Embed Size (px)
Citation preview

Synthese und Struktur von
Eisen(II)-Spin-Crossover-Verbindungen
vorgelegt von
Diplom-Chemiker
Holger Christian Kämpf
aus Erlangen
Von der Fakultät II – Mathematik und Naturwissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften
– Dr. rer. nat. –
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. rer. nat. R. Süssmuth
1. Berichter: Prof. Dr. rer. nat. A. Grohmann
2. Berichter: Prof. Dr. rer. nat. H. Schumann
Tag der wissenschaftlichen Aussprache: 02.02.2007
Berlin 2007
D83

Es ist schwieriger, eine vorgefasste Meinung zu zertrümmern als ein Atom.
Albert Einstein
14.03.1879 - 18.04.1955
Physiker und Nobelpreisträger
Für meine lieben Eltern

Abstract:
Die Arbeit umfasst die Synthese und Charakterisierung neuer FeII-Spin-Crossover-
Verbindungen. Ausgehend von 2,6-Bis(1’,3’-diamino-2’-methylprop-2’-yl)pyridin · 4 HBr (1
· 4 HBr) sowie Fe(CH3CN)2(OTf)2 bzw. FeCl2 und dem entsprechenden einzähnigen
Liganden wurden die Verbindungen [(1)FeII(1-Methyl-1H-imidazol)](OTf)2 (5), [(1)FeII-
(Pyridin-4-carbonsäuremethylester)]Br2 (8) und [(1)FeII((E)-4-(Phenyldiazenyl)pyridin)]-
(OTf)2 (12) synthetisiert. Der Spinübergang wurde mittels magnetischer Messungen (SQUID)
(5) und temperaturabhängiger NMR- und UV/Vis-Spektroskopie (5, 8, 12) verfolgt. 5, 8 und
[(1)FeII((E)-4-(Phenyldiazenyl)pyridin)]Br2 (12a/b) wurden röntgenstrukturanalytisch charak-
terisiert. Von 5 und 8 wurden Cyclovoltammogramme aufgenommen. Die Verbindungen
[(1)FeIII(OOCC5H4N)]Br2 · MeOH (6 · MeOH), [(1)FeII(NC5H3(CO2)(COO))FeII(1)]Br2 · 5.5
MeOH (7 · 5.5 MeOH), [(1)FeII (NC5H4S) {FeII(1)}2]Br5 (10), [(1)FeII(Pyridin-4-thiolat)]Br ·
MeOH (9 · MeOH) und [(1)2(FeIII)2(µ2-S2)]Br4 · 4 MeOH (11 · 4 MeOH) wurden mittels
Röntgenstrukturanalyse untersucht. Von [(1)FeII(tert-Butylisonitril)]Br2 (13), [(1)FeII(Cyclo-
hexylisonitril)]Br2 (14) und [(1)FeII(p-Methoxyphenylisonitril)]Br2 (15) wurden die
Festkörperstrukturen, IR- und NMR-Spektren bestimmt.
Ausgehend von 2-(2-Methyl-1,3-dimethansulfonyl-propan-2-yl)-6-(2-methyl-1-methan-
sulfonyl-propan-2-yl)pyridin wurde, mit 2-(1,3-diazido-2-methylpropan-2-yl)-6-(1-azido-2-
methylpropan-2-yl)pyridin (16) als Zwischenstufe, der neue Ligand 2-(1,3-diamino-2-
methylpropan-2-yl)-6-(1-amino-2-methylpropan-2-yl)pyridin (2) synthetisiert. Die Koordina-
tionseigenschaften von 2 gegenüber FeII, CuII, NiII und ZnII wurden untersucht. Weiterhin
wurden die Verbindungen [(2)CuII(dmf)](ClO4)2 (20), [(2)NiIIBr]Br (21), [(2)ZnII](ClO4)2
(23) und [(2)2ZnII2Br2]Br0.21(ClO4)1.79 (24) synthetisiert. Die NMR-Spektren von 23 und 24
und die Festkörperstrukturen von 21 und 24 wurden bestimmt. Verbindung 2 wurde durch
Schiffbasenkondensation mit Pyridin-2-carbaldehyd bzw. 1-Methyl-1H-imidazol-2-carb-
aldehyd und anschließende Reduktion derivatisiert. Bei der Umsetzung mit Fe(CH3CN)2-
(OTf)2 entstand der gemischtvalente, siebenkernige Cluster [FeIII3FeII
4L12](OTf)5 · 2 MeOH
(19) (L = (1-Methyl-1H-imidazol-2-yl)methanolat). 2,2'-(Pyridin-2,6-diyl)bis(N,N-dimethyl-
propan-1-amin) · 3 HBr (25) wurde aus 1 unter Eschweiler-Clarke-Bedingungen erhalten. Um
einen in para-Postion des Pyrdinringes modifizierbaren, zu 1 analogen Liganden zu erhalten,
wurde 4-Brom-2,6-diethylpyridin (26) synthetisiert.

Die vorliegende Arbeit entstand in der Zeit von Semptember 2003 bis Semptember 2006 am
Institut für Chemie
der Technischen Universität Berlin
An dieser Stelle möchte ich mich bedanken bei
Prof. Dr. Andreas Grohmann für die herzliche Betreuung und für das überaus große Interesse,
mit dem er meine Arbeit begleitet hat. Die gewinnbringenden Diskussionen und wertvollen
Anregungen haben maßgeblich zum Gelingen beigetragen;
Prof. Dr. H. Schumann Übernahme der zweiten Berichterstattung und bei Prof. Dr. R.
Süssmuth für die Übernahme des Prüfungsvorsitzes;
meinem langjährigen Laborkollegen Herrn Dr. Stefan Leopold für die zahlreichen
wissenschaftlichen Diskussionen und das gute Miteinander im Labor;
Prof. Dr. Walter Bauer (Universität Erlangen-Nürnberg), Herrn Manfred Dettlaff und Dr.
Heinz-Jürgen Kroth für die Aufnahme der Kernresonanzspektren;
Frau Sigrid Imme für die Ausführung der Elementaranalysen und der IR-Messungen;
Dr. M. Grunert und Prof. P. Gütlich (Universität Mainz) für die Durchführung von
magnetischen Messungen und Aufnahme von Mößbauerspektren;
Dr. Claudia Trage und Frau Alice Stöckel für die Aufnahme der Massenspektren und bei Frau
Marina Borowski, Dr. Markus Hummert, Herrn Panagiotis Bakatselos und Dr. Frank W.
Heinemann (Universität Erlangen-Nürnberg) für die Durchführung der Röntgenstruktur-
analysen;
Ferner danke ich allen Mitarbeitern und „Ehemaligen“ des Arbeitskreises Grohmann für ihre
stete Hilfsbereitschaft und das überaus angenehme Arbeitsklima.

5
Inhalt: 1 Einleitung ...........................................................................................................................8
1.1 Das Spin-Crossover-Phänomen..................................................................................8
1.1.1 Spinübergang im Festkörper und in Lösung ......................................................9
1.1.2 Charakterisierungsmethoden ............................................................................10
1.2 Biologische Aspekte .................................................................................................12
1.3 Stand der Forschung .................................................................................................13
1.4 Literatur ....................................................................................................................14
2 Motivation ........................................................................................................................16
2.1 Literatur ....................................................................................................................17
3 Eisenkomplexe des pyN4-Liganden .................................................................................18
3.1 Einleitung .................................................................................................................18
3.2 Reaktionen in Anwesenheit von H2O.......................................................................20
3.3 1-Methyl-1H-imidazol..............................................................................................23
3.3.1 NMR-Spektren von [FeII(pyN4)(C4H6N2)](OTf)2 ............................................25
3.3.2 Temperaturabhängige UV/Vis-Spektren von [FeII(pyN4)(C4H6N2)](OTf)2 .....26
3.3.3 Magnetische Messung und Mößbauerspektrum von
[FeII(pyN4)(C4H6N2)](OTf)2 .............................................................................27
3.3.4 Elektrochemie von [FeII(pyN4)(C4H6N2)](OTf)2..............................................29
3.4 Pyridincarbonsäuren .................................................................................................31
3.4.1 Pyridin-4-carbonsäure ......................................................................................31
3.4.2 Pyridin-3,5-dicarboxylat...................................................................................34
3.5 Pyridin-4-carbonsäuremethylester (Methylisonicotinat)..........................................37
3.5.1 NMR-Spektren von [FeII(pyN4)(C7H7NO2)]Br2...............................................39
3.5.2 Temperaturabhängige UV/Vis-Spektren von [FeII(pyN4)(C7H7NO2)]Br2 .......40
3.5.3 Elektrochemie von [FeII(pyN4)(C7H7NO2)]Br2 ................................................41
3.6 4-Mercaptopyridin und 4-(Methyldisulfanyl)-pyridin .............................................41
3.7 (E)-4-(Phenyldiazenyl)pyridin .................................................................................48
3.7.1 NMR-Spektren von [FeII(pyN4)(4-PAPY)](OTf)2 ...........................................51
3.7.2 Temperaturabhängige UV/Vis-Spektren von [FeII(pyN4)(4-PAPY)](OTf)2....51
3.8 Isonitrile....................................................................................................................53
3.9 Experimenteller Teil .................................................................................................58
3.9.1 Allgemeines ......................................................................................................58

6
3.9.2 Synthesen..........................................................................................................60
3.9.2.1 [FeII(pyN4)(H2O)](BF4)2 (4) .........................................................................60
3.9.2.2 Umsetzung von pyN4 mit Fe(ClO4)2 · x H2O ...............................................61
3.9.2.3 [FeII(pyN4)(C4H6N2)](OTf)2 (5) ...................................................................62
3.9.2.4 [FeIII(pyN4)(C5H4NCO2]Br2 · MeOH (6 · MeOH) .......................................63
3.9.2.5 [(pyN4)FeII–NC5H3(CO2)(COO–)FeII(pyN4)]Br2 · 5.5 MeOH;
7 · 5.5 MeOH ................................................................................................64
3.9.2.6 [FeII(pyN4)(C7H7NO2)]Br2 (8)......................................................................65
3.9.2.7 [FeII(pyN4)(C5H4NS)]Br · MeOH; 9 · MeOH...............................................66
3.9.2.8 [(pyN4)FeII–(NC5H4S)–{FeII(pyN4)}2]Br5 (10) ............................................67
3.9.2.9 [(pyN4)2(FeIII)2(µ2-S2)]Br4 · 4 MeOH (11 · 4 MeOH) ..................................68
3.9.2.10 [FeII(pyN4)(4-PAPY)](OTf)2 (12) ................................................................68
3.9.2.11 [FeII(pyN4)(CNtBu)]Br2 (13), [FeII(pyN4)(CNcy)]Br2 (14) und
[FeII(pyN4)(CNC6H4OCH3)]Br2 (15)............................................................69
3.9.3 Röntgenstrukturanalysen ..................................................................................73
3.10 Literatur ....................................................................................................................76
4 Übergangsmetallkomplexe des pyN3-Liganden ...............................................................79
4.1 Einleitung .................................................................................................................79
4.2 Synthese des pyN3-Liganden....................................................................................80
4.3 Umsetzungen von pyN3 mit FeII ...............................................................................82
4.4 Umsetzungen von pyN3 mit CuII, NiII und ZnII ........................................................90
4.5 Experimenteller Teil .................................................................................................95
4.5.1 Allgemeines ......................................................................................................95
4.5.2 Synthesen..........................................................................................................96
4.5.2.1 2-(1,3-diazido-2-methylpropan-2-yl)-6-(1-azido-2-methylpropan-2-
yl)pyridin (16) ..............................................................................................96
4.5.2.2 2-(1,3-diamino-2-methylpropan-2-yl)-6-(1-amino-2-methylpropan-2-
yl)pyridin (2) ................................................................................................97
4.5.2.3 [FeII(C4H6N2)6]2+ ..........................................................................................98
4.5.2.4 2-Methyl-2-(6-(2-methyl-1-(pyridin-2-ylmethylamino)propan-2-yl)-
pyridin-2-yl)-N1,N3-bis(pyridin-2-ylmethyl)propan-1,3-diamin (17)...........99
4.5.2.5 2-Methyl-2-(6-(2-methyl-1-((1-methyl-1H-imidazol-2-
yl)methylamino)propan-2-yl)pyridin-2-yl)-N1,N3-bis((1-methyl-1H-
imidazol-2-yl)methyl)propan-1,3-diamine (18) .........................................100

7
4.5.2.6 [FeIII3FeII
4L12](OTf)5 · 2 MeOH (19)..........................................................101
4.5.2.7 [Cu(pyN3)(dmf)](ClO4)2 (20) .....................................................................101
4.5.2.8 [Ni(pyN3)Br]Br · MeOH (21).....................................................................102
4.5.2.9 Umsetzung von pyN3 mit Zn(OAc)2 (22)...................................................103
4.5.2.10 [Zn(pyN3)](ClO4)2 (23) ..............................................................................103
4.5.2.11 [Zn(pyN3)Br]2Br0.21(ClO4)1.79 (24) .............................................................104
4.5.3 Röntgenstrukturanalysen ................................................................................105
4.5.4 BVS-Rechnungen an [FeIII3FeII
4L12](OTf)5 · 2 MeOH (19) ...........................106
4.6 Literatur ..................................................................................................................107
5 Versuche zur Derivatisierung des pyN4-Liganden .........................................................109
5.1 Versuche zur Permethylierung des pyN4-Liganden ...............................................109
5.1.1 Allgemeines ....................................................................................................109
5.1.2 Synthesen........................................................................................................109
5.2 Modifizierung des pyN4-Liganden in para-Postion ...............................................111
5.2.1 Allgemeines ....................................................................................................111
5.3 Experimenteller Teil ...............................................................................................114
5.3.1 Allgemeines ....................................................................................................114
5.3.1.1 2,2'-(Pyridin-2,6-diyl)bis(N,N-dimethylpropan-1-amin) · 3 HBr (25).......115
5.3.1.2 4-Brom-2,6-diethylpyridin (26)..................................................................116
5.4 Literatur ..................................................................................................................116
6 Zusammenfassung ..........................................................................................................118
7 Anhang ...........................................................................................................................124
7.1 Verwendete Abkürzungen ......................................................................................124

Einleitung 8
1 Einleitung
1.1 Das Spin-Crossover-Phänomen
Das Spin-Crossover-(SCO)-Phänomen wurde von Cambi et al. 1932 an einer Reihe von FeIII-
N,N-dialkyl-dithiocarbamat-Komplexen erstmals beobachtet.[1] Dreißig Jahre später wurden
die ersten FeII-SCO-Verbindungen [Fe(phen)2(SCN)2] und [Fe(bipy)2(SCN)2] synthetisiert.[2]
Obwohl Spin Crossover in oktaedrischen Komplexen mit Elektronenkonfigurationen von d4
bis d7 auftritt, sind mittlerweile FeII-Verbindungen die häufigsten und am umfassendsten
untersuchten Vertreter dieser Verbindungsklasse.[3][4]
Für die Elektronenkonfiguration von FeII (d6) gibt es zwei Möglichkeiten:
Maximale Spin-Paarung: low-spin (LS) starkes Ligandenfeld (t2g)6 (1A1g)
Maximale Multiplizität: high-spin (HS) schwaches Ligandenfeld (t2g)4(eg)2 (5T2g)
Abb. 1.1: Elektronenkonfiguration des high-spin- (links) und low-spin-Zustandes (rechts),
(∆: Ligandenfeldstärke, P: mittlere Spin-Paarungsenergie).[9]
Diese beiden Zustände unterscheiden sich durch unterschiedliche Besetzung der
antibindenden eg*-Orbitale und der in erster Näherung nichtbindenden t2g-Orbitale.[5][6] Dies
hat drastische Änderungen der geometrischen Parameter (v. a. Bindungslängen) zur Folge.
Beim LS-HS-Übergang nimmt der Ionenradius um bis zu 0.2 Å zu, die entsprechenden
Komplexe „schwellen“.[7]
Ligandenfeldstärke
E
∆ > P ∆ < P
(t2g)4(eg)2 (t2g)6
Einleitung 9
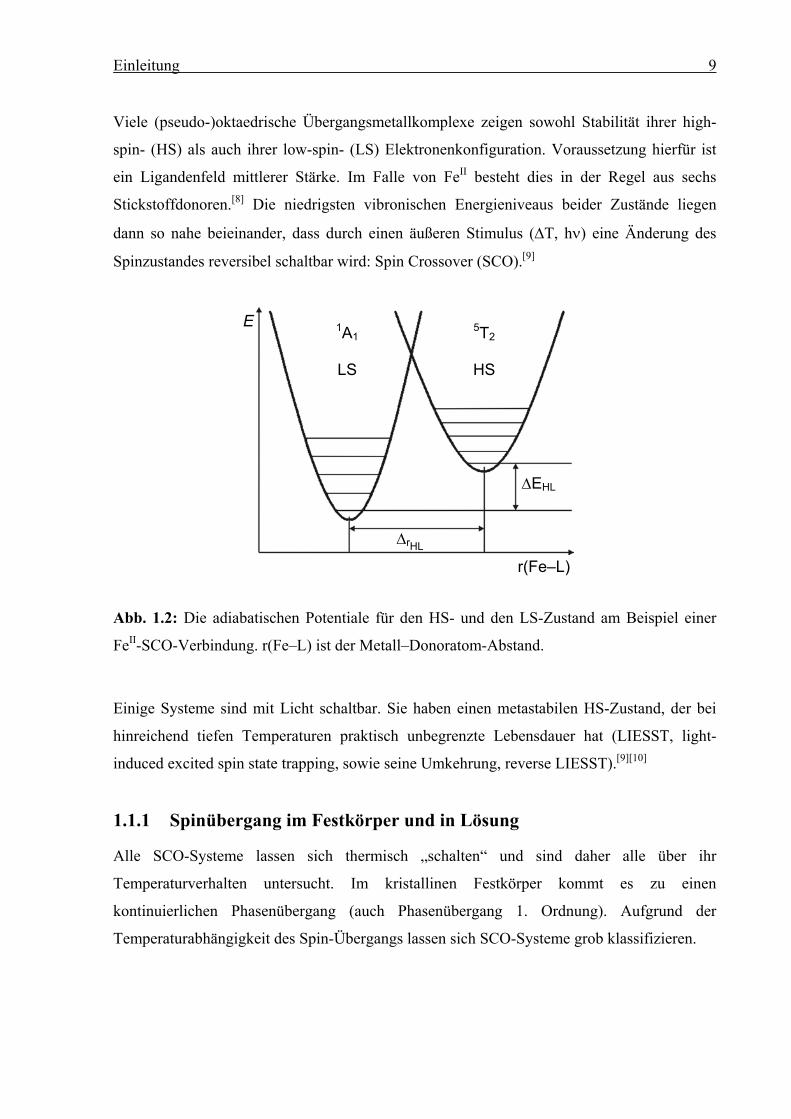
Viele (pseudo-)oktaedrische Übergangsmetallkomplexe zeigen sowohl Stabilität ihrer high-
spin- (HS) als auch ihrer low-spin- (LS) Elektronenkonfiguration. Voraussetzung hierfür ist
ein Ligandenfeld mittlerer Stärke. Im Falle von FeII besteht dies in der Regel aus sechs
Stickstoffdonoren.[8] Die niedrigsten vibronischen Energieniveaus beider Zustände liegen
dann so nahe beieinander, dass durch einen äußeren Stimulus (∆T, hν) eine Änderung des
Spinzustandes reversibel schaltbar wird: Spin Crossover (SCO).[9]
Abb. 1.2: Die adiabatischen Potentiale für den HS- und den LS-Zustand am Beispiel einer
FeII-SCO-Verbindung. r(Fe–L) ist der Metall–Donoratom-Abstand.
Einige Systeme sind mit Licht schaltbar. Sie haben einen metastabilen HS-Zustand, der bei
hinreichend tiefen Temperaturen praktisch unbegrenzte Lebensdauer hat (LIESST, light-
induced excited spin state trapping, sowie seine Umkehrung, reverse LIESST).[9][10]
1.1.1 Spinübergang im Festkörper und in Lösung
Alle SCO-Systeme lassen sich thermisch „schalten“ und sind daher alle über ihr
Temperaturverhalten untersucht. Im kristallinen Festkörper kommt es zu einen
kontinuierlichen Phasenübergang (auch Phasenübergang 1. Ordnung). Aufgrund der
Temperaturabhängigkeit des Spin-Übergangs lassen sich SCO-Systeme grob klassifizieren.
E 1A1
LS
5T2
HS
∆rHL
∆EHL
r(Fe–L)

Einleitung 10
a) b) c) d) e)
Abb. 1.3: Der Verlauf der Spinübergangs- (spin transition, ST) Kurven für SCO-Systeme im
Festkörper: a) allmählich, b) abrupt, c) mit Hysterese, d) stufenweise, e) unvollständig
(relativer LS/HS-Anteil γHS als Funktion von T).
Systeme in Lösung zeigen typischerweise das Verhalten in a); bei Systemen im Festkörper
existieren Beispiele für alle Charakteristiken (a - e). Je sprunghafter der Übergang, desto
höher das Maß an sterischer und/oder elektronischer Kooperativität (Kopplung) der
Metallzentren im Gitter. Zeigt sich Hysterese (c), d. h. hängt der Spin-Zustand von der
Vorgeschichte der Probe ab, so ist das System bistabil.[9][11]
1.1.2 Charakterisierungsmethoden
Ein thermisch angeregter Spinübergang ändert die elektronische Struktur und damit mehrere
Eigenschaften des Systems. Die drei wichtigsten sind Struktur, Magnetismus und Farbe.[9] Die
Struktur ändert sich aufgrund der unterschiedlichen Besetzung der eg*-Orbitale (vgl. S. 8). Das
magnetische Moment ändert sich von ca. 5 BM (HS) zu ca. 0 BM (LS).
Die 57Fe-Mößbauerspektren von FeII-HS und FeII-LS weisen signifikante Unterschiede auf.
Abb. 1.4 zeigt dies anhand der Beispiele [Fe(H2O)6]2+ in Fe(SO4) · 7 H2O (HS) und
K4[Fe(CN)6] (LS).[12]
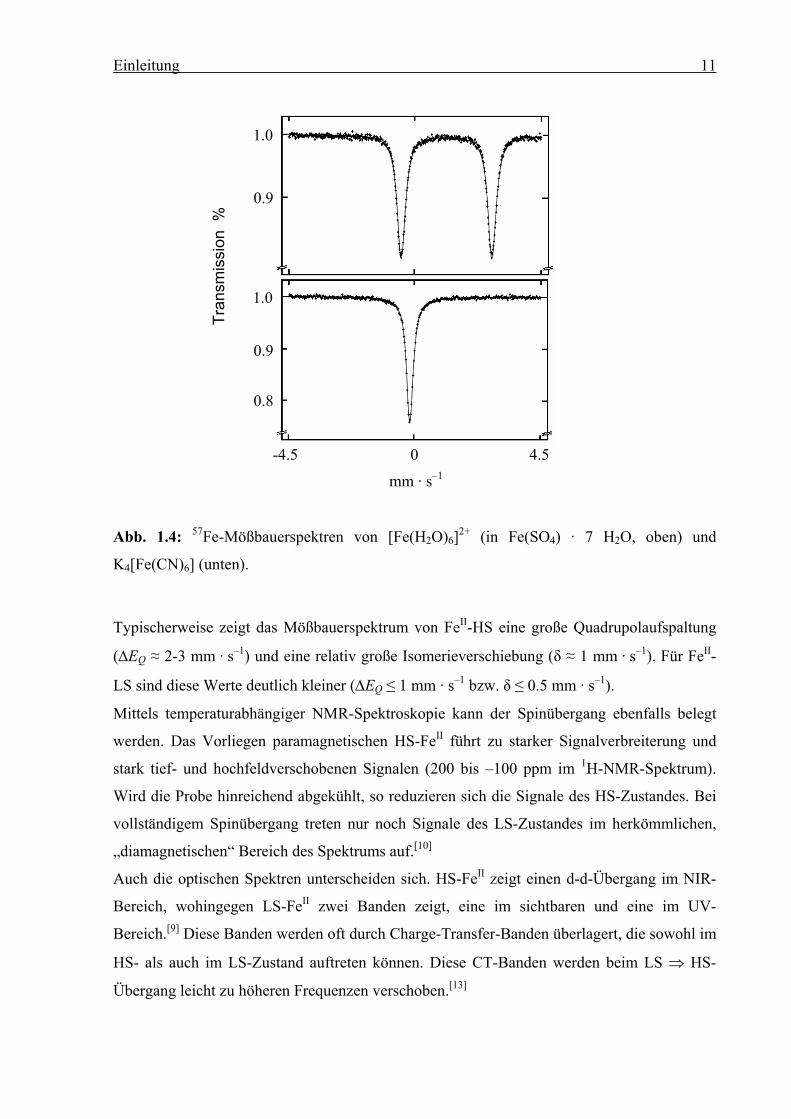
Einleitung 11
Abb. 1.4: 57Fe-Mößbauerspektren von [Fe(H2O)6]2+ (in Fe(SO4) · 7 H2O, oben) und
K4[Fe(CN)6] (unten).
Typischerweise zeigt das Mößbauerspektrum von FeII-HS eine große Quadrupolaufspaltung
(∆EQ ≈ 2-3 mm · s–1) und eine relativ große Isomerieverschiebung (δ ≈ 1 mm · s–1). Für FeII-
LS sind diese Werte deutlich kleiner (∆EQ ≤ 1 mm · s–1 bzw. δ ≤ 0.5 mm · s–1).
Mittels temperaturabhängiger NMR-Spektroskopie kann der Spinübergang ebenfalls belegt
werden. Das Vorliegen paramagnetischen HS-FeII führt zu starker Signalverbreiterung und
stark tief- und hochfeldverschobenen Signalen (200 bis –100 ppm im 1H-NMR-Spektrum).
Wird die Probe hinreichend abgekühlt, so reduzieren sich die Signale des HS-Zustandes. Bei
vollständigem Spinübergang treten nur noch Signale des LS-Zustandes im herkömmlichen,
„diamagnetischen“ Bereich des Spektrums auf.[10]
Auch die optischen Spektren unterscheiden sich. HS-FeII zeigt einen d-d-Übergang im NIR-
Bereich, wohingegen LS-FeII zwei Banden zeigt, eine im sichtbaren und eine im UV-
Bereich.[9] Diese Banden werden oft durch Charge-Transfer-Banden überlagert, die sowohl im
HS- als auch im LS-Zustand auftreten können. Diese CT-Banden werden beim LS ⇒ HS-
Übergang leicht zu höheren Frequenzen verschoben.[13]
Tran
smis
sion
%
-4.5 4.5 0
1.0
0.9
0.8
1.0
0.9
mm · s–1

Einleitung 12
Der Spinübergang ist somit häufig von einem sehr deutlichen Farbwechsel begleitet, was
zumeist der erste Hinweis auf mögliches SCO-Verhalten eines Systems ist.
1.2 Biologische Aspekte
Spin Crossover spielt eine Schlüsselrolle für die Funktion einer Reihe biologischer Systeme,
vor allem in Häm-Proteinen.[14] Er ist wesentlich für die Funktion von Enzymen wie
Peroxidasen, Cytochromen, Cytochrom P-450 und Metalloproteinen wie Hämoglobin und
Myoglobin. Desoxy-Hämoglobin hat Eisen(II)-Zentren im HS-Zustand, die nach
Koordination von Disauerstoff (Bildung von Oxy-Hämoglobin) in den LS-Zustand
übergehen. Das Eisen-Ion wird „kleiner“, was weitreichende Konsequenzen für die
Proteinstruktur des Hämoglobin-Tetrameren hat. Diese Neuordnung der Proteinstruktur
erleichtert die Aufnahme weiterer Sauerstoffmoleküle („Kooperativität der O2-Bindung“).[15]
Trotz bereits langjähriger Arbeiten sind sowohl die Synthese funktioneller Modellkomplexe,
die zur Aufklärung der Mechanismen von Sauerstoffspeicherung bzw. -aktivierung beitragen,
als auch die Charakterisierung der biologischen Systeme selbst, weiterhin Gegenstand
aktueller Forschung.[16]
Abb. 1.5: Desoxy- (links) und Oxyform (rechts) von Hämoglobin.[17]

Einleitung 13
1.3 Stand der Forschung
Bis heute sind mehrere hundert FeII-SCO-Systeme bekannt, mit einfachen einzähnigen
Liganden wie Isoxazolen oder Tetrazolen über polydentate Liganden bis hin zu mehrkernigen
Systemen. Auf eine detaillierte Auflistung wird daher verzichtet. Bis heute ist es praktisch
unmöglich, einen Merkmalkatalog für die Koordinationssphäre des Eisenzentrums anzugeben,
dessen Realisierung SCO-Verhalten des Komplexes sicher zur Folge hat. Die SCO-
Temperatur ist sowohl in Lösung als auch im Festkörper von zahlreichen Faktoren
abhängig.[18] Eine höhere π-Rückbindung der N-Donor-Liganden vergrößert die
Ligandenfeldaufspaltung. So liegt etwa [Fe(bipy)3]2+ ausschließlich im LS-Zustand vor.
Ersetzt man aber einen der beiden Pyridinringe des 2,2’-Bipyridins durch eine
Aminomethyleinheit (⇒ 2-Picolylamin) oder durch einen Imidazolring (⇒ 2-(1H-Imidazol-2-
yl)pyridin), so zeigen die resultierenden Komplexe SCO-Verhalten.[19] Die Einführung
sterisch anspruchsvoller Substituenten verhindert eine einfache Annährung der Donoratome
an das FeII-Zentrum, wodurch längere Fe–N-Bindungsabstände favorisiert werden. Damit
wird die Besetzung der antibindenden eg*-Orbitale erleichtert. [Fe(phen)3]2+ ist ein LS-
Komplex und zeigt keinen Spinübergang im Gegensatz zu dem sterisch anspruchsvollerem
[Fe(mephen)3]2+ (mephen = 2-Methyl-1,10-phenantrolin).[20] Auch die Größe von
Chelatringen und Verzerrungen der Oktaedersymmetrie spielen eine Rolle.[18] Im kristallinen
Festkörper kommt Kristallpackungseffekten und den Gegenionen eine wichtige Rolle zu.
Selbst die Art eventuell eingeschlossener Solvatmoleküle kann einen Einfluss auf die
Temperatur des Spinüberganges haben, ja diesen sogar völlig verhindern.[5][21]
Die Untersuchung von SCO-Verbindungen beschränkte sich bislang fast ausschließlich auf
die Lösung und den kristallinen Festkörper. In wenigen Fällen wurde die Übertragung von
SCO-Verhalten auf dünne Schichten mittels Langmuir-Blodgett-Techniken oder Einbettung
von SCO-Komplexen in Polymer-Matrices versucht. Hintergrund dieser Versuche ist das
gestiegene Interesse an der technischen Anwendung von SCO-Verbindungen.[22] Bisher haben
diese Versuche allerdings nicht zu schlüssigen Resultaten geführt.[23]
Das dreidimensionale Koordinationspolymer {FeII(Pyrazin)[(M(CN)4]} (M = NiII, PdII, PtII)
zeigt im Festkörper SCO-Verhalten. Dieses Polymer wurde auf einer zuvor mit 4-
Mercaptopyridin funktionalisierten Goldoberfläche schrittweise als dünner Film aufgebaut.[24]
Die Abscheidung von Thiolen aus Lösung auf Goldoberflächen ist seit ca. 25 Jahren
Gegenstand der Forschung.[25]

Einleitung 14
Die Abscheidung einer molekularen SCO-Verbindung in einer Monolage (self assembled
monolayer = SAM) wurde bisher nicht durchgeführt. Solche Ensembles markieren die
Scheidelinie zwischen Einzelmolekül- und Volumenverhalten und sind für das Verständnis
kooperativer SCO-Prozesse von fundamentaler Bedeutung.
1.4 Literatur
[1] a) L. Cambi, A. Cagnasso, Atti. Accad. Naz. Lincei, Rend., CI. Sci. Fis. Mat. Nat.
1931, 13, 809. b) L. Cambi, L. Szego, Ber. Dtsch. Chem. Ges. B 1931, 64, 2591.
[2] W. A. Baker, H. M. Bobonich, Inorg. Chem. 1964, 3, 1184.
[3] A. Hauser, Topics Curr. Chem. 2004, 233, 49.
[4] A. Bousseksou, G. Molnar, G. Matouzenko, Eur. J. Inorg. Chem. 2004, 4353-4369.
[5] P. Gütlich, H. A. Goodwin, Topics in Current Chemistry, Spin-Crossover in
Transition Metal Compounds I.-III. 2004, Vol. 233-235.
[6] a) P. Gütlich, A. Hauser, H. Spiering, Angew. Chem. Int. Ed. Engl. 1994, 33, 2024. b)
H. A. Goodwin, Coord. Chem. Rev. 1976, 18, 83. c) H. Toftlund, Coord. Chem. Rev.
1989, 94, 67.
[7] A. F. Holleman, E. Wiberg, Lehrbuch der Anorganischen Chemie, 101. Auflage,
Walter de Gruyter, Berlin, New York, 1995, 1839.
[8] K. S. Murray, C. J. Kepert, Topics Curr. Chem. 2004, 233, 195.
[9] P. Gütlich, Y. Garcia, H. A. Goodwin, Chem. Soc. Rev. 2000, 29, 419–427.
[10] a) A. Hauser, J. Chem. Phys. 1991, 94, 2741-2748. b) S. Decurtius, P. Gütlich, K. M.
Hasselbach, A. Haus, H. Spiering, Inorg. Chem. 1985, 24, 2174-2178.
[11] V. Ksenofontov, A. B. Gaspar, V. Niel, S. Reiman, J. A. Real, P. Gütlich, Chem. Eur.
J. 2004, 10, 1291-1298.
[12] P. Gütlich, Mößbauer Spectroscopy: Principles and Applications, Vorlesung an der
Universität Mainz, September 2006.
http://ak-guetlich.chemie.uni-mainz.de/Moessbauer_Lectures_web.pdf
[13] A. Hauser, J. Adler, P. Gütlich, Chem. Phys. Lett. 1988, 152, 468-472.
[14] W. A. Scheidt, C. A. Reed, Chem. Rev. 1981, 81, 543-555.
[15] M. F. Perutz, G. Fermi, and B. Luisi, Acc. Chem. Res. 1987, 20, 309-321.
[16] a) S. V. Kryatov, E. V. Rybak-Akimova, S. Schindler, Chem. Rev. 2005, 105, 2175-
2226. b) M. Costas, M. P. Mehn, M. P. Jensen, L. Que, Jr., Chem. Rev. 2004, 104,

Einleitung 15
939-986. c) C. E. MacBeth, R. Gupta, K. R. Michell-Koch, V. G. Young, G. H.
Lushington, W. H. Thompson, M. P. Hendrich, A. S. Borovik, J. Am. Chem. Soc.
2004, 126, 2556-2567. d) I. G. Denisov, T. M. Makris, S. G. Sligar, I. Schlichting,
Chem. Rev. 2005, 105, 2253-2277.
[17] Bildquelle: http://de.wikipedia.org/wiki/H %C3 %A4moglobin, September 2006.
[18] H. Toftlund, Coord. Chem. Rev. 1989, 94, 67-108.
[19] a) H. L. Chum, J. A. Vanin, M. I. D. Holanda, Inorg. Chem. 1982, 21, 1146-1152. b)
R. J. Dosser, W. J. Eilbeck, A. E. Underhill, P. R. Edwards and C. E. Johnson, J.
Chem. Soc. A, 1969, 810.
[20] H. A. Goodwin, E. S. Kucharski and A. H. White, Aust. J. Chem. 1983, 36, 1115.
[21] M. Hostettler, K. W. Törnroos, D. Chernyshov, B. Vangdal, H.-B. Bürgi, Angew.
Chem. Int. Ed. Engl. 2004, 43, 4589-4594.
[22] J. A. Real, A. B. Gaspar, M. C. Munoz, Dalton Trans. 2005, 2062–2079.
[23] M.-L. Boillot, J. Zarembowitch, A. Sour, Topics Curr. Chem. 2004, 234, 270.
[24] S. Cobo, G. Molnár, J. A. Real, A. Bousseksou, Angew. Chem. Int. Ed. 2006, 45,
5786–5789.
[25] a) J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo, G. M. Whitesides, Chem. Rev.
2005, 105, 1103-1169 und enthaltene Referenzen. b) J. K. Whitesell, H. K. Chang,
Science, 1993, 261, 73-76.

Motivation 16
2 Motivation
Aliphatische, vor allem primäre Amine als Donoratome in offenkettigen Liganden spielen in
der Koordinationschemie von FeII und FeIII kaum eine Rolle.[1] Offenkettige oder
heterocyclische Imine sind in der Lage, durch π-Rückbindung Elektronendichte vom Metall
aufzunehmen, während primäre Amine reine σ-Donoren sind. Einige dieser Komplexe sind
sehr empfindlich gegenüber metall- und/oder ligandzentrierter Oxidation.[2]
Der von Grohmann konzipierte tetrapodal-fünfzähnige pyN4-Ligand hingegen bildet mit FeII
stabile einkernige Komplexe.[3] Dabei bildet der Chelatligand in diesen oktaedrischen
Komplexen eine quadratisch-pyramidale Koordinationskappe, die sechste Koordinationsstelle
steht für die Bindung eines einzähnigen Liganden (L) zur Verfügung. Mit molekularem
Sauerstoff reagiert {(1)FeII} zu dem zweikernigen oxo-verbrückten FeIII-Komplex
[(1)Fe–O–Fe(1)]4+.[4] Eine Zersetzung des Liganden infolge Oxidation wird nicht beobachtet.
N
NH2
NH2NH2
NH2
1
N
H2NH2N FeII
NH2
NH2
L
Abb. 2.1: links: pyN4-Ligand (1); rechts: Struktur der [(1)FeIIL]-Komplexe.
Ziel der vorliegenden Arbeit war die Synthese und Charakterisierung von [(1)FeIIL]-SCO-
Komplexen. Das SCO-Verhalten dieser Verbindungen sollte sowohl in Lösung als auch im
kristallinen Festkörper untersucht werden. Zusätzliches Interesse galt der Aufbringung dieser
SCO-Moleküle auf Festkörperoberflächen als Teilprojekt im Rahmen des Sonderforschungs-
bereiches 658: „Elementarprozesse in molekularen Schaltern an Oberflächen“ der DFG.
Hierzu wurden die benötigten Vorarbeiten geleistet.
Die Synthese eines Liganden, der ein basales Donoratom weniger aufweist als „pyN4“ wurde
durch Arbeiten an einem zu 1 analogen tetrapodalen Phosphanliganden ermöglicht, der
Dimethylphosphansubstituenten statt primärer Aminogruppen trägt.[5]

Motivation 17
Die Synthese des neuen Liganden 2-(1,3-diamino-2-methylpropan-2-yl)-6-(1-amino-2-
methylpropan-2-yl)pyridin (2), im Folgenden pyN3 genannt, wurde ausgearbeitet. Von
besonderem Interesse war das Koordinationsverhalten gegenüber FeII, aber auch CuII-, NiII-
und ZnII-Salze wurden untersucht.
N
NH2
NH2NH2
2
Abb. 2.2: Struktur des von 1 abgeleiteten pyN3-Liganden (2).
2.1 Literatur
[1] M. V. Twigg, J. Burgess, Iron, in: Comprehensive Coordination Chemistry II, J. A.
McCleverty, T. J. Meyer (eds.) Elsevier Pergamon, Amsterdam 2003, vol. 5, p. 432.
[2] a) N. Sadasivan, J. F. Endicott, J. Am. Chem. Soc. 1966, 88, 5468-5472. b) L. R.
Melby, Inorg. Chem. 1970, 9, 2186-2188.
[3] J. Pitarch López, F. W. Heinemann, R. Prakash, B. A. Hess, O. Horner, C. Jeandey, J.-
L. Oddou, J.-M. Latour, A. Grohmann, Chem. Eur. J. 2002, 8, 5709-5722.
[4] J. Pitarch López, F. W. Heinemann, A. Grohmann, O. Horner, J.-M. Latour, G.
Ramachandraiah, Inorg. Chem. Comm. 2004, 7, 773-776.
[5] Stephan Kohl, Dissertation, Technische Universität Berlin, 2006.

Eisenkomplexe des pyN4-Liganden 18
3 Eisenkomplexe des pyN4-Liganden
3.1 Einleitung
Der Spinzustand in Komplexen des Typs [(1)FeIIL] ist von der Art des sechsten Liganden (L)
abhängig. Während [(1)FeIIBr]Br bei Raumtemperatur im high-spin-Zustand vorliegt (4
ungepaarte Elektronen), liegt der Carbonylkomplex [(1)FeII(CO)]Br2 im low-spin-Zustand vor
und ist diamagnetisch.[1] Die beiden einzähnigen Liganden markieren die jeweiligen
Endpunkte der spektrochemischen Reihe.[2] Bei Verwendung stickstoffhaltiger Liganden (z.
B. heterocyclischer Iminliganden), die sich in der Mitte der spektrochemischen Reihe finden,
werden SCO-Verbindungen erwartet. Von den mehr als zweihundert heute bekannten FeII-
SCO-Systemen weisen die meisten eine N6-Koordinationsumgebung um das FeII-Zentrum
auf.[3] Keine dieser Verbindungen enthält vier primäre Aminofunktionen. Diese ermöglichen
eine einfache und vielseitige Derivatisierung, um die Donoreigenschaften des Liganden und
damit die Ligandenfeldaufspaltung zu modifizieren.[4] Die Abscheidung auf einer
Festkörperoberfläche als SAM (self assembled monolayer) ist das langfristige Ziel dieser
Arbeit. Die C2v-Symmetrie des Liganden verhindert die Bildung verschiedener
Koordinationsisomere, wie sie vor allem bei zweizähnigen Liganden auftritt.[5] Die primären
Aminogruppen können darüber hinaus durch Wasserstoffbrückenbindungen ordnend wirken.
Diese Derivatisierungsmöglichkeiten und strukturbildenden Eigenschaften fehlen dem von
der Gruppe um Stack verwendeten PY5-Liganden. Er ist ein nach dem Modell von 1
geschaffenes Pentaimin und ist der einzige C2v-symmetrische fünfzähnige Ligand, von dem
ebenfalls FeII-SCO-Komplexe bekannt sind.[6]
N
NH2
NH2NH2
NH2
1
NOMeMeO
N N NN
Abb. 3.1: Das Tetraaminimin (1) und der Pentaimin-Ligand PY5.

Eisenkomplexe des pyN4-Liganden 19
Bei der Bildung von SAMs (= self assembled monolayers) werden Moleküle mittels
oberflächenspezifischer Ankergruppen chemisch auf dem Substrat gebunden.[7] Bei
organischen Molekülen wird durch van-der-Waals- und π-π-Stapelwechselwirkungen die
gleichmäßige Anordnung der Moleküle erreicht. Der Abstand paralleler Molekülketten
beträgt hier also ca. 2 - 4 Ǻ. Darüber hinaus stehen die Moleküle meist nicht senkrecht auf der
Oberfläche, sondern sind leicht abgewinkelt. Abb. 3.2 zeigt den schematischen Aufbau einer
idealen SAM.
AG
FG
AG
FG
AG
FG
AG
FG
AG
FG
AG
FG
AG
FG
AG
FG
Oberfläche
Abb. 3.2: Schema einer idealen Monolage auf einer Oberfläche.
Die [(1)FeII]-Komplexfragmente haben jedoch eine Ausdehnung von ca. 9 × 5 Ǻ (B × T).
Wenn die funktionelle Gruppe ein Donoratom ist, kann das [(1)FeII]-Fragment aus sterischen
Gründen nicht koordinieren. Eine Möglichkeit, dies zu umgehen, wäre die Abscheidung des
gesamten Moleküls in einem Schritt.
N
H2NH2N FeII
NH2
NH2
L
Oberfläche
AG
N
H2NH2N FeII
NH2
NH2
L
AG
Abb. 3.3: Geplante Abscheidung der [(1)FeII(L)]-Komplexe.(L = Ligand koordiniert an FeII)
FG: terminale funktionelle Gruppe
AG: Ankergruppe
Molekülkette (aliphatisch/aromatisch)

Eisenkomplexe des pyN4-Liganden 20
Alle Synthesen gehen nicht vom freien pyN4-Liganden 1, sondern von seinem Addukt mit
Bromwasserstoffsäure: 1 · 4 HBr aus. Dieses ist als Pulver besser handhabbar und weit
weniger hygroskopisch als der reine Ligand, der als Öl anfällt. Um Reaktionen
durchzuführen, muss zuvor mit vier Äquivalenten Base (z. B. LiOMe) umgesetzt werden.
Da die Mehrzahl der bekannten FeII-SCO-Verbindungen eine N6-Koordinationsumgebung um
das Metallzentrum enthalten, lag es nahe, N-Donorliganden mit dem [(1)FeII]-Fragment
umzusetzen. Alle Versuche, aliphatische Amine—reine σ-Donoren—an das ohnehin schon
sehr elektronenreiche [(1)FeII]-Fragment (vgl. Kap. 2; S. 16) zu koordinieren, führten bislang
zu keinem verwertbaren Ergebnis. Deshalb wurden die folgenden Versuche mit aromatischen
Iminen durchgeführt. Durch π-Rückbindung können diese Elektronendichte vom Metall
aufnehmen und so zu einer Stabilisierung des zu erwarteten Komplexes beitragen.
3.2 Reaktionen in Anwesenheit von H2O
Erste Versuche wurden von J. Pitarch López mit 1-Methyl-1H-imidazol gemäß folgender
Gleichung durchgeführt.[8]
1 · 4 HBr + 4 LiOMe + Fe(ClO4)2 · x H2O +
N
N MeOH
i. D. Et2O
Schema 3.1: Versuch zur Synthese des Komplexes [(1)FeII(C4H6N2)]X2.
Nach vier Tagen wurden orange Kristalle erhalten, die röntgenstrukturanalytisch als der
zweikernige Komplex [(1)2(FeII)2(µ2-OH)]Br(ClO4)2 identifiziert wurden.

