Embed Size (px)
Citation preview

Imagerie en ORL© 2010 Elsevier Masson SAS. Tous droits réservés
Chapitre 25
Généralités
La connaissance et la compréhension des malfor-mations de l’os temporal ont largement bénéfi-cié des progrès de l’imagerie et en particulier du scanner [1, 2]. Les techniques d’imagerie IRM en haute résolution apportent quant à elles des élé-ments particulièrement intéressants sur les anoma-lies du labyrinthe.L’os temporal résulte de deux précurseurs pen-dant le développement embryologique (DE).Les premiers et deuxièmes arcs branchiaux, la première fente branchiale et le mésenchyme adja-cent vont constituer l’oreille externe et l’oreille moyenne.L’oreille interne est formée à partir de la cap-sule auditive cartilagineuse qui trouve son ori-gine dans la vésicule auditive et le mésenchyme adjacent.Cette double origine explique que l’on différen-cie en deux grands groupes les malformations de l’oreille externe et de l’oreille moyenne et les mal-formations de l’oreille interne qui peuvent être observées de façon indépendante. Cependant, le mésenchyme étant commun aux deux précurseurs, les malformations associées de l’oreille externe, de l’oreille moyenne et de l’oreille interne ne sont pas exceptionnelles.Le développement du méat acoustique interne est, quant à lui, lié à la progression des structures ner-
veuses dans l’angle pontocérébelleux. Cependant les anomalies du conduit auditif interne sont assez souvent associées aux malformations de l’oreille interne sans que la raison en soit connue.
Malformations de l’oreille externe
Origine embryologique
Le pavillon de l’oreille résulte de six bourgeons auriculaires issus du premier et du deuxième arc branchial et qui vont constituer l’hélix, l’anthélix, la conche, le tragus et le lobe de l’oreille.Le conduit auditif externe se développe à partir de la première fente branchiale qui s’invagine dès la 6e semaine du développement embryonnaire (DE). Il se constitue un bourgeon qui se creuse à partir de la 26e semaine du DE. Sa persistance est responsable des perforations incomplètes et des sténoses du conduit auditif externe.Parallèlement, la poche pharyngée s’étend à partir de la 4e semaine du DE pour constituer la cavité tympanique. La trompe d’Eustache résulte de la communication entre la cavité tympanique et le pharynx embryonnaire.Les structures ossiculaire sont formées à partir des trois dérivés du cartilage du premier arc branchial (marteau ou malleus et enclume ou incus) et du dérivé du 2e arc branchial (étrier ou stapes).
Imagerie des malformations du rocher
A. Varoquaux, G. Moulin, P. Cassagneau, N. Girard, B. Bourelière, F. Desmots, H. Brunel,O. Monnet, C. Muller, S. Marciano-Chagnaud, J.-M. Thomassin,
A. Deveze, J. Magnan, J.-P. Lavieille, J.-M. Bartoli
420 Os temporal et angle pontocérébelleux
Malformations du pavillon
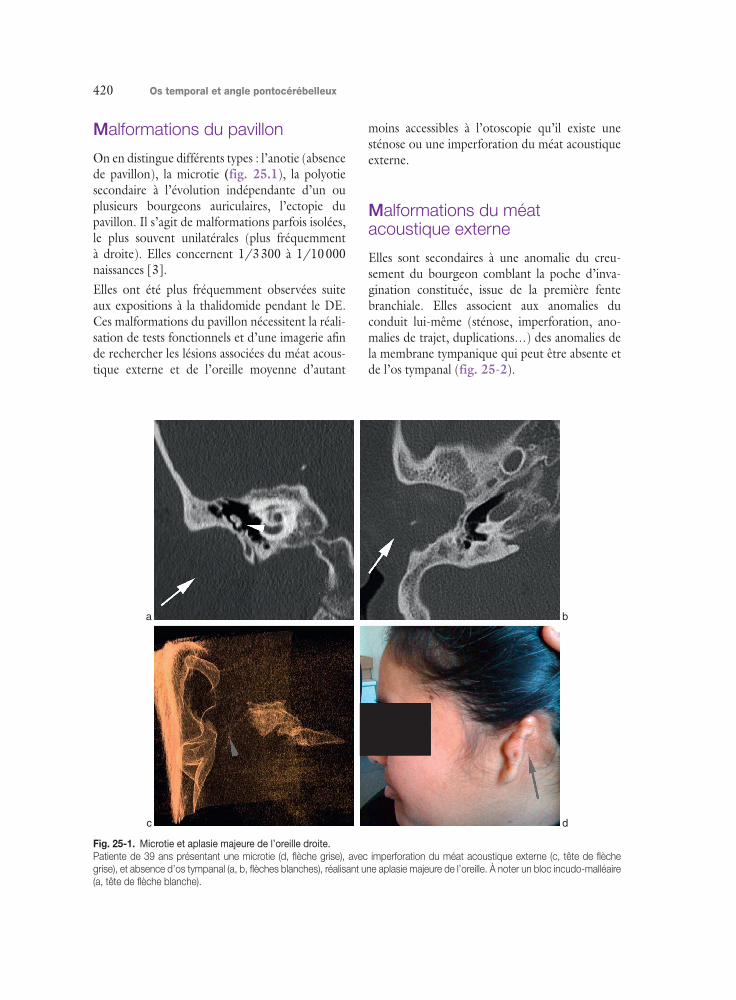
On en distingue différents types : l’anotie (absence de pavillon), la microtie (fig. 25.1), la polyotie secondaire à l’évolution indépendante d’un ou plusieurs bourgeons auriculaires, l’ectopie du pavillon. Il s’agit de malformations parfois isolées, le plus souvent unilatérales (plus fréquemment à droite). Elles concernent 1/3 300 à 1/10 000 naissances [3].Elles ont été plus fréquemment observées suite aux expositions à la thalidomide pendant le DE. Ces malformations du pavillon nécessitent la réali-sation de tests fonctionnels et d’une imagerie afin de rechercher les lésions associées du méat acous-tique externe et de l’oreille moyenne d’autant
moins accessibles à l’otoscopie qu’il existe une sténose ou une imperforation du méat acoustique externe.
Malformations du méat acoustique externe
Elles sont secondaires à une anomalie du creu-sement du bourgeon comblant la poche d’inva-gination constituée, issue de la première fente branchiale. Elles associent aux anomalies du conduit lui-même (sténose, imperforation, ano-malies de trajet, duplications…) des anomalies de la membrane tympanique qui peut être absente et de l’os tympanal (fig. 25-2).
Fig. 25-1. Microtie et aplasie majeure de l’oreille droite.
Patiente de 39 ans présentant une microtie (d, flèche grise), avec imperforation du méat acoustique externe (c, tête de flèche
grise), et absence d’os tympanal (a, b, flèches blanches), réalisant une aplasie majeure de l’oreille. À noter un bloc incudo-malléaire
(a, tête de flèche blanche).
a b
c d

