Embed Size (px)
Citation preview

A data intensive future:How can biology take full advantage of the
coming data deluge?
C. Titus BrownSchool of Veterinary Medicine;
Genome Center & Data Science Initiative1/22/16
#titusplantz
Slides are on slideshare.net/c.titus.brown/

(My one plant collaboration)
Helping Shamoni Maheshwari (Comai Lab) w/analysis of ChIP-seq data from
Arabidopsis.

Outline
0. Background: what’s coming?1. Research: what do we do with infinite data?2. Development: software and infrastructure.3. Open science & reproducibility.4. Training

0. Background
What is going to be happening in the next 5 years with biological data
generation?

DNA sequencing rates continues to grow.
Stephens et al., 2015 - 10.1371/journal.pbio.1002195

(2015 was a good year)

Oxford Nanopore sequencing
Slide via Torsten Seeman
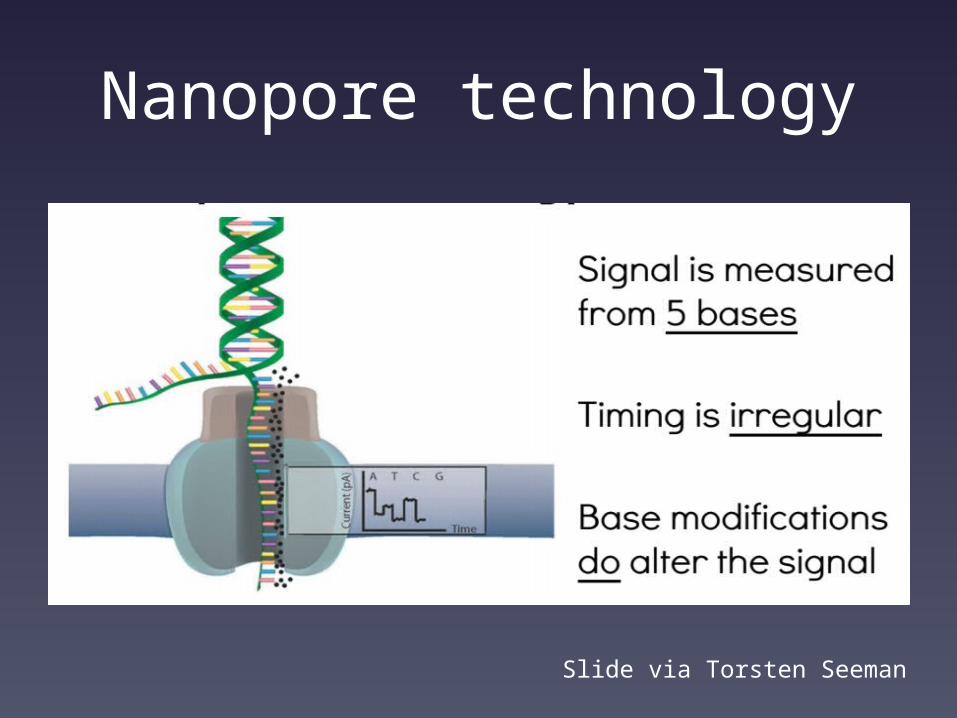
Nanopore technology
Slide via Torsten Seeman

Scaling up --

Scaling up --

Slide via Torsten Seeman

http://ebola.nextflu.org/

“Fighting Ebola With a Palm-Sized DNA Sequencer”
See: http://www.theatlantic.com/science/archive/2015/09/
ebola-sequencer-dna-minion/405466/

