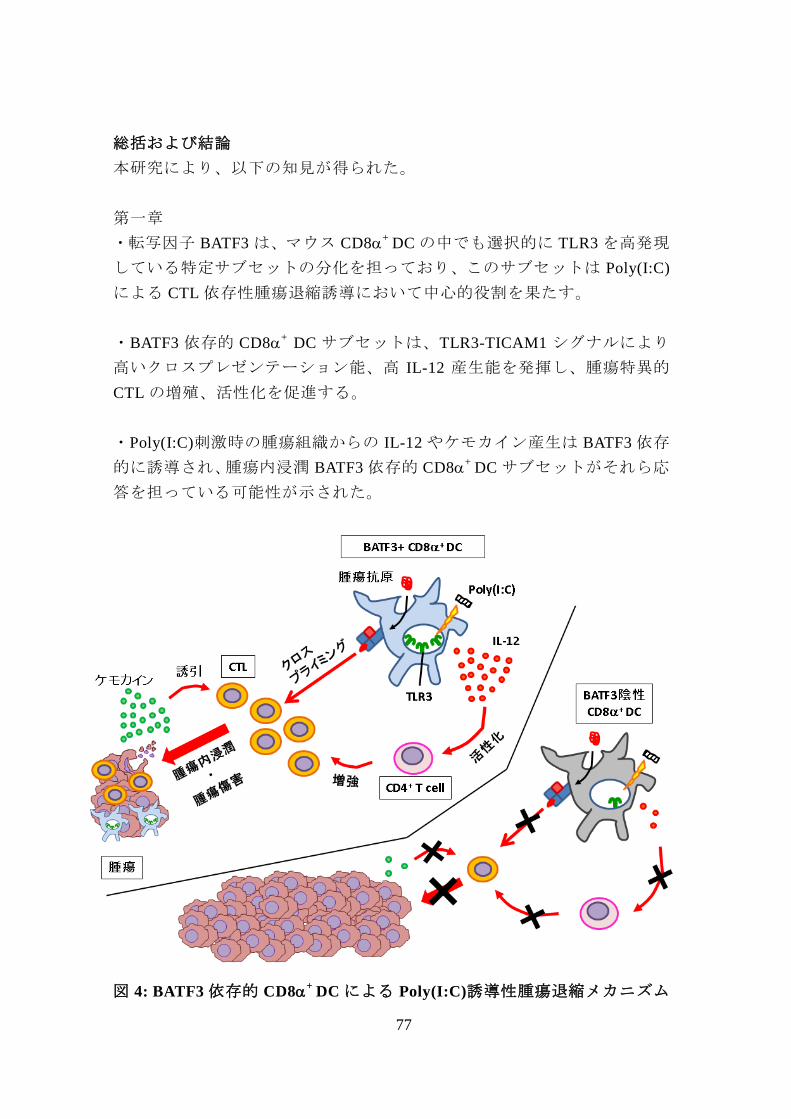
・転写因子 BATF3 は、マウス CD8+
DC の中でも選択的に TLR3 を高発現
している特定サブセットの分化を担っており、このサブセットは Poly(I:C)
による CTL 依存性腫瘍退縮誘導において中心的役割を果たす。
・BATF3 依存的 CD8+
DC サブセットは、TLR3-TICAM1 シグナルにより
高いクロスプレゼンテーション能、高 IL-12 産生能を発揮し、腫瘍特異的
CTL の増殖、活性化を促進する。
・Poly(I:C)刺激時の腫瘍組織からの IL-12 やケモカイン産生は BATF3 依存
的に誘導され、腫瘍内浸潤 BATF3 依存的 CD8+
DC サブセットがそれら応
答を担っている可能性が示された。
図 4: BATF3 依存的 CD8+
DC による Poly(I:C)誘導性腫瘍退縮メカニズム
78
第二章
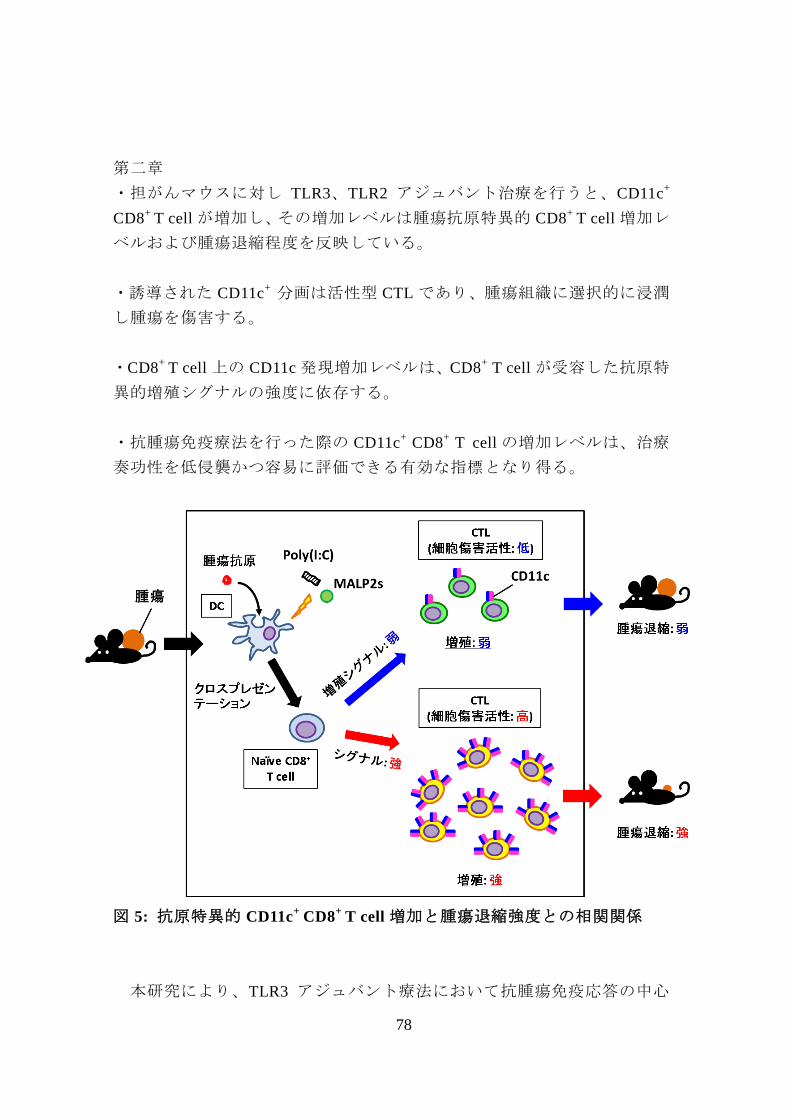
・担がんマウスに対し TLR3、TLR2 アジュバント治療を行うと、CD11c+
CD8+
T cell が増加し、その増加レベルは腫瘍抗原特異的 CD8+
T cell 増加レ
ベルおよび腫瘍退縮程度を反映している。
・誘導された CD11c+ 分画は活性型 CTL であり、腫瘍組織に選択的に浸潤
し腫瘍を傷害する。
・CD8+
T cell 上の CD11c 発現増加レベルは、CD8+ T cell が受容した抗原特
異的増殖シグナルの強度に依存する。
・抗腫瘍免疫療法を行った際の CD11c+
CD8+
T cell の増加レベルは、治療
奏功性を低侵襲かつ容易に評価できる有効な指標となり得る。
図 5: 抗原特異的 CD11c+
CD8+
T cell 増加と腫瘍退縮強度との相関関係
本研究により、TLR3 アジュバント療法において抗腫瘍免疫応答の中心

79
的役割を担う樹状細胞サブセットの同定、およびその腫瘍退縮制御メカニ