Chapitre 25. Imagerie des malformations du rocher 421
On distingue ainsi :• les aplasies mineures dans lesquelles l’os tym-
panal est présent et où le conduit est le siège d’une sténose plus ou moins sévère, ou d’une imperforation totale (fig. 25-3) ;
• les aplasies majeures où l’os tympanal est absent et le conduit auditif externe n’est pas perforé (fig. 25-4).
Le bilan des malformations du méat acousti-que externe doit comprendre la réalisation d’un scanner afin de classer les anomalies (présence ou absence d’os tympanal) et de rechercher des anomalies ossiculaire associées. S’il existe une mal-formation de la chaîne ossiculaire, le bilan radio-
logique doit aussi rechercher les anomalies de la fenêtre ovale liées à une absence ou à une dysplasie de l’étrier.Le trajet du nerf facial, essentiellement ses por-tions tympanique et mastoïdienne, doit être pré-cisé surtout s’il existe une indication chirurgicale de réfection du conduit auditif externe.L’association d’anomalies congénitales de l’oreille interne ou du méat acoustique interne est obser-vée dans 12 % des cas et justifie là aussi la réalisa-tion d’un scanner.Enfin l’examen TDM doit mettre en évidence un éventuel cholestéatome primitif associé lié au trappage de tissu ectodermique pendant le développement.
Malformations de l’oreille moyenne
Elles peuvent concerner la cavité tympanique elle-même mais aussi son contenu (chaîne ossiculaire, nerf facial, trajet vasculaire aberrant).
Anomalies ossiculaires
Elles peuvent être isolées, ne concerner qu’un seul, deux, voire les trois osselets. Elles peuvent aller de l’absence totale de chaîne ossiculaire à des anomalies morphologiques sur l’un des osselets
Fig. 25-2. Duplication (flèche) du méat acoustique gauche.
TDM de l’oreille gauche chez un nourrisson. Duplication (flèche)
du méat acoustique externe gauche (conduit membraneux).
Fig. 25-3. Imperforation du méat acoustique externe droit.
Scanner de l’oreille droite chez un enfant de 12 ans.
Imperforation du conduit membraneux (flèche) du méat
acoustique externe droit. Le conduit osseux, l’oreille moyenne
et la chaîne ossiculaire sont normaux.
Fig. 25-4. Aplasie bilatérale de l’oreille externe : absence
d’os tympanal.
Scanner, coupe transversale du crâne. Absence d’os tym-
panal et imperforation bilatérale des conduits membraneux
(astérisques).

422 Os temporal et angle pontocérébelleux
(anomalies de taille, non-ossification d’une por-tion d’un osselet, duplication, etc.). Il peut s’agir aussi de blocs ossiculaires (fig. 25-5) correspon-dant à la fusion totale ou partielle des osselets.L’examen scanographique est absolument indispen-sable pour faire le bilan de ces anomalies, d’autant qu’il existe des anomalies associées de l’oreille externe empêchant l’otoscopie (aplasie, hypoplasie, etc.). Il doit visualiser les autres anomalies qui peu-vent être associées et préciser en particulier le trajet du nerf facial, la morphologie des fenêtres (surtout s’il existe une anomalie de l’étrier) et la présence ou non de structures vasculaires intratympaniques.
Anomalies du marteau (malleus)
Les anomalies du marteau, isolées ou associées, sont observées dans 50 % des anomalies de la
chaîne ossiculaires. Les aplasies totales sont obser-vées en cas d’absence de membrane tympanique et sont donc plus fréquemment associées aux ano-malies de l’oreille externe (fig. 25-6). L’absence de manche ou de tête du marteau traduit une aplasie partielle. La tête du marteau et le corps de l’enclume peuvent être fusionnés et remplacés par un bloc ossiculaire unique.D’autres anomalies mineures peuvent être obser-vées et leur bilan peut être réalisé par examen scanographique. Il peut s’agir de déformations de la tête du marteau avec perte de son caractère arrondi, de fusion de la tête du marteau à une des parois de la cavité tympanique et plus particulière-ment au mur de l’attique.
Anomalies de l’enclume (incus)
Ce sont les anomalies les plus fréquentes de la chaîne ossiculaire. Elles sont observées dans 80 % des cas qu’elles soient isolées ou associées à des anomalies des autres osselets. L’enclume peut être totalement absente ou réduite à l’état d’ébauche fusionnée ou non à la tête du marteau. L’anomalie peut ne concerner qu’une portion de l’enclume ; les hypo-plasies du corps ou de la courte apophyse (proces-sus court de l’incus) sont moins fréquentes que les hypoplasies de la longue apophyse (processus long) qui est alors très courte, incurvée vers le dedans, voire absente et remplacée par un tractus fibreux à peine visible sur les reconstructions en UHR (fig. 25-6 et 25-7). Ces anomalies du processus long de l’enclume, lorsqu’elles sont isolées, peuvent être
Fig. 25-5. Aplasie de l’oreille externe : bloc incudo-malléaire.
Scanner du rocher droit en coupe transversale. Présence d’un
bloc incudo-malléaire complet (flèche).
a b
Fig. 25-6. Aplasie de l’oreille externe : hypoplasie ossiculaire.
Scanner du rocher gauche, coupe transversale (a), coupe coronale (b). Absence d’os tympanal et de méat acoustique externe. L’absence
de membrane tympanique s’accompagne d’une aplasie du manche du marteau (flèche noire). Il existe une hypoplasie de la longue apo-
physe de l’enclume (flèche blanche) qui est courte et partiellement non ossifiée (bandelette membraneuse). L’étrier est normal.

