Embed Size (px)
DESCRIPTION
Carcinogénesis
Citation preview

“AÑO DE LA CONSOLIDACIÓN DEL MAR DE GRAU”
“UNIVERSIDAD NACIONAL DE PIURA”
FACULTAD DE INGENIERÍA INDUSTRIAL
ESCUELA PROFESIONAL DE INGENIERÍA AGROINDUSTRIAL E INDUSTRIAS
ALIMENTARIAS
DEPARTAMENTO DE AGROINDUSTRIA E INDUSTRIAS ALIMENTARIAS
CURSO: TOXICOLOGÍA DE ALIMENTOS
TEMA: CARCINOGÉNESIS
ALUMNOS: DIEGO ABRAHAM SANDOVAL TOCTO
DOCENTE: MSC. NELLY LUZ LEYVA POVIS
FECHA: 11 DE MAYO DEL 2016
PIURA – PERU2016
1CARCINOGENESIS

CARCINOGÉNESIS
Es el proceso por el cual una célula normal se convierte en una célula cancerosa. Se caracteriza por la progresión de varios cambios celulares a nivel del material genético que finalmente desemboca en la reprogramación de la célula provocando que se reproduzca de manera descontrolada, formando de esta forma masa maligna.
Proceso por el cual una célula adquiere la capacidad de multiplicarse incontroladamente, llegando a invadir otros órganos del cuerpo. Este proceso de transformación de una célula normal en una célula cancerígena puede durar años. Se produce una alteración en los genes de la célula, en su ADN.
Las células tienen mecanismos para reparar el daño provocado en su ADN. Cuando estos mecanismos fallan por una mutación o por la presencia de agentes cancerígenos, puede producirse el cáncer.
Se llama carcinógeno al referirse a cualquier factor que favorece la aparición o la agravación de una enfermedad cancerosa. Puede tratarse de un agente químico o radiológico (rayos X), un factor de riesgo (de carácter familiar), una exposición a ciertos agentes como parte de una práctica profesional... Hay una amplia variedad de factores carcinógenos. Sin embargo, es importante distinguir los factores externos (como los productos químicos) de los factores internos como los fenómenos hereditarios, las alteraciones y los desarreglos en el seno del organismo.
La capacidad de un agente de producir una neoplasia se denomina carcinogénesis. En el proceso de transformación progresiva de las células normales en células malignas, se produce la adquisición de autonomía por las mismas, lo que es un reflejo de una regulación y expresión anormal de su carga génica. Como consecuencia final se induce una neoplasia (crecimiento autónomo de un tejido o de una parte de las células del mismo).
Este proceso puede ser resultado de eventos endógenos como errores en la replicación del ADN, la inestabilidad intrínseca de ciertas bases del ADN o el ataque de radicales libres generados durante el metabolismo celular. También puede ser resultado de procesos exógenos como radiaciones ionizantes, radiaciones ultravioletas (UV) y carcinógenos químicos. Aunque las células tienen mecanismos para reparar las alteraciones producidas, a veces hay errores en dichos mecanismos y los cambios introducidos en el genoma permanecen. Durante esta transformación hay una serie de procesos en los que se alteran los genes implicados en el funcionamiento normal de la célula, responsables de mantener el equilibrio entre la proliferación y la muerte celular.
2CARCINOGENESIS

El resultado consiste en la activación de estos genes, estimulando la proliferación o la protección contra la muerte celular (oncogenes) y en la inactivación de genes que normalmente actuarían inhibiendo la proliferación (genes supresores de tumor). Finalmente, una vez superados los controles de nacimiento y muerte celular el proceso de malignidad de la célula se "fija" cuando ésta es capaz de inmortalizarse y de obtener suficiente cantidad de oxígeno y nutrientes para mantener su elevado rango de proliferación. La célula maligna formará una nueva población de células genéticamente diferentes a la original, constituyendo el tumor.
Mientras que el término neoplasia implica crecimiento celular descontrolado, ya sea clínicamente benigno o maligno, el término tumor describe lesiones ocupantes de espacio ya sean o no neoplasias. Por contra el término cáncer se utiliza, generalmente, para definir neoplasias malignas. En este tema vamos a considerar especialmente la carcinogenicidad inducida por sustancias químicas, haciendo especial mención a la carcinogénesis hormonal, ya que los principios generales de la carcinogénesis química son aplicables a cualquier proceso carcinogénico, sea cual sea su etiología, siendo la etiología hormonal la más vinculada a la patología oncológica del aparato reproductor femenino.
La división celular es un proceso fisiológico que ocurre en casi todos los tejidos y bajo diversas circunstancias. Bajo circunstancias normales, el balance entre la proliferación y la muerte celular programada, usualmente por medio del mecanismo de apoptosis se mantiene estrechamente regulado asegurando de esta forma la integridad de órganos y tejidos. Las mutaciones en el ADN que pueden conducir a una transformación cancerosa interfieren con este proceso de control interfiriendo con el programa que lo controla.
El proceso de carcinogénesis es causado por estas mutaciones en el material genético de las células normales, que alteran el balance normal entre proliferación y muerte celular. Como resultado se produce una división celular descontrolada y un proceso evolutivo de estas células por medio de selección natural dentro del organismo. La reproducción rápida y descontrolada de células pueden producir tumores benignos y algunos tipos de estos tumores pueden convertirse en malignos que es lo que se conoce como cáncer. Los tumores benignos no se esparcen a lugares lejanos del organismo ni invaden otros tejidos, y por lo general no representan una amenaza para la vida a menos que compriman estructuras vitales o tengan alguna actividad fisiológica (por ejemplo, que sean capaces de producir alguna hormona). Los tumores malignos son capaces de invadir otros órganos, esparcirse a lugares distantes (proceso conocido como metástasis) y convertirse en una amenaza para la vida.
3CARCINOGENESIS

Para que una célula normal cambie su fenotipo y se convierta en una célula neoplásica, se requieren varias mutaciones en varios genes y eso ocurre a través de mucho tiempo, a veces de años, de estar expuesto a un agente carcinogenético.
El cáncer comienza en una célula, es decir que es de origen monoclonal. Esa célula alterada, escapa a los controles que anteriormente habíamos mencionado y se vuelve “anárquica”, iniciando una generación de más “células anárquicas”, que a su vez pueden inducir a cambios similares, en las células vecinas. Pero no sólo afectan a la célula las mutaciones inducidas por los carcinógenos, sino que a lo largo de cada división celular (recordemos que pueden llegar a 50 divisiones) se producen errores espontáneos en cada duplicación y los mismos se van acumulando constituyendo un factor intrínseco de riesgo; aquí vale la pena señalar, que los radicales libres, son productos normales del metabolismo celular pero un exceso de los mismos, pueden acarrear efectos genotóxicos por lo que toma vigencia el valor de los suplementos dietarios con antioxidantes.
La mutación genética conduce a la modificación de los productos que codificaría el gen normal y en la vía de la carcinogénesis darán origen a:
A) Los cánceres heredables por mutaciones en uno o ambos alelos de las células germinales El análisis citogenético ha permitido individualizar algunos genes cuyas mutaciones han demostrado ser de predisposición familiar.
B) Los cánceres esporádicos, donde las alteraciones genéticas dependen de los mutágenos ambientales (virus, radiaciones o substancias químicas) En la predisposición no heredable, la mutación de algunos genes, conduce a consecuencias metabólicas que podría significar una ruta hacia la carcinogénesis. El 80% de los cánceres esporádicos, se deben a exposición ambiental, esto sustentado por la gran cantidad de carcinógenos químicos existentes y los distintos tipos de cánceres que promueven. Por ejemplo: el cigarrillo predispone al cáncer de pulmón y de vejiga; las aminas aromáticas al cáncer de vejiga; la aflatoxina al cáncer de hígado; el benceno a las leucemias. Los carcinógenos químicos pueden actuar como inhibidores o activadores de enzimas que
4CARCINOGENESIS