ズムの一端を明らかにした。また、上記メカニズムにより誘導される腫瘍
抗原特異的 CTL に特徴的な CD11c 分子の発現増加という新たな表現型を
見出し、有用性の高い治療奏功性評価の指標となり得る可能性を示した。
Poly(I:C)は抗腫瘍応答を誘導する物質として 1960 年代に合成された後、
盛んにがん患者に対する臨床治験が行われた。その際、効果的な腫瘍退縮
が認められる症例が多く確認されたが、同時に強い副作用を伴うことが明
らかとなり現在では殆どの症例で適応外となっている 15。しかし、今なお
Poly(I:C)を用いた研究が数多く行われているのは、Poly(I:C)が強力な腫瘍
退縮誘導能を有するために他ならない。その後の研究により、Poly(I:C)が
TLR3 と MAVS を介した 2 つの経路を活性化することが明らかとなり、そ
して瀬谷研究室のこれまでの研究に加え、本研究により TLR3 経路の重要
性、またその抗腫瘍免疫誘導メカニズムが明らかにされつつある。そのよ
うな知見のもと、瀬谷研究室では TLR3 特異的アジュバント開発や、副作
用を最小限に抑えつつ抗腫瘍効果を最大限に発揮する様な治療戦略の研究
を行っている。本研究第一章で得られた知見は、より優れた TLR3 アジュ
バント療法の確立に寄与するものであり、また第二章で得られた知見は治
療の際の応答性評価に利用でき、治療継続の可否や予後の予測に有用であ
る。更に、その評価は TLR3 アジュバント療法に限定されず、様々な治療
症例に利用出来る可能性も示された。
この様に、本研究で得られた知見は TLR3 アジュバント療法を始めとし
た抗腫瘍免疫療法の更なる発展に寄与し得るものであるが、実際の臨床症
例に応用してゆくためには、マウス実験モデルと同様の現象がヒトにおい
ても認められるかの評価が必要である。そのため、今後は他の組織と連携
し、ヒトにおける知見を蓄積しつつ、より有用性の高い TLR3 アジュバン
ト療法の実用化を目指した橋渡し研究に繋げてゆきたいと考える。
