Embed Size (px)
DESCRIPTION
定量聚合酶链反应 (Q-PCR) 测定技术 及其临床应用. 卫生部临床检验中心 李金明. PCR?. PCR 只是一个简单的不起眼玩艺 —— 凯利 · 穆利斯( Kary Mullis) PCR 只是一个概念,所发生的奇迹是: PCR 概念变成了可操作的实验系统,变成了一项成熟的技术,后者又上升成为新的概念。 … 它最大的特点就是能不断推出新形式。 —— 摘自 [Making PCR: A Story of Biotechnology by Paul Rabinow]. ( Kary Mullis). 什么是 PCR?. - PowerPoint PPT Presentation
Citation preview
定量聚合酶链反应 (Q-PCR) 测定技术及其临床应用卫生部临床检验中心 李金明

PCR? PCR 只是一个简单的不起眼玩艺 ——凯利 · 穆利斯( Kary Mullis)
PCR 只是一个概念,所发生的奇迹是: PCR 概念变成了可操作的实验系统,变成了一项成熟的技术,后者又上升成为新的概念。 … 它最大的特点就是能不断推出新形式。 —— 摘自 [Making PCR: A Story of Biotechnology by Paul Rabinow]

( Kary Mullis)

什么是 PCR?
我不认为 PCR 能够用两种“革命” ( 政治革命和科学革命 ) 加以说明。……它的发明并没有改变基因操作的本质。有了 PCR ,我们能在更广的范围内更快、更容易地进行基因操作,学术界也完全不用等到某人死去或退休后才能接受 PCR 。它只不过是一种新工具。 ——凯利 · 穆利斯( Kary Mullis)
PCR 及有关技术的发展历史( 1 )1983 Cetus 公司的 K. Mullis 在这年底( 12 月 16
日第一次成功实验)发明了 PCR 。 “这种简单得令人惊奇,可以无限量地拷贝 DNA
片段的方法是在不可想象的情况下,即驾车行驶在月色下的加利福尼亚山间公路上时想出来的”。
--KB Mullis, Scientific American, april 1990

PCR 发明过程的简单回顾



PCR 及有关技术的发展历史( 2 )1985 关于 PCR 的文章首次由 Cetus 公司 Saiki 等人在 Science 上 发 表 (R. Saiki, S. Scharf, F. F
aloona, K. Mullis, G. Horn, H. Erlich and N. Arnheim)
1987 KB Mullis et al. ”Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods in Enzymology 155:335
PCR 及有关技术的发展历史( 3 )1987 当年 7 月 Cetus 公司在美国获得 PCR 基本技术专利批准。 1985 年底成立的 Perkin-Elmer Cetus 仪器公司 ( PE
CI ) 于这一年的 11 月推出了第一 个 PCR 试剂产品及第一台热循环仪。1989 Science 报道了耐热性 DNA 多聚酶 Taq 酶的发现 , 预示着分子时代的到来。 12 月《 Science 》杂志将 PCR 和它所使用的聚合酶命名为第一个“年度分子”。 Hoffmann-La Roche 公司和 Cetus 开 始合作将 PCR应用于临床诊断 。 RK Saiki, … KB Mullis et al. (1988) ”Primer-directed
enzymatic amplification of DNA with a thermo-stable DNA polymerase”, Science 239:487
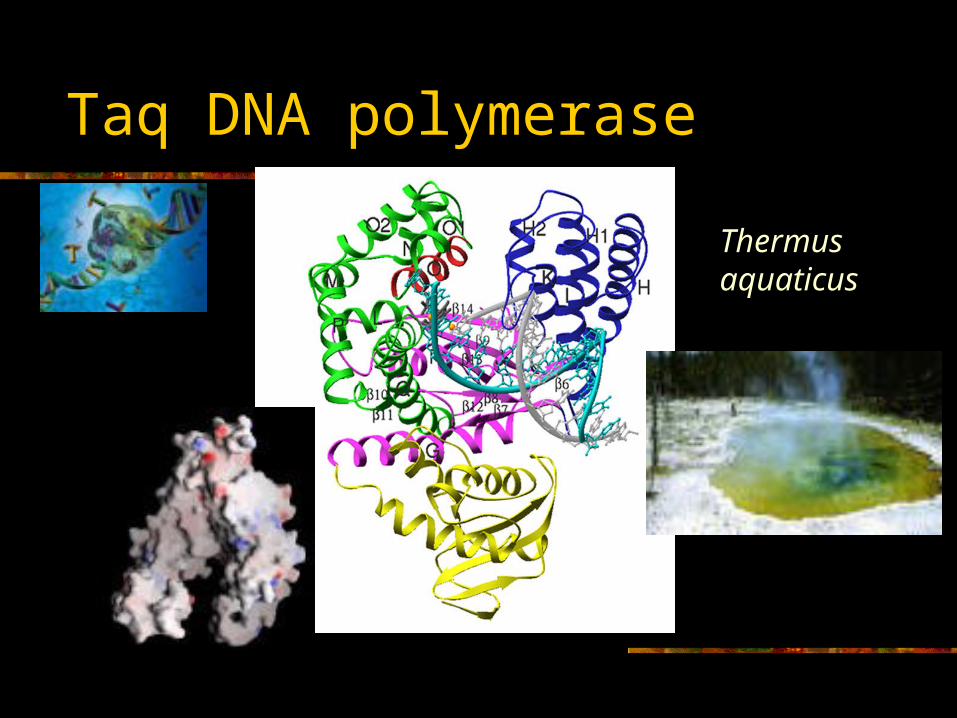
Taq DNA polymerase
Thermus aquaticus

Taq DNA polymerase –A thermostable DNA polymerase from Thermus
aquaticus.
PCR 及有关技术的发展历史( 4 ) DuPont 理由是, 1971 年 PCR � 的雏形已由 Khorana 一系列文章阐明,并公布于众,应当是公共产权。斯坦福大学 Kornberg (研究 DNA 聚合酶获诺贝尔奖)也认为,任何具备生物技术知识的人都可以从 Khorana 等的文章中推知如何操作 PCR 。

PCR 及有关技术的发展历史( 5 )
H.G. Khorana 美国MIT 教授、 1968年诺贝尔医学奖得主
Study on polynucleotides: repair replication of short synthetic DNA’s as catalyzed by DNA polymerases. J Mol Biol, 1971, 56:341 “经过 DNA 变性,与合适引物杂交,用DNA 聚合酶延伸引物,并不断重复该过程便可克隆 tRNA 基因”。核酸体外扩增最早设想不能实现的原因 : 当时很难进行测序和合成寡核苷酸引物; 当时 (1970)Smith 等发现了内切酶,体外克隆基因成为可能