a su vez podrían facilitar la acción de esos carcinógenos en el daño genómico y activar algunos oncogenes. En la dieta diaria se ingieren diferentes tóxicos y mutágenos y los organismos han desarrollado un sistema de adaptación relacionado a su capacidad individual de detoxificación.
Cabe señalar que existen dos mecanismos por los cuales los genes pueden alterarse:
A) GENÉTICO: Donde se producen alteraciones estructurales del genoma por cambios en la disposición de los propios genes o de sus bases, como ser las mutaciones, translocaciones o deleciones,
B) EPIGENÉTICO: En acciones moleculares por alteraciones de las enzimas o de los sustratos de las mismas, tal el caso de la metilación de las bases. Este mecanismo generalmente compromete simultáneamente los dos alelos y la hipometilación conduce a la mayor expresión de los genes, por lo tanto, una mayor cantidad de la enzima metiltransferasa que inhibe la metilación, puede conducir a la mayor expresión de oncogenes. Esta enzima se encuentra elevada en los tejidos tumorales. Para una mejor comprensión de los mecanismos epigenéticos deben tenerse en cuenta tres enzimas que juegan un rol importante en la susceptibilidad al cáncer: Citocromo p 450 mono-oxigenasa; Glutatión- transferasa y Acetil-transferasa. No nos referiremos a sus acciones moleculares, considerando que ese aspecto escapa a la intención genérica de este trabajo.
MECANISMOS MOLECULARES DE DEFENSA
La célula expuesta a tantos factores que pueden dañar el genoma, cuenta sin embargo con mecanismos de defensa, de los cuales dependen la normal replicación celular.
Las proteínas anticiclinas que “enlentecen” el ciclo celular y dan tiempo a actuar a los mecanismos reparadores del genoma.
Las proteínas del complejo NER (nucleotide-excisión-repair) también denominadas MMR (mismatch repair genes) que localizan los sectores dañados, devanan la hélice, excluyen el segmento de ADN afectado e incorporan las secuencias correctas.
El acortamiento fisiológico de los telómeros. Los telómeros, son secuencias del genoma que se encuentran en los extremos de los cromosomas y no se ha constatado hasta ahora, que codifiquen señales
5CARCINOGENESIS

de proliferación. Impiden la pérdida y alteración espontánea de las secuencias de ADN y al mismo tiempo son marcadores del envejecimiento celular, puesto que se van acortando con cada división y llega un punto que ellos mismos inducen el “suicidio” de la célula. Cuando los telómeros son “repuestos” por acción de una telomerasa , no llega ese final conveniente para el porvenir del “clon”, de ahí, que la presencia de telomerasas, constituya un signo desfavorable. Las telomerasas se sobreexpresan en los tejidos neoplásicos.
Sin embargo, esta enzima puede estar aumentada también en células normales que requieren reproducción activa útil, como son las germinales y algunas hematopoyéticas.
EVOLUCIÓN DEL CANCER
Galeno consideraba que el cáncer aparecía a consecuencia de un desequilibrio entre los humores, acumulación de bilis negra, tumor, estado de melancolia.
Theodor boveri en (1914): reconoció que el defecto fundamental del cáncer residía en el material genético de la célula cromosomas.
En la actualidad se considera al cáncer como una enfermedad genética que se produce por alteración de los genes celulares.
El desarrollo de un tumor requiere interacciones entre: Factores exógenos y factores endógenos-
Snabick 1974 demostró que la carcinogénesis podía dividirse en 2 etapas: iniciación y promoción.
Actualmente se acepta una tercera etapa: Progresión tumoral.
LA CARCINOGÉNESIS CONSTA DE TRES ETAPAS:
La carcinogénesis consta de tres etapas: iniciación, promoción y progresión. La última de estas etapas, progresión, es exclusiva de la transformación maligna e implica la capacidad de invadir tejidos vecinos o a distancia. Para que se lleve a cabo el proceso metastásico, se requiere de una serie de mecanismos: angiogénesis, degradación de matrices, migración celular, evasión de la respuesta inmune del hospedero y colonización metastásica.
6CARCINOGENESIS

1. INICIACIÓN
Ocurre a nivel del genoma y las alteraciones pueden darse en los tumores benignos y malignos al igual que la segunda etapa, pero la tercera, o sea la de progresión, es exclusiva de la transformación maligna.
Los agentes que actúan en la primera etapa pueden ser: Físicos – Químicos o Virales.
Los carcinógenos físicos Están constituidos por las radiaciones que dañan, ionizando las bases, deprimen el gen de la proteína p53, pueden estimular citoquinas como la IL 1 y 6, que actúan como verdaderos factores de crecimiento, facilitan la formación de radicales libres y pueden lesionar el gen que codifica para el Complejo Mayor de Histocompatibilidad (CMH) el cual se encuentra en el Cr 6. Las fuentes radiantes pueden surgir de la metodología diagnóstica o terapéutica como así también por exposición a los rayos solares en forma persistente o por emanaciones de radón de los suelos.
Los carcinógenos químicos Tienen como blanco preferencial al nitrógeno de la guanina (alquilantes, aminas aromáticas, nitrosaminas y grasas poliinsaturadas) produciendo mutaciones irreversibles. La aflatoxina (aislada de alimentos contaminados con un tipo de hongo) se considera oncogénica para la célula hepática. Se atribuyen afectos genotóxicos a los compuestos policlorados contenidos en insecticidas y plaguicidas, así como también productos de la manufactura de materiales eléctricos y plásticos formando parte de los contaminantes ambientales, que llegan a los seres vivos a través del aire, del agua y de los alimentos.
Los carcinógenos virales Actúan introduciendo sus propias oncoproteinas al genoma de la célula afectada con lo que la misma cambiará su código normal, por el que le imponen los oncogenes virales. Tal es el caso del papiloma virus humano, del Epstein Bar y de las hepatitis B y C. Los oncogenes virales se ubican generalmente en las proximidades de proto-oncogenes o de oncogenes supresores, activando a los primeros y desactivando a los segundos.
7CARCINOGENESIS

Representación esquemática de la evolución espontánea de un cáncer estándar
2. LA PROMOCIÓN
Representa la etapa de crecimiento tisular con la formación del tumor. Participan: los factores de crecimiento y los receptores a los factores de crecimiento, como así también la angiogénesis y degradación de las matrices extracelulares.
Los factores de crecimiento (FC)Son péptidos producidos por las mismas células o por las vecinas y actúan como facilitadores de la mitosis incorporando en fase S, a algunas células que se encuentran en fase G0 o G1 prolongada.
Los FC se sintetizan en una célula y luego migran al espacio intercelular, ejerciendo sus acciones sobre las células vecinas. Los primeros FC descubiertos fueron el de crecimiento neuronal ( NGF) y el epidérmico (EGF), a los que se sumaron muchos más, entre ellos el derivado de plaquetas( PDGF) ,el de hepatocitos (HGF), el de crecimiento de fibroblastos (FGF) el estimulante de crecimiento de colonias, el simil insulina IGF-1).
Algunas hormonas ejercen acciones similares a éstos factóres peptídicos una vez que fueron captadas por los receptores de membrana o intracitoplasmáticos. Es reconocido el efecto
8CARCINOGENESIS

proliferativo de los estrógenos sobre los epitelios mamarios y del tracto genital; las gonadotrofinas hipofisarias, estimulan especialmente al epitelio ovárico; la prolactina ejerce su acción en el ámbito de la mama y también del ovario; la insulina de origen pancreático y el factor símil insulina, de origen hepático, son verdaderos factores de crecimiento.Merece un comentario aparte la acción de los estrógenos, tan vinculados a cánceres hormono dependientes y su uso como terapia hormonal de reemplazo, a originadas controversias al respecto.
a) estimulan algunos proto-oncogenes como el FOS b) estimulan la acción de otros factores de crecimiento (FC) y los receptores para FC. c) Facilitan la síntesis y liberación de prolactina d) Estimulan la síntesis de receptores de PG e) Aumentan el AMP cíclico que participa en la transducción de señales activando la replicación celular. f) Eleva los niveles de ciclinas (especialmente la E) posiblemente por mecanismos indirectos.(vía estimulación de proto-oncogenes por el AMPc responsive elements)
Los receptores de membranaSon compuestos gluco-proteicos, que se unen a los factores de crecimiento y transmiten los mensajes proliferativos por intermedio de sus conexiones transmembrana. Algunas veces, la sobreexpresión de éstos receptores los hace autoinducibles es decir, que se encuentran en acción permanente aún en ausencia del factor de crecimiento. Algunas citocinas, que son productos de distintos tipos de células, pueden ejercer efectos modulatorios o inhibitorios de la proliferación; tal es el caso del factor TGF beta, del interferón y del TNF o factor de necrosis tumoral que antagonizan a los factores de crecimiento.
9CARCINOGENESIS

