Embed Size (px)
Citation preview

Les Hémoglobinopathies
Nicole COUPRIELaboratoire Marcel Mérieux – Hématologie Spécialisée

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
Sommaire
1- Généralités et définitions
1-1. L’hémoglobine1-2. Les hémoglobinopathies� les anomalies de structure de la protéine� les anomalies de synthèse des chaînes de globine
2- Rappels structuraux
2-1. Structure de l’hémoglobine2-2. La structure des gènes de l’hémoglobine
3- Composantes du diagnostic clinico-biologique
3-1. Circonstances de diagnostic d’une hémoglobinopathie3-2. Prélèvement3-3. Approche clinique3-3.1. Données anamnestiques3-3.2. Examen clinique3-4. Les techniques biologiques d’exploration des hémoglobinopathies3-4.1. Données biologiques de base3-4.2. Techniques spécifiques d’exploration des hémoglobinopathies
a/ Techniques électrophorétiques et chromatographiquesa1/ Electrophorèse sur acétate de cellulose à pH alcalina2/ Electrophorèse sur agar à pH acidea3/ Chromatographie liquide à haute performancea4/ Focalisation isoélectrique ou isoélectrofocalisationa5/ Electrophorèse des chaînes de globinea6/ Etude de la biosynthèse in vitro des chaînes de globine
b/ Techniques hématologiques et biochimiques complémentairesb1/ Test de falciformation d’Emmelb2/ Test de Kleihauerb3/ Etude de la distribution de la densité des globules rougesb4/ Test de solubilité de l’hémoglobine S ou test d’Itanob5/ Dosage de l’hémoglobine foetaleb6/ Test de stabilité de l’hémoglobine à l’isopropanol
c/ Explorations génomiquesc1/ Haplotypesc2/ Polymerase Chain Reaction

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
4- Quelques tableaux clinico-biologiques d’hémoglobinopathies
4-1. Les thalassémies
4-1.1. Les ββββ-thalassémiesa/ Répartition géographique
b/ Physiopathologie des β-thalassémies
c/ Classification des lésions moléculaires des β-thalassémies
d/ Classification clinique des β-thalassémiesd1/ β-thalassémie homozygoted2/ β-thalassémie intermédiaired3/ β-thalassémie mineured4/ β-thalassémie silencieuse
e/ Diagnostic biologique des β-thalassémiese1/ Anomalies hématologiquese2/ Explorations phénotypiques et moléculaires de l’hémoglobine
4-1.2. Les αααα-thalassémiesa/ Répartition géographique
b/ Physiopathologie et classification des lésions moléculairesb1/ Les α-thalassémies délétionnellesb2/ Les α-thalassémies non délétionnellesb3/ Les α-thalassémies acquises
c/ Classification clinique des α-thalassémiesc1/ Anasarque fœtal de Bartc2/ Hémoglobinose Hc3/ α-thalassémie-1 ou α-thalassémie mineurec4/ α-thalassémie-2 ou α-thalassémie silencieuse
d/ Diagnostic biologique des α-thalassémiesd1/ Anomalies hématologiquesd2/ Explorations phénotypiques et moléculaires de l’hémoglobine
4-1.3. Les δβδβδβδβ-thalassémiesa/ Définition moléculaireb/ Tableaux cliniquesc/ Explorations biologiques
4-1.4. Les hémoglobines Leporea/ Définition moléculaire
b/ Tableaux cliniques
c/ Explorations biologiques
4-2. Les persistances héréditaires de l’hémoglobine fœtale4-2.1. Définition moléculaire4-2.2. Aspect clinique4-2.3. Explorations biologiques

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
4-3. Quelques hémoglobines anormales4-3.1. La drépanocytose
a/ Répartition géographique
b/ Définition moléculaire
c/ Physiopathologiec1/ Physiopathologie au niveau moléculairec2/ Physiopathologie au niveau cellulaire
d/ Aspects cliniquesd1/ Drépanocytose homozygoted2/ Drépanocytose hétérozygoted3/ Autres syndromes drépanocytaires
e/ Diagnostic biologiquee1/ Anomalies hématologiquese2/ Explorations phénotypiques
4-3.2. L’hémoglobine Ca/ Répartition géographique et définition moléculaire
b/ Physiopathologie
c/ Aspects cliniques
d/ Diagnostic biologiqued1/ Anomalies hématologiquesd2/ Explorations phénotypiques
4-3.3. L’hémoglobine Ea/ Répartition géographique et définition moléculaire
b/ Physiopathologie
c/ Aspects cliniques
d/ Diagnostic biologiqued1/ Anomalies hématologiquesd2/ Explorations phénotypiques
4-3.4. Une hémoglobine instable : l’hémoglobine Djelfaa/ Définition
b/ Répartition géographique et définition moléculaire
c/ Physiopathologie
d/ Aspects cliniques
e/ Diagnostic biologiquee1/ Anomalies hématologiquese2/ Explorations biologiques spécifiques
5- En pratique biologique courante
6- Pour en savoir plus

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
1/24
Les hémoglobinopathies
1- Généralités et définitions
1-1. L’hémoglobine
L’hémoglobine appartient à la famille des pigments respiratoires qui sont des macromoléculesprotéiques fixant réversiblement l’oxygène et qui se sont développées aussi bien chez lesvertébrés que chez les invertébrés. Outre son rôle principal dans le transport de l’oxygène despoumons aux tissus, l’hémoglobine est impliquée dans l’élimination du gaz carbonique et lemaintien du pH intra-érythrocytaire.