Chapitre 25. Imagerie des malformations du rocher 423
a cb
Fig. 25-7. Aplasie de l’oreille externe.
Scanner du rocher gauche, coupes transversales (a et b), coupe coronale (b). Absence d’os tympanal et de méat acoustique
externe. Présence d’un bloc incudo-malléaire avec ébauche d’articulation (a, flèche). La longue apophyse de l’enclume est réduite
à une ébauche et se poursuit par une bandelette non ossifiée (b, c, flèches).
Fig. 25-8. Aplasie de l’étrier.
Scanner du rocher droit en reconstructions transversales passant par la chaîne ossiculaire et la fosse ovale (a), du bas (a1) vers
le haut (a4) et reconstructions coronales (b) passant par la fosse ovale, de l’avant (b1) vers l’arrière (b4). Hypoplasie de l’enclume
avec une longue apophyse partiellement minéralisée (têtes de flèches). L’étrier est réduit à sa branche antérieure (a3, flèche). La
platine est présente sans anomalie. Absence d’articulation incudo-stapédienne (b2, flèche large).
a1 a2a3 a4
b1 b2b3 b4
responsables de surdités de transmission à tympan normal et justifient la réalisation d’un scanner.La fusion de l’enclume aux structures adjacentes est possible, il peut s’agir de fusion à la tête du marteau avec formation d’un bloc incudo-malléaire articulé ou non au bouton de l’étrier, de fusion du corps de l’enclume au bouton de l’étrier ou de fusion de l’enclume aux parois de la caisse et en particulier à la paroi du canal semicirculaire latéral.
Anomalies de l’étrier (stapes)
L’étrier présente la particularité d’avoir une dou-ble origine embryologique. Le bouton et les deux branches dérivent du 2e arc aortique alors que la platine est un dérivé de la capsule otique. C’est le contact des branches de l’étrier avec la capsule otique qui va induire la formation de la platine et le creusement de la fosse ovale. Tout obstacle à ce contact entraîne donc des anomalies de dévelop-pement de la fosse ovale.
Là encore, tous les types d’anomalie de l’étrier peu-vent être observés depuis l’aplasie associée à une fosse ovale plus ou moins hypoplasique, à l’absence d’une portion de l’étrier (absence de bouton avec absence d’articulation incudo-stapédienne, absence d’une branche ou branches incomplètes) (fig. 25-8).Les dysplasies stapédiennes sont de différents types. La déformation « en sabre », avec incurvation à concavité antérieure des deux branches est l’ano-malie le plus fréquemment observée. Ces anoma-lies sont souvent associées aux procidences de la seconde portion du nerf facial (fig. 25-9). Elles n’ont pas obligatoirement de traduction clinique.Il peut exister des étriers multiples plus ou moins complets, doubles, voire triples.Des fixations au vestibule, au canal du nerf facial, au promontoire ou à la pyramide de l’étrier peu-vent être observées. Parmi les anomalies associées, l’absence de pyramide et de muscle de l’étrier est relativement rare mais peut s’observer.

424 Os temporal et angle pontocérébelleux
Toutes ces anomalies doivent être analysées en scan-ner ultrahaute définition avec des reconstructions dans l’axe de la fenêtre ovale et des reconstructions transversales-obliques dans l’axe de l’étrier afin de mieux comprendre la morphologie de celui-ci.Les anomalies associées doivent, là encore, être soigneusement recherchées, en particulier les ano-malies de trajet de la 2e portion du nerf facial. Il peut s’agir de trajets aberrants qui impactent sur le développement de l’étrier pendant le dévelop-pement embryonnaire. Le nerf facial peut être très saillant dans la fosse ovale et être respon-sable d’une sténose au niveau de son ouverture dans l’oreille moyenne (fig. 25-10). L’étrier est alors volontiers abaissé ou déformé en sabre. Plus rarement, le nerf peut obturer complètement la fosse ovale et cheminer dans le fond de celle-ci (fig. 25-11). Dans les formes plus avancées, le
nerf facial peut traverser la cavité tympanique à distance de sa paroi médiale, la corde du tympan peut être hypertrophiée, le nerf facial peut être double, voire triple avec un contingent traversant la cavité tympanique ou barrant la fenêtre ovale.
ab1 b2b3 b4
Fig. 25-9. Hypoplasie de la fenêtre ovale et dysplasie « en sabre » de l’étrier.
Scanner du rocher gauche, coupe transversale (a) et reconstructions coronales (b) passant par la fosse ovale, de l’avant (b1) vers
l’arrière (b4). Il existe une hypoplasie importante de la fosse ovale dont le diamètre craniocaudal est très réduit, avec procidence
marquée de la portion tympanique du VII (flèche). L’étrier (tête de flèche) est au contact du VII2. Il est déformé avec une concavité
marquée vers l’avant.
Fig. 25-10. Atrésie de la fenêtre ovale droite.
Scanner, coupe coronale passant par la fosse ovale droite.
Atrésie de la fenêtre ovale, avec hypoplasie de la platine et proci-
dence du VII2, dont le canal est partiellement déhiscent (flèche).
a cb
Fig. 25-11. Hypoplasie de la fenêtre ovale, procidence du VII et dysplasie de la platine gauche.
Scanner du rocher gauche. Procidence du VII2 devant la platine et dysplasie platinaire. Scanner, reconstructions transversale (a),
coronale (b) et sagittale (c) passant par la fosse ovale gauche. Le nerf facial (c, flèche) vient au fond de la fenêtre ovale au contact
de la platine ; celle ci est hypoplasique (a, b, flèches).