“DeepDOM” cruise: examination of dissolved organic matter & microbial metabolism vs physical parameters – potential collab.
Via Elizabeth Kujawinski
Another challenge beyond volume and velocity – variety.
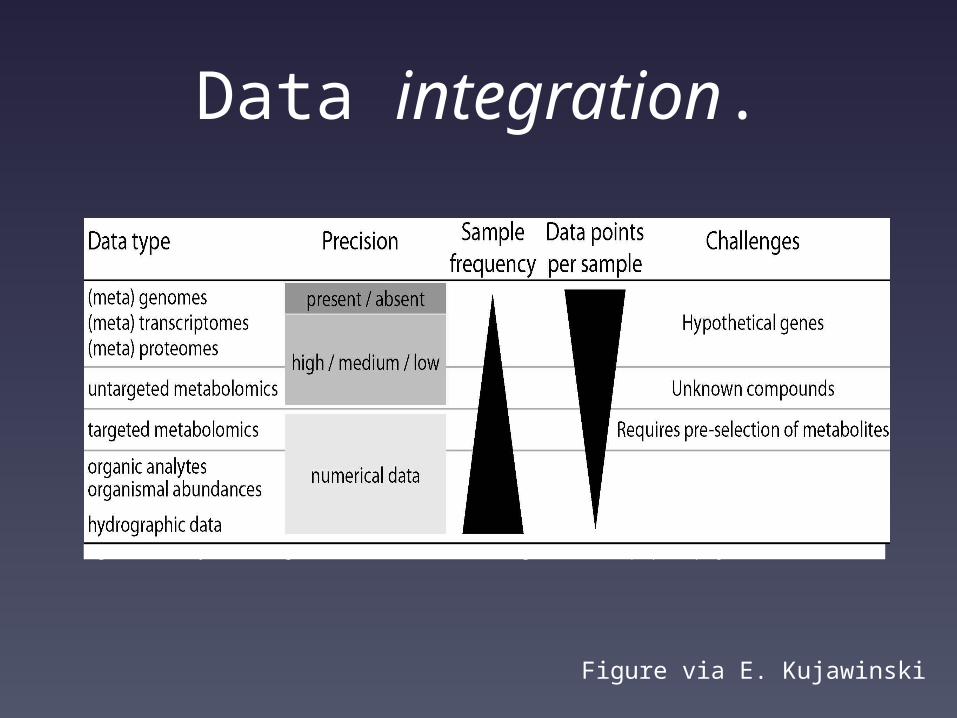
Data integration.
Figure 2. Summary of challenges associated with the data integration in the proposed project.
Figure via E. Kujawinski

CRISPRThe challenge with genome editing is
fast becoming what to edit rather than how to do.

A point for reflection…
Increasingly, the best guide to the next 10 years of biology is science fiction ...

1. Research
Working with ~infinite amounts of data, and doing something effective
with it.

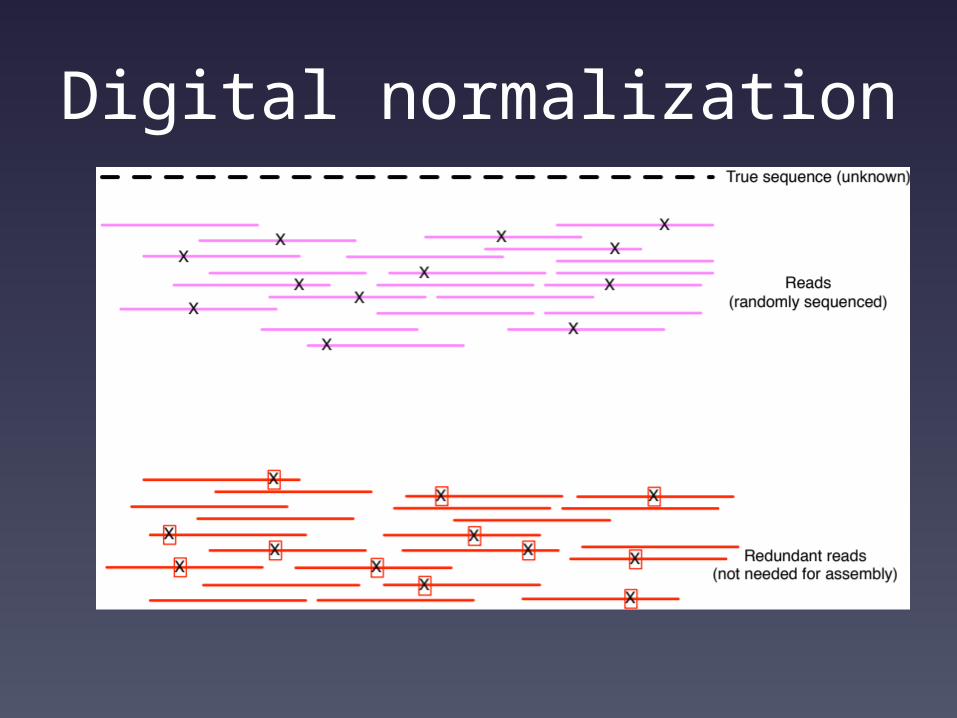
Shotgun sequencing and coverage
“Coverage” is simply the average number of reads that overlap
each true base in genome.
Here, the coverage is ~10 – just draw a line straight down from the top through all of the reads.