1-2. Les hémoglobinopathies
Ce sont des maladies génétiquement déterminées qui constituent un problème de santé publiquedans de vastes parties du monde. Les praticiens sont confrontés de plus en plus souvent à cesaffections en raison des migrations de populations. Ils doivent poser un diagnostic précis pourprendre en charge les patients, donner un conseil génétique et, si nécessaire, porter un diagnosticprénatal.
Les anomalies de l’hémoglobine se répartissent en deux grands groupes :
� les anomalies de structure de la protéine, responsables d’anémies, plus rarement depolyglobulie ou de cyanose. C’est dans cette catégorie que se situe la drépanocytose par exemple.� les anomalies de synthèse des chaînes de globine : ce sont d’une part les syndromesthalassémiques (α-thalassémies dues à un déficit de synthèse des chaînes α, et les β–thalassémies résultant d’un déficit de synthèse des chaînes β) et d’autre part les persistanceshéréditaires de l’hémoglobine fœtale.
Les deux types d’anomalies peuvent être intriqués : ce sont par exemple les syndromes thalasso-drépanocytaires.
Les mécanismes moléculaires responsables de ces anomalies sont multiples. Il existe une grandediversité des syndromes cliniques.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
2/24
2- Rappels structuraux
2-1. Structure de l’hémoglobine
L’hémoglobine humaine comporte une partie protéique : les chaînes de globine, au nombre dequatre et identiques deux à deux (deux chaînes de type α et deux chaînes de type β), unies pardes liaisons non covalentes et une partie non protéique : l’hème.
Dans la structure tétramérique, les dimères sont disposés de telle sorte que la sous-unité α1 soiten contact avec la sous-unité β2 et α2 de β1. Le contact est rigide entre les sous-unités d’un mêmedimère (α1β1 ou α2β2). Le contact entre les chaînes β, au niveau de la cavité centrale, est établi parl’intermédiaire d’une molécule de 2,3-diphosphoglycérate (2,3-DPG) et stabilise la configurationdésoxygénée. Lors de la transition de l’état désoxygéné, le 2,3-DPG est expulsé de la cavitécentrale.Les sous-unités protéiques ont une surface externe, en contact avec le milieu aqueux ambiant,constituée de résidus hydrophiles alors que les régions internes comprennent des résidushydrophobes qui, en échangeant un très grand nombre de liaisons de faible énergie, stabilisentl’édifice moléculaire. Les hélices constituant chaque sous-unité de globine sont désignées par unelettre de A à H.
La molécule d’hème est logée dans une cavité en forme de V de chaque sous-unité de globine.C’est une protoporphyrine ayant à son centre un atome de fer sous forme réduite, qui peut fixer defaçon réversible un atome d’oxygène.
2-2. La structure des gènes de l’hémoglobine
Les gènes de la famille α sont situés sur le chromosome 16 et ceux de la famille β sur lechromosome 11. La famille α comporte trois gènes et la famille β cinq. Il existe de plus desséquences similaires à celles des gènes ne codant pour aucune chaîne polypeptidique(pseudogènes). Comme d’autres gènes, ceux de la globine sont constitués par des zonescodantes et des zones non codantes.L’ordre des gènes de 5’ en 3’ sur le chromosome correspond à l’ordre de leur expression au coursdu développement. En effet le profil électrophorétique des hémoglobines varie au cours de la vie :� chez l’embryon, trois hémoglobines coexistent :
Hb Gower 1 (ζ2ε2)Hb Gower 2(α2ε2)Hb Portland (ζ2γ2)
� chez le fœtus apparaît l’hémoglobine fœtale (α2γ2). Les chaînes γ sont produites par deux gènesdupliqués : Gγ et Aγ ; le rapport Gγ / Aγ = 3/1.� après la naissance, l’hémoglobine F diminue rapidement ; le rapport Gγ / Aγ = 2/3.� le statut hémoglobinique adulte est atteint, en principe, entre l’âge de 1 et 2 ans, et comporte :
Hb A ≈ 97 %Hb A2 ≤ 3,1 %Hb F ≤ 1 %

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
3/24
3- Composantes du diagnostic clinico-biologique
3-1. Circonstances de diagnostic d’une hémoglobinopathie
Les situations amenant à étudier une hémoglobinopathie sont multiples et comprennent :
� le diagnostic étiologique d’anomalies biologiques :- anomalies hématologiques au niveau d’un frottis sanguin,- signes d’hémolyse,- fraction hémoglobinique anormale sur le tracé chromatographique pour dosage de l’hémoglobineglyquée� le diagnostic étiologique d’anomalies cliniques :- anémie hémolytique (lithiase vésiculaire),- polyglobulie,- cyanose,� l’enquête familiale, suite à la découverte d’une hémoglobinopathie� la recherche systématique chez les nouveau-nés de certaines maternités.