The article from 1971 in which Kleppe et al. describe Repair replication – basically the same principle as PCR!
J. Mol. Biol. (1971) 56 341-361
PCR 及有关技术的发展历史( 6 )1990 Cetus 科学家 D.Gelfand 和 S.Stoffel 发明了纯 化 Taq
DNA 多聚酶的方法。 Cetus 科学家 H. Erlich 和 K. Mullis 在德国临床化学领域获得生化分析奖。1991 TaqMan 技 术 发 表。 当年 11 月 Hoffmann-La Roche 公司从 Cetus 获得全球 的 PCR 专利权。 Roche Molecular Systems 创建了单一耐热多聚酶 rTth 进行 RT-PCR 技术,并用于临床 RNA病毒检测。 耐热逆转录酶首次由 Cetus 科学家发现并报道。
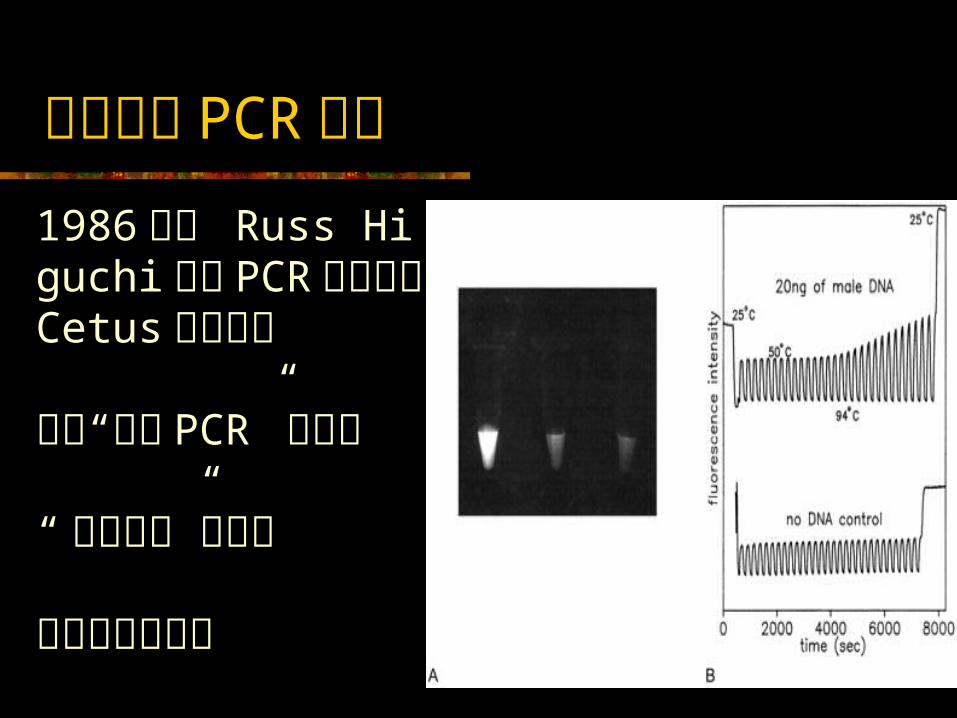
实时荧光 PCR 技术1986 年末 Russ Higuchi 来到 PCR 的诞生地 Cetus 公司工作 探讨“闭管 PCR” 的可能“ 溴化乙锭”的使用光导纤维的使用

PCR 及有关技术的发展历史( 7 )1992 当年 3 月 Cetus 公司获得 Taq 酶、热循环扩增仪等专利; 1993 AMPLICOR HCV*首次用于临床 RNA PCR 标 准试剂盒。 Kary Mullis 因 PCR的发明获得诺贝尔化学奖。

PCR 及有关技术的发展历史( 8 ) 九十年代中期 PCR 临床应用在国内全面展开 1998 年 实时荧光 PCR 技术开始在中国较广泛的应用于临床检测 1999 年 西方发达国家尝试将 PCR 应用于血液筛查
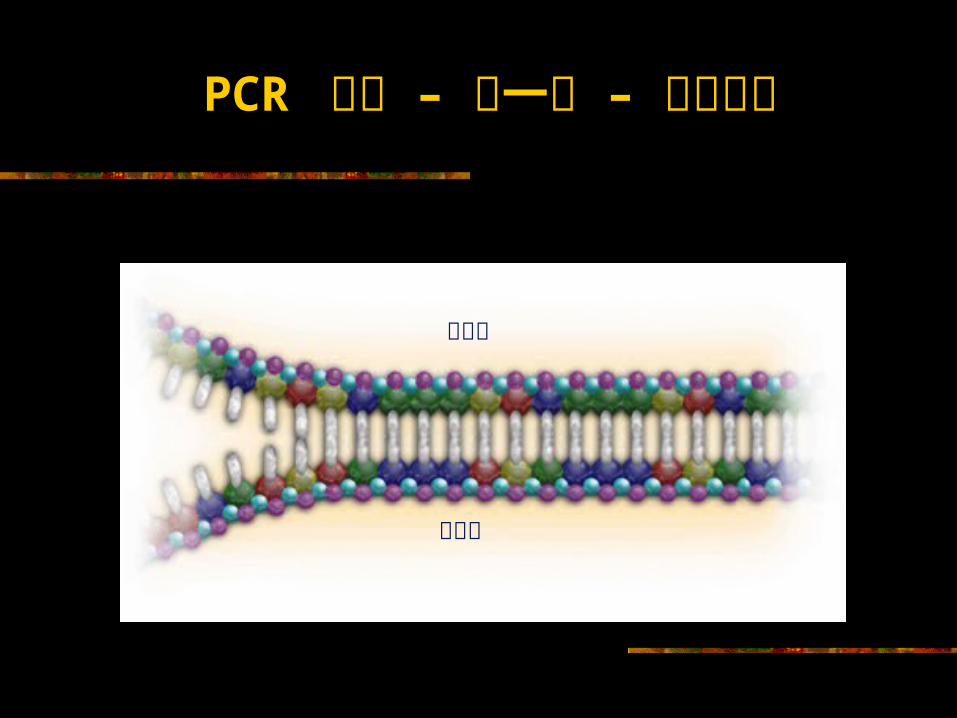
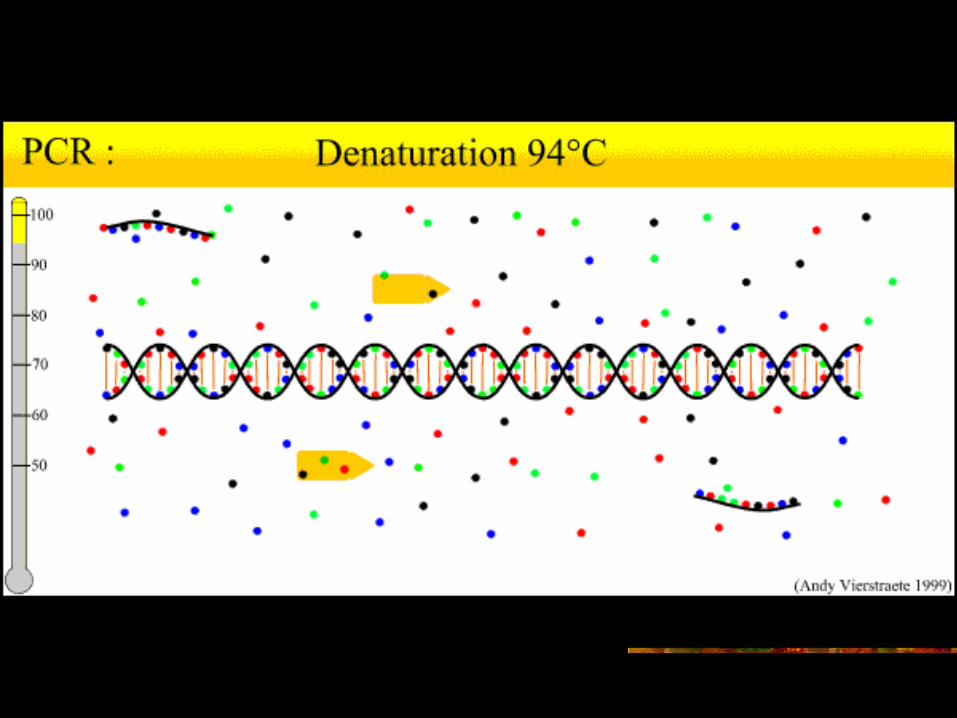
PCR 循环 – 第一步 – 加热变性
靶序列
靶序列