Random sampling => deep sampling needed
Typically 10-100x needed for robust recovery (30-300 Gbp for human)

Digital normalization

Digital normalization
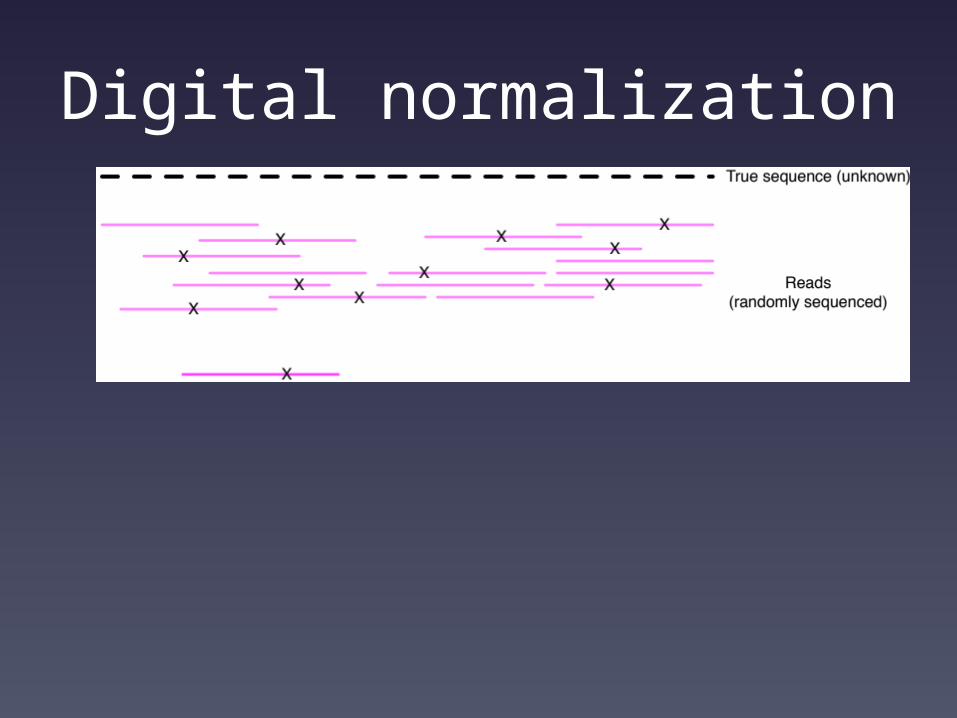
Digital normalization

Digital normalization
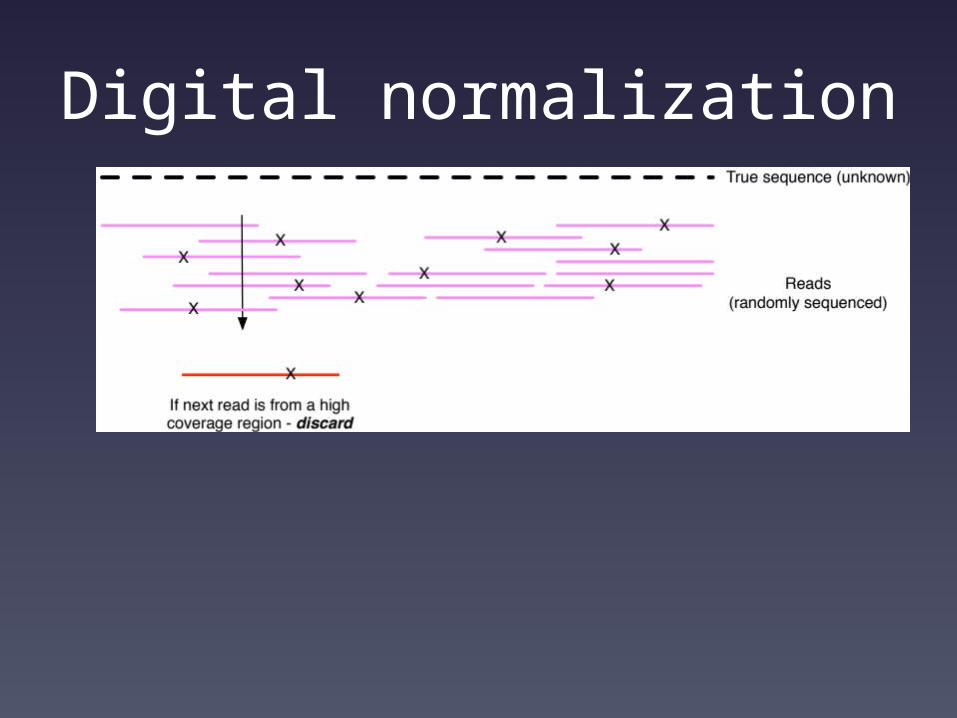
Digital normalization
Digital normalization