3-2. Prélèvement
Pour la plupart des explorations d’une hémoglobinopathie, un tube de sang total recueilli sur EDTAsuffit. Le diagnostic d’une drépanocytose chez un nouveau-né peut même être réalisé sur unéchantillon d’une centaine de microlitres de sang.L’idéal est de travailler sur du sang frais. Le délai de conservation au-delà de trois ou quatre jourspeut faire apparaître des fractions hémoglobiniques dénaturées qui peuvent poser des problèmesdiagnostiques avec certaines hémoglobines en très petite quantité ; de même, la méthémoglobine,qui représente moins de 1 % de l’hémoglobine à l’état normal, peut augmenter dans lesprélèvements vieillis.
3-3. Approche clinique
3-3.1. Données anamnestiques
� origine ethno-géographique,� contexte familial,� données physiologiques : âge, grossesse� données pathologiques : traitement cytostatique par l’hydroxyurée (Hydrea )
3-3.2. Examen clinique
� signes d’anémie : pâleur cutanéo-muqueuse, asthénie, souffle cardiaque…� signes d’hémolyse : splénomégalie, lithiase vésiculaire…

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
4/24
3-4. Les techniques biologiques d’exploration des hémoglobinopathies
3-4.1. Données biologiques de base
� Hémogrammehémoglobinenombre d’hématiesvolume globulaire moyenteneur hémoglobinique corpusculairemorphologie érythrocytaire : cellules cibles, drépanocytes, poïkilocytose, microcytose,hypochromieinclusions érythrocytaires : corps de Jolly, ponctuations basophiles, corps de Heinzréticulocytes, érythroblastesleucocytesplaquettes
� bilan martial : fer sérique, coefficient de saturation de la transferrine, ferritine� paramètres d’hémolyse : bilirubine libre et conjuguée, haptoglobine
3-4.2. Techniques spécifiques d’exploration des hémoglobinopathies
a/ Techniques électrophorétiques et chromatographiques
a1/ Electrophorèse sur acétate de cellulose à pH alcalin
C’est la technique standard la plus simple à mettre en œuvre. Elle sépare les différenteshémoglobines en fonction de leur charge et de la position de l’acide aminé muté dans la molécule.Elle permet une bonne séparation des différentes fractions hémoglobiniques normales : A, F, A2 etle dépistage des syndromes thalassémiques.Cependant il faut doser l’hémoglobine A2 pour confirmer le diagnostic de β-thalassémie et utiliserun autre système de séparation pour identifier les hémoglobines anormales. Un tracé normaln'exclut pas une hémoglobinopathie.
a2/ Electrophorèse sur agar à pH acide
Cette technique complète l’électrophorèse à pH alcalin. La migration d’une hémoglobine anormaleen agar dépend d’abord de la localisation de la mutation et secondairement du changement decharge ; cette migration résulte de l’électroendosmose, de la liaison à l’agaropectine et de l’effet del’ion citrate.En effet, elle permet de séparer les variants ayant la même mobilité que les hémoglobines A, S ouC sur acétate de cellulose. Elle permet une très bonne séparation des hémoglobines A et F, ce quin’est pas le cas dans l’électrophorèse à pH alcalin.Cependant la mise en évidence de mutants de même mobilité que l’hémoglobine A n’est paspossible par cette seule technique. De plus, les anomalies qualitatives observées sur les tracésdoivent être précisées par dosage.