Eisenkomplexe des pyN4-Liganden 21
3
Abb. 3.4: Molekülstruktur des Trikations in 3 (50 % Wahrscheinlichkeitsellipsoide; ohne H-
Atome (außer verbrückendes OH), Anionen und Solvensmoleküle).
Die elementaranalytische Untersuchung bestätigt die Zusammensetzung [(1)2(FeII)2(µ2-
OH)]Br(ClO4)2. Da Fe(ClO4)2 · x H2O kein definiertes Hydrat ist, wurde in den ersten
Reaktionen die molare Masse wasserfreien Eisen(II)-Perchlorates (M = 254.75 gmol–1)
angesetzt. Es handelt sich jedoch mindestens um ein Heptahydrat (M = 380.85 gmol–1).[9] Das
Verhältnis 1 : FeII betrug somit 1.5 : 1. Der Überschuss an basischem 1 und/oder 1-Methyl-
1H-imidazol deprotoniert H2O. Die Bildung von 3 wird so erst ermöglicht. Alle Versuche, 3
aus einer stöchiometrischen Reaktion nur durch Zugabe von 1-Methyl-1H-imidazol bzw.
LiOMe zu erhalten, schlugen fehl.
Verbindung 3 kristallisiert in der tetragonalen Raumgruppe P 4 21m. Der untersuchte Kristall
erwies sich als Inversionszwilling mit einem Verhältnis der Zwillingsindividuen von etwa 1:1.
Das zweikernige Komplexkation befindet sich auf der vierzähligen Lage 2c der Raumgruppe
P 4 21m und hat dementsprechend D2d-Molekülsymmetrie. Die Eisenzentren sind verzerrt
oktaedrisch koordiniert Die Fe–N-Abstände liegen im Bereich zwischen 2.18 und 2.23 Ǻ,
typisch für HS-FeII-Zentren.[10] Der Fe–O–Fe-Winkel beträgt 146.7(2)°, in Übereinstimmung
mit einer Hydroxylgruppe. Das IR-Spektrum (KBr-Pressling) zeigt eine deutliche Bande bei
3459 cm–1, die der OH-Streckschwingung zugeordnet wird. Die deutliche Aufweitung über
109° bzw. 120° für eine regelmäßige tetraedrische bzw. trigonal planare Koordination ist auf
den sterischen Anspruch der zwei [(1)FeII]-Fragmente zurückzuführen. Orange Kristalle von 3

Eisenkomplexe des pyN4-Liganden 22
verfärben sich an Luft innerhalb von wenigen Stunden braun, unter Stickstoff sind sie
mehrere Wochen stabil. Soweit bekannt, ist dies das erste Beispiel für eine µ2-Hydroxobrücke
zwischen zwei FeII-Zentren, die nicht durch zusätzliche verbrückende Liganden stabilisiert ist.
Die [µ2-OH(FeII)2]-Struktureinheit kommt in einer Reihe biologischer Systeme vor (z. B.
Hämerythrin).[11]
Verbindung 3 ist in MeOH, DMSO und DMF nahezu unlöslich. Ein Austausch der Anionen
gegen NaBArF (BArF = Tetrakis[3,5-bis(trifluormethyl)phenyl]borat) in CH2Cl2, wie bei
[(1)FeIIBr]Br erfolgreich durchführbar, führte zu keinem verwertbaren Ergebnis.[12] Die
erhaltene gelbe Lösung zeigt im IR-Spektrum keine OH-Bande, der nach Abziehen des
Lösemittels erhaltene Feststoff ebenfalls nicht. Der Versuch, die Verbindung ausgehend von
Fe(BF4)2 · 6 H2O in analoger Weise zu synthetisieren, führt zu keinem einheitlichen Produkt.
Bei den o. g. Versuchen wurde auch 1 mit Fe(BF4)2 · 6 H2O in THF umgesetzt. Hierzu wird 1
· 4 HBr mit 4.4 eq Natriummethanolat in Methanol umgesetzt und anschließend das
Lösemittel entfernt. Der ölige Feststoff, bestehend aus NaBr und 1, wird anschließend mit
THF ausgerührt und das feste Natriumbromid abfiltriert. Bei der Zugabe zu einer Lösung von
Fe(BF4)2 · 6 H2O fällt ein zitronengelber Feststoff aus, der nach Filtration und Reinigung mit
Diethylether im Stickstoffstrom getrocknet wird. Die verbrennungsanalytische Untersuchung
ergibt die Zusammensetzung [(1)FeII(H2O)](BF4)2, und das IR-Spektrum (KBr-Pressling)
zeigt eine Bande bei 3589 cm–1.
1 + Fe(BF4)2 · 6 H2OTHF N
H2NH2N Fe NH2
NH2
OH H
2+
(BF4–)2
4
Schema 3.2: Synthese des Aqua-Komplexes [(1)FeII(OH2)](BF4)2 (4).
Gegenüber Luft ist 4 nicht stabil, der gelbe Feststoff verfärbt sich innerhalb von einer Minute
orangebraun. Die Bande bei 3589 cm–1 im KBr-IR-Spektrum ist zu 3548 cm–1 verschoben. In
DMSO-d6 und entgastem D2O bildet sich zunächst eine gelbe Lösung, innerhalb von 15
Minuten fällt jedoch ein brauner Feststoff aus, der nicht näher charakterisiert werden konnte.

Eisenkomplexe des pyN4-Liganden 23
Unter Stickstoff nimmt 4 innerhalb von drei Wochen dieselbe rotbraune Farbe an. Der
Versuch Kristalle von 4 durch langsame Diffusion der Reaktionspartner zu erhalten, führt zu
braunem, öligem Niederschlag. Die analoge Reaktion mit Fe(ClO4)2 · x H2O führt zu einem
gelben Niederschlag. Bei dem nach 30 Minuten Reaktionszeit isolierten gelben Feststoff zeigt
das IR-Spektrum (KBr-Pressling) eine Bande bei 3549 cm–1. Dies entspricht dem nach
Luftzutritt zu 4 erhaltenen Produkt. Bei längerer Reaktionszeit (18 h) färbt sich die
Suspension schwarzbraun. Die Aufklärung der nach Luftzutritt zu 4 erhaltenen Spezies gelang
nicht. Wahrscheinlich ist eine {(1)FeIII(OH)}2+- oder {(1)FeIII(H2O)}3+-Spezies entstanden.
Diese könnten in Lösung zum zweikernigen [(1)2(FeIII)2(µ-O)]4+-Kation reagieren.[12]
Offenbar ermöglicht nicht primär die Stabilität, sondern die schlechte Löslichkeit von 4
dessen Isolierung. Aufgrund der gemachten Erfahrungen wurden bei den folgenden
Reaktionen ausschließlich wasserfreie FeII-Vorläuferverbindungen verwendet.
3.3 1-Methyl-1H-imidazol
Durch Umsetzung äquimolarer Mengen von 1 mit Fe(CH3CN)2(OTf)2 in Methanol entsteht
eine hellrote Lösung, die sich nach Zugabe von 1-Methyl-1H-imidazol dunkelrot verfärbt.
Durch Eindiffusion von Diethylether bei Raumtemperatur entstehen innerhalb von vier Tagen
dunkelrote Kristalle, die abfiltriert und getrocknet werden. Die Elementaranalyse ergibt die
Zusammensetzung [(1)FeII(C4H6N2)](OTf)2 (5). Die erhaltenen Kristalle zerfallen nicht beim
Trocknen und sind unter N2-Atmosphäre mehrere Monate stabil. In methanolischer Lösung ist
5 unter Inertgasbedingungen mehrere Tage stabil, an Luft tritt innerhalb von einer Minute
Zersetzung ein. Die rote Lösung färbt sich gelb.
1 + Fe(MeCN)2(OTf)2 +MeOH
N
H2NH2N Fe NH2
NH2
N
2+
(OTf–)2
N
N
N
i.D. Et2O
5
Schema 3.3: Synthese des Komplexes [(1)FeII(1-Methyl-1H-imidazol)](OTf)2 (5).
Eisenkomplexe des pyN4-Liganden 24
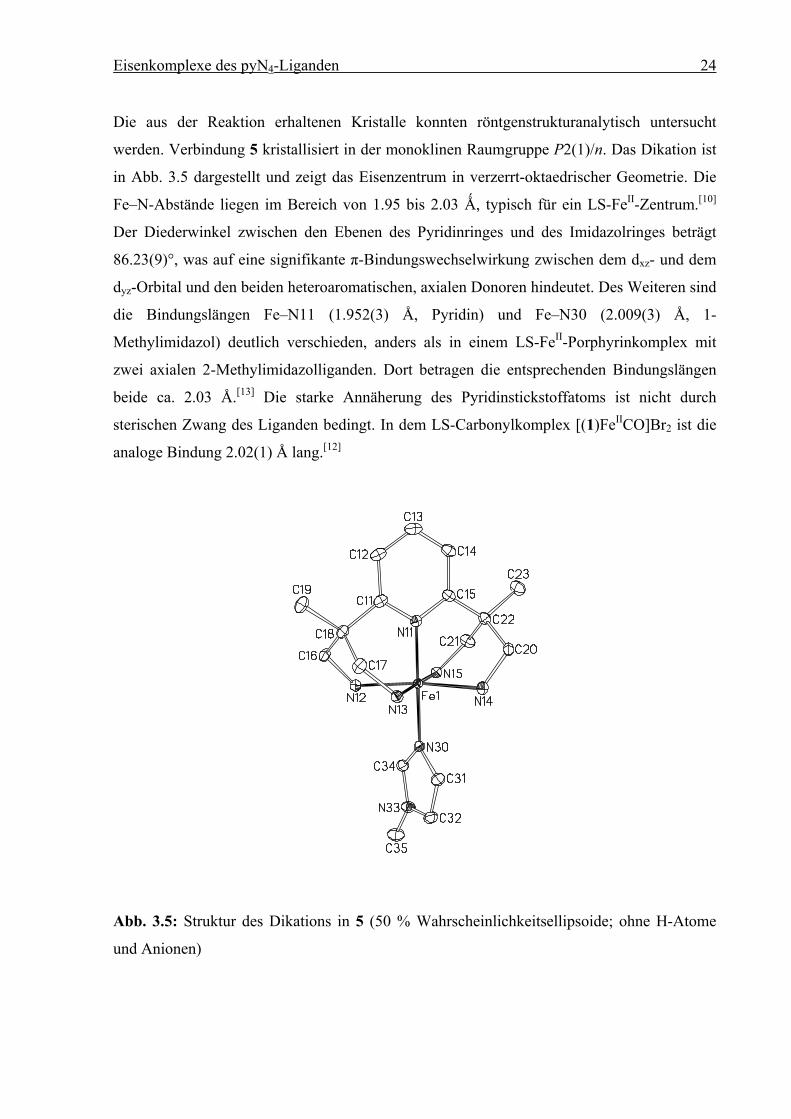
Die aus der Reaktion erhaltenen Kristalle konnten röntgenstrukturanalytisch untersucht
werden. Verbindung 5 kristallisiert in der monoklinen Raumgruppe P2(1)/n. Das Dikation ist
in Abb. 3.5 dargestellt und zeigt das Eisenzentrum in verzerrt-oktaedrischer Geometrie. Die
Fe–N-Abstände liegen im Bereich von 1.95 bis 2.03 Ǻ, typisch für ein LS-FeII-Zentrum.[10]
Der Diederwinkel zwischen den Ebenen des Pyridinringes und des Imidazolringes beträgt
86.23(9)°, was auf eine signifikante π-Bindungswechselwirkung zwischen dem dxz- und dem
dyz-Orbital und den beiden heteroaromatischen, axialen Donoren hindeutet. Des Weiteren sind
die Bindungslängen Fe–N11 (1.952(3) Å, Pyridin) und Fe–N30 (2.009(3) Å, 1-
Methylimidazol) deutlich verschieden, anders als in einem LS-FeII-Porphyrinkomplex mit
zwei axialen 2-Methylimidazolliganden. Dort betragen die entsprechenden Bindungslängen
beide ca. 2.03 Å.[13] Die starke Annäherung des Pyridinstickstoffatoms ist nicht durch
sterischen Zwang des Liganden bedingt. In dem LS-Carbonylkomplex [(1)FeIICO]Br2 ist die
analoge Bindung 2.02(1) Å lang.[12]
Abb. 3.5: Struktur des Dikations in 5 (50 % Wahrscheinlichkeitsellipsoide; ohne H-Atome
und Anionen)
Eisenkomplexe des pyN4-Liganden 25
Tabelle 3.1: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 5. Die Standardabweichun-
gen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N11 1.952(2) N11–Fe1–N12 92.60(8)
Fe1–N12 2.026(2) N11–Fe1–N13 90.24(9)
Fe1–N13 2.033(2) N11–Fe1–N14 93.41(9)
Fe1–N14 2.030(2) N11–Fe1–N15 90.18(8)
Fe1–N15 2.028(2) N11–Fe1–N30 178.97(9)
Fe1–N30 2.009(2)
3.3.1 NMR-Spektren von [FeII(pyN4)(C4H6N2)](OTf)2
Das 1H-NMR-Spektrum bei Raumtemperatur zeigt paramagnetisch verbreiterte Signale in
einem Bereich von 0 ppm bis 143 ppm. Bei –80 °C hat ein vollständiger Spinübergang
stattgefunden, das 1H-NMR-Spektrum zeigt ausschließlich gut aufgelöste Signale, die einen
diamagnetischen LS-Komplex 5 nahelegen (vgl. Kap. 3.5.1; S. 39 und Kap. 3.7.1; S. 51).
Abb. 3.6: 1H-NMR-Spektren von 5 in Methanol-d4.
27 °C –80 °C
0 0 140 100 50 0 140 100 50
Eisenkomplexe des pyN4-Liganden 26
Im 13C-NMR-Spektrum sind die Signale der meta-Kohlenstoffatome des Pyridinringes um 0.5
ppm separiert, alle anderen Signale entsprechen einer C2v-symmetrischen Verbindung. Das 1H-NMR-Spektrum zeigt die aromatischen Protonen (Pyridin-AB2 und 1-Methyl-1H-
imidazol) zwischen 8.0 und 7.1 ppm und die Methylgruppe von 1-Methylimidazol als
Singulett bei 3.95 ppm. Die Signale der diasterotopen Protonen der Methylengruppen sind bei
2.75 bzw. 2.59 ppm und die der Methylgruppen von 1 als Singulett bei 1.45 ppm. Dies deutet
auf schnelle Rotation (auf der NMR-Zeitskala) des 1-Methyl-1H-imidazol-Liganden um die
Fe–N-Bindungsachse hin.
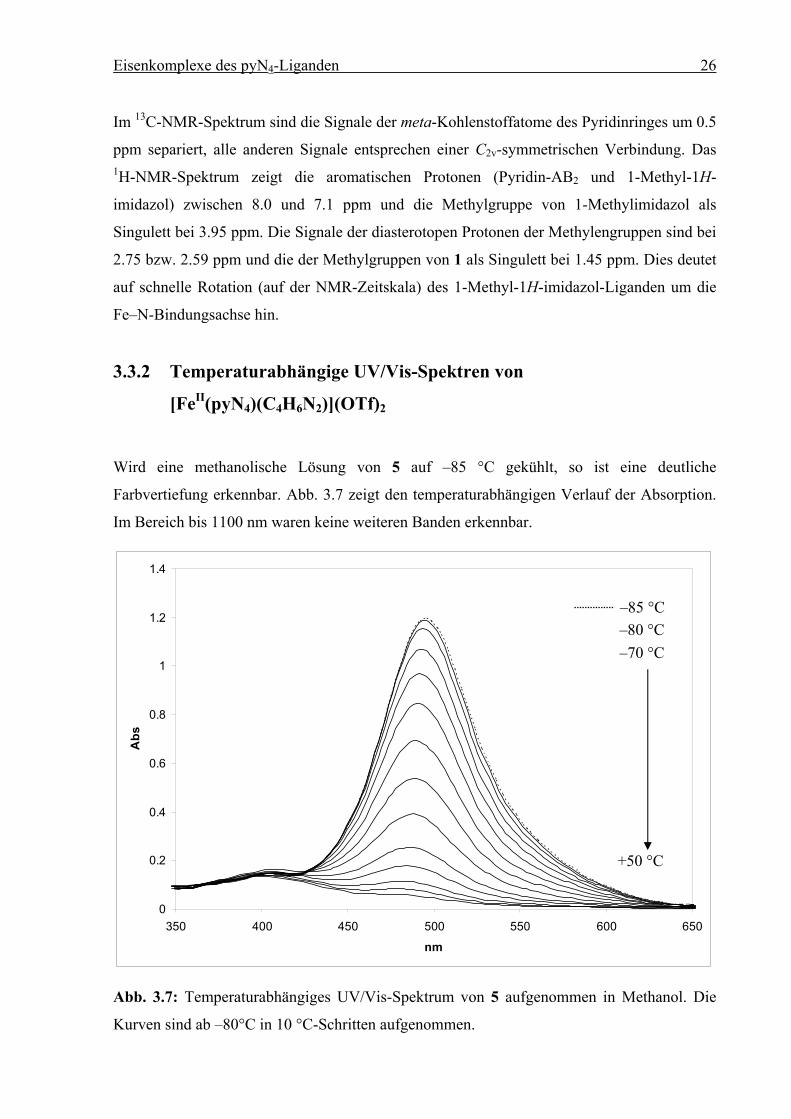
3.3.2 Temperaturabhängige UV/Vis-Spektren von
[FeII(pyN4)(C4H6N2)](OTf)2
Wird eine methanolische Lösung von 5 auf –85 °C gekühlt, so ist eine deutliche
Farbvertiefung erkennbar. Abb. 3.7 zeigt den temperaturabhängigen Verlauf der Absorption.
Im Bereich bis 1100 nm waren keine weiteren Banden erkennbar.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
350 400 450 500 550 600 650
nm
Abs
Abb. 3.7: Temperaturabhängiges UV/Vis-Spektrum von 5 aufgenommen in Methanol. Die
Kurven sind ab –80°C in 10 °C-Schritten aufgenommen.
–85 °C
+50 °C
–80 °C –70 °C
Eisenkomplexe des pyN4-Liganden 27
Das Absorptionsmaximum liegt bei 496 nm (–80°C) und verschiebt sich erwartungsgemäß
leicht zu 484 nm (10 °C).[5a][10][14] Es handelt sich um einen CT-Übergang mit ε = 5900
lmol–1cm–1. Aus dem Spektrum wird ersichtlich, dass der Spinübergang erst oberhalb von +50
°C vollständig ist (vgl. Kap. 3.5.2; S. 40 und Kap. 3.7.2; S. 51).
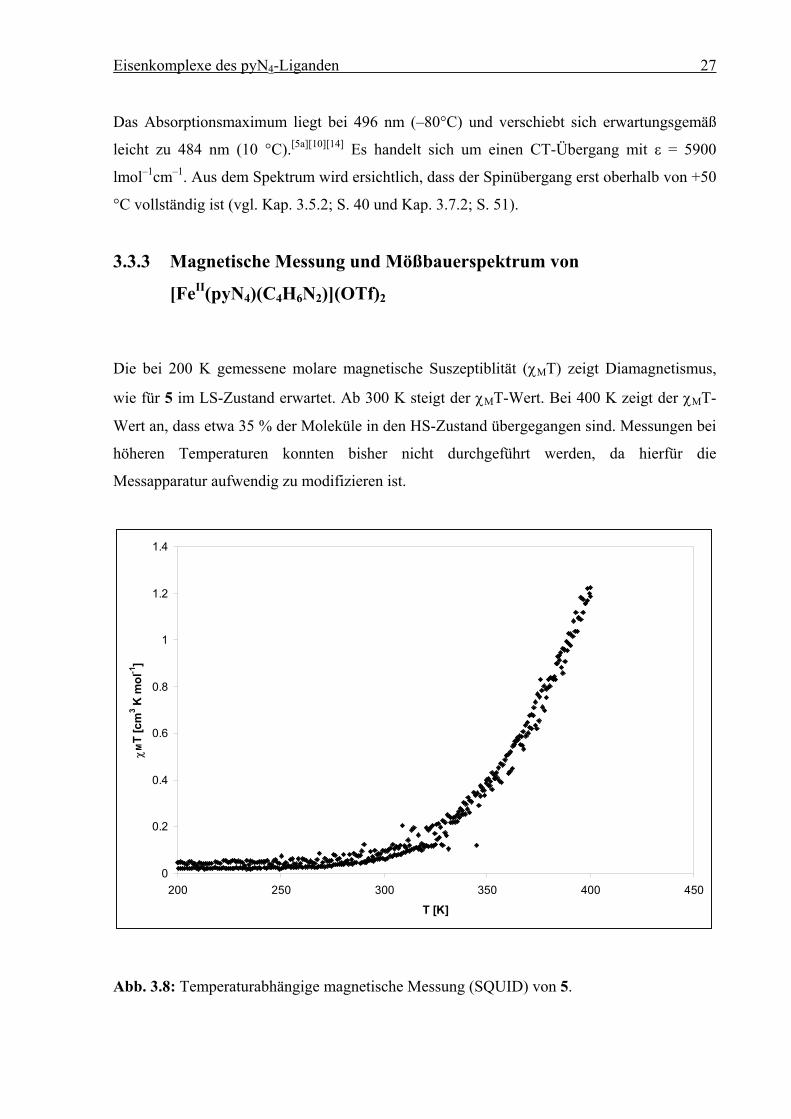
3.3.3 Magnetische Messung und Mößbauerspektrum von
[FeII(pyN4)(C4H6N2)](OTf)2
Die bei 200 K gemessene molare magnetische Suszeptiblität (χMT) zeigt Diamagnetismus,
wie für 5 im LS-Zustand erwartet. Ab 300 K steigt der χMT-Wert. Bei 400 K zeigt der χMT-
Wert an, dass etwa 35 % der Moleküle in den HS-Zustand übergegangen sind. Messungen bei
höheren Temperaturen konnten bisher nicht durchgeführt werden, da hierfür die
Messapparatur aufwendig zu modifizieren ist.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
200 250 300 350 400 450
T [K]
χM
T [c
m3 K
mol
-1]
Abb. 3.8: Temperaturabhängige magnetische Messung (SQUID) von 5.
Eisenkomplexe des pyN4-Liganden 28
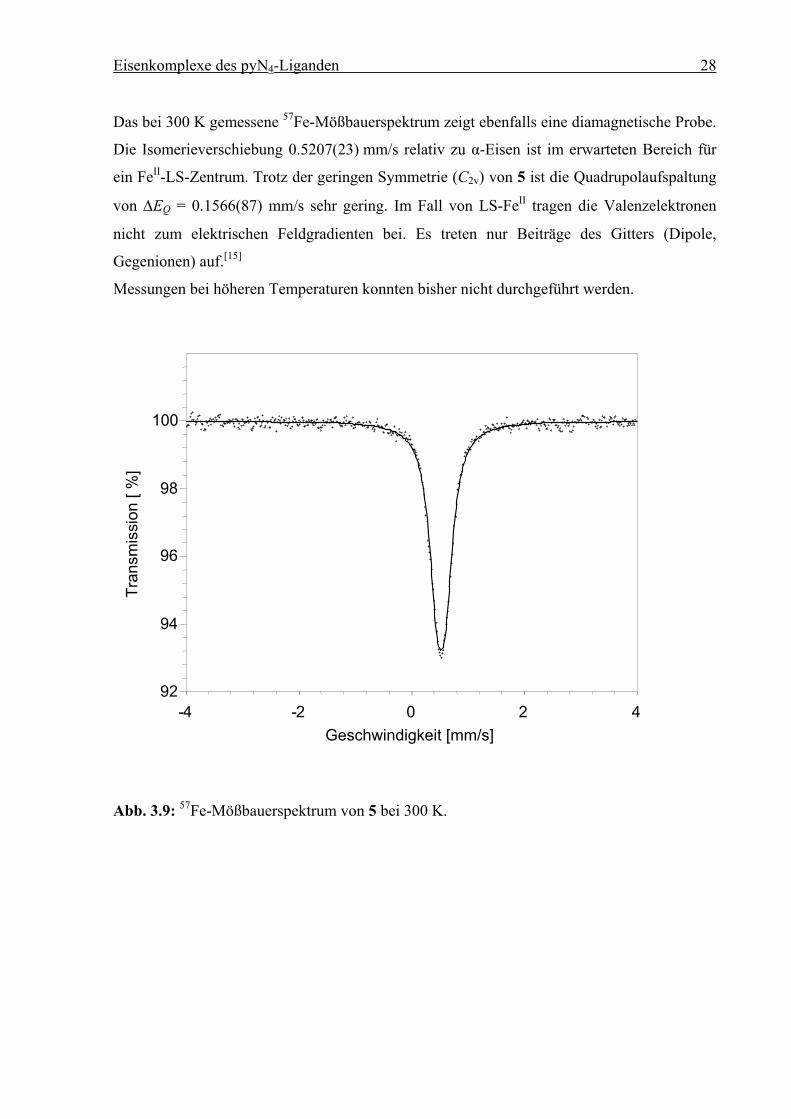
Das bei 300 K gemessene 57Fe-Mößbauerspektrum zeigt ebenfalls eine diamagnetische Probe.
Die Isomerieverschiebung 0.5207(23) mm/s relativ zu α-Eisen ist im erwarteten Bereich für
ein FeII-LS-Zentrum. Trotz der geringen Symmetrie (C2v) von 5 ist die Quadrupolaufspaltung
von ∆EQ = 0.1566(87) mm/s sehr gering. Im Fall von LS-FeII tragen die Valenzelektronen
nicht zum elektrischen Feldgradienten bei. Es treten nur Beiträge des Gitters (Dipole,
Gegenionen) auf.[15]
Messungen bei höheren Temperaturen konnten bisher nicht durchgeführt werden.
Abb. 3.9: 57Fe-Mößbauerspektrum von 5 bei 300 K.
4 20- 2 - 4
Tran
smis
sion
[ %
]
100
98
96
94
92
Geschwindigkeit [mm/s]
Eisenkomplexe des pyN4-Liganden 29
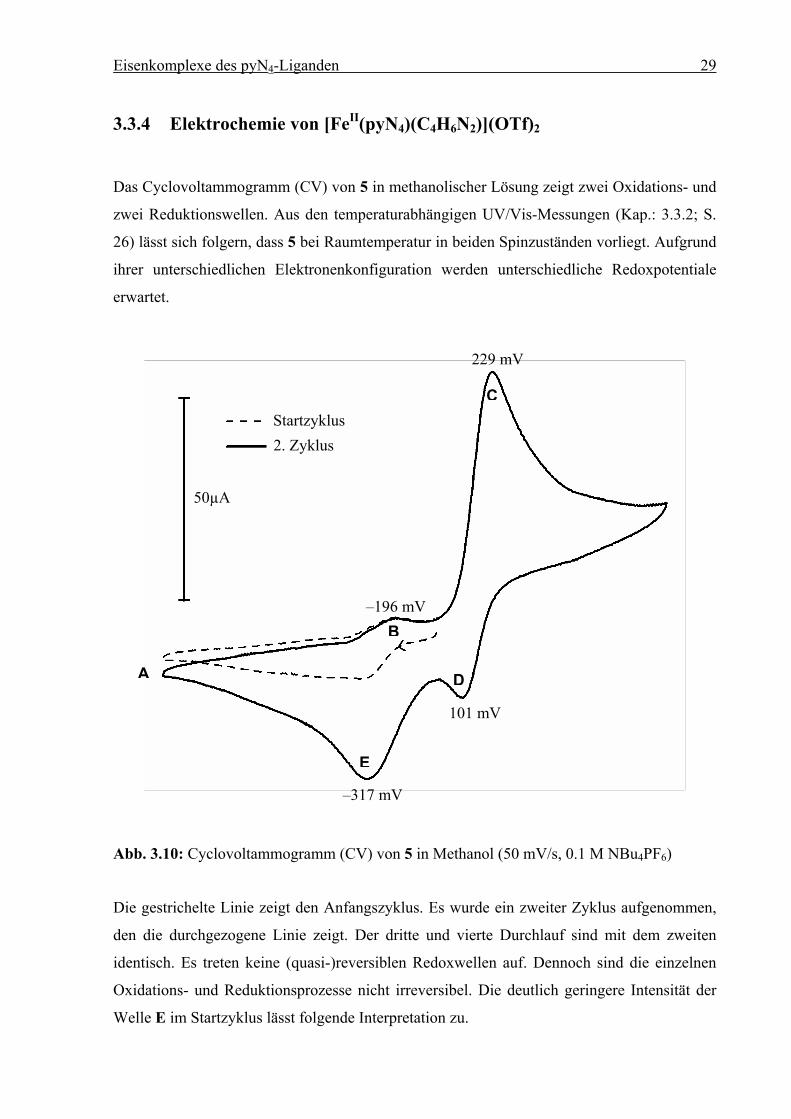
3.3.4 Elektrochemie von [FeII(pyN4)(C4H6N2)](OTf)2
Das Cyclovoltammogramm (CV) von 5 in methanolischer Lösung zeigt zwei Oxidations- und
zwei Reduktionswellen. Aus den temperaturabhängigen UV/Vis-Messungen (Kap.: 3.3.2; S.
26) lässt sich folgern, dass 5 bei Raumtemperatur in beiden Spinzuständen vorliegt. Aufgrund
ihrer unterschiedlichen Elektronenkonfiguration werden unterschiedliche Redoxpotentiale
erwartet.
Abb. 3.10: Cyclovoltammogramm (CV) von 5 in Methanol (50 mV/s, 0.1 M NBu4PF6)
Die gestrichelte Linie zeigt den Anfangszyklus. Es wurde ein zweiter Zyklus aufgenommen,
den die durchgezogene Linie zeigt. Der dritte und vierte Durchlauf sind mit dem zweiten
identisch. Es treten keine (quasi-)reversiblen Redoxwellen auf. Dennoch sind die einzelnen
Oxidations- und Reduktionsprozesse nicht irreversibel. Die deutlich geringere Intensität der
Welle E im Startzyklus lässt folgende Interpretation zu.
229 mV
101 mV
–196 mV
–317 mV
50µA
B
C
D
E
Startzyklus 2. Zyklus
A

Eisenkomplexe des pyN4-Liganden 30
Schema 3.4: Interpretation des Cyclovoltammogramms von 5.
Nach Durchlaufen der Oxidationswelle C liegt ausschließlich FeIII vor. Die
Ligandenfeldaufspaltung steigt bei Oxidation des Zentralmetalles.[16] Deshalb liegen für FeIII
mehr Teilchen im LS-Zustand vor als für FeII. Die zu C korrespondierende Reduktionswelle
D ist daher kleiner. Die zu B korrespondierende Reduktionswelle E ist folglich größer. Die
Redoxpotentiale betragen etwa 160 mV [HS-5] bzw. -257 mV [LS-5]. Da es sich nicht um
(quasi-)reversible Redoxwellen handelt, kann keine exakte Auswertung vorgenommen
werden. Der Bromokomplex [(1)FeIIBr]Br zeigt eine quasireversible Redoxwelle bei +0.17 V
vs. NHE. Das entsprechende Potential von HS-5 liegt im gleichen Bereich. Der LS-
Carbonylkomplex [(1)FeII(CO)]Br2 zeigt eine irreversible Welle bei +0.79 V vs. NHE.
Der Carbonylligand stabilisiert durch starke π-Rückbindung FeII, 1-Methylimidazol ist ein
deutlich schwächerer π-Akzeptorligand. Das Potential der Oxidation ist folglich niedriger.
Getrennte quasireversible Redoxwellen treten in dem zweikernigen SCO-Komplex
[L3FeIII2](ClO4)3 auf (L = 2,6-Di(aminomethyl)-4-tert-butyl-thiophenolat).[17] Ein Spin-
übergang findet hier auf der CV-Zeitskala nicht statt. Das Redoxpotential der LS-Verbindung
ist hier auch niedriger als das des HS-Zustandes.
Das Cyclovoltammogramm bestätigt das Vorliegen beider Spinzustände bei Raumtemperatur.
Einstellung der FeII-Spinverteilung
A nur FeII
vorwiegend im HS-Zustand
B Oxidation von
LS-FeII
C Oxidation von
HS-FeII
D Reduktion von
HS-FeIII
E Reduktion von
LS-FeIII
Neuverteilung der Spinzustände: LS-Anteil bei
FeIII höher als bei FeII

Eisenkomplexe des pyN4-Liganden 31
3.4 Pyridincarbonsäuren
Carbonsäuren und Carboxylate sind geeignete Ankergruppen zur Chemisorption auf α-Al2O3,
FexOy, Ni und Ti/TiO2. Ebenso können z. B. durch Peptidbindung oder Veresterung
Ankergruppen für beliebige Substrate eingeführt werden.[7b]
3.4.1 Pyridin-4-carbonsäure
Zunächst wurde versucht, Pyridin-4-carbonsäure (Isonicotinsäure) an das [(1)FeII]-Fragment
zu koordinieren. Durch Umsetzung äquimolarer Mengen von 1 mit FeCl2 in Methanol
entsteht eine orange Lösung, die sich nach Zugabe von Pyridin-4-carbonsäure dunkelrot
verfärbt. Durch Eindiffusion von Diethylether bei Raumtemperatur entstehen innerhalb von
vier Tagen hellrote Kristalle, die elementaranalytisch und röntgendiffraktometrisch
charakterisiert werden konnten.
1·4 HBr + 4 LiOMe + FeCl2 +
N
H2NH2N Fe NH2
NH2
O
2+
(Br–)2
N
OHO
1. MeOH2. Et2O
- 1/2 H2
N
O
6
Schema 3.5: Synthese von [(1)FeIII(C5H4NCO2)]Br2 (6).
Das FeII-Zentrum wurde zu FeIII oxidiert. Es liegen keine Hinweise auf den zweikernigen
oxo-verbrückten FeIII-Komplex vor, der unter Luftzutritt oder bei Anwesenheit von Wasser
entsteht.[12] Als Oxidationsmittel kommen daher nur die aciden Protonen der Isonicotinsäure
in Frage. Vorangegangene Versuche haben gezeigt, dass bereits Phenole in der Lage sind, auf
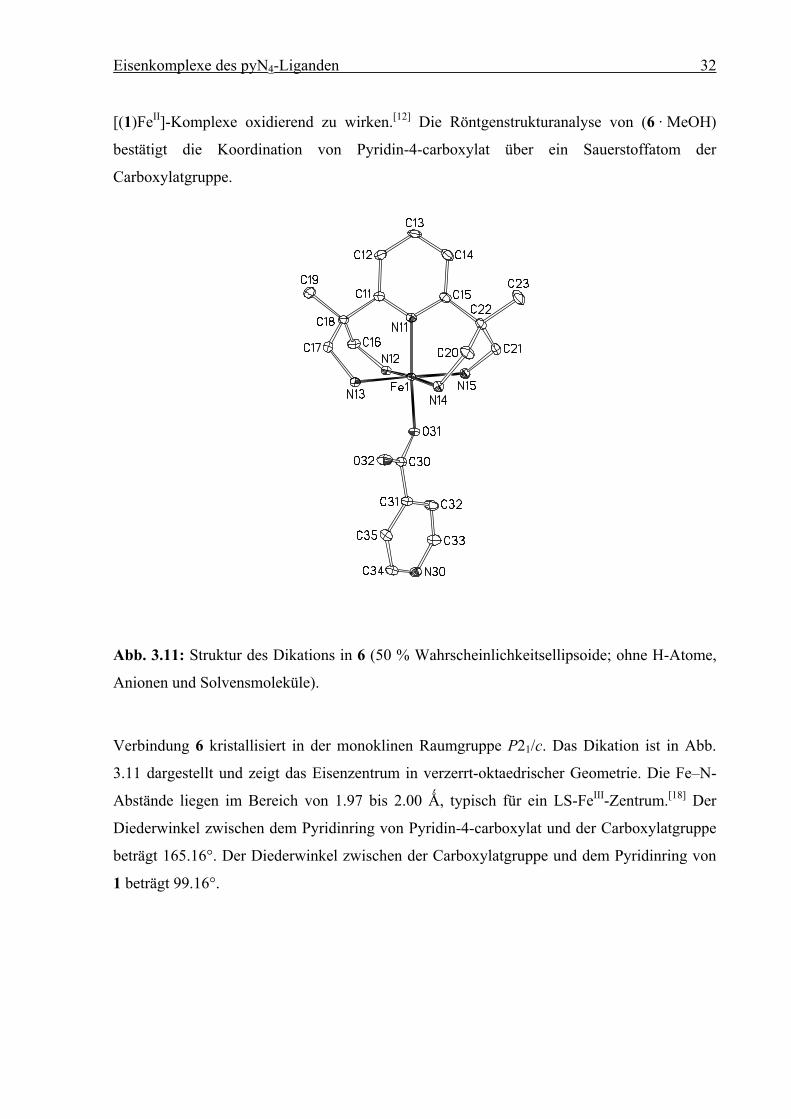
Eisenkomplexe des pyN4-Liganden 32
[(1)FeII]-Komplexe oxidierend zu wirken.[12] Die Röntgenstrukturanalyse von (6 · MeOH)
bestätigt die Koordination von Pyridin-4-carboxylat über ein Sauerstoffatom der
Carboxylatgruppe.
Abb. 3.11: Struktur des Dikations in 6 (50 % Wahrscheinlichkeitsellipsoide; ohne H-Atome,
Anionen und Solvensmoleküle).
Verbindung 6 kristallisiert in der monoklinen Raumgruppe P21/c. Das Dikation ist in Abb.
3.11 dargestellt und zeigt das Eisenzentrum in verzerrt-oktaedrischer Geometrie. Die Fe–N-
Abstände liegen im Bereich von 1.97 bis 2.00 Ǻ, typisch für ein LS-FeIII-Zentrum.[18] Der
Diederwinkel zwischen dem Pyridinring von Pyridin-4-carboxylat und der Carboxylatgruppe
beträgt 165.16°. Der Diederwinkel zwischen der Carboxylatgruppe und dem Pyridinring von
1 beträgt 99.16°.
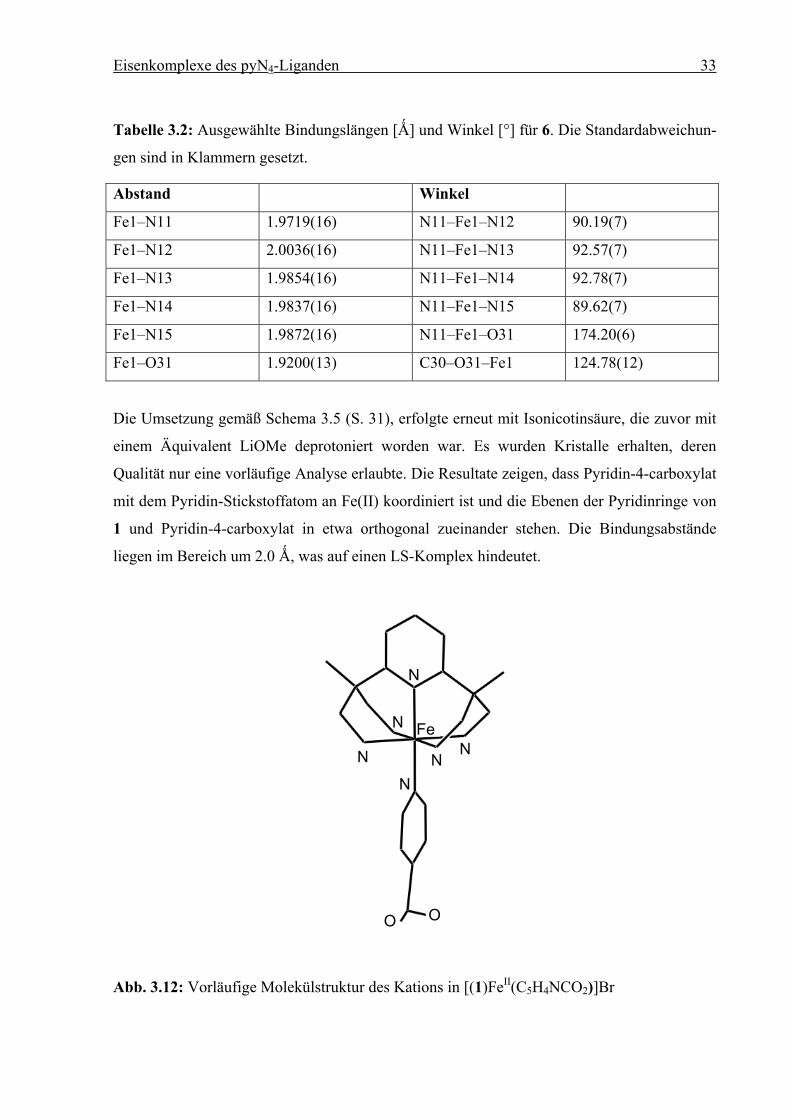
Eisenkomplexe des pyN4-Liganden 33
Tabelle 3.2: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 6. Die Standardabweichun-
gen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N11 1.9719(16) N11–Fe1–N12 90.19(7)
Fe1–N12 2.0036(16) N11–Fe1–N13 92.57(7)
Fe1–N13 1.9854(16) N11–Fe1–N14 92.78(7)
Fe1–N14 1.9837(16) N11–Fe1–N15 89.62(7)
Fe1–N15 1.9872(16) N11–Fe1–O31 174.20(6)
Fe1–O31 1.9200(13) C30–O31–Fe1 124.78(12)
Die Umsetzung gemäß Schema 3.5 (S. 31), erfolgte erneut mit Isonicotinsäure, die zuvor mit
einem Äquivalent LiOMe deprotoniert worden war. Es wurden Kristalle erhalten, deren
Qualität nur eine vorläufige Analyse erlaubte. Die Resultate zeigen, dass Pyridin-4-carboxylat
mit dem Pyridin-Stickstoffatom an Fe(II) koordiniert ist und die Ebenen der Pyridinringe von
1 und Pyridin-4-carboxylat in etwa orthogonal zueinander stehen. Die Bindungsabstände
liegen im Bereich um 2.0 Ǻ, was auf einen LS-Komplex hindeutet.
Abb. 3.12: Vorläufige Molekülstruktur des Kations in [(1)FeII(C5H4NCO2)]Br
FeN
N
N NN
N
O O

Eisenkomplexe des pyN4-Liganden 34
Die elementaranalysenreine Darstellung dieser Verbindung gelang bislang nicht.
Ohne Etherdiffusion entstand unter ansonsten analogen Bedingungen ein pyN4-freies
Koordinationspolymer der Zusammensetzung [Fe(C5H4NCO2)2]n.
3.4.2 Pyridin-3,5-dicarboxylat
Der ambidentate Charakter von Pyridincarboxylaten wurde auch bei der parallel
durchgeführten Umsetzung mit Pyridin-3,5-dicarboxylat beobachtet. Die Reaktion sollte zu
einem Neutralkomplex gemäß Schema 3.6 führen. Durch Umsetzung äquimolarer Mengen
von 1 mit FeCl2 in Methanol entsteht eine orange Lösung, die sich nach Zugabe von Pyridin-
3,5-dicarboxylat violett verfärbt. Durch Eindiffusion von Diethylether bei Raumtemperatur
entstehen nach zwei Wochen violette Kristalle, die röntgendiffraktometrisch charakterisiert
werden konnten.
1·4 HBr + 4 LiOMe + FeCl2 + Li2
N
H2NH2N FeII
NH2
NH2
N
1. MeOH2. Et2O
O
O
O
O
N
OO
OO
Schema 3.6: Versuchte Umsetzung mit Pyridin-3,5-carboxylat.
Es hat sich der zweikernige Komplex [(1)FeII–NC5H3(CO2)(COO–)FeII(1)]Br2 (7) gebildet.
Verbindung 7 kristallisiert in der monoklinen Raumgruppe P21/n. Das Dikation ist in Abb.
3.13 dargestellt und zeigt beide Eisenzentren in verzerrt-oktaedrischer Geometrie. Die Fe–N-
Abstände liegen im Bereich von 1.93 bis 2.05 Ǻ, typisch für LS-FeII-Zentren.[10] Der
Diederwinkel zwischen den beiden Pyridinringen an Fe1 beträgt 88.73°. Der Diederwinkel
zwischen dem Pyridinring von Pyridin-3,5-carboxylat und der an Fe2 koordinierten
Carboxylatgruppe beträgt 169.57°. Der Diederwinkel zwischen der Carboxylatgruppe und
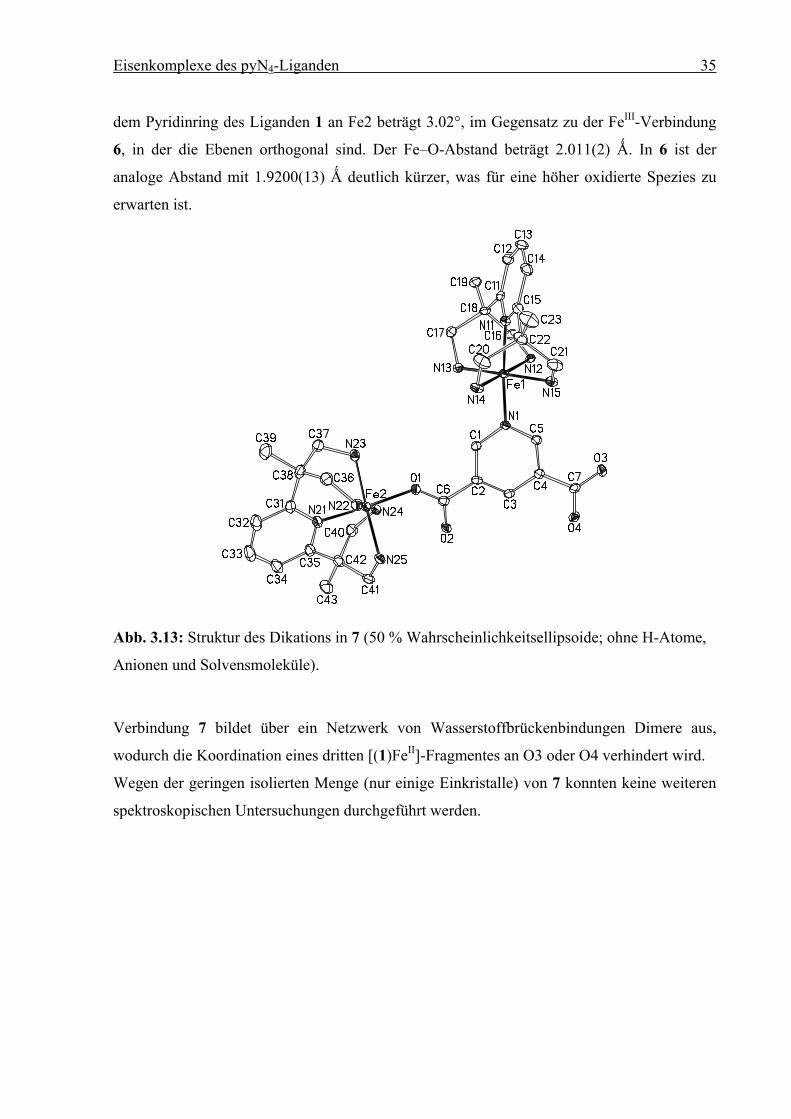
Eisenkomplexe des pyN4-Liganden 35
dem Pyridinring des Liganden 1 an Fe2 beträgt 3.02°, im Gegensatz zu der FeIII-Verbindung
6, in der die Ebenen orthogonal sind. Der Fe–O-Abstand beträgt 2.011(2) Ǻ. In 6 ist der
analoge Abstand mit 1.9200(13) Ǻ deutlich kürzer, was für eine höher oxidierte Spezies zu
erwarten ist.
Abb. 3.13: Struktur des Dikations in 7 (50 % Wahrscheinlichkeitsellipsoide; ohne H-Atome,
Anionen und Solvensmoleküle).
Verbindung 7 bildet über ein Netzwerk von Wasserstoffbrückenbindungen Dimere aus,
wodurch die Koordination eines dritten [(1)FeII]-Fragmentes an O3 oder O4 verhindert wird.
Wegen der geringen isolierten Menge (nur einige Einkristalle) von 7 konnten keine weiteren
spektroskopischen Untersuchungen durchgeführt werden.