謝辞
本研究の推進を医学研究科としてサポートして下さった北海道大学大学
院医学研究科 分子病理学分野 笠原正典教授に感謝致します。
本研究の遂行において終始適切なご指導、ご討論を賜り、本論文作成に
あたって惜しみないご助力をいただきました北海道大学大学院医学研究科
分子病理学分野 瀬谷司客員教授ならびに松本美佐子客員教授に深く感謝

80
致します。
実験の基礎的手技から丁寧にご指導頂き、また研究の素晴らしさを教え
て下さいました元北海道大学大学院医学研究科 免疫学分野 東正大助教
に心より感謝致します。また、本論文作成にあたり多大なご協力を賜りま
した、理化学研究所統合生命医学研究センター 免疫転写制御研究グルー
プ 海老原敬上級研究員ならびに大阪大学大学院医学系研究科 中島博子
特任准教授に厚く御礼申し上げます。
本論文を審査して頂きました、北海道大学遺伝子病制御研究所 免疫機
能学分野 北村秀光准教授、北海道大学大学院医学研究科 病原微生物学分
野 有川二郎教授、北海道大学大学院医学研究科 腫瘍病理学分野 田中
伸哉教授、北海道大学遺伝子病制御研究所 免疫生物分野 清野研一郎教
授に深く感謝いたします。
研究生活のあらゆる面でお世話になりました、北海道大学大学院医学研
究科 免疫学分野の皆様に心より感謝致します。
最後に、長い学生生活を見守り、精神的、経済的に支えて下さいました
両親に深く感謝致します。
2017 年 3 月
引用文献
1. Iwasaki, A. & Medzhitov, R. Control of adaptive immunity by the innate
immune system. Nat. Immunol. 16, 343-353 (2015).
2. Gasteiger, G. & Rudensky, A. Y. Interactions between innate and adaptive
lymphocytes. Nat. Rev. Immunol. 14, 631-639 (2014).