PCR 循环 - 第二步 – 引物与靶序列退火靶序列
靶序列
Primer 1Primer 2
Biotin
Biotin
5’
3’
5’
5’
3’
5’
3’
3’
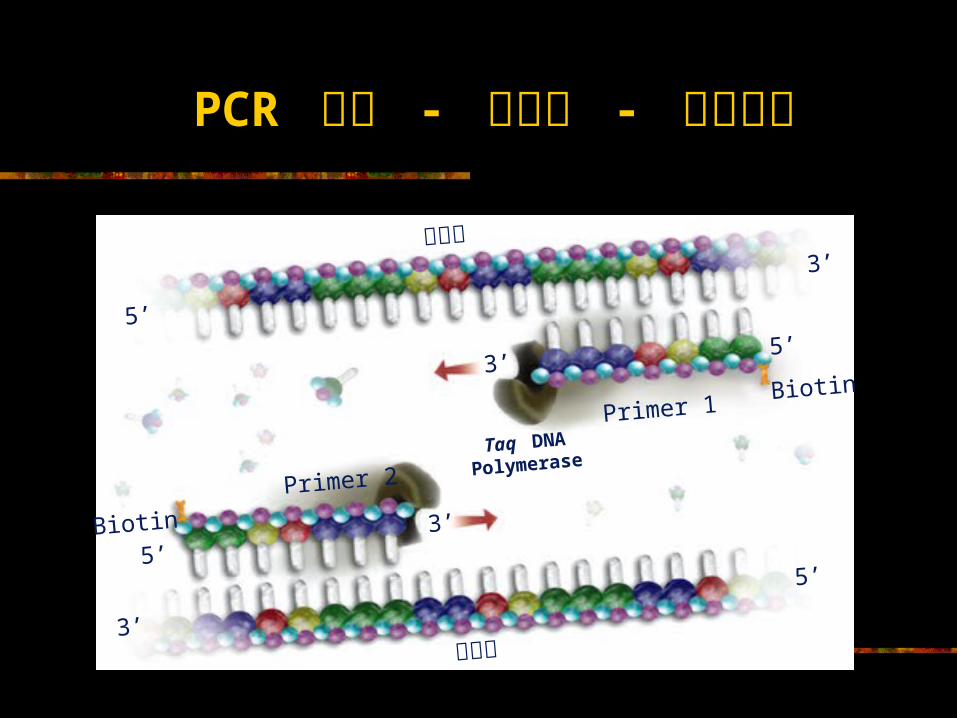
PCR 循环 - 第三步 - 引物延伸靶序列
靶序列
Primer 1
Primer 2
Biotin
Biotin
5’
3’
5’
5’
3’
5’
3’
3’
Taq DNAPolymerase
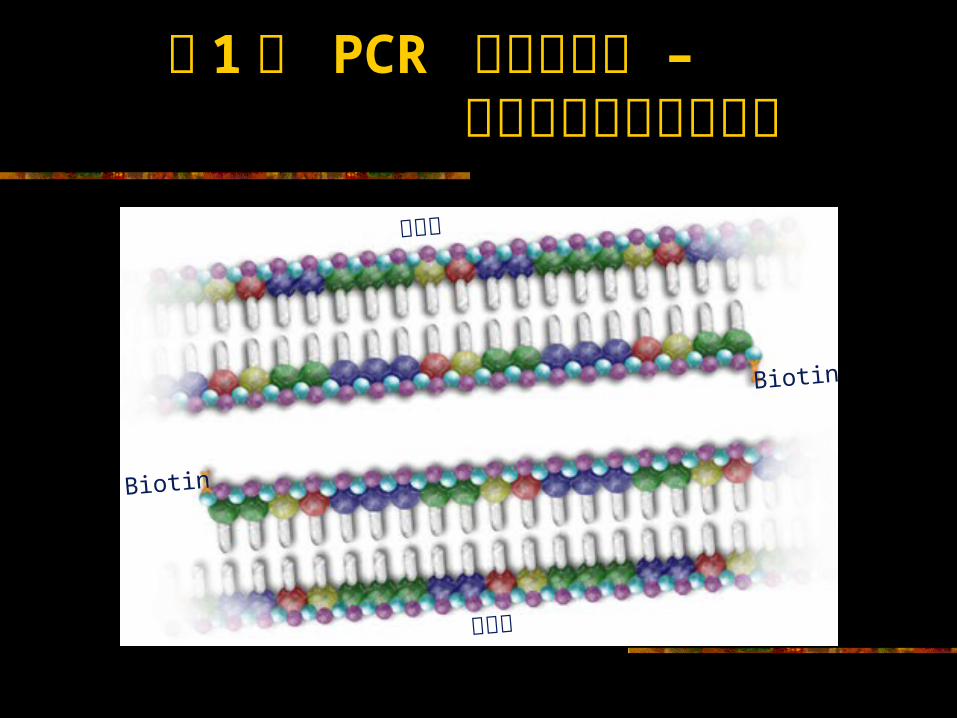
第 1个 PCR 循环完成后 – 得到两个拷贝的靶序列靶序列
靶序列
Biotin
Biotin
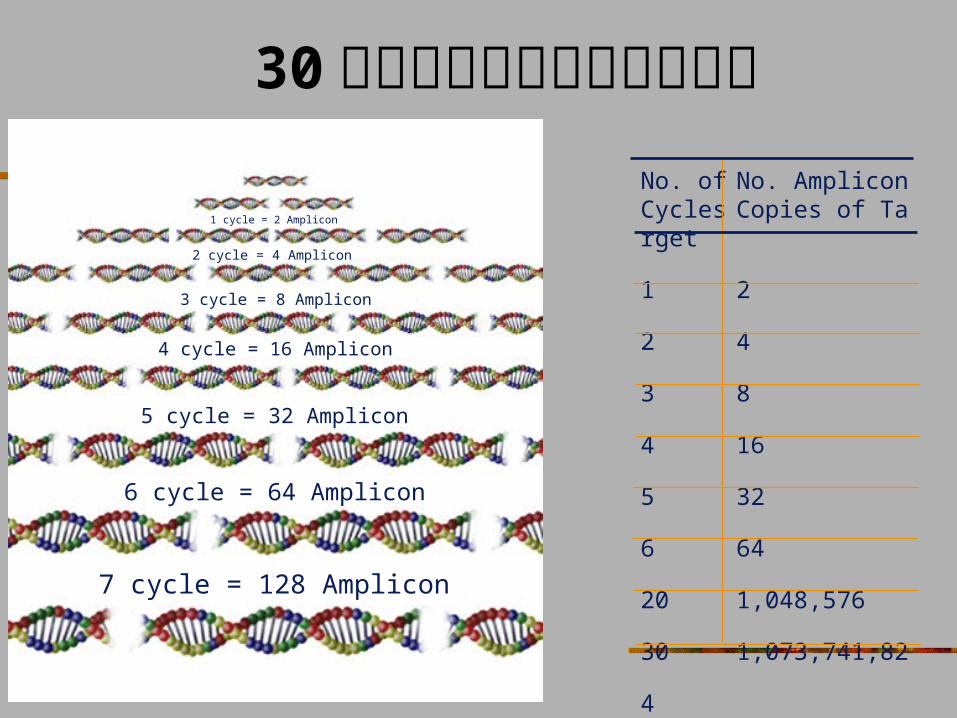