Eisenkomplexe des pyN4-Liganden 36
Abb. 3.14: Struktur des durch Wasserstoffbrückenbindungen ausgebildeten Dimers des
Dikations von 7 (ohne Anionen und Solvensmoleküle; Kugel-Stab-Darstellung).
Tabelle 3.3: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 7. Die Standardabweichun-
gen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N11 1.976(3) N11–Fe1–N12 85.20(11)
Fe1–N12 2.034(3) N11–Fe1–N13 92.73(11)
Fe1–N13 2.016(3) N11–Fe1–N14 92.30(11)
Fe1–N14 2.017(3) N11–Fe1–N15 88.62(11)
Fe1–N15 2.047(3) N11–Fe1–N1 175.85(12)
Fe1–N1 2.002(3)
Fe2–N21 1.935(3) N21–Fe2–N22 86.97(12)
Fe2–N22 2.037(3) N21–Fe2–N23 94.29(12)
Fe2–N23 1.999(3) N21–Fe2–N24 94.89(12)
Fe2–N24 2.011(3) N21–Fe2–N25 88.22(12)
Fe2–N25 2.032(3) N21–Fe2–O1 174.59(11)
Fe2–O1 2.011(2) C6–O1–Fe2 128.5(2)
Eisenkomplexe des pyN4-Liganden 37
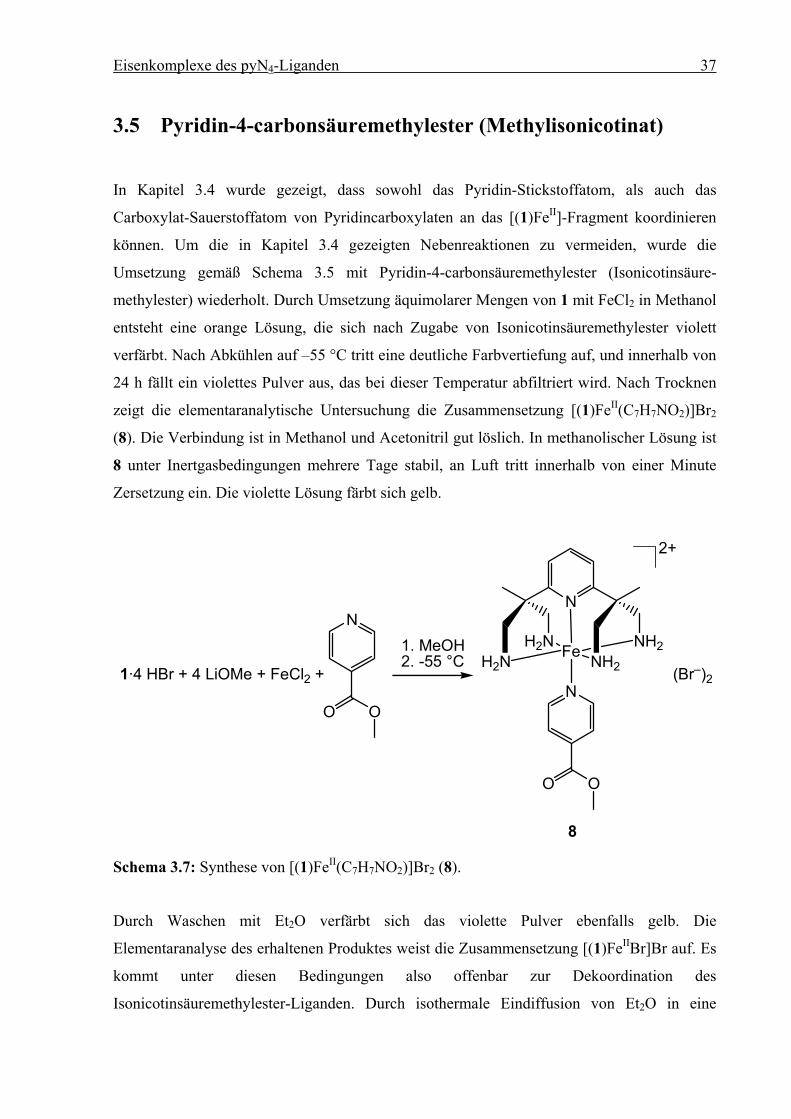
3.5 Pyridin-4-carbonsäuremethylester (Methylisonicotinat)
In Kapitel 3.4 wurde gezeigt, dass sowohl das Pyridin-Stickstoffatom, als auch das
Carboxylat-Sauerstoffatom von Pyridincarboxylaten an das [(1)FeII]-Fragment koordinieren
können. Um die in Kapitel 3.4 gezeigten Nebenreaktionen zu vermeiden, wurde die
Umsetzung gemäß Schema 3.5 mit Pyridin-4-carbonsäuremethylester (Isonicotinsäure-
methylester) wiederholt. Durch Umsetzung äquimolarer Mengen von 1 mit FeCl2 in Methanol
entsteht eine orange Lösung, die sich nach Zugabe von Isonicotinsäuremethylester violett
verfärbt. Nach Abkühlen auf –55 °C tritt eine deutliche Farbvertiefung auf, und innerhalb von
24 h fällt ein violettes Pulver aus, das bei dieser Temperatur abfiltriert wird. Nach Trocknen
zeigt die elementaranalytische Untersuchung die Zusammensetzung [(1)FeII(C7H7NO2)]Br2
(8). Die Verbindung ist in Methanol und Acetonitril gut löslich. In methanolischer Lösung ist
8 unter Inertgasbedingungen mehrere Tage stabil, an Luft tritt innerhalb von einer Minute
Zersetzung ein. Die violette Lösung färbt sich gelb.
1·4 HBr + 4 LiOMe + FeCl2 +
N
H2NH2N Fe NH2
NH2
N
2+
(Br–)2
N
OO
1. MeOH2. -55 °C
OO
8
Schema 3.7: Synthese von [(1)FeII(C7H7NO2)]Br2 (8).
Durch Waschen mit Et2O verfärbt sich das violette Pulver ebenfalls gelb. Die
Elementaranalyse des erhaltenen Produktes weist die Zusammensetzung [(1)FeIIBr]Br auf. Es
kommt unter diesen Bedingungen also offenbar zur Dekoordination des
Isonicotinsäuremethylester-Liganden. Durch isothermale Eindiffusion von Et2O in eine
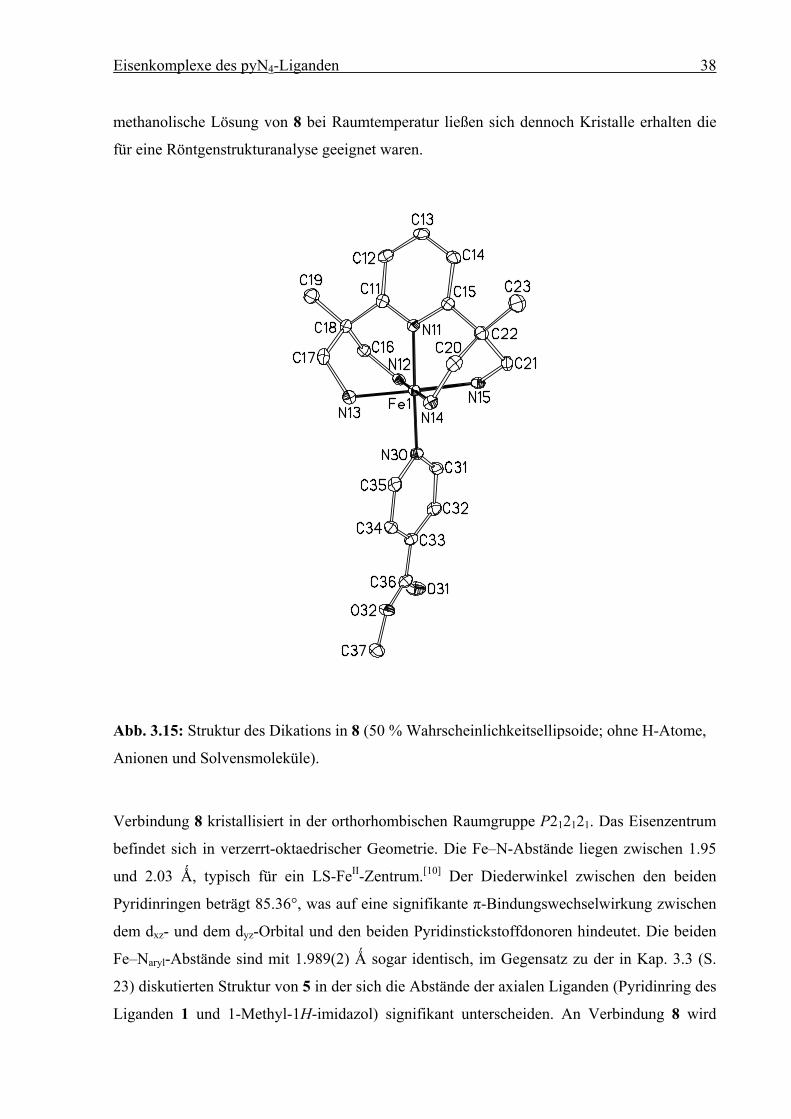
Eisenkomplexe des pyN4-Liganden 38
methanolische Lösung von 8 bei Raumtemperatur ließen sich dennoch Kristalle erhalten die
für eine Röntgenstrukturanalyse geeignet waren.
Abb. 3.15: Struktur des Dikations in 8 (50 % Wahrscheinlichkeitsellipsoide; ohne H-Atome,
Anionen und Solvensmoleküle).
Verbindung 8 kristallisiert in der orthorhombischen Raumgruppe P212121. Das Eisenzentrum
befindet sich in verzerrt-oktaedrischer Geometrie. Die Fe–N-Abstände liegen zwischen 1.95
und 2.03 Ǻ, typisch für ein LS-FeII-Zentrum.[10] Der Diederwinkel zwischen den beiden
Pyridinringen beträgt 85.36°, was auf eine signifikante π-Bindungswechselwirkung zwischen
dem dxz- und dem dyz-Orbital und den beiden Pyridinstickstoffdonoren hindeutet. Die beiden
Fe–Naryl-Abstände sind mit 1.989(2) Ǻ sogar identisch, im Gegensatz zu der in Kap. 3.3 (S.
23) diskutierten Struktur von 5 in der sich die Abstände der axialen Liganden (Pyridinring des
Liganden 1 und 1-Methyl-1H-imidazol) signifikant unterscheiden. An Verbindung 8 wird
Eisenkomplexe des pyN4-Liganden 39
erneut deutlich, dass die starke Annäherung von N11 nicht durch sterischen Einfluss von 1
erzwungen wird.
Tabelle 3.4: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 8. Die Standardabweichun-
gen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N11 1.989(2) N11–Fe1–N12 87.56(8)
Fe1–N12 2.053(2) N11–Fe1–N13 93.67(8)
Fe1–N13 2.019(2) N11–Fe1–N14 92.25(8)
Fe1–N14 2.023(2) N11–Fe1–N15 85.82(8)
Fe1–N15 2.029(2) N11–Fe1–N30 177.90(8)
Fe1–N30 1.989(2)
3.5.1 NMR-Spektren von [FeII(pyN4)(C7H7NO2)]Br2
Das 1H-NMR-Spektrum bei Raumtemperatur zeigt paramagnetisch verbreiterte Signale in
einem Bereich von 0 ppm bis 134 ppm. Bei –80 °C hat ein vollständiger Spinübergang
stattgefunden: das 1H-NMR-Spektrum zeigt ausschließlich Signale, die einen diamag-
netischen LS-Komplex 8 nahelegen (vgl. Kap. 3.3.1; S. 25 und Kap. 3.7.1; S. 51). Im 13C-
NMR-Spektrum entsprechen alle Signale einer C2v-symmetrischen Verbindung. Das 1H-
NMR-Spektrum zeigt die aromatischen Protonen beider Pyridinringe zwischen 9.0 und 7.6
ppm, die diasterotopen Protonen der Methylengruppen bei 2.46 bzw. 2.40 ppm und die
Methylgruppen von 1 als Singulett bei 1.44 ppm. Das Signal der Methylestergruppe ist im 1H-
NMR-Spektrum nicht erkennbar, da es offenbar unter dem Solvenssignal liegt. Dies deutet
auf schnelle Rotation (auf der NMR-Zeitskala) des Isonicotinsäuremethylester-Liganden um
die Fe–N-Bindungsachse hin.
Eisenkomplexe des pyN4-Liganden 40
3.5.2 Temperaturabhängige UV/Vis-Spektren von
[FeII(pyN4)(C7H7NO2)]Br2
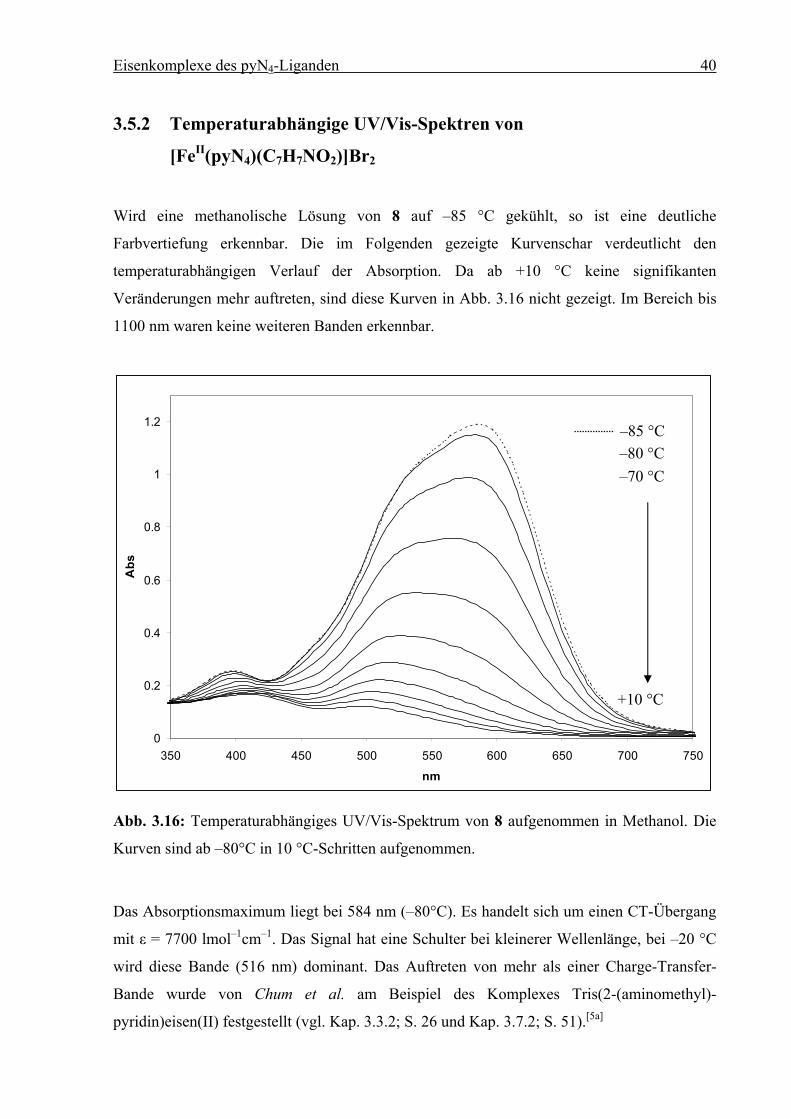
Wird eine methanolische Lösung von 8 auf –85 °C gekühlt, so ist eine deutliche
Farbvertiefung erkennbar. Die im Folgenden gezeigte Kurvenschar verdeutlicht den
temperaturabhängigen Verlauf der Absorption. Da ab +10 °C keine signifikanten
Veränderungen mehr auftreten, sind diese Kurven in Abb. 3.16 nicht gezeigt. Im Bereich bis
1100 nm waren keine weiteren Banden erkennbar.
0
0.2
0.4
0.6
0.8
1
1.2
350 400 450 500 550 600 650 700 750
nm
Abs
Abb. 3.16: Temperaturabhängiges UV/Vis-Spektrum von 8 aufgenommen in Methanol. Die
Kurven sind ab –80°C in 10 °C-Schritten aufgenommen.
Das Absorptionsmaximum liegt bei 584 nm (–80°C). Es handelt sich um einen CT-Übergang
mit ε = 7700 lmol–1cm–1. Das Signal hat eine Schulter bei kleinerer Wellenlänge, bei –20 °C
wird diese Bande (516 nm) dominant. Das Auftreten von mehr als einer Charge-Transfer-
Bande wurde von Chum et al. am Beispiel des Komplexes Tris(2-(aminomethyl)-
pyridin)eisen(II) festgestellt (vgl. Kap. 3.3.2; S. 26 und Kap. 3.7.2; S. 51).[5a]
–85 °C
+10 °C
–80 °C –70 °C
Eisenkomplexe des pyN4-Liganden 41
3.5.3 Elektrochemie von [FeII(pyN4)(C7H7NO2)]Br2
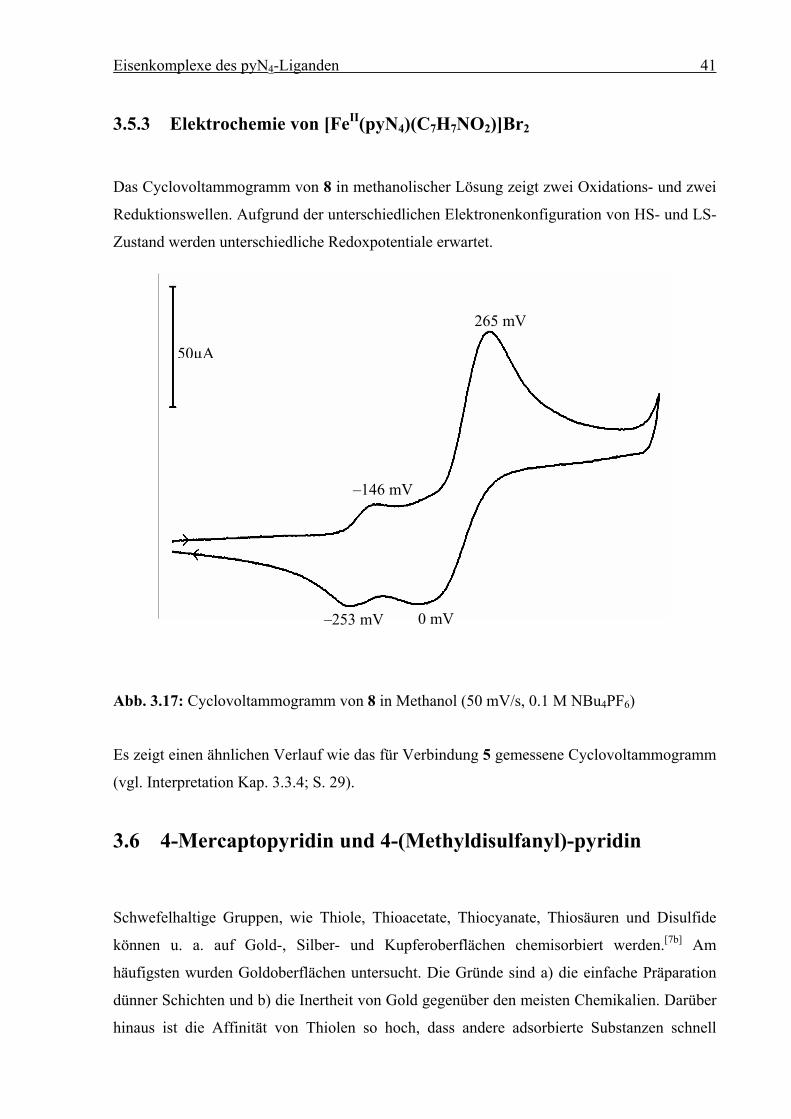
Das Cyclovoltammogramm von 8 in methanolischer Lösung zeigt zwei Oxidations- und zwei
Reduktionswellen. Aufgrund der unterschiedlichen Elektronenkonfiguration von HS- und LS-
Zustand werden unterschiedliche Redoxpotentiale erwartet.
Abb. 3.17: Cyclovoltammogramm von 8 in Methanol (50 mV/s, 0.1 M NBu4PF6)
Es zeigt einen ähnlichen Verlauf wie das für Verbindung 5 gemessene Cyclovoltammogramm
(vgl. Interpretation Kap. 3.3.4; S. 29).
3.6 4-Mercaptopyridin und 4-(Methyldisulfanyl)-pyridin
Schwefelhaltige Gruppen, wie Thiole, Thioacetate, Thiocyanate, Thiosäuren und Disulfide
können u. a. auf Gold-, Silber- und Kupferoberflächen chemisorbiert werden.[7b] Am
häufigsten wurden Goldoberflächen untersucht. Die Gründe sind a) die einfache Präparation
dünner Schichten und b) die Inertheit von Gold gegenüber den meisten Chemikalien. Darüber
hinaus ist die Affinität von Thiolen so hoch, dass andere adsorbierte Substanzen schnell
50µA
265 mV
0 mV
–146 mV
–253 mV

Eisenkomplexe des pyN4-Liganden 42
verdrängt werden.[19] Dies ermöglicht eine einfache Präparation unter normalen
Laborbedingungen.
Vorangegangene Arbeiten mit 4,4’-Dipyridyldisulfid und 4-Mercaptopyridin auf
Goldoberflächen wurden vorrangig zu Proteinstudien durchgeführt.[20] Inspiriert durch diese
Arbeiten wurden Versuche mit 4-Mercaptopyridin als Brückenligand für die Adsorption von
[(1)FeII]-Fragmenten auf Au unternommen. 4,4’-Dipyridyldisulfid wurde nicht verwendet, da
hier ein zweikerniger, tetrakationischer Komplex zu erwarten ist. Dieser wäre einerseits
schlecht löslich, andererseits wäre die Abscheidung auf Gold aufgrund der Größe der
[(1)FeII]-Fragmente schwieriger. 4-Mercaptopyridin kann in zwei tautomeren Formen
vorliegen, 4-Thiopyridon und Pyridin-4-thiol. Die Acidität des Thiols ist außerdem sehr hoch.
Die Verschiebung des SH-Protons im 1H-NMR-Spektrum in DMSO-d6 liegt bei 12.5 ppm.
Um Oxidationen wie bei 6 auszuschließen, wird das Thiol deprotoniert und anschließend in
folgender Reaktion eingesetzt.
(1)·4 HBr + 4 LiOMe + FeCl2 +
N
S-
1. MeOH2. Et2O
Li+
Schema 3.8: Stöchiometrische Umsetzung mit Pyridin-4-thiolat.
Die nach Zugabe von FeCl2 orange Lösung färbt sich nach Zugabe von Pyridin-4-thiolat rot.
Nach Eindiffusion von Diethylether wurden orange und rotbraune Kristalle erhalten. Die
rotbraunen Kristalle konnten röntgenstrukturanalytisch charakterisiert werden. Es handelt sich
um den erwarteten Komplex [(1)FeII(NC5H4S)]Br (9).
Eisenkomplexe des pyN4-Liganden 43
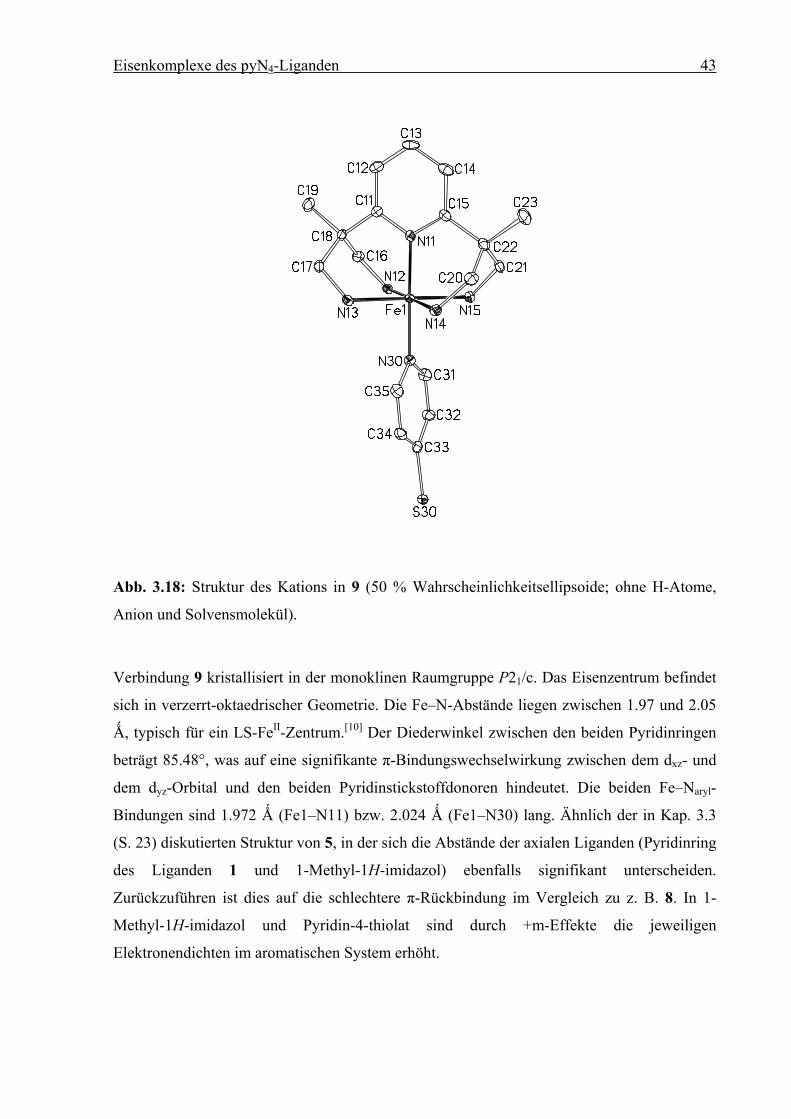
Abb. 3.18: Struktur des Kations in 9 (50 % Wahrscheinlichkeitsellipsoide; ohne H-Atome,
Anion und Solvensmolekül).
Verbindung 9 kristallisiert in der monoklinen Raumgruppe P21/c. Das Eisenzentrum befindet
sich in verzerrt-oktaedrischer Geometrie. Die Fe–N-Abstände liegen zwischen 1.97 und 2.05
Ǻ, typisch für ein LS-FeII-Zentrum.[10] Der Diederwinkel zwischen den beiden Pyridinringen
beträgt 85.48°, was auf eine signifikante π-Bindungswechselwirkung zwischen dem dxz- und
dem dyz-Orbital und den beiden Pyridinstickstoffdonoren hindeutet. Die beiden Fe–Naryl-
Bindungen sind 1.972 Ǻ (Fe1–N11) bzw. 2.024 Ǻ (Fe1–N30) lang. Ähnlich der in Kap. 3.3
(S. 23) diskutierten Struktur von 5, in der sich die Abstände der axialen Liganden (Pyridinring
des Liganden 1 und 1-Methyl-1H-imidazol) ebenfalls signifikant unterscheiden.
Zurückzuführen ist dies auf die schlechtere π-Rückbindung im Vergleich zu z. B. 8. In 1-
Methyl-1H-imidazol und Pyridin-4-thiolat sind durch +m-Effekte die jeweiligen
Elektronendichten im aromatischen System erhöht.
Eisenkomplexe des pyN4-Liganden 44
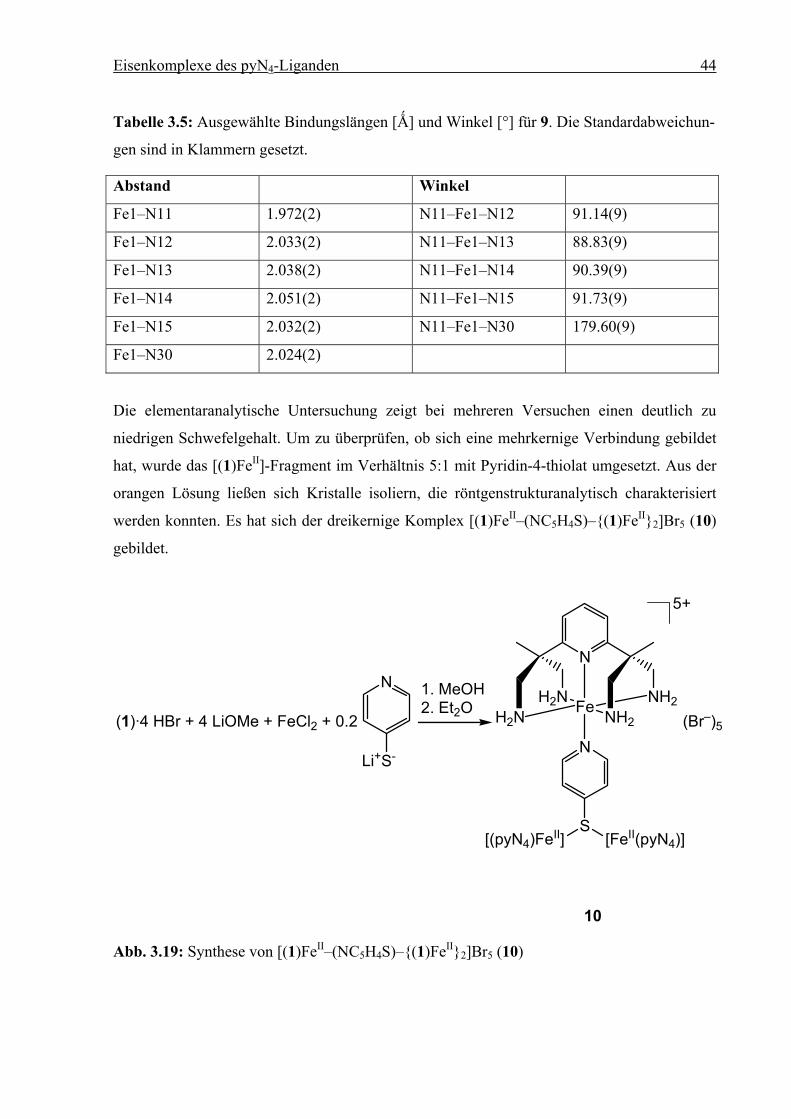
Tabelle 3.5: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 9. Die Standardabweichun-
gen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N11 1.972(2) N11–Fe1–N12 91.14(9)
Fe1–N12 2.033(2) N11–Fe1–N13 88.83(9)
Fe1–N13 2.038(2) N11–Fe1–N14 90.39(9)
Fe1–N14 2.051(2) N11–Fe1–N15 91.73(9)
Fe1–N15 2.032(2) N11–Fe1–N30 179.60(9)
Fe1–N30 2.024(2)
Die elementaranalytische Untersuchung zeigt bei mehreren Versuchen einen deutlich zu
niedrigen Schwefelgehalt. Um zu überprüfen, ob sich eine mehrkernige Verbindung gebildet
hat, wurde das [(1)FeII]-Fragment im Verhältnis 5:1 mit Pyridin-4-thiolat umgesetzt. Aus der
orangen Lösung ließen sich Kristalle isoliern, die röntgenstrukturanalytisch charakterisiert
werden konnten. Es hat sich der dreikernige Komplex [(1)FeII–(NC5H4S)–{(1)FeII}2]Br5 (10)
gebildet.
N
H2NH2N Fe NH2
NH2
N
5+
(Br–)5
S[FeII(pyN4)][(pyN4)FeII]
(1)·4 HBr + 4 LiOMe + FeCl2 + 0.2
N
S-
1. MeOH2. Et2O
Li+
10
Abb. 3.19: Synthese von [(1)FeII–(NC5H4S)–{(1)FeII}2]Br5 (10)
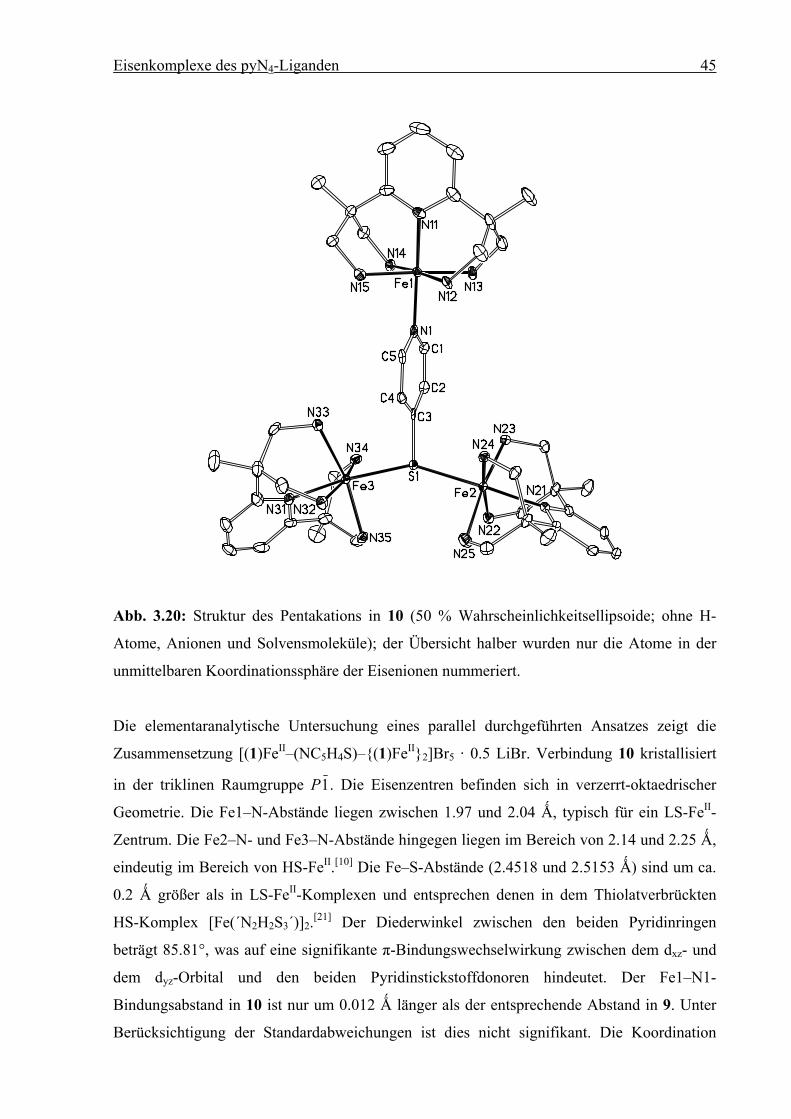
Eisenkomplexe des pyN4-Liganden 45
Abb. 3.20: Struktur des Pentakations in 10 (50 % Wahrscheinlichkeitsellipsoide; ohne H-
Atome, Anionen und Solvensmoleküle); der Übersicht halber wurden nur die Atome in der
unmittelbaren Koordinationssphäre der Eisenionen nummeriert.
Die elementaranalytische Untersuchung eines parallel durchgeführten Ansatzes zeigt die
Zusammensetzung [(1)FeII–(NC5H4S)–{(1)FeII}2]Br5 · 0.5 LiBr. Verbindung 10 kristallisiert
in der triklinen Raumgruppe P1. Die Eisenzentren befinden sich in verzerrt-oktaedrischer
Geometrie. Die Fe1–N-Abstände liegen zwischen 1.97 und 2.04 Ǻ, typisch für ein LS-FeII-
Zentrum. Die Fe2–N- und Fe3–N-Abstände hingegen liegen im Bereich von 2.14 und 2.25 Ǻ,
eindeutig im Bereich von HS-FeII.[10] Die Fe–S-Abstände (2.4518 und 2.5153 Ǻ) sind um ca.
0.2 Ǻ größer als in LS-FeII-Komplexen und entsprechen denen in dem Thiolatverbrückten
HS-Komplex [Fe(´N2H2S3´)]2.[21] Der Diederwinkel zwischen den beiden Pyridinringen
beträgt 85.81°, was auf eine signifikante π-Bindungswechselwirkung zwischen dem dxz- und
dem dyz-Orbital und den beiden Pyridinstickstoffdonoren hindeutet. Der Fe1–N1-
Bindungsabstand in 10 ist nur um 0.012 Ǻ länger als der entsprechende Abstand in 9. Unter
Berücksichtigung der Standardabweichungen ist dies nicht signifikant. Die Koordination
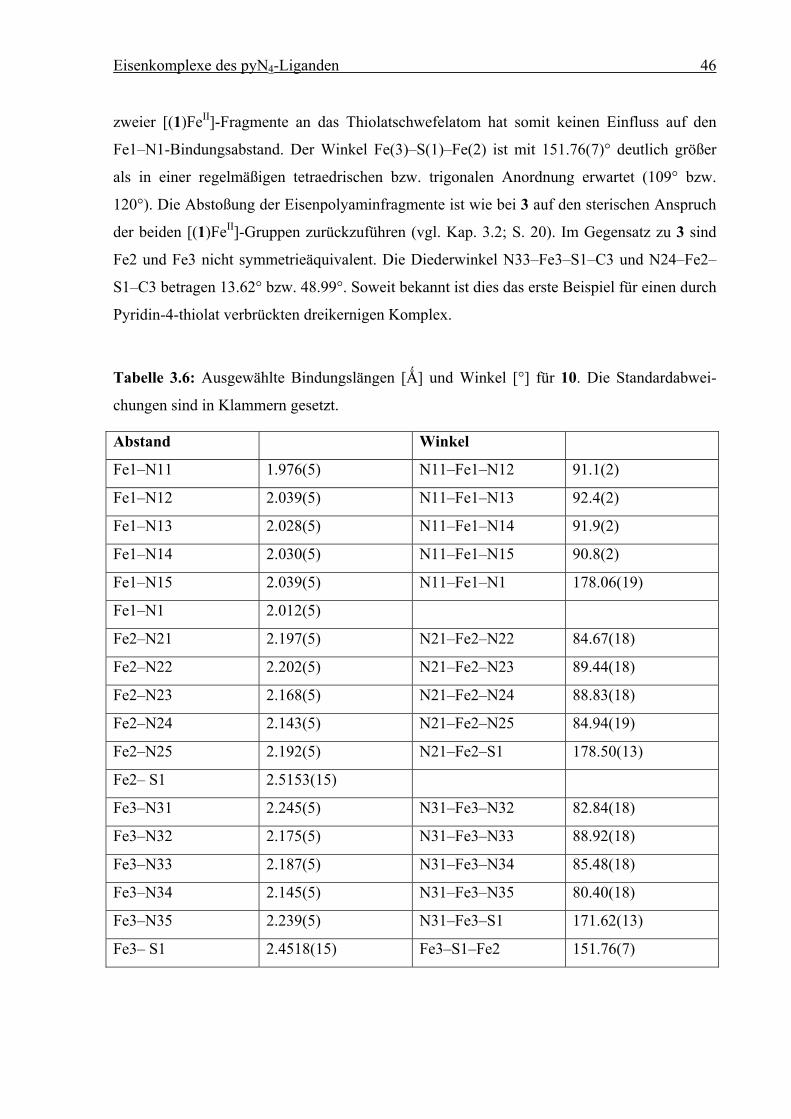
Eisenkomplexe des pyN4-Liganden 46
zweier [(1)FeII]-Fragmente an das Thiolatschwefelatom hat somit keinen Einfluss auf den
Fe1–N1-Bindungsabstand. Der Winkel Fe(3)–S(1)–Fe(2) ist mit 151.76(7)° deutlich größer
als in einer regelmäßigen tetraedrischen bzw. trigonalen Anordnung erwartet (109° bzw.
120°). Die Abstoßung der Eisenpolyaminfragmente ist wie bei 3 auf den sterischen Anspruch
der beiden [(1)FeII]-Gruppen zurückzuführen (vgl. Kap. 3.2; S. 20). Im Gegensatz zu 3 sind
Fe2 und Fe3 nicht symmetrieäquivalent. Die Diederwinkel N33–Fe3–S1–C3 und N24–Fe2–
S1–C3 betragen 13.62° bzw. 48.99°. Soweit bekannt ist dies das erste Beispiel für einen durch
Pyridin-4-thiolat verbrückten dreikernigen Komplex.
Tabelle 3.6: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 10. Die Standardabwei-
chungen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N11 1.976(5) N11–Fe1–N12 91.1(2)
Fe1–N12 2.039(5) N11–Fe1–N13 92.4(2)
Fe1–N13 2.028(5) N11–Fe1–N14 91.9(2)
Fe1–N14 2.030(5) N11–Fe1–N15 90.8(2)
Fe1–N15 2.039(5) N11–Fe1–N1 178.06(19)
Fe1–N1 2.012(5)
Fe2–N21 2.197(5) N21–Fe2–N22 84.67(18)
Fe2–N22 2.202(5) N21–Fe2–N23 89.44(18)
Fe2–N23 2.168(5) N21–Fe2–N24 88.83(18)
Fe2–N24 2.143(5) N21–Fe2–N25 84.94(19)
Fe2–N25 2.192(5) N21–Fe2–S1 178.50(13)
Fe2– S1 2.5153(15)
Fe3–N31 2.245(5) N31–Fe3–N32 82.84(18)
Fe3–N32 2.175(5) N31–Fe3–N33 88.92(18)
Fe3–N33 2.187(5) N31–Fe3–N34 85.48(18)
Fe3–N34 2.145(5) N31–Fe3–N35 80.40(18)
Fe3–N35 2.239(5) N31–Fe3–S1 171.62(13)
Fe3– S1 2.4518(15) Fe3–S1–Fe2 151.76(7)
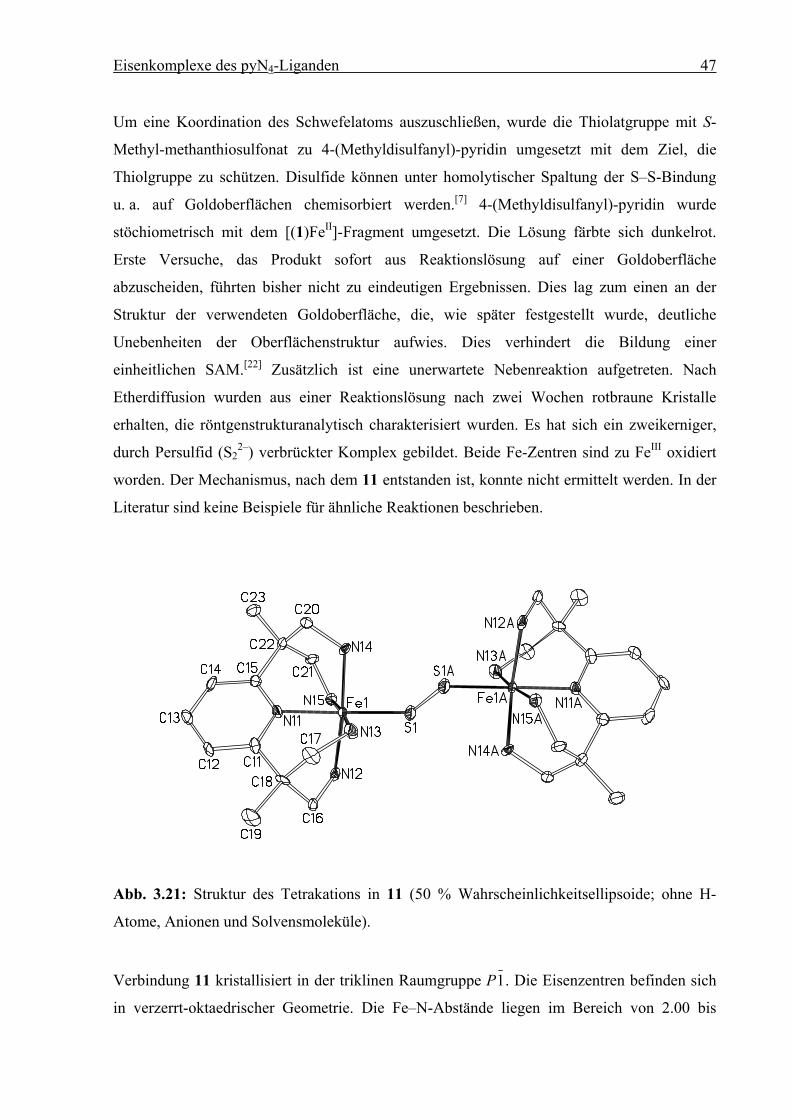
Eisenkomplexe des pyN4-Liganden 47
Um eine Koordination des Schwefelatoms auszuschließen, wurde die Thiolatgruppe mit S-
Methyl-methanthiosulfonat zu 4-(Methyldisulfanyl)-pyridin umgesetzt mit dem Ziel, die
Thiolgruppe zu schützen. Disulfide können unter homolytischer Spaltung der S–S-Bindung
u. a. auf Goldoberflächen chemisorbiert werden.[7] 4-(Methyldisulfanyl)-pyridin wurde
stöchiometrisch mit dem [(1)FeII]-Fragment umgesetzt. Die Lösung färbte sich dunkelrot.
Erste Versuche, das Produkt sofort aus Reaktionslösung auf einer Goldoberfläche
abzuscheiden, führten bisher nicht zu eindeutigen Ergebnissen. Dies lag zum einen an der
Struktur der verwendeten Goldoberfläche, die, wie später festgestellt wurde, deutliche
Unebenheiten der Oberflächenstruktur aufwies. Dies verhindert die Bildung einer
einheitlichen SAM.[22] Zusätzlich ist eine unerwartete Nebenreaktion aufgetreten. Nach
Etherdiffusion wurden aus einer Reaktionslösung nach zwei Wochen rotbraune Kristalle
erhalten, die röntgenstrukturanalytisch charakterisiert wurden. Es hat sich ein zweikerniger,
durch Persulfid (S22–) verbrückter Komplex gebildet. Beide Fe-Zentren sind zu FeIII oxidiert
worden. Der Mechanismus, nach dem 11 entstanden ist, konnte nicht ermittelt werden. In der
Literatur sind keine Beispiele für ähnliche Reaktionen beschrieben.
Abb. 3.21: Struktur des Tetrakations in 11 (50 % Wahrscheinlichkeitsellipsoide; ohne H-
Atome, Anionen und Solvensmoleküle).
Verbindung 11 kristallisiert in der triklinen Raumgruppe P1. Die Eisenzentren befinden sich
in verzerrt-oktaedrischer Geometrie. Die Fe–N-Abstände liegen im Bereich von 2.00 bis