3. LA PROGRESIÓN
Implica la capacidad de invadir tejidos vecinos o a distancia, por parte de la célula tumoral maligna. Esa capacidad está codificada también en los genes de la misma con modificaciones estructurales y funcionales.(29) Las células normales, se encuentran “ancladas” en un habitat que le es propio. El contacto con las células vecinas controla su propia división celular y existen moléculas de adhesión que las mantienen próximas y permiten la transmisión de señales de una a otra; las células normales son incapaces de atravesar la membrana basal que las separa del tejido conjuntivo sub-basal de donde obtiene los materiales que la nutren; tampoco tienen capacidad de introducirse a los capilares sanguíneos o linfáticos, aunque los linfocitos, hacen excepción a esta particularidad.
En algunas ocasiones tras estas primeras etapas en el proceso carcinogénico es posible objetivar lesiones tisulares (evidenciables microscópicamente) denominadas precancerosas (situaciones patológicas que, de manera estadísticamente significativa, preceden o favorecen la aparición de procesos tumorales).
10CARCINOGENESIS

TIPOS DE CARCINÓGENOS
Los agentes con capacidad carcinogenética se clasifican atendiendo a que actúen o no a afectando al ADN celular, diferenciándose entre agentes genotóxicos y no genotóxicos o epigenéticos.
1. Carcinógenos genotóxicoAlgunos agentes carcinógenos, en especial los agentes iniciadores y progresores se caracterizan por su capacidad de alterar la estructura del ADN y/o de los cromosomas. Estos efectos genotóxicos inducen directamente la aparición de células neoplásicas (transformadas o malignas). Tales efectos genotóxicos podríamos sumarizarlos en mutaciones, formación de aductos y aberraciones cromosómicas. Sin embargo, en la mayor parte de los casos, la acción carcinógena de estos agentes consiste en un aumento del potencial oxidativo de las células lo cual resulta en modificaciones en el ADN (oxidación del ADN) o formación de uniones covalentes de los agentes o sus metabolitos a las cadenas de ADN (aductos). En la acción de este tipo de sustancias juega un papel fundamental el metabolismo celular, a través del cual se produce la biotransformación de sustancias en principio inocuas a compuestos (generalmente reactivos) que presentan capacidad genotóxica y que son llamados (carcinógenos finales). En todo caso, la acción de un agente carcinogénico debe acompañarse, para que ésta sea efectiva, de un disbalance en los mecanismos de reparación de ADN.
1.1 Carcinógenos endógenosLos mecanismos implicados en la carcinogénesis endógena son: oxidación por especies reactivas del oxígeno, reducción con antioxidantes, reacción con radicales libres e inhibición en la reparación de la oxidación del ADN. Los carcinógenos endógenos son especies reactivas del oxígeno. Las más importantes son los radicales hidroxilo (OH·) y el oxígeno singlete (O2), destacando también el anión superóxido (O´2), peróxido de hidrógeno (H2O2), especies peroxiladas (RO·2) y especies alkoxiladas (RO·). Las especies reactivas del oxígeno se producen a partir de las reacciones celulares como la respiración celular (transporte electrónico mitocondrial), procesos de síntesis y degradación del metabolismo (metabolismo del ácido araquidónico, $-oxidación de los ácidos grasos de alto peso molecular, oxidación de aminoácidos, síntesis del ácido
11CARCINOGENESIS

ascórbico, oxidación de poliaminas, esteroidogénesis, oxidación de purinas), biotransformación de xenobióticos (transporte electrónico microsomal, oxidación peroxidativa, funciones de las oxidasas) y activación de células fagocíticas (leucocitos periféricos, macrófagos, células de Kupffer del hígado y células Clara del pulmón).
1.2 Carcinógenos exógenosLos carcinógenos exógenos son aquellos que incrementan la oxidación del ADN como, por ejemplo, agentes de proliferación de peroxisomas, benceno, arsénico, estradiol, nitrosaminas, bromuro de potasio, radiaciones ultravioletas (UVA y UVB) e ionizantes (rayos X). Muchas sustancias inorgánicas, particularmente hierro, cromo, cobalto (II) y sales de níquel en presencia de H2O2 producen la oxidación de las bases del ADN. Por otro lado, la exposición a la polución de los ambientes urbanos produce también altos niveles de ADN oxidado. La exposición a altos niveles de agentes contaminantes como el ozono, materia particulada, aldehídos, metales y óxidos de nitrógeno incrementa también los niveles de 8-oxo-dG. La luz UVB (290-320 nm) produce mutaciones en el ADN y como consecuencia tumores de piel. La luz UVA (320-400 nm) es menos carcinógena y produce mayormente oxidación del ADN mediante la activación fotodinámica de especies reactivas del oxígeno. Hay compuestos químicos que pueden ser afectados por la radiación UV y producir la oxidación del ADN, principalmente mediante la generación de especies reactivas del oxígeno. Por ejemplo, el azul de metileno produce oxígeno singlete en presencia de UV. El oxígeno singlete formado reacciona con residuos de guanina produciéndose 8-oxo-dG. Otro ejemplo son ciertos antibióticos (tetraciclina, fluoroquinolonas) con capacidad fototóxica.
2. Carcinógenos no genotóxicos o epigenéticos
Son aquellos compuestos químicos que actúan por mecanismos que no incluyen la modificación directa del ADN, dando lugar finalmente a células genéticamente inestables (tumor). Estos agentes parece ser que modulan el crecimiento y la muerte celular. En general, estos compuestos actúan modificando la fisiología normal de órganos y sistemas específicos produciéndose una sobreestimulación persistente cuyo resultado es una replicación celular intensificada (alteración del ciclo celular con efecto mitogénico). Esto puede dar lugar a un incremento de mutaciones espontáneas y de las probabilidades de
12CARCINOGENESIS

alterar el ADN tanto por factores endógenos como exógenos antes de que haya posibilidad de reparación. Normalmente se trata de compuestos exógenos, aunque en determinadas circunstancias compuestos endógenos (hormonas) podrían considerarse como carcinógenos epigenéticos. La acción carcinógena de los compuestos puede tener diferentes mecanismos pero todos ellos comparten las siguientes características principales:
EspecificidadLos compuestos epigenéticos, al contrario que los genotóxicos, pueden ser más específicos en su capacidad de inducir carcinogénesis ya que frecuentemente inducen la formación de tumores en una especie animal, un sexo determinado y en la mayor parte de los casos en uno o varios órganos determinados dentro de una especie. Esta especificidad puede ser explicada por diferencias fisiológicas, metabólicas y de sensibilidad inter especies.
Existencia de un umbral en el desarrollo del tumorEn la mayoría de los casos el efecto carcinógeno se produce solamente cuando se administran altas dosis de los compuestos por lo que la carcinogénesis no aparecerá hasta que se alcance un determinado umbral. Según estos datos, se pueden construir curvas de dosis-respuesta para correlacionar qué dosis son perjudiciales. El análisis de esta curvas dosis- respuestas son de especial utilidad para determinar a qué niveles de un compuesto determinado no se produce efecto adverso y cuales constituyen factores de riesgo para el desarrollo del tumor en humanos.
ReversibilidadLos carcinógenos epigenéticos actúan generalmente como promotores del tumor cuando son administrados continua y prolongadamente. Los efectos producidos pueden revertir parcialmente cuando se interrumpe la administración del compuesto.
CitotoxicidadLos agentes epigenéticos son citotóxicos, produciendo un perjuicio crónico en las células que resulta en un aumento en la proliferación celular. Este incremento en la proliferación celular puede ser responsable del desarrollo neoplásico, ya que el ADN es cada vez más sensible a mutaciones a lo largo de sucesivas divisiones celulares. Por otro lado, la modificación producida en el ADN ya sea de forma endógena o exógena tiene posibilidades muy altas de convertirse en mutaciones heredables puesto que las posibilidades de reparación disminuyen.
En general, los agentes epigenéticos se pueden considerar como promotores en la expansión de células espontáneamente iniciadas. Algunos de estos agentes químicos son el benceno, cloroformo, tricloroetileno, furfural, metapirileno, lindano y bifenilos policlorinados. Un ejemplo clásico de carcinogénesis epigenética es la aparición de hepatomas o
13CARCINOGENESIS