Computational problems now scale with information content rather than data set size.
Most samples can be reconstructed via de novo assembly on commodity computers.
A local collaboration:The horse genome & transcriptome
Tamer Mansour w/Bellone, Finno, Penedo, & Murray labs.
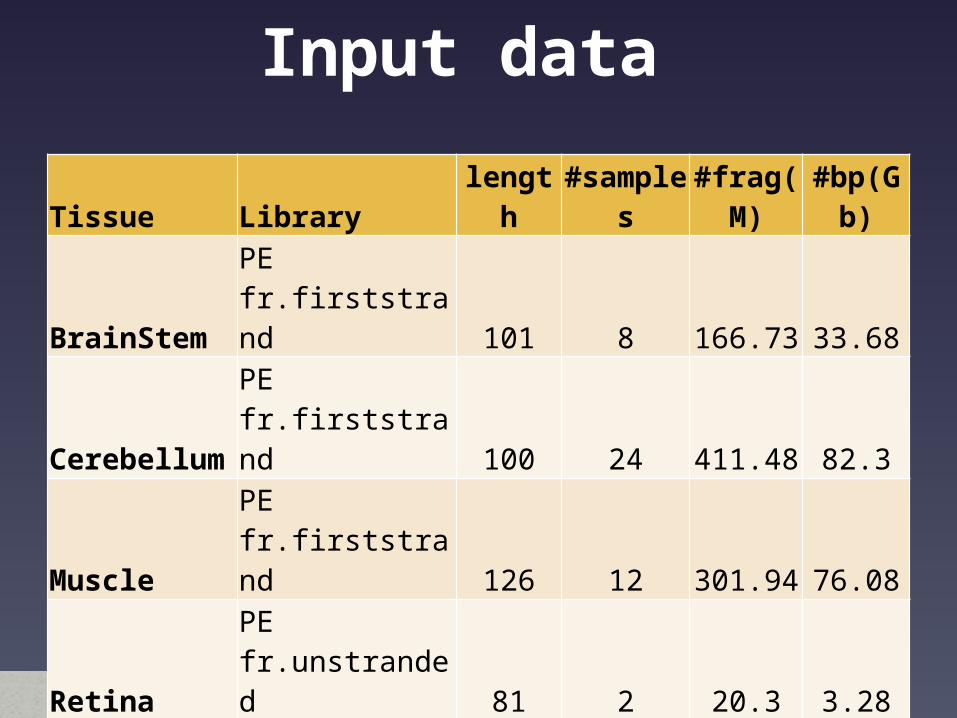
Input data Tissue Library length #samples #frag(M) #bp(Gb)BrainStem PE fr.firststrand 101 8 166.73 33.68Cerebellum PE fr.firststrand 100 24 411.48 82.3Muscle PE fr.firststrand 126 12 301.94 76.08Retina PE fr.unstranded 81 2 20.3 3.28SpinalCord PE fr.firststrand 101 16 403 81.4Skin PE fr.unstranded 81 2 18.54 3