Chapitre 25. Imagerie des malformations du rocher 425
Toutes ces anomalies peuvent s’associer à des anoma-lies de la platine. En effet, les anomalies de la platine isolées (dysplasies platinaires) sont relativement rares. Elles sont plus fréquemment associées ou secondaires à des anomalies de trajet du nerf facial qui ont empê-ché l’induction de la formation de la platine (apla-sie complète) et du creusement de la fosse ovale par perte du contact de l’étrier avec la capsule otique.Les anomalies associées de la fosse sont donc fréquen-tes, elles doivent être précisées, il peut s’agir d’atrésie avec une importante procidence du nerf facial, une absence totale de fosse ovale avec, dans ces cas, un nerf facial obturant complètement la fosse ovale et entraînant une absence de platine (fig. 25-12)…
Autres anomalies de l’oreille moyenne
Elles sont nombreuses, elles peuvent être isolées ou associées à des anomalies ossiculaires. Lorsqu’elles sont isolées, elles n’ont la plupart du temps pas de traduction clinique et sont de découverte fortuite, elles n’ont alors d’incidence qu’en cas d’abord chirurgical de la cavité tympanique réalisé pour une autre raison.Leur bilan doit être réalisé par examen scannogra-phique :• diminution de volume, isolée (avec ou sans tra-
duction clinique) ou associée à une hypoplasie de l’oreille externe ;
• hyperpneumatisation avec un sinus tympani très profond ou une fissure de Hyrtl passant sous le labyrinthe osseux ;
• cloisonnements osseux prolongeant les structu-res ligamentaires intratympaniques (cloisonne-ment osseux sur le bec-de-cuiller, etc.) ;
• déhiscence des parois : tegmen tympani, hypo-tympanum avec procidence jugulaire ;
• les anomalies de l’oreille moyenne peuvent inclure des anomalies de trajets nerveux (VII2) ou de trajets artériels (cf. infra).
Malformations de l’oreille interne
L’oreille interne résulte d’une invagination de la placode qui va constituer la vésicule otique à l’ori-gine du labyrinthe membraneux. Le mésenchyme adjacent participe à la formation de la capsule labyrinthique.Le type et le degré des malformations de l’oreille interne dépendent directement du stade du déve-loppement auquel l’anomalie est apparue.Ces malformations entraînent des surdités de per-ception endocochléaires, plus rarement des surdi-tés de perception rétrocochléaires, ou des surdités mixtes. Vingt pour-cent seulement des surdités neurosensorielles congénitales s’accompagnent d’anomalies radiologiques.Ces malformations sont extrêmement nom-breuses :• cavité cochléovestibulaire commune, l’anomalie
est apparue à la 4e semaine du DE ;• agénésie cochléaire, anomalie du DE à la
5e semaine ;
a cb
Fig. 25-12. Aplasie stapédienne gauche et absence de fosse ovale.
Scanner du rocher gauche en reconstructions transversales (a et b), et coronale (c). Absence de platine et de fosse ovale. Le VII2
(flèche) vient barrer la zone de la platine en passant le long du promontoire. Absence d’étrier. Le marteau et l’enclume sont par
contre normaux.

426 Os temporal et angle pontocérébelleux
• hypoplasie cochléaire, anomalie du DE à la 6e semaine ;
• partitions incomplètes, anomalie du DE à la 7e semaine. La malformation cochléovestibulaire kystique constitue la partition incomplète de type 1 où la cochlée et le vestibule sont kysti-ques. La malformation majeure du sac endolym-phatique où l’aqueduc et le sac endolymphatique sont dilatés constitue le type 2 ; il peut s’y asso-cier une dysplasie modérée de la cochlée.
Ces anomalies s’associent dans de nombreux syn-dromes dysplasiques [4]. On retiendra :les dysplasies de Bing-Siebenmann qui n’ont pas
de traduction scanographique et qui correspon-dent à une absence d’organe de Corti ;
• les dysplasies de Scheibe correspondent à des anomalies du labyrinthe membraneux au niveau du saccule et de la cochlée ;
• les dysplasies de Mondini sont les plus connues, elles correspondent à toutes les anomalies des tours supérieurs de la cochlée. Le tour basal de la cochlée est conservé. Ces anomalies sont liées à une hypoplasie du modiolus [5] ;
• la dysplasie d’Alexander est une dysplasie plus grave avec l’absence de développement du tour basal de la cochlée ;
• dans la dysplasie de Michel, il n’y a pas de laby-rinthe ni de structure neurosensorielle ;
• les syndromes de Uscher, de Refsum et Cockayne sont des anomalies intéressant uni-quement le labyrinthe membraneux sans tra-duction radiologique.
Elles peuvent s’inscrire dans des syndromes mal-formatifs généraux à déterminisme génétique dont le plus représentatif est le syndrome CHARGE (colobome, malformation cardiaque [heart mal-formation], atrésie choanale, retard de croissance et/ou retard mental, hypoplasie génitale, surdité et malformations des oreilles).Les anomalies profondes de l’oreille interne néces-sitent un bilan scanographique systématique afin de les classer. Le bilan doit associer une imagerie par résonance magnétique en haute résolution et à haut champ afin de visualiser les tours de spire de la cochlée en cas de doute sur le scanner et en cas d’insuffisance du modiolus. L’IRM est indispensa-ble si une implantation cochléaire est envisagée, à la recherche d’anomalie associée du nerf cochléaire.
Il existe en effet des surdités profondes d’origine génétique qui n’ont pas toujours de traduction tomodensitométrique. Elles peuvent par contre s’accompagner parfois d’anomalies à l’IRM :• dilatation du sac endolymphatique ;• insuffisance de modiolus ;• hypoplasie du canal semicirculaire latéral et de la
cochlée dans les mutations GJB2 ;• hypoplasie du nerf cochléaire dans les mutations
CX26…
Malformations du labyrinthe postérieur
Le canal semicirculaire latéral qui est le dernier à se former est le plus fréquemment atteint [6]. Les anomalies isolées du canal semicirculaire supérieur ou du canal semicirculaire postérieur sont rares.Dans tous les cas, il peut s’agir d’une diminution du calibre, ou au contraire d’un élargissement du canal considéré (fig. 25-13). Le rapport normal entre le diamètre de l’arche interne du canal semicirculaire latéral et le calibre transversal du vestibule doit être supérieur à 0,5. Il peut s’agir aussi de cavité unique dans laquelle sont fusionnés les cavités utriculo-sacculaires et le canal semicirculaire latéral lorsque le vestibule est intéressé (fig. 25-14). Les aplasies complètes des canaux semicirculaires sont relative-ment rares, elles sont observées dans les syndromes CHARGE (fig. 25-15). On peut enfin observer,
Fig. 25-13. Dilatation du canal semicirculaire latéral.
Scanner du rocher gauche en coupe transversale.
Comparaison du diamètre interne de l’arche du canal semi-
circulaire latéral (double flèche noire) et du diamètre du ves-
tibule (double flèche blanche). Le canal semicirculaire latéral
est large et son diamètre interne est inférieur à la moitié de
celui du vestibule.

