Embed Size (px)
Citation preview

Estudio generalizado de sistemas miniaturizados de
extracción para la preconcentración y determinación de
metales pesados en aguas en concentraciones a nivel de
subtrazas
Trabajo de grado presentado para optar al título de Químico de la
Universidad de los Andes
Ana María Jiménez Fino
Director:
Dr. Ricardo Eusebio Rivas Hernández
Grupo de Investigación en Analítica Aplicada
Departamento de Química
Facultad de Ciencias
Universidad de Los Andes
Mayo del 2020

~ 2 ~

~ 3 ~
Hoy al finalizar esta etapa me doy cuenta de lo afortunada que he sido, en primer
lugar, quiero agradecer a Dios por acompañarme durante estos años y por
rodearme de personas increíbles que me han brindado su conocimiento, apoyo y
amor.
A mis padres, puesto que sin ellos nada de esto sería posible.
Al Dr. Ricardo Rivas, por su dirección, acompañamiento y ayuda brindada en mi
formación académica.
Al Departamento de Química de la Universidad de los Andes que acoge a cada
uno de sus estudiantes y los hace sentir como en casa. A cada uno de los
profesores que contribuyeron a enriquecer mi aprendizaje y con especial afecto al
Dr. Edgar Vargas por sus enseñanzas, sus consejos y su apoyo.
De igual forma, debo expresar mi profundo agradecimiento a mis compañeros y
amigos, especialmente a Sonia, que a lo largo del pregrado crecimos y
aprendimos juntas. También a Alejandra, Sergio, Santiago, Laura, Daniel y Elkin
por su compañía, amistad y apoyo.
Finalmente, pero por supuesto no menos importante, quiero agradecer a María
Cristina por su alegría, sus consejos, su motivación y su cariño siempre
incondicional.

~ 4 ~
Acrónimos .................................................................................................................................... 6
Resumen ....................................................................................................................................... 7
Objetivos ...................................................................................................................................... 8
Capítulo 1: Introducción ............................................................................................................ 9
Capítulo 2: Microextracción en fase sólida ............................................................................. 13
2.1 Microextracción en fase sólida en fibra (SPME) .............................................................. 13
2.2 Materiales empleados como sorbentes en SPME .............................................................. 15
2.3 Termodinámica de la microextracción en fase sólida ....................................................... 17
2.4 Optimización de las condiciones de extracción de SPME ................................................ 19 2.4.1 Temperatura .............................................................................................................................. 19 2.4.2 Tiempo de extracción................................................................................................................ 19 2.4.3 Agitación de muestra ................................................................................................................ 20 2.4.4 pH ............................................................................................................................................. 21 2.4.5 Efecto salino (“Salting-out”) .................................................................................................... 21 2.4.6 Derivatización ........................................................................................................................... 21
2.5 Aplicaciones de SPME en fibra en la determinación de metales pesados en agua ........... 22
2.6 Métodos alternos a SPME en fibra .................................................................................... 23 2.6.1 IT-SPME: Microextracción en fase sólida en tubo ................................................................... 24 2.6.2 Microextracción en fase sólida en aguja ................................................................................... 25 2.6.3 TFME: Microextracción de película delgada ............................................................................ 25 2.6.4 MEPS: Microextracción por sorbente empaquetado ................................................................. 26 2.6.5 SBSE: Microextracción por sorción sobre barra agitadora ....................................................... 27 2.6.6 SCSE: Extracción adsorbente con disco agitador ..................................................................... 28 2.6.7 RDSE: Extracción adsorbente con disco giratorio .................................................................... 28 2.6.8 DµSPE (DMSPE): Extracción en microfase sólida dispersiva ................................................. 29 2.6.8.1 Termodinámica de la extracción en fase sólida microdispersiva ........................................... 30 2.6.8.2 Ventajas y desventajas de DµSPE ......................................................................................... 32 2.6.8.3 Enfoques de DµSPE .............................................................................................................. 32 2.6.8.3.1 DµSPE tradicional .............................................................................................................. 32 2.6.8.3.2 DµSPE asistido por vórtex (VA- DµSPE) .......................................................................... 34 2.6.8.3.3 DµSPE asistido por ultrasonido (UA- DµSPE) .................................................................. 34 2.6.8.4 Aplicaciones de DµSPE en la determinación de metales pesados en agua ............................ 35
Capítulo 3: Microextracción en fase líquida ........................................................................... 39
3.1 Técnicas de microextracción en fase líquida ..................................................................... 39 3.1.1 SDME: Microextracción en gota simple ................................................................................... 39 3.1.1.1 DSDME: Microextracción con gota directa suspendida ........................................................ 40 3.1.1.2 LLLME: Microextracción líquido-liquido-líquido ................................................................ 41 3.1.2 DLLME: Microextracción líquido-líquido dispersiva .............................................................. 41 3.1.2.1 SFODME: Microextracción de gota orgánica solidificada .................................................... 42

~ 5 ~
3.1.3 HF-LPME: Microextracción en fase líquida con fibra hueca ................................................... 43
3.2 Fases extractantes en LPME ............................................................................................. 43 3.2.1 Solventes orgánicos tradicionales ............................................................................................. 44 3.2.2 Líquidos Iónicos (IL) ................................................................................................................ 47 3.2.3 Solventes eutécticos profundos (DES) ...................................................................................... 47 3.2.4 Solventes intercambiables (SHS) .............................................................................................. 48
3.3 Aplicación de técnicas de LPME para el análisis de metales pesados en agua ................. 49
Capítulo 4: Metodología para la optimización de un sistema de microextracción en fase
líquida para la determinación de cobre, plomo y cadmio inorgánicos ................................. 51
4.1 Evaluación de las potencialidades preconcentrativas de la técnica de microextracción con
gota directa suspendida (DSDME) para la determinación colorimétrica de Cu2+
, Pb2+
y Cd2+
................................................................................................................................................. 51 4.1.1 Reactivos .................................................................................................................................. 51 4.1.2 Instrumentación ........................................................................................................................ 51 4.1.3 Evaluación de factores que afectan la eficiencia de la extracción……………………….……52
4.1.3.1 Selección de agente quelante…………………………………………………………..........52
4.1.3.2 Efecto del pH ......................................................................................................................... 52 4.1.3.3 Selección del solvente extractante…………………………………………………………..53
4.1.3.4 Efecto del tiempo de extracción ............................................................................................. 54
4.2 Evaluación de la potencialidad preconcentrativa con fases sólidas como extractantes de
Cu2+, Pb2+ y Cd2+ con detección analítica no colorimétrica ................................................ 55 4.2.1 Síntesis de un material magnético a base de carbón activado ................................................... 55
4.2.2 Atomización electrotérmica (ETAAS) como método de detección luego de la preconcentración
con una técnica SPME ....................................................................................................................... 56
Capítulo 5: Resultados y discusión .......................................................................................... 58
5.1 Optimización de parámetros del método DSDME para la determinación colorimétrica de
Cu2+
, Pb2+
y Cd2+
..................................................................................................................... 58 5.1.1 Selección de agente quelante .................................................................................................... 58 5.1.2 Efecto del pH ............................................................................................................................ 60 5.1.3 Efecto del tiempo de extracción ................................................................................................ 62
5.2 Comparación de métodos reportados para la microextracción de cadmio en agua ........... 63
5.3 Evaluación de la potencialidad preconcentrativa con fases sólidas como extractantes de
metales con detección analítica no colorimétrica .................................................................... 63
5.4 Condiciones que se deben optimizar en DµSPE para la preconcentración y determinación
de iones metálicos en matrices acuosas ................................................................................... 67 5.4.1 Selección del adsorbente ........................................................................................................... 67 5.4.2 Concentración inicial del analito en la matriz acuosa ............................................................... 67 5.4.3 Método y velocidad de agitación .............................................................................................. 68 5.4.4 Tiempo de extracción................................................................................................................ 68 5.4.5 Temperatura .............................................................................................................................. 68 5.4.6 pH ............................................................................................................................................. 68
5.5 Atomización electrotérmica (ETAAS) como método de detección luego de la
preconcentración con una técnica SPME ................................................................................ 68
Conclusiones .............................................................................................................................. 71
Bibliografía ................................................................................................................................ 72

~ 6 ~
AµE Adsorptive microextraction Microextracción adsorbente
BAµE Bar adsorptive microextraction Microextracción con adsorción en barra
DµSPE Dispersive micro solid-phase extraction Extracción en microfase sólida dispersiva
DES Deep eutectic solvent Solvente eutéctico profundo
DI-SDME Direct inmersion single drop microextraction Microextracción en gota simple en el modo de inmersión directa
DLLME Dispersive Liquid-Liquid Microextraction Microextracción líquido-líquido dispersiva
DSDME Directly-suspended droplet microextraction Microextracción con gota directa suspendida
ETAAS Electrothermal atomic absorption spectrometry Espectrometría de absorción atómica electrotérmica
FAAS Flame atomic absorption spectrometry Espectrometría de absorción atómica con llama
GC Gas Chromatography Cromatografía de gases
HF-LPME Hollow fibre liquid-phase microextraction Microextracción en fase líquida con fibra hueca
HPLC High performance liquid chromatography Cromatografía líquida de alta resolución
HS-SDME Headspace Solid Phase Microextraction Microextracción en gota simple en el modo de espacio de cabeza
ICP-MS Inductively coupled plasma mass spectrometry Espectrometría de masas con plasma acoplado inductivamente
ICP-OES Inductively coupled plasma optical emission spectrometry Espectrometría de emisión óptica con plasma acoplado inductivamente
IL Ionic liquids Líquido iónico
IT-SPME In tube- Solid phase microextraction Microextracción en fase sólida en tubo
LLE Liquid-Liquid extraction Extracción líquido-líquido
LLLME Liquid-Liquid-Liquid microextraction Microextracción líquido-líquido-líquido
LPME Liquid phase microextraction Microextracción en fase líquida
MDµSPE Magnetic dispersive micro solid-phase extraction Extracción en microfase sólida magnética dispersiva
MEPS Micro Extraction by Packed Sorbent Microextracción por sorbente empaquetado
MIP Molecularly imprinted polymer Polímeros impresos molecularmente
MOFs Metal–organic framework Metal–organic framework
MSAµE Multi-Spheres Adsorptive Microextraction Microextracción con adsorción en multi esféras
NTD Needle trap device Trampa de aguja (Microextracción en fase sólida en aguja)
RDSE Rotating-disk sorptive extraction Extracción adsorbente con disco giratorio
SBSE Stir bar sorptive extraction Microextracción por sorción sobre barra agitadora
SCSE Stir cake sorptive extraction Extracción adsorbente con disco agitador
SDME Single drop microextraction Microextracción en gota simple
SFODME Solidified floating organic drop microextraction Microextracción de gota orgánica solidificada
SHS Switchable hydrophilicity solvent Solvente intercambiable
SPE Solid phase extraction Extracción en fase sólida
SPME Solid phase microextraction Microextracción en fase sólida
TFME Thin film microextraction Microextracción de película delgada
UA-DµSPE Ultrasound assisted- Dispersive micro solid-phase extraction Extracción en microfase sólida dispersiva asistida por ultrasonido
VA-DµSPE Vortex assisted- Dispersive micro solid-phase extraction Extracción en microfase sólida dispersiva asistida por vórtex

~ 7 ~
Hoy en día es de gran interés y preocupación la contaminación de las fuentes de agua por
metales pesados debido a su toxicidad, incluso a bajas concentraciones, y a su carácter no
biodegradable que conduce a que se acumulen en los tejidos vivos causando diversas
enfermedades y trastornos. Como consecuencia de lo anterior, es de gran importancia
monitorear y detectar oportunamente bajos niveles de metales pesados en las fuentes de agua
con el fin de prevenir posibles riesgos para la salud humana y para el medio ambiente.
En los últimos años se han empleado diferentes técnicas analíticas para la determinación de
iones metálicos como por ejemplo espectrometría de absorción atómica de llama (FAAS),
espectrometría de absorción atómica electrotérmica (ETAAS), cromatografía líquida de alta
resolución (HPLC), espectrometría de emisión óptica y espectrometría de masas con plasma
acoplado inductivamente (ICP-OES, ICP-MS), entre otras. No obstante, pese a las ventajas que
poseen estas técnicas, el análisis de metales en muestras de agua presenta un reto debido a la
complejidad de la matriz y a las bajas concentraciones que pueden estar por debajo del límite de
detección del instrumento analítico.
Con el propósito de superar estas dificultades mencionadas, se emplearon inicialmente métodos
exhaustivos de extracción como LLE y SPE. Sin embargo, ya que estos métodos tradicionales
de preparación de muestras presentan ciertas desventajas, actualmente se ha desarrollado un
interés en nuevos métodos no exhaustivos de microextracción con el fin de separar y
preconcentrar los iones metálicos previamente a su determinación.
En este documento se realiza inicialmente una revisión de los principios, configuraciones,
ventajas y limitaciones de las técnicas actuales de microextracción desarrolladas para la
preconcentración y determinación de metales pesados en concentraciones a nivel de subtrazas,
centrándose específicamente en la determinación de cadmio, plomo y cobre en matrices de
naturaleza acuosa. Posteriormente se busca optimizar las condiciones experimentales para llevar
a cabo una preconcentración de diferentes iones metálicos divalentes (Cu2+
, Pb2+
, Cd2+
) por
medio de una técnica de microextracción en fase líquida acoplada a un sistema de detección
colorimétrico. Finalmente, se estudia la factibilidad de llevar a cabo la microextracción en fase
sólida de los ya mencionados iones metálicos para su eventual determinación analítica mediante
técnicas atómicas de detección no colorimétrica.

~ 8 ~
Objetivo General
Llevar a cabo un estudio documental generalizado para abstraer los aspectos más comunes de
las diferentes técnicas miniaturizadas de extracción en fase líquida y sólida, complementado
experimentalmente con algunos estudios de laboratorio aplicables para dichas técnicas en la
determinación de metales pesados a niveles de subtrazas.
Objetivos específicos
i. Llevar a cabo un estudio monográfico a cerca de las diferentes técnicas miniaturizadas
de extracción, con una exposición generalizada de sus modalidades características, de
su proceso de su optimización, de las aplicaciones más habituales, al igual que de sus
ventajas y limitaciones.
ii. Optimizar las condiciones experimentales para llevar a cabo una preconcentración
elevada de diferentes iones metálicos divalentes (Cu2+
, Pb2+
, Cd2+
) haciendo uso de una
técnica de microextracción líquido-líquido (en gota directa suspendida, DSDME)
acoplada a la técnica de UV-Visible como sistema de detección colorimétrica.
iii. Estudiar la factibilidad para llevar a cabo la microextracción en fase sólida de metales
(Cu2+
, Pb2+
, Cd2+
) para su eventual determinación analítica en matrices de naturaleza
acuosa mediante técnicas atómicas de detección no colorimétrica.
iv. Poner a punto los parámetros instrumentales de la técnica espectroscopía de absorción
atómica con calentamiento electroquímica (ETAAS) para la medición de cadmio.

~ 9 ~
La contaminación por metales pesados se ha convertido en un importante problema ambiental y
una amenaza para el bienestar de los organismos vivos ya que a diferencia de muchos
contaminantes orgánicos no son biodegradables y pueden acumularse en los organismos
causando diversas enfermedades y trastornos.1 Es bien sabido que algunos de estos metales
pesados son elementos esenciales para el correcto funcionamiento de los organismos como por
ejemplo el cobalto, el níquel, el cobre y el zinc, sin embargo, hay otros como el cadmio, el
mercurio y el plomo que no son esenciales y no desempeñan papeles biológicos significativos
pero que si tienen un efecto adverso sobre la salud incluso a nivel de trazas.2–4
Estos efectos tóxicos agudos son conocidos desde hace más de cien años, cuando las sales de
estos metales se empleaban como pigmentos, no obstante, tras el reconocimiento de sus valiosas
propiedades metalúrgicas, se generó una mayor producción de metales pesados después de 1910
de aproximadamente 100,000 lb por año a 31, 000,000 lb por año en 1968.5 Como consecuencia
del desarrollo de actividades industriales, la presencia de metales pesados en las fuentes
naturales de agua ha aumentado rápidamente.6 Además de los procesos antropogénicos como la
minería, la fundición y la agricultura, la contaminación del agua también se produce por
procesos naturales como la meteorización, la erosión de las rocas y los depósitos de minerales.7
Entre los diversos iones metálicos, el cadmio, el mercurio y el plomo presentan una mayor
toxicidad para los seres humanos.7 Pese a la alta toxicidad del cadmio, este elemento es
ampliamente utilizado en la industria, especialmente en galvanoplastia y en la producción de
pigmentos, esmaltes, vidrios, plásticos, tintas de impresión, caucho, lacas, aleaciones, textiles,
fertilizantes fosfatados, baterías de Ni-Cd., fotoconductores, celdas solares fotoeléctricas y
neumáticos de automóviles, entre otros.8,9
Las principales fuentes de exposición al cadmio en
los seres humanos son las fuentes de agua, los alimentos y vegetales contaminados y el
consumo de tabaco.10,11
Los efectos a corto plazo incluyen náuseas, vómitos, diarrea y
calambres y se ha demostrado que concentraciones relativamente bajas de cadmio absorbido
tiende a retenerse por largos periodos, en general, la vida media de este metal es de 4 a 19
años.9,12
Esta bioacumulación conlleva a que la exposición del cadmio a largo plazo cause
graves problemas de salud como hipertensión, enfermedades óseas, daños hepáticos y renales e
incluso cáncer.6,13,14
La ingesta semanal tolerable de Cd es de 0.8 µg / kg de peso corporal15
por
lo cual la Organización Mundial de la Salud (OMS) estableció en el 2010 que la concentración
máxima tolerable de cadmio en agua potable es de 3.0 µg/ L.16
De igual forma, el mercurio es uno de los metales pesados tóxicos con el mayor riesgo
ambiental. La exposición excesiva de mercurio al cuerpo humano genera daños en el ADN,
insuficiencia respiratoria, hipertensión, trastornos del sistema nervioso, desajuste en la secreción
hormonal del organismo, colapso cardiovascular, insuficiencia renal aguda, trastornos
gastrointestinales, daños cerebrales y defectos del sistema nervioso, como la enfermedad de
Minamata.17,18
En este caso, el nivel permisible de mercurio inorgánico en el agua potable es de
1.0 µg/ L.19
Asimismo, el plomo es un elemento tóxico de carácter acumulativo que representa un riesgo
para la salud humana y para el medio ambiente. Debido a sus propiedades físicas como bajo
punto de fusión y fácil maleabilidad se ha empleado en la fabricación de diferentes productos
como tuberías, alambres, soldaduras, baterías, pinturas y cerámicas.20,21
La exposición a corto
plazo genera dolores de cabeza y trastornos en el sueño mientras que a largo plazo conduce a

~ 10 ~
efectos nocivos sobre la salud humana como disfunción cerebral y renal, infertilidad, reducción
de la actividad enzimática y dificultades neuromusculares, entre otras.21,22
Adicionalmente, la
Agencia de Protección Ambiental de EE.UU. (EPA) clasificó el plomo como un carcinógeno
humano del Grupo B2. Como consecuencia de su carácter tóxico, se estableció que el límite
tolerable para la ingesta semanal de Pb (II) es de 25 µg / kg de peso corporal y de acuerdo a la
Organización Mundial de la Salud (OMS) la concentración máxima tolerable de plomo en agua
para el consumo humano es de 10 µg/ L.16,23
El cobre a diferencia de los anteriores metales es un elemento esencial que se requiere para la
síntesis de glóbulos rojos, enzimas, tejidos y para el desarrollo óseo, sin embargo, si es ingerido
en grandes cantidades es tóxico y cancerígeno.24
Debido a su alta conductividad térmica y
eléctrica se ha utilizado como conductor térmico, conductor eléctrico y como componente de
varias aleaciones metálicas.25
Las diferentes aplicaciones industriales pueden llevar a la
exposición excesiva de este metal lo que puede resultar en dolor de cabeza, vómitos, náuseas,
insuficiencia hepática y renal, problemas respiratorios, dolor abdominal y diversas y graves
enfermedades como la esclerosis lateral amiotrófica, Alzheimer, Parkinson, la enfermedad de
Wilson y el síndrome de Menkes.4,25
La ingesta diaria máxima tolerable de cobre es de 0,5 mg /
kg de peso corporal26
y el límite permisible dado por la OMS para Cu (II) en el agua potable es
de 1.5–2 mg / L.27
En este contexto, es evidente la importancia de la determinación oportuna de bajos niveles de
metales pesados en aguas naturales con el fin de prevenir efectos adversos en la salud humana.
Las técnicas analíticas comunes para la detección de iones de metales pesados son la
espectroscopía de absorción atómica (AAS), la espectrometría de masas con plasma acoplado
inductivamente (ICP-MS), la cromatografía líquida de alta resolución (HPLC), la espectrometría
de emisión atómica con plasma acoplado inductivamente (ICP-AES), el análisis de
activación por neutrones (NAA) y la espectroscopía de emisión óptica con plasma acoplado
inductivamente (ICP-OES).8,9,13
Estas técnicas son confiables, sensibles y permiten una
detección cuantitativa de los iones metálicos, sin embargo, no son adecuadas para el análisis in
situ debido al alto costo de los instrumentos, la necesidad de procedimientos de
preconcentración extensos y el control de las condiciones experimentales.25,28
Una alternativa
fácil y rápida empleada como método analítico tradicional es el análisis colorimétrico, en el cual
diferentes agentes quelantes inducen afinidades diferentes entre iones metálicos generando
complejos coloreados, el cambio de color evidente de la muestra no solo hace que la prueba sea
directa y conveniente, sino que también es posible establecer una relación lineal entre la
concentración de los iones metálicos y la absorbancia.29
Ahora bien, de no ser posible la detección colorimétrica es necesario emplear métodos atómicos
espectrométricos instrumentales. La espectrometría de absorción atómica en llama es una
técnica analítica comúnmente utilizada para la determinación de metales pesados30
, sin
embargo, una desventaja importante de esta técnica es su baja sensibilidad para la mayoría de
los metales debido a la baja eficiencia de nebulización del sistema.14
Como consecuencia de
ello, se han desarrollado otras técnicas como la espectrometría de absorción atómica de horno
de grafito también conocida como espectrometría de absorción atómica electrotérmica (ETAAS)
la cual presenta límites de detección sustancialmente inferiores en comparación con la
espectroscopía de absorción atómica de llama.31
Por ejemplo, para la determinación de cadmio,
el límite de detección es de 5 µg / L con el método de llama y 0.1 µg / L con el procedimiento
de horno.32
Esta mejora significativa del límite de detección permite que ETAAS sea una

~ 11 ~
técnica alternativa a la técnica de espectrometría de masas por plasma de acoplamiento
inductivo (ICP-MS) cuando la concentración de los analitos se encuentra en el nivel de ppb.33,34
No obstante, el análisis de metales pesados en muestras de agua es un trabajo desafiante puesto
que estos generalmente existen a niveles de concentración extremadamente bajos y en ocasiones
la matriz de la muestra es muy compleja.35
En estos casos, puede ser necesario un
pretratamiento de la muestra con el fin de aislar o enriquecer los analitos de la matriz. Este paso
de preparación es considerado uno de los pasos más críticos en el proceso analítico ya que
representa el mayor consumo de tiempo y costos e influye directamente en los rendimientos de
extracción.36
La selección del procedimiento de preparación de la muestra depende de factores
como el tamaño de la muestra, el sistema de medición o detección, el tipo de análisis
(cualitativo o cuantitativo), la naturaleza del analito y la concentración inicial del analito en la
muestra. 37
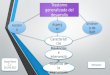
Dentro de los procedimientos para la preparación de la muestra se encuentran los métodos de
extracción que pueden ser exhaustivos o no exhaustivos (Fig. 1). En los métodos tradicionales
de extracción exhaustivos, como extracción líquido-líquido (LLE) y extracción en fase sólida
(SPE), todo el analito se transfiere de la fase “donora” a la fase aceptora. Pese a la alta
recuperación de extracción, los enfoques de extracción exhaustivos presentan desventajas como
la complejidad inherente del método con múltiples pasos y tiempos extensos, el alto consumo de
muestra y solventes, la alta contaminación ambiental, los altos costos y la dificultad de
automatización.37
Figura 1. Clasificación general de las técnicas de extracción.
Probablemente, entre estos métodos de pretratamiento mencionados anteriormente, la extracción
LLE es el más utilizado5 pese a tener claras desventajas como el consumo y la eliminación de
cantidades significativas de solventes tóxicos, la complejidad y el uso de protocolos largos.38,39
Otra técnica comúnmente empleada es SPE, en comparación con LLE, esta ofrece una
operación más simple y la posibilidad de automatización, aun así en muchas ocasiones se
requieren de grandes cantidades de solventes para la elución de los analitos.35
Por tal motivo,
con el objetivo de simplificar y mejorar el proceso de pretratamiento, en los últimos años se han
desarrollado una serie de técnicas miniaturizadas no-exhaustivas basadas en LLE y SPE
conocidas como técnicas de microextracción cuyas ventajas incluyen alta velocidad de análisis,

~ 12 ~
gran eficiencia, bajos costos operativos, procedimientos analíticos amigables con el medio
ambiente y análisis altamente selectivos.40–42
Además, estas técnicas de microextracción no exhaustivas poseen ventajas únicas puesto que
solo una pequeña porción del analito objetivo se extrae de la matriz. Esta característica permite
el monitoreo de cambios químicos, equilibrios de partición y especiación en el sistema
investigado porque el muestreo causa una perturbación mínima en el sistema. Por lo tanto, el
uso de estrategias basadas en microextracción da como resultado una mejor caracterización y
una información más precisa sobre el sistema o proceso investigado en comparación con las
técnicas exhaustivas. Sin embargo, este tipo de técnicas requiere una cuidadosa calibración y
optimización por lo cual el desarrollo de métodos analíticos cuantitativos basados en
microextracción requieren más tiempo, pero una vez los procedimientos se optimizan, son más
convenientes y rentables en comparación con los enfoques de extracción exhaustiva
convencionales. 43
Figura 2. Técnicas de microextracción en fase sólida y en fase líquida. *Nota: Acrónimos
en la página 6.
Como se observa en el esquema anterior (Fig. 2), en general, las técnicas de microextracción se
pueden clasificar en dos categorías principales: técnicas basadas en extracción en fase sólida y
técnicas basadas en extracción en fase líquida.44
En los siguientes dos capítulos (capítulos 2 y 3)
del presente manuscrito se discutirán monográficamente algunas de estas técnicas generales de
microextracción desarrolladas para el análisis y determinación de metales pesados con el fin de
describir sus potencialidades extractivas con base a sus características, su proceso de
optimización, sus aplicaciones más habituales, al igual que a sus ventajas y limitaciones.