a3/ Chromatographie liquide à haute performance
Des systèmes complètement automatisés peuvent être utilisés, comme le Variant qui est uneCLHP par échange de cation avec détection des composés élués à double longueur d’onde (415 /690 nm).Des fenêtres sont établies pour les hémoglobines les plus fréquentes en fonction de leur temps derétention. L’identification de ces variants spécifiques nécessite cependant des techniquesadditionnelles.Un étalon avec des valeurs connues (exprimées en pourcentage de surface) pour leshémoglobines A2 et F permet de quantifier ces fractions.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
5/24
Il est ainsi possible de détecter des anomalies quantitatives du chromatogramme qui ne sont pasen rapport avec des hémoglobinopathies :
� L’Hb F peut s’élever quand il y a expansion de l’érythropoïèse : après une hémorragie ou unehémolyse aiguë par exemple, au cours du deuxième trimestre de la grossesse, ou lors d’un stressérythropoïétique aigu qui s’accompagne d’une régénération médullaire rapide.Des augmentations de l’Hb F se rencontrent dans diverses pathologies comme : les syndromesmyélomonocytaires de l’enfant, certains cancers métastasés, la myélofibrose, certaines anémies(anémies inflammatoires chroniques, anémie de Biermer, microsphérocytose, maladie de Fanconi,maladie de Blackfan-Diamond), l’hémoglobinurie nocturne paroxystique…
� Une élévation du taux de l’Hb A2 peut s’observer en dehors de toute hémoglobinopathie, dans lecadre de maladies virales (hépatites, infection par le VIH…), hyperthyroïdies, dysérythropoïèses…
a4/ Focalisation isoélectrique ou isoélectrofocalisation
L’isoélectrofocalisation est une technique d’électrophorèse sur gel de polyacrylamide en gradientde pH, sous voltage élevé. Les hémoglobines sont séparées grâce à leur point isoélectrique. Lavisualisation définitive des fractions hémoglobiniques est réalisée par une brève fixation par l’acidetrichloracétique.Le pouvoir de résolution de cette technique est proche de celui des meilleures techniqueschromatographiques. Elle permet d’identifier les hémoglobines anormales chez l’adulte parcomparaison de la position isoélectrique du mutant inconnu avec celle d’un mutant de référence.Elle permet de détecter les hémoglobines anormales chez le nouveau-né alors que l’hémoglobineF est le composant hémoglobinique majeur dans les premiers mois de la vie.
a5/ Electrophorèse des chaînes de globine
Cette électrophorèse est réalisée en gel de polyacrylamide en milieu acide dissociant, c’est-à-direen présence d’urée 8M et d’un détergent le Triton-X100Cette technique met en évidence toute substitution d’acides aminés impliquant une différenced’hydrophobicité de la chaîne latérale de l’acide aminé concerné, même en l’absence de différencede charge. Elle permet de séparer les deux chaînes γ qui ne diffèrent entre elles que par un résiduméthyl (γ136 Ala ou Gly).
a6/ Etude de la biosynthèse in vitro des chaînes de globineLes globules rouges (fraction enrichie en réticulocytes) sont mis en incubation en présence deleucine tritiée. Après incorporation de cet acide aminée radio-marqué, la séparation des chaînesde globine synthétisées est réalisée par chromatographie liquide à haute performance en phaseinverse par exemple. La radioactivité de chaque pic est déterminée. Les rapports de biosynthèsedes chaînes α / non α sont ainsi déterminés.
b/ Techniques hématologiques et biochimiques complémentaires
b1/ Test de falciformation d’Emmel
Il permet de rechercher une drépanocytose. La technique consiste à mettre en contact, sur unelame, une goutte de sang avec une goutte de métabisulfite de sodium. L’hémoglobine S et d’autreshémoglobines (comme l’hémoglobie C Harlem) ont une solubilité très diminuée quand elles sontdésoxygénées. Les hématies qui les contiennent changent de forme et deviennent incurvées. Ondécèle alors rapidement au microscope la falciformation.C’est une technique très grossière qui ne permet pas de faire la différence entre drépanocytosehétérozygote et homozygote.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
6/24
b2/ Test de Kleihauer
Il s’agit d’une technique cytologique qui permet de repérer les hématies contenant del’hémoglobine F et de connaître la distribution de cette hémoglobine au sein de la populationérythrocytaire.Cette technique permet de préciser le caractère pancellulaire ou hétérocellulaire d’une PersistanceHéréditaire de l’Hémoglobine Fœtale.
b3/ Etude de la distribution de la densité des globules rouges
La densité des globules rouges est étudiée par la méthode des phtalates.La densité des globules rouges se répartit selon une distribution gaussienne caractérisée par unedensité médiane ; on peut déterminer aussi le pourcentage de cellules denses.Chez le drépanocytaire homozygote, il semble que la densité médiane et le pourcentage decellules denses varient d’un malade à l’autre en restant constants dans le temps pour un mêmepatient. Cela permettrait d’appréhender l’hétérogénéité de la maladie drépanocytaire en comparantles malades.
b4/ Test de solubilité de l’hémoglobine S ou test d’Itano
L’hémoglobine S, sous forme désoxygénée, précipite quand elle se trouve en solution salineconcentrée. Seule l’hémoglobine H, instable, précipite dans les mêmes conditions.