hepatocarcinomas inducidos en modelos animales y en humanos tras la exposición prolongada a estrógenos (hepatocarcinogénesis).
CLASIFICACIÓN DE LOS CARCINÓGENOS
Desde hace ya muchos años, los estudios de toxicidad de cualquier sustancia química incluyen la evaluación del riesgo de carcinogenicidad. La necesidad de este tipo de pruebas descansa tanto en razones científicas como en razones legales y reglamentarias. La lista de sustancias químicas (derivados industriales, plaguicidas, herbicidas, insecticidas, aditivos alimentarios, drogas cosméticas, sustancias originadas en la naturaleza) que por exposición accidental, médica, ocupacional o industrial suponen un riesgo de carcinogénesis es muy numerosa.
La evidencia más relevante para asegurar que cualquier sustancia química supone un riesgo de carcinogenicidad para el hombre deriva de los estudios epidemiológicos realizados en la especie humana, pero la asunción de riesgos que supondría el uso indiscriminado de nuevas sustancias químicas sin unos previos estudios del potencial carcinogénico de esas sustancias no podría ser socialmente aceptable. A pesar de los importantes avances en el desarrollo de pruebas para detectar precozmente la actividad carcinogénica sin necesidad de tener que recurrir al concurso de seres vivos complejos, que, exigen estudios largos, pesados y muy costosos, una parte importante de los estudios debe realizarse todavía en animales porque, tal como señala la Organización Mundial de la Salud, la única prueba definitiva de actividad carcinogénica continúa siendo el desarrollo de un tumor histológicamente evidenciable en un animal. De todos modos, no se puede afirmar con precisión absoluta que una sustancia que ha sido comprobada como carcinogénica en animales lo vaya a ser también en los humanos; sin embargo, en la mayoría de los casos, los carcinógenos comprobados en los humanos también lo son al menos en una especie animal y, a menudo, en varias. En este sentido, se puede asegurar que existe una clara correlación positiva entre las observaciones realizadas en el hombre y los índices de carcinogenicidad en los animales, que, además, a menudo han precedido a las observaciones humanas. Por ello, la observación de carcinogenicidad de una determinada sustancia en una especie animal debería, al menos, ser interpretada como una señal de atención para estudiar la adopción de medidas preventivas.
La utilización de los animales en la evaluación de la carcinogenicidad de las sustancias químicas resulta obligatoria ya que la misma clasificación de las sustancias carcinogénicas, se basa, entre otros criterios, en la existencia o no de suficiente evidencia de carcinogenicidad en animales. A este respecto no citaremos aquí más que la que probablemente es la de mayor prestigio internacional actualmente, la de la Agencia Internacional para la Investigación sobre el Cáncer (International Agency for Research on Cancer, IARC) (En la siguiente tabla), en la que (aunque domina el criterio de las pruebas en humana) la existencia de pruebas en los animales
14CARCINOGENESIS

permite ubicar (con carácter habitual, ocasional o excepcional) a las sustancias en los distintos grupos de riesgo carcinogénico.
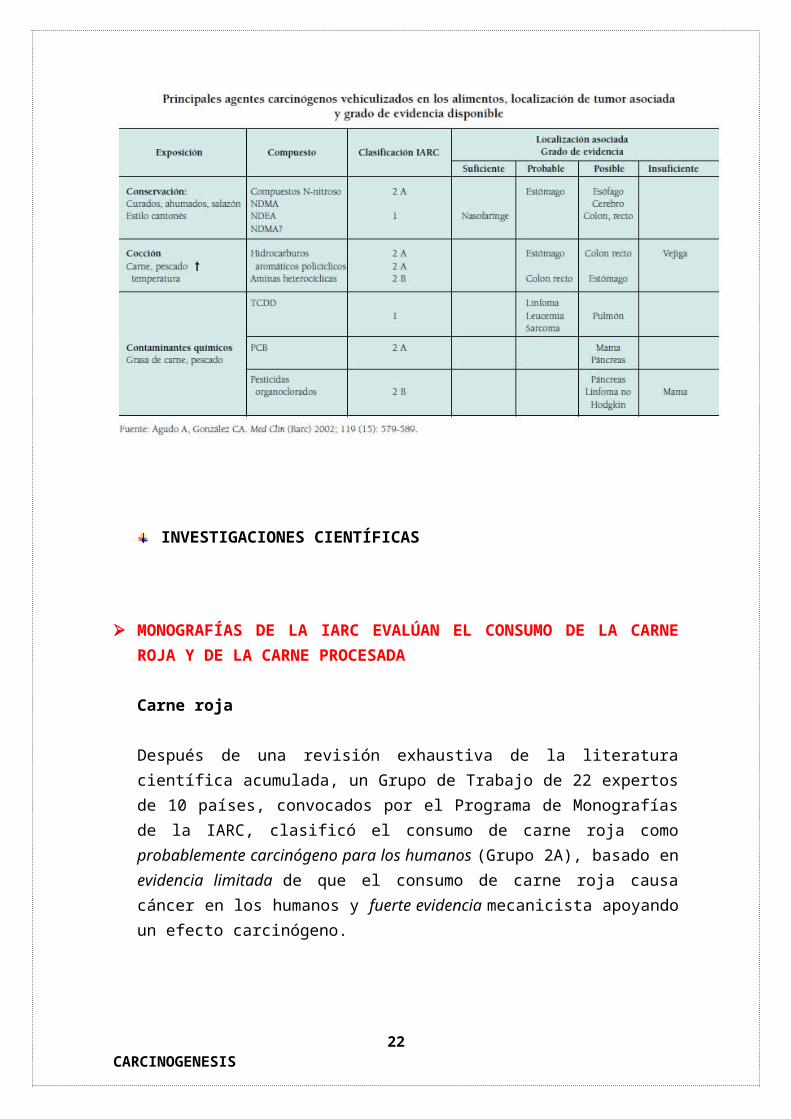
Clasificación de las sustancias químicas cancerígenas (IARC)
15CARCINOGENESIS

INVESTIGACIONES CIENTÍFICAS
MONOGRAFÍAS DE LA IARC EVALÚAN EL CONSUMO DE LA CARNE ROJA Y DE LA CARNE PROCESADA
Carne roja
Después de una revisión exhaustiva de la literatura científica acumulada, un Grupo de Trabajo de 22 expertos de 10 países, convocados por el Programa de Monografías de la IARC, clasificó el consumo de carne roja como probablemente carcinógeno para los humanos (Grupo 2A), basado en evidencia limitada de que el consumo de carne roja causa cáncer en los humanos y fuerte evidencia mecanicista apoyando un efecto carcinógeno.
Esta asociación se observó principalmente con el cáncer colorrectal, pero también se han visto asociaciones con el cáncer de páncreas y el cáncer de próstata.
Carne procesada
16CARCINOGENESIS

La carne procesada se clasificó como carcinógena para los humanos (Grupo1), basada en evidencia suficiente en humanos de que el consumo de carne procesada causa cáncer colorrectal.
Consumo de la carne y sus efectos
El consumo de la carne varía mucho entre los países, desde un pequeño porcentaje hasta un 100% de las personas que comen carne roja, dependiendo del país, y proporciones algo más bajas en el consumo de carnes procesadas.
Los expertos concluyeron que cada porción de 50 gramos de carne procesada consumida diariamente aumenta el riesgo de cáncer colorrectal en un 18%.
“Para un individuo, el riesgo de desarrollar cáncer colorrectal por su consumo de carne procesada sigue siendo pequeño, pero este riesgo aumenta con la cantidad de carne consumida”, dijo el doctor Kurt Straif, Jefe del Programa de Monografías de la IARC. “En vista del gran número de personas que consumen carne procesada, el impacto global sobre la incidencia del cáncer es de importancia para la salud pública”, añadió. El Grupo de Trabajo de la IARC consideró más de 800 estudios que investigaron asociaciones para más de una docena de tipos de cáncer con el consumo de carne roja y de carne procesada en muchos países y poblaciones con dietas diversas. La evidencia más influyente provino de grandes estudios de cohorte prospectivos realizados en los últimos 20 años.
Salud Pública
“Estos hallazgos apoyan aún más las actuales recomendaciones de salud pública acerca de limitar el consumo de carne”, dijo el doctor Christopher Wild, director de la IARC. "Al mismo tiempo, la carne roja tiene un valor nutricional. Por lo tanto, estos resultados son importantes para permitir a los gobiernos y a las agencias reguladoras internacionales realizar evaluaciones de riesgo, a fin de balancear los riesgos y beneficios de consumir carne roja y carne procesada, y poder brindar las mejores recomendaciones dietéticas posibles”, indicó.
PAPEL PROTECTOR DE P53 EN EL CÁNCER DE PIEL: LOS ESTUDIOS DE CARCINOGÉNESIS EN RATONES QUE CARECEN DE P53 EPIDÉRMICA
17CARCINOGENESIS