Eisenkomplexe des pyN4-Liganden 48
2.03 Ǻ, typisch für ein LS-FeIII-Zentrum.[18] Das Komplexkation liegt auf einem
kristallographischen Inversionszentrum und hat folglich Ci-Symmetrie. Die Abstände und
Winkel der Fe1–S1–S1A–Fe1A-Einheit entsprechen denen analoger Verbindungen.[23] Wegen
der geringen isolierten Menge (nur einige Einkristalle) von 11 konnten keine weiteren
spektroskopischen Untersuchungen durchgeführt werden.
Tabelle 3.7: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 11. Die Standardab-
weichungen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N11 2.020(7) N11–Fe1–N12 85.3(3)
Fe1–N12 2.026(8) N11–Fe1–N13 92.5(3)
Fe1–N13 2.004(8) N11–Fe1–N14 89.6(3)
Fe1–N14 2.012(8) N11–Fe1–N15 84.1(3)
Fe1–N15 2.028(8) N11–Fe1–S1 175.5(2)
Fe1–S1 2.163(3) Fe1–S1–S1A 108.01(18)
S1–S1A 2.033(5)
3.7 (E)-4-(Phenyldiazenyl)pyridin
Neben dem LIESST-Effekt (vgl. Einleitung), der nur im Festkörper auftritt, kann in Lösung
der LD-LISC-Effekt auftreten (ligand-driven light induced spin-state change). Voraussetzung
hierfür sind Liganden, die Photoreaktionen zeigen, wie cis-trans-schaltbare Liganden.[24] Dazu
zählt auch (E)-4-(Phenyldiazenyl)pyridin (4-PAPY).[25] Beim Übergang von der planaren,
vollständig π-konjugierten trans- zur cis-Form ändert sich die chemische Verschiebung im 1H-NMR-Spektrum (CDCl3) der Pyridinprotonen. Das Signal der orthoständigen H-Atome
verschiebt sich von 8.7 ppm nach 8.4 ppm, das der metaständigen von 7.65 nach 6.61 ppm.
Infolge der Isomerisierung ändern sich die σ-Donor und π-Donor-/Akzeptoreigenschaften.
Zuvor aus dem Hydrobromid freigesetzter Ligand 1 wurde mit einer äquimolaren Menge
Fe(CH3CN)2(OTf)2 in Methanol umgesetzt. Die rote Lösung wurde mit einer
stöchiometrischen Menge 4-PAPY versetzt. Bei Zutropfen der schwach orangen,
methanolischen Lösung von 4-PAPY färbt sich die Reaktionslösung sofort grün. Nach
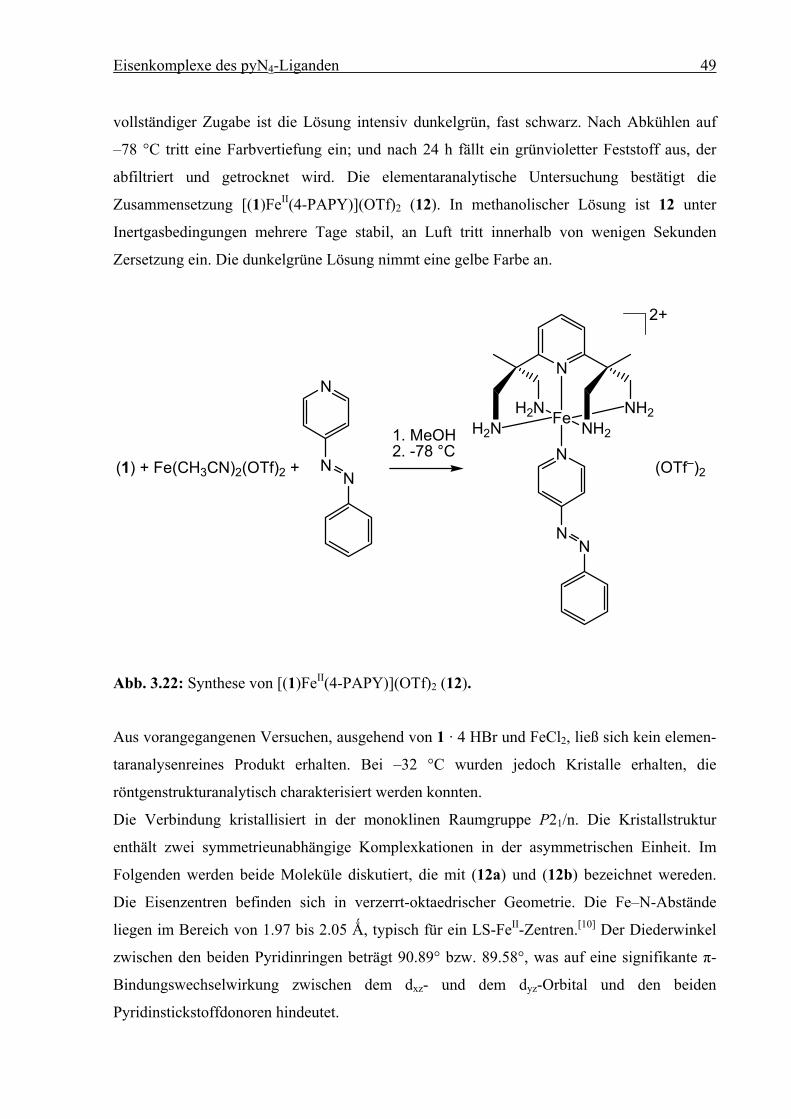
Eisenkomplexe des pyN4-Liganden 49
vollständiger Zugabe ist die Lösung intensiv dunkelgrün, fast schwarz. Nach Abkühlen auf
–78 °C tritt eine Farbvertiefung ein; und nach 24 h fällt ein grünvioletter Feststoff aus, der
abfiltriert und getrocknet wird. Die elementaranalytische Untersuchung bestätigt die
Zusammensetzung [(1)FeII(4-PAPY)](OTf)2 (12). In methanolischer Lösung ist 12 unter
Inertgasbedingungen mehrere Tage stabil, an Luft tritt innerhalb von wenigen Sekunden
Zersetzung ein. Die dunkelgrüne Lösung nimmt eine gelbe Farbe an.
N
H2NH2N Fe NH2
NH2
N
2+
(OTf–)2
N
(1) + Fe(CH3CN)2(OTf)2 +
N
N
1. MeOH2. -78 °C
N
N
Abb. 3.22: Synthese von [(1)FeII(4-PAPY)](OTf)2 (12).
Aus vorangegangenen Versuchen, ausgehend von 1 · 4 HBr und FeCl2, ließ sich kein elemen-
taranalysenreines Produkt erhalten. Bei –32 °C wurden jedoch Kristalle erhalten, die
röntgenstrukturanalytisch charakterisiert werden konnten.
Die Verbindung kristallisiert in der monoklinen Raumgruppe P21/n. Die Kristallstruktur
enthält zwei symmetrieunabhängige Komplexkationen in der asymmetrischen Einheit. Im
Folgenden werden beide Moleküle diskutiert, die mit (12a) und (12b) bezeichnet wereden.
Die Eisenzentren befinden sich in verzerrt-oktaedrischer Geometrie. Die Fe–N-Abstände
liegen im Bereich von 1.97 bis 2.05 Ǻ, typisch für ein LS-FeII-Zentren.[10] Der Diederwinkel
zwischen den beiden Pyridinringen beträgt 90.89° bzw. 89.58°, was auf eine signifikante π-
Bindungswechselwirkung zwischen dem dxz- und dem dyz-Orbital und den beiden
Pyridinstickstoffdonoren hindeutet.
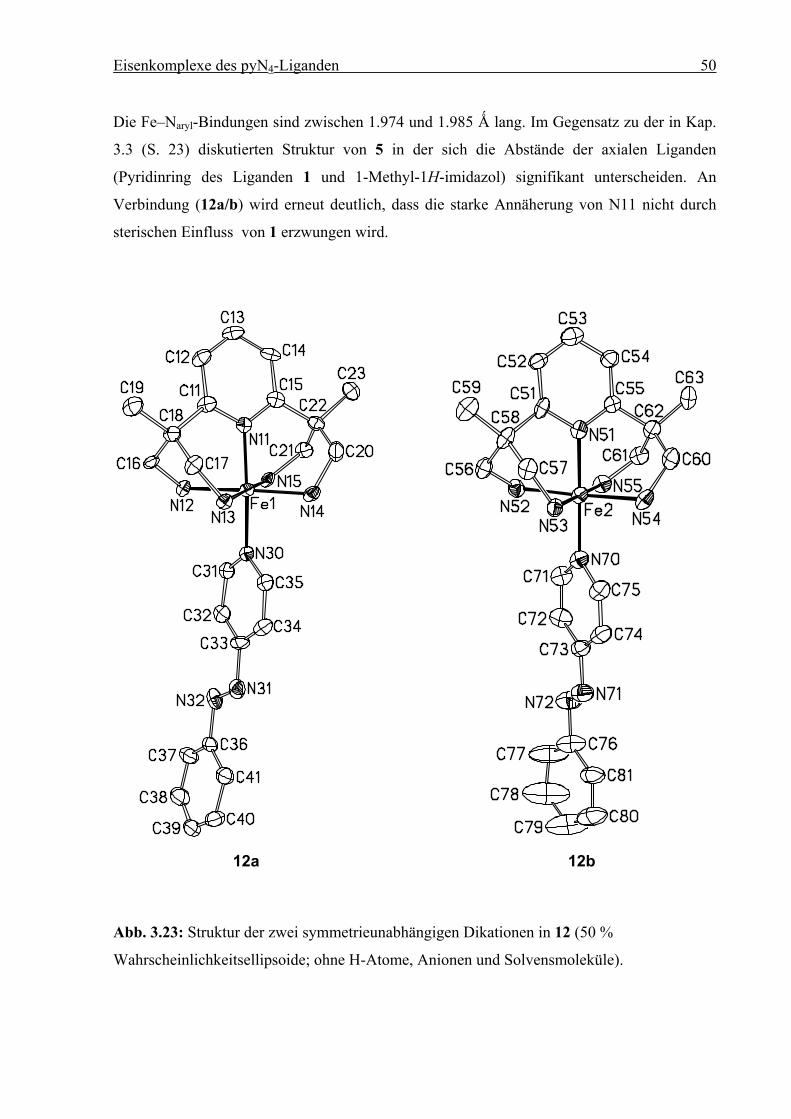
Eisenkomplexe des pyN4-Liganden 50
Die Fe–Naryl-Bindungen sind zwischen 1.974 und 1.985 Ǻ lang. Im Gegensatz zu der in Kap.
3.3 (S. 23) diskutierten Struktur von 5 in der sich die Abstände der axialen Liganden
(Pyridinring des Liganden 1 und 1-Methyl-1H-imidazol) signifikant unterscheiden. An
Verbindung (12a/b) wird erneut deutlich, dass die starke Annäherung von N11 nicht durch
sterischen Einfluss von 1 erzwungen wird.
12a 12b
Abb. 3.23: Struktur der zwei symmetrieunabhängigen Dikationen in 12 (50 %
Wahrscheinlichkeitsellipsoide; ohne H-Atome, Anionen und Solvensmoleküle).

Eisenkomplexe des pyN4-Liganden 51
Tabelle 3.8: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für (12a)/(12b). Die Standard-
abweichungen sind in Klammern gesetzt.
Abstand Winkel
Fe1–N11 1.984(6) / 1.974(6) N11–Fe1–N12 87.9(2) / 85.8(3)
Fe1–N12 2.028(6) / 2.044(6) N11–Fe1–N13 91.5(2) / 92.8(2)
Fe1–N13 2.009(5) / 2.012(6) N11–Fe1–N14 93.9(2) / 92.2(3)
Fe1–N14 2.019(6) / 2.023(6) N11–Fe1–N15 88.7(2) / 87.7(2)
Fe1–N15 2.022(5) / 2.052(6) N11–Fe1–N30 177.2(2) / 177.9(3)
Fe1–N30 1.975(6) / 1.985(6)
3.7.1 NMR-Spektren von [FeII(pyN4)(4-PAPY)](OTf)2
Das 1H-NMR-Spektrum bei Raumtemperatur zeigt paramagnetisch verbreiterte Signale in
einem Bereich von 0 ppm bis 140 ppm. Bei –60 °C hat ein vollständiger Spinübergang
stattgefunden, das 1H-NMR-Spektrum zeigt ausschließlich Signale, die einen
diamagnetischen LS-Komplex 12 nahelegen (vgl. Kap. 3.3.1; S. 25 und Kap. 3.5.1; S. 39). Im 13C-NMR-Spektrum treten 14 Signale für die aromatischen Kohlenstoffatome auf. Das 1H-
NMR-Spektrum zeigt die zwölf aromatischen Protonen im Bereich zwischen 9.0 und 7.5
ppm, die diasterotopen Protonen der Methylengruppen im Bereich von 3.0 bis 2.2 ppm und
die Methylgruppen von 1 als Singulett bei 1.46 ppm. Die Signale sind leicht verbreitert, beim
Abkühlen auf –80 °C nimmt diese Verbreiterung wieder zu. Die NMR-Daten zeigen, dass die
Rotation des 4-PAPY-Liganden um die Fe–N-Bindungsachse auf der NMR-Zeitskala deutlich
langsamer ist, als die in 5 und 8.
3.7.2 Temperaturabhängige UV/Vis-Spektren von [FeII(pyN4)(4-
PAPY)](OTf)2
Wird eine methanolische Lösung von 12 auf –80 °C gekühlt, so ist eine deutliche
Farbvertiefung wahrzunehmen. Abb. 3.24 zeigt den temperaturabhängigen Verlauf der
Absorption. Im Bereich bis 1100 nm waren keine weiteren Banden erkennbar.
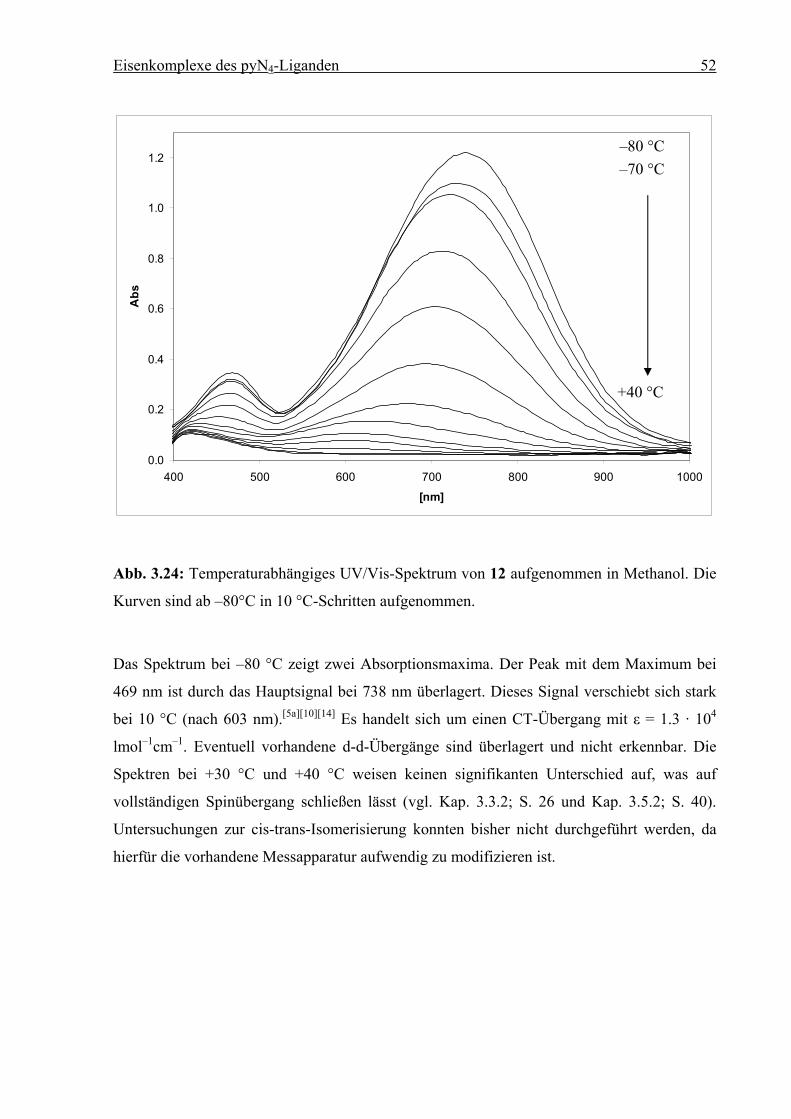
Eisenkomplexe des pyN4-Liganden 52
0.0
0.2
0.4
0.6
0.8
1.0
1.2
400 500 600 700 800 900 1000
[nm]
Abs
Abb. 3.24: Temperaturabhängiges UV/Vis-Spektrum von 12 aufgenommen in Methanol. Die
Kurven sind ab –80°C in 10 °C-Schritten aufgenommen.
Das Spektrum bei –80 °C zeigt zwei Absorptionsmaxima. Der Peak mit dem Maximum bei
469 nm ist durch das Hauptsignal bei 738 nm überlagert. Dieses Signal verschiebt sich stark
bei 10 °C (nach 603 nm).[5a][10][14] Es handelt sich um einen CT-Übergang mit ε = 1.3 · 104
lmol–1cm–1. Eventuell vorhandene d-d-Übergänge sind überlagert und nicht erkennbar. Die
Spektren bei +30 °C und +40 °C weisen keinen signifikanten Unterschied auf, was auf
vollständigen Spinübergang schließen lässt (vgl. Kap. 3.3.2; S. 26 und Kap. 3.5.2; S. 40).
Untersuchungen zur cis-trans-Isomerisierung konnten bisher nicht durchgeführt werden, da
hierfür die vorhandene Messapparatur aufwendig zu modifizieren ist.
+40 °C
–80 °C –70 °C

Eisenkomplexe des pyN4-Liganden 53
3.8 Isonitrile
Der elektronenreiche Charakter des FeII-Zentrums in den [(1)FeII]-Komplexen wurde anhand
der CO-Streckschwingung (1960 cm-1) und der chemischen Verschiebung des
Carbonylliganden im 13C-NMR-Spektrum (220.7 ppm) nachgewiesen. Eine Komplexierung
der zu CO isolobalen Isonitrile sollte also möglich sein.
Die Umsetzung dreier Isonitrile CN–R (R = tButyl, Cyclohexyl und p-Methoxyphenyl) wird
gemäß Schema 3.9 durchgeführt. Die nach Zugabe von FeCl2 erhaltene orange Lösung hellt
sich bei Zugabe des Isonitrils nur wenig auf.
N
H2NH2N Fe NH2
NH2
C
2+
(Br–)2(1)·4 HBr + 4 LiOMe + FeCl2 +
1. MeOH2. Et2OC
NR
NR
Schema 3.9: Synthese der Komplexe des Typs [(1)FeII(CN–R)]Br2.
Die IR-spektroskopische Reaktionsverfolgung zeigt eine deutliche Verschiebung der ν(C≡N)-
Bande zu niedrigeren Wellenzahlen infolge Koordination. Die Ergebnisse sind in Tabelle 3.9
zusammengefasst. Die Verbindungen 13, 14 und 15 fallen bei Einengen des Lösemittels als
oranger Niederschlag aus der methanolischen Lösung aus. Alternativ lassen sich die
Verbindungen durch Eindiffusion von Diethylether auskristallisieren. Dabei entstand in allen
Versuchen einkristallines Material. Die Kristalle zerfallen beim Trocknen im Hochvakuum.
Die elementaranalytische Untersuchung nach entsprechender Trocknung bestätigt die
Zusammensetzung [(1)FeII(CN–R)]Br2. Die Verbindungen sind in DMSO und Acetonitril gut
und in MeOH mäßig löslich. Sie sind gegenüber Luft und Wasser deutlich stabiler als die, in
den vorangegangenen Kapiteln beschriebenen Verbindungen. Offensichtlich stabilisiert die π-
Rückbindung wie in [(1)FeII(CO)]Br2 die Oxidationsstufe +II.
R = tert-Butyl 13 Cyclohexyl 14 p-Methoxyphenyl 15

Eisenkomplexe des pyN4-Liganden 54
Tabelle 3.9: Lage der ν(C≡N)-Streckschwingung [cm–1] in MeOH.
R freies Isonitril* [(1)FeII(CN–R)]Br2 Differenz tButyl (13) 2157; 2135 2113 44; 22
Cyclohexyl (14) 2163; 2141 2120 43; 21
p-Methoxyphenyl (15) 2144; 2125 2094 50; 31
* Die freien Isonitrile zeigten aufgrund von Fermi-Resonanz zwei Banden.[26]
Die 1H- und 13C-NMR-Spektren von 13, 14 und 15 belegen den diamagnetischen Charakter
und zeigen C2v-symmetrische Verbindungen (auf der NMR-Zeitskala). 13 zeigt ein Triplett
bei 8.20 ppm und ein Dublett bei 7.76 ppm für die aromatischen Protonen. Das für 1
charakteristische Dublett vom Dublett der diastereotopen Methylenprotonen liegt zwischen
2.22 und 2.02 ppm. Die tert-Butyl-Gruppe und die Methylprotonen ergeben Singuletts bei
1.71 bzw. 1.32 ppm. Das 13C-NMR-Spektrum zeigt das Isonitiril-Kohlenstoffatom bei
175.7 ppm. Die NMR-Spektren von 14 und 15 sind zu denen von 13 analog. In 14 liegt das
Multiplett für das Methinproton bei 4.3 ppm, die restlichen Signale treten im Bereich von 2.6
bis 1.1 ppm. Das Signal im 13C-NMR-Spektrum für das Isonitiril-Kohlenstoffatom befindet
sich bei 176.6 ppm. Der auffälligste Unterschied bei Verbindung 15 ist das 13C-Signal des
Isonitril-Kohlenstofffatomes, welches zu 185.83 ppm tieffeldverschoben ist. Durch den para-
Methoxyphenylrest tritt eine stärkere π-Rückbindung auf. Dies lässt sich auch anhand der IR-
Spektren feststellen. In 15 ist die ν(C≡N)-Bande des komplexierten Isonitrils gegenüber
freiem Isonitril stärker zu kleineren Wellenzahlen verschoben als bei 13 und 14.
Durch Eindiffusion von Et2O in eine methanolische Lösung von 13, 14 und 15 wurden
Kristalle erhalten, die röntgenstrukturanalytisch untersucht werden konnten.
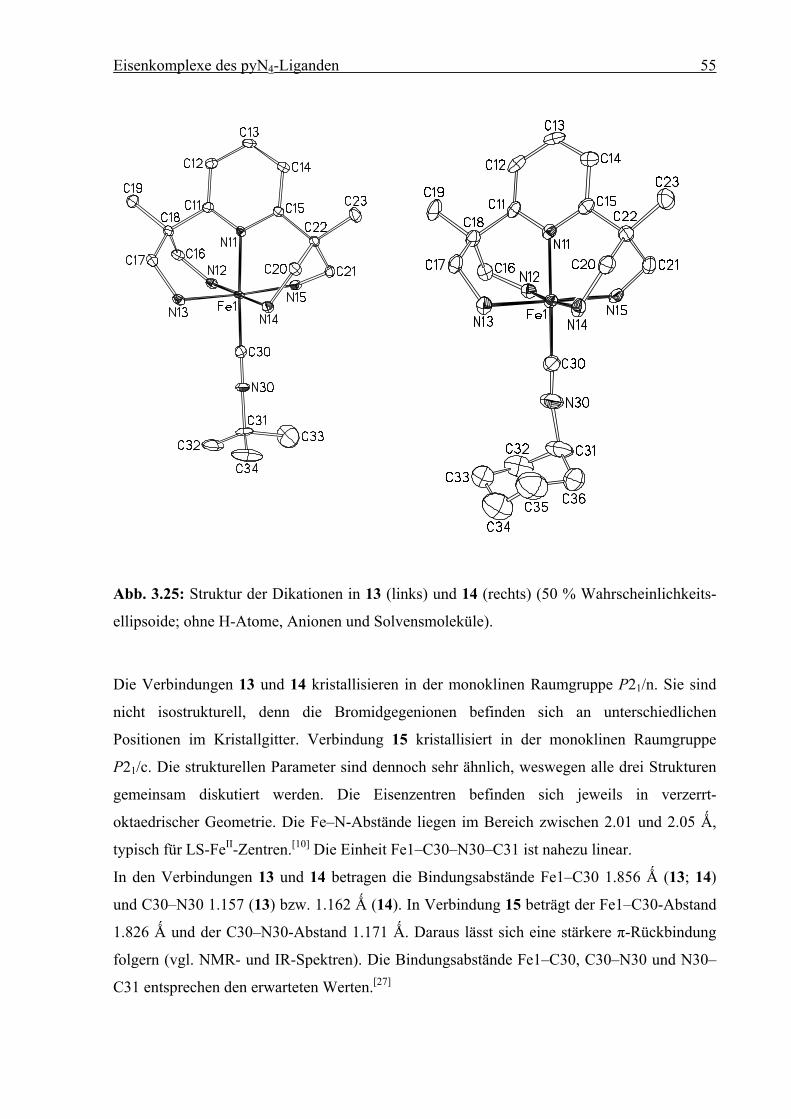
Eisenkomplexe des pyN4-Liganden 55
Abb. 3.25: Struktur der Dikationen in 13 (links) und 14 (rechts) (50 % Wahrscheinlichkeits-
ellipsoide; ohne H-Atome, Anionen und Solvensmoleküle).
Die Verbindungen 13 und 14 kristallisieren in der monoklinen Raumgruppe P21/n. Sie sind
nicht isostrukturell, denn die Bromidgegenionen befinden sich an unterschiedlichen
Positionen im Kristallgitter. Verbindung 15 kristallisiert in der monoklinen Raumgruppe
P21/c. Die strukturellen Parameter sind dennoch sehr ähnlich, weswegen alle drei Strukturen
gemeinsam diskutiert werden. Die Eisenzentren befinden sich jeweils in verzerrt-
oktaedrischer Geometrie. Die Fe–N-Abstände liegen im Bereich zwischen 2.01 und 2.05 Ǻ,
typisch für LS-FeII-Zentren.[10] Die Einheit Fe1–C30–N30–C31 ist nahezu linear.
In den Verbindungen 13 und 14 betragen die Bindungsabstände Fe1–C30 1.856 Ǻ (13; 14)
und C30–N30 1.157 (13) bzw. 1.162 Ǻ (14). In Verbindung 15 beträgt der Fe1–C30-Abstand
1.826 Ǻ und der C30–N30-Abstand 1.171 Ǻ. Daraus lässt sich eine stärkere π-Rückbindung
folgern (vgl. NMR- und IR-Spektren). Die Bindungsabstände Fe1–C30, C30–N30 und N30–
C31 entsprechen den erwarteten Werten.[27]
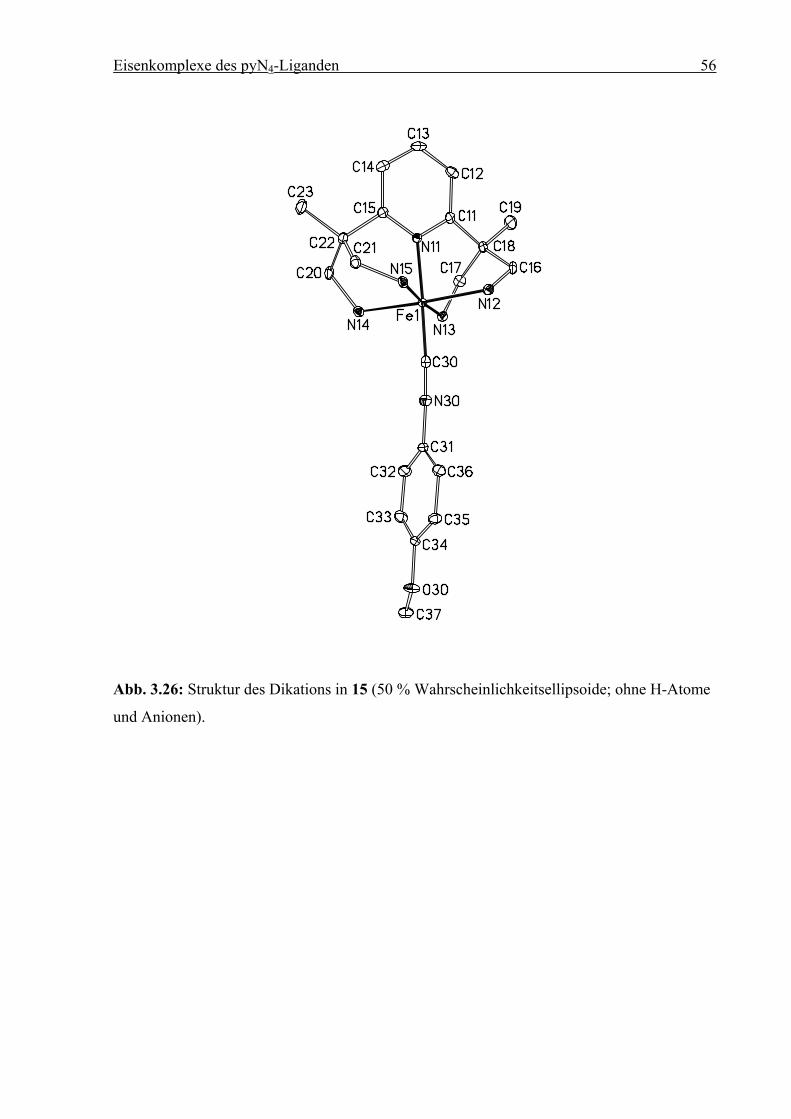
Eisenkomplexe des pyN4-Liganden 56
Abb. 3.26: Struktur des Dikations in 15 (50 % Wahrscheinlichkeitsellipsoide; ohne H-Atome
und Anionen).
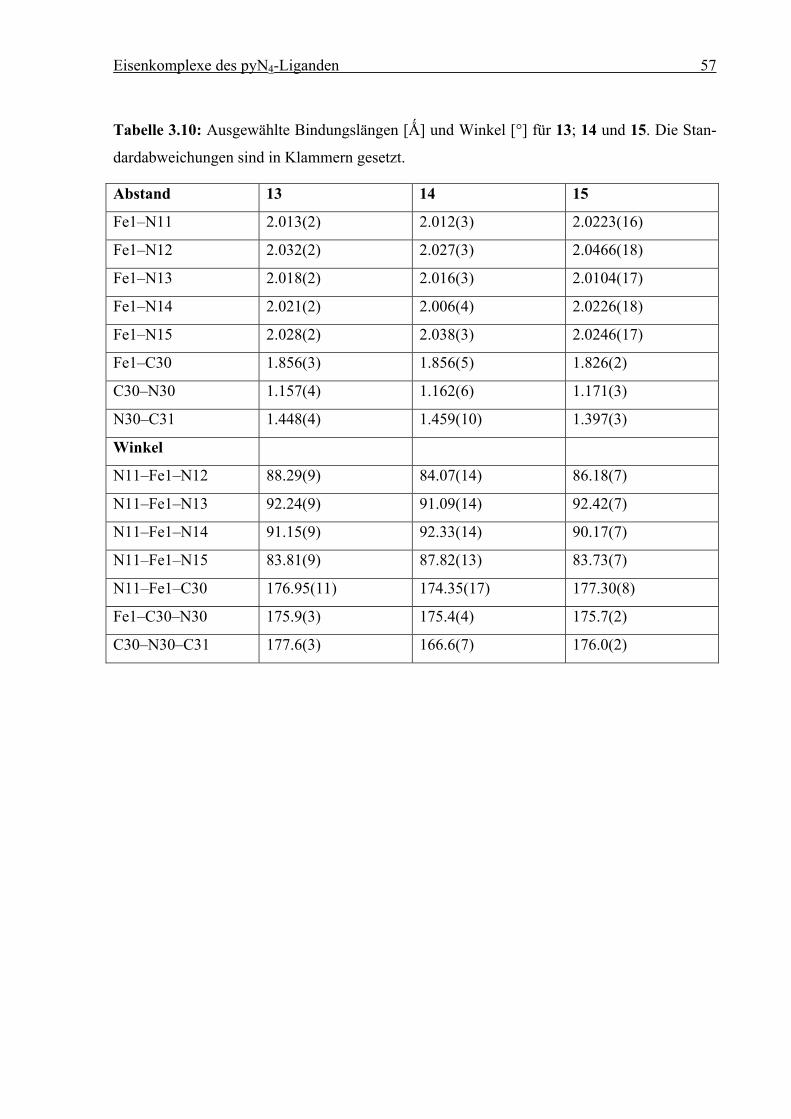
Eisenkomplexe des pyN4-Liganden 57
Tabelle 3.10: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 13; 14 und 15. Die Stan-
dardabweichungen sind in Klammern gesetzt.
Abstand 13 14 15
Fe1–N11 2.013(2) 2.012(3) 2.0223(16)
Fe1–N12 2.032(2) 2.027(3) 2.0466(18)
Fe1–N13 2.018(2) 2.016(3) 2.0104(17)
Fe1–N14 2.021(2) 2.006(4) 2.0226(18)
Fe1–N15 2.028(2) 2.038(3) 2.0246(17)
Fe1–C30 1.856(3) 1.856(5) 1.826(2)
C30–N30 1.157(4) 1.162(6) 1.171(3)
N30–C31 1.448(4) 1.459(10) 1.397(3)
Winkel
N11–Fe1–N12 88.29(9) 84.07(14) 86.18(7)
N11–Fe1–N13 92.24(9) 91.09(14) 92.42(7)
N11–Fe1–N14 91.15(9) 92.33(14) 90.17(7)
N11–Fe1–N15 83.81(9) 87.82(13) 83.73(7)
N11–Fe1–C30 176.95(11) 174.35(17) 177.30(8)
Fe1–C30–N30 175.9(3) 175.4(4) 175.7(2)
C30–N30–C31 177.6(3) 166.6(7) 176.0(2)

Eisenkomplexe des pyN4-Liganden 58
3.9 Experimenteller Teil
3.9.1 Allgemeines
Sofern nicht anders vermerkt, wurden alle Reaktionen unter N2-Atmosphäre in absoluten
Lösemitteln in Standard-Schlenk-Gefäßen durchgeführt. Die verwendeten absoluten
Lösemittel (Restgehalt H2O ≤ 50 ppm) sind kommerziell über Molsieb getrocknet
erhältlich.[28] Organische und anorganische Reagenzien wurden von den Firmen Aldrich,
Acros, Strem, Alfa Aesar oder Air Liquide bezogen und ohne weitere Reinigung benutzt. Die
Verbindungen 1 · 4 HBr · MeOH[29], Fe(CH3CN)2(OTf)2[30], Natriumtetrakis-bis(3,5-trifluor-
methyl-1-phenyl)borat (NaBArF)[31] und 4-(Methyldisulfanyl)-pyridin[32] wurden nach Litera-
turangaben hergestellt.
Zur Aufnahme der Daten wurden folgende Geräte verwendet:
Kernresonanzspektren: Die NMR-Spektren wurden, soweit nicht anders vermerkt, bei
Raumtemperatur in einem 5 mm-Röhrchen an Spektrometern der Firma Bruker
aufgenommen: ARX 200 (1H, 200 MHz; 13C, 50.32 MHz), ARX 400 (1H, 400 MHz; 13C,
100.64 MHz). Die temperaturabhängigen NMR-Spektren von 5, 8 und 12 wurden an einem
Alpha 500 der Firma Jeol gemessen (1H, 500 MHz; 13C, 125.78 MHz). Luftempfindliche
Proben wurden in einem unter Vakuum abgeschmolzenen Röhrchen vermessen. Alle
chemischen Verschiebungen sind in ppm relativ zur Restprotonen- bzw. 13C-Absorption des
verwendeten Lösemittels (interner Standard) angegeben. Die 13C-NMR-Spektren wurden
breitbandentkoppelt aufgenommen. Die Vorzeichen der Kopplungskonstanten wurden bei den 1H- und 13C-Spektren nicht bestimmt.
Elementaranalysen: Die quantitative Bestimmung von Kohlenstoff, Wasserstoff, Stickstoff
und Schwefel erfolgte verbrennungsanalytisch an einem Gerät des Typs Thermo Finnigan
EAGER 300 (Flash 1112 Series) Gerät.
Infrarotspektren: Die IR-Spektren von wurden an einem Nicolet Magna System 750-
Spektrometer aufgenommen. Feststoffe wurden als Kaliumbromid-Presslinge, Lösungen in
CaF2-Küvetten unter Kompensation der Solvensabsorption vermessen. Die Wellenzahlen ν̃
sind in cm–1 angegeben. Die Zuordnung der Banden erfolgte anhand einschlägiger
Literatur.[33]

Eisenkomplexe des pyN4-Liganden 59
Massenspektren: Die Aufnahme der Massenspektren erfolgte unter Verwendung von
folgendem Gerät: Varian 311A (EI-MS, 70 eV).
Einkristallröntgenstrukturanalysen: Die Datensammlung erfolgte an folgendem Gerät:
Oxford Diffraction Xcalibur S Sapphire-Diffraktometer (Siemens) unter Verwendung von
Mo-Kα-Strahlung (λ = 0.71073 Ǻ) bei 100 K. Zur empirischen Absorptionskorrektur wurde,
wenn erforderlich, das Programm SADABS (Bruker AXS, 2002) verwendet. Die Strukturen
wurden mit direkten Methoden gelöst und anschließend an F2 verfeinert (SHELXT NT 6.12,
Bruker AXS, 2002). Sofern nicht anders angegeben, wurden alle Nichtwasserstoffatome
anisotrop verfeinert. Sämtliche Wasserstoffatome befinden sich in nach geometrischen
Gesichtspunkten berechneten Lagen und wurden mit einem isotropen Auslenkungsparameter
versehen, der dem 1.2- bzw. 1.5 fachen des äquivalenten isotropen Auslenkungsparameters
des die Wasserstoffatome tragenden C-, N- bzw. O-Atomes entspricht.
UV/Vis-Spektroskopie: Gemessen wurde an einem Cary 50-Spektrometer der Firma Varian
mit einer UV/Vis-Quarz-Tauchsonde (Schichtdicke 1 mm, Fa. Hellma). Die Messzelle wurde
eigens konstruiert. Die Temperatur wurde direkt in Lösung gemessen. Vor der Aufnahme
jedes Spektrums wurde bei eingestellter Temperatur 5 min equilibriert. Es wurden folgende
Konzentrationen vermessen: 5: 2.0·10–3 mol/l; 8: 1,5·10–3 mol/l und 12: 9.7·10-4 mol/l.
Cyclische Voltammetrie: Gemessen wurde mit einem PAR Model 263A-Potentiostat mit
zwei Platinelektroden als Referenz- und Gegenelektrode. Als Arbeitselektrode diente eine
Glas-Kohlenstoffelektrode (Durchmesser 3 mm). Als Leitsalz wurde NBu4PF6 (0.1 M)
verwendet. Die gemessenen Potentiale wurden über Fc/Fc+ als internen Standard auf NHE
bezogen mit E(Fc/Fc+) = +0.41 V gegen NHE.[34]

Eisenkomplexe des pyN4-Liganden 60
3.9.2 Synthesen
3.9.2.1 [FeII(pyN4)(H2O)](BF4)2 (4)
C13H27B2F8FeN5O
M = 498.84
gelbes Pulver
Zu einer Suspension von 1 · 4 HBr · MeOH (88 mg, 0.145 mmol) in MeOH (5 ml) wird
NaOMe (0.58 ml einer 1M Lösung in MeOH, 0.58 mmol) gegeben. Die erhaltene Lösung
wird 5 min gerührt, das Lösemittel abgezogen und die verbliebene Substanz 30 min im
Hochvakuum getrocknet. Der erhaltene ölige Feststoff wird in THF (10 ml) suspendiert und
30 min gerührt. Mittels Kannülenfiltration wird vom NaBr abgetrennt. Fe(BF4)2 · 6 H2O (44
mg, 0.130 mmol), gelöst in 5 ml THF, wird durch einen 0.2 µm-Spritzenfilter zugetropft.
Zunächst bildet sich ein rötlicher Niederschlag, dessen Farbe nach vollständiger Zugabe in
gelb übergeht. Die Mischung wird 18 h gerührt und anschließend abfiltriert. Der erhaltene
Feststoff wird mit THF (5 ml) und Et2O (3 × 5 ml) gewaschen und 30 min im Stickstoffstrom
getrocknet. Das Produkt ist ein gelber Feststoff (20 mg, 31 % nach Umfüllen).
IR (KBr): ν̃ = 3589s (ν(O–H)), 3330vs, 2978vs, 2887s, 1603vs, 1579s, 1525m, 1473s,
1418m, 1400m, 1372m, 1292m, 1261m, 1062vs (BF4–), 879m, 829s, 819s, 768m, 708w,
611w, 572m, 492m, 468m, 404m cm–1.
Elementaranalyse: Ber. ( %) für C13H27B2F8FeN5O (498.84): C 31.30, H 5.46, N 14.04; gef.
C 31.58, H 5.21, N 14.07.
N
H2NH2N Fe NH2
NH2
OH H
2+
(BF4–)2

Eisenkomplexe des pyN4-Liganden 61
3.9.2.2 Umsetzung von pyN4 mit Fe(ClO4)2 · x H2O; versuchte Erzeugung von
[FeII(pyN4)(H2O)](ClO4)2
C13H27Cl2FeN5O9
M = 524.13
Das erhaltene Material ist ein gelbes Pulver. Es ist nicht stabil und zersetzt sich innerhalb von
12 h auch unter N2-Atmosphäre; wegen der extremen Empfindlichkeit der Substanz war eine
Charakterisierung bisher nicht möglich.
Verfahren: Zu einer Suspension von 1 · 4 HBr · MeOH (55 mg, 0.091 mmol) in MeOH (4 ml)
wird NaOMe (0.36 ml einer 1M Lösung in MeOH) gegeben. Die erhaltene Lösung wird 5
min gerührt, das Lösemittel abgezogen und der Rückstand 1.5 h im Hochvakuum getrocknet.
Der erhaltene ölige Feststoff wird in THF (3 ml) suspendiert und 30 min gerührt. Mittels
Kannülenfiltration wird vom NaBr abgetrennt. Die erhaltene Lösung wird zu Fe(ClO4)2 · x
H2O (27.5 mg, 0.082 mmol, x = 4.5 angenommen), gelöst in THF (3 ml), zugetropft. Es bildet
sich ein gelber Niederschlag, der nach 30 min Rühren abfiltriert wird. Der erhaltene Feststoff
wird mit THF (5 ml) und Et2O (3 × 5 ml) gewaschen und 30 min im Stickstoffstrom
getrocknet. Das Produkt ist ein gelber Feststoff (15 mg, 31 % nach Umfüllen, berechnet auf
den Aquaeisen(II)komplex als Perchloratsalz).
N
H2NH2N Fe NH2
NH2
OH H
2+
(ClO4–)2
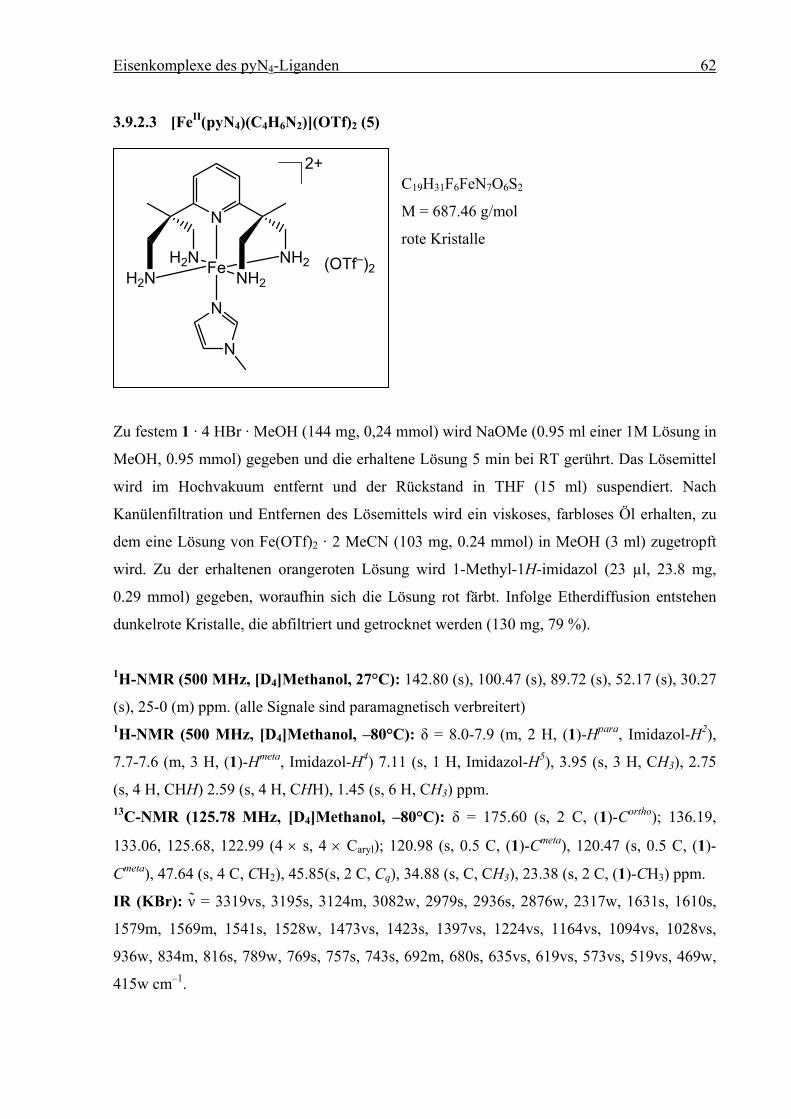
Eisenkomplexe des pyN4-Liganden 62
3.9.2.3 [FeII(pyN4)(C4H6N2)](OTf)2 (5)
C19H31F6FeN7O6S2
M = 687.46 g/mol
rote Kristalle
Zu festem 1 · 4 HBr · MeOH (144 mg, 0,24 mmol) wird NaOMe (0.95 ml einer 1M Lösung in
MeOH, 0.95 mmol) gegeben und die erhaltene Lösung 5 min bei RT gerührt. Das Lösemittel
wird im Hochvakuum entfernt und der Rückstand in THF (15 ml) suspendiert. Nach
Kanülenfiltration und Entfernen des Lösemittels wird ein viskoses, farbloses Öl erhalten, zu
dem eine Lösung von Fe(OTf)2 · 2 MeCN (103 mg, 0.24 mmol) in MeOH (3 ml) zugetropft
wird. Zu der erhaltenen orangeroten Lösung wird 1-Methyl-1H-imidazol (23 µl, 23.8 mg,
0.29 mmol) gegeben, woraufhin sich die Lösung rot färbt. Infolge Etherdiffusion entstehen
dunkelrote Kristalle, die abfiltriert und getrocknet werden (130 mg, 79 %).
1H-NMR (500 MHz, [D4]Methanol, 27°C): 142.80 (s), 100.47 (s), 89.72 (s), 52.17 (s), 30.27
(s), 25-0 (m) ppm. (alle Signale sind paramagnetisch verbreitert) 1H-NMR (500 MHz, [D4]Methanol, –80°C): δ = 8.0-7.9 (m, 2 H, (1)-Hpara, Imidazol-H2),
7.7-7.6 (m, 3 H, (1)-Hmeta, Imidazol-H4) 7.11 (s, 1 H, Imidazol-H5), 3.95 (s, 3 H, CH3), 2.75
(s, 4 H, CHH) 2.59 (s, 4 H, CHH), 1.45 (s, 6 H, CH3) ppm. 13C-NMR (125.78 MHz, [D4]Methanol, –80°C): δ = 175.60 (s, 2 C, (1)-Cortho); 136.19,
133.06, 125.68, 122.99 (4 × s, 4 × Caryl); 120.98 (s, 0.5 C, (1)-Cmeta), 120.47 (s, 0.5 C, (1)-
Cmeta), 47.64 (s, 4 C, CH2), 45.85(s, 2 C, Cq), 34.88 (s, C, CH3), 23.38 (s, 2 C, (1)-CH3) ppm.
IR (KBr): ν̃ = 3319vs, 3195s, 3124m, 3082w, 2979s, 2936s, 2876w, 2317w, 1631s, 1610s,
1579m, 1569m, 1541s, 1528w, 1473vs, 1423s, 1397vs, 1224vs, 1164vs, 1094vs, 1028vs,
936w, 834m, 816s, 789w, 769s, 757s, 743s, 692m, 680s, 635vs, 619vs, 573vs, 519vs, 469w,
415w cm–1.
N
H2NH2N Fe NH2
NH2
N
2+
(OTf–)2
N