b5/ Dosage de l’hémoglobine foetale
Plusieurs techniques existent.Cependant seule la CLHP permet de doser l’hémoglobine F quel que soit son taux.
b6/ Test de stabilité de l’hémoglobine à l’isopropanol
Les forces de cohésion internes de la molécule d’hémoglobine diminuent dans un milieu apolaireque constitue l’isopropanol. Quand celui-ci est à la concentration de 17 % V/V, l’hémoglobine Aprécipite après 50 à 60 minutes à 37°C.L’instabilité d’une hémoglobine se manifeste par une précipitation précoce (de 5 à 20 minutes).
b7/ Exploration de la fixation de l’oxygène (mesure de la P50 et du 2, 3-diphosphoglycérateintra-érythrocytaire)
L’affinité des hématies pour l’oxygène est définie par la pression partielle d’oxygène à laquellel’hémoglobine contenue dans les globules rouges est à mi-saturation (P50). Cette déterminationnécessite un équipement particulier et ne peut être réalisée que sur un prélèvement extemporané.
c/ Explorations génomiques
c1/ Haplotypes
Il existe dans le génome de nombreuses substitutions ou insertions nucléotidiques sansexpression phénotypique mais détectables par les enzymes de restriction. Ces enzymes cliventl’ADN quand elles rencontrent une séquence de bases spécifiques (d’où leur caractère derestriction). Elles donnent lieu à des différences (« polymorphismes ») de la longueur desfragments de l’ADN générés par les clivages. En effet une mutation sur l’ADN peut abolir (-) ougénérer (+) un site de clivage.Actuellement une vingtaine de sites de restriction ont été identifiés le long du complexe β. Lacombinaison des sites de restriction polymorphes observés sur chaque chromosome permet dedéfinir un haplotype chromosomique.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
7/24
c2/ Polymerase Chain Reaction
Des techniques d’amplification de l’ADN à l’aide d’amorces judicieusement choisies permettent dufaire apparaître sur l’électrophorèse en gel d’agarose ou de polyacrylamide, des produitsd’amplification de tailles différentes selon la présence ou l’absence d’une mutation ou d’unedélétion.
4- Quelques tableaux clinico-biologiques d’hémoglobinopathies
4-1. Les thalassémies
Les thalassémies sont définies par un déficit total ou partiel de synthèse de chaînes del’hémoglobine. Elles peuvent concerner toutes les chaînes de l’hémoglobine. Nous nous limiteronsaux thalassémies suivantes : α, β, et δβ-thalassémies.
4-1.1. Les ββββ-thalassémies
a/ Répartition géographique
Les plus fortes densités de β-thalassémies sont décrites sur le pourtour méditerranéen : Italie,Sardaigne, Sicile, Grèce (la fréquence du trait est de l’ordre de 8 % de la population), Afrique duNord, Proche et Moyen-Orient sauf le Japon. En Afrique intertropicale, la fréquence varie de 1 à 5% selon les régions.En France, 36,5 % des sujets originaires de pays à haut risque pour les β-thalassémies résidentdans la région parisienne et 63,5 % en province (d’après D. Lena-Russo, 1992).
b/ Physiopathologie des ββββ-thalassémies
Les β-thalassémies sont définies par un déficit total ou partiel en synthèse de chaînes β del’hémoglobine. On distingue les β0-thalassémies (absence de synthèse) et les β+-thalassémies(présence d’une synthèse diminuée).
En raison de l’absence ou de la diminution de synthèse des chaînes β, le tétramèrehémoglobinique normal est peu ou pas formé et les chaînes α non appariées, moins solubles,précipitent et altèrent l’érythroblaste provoquant sa destruction.Les troubles sont davantage liés à la dysérythropoïèse qu’à l’hémolyse. La précipitation deschaînes libres s’effectue dès les étapes précoces de l’érythrogenèse. Il s’en suit une érythropoïèseinefficace ainsi qu’une prolifération du tissu érythroïde.Cependant dans les hématies du sang périphérique, la précipitation des chaînes α libres conduit àdes altérations membranaires et à une hémolyse.La chaîne δ n’étant synthétisée qu’en faible quantité, la seule compensation possible est uneproduction accrue des sous-unités γ, qui constituent, associées aux sous-unités α, l’hémoglobineF. L’hémoglobine F circulante, quantitativement insuffisante et ayant une affinité élevée pourl’oxygène, ne peut corriger le défaut d’oxygénation des tissus. L’anoxie tissulaire, à l’origine d’unestimulation de la synthèse de l’érythropoïétine, accroît encore le processus érythropoïétique.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
8/24
c/ Classification des lésions moléculaires des ββββ-thalassémies
Les lésions moléculaires à l’origine des β-thalassémies sont nombreuses et la liste ne peut en êtreexhaustive.