SE fr.unstranded 81 2 16.57 1.34SE fr.unstranded 95 3 105.51 10.02
Embryo ICM PE fr.unstranded 100 3 126.32 25.26SE fr.unstranded 100 3 115.21 11.52
Embryo TE PE fr.unstranded 100 3 129.84 25.96SE fr.unstranded 100 3 102.26 10.23
Total 81 1917.7 364.07
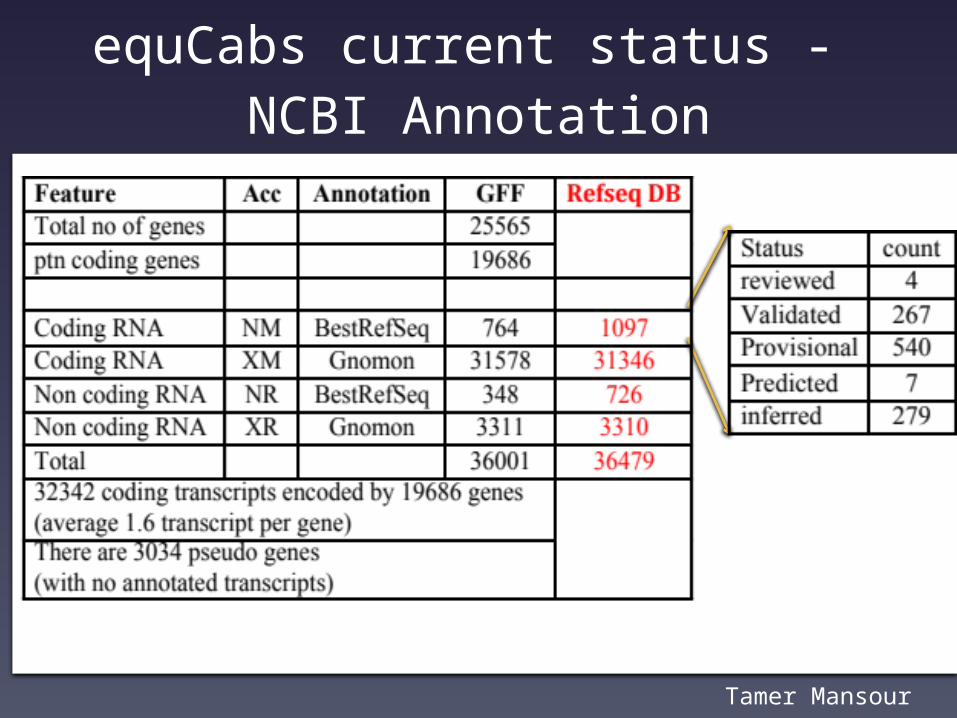
equCabs current status - NCBI Annotation
Tamer Mansour
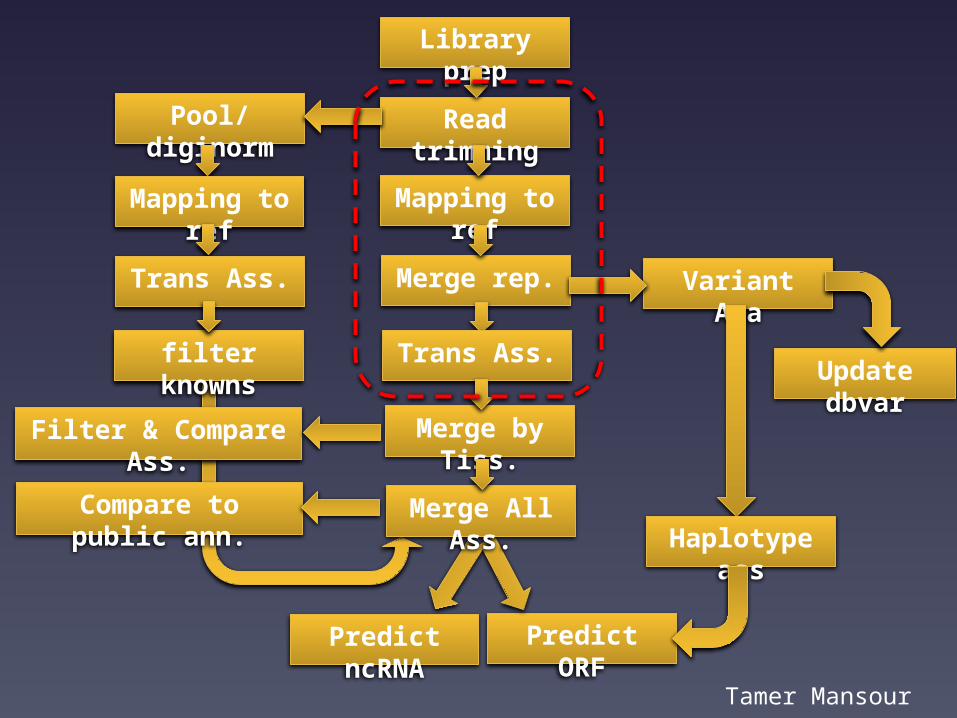
Library prepRead
trimmingMapping
to refMerge rep.
Trans Ass.
Merge by Tiss.
Predict ORF
Variant Ana
Update dbvar
Haplotype ass
Pool/diginorm
Predict ncRNA
Filter & Compare Ass.
filter knowns
Compare to public ann.
Merge All Ass.
Mapping to ref
Trans Ass.
Tamer Mansour