3. Blander, J. M. & Sander, L. E. Beyond pattern recognition: five immune
checkpoints for scaling the microbial threat. Nat. Rev. Immunol. 12, 215-225
(2012).
4. Kono, H. & Rock, K. L. How dying cells alert the immune system to danger.
Nat. Rev. Immunol. 8, 279-289 (2008).
5. Takeuchi, O. & Akira, S. Pattern Recognition Receptors and Inflammation.
Cell 140, 805-820 (2010).
6. Kawai, T. & Akira, S. The roles of TLRs, RLRs and NLRs in pathogen

81
recognition. Int. Immunol. 21, 317-337 (2009).
7. Desmet, C. J. & Ishii, K. J. Nucleic acid sensing at the interface between
innate and adaptive immunity in vaccination. Nat. Rev. Immunol. 12, 479-491
(2012).
8. Oshiumi, H., Matsumoto, M., Funami, K., Akazawa, T. & Seya, T. TICAM-1,
an adaptor molecule that participates in Toll-like receptor 3-mediated
interferon-beta induction. Nat. Immunol. 4, 161-167 (2003).
9. Kato, H., Takeuchi, O., Sato, S., Yoneyama, M., Yamamoto, M., Matsui, K.,
Uematsu, S., Jung, A., Kawai, T., Ishii, K.J., Yamaguchi, O., Otsu, K., Tsujimura,
T., Koh, C.S., Reis e Sousa, C., Matsuura, Y., Fujita, T. & Akira, S. Differential
roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature
441, 101-105 (2006).
10. Yoshida, R., Takaesu, G., Yoshida, H., Okamoto, F., Yoshioka, T., Choi, Y.,
Akira, S., Kawai, T., Yoshimura, A. & Kobayashi, T. TRAF6 and MEKK1 play a
pivotal role in the RIG-I-like helicase antiviral pathway. J. Biol. Chem. 283,
36211-36220 (2008).
11. Kawai, T., Takahashi, K., Sato, S., Coban, C., Kumar, H., Kato, H., Ishii, K.J.,
Takeuchi, O. & Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated
type I interferon induction. Nat. Immunol. 6, 981-988 (2005).
12. Seth, R. B., Sun, L., Ea, C. K. & Chen, Z. J. Identification and
characterization of MAVS, a mitochondrial antiviral signaling protein that
activates NF-kappaB and IRF 3. Cell 122, 669-682 (2005).
13. Kawai, T. & Akira, S. Signaling to NF-κB by Toll-like receptors. Trends Mol.
Med. 13, 460-469 (2007).
14. Joffre, O. P., Segura, E., Savina, A. & Amigorena, S. Cross-presentation by
dendritic cells. Nat. Rev. Immunol. 12, 557-569 (2012).
15. Seya, T., Shime, H., Takeda, Y., Tatematsu, M., Takashima, K. & Matsumoto,
M. Adjuvant for vaccine immunotherapy of cancer--focusing on Toll-like
receptor 2 and 3 agonists for safely enhancing antitumor immunity. Cancer Sci.
106, 1659-1668 (2015).
16. Dalod, M., Chelbi, R., Malissen, B. & Lawrence, T. Dendritic cell
maturation: functional specialization through signaling specificity and
transcriptional programming. EMBO. J. 33, 1104-1116 (2014).
17. Galluzzi, L., Vacchelli, E., Eggermont, A., Fridman, W. H., Galon, J.,
Sautès-Fridman, C., Tartour, E., Zitvogel, L. & Kroemer, G. Trial Watch:

82
Experimental Toll-like receptor agonists for cancer therapy. Oncoimmunology 1,
699-716 (2012).
18. Azuma, M., Ebihara, T., Oshiumi, H., Matsumoto, M. & Seya, T.
Cross-priming for antitumor CTL induced by soluble Ag + polyI:C depends on
the TICAM-1 pathway in mouse CD11c(+)/CD8α(+) dendritic cells.
Oncoimmunology 1, 581-592 (2012).