~ 13 ~
2.1 Microextracción en fase sólida en fibra (SPME)
A principios de la década de 1990, Pawliszyn et al.45
introdujeron por primera vez la técnica
SPME para abordar la necesidad de una preparación de muestra rápida con menor impacto sobre
la salud humana y el medio ambiente. Esta técnica no exhaustiva se basa en la exposición de
una pequeña cantidad de fase sólida extractante a la muestra para la extracción y
preconcentración de compuestos tanto orgánicos como inorgánicos, así como de compuestos
que se caracterizan por diferente polaridad y volatilidad. SPME no solo permite la integración
de muestreo y preparación de muestras, simplificando el proceso analítico sino que gracias a su
tamaño reducido es muy conveniente para el análisis y monitoreo in situ.46
Figura 3. Dispositivo SPME en fibra. Adaptado de: 47
En el enfoque tradicional, el dispositivo SPME está constituido por una delgada fibra de sílice
fundida recubierta con una capa de adsorbente como fase de extracción (Fig. 3).48
Por otro lado,
y como se observa en la figura 4, existen tres modos básicos para la extracción por SPME en
fibra. El primero de ellos es el modo de extracción directo DI-SPME (Fig. 4a), en este la fibra
recubierta se sumerge en la solución de la muestra y los analitos se transfieren directamente
desde la matriz al recubrimiento extractante. La agitación de la muestra puede acelerar el
proceso de transferencia de masa de los analitos de la muestra a la fase de extracción
disminuyendo así el tiempo de extracción. En el segundo modo conocido como HS-SPME (Fig.
4b) la fibra se coloca en el espacio superior sobre la matriz de la muestra, este modo es de gran
utilidad para compuestos volátiles y muestras complejas con interferentes de alto peso
molecular como por ejemplo muestras biológicas. Para el análisis de compuestos no volátiles
con la interferencia de macromoléculas, ninguno de los dos modos presentados anteriormente es
útil por lo cual en tales ocasiones es oportuna la extracción protegida con membrana. Con la
protección de membranas de acceso selectivo, las macromoléculas no deseadas pueden
impedirse y el recubrimiento se mantiene aislado por lo cual la vida útil de la fibra se puede
extender significativamente.46,49,50

~ 14 ~
Figura 4. Modos de SPME: a) inmersión directa, b) en espacio de cabeza o “headspace”, c)
protegido por membrana. Adaptado de 49
La técnica SPME en fibra es probablemente uno de los enfoques de microextracción en fase
sólida más empleados debido a su característica simplicidad, alta eficiencia de extracción y alto
factor de preconcentración. Sin embargo, con todos los beneficios y aplicaciones, existen varias
limitaciones inherentes de este método como se muestra a continuación en la tabla 1:37,46
Tabla 1. Ventajas y desventajas de SPME en fibra
Con el objetivo de superar estas limitaciones de SPME en fibra se desarrollaron diferentes
enfoques. En primer lugar, la preparación de fibras con técnicas como sol-gel que proporcionan
uniones covalentes entre el sorbente y el núcleo de fibra para contribuir a la resistencia y
estabilidad de la fibra formada. En segundo lugar, el uso de membranas con el fin de crear una
barrera entre el extractor y la muestra de forma tal que la vida útil de la fibra se extienda
significativamente. Y en tercer lugar, el desarrollo de métodos alternos de microextracción en
fase sólida sin fibra.37,46

~ 15 ~
Adicionalmente, es importante la optimización de ciertos parámetros experimentales de SPME,
como se observa en la figura 5 puesto que estos influyen directamente en la eficiencia de la
extracción.
Figura 5. Parámetros a considerar en el desarrollo de un método SPME. Adaptado de:43
2.2 Materiales empleados como sorbentes en SPME
La elección del adsorbente apropiado es un factor crítico para obtener una buena precisión y una
alta recuperación en el procedimiento de microextracción en fase sólida. En este sentido, existen
cuatro criterios principales que se usan comúnmente para seleccionar el recubrimiento de la
fibra: (a) el peso molecular y el tamaño de los analitos, (b) la polaridad de los analitos, (c) los
niveles de concentración de los analitos, y (d) la complejidad de la muestra. 50
Dentro de los recubrimientos de fibra comerciales más comunes se encuentran
polidimetilsiloxano (PDMS), divinilbenceno (DVB), carboxeno (CAR), polietilenglicol (PEG) y
Carbowax (CW). Estos recubrimientos sólidos generalmente son materiales porosos con una
gran área superficial y los analitos quedan atrapados en la superficie del sorbente por fuerzas de
Van der Waals o interacciones iónicas. Aunque estos recubrimientos sólidos se caracterizan por
una alta eficiencia de extracción, algunos inconvenientes inherentes limitan su utilidad en
algunas aplicaciones. Adicionalmente, a medida que la tecnología de preparación de muestras
ha progresado, se han desarrollado varios materiales novedosos como por ejemplo materiales a
base de carbono, polímeros impresos molecularmente (MIP), nanopartículas metálicas, MOFs y
aerogeles.44,51–53
Materiales a base de carbono
Los adsorbentes a base de carbono han despertado una considerable atracción en la
determinación y análisis de iones metálicos ya que estos adsorbentes presentan características
como una alta capacidad de sorción, la facilidad de modificación de la superficie y excelentes
propiedades eléctricas, químicas y térmicas.54

~ 16 ~
Uno de estos materiales es el carbón activado el cual ha demostrado ser un adsorbente eficiente
para la eliminación de una amplia variedad de contaminantes orgánicos e inorgánicos presentes
en el medio ambiente. Se caracteriza por una gran área superficial (entre 500 a 1500 m2)
relacionada con su forma estructural y una alta estabilidad fisicoquímica, porosidad, y capacidad
de sorción. Debido a estas propiedades, al bajo costo y alta accesibilidad, el carbón activado es
visto como el sorbente más utilizado en el tratamiento de agua.55,56
Adicionalmente, hoy en día el uso de nanomateriales como adsorbentes genera gran interés
debido a sus prometedoras propiedades. Dentro de los nanomateriales de carbono (Fig. 6) se
encuentra el grafeno, un nanomaterial bidimensional que se puede sintetizar mediante métodos
simples a partir de grafito. Esta facilidad de síntesis aumenta en gran medida su aplicación en
diferentes campos y permite la producción de grafeno a bajo costo. Como sorbente, este
nanomaterial es de gran utilidad debido a su superficie específica alta y a su excelente
estabilidad química.57
Otro tipo de nanomaterial de carbono son los nanotubos de carbono (CNT) los cuales han
despertado una gran atención como adsorbentes debido a su excelente capacidad para la
eliminación de diversos contaminantes orgánicos e inorgánicos puesto que presentan
propiedades como alta resistencia, gran área superficial, conductividad térmica, y estabilidad.58
Figura 6. Nanomateriales usados en SPME. Tomado de:59
Polímeros impresos molecularmente (MIP)
Los polímeros impresos molecularmente (MIP) son polímeros sintéticos que poseen sitios de
unión selectivos para analitos de interés, éstos se forman a partir de un proceso de
copolimerización en presencia de una molécula de impresión o plantilla. Una vez la plantilla se
retira del polímero, se convierten en sitios de reconocimiento lo que los hace adecuados para su
uso en procesos de separación.44
Metal organic frameworks (MOFs)
Los materiales organometálicos conocidos como MOFs, por sus siglas en inglés, construidos a
partir de la coordinación de ligandos orgánicos y grupos/iones metálicos, han surgido como un
nuevo tipo de adsorbente debido a sus propiedades como alta porosidad, gran área superficial,
flexibilidad, tamaño de poro ajustable, además de su estabilidad térmica y mecánica.60

~ 17 ~
Aerogeles
Los aerogeles se sintetizan mediante la técnica sol-gel, en la que se utiliza la hidrólisis para
convertir los precursores en una suspensión coloidal que finalmente se convierte en una red
tridimensional. Estos materiales tienen propiedades únicas, que incluyen alta área superficial,
baja densidad, alta porosidad y estabilidad térmica, lo que los hace viables como adsorbentes en
la extracción de analitos polares y no polares en muestras acuosas.61
2.3 Termodinámica de la microextracción en fase sólida
Independientemente del tipo de fase extractante, el fenómeno fundamental que ocurre en el
sistema de extracción es la partición de analitos entre todas las fases del sistema. La
comprensión de la teoría de SPME proporciona información y dirección al desarrollar y
optimizar un método de extracción ya que permite identificar qué parámetros (temperatura,
volumen de las fases del sistema, recubrimiento de fibra, pH, fuerza iónica, entre otros) son
termodinámicamente significativos y decisivos con respecto a la eficiencia de extracción.43,46
Figura 7. Proceso de extracción por SPME: Vm (volumen de la muestra); Vf (volumen del
recubrimiento de la fibra); Kfm (coeficiente de distribución fibra / muestra; C0
(concentración inicial del analito en la muestra). Adaptado de:43
Por lo general, el proceso de microextracción se considera completo cuando la concentración de
analito alcanza el equilibrio entre la matriz de la muestra y el recubrimiento de la fibra (Fig. 7).
Cuando solo se consideran dos fases (por ejemplo, en el modo de inmersión directa, la matriz de
muestra y el recubrimiento de la fibra) las condiciones de equilibrio se pueden describir
mediante la ecuación (1) de acuerdo con la ley de conservación de masas.
𝑛0 = 𝑛𝑚 + 𝑛𝑓 (1)
En la ecuación anterior se expresa la cantidad molar de analito antes y después del equilibrio. Si
se expresa la cantidad molar en términos de concentraciones y volúmenes es posible reescribir
la ecuación (1):
𝐶0𝑉𝑚 = 𝐶𝑚∞𝑉𝑚 + 𝐶𝑓
∞𝑉𝑓 (2)
Donde Cf∞ y Cm
∞ son concentraciones del analito en el equilibrio en el recubrimiento de fibra y
la muestra, respectivamente.

~ 18 ~
El coeficiente de distribución Kfm del analito entre el recubrimiento de la fibra y la matriz de la
muestra se define como:
𝐾𝑓𝑚 = 𝐶𝑓
∞
𝐶𝑚∞ (3)
Las ecuaciones (2) y (3) se pueden combinar y reorganizar para obtener (4)
𝐶𝑚∞ =
𝐶𝑓∞
𝐾𝑓𝑚
𝐶0𝑉𝑚 = 𝐶𝑓
∞
𝐾𝑓𝑚𝑉𝑚 + 𝐶𝑓
∞𝑉𝑓
𝐶0𝑉𝑚 = 𝐶𝑓∞ (
𝑉𝑚
𝐾𝑓𝑚+ 𝑉𝑓)
𝐶𝑓∞ =
𝐶0𝑉𝑚
( 𝑉𝑚 + 𝐾𝑓𝑚𝑉𝑓
𝐾𝑓𝑚)
𝐶𝑓∞ = 𝐶0
𝐾𝑓𝑚𝑉𝑚
𝐾𝑓𝑚𝑉𝑓+𝑉𝑚 (4)
Finalmente, el número de moles de analito n extraído por el recubrimiento puede calcularse a
partir de la ecuación. (5):
𝑛 = 𝐶𝑓∞𝑉𝑓 = 𝐶0
𝐾𝑓𝑚𝑉𝑚𝑉𝑓
𝐾𝑓𝑚𝑉𝑓+𝑉𝑚 (5)
Cuando el volumen de la muestra es muy grande, es decir, Vm >>KfmVf, la ecuación (5) se puede
simplificar a:
𝑛 = 𝐶𝑓∞𝑉𝑓 = 𝐶0
𝐾𝑓𝑚𝑉𝑚𝑉𝑓
𝑉𝑚
𝑛 = 𝐶0𝐾𝑓𝑚𝑉𝑓 (6)
La ecuación (6) indica que la cantidad de analito extraído en el recubrimiento (n) es linealmente
proporcional a la concentración de analito en la muestra (C0), ésta es la base analítica para la
cuantificación usando SPME.
Ahora bien, si se considera un sistema de tres fases (por ejemplo, en el modo de espacio de
cabeza o “headspace”), las condiciones de equilibrio se pueden describir mediante la ecuación
(7) de acuerdo con la ley de conservación de masas.
𝐶0𝑉𝑚 = 𝐶𝑚∞𝑉𝑚 + 𝐶𝑓
∞𝑉𝑓 + 𝐶ℎ∞𝑉ℎ (7)
Donde C0 es la concentración inicial del analito en la matriz; Cf∞, Ch
∞ y Cm
∞ son las
concentraciones de equilibrio del analito en la fibra, el espacio de cabeza y la matriz de la
muestra, respectivamente; y Vf, Vh y Vm son los volúmenes del recubrimiento, el espacio
superior y la matriz, respectivamente. Si se define la constante de distribución entre fibra /
espacio de cabeza como 𝐾𝑓ℎ = 𝐶𝑓
∞
𝐶ℎ∞ y la constante de distribución entre espacio de cabeza/

~ 19 ~
muestra como 𝐾ℎ𝑚 = 𝐶ℎ
∞
𝐶𝑚∞ la cantidad del analito absorbida por el recubrimiento 𝑛 = 𝐶𝑓
∞𝑉𝑓
puede expresarse como
𝑛 = 𝐶𝑓∞𝑉𝑓 =
𝐾𝑓ℎ𝐾ℎ𝑚𝑉𝑓𝐶0 𝑉𝑚
𝐾𝑓ℎ𝐾ℎ𝑚𝑉𝑓+𝐾ℎ𝑚𝑉ℎ+𝑉𝑚 (8)
Ya que 𝐾𝑓𝑚 = 𝐾𝑓ℎ𝐾ℎ𝑚
𝑛 = 𝐶𝑓∞𝑉𝑓 =
𝐾𝑓𝑚𝑉𝑓𝐶0 𝑉𝑚
𝐾𝑓𝑚𝑉𝑓+𝐾ℎ𝑚𝑉ℎ+𝑉𝑚 (9)
La ecuación anterior establece que bajo condiciones de equilibrio la cantidad de analito extraído
es independiente de la ubicación de la fibra en el sistema. Se puede colocar en el espacio
superior o directamente en la muestra siempre que los volúmenes del revestimiento de fibra, el
espacio superior y la muestra se mantengan constantes.
2.4 Optimización de las condiciones de extracción de SPME
La termodinámica del proceso que está definida por la constante de distribución de la fase de
extracción y la matriz de la muestra predice los efectos de modificar ciertas condiciones
experimentales.43,46
2.4.1 Temperatura
La temperatura a la que la se da el proceso influye tanto en la termodinámica como en la
cinética del aislamiento del analito. Si tanto la temperatura de la muestra como la de la fibra
cambian de T0 a T, la constante de distribución cambia de acuerdo con la siguiente ecuación:43
𝐾𝑓𝑚 = 𝐾0𝑒𝑥𝑝 [−∆𝐻
𝑅(
1
𝑇−
1
𝑇0)] (10)
Donde K0 es la constante de distribución cuando la fibra y la muestra están a la temperatura T0
(en grados Kelvin), ΔH es el cambio de la entalpía molar del analito cuando se transfiere de la
muestra al recubrimiento de la fibra y R es la constante de los gases. El cambio de entalpía, ΔH,
se considera constante sobre los rangos de temperatura típicos de los experimentos SPME. Se
puede determinar midiendo 𝐾𝑓𝑚 a dos temperaturas diferentes.
Los efectos de la temperatura deben tenerse en cuenta cuando se producen variaciones de
temperatura durante el muestreo o cuando se usa calentamiento para aumentar las tasas de
extracción. Cuando el valor de 𝐾𝑓𝑚 para un analito es mayor que 1, el proceso es exotérmico lo
que significa que el cambio molar en la entalpía del analito cuando se transfiere de la muestra al
recubrimiento de fibra es mayor que 0. Por lo tanto, de acuerdo a la ecuación (10) un aumento
en la temperatura conduce a una disminución en el coeficiente de partición 𝐾𝑓𝑚.43,46
2.4.2 Tiempo de extracción
El tiempo de extracción es un parámetro crítico en el proceso de SPME. Antes del equilibrio
entre la fibra y la muestra el tiempo es un factor decisivo, sin embargo, una vez se alcanza el
equilibrio los pequeños cambios en los tiempos de extracción no tienen influencia crítica en los
resultados cuantitativos.62

~ 20 ~
Este parámetro depende entonces del objetivo del análisis, por ejemplo, si se requiere realizar
una gran cantidad de mediciones reproducibles es posible trabajar en condiciones de
“preequilibrio”. No obstante, en este caso es imprescindible mantener el tiempo de extracción y
la agitación exactamente iguales. Actualmente, la posibilidad de realizar el aislamiento del
analito en condiciones de no equilibrio se ha vuelto bastante atractiva gracias a la facilidad de
automatización de SPME. Vale la pena mencionar que, en condiciones de no equilibrio, algunos
otros parámetros, como el coeficiente de partición, los volúmenes de fases o la concentración de
analitos, influyen en gran medida sobre el tiempo puesto que afectan el flujo de analitos en el
sistema.
Figura 8. Efecto del tiempo de extracción sobre la concentración de analito extraído en el
sorbente Adaptado de: 62
Ahora bien, como se observa en la figura 8, se puede hacer la suposición de que en la sección
inicial del preequilibrio, la cantidad de analitos retenidos en la fibra aumenta linealmente a
medida que aumenta el tiempo. El supuesto mencionado anteriormente se refiere solo a tiempos
de extracción muy cortos, en relación a la cuantificación esto hace posible realizar una
calibración controlada cinéticamente solo si los parámetros del proceso son constantes.46
Por otra parte, la extracción en condiciones de equilibrio se realiza en la práctica solo cuando es
necesario, es decir, cuando la sensibilidad es un factor importante. Por ejemplo, si la cantidad de
analitos retenidos calculada a partir de la ecuación (5) está cerca de los límites de detección del
detector que se va a emplear. En este caso se usa un tiempo de extracción después del cual la
sorción del analito en la fibra permanece constante. Adicionalmente, el equilibrio se puede
cambiar para proporcionar mayores eficiencias de extracción por variación de temperatura y
modificación de parámetros de la muestra como la fuerza iónica y el pH.46,63
2.4.3 Agitación de muestra
La agitación de la muestra facilita el transporte de masa en el sistema, lo que resulta en una
disminución del tiempo de extracción necesario para alcanzar el estado de equilibrio. Desde el
punto de vista práctico, es necesario asegurar una mezcla homogénea de la muestra para lograr
la repetibilidad de los resultados por lo cual es esencial mantener el mismo método e intensidad
de agitación. Hay varios métodos de agitación disponibles en SPME, los más comunes son
agitación magnética, agitación con ultrasonido y agitación con vórtex (Tabla 2).

~ 21 ~
En el caso de la agitación magnética, con el fin de lograr una alta repetibilidad de las
mediciones es imprescindible tener en cuenta aspectos como: a) las dimensiones del agitador,
las cuales deben ser siempre las mismas, b) la velocidad de agitación que debe permanecer
constante (rpm), c) en el caso de la fibra, esta debe exponerse siempre en la misma posición con
respecto al eje del contenedor de la muestra.46
Tabla 2. Ventajas y desventajas de los diferentes métodos de agitación en SPME
2.4.4 pH
La sensibilidad de SPME es máxima cuando se extraen analitos neutros que no están disociados.
Por lo tanto, la modificación del pH puede mejorar la sensibilidad del método para compuestos
básicos o ácidos. Cuando se trata de compuestos ácidos, se sugiere seleccionar un pH dos
unidades más bajo que el pKa del compuesto. Al tratar con compuestos básicos, se sugiere
seleccionar un pH que sea mayor que el pKa del compuesto (dos unidades o más). Al realizar
SPME en el modo de “headspace”, la matriz se puede ajustar a cualquier valor de pH sin dañar
la fibra. Pero si se realiza DI-SPME se debe tener precaución al ajustar el pH puesto que se
puede afectar el recubrimiento de la fibra.43,63
2.4.5 Efecto salino (“Salting-out”)
En ciertas aplicaciones, la adición de una sal a la solución acuosa minimiza la variabilidad en la
fuerza iónica de la muestra mejorando la eficiencia de extracción. Los analitos orgánicos,
particularmente aquellos con alta polaridad, disminuyen su solubilidad en agua, lo que da como
resultado una mayor cantidad de moléculas de analito en el recubrimiento de la fibra. El
aumento en la eficiencia de extracción al emplear salting-out depende en gran medida de la
concentración de analito y de la concentración de la sal. Es importante tener en cuenta que si
bien la sal puede mejorar la extracción de los analitos deseados, también puede causar la
extracción conjunta de otros compuestos no deseados de la matriz de la muestra.46,63
2.4.6 Derivatización
La baja eficiencia de la técnica SPME es particularmente visible cuando se analizan analitos
presentes en niveles de trazas, así como durante la extracción de analitos de muestras con
matrices complejas. Para algunos compuestos, la conversión de analitos en sus derivados
caracterizados por diferentes estructuras y propiedades químicas, seguida del proceso de
extracción, es una buena solución para mejorar la eficiencia.46

~ 22 ~
2.5 Aplicaciones de SPME en fibra en la determinación de metales
pesados en agua
Realizando una revisión bibliográfica se observa que la aplicación de SPME en fibra para la
determinación de iones metálicos es limitada debido a la disponibilidad de adsorbentes. Por lo
general es necesaria una derivatización para obtener un compuesto organometálico hidrófobo
con el fin de lograr la adsorción sobre las fibras.
Beceiro-González et al.64
reportan un método de HS-SPME acoplado a cromatografía de gases
(SPME-GC) para el análisis de iones metálicos. Si bien este método es bastante sensible, se
limita solo a complejos metálicos volátiles por lo cual este estudio se centra únicamente en la
determinación de especies organometálicas en muestras de agua.
SPME también se ha combinado con cromatografía líquida de alto rendimiento (HPLC) con
detección UV para la determinación de metales. Kaur et al.65
proponen un método rápido,
sensible, selectivo y económico para la preconcentración y análisis de Co (II), Ni (II), Cu (II) y
Pd (II). Sin embargo, en este método se requiere un paso de derivatización mediante el
acomplejamiento con TPTS (2-tiofenaldehído-3-tiosemicarbazona) para generar compuestos
organometálicos hidrófobos que puedan adsorberse en una fibra de PDMS.
Por último, solo se encontró un método para la determinación sensible de metales traza sin
derivatización o volatilización. En 2018, Rohanifar y su grupo1 reportaron un método simple y
sensible para la preconcentración y determinación de Ag, Cd, Co, Ni, Pb y Zn por medio de DI-
SPME con detección por ICP-MS. Como se observa en la figura 9, se empleó una fibra
recubierta con un nuevo compuesto formado por polipirrol, un material poroso y económico con
excelente estabilidad química y térmica, con nanotubos de carbono CNT y 1,10-fenantrolina
como agente quelante.
Figura 9. Esquema del método de DI-SPME desarrollado para la preconcentración y análisis
de plata, cadmio, cobalto, hierro, níquel, plomo y zinc en muestras de agua. Tomado de: 1
La tabla 3 resume algunas de las características y figuras de mérito de los métodos mencionados
anteriormente. Como se mencionó inicialmente, este documento se enfocará en describir las
potencialidades extractivas de sistemas generalizados de microextracción en el análisis y
determinación de cadmio, plomo y cobre.