� les délétions étenduesElles sont rares (contrairement au cas des α-thalassémies)
� les mutations non-sensElles aboutissent à l’apparition d’un codon de terminaison prématurée (par exemple : apparitiond’un codon-stop β0 39).
� les mutations décalantes du cadre de lecture par insertion ou délétion d’une ou de plusieursbases (mutations « frame-shift »).
� les mutations affectant l’épissage normal du transcrit primaire en ARN messager : anomalie de lacharnière exon-intron, anomalies de séquences consensus, anomalies à l’intérieur d’un intron…
� les mutations dans le promoteur du gène
� les mutations dans le site de polyadénylation (extrémité 3’ de l’ARN messager)
� les mutations du site CAP (extrémité 5’ de l’ARN messager)
� les mutations du codon d’initiation
� des hémoglobines hyper-instables peuvent entraîner une β-thalassémie commel’hémoglobine Cagliari β 60 [E4] Val � Glu, par exemple.
� certaines hémoglobines anormales comme l’hémoglobine E (β 26 [B8] Glu � Lys) oul’hémoglobine Knossos (β 27 [B9] Ala � Ser) s’accompagnent d’un syndrome thalassémique.
d/ Classification clinique des ββββ-thalassémies
d1/ ββββ-thalassémie homozygote
La forme homozygote habituelle est l’anémie de Cooley.La maladie apparaît au cours des premiers mois de la vie lorsque s’effectue la commutation entrehémoglobine fœtale et hémoglobines adultes. Elle se traduit par un syndrome anémique trèssévère, une hépato-splénomégalie, un retard de croissance.Non traitée, la maladie évolue spontanément vers le décès en quelques mois ou années dans untableau d’anémie parfois aggravé par des infections intercurrentes. Cette pathologie nécessite destransfusions sanguines régulières accompagnées d’un traitement chélateur du fer et généralementd’une splénectomie en attendant une greffe de moelle.
d2/ ββββ-thalassémie intermédiaire
La définition de cette entité est purement clinique : elle est caractérisée par une bonne tolérance àl’anémie sans asthénie. Le retentissement sur l’état général est le plus souvent modéré.Cependant la puberté est souvent retardée, mais généralement complète. La splénomégalie esthabituelle dans ces formes de thalassémie ; elle peut évoluer vers un hypersplénisme et rendrecompte des besoins transfusionnelles.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
9/24
Sur le plan génétique, il peut s’agir d’une anomalie homozygote dans laquelle le déséquilibre desynthèse des chaînes de globine est plus modéré que dans les β0-thalassémies. Mais certaines β-thalassémies intermédiaires surviennent chez des hétérozygotes dans le cadre de certainesmutations du troisième exon en particulier.
d3/ ββββ-thalassémie mineure
C’est la forme le plus souvent observée chez les sujets hétérozygotes.La traduction en est essentiellement hématologique avec une discrète anémie microcytairepseudo-polyglobulique. Toutes les complications des β-thalassémies intermédiaires peuvent êtreobservées mais leur fréquence et leur gravité sont infiniment moindres : splénomégalie, ulcères dejambes, lithiase vésiculaire.
d4/ ββββ-thalassémie silencieuse
Il s’agit d’une forme inapparente de β-thalassémie, même sur le plan hématologique etl’hémoglobine A2 peut être normale. Cette forme est déduite de l’étude familiale. Sur le planmoléculaire, il s’agit de certaines mutations du promoteur diminuant faiblement la transcription dugène β.
e/ Diagnostic biologique des ββββ-thalassémies
e1/ Anomalies hématologiques
Selon l’anomalie génétique, on observe :
Anomalie Numération Aspect sur lame
β0 thal homozygote Hb : 2-10 g/dlVGM : 60-75 fl
anisocytosehypochromiepoïkilocytoseérythroblastes
β+ thal homozygote Hb : 2-10 g/dlVGM : 60-75 fl
anisocytosehypochromiepoïkilocytoseérythroblastes
β thal hétérozygote Hb : 10-13 g/dlVGM : 65-75 fl
anisocytose± poïkilocytose
e2/ Explorations phénotypiques et moléculaires de l’hémoglobine
Dans la forme hétérozygote, la chromatographie de l’hémoglobine est essentielle et montre untaux d’hémoglobine A2 supérieur à 3,4 % ; le taux d’hémoglobine F est supérieur à 1 % dansenviron la moitié des cas. Le diagnostic est difficile quand sidéropénie et β-thalassémie mineurecoexistent chez un même patient car la carence martiale provoque une diminution du taux del’hémoglobine A2. Il convient alors de renouveler l’exploration biologique après un traitementmartial suffisant.Dans la β-thalassémie homozygote, la chromatographie montre exclusivement de l’hémoglobineA2 et de l’hémoglobine F. Il convient de confirmer ces fractions hémoglobiniques dans undeuxième système de migration comme la focalisation isoélectrique. L’étude phénotypiquefamiliale est importante à ce stade.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
10/24
C’est la biosynthèse in vitro des chaînes de globine qui assure le diagnostic de certitude. Lediagnostic précis de la mutation au niveau de l’ADN est réalisé par des techniques d’amplificationPCR, hybridation avec des sondes oligonucléotidiques spécifiques, analyse électrophorétiquedes produits d’amplification et éventuellement séquençage.