Digital normalization & (e.g.) horse transcriptome
The computational demands for cufflinks- Read binning (processing time)- Construction of gene models (no of genes, no of splicing junctions, no of reads per locus, sequencing errors, complexity of the locus like gene overlap and multiple isoforms (processing time & Memory utilization)
Diginorm- Significant reduction of binning time
- Relative increase of the resources required for gene model construction with merging more samples and tissues- ? false recombinant isoformsTamer Mansour

Effect of digital normalization
** Should be very valuable for detection of ncRNA
Tamer Mansour

The ORF problem
Hestand et al 2014: “we identified 301,829 positions with SNPs or small indels within these transcripts relative to EquCab2. Interestingly, 780 variants extend the open reading frame of the transcript and appear to be small errors in the equine reference genome”
Tamer Mansour

We merged the assemblies into six tissue-specific transcription profiles for cerebellum, brainstem, spinal cord, retina, muscle and skin. The final merger of all
assemblies overlaps with 63% and 73% of NCBI and Ensembl loci, respectively, capturing about 72% and 81% of their coding bases. Comparing our assembly to the most recent transcriptome annotation shows ~85% overlapping
loci. In addition, at least 40% of our annotated loci represent novel transcripts.
Tamer Mansour

2. Software and infrastructure
Alas, practical data analysis depends on software and computers, which
leads to depressingly practical considerations for gentleperson
scientists.

SoftwareIt’s all well and good to develop new data
analysis approaches, but their utility is greater when they are implemented in usable
software.
Writing, maintaining, and progressing research software is hard.

The khmer software package
• Demo implementation of research data structures & algorithms;
• 10.5k lines of C++ code, 13.7k lines of Python code;• khmer v2.0 has 87% statement coverage under test;• ~3-4 developers, 50+ contributors, ~1000s of users
(?)• Developed as a “true” open source project.
The khmer software package, Crusoe et al., 2015. http://f1000research.com/articles/4-900/v1

Challenges:
Research vs stability!Stable software for users, & platform
for future research;vs research “culture”(funding and careers)