~ 23 ~
Tabla 3. Métodos reportados de SPME en fibra para la preconcentración y determinación
de Cd, Pb y Cu en agua. (LOD: límite de detección; RR%: Recuperación relativa; RSD%:
Desviación estándar relativa)
2.6 Métodos alternos a SPME en fibra
Posteriormente se desarrollaron varias configuraciones alternas al enfoque tradicional como
consecuencia a las limitaciones de SPME en fibra anteriormente descritas. Cabe señalar que si
bien el nombre SPME inicialmente se refería solo a este enfoque en fibras hoy en día se refiere
en general a todos los métodos de microextracción que implican una fase sólida como fase
extractante.
A continuación, en la figura 10, se muestran de forma descriptiva algunos de estos métodos
alternativos al enfoque tradicional de SPME propuestos en los últimos años.
Figura 10. Algunas configuraciones de SPME. *Nota: Ver acrónimos en la página 6.

~ 24 ~
2.6.1 IT-SPME: Microextracción en fase sólida en tubo
Pawliszyn et al., que inicialmente presentaron SPME en fibra, también introdujeron la técnica
de microextracción en fase sólida en tubo (IT-SPME)66
en 1997 para su aplicación en
cromatografía líquida de alto rendimiento (HPLC) (Fig. 11).
Figura 11. Representación esquemática de SPME en tubo. Adaptado de:67
Esta técnica capilar tiene la ventaja adicional de automatización y acoplamiento en línea entre el
método de separación y el método de detección. En este modo de SPME, los analitos se extraen
y se preconcentran por sorción en la superficie interna de una columna capilar recubierta
internamente con fase extractiva. Esta columna capilar a su vez se conecta en línea con un
sistema cromatográfico y un detector.68
IT-SPME se ha aplicado ampliamente para extraer diversos compuestos orgánicos, aunque rara
vez se utiliza para preconcentrar iones de metales pesados. En 2019, Mei et al.69
desarrollaron
un método de microextracción en fase sólida en tubo reforzado con magnetismo (MR/IT-
SPME) para la preconcentración y determinación de Cu (II), Co (II) y Hg (II) en muestras de
agua. Los parámetros analíticos respectivos de este método se resumen en la tabla 4. La
columna de microextracción capilar empleada se preparó con nanopartículas magnéticas Fe3O4
modificadas (MCEN) y se empleó dietilditiocarbamato de sodio trihidratado (DDTC) para
formar compuestos de coordinación metálicos que fácilmente se pudieran retener en la fase
extractante. Luego del paso de preconcentración, los complejos se determinaron en línea
mediante cromatografía líquida de alto rendimiento con detección de arreglo de diodos (HPLC-
DAD).
Tabla 4. Método reportado de SPME en tubo por Mei et al. para la preconcentración y
determinación de Cu en agua. (LOD: límite de detección; LOQ: Límite de cuantificación;
RSD%: Desviación estándar relativa)

~ 25 ~
2.6.2 Microextracción en fase sólida en aguja
En las técnicas de microextracción de flujo de muestra (SFME), el muestreo dinámico se realiza
mediante una jeringa o bomba que introduce la muestra a través de los recubrimientos o
sorbentes que se encuentran ya sea en una aguja, un tubo/ capilar o una punta. Entre este tipo de
técnicas SPME con aguja ha atraído mucha atención en los últimos años. El método inicial
conocido como extracción dinámica en fase sólida (SPDE) usaba una jeringa con una aguja
recubierta internamente como dispositivo de extracción (Fig. 12a). El volumen del
recubrimiento en la pared interna de la aguja es de varios µL, en comparación con ≈ 0.5 µL para
una fibra SPME, por lo que SPDE logra un mayor factor de enriquecimiento. Además, el
dispositivo es más robusto que las fibras SPME que son frágiles. El principal inconveniente de
SPDE es que los analitos tienden a permanecer en la pared interna de la aguja después de la
desorción térmica.70
Figura 12. Técnicas de SPME en aguja. Adaptado de:44,53
Otro tipo de SPME en aguja fue introducida en 2001 por Pawliszyn et al71
en el que se emplea
un dispositivo conocido como trampa de aguja (NTD) (Fig. 12b) en el que los sorbentes o fibras
se empaquetan en una aguja. Para la extracción, la muestra se succiona dentro al interior de la
aguja haciéndola pasar por el lecho sorbente. La desorción directa se facilita insertando la aguja
dentro del puerto de inyección GC.72
2.6.3 TFME: Microextracción de película delgada
La microextracción de película delgada (TFME) es una técnica de preparación de muestras
introducida inicialmente en 2001. La principal diferencia entre esta técnica y la microextracción
tradicional en fase sólida SPME es que se utiliza una lámina de película plana en lugar de una
fibra que permite la exposición de un volumen mayor de fase extractiva con un área superficial
mayor.
Figura 13. Representación esquemática de TFME. Adaptado de:73

~ 26 ~
Esta configuración, esquematizada en la figura 13, permite mejorar la sensibilidad sin afectar
significativamente el tiempo de extracción.73,74
En 2019 de la Calle et al.75
informaron sobre el desarrollo de un procedimiento de
microextracción con inmersión directa de película delgada para la preconcentración simultánea
de diferentes iones metálicos presentes en muestras de agua utilizando membranas de grafeno
como nuevas plataformas de absorción. El método se basa en la sorción de iones metálicos
acomplejados con pirrolidina ditiocarbamato de amonio (APDC) antes de la determinación por
la técnica de fluorescencia de rayos X por reflexión total (TXRF). Los parámetros analíticos
respectivos de este método se resumen en la tabla 5.
Tabla 5. Método reportado de TFME por de la Calle et al. para la preconcentración y
determinación de Pb en agua. (LOD: límite de detección; LOQ: Límite de cuantificación;
RR%: Recuperación relativa; RSD%: Desviación estándar relativa)
2.6.4 MEPS: Microextracción por sorbente empaquetado
En 2003, Abdel-Rehim76
introdujo una nueva técnica de preparación de muestras, conocida
como microextracción por sorbente empaquetado (MEPS). En esta técnica tanto el volumen de
muestra como el eluyente se reducen de 10 a 100 veces, es decir, de mL a μL. MEPS es
adecuado tanto para pequeños volúmenes de muestra (muestras biológicas) como también para
volúmenes de muestra más grandes (muestras ambientales). En esta técnica, una microcolumna
se llena con 1–4 mg de material adsorbente y se conecta a la aguja de una microjeringa (Fig.
14). Posteriormente, La muestra se succiona y expulsa a través de la microjeringa en repetidas
ocasiones hasta alcanzar un equilibrio. Finalmente, los analitos extraídos son eluídos con un
pequeño volumen de disolvente (10-50 µL) directamente en el inyector de GC o LC.77,78
Figura 14. Esquema general del procedimiento de microextracción por sorbente
empaquetado (MEPS). Adaptado de: 47

~ 27 ~
2.6.5 SBSE: Microextracción por sorción sobre barra agitadora
La extracción sorptiva con barra de agitación (SBSE), desarrollada en 1999 por Baltussen et al. 79
, es una técnica de preparación de muestras en la que un tubo cilíndrico de vidrio se recubre
con una capa de polidimetilsiloxano (PDMS) y adicionalmente, al interior del soporte de vidrio
se encuentra un componente magnético que permite la agitación durante el procedimiento de
extracción (Fig. 15). Después de la etapa de extracción, generalmente se emplea una unidad de
desorción térmica (TD) para llevar los analitos extraídos al cromatógrafo de gases.36
En
comparación con la microextracción en fase sólida tradicional, SBSE posee una mejor
recuperación ya que tiene mayor área superficial.
Figura 15. Esquema general del procedimiento de microextracción por sorción sobre barra
agitadora. Adaptado de:47
En 2010, se desarrolló una técnica alternativa a SBSE conocida como microextracción
adsorbente (AμE ) ya que se observó que el contacto de la barra de agitación recubierta con el
fondo del recipiente donde se contiene la muestra puede dañar la superficie del polímero y
reducir significativamente la eficiencia de la extracción. El primer enfoque de esta modificación
se conoce como microextracción con adsorción en barra (BAμE). En este caso la extracción se
realiza con barras cilíndricas de polipropileno. Esta barra de polipropileno se introduce en el
recipiente que contiene la muestra acuosa, lo que permite una configuración flotante debido a la
baja densidad del polímero. Esto mejora la vida útil de la fase extractante al evitar el contacto
directo con las paredes del recipiente. Al mismo tiempo, se inserta una barra de agitación
magnética en la muestra que permite altas velocidades de rotación, contribuyendo a una
transferencia efectiva de masa de analito desde la muestra acuosa a la fase sorbente. Después de
la extracción, la barra de polipropileno recubierta se inserta en un vial que contiene unos pocos
microlitros de un disolvente orgánico para la desorción de los analitos. Finalmente, el disolvente
orgánico que contiene los analitos se transfiere a un instrumento analítico. 36 Posteriormente, como se observa en la figura 16, se propuso otra configuración de
microextracción adsorbente AμE en la cual se emplean multi esferas como medio extractante
(MSAμE).53
Figura 16. Configuraciones de microextracción adsorbente AμE: (A) BAμE; (B) MSAμE.
Adaptado de:53

~ 28 ~
Este tipo de técnicas se emplea principalmente para la determinación de especies orgánicas, sin
embargo, en 2019, Hashemi et al.80
emplearon una barra de agitación recubierta con
nanopartículas de plata para la extracción selectiva de metales pesados (Fe+3
, Cu+2
, Pb+2
y Zn+2
)
de muestras de agua potable, seguido de su análisis por HPLC. Los parámetros analíticos
respectivos de este método se resumen en la tabla 6.
Tabla 6. Método reportado de SBSE por Hashemi et al.80
para la preconcentración y
determinación de Cu y Pb en agua. (LOD: límite de detección; RR%: Recuperación
relativa; RSD%: Desviación estándar relativa)
2.6.6 SCSE: Extracción adsorbente con disco agitador
Esta técnica es una versión mejorada de SBSE que se caracteriza por bajos límites de detección,
alto factor de recuperación, alta selectividad y amplio rango lineal. La fase extractante que es un
material microporoso de gran superficie conocido como disco monolítico se dispone sobre un
soporte que es su parte inferior posee una barra de hierro recubierta con una capa de protección
de vidrio (Fig. 17). Este soporte actúa como agitador magnético para aumentar la velocidad de
extracción, sin embargo, a diferencia de SBSE el medio de extracción posee una mayor vida
media puesto que no hay interacción entre el sorbente y el fondo del recipiente de la muestra.53
Figura 17. Esquema del disco monolítico empleado en SCSE. Adaptado de:53
2.6.7 RDSE: Extracción adsorbente con disco giratorio
En 2009, Richter y su grupo de investigación propusieron RDSE como otra herramienta
analítica útil para la extracción y la concentración previa de analitos de baja polaridad.81
El
dispositivo de extracción consiste en un disco de teflón sobre el cual se deposita la fase
estacionaria. En la parte inferior del disco, como en SCSE, posee un agitador magnético (Fig.

~ 29 ~
18). Después de la extracción, los discos se secan y los analitos se desorben con una pequeña
cantidad de solvente orgánico y posteriormente son determinados por cromatografía o
espectrofotometría. Una ventaja indudable de este tipo de disco es que puede usarse varias
veces.
Figura 18. Representación esquemática de las posibles configuraciones de RDSE. Adaptado de: 82
2.6.8 DµSPE (DMSPE): Extracción en microfase sólida dispersiva
La extracción en microfase sólida dispersiva (DµSPE) también es un método SPME sin fibra
que se desarrolló en 200983
con el objetivo de eliminar las limitaciones inherentes a SPME
tradicional. En este procedimiento, el sorbente no se deposita sobre un soporte, sino que se
vierte directamente en la solución de muestra para extraer el analito. El sorbente se dispersa de
forma tal que se proporciona un área superficial mayor al tener mayor contacto el sorbente con
la muestra lo cual conduce a un menor tiempo de extracción y a una mejor capacidad de sorción.
Después de la extracción, el sorbente que contiene los analitos se aísla por centrifugación o
filtración y los analitos contenidos en el sorbente sólido se eluyen con una pequeña cantidad de
solvente orgánico, se desorben térmicamente o se determinan directamente mediante una técnica
espectroscópica adecuada (Fig. 19).37,84,85
Por lo general este método ha sido llamado microextracción en fase sólida dispersiva
(DSPME), sin embargo, este nombre causa ambigüedad debido a que el procedimiento
convencional de SPME se lleva a cabo utilizando una fibra y este término puede indicar la
dispersión de la fibra dentro de la solución de muestra. 37
Figura 19. Representación esquemática del procedimiento de DµSPE. Adaptado de:85

~ 30 ~
La naturaleza y las propiedades del sorbente sólido son de primordial importancia en DµSPE.
En la práctica, los requisitos principales para un sorbente sólido son:84
(a) Rápida elución y sorción
(b) Gran área superficial y alta capacidad de sorción
(c) Alta dispersión de la fase extractante en muestras líquidas.
En este contexto, los sorbentes a base de carbono han despertado una considerable atracción en
la determinación y análisis de iones metálicos ya que estos presentan características como la alta
capacidad de sorción, la facilidad de modificación de la superficie y sus excelentes propiedades
eléctricas, químicas y térmicas.54
De igual forma, la incorporación de nanomateriales como
fases de extracción son de gran interés. Este hecho se apoya en sus excelentes propiedades como
sorbentes, que se derivan de su tamaño reducido y su alta área superficial, lo que maximiza la
interacción con los analitos. Hoy en día, varios nanomateriales comerciales o sintéticos que
incluyen sílice funcionalizada, nanotubos de carbono, grafeno, óxido de grafeno y
nanopartículas magnéticas modificadas se han aplicado como sorbentes en DµSPE.84,86
2.6.8.1 Termodinámica de la extracción en fase sólida microdispersiva
Para comprender los factores efectivos en DµSPE y su optimización es posible revisar los
parámetros termodinámicos al igual que como se hizo para SPME. Para esto, comencemos
describiendo el proceso esquematizado en la figura 20.
Figura 20. Proceso de extracción por DµSPE: Vm (volumen de la muestra); C0
(concentración inicial del analito en la muestra); Ci (concentración de equilibrio del analito
en la muestra ); Vfi (volumen de cada partícula de sorbente disperso); Cfi (concentración de
equilibrio del analito en cada partícula de sorbente disperso). Adaptado de:37
Cuando solo se consideran dos fases las condiciones de equilibrio se pueden describir mediante
la ecuación (11) de acuerdo con la ley de conservación de masas.
𝑛0 = 𝑛𝑖 + 𝑛𝑓 (11)
Donde no, ni y nf son la cantidad molar del analito en la solución de muestra antes y después del
equilibrio, y la cantidad molar del analito en el sorbente después del equilibrio, respectivamente.
Si se expresa la cantidad molar en términos de concentraciones y volúmenes es posible
reescribir la ecuación (11):
𝐶0𝑉𝑚 = 𝐶𝑖𝑉𝑚 + ∑ 𝐶𝑓𝑖𝑉𝑓𝑖∞𝑖=1 (12)

~ 31 ~
Donde Vm es el volumen de la muestra, C0 y Ci es la concentración inicial y de equilibrio del
analito en la muestra, Vfi es el volumen de cada partícula de sorbente disperso y Cfi es la
concentración de equilibrio del analito en cada partícula de sorbente disperso.
Suponiendo que el tamaño de las partículas sorbentes y las condiciones de adsorción del analito
son las mismas en todas las partículas, se puede escribir:
𝐶𝑓1 = 𝐶𝑓2 = 𝐶𝑓3 = ⋯ = 𝐶𝑓𝑚
𝑉𝑓1 = 𝑉𝑓2 = 𝑉𝑓3 = ⋯ = 𝑉𝑓𝑚
∑ 𝐶𝑓𝑖𝑉𝑓𝑖𝑚𝑖=1 = 𝑚𝐶𝑓𝑚𝑉𝑓𝑚 (13)
Donde 𝑚 es el número de partículas de sorbente dispersas. Las ecuaciones (12) y (13) se pueden
combinar y reorganizar en:
𝐶0𝑉𝑚 = 𝐶𝑖𝑉𝑚 + 𝑚𝐶𝑓𝑚𝑉𝑓𝑚 (14)
El coeficiente de distribución (Kfm) del analito entre las partículas de sorbente dispersas y la
solución de muestra se define como:
𝐾𝑓𝑚 = 𝐶𝑓𝑚
𝐶𝑖 (15)
La ecuación (18) se obtiene dividiendo la ecuación (14) en Cfm y combinando y reorganizando
la ecuación resultante (16) con la Ecuación (15):
𝐶0𝑉𝑚
𝐶𝑓𝑚=
𝐶𝑖𝑉𝑚
𝐶𝑓𝑚+ 𝑚
𝐶𝑓𝑚𝑉𝑓𝑚
𝐶𝑓𝑚 (16)
𝐶0𝑉𝑚
𝐶𝑓𝑚=
𝑉𝑚
𝐾𝑓𝑚+ 𝑚𝑉𝑓𝑚 (17)
𝐶0𝑉𝑚
𝐶𝑓𝑚=
𝑉𝑚+𝑚𝑉𝑓𝑚𝐾𝑓𝑚
𝐾𝑓𝑚
𝐶𝑓𝑚 = 𝐶0 (𝑉𝑓𝑚𝐾𝑓𝑚
𝑉𝑚+𝑚𝑉𝑓𝑚𝐾𝑓𝑚) (18)
La cantidad molar de analito extraído en cada dispersión de partículas sorbentes se puede
determinar usando la ecuación (19).
𝐶𝑓𝑚𝑉𝑓𝑚 = 𝐶0 (𝑉𝑚𝑉𝑓𝑚𝐾𝑓𝑚
𝑉𝑚 + 𝑚𝑉𝑓𝑚𝐾𝑓𝑚)
𝑛𝑓𝑚 = 𝐶0 (𝑉𝑚𝑉𝑓𝑚𝐾𝑓𝑚
𝑉𝑚+𝑚𝑉𝑓𝑚𝐾𝑓𝑚) (19)
La cantidad molar total del analito extraído (nf) se puede obtener multiplicando el número de
partículas de sorbente dispersas (𝑚) por la cantidad molar del analito extraído (nfm) en cada
partícula de sorbente dispersa (Ecuación (20)). Además, el volumen del sorbente (vf) es igual a
multiplicar el número de partículas de sorbente dispersas (𝑚) por el volumen de cada partícula
de sorbente disperso (Vfm) (Ecuación (21)).
𝑛𝑓 = 𝑚𝑛𝑓𝑚 (20)
𝑣𝑓 = 𝑚𝑣𝑓𝑚 (21)

~ 32 ~
Reemplazando en (19)
𝑛𝑓
𝑚= 𝐶0 (
𝑉𝑚𝑉𝑓𝑚𝐾𝑓𝑚
𝑉𝑚+𝑉𝑓𝐾𝑓𝑚)
𝑛𝑓 = 𝑚𝐶0 (𝑉𝑚𝑉𝑓𝑚𝐾𝑓𝑚
𝑉𝑚+𝑉𝑓𝐾𝑓𝑚) (22)
𝑛 = 𝐶0𝐾𝑓𝑚𝑉𝑚𝑉𝑓
𝐾𝑓𝑚𝑉𝑓+𝑉𝑚 (5)
Si se realiza una comparación con el número de moles de analito extraídos por la técnica de
SPME tradicional (Ec. 5). Se observa que la cantidad molar de analito extraído aumenta con el
número de partículas dispersadas en la solución de muestra. Por lo tanto, el uso del método
DµSPE puede aumentar la eficiencia de extracción del analito en comparación con el método
tradicional SPME.37
2.6.8.2 Ventajas y desventajas de DµSPE
La siguiente tabla (Tabla 7) resume las ventajas y desventajas de la técnica DµSPE desde un
punto de vista operativo.
Tabla 7. Ventajas y desventajas de la técnica DµSPE.
2.6.8.3 Enfoques de DµSPE
El método DµSPE se puede clasificar en tres categorías dependiendo de la forma de dispersión
del sorbente en la solución de muestra:
i. DµSPE tradicional
ii. DµSPE asistido por vórtex (VA- DµSPE)
iii. DµSPE asistido por ultrasonido (UA- DµSPE)
2.6.8.3.1 DµSPE tradicional
En DµSPE tradicional, se vierte un sorbente adecuado en la solución de muestra y se agita
durante un tiempo específico hasta que el analito se extrae en el sorbente. Posteriormente, con el
fin de separar el sorbente de la solución de la muestra se centrifuga y la solución sobrenadante

~ 33 ~
se desecha completamente con una jeringa. Para separar el analito es posible adicionar un
pequeño volumen de solvente orgánico que luego es llevado al sistema de detección para la
determinación del analito.
Como se mencionó anteriormente, una de las limitaciones más críticas del método que afecta
drásticamente la eficiencia de extracción y la reproducibilidad de este es la separación del
sorbente de la solución de la muestra luego de la extracción. Con el fin de superar esta dificultad
se desarrolló un nuevo método, la extracción en microfase sólida magnética dispersiva
(MDµSPE). En este procedimiento, se emplea un sorbente con propiedades magnéticas (por ej.,
magnetita Fe3O4) que se aísla fácilmente de la solución de muestra empleando un campo
magnético externo (Fig. 21).37
Figura 21. Representación esquemática de MDµSPE. Adaptado de: 53
El procedimiento de extracción puede realizarse mediante nanopartículas magnéticas
funcionalizadas o sus compuestos con otros materiales como nanotubos de carbono, grafeno,
óxido de grafeno, entre otros.54,87
Los nanomateriales magnéticos son de gran utilidad ya que
presentan varias ventajas como:
Fácil síntesis
Alta capacidad de adsorción debido a su gran área superficial
Reducción del tiempo de extracción al eliminar la etapa de centrifugación
Es bien sabido que la automatización del tratamiento de muestras reduce los errores asociados
con el manejo de los operadores y permite el análisis de un mayor número de muestras por
unidad de tiempo. Gracias a las ventajas que proveen los materiales magnéticos en la extracción
en microfase sólida dispersiva ha sido posible implementar la automatización del método. Por
ejemplo, en 2018 Vakh et al. desarrollaron un procedimiento de extracción en microfase sólida
dispersiva magnética automatizada en un reactor fluídico (Fig. 22) para la determinación de
fármacos antimicrobianos de fluoroquinolona en muestras de alimentos para bebés.88

~ 34 ~
Figura 22. Automatización de MDµSPE. Tomado de: 88
2.6.8.3.2 DµSPE asistido por vórtex (VA- DµSPE)
Un factor esencial en la eficiencia de extracción del analito con DµSPE es la dispersión
uniforme y completa de las partículas sorbentes en la solución de muestra. Al aumentar el
número de partículas sorbentes y disminuir su tamaño en la solución de muestra, la superficie de
contacto entre el sorbente y el analito aumenta disminuyendo así el tiempo de extracción. En
este sentido, se ha evidenciado que al emplear vórtex (Fig. 23) con el fin de mejorar la
dispersión de las nanopartículas, tanto el tiempo de extracción como la cantidad de sorbente
requerido se reducen significativamente.37
La aplicación de esta energía externa acelera la
extracción no solo por la dispersión del sorbente sino que también favorece la difusión de los
analitos. Sin embargo, es importante mencionar que la aplicación de una fuente de energía que
no se controla, induce a un aumento en la temperatura del sistema lo cual puede afectar
negativamente a la termodinámica de la extracción e incluso llegar a degradar los analitos.89
Figura 23. Representación esquemática de VA-DµSPE. Adaptado de:90
2.6.8.3.3 DµSPE asistido por ultrasonido (UA- DµSPE)
Al igual que el vórtex, el baño de ultrasonido es una herramienta útil para dispersar
nanopartículas y para eliminar la aglomeración en suspensiones acuosas. Los efectos mecánicos

~ 35 ~
resultantes de la cavitación conducen a un aumento de las colisiones entre las partículas y
eliminan las partículas aglomeradas, lo cual es especialmente útil para la dispersión de
nanopartículas en suspensiones acuosas.37
Si bien se reduce drásticamente el tiempo de extracción y la cantidad de sorbente requerido, se
pueden encontrar dos limitaciones inherentes al uso de ondas de ultrasonido en la extracción en
microfase sólida dispersiva:91
Degradación de analitos o sorbentes y formación de radicales libres durante la
cavitación debido a las altas temperaturas producidas.
Dificultad en la separación del analito cuando se da la formación de suspensiones
termodinámicamente estables con el sorbente luego de la extracción.
2.6.8.4 Aplicaciones de DµSPE en la determinación de metales pesados en agua
De acuerdo con la revisión bibliográfica, dentro de las técnicas de microextracción en fase
sólida, la extracción en microfase sólida dispersiva es probablemente la más empleada en la
preconcentración y determinación de iones metálicos. En la tabla 8 se puede encontrar un
resumen de algunos estudios que se han desarrollado en los últimos años empleando DµSPE en
el análisis de Cu(II), Pb(II) y Cd(II) en muestras de agua.
En 2014, Kocot et al.92
emplearon DµSPE con nanohojas de grafeno para la preconcentración
simultánea de iones de cobalto, níquel, cobre y plomo en muestras de agua. Inicialmente los
metales se acomplejaron con pirrolidinditiocarbamato de amonio (APDC) y luego de la
extracción se realizó una filtración del sorbente para finalmente detectar los analitos por medio
de espectrometría de fluorescencia de rayos X dispersiva en energía (EDXRF). En este caso la
presencia de un agente quelante es esencial, puesto que los iones metálicos en forma de
complejos hidrofílicos no se adsorben en la superficie del grafeno. El empleo de un agente
quelante permite la formación de complejos hidrofóbicos que pueden ser adsorbidos en la
superficie del nanomaterial a través de fuerzas de Van der Waals y de interacciones
hidrofóbicas.
Figura 24. Configuración esquemática de la extracción en microfase sólida dispersiva asistida
por jeringa acoplada con espectrometría de absorción atómica de llama. Tomado de: 92