Chapitre 25. Imagerie des malformations du rocher 427
soit de façon isolée, soit en association à d’autres malformations, des canaux semicirculaires incom-plets, parfois réduits à l’état d’ébauche.Les anomalies des canaux semicirculaires peu-vent être des anomalies plus modestes avec des
déhiscences des parois du canal semicirculaire supérieur (fig. 25-16) ou plus rarement du canal semicirculaire postérieur. Ces anomalies identi-fiées par Minor entraînent le phénomène de Tullio (vertiges et nystagmus induit par les sons) [7, 8].
a cb
Fig. 25-14. Dysplasie du canal semicirculaire latéral.
Scanner du rocher droit en reconstructions transversales (a et b) et coronale (c). Cavité commune entre le canal semicirculaire
latéral et le vestibule qui ne sont pas différenciés (flèche blanche). Les canaux semicirculaires postérieur (tête de flèche blanche) et
supérieur (tête de flèche noire) sont normalement différenciés.
Fig. 25-15. Syndrome CHARGE.
Patient présentant un colobome (malformation oculaire), une malformation cardiaque, une atrésie des choanes nasales, un retard
de croissance, une hypoplasie génitale et des malformations de l’oreille. Scanner du rocher gauche en coupes transversales (a, b
et c de haut en bas) et reconstruction coronale (d). Dysplasie de l’oreille interne avec ébauches de canaux semicirculaires latéral
(tête de flèche noire) et supérieur (flèche noire). Absence de fosse ovale et de platine de l’étrier (flèche blanche) avec procidence
du VII2 (tête de flèche blanche).
a
c
b
d

428 Os temporal et angle pontocérébelleux
Les anomalies isolées du vestibule, du saccule et de l’utricule sont relativement exceptionnelles (exposition à la thalidomide). En réalité, elles sont le plus souvent associées aux anomalies du canal semicirculaire latéral.On peut estimer qu’il existe une anomalie du ves-tibule ou du canal semicirculaire latéral lorsque le rapport diamètre du vestibule/diamètre de l’arche interne du canal semicirculaire latéral est supérieur ou égal à 1,2 [9].
Malformations de la cochlée
Les anomalies de la cochlée peuvent, rarement, résulter d’une aberration du développement, plus souvent d’un arrêt de maturation.Les aberrations du développement peuvent être des tours supplémentaires (4e tour de spire, fig. 25-17), des duplications cochléaires ou des cochlées naines hypoplasiques mais dont la parti-tion est respectée.
Les arrêts des maturations sont responsables de tous les stades de malformation depuis l’anomalie d’un ou plusieurs tours de spire jusqu’à l’absence de cochlée, que cela s’inscrive ou non dans des anomalies de l’ensemble du labyrinthe.Il existe donc de nombreuses anomalies qui résul-tent d’un arrêt de la maturation :• à la 3e semaine du DE : aplasie du labyrinthe
(dysplasie de Michel) [10] ;• à la 4e semaine du DE : cavité cochléovestibu-
laire commune plus ou moins associée à des ano-malies des canaux semicirculaires (fig. 25-18 et 25-19) ;
• à la 5e semaine du DE : aplasie cochléaire isolée ou non ;
• à la 6e semaine du DE : hypoplasie cochléaire avec une cochlée rudimentaire ;
• à partir de la 7e semaine du DE : partitions incomplètes avec dilatations localisées (anoma-lies de Mondini [5]).
Les partitions incomplètes et les dilatations localisées forment un groupe de malformations dont l’ex-
a b
Fig. 25-17. Cochlée aberrante.
Rocher gauche en reconstructions transversales (a et b) : aberration du développement de la cochlée avec présence d’un qua-
trième tour de spire.
a cb
Fig. 25-16. Anomalie de Minor.
Déhiscence de la convexité du canal semicirculaire supérieur responsable d’un phénomène de Tullio. Rocher droit en reconstruc-
tions sagittale oblique dans l’axe du canal semicirculaire supérieur (a) et coronales (b et c).

Chapitre 25. Imagerie des malformations du rocher 429
pression clinique est variable à la fois dans l’inten-sité et dans le temps avec des surdités d’apparition et d’aggravation progressive [5].Elles sont liées à une anomalie morphologique du modiolus.Le modiolus est une structure osseuse en sablier, triangulaire, rectangulaire ou trapézoïdale qui représente l’axe central du tour basal et du tour médian de la cochlée et sur lequel sont insérées la lame spirale qui sépare les tours de spire les uns des autres, et la cloison spirale qui sépare au sein du limaçon la chambre antérieure (rampe vestibulaire et rampe cochléaire) de la chambre postérieure. Le modiolus a un diamètre de 4 mm pour une hauteur de 3 mm et une surface de 4 à 6 mm2 ; cette surface peut être calculée sur les examens en très haute résolution en IRM.Les anomalies du modiolus peuvent s’accompa-gner d’une absence totale ou partielle de lame spi-rale. Le tour basal est alors respecté mais les tours supérieurs de la cochlée sont fusionnés en une
cavité unique. L’absence de cloison spirale impli-que la constitution d’une cavité commune avec une rampe cochléaire commune. Cette absence de cloison spirale peut être isolée, la lame spi-rale étant présente, ou au contraire accompagnée d’une absence de lame spirale.Les anomalies du modiolus peuvent être de degrés très variables, depuis l’absence totale avec absence de lame spirale et de cloison spirale, jusqu’aux anomalies mineures, où lames et cloisons spirales sont présentes mais où le modiolus est de petite taille. Dans tous les cas, l’insuffisance de modio-lus peut entraîner une hyperpression des liquides intralabyrinthiques, une dilatation des canaux et un risque d’oreille geyser.Ces anomalies peuvent être isolées ou associées à d’autres anomalies cochléaires ou du vestibule (fig. 25-20) et des canaux semicirculaires. Elles accompagnent volontiers les dilatations de l’aque-duc du vestibule qui doivent être recherchées systématiquement.
Fig. 25-18. Cavités cochléo-vestibulaires communes.
Anomalie du développement embryonnaire de la 4e semaine avec cavité cochléovestibulaire commune bilatérale. Scanner, coupes
transversales. Rocher droit (a et b) : présence d’une cavité labyrinthique unique (tête de flèche noire). Rocher gauche (c et d) : volu-
mineuse cavité commune postérieure (tête de flèche blanche), le canal semicirculaire supérieur est présent (d, flèche noire). Des deux
côtés, on note une dilatation du fond du méat acoustique interne et de la portion labyrinthique du nerf facial (b, c, flèches blanches).