19. Akazawa, T., Ebihara, T., Okuno, M., Okuda, Y., Shingai, M., Tsujimura, K.,
Takahashi, T., Ikawa, M., Okabe, M., Inoue, N., Okamoto-Tanaka, M., Ishizaki,
H., Miyoshi, J., Matsumoto, M. & Seya, T. Antitumor NK activation induced by
the Toll-like receptor 3-TICAM-1 (TRIF) pathway in myeloid dendritic cells.
Proc. Natl. Acad. Sci. U. S. A. 104, 252-257 (2007).
20. Kasamatsu, J., Azuma, M., Oshiumi, H., Morioka, Y., Okabe, M., Ebihara, T.,
Matsumoto, M., & Seya, T. INAM plays a critical role in IFN-γ production by
NK cells interacting with polyinosinic-polycytidylic acid-stimulated accessory
cells. J. Immunol. 193, 5199-5207 (2014).
21. Temizoz, B., Kuroda, E. & Ishii, K. J. Vaccine adjuvants as potential cancer
immunotherapeutics. Int. Immunol. 28, 329-338 (2016).
22. Levine, A. S., Sivulich, M., Wiernik, P. H. & Levy, H. B. Initial clinical trials
in cancer patients of polyriboinosinic-polyribocytidylic acid stabilized with
poly-L-lysine, in carboxymethylcellulose [poly(ICLC)], a highly effective
interferon inducer. Cancer Res. 39, 1645-1650 (1979).
23. Matsumoto, M., Tatematsu, M., Nishikawa, F., Azuma, M., Ishii, N.,
Morii-Sakai, A., Shime, H. & Seya, T. Defined TLR3-specific adjuvant that
induces NK and CTL activation without significant cytokine production in vivo.
Nat. Commun. 6, 6280 (2015).
24. Pardoll, D. M. The blockade of immune checkpoints in cancer
immunotherapy. Nat. Rev. Cancer 12, 252-264 (2012).
25. Hamanishi, J., Mandai, M., Matsumura, N., Abiko, K., Baba, T. & Konishi, I.
PD-1/PD-L1 blockade in cancer treatment: perspectives and issues. Int. J. Clin.
Oncol. 21, 462-473 (2016).
26. Kataoka, K., Shiraishi, Y., Takeda, Y., Sakata, S., Matsumoto, M., Nagano, S.,
Maeda, T., Nagata, Y., Kitanaka, A., Mizuno, S., Tanaka, H., Chiba, K., Ito, S.,
Watatani, Y., Kakiuchi, N., Suzuki, H., Yoshizato, T., Yoshida, K., Sanada, M.,
Itonaga, H., Imaizumi, Y., Totoki, Y., Munakata, W., Nakamura, H., Hama, N.,

83
Shide, K., Kubuki, Y., Hidaka, T., Kameda, T., Masuda, K., Minato, N.,
Kashiwase, K., Izutsu, K., Takaori-Kondo, A., Miyazaki, Y., Takahashi, S.,
Shibata, T., Kawamoto, H., Akatsuka, Y., Shimoda, K., Takeuchi, K., Seya, T.,
Miyano, S. & Ogawa, S. Aberrant PD-L1 expression through 3'-UTR disruption
in multiple cancers. Nature 534, 402-406 (2016).
27. Hashimoto, D., Miller, J. & Merad, M. Dendritic cell and macrophage
heterogeneity in vivo. Immunity 35, 323-335 (2011).
28. Bachem, A., Güttler, S., Hartung, E., Ebstein, F., Schaefer, M., Tannert, A.,
Salama, A., Movassaghi, K., Opitz, C., Mages, H. W., Henn, V., Kloetzel, P. M.,
Gurka, S. & Kroczek, R. A. Superior antigen cross-presentation and XCR1
expression define human CD11c+CD141+ cells as homologues of mouse CD8+
dendritic cells. J. Exp. Med. 207, 1273-1281 (2010).