Eisenkomplexe des pyN4-Liganden 63
Elementaranalyse: Ber. ( %) für C19H31F6FeN7O6S2 (687.46): C 33.20, H 4.55, N 14.26, S
9.33; gef. C 33.33, H 4.64, N 14.21, S 9.53.
3.9.2.4 [FeIII(pyN4)(C5H4NCO2]Br2 · MeOH (6 · MeOH)
C20H33Br2FeN6O3
M = 621.17 g/mol
rote Kristalle
Zu einer Suspension von 1 · 4 HBr · MeOH (100 mg, 0.16 mmol) in MeOH (3 ml) wird
LiOMe (0.66 ml einer 1M Lösung in MeOH, 0.66 mmol) gegeben. Die erhaltene Lösung wird
5 min bei RT gerührt und danach eine gelbe Lösung von FeCl2 (19 mg, 0.15 mmol) in MeOH
(1.5 ml) zugegeben, woraufhin die Lösung eine orange Farbe annimmt. Nach Zugabe von
Isonicotinsäure (19 mg, 0.15 mmol) gelöst in MeOH (15 ml) wird die Lösung rot. Die Lösung
wird auf die Hälfte eingeengt und filtriert. Nach 4 d bilden sich größere hellrote Kristalle, die
röntgenstrukturanalytisch charakterisiert werden konnten.
IR (KBr): ν̃ = 3180s, 3074s, 1644s, 1599s, 1575s, 1555s, 1494m, 1470s, 1456s, 1410s,
1402s, 1372s, 1341s, 1316s, 1304s, 1265s, 1221s, 1191s, 1162s, 1141s, 1103s, 1093s, 1060s,
1040s, 1021s, 997s, 983w, 947w, 902w, 882w, 860w, 845w, 830m, 775m, 762s, 747s, 707s,
685s, 591m, 542m, 493m, 455m, 425m, 419, 408m cm–1.
Elementaranalyse: Ber. ( %) für C20H33Br2FeN6O3 (621,17): C 38.67, H 5.35, N 13.53; gef.
C 38.84, H 5.13, N 13.53.
N
H2NH2N Fe NH2
NH2
O
2+
(Br–)2 · MeOH
N
O

Eisenkomplexe des pyN4-Liganden 64
3.9.2.5 [(pyN4)FeII–NC5H3(CO2)(COO–)FeII(pyN4)]Br2 · 5.5 MeOH; 7 · 5.5 MeOH
C38.5H75Br2Fe2N11O9.5
M = 1115.57 g/mol
rote irreguläre Kristalle
Zu einer Suspension von 1 · 4 HBr · MeOH (107 mg, 0.18 mmol) in MeOH (10 ml) wird
LiOMe (0.70 ml einer 1M Lösung in MeOH, 0.70 mmol) gegeben. Die erhaltene Lösung wird
5 min bei RT gerührt und danach eine gelbe Lösung von FeCl2 (22 mg, 0.18 mmol) in MeOH
(2 ml) zugegeben, woraufhin die Lösung eine orange Farbe annimmt. Pyridin-3,5-
dicarbonsäure (29 mg, 0.17 mmol) wird in MeOH (3 ml) gelöst und mit LiOMe (0.35 ml
einer 1M Lösung in MeOH, 0.35 mmol) deprotoniert. Diese Lösung wird zur [(1)FeII]-Lösung
zugetropft, die daraufhin eine dunkelrote Farbe annimmt. Nach 4 d isothermaler Eindiffusion
von Et2O bilden sich tiefrote Kristalle, die röntgenstrukturanalytisch charakterisiert werden
konnten.
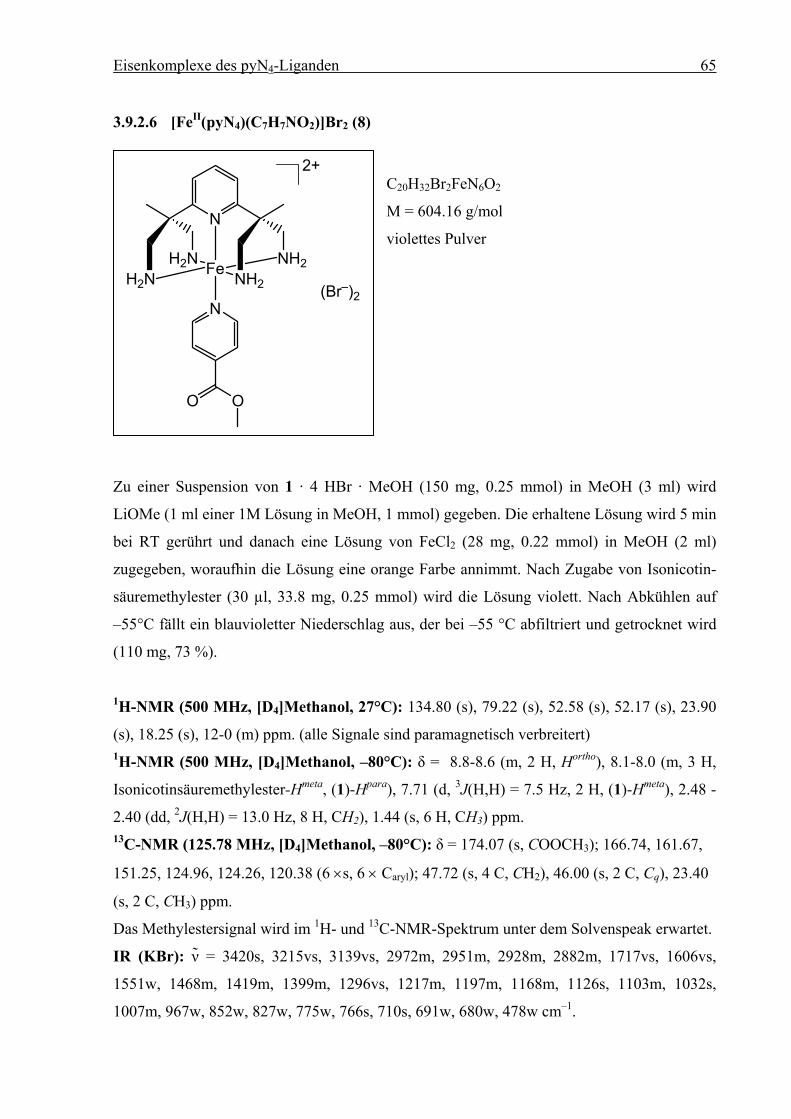
Eisenkomplexe des pyN4-Liganden 65
3.9.2.6 [FeII(pyN4)(C7H7NO2)]Br2 (8)
C20H32Br2FeN6O2
M = 604.16 g/mol
violettes Pulver
Zu einer Suspension von 1 · 4 HBr · MeOH (150 mg, 0.25 mmol) in MeOH (3 ml) wird
LiOMe (1 ml einer 1M Lösung in MeOH, 1 mmol) gegeben. Die erhaltene Lösung wird 5 min
bei RT gerührt und danach eine Lösung von FeCl2 (28 mg, 0.22 mmol) in MeOH (2 ml)
zugegeben, woraufhin die Lösung eine orange Farbe annimmt. Nach Zugabe von Isonicotin-
säuremethylester (30 µl, 33.8 mg, 0.25 mmol) wird die Lösung violett. Nach Abkühlen auf
–55°C fällt ein blauvioletter Niederschlag aus, der bei –55 °C abfiltriert und getrocknet wird
(110 mg, 73 %).
1H-NMR (500 MHz, [D4]Methanol, 27°C): 134.80 (s), 79.22 (s), 52.58 (s), 52.17 (s), 23.90
(s), 18.25 (s), 12-0 (m) ppm. (alle Signale sind paramagnetisch verbreitert) 1H-NMR (500 MHz, [D4]Methanol, –80°C): δ = 8.8-8.6 (m, 2 H, Hortho), 8.1-8.0 (m, 3 H,
Isonicotinsäuremethylester-Hmeta, (1)-Hpara), 7.71 (d, 3J(H,H) = 7.5 Hz, 2 H, (1)-Hmeta), 2.48 -
2.40 (dd, 2J(H,H) = 13.0 Hz, 8 H, CH2), 1.44 (s, 6 H, CH3) ppm. 13C-NMR (125.78 MHz, [D4]Methanol, –80°C): δ = 174.07 (s, COOCH3); 166.74, 161.67,
151.25, 124.96, 124.26, 120.38 (6 ×s, 6 × Caryl); 47.72 (s, 4 C, CH2), 46.00 (s, 2 C, Cq), 23.40
(s, 2 C, CH3) ppm.
Das Methylestersignal wird im 1H- und 13C-NMR-Spektrum unter dem Solvenspeak erwartet.
IR (KBr): ν̃ = 3420s, 3215vs, 3139vs, 2972m, 2951m, 2928m, 2882m, 1717vs, 1606vs,
1551w, 1468m, 1419m, 1399m, 1296vs, 1217m, 1197m, 1168m, 1126s, 1103m, 1032s,
1007m, 967w, 852w, 827w, 775w, 766s, 710s, 691w, 680w, 478w cm–1.
N
H2NH2N Fe NH2
NH2
N
2+
(Br–)2
OO

Eisenkomplexe des pyN4-Liganden 66
Elementaranalyse: Ber. ( %) für C20H32Br2FeN6O2 (604.16): C 39.76, H 5.34, N 13.91; gef.
C 39.84, H 5.27, N 13.85.
3.9.2.7 [FeII(pyN4)(C5H4NS)]Br · MeOH; 9 · MeOH
C19H33BrFeN6OS
M = 529.29 g/mol
rotbraune Prismen
Zu einer Suspension von 1 · 4 HBr · MeOH (172 mg, 0.28 mmol) in MeOH (3 ml) wird
LiOMe (1.1 ml einer 1M Lösung in MeOH, 1.1 mmol) gegeben. Die erhaltene Lösung wird
5 min bei RT gerührt und danach eine Lösung von FeCl2 (36 mg, 0.28 mmol) in MeOH (2 ml)
zugegeben, woraufhin die Lösung eine orange Farbe annimmt. 4-Mercaptopyridin (27 mg,
0.24 mmol) wird in MeOH (3 ml) gelöst und mit LiOMe (0.24 ml einer 1M Lösung in MeOH,
0.24 mmol) deprotoniert. Diese Lösung wird zur [(1)FeII]-Lösung zugetropft, die daraufhin
eine dunkelrote Farbe annimmt. Nach 4 d isothermaler Eindiffusion von Et2O bilden sich
rotbraune Kristalle, die röntgenstrukturanalytisch charakterisiert werden konnten.

Eisenkomplexe des pyN4-Liganden 67
3.9.2.8 [(pyN4)FeII–(NC5H4S)–{FeII(pyN4)}2]Br5 (10)
C44H79Br5Fe3N16S
M = 1431.33 g/mol
rotes Pulver
Zu einer Suspension von 1 · 4 HBr · MeOH (359 mg, 0.60 mmol) in MeOH (7 ml) wird
LiOMe (2.4 ml einer 1M Lösung in MeOH, 2.4 mmol) gegeben. Die erhaltene Lösung wird
5 min bei RT gerührt und danach eine Lösung von FeCl2 (75 mg, 0.59 mmol) in MeOH (5 ml)
zugegeben, woraufhin die Lösung eine orange Farbe annimmt. 4-Mercaptopyridin (13 mg,
0.12 mmol) wird in MeOH (4 ml) gelöst und mit LiOMe (0.12 ml einer 1M Lösung in MeOH,
0.12 mmol) deprotoniert. Diese Lösung wird zur [(1)FeII]-Lösung zugetropft, die daraufhin
eine dunkelrote Farbe annimmt. Die erhaltene Lösung wird 18 h gerührt und anschließend
filtriert. Nach 4 d isothermaler Eindiffusion von Et2O bilden sich rotbraune Kristalle, die
abfiltriert und getrocknet werden (137 mg, 77 % berechnet auf 4-Mercaptopyridin).
Elementaranalyse: Ber. ( %) für C44H79Br5Fe3N16S · 0.5 LiBr (1474.75): C 35.83, H 5.40, N
15.20, S 2.17; gef. C 36.07, H 5.39, N 15.00, S 1.74.
N
H2NH2N Fe NH2
NH2
N
5+
(Br–)5
S[FeII(pyN4)][(pyN4)FeII]
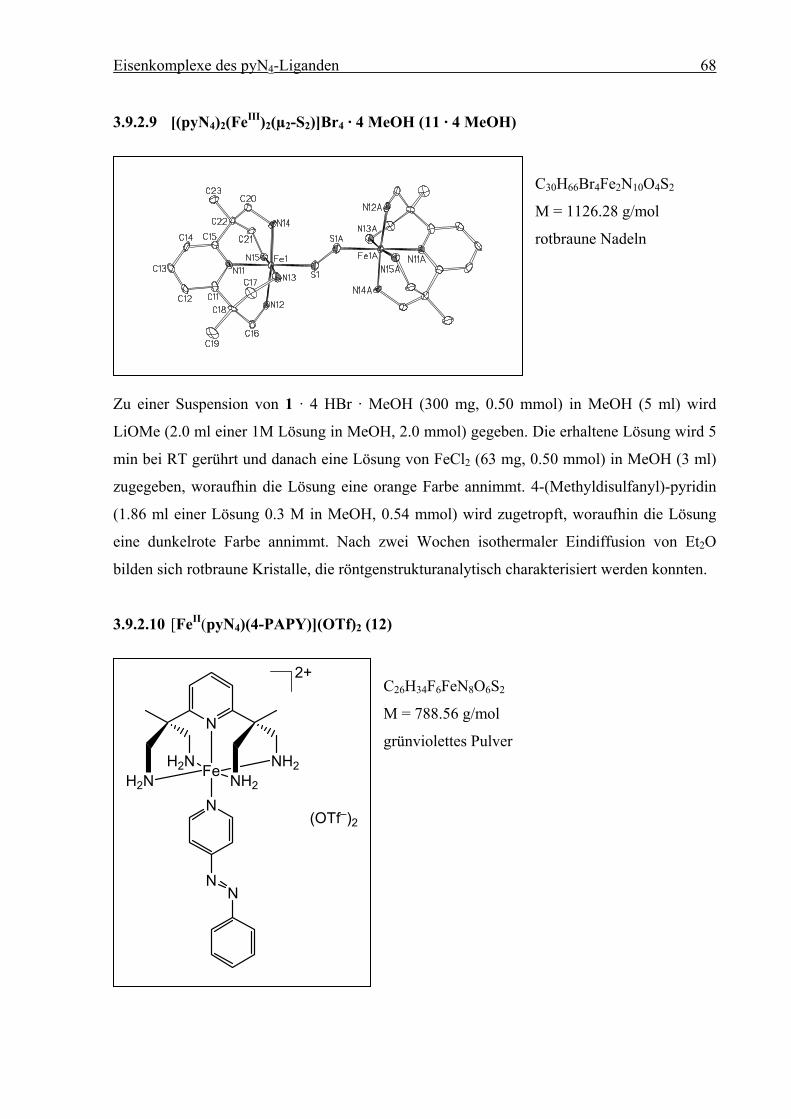
Eisenkomplexe des pyN4-Liganden 68
3.9.2.9 [(pyN4)2(FeIII)2(µ2-S2)]Br4 · 4 MeOH (11 · 4 MeOH)
C30H66Br4Fe2N10O4S2
M = 1126.28 g/mol
rotbraune Nadeln
Zu einer Suspension von 1 · 4 HBr · MeOH (300 mg, 0.50 mmol) in MeOH (5 ml) wird
LiOMe (2.0 ml einer 1M Lösung in MeOH, 2.0 mmol) gegeben. Die erhaltene Lösung wird 5
min bei RT gerührt und danach eine Lösung von FeCl2 (63 mg, 0.50 mmol) in MeOH (3 ml)
zugegeben, woraufhin die Lösung eine orange Farbe annimmt. 4-(Methyldisulfanyl)-pyridin
(1.86 ml einer Lösung 0.3 M in MeOH, 0.54 mmol) wird zugetropft, woraufhin die Lösung
eine dunkelrote Farbe annimmt. Nach zwei Wochen isothermaler Eindiffusion von Et2O
bilden sich rotbraune Kristalle, die röntgenstrukturanalytisch charakterisiert werden konnten.
3.9.2.10 [FeII(pyN4)(4-PAPY)](OTf)2 (12)
C26H34F6FeN8O6S2
M = 788.56 g/mol
grünviolettes Pulver
N
H2NH2N Fe NH2
NH2
N
2+
(OTf–)2
NN

Eisenkomplexe des pyN4-Liganden 69
Zu festem 1 · 4 HBr · MeOH (783 mg, 1.29 mmol) wird NaOMe (5.68 ml einer 1M Lösung in
MeOH, 5.68 mmol) gegeben und die erhaltene Lösung 15 min bei RT gerührt. Das Lösemittel
wird im Hochvakuum entfernt und der Rückstand in THF (25 ml) suspendiert. Nach
Kanülenfiltration und Entfernen des Lösemittels wird ein viskoses, farbloses Öl erhalten, zu
dem eine Lösung von Fe(OTf)2 · 2 MeCN (563 mg, 1.29 mmol) in MeOH (3 ml) gegeben
wird. Zu der erhaltenen roten Lösung wird (E)-4-(Phenyldiazenyl)pyridin (262 mg, 1.43
mmol) gegeben, woraufhin die Lösung eine tiefdunkelgrüne Farbe annimmt. Die Lösung wird
am Vakuum getrocknet und der entstandene blaue Feststoff in wenig Methanol
aufgenommen. Innerhalb von 24 h entsteht bei –78 °C ein grüner Niederschlag, der bei
–78 °C abfiltriert und getrocknet wird (773 mg, 76 %).
1H-NMR (500 MHz, [D4]Methanol, 27°C): 140.37 (s), 98.39 (s), 51.52 (s), 24.63 (s), 13-0
(m) ppm. (alle Signale sind paramagnetisch verbreitert) 1H-NMR (500 MHz, [D4]Methanol, –60°C): δ = 9.1-9.0 (m, 2 H, py-Hortho), 8.5-7.5 (m, 10
H, 4-PAPY, (1)-Hpara), 3.0 - 2.2 (m, 8 H, CH2), 1.46 (s, 6 H, CH3) ppm. 13C-NMR (125.78 MHz, [D4]Methanol, –80°C): δ = 173.92 (s, 2 C, 4-PAPY, py-Cortho),
162.06, 157.09, 153.68, 152.10, 137.97, 134.52, 130.97, 125.50, 124.93, 122.98, 120.45,
118.43, 117.97 (13 × s, 13 C, 13 × Caryl), 47.80 (s, 4 C, CH2), 46.02 (s, 2 C, Cq), 23.36 (s, 2 C,
CH3) ppm.
Elementaranalyse: Ber. ( %) für C26H34F6FeN8O6S2 (788.56): C 39.60, H 4.35, N 14.21, S
8.13; gef. C 39.26, H 4.42, N 14.29, S 8.04.
3.9.2.11 [FeII(pyN4)(CNtBu)]Br2 (13), [FeII(pyN4)(CNcy)]Br2 (14) und
[FeII(pyN4)(CNC6H4OCH3)]Br2 (15)
Allgemeine Arbeitsvorschrift für die Darstellung der Komplexe des Typs:[(1)FeII(CN–R)]Br2:
Zu einer Suspension von 1 · 4 HBr · MeOH (620 mg, 1.02 mmol) in MeOH (10 ml) wird
LiOMe (4.08 ml einer 1M Lösung in MeOH, 4.08 mmol) gegeben. Die erhaltene Lösung wird
5 min bei RT gerührt und danach eine Lösung von FeCl2 (130 mg, 1.02 mmol) in MeOH
(13 ml) zugegeben, woraufhin die Lösung eine orange Farbe annimmt. Nach Zugabe von 1.1
Äquivalenten Isonitril hellt die Lösung leicht auf. Das Lösemittel wird auf 1/5 eingeengt,
woraufhin sich ein oranger Niederschlag bildet, der abfiltriert und getrocknet wird (86 %).
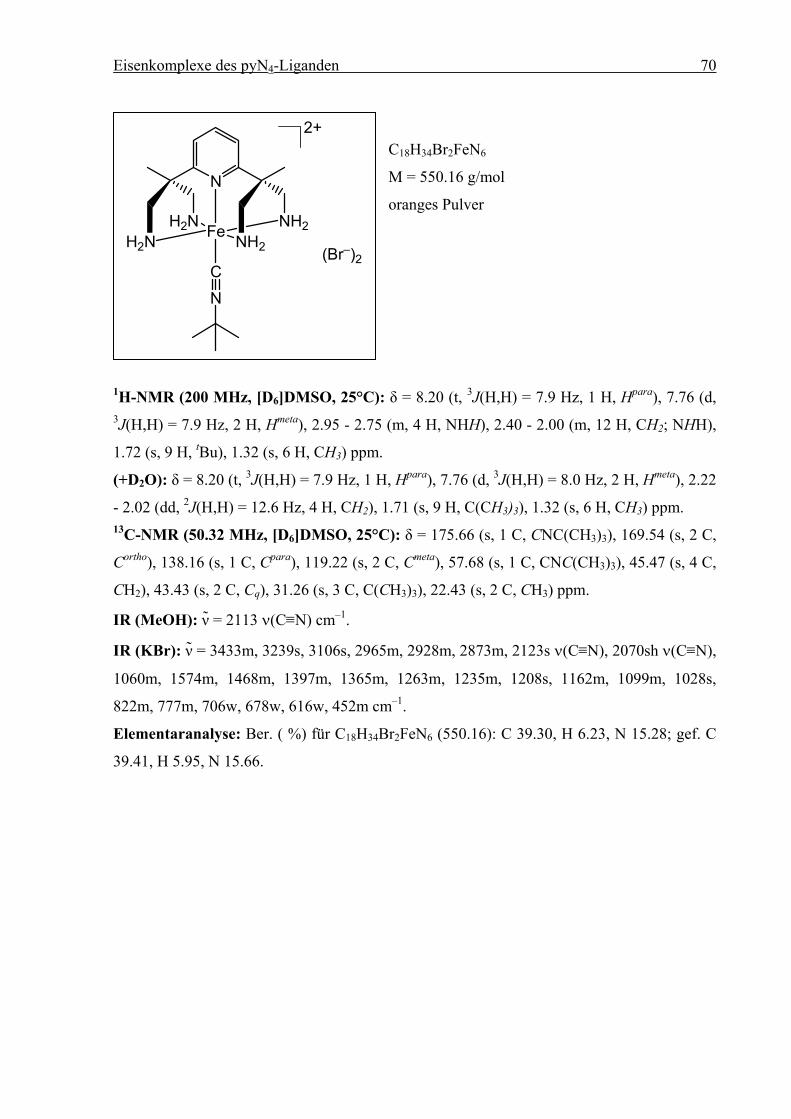
Eisenkomplexe des pyN4-Liganden 70
C18H34Br2FeN6
M = 550.16 g/mol
oranges Pulver
1H-NMR (200 MHz, [D6]DMSO, 25°C): δ = 8.20 (t, 3J(H,H) = 7.9 Hz, 1 H, Hpara), 7.76 (d, 3J(H,H) = 7.9 Hz, 2 H, Hmeta), 2.95 - 2.75 (m, 4 H, NHH), 2.40 - 2.00 (m, 12 H, CH2; NHH),
1.72 (s, 9 H, tBu), 1.32 (s, 6 H, CH3) ppm.
(+D2O): δ = 8.20 (t, 3J(H,H) = 7.9 Hz, 1 H, Hpara), 7.76 (d, 3J(H,H) = 8.0 Hz, 2 H, Hmeta), 2.22
- 2.02 (dd, 2J(H,H) = 12.6 Hz, 4 H, CH2), 1.71 (s, 9 H, C(CH3)3), 1.32 (s, 6 H, CH3) ppm. 13C-NMR (50.32 MHz, [D6]DMSO, 25°C): δ = 175.66 (s, 1 C, CNC(CH3)3), 169.54 (s, 2 C,
Cortho), 138.16 (s, 1 C, Cpara), 119.22 (s, 2 C, Cmeta), 57.68 (s, 1 C, CNC(CH3)3), 45.47 (s, 4 C,
CH2), 43.43 (s, 2 C, Cq), 31.26 (s, 3 C, C(CH3)3), 22.43 (s, 2 C, CH3) ppm.
IR (MeOH): ν̃ = 2113 ν(C≡N) cm–1.
IR (KBr): ν̃ = 3433m, 3239s, 3106s, 2965m, 2928m, 2873m, 2123s ν(C≡N), 2070sh ν(C≡N),
1060m, 1574m, 1468m, 1397m, 1365m, 1263m, 1235m, 1208s, 1162m, 1099m, 1028s,
822m, 777m, 706w, 678w, 616w, 452m cm–1.
Elementaranalyse: Ber. ( %) für C18H34Br2FeN6 (550.16): C 39.30, H 6.23, N 15.28; gef. C
39.41, H 5.95, N 15.66.
N
H2NH2N Fe NH2
NH2
C
2+
(Br–)2
N

Eisenkomplexe des pyN4-Liganden 71
C20H36Br2FeN6
M = 576.19 g/mol
oranges Pulver
1H-NMR (200 MHz, [D6]DMSO, 25°C): δ = 8.19 (t, 3J(H,H) = 8.0 Hz, 1 H, Hpara), 7.75 (d, 3J(H,H) = 8.0 Hz, 2 H, Hmeta), 4.45 - 4.25 (m, 1 H, CHNC), 3.05 - 2.75 (m, 4 H, NHH), 2.60 -
1.15 (m, 22 H, CH2 (1) und Cyclohexylrest), NHH), 1.31 (s, 6 H, CH3) ppm.
(+D2O): δ = 8.18 (t, 3J(H,H) = 8.0 Hz, 1 H, Hpara), 7.75 (d, 3J(H,H) = 8.0 Hz, 2 H, Hmeta), 4.40
- 4.20 (m, 1 H, CHNC), 2.25 - 1.95 (dd, 2J(H,H) = 12.7 Hz, 4 H, CH2), 2.10 - 1.15 (m, 10 H,
Cyclohexyl-CH2), 1.31 (s, 6 H, CH3) ppm. 13C-NMR (50.32 MHz, [D6]DMSO, 25°C): δ = 176.66 (s, 1 C, C≡N), 169.50 (s, 2 C, Cortho),
138.14 (s, 1 C, Cpara), 119.21 (s, 2 C, Cmeta), 55.08 (s, 1 C, C≡N–C), 45.41 (s, 4 C, CH2),
43.40 (s, 2 C, Cq), 33.23 (s, 2 C, CH2), 24.84 (s, 1 C, CH2), 22.88 (s, 2 C, CH2), 22.42 (s, 2 C,
CH3) ppm.
IR (MeOH): ν̃ = 2120 ν(C≡N) cm–1.
IR (KBr): ν̃ = 3479m, 3424m, 3216s, 3138s, 2930s, 2882m, 2857m, 2118vs ν(C≡N), 1604m,
1572m, 1470s, 1397m, 1366m, 1321w, 1262w, 1169m, 1092w, 1025m, 820w, 766w, 656w,
562w, 444w cm–1.
Elementaranalyse: Ber. ( %) für C20H36Br2FeN6 (576.19): C 41.69, H 6.30, N 14.59; gef. C
40.79, H 6.06, N 14.59.
N
H2NH2N Fe NH2
NH2
C
2+
(Br–)2
N
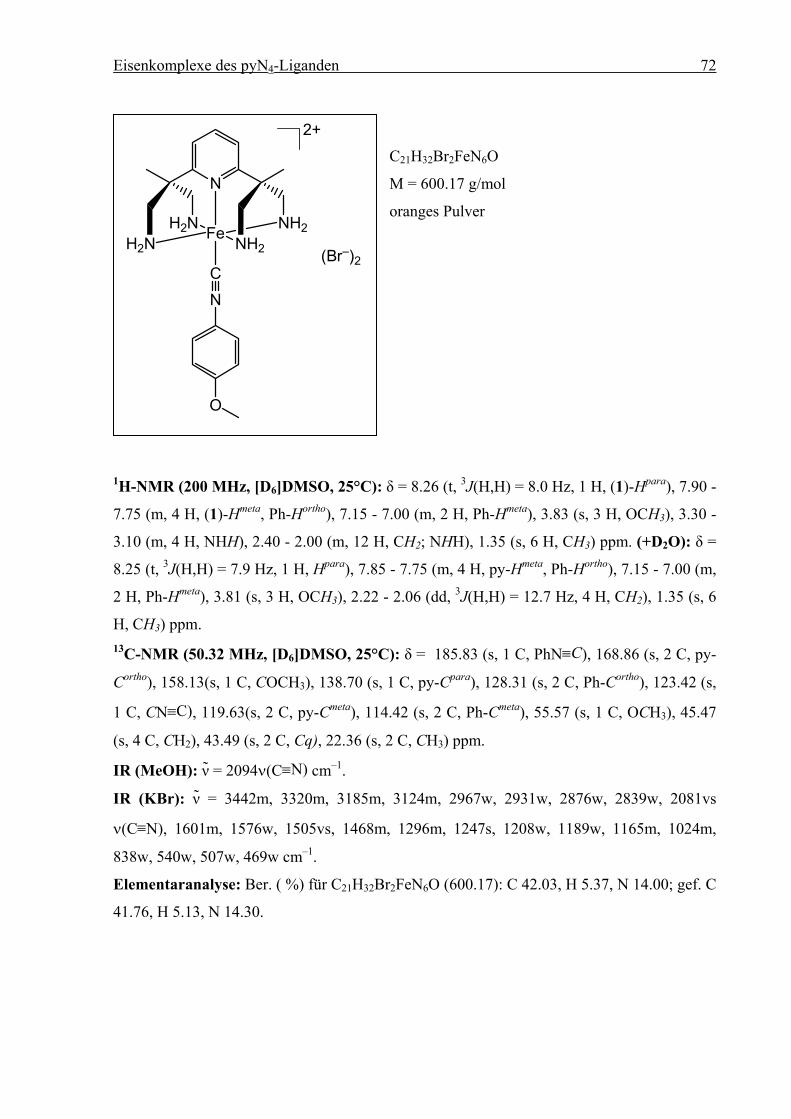
Eisenkomplexe des pyN4-Liganden 72
C21H32Br2FeN6O
M = 600.17 g/mol
oranges Pulver
1H-NMR (200 MHz, [D6]DMSO, 25°C): δ = 8.26 (t, 3J(H,H) = 8.0 Hz, 1 H, (1)-Hpara), 7.90 -
7.75 (m, 4 H, (1)-Hmeta, Ph-Hortho), 7.15 - 7.00 (m, 2 H, Ph-Hmeta), 3.83 (s, 3 H, OCH3), 3.30 -
3.10 (m, 4 H, NHH), 2.40 - 2.00 (m, 12 H, CH2; NHH), 1.35 (s, 6 H, CH3) ppm. (+D2O): δ =
8.25 (t, 3J(H,H) = 7.9 Hz, 1 H, Hpara), 7.85 - 7.75 (m, 4 H, py-Hmeta, Ph-Hortho), 7.15 - 7.00 (m,
2 H, Ph-Hmeta), 3.81 (s, 3 H, OCH3), 2.22 - 2.06 (dd, 3J(H,H) = 12.7 Hz, 4 H, CH2), 1.35 (s, 6
H, CH3) ppm. 13C-NMR (50.32 MHz, [D6]DMSO, 25°C): δ = 185.83 (s, 1 C, PhN≡C), 168.86 (s, 2 C, py-
Cortho), 158.13(s, 1 C, COCH3), 138.70 (s, 1 C, py-Cpara), 128.31 (s, 2 C, Ph-Cortho), 123.42 (s,
1 C, CN≡C), 119.63(s, 2 C, py-Cmeta), 114.42 (s, 2 C, Ph-Cmeta), 55.57 (s, 1 C, OCH3), 45.47
(s, 4 C, CH2), 43.49 (s, 2 C, Cq), 22.36 (s, 2 C, CH3) ppm.
IR (MeOH): ν̃ = 2094ν(C≡N) cm–1.
IR (KBr): ν̃ = 3442m, 3320m, 3185m, 3124m, 2967w, 2931w, 2876w, 2839w, 2081vs
ν(C≡N), 1601m, 1576w, 1505vs, 1468m, 1296m, 1247s, 1208w, 1189w, 1165m, 1024m,
838w, 540w, 507w, 469w cm–1.
Elementaranalyse: Ber. ( %) für C21H32Br2FeN6O (600.17): C 42.03, H 5.37, N 14.00; gef. C
41.76, H 5.13, N 14.30.
N
H2NH2N Fe NH2
NH2
C
2+
(Br–)2
N
O
Eisenkomplexe des pyN4-Liganden 73
3.9.3 Röntgenstrukturanalysen
Röntgenfähige Kristalle von 5, 8 · MeOH, 10 · 5 MeOH, 13 · 2 MeOH, 14 · 2 MeOH und 15
wurden durch Eindiffusion von Diethylether direkt aus der methanolischen Reaktionslösung
erhalten. Die Kristallisationen von 6 · MeOH, 7 · 5.5 MeOH, 9 · MeOH und 11 · 4 MeOH
sind im Experimentellen Teil beschrieben. Die Kristalle von 12a/b · 1.5 MeOH wurden aus
einer grünen Reaktionslösung von 1 · 4 HBr + 4 LiOMe + FeCl2 + 4-PAPY nach Abkühlen
auf –32 °C erhalten.
Tabelle 3.11: Kristallographische Daten und Angaben zur Strukturbestimmung.
Verbindung 5 6 · MeOH 7 · 5.5 MeOH 8 · MeOH Summenformel C19H31F6FeN7O6
S2 C20H33Br2FeN6O3
C38.50H75Br2Fe2N11O9.50
C21H36Br2FeN6O3
Mr [gmol–1] 687.48 621.19 1115.61 636.23 Kristallgröße [mm]
0.18 × 0.18 × 0.14
0.28 × 0.25 × 0.23
0.16 × 0.15 × 0.08
0.18 × 0.14 × 0.14
Farbe rotbraun rot rot rot F (000) 1416 1260 2324 1296 Kristallsystem monoklin monoklin monoklin orthorhombisch Raumgruppe P21/n (Nr. 14) P21/c (Nr. 14) P21/n (Nr. 14) P212121 (Nr. 19) a [Å] 10.1099(6) 11.2961(5) 18.529(2) 10.3279(6) b [Å] 11.2823(9) 11.5781(8) 12.904(1) 11.5295(7) c [Å] 24.199(3) 18.848(2) 21.872(2) 21.903(2) α [°] 90 90 90 90 β [°] 90.774(7) 92.782(5) 100.631(7) 90 γ [°] 90 90 90 90 V [Å 3] 2760.0(4) 2462.2(3) 5139.8(8) 2608.1(3) Z 4 4 4 4 ρber [gcm–3] 1.654 1.676 1.442 1.620 µ [mm–1] 0.787 3.892 2.179 3.676 Absorptions- korrektur
numerisch (Gauss-Integration)
SADABS SADABS SADABS
Temperatur [K] 100(2) 100(2) 100(2) 100(2) Tmin/Tmax 0.881/0.916 0.783/1.000 0.787/1.000 0.863/1.000 Scanmethode φ und ω-
rotations φ und ω-rotations
φ und ω-rotations
φ und ω-rotations
2 Θ Bereich [°] 3.10 - 27.10 5.7 - 57.4 6.5 - 54.2 6.4 - 57.0 gemessene Reflexe
80280 74665 61147 37330
Unabhängige Reflexe
6085 6347 11152 6532
beobachtete Reflexe[a]
5220 4990 7456 5525
Verfeinerte 373 388 610 334
Eisenkomplexe des pyN4-Liganden 74
Parameter wR2 (alle Daten) [b]
0.0856 0.0518 0.0980 0.0566
R1 (beob. Daten) [c]
0.0391 0.0256 0.0471 0.0282
ρfin (max/min) [e Å–3]
0.439/–0.520 0.503/–0.399 0.685/–0.459 0.291/–0.331
Gewichtungs-schema[d]
k = 0.0103 / l = 6.1909
k = 0.215 / l = 1.5315
k = 0.0247 / l = 11.7744
k = 0.0285 / l = 0.2519
Abs. Struktur- parameter
- - - 0.036(6)
Verbindung 9 · MeOH 10 · 5 MeOH 11 · 4 MeOH (12a/b) · 1.5 MeOH
Summenformel C19H33BrFeN6OS
C49H99Br5Fe3N16O5S
C30H66Br4Fe2N10O4S2
C25.5H40Br2FeN8O1.5
Mr [gmol–1] 529.29 1591.60 1126.39 698.33 Kristallgröße [mm]
0.25 × 0.25 × 0.23
0.23 × 0.15 × 0.08
0.14 × 0.03 × 0.02
0.20 × 0.13 × 0.02
Farbe rotbraun orange rotbraun braun F (000) 1096 1628 572 2856 Kristallsystem monoklin triklin triklin monoklin Raumgruppe P21/c (Nr. 14) P1 (Nr. 2) P1 (Nr. 2) P21/n (Nr. 14) a [Å] 8.4809(4) 15.563(2) 7.966(2) 19.372(7) b [Å] 20.770(1) 16.595(2) 10.432(2) 11.820(1) c [Å] 12.9507(7) 16.645(1) 13.577(2) 27.894(4) α [°] 90 111.455(6) 86.86(1) 90 β [°] 97.253(5) 101.920(7) 88.29(2) 105.87(2) γ [°] 90 113.011(7) 86.61(2) 90 V [Å 3] 2263.0(2) 3362.0(6) 1124.2(4) 6144(3) Z 4 2 1 8 ρber [gcm–3] 1.554 1.572 1.664 1.510 µ [mm–1] 2.547 3.694 4.337 3.127 Absorptions- korrektur
SADABS SADABS SADABS SADABS
Temperatur [K] 100(2) 100(2) 100(2) 100(2) Tmin/Tmax 0.952/1.000 0.546/0.740 0.652/0.920 0.535/1.000 Scanmethode φ und ω-
rotations ω-rotations ω-rotations φ und ω-
rotations 2 Θ Bereich [°] 6.2 - 55.8 6.7 - 52.8 6.0 - 50.1 6.4 - 52.1 gemessene Reflexe
63229 54965 16055 55103
Unabhängige Reflexe
5390 13667 3955 12013
beobachtete Reflexe[a]
4370 10130 2446 7468
Verfeinerte Parameter
266 729 241 747
Eisenkomplexe des pyN4-Liganden 75
wR2 (alle Daten) [b]
0.0810 0.1636 0.1835 0.1609
R1 (beob. Daten) [c]
0.0352 0.0539 0.0685 0.0726
ρfin (max/min) [e Å–3]
0.455/–0.736 1.531/–3.209 1.525/–1.084 1.117/–0.734
Gewichtungs-schema[d]
k = 0.0290 / l = 4.6552
k = 0.0700 / l = 33.3010
k = 0.0770 / l = 7.4831
k = 0.0374 / l = 46.9873
Abs. Struktur- parameter
- - - -
Verbindung 13 · 2 MeOH 14 · 2 MeOH 15 Summenformel C20H42Br2FeN6
O2 C22H44Br2FeN6O2
C21H32Br2FeN6O
Mr [gmol–1] 614.27 640.30 600.20 Kristallgröße [mm]
0.28 × 0.14 × 0.17
0.25 × 0.23 × 0.21
0.25 × 0.21 × 0.18
Farbe rotbraun rot rot F (000) 1264 1320 1216 Kristallsystem monoklin monoklin monoklin Raumgruppe P21/n (Nr. 14) P21/n (Nr. 14) P21/c (Nr. 14) a [Å] 12.0765(2) 10.0364(5) 10.1680(6) b [Å] 11.7492(5) 11.8961(8) 12.5091(8) c [Å] 19.7937(12) 23.432(2) 19.1339(12) α [°] 90 90 90 β [°] 103.429(4) 90.055(5) 100.659(4) γ [°] 90 90 90 V [Å 3] 2731.7(2) 2797.6(3) 2391.7(3) Z 4 4 4 ρber [gcm–3] 1.494 1.520 1.667 µ [mm–1] 3.504 3.425 3.997 Absorptions- korrektur
SADABS SADABS SADABS
Temperatur [K] 100(2) 100(2) 100(2) Tmin/Tmax 0.411/0.551 0.489/0.429 0.369/ 0.487 Scanmethode ω-rotations φ und ω-
rotations ω-rotations
2 Θ Bereich [°] 2.96 - 28.00 3.43 - 27.10 3.43 - 27.88 gemessene Reflexe
49195 57599 31091
Unabhängige Reflexe
6581 6153 5695
beobachtete Reflexe[a]
5549 5122 4624
Verfeinerte Parameter
319 380 283
wR2 (alle Daten) [b]
0.0932 0.1224 0.0547

Eisenkomplexe des pyN4-Liganden 76
R1 (beob. Daten) [c]
0.0342 0.0476 0.0252
ρfin (max/min) [e Å–3]
0.777/–1.123 0.874/–1.033 0.442/ –0.410
Gewichtungs-schema[d]
k = 0.0362 / l = 7.4833
k = 0.0468 / l = 11.7516
k = 0.0200 / l = 2.1174
Abs. Struktur- parameter
- - -
[a] mit Fo ≥ 4 σ(F); [b] wR2 = ({Σ [w(Fo
2 – Fc2)2]}/{Σ [w(Fo
2)2]})0.5; [c] R1 = Σ ||Fo| – |Fc||/Σ |Fo| for Fo > 4 σ(F); [d] w = 1/[σ2(Fo
2) + (k · P)2 + l · P] and P = (Fo2 + 2 · Fc
2)/3
3.10 Literatur
[1] J. Pitarch López, F. W. Heinemann, R. Prakash, B. A. Hess, O. Horner, C. Jeandey, J.-
L. Oddou, J.-M. Latour, A. Grohmann, Chem. Eur. J. 2002, 8, 5709-5722.
[2] A. F. Holleman, E. Wiberg, Lehrbuch der Anorganischen Chemie, 101. Auflage,
Walter de Gruyter, Berlin, New York, 1995, 1252.
[3] a) A. Bousseksou, G. Molnar, G. Matouzenko, Eur. J. Inorg. Chem. 2004, 4353-4369.
b) K. S. Murray, C. J. Kepert, Topics Curr. Chem. 2004, 233, 195.
[4] Stefan Leopold, Dissertation, Technische Universität Berlin, 2006.
[5] a) H. L. Chum, J. A. Vanin, M. I. D. Holanda, Inorg. Chem. 1982, 21, 1146-1152. b)
W. A. Baker, H. M. Bobonich, Inorg. Chem. 1964, 3, 1184.
[6] a) D. J. Rudd, C. R. Goldsmith, A. P. Cole, T. D. P. Stack, K. O. Hodgson, B.
Hedman, Inorg. Chem. 2005, 44, 1221-1229. b) C. R. Goldsmith, T. D. P. Stack,
Inorg. Chem. 2006, 45, 6048-6055.
[7] a) Y. Tai, A. Shaporenko, H.-T. Rong, M. Buck, W. Eck, M. Grunze, M. Zharnikov, J.
Phys. Chem. B 2004, 108, 16806-16810. b) J. C. Love, L. A. Estroff, J. K. Kriebel, R.
G. Nuzzo, G. M. Whitesides, Chem. Rev. 2005, 105, 1103-1169 und enthaltene
Referenzen. c) S. Onclin, B. J. Ravoo, D. N. Reinhoudt, Angew. Chem. Int. Ed. 2005,
44, 6282–6304.
[8] J. Pitarch Lopéz, H. Kämpf, M. Grunert, P. Gütlich, F. W. Heinemann, R. Prakasch,
A. Grohmann, Chem. Comm. 2006, 16, 1718-1720.