30 次循环后靶序列扩增的数量No. of No. Amplicon Cycles Copies of Target
1 2
2 4
3 8
4 16
5 32
6 64
20 1,048,576
30 1,073,741,824
1 cycle = 2 Amplicon
2 cycle = 4 Amplicon
3 cycle = 8 Amplicon
4 cycle = 16 Amplicon
5 cycle = 32 Amplicon
6 cycle = 64 Amplicon
7 cycle = 128 Amplicon
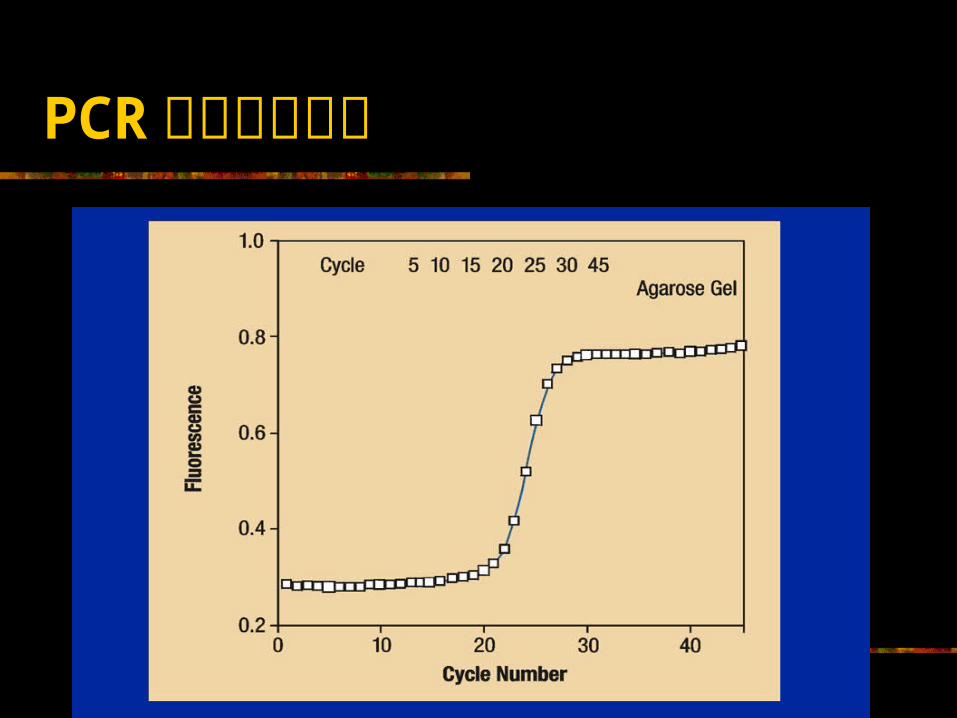
PCR 扩增过程图示