29. Jongbloed, S. L., Kassianos, A. J., McDonald, K. J., Clark, G. J., Ju, X.,
Angel, C. E., Chen, C. J., Dunbar, P. R., Wadley, R. B., Jeet, V., Vulink, A. J.,
Hart, D. N. & Radford, K. J. Human CD141+ (BDCA-3)+ dendritic cells (DCs)
represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J.
Exp. Med. 207, 1247-1260 (2010).
30. Poulin, L. F., Salio, M., Griessinger, E., Anjos-Afonso, F., Craciun, L., Chen,
J. L., Keller, A. M., Joffre, O., Zelenay, S., Nye, E., Le Moine, A., Faure, F.,
Donckier, V., Sancho, D., Cerundolo, V., Bonnet, D. & Reis e Sousa, C.
Characterization of human DNGR-1+ BDCA3+ leukocytes as putative
equivalents of mouse CD8alpha+ dendritic cells. J. Exp. Med. 207, 1261-1271
(2010).
31. Edwards, A. D., Diebold, S. S., Slack, E. M., Tomizawa, H., Hemmi, H.,
Kaisho, T., Akira, S. & Reis e Sousa, C. Toll-like receptor expression in murine
DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with
unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33, 827-833 (2003).
32. Hildner, K., Edelson, B.T., Purtha, W. E., Diamond, M., Matsushita, H.,
Kohyama, M., Calderon, B., Schraml, B. U., Unanue, E. R., Diamond, M. S.,
Schreiber, R. D., Murphy, T. L. & Murphy, K. M. Batf3 deficiency reveals a
critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science
322, 1097-1100 (2008).
33. Tussiwand, R., Lee, W. L., Murphy, T. L., Mashayekhi, M., KC, W., Albring,
J. C., Satpathy, A. T., Rotondo, J. A., Edelson, B. T., Kretzer, N. M., Wu, X.,
Weiss, L. A., Glasmacher, E., Li, P., Liao, W., Behnke, M., Lam, S. S., Aurthur, C.

84
T., Leonard, W. J., Singh, H., Stallings, C. L., Sibley, L. D., Schreiber, R. D. &
Murphy, K. M. Compensatory dendritic cell development mediated by BATF-IRF
interactions. Nature 490, 502-507 (2012).
34. Murphy, T. L., Tussiwand, R. & Murphy, K. M. Specificity through
cooperation: BATF-IRF interactions control immune-regulatory networks. Nat.
Rev. Immunol. 13, 499-509 (2013).
35. Mashayekhi, M., Sandau, M. M., Dunay, I. R., Frickel, E. M., Khan, A.,
Goldszmid, R. S., Sher, A., Ploegh, H. L., Murphy, T. L., Sibley, L. D. & Murphy,
K. M. CD8α(+) dendritic cells are the critical source of interleukin-12 that
controls acute infection by Toxoplasma gondii tachyzoites. Immunity 35, 249-259
(2011).
36. Ashok, D., Schuster, S., Ronet, C., Rosa, M., Mack, V., Lavanchy, C.,
Marraco, S. F., Fasel, N., Murphy, K. M., Tacchini-Cottier, F. & Acha-Orbea, H.
Cross-presenting dendritic cells are required for control of Leishmania major
infection. Eur. J. Immunol. 44, 1422-1432 (2014).
37. Nakajima, H., Oka, Y., Tsuboi, A., Tatsumi, N., Yamamoto, Y., Fujiki, F., Li,
Z., Murao, A., Morimoto, S., Hosen, N., Shirakata, T., Nishida, S., Kawase, I.,
Isaka, Y., Oji, Y. & Sugiyama, H Enhanced tumor immunity of WT1 peptide
vaccination by interferon-β administration. Vaccine 30, 722-729 (2012).
38. Bachem, A., Hartung, E., Güttler, S., Mora, A., Zhou, X., Hegemann, A.,
Plantinga, M., Mazzini, E., Stoitzner, P., Gurka, S., Henn, V., Mages, H. W. &
Kroczek, R. A. Expression of XCR1 Characterizes the Batf3-Dependent Lineage
of Dendritic Cells Capable of Antigen Cross-Presentation. Front. Immunol. 3,
214 (2012).