430 Os temporal et angle pontocérébelleux
L’imagerie est essentielle. Le scanner et l’IRM (en ultrahaute définition, reconstructions inframillimé-triques de séquences de liquides labyrinthiques), si possible à haut ou très haut champ (3T), sont indis-
pensables pour dénombrer les tours de la cochlée, visualiser le modiolus, rechercher une dilatation localisée et mesurer (sur l’IRM à très haut champ) les rampes antérieures et postérieures.
a b
c d
Fig. 25-19. Cavité commune droite avec CSC postérieurs et supérieurs normaux.
Anomalie du DE (4e semaine) avec cavité cochléovestibulaire commune. Scanner, coupes transversales (a, b et c) et coronales
(d) du rocher droit : présence d’une cavité labyrinthique unique (têtes de flèches noires). Les canaux semicirculaires postérieur et
supérieur sont présents (flèches blanches).
a b
Fig. 25-20. Partition incomplète de type 2.
Scanner du rocher droit en coupes transversales (a et b). Le tour basal est présent (tête de flèche noire). Les deux tours supérieurs sont
réunis dans une cavité unique globuleuse (flèche noire), la lame spirale et la cloison spirale ne sont pas visibles. Le modiolus est réduit à une
fine structure lamellaire (flèche blanche). Dilatation de l’aqueduc du vestibule (tête de flèche blanche) et dilatation modérée du vestibule.

Chapitre 25. Imagerie des malformations du rocher 431
Absence de modiolus
Elle peut être observée dans certaines surdités d’ori-gine génétique, comme la surdité progressive liée à l’X. En l’absence totale de modiolus, le fond du méat acoustique interne communique à plein canal avec le tour basal de la cochlée (fig. 25-21). Il n’y a pas de cloison spirale et les rampes antérieure et postérieure sont indistinctes. Le tour central et le tour médian de la cochlée peuvent être différenciés. Il existe de plus une hypoplasie de la platine et un élargissement associé du VII et du conduit endo-lymphatique. Le risque d’oreille geyser est majeur.
Hypoplasies du modiolus
S’il n’y a pas de cloison spirale, les chambres anté-rieure (rampes vestibulaire et moyenne ou cochléaire) et postérieure (rampe tympanique) sont fusionnées.Les hypoplasies « modérées » (simple diminution du volume du modiolus) peuvent s’accompagner d’une asymétrie des rampes avec un rapport chambre anté-rieure sur chambre postérieure > à 1,45 [9].
L’hypoplasie du modiolus est souvent responsable d’une hyperpression du liquide endolymphatique qui s’exprime par une dilatation de l’aqueduc du vestibule [11].
Dilatations du conduit endolymphatique (aqueduc du vestibule)
On considère que l’aqueduc du vestibule est anormal et dilaté si son diamètre en scanner ou en IRM est supérieur à 1,5 mm à mi-distance entre la fossette unguéale (ouverture dans la fosse posté-rieure) et le vestibule.Ces dilatations peuvent être isolées dans 14 % des cas.Elles sont très fréquemment associées aux ano-malies de la cochlée (fig. 25-22). On observe en effet une dilatation du conduit endolymphatique dans 94 % des cas d’anomalies du modiolus [12],
Fig. 25-21. Partition incomplète de type 1 dans le cadre d’une surdité liée à l’X.
Scanner, coupes coronale (a) et transversale (b) du rocher droit. Coupe transversale du rocher gauche (c). IRM, séquence pondérée
en T2, coupe transversale du rocher gauche (d). Aplasie bilatérale du modiolus et de la lame spirale, dilatation du canal cochléaire
(flèches blanches) et oreille geyser. Hypoplasie vestibulaire (flèche noire), absence de platine et de fosse ovale (tête de flèche noire).

432 Os temporal et angle pontocérébelleux
dans 71 % des cas de dysplasie cochléaire et dans 65 % des cas d’asymétries des rampes.Ces dilatations pourraient être responsables d’une surdité progressive, en particulier s’il existe une hypoplasie associée du modiolus. Les mécanismes invoqués pourraient être soit l’hyperpression elle-même, soit la toxicité pour le neuroépithélium de l’excès de concentration protéique dans le liquide endolymphatique [13].
Dilatations du conduit périlymphatique (aqueduc de la cochlée)
Elles sont exceptionnelles et rarement isolées.
Anomalies du méatacoustique interne et du nerf cochléovestibulaire
Le diamètre du méat acoustique interne va de 2 à 8 mm. Il peut exister des sténoses (si le dia-mètre est inférieur à 2 mm) ou des atrésies du méat (fig. 25-23), des déhiscences d’une paroi (fig. 25-24), des cloisonnements intraméatiques. Des sténoses isolées ainsi que des duplications du CAI sont rares ; elles sont généralement décrites dans les syndromes polymalformatifs, en particu-lier dans la trisomie 21 [14].
Dans tous les cas, s’il existe une surdité rétroco-chléaire, un bilan par IRM est nécessaire afin de vérifier la présence ou l’absence de nerf acoustico-vestibulaire [15].
Fig. 25-22. Partition incomplète de type 2 : dilatation de l’aqueduc du vestibule gauche associée à une dysplasie cochléaire.
Scanner du rocher gauche en coupes transversales (a et b). Dilatation du canal endolymphatique sur toute sa longueur (tête
de flèche noire). Partition incomplète de la cochlée, avec dilatation du canal cochléaire et absence de modiolus visible (flèche
blanche).
a b
Fig. 25-23. Hypoplasie du méat acoustique interne.
Scanner, coupe coronale du rocher gauche montrant l’étroi-
tesse du méat auditif interne (flèches).
Fig. 25-24. Anomalie de recouvrement osseux du méat
auditif interne.
Scanner du rocher gauche en coupe coronale : absence de
paroi supérieure du méat acoustique interne (têtes de flèches).