4-1.2. Les αααα-thalassémies
a/ Répartition géographique
Elles ont une fréquence encore plus importante que les β-thalassémies en Afrique, en Asie etautour de la Méditerranée. En général, elles n’ont de conséquence clinique que dans les formesoù trois ou quatre gènes α sont anormaux ou absents. La délétion des quatre gènes α concerneparticulièrement les populations du Sud-Est asiatique.
b/ Physiopathologie et classification des lésions moléculaires
Les α-thalassémies traduisent un défaut d’expression d’un ou de plusieurs gènes codant pour leschaînes α de globine. Le mécanisme moléculaire le plus souvent incriminé est la délétion desgènes α. Des mutations ponctuelles affectant la transcription ou la traduction de la chaîne α ontété également décrites.
b1/ Les αααα-thalassémies délétionnelles
Elles sont liées à une perte de matériel génétique. Celles-ci sont secondaires à des phénomènesde recombinaison génétique inégale entre les chromosomes homologues 16 (exemple de lathalassémie α -3.7 ).
Phénotype Nombre de gènes α fonctionnels GénotypeNormal 4 αα / ααα-thalassémie –1α+-thalassémie hétérozygote
3 αα / −α
α-thalassémie –2α+-thalassémie homozygoteα0-thalassémie hétérozygote
2−α / −ααα / −−
Hémoglobinose H 1 −α / −−Hydrops fœtalis 0 −−=/ −−
b2/ Les αααα-thalassémies non délétionnelles
Les causes en sont multiples :- mutation décalante du cadre de lecture (mutations « frameshift »)- épissage aberrant par délétion de nucléotides au niveau d’introns- perturbation de l’extrémité 3’ de l’ARN messager au niveau du site de polyadénylation- mutation dans le codon de terminaison avec élongation de chaîne et production de globine
hyperinstable (par exemple : hémoglobine Constant Spring).
b3/ Les αααα-thalassémies acquises
Il a été décrit quelques cas d’ α-thalassémie acquise : hémoglobinose H associée à un syndromede prolifération leucémique, le plus souvent chez des sujets âgés de sexe masculin. Lemécanisme moléculaire responsable de l’absence de synthèse des chaînes α dans cesérythrocytes anormaux est encore totalement inconnu.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
11/24
c/ Classification clinique des αααα-thalassémies
Les α-thalassémies s’expriment selon quatre formes cliniques, en fonction du nombre de gènesdéfectueux ou absents. En pratique, cette classification doit être nuancée par la différenced’expression des gènes α1 et α2.
c1/ Anasarque fœtal de Bart
Il s’agit de l’absence totale de gène α fonctionnel. Cette anomalie est à l’origine d’une anémiehémolytique extrêmement sévère durant la vie fœtale, conduisant à la mort in utero dans untableau d’hydrops fœtalis.
c2/ Hémoglobinose H
Elle se rencontre le plus souvent chez des patients d’origine asiatique ou méditerranéenne etexceptionnellement dans la race noire. Elle se présente comme une anémie hémolytique assezgrave, accompagnée de pâleur, d’ictère cutanéo-muqueux et de splénomégalie ; la survenue d’unelithiase en est une complication fréquente.Des épisodes hyperhémolytiques avec séquestration splénique sont une indication desplénectomie.
c3/ αααα-thalassémie 1 ou αααα-thalassémie mineure
Deux gènes sont délétés et seule existe une discrète anémie microcytaire.
c4/ αααα-thalassémie 2 ou αααα-thalassémie silencieuse
Quand il reste trois gènes normaux, l’α-thalassémie est pratiquement asymptomatique en dehorsde la présence d’hémoglobine Bart (tétramères γ4) présente à un taux de 1 à 2 %, à la naissance.Elle peut cependant être suspectée quand elle est associée à une hémoglobine anormale, parexemple : hémoglobine S, C ou E, ou en cas de β-thalassémie de sévérité intermédiaire chez unhomozygote β0-thalassémique.
d/ Diagnostic biologique des αααα-thalassémies
d1/ Anomalies hématologiques� Dans les α-thalassémies 2, le volume globulaire moyen est souvent normal.