39. Crozat, K., Guiton, R., Contreras, V., Feuillet, V., Dutertre, C. A., Ventre, E.,
Vu Manh, T. P., Baranek, T., Storset, A. K., Marvel, J., Boudinot, P., Hosmalin,
A., Schwartz-Cornil, I. & Dalod, M. The XC chemokine receptor 1 is a conserved
selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic
cells. J. Exp. Med. 207, 1283-1292 (2010).
40. McNally, B., Willette, M., Ye, F., Partida-Sanchez, S. & Flaño, E. Intranasal
administration of dsRNA analog poly(I:C) induces interferon-α
receptor-dependent accumulation of antigen experienced T cells in the airways.
PLoS One 7, e51351 (2012).
41. Woo, S. R., Fuertes, M. B., Corrales, L., Spranger, S., Furdyna, M. J., Leung,
M. Y., Duggan, R., Wang, Y., Barber, G. N., Fitzgerald, K. A., Alegre, M. L. &

85
Gajewski, T. F. STING-dependent cytosolic DNA sensing mediates innate
immune recognition of immunogenic tumors. Immunity 41, 830-842 (2014).
42. Coulie, P. G., Van den Eynde, B. J., van der Bruggen, P. & Boon, T. Tumour
antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat.
Rev. Cancer 14, 135-146 (2014).
43. Chen, Z., Han, Y., Gu, Y., Liu, Y., Jiang, Z., Zhang, M. & Cao, X.
CD11c(high)CD8+ regulatory T cell feedback inhibits CD4 T cell immune
response via Fas ligand-Fas pathway. J. Immunol. 190, 6145-6154 (2013).
44. Cooney, L. A., Gupta, M., Thomas, S., Mikolajczak, S., Choi, K. Y., Gibson,
C., Jang, I. K., Danziger, S., Aitchison, J., Gardner, M. J., Kappe, S. H. & Wang,
R. Short-lived effector CD8 T cells induced by genetically attenuated malaria
parasite vaccination express CD11c. Infect. Immun. 81, 4171-4181 (2013).
45. Beyer, M., Wang, H., Peters, N., Doths, S., Koerner-Rettberg, C., Openshaw,
P. J. & Schwarze, J. The beta2 integrin CD11c distinguishes a subset of cytotoxic
pulmonary T cells with potent antiviral effects in vitro and in vivo. Respir. Res. 6,
70 (2005).
46. Lin, Y., Roberts, T. J., Sriram, V., Cho, S. & Brutkiewicz, R. R. Myeloid
marker expression on antiviral CD8+ T cells following an acute virus infection.
Eur. J. Immunol. 33, 2736-2743 (2003).
47. Prajeeth, C. K., Jirmo, A. C., Krishnaswamy, J. K., Ebensen, T., Guzman, C.
A., Weiss, S., Constabel, H., Schmidt, R. E. & Behrens, G. M. The synthetic
TLR2 agonist BPPcysMPEG leads to efficient cross-priming against
co-administered and linked antigens. Eur. J. Immunol. 40, 1272-1283 (2010).
48. Shen, K. Y., Song, Y. C., Chen, I. H., Leng, C. H., Chen, H. W., Li, H. J.,
Chong, P. & Liu, S. J. Molecular mechanisms of TLR2-mediated antigen
cross-presentation in dendritic cells. J. Immunol. 192, 4233-4241 (2014).
49. Santone, M., Aprea, S., Wu, T. Y., Cooke, M. P., Mbow, M. L., Valiante, N.
M., Rush, J. S., Dougan, S., Avalos, A., Ploegh, H., De Gregorio, E., Buonsanti,
C. & D'Oro, U. A new TLR2 agonist promotes cross-presentation by mouse and
human antigen presenting cells. Hum. Vaccin. Immunother. 11, 2038-2050 (2015).