Infrastructure issuesSuppose that we have a nice ecosystem of bioinformatics &
data analysis tools.Where and how do we run them?
Consider:1. Biologists hate funding computational infrastructure.2. Researchers are generally incompetent at building and
maintaining usable infrastructure.3. Centralized infrastructure fails in the face of infinite data.
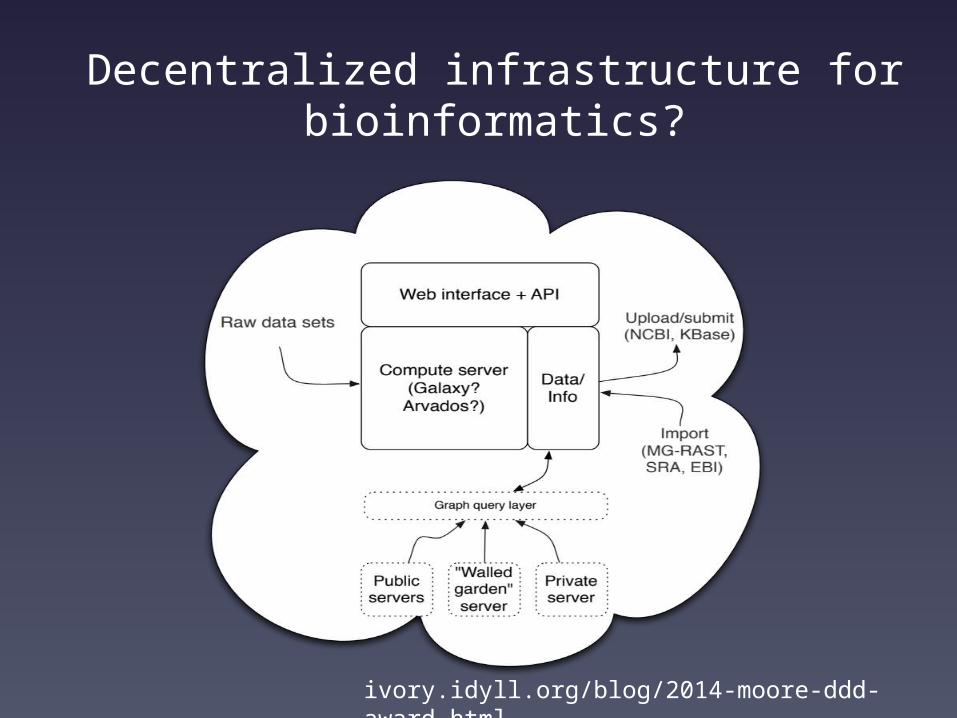
Decentralized infrastructure for bioinformatics?
ivory.idyll.org/blog/2014-moore-ddd-award.html

3. Open science and reproducibility
In my experience, most researchers* cannot replicate their own computational
analyses, much less reproduce those published by anyone else.
* This doesn’t apply to anyone in this
audience; you’re all outliers!
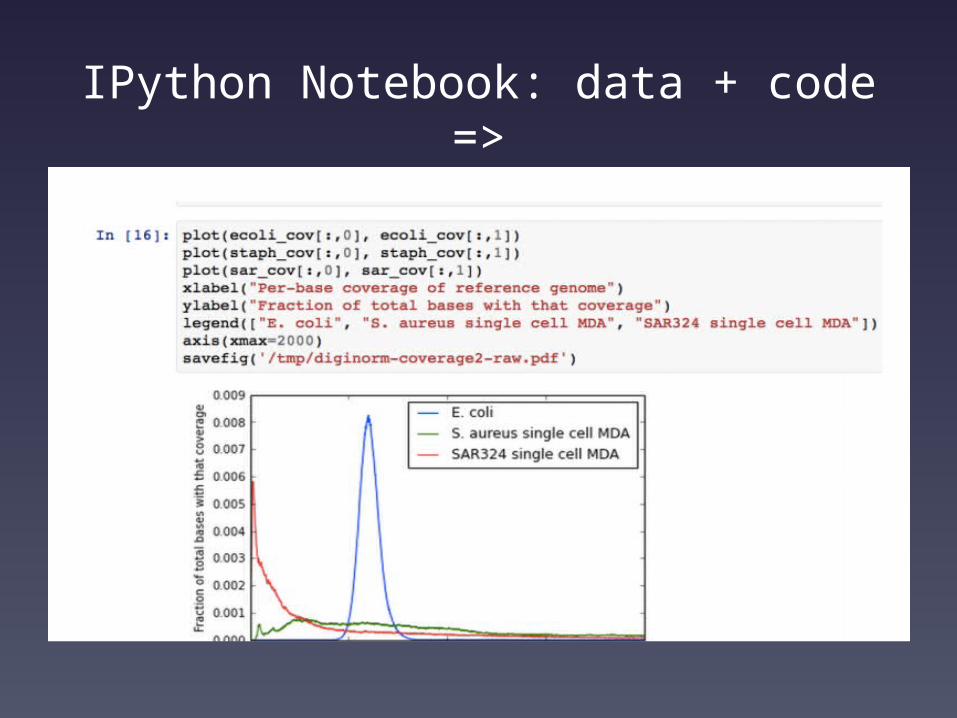
IPython Notebook: data + code =>