p53 es una proteína que causa la detención del ciclo celular, apoptosis o senescencia, siendo crucial en el proceso de supresión de tumores en varios tipos de células. Diferente in vitro modelos animales y han sido diseñados para el estudio de la función de p53 en el cáncer de piel. Estos modelos han revelado resultados opuestos, ya que en algunos parámetros experimentales, parece que p53 protege contra el cáncer de piel, pero en otros, surge la conclusión opuesta. Hemos generado cohortes de ratones con deleción p53 eficiente restringidas a epitelios estratificados y de control de los compañeros de camada que expresan p53 de tipo salvaje y estudiado su sensibilidad tanto a la transformación tumoral químicamente inducida y espontánea, así como el tumor tipos originaron en cada grupo experimental. Nuestros resultados indican que la ausencia de p53 en el epitelio estratificado conduce a la aparición, en dos etapas experimentos de carcinogénesis de la piel, de un mayor número de tumores que crecen más rápido y se vuelven malignas más frecuentes que los tumores surgido en ratones con genotipo de tipo salvaje p53.Además, la diversidad histológico del tipo de tumor es mayor en ratones con pérdida epidérmica p53, lo que indica el papel de supresión tumoral de p53 en diferentes tipos de células de la epidermis. Los ratones con envejecimiento inactivación de p53 en el epitelio estratificado desarrollaron carcinomas espontáneos en la piel y otros epitelios. En general, estos resultados ponen de manifiesto la naturaleza verdaderamente protectora de las funciones p53 en el desarrollo del cáncer en la piel y en otros epitelios estratificados.
INTRODUCCIÓN
El gen que codifica la proteína p53 tumor celular, TP53 , es el gen más frecuentemente mutado en los cánceres humanos, siendo alterado en aproximadamente el 50% de los tumores malignos. p53 es un factor de transcripción capaz de interactuar tanto con el ADN, la regulación de la expresión de un gran número de genes diana, y con numerosas proteínas, mutuamente la modificación de su actividad. p53 se encuentra generalmente en niveles muy bajos en las células, pero se acumula en caso de daño genético. p53 promueve múltiples funciones de las células asociadas a la supresión de tumores, como la detención del ciclo celular, la apoptosis y senescencia celular, siendo fundamental en la prevención de la división de las células que han sufrido daños en el ADN; para todas estas funciones, p53 ha sido apodado como "guardián del genoma". p53 también se opone a la formación de tumores mediante la regulación de mecanismos adicionales, como ratones modificados genéticamente que expresa una forma mutante de p53 incapaz de detención del ciclo celular directa, la apoptosis y la senescencia no sufren de formación temprana tumor inicio. Entre estos mecanismos adicionales de la prevención del cáncer mediados por p53 son la regulación de la estabilidad del ADN, el
18CARCINOGENESIS

metabolismo celular, la autofagia, el vástago de mantenimiento celular y la metástasis . La actividad preventiva del cáncer de p53 también podría estar mediada por otras vías; por ejemplo, se ha demostrado que la rapamicina, un conocido inhibidor de la vía de mTOR, disminuye el número de tumores se originó en p53 - / - o p53 +/- ratones , lo que sugiere un papel causal de la inhibición de mTOR en la prevención del tumor. Como p53 induce la expresión de varios reguladores negativos de la vía de mTOR, parte del efecto antitumoral de p53 podría estar mediada por mTOR regulación a la baja.
A pesar de los intensos estudios realizados en los últimos 35 años, estamos muy lejos de tener un conocimiento preciso de las funciones de p53. La comprensión completa de las funciones de p53 se ve obstaculizada por la multiplicidad y complejidad de los procesos biológicos que regula. Así, además de su papel en el cáncer, p53 está implicado en el aclaramiento de las células apoptóticas y la prevención de las enfermedades autoinmunes, así como en el desarrollo embrionario, lo que resulta tanto su inactivación y su hiperactivación en defectos de desarrollo. Otra fuente de complejidad viene del hecho de que algunas funciones de tipo salvaje (wt) o mutantes de p53 no son igualmente reguladas en diferentes tejidos o tipos de células. Además, hay más de diez isoformas de transcritos de la TP53 gen, con diferencias importantes en la localización subcelular y la actividad biológica entre ellos, y apenas conocidos relaciones de regulación de cruz. Son ilustrativos de esta complejidad es el reciente descubrimiento de una isoforma de p53 con un papel sorprendente en la promoción de metástasis. Así se requieren nuevos estudios y modelos experimentales para profundizar en la comprensión de las funciones p53 en la fisiología celular y la transformación tumoral.
La mutación de p53 puede resultar en cáncer de los efectos asociados a la pérdida de sus funciones normales o por la adquisición de nuevas capacidades de transformación, en calidad de un mutante dominante negativo de la p53 de tipo salvaje o como de buena fe oncogén con potencial de transformación. La presencia y el tipo de mutaciones de p53 en tumores son clínicamente importantes, como versiones mutadas de p53 puede modificar en gran medida la respuesta a terapias.
Muchos modelos animales han sido desarrollados para el estudio de la p53. Ratones knockout que carecen de p53 mueren a una edad temprana y se desarrollan una serie de tumores espontáneos, principalmente linfomas y sarcomas, con distinta frecuencia en función de los antecedentes genéticos. Más recientemente, después de la generación de p53 floxed (fl) ratones, una gran cantidad de estudios se han realizado mediante el uso de modelos
19CARCINOGENESIS

animales con la inactivación de p53 restringido a tipos celulares específicos. Transgénicos (Tg) modelos de ratón que llevan diferentes formas mutantes de p53, solo o en asociación con mutaciones en otros genes, también han hecho que la información pertinente sobre el papel de p53 en la transformación tumoral en diferentes tipos de cáncer.
El cáncer de piel es la lesión oncogénica humano más abundante, y TP53 es a menudo mutado en pacientes con cáncer de piel. Diferentes espectros de TP53 mutaciones se han encontrado en diferentes tipos de tumores de la piel (es decir, carcinoma de células basales (BCC), carcinoma de células escamosas (SCC), y melanoma).
Los datos de la literatura indican que p53 coopera con una gran cantidad de insultos oncogénicas, tales como la activación de Ras, la inactivación Rb, la pérdida de alpha v integrina o Snail sobreexpresión, en el desarrollo de carcinomas de piel. De hecho, un número de experimentos llevados a cabo en diferentes modelos experimentales indican un papel supresor de tumor de p53 en cáncer de piel. Así p53 ratones nulos son propensos al desarrollo de los CE de la piel después de la radiación de luz UV , y un requisito para la pérdida de p53 para la conversión maligna de tumores de la piel se ha descrito, incluso en el contexto de diversos otros insultos oncogénicos genéticos. Además, los ratones que carecen de p53 en la epidermis desarrollan tumores espontáneos en la piel; Curiosamente, la piel y los tumores que carecen de p53 mostraron mayor inestabilidad cromosómica que aquellos con p53 en peso de fondo. En un modelo de cáncer diferente (es decir, los queratinocitos de ratón injertadas en ratones desnudos), con el objetivo de estudiar la cooperación de la pérdida de p53 y v-ras Ha de iniciación mediada, el tumor se originaron indicaron un aumento en el crecimiento y la malignidad en la ausencia de uno o dos alelos de p53. En conjunto, estos resultados parecen demostrar un papel protector de p53 en la carcinogénesis de la piel y malignización.
Pero también hay informes que indican un papel protumoral parecer discrepante de p53 en la carcinogénesis de piel. Así experimentos de dos etapas de la piel de carcinogénesis muestran que los ratones nulos p53 desarrollan menos y de la piel más pequeña tumores de p53 ratones de tipo salvaje, lo que indica que la presencia de p53 en células de la piel provoca la aparición de más tumores, a diferencia con los resultados encontrado en las células epiteliales del intestino y otros tipos de células. Otros parámetros experimentales también han dado lugar a la idea de un papel oncogénico de p53 en el cáncer de piel; lo que la pérdida de p53 se opone a la formación de tumores en ratones transgénicos que sobreexpresan oncogenes activados o factores de crecimiento, como v-ras Ha , v-fos o TGFa humano
20CARCINOGENESIS