Eisenkomplexe des pyN4-Liganden 77
[9] http://www.sigmaaldrich.com/catalog/search/SpecificationSheetPage/ALDRICH/
334081, September 2006.
[10] P. Gütlich, H. A. Goodwin, Topics Curr. Chem. 2004, 233, 14.
[11] E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F.
Neese, A. J. Skulan, Y.-S. Yang, J. Zhou, Chemical Reviews, 2000, 100, 235-349.
[12] J. Pitarch Lopéz, Dissertation, Universität Erlangen-Nürnberg, 2003.
[13] C. Hu, B. C. Noll, C. E. Schulz, and W. R. Scheidt, Inorg. Chem., 2005, 44, 4346-
4358.
[14] A. Hauser, J. Adler, P. Gütlich, Chem. Phys. Lett. 1988, 152, 468-472.
[15] P. Gütlich, Mößbauer-Spectroscopy: Principles and Applications, Vorlesung an der
Universität Mainz, September 2006.
http://ak-guetlich.chemie.uni-mainz.de/Moessbauer_Lectures_web.pdf
[16] A. F. Holleman, E. Wiberg, Lehrbuch der Anorganischen Chemie, 101. Auflage,
Walter de Gruyter, Berlin, New York, 1995, 1252-1253.
[17] B. Kersting, M. J. Kolm, C. Janiak, Z. anorg. allg. Chem. 1998, 624, 775-780.
[18] a) C. A. Grapperhaus, B. Mienert, E. Bill, T. Weyermüller, K. Wieghardt, Inorg.
Chem. 2000, 39, 5306-5317. b) K. Meyer, E. Bill, B. Mienert, T. Weyhermüller, K.
Wieghardt, J. Am. Chem. Soc. 1999, 121, 4859-4876.
[19] R. G: Nuzzo, D. L. Allara, J. Am. Chem. Soc. 1983, 105, 4481-4483.
[20] a) K. Ataka, J. Heberle, J. Am. Chem. Soc. 2004, 126, 9445-9457. b) W. Zhou, T.
Baunach, V. Ivanova, D. M. Kolb, Langmuir 2004, 20, 4590-4595. c) I. Taniguchi, S.
Yoshimoto, K. Nishiyama, Chem. Let. 1997, 353-354. d) B. D. Lamp, D. Hobara, M.
D. Porter, K. Niki, T. M. Cotton, Langmuir 1997, 13, 736-741.
[21] a) D. Sellmann, G. Mahr, F. Knoch, Angew. Chem. Int. Ed. Engl. 1991, 30, 1477-
1479. b) D. Sellmann, J. Utz, F. W. Heinemann, Inorg.Chem. 1999, 38, 459 466.
[22] M. Bernien, W. Kuch, Freie Universität Berlin, unveröffentliche Ergebnisse.
[23] a) J. D. Franolic, M. Millar, S. A. Koch, Inorg. Chem. 1995, 34, 1981-1982. b) G. T.
Kubas, T. G. Spiro, J. Am. Chem. Soc. 1973, 95, 273-274. c) D. Seyferth, R. S.
Henderson, L.-C. Song, Organometallics 1982, 1, 125-133.
[24] C. Roux, J. Zarembowitch, B. Gallois, T. Granier and R. Claude, Inorg. Chem. 1994,
33, 2273-2279.
[25] E. V. Brown, G. R. Granneman, J. Am. Chem. Soc. 1975, 97, 621-627.

Eisenkomplexe des pyN4-Liganden 78
[26] a) I. S. Zavarine, C. P. Kubiak, T. Yamaguchi, K. Ota, T. Matsui, T. Ito, Inorg. Chem.,
2000, 39, 269-2698. b) M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in
der organischen Chemie, 5. Auflage, Georg Thieme Verlag Stuttgart, 1995, S. 38. c)
F. A. Cotton, chemical applications of group theory, interscience publishers a division
of John Wiley & sons, inc., New York, 1963, 266. d) D. A. Morgenstern, G.M.
Ferrence, J. Washington, J. I. Henderson, L. Rosenhein, J. D. Heise, P.E. Fanwick, C.
P. Kubiak, J. Am. Chem. Soc., 1996, 118, 2198.
[27] a) P. P. M. de Lange, M. v. Wijnkoop, H.-W. Frühauf, K. Vrieze, Organometallics,
1993, 12, 428-439. b) B.-S. Yoo, N.-S. Choi, C. Y. Shim, Y. Son, S.W. Lee, Inorg.
Chim. Acta, 2000, 309, 137.
[28] Acros Organics, Handbuch für Feinchemikalien, Schwerte, Deutschland, 2006-2007.
[29] S. Schmidt, L. Omnès, F. W. Heinemann, J. Kuhnigk, C. Krüger, A. Grohmann, Z.
Naturforsch. Teil B 1998, 53, 946-954.
[30] K. S. Hagen, Inorg. Chem. 2000, 39, 5867-5869.
[31] a) M. Brookhart, B. Grant, A. F. Volpe, Organometallics 1992, 11, 3920-3922. b) N.
A. Yakelis, R. G. Bergman, Organometallics 2005, 24, 3579-3581.
[32] T. M. Kitson, K. M. Loomes, Analytical biochemistry 1985, 146, 429-430.
[33] G. Socrates, Infrared Characteristic Group Frequencies, 2. ed., Wiley, New York
1994.
[34] H. M. Koepp, H. Wendt, H. Strehlow, Z. Elektrochem. 1960, 64, 483-491.

Übergangsmetallkomplexe des pyN3-Liganden 79
4 Übergangsmetallkomplexe des pyN3-Liganden
4.1 Einleitung
Es sind eine große Zahl vierzähniger Aminliganden und ihrer FeII-Komplexe beschrieben.
Zwei mit 2 (s. Kap. 2) vergleichbare Vertreter sind tren (Tris(2-aminomethyl)amin) und tpa
(Tris(pyridin-2-ylmethyl)amin). Der tren-Ligand bildet mit FeII keine einkernigen Komplexe.
Werden die primärem Amine derivatisiert oder die aliphatischen Amindonoren formal durch
Pyridindonoren ersetzt, so werden einkernige FeII-Komplexe erhalten.[1]
NNH2
H2NH2N
NN
NN
tren tpa
Abb. 4.1: Molekülstrukturen der Liganden tren und tpa.
Die Umsetzung des zu 1 analogen Tetraphosphanliganden (1Me, rationeller Name: 2,6-Bis(2-
methyl-1,3-bis(dimethylphosphino)propan-2-yl)pyridin) mit FeII-Salzen führt zu einer
selektiven P–C-Bindungsspaltung. Die rationelle Synthese des daraus resultierenden
vierzähnigen Triphosphanliganden py(PMe2)3 wurde von S. W. Kohl durchgeführt.[2]
N
PMe2
PMe2PMe2
Abb. 4.2: Molekülstruktur des py(PMe2)3-Liganden.

Übergangsmetallkomplexe des pyN3-Liganden 80
Dadurch eröffnet sich der Weg zur Synthese des tetradentaten Liganden pyN3 (Abb. 4.3). Es
ist zu erwarten, dass dieser an einem sechsfach koordinierten Metallzentrum zwei cis-ständige
Koordinationsstellen zur Verfügung stellt. Die höhere Flexibilität des Liganden gibt keine
Oktaedergeometrie vor, sondern sollte auch Koordinationsgeometrien geringerer Symmetrie
ermöglichen, wie sie u. a. in biologischen Systemen auftreten.[3]
N
NH2
NH2NH2
2
N
H2NH2N M NH2
LL
Abb. 4.3: links: pyN3-Ligand (2); rechts: denkbare Struktur von Komplexen des Chelat-
liganden 2 mit zwei einzähnigen Liganden in Nachbarstellung.
Das Koordinationsverhalten dieses neuen Liganden gegenüber Übergangsmetallen, vor allem
FeII, sollte untersucht werden. Die Reaktionen mit CuII, NiII und ZnII wurden ebenfalls
untersucht. Diese Metalle bilden mit 1 einkernige, gut charakterisierte Komplexe und eignen
sich daher gut für einen Vergleich beider Ligandtypen.[4]
4.2 Synthese des pyN3-Liganden
Die Synthese des zu py(PMe2)3 analogen pyN3-Liganden wurde ausgehend von der
gemeinsamen Vorstufe py(OMes)3 (rationeller Name: 2-(2-Methyl-1,3-dimethansulfonyl-
propan-2-yl)-6-(2-methyl-1-methansulfonyl-propan-2-yl)pyridin) durchgeführt. Die Synthese
erfolgt völlig analog zu der Synthese von 1.

Übergangsmetallkomplexe des pyN3-Liganden 81
1. PPh3/Pyridin2. NH3 cc/140°C/18h
DMSO70 °C, 2 d
NaN312
34
6
57
8 9 10111213
N
OMesOMes
OMes
N
N3
N3N3
N
NH2
NH2NH2
16 2
Schema 4.1: Synthese von 2.
py(OMes)3 wurde in DMSO mit sechs Äquivalenten NaN3 umgesetzt. Das nach Aufarbeitung
erhaltene 2-(1,3-diazido-2-methylpropan-2-yl)-6-(1-azido-2-methylpropan-2-yl)pyridin (16),
nachfolgend als Triazid (py(N3)3) bezeichnet, wurde aus Sicherheitsgründen aus der
etherischen Lösung nicht vollständig isoliert. Eine kleine Menge wurde zur Aufnahme von
NMR-Spektren und IR-Spektren getrocknet. Die Signale der drei Pyridinprotonen ergeben im 1H-NMR-Spektrum ein ABC-Spinsystem. Es zeigt bei 7.68 ppm ein Triplett für das para-
ständige Wasserstoffatom und bei 7.25 und 7.17 ppm die beiden Dubletts für die zum
Pyridinstickstoffatom meta-ständigen H-Atome. Die Kopplungskonstanten betragen jeweils
7.9 Hz. Die Verbindung ist Cs-symmetrisch. Die Methylgruppen 11 und 12 erscheinen in den 1H- und 13C-NMR-Spektren als ein Signal. Das IR-Spektrum (Film auf KBr-Pressling) zeigt
eine Bande bei 2101 cm–1 für die Azidgruppen.
Das Lösemittel wird durch Pyridinzugabe und Entfernen des Diethylethers im Vakuum
ausgetauscht. Nach Staudinger-Reduktion mit Triphenylphosphan kann 2-(1,3-diamino-2-
methylpropan-2-yl)-6-(1-amino-2-methylpropan-2-yl)pyridin (2) in sehr guter Ausbeute (95
%) erhalten werden. Das Produkt ist noch mit Spuren von Triphenylphosphanoxid
verunreinigt. Die 1H- und 13C-NMR-Spektren sind denen von Verbindung 16 ähnlich. Die
Zuordnung erfolgte durch Vergleich mit den Spektren der analogen py(PR2)3-Liganden.[5]
Das so erhaltene 2 ist ein gelbliches, zähes Öl. Analog zur pyN4-Synthese wurde mit 48 %er
HBr umgesetzt und getrocknet. Da sich die erhaltene Verbindung deutlich besser in MeOH
löst als 1 · 4 HBr, wurde statt mit MeOH mit Diethylether ausgerührt. Man erhält ein weißes
hygroskopisches Pulver, welches laut 1H-NMR-Spektrum und Elementaranalyse die
Zusammensetzung 2 · 4 HBr aufweist. Es sind folglich sowohl die drei primären
Aminofunktionen als auch das Pyridinstickstoffatom protoniert, was den hygroskopischen
Charakter des Produkts erklärt. Das Pulver ist noch mit Spuren von Triphenylphosphanoxid
verunreinigt, die sich auch nach mehrmaligem Waschen mit Diethylether und THF nicht
vollständig entfernen lassen.

Übergangsmetallkomplexe des pyN3-Liganden 82
Parallele Versuche, 2 durch Lösen in THF und anschließende Filtration zu reinigen
scheiterten, da 2 nach THF-Zugabe eine weiße gallertartige Masse bildet, sich jedoch nicht in
THF löst. Nach Entfernen von THF bildet sich wieder das zuvor eingesetzte Öl. Mit
Diethylether werden dieselben Beobachtungen gemacht.
Eine Reaktion analog der Freisetzung von 1 aus 1 · 4 HBr (Schema 4.2), um 2 in freier Form
zu erhalten, kann also nicht durchgeführt werden.
pyN3 · 4 HBr + 4.4 NaOMe
1. MeOH2. Trocknen3. Extraktion mit THF
pyN3
Schema 4.2: Versuch der Freisetzung von 2 aus dem Hydrobromid.
Die versuchte Extraktion mit CH2Cl2 führt wie im Falle der Freisetzung von 1 aus seinem
Hydrobromid zu nicht näher charakterisierten Reaktionsprodukten.
Schließlich konnte wasserfreies 2 durch viertägiges Trocknen des nach Staudinger-Reduktion
erhaltenen Öls im Hochvakuum bei 40°C erhalten werden.
4.3 Umsetzungen von pyN3 mit FeII
Die ersten Umsetzungen mit nicht wasserfreiem 2 und FeII-Salzen führten zu grünlichen
Lösungen und der Bildung eines weißen Niederschlags, was auf Bildung von Fe(OH)2
hindeutet. Mit getrocknetem 2 bzw. 2 · 4 HBr bleiben diese Niederschläge aus.
Die Umsetzung von 2 mit Fe(CH3CN)2(OTf)2 und zwei Äquivalenten 1-Methyl-1H-imidazol
in Acetonitril wurde gemäß Schema 4.3 durchgeführt. Nach Eindiffusion von Diethylether
wurden Kristalle erhalten, die röntgenstrukturanalytisch charakterisiert wurden. Es hat sich
jedoch nicht der gewünschte Komplex des Fragmentes [(2)FeII] gebildet, sondern—laut
vorläufigen Röntgenbeugungsdaten—der Hexakis(methylimidazol)eisen(II)-Komplex
[FeII(C4H6N2)6]2+. Eine genaue Auswertung der Kristallstruktur wurde nicht durchgeführt, es
war jedoch möglich, eine vorläufige Molekülstruktur zu erhalten.
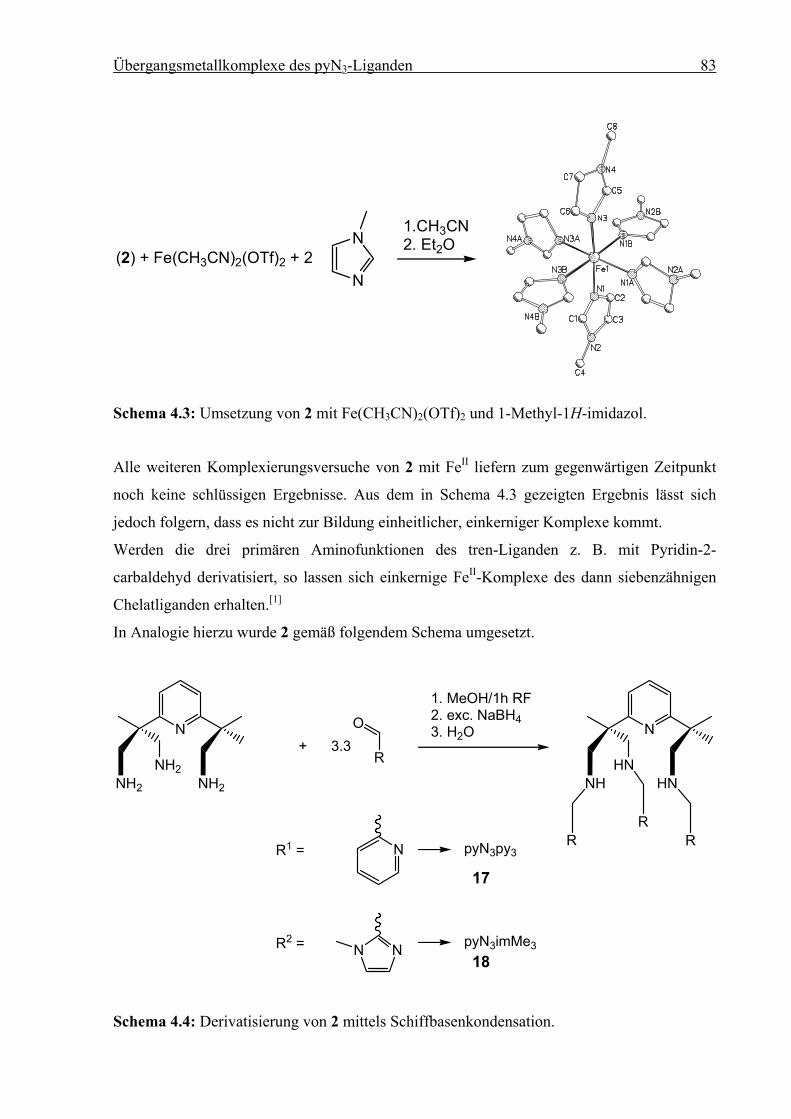
Übergangsmetallkomplexe des pyN3-Liganden 83
(2) + Fe(CH3CN)2(OTf)2 + 2
1.CH3CN2. Et2O
N
N
Schema 4.3: Umsetzung von 2 mit Fe(CH3CN)2(OTf)2 und 1-Methyl-1H-imidazol.
Alle weiteren Komplexierungsversuche von 2 mit FeII liefern zum gegenwärtigen Zeitpunkt
noch keine schlüssigen Ergebnisse. Aus dem in Schema 4.3 gezeigten Ergebnis lässt sich
jedoch folgern, dass es nicht zur Bildung einheitlicher, einkerniger Komplexe kommt.
Werden die drei primären Aminofunktionen des tren-Liganden z. B. mit Pyridin-2-
carbaldehyd derivatisiert, so lassen sich einkernige FeII-Komplexe des dann siebenzähnigen
Chelatliganden erhalten.[1]
In Analogie hierzu wurde 2 gemäß folgendem Schema umgesetzt.
R
O+ 3.3
1. MeOH/1h RF2. exc. NaBH43. H2ON
NH2
NH2NH2
N
NHHN
HN
RRR
R1 = N
NNR2 =
pyN3py3
pyN3imMe3
Schema 4.4: Derivatisierung von 2 mittels Schiffbasenkondensation.
17
18
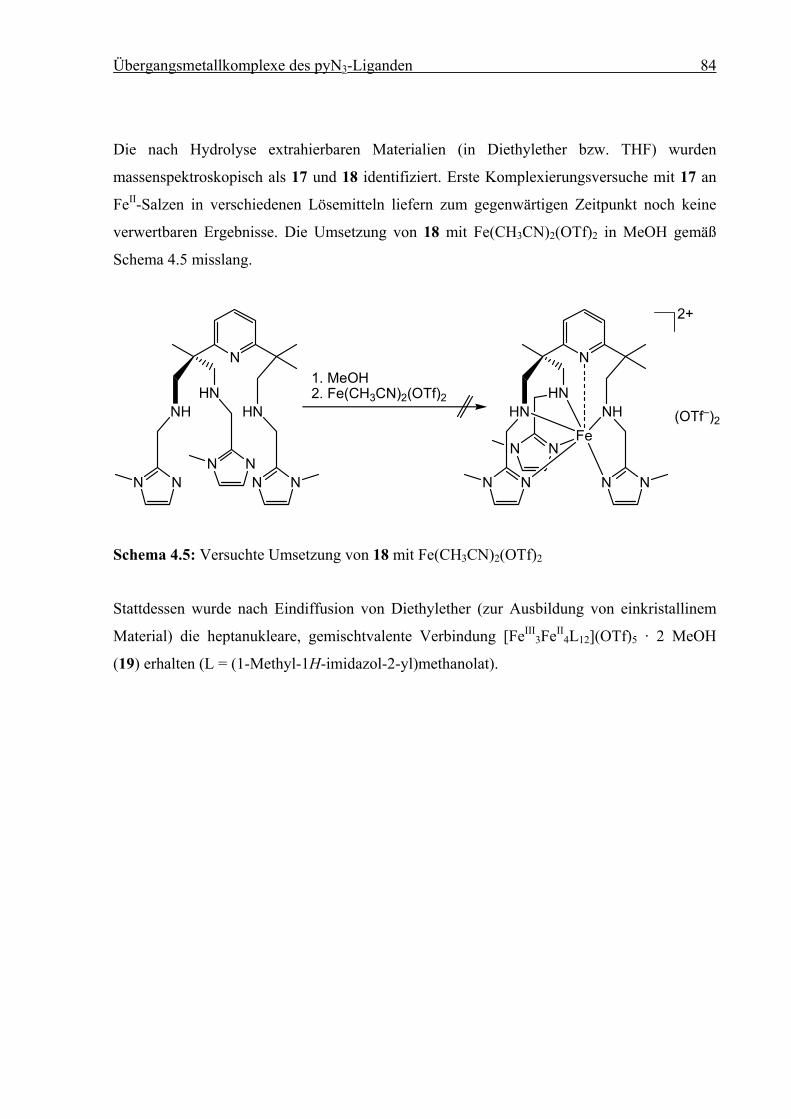
Übergangsmetallkomplexe des pyN3-Liganden 84
Die nach Hydrolyse extrahierbaren Materialien (in Diethylether bzw. THF) wurden
massenspektroskopisch als 17 und 18 identifiziert. Erste Komplexierungsversuche mit 17 an
FeII-Salzen in verschiedenen Lösemitteln liefern zum gegenwärtigen Zeitpunkt noch keine
verwertbaren Ergebnisse. Die Umsetzung von 18 mit Fe(CH3CN)2(OTf)2 in MeOH gemäß
Schema 4.5 misslang.
N
NHHN
HN
N NN N
N N
N
HNHN
NH
N N
N N
N N
Fe
2+
(OTf–)2
1. MeOH2. Fe(CH3CN)2(OTf)2
Schema 4.5: Versuchte Umsetzung von 18 mit Fe(CH3CN)2(OTf)2
Stattdessen wurde nach Eindiffusion von Diethylether (zur Ausbildung von einkristallinem
Material) die heptanukleare, gemischtvalente Verbindung [FeIII3FeII
4L12](OTf)5 · 2 MeOH
(19) erhalten (L = (1-Methyl-1H-imidazol-2-yl)methanolat).
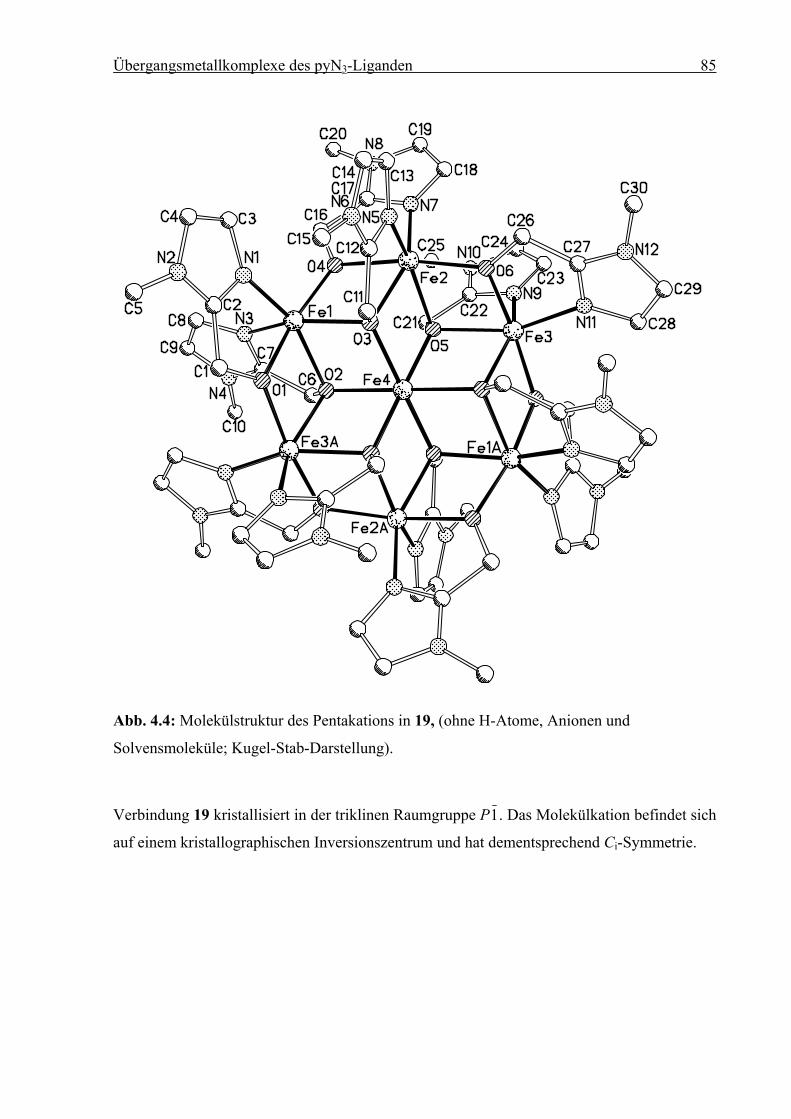
Übergangsmetallkomplexe des pyN3-Liganden 85
Abb. 4.4: Molekülstruktur des Pentakations in 19, (ohne H-Atome, Anionen und
Solvensmoleküle; Kugel-Stab-Darstellung).
Verbindung 19 kristallisiert in der triklinen Raumgruppe P1. Das Molekülkation befindet sich
auf einem kristallographischen Inversionszentrum und hat dementsprechend Ci-Symmetrie.
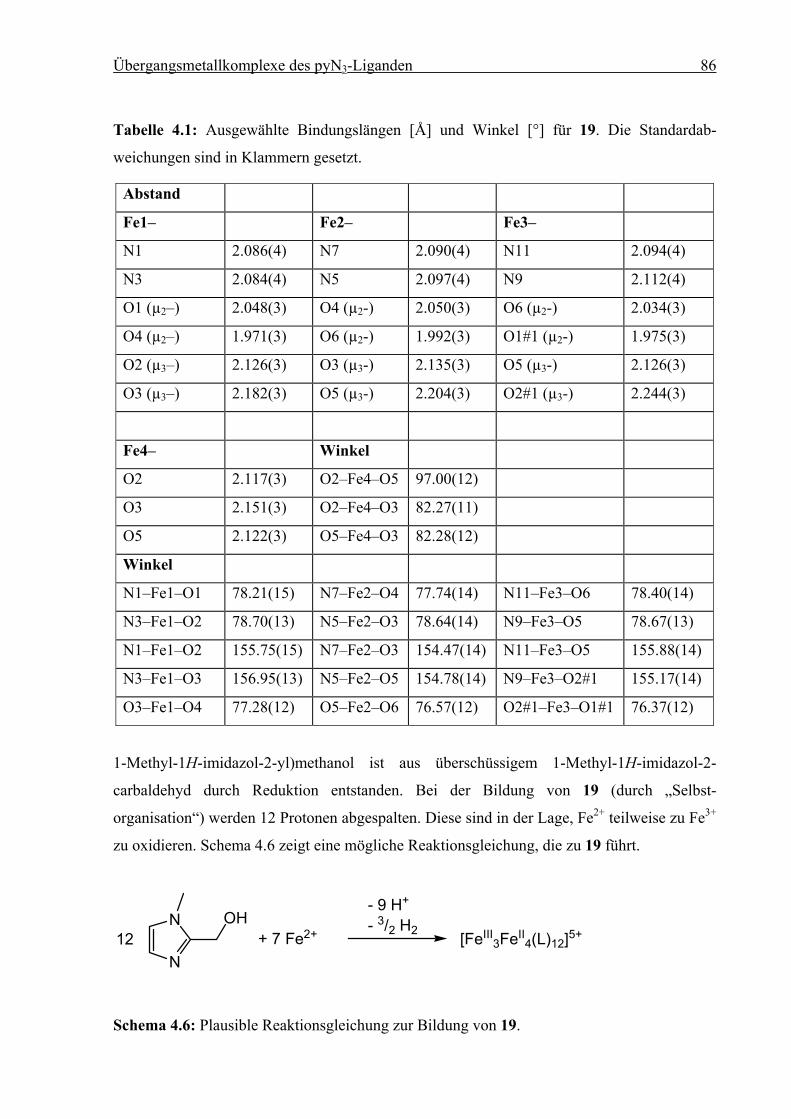
Übergangsmetallkomplexe des pyN3-Liganden 86
Tabelle 4.1: Ausgewählte Bindungslängen [Å] und Winkel [°] für 19. Die Standardab-
weichungen sind in Klammern gesetzt.
Abstand
Fe1– Fe2– Fe3–
N1 2.086(4) N7 2.090(4) N11 2.094(4)
N3 2.084(4) N5 2.097(4) N9 2.112(4)
O1 (µ2–) 2.048(3) O4 (µ2-) 2.050(3) O6 (µ2-) 2.034(3)
O4 (µ2–) 1.971(3) O6 (µ2-) 1.992(3) O1#1 (µ2-) 1.975(3)
O2 (µ3–) 2.126(3) O3 (µ3-) 2.135(3) O5 (µ3-) 2.126(3)
O3 (µ3–) 2.182(3) O5 (µ3-) 2.204(3) O2#1 (µ3-) 2.244(3)
Fe4– Winkel
O2 2.117(3) O2–Fe4–O5 97.00(12)
O3 2.151(3) O2–Fe4–O3 82.27(11)
O5 2.122(3) O5–Fe4–O3 82.28(12)
Winkel
N1–Fe1–O1 78.21(15) N7–Fe2–O4 77.74(14) N11–Fe3–O6 78.40(14)
N3–Fe1–O2 78.70(13) N5–Fe2–O3 78.64(14) N9–Fe3–O5 78.67(13)
N1–Fe1–O2 155.75(15) N7–Fe2–O3 154.47(14) N11–Fe3–O5 155.88(14)
N3–Fe1–O3 156.95(13) N5–Fe2–O5 154.78(14) N9–Fe3–O2#1 155.17(14)
O3–Fe1–O4 77.28(12) O5–Fe2–O6 76.57(12) O2#1–Fe3–O1#1 76.37(12)
1-Methyl-1H-imidazol-2-yl)methanol ist aus überschüssigem 1-Methyl-1H-imidazol-2-
carbaldehyd durch Reduktion entstanden. Bei der Bildung von 19 (durch „Selbst-
organisation“) werden 12 Protonen abgespalten. Diese sind in der Lage, Fe2+ teilweise zu Fe3+
zu oxidieren. Schema 4.6 zeigt eine mögliche Reaktionsgleichung, die zu 19 führt.
12N
N OH+ 7 Fe2+
- 9 H+
- 3/2 H2 [FeIII3FeII
4(L)12]5+
Schema 4.6: Plausible Reaktionsgleichung zur Bildung von 19.
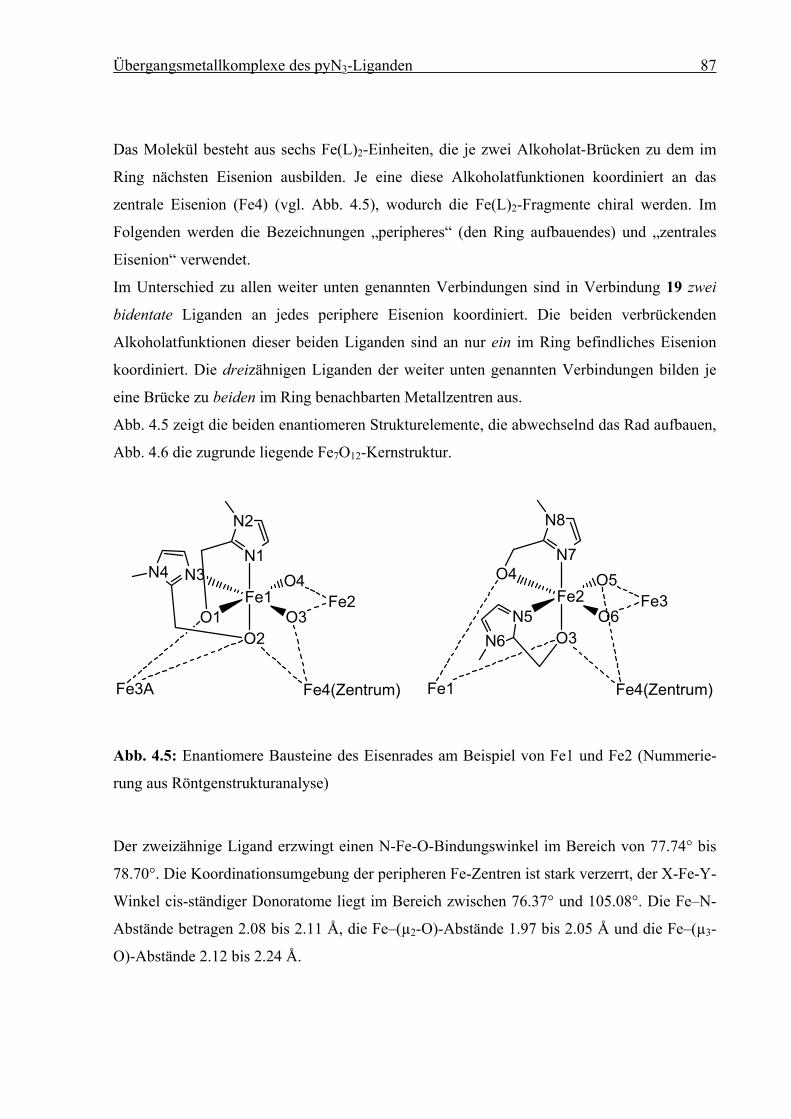
Übergangsmetallkomplexe des pyN3-Liganden 87
Das Molekül besteht aus sechs Fe(L)2-Einheiten, die je zwei Alkoholat-Brücken zu dem im
Ring nächsten Eisenion ausbilden. Je eine diese Alkoholatfunktionen koordiniert an das
zentrale Eisenion (Fe4) (vgl. Abb. 4.5), wodurch die Fe(L)2-Fragmente chiral werden. Im
Folgenden werden die Bezeichnungen „peripheres“ (den Ring aufbauendes) und „zentrales
Eisenion“ verwendet.
Im Unterschied zu allen weiter unten genannten Verbindungen sind in Verbindung 19 zwei
bidentate Liganden an jedes periphere Eisenion koordiniert. Die beiden verbrückenden
Alkoholatfunktionen dieser beiden Liganden sind an nur ein im Ring befindliches Eisenion
koordiniert. Die dreizähnigen Liganden der weiter unten genannten Verbindungen bilden je
eine Brücke zu beiden im Ring benachbarten Metallzentren aus.
Abb. 4.5 zeigt die beiden enantiomeren Strukturelemente, die abwechselnd das Rad aufbauen,
Abb. 4.6 die zugrunde liegende Fe7O12-Kernstruktur.
Fe1O1
N3 O4
O3
N1
O2
N2
Fe3A
N4
Fe4(Zentrum)
Fe2
Fe2N5
O4 O5
O6
N7
O3
N8
Fe1 Fe4(Zentrum)
Fe3
N6
Abb. 4.5: Enantiomere Bausteine des Eisenrades am Beispiel von Fe1 und Fe2 (Nummerie-
rung aus Röntgenstrukturanalyse)
Der zweizähnige Ligand erzwingt einen N-Fe-O-Bindungswinkel im Bereich von 77.74° bis
78.70°. Die Koordinationsumgebung der peripheren Fe-Zentren ist stark verzerrt, der X-Fe-Y-
Winkel cis-ständiger Donoratome liegt im Bereich zwischen 76.37° und 105.08°. Die Fe–N-
Abstände betragen 2.08 bis 2.11 Å, die Fe–(µ2-O)-Abstände 1.97 bis 2.05 Å und die Fe–(µ3-
O)-Abstände 2.12 bis 2.24 Å.

Übergangsmetallkomplexe des pyN3-Liganden 88
= Fe2+/3+
= O
Abb. 4.6: Fe7O12-Kernstruktur von 19.
Das zugrunde liegende Strukturelement [M7O12] kommt in einer Reihe gemischtvalenter
Manganverbindungen bzw. heterobimetallischer [Fe-Mn]- und [Mn-In]-Cluster vor.[6] Es sind
nur zwei gemischtvalente siebenkernige Eisencluster beschrieben, die eine zu Verbindung 19
analoge Struktur haben. In [FeII3FeIII
4(L)6Cl6] · 2 CH3CN · H2O (L = 2-Amino-2-methyl-1,3-
propandiolat) verbrückt der dreizähnige Ligand mit je einer Alkoholatfunktion zum im Ring
vorher und nachher gelegenen Eisenion. Ein Alkoholatdonor koordiniert zusätzlich an das
zentrale Eisenion. Dessen Oxidationstufe wurde mittels BVS-Rechnungen (BVS = bond
valence sum) mit Parametern von H. H. Thorp auf zwei bestimmt.[7][8] Es liegen keine
weiteren experimentellen Daten vor.
In der Verbindung [FeIIIFeII6(OCH3)6(L)6]Cl3 (L = (E)-2-((2-hydroxyethylimino)methyl)-6-
methoxyphenolat) sind sechs FeII-Zentren um ein FeIII-Zentrum angeordnet.[9] Die
Koordinationsumgebung des Zentrums besteht aus sechs dreifach verbrückenden
Methanolatliganden. Das Mößbauerspektrum zeigt je ein Dublett für high-spin-FeII und high-
spin-FeIII im Verhältnis 6 : 1. Bei statistischer Verteilung der Oxidationsstufen würde ein
Mößbauerspektrum mit vier Dubletts resultieren. Die von Oshio et al. ebenfalls
durchgeführten BVS-Rechnungen konnten jedoch nicht reproduziert werden, sondern ergaben
statt eines FeIII- ein FeII-Zentrum. Zur Berechnung wurden auch überarbeitete Parameter
verwendet.[10]
Die Verbindung [PPh4][FeIII3MnII
4Cl6(L)6] (H2L = N-benzyldiethanolamin) enthält ein HS-
FeIII-Zentrum in oktaedrischer Umgebung von sechs µ3-Alkoholatdonoren.[6] Die
Bindungsabstände Fe–O betragen 2.204 Å (4×) bzw. 2.213 Å (2×). Eine mit diesen Werten

Übergangsmetallkomplexe des pyN3-Liganden 89
durchgeführte BVS-Rechnung kommt aber zu dem Ergebnis, das es sich um ein FeII-Zentrum
handeln müsste.
In einem aus 13 FeIII-Zentren aufgebauten Cluster, der ebenfalls das [Fe7O12]-Strukturelement
enthält, ist das zentrale Eisenion von sechs Hydroxidionen umgeben.[11] Im Gegensatz zu
allen anderen o. g. Verbindungen besteht der Kern aus einer [Fe7(µ3-OH)6(µ3-O)6]-Einheit,
also nur aus einzähnigen Liganden. Der Fe–O-Bindungsabstand beträgt 2.032 Å (6×). Eine
BVS-Rechnung ergibt hier den Wert von 2.92, der dem erwarteten Wert von drei entspricht.
Die BVS-Rechnung des zentralen Eisenions von 19 ergibt die Oxidationsstufe +II. Die
Berechnung der Oxidationsstufe der peripheren Eisenionen ergibt in sehr guter
Übereinstimmung den erwarteten Wert von 2.5 (siehe S. 106). Da an obigen Beispielen
gezeigt wurde, dass die BVS-Methode bei dieser Art von Verbindungen für Fe-Zentren keine
verlässlichen Werte liefert, sind weitere Untersuchungen (u. a. Mößbauerspektroskopie)
notwendig.
Es gibt mehrere mögliche Ursachen für die starken Ungenauigkeiten der BVS-Rechnungen.
Die Bindungslängenunterschiede zwischen FeII und FeIII bei ähnlicher
Koordinationsumgebung sind geringer als die Bindungslängenunterschiede zwischen high-
spin und low-spin-Zustand derselben Oxidationstufe.[12] Bei Oxidation von FeII zu FeIII wird,
im high-spin- wie im low-spin-Fall, ein Elektron aus einem nur schwach bindenden oder
antibindenden t2g-Orbital entfernt. Bei Übergang von low-spin zu high-spin werden die stark
antibindenden eg*-Orbitale mit zwei Elektronen besetzt. Die BVS-Methode unterscheidet
nicht zwischen den verschiedenen spin-Zuständen.
Trotz eines Überschusses an Oxidationsmittel (H+) ist keine Oxidation aller sieben FeII-Ionen
zu FeIII aufgetreten. In den o. g. gemischtvalenten Mangan- und Eisenclustern tritt ebenfalls
trotz eines Überschusses der Oxidationsmittel keine vollständige Reaktion zu Mn(III) bzw.
FeIII auf. Dies spricht dafür, dass die Redoxpotentiale der einzelnen Metallionen voneinander
abhängig sind. Diese Annahme wird durch die Cyclovoltammogramme einer der o.g.
gemischtvalenten MnIIMnIII-verbindungen bzw. des heterobimetallischen [Fe-Mn]-Clusters
gestützt. Diese zeigen mehrere deutlich voneinander abgesetzte quasireversible
Redoxwellen.[6] Die rationelle Synthese von 19 zur weiteren Untersuchung (insbesondere des
elektrochemischen Verhaltens) sollte also möglich sein.

Übergangsmetallkomplexe des pyN3-Liganden 90
4.4 Umsetzungen von pyN3 mit CuII, NiII und ZnII
Da die Komplexierungsversuche mit pyN3 an FeII bisher keine schlüssigen Ergebnisse
lieferten, wurden die Untersuchungen auf CuII, NiII und ZnII ausgeweitet. Zunächst wurde 2
mit Cu(dmf)6(ClO4)2 in Methanol im Verhältnis 1 : 1 gemäß Schema 4.7 umgesetzt. Nach
Etherdiffusion wurden blaue Kristalle erhalten, die in MeOH gut löslich waren. Die
elementaranalytische Untersuchung zeigt die Zusammensetzung [(2)Cu(dmf)](ClO4)2 (20).
Das IR-Spektrum (KBr-Pressling) zeigt eine Bande bei 1657 cm–1, die der ν(C=O)-
Valenzschwingung von koordiniertem DMF zugeordnet wurde. Diese Bande tritt im freien
Liganden und den im Folgenden genannten pyN3-Komplexen nicht auf. Es gibt keine
Hinweise darauf, dass ein Perchloration am Metall koordiniert ist. Eine Koordination hätte
eine Erniedrigung der lokalen Symmetrie von Td nach C3v zur Folge, was durch eine
Aufspaltung der Anionenbande angezeigt werden würde.[18]
Daher zeigt Schema 4.7 einen Strukturvorschlag, der aufgrund der zur Verfügung stehenden
Daten gemacht wurde.
Cu(dmf)6(ClO4)2 + (2)
1. MeOH2. Et2O-Diffusion N
H2NH2N Cu NH2
dmf
2+
(ClO4–)2
Schema 4.7: Umsetzung von 2 mit Cu(dmf)6(ClO4)2
Die Reaktion von 2 nach Freisetzung aus seinem HBr-Addukt durch vier Äquivalente LiOMe
und anschließende Umsetzung mit Ni(dmf)6(BF4)2 ergab eine violette Lösung, aus der sich
nach Etherdiffusion violette Kristalle isolieren ließen. Das IR-Spektrum (KBr-Pressling) zeigt
keine Bande für BF4–-Ionen, und die Elementaranalyse zeigt die Zusammensetzung
[(2)NiBr]Br · MeOH, 21 · MeOH.
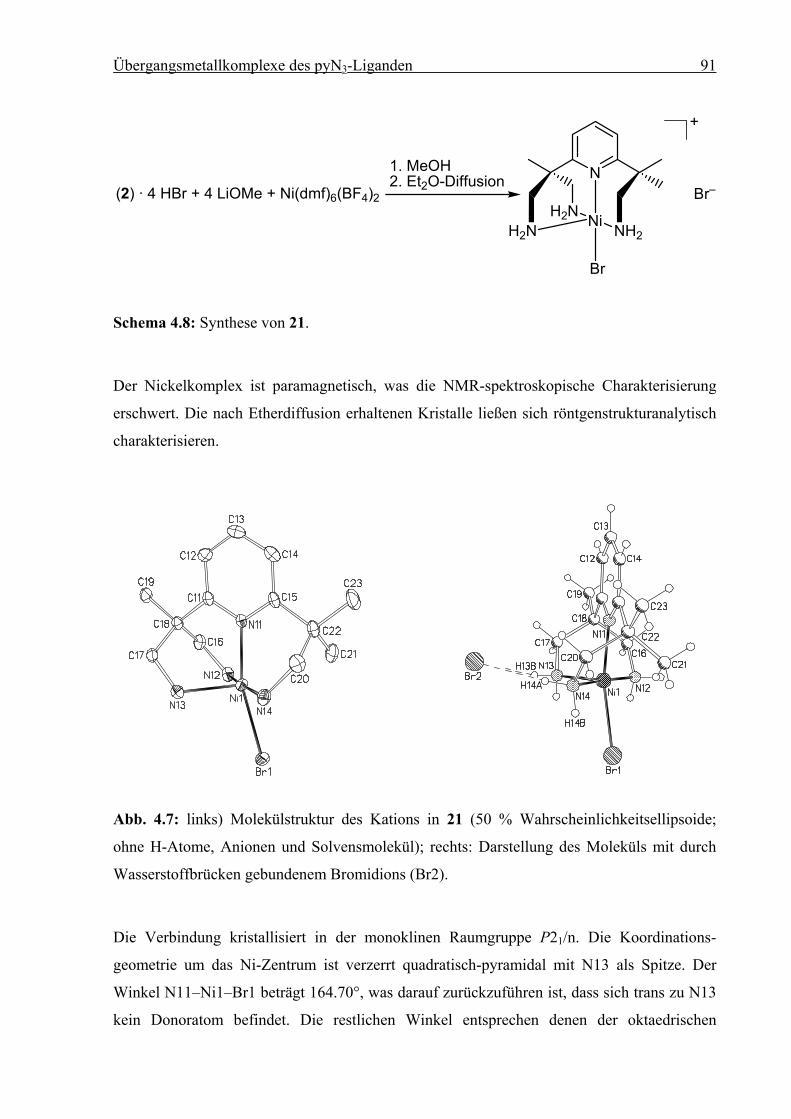
Übergangsmetallkomplexe des pyN3-Liganden 91
(2) · 4 HBr + 4 LiOMe + Ni(dmf)6(BF4)2
1. MeOH2. Et2O-Diffusion N
H2NH2N Ni NH2
Br
+
Br–
Schema 4.8: Synthese von 21.
Der Nickelkomplex ist paramagnetisch, was die NMR-spektroskopische Charakterisierung
erschwert. Die nach Etherdiffusion erhaltenen Kristalle ließen sich röntgenstrukturanalytisch
charakterisieren.
Abb. 4.7: links) Molekülstruktur des Kations in 21 (50 % Wahrscheinlichkeitsellipsoide;
ohne H-Atome, Anionen und Solvensmolekül); rechts: Darstellung des Moleküls mit durch
Wasserstoffbrücken gebundenem Bromidions (Br2).
Die Verbindung kristallisiert in der monoklinen Raumgruppe P21/n. Die Koordinations-
geometrie um das Ni-Zentrum ist verzerrt quadratisch-pyramidal mit N13 als Spitze. Der
Winkel N11–Ni1–Br1 beträgt 164.70°, was darauf zurückzuführen ist, dass sich trans zu N13
kein Donoratom befindet. Die restlichen Winkel entsprechen denen der oktaedrischen
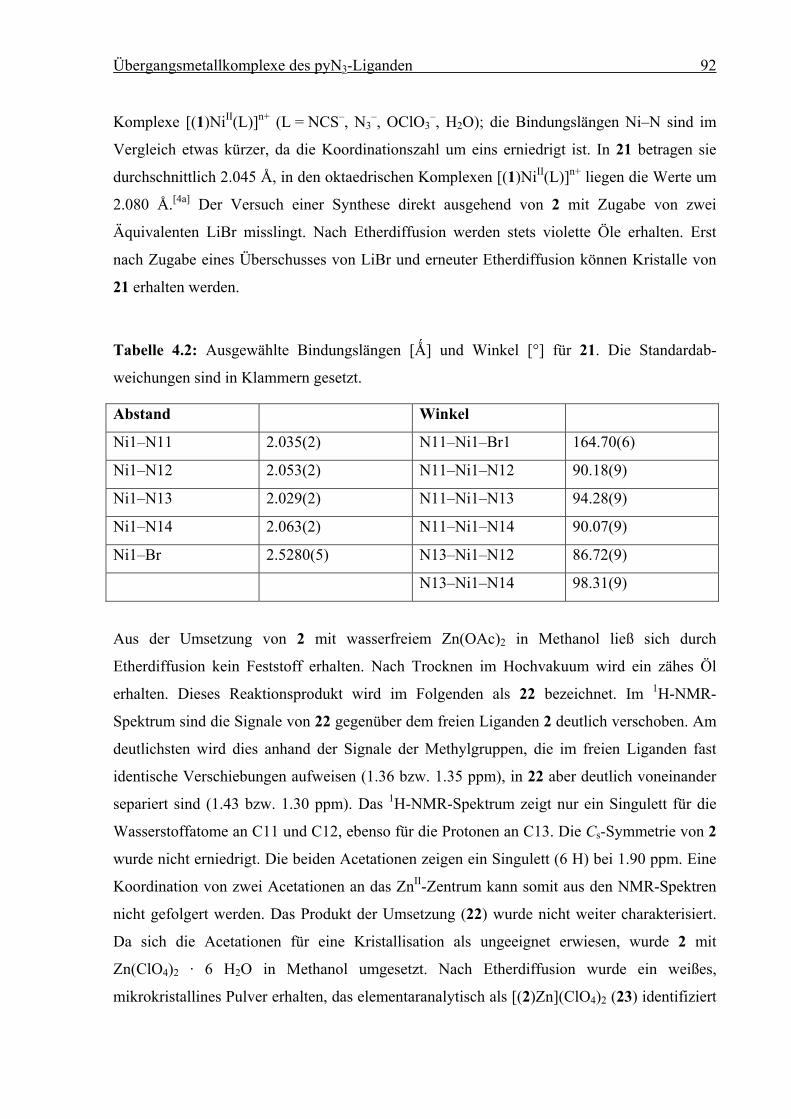
Übergangsmetallkomplexe des pyN3-Liganden 92
Komplexe [(1)NiII(L)]n+ (L = NCS–, N3–, OClO3
–, H2O); die Bindungslängen Ni–N sind im
Vergleich etwas kürzer, da die Koordinationszahl um eins erniedrigt ist. In 21 betragen sie
durchschnittlich 2.045 Å, in den oktaedrischen Komplexen [(1)NiII(L)]n+ liegen die Werte um
2.080 Å.[4a] Der Versuch einer Synthese direkt ausgehend von 2 mit Zugabe von zwei
Äquivalenten LiBr misslingt. Nach Etherdiffusion werden stets violette Öle erhalten. Erst
nach Zugabe eines Überschusses von LiBr und erneuter Etherdiffusion können Kristalle von
21 erhalten werden.
Tabelle 4.2: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 21. Die Standardab-
weichungen sind in Klammern gesetzt.
Abstand Winkel
Ni1–N11 2.035(2) N11–Ni1–Br1 164.70(6)
Ni1–N12 2.053(2) N11–Ni1–N12 90.18(9)
Ni1–N13 2.029(2) N11–Ni1–N13 94.28(9)
Ni1–N14 2.063(2) N11–Ni1–N14 90.07(9)
Ni1–Br 2.5280(5) N13–Ni1–N12 86.72(9)
N13–Ni1–N14 98.31(9)
Aus der Umsetzung von 2 mit wasserfreiem Zn(OAc)2 in Methanol ließ sich durch
Etherdiffusion kein Feststoff erhalten. Nach Trocknen im Hochvakuum wird ein zähes Öl
erhalten. Dieses Reaktionsprodukt wird im Folgenden als 22 bezeichnet. Im 1H-NMR-
Spektrum sind die Signale von 22 gegenüber dem freien Liganden 2 deutlich verschoben. Am
deutlichsten wird dies anhand der Signale der Methylgruppen, die im freien Liganden fast
identische Verschiebungen aufweisen (1.36 bzw. 1.35 ppm), in 22 aber deutlich voneinander
separiert sind (1.43 bzw. 1.30 ppm). Das 1H-NMR-Spektrum zeigt nur ein Singulett für die
Wasserstoffatome an C11 und C12, ebenso für die Protonen an C13. Die Cs-Symmetrie von 2
wurde nicht erniedrigt. Die beiden Acetationen zeigen ein Singulett (6 H) bei 1.90 ppm. Eine
Koordination von zwei Acetationen an das ZnII-Zentrum kann somit aus den NMR-Spektren
nicht gefolgert werden. Das Produkt der Umsetzung (22) wurde nicht weiter charakterisiert.
Da sich die Acetationen für eine Kristallisation als ungeeignet erwiesen, wurde 2 mit
Zn(ClO4)2 · 6 H2O in Methanol umgesetzt. Nach Etherdiffusion wurde ein weißes,
mikrokristallines Pulver erhalten, das elementaranalytisch als [(2)Zn](ClO4)2 (23) identifiziert