PCR 扩增的理论模式 PCR 扩增为指数扩增,每一扩增周期后产物的量可以下式表达: Yn=Yn-1·(1+E) = Yn-2·(1+E)2=…= X·(1+E)n
0≤E≤1
其中 E 表示扩增效率, Yn表示在 n 个周期后 PCR 产物的分子数量, Yn-1为 n-1 个周期后 PCR 产物的分子数量。
等式仅在限定的扩增周期数(通常为 20 或 30 )内成立。超过此周期数,扩增过程即由指数扩增降低至稳定的扩增速率,最终达到平台,不再扩增 .

影响 PCR 扩增效率的因素 : PCR定量的困难 ?
引物 /靶的杂交 反应试剂的相对量 样本在扩增仪中的位置的不同 临床样本中 DNA 聚合酶抑制物的存在 PCR 扩增模板的量 靶核酸链的再退火及酶过量可致 E降低
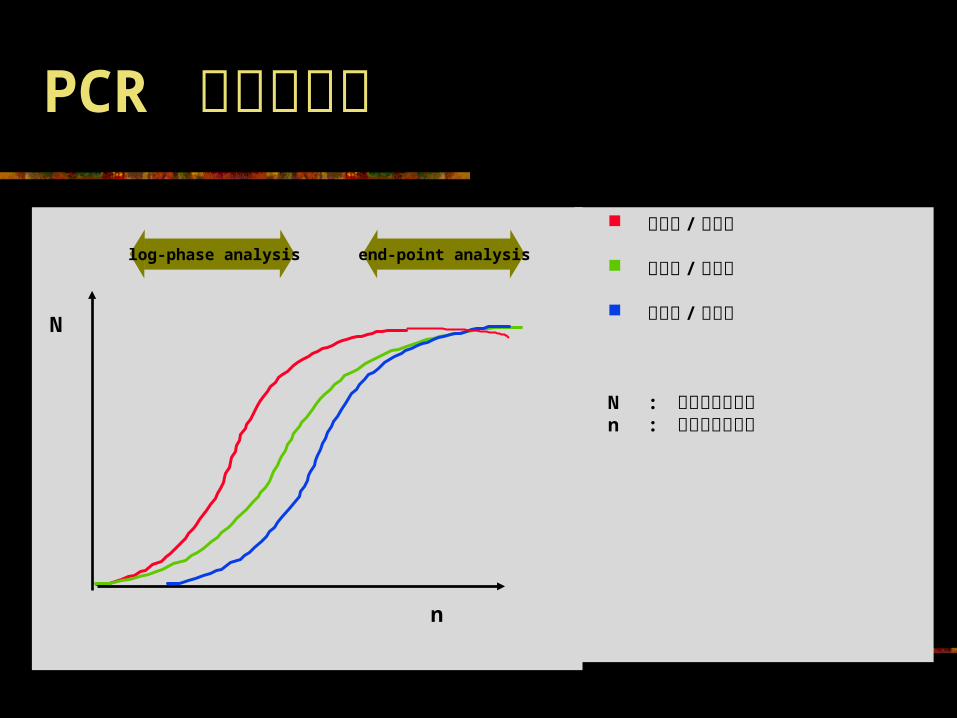
PCR 和定量问题
高浓度 / 高效率 高浓度 / 低效率 低浓度 / 高效率
N : 扩增分子的数目n : 扩增循环的次数
log-phase analysis
N
n
end-point analysis
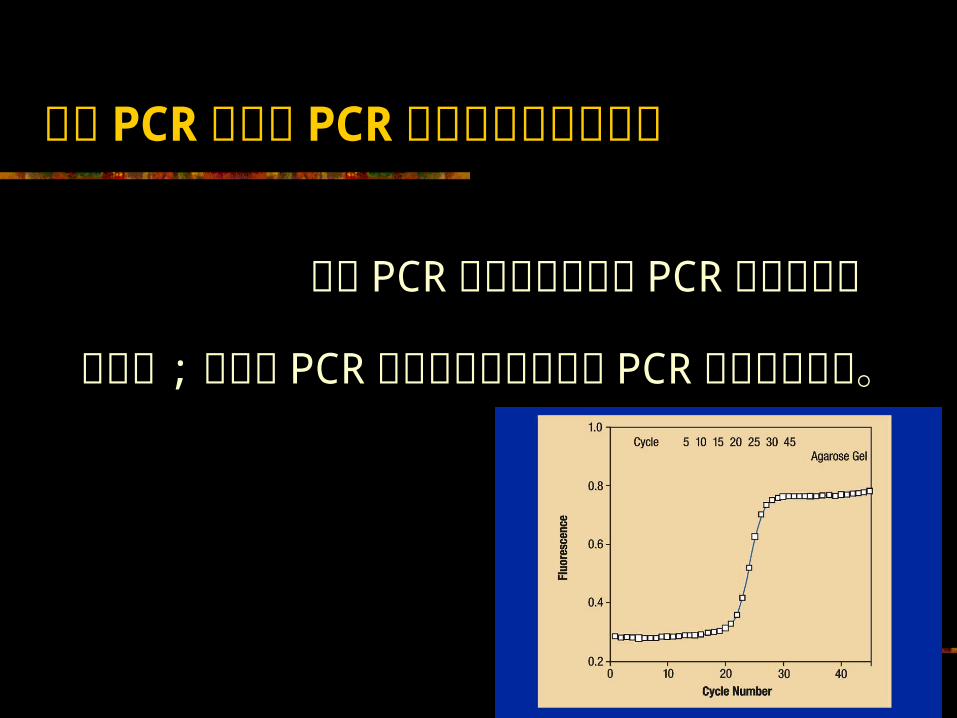
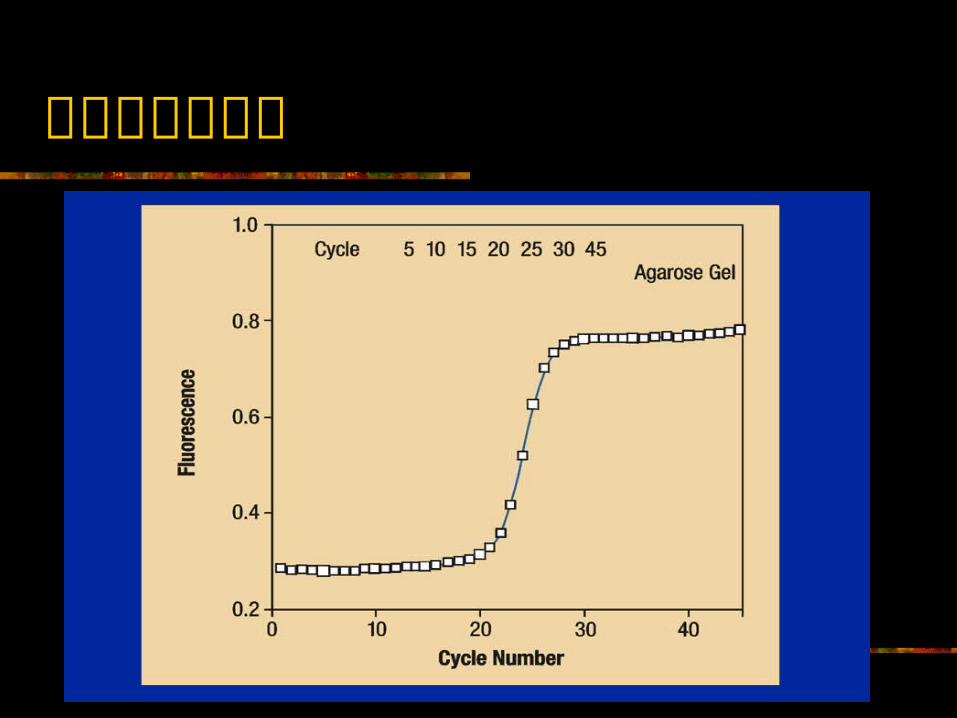
定量 PCR 与定性 PCR 测定的最根本的区别 定量 PCR测定的测定点为 PCR 扩增的指数扩增期 ;而定性 PCR测定的测定点则多为 PCR 扩增的平台期。