~ 36 ~
Un año más tarde, Barfi et al.86
desarrollaron un nuevo método al que nombraron extracción en
microfase sólida dispersiva asistida por jeringa (SA-DµSPE) para la determinación de Pb2 +
, Cd2
+, Co
2 +, Ni
2 + y Cr
3+ empleando como adsorbente nanotubos de carbono de pared múltiple
(MWCNT) con detección por espectrometría de absorción atómica de llama. Como se observa
en la figura 24, este método se basa en la succión y expulsión repetida de la muestra acuosa que
incluye los iones metálicos acomplejados y un bajo nivel del adsorbente.
En 2016, Khan et al.93
desarrollaron un método de extracción en microfase sólida magnética
dispersiva asistida por vórtex (VA- MDµSPE) para la determinación simultánea y separación y
preconcentración de plomo(II), cadmio(II), cobre (II), níquel (II) y cobalto (II) de muestras
ambientales y de alimentos empleando como sorbente óxido de grafeno magnético modificado
con poli(vinilacetato-divinilbenceno)(MGO-DVB-VA). De igual forma Behbahani94
empleó
una energía externa, en este caso ultrasonido, con el fin de mejorar la dispersión de
nanopartículas magnéticas recubiertas de sílice y politiofeno para la extracción rápida y la
preconcentración de iones de cadmio que luego se detectaron utilizando espectrometría de
absorción atómica electrotérmica (ETAAS).
Posteriormente, Baghban et al.87
llevaron a cabo la separación y preconcentración de Pb (II) y
Cu (II) por medio de una extracción en microfase sólida magnética con un nuevo adsorbente
magnético de nanopartículas de disulfuro de molibdeno magnético (mag-MoS2-Fe3O4) mediante
detección por FAAS. Lo interesante de este adsorbente es que similar al grafeno, las capas de
MoS2 se mantienen unidas por débiles interacciones de van der Waals lo cual proporciona
suficientes espacios entre las capas de este material que pueden alojar analitos.
En 2018, Ruíz y su grupo de investigación95
evaluaron la extracción en microfase sólida
dispersiva (DµSPE) con detección por espectroscopía de ruptura inducida por láser (LIBS) para
la preconcentración y determinación simultánea de Zn, Cd, Mn, Ni, Cr y Pb en muestras
acuosas. En este estudio se analizaron dos materiales adsorbentes en la etapa de
microextracción, óxido de grafeno y carbón activado.
Feist et al.96
también emplearon óxido de grafeno como adsorbente en un procedimiento de
extracción en microfase sólida dispersiva para la preconcentración de trazas de Pb (II), Cd (II),
Zn (II), Cr (III), Mn (II) y Fe (III). En este caso, debido a las propiedades no selectivas del
óxido de grafeno se empleó batofenantrolina, un derivado de la 1,10-fenantrolina, para
acomplejar los metales previamente a la extracción.
Asimismo, en 2019, López-García et al.97
emplearon un método de DµSPE usando
nanopartículas de óxido de grafeno y plata (Ag@rGO) para la determinación de plomo y
cadmio en agua. Debido al bajo tamaño de las nanopartículas obtenidas, no fue fácil la
separación después del proceso de extracción, incluso después de una etapa de centrifugación
prolongada. Por lo tanto, la separación de la fase sólida se llevó a cabo mediante un proceso de
extracción en punto de nube (CPE) utilizando Triton X-114.
Más recientemente, Montoro-Leal y su grupo98
emplearon nanoesferas magnéticas de óxido de
grafeno (Fe3O4@GO) para la preconcentración y detección simultánea de Cd y Pb en muestras
de agua. Nabavi et al.99
también emplearon nanopartículas magnéticas (en este caso CoO-
Fe2O3@SiO2@TiO2) para la extracción en microfase sólida asistida por ultrasonido con el
objetivo de determinar simultáneamente Cu, Ni, Pb, Co y Cd.

~ 37 ~

~ 38 ~
Tabla 8. Métodos reportados de DµSPE para la preconcentración y determinación de Cd,
Pb y Cu en agua. (LOD: límite de detección; LOQ: Límite de cuantificación; RR%:
Recuperación relativa; RSD%: Desviación estándar relativa)

~ 39 ~
La microextracción en fase líquida (LPME) ha despertado el mayor interés en la química
analítica desde que fue desarrollada por Jeannot y Cantwell100
y Liu y Dasgupta101
en la década
de 1990. Normalmente, LPME emplea un pequeño volumen de solvente (dentro del rango de
microlitros) como fase extractante para concentrar analitos que se encuentran en muestras
acuosas.38,102
En general como se muestra en la figura 25, las técnicas de microextracción en
fase líquida se pueden clasificar en tres categorías principales: microextracción en gota simple
(SDME), microextracción líquido-líquido dispersiva (DLLME) y microextracción en fase
líquida con fibra hueca (HF-LPME). Todos estos modos de LPME se caracterizan por ser
simples, rápidos y con un bajo consumo de disolventes orgánicos.35,77
Figura 25. Algunas configuraciones de LPME. *Nota: Ver acrónimos de la página 6.
3.1 Técnicas de microextracción en fase líquida
3.1.1 SDME: Microextracción en gota simple
En 1996 se introdujo por primera vez esta técnica de microextracción en fase líquida la cual es
una técnica simple, de bajo costo y rápida basada en la reducción del volumen de la fase
extractante en comparación con el volumen de la muestra.40
De forma general, el principio de
esta técnica es la distribución de los analitos entre una gota de disolvente orgánico y la matriz
acuosa.36
SDME puede llevarse a cabo en dos modos diferentes: microextracción en gota simple
directa (DI-SDME) y microextracción en gota simple en espacio de cabeza (HS-SDME), en la
(Fig. 26) puede observarse una representación esquemática de estos. En el modo de inmersión

~ 40 ~
directa, la gota de la fase extractora inmiscible con el agua que se encuentra suspendida en la
punta de una microjeringa es sumergida en la muestra acuosa. En este caso, aumentar la
velocidad de agitación de la fase donora o acuosa aumenta la velocidad de extracción, sin
embargo, esto puede hacer caer la gota del soporte.103
Ahora bien, en cuanto al modo de espacio
de cabeza en este no se sumerge la gota de solvente extractante sino que se suspende en el
espacio superior sobre la muestra.38
En comparación con el modo de inmersión directa (DI), el
modo de espacio de cabeza (HS) evita posibles interferencias de la matriz de la muestra, este
método ha sido ampliamente utilizado para la extracción de compuestos volátiles.104
En ambos
casos, una vez finaliza la extracción, la gota es succionada con la microjeringa y finalmente se
inyecta en el detector para obtener la señal analítica correspondiente.77
Figura 26. Diagrama esquemático de la microextracción en gota simple SDME: (a) DI-
SDME, (b) HS-SDME. Adaptado de: 105
Como ya se había mencionado, el bajo costo, la simplicidad de operación y el uso de equipos
comunes son ventajas de SDME. Sin embargo, también tiene algunas desventajas, la principal
se presenta especialmente en el modo de inmersión directa (DI-SDME) y es la inestabilidad de
la microgota suspendida durante el proceso de extracción. Con el fin de superar este
inconveniente, en 2006 se propuso una técnica alternativa a SDME conocida como
microextracción con gota directa suspendida (DSDME).106
3.1.1.1 DSDME: Microextracción con gota directa suspendida
En este método, se suspende una microgota de disolvente libre (con menor densidad que el
agua) sobre la superficie de la muestra acuosa (Fig. 27) mientras esta es agitada con una barra
de agitación. En condiciones adecuadas, la microgota se puede suspender en la posición central
superior de la solución de muestra y no se romperá incluso en ausencia de un soporte.107
Figura 27. Esquema de la microextracción con gota directa suspendida DSDME. Adaptado de:40

~ 41 ~
3.1.1.2 LLLME: Microextracción líquido-liquido-líquido
La microextracción líquido-líquido-líquido (LLLME) también se puede incluir dentro de los
modos de SDME. Estas configuraciones diferentes se pueden clasificar en términos generales
como técnicas de dos o tres fases, de acuerdo con el número de fases de la extracción.103
La modalidad LLLME (Fig. 28), es un proceso de microextracción con tres fases. Inicialmente
los analitos se extraen de una fase acuosa a una fase orgánica. En este primer paso se debe
emplear un solvente orgánico inmiscible en agua y de menor densidad (por ej. tolueno, n-
octano, 1-octanol, n-hexano, entre otros). Posteriormente, se realiza una nueva extracción en
una gota de fase acuosa. Para lograr un alto nivel de enriquecimiento de analito, se seleccionan
las fases aceptoras y donoras con un pH adecuado, mientras que también se consideran las
propiedades básicas y ácidas de los analitos extraídos. Esta técnica presenta una gran
selectividad y es de gran utilidad para la preconcentración de analitos ionizables.108
Figura 28. Diagrama esquemático de LLLME. Tomado de:109
3.1.2 DLLME: Microextracción líquido-líquido dispersiva
Todas las técnicas que utilizan soporte para la fase extractante tienen el inconveniente de que la
cinética del proceso de extracción es lenta.38
Una forma de acelerar esta etapa es distribuir la
fase del extractante en toda la fase acuosa para mejorar la transferencia de masa del analito de
interés. Teniendo en cuenta esto, se desarrolló en 2006 el método dispersivo de microextracción
líquido-líquido (DLLME).
En esta técnica se introduce rápidamente en la muestra una mezcla de un extractante (solvente
de extracción) con una densidad mayor que el agua y un solvente dispersante con alta
miscibilidad tanto en la fase extractante como acuosa con el fin de promover la formación de
microgotas con un área superficial mayor.36,41
Los analitos en solución acuosa se extraen en un
pequeño volumen de extractante y la posterior separación de fases se logra mediante
centrifugación (Fig. 29).

~ 42 ~
Figura 29. Ilustración esquemática de DLLME. Adaptado de: 105
Esta técnica de microextracción ha sido ampliamente utilizada para la preconcentración de iones
metálicos y compuestos orgánicos en muestras ambientales y biológicas ya que posee ventajas
como operación simple, velocidad y fácil vinculación con la mayoría de los métodos
analíticos.110
Sin embargo, la microextracción líquido-líquido dispersiva presenta algunas
desventajas, la primera de ellas es que la mayoría de los solventes que tienen una densidad más
alta que el agua son peligrosos, como los solventes clorados que incluyen clorobenceno,
cloroformo y tetracloruro de carbono.107
Adicionalmente, otro inconveniente de DLLME es la
dificultad para recuperar la fase orgánica enriquecida lo cual puede resultar en la pérdida de
solvente y en porcentajes de recuperación de los analitos relativamente bajos. Para superar los
inconvenientes anteriores, Zanjani et al.111
introdujeron una nueva técnica de LPME
denominada microextracción de gota orgánica flotante solidificada (SFODME).35
3.1.2.1 SFODME: Microextracción de gota orgánica solidificada
Figura 30. Ilustración esquemática del procedimiento experimental de la microextracción
dispersiva líquido-líquido basada en la solidificación de gota orgánica flotante (DLLME-SFO).
Adaptado de:112

~ 43 ~
Como se ilustra en la figura 30, en este método se mezcla un volumen de disolvente extractante
de baja densidad, baja toxicidad y un punto de fusión cercano a la temperatura ambiente con un
disolvente dispersivo miscible en agua. Al inyectar esta mezcla de disolventes en una muestra
acuosa, se forma una solución turbia y los analitos se enriquecen en el disolvente de extracción
que se dispersa por toda la solución acuosa. Posteriormente, la mezcla se centrifuga y la gota de
la fase orgánica flota sobre la superficie de la fase acuosa. La gota extractora flotante se
solidifica rápidamente en un baño de hielo para que pueda extraerse con facilidad para su previo
análisis.26,113
3.1.3 HF-LPME: Microextracción en fase líquida con fibra hueca
La microextracción líquido-líquido basada en membranas, generalmente conocida como
microextracción de fase líquida basada en fibra hueca (HF-LPME), fue introducida por primera
vez por Pedersen-Bjergaard114
en 1999 también con el fin de abordar los inconvenientes
asociados con la inestabilidad de la gota del método SDME. En HF-LPME (Fig. 31), el
disolvente orgánico se inmoviliza y se mantiene en los poros de una fibra hueca porosa
hidrofóbica por fuerzas capilares. Luego, el sistema se ubica en la solución de muestra y la
transferencia de masa se logra mediante la agitación de la solución acuosa.115
Las principales
ventajas de esta técnica son la alta versatilidad, selectividad, economía y especialmente un alto
factor de preconcentración.116
Figura 31. Representación de un sistema de microextracción en fase líquida con fibra
hueca (HF-LPME).Adaptado de:117
3.2 Fases extractantes en LPME
Con el fin de lograr una buena sensibilidad, precisión y selectividad es imprescindible realizar
una correcta selección del solvente extractante. Dentro de las propiedades que se deben tener en
cuenta al momento de elegir el solvente están: el punto de fusión y ebullición, la presión de
vapor, la solubilidad en agua, la densidad, la viscosidad, la tensión superficial, el momento
dipolar, la constante dieléctrica, la constante de distribución del analito, la compatibilidad con el
sistema de detección y en lo posible, una baja toxicidad.118

~ 44 ~
3.2.1 Solventes orgánicos tradicionales
Inicialmente, los solventes más empleados para los modos de LPME como extractantes eran
éter dietílico, metil tert-butil éter (MTBE), acetato de etilo, benceno, pentano, hexano,
ciclohexano, diclorometano (CH2Cl2), cloroformo (CHCl3), 1,1,1-tricloroetano y tetracloruro de
carbono (CCl4). Algunos de estos, como el éter dietílico y el acetato de etilo, se dejaron de
emplear puesto que son demasiado solubles en agua y demasiado volátiles. Algunos, como el
benceno, ya no se usan debido a su toxicidad y a su carácter carcinogénico. Adicionalmente, se
ha descubierto que otros solventes, como el 1,1,1-tricloroetano, tienen efectos ambientales
perjudiciales para el medio ambiente.
Por otro lado, algunos solventes, que son demasiado solubles en agua, como CH2Cl2 y CHCl3, y
otros, como CCl4, que son claramente tóxicos, continúan utilizándose como solventes de
extracción, especialmente para la microextracción dispersiva líquido-líquido (DLLME ), debido
a que hay un número limitado de solventes de alta densidad disponibles para este modo de
microextracción en fase líquida.119
Algunos de los solventes comúnmente empleados en LPME y algunas de sus propiedades
físicas importantes se enumeran en la tabla 9.

~ 45 ~

~ 46 ~
Tabla 9. Propiedades físicas de algunos extractantes empleados en las técnicas de LPME.
Adaptado de: 119

~ 47 ~
3.2.2 Líquidos Iónicos (IL)
En los últimos años se ha buscado reemplazar los solventes orgánicos con el objetivo de que las
técnicas de microextracción tengan un menor impacto ambiental. Una de las alternativas que
hoy en día se emplea son los líquidos iónicos (Fig. 32), los cuales son disolventes no
moleculares de naturaleza iónica, que tienen bajos puntos de fusión (por debajo de 100 °C),
viscosidad variable, estabilidad térmica y baja presión de vapor a 25 °C.120
Figura 32. Estructuras comunes en los líquidos iónicos. Adaptado de:120
Estos tipos de solventes debido a su carácter iónico poseen temperaturas de descomposición
muy altas (~ 400 °C) y muy baja volatilidad, no son buenos candidatos para su uso con el
análisis instrumental de cromatografía de gases (GC). Adicionalmente, los líquidos iónicos
generalmente tienen tensiones y viscosidades superficiales muy grandes, y densidades mayores
que el agua. La gran tensión superficial y la baja volatilidad hacen que estos solventes sean una
buena elección para usar en los modos de microextracción de gota (SDME). Por otro lado, la
alta viscosidad afecta la transferencia de masa del analito por lo cual se requiere de tiempos de
extracción más largos. Si bien sigue habiendo un gran esfuerzo en la aplicación de IL en LPME,
en los últimos años se han empleado con precaución debido a su alto costo, compleja síntesis,
larga biodegradación debido a su alta estabilidad química que conduce a la bioacumulación y la
toxicidad de algunos de ellos dependiendo de su composición.119,121
3.2.3 Solventes eutécticos profundos (DES)
Los disolventes eutécticos profundos (DES) se han introducido como alternativa a los líquidos
iónicos. Los DES se forman típicamente mezclando una cantidad apropiada de un aceptor de
enlaces de hidrógeno como la sal de cloruro de colina (ChCl, Vitamina B4) con donantes de
enlaces de hidrógeno (por ejemplo, urea, glicerol, azúcares, ácidos carboxílicos, etc.).122
La

~ 48 ~
formación de enlaces de hidrógeno es responsable de la disminución del punto de fusión de la
mezcla en relación con los puntos de fusión de los componentes individuales. En comparación
con los líquidos iónicos, los disolventes eutécticos profundos pueden producirse fácilmente a
partir de reactivos económicos, no tóxicos y biocompatibles. El desarrollo de estos solventes se
ha utilizado para la extracción de diferentes analitos. Los DES disponibles son en su mayoría de
naturaleza hidrofílica que restringen los métodos de microextracción a solo compuestos polares.
Recientemente, se ha buscado sintetizar DES hidrofóbicos para ser empleados como disolventes
alternativos en la extracción de analitos orgánicos e inorgánicos no polares de muestras
acuosas.123
3.2.4 Solventes intercambiables (SHS)
Se han buscado también nuevas opciones puesto que los disolventes eutécticos profundos por lo
general son solubles en agua y en ocasiones se dificulta su separación luego de la extracción.
Recientemente ha aumentado el interés por los solventes intercambiables. Un SHS (que puede
ser una amidina o una amina secundaria o terciaria) es un solvente que puede ser hidrofóbico e
hidrofílico mediante la adición o eliminación de dióxido de carbono del sistema. El cambio en
la miscibilidad se debe a una reacción ácido-base entre el SHS y el dióxido de carbono
generando una especie protonada hidrofílica del SHS y ácido carbónico.124,125
Como se observa
en la figura 33, la amina/ amidina nativa, que es inmiscible en agua, se convierte en una especie
hidrofílica luego de ser tratada con CO2. Una vez se permite la transferencia de masa del analito
de la matriz de la muestra al SHS hidrofílico, la solubilización se revierte induciendo la
separación de fases mediante la adición de una base fuerte como por ejemplo hidróxido de
sodio.126
Figura 33. Representación esquemática del uso de un solvente intercambiable en LPME.
Adaptado de:112

~ 49 ~
3.3 Aplicación de técnicas de LPME para el análisis de metales pesados
en agua
Los métodos de microextracción en fase líquida para la preparación de muestras son rápidos,
selectivos y relativamente económicos. Aunque la gran mayoría de los trabajos de investigación
sobre la microextracción se concentran en la determinación de analitos orgánicos, se ha
reconocido también su potencial para la preconcentración de metales traza.127
Aguirre et al128
. realizaron una revisión de las técnicas de microextracción empleadas para el
análisis de trazas y ultratrazas de metales entre 2003 y 2017. Como se observa en la (Fig. 34a)
dentro de las técnicas de microextracción en fase líquida, DLLME es el procedimiento más
empleado, se pueden encontrar alrededor de 200 publicaciones con este método desde 2007. La
preferencia por este método puede deberse a su rapidez, alto factor de enriquecimiento y a su
fácil acoplamiento con las técnicas de detección. Por otra parte, en la (Fig. 34a) donde se
muestra las diferentes técnicas de detección empleadas con los procedimientos LPME se puede
ver que ETAAS es la técnica más empleada (Fig. 34b). Esta técnica de detección brinda
ventajas como una alta sensibilidad, con niveles de detección por debajo de los µg/L y además
requiere de volúmenes muy bajos de muestra para el análisis.
Figura 34. a) Diagrama que muestra el porcentaje de publicaciones que utilizan diferentes
procedimientos LPME en el análisis elemental de trazas de 2003 a 2017. b) Diagrama que
muestra el porcentaje de publicaciones que utilizan diferentes sistemas de detección de
2003 a 2017. Adaptado de:128
A continuación, en la tabla 10 se resume la aplicación de las diferentes técnicas de LPME en la
determinación de Cu(II), Pb(II) y Cd(II) en muestras de agua. En general, se observa que las
técnicas más empleadas con este fin son DLLME24,129,130
y SDME131–135
y como se mencionó
anteriormente, los métodos de espectrometría de absorción atómica son los que por lo general se
emplean para la detección de los iones metálicos luego de la extracción. Recientemente,
también se encuentran estudios en los que se emplean otros solventes como IL13
, DES10
y
SHS124
.

~ 50 ~
Tabla 10. Algunos métodos reportados de LPME para la preconcentración y determinación de Cd, Pb y
Cu en agua. (LOD: límite de detección; LOQ: Límite de cuantificación; EF: Factor de enriquecimiento;
RR%: Recuperación relativa; RSD%: Desviación estándar relativa)

~ 51 ~
4.1 Evaluación de las potencialidades preconcentrativas de la técnica
de microextracción con gota directa suspendida (DSDME) para la
determinación colorimétrica de Cu2+
, Pb2+
y Cd2+
4.1.1 Reactivos
Se prepararon soluciones a dos concentraciones (500 y 53.5 ppb) de los metales de interés
(Cu2+
, Pb2+
, Cd2+
) a partir de soluciones estándar (1000 mg / L) en agua desionizada (18
MΩ ∙ 𝑐𝑚). Se prepararon también soluciones con etanol de los complejantes, 1-(2-Piridilazo)-2-
naftol (PAN), ditizona y 1,10-fenentrolina a una concentración de 3.934 𝑥 10−3𝑀. Por último,
se prepararon ocho diferentes soluciones amortiguadoras con valores de pH comprendidos entre
3 y 10, partiendo de soluciones 0.1M de acetato de sodio (NaC2H3O2) y fosfato monopotásico
(KH2PO4), regulando el pH con ácido acético (C2H4O2) y amoniaco (NH3) respectivamente.
4.1.2 Instrumentación
La espectrofotometría molecular UV-Vis es un método de detección ampliamente utilizado en
diversos análisis debido a su disponibilidad, flexibilidad, bajo costo y conveniencia. En el
análisis de trazas, sin embargo, no suele emplearse frecuentemente ya que en comparación con
las otras técnicas de detección, como las atómicas, presenta baja sensibilidad. Durante los
últimos años se ha evidenciado un progreso en los procesos de detección, con el fin de mejorar
su sensibilidad, precisión y exactitud y al mismo tiempo para minimizar el costo y el tiempo de
análisis. Estos avances han permitido implementar métodos colorimétricos junto con técnicas de
LPME para la preconcentración y determinación de iones metálicos.129,136
Figura 35. Espectrofotómetro Multiskan Sky Microplate (Thermo Fisher, Waltham MA,
USA) y placa µDropTM
.