Übergangsmetallkomplexe des pyN3-Liganden 93
wurde. 23 ist gut in MeOH löslich. Die 1H- und 13C-NMR-Spektren zeigen ebenfalls keine
Erniedrigung der Cs-Symmetrie von 2.
(2) + Zn(ClO4) · 6 H2O
1. MeOH2. Et2O-Diffusion
[(2)ZnII](ClO4)2
Schema 4.9: Umsetzung von 2 mit Zn(ClO4)2 · 6 H2O (22).
Auch hier konnten durch mehrmaliges Umkristallisieren keine für die Röntgenstrukturanalyse
geeigneten Kristalle erhalten werden. Nach Zugabe von sechs Äquivalenten LiBr und erneuter
Etherdiffusion wurde ein Pulver erhalten. Dies war unlöslich selbst in heißem Methanol. Es
löste sich erst in Methanol/Acetonitril 1:1 in der Siedehitze. Nach Abkühlen auf RT und
erneuter Etherdiffusion wurden Kristalle der Zusammensetzung [{(2)ZnBr}2]Br0.21(ClO4)1.79
(24) erhalten, die röntgenstrukturanalytisch charakterisiert wurden.
Abb. 4.8: Molekülstruktur des Dikations in 24 (50 % Wahrscheinlichkeitsellipsoide; ohne H-
Atome und Anionen).
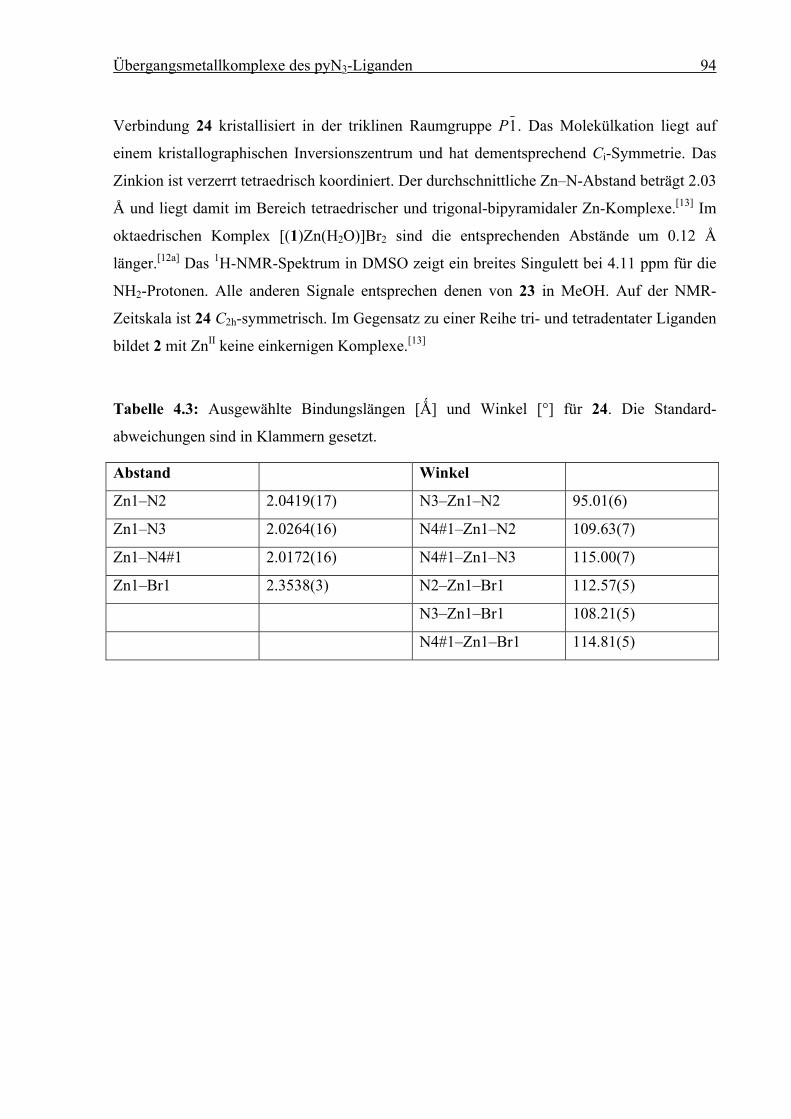
Übergangsmetallkomplexe des pyN3-Liganden 94
Verbindung 24 kristallisiert in der triklinen Raumgruppe P1. Das Molekülkation liegt auf
einem kristallographischen Inversionszentrum und hat dementsprechend Ci-Symmetrie. Das
Zinkion ist verzerrt tetraedrisch koordiniert. Der durchschnittliche Zn–N-Abstand beträgt 2.03
Å und liegt damit im Bereich tetraedrischer und trigonal-bipyramidaler Zn-Komplexe.[13] Im
oktaedrischen Komplex [(1)Zn(H2O)]Br2 sind die entsprechenden Abstände um 0.12 Å
länger.[12a] Das 1H-NMR-Spektrum in DMSO zeigt ein breites Singulett bei 4.11 ppm für die
NH2-Protonen. Alle anderen Signale entsprechen denen von 23 in MeOH. Auf der NMR-
Zeitskala ist 24 C2h-symmetrisch. Im Gegensatz zu einer Reihe tri- und tetradentater Liganden
bildet 2 mit ZnII keine einkernigen Komplexe.[13]
Tabelle 4.3: Ausgewählte Bindungslängen [Ǻ] und Winkel [°] für 24. Die Standard-
abweichungen sind in Klammern gesetzt.
Abstand Winkel
Zn1–N2 2.0419(17) N3–Zn1–N2 95.01(6)
Zn1–N3 2.0264(16) N4#1–Zn1–N2 109.63(7)
Zn1–N4#1 2.0172(16) N4#1–Zn1–N3 115.00(7)
Zn1–Br1 2.3538(3) N2–Zn1–Br1 112.57(5)
N3–Zn1–Br1 108.21(5)
N4#1–Zn1–Br1 114.81(5)

Übergangsmetallkomplexe des pyN3-Liganden 95
4.5 Experimenteller Teil
4.5.1 Allgemeines
Sofern nicht anders vermerkt, wurden alle Reaktionen unter N2-Atmosphäre in absoluten
Lösemitteln in Standard-Schlenk-Gefäßen durchgeführt. Die verwendeten absoluten
Lösemittel (Restgehalt H2O ≤ 50 ppm) sind kommerziell über Molsieb getrocknet
erhältlich.[14] Organische und anorganische Reagenzien wurden von den Firmen Aldrich,
Acros, Strem oder Alfa Aesar bezogen und ohne weitere Reinigung benutzt. Die
Verbindungen py(OMes)3[15], Ni(dmf)6(BF4)2
[16] und Cu(dmf)6(ClO4)2[17] wurden nach
Literaturangaben hergestellt.
Zur Aufnahme der Daten wurden folgende Geräte verwendet:
Kernresonanzspektren: Die NMR-Spektren wurden, soweit nicht anders vermerkt, bei
Raumtemperatur in einem 5 mm-Röhrchen an Spektrometern der Firma Bruker
aufgenommen: ARX 200 (1H, 200 MHz; 13C, 50.32 MHz), ARX 400 (1H, 400 MHz; 13C,
100.64 MHz). Die temperaturabhängigen NMR-Spektren von 5, 8 und 12 wurden an einem
Alpha 500 der Firma Jeol gemessen (1H, 500 MHz; 13C, 125.78 MHz). Luftempfindliche
Proben wurden in einem unter Vakuum abgeschmolzenen Röhrchen vermessen. Alle
chemischen Verschiebungen sind in ppm relativ zur Restprotonen- bzw. 13C-Absorption des
verwendeten Lösemittels (interner Standard) angegeben. Die 13C-NMR-Spektren wurden
breitbandentkoppelt aufgenommen. Die Vorzeichen der Kopplungskonstanten wurden bei den 1H- und 13C-Spektren nicht bestimmt.
Elementaranalysen: Die quantitative Bestimmung von Kohlenstoff, Wasserstoff, Stickstoff
und Schwefel erfolgte verbrennungsanalytisch an einem Gerät des Typs Thermo Finnigan
EAGER 300 (Flash 1112 Series) Gerät.
Infrarotspektren: Die IR-Spektren von wurden an einem Nicolet Magna System 750-
Spektrometer aufgenommen. Feststoffe wurden als Kaliumbromid-Presslinge, Lösungen in
CaF2-Küvetten unter Kompensation der Solvensabsorption vermessen. Die Wellenzahlen ν̃
sind in cm–1 angegeben. Die Zuordnung der Banden erfolgte anhand einschlägiger
Literatur.[18]

Übergangsmetallkomplexe des pyN3-Liganden 96
Massenspektren: Die Aufnahme der Massenspektren erfolgte unter Verwendung von
folgendem Gerät: Varian 311A (EI-MS, 70 eV).
Einkristallröntgenstrukturanalysen: Die Datensammlung erfolgte an folgendem Gerät:
Oxford Diffraction Xcalibur S Sapphire-Diffraktometer (Siemens) unter Verwendung von
Mo-Kα-Strahlung (λ = 0.71073 Ǻ) bei 100 K. Zur empirischen Absorptionskorrektur wurde,
wenn erforderlich, das Programm SADABS (Bruker AXS, 2002) verwendet. Die Strukturen
wurden mit direkten Methoden gelöst und anschließend an F2 verfeinert (SHELXT NT 6.12,
Bruker AXS, 2002). Sofern nicht anders angegeben, wurden alle Nichtwasserstoffatome
anisotrop verfeinert. Sämtliche Wasserstoffatome befinden sich in nach geometrischen
Gesichtspunkten berechneten Lagen und wurden mit einem isotropen Auslenkungsparameter
versehen, der dem 1.2- bzw. 1.5 fachen des äquivalenten isotropen Auslenkungsparameters
des die Wasserstoffatome tragenden C-, N- bzw. O-Atomes entspricht.
4.5.2 Synthesen
4.5.2.1 2-(1,3-diazido-2-methylpropan-2-yl)-6-(1-azido-2-methylpropan-2-yl)pyridin (16)
C13H18N10
M = 314.35
gelbes Öl
Das Trimesylat (py(OMes)3) (18.67 g, 39.4 mmol) wird in DMSO (120 ml) gelöst und festes
NaN3 (11.5 g, 177.4 mmol) zugegeben. Die resultierende Suspension wird auf 70 °C erhitzt
und man erhält eine klare gelbe Lösung. Nach 1 h bildet sich ein weißer Niederschlag. Es
wird noch 48 h bei 70 °C gerührt. Nach Abkühlen auf RT wird die Suspension auf Eis/Wasser
(250 ml) gegeben, woraufhin sich eine trübe Lösung bildet, aus der sich nach 1 h rühren ein
gelbes Öl abscheidet. Dies wird mit Et2O (5 × 100 ml) ausgeschüttelt. Die organische Phase
wird im Vakuum auf ein Viertel eingeengt und 18 h über Na2SO4 getrocknet. Das
Trockenmittel wird abfiltriert Eine kleine Menge wird für NMR- und IR-Spektroskopie
N
N3
N3N3

Übergangsmetallkomplexe des pyN3-Liganden 97
getrocknet. Die verbleibende Lösung wird mit 60 ml Pyridin versetzt und der Diethylether bei
60°C und 200 mbar Enddruck am Rotationsverdampfer entfernt. Die nachfolgende
Berechnung bezieht sich auf 100 % Umsatz. 1H-NMR (200 MHz, [D1]Chloroform, 25°C): δ = 7.68 ppm (t, 3J(H,H) = 7.9 Hz, 1 H, H3),
7.25 ppm (d, 3J(H,H) = 7.9 Hz, 4J(H,H) = 0.8 Hz, 1 H, py-H2), 7.17 ppm (d, 3J(H,H) = 7.8 Hz, 4J(H,H) = 0.8 Hz, 1 H, 1 H, py-H4), 3.83 - 3.65 (dd, AB, 2J(H,H) = 11.9 Hz, 4 H, H8, 9), 3.65
(s, 2 H, H13), 1.44 (s, 3 H, H7), 1.40 (s, 6 H, H11, 12) ppm. 13C-NMR (50.32 MHz, [D1]Chloroform, 25°C): δ = 164.53 (s, 1 C, C1), 160,45 (s, 1 C, C5),
137.34 (s, 1 C, C3), 118.70 (s, 2 C, C2, 4), 62.30 (s, 1 C, C13), 58.35 (s, 2 C, C8, 9), 46.61 (s, 1
C, C6), 42.46 (s, 1 C, C10), 25.67 (s, 2 C, C11, 12), 21.00 (s, 1 C, C7) ppm.
IR (Film auf KBr): ν̃ = 2972m, 2934m, 2872m, 2101vs (–N3), 1584m, 1577m, 1473m,
1461m, 1450m, 1384m, 1362m, 1339m, 1302s, 1277s, 1216w, 1176m, 1094w, 995w, 937w,
814m, 751m cm–1.
4.5.2.2 2-(1,3-diamino-2-methylpropan-2-yl)-6-(1-amino-2-methylpropan-2-yl)pyridin (2)
C13H24N4
M = 236.36
gelbes Öl
Zu der auf 0 °C gekühlten Lösung von 16 (39.4 mmol) in Pyridin (60 ml) wird PPh3 (39.3 g,
150 mmol), gelöst in Pyridin (70 ml), innerhalb von 1 h zugetropft. Nach 30 min rühren bei
0°C wird das Kühlbad entfernt und weitere 24 h bei RT gerührt. Alle flüchtigen Bestandteile
werden im Hochvakuum entfernt, zurück bleibt eine schwach gelbliche amorphe Masse.
Diese wird mit NH3-Lösung (25 %, 150 ml) versetzt und 18 h auf 140 °C erhitzt. Nach
Abkühlen auf RT bildet sich eine gelbe Lösung über einem weißen Feststoff. Die Lösung
wird durch einen Filter abdekantiert. Der Feststoff wird noch dreimal mit NH3-Lösung (25 %,
50 ml) auf 140 °C erhitzt und nach Abkühlen auf RT die Lösung vom Feststoff abdekantiert.
Die vereinigten wässrigen Phasen werden zur Trockne gebracht. Das so erhaltene Produkt ist
ein gelbes Öl welches noch mit Spuren von OPPh3 verunreinigt ist (8.8 g, 95 %).
N
NH2
NH2NH2

Übergangsmetallkomplexe des pyN3-Liganden 98
1H-NMR (200 MHz, [D1]Chloroform, 25°C): δ = 7.59 (t, 3J(H,H) = 7.8 Hz, 1 H, H3), 7.13
(d, 3J(H,H) = 7.0 Hz, 1 H, py-H2), 7.10 (d, 3J(H,H) = 7.0 Hz, 1 H, py-H4), 3.16 - 2.89 (dd,
AB, 2J(H,H) = 12.8 Hz, 4 H, H8, 9), 2.91 (s, 2 H, H13), 1.95 (br, 6 H, NH2) 1.30 (s, 9 H, H7, 11,
12) ppm. 1H-NMR (200 MHz, [D2]Dichlormethan, 25°C): δ = 7.63 (t, 3J(H,H) = 7.9 Hz, 1 H, H3),
7.17 (d, 3J(H,H) = 7.2 Hz, 1 H, py-H2), 7.13 (d, 3J(H,H) = 7.2 Hz, 1 H, py-H4), 3.38 (br, 6 H,
NH2) 3.11 - 2.95 (dd, AB, 2J(H,H) = 12.9 Hz, 4 H, H8, 9), 2.91 (s, 2 H, H13), 1.29 (s, 6 H, H11,
12), 1.25 (s, 3 H, H7) ppm. 1H-NMR (200 MHz, [D4]Methanol, 25°C): δ = 7.74 (t, 3J(H,H) = 7.8 Hz, 1 H, H3), 7.30 (d, 3J(H,H) = 7.3 Hz, 1 H, py-H2), 7.26 (d, 3J(H,H) = 7.8 Hz, 1 H, py-H4), 3.15 - 2.86 (dd, AB, 2J(H,H) = 13.0 Hz, 4 H, H8, 9), 2.95 (s, 2 H, H13), 1.36 (s, 3 H, H7), 1.35 (s, 6 H, H11, 12) ppm. 13C-NMR (50.32 MHz, [D2]Dichlormethan, 25°C): δ = 165.60 (s, 1 C, C1), 162.97(s, 1 C,
C5), 139.38 (s, 1 C, C3), [133.39, 132.35, 132.26, 132.22, 132.12, 128.87, 128.75, 128.68
(OPPh3-Verunreinigung)], 118.80 (s, 1 C, C2), 118.30 (s, 1 C, C4), 52.71 (s, 1 C, C13), 49.67
(s, 2 C, C8, 9), 45.86 (s, 1 C, C6), 41.86 (s, 1 C, C10), 25.93 (s, 2 C, C11, 12), 21.76 (s, 1 C, C7)
ppm.
IR (Film auf KBr): ν̃ = 3369s, 3290s, 3057vs, 2962vs, 2910vs, 2865vs, 2191sh, 1575vs,
1452vs, 1384vs, 1215s, 1183s, 1120s, 1040s, 851s, 813s, 754s, 723s, 696s, 671m, 613w cm–1.
4.5.2.3 [FeII(C4H6N2)6]2+
gelbe, stark fehlgeordnete Kristalle

Übergangsmetallkomplexe des pyN3-Liganden 99
2 (30 mg, 0.13 mmol) wird in Acetonitril (5 ml) gelöst. Eine farblose Lösung von
Fe(CH3CN)2(OTf)2 (55 mg, 0.13 mmol) in Acetonitril (5 ml) wird zugegeben woraufhin die
Lösung eine gelbe Farbe annimmt. Die Farbe ändert sich nach Zugabe von 1-Methyl-1H-
imidazol (23 µl, 23.8 mg, 0.29 mmol) nicht. Durch Eindiffusion von Et2O wurden Kristalle
erhalten, von denen eine vorläufige Röntgenstrukturanalyse erstellt werden konnte.
4.5.2.4 2-Methyl-2-(6-(2-methyl-1-(pyridin-2-ylmethylamino)propan-2-yl)pyridin-2-yl)-
N1,N3-bis(pyridin-2-ylmethyl)propan-1,3-diamin (17)
C31H39N7
M = 509.69
orange-rotes Öl
2 (0.6 g, 2.55 mmol) wird in MeOH (5 ml) gelöst. Zu der gelben Lösung wird innerhalb von 5
min Pyridin-2-carbaldehyd (0.9 g, 8.4 mmol) zugetropft. Während 1 h zum RF erhitzt wird
die Lösung schwarz. Nach Abkühlen auf RT wird NaBH4 (0.29 g, 7.6 mmol) langsam
zugegeben, woraufhin eine klare orange Lösung resultiert. Nach 15 min rühren wird
überschüssiges NaBH4 hydrolysiert. Das Methanol wird bei 15 mbar entfernt, woraufhin sich
aus der wässrigen Phase ein orangenes Öl abscheidet, dass mit Et2O (6 × 20 ml) extrahiert
wird. Die vereinigten organischen Phasen werden 18 h über Na2SO4 getrocknet, das
Trockenmittel abfiltriert und im Hochvakuum getrocknet. Das Produkt wird
massenspektroskopisch identifiziert und ohne weitere Reinigung eingesetzt.
N
NHHN
HN
NN
N

Übergangsmetallkomplexe des pyN3-Liganden 100
4.5.2.5 2-Methyl-2-(6-(2-methyl-1-((1-methyl-1H-imidazol-2-yl)methylamino)propan-2-
yl)pyridin-2-yl)-N1,N3-bis((1-methyl-1H-imidazol-2-yl)methyl)propan-1,3-
diamine (18)
C28H42N10
M = 518.70
gelbes Öl
2 (0.62 g, 2.6 mmol) wird in MeOH (5 ml) gelöst. Zu der gelben Lösung wird innerhalb von 5
min 1-Methyl-1H-imidazol-2-carbaldehyd (1.0 g, 8.9 mmol) zugetropft. Es wird 18 h gerührt
und danach 1 h zum RF erhitzt. Die Lösung nimmt eine dunkelorange Farbe an. Nach
Abkühlen auf RT wird noch 18 h gerührt. Danach wird NaBH4 (0.30 g, 7.8 mmol) langsam
zugegeben, woraufhin eine klare hellorange Lösung resultiert. Nach 30 min rühren wird
überschüssiges NaBH4 hydrolysiert. Das Methanol wird bei 15 mbar entfernt, woraufhin sich
aus der wässrigen Phase ein zähes gelbes Öl abscheidet. Es wird THF (20 ml)zugegeben und
die wässrige Phase mit NaCl gesättigt. Die organische Phase wird abgetrennt und die wässrige
Phase mit THF (3 × 20 ml) extrahiert. Die vereinigten organischen Phasen werden 18 h über
Na2SO4 getrocknet, das Trockenmittel abfiltriert und im Hochvakuum getrocknet. Das
Produkt wird massenspektroskopisch identifiziert und ohne weitere Reinigung eingesetzt.
(1.41 g Rohprodukt, 105 %).
N
NHHN
HN
N NN N
N N
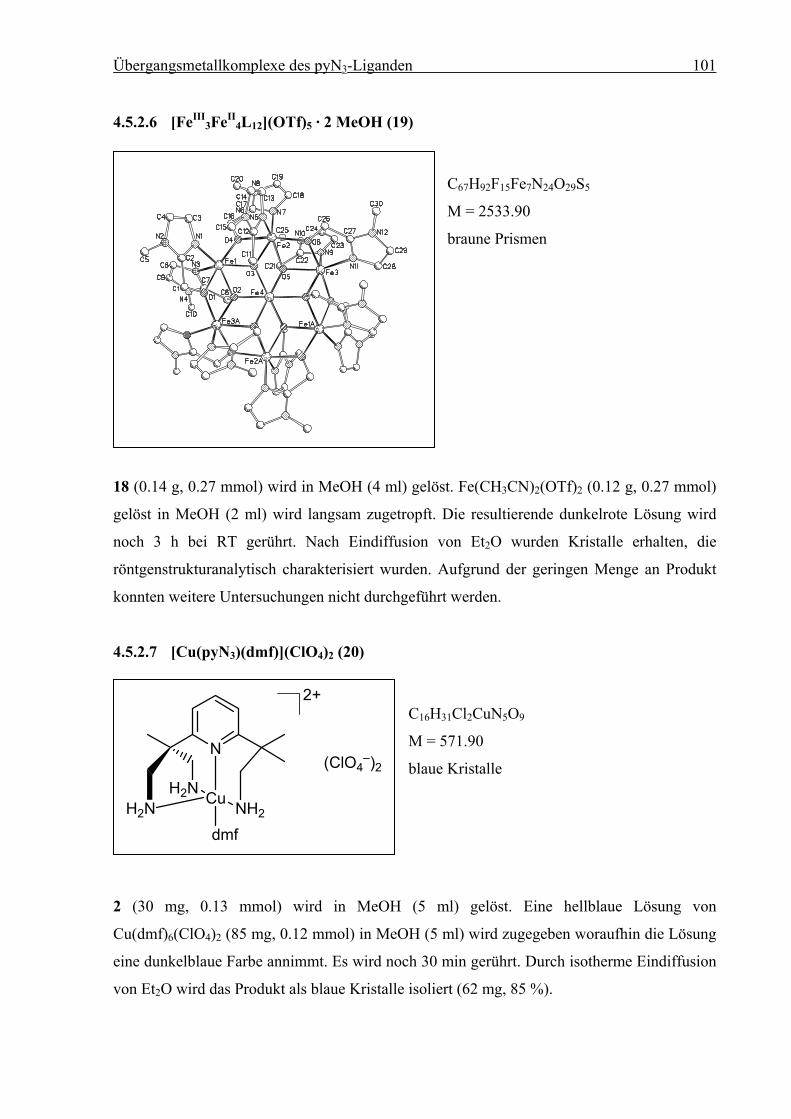
Übergangsmetallkomplexe des pyN3-Liganden 101
4.5.2.6 [FeIII3FeII
4L12](OTf)5 · 2 MeOH (19)
C67H92F15Fe7N24O29S5
M = 2533.90
braune Prismen
18 (0.14 g, 0.27 mmol) wird in MeOH (4 ml) gelöst. Fe(CH3CN)2(OTf)2 (0.12 g, 0.27 mmol)
gelöst in MeOH (2 ml) wird langsam zugetropft. Die resultierende dunkelrote Lösung wird
noch 3 h bei RT gerührt. Nach Eindiffusion von Et2O wurden Kristalle erhalten, die
röntgenstrukturanalytisch charakterisiert wurden. Aufgrund der geringen Menge an Produkt
konnten weitere Untersuchungen nicht durchgeführt werden.
4.5.2.7 [Cu(pyN3)(dmf)](ClO4)2 (20)
C16H31Cl2CuN5O9
M = 571.90
blaue Kristalle
2 (30 mg, 0.13 mmol) wird in MeOH (5 ml) gelöst. Eine hellblaue Lösung von
Cu(dmf)6(ClO4)2 (85 mg, 0.12 mmol) in MeOH (5 ml) wird zugegeben woraufhin die Lösung
eine dunkelblaue Farbe annimmt. Es wird noch 30 min gerührt. Durch isotherme Eindiffusion
von Et2O wird das Produkt als blaue Kristalle isoliert (62 mg, 85 %).
N
H2NH2N Cu NH2
dmf
2+
(ClO4–)2

Übergangsmetallkomplexe des pyN3-Liganden 102
IR (KBr): ν̃ = 3353m, 3310m, 2931m, 1657s (ν(C=O)), 1601m, 1578m, 1462m, 1417m,
1382m, 1255m, 1087vs, 1001sh, 820m, 762m, 625m cm–1.
Elementaranalyse: Ber. ( %) für C16H31Cl2CuN5O9 (571.90): C 33.60, H 5.46, N 12.25; gef.
C 33.69, H 5.59, N 12.24.
4.5.2.8 [Ni(pyN3)Br]Br · MeOH (21)
C17H35Br2N4NiO
M = 530.00
violette Nadeln
a) 2 · 4 HBr (120 mg, 0.21 mmol) wird in MeOH (3 ml) gelöst und mit LiOMe (0.84 ml einer
Maßlösung 1M in MeOH, 0.84 mmol) versetzt. Nach Zugabe von Ni(dmf)6(BF4)2 (141 mg,
0.21 mmol) nimmt die Lösung eine violette Farbe an. Nach Etherdiffusion bei
Raumtemperatur werden violette Kristalle erhalten, die röntgenstrukturanalytisch
charakterisiert werden konnten. Eine Ausbeute wurde nicht bestimmt.
b) 2 (72 mg, 0.31 mmol) wird in MeOH (1.2 ml) gelöst und mit Ni(dmf)6(BF4)2 (200 mg, 0.30
mmol) gelöst in MeOH (3 ml) versetzt. Die Lösung nimmt eine violette Farbe an. Festes LiBr
(164 mg, 1.9 mmol, 6.3 eq.) wird zugegeben, welches sich nach 5 min Rühren aufgelöst hat.
Durch isotherme Eindiffusion von Diethylether kristallisiert das Produkt als violette Nadeln
(146 mg, 95 %).
IR (KBr): ν̃ = 3313s, 3280s, 3217s, 3145s, 3087sh, 2970m, 2934m, 2870m, 2815m, 1600m,
1577m, 1463m, 1398w, 1375w, 1161w, 1143w, 1121w, 1109w, 1092m, 1023vs, 971w, 957w,
818m, 760w, 687w, 623w, 579w cm–1.
Elementaranalyse: Ber. ( %) für C16H31Br2N4Ni (497.96): C 34.53, H 5.80, N 11.51; gef. C
34.58, H 5.48, N 11.45.
N
H2NH2N Ni NH2
Br
+
Br–

Übergangsmetallkomplexe des pyN3-Liganden 103
4.5.2.9 Umsetzung von pyN3 mit Zn(OAc)2 (22)
C13H24Cl2N4O8Zn
M = 500.65
weißes Öl
2 (66 mg, 0.28 mmol) wird in MeOH (1 ml) gelöst und mit einer farblosen Lösung von
Zn(OAc)2 (49.5 mg, 0.27 mmol) gelöst in MeOH (8 ml) versetzt. Es wird noch 1 h gerührt.
Durch Etherdiffusion konnte kein Produkt isoliert werden. Nach Entfernen des Lösemittels
wird ein noch mit Et2O verunreinigtes Öl erhalten. Eine Ausbeute wurde nicht bestimmt. 1H-NMR (200 MHz, [D4]Methanol, 25°C): δ = 7.90 (t, 3J(H,H) = 8.0 Hz, 1 H, H3), 7.54 -
7.45 (m, 2 H, H2, 4), 3.40 - 3.10 (m, 6 H, H8, 9, 13), 1.90 (s, 6 H, H3CCO2– H11, 12), 1.44 (s, 6 H,
H11, 12), 1.30 (s, 3 H, H7) ppm. Das Solvenssignal von [D4]Methanol überlagert die Signale
von H8, 9, 13.
4.5.2.10 [Zn(pyN3)](ClO4)2 (23)
C13H24Cl2N4O8Zn
M = 500.65
weißes Pulver
2 (30 mg, 0.13 mmol) wird in MeOH (3 ml) gelöst und mit einer farblosen Lösung von
Zn(ClO4)2 · 6 H2O (48 mg, 0.13 mmol) gelöst in MeOH (3 ml) versetzt. Es wird noch 1 h
gerührt. Durch Eindiffusion von Et2O bildet sich ein weißes Pulver, das abfiltriert und
getrocknet wird. (56 mg, 86 %). 1H-NMR (200 MHz, [D4]Methanol, 25°C): δ = 8.02 (t, 3J(H,H) = 8.1 Hz, 1 H, H3), 7.66 (d, 3J(H,H) = 8.2 Hz, 4J(H,H) = 0.9 Hz, 1 H, H2), 7.58 (d, 3J(H,H) = 8.0 Hz, 4J(H,H) = 0.9 Hz, 1
N
H2NH2N Zn NH2
2+
(ClO4–)2
Strukturvorschlag
N
H2NH2N Zn NH2
2+
(OAc–)2
Strukturvorschlag

Übergangsmetallkomplexe des pyN3-Liganden 104
H, H4), 3.33 - 3.03 (dd, AB, 2J(H,H) = 13.5 Hz, 4 H, H8, 9), 3.20 (s, 2 H, H13), 1.47 (s, 6 H,
H11, 12), 1.37 (s, 3 H, H7) ppm. 13C-NMR (50.32 MHz, [[D4]Methanol, 25°C): δ = 168.16 (s, 1 C, C1), 164.01(s, 1 C, C5),
142.00 (s, 1 C, C3), 121.85 (s, 1 C, C2), 121.73 (s, 1 C, C4), 51.75 (s, 1 C, C13), 51.61 (s, 2 C,
C8, 9), 40.83 (s, 1 C, C6), 39.96 (s, 1 C, C10), 27.63 (s, 2 C, C11, 12), 24.07 (s, 1 C, C7) ppm.
IR (KBr): ν̃ = 3354vs, 3326vs, 3298vs, 3244vs, 3163w, 2977m, 2968m, 2945m, 2903sh,
2877w, 1603vs, 195vs, 1579vs, 1477s, 1464s, 1457s, 1423w, 1402w, 1384w, 1368w, 1348w,
1108vs, 1046vs, 1016 vs, 974m, 820m, 763m, 694w, 664m, 625vs, 567m cm–1.
Elementaranalyse: Ber. ( %) für C13H24Cl2N4O8Zn (500.65): C 31.19, H 4.83, N 11.19; gef.
C 30.84, H 4.67, N 10.98.
4.5.2.11 [Zn(pyN3)Br]2Br0.21(ClO4)1.79 (24)
C26H48Br2.21Cl1.79N8O7.17Zn2
M = 958.21
weiße Kristalle
2 (102 mg, 0.43 mmol) wird in MeOH (1.5 ml) gelöst und mit einer farblosen Lösung von
Zn(ClO4)2 · 6 H2O (159 mg, 0.43 mmol) gelöst in MeOH (3 ml) versetzt. Nach Zugabe von
LiBr (37 mg, 0.43 mmol) wird noch 1 h gerührt. Durch Eindiffusion von Et2O entsteht ein
weißes Pulver. Das Lösemittel wird im Vakuum entfernt und der Rückstand in MeOH (5 ml)
aufgelöst. Nach Zugabe von LiBr (185 mg, 2.15 mmol) und erneuter Etherdiffusion wird nach
2 d ein weißes Pulver erhalten. Das Lösemittel wird erneut im Vakuum entfernt und der
erhaltene Rückstand in MeOH : CH3CN 1 : 1 (20 ml) in der Siedehitze gelöst. Nach erneuter
Etherdiffusion werden Kristalle von 24 erhalten. 1H-NMR (200 MHz, [D6]DMSO, 25°C): δ = 7,85 (t, 3J(H,H) = 7.9 Hz, 1 H, H3), 7.46 (d, 3J(H,H) = 7.9 Hz, 1 H, H2), 7.37 (d, 3J(H,H) = 7.8 Hz, 1 H, H4), 4.11 (br, 6 H, –NH2) 3.15 -
3.00 (m, 6 H, H8, 9, 13, vom HOD-Signal überlagert), 1.31 (s, 6 H, H11, 12), 1.21 (s, 3 H, H7)
ppm.
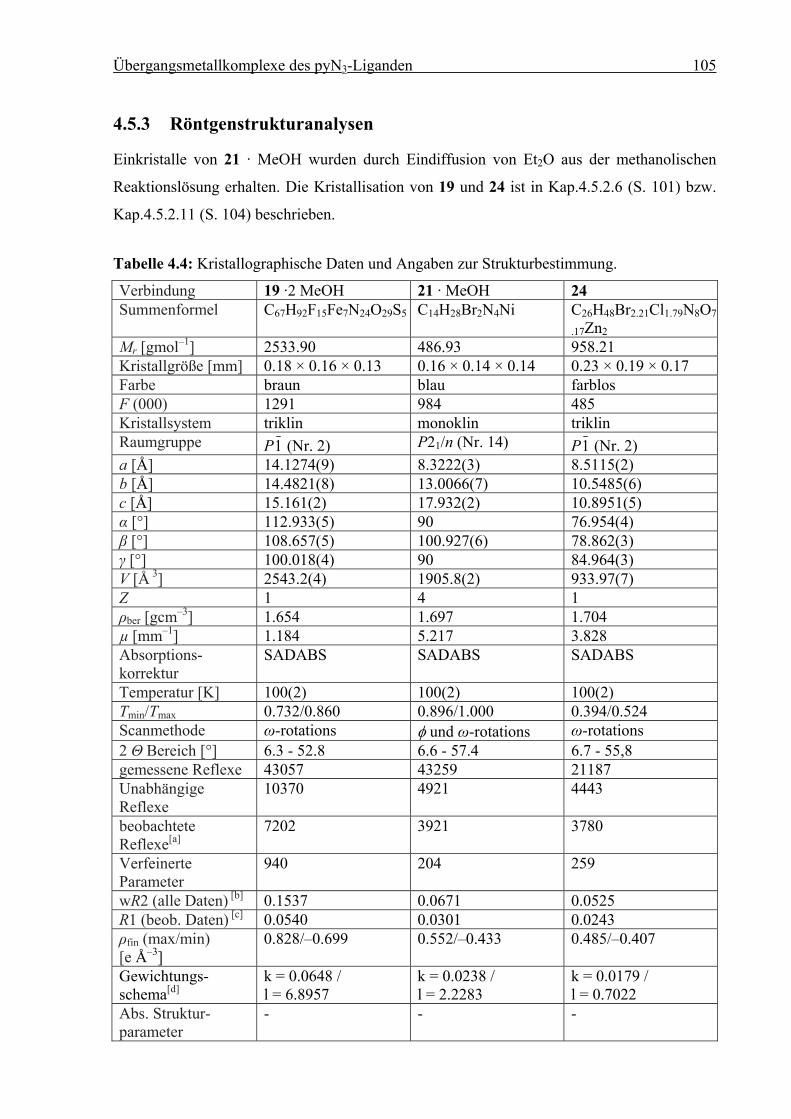
Übergangsmetallkomplexe des pyN3-Liganden 105
4.5.3 Röntgenstrukturanalysen
Einkristalle von 21 · MeOH wurden durch Eindiffusion von Et2O aus der methanolischen
Reaktionslösung erhalten. Die Kristallisation von 19 und 24 ist in Kap.4.5.2.6 (S. 101) bzw.
Kap.4.5.2.11 (S. 104) beschrieben.
Tabelle 4.4: Kristallographische Daten und Angaben zur Strukturbestimmung.
Verbindung 19 ·2 MeOH 21 · MeOH 24 Summenformel C67H92F15Fe7N24O29S5 C14H28Br2N4Ni C26H48Br2.21Cl1.79N8O7
.17Zn2 Mr [gmol–1] 2533.90 486.93 958.21 Kristallgröße [mm] 0.18 × 0.16 × 0.13 0.16 × 0.14 × 0.14 0.23 × 0.19 × 0.17 Farbe braun blau farblos F (000) 1291 984 485 Kristallsystem triklin monoklin triklin Raumgruppe P1 (Nr. 2) P21/n (Nr. 14) P1 (Nr. 2) a [Å] 14.1274(9) 8.3222(3) 8.5115(2) b [Å] 14.4821(8) 13.0066(7) 10.5485(6) c [Å] 15.161(2) 17.932(2) 10.8951(5) α [°] 112.933(5) 90 76.954(4) β [°] 108.657(5) 100.927(6) 78.862(3) γ [°] 100.018(4) 90 84.964(3) V [Å 3] 2543.2(4) 1905.8(2) 933.97(7) Z 1 4 1 ρber [gcm–3] 1.654 1.697 1.704 µ [mm–1] 1.184 5.217 3.828 Absorptions- korrektur
SADABS SADABS SADABS
Temperatur [K] 100(2) 100(2) 100(2) Tmin/Tmax 0.732/0.860 0.896/1.000 0.394/0.524 Scanmethode ω-rotations φ und ω-rotations ω-rotations 2 Θ Bereich [°] 6.3 - 52.8 6.6 - 57.4 6.7 - 55,8 gemessene Reflexe 43057 43259 21187 Unabhängige Reflexe
10370 4921 4443
beobachtete Reflexe[a]
7202 3921 3780
Verfeinerte Parameter
940 204 259
wR2 (alle Daten) [b] 0.1537 0.0671 0.0525 R1 (beob. Daten) [c] 0.0540 0.0301 0.0243 ρfin (max/min) [e Å–3]
0.828/–0.699 0.552/–0.433 0.485/–0.407
Gewichtungs-schema[d]
k = 0.0648 / l = 6.8957
k = 0.0238 / l = 2.2283
k = 0.0179 / l = 0.7022
Abs. Struktur- parameter
- - -

Übergangsmetallkomplexe des pyN3-Liganden 106
[a] mit Fo ≥ 4 σ(F); [b] wR2 = ({Σ [w(Fo2 – Fc
2)2]}/{Σ [w(Fo2)2]})0.5;
[c] R1 = Σ ||Fo| – |Fc||/Σ |Fo| for Fo > 4 σ(F); [d] w = 1/[σ2(Fo
2) + (k · P)2 + l · P] and P = (Fo2 + 2 · Fc
2)/3
4.5.4 BVS-Rechnungen an [FeIII
3FeII4L12](OTf)5 · 2 MeOH (19)
Zur Berechnung wurde folgende Gleichung verwendet:[8]
Brr
es−
=0
s: Bindungsvalenz einer Metall-Ligand-Bindung
r0: tabellierter Bindungsvalenzparameter
r: Bindungslänge aus Röntgenstrukturanalyse
B: empirischer Parameter, festgelegt auf 0.37.[8][10]
Verwendete Werte für r0:[10]
Ergebnisse für 19 (in Klammern die Abweichung [ %] Ist- durch Sollwert):
FeII FeIII Fe1 2.31 (16) 2.70 (10) Fe2 2.24 (12) 2.62 (13) Fe3 2.24 (12) 2.62 (13) Fe4 (zentrales Ion) 1.88 (6) 2.24 (25)
Mittelwert der BVS von Fe1; Fe2 und Fe3: 2.46
Erwarteter Wert 2.5.
FeII FeIII N 1.769 1.815 O 1.700 1.765

Übergangsmetallkomplexe des pyN3-Liganden 107
4.6 Literatur
[1] a) I. Morgenstern-Badarau, F. Lambert, J. P. Renault, M. Cesario, J.-D. Maréchal, F.
Maseras, Inorg. Chim. Acta 2000, 297, 338–350. b) M. J. Hardie, C. A. Kilner, M, A.
Halcrow, Acta Cryst. 2004, C60, m177-m179. c) G. J. P. Britovsek, J. England, A. J.
P. White, Inorg. Chem. 2005, 44, 8125-8134. d) M. K. Zart, T. N. Sorrell, D. Powell,
A. S. Borovik, Dalton Trans. 2003, 10, 1986-1992. e) F. Hojland, H. Toftlund, S. Yde
Andersen, Acta Chem. Scan. 1983, A37, 251-257. f) G. Brehm, M. Reiher, B. Le
Guennic, M. Leibold, S. Schindler, F. W. Heinemann, S. Schneider, J. Ram. Spec.
2006, 37, 108-122.
[2] S. W. Kohl, F. W. Heinemann, M. Hummert, W. Bauer, A. Grohmann, Chem. Eur. J.
2006, 12, 4313-4320.
[3] E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kemsley, S.-K. Lee, N. Lehnert, F.
Neese, A. J. Skulan, Y.-S. Yang, J. Zhou, Chemical Reviews, 2000, 100, 235-349.
[4] a) C. Dietz, F.W. Heinemann, J. Kuhnigk, C. Krüger, M. Gerdan, A.X. Trautwein, A.
Grohmann, Eur. J. Inorg. Chem. 1998, 1041 - 1049. b) C. Dietz, F.W. Heinemann, A.
Grohmann, Eur. J. Inorg.Chem. 1999, 2147 - 2156. c) C. Zimmermann, F.W.
Heinemann, A. Grohmann, Eur. J. Inorg. Chem. 2001 , 547 - 555. d) J. Pitarch López,
Dissertation, Universität Erlangen-Nürnberg 2003.
[5] Stephan Kohl, Dissertation, Technische Universität Berlin, 2006.
[6] a) R. W. Saalfrank, T. Nakajima, N. Mooren, A. Scheurer, H. Maid, F. Hampel, C.
Trieflinger, J. Daub, Eur. J. Inorg. Chem. 2005, 1149–1153. b) R. W. Saalfrank, R.
Prakash, H. Maid, F. Hampel, F. W. Heinemann, A. X.Trautwein, L. H. Böttger,
Chem. Eur. J. 2006, 12, 2428–2433.
[7] G. Labat, C. Boskovic, H. U. Gündel, Acta Cryst. 2005, E61, m611-m613.
[8] H. H. Thorp, Inorg. Chem. 1992, 31, 1585-1588.
[9] H. Oshio, N. Hoshino, T. Ito, M. Nakano, F. Renz, P. Gütlich, Angew. Chem. Int. Ed.
2003, 43(3), 223-225.
[10] W. Liu and H. H. Thorp, Inorg. Chem. 1993, 32, 4102-4105.
[11] M. Murugesu, R. Clérac, W. Wernsdorfer, C. E. Anson, A. K. Powell, Angew. Chem.
Int. Ed. 2005, 44, 6678-6682.
[12] a) J. Pitarch López, Dissertation, Universität Erlangen-Nürnberg, 2003. b) Beispiele in
dieser Arbeit.