Chapitre 25. Imagerie des malformations du rocher 433
Les malformations du nerf cochléovestibulaire sont relativement rares, il en existe trois types :• type I : absence de nerf cochléovestibulaire
(fig. 25-25) ;• type 2A : aplasie du VIII cochléaire et présence
d’un nerf commun, associé à des anomalies du labyrinthe ;
• type 2B : aplasie du VIII cochléaire et pré-sence d’un nerf commun, sans anomalie du labyrinthe.
Il peut exister des variations anatomiques des canaux du fond du méat, en particulier des dédoublements ou triplements du canal du nerf de Morgagni, des canaux autonomes cheminant parallèlement à la paroi postérieure du MAI (fig. 25-26).
Anomalies du nerf facial
Les hypoplasies et les aplasies du nerf facial sont très rares. Elles sont responsables de parésie et paralysie faciale néonatale uni- ou bilatérales (syn-drome de Moebius).Les anomalies du trajet du nerf facial peuvent intéresser tout ou partie de l’aqueduc de Fallope (trajet intrapétreux). Elles peuvent n’avoir aucune traduction clinique et n’être alors que des varia-tions anatomiques ou être associées à des malfor-mations plus complexes du massif pétreux. Il peut s’agir de canaux séparés du MAI, d’anomalies de direction de l’une des portions, etc.Il peut s’agir aussi de doublement voire d’un tri-plement de tout ou partie du nerf facial :
a b
Fig. 25-25. Aplasie du nerf cochléaire.
Scanner, coupe transversale du rocher gauche (a). IRM, séquence pondérée en T2, coupe transversale (b) du rocher gauche. Dysplasie
cochléaire avec partition incomplète de type 1 et absence de modiolus. Absence de nerf cochléaire gauche visible en IRM.Remerciements au professeur Nadine GIRARD.
a cb
Fig. 25-26. Anomalies du canal du nerf de Morgagni.
Scanner en coupe transversale du rocher droit (a) : doublement du nerf de Morgagni ; scanner en coupes transversales du
rocher gauche (b, c) : canal du nerf de Morgagni séparé cheminant parallèlement à la paroi postérieure du méat acoustique
interne.

434 Os temporal et angle pontocérébelleux
• doublement de la première portion du nerf facial (fig. 25-27) avec un canal séparé pour les nerfs grand et petit pétreux ;
• doublement ou triplement de la portion tym-panique (fig. 25-28) et/ou de la portion mastoïdienne ;
• doublement ou triplement des trois portions… ;• variation de l’origine de la corde du tympan…Les procidences du nerf facial dans la cavité tym-panique sont fréquentes. Il peut s’agir de proci-dences simples de la portion tympanique du nerf qui fait saillie sous la paroi inférieure du canal semicirculaire latéral. L’importance de cette saillie va définir dans quelle mesure le nerf va plus ou moins fermer l’ouverture latérale de la fosse ovale. Le canal du nerf peut être plus ou moins ossifié, parfois franchement déhiscent… Le nerf facial peut avoir un trajet aberrant au fond de la fosse ovale, il n’y alors en général pas de platine (en raison du défaut d’induction de celle-ci). Il
peut enfin passer sous la fosse ovale le long du promontoire…La procidence peut aussi concerner la portion mas-toïdienne du nerf. Elle est liée à la présence d’un sinus tympani et d’un récessus facial très profonds qui creusent la paroi postérieure de la caisse de part et d’autre du canal facial. Dans les cas extrêmes, les deuxième et troisième portions du nerf traversent complètement la cavité tympanique, en passant en dedans du marteau et de l’enclume et au-dessus du plan de l’étrier, voire en empruntant le trajet de la corde du tympan entre marteau et enclume.
Anomalies vasculaires du rocher
Les anomalies vasculaires sont très rarement symptomatiques. On peut parfois imputer des acouphènes pulsatiles à un trajet artériel aberrant
a
c
b
d
Fig. 25-27. Dédoublement du nerf facial.
Scanner, coupes transversales du rocher gauche (a et b), coupes coronales (c et d). Présence de deux portions labyrinthiques
avec deux canaux séparés. Le contingent principal chemine en avant (flèches noires), le contingent accessoire en arrière et en haut
(têtes de flèches). Les flèches blanches pointent la portion labyrinthique du nerf issue du contingent antérieur.

Chapitre 25. Imagerie des malformations du rocher 435
s’il existe une érosion du promontoire au contact du vaisseau.Elles doivent cependant être reconnues dans tous les cas sur les examens d’imagerie préopératoire en raison du risque chirurgical majeur qu’elles représentent.
Anomalies de l’artère carotide interne
Il peut s’agir soit d’absence totale de l’artère carotide interne, ce qui est extrêmement rare, soit de trajet aberrant de l’artère carotide interne qui passe dans la cavité tympanique en emprun-tant le trajet de l’artère tympanique inférieure et l’artère carotico-tympanique (fig. 25-29). Dans ce cas, il n’y pas de canal pétreux habituel et la cavité tympanique est traversée, le long du pro-montoire, par l’artère qui pénètre dans celle-ci par sa face inférieure et en sort par sa face anté-rieure [3].
a
c
b
d
Fig. 25-28. Dédoublement de la portion tympanique du nerf facial.
Scanner, coupes transversales du rocher gauche (a et b), coupes coronales (c et d). Présence de deux portions tympaniques avec
deux canaux séparés. Le contingent principal chemine en bas (flèches longues). Il traverse la cavité tympanique dans laquelle il est
largement procident. Il n’y a pas de portion mastoïdienne, le nerf sortant du rocher à partir du plancher de la caisse. Le contingent
accessoire chemine en dedans et au-dessus (flèches courtes). Il barre la fosse ovale (a) et il n’y a pas de platine de l’étrier. Il rejoint
le contingent principal dans la cavité tympanique.
a
b
Fig. 25-29. Artère carotide interne droite intratympanique.
Scanner, coupe transversale (a) et reconstruction coronale (b)
du rocher droit. L’hypotympanum est occupé par une masse
arrondie, régulière, pénétrant dans la caisse par sa face infé-
rieure, cheminant le long du promontoire (qui est très légère-
ment érodé, flèches) et rejoignant en avant le canal carotidien.