~ 52 ~
En este caso se empleó el espectrofotómetro de última generación (Fig. 35) Multiskan Sky
Microplate (Thermo Fisher, Waltham MA, USA). Este tipo de instrumentos resultó de la
necesidad de minimizar la cantidad de muestra analizada y de disminuir el tiempo de análisis
cuando se tiene una gran cantidad de muestras. Inicialmente, para el estudio del agente quelante
y el efecto del pH se empleó la microplaca de 96 pocillos la cual permite medir en un corto
tiempo (unos cuantos segundos) muestras con un volumen entre 200-300 µL. Posteriormente,
para el estudio del tiempo de extracción se empleó la placa µDropTM
que permite analizar
simultáneamente hasta 16 muestras con un volumen inferior a 2 µL.
4.1.3 Evaluación de factores que afectan la eficiencia de la extracción
4.1.3.1 Selección de agente quelante
La eficiencia de las microextracciones en fase líquida depende de la afinidad entre el analito y la
fase extractiva, porque cuanto mayor es la afinidad, mayor es el factor de preconcentración. En
este contexto, es bien sabido que los metales divalentes poseen baja afinidad por el medio
orgánico, lo que dificulta su extracción por DSDME. Por lo tanto, en la mayoría de
metodologías de LPME reportadas es importante una previa formación de complejos metálicos
hidrofóbicos con agentes quelantes.2 Para la preconcentración y determinación de trazas de
cobre, plomo y cadmio por este método de microextracción se encuentra en la literatura el uso
de agentes quelantes como OVAC (N-o-vanillidine-2-amino-p-cresol)137
, 3-Amino-7-
dimetilamino-2-metilfenazina clorhidrato (rojo neutro, NR)138
, 1,5 difenil carbazida (DPC)139
,
1,2-tiazolilazo-2-naftol (TAN)140
, 1-(2-piridilazo)-2-naftol (PAN)141–143
, yoduro de potasio5,
4,4’-dimetil-2,2’ bipiridina144
, 2-(4-metilfenil)-1H-imidazo-[4,5,f] [1,10]-fenantrolina11
, dietil
ditiocarbamato de sodio (DDTC), ditizona145
, entre otros.
En este estudio, se evaluaron tres diferentes complejantes con el propósito de seleccionar el más
adecuado para la determinación colorimétrica de los iones metálicos: Ditizona, PAN y 1,10-
fenantrolina, cuyas estructuras se muestran en la figura 36.
Figura 36. Agentes quelantes estudiados para el acomplejamiento de los iones metálicos. a)
Ditizona (1,5-difeniltiocarbazona) b) PAN (1-(2-piridilazo)-2-naftol) c) 1,10-fenantrolina.
4.1.3.2 Efecto del pH
Inicialmente, se realizó una prueba preliminar con el propósito de determinar el volumen de
buffer necesario para ajustar el pH de las soluciones estándar de los respectivos elementos
metálicos. Para esto se le adicionaron sucesivamente pequeñas fracciones de 50 µL de buffer a
un volumen de 10 mL de la solución de Cu2+
(500 ppb) y tras el seguimiento del pH se
determinó un volumen requerido de 30 µL de buffer / mL de solución M2+
.

~ 53 ~
El pH tiene un papel fundamental en la formación de los complejos metálicos para su posterior
extracción. La separación de iones metálicos mediante un método LPME implica la formación
de un complejo con suficiente hidrofobicidad para extraerse en el pequeño volumen de
disolvente de extracción. Debido a lo anterior, se estudió el efecto del pH en el acomplejamiento
de los iones metálicos en el rango de pH de 3 a 10. Como se observa en la figura 37, se empleó
la microplaca de 96 pocillos para analizar soluciones de 200 µL de los complejos de Cu2+
, Pb2+
y Cd2+
con los tres agentes quelantes estudiados. En principio se empleó una concentración de
2 𝑥 10−3 𝑀 de los acomplejantes, sin embargo, posteriormente como se mostrará más adelante
fue necesario disminuir esta concentración. La formación de estos complejos se estudió en la
región visible del espectro entre 380 – 800 nm.
Figura 37. Estudio colorimétrico de los complejos a diferentes pH. A: Complejante; B:
Acomplejante-Cu2+
; C: Acomplejante-Pb2+
; D: Acomplejante-Cd2+
.
4.1.3.3 Selección del solvente extractante
Otro aspecto importante que se debe considerar es la correcta selección del solvente extractante
puesto que no solo contribuye a una alta eficiencia de extracción sino que también facilita la
recuperación de la fase orgánica enriquecida.107
El extractante utilizado en este método debe
cumplir ciertos requisitos, como una baja solubilidad en agua y baja volatilidad en aras de evitar
un posible cambio de concentración por efectos de cambios en el volumen de este extracto. De
acuerdo con estas consideraciones, se podrían usar varios solventes de extracción que incluyen
1-undecanol, 1-dodecanol, 2-dodecanol, 1-bromohexadecano, n-hexadecano, 1,10-
diclorodecano y 1-clorooctadecano.139
En este estudio, se seleccionó 1-dodecanol como solvente
de extracción, no solo por disponibilidad sino por su alto coeficiente de reparto octanol-agua
(Krep=5.13), una baja presión de vapor (2.9 𝑥 10−3 𝑘𝑃𝑎), una densidad inferior al agua

~ 54 ~
(0.833 𝑔/𝑐𝑚3) y una baja toxicidad.119,146
El volumen del solvente extractante también es un
factor importante a considerar puesto que afecta tanto la eficiencia como el tiempo de
extracción. Por lo general, se emplean volúmenes entre 25-100 µL, en este caso se escogió un
volumen de 50 µL, en vista de su mejor operatividad al momento de recuperar el extracto.
4.1.3.4 Efecto del tiempo de extracción
El tiempo de extracción es uno de los factores más importantes en la mayoría de los
procedimientos de preconcentración. Para aumentar la repetibilidad de la extracción, es
necesario elegir un tiempo durante el cual se alcanza el equilibrio entre la fase acuosa y la
orgánica, sin la necesidad de una exhaustividad en la extracción a razón de que no es un
requerimiento para la cuantificación. El tiempo para alcanzar el equilibrio determina la cantidad
máxima de analito que puede extraerse por medio de la gota de solvente orgánico. El efecto del
tiempo de extracción se estudió dentro de un rango de 5-80 min. Inicialmente se ajustó el pH de
una solución que contenía 24 mL de una solución 53.5 ppb del metal divalente y 195 µL de la
solución de ditizona (3.934 𝑥 10−3𝑀). Posteriormente, se adicionó a esta solución 50 µL del
solvente extractante, 1-dodecanol y se llevó a cabo la extracción a 40 °C con agitación durante
diferentes tiempos (procedimiento completo ilustrado en la figura 38).
Figura 38. Ilustración del procedimiento empleado para el estudio del efecto del tiempo de extracción.

~ 55 ~
4.2 Evaluación de la potencialidad preconcentrativa con fases sólidas
como extractantes de Cu2+
, Pb2+
y Cd2+
con detección analítica no
colorimétrica
A partir de una previa revisión bibliográfica, se discutirá las potencialidades preconcentrativas
de diferentes fases sólidas (carbón activado, magnetita y carbón activado magnético) para su
posible aplicación en D-μSPE con el fin de preconcentrar y determinar metales pesados a nivel
de subtrazas. Como se vio anteriormente, en esta técnica el adsorbente se dispersa en la matriz
de la muestra y como consecuencia del contacto cercano entre las partículas sorbentes y las
partículas del analito se favorece la cinética de la sorción, lo que aumenta la eficiencia de la
microextracción en fase sólida y a su vez disminuye el tiempo de extracción.147
Una vez expuestas las ventajas y desventajas de las fases sólidas mencionadas, se discutirán
varios factores experimentales que se deben evaluar con el fin de maximizar el porcentaje de
recuperación. Dentro de estos parámetros a considerar se encuentran la concentración inicial del
ion metálico, la concentración de adsorbente, el pH y el tiempo de extracción.
4.2.1 Síntesis de un material magnético a base de carbón activado
Para demostrar el poder extractante de estos materiales carbonosos se sintetizó un material
compuesto de carbón activado recubierto de magnetita, para lo cual se tomaron 3.0 gramos de
un carbón activado y se tamizó hasta un tamaño máximo de partícula equivalente a 100 micras.
En primera instancia se tomó 1.0 gramo del carbón tamizado y se mantuvo durante 6 horas en
agitación en un medio 1.0 M de ácido nítrico a 50 °C con el fin de funcionalizar la superficie del
mismo. Se supone que la superficie del carbón queda funcionalizada con grupos carboxilos que
pudiesen retener satisfactoriamente los metales en estudio. Luego de la funcionalización, se
filtró y lavó numerosas veces el sólido hasta obtener un sobrenadante de lavado con un pH
cercano a 5. El recubrimiento del carbón con magnetita se hizo acorde al procedimiento de
Pastor-Belda et al148
con ligeras modificaciones, para encontrar un material carbonoso (fase
sólida extractante) con propiedades magnéticas que faciliten su separación desde la solución
acuosa. Para llevar a cabo la síntesis del material extractante, se tomaron 0.5 g del carbón ya
lavado y seco, y se suspendieron en 250 mL de agua desionizada (18 MΩ ∙ 𝑐𝑚) con una
agitación constante a 50 °C. Posteriormente, se agregaron simultáneamente 0.43 g de
FeCl2∙4H2O y 1.20 g de FeCl3∙6H2O a la suspensión de carbón, de manera que la relación molar
de Fe(II) a Fe(III) en la mezcla corresponda a una relación 1:2. Se sonicó la suspensión por 30
minutos y luego se agregaron 20 mL de amoniaco (NH3) 8 M gota a gota, en constante agitación
y sonicado, a una temperatura de 50 °C. Una vez se agregaron los 20 mL del amoniaco, el pH
de la mezcla era aproximadamente 10. Se mantuvo la mezcla en las mismas condiciones por
otros 30 minutos adicionales para favorecer el completo crecimiento de los cristales de
magnetita sobre el carbón activado. Por último, se lavó el material a temperatura ambiente con
abundante agua desionizada hasta obtener un sobrenadante con un pH aproximado de 9 y se
hizo un último lavado con etanol. Cabe destacar que la naturaleza magnética del carbón
resultante favoreció el proceso de lavado, ya que era fácilmente separable de los sobrenadantes
con la ayuda de un imán de neodimio sostenido en el exterior de las paredes del recipiente
contenedor.

~ 56 ~
Figura 39. Montaje empleado en la síntesis del carbón magnético.
4.2.2 Atomización electrotérmica (ETAAS) como método de detección luego de la
preconcentración con una técnica SPME
Se discutirá también las características de la atomización electrotérmica (ETAAS) para la
detección de metales pesados a nivel de subtrazas y se realizará una comparación con los otros
métodos de detección.
Es importante mencionar que en los análisis realizados con este método de detección la
optimización del programa de tiempo / temperatura propios de la técnica de absorción atómica
con atomización electrotérmica es un paso fundamental para asegurar una medición de los
analitos de interés, mejorada en términos de sensibilidad.
Figura 40. Pasos de un programa de temperatura para el análisis por absorción atómica
electrotérmica. (1) Secado; (2) Calcinación; (3) Atomización; (4) Limpieza y
acondicionamiento. Adaptado de:149
Una determinación por ETAAS comienza por dispensar un volumen conocido de muestra en el
horno (por lo general unos 20 µL). Luego, la muestra se somete a un programa de calentamiento
con varios pasos o etapas aumentando la temperatura por efecto de un cambio de corriente
eléctrica a través del cuerpo del atomizador. Como se puede ver en la figura 40, estas etapas de

~ 57 ~
calentamiento del horno incluyen por lo general: secado, calcinación, atomización y limpieza.
Por último, el horno se enfría para dar paso a una nueva medición. Los parámetros importantes
que deberán controlarse en cada etapa son la temperatura final, el tiempo de rampa o velocidad
con que se alcanzar esa temperatura, y el tiempo de retención, es decir, el tiempo que se
mantiene en calentamiento continuo una vez se alcanza esa temperatura. Además, la velocidad
de flujo de gas inerte (Argón de alta pureza) también debe controlarse en cada paso.149
Un ejemplo típico de programa de calentamiento se puede observar en la tabla 11, donde se
distingue cada una de las eventuales etapas con sus correspondientes temperaturas, rampas y
tiempos de mantenido. Este es el caso de las condiciones instrumentales preestablecidas por
defecto por el software del equipo CONTRAA800 (Analytik Jena, Alemania) para las
determinaciones de cadmio.
Etapa Temperatura / °C Rampa/ °Cs
-1 Mantenido/ s Flujo Argón / Lmin
-1
Secado 80 6 20 2
Secado 90 3 20 2
Secado 110 5 10 2
Calcinación 350 50 20 2
Calcinación 600 300 10 2
Atomización 1200 1400 3 0
Limpieza 2450 500 4 2
Tabla 11. Condiciones preestablecidas para la medición de cadmio. (ASpect CS 2.2.1.0.)

~ 58 ~
5.1 Optimización de parámetros del método DSDME para la
determinación colorimétrica de Cu2+
, Pb2+
y Cd2+
5.1.1 Selección de agente quelante
Se evaluaron tres diferentes complejantes: Ditizona, PAN y 1,10-fenantrolina con el objetivo de
seleccionar el más adecuado para la determinación colorimétrica de los iones metálicos.
La 1,10-fenantrolina es un ligando bidentado que ha desempeñado un papel importante en el
desarrollo de la química de coordinación, tradicionalmente se ha empleado como agente
quelante de iones metálicos. Las características de esta molécula como su hidrofobicidad, su
estructura plana y la presencia de heteroátomos le permiten actuar cooperativamente en la
coordinación de cationes metálicos. En la figura 37 es posible observar que por medio la
coordinación de los metales divalentes con la 1,10 fenantrolina no se obtiene complejos
coloreados. La fenantrolina presenta transiciones 𝜋 → 𝜋∗ permitidas por espín, la energía
requerida para esta transición cae en la región UV (200–400 nm), en el caso de este agente
quelante en específico solo presenta absorciones a 229 y 265 nm.150
En este punto los siguientes
experimentos solo se continuaron con ditizona y PAN.
Figura 41. Transiciones electrónicas. Tomado de: 151
Las transiciones responsables de impartir colores a los complejos metálicos son las transiciones
de electrones de un orbital d a otro puesto que la energía se absorbe en la región visible (Fig.
41).151
El agente quelante 1-(2-piridilazo)-2-naftol (PAN) reacciona con muchos iones
metálicos, produciendo complejos coloreados que generalmente son insolubles en agua y
solubles en reactivos orgánicos. Se caracteriza por su alta sensibilidad, la estabilidad de sus
complejos y el cambio de color característico producido en el acomplejamiento152
El PAN
presenta una absorción máxima λmax. (Etanol) entre 461 y 465 nm. De acuerdo con una
caracterización estructural de un complejo de cobre, se estableció que el ligando tridentado se
une al centro metálico a través de la disociación del protón naftólico y como resultado los
átomos N, N, O actúan como donores de carga formando dos quelatos contiguos de cinco
miembros como se muestra en la figura 42.

~ 59 ~
Figura 42. Formación de complejos metálicos con PAN. Tomado de:153
La 1,5-difeniltiocarbazona (ditizona) se seleccionó como posible agente quelante puesto que
contiene átomos donores de carga que de acuerdo al principio de ácido-base de Pearson le
permiten interactuar con metales blandos (como Cu (II), Pb (II), Cd (II)) obteniendo así
complejos coloreados insolubles en agua.154,155
En el acomplejamiento se ha establecido que el
átomo de hidrazina N-H, adyacente al grupo tiocarbonilo, se desprotona formando el ligando
monoaniónico (HDZ-) que luego se acompleja a través de los átomos de azufre y nitrógeno azo
terminal, como se observa en la figura 43. En el caso de los metales divalentes esta especie
aniónica forma complejos neutros del tipo [(HDZ)2M].156
Figura 43. Formación de complejos metálicos neutros con ditizona.
La ley de Lambert Beer (23) indica la relación lineal entre la absorbancia y la concentración de
una especie absorbente.157
Esta ley se basa en el supuesto de que cada cromóforo o molécula
absorbente es independiente de la otra y no interactúan entre sí, de esta forma cada molécula en
solución tiene la misma probabilidad de absorber energía en forma de fotones al hacerle incidir
un haz de luz. A concentraciones altas no se cumple esta relación lineal debido a interacciones
electrostáticas entre moléculas cercanas.
𝐴 = − log(𝑇) = − log (𝐼
𝐼0) = 𝜀 ∙ 𝑏 ∙ 𝑐 (23)
Inicialmente en la formación de los complejos metálicos se empleó una concentración de
2 𝑥 10−3 𝑀 de los acomplejantes, sin embargo, se determinó que las soluciones estaban muy
concentradas por lo cual se emplearon tres nuevas concentraciones 3.026 𝑥 10−4 𝑀,

~ 60 ~
1.512 𝑥 10−4 𝑀 y 2.985 𝑥 10−5 𝑀. En el caso de las soluciones con PAN, estas
concentraciones seguían siendo inadecuadas para la medición espectrofotométrica por lo cual se
seleccionó la ditizona a una concentración de 2.985 𝑥 10−5 𝑀 como el agente quelante que se
emplearía en la microextracción (Fig 44).
Figura 44 Estudio colorimétrico de los complejos metálicos con PAN y ditizona.
5.1.2 Efecto del pH
El pH tiene un papel importante en la formación de los complejos metálicos para su posterior
extracción. La separación de los iones metálicos implica la formación de un complejo con
suficiente hidrofobicidad para ser extraído en la gota de solvente orgánico. En consecuencia, la
eficiencia de la extracción también depende del pH al que se produce la formación del
complejo. Para encontrar el pH óptimo, se investigó el efecto del este en el rango de 3 a 10
como se observa en la figura anterior. Se seleccionó el pH 6 para los experimentos posteriores,
visualmente se puede observar una notable diferencia colorimétrica en los tres complejos
metálicos a este pH. Por otra parte, en diferentes estudios en los que se emplea ditizona como
complejante de Cu (II), Pb (II) y Cd (II) se reporta un valor de pH cercano a 6 como valor
óptimo para llevar a cabo la extracción cuantitativa de estos complejos metálicos.131,134,158

~ 61 ~
En la figura 45, se presentan los espectros de absorción de la ditizona y de los complejos
metálicos al pH seleccionado. La ditizona presenta dos bandas de absorción a 463 y 602 nm,
esto se debe al cromismo de este agente quelante (Fig. 46). La presencia de estas bandas se
atribuye a la tautomería entre la especie A y B que se da por la transferencia intramolecular de
protones.159
En cuanto a los complejos con Cu2+
, Pb2+
y Cd2+
se observa una longitud de onda de
máxima absorbancia de 480, 472 y 450 nm, respectivamente.
Figura 45. Espectro de absorción de la ditizona y sus complejos con Cu2+
, Pb2+
, Cd2+
.
Ditizona: 0.02985 mmol/L; M2+
: 0.0535 mg/ L; pH = 6
Figura 46. Cromismo en el ligando de ditizona atribuido a la transferencia intramolecular
de protones. Adaptado de: 159

~ 62 ~
5.1.3 Efecto del tiempo de extracción
El efecto del tiempo en la eficiencia de extracción se estudió dentro de un rango entre 5 y 80
minutos, cuyos resultados experimentales se muestran en la figura 47. Como se puede ver, la
absorbancia para los tres complejos aumenta con el aumento del tiempo de extracción, sin
embargo, no se alcanzó el equilibrio en todo el rango de tiempo examinado. Una de las posibles
razones para esto puede haberse atribuido a una insuficiente velocidad de agitación con una
notable reducción del contacto requerido para alcanzar el enriquecimiento esperado de los
analitos por difusión desde la solución acuosa hasta la gota extractante (fase orgánica).
Figura 47. Efecto del tiempo de extracción. Solvente extractante: 1-dodecanol (50 µL);
Solución acuosa del metal: 24 mL (53.5 ppb); Complejante: ditizona 195 µL
(3.934 𝑥 10−3𝑀); Temperatura 40ºC
Si bien, se alcanza la máxima sensibilidad después de que se haya logrado el equilibrio entre la
fase acuosa y orgánica, se debe recordar que las técnicas de microextracción no son exhaustivas.
En este caso no es necesario ni mucho menos práctico que el tiempo de extracción sea
demasiado prolongado, por lo que es necesario optimizar entonces otros parámetros como la
intensidad de la agitación, la temperatura y la relación volumétrica fase acuosa/fase orgánica. El
volumen de fase extractante, por ejemplo, tiene una gran influencia sobre la extracción de los
analitos ya que la cinética de la extracción depende del área interfacial (𝐴) y el volumen del
disolvente orgánico (𝑉0) de acuerdo con la siguiente ecuación:
𝑘 = 𝐴
𝑉𝑜𝛽0̅̅ ̅ (1 + 𝐾
𝑉0
𝑉𝑎𝑐) (24)
Donde 𝑘 es la constante de velocidad observada, 𝑉𝑎𝑐 es el volumen de la muestra y 𝛽0 el
coeficiente de transferencia de masa global con respecto a la fase extractora.
Además, el volumen del solvente orgánico define el factor de enriquecimiento en un volumen
de muestra fijo, de acuerdo con la ecuación:
𝐸𝐹 = 𝐶𝑜
𝐶𝑖𝑎𝑐 =
𝐾
1+𝐾(𝑉𝑜
𝑉𝑎𝑐) (25)
0 10 20 30 40 50 60 70 80 90
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,1
1,2
1,3
1,4
Ab
sorb
an
cia
Tiempo (minutos)
Ditizona-Cd
Ditizona Cu
Ditizona-Pb
Ditizona

~ 63 ~
5.2 Comparación de métodos reportados para la microextracción de
cadmio en agua
En la sección 3.3 se mencionan algunos estudios reportados de técnicas de LPME para el
análisis de cobre, plomo y cadmio en agua. En la tabla 12 se presentan algunos métodos
establecidos para la determinación de cadmio. Se puede observar entonces que por medio de las
técnicas LPME junto con la espectrofotometría UV-Vis que se caracteriza por su simplicidad,
bajo costo y rapidez es posible lograr la detección de trazas y ultratrazas de metales incluso con
mejores límites de detección en comparación con ETAAS. Por otra parte, comparando el tiempo
de extracción es evidente que DLLME es mucho más rápida que SDME ya que al distribuir la
fase del extractante en toda la fase acuosa se mejora significativamente la transferencia de masa
del analito.
Tabla 12. Datos característicos obtenidos mediante el uso de métodos de microextracción
en fase líquida en la determinación de cadmio.
5.3 Evaluación de la potencialidad preconcentrativa con fases sólidas
como extractantes de metales con detección analítica no colorimétrica
La microextracción en fase sólida y la microextracción en fase líquida son muy similares tanto
en la teoría como en el desarrollo de los métodos prácticos, y potencialmente pueden reemplazar
o complementar los métodos tradicionales de preparación de muestras. En muchos sentidos,
estas dos técnicas son complementarias.160
Como vimos en el capítulo 2, SPME es un método importante de preparación de muestras con
muchas ventajas y algunas limitaciones. El método tradicional de SPME se desarrolló con el
objetivo de que se pudiera acoplar a la técnica de cromatografía. En este método como ya se
mencionó, una fibra de sílice fundido (100 – 250 µm) se recubre con un sorbente (7 – 100 µm)
con el fin de extraer analitos de una muestra acuosa. Una de las principales ventajas de SPME
frente a LPME es que en esta se elimina por completo el uso de solventes orgánicos (en los

~ 64 ~
casos en los que la desorción no se lleva a cabo por elución). La técnica también permite extraer
una muestra e inyectar el extracto en el instrumento de detección utilizando un solo dispositivo,
simplificando así el proceso analítico. Adicionalmente, puede llevarse a cabo manualmente
usando un soporte especial tipo jeringa o automatizado completamente para GC o HPLC.
Finalmente, otra ventaja significativa de SPME en fibra es que puede emplearse como un
dispositivo de muestreo de campo, actualmente es bastante empleado en el análisis atmosférico,
de agua, agrícola y forense.43,46,160
Sin embargo, también presenta algunas limitaciones importantes, una de ellas que en ocasiones
no se tiene en consideración es el volumen limitado del sorbente. Se calcula que un cm de un
recubrimiento PDMS de 100 µm de espesor en una fibra de 11 µm tiene un volumen de
aproximadamente 0.628 µL, esto es algo que se debe tener en cuenta al momento de diseñar el
experimento. Una segunda limitación resulta del hecho de que el polímero es un material muy
viscoso. Esto significa que se necesita un período de tiempo prolongado (hasta una hora o más)
para que las concentraciones de analito en la muestra y el polímero lleguen al equilibrio. Este no
es un problema importante si se utiliza un muestreo manual muy cuidadoso o un
automuestreador automático para el procedimiento, ya que el método no depende de una
extracción exhaustiva, y se pueden lograr resultados reproducibles si las condiciones de
extracción siguen siendo las mismas.160
Una de las principales limitaciones que se ha evidenciado es la fragilidad de la fibra y las
diferencias de extracción entre fibras individuales. No obstante, estos problemas hoy en día se
han abordado mediante técnicas de preparación como sol-gel que contribuyen a la resistencia y
estabilidad de la fibra formada o mediante el uso de fibras metálicas. Sin embargo, la vida útil
de la fibra puede variar de un solo uso a aproximadamente 1000. La vida útil limitada de las
fibras debe tenerse en cuenta debido a sus elevados costos, a la fecha una fibra cuesta alrededor
de unos 170 dólares (https://www.sigmaaldrich.com/, mayo de 2020).
En cualquier método de microextracción la fase extractante empleada debe ser altamente
eficiente en la extracción de los analitos teniendo en cuenta la cantidad reducida utilizada. En
este contexto, los procedimientos basados en dispersión han ganado relevancia en los últimos
años a medida que mejoran el área de superficie de contacto entre la muestra y el extractante y,
por lo tanto, la cinética del procedimiento de extracción.
La extracción en microfase sólida dispersiva (DµSPE) ha sido ampliamente utilizada en el
análisis de diversas especies orgánicas e inorgánicas ya que además de reducir el tiempo de
extracción se evitan algunas de las limitaciones inherentes a la fibra. Al igual que en los otros
métodos SPME, la naturaleza y las propiedades del adsorbente son de primordial importancia.
Los adsorbentes a base de carbono se han empleado ampliamente en la determinación y análisis
de iones metálicos. Dentro de estos materiales el carbón activado (CA) ha demostrado ser un
adsorbente eficiente ya que se caracteriza por una gran área superficial, estabilidad
fisicoquímica, porosidad, capacidad de sorción y bajo costo.55,56
Wahid et al.161
reportan el uso
de carbón activado para la eliminación de cobre, plomo y níquel de muestras acuosas mediante
el método de sorción mientras que Dong et al. emplean CA para la extracción de Pb(II) y As(V)
por intercambio iónico.162
Una desventaja que se presenta al usar este adsorbente es la eliminación y recuperación de la
fase enriquecida tras la extracción. Por lo cual se ha optado por el uso de nanopartículas de
óxido de hierro como adsorbentes para el tratamiento del agua debido a que presentan una gran
área superficial, excelentes propiedades magnéticas, gran biocompatibilidad, facilidad de