Übergangsmetallkomplexe des pyN3-Liganden 108
[13] a) T. Brandsch, F. Schell, K. Weiss, M. Ruf, B. Müller, H. Vahrenkamp, Chem.
Ber./Recueil 1997, 130, 283-289. b) M. M. Ibrahim, N. Shimoura, K. Ichikawa, M.
Shiro, Inorg. Chim. Acta. 2001, 313, 125-136.
[14] Acros Organics, Handbuch für Feinchemikalien, Schwerte, Deutschland, 2006-2007.
[15] S. W. Kohl, F. W. Heinemann, M. Hummert, W. Bauer, A. Grohmann, Chem. Eur. J.
2006, 12, 4313-4320.
[16] R. C. Paul, P. Kapila,A. S. Bedi, S. K. Vasisht, J. Ind. Chem. Soc. 1976, 53, 768-772.
[17] J. R. Röper, Dissertation , Universität Darmstadt, 1990.
[18] G. Socrates, Infrared Characteristic Group Frequencies, 2. ed., Wiley, New York
1994.

Versuche zur Derivatisierung des pyN4-Liganden 109
5 Versuche zur Derivatisierung des pyN4-Liganden
5.1 Versuche zur Permethylierung des pyN4-Liganden
5.1.1 Allgemeines
Die Derivatisierung der primären Aminofunktionen von 1 zu NHMe-Gruppen wurde von S.
Leopold in einer zweistufigen Synthese erreicht.[1] Ziel dieser Arbeit war die Deprotonierung
dieser sekundären Aminfunktionen, gefolgt von der Komplexierung des so erhaltenen
Tetraamidoiminliganden. Bei Komplexierung des nicht deprotonierten Liganden ist die
Bildung eines Diastereomerengemisches zu erwarten, da die Stickstoffatome der sekundären
Aminfunktionen infolge Metallkoordination zu stereogenen Zentren werden. Dies Problem
stellt sich bei Vorliegen von NMe2-Gruppen nicht. Der +I-Effekt von acht Methylgruppen
erhöht die Elektronendichte des koordinierten Metallions. Dies hätte unter anderem einen
Einfluss auf das Spin-Crossover-Verhalten von FeII-Komplexen (vgl. Kap. 1 und 3). Überdies
ist zu erwarten, dass die Oxidationsempfindlichkeit des Liganden durch Einführung von
NMe2-Gruppen herabgesetzt wird (Vermeidung der Iminbildung).
N
NN
NN
Abb. 5.1: Molekülstruktur des Zielliganden py(NMe2)4.
5.1.2 Synthesen
Die Methylierung von primären und sekundären Aminen zu tertiären Aminen kann mittels
Eschweiler-Clarke-Reaktion erreicht werden.[2] Bei Verbindungen, die mehrere primäre
Aminofunktionen enthalten, sinkt jedoch die Ausbeute.[3] Daher wurde zunächst der folgende
Syntheseweg verfolgt.

Versuche zur Derivatisierung des pyN4-Liganden 110
Der zu py(NMe2)4 analoge tetrapodale Polyphosphan-Ligand py(PMe2)4 wurde ausgehend
von dem Tetrabromid pyBr4 (rationeller Name: 2,6-Bis(2-methyl-1,3-dibromo-propan-2-
yl)pyridin) durch Umsetzung mit vier Äquivalenten LiPMe2 in Diethylether synthetisiert (S.
W. Kohl). [4] Das Produkt wird in 83 % Ausbeute erhalten.
Analog wird eine etherische Lösung von pyBr4 bei –78 °C zu einer Suspension von LiNMe2
in Et2O getropft. Die Mischung wird über Nacht ohne weitere Kühlung gerührt. Die
Suspension nimmt eine intensiv rote Farbe an. Nach Hydrolyse und Entfernen des Lösemittels
wurde ein 1H-NMR-Spektrum aufgenommen. Es haben sich mehrere Produkte gebildet, es
gibt jedoch keinen Hinweis auf die Bildung des gewünschten Liganden py(NMe2)4. Es kommt
hier offensichtlich zu Nebenreaktionen, die eine Bildung des gewünschten Produktes
verhindern. Deshalb wurde dieser Weg nicht weiter verfolgt.
Stattdessen wurde eine Synthese unter Eschweiler-Clarke-Bedingungen versucht. Hierzu wird
1 · 4 HBr mit einem Überschuss an Formaldehydlösung (37 %) und Ameisensäure acht
Stunden zum Rückfluss erhitzt. Nach Abkühlen auf Raumtemperatur erfolgt die Extraktion
und Umsetzung mit Bromwasserstoffsäure (48 %; Details im Experimentellen Teil).
Das 1H-NMR-Spektrum zeigt drei charakteristische Merkmale. Zum einem finden sich zwei
stark verbreiterte Signale zwischen 10.0 und 8.5 ppm im Verhältnis 1 : 2. Diese werden
protonierten Stickstoffatomen zugeordnet. Bei 7.8 ppm erscheinen zwei übereinander
liegende Tripletts für das Wasserstoffatom in para-Position. Statt eines Singuletts für die
Methylgruppen sind im Bereich um 1.3 ppm zwei Dubletts zu erkennen. Diese und weitere
spektroskopische Befunde belegen die Bildung des Produktes 25 (Abb. 5.2).
N
NH HN
* *N
NH2
NH2NH2
NH2
1. H2CO/HCOOH2. 8 h RF3. HBr (exc.)
H(Br–)3
3+
25
Abb. 5.2: Reaktion von 1 unter Eschweiler-Clarke-Bedingungen.

Versuche zur Derivatisierung des pyN4-Liganden 111
Verbindung 25 enthält zwei Stereozentren. Es entstehen sowohl das R,R’- und S, S’-
Enantiomerenpaar als auch die meso-Form (R,S). Dies erklärt das Auftreten des doppelten
Signalsatzes.
Die selektive Spaltung dieser zwei C–C-Bindungen kann auf eine Retro-Mannich-Reaktion
zurückzuführen sein, wie sie auch bei ähnlichen, eine benzylische Position aufweisenden
Systemen und bei Systemen in α-Stellung von Ketonen auftritt.[5] Ein schneller Zugang zum
permethylierten Liganden py(NMe2)4 ist auf diesem Weg also nicht möglich. Weitere
Syntheseversuche wurden nicht durchgeführt.
5.2 Modifizierung des pyN4-Liganden in para-Postion
5.2.1 Allgemeines
In Kap. 3 ist die Synthese mehrerer SCO-Verbindungen des Typs [(1)FeII(L)] beschrieben.
Zur Chemisorption auf Oberflächen muss der sechste Ligand L eine geeignete Ankergruppe
(AG) enthalten. Ein Weg zu erwartungsgemäß stabileren Schichten wäre, den pyN4-Liganden
über die para-Position seines Pyridinringes direkt an die Oberfläche zu binden.
Oberfläche
N
H2NH2N FeII
NH2
NH2
L
AG
N
NH2NH2
FeIIH2NH2N
L
Oberfläche
AG
Abb. 5.3: Mögliche Chemisorption von Komplexen des Typs [(1)FeII(L)] auf Oberflächen.
Den pyN4-Liganden an sich zu derivatisieren ist aufgrund seines polyfunktionellen Charakters
schwierig (Reaktivität der primären Aminofunktionen) und würde aufwendige Schutz-
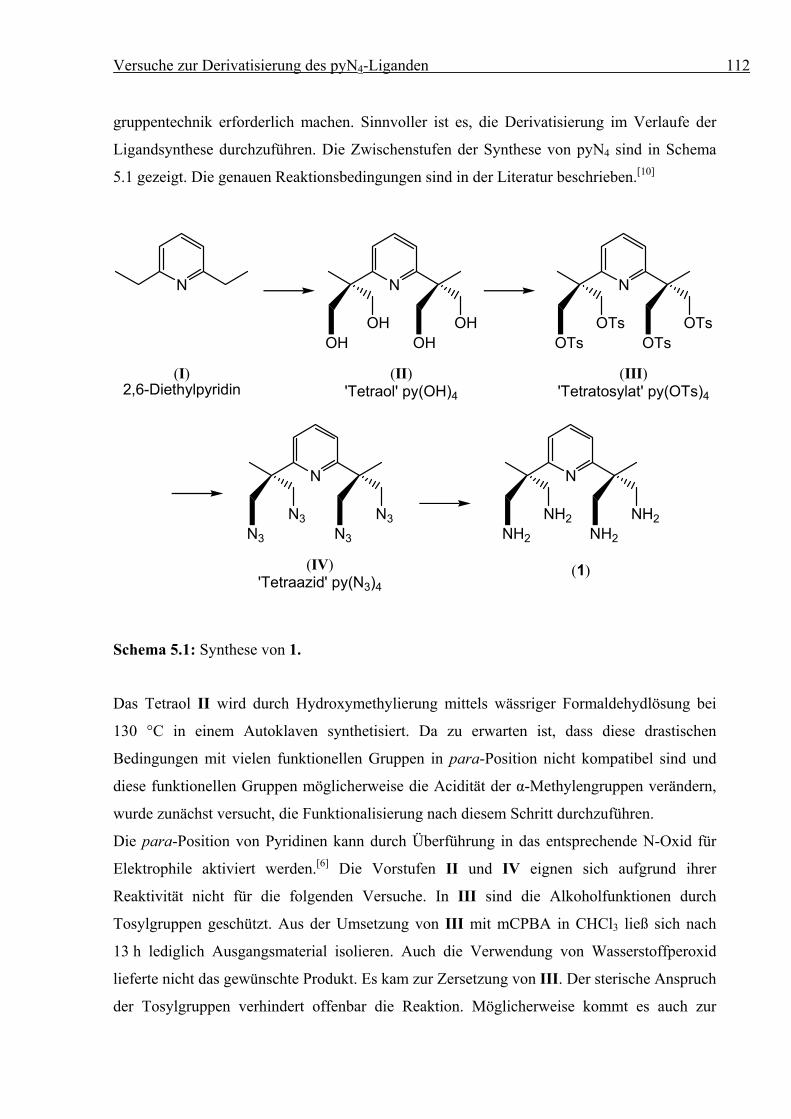
Versuche zur Derivatisierung des pyN4-Liganden 112
gruppentechnik erforderlich machen. Sinnvoller ist es, die Derivatisierung im Verlaufe der
Ligandsynthese durchzuführen. Die Zwischenstufen der Synthese von pyN4 sind in Schema
5.1 gezeigt. Die genauen Reaktionsbedingungen sind in der Literatur beschrieben.[10]
N N
OHOH
OHOH
N
OTsOTs
OTsOTs
N
N3
N3N3
N3
N
NH2
NH2NH2
NH2
(I)2,6-Diethylpyridin
(II)'Tetraol' py(OH)4
(III)'Tetratosylat' py(OTs)4
(IV)'Tetraazid' py(N3)4
(1)
Schema 5.1: Synthese von 1.
Das Tetraol II wird durch Hydroxymethylierung mittels wässriger Formaldehydlösung bei
130 °C in einem Autoklaven synthetisiert. Da zu erwarten ist, dass diese drastischen
Bedingungen mit vielen funktionellen Gruppen in para-Position nicht kompatibel sind und
diese funktionellen Gruppen möglicherweise die Acidität der α-Methylengruppen verändern,
wurde zunächst versucht, die Funktionalisierung nach diesem Schritt durchzuführen.
Die para-Position von Pyridinen kann durch Überführung in das entsprechende N-Oxid für
Elektrophile aktiviert werden.[6] Die Vorstufen II und IV eignen sich aufgrund ihrer
Reaktivität nicht für die folgenden Versuche. In III sind die Alkoholfunktionen durch
Tosylgruppen geschützt. Aus der Umsetzung von III mit mCPBA in CHCl3 ließ sich nach
13 h lediglich Ausgangsmaterial isolieren. Auch die Verwendung von Wasserstoffperoxid
lieferte nicht das gewünschte Produkt. Es kam zur Zersetzung von III. Der sterische Anspruch
der Tosylgruppen verhindert offenbar die Reaktion. Möglicherweise kommt es auch zur

Versuche zur Derivatisierung des pyN4-Liganden 113
Oxidation in der Seitenkette, wie bei der Umsetzung von 2-Cyanopyridin mit
Wasserstoffperoxid.[7]
Diese Ergebnisse zeigen, dass die Derivatisierung vorher erfolgen muss. Um die Reaktivität
von I nicht wesentlich zu verändern, soll in para-Position ein Bromsubstituent eingeführt
werden. Eine direkte Synthese ausgehend von I kann über folgende Zwischenstufen
verlaufen.
N+
O-
N+
O-
NO2
N+
O-
Br
N
Br
N
26
Schema 5.2: Zwischenstufen der Synthese von 26 ausgehend von 2,6-Diethylpyridin.
Erste Versuche haben gezeigt, dass die säulenchromatische Reinigung der Zwischenstufen
erforderlich ist. Die quantitative Trennung der N-Oxide erweist sich aufgrund ihrer Polarität
als schwierig, was den schnellen und einfachen Zugang zu größeren Mengen 26 erschwert.
Die Synthese von 4-Amino-2,6-diethylpyridin im Kilogrammmaßstab ist in der Literatur
beschrieben.[8] Die Aminogruppe kann durch Umsetzung mit Natriumnitrit in 48 %iger
Bromwasserstoffsäure durch Brom substituiert werden.
N
NH2
N
Br1. HBr (48%)2. NaNO2
26
Schema 5.3: Synthese von 26 ausgehend von 4-Amino-2,6-diethylpyridin.
Verbindung 26 wurde 1H-NMR- und massenspektroskopisch charakterisiert. Das
Massenspektrum zeigt das typische Isotopenmuster für Monobrom-Substitution (1 : 1). In
einem nächsten Schritt ist die Reaktivität von 26 gegenüber Formaldehyd zu untersuchen
(Aubau eines zu II analogen Tetraalkohols).

Versuche zur Derivatisierung des pyN4-Liganden 114
5.3 Experimenteller Teil
5.3.1 Allgemeines
Sofern nicht anders vermerkt, wurden alle Reaktionen unter N2-Atmosphäre in absoluten
Lösemitteln in Standard-Schlenk-Gefäßen durchgeführt. Die verwendeten absoluten
Lösemittel (Restgehalt H2O ≤ 50 ppm) sind kommerziell über Molsieb getrocknet
erhältlich.[9] Organische und anorganische Reagenzien sind von den Firmen Aldrich, Acros,
Strem oder Alfa Aesar und wurden ohne weitere Reinigung benutzt. Die Verbindungen 1 · 4
HBr · MeOH[10] und 4-Amino-2,6-diethylpyridin[8] wurden nach Literaturangaben hergestellt.
Zur Aufnahme der Daten wurden folgende Geräte verwendet:
Kernresonanzspektren: Die NMR-Spektren wurden, soweit nicht anders vermerkt, bei
Raumtemperatur in einem 5 mm-Röhrchen an Spektrometern der Firma Bruker
aufgenommen: ARX 200 (1H, 200 MHz; 13C, 50.32 MHz), ARX 400 (1H, 400 MHz; 13C,
100.64 MHz). Die temperaturabhängigen NMR-Spektren von 5, 8 und 12 wurden an einem
Alpha 500 der Firma Jeol gemessen (1H, 500 MHz; 13C, 125.78 MHz). Luftempfindliche
Proben wurden in einem unter Vakuum abgeschmolzenen Röhrchen vermessen. Alle
chemischen Verschiebungen sind in ppm relativ zur Restprotonen- bzw. 13C-Absorption des
verwendeten Lösemittels (interner Standard) angegeben. Die 13C-NMR-Spektren wurden
breitbandentkoppelt aufgenommen. Die Vorzeichen der Kopplungskonstanten wurden bei den 1H- und 13C-Spektren nicht bestimmt.
Elementaranalysen: Die quantitative Bestimmung von Kohlenstoff, Wasserstoff, Stickstoff
und Schwefel erfolgte verbrennungsanalytisch an einem Gerät des Typs Thermo Finnigan
EAGER 300 (Flash 1112 Series) Gerät.
Infrarotspektren: Die IR-Spektren von wurden an einem Nicolet Magna System 750-
Spektrometer aufgenommen. Feststoffe wurden als Kaliumbromid-Presslinge, Lösungen in
CaF2-Küvetten unter Kompensation der Solvensabsorption vermessen. Die Wellenzahlen ν̃
sind in cm–1 angegeben. Die Zuordnung der Banden erfolgte anhand einschlägiger
Literatur.[11]

Versuche zur Derivatisierung des pyN4-Liganden 115
Massenspektren: Die Aufnahme der Massenspektren erfolgte unter Verwendung von
folgendem Gerät: Varian 311A (EI-MS, 70 eV).
5.3.1.1 2,2'-(Pyridin-2,6-diyl)bis(N,N-dimethylpropan-1-amin) · 3 HBr (25)
C15H30Br3N3
M = 492.13 g/mol
weißes, hygroskopisches Pulver
In einer Mischung aus wässriger, 37%iger Formaldehydlösung (10-15% MeOH) (4.0 ml, 4.3
g, 53 mmol), 99%iger Ameisensäure (5 ml, 6.1 g, 131 mmol) und H2O (1 ml) werden 600 mg
1 · 4 HBr · MeOH (600 mg, 1 mmol) gelöst und anschließend 8 h unter Rückfluss erhitzt.
Im Vakuum werden alle flüchtigen Bestandteile entfernt und der weiße Rückstand in
wässriger KOH-Lösung (10 ml, 10 %) gelöst. Es wird mit Et2O und CH2Cl2 (3 × 10 ml)
extrahiert. Die vereinigten organischen Phasen werden getrocknet und mit HBr (48 %; 2 ml)
versetzt. Anschließend wird im Hochvakuum getrocknet und 1 h in MeOH (5 ml) ausgerührt.
Der weiße Niederschlag wird abfiltriert, mit Et2O (3 x 5 ml) gewaschen und im Hochvakkum
getrocknet.
Die Zuordnung erfolgt anhand von 1H-1H- und 1H-13C-korrelierten NMR-Spektren. 1H-NMR (200 MHz, [D6]DMSO, 25°C): δ = 10.0 - 9.4 (s, br, 2 H, Pyridinium-H), 9.1 - 8.7
(s, br, 4 H, NHMe2), 7.9 - 7.7 (m, 2 H, Hpara), 7.34 (d, 4 H, 3J(H,H) = 7.7 Hz), 3.8 - 3.5 (m, 4
H, CHH), 3.5 - 3.2 (m, 8 H , CHH/CH), 2.9 - 2.6 (m, 24 H, N(CH3)2), 1.30 - 1.15 (m, 12 H,
CH3) ppm. 13C-NMR (50.32 MHz, [D6]DMSO, 25°C): δ = 160.86 (s, 4 C, Cortho), 139.16 (s, 2 C, py-
Cpara), 122.00 (s, 2 C, Cmeta), 121.76 (s, 2 C, Cmeta), 61.32 (s, 2 C, CH2), 61.21 (s, 2 C, CH2),
43.82 (s, 2 C, NCH3), 43.53 (s, 2 C, NCH3), 43.13 (s, 2 C, NCH3), 42.59 (s, 2 C, NCH3),
36.48 (s, 2 C, CH), 36.35 (s, 2 C, CH), 19.78 (s, 2 C, CH3), 19.76 (s, 2 C, CH3) ppm.
IR (KBr): ν̃ = 3061m, 3012m, 2970m, 2885m, 2618s, 2510m, 2466m, 1637m, 1623m,
1543w, 1464m, 1415m, 1162m, 1002w, 980m, 829m, 754w cm–1.
N
NH HN
* *
H(Br–)3
3+

Versuche zur Derivatisierung des pyN4-Liganden 116
Elementaranalyse: Ber. ( %) für C15H30Br3N3 (492.13): C 36.61, H 6.14, N 8.54; gef. C
36.67, H 6.13, N 8.11.
EI-MS (70 eV): m/z ( %) = 212 (100) [M-H]+, 214 (100) [M-H]+.
5.3.1.2 4-Brom-2,6-diethylpyridin (26)
C9H12BrN
M = 214.10 g/mol
braune Kristalle
4-Amino-2,6-diethylpyridin (0.15 g, 1.0 mmol) wird in wässriger Bromwasserstoffsäure (48
%; 5 ml) bei 0 °C gelöst. Wässrige Natriumnitritlösung (50 %; 4 ml) wird bei 0°C unter
Rühren langsam hinzugetropft und die Mischung 2 Stunden gerührt. Anschließend wird das
Reaktionsgemisch mit Chloroform (4 × 25 ml) extrahiert. Die organische Phase wird
abgetrennt und mit Wasser (4 × 50 ml), mit gesättigter Natriumthiosulfatlösung (4 × 25 ml)
und ein zweites mal mit Wasser (2 × 50 ml) gewaschen. Die organischen Phasen werden über
Natriumsulfat getrocknet. Das Lösemittel wird im Vakuum entfernt und das Produkt im
Hochvakuum getrocknet. (0.1 g, 46 %). 1H-NMR (200 MHz, [D1]Chloroform, 25°C): δ = 7.64 (s, 2 H, Hmeta), 3.19 (q, 4 H, 3J(H,H)
= 7.6 Hz, CH2), 1.42 (t, 6 H, 3J(H,H) = 7.6 Hz, CH3) ppm.
EI-MS (70 eV): m/z ( %) = 212 (100) [M-H]+, 214 (100) [M-H]+.
5.4 Literatur
[1] Stefan Leopold, Dissertation, Technische Universität Berlin, 2006.
[2] a) F. G. Riddell, M. Rogerson, J. Chem. Soc., Perkin Trans. 1997, 2, 249-255. b) J. A.
Sintas, A. A. Vitale, J. Lab. Comp. Radiopharm. 1998, 41, 53-68. c) M. Taki, S.
Teramae, S. Nagatomo, Y. Tachi, T. Kitagawa, S. Itoh, S. Fukuzumi, J. Am. Chem.
Soc. 2002, 124, 6367-6377.
N
Br

Versuche zur Derivatisierung des pyN4-Liganden 117
[3] a) M. Ciampolini, N. Nardi, Inorg. Chem. 1966, 5, 41-44. b) R. H. Mizzoni, M. A.
Hennessey, C. R. Scholz, J. Am. Chem. Soc. 1954, 76, 2414-2417.
[4] S. W. Kohl, F. W. Heinemann, M. Hummert, W. Bauer, A. Grohmann, Chem. Eur. J.
2006, 12, 4313-4320.
[5] a) H. Möhrle, M. Pycior, J. Lessel, Sci. Pharm. 1996, 64, 569-575. b) H. Möhrle, M.
Pycior, Arch. Pharm., 1995, 293-296. c) G. L. Buchanan, A. C. W. Curran, Chem.
Commun. (London), 1966, 21, 773.
[6] a) E. Ochiai, J. Org. Chem. 1954, 18, 534-551. b) K. Yamaguchi, T. Mizugaki, K.
Ebitani, K. Kaneda, New J. Chem., 1999, 23, 799-801. c) D. Wenkert, R. B.
Woodward, J. Org. Chem. 1983, 48, 284-289.
[7] G. B. Payne, J. Org. Chem. 1961, 26, 668-670.
[8] W. J. Watkins, G. E. Robinson, P. J. Hogan, D. Smith, Synth. Comm. 1994, 24, 1709-
1714.
[9] Acros Organics, Handbuch für Feinchemikalien, Schwerte, Deutschland, 2006-2007.
[10] S. Schmidt, L. Omnès, F. W. Heinemann, J. Kuhnigk, C. Krüger, A. Grohmann, Z.
Naturforsch. Teil B 1998, 53, 946-954.
[11] G. Socrates, Infrared Characteristic Group Frequencies, 2. ed., Wiley, New York
1994.

Zusammenfassung 118
6 Zusammenfassung
Die Koordinationschemie von Eisen(II) wird von Iminliganden dominiert, die durch π-
Rückbindung Elektronendichte vom FeII-Zentrum aufnehmen können. Demgegenüber sind
aliphatische Amine reine σ-Donoren. Eisen(II)-Komplexe offenkettiger Liganden dieses Typs
sind sehr empfindlich gegenüber metall- und/oder ligandzentrierter Oxidation. Der tetrapodal-
fünfzähnige Ligand 1 („pyN4“) hingegen bildet mit Eisen(II) einkernige Komplexe hoher
Stabilität, die sich offenbar einem starken Chelateffekt verdankt.
N
NH2
NH2NH2
NH2
1
N
H2NH2N FeII
NH2
NH2
L
Abb. 6.1: links: Ligand 1 („pyN4“); rechts: Struktur der Komplexe des Typs [(1)FeIIL].
Vorangegangene Arbeiten (J. Pitarch López) zeigten, dass der Spinzustand des FeII-Zentrums
von sechsten Liganden L abhängig ist. Bei L = Br– (Schwachfeldligand) wird ein high-spin-
Komplex erhalten, bei L = CO (Starkfeldligand) ein low-spin-Komplex. Dies legte die
Vermutung nahe, dass Liganden mittlerer Position in der spektrochemischen Reihe spin-
schaltbare, d. h. bei Anwendung eines äußeren Stimulus (z. B. Variation der Temperatur)
einen Spinübergang (Spin Crossover, SCO) zeigende Komplexe ergeben sollten.
Kapitel 3 beschreibt die Synthese und Charakterisierung der SCO-Verbindungen [(1)FeII(1-
Methyl-1H-imidazol)](OTf)2 (5), [(1)FeII(Pyridin-4-carbonsäuremethylester)]Br2 (8) und
[(1)FeII((E)-4-(Phenyldiazenyl)pyridin)](OTf)2 (12). Durch Umsetzung von FeCl2 (Synthese
von 8) bzw. Fe(CH3CN)2(OTf)2 (Synthese von 5 und 12) mit pyN4 (1) und dem
entsprechenden Koliganden in Methanol lassen sich die Verbindungen in Ausbeuten von
jeweils über 70 % erhalten. Die Darstellung elementaranalysenreiner Verbindungen ist
aufwendiger als bei den reinen LS-Verbindungen [FeII(pyN4)(CNtBu)]Br2 (13),
[FeII(pyN4)(CNcy)]Br2 (14) und [FeII(pyN4)(CNC6H4OCH3)]Br2 (15). Bei den Verbindungen
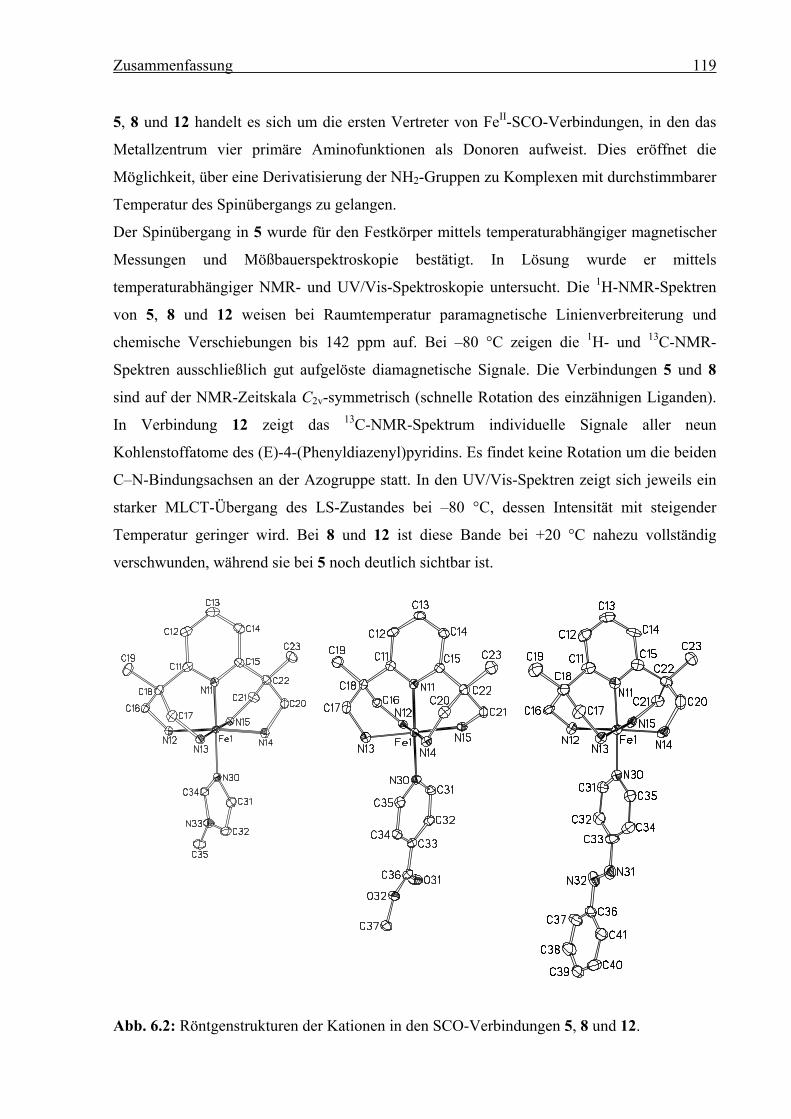
Zusammenfassung 119
5, 8 und 12 handelt es sich um die ersten Vertreter von FeII-SCO-Verbindungen, in den das
Metallzentrum vier primäre Aminofunktionen als Donoren aufweist. Dies eröffnet die
Möglichkeit, über eine Derivatisierung der NH2-Gruppen zu Komplexen mit durchstimmbarer
Temperatur des Spinübergangs zu gelangen.
Der Spinübergang in 5 wurde für den Festkörper mittels temperaturabhängiger magnetischer
Messungen und Mößbauerspektroskopie bestätigt. In Lösung wurde er mittels
temperaturabhängiger NMR- und UV/Vis-Spektroskopie untersucht. Die 1H-NMR-Spektren
von 5, 8 und 12 weisen bei Raumtemperatur paramagnetische Linienverbreiterung und
chemische Verschiebungen bis 142 ppm auf. Bei –80 °C zeigen die 1H- und 13C-NMR-
Spektren ausschließlich gut aufgelöste diamagnetische Signale. Die Verbindungen 5 und 8
sind auf der NMR-Zeitskala C2v-symmetrisch (schnelle Rotation des einzähnigen Liganden).
In Verbindung 12 zeigt das 13C-NMR-Spektrum individuelle Signale aller neun
Kohlenstoffatome des (E)-4-(Phenyldiazenyl)pyridins. Es findet keine Rotation um die beiden
C–N-Bindungsachsen an der Azogruppe statt. In den UV/Vis-Spektren zeigt sich jeweils ein
starker MLCT-Übergang des LS-Zustandes bei –80 °C, dessen Intensität mit steigender
Temperatur geringer wird. Bei 8 und 12 ist diese Bande bei +20 °C nahezu vollständig
verschwunden, während sie bei 5 noch deutlich sichtbar ist.
Abb. 6.2: Röntgenstrukturen der Kationen in den SCO-Verbindungen 5, 8 und 12.

Zusammenfassung 120
Die cyclovoltammetrischen Untersuchungen von 5 und 8 zeigen, dass sich beide
Verbindungen reversibel oxidieren lassen. Die HS- und die LS-Spezies haben erwartungs-
gemäß unterschiedliche Redoxpotentiale. Nach Oxidation beider Spezies ändert sich die
Spinverteilung zugunsten von LS-FeIII, weswegen die Reduktionswellen nicht komplementär
zu den Oxidationswellen sind. Daraus lässt sich folgern, dass die zu 5 und 8 analogen FeIII-
Verbindungen stabil sind und es sich ebenfalls um SCO-Verbindungen handelt.
Die Synthese einer ersten dreikernigen, durch Pyridin-4-thiolat verbrückten Verbindung 10
ergab sich beim Versuch, die korrespondierende einkernige Verbindung (9) zu synthetisieren.
Die Verbindung 9 ließ sich bisher nicht elementaranalysenrein gewinnen. Aus den
analytischen Daten lässt sich folgern, dass in einer Konkurrenzreaktion auch Verbindung 10
entsteht. Deren Synthese gelingt in einer Ausbeute von 70%. Soweit bekannt, ist 10 der erste
Vertreter von durch Pyridin-4-thiolat verbrückten dreikernigen Übergangsmetallkomplexen.
Aus der Festkörperstruktur lässt sich folgern, dass in 10 zwei Eisen(II)-Zentren im high spin-
und ein Eisen(II)-Zentrum im low spin-Zustand vorliegen. Ob eine antiferromagnetische
Kopplung der beiden HS-FeII-Zentren vorliegt, müssen weitere Untersuchungen klären.
Abb. 6.3: Röntgenstrukturen der Kationen in 9 und 10.
In den Verbindungen 5, 7, 8, 9, 10 und 12 befinden sich an den beiden axialen Positionen
aromatische Imine. In den Röntgenstrukturanalysen stehen die Ebenen der aromatischen
Ringe orthogonal zueinander. Die Fe–Naliph-Abstände sind in allen Verbindungen signifikant
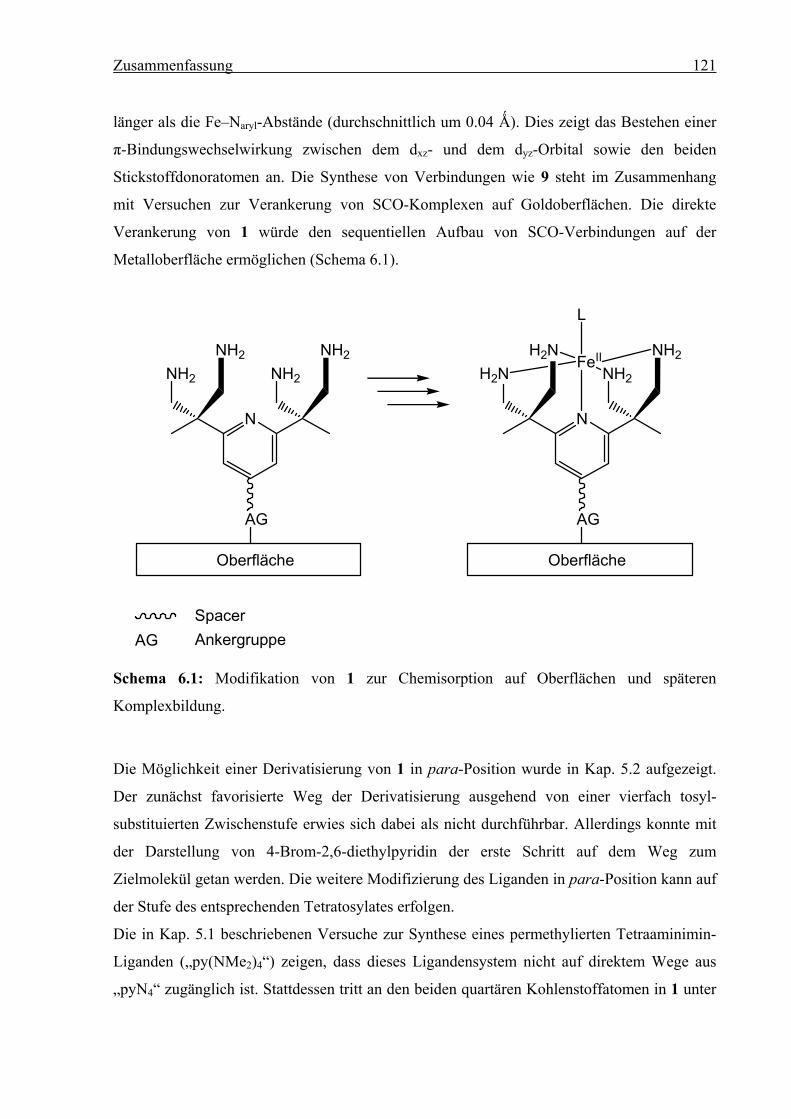
Zusammenfassung 121
länger als die Fe–Naryl-Abstände (durchschnittlich um 0.04 Ǻ). Dies zeigt das Bestehen einer
π-Bindungswechselwirkung zwischen dem dxz- und dem dyz-Orbital sowie den beiden
Stickstoffdonoratomen an. Die Synthese von Verbindungen wie 9 steht im Zusammenhang
mit Versuchen zur Verankerung von SCO-Komplexen auf Goldoberflächen. Die direkte
Verankerung von 1 würde den sequentiellen Aufbau von SCO-Verbindungen auf der
Metalloberfläche ermöglichen (Schema 6.1).
N
NH2
NH2
NH2
NH2
Oberfläche
AG
N
NH2
NH2FeIIH2N
H2N
L
Oberfläche
AG
SpacerAG Ankergruppe
Schema 6.1: Modifikation von 1 zur Chemisorption auf Oberflächen und späteren
Komplexbildung.
Die Möglichkeit einer Derivatisierung von 1 in para-Position wurde in Kap. 5.2 aufgezeigt.
Der zunächst favorisierte Weg der Derivatisierung ausgehend von einer vierfach tosyl-
substituierten Zwischenstufe erwies sich dabei als nicht durchführbar. Allerdings konnte mit
der Darstellung von 4-Brom-2,6-diethylpyridin der erste Schritt auf dem Weg zum
Zielmolekül getan werden. Die weitere Modifizierung des Liganden in para-Position kann auf
der Stufe des entsprechenden Tetratosylates erfolgen.
Die in Kap. 5.1 beschriebenen Versuche zur Synthese eines permethylierten Tetraaminimin-
Liganden („py(NMe2)4“) zeigen, dass dieses Ligandensystem nicht auf direktem Wege aus
„pyN4“ zugänglich ist. Stattdessen tritt an den beiden quartären Kohlenstoffatomen in 1 unter

Zusammenfassung 122
Eschweiler-Clarke-Bedingungen jeweils selektive C–C-Bindungsspaltung ein (Retro-
Mannich-Reaktion; Schema 6.2).
N
NH HN
* *N
NH2
NH2NH2
NH2
1. H2CO/HCOOH2. 8 h RF3. HBr (exc.)
H(Br–)3
3+
Schema 6.2: Reaktion des pyN4-Liganden unter Eschweiler-Clarke-Bedingungen.
Der Ligand 2 („pyN3“), der ein basales Donoratom als 1 („pyN4“) aufweist gibt durch höhere
Flexibilität keine Oktaedergeometrie vor.
N
NH2
NH2NH2
Abb. 6.4: pyN3-Ligand (2).
Kapitel 4 beschreibt die zweistufige Synthese dieses Liganden ausgehend von einer aus 2-
Ethyl-6-isopropylpyridin gewinnbaren dreifach mesylierten Zwischenstufe. Durch
nucleophile Substitution der Methylsulfonylgruppen mit Natriumazid in DSMO bei 70 °C
wird das entsprechende Triazid erhalten. Die Azidgruppen werden anschließend mittels
Staudinger-Reduktion zu primären Aminen reduziert, und es wird 2 erhalten (vgl. Schema
4.1; S. 81). Komplexierungsversuche mit FeII-Salzen führten bislang nicht zur Bildung
einkerniger Komplexe. Die Festkörperstrukturen von [FeII(C4H6N2)6]X2—die
Röntgenstrukturanalyse konnte nicht vollständig gelöst werden—und die des zweikernigen
Zink(II)-Komplexes 24 zeigen, dass sich 2 in seinem Komplexierungsverhalten gegenüber
FeII und ZnII grundlegend anders verhält als 1. Mit CuII und NiII werden die erwarteten
einkernigen Verbindungen 20 und 21 erhalten. Beide Verbindungen weisen einen
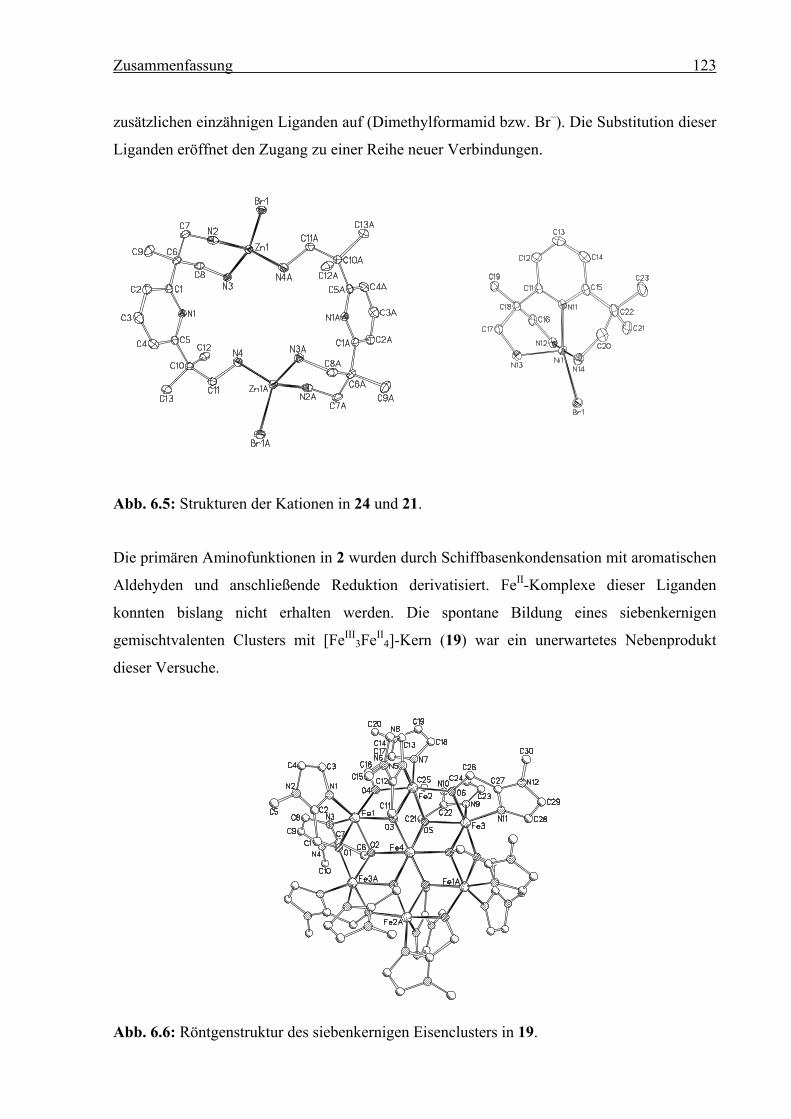
Zusammenfassung 123
zusätzlichen einzähnigen Liganden auf (Dimethylformamid bzw. Br–). Die Substitution dieser
Liganden eröffnet den Zugang zu einer Reihe neuer Verbindungen.
Abb. 6.5: Strukturen der Kationen in 24 und 21.
Die primären Aminofunktionen in 2 wurden durch Schiffbasenkondensation mit aromatischen
Aldehyden und anschließende Reduktion derivatisiert. FeII-Komplexe dieser Liganden
konnten bislang nicht erhalten werden. Die spontane Bildung eines siebenkernigen
gemischtvalenten Clusters mit [FeIII3FeII
4]-Kern (19) war ein unerwartetes Nebenprodukt
dieser Versuche.
Abb. 6.6: Röntgenstruktur des siebenkernigen Eisenclusters in 19.

Anhang 124
7 Anhang
7.1 Verwendete Abkürzungen
Ǻ Angström (10–10 m)
B Breite (Abmessungen)
BArF Tetrakis[3,5-bis(trifluormethyl)phenyl]borat
bipy 2,2’-Bipyridin
BM Bohr’sches Magneton
br breit (IR und NMR) tBu tert-Butyl
cat Catecholat-Dianion
CHN(S)-Analyse Elementaranalyse
COSY Correlated Spectroscopy
Cq quartäres Kohlenstoffatom (13C-NMR)
d Dublett (NMR); Tag
dd Dublett vom Dublett (NMR)
DCM Dichlormethan
δ Chemische Verschiebung (NMR)
DMSO Dimethylsulfoxid
EI Elektronenstoß-Ionisation
eV Elektronenvolt
g Gramm
h Stunde
IR Infrarot
IUPAC International Union of Pure and Applied Chemistry
l Liter
λ Wellenlänge
m Multiplett (NMR); mittelstark (IR); Masse
M molar
mCPBA meta-Chlorperbenzoesäure
mm Millimeter
Me Methyl
Mes Mesyl (Methylsulfonyl; –SO2–CH3)

Anhang 125
MHz Megahertz
mg Milligramm
min Minute
ml Milliliter
MS Massenspektrum
m/z Peakposition im Massenspektrum
N2H2S3 2,2'-Bis(2-mercaptophenylamino)diethylsulfid(2–)
NHE Normal-Wasserstoffelektrode (Cyclische Voltammetrie)
nm Nanometer (10–9 m)
NMR Nuclear Magnetic Resonance
ν̃ Wellenzahl (IR)
OAc Acetatrest
p Druck
4-PAPY (E)-4-(Phenyldiazenyl)pyridin
Ph Phenyl
phen 1,10-Phenantrolin
pm Pikometer (10–12 m)
ppm parts per million
pyN3 2-(1,3-diamino-2-methylpropan-2-yl)-6-(1-amino-2-methylpropan-2-
yl)pyridin
pyN4 2,6-Bis(1’,3’-diamino-2’methylprop-2’yl)pyridin
q Quartett (NMR)
quint. Quintett (NMR)
R nicht definierter organischer Rest
RT Raumtemperatur
s Singulett (NMR); stark (IR)
t Triplett (NMR)
T Temperatur, Tiefe (Abmessungen)
THF Tetrahydrofuran
TPA Tris(pyridin-2-ylmethyl)amin
Ts Tosyl (p-Toluolsulfonyl; –SO2–C6H4–CH3)
UV Ultraviolett
vs sehr stark (IR)
X-Ray Röntgenstrahlung

Begutachtete Publikationen in Fachzeitschriften: Communication: Jesús Pitarch López, Holger Kämpf, Matthias Grunert, Philipp Gütlich, Frank W. Heinemann, Raju Prakash, Andreas Grohmann „A non-heme dinuclear iron(II) complex containing a single, unsupported hydroxo bridge“ Chem. Comm. 2006, 16, 1718-1720. Berlin, 11.12.2006