定性 PCR 测定方法 荧光 PCR ( Taq Man 和分子信标等) PCR- 膜上杂交 PCR-ELISA

定量 PCR 的方法 定量 PCR 方法可归为三大类,即外标方法、动力学方法和内标共扩增方法 定量还可分为绝对定量(即测定 X 的分子数量)和相对定量(即测定不同样本中 X的比率)
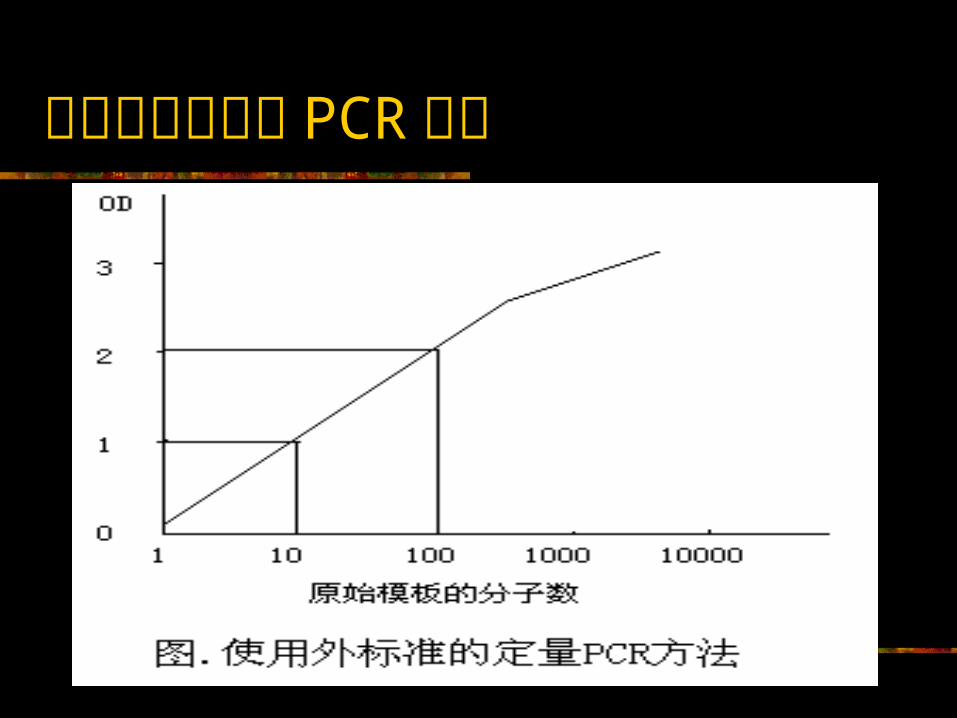
用外标准的定量 PCR 方法

使用外标的定量 PCR 方法的优缺点 优点:( 1 )方法简便,容易建立;( 2 )使用双孔重复测定,这种方法可得到非常准确的结果,甚至可排除管间的差异,但不能排除样本间的差异; 缺点:( 1 ) PCR反应体系中小的区别也会对测定造成较大的影响。由于最后在扩增效率上的差异,从而使得定量测定精密度和重复性不佳。 因此,如果建立一个使用外标准的 PCR定量方法,则必须进行方法的精密度(批内变异)和重复性(批间变异)分析,以确定其应用的局限性。
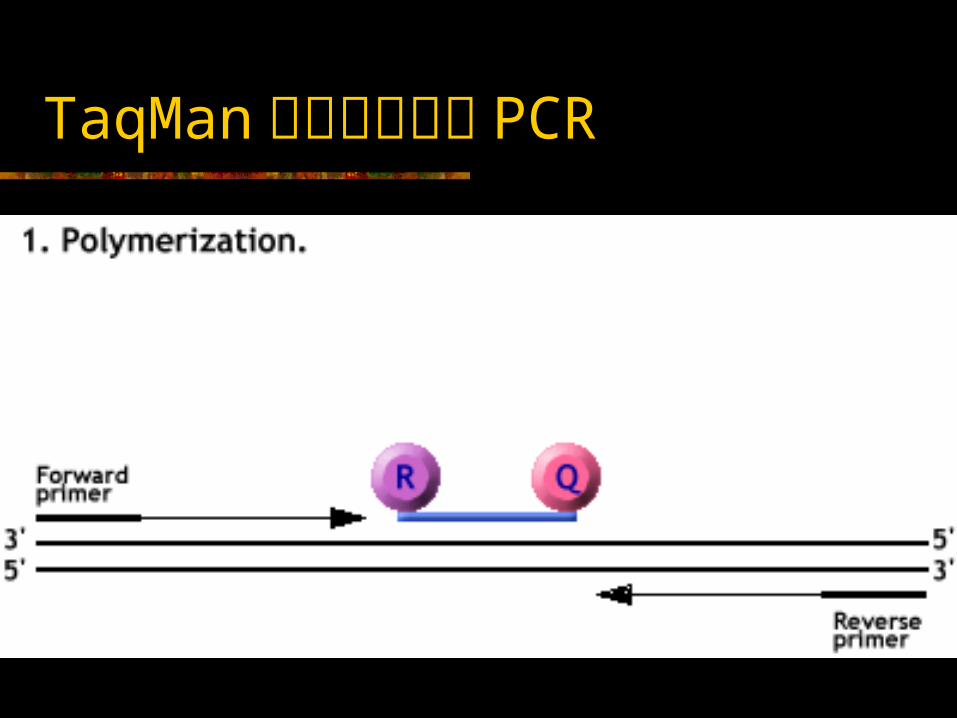
TaqMan 实时荧光定量 PCR
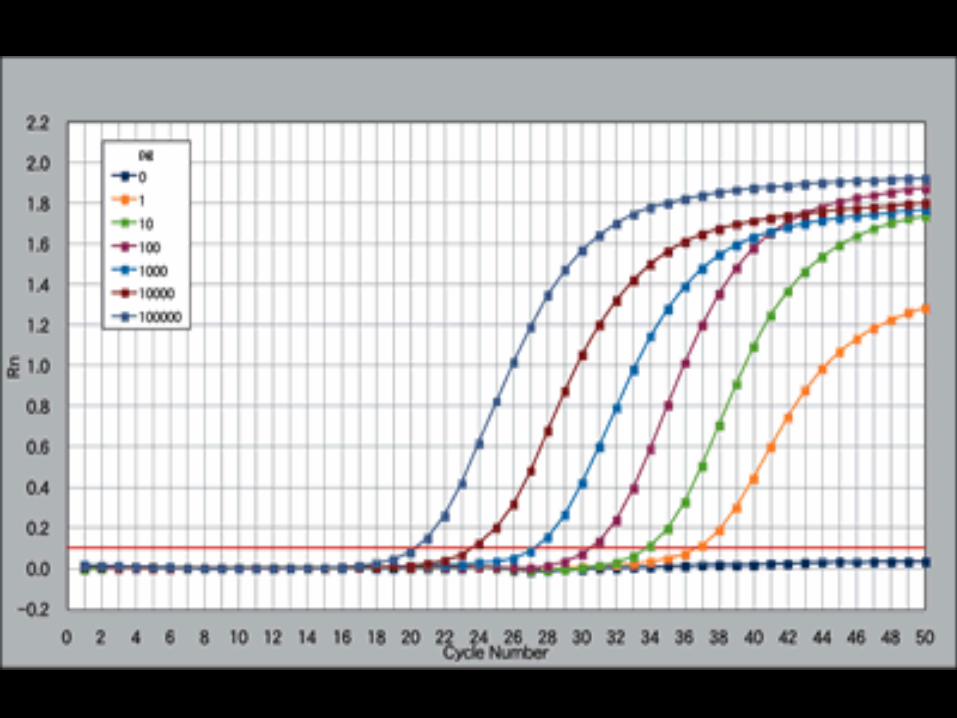