en epidermis. En resumen, existen algunas discrepancias sobre el papel específico de p53 en el cáncer de piel, lo que podría estar relacionado con la estimación inexacta de la inactivación de p53 en los experimentos con cre-mediada por el tejido específico de p53 a cabo ronda, a las diferencias entre los experimentos sobre la influencia genética fondo de los modelos animales o a diferencias en el estado de p53 en las células dérmicas y la posible existencia de células efectos no autónomas de p53.
Teniendo en cuenta el papel protector generalmente aceptada de p53 en la carcinogénesis, nuestra hipótesis es que p53 en los queratinocitos ausencia causará tanto espontánea y la carcinogénesis inducida químicamente. En este trabajo, hemos generado ratones que carecen de p53 específicamente en los queratinocitos (p53 EKO ratones) y hemos evaluado la eficacia de esta supresión.Entonces, tratamos de dilucidar el papel de p53 en el cáncer de piel epidérmica mediante el estudio de carcinogénesis de piel de dos etapas, tanto espontánea e inducida químicamente en ratones de antecedentes genéticos homogénea. Se concluye que la p53 en los queratinocitos epidérmicos verdaderamente protege contra la promoción tumoral, la progresión y malignidad en la piel, tanto en químicamente inducida y en la carcinogénesis espontánea, actuando así como una buena fe gen supresor de tumor, de acuerdo con el papel generalmente admitida de p53 en otra tipos de células
RESULTADOS Y DISCUSIÓN
La inactivación de p53 eficaz en la piel de p53 fl / fl ; Ratón K14-Cre
Todas las células de la epidermis se derivan de las células epidérmicas basales expresan la queratina K14. Por lo tanto, se espera que la expresión de la recombinasa Cre de un transgén K14-Cre dará lugar a la recombinación de alelos floxed en las células del epitelio estratificado, incluyendo epidermis, aunque muchas de estas células realmente dejan de expresar K14 a medida que avanzan a través del proceso de diferenciación. Hemos generado cohortes de p53 fl / fl ratones / K14-Cre para estudiar la importancia de p53 en el cáncer de piel. No observamos desviaciones significativas entre la frecuencia obtenida para cada genotipo y las proporciones esperadas (no mostrado). Se evaluó la eficiencia de Trp53 recombinación en epidermis de p53 fl / fl ratones / K14-Cre por análisis de PCR usando combinaciones de cebadores capaces de distinguir entre la floxed y las formas eliminados del Trp53gen ( Figura 1A y 1B ). Simultánea análisis de PCR para el transgen Cre como para la forma floxed de Trp53 en el ADN de la epidermis y la cola dérmica de la piel mostró la característica banda del
21CARCINOGENESIS
alelo FL (584 pb) en ambos dermis y la epidermis en ratones que carecían del transgén K14-Cre ( Figura 1B , panel superior); Esta banda también es claramente detectable en el ADN dérmica, pero no en el ADN de epidermis de p53 fl / fl ratones / K14-Cre ( Figura 1B , panel superior). En consecuencia, el análisis de PCR con cebadores específicos para el alelo suprimido reveló una banda predominante 612 pb en la epidermis de p53 fl / fl ratones / K14-Cre.También se detectó esta banda, aunque con menor intensidad, en la dermis de estos ratones, probablemente debido a la presencia de los folículos pilosos que expresan el transgén K14-Cre en la dermis ( Figura 1B , panel inferior). Como era de esperar, la banda 612 pb supresión específica no se detectó en los ratones que carecen del transgén K4-Cre ( Figura 1B , panel inferior).
Figura 1: la recombinación mediada por Cre de floxed Trp53 alelos afecta a la mayoría de las células epidérmicas. A. estructura esquemática de floxed Trp53 locus y del locus eliminado exones que carecen de 2 a 10 (del 2-10) una vez que la recombinación mediada por Cre ha tenido lugar . Triángulos representan sitios loxP, y las flechas representan los cebadores utilizados para la detección específica de alelo. Los números entre paréntesis indican la longitud del fragmento amplificado en pares de bases (pb). B. Ejemplo representativo de análisis específico de alelo de p53 en muestras de ADN obtenidas a partir de epidermis (E) y la dermis (D) de un ratón transgénico con alelos p53 floxed (carriles marcados p53 fl / fl ) y de ratones transgénicos dobles que llevan
22CARCINOGENESIS

también el transgén K14-Cre. También se incluyeron los cebadores específicos de K14-Cre transgén en la reacción de amplificación del panel superior. MWM:. ADN marcador de peso molecular C. La competencia por PCR de las mezclas indicadas de forma de ADN dermis de un p53 no recombinado fl / fl del ratón y de la epidermis de doble p53 fl / fl / K14-Cre ratones transgénicos (que presumiblemente han recombinado completamente los alelos floxed). ns: bandas no específicas.
PCR de competición usando ADN mixtas de epidermis de una p53 fl / fl / K14-Cre ratón (presumiblemente contiene principalmente recombinados Trp53 alelos) y la dermis de un p53 fl / fl ratón (que contiene sólo floxed Trp53 alelos) confirmó además que la recombinación afecta a la gran mayoría de las células de la epidermis en ratones de la p53 fl / fl / K14-Cre genotipo, como la adición de sólo el 2% o 5% de dérmicos no recombinado resultados de ADN en un aumento detectable en la banda indicativa del alelo floxed y una reducción en la intensidad de las bandas no específicas ( Figura 1C ). Razonó que si la adición de estas pequeñas cantidades de ADN que contienen alelos p53 floxed es detectable, la cantidad inicial de alelos floxed en la epidermis de p53 fl / flratones / K14-Cre debe ser muy baja y la eficacia de recombinación en la epidermis que estar muy alto.A partir de estos resultados, se concluye que la supresión de p53 es eficiente, que afecta a la mayoría de las células epidérmicas de p53 fl /
fl ratones / K14-Cre (en adelante, p53 EKO ratones), y que la supresión de la epidermis p53 no da lugar a efectos nocivos brutos más embrionaria el desarrollo o en la vida postnatal antes de la edad de genotipado y el destete (3 semanas de edad).
p53 EKO ratones son más susceptibles a la carcinogénesis de la piel de dos etapas de p53 WT ratonesComo primera aproximación para estudiar el efecto de p53 epidérmico sobre el cáncer, se estudió la susceptibilidad de p53 EKOratones de desarrollar cáncer de piel en un protocolo de la carcinogénesis química de dos etapas; los tumores murinos así generados presente un paisaje mutacional muy similar a la encontrada en los CE humanos. Se realizó el tratamiento tópico de la piel de la espalda con el carcinógeno DMBA y el agente hiperplásico TPA como se indica en la figura 2A , registradas semanalmente tanto el número y tamaño de los tumores de piel. El experimento terminó en la semana 18, debido al alto número y el gran tamaño de algunos de los tumores que surgieron en p53 EKO ratones ( Figura 2B ). Aunque los tumores comenzaron a surgir en semana 6 en ambos genotipos, y por semana 9 todos los animales habían
23CARCINOGENESIS

desarrollado al menos un tumor de la piel, tumores surgieron más rápido en p53 EKO ratones que en p53 en peso de los ratones, como se indica por el mayor porcentaje de ratones con tumores en p53 EKO comparación con p53 en peso de los ratones en este periodo de tiempo ( Figura 2C ). Los ejemplos representativos de desarrollo de tumores en p53 en peso y p53 EKO ratones en la semana 14 se muestran en la Figura 2B . p53 en peso
de los ratones de control desarrollaron tumores pequeños de coliflor pedunculados exofíticos típicos con apariencia papilomatosa bruto, duro (hiperqueratosis) en la consistencia, con una superficie seca y un suministro de sangre pobre (puntas de flecha en la fotografía izquierda de la Figura 2B ). Por el contrario, los tumores desarrollados en p53 EKO ratones eran generalmente más grandes, sésiles y firmemente infiltrados en la dermis (por ejemplo, véase puntas de flecha en la fotografía derecha de la Figura 2B ), algunos de ellos que muestra la discontinuidad de la barrera epidérmica, con una superficie sangrante ulcerada que no curarse con el tiempo; macroscópicamente, estos tumores tenían un aspecto más maligno, se asemeja a la piel carcinomas más de papilomas (ver flechas en la Figura 2B ). Además, aunque los tumores se hicieron visibles en ambos genotipos al mismo tiempo, p53EKO ratones desarrollaron más tumores en promedio que los ratones de control de semana 6; estas diferencias fueron estadísticamente significativas entre las semanas 8 y 11 ( Figura 2D ). Medición del tamaño del tumor confirmó que las lesiones de p53 EKO ratones eran más grandes que los de p53 en peso de los ratones ( Figura 2E ). Tanto la cantidad total y el porcentaje de tumores más grandes que 2 mm de diámetro fueron mayores en p53 EKO que en p53 WT ratones. También notable es la ausencia de tumores de más de 5 mm de diámetro en p53 en peso de los ratones incluso a semana 14; Por el contrario, algunos de los tumores en la población de p53 EKO ratones alcanzaron este tamaño en la semana 10, y por semana 14, el 17% de p53 EKO tumores (33 de los 196 tumores) habían alcanzado este tamaño. En resumen, se observó en los animales tratados con DMBA y TPA que carecen de p53 en las células epidérmicas conduce a una mayor tasa de transformación tumoral de la piel. Estos resultados contrastan con el rendimiento reducido publicada de papilomas en ratones nulos p53 en comparación con p53 WT ratones [ 31 , 32 ], y podrían ser explicados por las diferencias en el fondo genético o tal vez por otras diferencias entre los diferentes modelos animales, tales como la diferente momento de p53 eliminación [de la concepción en ratones nulos p53, y alrededor de día embrionario 12,5 en nuestro modelo experimental o la presencia de p53 en las células dérmicas en p53 EKO , que no se producen en ratones nulos p53. De hecho, un efecto no celular autónoma de p53 dérmica sobre el crecimiento tumoral en xenoinjertos de líneas de la glándula de la próstata o de mama
24CARCINOGENESIS