~ 65 ~
separación mediante campo magnético externo, reutilización y costo relativamente bajo.4 En la
literatura es posible encontrar investigaciones en las que se emplean también otros diferentes
adsorbentes como la nanopartículas de óxido de hierro para la extracción de metales pesados.
Por ejemplo, Pashai Gatabi et al.163
estudian la eficiencia de adsorción y la viabilidad del uso de
nanotubos de carbono (MWCNT) con maghemita para la recuperación de iones de cadmio a
partir de soluciones acuosas; Wannahari et al.164
sintetizaron un material adsorbente a partir de
caña de azúcar y nanopartículas magnéticas con el fin de evaluar su capacidad adsorbente de
Cu(II); Shen et al.165
sintetizaron partículas de magnetita, una mezcla de óxidos de hierro(II) y
hierro(III) (Fe3O4) para el tratamiento de aguas residuales contaminadas con Ni(II), Cu(II),
Cd(II) y Cr(VI); Kakavandi et al.166
depositaron nanopartículas bimetálicas de hierro y plata
sobre carbón activado para la preparación de un nuevo adsorbente (AC-Fe0/ Ag) que
posteriormente se empleó para la adsorción de Cr(VI) y Jain et al.4 sintetizaron un adsorbente de
óxido de hierro / carbón activado (Fe3O4 / CA) para la eliminación de iones Cr (VI), Cu (II) y
Cd (II) de muestras acuosas.
En las técnicas de microextracción en fase sólida, sin embargo, no es común que se emplee
carbón activado como fase extractante. Ruíz et al.95
por ejemplo, reportan un estudio para la
preconcentración y detección de Zn, Cd, Mn, Ni, Cr y Pb en muestras acuosas por medio de
DµSPE mediante detección con espectroscopía de ruptura inducida por láser (LIBS). En este
caso se probaron dos materiales adsorbentes: carbón activado (CA) y óxido de grafeno (OG),
como se observa en la figura 48.
Figura 48. DµSPE para la detección de Zn, Cd, Mn, Ni, Cr y Pb empleando CA y OG
como adsorbentes. Tomado de:95
Luego de examinar la morfología de ambos adsorbentes por medio de microscopía electrónica
de barrido (SEM) y espectroscopía electrónica de transmisión (TEM) y de analizar el área
superficial y el tamaño de poro mediante estudios de adsorción-desorción se determinó que el
área de superficie específica del carbón activado (1707 𝑚2 𝑔⁄ ) era aproximadamente doce
veces mayor que la del óxido de grafeno (144 𝑚2 𝑔⁄ ) lo cual atribuyen al apilamiento de las
láminas de grafeno. A pesar del área superficial específica baja del OG, en el proceso de DµSPE

~ 66 ~
el adsorbente es dispersado en la muestra acuosa con la ayuda de ultrasonido de forma tal que se
evitan los agregados y se aumenta el área superficial del adsorbente.
Adicionalmente, se identificaron los grupos funcionales de la superficie de los adsorbentes
mediante FTIR-ATR.
Figura 49. Espectro FTIR-ATR de CA y OG. Tomado de:95
En el espectro del óxido de grafeno (Fig. 49) se evidencian diferentes grupos funcionales a
diferencia de la superficie del carbón activado. Para poder emplear CA como adsorbente, es
necesario realizar un tratamiento previo de activación o emplear un agente quelante ya que se
encuentra libre de grupos funcionales. En este caso se empleó DDTC como agente quelante en
el procedimiento de DµSPE-CA con el objetivo de mejorar la eficiencia de adsorción para los
analitos en forma de complejos metálicos. El óxido de grafeno, por el contrario, puede
caracterizarse por su naturaleza ambivalente hidrófoba e hidrófila, lo que permite la adsorción
de compuestos orgánicos y de iones metálicos. Debido a la presencia de los grupos funcionales
que contienen oxígeno en la superficie del OG, muchos autores han informado la extracción
exitosa de iones metálicos de soluciones acuosas sin la necesidad de un paso previo para el
acomplejamiento de los metales. En este estudio se estableció que el óxido de grafeno era mejor
adsorbente puesto que se obtuvieron límites de detección por debajo de 100 ppb y 50 ppb con el
uso de CA y OG, respectivamente.
Barfi et al.86
reportan la determinación de Pb2 +
, Cd2 +
, Co2 +
, Ni2 +
y Cr3 +
por DµSPE con
detección por FAAS. En este caso se estudian las propiedades preconcentrativas de óxido de
zinc (ZnO), carbón activado (CA), nanotubos de carbono de pared múltiple (MWCNT) y
nanopartículas de oro (Au). Según los resultados obtenidos se establece que los MWCNT
proporcionan una mejor eficiencia de adsorción para los analitos en comparación con los otros
adsorbentes.
El carbón activado como adsorbente de iones metálicos presenta entonces dos limitaciones, la
primera de ellas es su superficie sin grupos funcionales, sin embargo, es posible modificar
químicamente la superficie de este material mediante tratamientos con agentes oxidantes como

~ 67 ~
HNO3 o H2O2167
o también puede emplearse un agente quelante para generar complejos
hidrofóbicos que puedan ser adsorbidos en la superficie con mayor facilidad. La segunda
limitación es su difícil separación y recuperación tras la extracción, en este caso hay la
posibilidad de facilitar la separación por medio de un imán empleando un carbón magnético que
puede sintetizarse mediante un proceso de coprecipitación con un nanomaterial magnético.
Estos nanomateriales magnéticos pueden ser compuestos por metales puros (Fe, Co y Ni),
óxidos metálicos (Fe3O4 y γ-Fe2O3), ferritas (MFe2O4 (M = Cu, Ni, Mn, Mg, etc.)) y aleaciones
metálicas (FePt, CoPt). Debido a la relativamente buena estabilidad, el momento magnético, la
menor toxicidad y su fácil síntesis, las nanopartículas de óxido de hierro como la magnetita
(Fe3O4) y la maghemita (γ-Fe2O3) son los que se emplean con más frecuencia.168
Tomando esto en consideración, para la microextracción de metales pesados en matrices
acuosas es posible evaluar la capacidad preconcentrativa de un adsorbente híbrido que combine
las características del carbón activado como su gran área superficial (entre 500 a 1800 m2), su
alta estabilidad fisicoquímica, porosidad, y capacidad de sorción junto con las características
magnéticas de las nanopartículas de óxido de hierro como la magnetita permitiendo una
considerable simplificación del proceso de microextracción.
5.4 Condiciones que se deben optimizar en DµSPE para la
preconcentración y determinación de iones metálicos en matrices
acuosas
Al igual que en todos los métodos de extracción, en DµSPE es crucial realizar la optimización
de las condiciones de extracción para lograr la máxima sensibilidad. Los parámetros principales
que se deben considerar son la cantidad de adsorbente, la concentración inicial del analito, el
método y la velocidad de agitación, el tiempo de extracción, la temperatura, y el pH.
5.4.1 Selección del adsorbente
La naturaleza y las propiedades del sorbente sólido son fundamentales en DµSPE. Se debe
seleccionar entonces un material que tenga una alta selectividad para la extracción de los
analitos de interés, una gran área superficial y alta capacidad de sorción y una facilidad de
separación. Adicionalmente, se debe determinar la cantidad óptima de extractante, por lo
general, se espera que un aumento de la cantidad de adsorbente genere un aumento en el
porcentaje de recuperación del analito de interés, sin embargo, se ha encontrado que llega un
punto en el que el porcentaje de recuperación no varía significativamente.169
Si por ejemplo se
emplea como método de detección una técnica como ETAAS con medición directa del sólido,
es imprescindible determinar la cantidad óptima de adsorbente puesto que la cantidad de
muestra introducida en el tubo de grafito es por lo general de 5–50 µL para líquidos y del orden
de µg para sólidos.
5.4.2 Concentración inicial del analito en la matriz acuosa
La concentración inicial (Co) de analito es un parámetro importante que proporciona
información sobre la capacidad de extracción del material utilizando una cantidad fija del
adsorbente. Generalmente, aumentar la concentración inicial de adsorbato conduciría a un
aumento en la cantidad adsorbida, hasta que el adsorbente se sature. En la literatura se informa
que las capacidades de sorción de algunos adsorbentes, como por ejemplo los nanotubos de

~ 68 ~
carbono de paredes múltiples (MWCNT) aumentan con un aumento de la concentración de
adsorbato, sin embargo, un aumento en la concentración inicial conduce a un aumento en el
tiempo necesario para alcanzar el equilibrio 169
5.4.3 Método y velocidad de agitación
La agitación de la muestra favorece el transporte de masa del analito entre la muestra y el
adsorbente. El tiempo requerido para alcanzar el equilibrio puede reducirse utilizando un
método de agitación. Cuanto más efectiva es la agitación, más cortos son los tiempos de
extracción necesarios para lograr el equilibrio o una sensibilidad satisfactoria en las extracciones
no exhaustivas.43
Se pueden usar varios métodos de agitación en SPME, dependiendo del tipo
de aplicación entre estos se encuentra la agitación magnética, la agitación asistida por vórtex y
la agitación asistida por ultrasonido, cada una de estas tiene ventajas y desventajas particulares
como ya se vio anteriormente.
5.4.4 Tiempo de extracción
La selección del tiempo de extracción óptimo es uno de los pasos críticos en el desarrollo del
método SPME. La selección del tiempo de extracción es siempre un compromiso entre la
longitud, la sensibilidad y la repetibilidad del método analítico. La extracción hasta el punto de
equilibrio proporciona la mayor sensibilidad, pero en la mayoría de las aplicaciones SPME, se
utilizan condiciones de preequilibrio porque los tiempos de extracción de equilibrio tienden a
ser bastante altos y, por lo tanto, no son prácticos.43
5.4.5 Temperatura
El aumento de la temperatura de extracción puede reducir significativamente el tiempo de
equilibrio y hacer que todo el procedimiento sea más rápido. Sin embargo, también debe
considerarse que el coeficiente de partición puede variar.
5.4.6 pH
La conversión completa de los analitos en formas neutras mediante el ajuste del pH puede
mejorar significativamente la sensibilidad del método.
5.5 Atomización electrotérmica (ETAAS) como método de detección
luego de la preconcentración con una técnica SPME
Las técnicas más empleadas para la determinación de trazas de iones de metales pesados en
muestras ambientales son la espectrometría de absorción atómica de llama (FAAS), la
espectrometría de absorción atómica electrotérmica (ETAAS), la espectrometría de masas con
plasma acoplado inductivamente (ICP-MS) y la espectroscopía de emisión óptica con plasma
acoplado inductivamente (ICP-OES).
ETAAS proporciona la ventaja de poder medir tanto en líquidos como en sólidos, es decir que
en el caso de emplear una fase sólida para la extracción no se tiene la necesidad de utilizar
solventes para eluir el analito. Este método cuenta además con niveles de detección muy bajos,
por debajo de los µg/L. Hoy en día es posible encontrar dispositivos para el análisis
multielemental altamente eficientes y sensibles como por ejemplo el espectrofotómetro de

~ 69 ~
absorción atómica CONTRAA800 (Analytik Jena, Alemania) utilizado para las mediciones del
cadmio en este trabajo de investigación.
Cuando se emplean técnicas de microextracción para la preconcentración de la muestra en
conjunción con la espectrometría de absorción atómica electrotérmica es posible obtener
mejores límites de detección en comparación con los otros métodos de detección. En la tabla 8
(Sección 2.6.8.4) donde se presentan algunos métodos reportados de DµSPE para la
preconcentración y determinación de Cd, Pb y Cu es posible observar que aquellos en donde se
emplea ETAAS como técnica de detección presentan una mayor sensibilidad.
Para demostrar esto se tomaron 10 mL de una solución de 2 ppb de cadmio y se le agregaron
cerca de 2 miligramos de un carbón magnético sintetizado (sección 4.2.1.), para luego agitar
vigorosamente por unos minutos y medir la señal de cadmio en el sobrenadante posterior a la
extracción. La figura 50b muestra cómo fue posible una retención total del analito sobre la
pequeña cantidad del sólido utilizado como fase extractante de los 10 mL de solución 2 ppb de
cadmio (Fig. 50a).
(a) (b)
(c)
Figura 50. Señal atómica de cadmio medida mediante la técnica de absorción atómica de
fuente continua HR-CS-AAS en modalidad de calentamiento electrotérmico (ETAAS). (a)
antes de la extracción (solución de Cd de 2 ppb) y (b) en el sobrenadante, luego de la
extracción con 2 mg de material carbonoso magnético. (c) Señal atómica en la suspensión
regenerada equivalente a 2 ppb de Cd.

~ 70 ~
Ahora bien, para poner a prueba la capacidad de la técnica de absorción atómica para llevar a
cabo las medidas de los elementos de interés en matrices sólidas, se regeneró la suspensión
resultante de la extracción agitando ligeramente la mezcla. Se inyecto en el horno el mismo
volumen que en el experimento anterior, pero esta vez de la suspensión carbonosa. La figura
50c muestra la señal de cadmio regenerada en señal atómica desde el sólido suspendido. Está
claro que no es una señal fidedigna de la concentración de cadmio inicial en vista de que para
las medidas en suspensión tendrá que ser optimizado una vez más el programa de
calentamiento, pero se demuestra la capacidad del método para ser optimizable en términos de
llevar a cabo las medidas del cadmio de forma directa en el mismo sólido. Sin embargo, ya que
ni las alturas de las señales ni las áreas de pico (medida fidedigna de la masa de elemento
inyectada en el horno de grafito) son similares, 0.28134 y 0.19074 respectivamente, se justifica
la optimización de la sensibilidad del cadmio para la medida directa del sólido extractante.
Por otro lado, este material carbonoso funciona de manera robusta en un amplio intervalo de
valores de pH. A pesar de no haberse hecho un estudio formalizado de la acidez del medio
donde se lleva a cabo la extracción, se puede observar una capacidad enorme para retener el
cadmio de una manera exhaustiva tanto a pHs ácidos como ligeramente alcalinos (Fig 51).
Figura 51. Perfil de atomización de la señal de cadmio en los sobrenadantes resultantes
luego de llevar a cabo extracciones sobre 10 mL de estándar acuoso de cadmio 2 ppb a
diferentes valores de pH. Masa de fase extractante, 2 mg del carbón magnético en cada caso.

~ 71 ~
Hoy en día las técnicas de microextracción para la preparación de muestras han sustituido en
gran medida las técnicas tradicionales como LLE o SPE. En general, estas técnicas ofrecen
ventajas como un menor impacto ambiental, una alta velocidad de análisis, gran eficiencia,
bajos costos operativos y análisis selectivos. Emplear estas técnicas de microextracción en
conjunción con métodos de detección como FAAS, ETAAS, ICP-MS, ICP-OES o UV-Vis
proporciona métodos altamente sensibles que pueden ser utilizados para la determinación de
metales pesados incluso a nivel de trazas.
Inicialmente, se realizó una revisión de los principios, configuraciones, ventajas y limitaciones
de las técnicas de SPME y LPME. Adicionalmente, se compararon algunos métodos reportados
de estas técnicas para la preconcentración y determinación de Cd2+
, Pb2+
y Cu2+
en agua y se
discutieron los parámetros que se deben considerar a la hora de desarrollar estos métodos de
extracción no exhaustivos.
Posteriormente, se discutieron los resultados experimentales obtenidos en el desarrollo de un
método de microextracción con gota directa suspendida para la determinación colorimétrica de
cobre, plomo y cadmio. Por otra parte, analizando diferentes métodos reportados se estableció
que por medio de las técnicas LPME junto con la espectrofotometría UV-Vis que se caracteriza
por su simplicidad, bajo costo y rapidez es posible lograr la detección de trazas y ultratrazas de
metales incluso con mejores límites de detección en comparación con otros métodos de
detección. Además, hoy en día existe la ventaja de encontrar espectrofotómetros que permiten
analizar pequeños volúmenes y múltiples muestras en corto tiempo.
Adicionalmente, se discutieron las potencialidades preconcentrativas de diferentes fases sólidas
como extractantes de metales. Dentro de los adsorbentes a base de carbono que se emplean con
más frecuencia se encuentran los nanotubos de carbono y el óxido de grafeno, estos se
comparan con el carbón activado que pese a sus limitaciones se considera que es posible
emplearse en un método de extracción en microfase sólida dispersiva para la preconcentración y
determinación de iones metálicos. Finalmente, se mencionaron las ventajas que posee la
espectrometría de absorción atómica electrotérmica como método de detección no colorimétrico
y con el objetivo de visualizar las ventajas de este método de detección en conjunción con un
método de microextracción se realizó una prueba preliminar para la preconcentración y
detección de trazas de cadmio empleando como adsorbente un carbón activado magnético
sintetizado.

~ 72 ~
(1) Rohanifar, A.; Rodriguez, L. B.; Devasurendra, A. M.; Alipourasiabi, N.; Anderson, J. L.; Kirchhoff, J. R.
Solid-Phase Microextraction of Heavy Metals in Natural Water with a Polypyrrole/Carbon Nanotube/1, 10–
Phenanthroline Composite Sorbent Material. Talanta 2018, 188, 570–577.
https://doi.org/10.1016/j.talanta.2018.05.100.
(2) Arpa Şahin, Ç.; Durukan, İ. Ligandless-Solidified Floating Organic Drop Microextraction Method for the
Preconcentration of Trace Amount of Cadmium in Water Samples. Talanta 2011, 85 (1), 657–661.
https://doi.org/10.1016/j.talanta.2011.04.044.
(3) Shaikh, R.; Kazi, T. G.; Afridi, H. I.; Akhtar, A.; Baig, J. A. An Environmental Friendly Enrichment Method
for Microextraction of Cadmium and Lead in Groundwater Samples: Impact on Biological Sample of
Children. Chemosphere 2019, 237, 124444. https://doi.org/10.1016/j.chemosphere.2019.124444.
(4) Jain, M.; Yadav, M.; Kohout, T.; Lahtinen, M.; Garg, V. K.; Sillanpää, M. Development of Iron
Oxide/Activated Carbon Nanoparticle Composite for the Removal of Cr(VI), Cu(II) and Cd(II) Ions from
Aqueous Solution. Water Resour. Ind. 2018, 20, 54–74. https://doi.org/10.1016/j.wri.2018.10.001.
(5) Dadfarnia, S.; Haji Shabani, A. M.; Kamranzadeh, E. Separation/Preconcentration and Determination of
Cadmium Ions by Solidification of Floating Organic Drop Microextraction and FI-AAS. Talanta 2009, 79
(4), 1061–1065. https://doi.org/10.1016/j.talanta.2009.02.004.
(6) Kasa, N. A.; Chormey, D. S.; Büyükpınar, Ç.; Turak, F.; Budak, T. B.; Bakırdere, S. Determination of
Cadmium at Ultratrace Levels by Dispersive Liquid-Liquid Microextraction and Batch Type Hydride
Generation Atomic Absorption Spectrometry. Microchem. J. 2017, 133, 144–148.
https://doi.org/10.1016/j.microc.2017.03.035.
(7) Jafari Kang, A.; Baghdadi, M.; Pardakhti, A. Removal of Cadmium and Lead from Aqueous Solutions by
Magnetic Acid-Treated Activated Carbon Nanocomposite. Desalination Water Treat. 2016, 57 (40), 18782–
18798. https://doi.org/10.1080/19443994.2015.1095123.
(8) Nazari, S. Determination of Trace Amounts of Cadmium by Modified Graphite Furnace Atomic Absorption
Spectrometry after Liquid Phase Microextraction. Microchem. J. 2008, 90 (2), 107–112.
https://doi.org/10.1016/j.microc.2008.04.002.
(9) Thongsaw, A.; Chaiyasith, W. C.; Sananmuang, R.; Ross, G. M.; Ampiah-Bonney, R. J. Determination of
Cadmium in Herbs by SFODME with ETAAS Detection. Food Chem. 2017, 219, 453–458.
https://doi.org/10.1016/j.foodchem.2016.09.177.
(10) Zounr, R. A.; Tuzen, M.; Deligonul, N.; Khuhawar, M. Y. A Highly Selective and Sensitive Ultrasonic
Assisted Dispersive Liquid Phase Microextraction Based on Deep Eutectic Solvent for Determination of
Cadmium in Food and Water Samples Prior to Electrothermal Atomic Absorption Spectrometry. Food
Chem. 2018, 253, 277–283. https://doi.org/10.1016/j.foodchem.2018.01.167.
(11) Fındıkoğlu, M. S.; Fırat, M.; Chormey, D. S.; Turak, F.; Şahin, Ç.; Bakırdere, S. Determination of Cadmium
in Tap, Sea and Waste Water Samples by Vortex-Assisted Dispersive Liquid-Liquid-Solidified Floating
Organic Drop Microextraction and Slotted Quartz Tube FAAS After Complexation with a Imidazole Based
Ligand. Water. Air. Soil Pollut. 2018, 229 (2). https://doi.org/10.1007/s11270-018-3689-1.
(12) Arpa Şahin, Ç.; Durukan, İ. Ligandless-Solidified Floating Organic Drop Microextraction Method for the
Preconcentration of Trace Amount of Cadmium in Water Samples. Talanta 2011, 85 (1), 657–661.
https://doi.org/10.1016/j.talanta.2011.04.044.
(13) Shaikh, R.; Kazi, T. G.; Afridi, H. I.; Akhtar, A.; Baig, J. A. An Environmental Friendly Enrichment Method
for Microextraction of Cadmium and Lead in Groundwater Samples: Impact on Biological Sample of
Children. Chemosphere 2019, 237, 124444. https://doi.org/10.1016/j.chemosphere.2019.124444.
(14) Fırat, M.; Bakırdere, S.; Fındıkoğlu, M. S.; Kafa, E. B.; Yazıcı, E.; Yolcu, M.; Büyükpınar, Ç.; Chormey, D.
S.; Sel, S.; Turak, F. Determination of Trace Amount of Cadmium Using Dispersive Liquid-Liquid
Microextraction-Slotted Quartz Tube-Flame Atomic Absorption Spectrometry. Spectrochim. Acta Part B At.
Spectrosc. 2017, 129, 37–41. https://doi.org/10.1016/j.sab.2017.01.006.
(15) Mandlate, J. S.; Soares, B. M.; Seeger, T. S.; Vecchia, P. D.; Mello, P. A.; Flores, E. M. M.; Duarte, F. A.
Determination of Cadmium and Lead at Sub-Ppt Level in Soft Drinks: An Efficient Combination between
Dispersive Liquid-Liquid Microextraction and Graphite Furnace Atomic Absorption Spectrometry. Food
Chem. 2017, 221, 907–912. https://doi.org/10.1016/j.foodchem.2016.11.075.
(16) World Health Organization. World Health Statistics 2010 Geneva.
(17) Adlnasab, L.; Ebrahimzadeh, H.; Asgharinezhad, A. A.; Aghdam, M. N.; Dehghani, A.; Esmaeilpour, S. A
Preconcentration Procedure for Determination of Ultra-Trace Mercury (II) in Environmental Samples
Employing Continuous-Flow Cold Vapor Atomic Absorption Spectrometry. Food Anal. Methods 2014, 7
(3), 616–628. https://doi.org/10.1007/s12161-013-9664-y.
(18) Shahat, A.; Ali, E. A.; El Shahat, M. F. Colorimetric Determination of Some Toxic Metal Ions in Post-
Mortem Biological Samples. Sens. Actuators B Chem. 2015, 221, 1027–1034.
https://doi.org/10.1016/j.snb.2015.07.032.
(19) Akramipour, R.; Golpayegani, M. R.; Gheini, S.; Fattahi, N. Speciation of Organic/Inorganic Mercury and
Total Mercury in Blood Samples Using Vortex Assisted Dispersive Liquid-Liquid Microextraction Based on
the Freezing of Deep Eutectic Solvent Followed by GFAAS. Talanta 2018, 186, 17–23.
https://doi.org/10.1016/j.talanta.2018.04.042.