分子信标实时荧光定量 PCR

动力学定量 PCR 方法 第一步:确定 PCR 扩增的效率;第二步:根据相应的公式计算原始模板数。

扩增效率的计算 (1) 如果在 PCR每一个扩增循环中扩增效率为常数,则可通过在 PCR 扩增的指数期的数个连续的或非连续的周期中取扩增样本,然后测定每份样本中的产物 Yn 的量来测定 E值。有必要收集 2份以上的样本,最好是尽可能多的样本以确证扩增效率保持恒定;换句话说,也就是 PCR尚未达到扩增效率开始降低时的阶段。如果取的是连续的样本,则可从公式( 1 )测得 E值;
扩增效率的计算

扩增效率的计算 (2)
如果取的样本不连续,则可使用下述将公式( 2 )重排后得到的更为通用的公式,用参数 j (取样中间隔的周期数)替代 n ,Yj(在较高循环周期数取样的产物量)替代 Yn, Yi-j(在较低循环周期数取样的产物量)替代 X:
E=- 1+( Yi/Yi-j) 1/j (3)
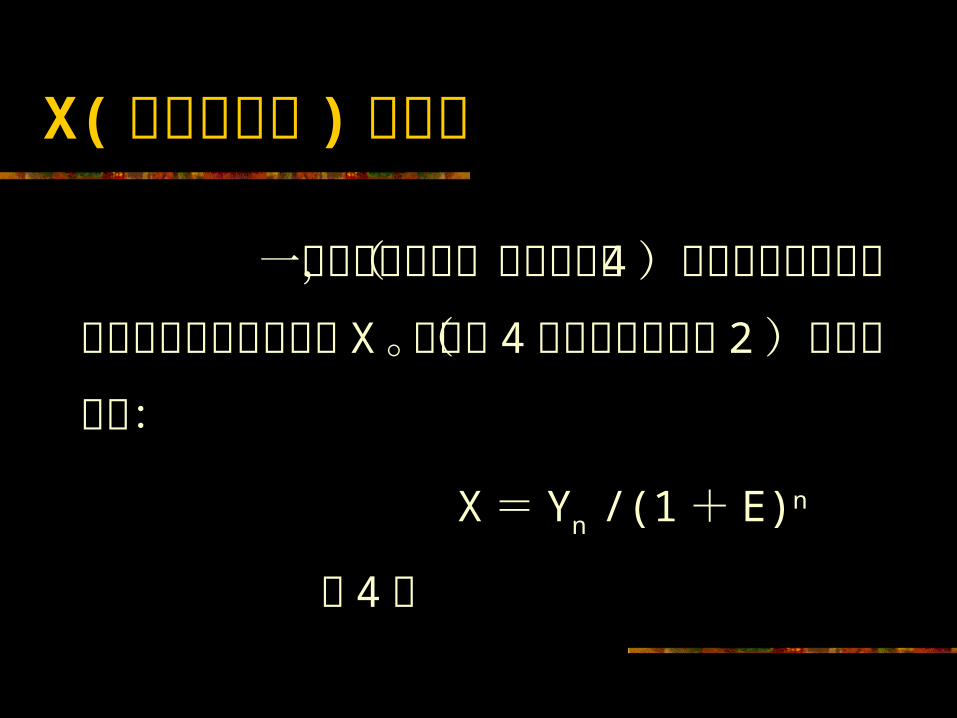
X( 原始模板数 ) 的计算 一旦效率确定后,根据公式( 4 )可从测定的产物量和循环周期数计算得到X 。公式( 4 )来源于公式( 2 )的重新排列:
X= Yn /(1+ E)n ( 4 )

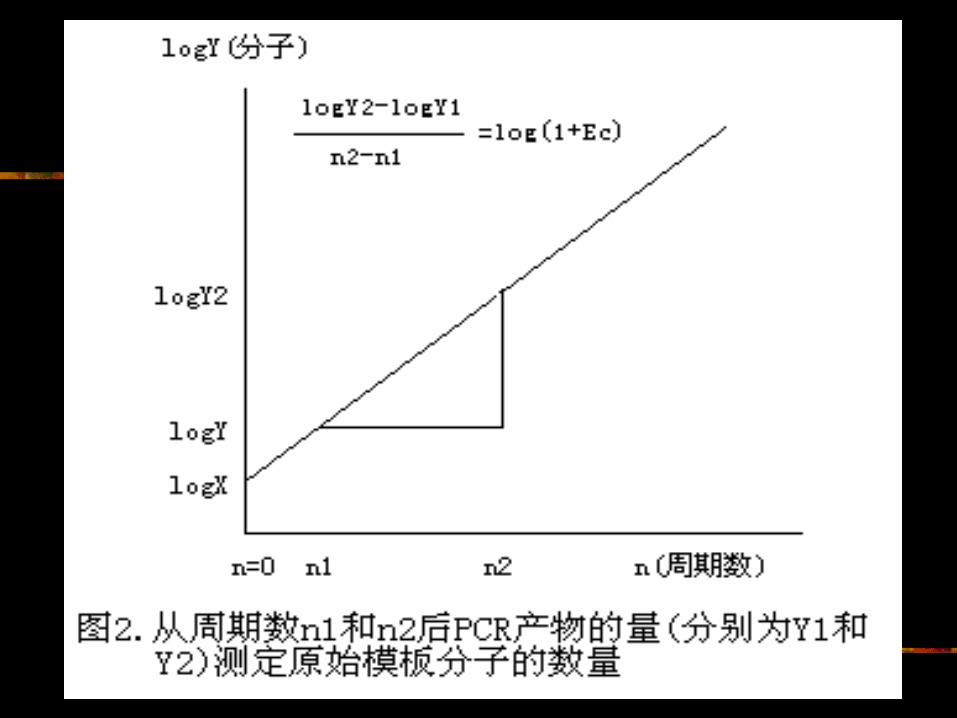
X 的另一种计算方式 (1) 另一种方式是,根据公式( 2 )的对数形式,以所测定的 Yn值的对数对函数
n绘图得到 X值: logYn = logX + n×log(1+E) (5)
当 n= 0 时,可在图上读出 X值,或通过对公式( 5 )的线性回归计算 X值(图 2 )。
动力学方法的特点 这种方法的优点 : (1) 不需要外标准;( 2 )如果能测定 PCR 产物分子的绝对数量,则可得到待测样本的不同的扩增效率( Ee) 。