tumorales humanos de células se ha descrito, aunque p53 en las células del estroma por lo general impide el crecimiento del tumor de células. En resumen, nuestros datos indican que, a diferencia del efecto descrito de la ausencia de p53 en ratones generales knock-out, p53 en las células epidérmicas en realidad protege en las primeras fases del desarrollo de tumores de piel, como p53 EKO mostraron aumento de ratones tasas de aparición de tumores y el crecimiento tumoral de ratones portadores de alelos no recombinado de p53 en sus células epidérmicas. Por lo tanto, la conclusión de que p53 es relevante en el proceso de iniciación de la carcinogénesis de la piel, al menos en el contexto de insultos químicos que activa la señalización de Ras
Figura 2: La ausencia de p53 en epidermis predisponen al desarrollo de tumores en un protocolo de la carcinogénesis química de dos etapas.
25CARCINOGENESIS

A. Línea de tiempo del experimento carcinogénesis de piel. 8 a 9 semanas de edad, los ratones fueron afeitados, y tres días más tarde trataron tópicamente con el carcinógeno DMBA. TPA se administró dos veces por semana durante 12 semanas. Toma de muestras de tumor se extendía hasta la semana 18 en p53 EKO ratones. B. Ejemplo representativo de un p53 en
peso (izquierda) o p53 EKO(derecha) del ratón en la semana 14. Nota los pequeños tumores de la piel con aspecto pediculados papilomatosa en el p53 en peso del ratón y el más grande tumores de piel en p53 EKO ratones (puntas de flecha); Además, algunos de los tumores en p53 EKO ratones son sésiles y ulcerada, se asemeja a carcinomas malignos (flechas). C. Los tumores surgen anteriormente en p53 EKO ratones que en p53 en peso de los ratones cuando se someten a un protocolo de la carcinogénesis química DMBA / TPA. D. tiempo de evolución de número medio de tumores por fenotipos. p53 EKO ratones desarrollaron más tumores que p53 wt ratones. Los asteriscos indican el valor de p <0,05 en el test de la t de Student. E. Distribución de tamaño de los tumores de piel en p53 EKO y p53 wt ratones. Tenga en cuenta la aparición anterior y el mayor número de medio (diámetro> 2 mm) y grandes (diámetro> 5 mm), los tumores en p53 EKO ratones en comparación con p53 en peso de los ratones.
Diferentes tipos de tumores de la piel emergen en p53 EKO ratonesComo se indica, p53 EKO tumores eran con frecuencia mayor que los obtenidos en p53 en peso de los ratones, lo que indica un crecimiento más rápido. Macroscópicamente, los tumores que surgen en ratones de tipo salvaje eran típicamente exofítico y papilomatosa. Por el contrario, varios p53 EKO tumores de la piel tenían un aspecto externo sugerente de un estado más maligno: eran sésiles, con una tendencia a infiltrarse profundamente en el tejido subcutáneo, mostrando frecuentemente ulceración persistente de la superficie. Se evaluaron histológicamente los 18 y los 51 tumores de p53 WT y p53 EKO ratones, respectivamente, recogidas en las semanas 14-18 del experimento. Microscópicamente, los tumores de p53 WT ratones fueron homogéneos, con alto nivel de diferenciación y queratinización del epitelio estratificado que cubre un estroma tejido conjuntivo ramificado, mal vascularizada, que ofrece el aspecto histológico clásico de papilomas escamosas ( Figura 3A ). Sólo dos de los papilomas mostraron los primeros signos de malignidad, en forma de pequeños focos de microcarcinoma, compuesta por nidos de células de la epidermis con la falta de diferenciación de una capa basal de invadir el estroma ( Figura 3B y Tabla 1 ). Sin embargo, p53 EKO ratones produce histotypes tumorales sean más variadas: encontramos papilomas escamosas ( Figura 3C ) y papilomas que
26CARCINOGENESIS

mostraron mayor atipia basal de p53 wt papilomas con múltiples focos de microcarcinoma ( Figura 3D , flechas). Algunos tumores p53 se encuentran en EKO ratones eran tricoepitheliomas, lesiones benignas de los folículos pilosos compuestas de quistes cornified, incapaces de formar cabello normal (flecha en la Figura 3E ); otros se clasificaron como tumores basosquamous ( Figura 3F ), lesiones derivadas de folículos pilosos que se asemeja carcinomas basosquamous, pero benigna debido a la integridad y continuidad de la membrana basal. Por último, también encontramos lesiones ulcerosas, que corresponden a tumores de la piel altamente malignos: la infiltración de los SCC pobremente diferenciados (18 de los 51 tumores de p53EKO ratones, ninguno de p53 en peso de los ratones), que mostró áreas sólidas de aumento de celularidad se infiltran desde la epidermis a la dermis y el tejido subcutáneo, compuestas por los queratinocitos con marcado pleomorfismo celular, la mayoría de ellos con fibroblastoide (husillo) forma de la célula, siendo común la presencia de células multinucleadas, células con núcleos gigantes aberrantes (por ejemplo, la flecha en la Figura 3H ) y figuras mitóticas (puntas de flecha en la Figura 3H ); estas características se parecen a los encontrados en los queratinocitos humanos con pérdida de p53 por medio de shRNAs contra p53 [ 38 ]. Estos SCC diferenciadas mal mostraron un patrón sólido muy homogénea con poca evidencia de diferenciación escamosa y / o deposición de queratina ( Figura 3G y3H ). Sorprendentemente, todo el p53 EKO ratones incluidos en este estudio desarrollado al menos un SCC pobremente diferenciado, lo que impide la prolongación del estudio para el análisis de la evolución de los otros tipos de tumores. La incidencia de cada tipo de tumor se indica en la Tabla 1 . A partir de los datos incluidos en la Figura 3 y la Tabla 1 , se concluye que p53 ausencia en las células epidérmicas conduce a una marcada aceleración del proceso de transformación maligna de los tumores surgido después del tratamiento con DMBA y TPA. Además, los tipos de tumores más variadas observadas en p53 EKO ratones sugiere que p53 actúa como un gen supresor de tumores en diferentes tipos de células de la epidermis; así que la falta de p53 daría lugar a diferentes tipos de tumores en función del grado de compromiso o la diferenciación del tipo de célula transformada.
27CARCINOGENESIS

Figura 3:. Aspecto histológico de los tumores de piel inducidos químicamente tumores desarrollados en p53 wt ratones eran típicas papilomas bien diferenciado A. que a veces mostró focos de microcarcinoma como grupos de células epidérmicas más profundas de la dermis ( B. , flecha). CH. Los tumores desarrollados en p53 EKO ratones eran histológicamente más diversa. Se extendieron de papilomas clásicos (C) y papilomas con abundantes focos de microcarcinoma (D), a tricoepithelioma (E), tumores basosquamous (F) y carcinomas pobremente diferenciados (G y H). Las barras de escala son iguales a 200 micras de AG y 50 micras de H.
MATERIALES Y MÉTODOS
Los ratones y tratamientosEl trabajo con animales se llevó a cabo siguiendo los protocolos aprobados por nuestro Comité Institucional de Ética de Experimentación Animal y según las regulaciones europeas, españolas y locales; los experimentos incluidos en esta publicación están bajo el número de permiso de BME 02/10 del Comité Ético de Experimentación Animal del CIEMAT. Se hicieron todos los esfuerzos para
28CARCINOGENESIS