To reproduce our papers:
git clone <khmer> && python setup.py install
git clone <pipeline>
cd pipeline
wget <data> && tar xzf <data>
make && cd ../notebook && make
cd ../ && make

This is standard process in lab --
Our papers now have:
• Source hosted on github;• Data hosted there or on
AWS;• Long running data
analysis => ‘make’• Graphing and data
digestion => IPython Notebook (also in github)
Zhang et al. doi: 10.1371/journal.pone.0101271
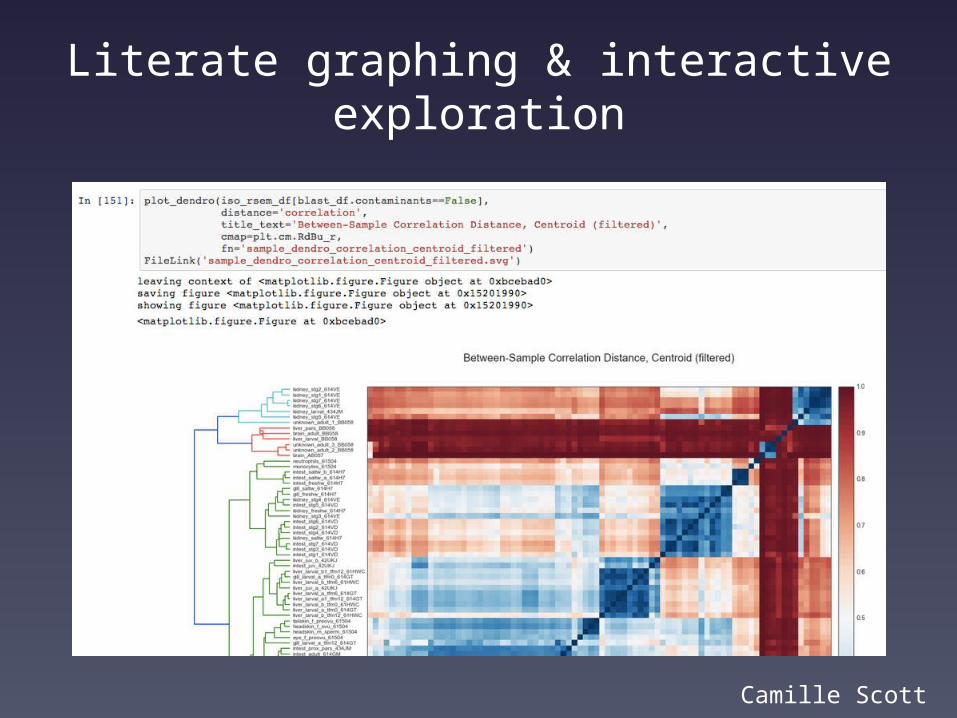
Literate graphing & interactive exploration
Camille Scott

Why do we do this?• We work faster and more reliably.• We can build on our own (and
others’) research.• Robust computational research,
released early, gives us a competitive advantage.

4. Training
Methods and tools do little without a trained hand wielding them, and a trained eye examining the results.

Perspectives on training• Prediction: The single biggest challenge
facing biology over the next 20 years is the lack of data analysis training (see: NIH DIWG report)
• Data analysis is not turning the crank; it is an intellectual exercise on par with experimental design or paper writing.
• Training is systematically undervalued in academia (!?)

UC Davis and trainingMy goal here is to support the
coalescence and growth of a local community of practice around “data
intensive biology”.

Summer NGS workshop (2010-2017)

General parameters:• Regular intensive workshops, half-day or longer.• Aimed at research practitioners (grad students &
more senior); open to all (including outside community).
• Novice (“zero entry”) on up.• Low cost for students.• Leveraging global training initiatives:

Thus far & near future~12 workshops on bioinformatics in 2015.
Trying out Q1 & Q2 2016:• Half-day intro workshops (27
planned);• Week-long advanced workshops;• Co-working hours (“data therapy”).
dib-training.readthedocs.org/

The End.• If you think 5-10 years out, we face
significant practical issues for data analysis in biology.
• We need new algorithms/data structures, AND good implementations, AND better computational practice, AND training.
(It’s a pretty good time to be doing biology.)






