使用非竞争性内标准的定量 PCR 方法 非竞争性内标准:( 1 )一般为与靶核酸无关的基因组;( 2 )既可是内源的也可是外源的;( 3 )与靶核酸无相同的引物结合位点和扩增序列。 对于 DNA 的扩增,非竞争性内标几乎所有的基因都可应用,最常用的有丙酮酸脱氢酶、前脑啡肽或 β肌动蛋白的基因。 而对 RNA 的扩增,则非竞争性内标较难寻找。理想的 m
RNA 内标应该在整个细胞周期中表达的水平变化不大,此外,其表达水平应接近于靶 RNA ,而且它们的扩增效率应相等,因此两者扩增线性区域是重合的。已有许多的 mRNA被尝试用来作为此种内标,如 HLA 、 β肌动蛋白、 GPDH 和组蛋白 H3.3 的 mRNA 或 14S rRNA 等。

以非竞争性内标定量的主要优点 内标的制备简单
复管测定可排除管间差异,并且在一定程度上,可排除样本间的差异。

以非竞争性内标定量的缺点 内标准和靶核酸的逆转录的效率有可能不同,并且有可能既使针对的是相同的靶核酸逆转录效率也会有很大差异。因此,使用这种方法进行 RNA 的定量 PCR 测定极为困难。 在扩增过程中的指数期测定可对原始模板相对定量,但如果没有验证在一定的扩增周期内靶核酸和内标有相同的扩增效率,则不能进行绝对定量。

使用竞争性内标准的定量 PCR 方法 “竞争性”内标:人为构建的可与原始靶核酸竞争酶、核苷酸和引物分子的核酸标准,其特点是,与靶核酸具有相同的引物结合位点、但引物之间的顺序需加以修饰,使得扩增产物在检测时能够很容易地通过诸如凝胶电泳、探针杂交或高效液相层析等方法区分。 对内标准的修饰方法包括对野生型基因组的点突变、部份缺失或插入外来顺序等,目前尚无通用的规则或程序来构建这种内标准。

使用竞争性内标准的定量 PCR 方法 使用竞争性内标既可进行相对定量也可进行绝对定量。 相对定量具有很好的精密度和重复性。而绝对定量,关键是要证明竞争性内标和野生型靶核酸的扩增效率相同。亦可将竞争性内标置于含已知量的野生型靶核酸的样本中同时扩增来评价。

使用竞争性内标准的定量 PCR 方法 要定量单份的临床样本,必须进行数管竞争性 PCR测定,每管中靶核酸的量不变,但改变竞争性内标的量,因为只有当待测靶核酸与竞争性内标等摩尔量时才能有可靠的定量。 由于竞争性 PCR 可排除管间和样本间的差异,因此,其可作为准确的 PCR定量方法,适用于绝对定量及含低拷贝数样本的定量。

定量和定性 PCR 测定的临床应用及应注意的问题为什么要做定量测定?定性和定量测定结果报告上的混淆。

为什么要做定量测定? 用于已知病毒感染患者抗病毒治疗效果的动态观察及抗病毒药物的临床研究; 用于基因表达方面的研究,如特定的
mRNA 的定量测定。

为什么要做定性测定? 用于未知患者的确认 用于血液筛检 突变检测

定量和定性测定的结果报告 定量测定测定的是量的“多”或“少”;定性测定则是测定某种物质的“有”或“无”,用于未知病人的确认诊断。 定量测定结果报告:根据所使用的测定方法的测定范围报告结果,如样本测定结果高出此范围,则可报告 > 多少,也可对样本稀释后再检测;如低于此范围,则报告 < 多少,如 <103拷贝数 /ml ,而不能报告为 0 拷贝数 /ml 或阴性。 定性测定结果报告 : 报告“阳性”或“阴性” ,
“ 检出”或“未检出” , “反应”或“未反应”

在感染性疾病病原体检测中的应用 病毒的检测 定量 定性 基因型及基因突变 细菌及其它微生物的检测 难培养菌 耐药基因 寄生虫的检测

在遗传病及其它基因相关疾病诊断中的应用 α 地贫 β 地贫 血友病 药物性耳聋

在亲子鉴定中的应用 每个人的遗传物质一半来自父亲,另一半则来自母亲。根据孟德尔遗传分离和自由组合定律,亲代基因型决定子代基因型,在没有基因突变和分型错误的前提下,孩子不可能带有双亲均没有的等位基因。 父系遗传的 Y染色体的标记,子代的分型必定与父亲的相同,而且同一父系的所有个体的分型一致。母系遗传的线粒体 DNA ,子代的分型与母亲的分型一致,并且同一母系的所有个体的分型一致。

在个体鉴别中的应用 基因指纹又称 DNA指纹,指的单基因座探针限制性片段长度多态性。 DNA 所含有的遗传信息是由遗传密码字母 A 、 C 、 G 和 T 的序列决定的。人类含有总数约 30亿个这种字母,它们在人体每个细胞的染色体上都以一定的次序排列,排列次序的不同使得一个个体与另一个个体完全不同。个体间的亲缘关系相距越远,基因组的核苷酸字母排列差异就越大。相反,遗传上相关的个体(如同胞、父子)相应地在其字母序列上有很大的相似性。

在恶性肿瘤诊断中的应用 肿瘤诊断 肿瘤疗效观察 肿瘤的转移

在血液筛检中的应用HCV和 HIV感染“窗口期”HBV DNA PCR检测筛检血液的意义

其 它 Immuno-PCR

临床 PCR 检验应用的发展趋势 核酸提取方法的简便化和自动化 项目的多样化:从病原体、到基因疾病的诊断、多标志物同时检测、疾病基因指纹 不同原理的实时基因扩增仪的普遍应用 扩增试剂的改进:标准化、防污染、有内标阴性质控 临床 PCR 实验室质量理念不断进入增强

总 结1.定量测定重在定量,即如何改善扩增指数期的线性及其范围。定性测定重在测定下限。2.定量测定的准确性较之测定下限要重要得多。3.非竞争性内标和竞争性内标对于使用内标的定量测定方法极为关键。合适的非竞争性内标应为在整个细胞周期中均匀表达的基因核酸,而竞争性内标则应尽可能近似原始待测模板。4.定量测定应在扩增的指数期进行,因此,更重要的是要使涉及整个扩增过程的每个参数都理想化,以便控制整个扩增,避开扩增“平台期” 。定性既可在扩增的平台期,也可在扩增的指数期测定。

总 结 5. 由样本制备(核酸提取)方法所引起的定量测定的差异应仔细研究加以避免。 6.对于绝对定量,必须确定靶核酸和内标(竞争的和非竞争的)的扩增效率,内标应以相同的效率与靶核酸一起扩增。为增加测定结果在统计学上的可信性,建议重复多次测定。






