minimizar el sufrimiento de los animales empleados. Los ratones se mantuvieron en autoclave con comida para roedores y agua estándar ad libitum y se mantuvieron bajo una luz -12 h oscuridad ciclo de 12 h. Floxed Trp53 ratones y ratones K14-Cre se han descrito anteriormente [ 17 , 47 ]. Para la obtención de ratones que carecen de p53 en la epidermis, cruzamos p53en peso / fl ; Ratones hembra K14-Cre con p53 fl peso / varones. En ratones heredar tanto el transgén K14-Cre y alelos floxed de Trp53, la actividad Cre sería eliminar los exones 2 a 10 de Trp53 en la epidermis y otros tipos de células que expresan el transgén K14-Cre ( Figura 1A ). Este alelo recombinado es incapaz de producir una proteína funcional, ya que carece de parte del dominio de activación transcripcional, el dominio de unión a ADN y el dominio de tetramerización de p53. Se seleccionaron para el estudio de carcinogénesis p53 fl / fl ; Ratón K14-Cre (denominado en este trabajo como p53 EKO ) y p53 p / p de camada como los ratones de control (denominado en este trabajo como p53 en peso ). Todos los ratones utilizados en los estudios de carcinogénesis eran del mismo fondo genético mixto (50% C57BL6JxDBA2J híbrido y 50% FVB). Cohortes de siete ratones de cada genotipo fueron sometidos a un protocolo de la carcinogénesis química de dos etapas cutánea. En este protocolo, los ratones se afeitaron usando una cortadora de cabello y se trataron tres días después con una única aplicación tópica de 100 g de 7,12-dimetilbenz [a] antraceno (DMBA, Sigma-Aldrich, referencia D3254) disuelto en 200 l de acetona ( semana 0). Siete días después de la aplicación DMBA, 5 g de se aplicó 12-O-tetradecanoilforbol 13-acetato (TPA, Sigma-Aldrich, P1585 referencia) en 200 l de acetona tópicamente dos veces por semana durante 12 semanas. El número y tamaño de los tumores por ratón se registró semanalmente.p53 EKO los ratones fueron sacrificados cuando los tumores tenían un diámetro mayor de 1 cm o en la semana 18, por razones humanitarias. Cuatro p53 wt ratones fueron sacrificados tiempo apareadas entre las semanas 13 y 18, y el resto fueron sacrificados entre las semanas 35 y 40, con el fin de obtener las lesiones de tamaño suficiente para permitir el análisis por Western blot. Las muestras fueron procesadas para histológico y análisis de Western blot, y los genotipos ratones fueron confirmados por volver a analizar nuevas muestras de ADN tomadas después de la muerte.
Para la carcinogénesis espontánea, se monitorizaron los ratones para el desarrollo del tumor durante 14 meses. Los ratones que muestran tumores o con signos evidentes de enfermedad fueron sacrificados para la necropsia y el análisis histológico.Para el estudio de la piel hiperplásico no tumoral, tres p53 EKO ratones y el mismo número de p53 en peso de los ratones se afeitaron y se trató por vía tópica dos veces con 5 g de TPA o vehículo en los días 3 y 5 después del afeitado.
Genotipificación y análisis de PCR
29CARCINOGENESIS

El ADN se obtiene a partir de biopsias de la cola de 2 semanas de edad, los ratones oa partir de muestras tomadas después de la muerte. Los ratones fueron genotipados por PCR. Los cebadores usados para la determinación de la Trp53 de estado (es decir: floxed, eliminado o de tipo salvaje) y para detectar la presencia del transgén K14-Cre se indican en [ 47 ].
Histología e inmunohistoquímicaTumores de ratón se diseccionaron e inmediatamente se fijaron en 10% de formalina tamponada o 70% de etanol y embebidos en parafina. 5 secciones micras de espesor se utilizaron para H & E tinción inmunohistoquímica o preparaciones. La mayoría de los tumores se fijaron y se clasificaron según su morfología después de la sección y tinción con H & E. Los anticuerpos utilizados en el análisis inmunohistoquímico estaban en contra de p53 (NCL-p53-CM5p, Novocastra, Leica Biosystems, Newcastle, Reino Unido); queratina K5 (PRB-160P), K10 (PRB-159P) (Covance, San Diego, CA); queratina K13 (ab6112), p19 ARF (AB80) (Abcam, Cambridge, Reino Unido); actina de músculo liso (C6198, Sigma-Aldrich, St Louis, MO); Ciclina D1 (RM-9104-CR, Thermo Fisher Scientific, MA, EE.UU.); Fosfo-Akt1 (Ser 473) (9277), Stat3 (4904) (Cell Signaling Technology, Danvers, MA, EE.UU.) y BrdU (11170376001, Roche, Mannheim, Alemania). La inmunorreactividad se reveló utilizando un sistema ABC avidina-biotina-peroxidasa y sustrato ABC (Vector Laboratories), y las secciones counterstained ligeramente con hematoxilina. Los experimentos de control omitiendo el anticuerpo primario no dieron señales.
Western blotEn aquellos tumores que eran lo suficientemente grande, que congelaba una parte del tumor en nitrógeno líquido en el momento del sacrificio para el análisis de transferencia Western. Extractos de proteínas de células enteras se sometieron a SDS / PAGE utilizando técnicas estándar. El contenido de proteína se determinó por el ensayo de proteína colorimétrico de Bradford (BioRad Laboratories; Hercules, CA; EE.UU.). Los anticuerpos utilizados en transferencias Western fueron contra p53 (NCL-p53-CM5p, Novocastra, Leica Biosystems, Newcastle, Reino Unido); Stat3 (tyr705 fosfo; 9131), Stat3 (4904); Akt (fosfo Ser473; 4058) (Cell Signaling Technology, Danvers, MA, EE.UU.); p19 ARF (AB80) (Abcam, Cambridge, Reino Unido); Akt1 / 2 (SC-1619), p16 INK4a(sc-1207), ciclina D1 (sc-753), y la actina (sc-1616) como control de carga (Santa Cruz Biotechnology, Santa Cruz, CA, EE.UU.).
ANÁLISIS ESTADÍSTICO
30CARCINOGENESIS

Los datos se expresan como media ± SEM. Los valores de p se determinaron mediante el uso de la, prueba t de Student para datos independientes de dos colas. Los valores de p <0,05 se consideraron significativos.
REFERENCIAS BIBLIOGRÁFICAS:
Maria Teresa Martin de Civetta, MC, Julio Domingo Civetta, MC. (2011), Carcinogénesis, Artículo de Revisión, Departamento de Oncología, Hospital Dr Jr Vidal, Corrientes, Argentina, 27 de septiembre de 2011.http://www.scielo.org.mx/pdf/spm/v53n5/a08v53n5.pdf
Luis Domínguez Boada (2004), Principios Generales De Carcinogénesis: Carcinogénesis Química y Hormonal, Deparatamendo de Ciencias Clínicas, Universidad de Las Palmas de Gran Canaria, Instituto Canario de Investigación del Cáncer, Biocancáncer 1, 2004.http://www.inen.sld.pe/portal/documentos/pdf/educacion/01102014_CARCINOGENESIS_III.pdf
31CARCINOGENESIS

IARC (2015), Monografias de la IARC evalúan el consumo de la carne roja y de la carne procesada, Agencia Internacional de Investigación sobre el cáncer, Organización Mundial de la Salud, 26 de octubre
https://www.iarc.fr/en/media-centre/pr/2015/pdfs/pr240_S.pdf
Manuel Navarro, Cristian Suarez Cabrera, Josefa P. Alameda, M. Llanos Casanova, Jesus M. Paramio, Ana Bravo, Angel Ramirez, (2016), PAPEL PROTECTOR DE P53 EN EL CÁNCER DE PIEL: LOS ESTUDIOS DE CARCINOGÉNESIS EN RATONES QUE CARECEN DE P53 EPIDÉRMICA “Protective role of p53 in skin cancer: Carcinogenesis studies in mice lacking epidermal p53”, Unidad de Oncología Molecular, Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT), Madrid España, Instituto de Investigaciones Biomédica, Hospital Universitario, Madrid, España, Departamenteo de Ciencias Clínicas Veterinarias de la Universida de Santiago de Compostela, Lugo, España, Publicado 3 de marzo de 2016http://www.impactjournals.com/oncotarget/index.php?journal=oncotarget&page=article&op=view&path%5B%5D=7897&path%5B%5D=23614
Mauricio Lema, (2012), El Proceso DE Carcinogénesis: Células Normales Vs Tumorales, Master en Oncología Molecular – CNIO; 2012-2014, (22.09.2012)http://mauriciolema.webhost4life.com/Moloncol2012/files/MOM_07_Carcinogenesis.pdf
https://www.ncbi.nlm.nih.gov/pubmed/21515812
https://www.ncbi.nlm.nih.gov/pubmed/22324939
32CARCINOGENESIS





























![Sarcoidosis-Associated Hepatocellular Carcinoma · in the carcinogenesis of sarcoidosis-associated HCC, as in the carcinogenesis associated with viral hepatitis [13]. Actually, cases](https://img.pdfslide.tips/doc/110x75/5e21d512d044f5667706527f/sarcoidosis-associated-hepatocellular-in-the-carcinogenesis-of-sarcoidosis-associated.jpg)