~ 73 ~
(20) Maratta, A.; Vázquez, S.; López, A.; Augusto, M.; Pacheco, P. H. Lead Preconcentration by Solid Phase
Extraction Using Oxidized Carbon Xerogel and Spectrophotometric Determination with Dithizone.
Microchem. J. 2016, 128, 166–171. https://doi.org/10.1016/j.microc.2016.04.017.
(21) Zounr, R. A.; Tuzen, M.; Khuhawar, M. Y. A Simple and Green Deep Eutectic Solvent Based Air Assisted
Liquid Phase Microextraction for Separation, Preconcentration and Determination of Lead in Water and
Food Samples by Graphite Furnace Atomic Absorption Spectrometry. J. Mol. Liq. 2018, 259, 220–226.
https://doi.org/10.1016/j.molliq.2018.03.034.
(22) Habila, M. A.; ALOthman, Z. A.; Yilmaz, E.; Alabdullkarem, E. A.; Soylak, M. A New Amine Based
Microextraction of Lead (II) in Real Water Samples Using Flame Atomic Absorption Spectrometry.
Microchem. J. 2019, 148, 214–219. https://doi.org/10.1016/j.microc.2019.04.078.
(23) Naseri, M. Rapid Determination of Lead in Water Samples by Dispersive Liquid–Liquid Microextraction
Coupled with Electrothermal Atomic Absorption Spectrometry. Talanta 2008, 75 (1), 56–62.
https://doi.org/10.1016/j.talanta.2007.10.029.
(24) Özzeybek, G.; Erarpat, S.; Chormey, D. S.; Fırat, M.; Büyükpınar, Ç.; Turak, F.; Bakırdere, S. Sensitive
Determination of Copper in Water Samples Using Dispersive Liquid-Liquid Microextraction-Slotted Quartz
Tube-Flame Atomic Absorption Spectrometry. Microchem. J. 2017, 132, 406–410.
https://doi.org/10.1016/j.microc.2017.02.031.
(25) Khalil, M. M. H.; Shahat, A.; Radwan, A.; El-Shahat, M. F. Colorimetric Determination of Cu(II) Ions in
Biological Samples Using Metal-Organic Framework as Scaffold. Sens. Actuators B Chem. 2016, 233, 272–
280. https://doi.org/10.1016/j.snb.2016.04.079.
(26) Karadaş, C.; Kara, D. Dispersive Liquid–Liquid Microextraction Based on Solidification of Floating Organic
Drop for Preconcentration and Determination of Trace Amounts of Copper by Flame Atomic Absorption
Spectrometry. Food Chem. 2017, 220, 242–248. https://doi.org/10.1016/j.foodchem.2016.09.005.
(27) World Health Organization. Copper in Drinking Water: Background Document for Development of WHO
Guidelines for Drinking-Water Quality, World Health Organization Geneva. 2004.
(28) Jia, F.; Liu, Q.; Chen, Z.; Wei, W. Colorimetric Determination of Nine Metal Ions Based on the De-
Aggregation of Papain-Functionalized Gold Nanoparticles and Using Three Chelating Agents. Microchim.
Acta 2019, 186 (12). https://doi.org/10.1007/s00604-019-4028-y.
(29) Li, M.; Li, D.-W.; Li, Y.-T.; Xu, D.-K.; Long, Y.-T. Highly Selective in Situ Metal Ion Determination by
Hybrid Electrochemical “Adsorption–Desorption” and Colorimetric Methods. Anal. Chim. Acta 2011, 701
(2), 157–163. https://doi.org/10.1016/j.aca.2011.06.016.
(30) Zurera Cosano, G.; Amaro López, M. A. CADMIUM | Properties and Determination. In Encyclopedia of
Food Sciences and Nutrition; Elsevier, 2003; pp 733–739. https://doi.org/10.1016/B0-12-227055-X/00142-5.
(31) Hill, S. J.; Fisher, A. S. Atomic Absorption, Methods and Instrumentation. In Encyclopedia of Spectroscopy
and Spectrometry; Elsevier, 2017; pp 37–43. https://doi.org/10.1016/B978-0-12-803224-4.00099-6.
(32) ISO International Organization for Standardization. Water Quality — Determination of Cadmium. Geneva,
International Organization for Standardization (ISO 5961:1985) Water Quality — Determination of
Cadmium — Flame Atomic Absorption Spectrometric Methods. 1985.
(33) Resano, M.; Vanhaecke, F.; de Loos-Vollebregt, M. T. C. Electrothermal Vaporization for Sample
Introduction in Atomic Absorption, Atomic Emission and Plasma Mass Spectrometry—a Critical Review
with Focus on Solid Sampling and Slurry Analysis. J. Anal. At. Spectrom. 2008, 23 (11), 1450.
https://doi.org/10.1039/b807756h.
(34) Sardans, J.; Montes, F.; Peñuelas, J. Determination of As, Cd, Cu, Hg and Pb in Biological Samples by
Modern Electrothermal Atomic Absorption Spectrometry. Spectrochim. Acta Part B At. Spectrosc. 2010, 65
(2), 97–112. https://doi.org/10.1016/j.sab.2009.11.009.
(35) Guo, X.; He, M.; Chen, B.; Hu, B. Solidified Floating Organic Drop Microextraction Combined with ETV-
ICP-MS for the Determination of Trace Heavy Metals in Environmental Water Samples. Talanta 2012, 94,
70–76. https://doi.org/10.1016/j.talanta.2012.02.053.
(36) Carasek, E.; Morés, L.; Merib, J. Basic Principles, Recent Trends and Future Directions of Microextraction
Techniques for the Analysis of Aqueous Environmental Samples. Trends Environ. Anal. Chem. 2018, 19,
e00060. https://doi.org/10.1016/j.teac.2018.e00060.
(37) Ghorbani, M.; Aghamohammadhassan, M.; Chamsaz, M.; Akhlaghi, H.; Pedramrad, T. Dispersive Solid
Phase Microextraction. TrAC Trends Anal. Chem. 2019, 118, 793–809.
https://doi.org/10.1016/j.trac.2019.07.012.
(38) Yan, Y.; Chen, X.; Hu, S.; Bai, X. Applications of Liquid-Phase Microextraction Techniques in Natural
Product Analysis: A Review. J. Chromatogr. A 2014, 1368, 1–17.
https://doi.org/10.1016/j.chroma.2014.09.068.
(39) Jalili, V.; Barkhordari, A.; Norouzian Baghani, A. The Role of Microextraction Techniques in Occupational
Exposure Assessment. A Review. Microchem. J. 2019, 150, 104086.
https://doi.org/10.1016/j.microc.2019.104086.
(40) Yangcheng, L.; Quan, L.; Guangsheng, L.; Youyuan, D. Directly Suspended Droplet Microextraction. Anal.
Chim. Acta 2006, 566 (2), 259–264. https://doi.org/10.1016/j.aca.2006.02.072.
(41) Giannoulis, K. M.; Giokas, D. L.; Zhu, Q.; Tsogas, G. Z.; Vlessidis, A. G.; Pan, Q. Surfactant-Enhanced
Liquid-Liquid Microextraction Coupled to Micro-Solid Phase Extraction onto Highly Hydrophobic Magnetic
Nanoparticles. Microchim. Acta 2013, 180 (9–10), 775–782. https://doi.org/10.1007/s00604-013-0987-6.

~ 74 ~
(42) Liu, D.; Min, S.; Ping, H.; Song, X. The Application of Directly Suspended Droplet Microextraction for the
Evaluation of Phthalic Acid Esters in Cow’s Milk by Gas Chromatography Mass Spectrometry. J.
Chromatogr. A 2016, 1443, 66–74. https://doi.org/10.1016/j.chroma.2016.03.062.
(43) Pawliszyn, J. Handbook of solid phase microextraction; Elsevier insights; Elsevier: London ;, 2012.
(44) Jalili, V.; Barkhordari, A.; Ghiasvand, A. New Extraction Media in Microextraction Techniques. A Review
of Reviews. Microchem. J. 2019, 104386. https://doi.org/10.1016/j.microc.2019.104386.
(45) Arthur, C. L.; Pawliszyn, Janusz. Solid Phase Microextraction with Thermal Desorption Using Fused Silica
Optical Fibers. Anal. Chem. 1990, 62 (19), 2145–2148. https://doi.org/10.1021/ac00218a019.
(46) Cases, M. Valcárcel.; Valcárcel Cases, M.; Aranzana, S. Cárdenas.; Rodríguez, R. Lucena. Analytical
Microextraction Techniques.; Bentham Science Publishers: Sharjah, 2017.
(47) Atta-ur-Rahman.; Ozkan, S. A.; Ahmed, Rida. Recent Advances in Analytical Techniques Volume 1.; Recent
Advances in Analytical Techniques ; v. 1; Bentham Science Publishers: Sharjah, 2017.
(48) Malik, A. K.; Kaur, V.; Verma, N. A Review on Solid Phase Microextraction—High Performance Liquid
Chromatography as a Novel Tool for the Analysis of Toxic Metal Ions. Talanta 2006, 68 (3), 842–849.
https://doi.org/10.1016/j.talanta.2005.06.005.
(49) Jalili, V.; Barkhordari, A.; Ghiasvand, A. A Comprehensive Look at Solid-Phase Microextraction
Technique: A Review of Reviews. Microchem. J. 2020, 152, 104319.
https://doi.org/10.1016/j.microc.2019.104319.
(50) Ouyang, Gangfeng.; Jiang, Ruifen. Solid Phase Microextraction : Recent Developments and Applications;
Springer Berlin Heidelberg: Berlin, Heidelberg, 2017. https://doi.org/10.1007/978-3-662-53598-1.
(51) Jalili, V.; Barkhordari, A.; Ghiasvand, A. A Comprehensive Look at Solid-Phase Microextraction
Technique: A Review of Reviews. Microchem. J. 2020, 152, 104319.
https://doi.org/10.1016/j.microc.2019.104319.
(52) Álvarez Méndez, J.; Barciela García, J.; García Martín, S.; Peña Crecente, R. M.; Herrero Latorre, C.
Determination of Cadmium and Lead in Urine Samples after Dispersive Solid–Liquid Extraction on
Multiwalled Carbon Nanotubes by Slurry Sampling Electrothermal Atomic Absorption Spectrometry.
Spectrochim. Acta Part B At. Spectrosc. 2015, 106, 13–19. https://doi.org/10.1016/j.sab.2015.01.008.
(53) Płotka-Wasylka, J.; Szczepańska, N.; de la Guardia, M.; Namieśnik, J. Miniaturized Solid-Phase Extraction
Techniques. TrAC Trends Anal. Chem. 2015, 73, 19–38. https://doi.org/10.1016/j.trac.2015.04.026.
(54) Hashemi, B.; Rezania, S. Carbon-Based Sorbents and Their Nanocomposites for the Enrichment of Heavy
Metal Ions: A Review. Microchim. Acta 2019, 186 (8), 578. https://doi.org/10.1007/s00604-019-3668-2.
(55) Karnib, M.; Kabbani, A.; Holail, H.; Olama, Z. Heavy Metals Removal Using Activated Carbon, Silica and
Silica Activated Carbon Composite. Energy Procedia 2014, 50, 113–120.
https://doi.org/10.1016/j.egypro.2014.06.014.
(56) Ibrahim, W. M.; Hassan, A. F.; Azab, Y. A. Biosorption of Toxic Heavy Metals from Aqueous Solution by
Ulva Lactuca Activated Carbon. Egypt. J. Basic Appl. Sci. 2016, 3 (3), 241–249.
https://doi.org/10.1016/j.ejbas.2016.07.005.
(57) Sitko, R.; Zawisza, B.; Malicka, E. Graphene as a New Sorbent in Analytical Chemistry. TrAC Trends Anal.
Chem. 2013, 51, 33–43. https://doi.org/10.1016/j.trac.2013.05.011.
(58) Ren, X.; Chen, C.; Nagatsu, M.; Wang, X. Carbon Nanotubes as Adsorbents in Environmental Pollution
Management: A Review. Chem. Eng. J. 2011, 170 (2–3), 395–410. https://doi.org/10.1016/j.cej.2010.08.045.
(59) Murtada, K. Trends in Nanomaterial-Based Solid-Phase Microextraction with a Focus on Environmental
Applications - A Review. Trends Environ. Anal. Chem. 2020, 25, e00077.
https://doi.org/10.1016/j.teac.2019.e00077.
(60) Duan, J.; Pan, Y.; Liu, G.; Jin, W. Metal-Organic Framework Adsorbents and Membranes for Separation
Applications. Curr. Opin. Chem. Eng. 2018, 20, 122–131. https://doi.org/10.1016/j.coche.2018.04.005.
(61) Zubair, N. A.; Abouzari-Lotf, E.; Mahmoud Nasef, M.; Abdullah, E. C. Aerogel-Based Materials for
Adsorbent Applications in Material Domains. E3S Web Conf. 2019, 90, 01003.
https://doi.org/10.1051/e3sconf/20199001003.
(62) Vas, G.; Vékey, K. Solid-Phase Microextraction: A Powerful Sample Preparation Tool Prior to Mass
Spectrometric Analysis. J. Mass Spectrom. 2004, 39 (3), 233–254. https://doi.org/10.1002/jms.606.
(63) Supelco: Bellefonte, PA. SPME for GC Analysis. 2018.
(64) Beceiro-González, E.; Guimaraes, A.; Alpendurada, M. F. Optimisation of a Headspace-Solid-Phase Micro-
Extraction Method for Simultaneous Determination of Organometallic Compounds of Mercury, Lead and
Tin in Water by Gas Chromatography–Tandem Mass Spectrometry. J. Chromatogr. A 2009, 1216 (29),
5563–5569. https://doi.org/10.1016/j.chroma.2009.05.056.
(65) Kaur, V.; Aulakh, J. S.; Malik, A. K. A New Approach for Simultaneous Determination of Co(II), Ni(II),
Cu(II) and Pd(II) Using 2-Thiophenaldehyde-3-Thiosemicarbazone as Reagent by Solid Phase
Microextraction–High Performance Liquid Chromatography. Anal. Chim. Acta 2007, 603 (1), 44–50.
https://doi.org/10.1016/j.aca.2007.09.031.
(66) Eisert, R.; Pawliszyn, J. Automated In-Tube Solid-Phase Microextraction Coupled to High-Performance
Liquid Chromatography. Anal. Chem. 1997, 69 (16), 3140–3147. https://doi.org/10.1021/ac970319a.
(67) Moein, M. M.; Said, R.; Bassyouni, F.; Abdel-Rehim, M. Solid Phase Microextraction and Related
Techniques for Drugs in Biological Samples. J. Anal. Methods Chem. 2014, 2014, 1–24.
https://doi.org/10.1155/2014/921350.

~ 75 ~
(68) Moliner-Martinez, Y.; Herráez-Hernández, R.; Verdú-Andrés, J.; Molins-Legua, C.; Campíns-Falcó, P.
Recent Advances of In-Tube Solid-Phase Microextraction. TrAC Trends Anal. Chem. 2015, 71, 205–213.
https://doi.org/10.1016/j.trac.2015.02.020.
(69) Mei, M.; Pang, J.; Huang, X.; Luo, Q. Magnetism-Reinforced in-Tube Solid Phase Microextraction for the
Online Determination of Trace Heavy Metal Ions in Complex Samples. Anal. Chim. Acta 2019, 1090, 82–90.
https://doi.org/10.1016/j.aca.2019.09.028.
(70) Duan, C.; Shen, Z.; Wu, D.; Guan, Y. Recent Developments in Solid-Phase Microextraction for on-Site
Sampling and Sample Preparation. TrAC Trends Anal. Chem. 2011, 30 (10), 1568–1574.
https://doi.org/10.1016/j.trac.2011.08.005.
(71) Koziel, J. A.; Odziemkowski, M.; Pawliszyn, J. Sampling and Analysis of Airborne Particulate Matter and
Aerosols Using In-Needle Trap and SPME Fiber Devices. Anal. Chem. 2001, 73 (1), 47–54.
https://doi.org/10.1021/ac000835s.
(72) Heidari, M.; Bahrami, A.; Ghiasvand, A. R.; Rafiei Emam, M.; Shahna, F. G.; Soltanian, A. R. Graphene
Packed Needle Trap Device as a Novel Field Sampler for Determination of Perchloroethylene in the Air of
Dry Cleaning Establishments. Talanta 2015, 131, 142–148. https://doi.org/10.1016/j.talanta.2014.07.043.
(73) Jiang, R.; Pawliszyn, J. Thin-Film Microextraction Offers Another Geometry for Solid-Phase
Microextraction. TrAC Trends Anal. Chem. 2012, 39, 245–253. https://doi.org/10.1016/j.trac.2012.07.005.
(74) Olcer, Y. A.; Tascon, M.; Eroglu, A. E.; Boyacı, E. Thin Film Microextraction: Towards Faster and More
Sensitive Microextraction. TrAC Trends Anal. Chem. 2019, 113, 93–101.
https://doi.org/10.1016/j.trac.2019.01.022.
(75) de la Calle, I.; Ruibal, T.; Lavilla, I.; Bendicho, C. Direct Immersion Thin-Film Microextraction Method
Based on the Sorption of Pyrrolidine Dithiocarbamate Metal Chelates onto Graphene Membranes Followed
by Total Reflection X-Ray Fluorescence Analysis. Spectrochim. Acta Part B At. Spectrosc. 2019, 152, 14–
24. https://doi.org/10.1016/j.sab.2018.12.005.
(76) Yang, L.; Said, R.; Abdel-Rehim, M. Sorbent, Device, Matrix and Application in Microextraction by Packed
Sorbent (MEPS): A Review. J. Chromatogr. B 2017, 1043, 33–43.
https://doi.org/10.1016/j.jchromb.2016.10.044.
(77) Kabir, A.; Locatelli, M.; Ulusoy, H. Recent Trends in Microextraction Techniques Employed in Analytical
and Bioanalytical Sample Preparation. Separations 2017, 4 (4), 36.
https://doi.org/10.3390/separations4040036.
(78) de Oliveira, H. L.; Teixeira, L. S.; Dinali, L. A. F.; Pires, B. C.; Simões, N. S.; Borges, K. B.
Microextraction by Packed Sorbent Using a New Restricted Molecularly Imprinted Polymer for the
Determination of Estrogens from Human Urine Samples. Microchem. J. 2019, 150, 104162.
https://doi.org/10.1016/j.microc.2019.104162.
(79) Baltussen, E.; Sandra, P.; David, F.; Cramers, C. Stir Bar Sorptive Extraction (SBSE), a Novel Extraction
Technique for Aqueous Samples: Theory and Principles. J. Microcolumn Sep. 1999, 11 (10), 737–747.
https://doi.org/10.1002/(SICI)1520-667X(1999)11:10<737::AID-MCS7>3.0.CO;2-4.
(80) Hashemi, S. H.; Kaykhaii, M.; Jamali Keikha, A.; Sajjadi, Z.; Mirmoghaddam, M. Application of Response
Surface Methodology for Silver Nanoparticle Stir Bar Sorptive Extraction of Heavy Metals from Drinking
Water Samples: A Box–Behnken Design. The Analyst 2019, 144 (11), 3525–3532.
https://doi.org/10.1039/C9AN00165D.
(81) Richter, P.; Cañas, A.; Muñoz, C.; Leiva, C.; Ahumada, I. Rotating Disk Sorbent Extraction for Pre-
Concentration of Chromogenic Organic Compounds and Direct Determination by Solid Phase
Spectrophotometry. Anal. Chim. Acta 2011, 695 (1–2), 73–76. https://doi.org/10.1016/j.aca.2011.04.009.
(82) Jachero, L.; Ahumada, I.; Richter, P. Rotating-Disk Sorptive Extraction: Effect of the Rotation Mode of the
Extraction Device on Mass Transfer Efficiency. Anal. Bioanal. Chem. 2014, 406 (12), 2987–2992.
https://doi.org/10.1007/s00216-014-7693-z.
(83) Tsai, W.-H.; Huang, T.-C.; Huang, J.-J.; Hsue, Y.-H.; Chuang, H.-Y. Dispersive Solid-Phase
Microextraction Method for Sample Extraction in the Analysis of Four Tetracyclines in Water and Milk
Samples by High-Performance Liquid Chromatography with Diode-Array Detection. J. Chromatogr. A
2009, 1216 (12), 2263–2269. https://doi.org/10.1016/j.chroma.2009.01.034.
(84) Kocot, K.; Zawisza, B.; Marguí, E.; Queralt, I.; Hidalgo, M.; Sitko, R. Dispersive Micro Solid-Phase
Extraction Using Multiwalled Carbon Nanotubes Combined with Portable Total-Reflection X-Ray
Fluorescence Spectrometry for the Determination of Trace Amounts of Pb and Cd in Water Samples. J.
Anal. At. Spectrom. 2013, 28 (5), 736. https://doi.org/10.1039/c3ja50047k.
(85) Khezeli, T.; Daneshfar, A. Development of Dispersive Micro-Solid Phase Extraction Based on Micro and
Nano Sorbents. TrAC Trends Anal. Chem. 2017, 89, 99–118. https://doi.org/10.1016/j.trac.2017.01.004.
(86) Barfi, B.; Asghari, A.; Rajabi, M.; Sabzalian, S.; Khanalipoor, F.; Behzad, M. Optimized Syringe-Assisted
Dispersive Micro Solid Phase Extraction Coupled with Microsampling Flame Atomic Absorption
Spectrometry for the Simple and Fast Determination of Potentially Toxic Metals in Fruit Juice and Bio-Fluid
Samples. RSC Adv. 2015, 5 (40), 31930–31941. https://doi.org/10.1039/C5RA03537F.
(87) Baghban, N.; Yilmaz, E.; Soylak, M. A Magnetic MoS2-Fe3O4 Nanocomposite as an Effective Adsorbent
for Dispersive Solid-Phase Microextraction of Lead(II) and Copper(II) Prior to Their Determination by
FAAS. Microchim. Acta 2017, 184 (10), 3969–3976. https://doi.org/10.1007/s00604-017-2384-z.
(88) Vakh, C.; Alaboud, M.; Lebedinets, S.; Korolev, D.; Postnov, V.; Moskvin, L.; Osmolovskaya, O.; Bulatov,
A. An Automated Magnetic Dispersive Micro-Solid Phase Extraction in a Fluidized Reactor for the