50. Weigt, H., Mühlradt, P. F., Emmendörffer, A., Krug, N. & Braun, A. Synthetic
mycoplasma-derived lipopeptide MALP-2 induces maturation and function of
dendritic cells. Immunobiology 207, 223-233 (2003).
51. Obar, J. J. & Lefrançois, L. Early events governing memory CD8+ T-cell
differentiation. Int. Immunol. 22, 619-625 (2010).

86
52. Myones, B. L., Dalzell, J. G., Hogg, N. & Ross, G. D. Neutrophil and
monocyte cell surface p150,95 has iC3b-receptor (CR4) activity resembling CR3.
J. Clin. Invest. 82, 640-651 (1988).
53. Metlay, J. P., Witmer-Pack, M. D., Agger, R., Crowley, M. T., Lawless, D. &
Steinman, R. M. The distinct leukocyte integrins of mouse spleen dendritic cells
as identified with new hamster monoclonal antibodies. J. Exp. Med. 171,
1753-1771 (1990).
54. Huleatt, J. W. & Lefrançois, L. Antigen-driven induction of CD11c on
intestinal intraepithelial lymphocytes and CD8+ T cells in vivo. J. Immunol. 154,
5684-5693 (1995).
55. Vinay, D. S., Kim, C. H., Choi, B. K. & Kwon, B. S. Origins and functional
basis of regulatory CD11c+CD8+ T cells. Eur. J. Immunol. 39, 1552-1563 (2009).
56. Fujiwara, D., Chen, L., Wei, B. & Braun, J. Small intestine CD11c+ CD8+ T
cells suppress CD4+ T cell-induced immune colitis. Am. J. Physiol. Gastrointest.
Liver. Physiol. 300, G939-947 (2011).
57. Vinay, D. S. & Kwon, B. S. CD11c+CD8+ T cells: two-faced adaptive
immune regulators. Cell. Immunol. 264, 18-22 (2010).
58. Seo, S. K., Choi, J. H., Kim, Y. H., Kang, W. J., Park, H. Y., Suh, J. H., Choi,
B. K., Vinay, D. S. & Kwon, B. S. 4-1BB-mediated immunotherapy of
rheumatoid arthritis. Nat. Med. 10, 1088-1094 (2004).
59. Choi, B. K., Asai, T., Vinay, D. S., Kim, Y. H. & Kwon, B. S.
4-1BB-mediated amelioration of experimental autoimmune uveoretinitis is
caused by indoleamine 2,3-dioxygenase-dependent mechanisms. Cytokine 34,
233-242 (2006).
60. Nagato, T., Lee, Y. R., Harabuchi, Y. & Celis, E. Combinatorial
immunotherapy of polyinosinic-polycytidylic acid and blockade of programmed
death-ligand 1 induce effective CD8 T-cell responses against established tumors.
Clin. Cancer Res. 20, 1223-1234 (2014).
61. Rosenberg, S. A., Yang, J. C. & Restifo, N. P. Cancer immunotherapy:
moving beyond current vaccines. Nat. Med. 10, 909-915 (2004).
62. Eggermont, A. M. Therapeutic vaccines in solid tumours: can they be
harmful? Eur. J. Cancer 45, 2087-2090 (2009).
63. Vici, P., Mariani, L., Pizzuti, L., Sergi, D., Di Lauro, L., Vizza, E., Tomao, F.,
Tomao, S., Cavallotti, C., Paolini, F. & Venuti, A. Immunologic treatments for
precancerous lesions and uterine cervical cancer. J. Exp. Clin. Cancer Res. 33, 29

87
(2014).
64. Van Driessche, A., Berneman, Z. N. & Van Tendeloo, V. F. Active specific
immunotherapy targeting the Wilms' tumor protein 1 (WT1) for patients with
hematological malignancies and solid tumors: lessons from early clinical trials.
Oncologist 17, 250-259 (2012).





