436 Os temporal et angle pontocérébelleux
Fig. 25-30. Variante anatomique vasculaire : persistance de l’artère stapédienne.
Patient de 20 ans, présentant un acouphène pulsatile de l’oreille droite. TDM en roconstructions axiales obliques du côté droit
(a, c et e) et du côté gauche (pour comprasion, b, d et f) : présence d’un élément nodulaire entre les deux banches de l’étrier droit
(a, flèche noire) ; absence de foramen spinosum du côté droit (c, flèches blanches), et un élargissement de la partie proximale de
la deuxième portion du canal du facial (segment tympanique) adjacent à la cochlée (e, flèche noire).
a b
dc
e f

Chapitre 25. Imagerie des malformations du rocher 437
Artère stapédienne persistante
Elle résulte de la persistance de l’artère carotico-tympanique (fig. 25-30). Elle naît soit de la bifur-cation aortique (trajet de l’artère tympanique), soit de la face postérieure de la carotide interne. Dans tous les cas, elle chemine le long du pro-montoire, passe entre les deux branches de l’étrier au contact de la platine et rejoint le canal du nerf facial dans une zone de « renflement » à la jonction ganglion géniculé-portion tympanique. Dans tous les cas, le foramen épineux est absent, témoignant de l’absence d’artère méningée moyenne [16].
Anomalies veineuses
Il peut s’agir de procidence jugulaire par déhiscence de l’hypotympan avec parfois présence d’un diver-ticule veineux qui remonte plus ou moins haut dans la cavité tympanique (fig. 25-29). Ces procidences représentent elles aussi un danger potentiel en cas d’abord chirurgical de la cavité tympanique.D’autres types de procidence veineuse peuvent s’observer, en particulier dans la paroi postérieure du méat acoustique interne. Des sinus latéraux procidents dans la mastoïde et/ou dans la paroi postérieure du méat acoustique externe peuvent aussi être observés et représentent eux aussi autant de facteurs de risque de la chirurgie de l’oreille moyenne ou de l’oreille interne [3].
Références
[1] Kadom N, Sze RW. Radiological reasoning : conge-nital sensorineural hearing loss. AJR Am J Roentgenol 2010 ; 194 : WS1-4.
[2] Park AH, Kou B, Hotaling A, Azar-Kia B, Leonetti J, Papsin B. Clinical course of pediatric congenital inner ear malformations. Laryngoscope 2000 ; 110 : 1715-9.
[3] Koesling S, Kunkel P, Schul T. Vascular anomalies, sutures and small canals of the temporal bone on axial CT. Eur J Radiol 2005 ; 54 : 335-43.
[4] Robson CD. Congenital hearing impairment. Pediatr Radiol 2006 ; 36 : 309-24.
[5] Sennaroglu L, Saatci I. A new classification for cochleovestibular malformations. Laryngoscope 2002 ; 112 : 2230-41.
[6] Casselman JW, Offeciers EF, De Foer B, Govaerts P, Kuhweide R, Somers T. CT and MR imaging of congential abnormalities of the inner ear and internal auditory canal. Eur J Radiol 2001 ; 40 : 94-104.
[7] Belden CJ, Weg N, Minor LB, Zinreich SJ. CT evaluation of bone dehiscence of the superior semi-circular canal as a cause of sound- and/or pressure-induced vertigo. Radiology 2003 ; 226 : 337-43.
[8] Branstetter BF 4th, Harrigal C, Escott EJ, Hirsch BE. Superior semicircular canal dehiscence : oblique reformatted CT images for diagnosis. Radiology 2006 ; 238 : 938-42.
[9] Davidson HC, Harnsberger HR, Lemmerling MM, Mancuso AA, White DK, Tong KA et al. MR eva-luation of vestibulocochlear anomalies associated with large endolymphatic duct and sac. AJNR Am J Neuroradiol 1999 : 1435-41.
[10] Lowe LH, Vézina LG. Sensorineural hearing loss in children. Radiographics 1997 ; 17 : 1079-93.
[11] Fatterpekar GM, Mukherji SK, Alley J, Lin Y, Castillo M. Hypoplasia of the bony canal for the cochlear nerve in patients with congenital sensori-neural hearing loss : initial observations. Radiology 2000 ; 215 : 243-6.
[12] Naganawa S, Ito T, Iwayama E, Fukatsu H, Ishigaki T, Nakashima T, Ichinose N. MR imaging of the cochlear modiolus : area measurement in heal-thy subjects and in patients with a large endolym-phatic duct and sac. Radiology 1999 ; 213 : 819-23.
[13] Wu CC, Chen YS, Chen PJ, Hsu CJ. Common clini-cal features of children with enlarged vestibular aque-
a cb
Fig. 25-31. Procidence du golfe de la jugulaire gauche dans la cavité tympanique.
Scanner en coupes transversales du rocher gauche (a et b), coupe coronale (c) montrant la déhiscence de la paroi du golfe
(a, flèche) et la remontée de la veine dans l’hypotympanum et comblant la fenêtre ronde (b, c flèches).

438 Os temporal et angle pontocérébelleux
duct and Mondini dysplasia. Laryngoscope 2005 ; 115 : 132-7.
[14] Blaser S, Propst EJ, Martin D, Feigenbaum A, James AL, Shannon P, Papsin BC. Inner ear dysplasia is common in children with Down syndrome (trisomy 21). Laryngoscope 2006 ; 116 : 2113-9.
[15] Casselman JW, Kuhweide R, Ampe W, D’Hont G, Offeciers EF, Faes WK, Pattyn G. Inner ear
malformations in patients with sensorineural hea-ring loss : detection with gradient-echo (3DFT-CISS) MRI. Neuroradiology 1996 ; 38 : 278-86.
[16] McEachen JC, Obrzut M, Bokhari SJ. A rare combination of carotid artery congenital abnor-malities : understanding the embryology and clinical associations. Emerg Radiol 2009 ; 16 : 411-4.





