~ 76 ~
Determination of Fluoroquinolones in Baby Food Samples. Anal. Chim. Acta 2018, 1001, 59–69.
https://doi.org/10.1016/j.aca.2017.11.065.
(89) Chisvert, A.; Cárdenas, S.; Lucena, R. Dispersive Micro-Solid Phase Extraction. TrAC Trends Anal. Chem.
2019, 112, 226–233. https://doi.org/10.1016/j.trac.2018.12.005.
(90) Nasrollahpour, A.; Moradi, S. E.; Baniamerian, M. J. Vortex-Assisted Dispersive Solid-Phase
Microextraction Using Ionic Liquid-Modified Metal-Organic Frameworks of PAHs from Environmental
Water, Vegetable, and Fruit Juice Samples. Food Anal. Methods 2017, 10 (8), 2815–2826.
https://doi.org/10.1007/s12161-017-0843-0.
(91) Andruch, V.; Burdel, M.; Kocúrová, L.; Šandrejová, J.; Balogh, I. S. Application of Ultrasonic Irradiation
and Vortex Agitation in Solvent Microextraction. TrAC Trends Anal. Chem. 2013, 49, 1–19.
https://doi.org/10.1016/j.trac.2013.02.006.
(92) Kocot, K.; Sitko, R. Trace and Ultratrace Determination of Heavy Metal Ions by Energy-Dispersive X-Ray
Fluorescence Spectrometry Using Graphene as Solid Sorbent in Dispersive Micro Solid-Phase Extraction.
Spectrochim. Acta Part B At. Spectrosc. 2014, 94–95, 7–13. https://doi.org/10.1016/j.sab.2014.02.003.
(93) Khan, M.; Yilmaz, E.; Sevinc, B.; Sahmetlioglu, E.; Shah, J.; Jan, M. R.; Soylak, M. Preparation and
Characterization of Magnetic Allylamine Modified Graphene Oxide-Poly(Vinyl Acetate-Co-
Divinylbenzene) Nanocomposite for Vortex Assisted Magnetic Solid Phase Extraction of Some Metal Ions.
Talanta 2016, 146, 130–137. https://doi.org/10.1016/j.talanta.2015.08.032.
(94) Behbahani, M.; Veisi, A.; Omidi, F.; Noghrehabadi, A.; Esrafili, A.; Ebrahimi, M. H. Application of a
Dispersive Micro‐solid‐phase Extraction Method for Pre‐concentration and Ultra‐trace Determination of
Cadmium Ions in Water and Biological Samples. Appl. Organomet. Chem. 2018, 32 (3), e4134.
https://doi.org/10.1002/aoc.4134.
(95) Ruiz, F. J.; Ripoll, L.; Hidalgo, M.; Canals, A. Dispersive Micro Solid-Phase Extraction (DµSPE) with
Graphene Oxide as Adsorbent for Sensitive Elemental Analysis of Aqueous Samples by Laser Induced
Breakdown Spectroscopy (LIBS). Talanta 2019, 191, 162–170.
https://doi.org/10.1016/j.talanta.2018.08.044.
(96) Feist, B.; Sitko, R. Fast and Sensitive Determination of Heavy Metal Ions as Batophenanthroline Chelates in
Food and Water Samples after Dispersive Micro-Solid Phase Extraction Using Graphene Oxide as Sorbent.
Microchem. J. 2019, 147, 30–36. https://doi.org/10.1016/j.microc.2019.03.013.
(97) López-García, I.; Marín-Hernández, J. J.; Hernández-Córdoba, M. Solid-Phase Dispersive Microextraction
Using Reduced Graphene Oxide for the Sensitive Determination of Cadmium and Lead in Waters. Anal.
Methods 2019, 11 (5), 635–641. https://doi.org/10.1039/C8AY02495B.
(98) Montoro-Leal, P.; García-Mesa, J. C.; Siles Cordero, M. T.; López Guerrero, M. M.; Vereda Alonso, E.
Magnetic Dispersive Solid Phase Extraction for Simultaneous Enrichment of Cadmium and Lead in
Environmental Water Samples. Microchem. J. 2020, 155, 104796.
https://doi.org/10.1016/j.microc.2020.104796.
(99) Nabavi, S. N.; Sajjadi, S. M.; Lotfi, Z. Novel Magnetic Nanoparticles as Adsorbent in Ultrasound-Assisted
Micro-Solid-Phase Extraction for Rapid Pre-Concentration of Some Trace Heavy Metal Ions in
Environmental Water Samples: Desirability Function. Chem. Pap. 2020, 74 (4), 1143–1159.
https://doi.org/10.1007/s11696-019-00954-z.
(100) Jeannot, M. A.; Cantwell, F. F. Solvent Microextraction into a Single Drop. Anal. Chem. 1996, 68 (13),
2236–2240. https://doi.org/10.1021/ac960042z.
(101) Liu, H.; Dasgupta, P. K. Analytical Chemistry in a Drop. Solvent Extraction in a Microdrop. Anal. Chem.
1996, 68 (11), 1817–1821. https://doi.org/10.1021/ac960145h.
(102) Lee, J.; Lee, H. K.; Rasmussen, K. E.; Pedersen-Bjergaard, S. Environmental and Bioanalytical Applications
of Hollow Fiber Membrane Liquid-Phase Microextraction: A Review. Anal. Chim. Acta 2008, 624 (2), 253–
268. https://doi.org/10.1016/j.aca.2008.06.050.
(103) Xu, L.; Basheer, C.; Lee, H. K. Developments in Single-Drop Microextraction. J. Chromatogr. A 2007, 1152
(1–2), 184–192. https://doi.org/10.1016/j.chroma.2006.10.073.
(104) Tang, S.; Qi, T.; Ansah, P. D.; Nalouzebi Fouemina, J. C.; Shen, W.; Basheer, C.; Lee, H. K. Single-Drop
Microextraction. TrAC Trends Anal. Chem. 2018, 108, 306–313. https://doi.org/10.1016/j.trac.2018.09.016.
(105) Płotka-Wasylka, J.; Namieśnik, J. Green analytical chemistry : past, present and perspectives; Green
chemistry and sustainable technology, 2196-6982; Springer: Singapore, 2019. https://doi.org/10.1007/978-
981-13-9105-7.
(106) Zarei, A. R.; Nedaei, M.; Ghorbanian, S. A. Ferrofluid of Magnetic Clay and Menthol Based Deep Eutectic
Solvent: Application in Directly Suspended Droplet Microextraction for Enrichment of Some Emerging
Contaminant Explosives in Water and Soil Samples. J. Chromatogr. A 2018, 1553, 32–42.
https://doi.org/10.1016/j.chroma.2018.04.023.
(107) Li, S.; Yang, X.; Hu, L.; Cui, X.; Zhang, S.; Lu, R.; Zhou, W.; Gao, H. Directly Suspended-Solidified
Floating Organic Droplets for the Determination of Fungicides in Water and Honey Samples. Anal Methods
2014, 6 (18), 7510–7517. https://doi.org/10.1039/C4AY00806E.
(108) Spietelun, A.; Marcinkowski, Ł.; de la Guardia, M.; Namieśnik, J. Green Aspects, Developments and
Perspectives of Liquid Phase Microextraction Techniques. Talanta 2014, 119, 34–45.
https://doi.org/10.1016/j.talanta.2013.10.050.
(109) Li, X.; Li, H.; Ma, W.; Guo, Z.; Li, X.; Li, X.; Zhang, Q. Determination of Patulin in Apple Juice by Single-
Drop Liquid-Liquid-Liquid Microextraction Coupled with Liquid Chromatography-Mass Spectrometry.
Food Chem. 2018, 257, 1–6. https://doi.org/10.1016/j.foodchem.2018.02.077.

~ 77 ~
(110) Wang, X.; Chen, J.; Zhou, Y.; Liu, X.; Yao, H.; Ahmad, F. Dispersive Liquid–Liquid Microextraction and
Micro-Solid Phase Extraction for the Rapid Determination of Metals in Food and Environmental Waters.
Anal. Lett. 2015, 48 (11), 1787–1801. https://doi.org/10.1080/00032719.2014.1002036.
(111) Khalili Zanjani, M. R.; Yamini, Y.; Shariati, S.; Jönsson, J. Å. A New Liquid-Phase Microextraction Method
Based on Solidification of Floating Organic Drop. Anal. Chim. Acta 2007, 585 (2), 286–293.
https://doi.org/10.1016/j.aca.2006.12.049.
(112) Chormey, D. S.; Zaman, B. T.; Kasa, N. A.; Bakırdere, S. Liquid Phase Microextraction Strategies and Their
Application in the Determination of Endocrine Disruptive Compounds in Food Samples. TrAC Trends Anal.
Chem. 2020, 115917. https://doi.org/10.1016/j.trac.2020.115917.
(113) Viñas, P.; Campillo, N.; Andruch, V. Recent Achievements in Solidified Floating Organic Drop
Microextraction. TrAC Trends Anal. Chem. 2015, 68, 48–77. https://doi.org/10.1016/j.trac.2015.02.005.
(114) Pedersen-Bjergaard, S.; Rasmussen, K. E. Liquid−Liquid−Liquid Microextraction for Sample Preparation of
Biological Fluids Prior to Capillary Electrophoresis. Anal. Chem. 1999, 71 (14), 2650–2656.
https://doi.org/10.1021/ac990055n.
(115) Esrafili, A.; Baharfar, M.; Tajik, M.; Yamini, Y.; Ghambarian, M. Two-Phase Hollow Fiber Liquid-Phase
Microextraction. TrAC Trends Anal. Chem. 2018, 108, 314–322. https://doi.org/10.1016/j.trac.2018.09.015.
(116) Ocaña-González, J. A.; Fernández-Torres, R.; Bello-López, M. Á.; Ramos-Payán, M. New Developments in
Microextraction Techniques in Bioanalysis. A Review. Anal. Chim. Acta 2016, 905, 8–23.
https://doi.org/10.1016/j.aca.2015.10.041.
(117) Khan, W. A.; Arain, M. B.; Yamini, Y.; Shah, N.; Kazi, T. G.; Pedersen-Bjergaard, S.; Tajik, M. Hollow
Fiber-Based Liquid Phase Microextraction Followed by Analytical Instrumental Techniques for Quantitative
Analysis of Heavy Metal Ions and Pharmaceuticals. J. Pharm. Anal. 2020, 10 (2), 109–122.
https://doi.org/10.1016/j.jpha.2019.12.003.
(118) Pena-Pereira, F.; Lavilla, I.; Bendicho, C. Miniaturized Preconcentration Methods Based on Liquid–Liquid
Extraction and Their Application in Inorganic Ultratrace Analysis and Speciation: A Review. Spectrochim.
Acta Part B At. Spectrosc. 2009, 64 (1), 1–15. https://doi.org/10.1016/j.sab.2008.10.042.
(119) Kokosa, J. M. Selecting an Extraction Solvent for a Greener Liquid Phase Microextraction (LPME) Mode-
Based Analytical Method. TrAC Trends Anal. Chem. 2019, 118, 238–247.
https://doi.org/10.1016/j.trac.2019.05.012.
(120) Marcinkowska, R.; Konieczna, K.; Marcinkowski, Ł.; Namieśnik, J.; Kloskowski, A. Application of Ionic
Liquids in Microextraction Techniques: Current Trends and Future Perspectives. TrAC Trends Anal. Chem.
2019, 119, 115614. https://doi.org/10.1016/j.trac.2019.07.025.
(121) Kokosa, J. M. Recent Trends in Using Single-Drop Microextraction and Related Techniques in Green
Analytical Methods. TrAC Trends Anal. Chem. 2015, 71, 194–204.
https://doi.org/10.1016/j.trac.2015.04.019.
(122) Soylak, M.; Koksal, M. Deep Eutectic Solvent Microextraction of Lead(II), Cobalt(II), Nickel(II) and
Manganese(II) Ions for the Separation and Preconcentration in Some Oil Samples from Turkey Prior to Their
Microsampling Flame Atomic Absorption Spectrometric Determination. Microchem. J. 2019, 147, 832–837.
https://doi.org/10.1016/j.microc.2019.04.006.
(123) Abdi, K.; Ezoddin, M.; Pirooznia, N. Temperature-Controlled Liquid–Liquid Microextraction Using a
Biocompatible Hydrophobic Deep Eutectic Solvent for Microextraction of Palladium from Catalytic
Converter and Road Dust Samples Prior to ETAAS Determination. Microchem. J. 2020, 157, 104999.
https://doi.org/10.1016/j.microc.2020.104999.
(124) Zhang, S.; Chen, B.; He, M.; Hu, B. Switchable Solvent Based Liquid Phase Microextraction of Trace Lead
and Cadmium from Environmental and Biological Samples Prior to Graphite Furnace Atomic Absorption
Spectrometry Detection. Microchem. J. 2018, 139, 380–385. https://doi.org/10.1016/j.microc.2018.03.017.
(125) Vanderveen, J. R.; Durelle, J.; Jessop, P. G. Design and Evaluation of Switchable-Hydrophilicity Solvents.
Green Chem 2014, 16 (3), 1187–1197. https://doi.org/10.1039/C3GC42164C.
(126) Lasarte-Aragonés, G.; Lucena, R.; Cárdenas, S.; Valcárcel, M. Use of Switchable Solvents in the
Microextraction Context. Talanta 2015, 131, 645–649. https://doi.org/10.1016/j.talanta.2014.08.031.
(127) Dadfarnia, S.; Haji Shabani, A. M. Recent Development in Liquid Phase Microextraction for Determination
of Trace Level Concentration of Metals—A Review. Anal. Chim. Acta 2010, 658 (2), 107–119.
https://doi.org/10.1016/j.aca.2009.11.022.
(128) Aguirre, M. Á.; Baile, P.; Vidal, L.; Canals, A. Metal Applications of Liquid-Phase Microextraction. TrAC
Trends Anal. Chem. 2019, 112, 241–247. https://doi.org/10.1016/j.trac.2018.11.032.
(129) Wen, X.; Yang, Q.; Yan, Z.; Deng, Q. Determination of Cadmium and Copper in Water and Food Samples
by Dispersive Liquid–Liquid Microextraction Combined with UV–Vis Spectrophotometry. Microchem. J.
2011, 97 (2), 249–254. https://doi.org/10.1016/j.microc.2010.09.010.
(130) Zeini Jahromi, E.; Bidari, A.; Assadi, Y.; Milani Hosseini, M. R.; Jamali, M. R. Dispersive Liquid–Liquid
Microextraction Combined with Graphite Furnace Atomic Absorption Spectrometry. Anal. Chim. Acta 2007,
585 (2), 305–311. https://doi.org/10.1016/j.aca.2007.01.007.
(131) Jiang, H.; Hu, B. Determination of Trace Cd and Pb in Natural Waters by Direct Single Drop
Microextraction Combined with Electrothermal Atomic Absorption Spectrometry. Microchim. Acta 2008,
161 (1–2), 101–107. https://doi.org/10.1007/s00604-007-0785-0.
(132) Wen, X.; Deng, Q.; Guo, J.; Yang, S. Ultra-Sensitive Determination of Cadmium in Rice and Water by UV–
Vis Spectrophotometry after Single Drop Microextraction. Spectrochim. Acta. A. Mol. Biomol. Spectrosc.
2011, 79 (3), 508–512. https://doi.org/10.1016/j.saa.2011.03.021.

~ 78 ~
(133) Durukan, İ.; Arpa Şahin, Ç.; Bektaş, S. Determination of Copper Traces in Water Samples by Flow
Injection-Flame Atomic Absorption Spectrometry Using a Novel Solidified Floating Organic Drop
Microextraction Method. Microchem. J. 2011, 98 (2), 215–219.
https://doi.org/10.1016/j.microc.2011.02.001.
(134) Dadfarnia, S.; Salmanzadeh, A. M.; Shabani, A. M. H. A Novel Separation/Preconcentration System Based
on Solidification of Floating Organic Drop Microextraction for Determination of Lead by Graphite Furnace
Atomic Absorption Spectrometry. Anal. Chim. Acta 2008, 623 (2), 163–167.
https://doi.org/10.1016/j.aca.2008.06.033.
(135) Neri, T. S.; Rocha, D. P.; Muñoz, R. A. A.; Coelho, N. M. M.; Batista, A. D. Highly Sensitive Procedure for
Determination of Cu(II) by GF AAS Using Single-Drop Microextraction. Microchem. J. 2019, 147, 894–
898. https://doi.org/10.1016/j.microc.2019.04.014.
(136) L.C. Passos, M.; M.F.S. Saraiva, M. L. Detection in UV-Visible Spectrophotometry: Detectors, Detection
Systems, and Detection Strategies. Measurement 2019, 135, 896–904.
https://doi.org/10.1016/j.measurement.2018.12.045.
(137) Karadaş, C.; Kara, D. Dispersive Liquid–Liquid Microextraction Based on Solidification of Floating Organic
Drop for Preconcentration and Determination of Trace Amounts of Copper by Flame Atomic Absorption
Spectrometry. Food Chem. 2017, 220, 242–248. https://doi.org/10.1016/j.foodchem.2016.09.005.
(138) Durukan, İ.; Arpa Şahin, Ç.; Bektaş, S. Determination of Copper Traces in Water Samples by Flow
Injection-Flame Atomic Absorption Spectrometry Using a Novel Solidified Floating Organic Drop
Microextraction Method. Microchem. J. 2011, 98 (2), 215–219.
https://doi.org/10.1016/j.microc.2011.02.001.
(139) Şahin, Ç. A.; Tokgöz, İ. A Novel Solidified Floating Organic Drop Microextraction Method for
Preconcentration and Determination of Copper Ions by Flow Injection Flame Atomic Absorption
Spectrometry. Anal. Chim. Acta 2010, 667 (1–2), 83–87. https://doi.org/10.1016/j.aca.2010.04.012.
(140) Neri, T. S.; Rocha, D. P.; Muñoz, R. A. A.; Coelho, N. M. M.; Batista, A. D. Highly Sensitive Procedure for
Determination of Cu(II) by GF AAS Using Single-Drop Microextraction. Microchem. J. 2019, 147, 894–
898. https://doi.org/10.1016/j.microc.2019.04.014.
(141) Erbas, Z.; Maulana, R.; Yilmaz, E.; Ozdemir, S.; Kilinc, E.; Soylak, M. Solid-Phase Extraction of Copper as
1-(2-Pyridylazo)-2-Naphthol (PAN) Chelates on Coprinus Atramentaria. Int. J. Environ. Anal. Chem. 2019,
1–12. https://doi.org/10.1080/03067319.2019.1646737.
(142) Yang, F.; Li, J.; Lu, W.; Wen, Y.; Cai, X.; You, J.; Ma, J.; Ding, Y.; Chen, L. Speciation Analysis of
Mercury in Water Samples by Dispersive Liquid-Liquid Microextraction Coupled to Capillary
Electrophoresis: Liquid Phase Separations. ELECTROPHORESIS 2014, 35 (4), 474–481.
https://doi.org/10.1002/elps.201300409.
(143) Jalbani, N.; Soylak, M. Determination of Cadmium and Lead in Water and Food by Organic Drop
Microextraction and Flame Atomic Absorption Spectrometry. Instrum. Sci. Technol. 2015, 43 (5), 573–587.
https://doi.org/10.1080/10739149.2015.1017768.
(144) Akkaya, E.; Chormey, D. S.; Bakırdere, S. Sensitive Determination of Cadmium Using Solidified Floating
Organic Drop Microextraction-Slotted Quartz Tube-Flame Atomic Absorption Spectroscopy. Environ.
Monit. Assess. 2017, 189 (10). https://doi.org/10.1007/s10661-017-6232-8.
(145) Sakanupongkul, A.; Sananmuang, R.; Udnan, Y.; Ampiah-Bonney, R. J.; Chaiyasith, W. C. Speciation of
Mercury in Water and Freshwater Fish Samples by a Two-Step Solidified Floating Organic Drop
Microextraction with Electrothermal Atomic Absorption Spectrometry. Food Chem. 2019, 277, 496–503.
https://doi.org/10.1016/j.foodchem.2018.10.131.
(146) 1-Dodecanol [MAK Value Documentation, 2006]. In The MAK-Collection for Occupational Health and
Safety; Deutsche Forschungsgemeinschaft, Commission for the Investigation of Health Hazards of Chemical
Compounds in the Work Area, Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012;
pp 117–132. https://doi.org/10.1002/3527600418.mb11253kske0022.
(147) Chisvert, A.; Cárdenas, S.; Lucena, R. Dispersive Micro-Solid Phase Extraction. TrAC Trends Anal. Chem.
2019, 112, 226–233. https://doi.org/10.1016/j.trac.2018.12.005.
(148) Pastor-Belda, M.; Navarro-Jiménez, T.; Garrido, I.; Viñas, P.; Campillo, N.; Fenoll, J.; Hernández-Córdoba,
M. Magnetic Solid-Phase Extraction or Dispersive Liquid-Liquid Microextraction for Pyrethroid
Determination in Environmental Samples. J. Sep. Sci. 2018, 41 (12), 2565–2575.
https://doi.org/10.1002/jssc.201800109.
(149) Sanz-Medel, A.; Pereiro, R. Atomic absorption spectrometry : an introduction, Second edition.; Momentum
Press: New York [New York] (222 East 46th Street, New York, NY 10017), 2014.
https://doi.org/10.5643/9781606504376.
(150) Bencini, A.; Lippolis, V. 1,10-Phenanthroline: A Versatile Building Block for the Construction of Ligands
for Various Purposes. Coord. Chem. Rev. 2010, 254 (17–18), 2096–2180.
https://doi.org/10.1016/j.ccr.2010.04.008.
(151) Sridharan, K. Spectral methods in transition metal complexes; Elsevier: Amsterdam, 2016.
(152) Safari, Z.; Gholivand, M. B.; Hosseinzadeh, L. Spectrophotometric Study of Complex Formations between
1-(2-Pyridylazo)-2-Naphthol (PAN) and Some Metal Ions in Organic Solvents and the Determination of
Thermodynamic Parameters. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2011, 78 (5), 1606–1610.
https://doi.org/10.1016/j.saa.2011.02.014.

~ 79 ~
(153) Pramanik, K.; Adhikari, B. 1-(2′-Pyridylazo)-2-Naphtholate (PAN) Complexes of Rhodium(III): Synthesis,
Structure and Spectral Studies. Polyhedron 2010, 29 (3), 1015–1022.
https://doi.org/10.1016/j.poly.2009.12.001.
(154) Ünlü, B.; Çakar, S.; Özacar, M. The Effects of Metal Doped TiO2 and Dithizone-Metal Complexes on
DSSCs Performance. Sol. Energy 2018, 166, 441–449. https://doi.org/10.1016/j.solener.2018.03.064.
(155) Costa, A. C. S.; Lopes, L.; Korn, M. das G. A.; Portela, J. G. Separation and Pre-Concentration of Cadmium,
Copper, Lead, Nickel and Zinc by Solid-Liquid Extraction of Their Cocrystallized Naphthalene Dithizone
Chelate in Saline Matrices. J. Braz. Chem. Soc. 2002, 13 (5), 674–678. https://doi.org/10.1590/S0103-
50532002000500022.
(156) Rice, C. R.; Faulkner, R. A.; Jewsbury, R. A.; Bullock, S.; Dunmore, R. A Structural Study of Dithizone
Coordination Chemistry. CrystEngComm 2017, 19 (25), 3414–3419. https://doi.org/10.1039/C7CE00580F.
(157) Wypych, G. PHOTOPHYSICS. In Handbook of Material Weathering; Elsevier, 2018; pp 1–26.
https://doi.org/10.1016/B978-1-927885-31-4.50003-0.
(158) Şahin, Ç. A.; Tokgöz, İ. A Novel Solidified Floating Organic Drop Microextraction Method for
Preconcentration and Determination of Copper Ions by Flow Injection Flame Atomic Absorption
Spectrometry. Anal. Chim. Acta 2010, 667 (1–2), 83–87. https://doi.org/10.1016/j.aca.2010.04.012.
(159) Ntoi, L. L. A.; Buitendach, B. E.; von Eschwege, K. G. Seven Chromisms Associated with Dithizone. J.
Phys. Chem. A 2017, 121 (48), 9243–9251. https://doi.org/10.1021/acs.jpca.7b09490.
(160) Kokosa, J. M.; Przyjazny, A. J.; Jeannot, M. A. Solvent Microextraction: Theory and Practice; Wiley:
Hoboken, N.J, 2009.
(161) Wahid, F.; Mohammadzai, I. U.; Khan, A.; Shah, Z.; Hassan, W.; Ali, N. Removal of Toxic Metals with
Activated Carbon Prepared from Salvadora Persica. Arab. J. Chem. 2017, 10, S2205–S2212.
https://doi.org/10.1016/j.arabjc.2013.07.054.
(162) Dong, L.; Hou, L.; Wang, Z.; Gu, P.; Chen, G.; Jiang, R. A New Function of Spent Activated Carbon in
BAC Process: Removing Heavy Metals by Ion Exchange Mechanism. J. Hazard. Mater. 2018, 359, 76–84.
https://doi.org/10.1016/j.jhazmat.2018.07.030.
(163) Pashai Gatabi, M.; Milani Moghaddam, H.; Ghorbani, M. Efficient Removal of Cadmium Using Magnetic
Multiwalled Carbon Nanotube Nanoadsorbents: Equilibrium, Kinetic, and Thermodynamic Study. J.
Nanoparticle Res. 2016, 18 (7). https://doi.org/10.1007/s11051-016-3487-x.
(164) Wannahari, R.; Sannasi, P.; Nordin, M. F. M.; Mukhtar, H. Sugarcane Bagasse Derived Nano Magnetic
Adsorbent Composite (SCB-NMAC) for Removal of Cu2+ from Aqueous Solution. ARPN J. Eng. Appl. Sci.
2018, 13, 1–9.
(165) Shen, Y. F.; Tang, J.; Nie, Z. H.; Wang, Y. D.; Ren, Y.; Zuo, L. Tailoring Size and Structural Distortion of
Fe3O4 Nanoparticles for the Purification of Contaminated Water. Bioresour. Technol. 2009, 100 (18), 4139–
4146. https://doi.org/10.1016/j.biortech.2009.04.004.
(166) Kakavandi, B.; Kalantary, R. R.; Farzadkia, M.; Mahvi, A. H.; Esrafili, A.; Azari, A.; Yari, A. R.; Javid, A.
B. Enhanced Chromium (VI) Removal Using Activated Carbon Modified by Zero Valent Iron and Silver
Bimetallic Nanoparticles. J. Environ. Health Sci. Eng. 2014, 12 (1). https://doi.org/10.1186/s40201-014-
0115-5.
(167) Guedidi, H.; Reinert, L.; Lévêque, J.-M.; Soneda, Y.; Bellakhal, N.; Duclaux, L. The Effects of the Surface
Oxidation of Activated Carbon, the Solution PH and the Temperature on Adsorption of Ibuprofen. Carbon
2013, 54, 432–443. https://doi.org/10.1016/j.carbon.2012.11.059.
(168) Hemmati, M.; Rajabi, M.; Asghari, A. Magnetic Nanoparticle Based Solid-Phase Extraction of Heavy Metal
Ions: A Review on Recent Advances. Microchim. Acta Anal. Sci. Based Micro- Nanomater. 2018, 185 (3),
1–32. https://doi.org/10.1007/s00604-018-2670-4.
(169) Bergmann, C. P.; Machado, F. Carbon nanomaterials as adsorbents for environmental and biological
applications; Carbon nanostructures, 2191-3013; Springer: Cham, 2015. https://doi.org/10.1007/978-3-319-
18875-1.