� Dans les α-thalassémies 1, le frottis sanguin montre une microcytose, une hypochromie et descellules cibles.
� Dans l’hémoglobinose H, on observe :- une microcytose importante (souvent inférieur à 50 fl)- des anomalies de la morphologie érythrocytaire : hypochromie, ponctuations basophiles,schizocytose, cellules cibles…- une réticulocytose supérieure à 5 %.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
12/24
Anomalie Numération Aspect sur lame
α+ thal hétérozygote : délétiond’1 gène α
Hb : 12-14 g/dlVGM : 62-78 fl Normal
α+ thal homozygoteα0 thal hétérozygote :délétion de 2 gènes α
Hb : 11-12 g/dlVGM : 62-78 fl
anisocytosehypochromiepoïkilocytose
Hémoglobinose H :délétion de 3 gènes α
Hb : 3-12 g/dlVGM : 50 fl
réticulocytose
anisocytosehypochromiepoïkilocytoseschizocytosecellules cibles
d2/ Explorations phénotypiques et moléculaires de l’hémoglobine
Les techniques d’électrophorèse à pH alcalin et à pH acide peuvent mettre en évidencel’hémoglobine Bart.La chromatographie liquide à haute performance des hémoglobines peut laisser suspecterl’existence d’une α-thalassémie 1 devant une diminution de l’hémoglobine A2. L’hémoglobine Bartest également mise en évidence et confirmée par la technique de focalisation isoélectrique.L’étude de la biosynthèse in vitro des chaînes de globine apporte un diagnostic de certitude.Les α-thalassémies délétionnelles peuvent également être détectées par l’utilisation de latechnique de PCR, avec choix opportun des amorces et digestion enzymatique des produitsd’amplification.
4-1.3. Les δβδβδβδβ-thalassémies
a/ Définition moléculaire
Les δβ-thalassémies se caractérisent par un atteinte concomitante des chaînes β et δ de globine.Elles résultent d’une délétion plus ou moins étendue des deux gènes.
b/ Tableaux cliniques
� La forme hétérozygote de δβ-thalassémie est asymptomatique.� La forme homozygote réalise un tableau d’anémie de Cooley.
c/ Explorations biologiques
� La δβ-thalassémie hétérozygote se traduit au niveau de l’hémogramme par une microcytose.La CLHP montre une absence d’élévation du pourcentage de l’hémoglobine A2 et une élévation del’hémoglobine fœtale (5 à 10 %).
� A l’état homozygote, dans la forme δβ0-thalassémie, il n’y a pas de synthèse d’hémoglobine A nid’hémoglobine A2 ; par contre il y a une synthèse d’hémoglobine fœtale. L’étude de la biosynthèsein vitro des chaînes de globine et la détermination du génotype assurent le diagnostic.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
13/24
4-1.4. Les hémoglobines Lepore
Les hémoglobines Lepore s’apparentent aux δβ-thalassémies ; il s’agit d’une forme avecexpression d’une hémoglobine structuralement anormale.
a/ Définition moléculaire
Les hémoglobines Lepore sont caractérisées par un gène de fusion δβ qui résulte d’un crossingover entre gènes δ et β= de deux chromosomes, dû à un mauvais alignement des gènes au coursde la méïose. Les différentes localisations du crossing over définissent plusieurs hémoglobinesLepore ; la plus fréquente est l’hémoglobine Boston.Par ailleurs, il existe également des hémoglobines anormales appelées anti-Lepore (hémoglobinesP-Congo…) dont la chaîne mutée commence par la séquence β et se termine par la séquence δ.
b/ Tableaux cliniques
Ces lésions sont responsables d’un syndrome thalassémique car la chaîne Lepore est expriméecinq fois moins que le gène β normal.Les formes hétérozygote et homozygote des hémoglobines Lepore se comportent comme des δβ-thalassémies hétérozygote ou homozygote, respectivement.
c/ Explorations biologiques
� L’hémoglobine Lepore à l’état hétérozygote constitue une bande supplémentaire anormaled’environ 10 % au niveau de l’hémoglobine S sur l’électrophorèse à pH alcalin ; cette fractionprésente un temps de rétention voisin de celui de l’hémoglobine A2 sur le tracé de CLHP.L’individualisation secondaire de cette fraction par la technique de focalisation isoélectrique permetd’affirmer l’existence de l’hémoglobine Lepore.
� Chez le sujet homozygote, la chromatographie, comme le tracé d’isoélectrofocalisation nemontre que cette bande anormale et de l’hémoglobine F.
4-2. Les persistances héréditaires de l’hémoglobine fœtale
4-2.1. Définition moléculaire
Les persistances héréditaires de l’hémoglobine fœtale (PHHF) correspondent à un état au coursduquel les gènes γ restent actifs pendant la vie adulte.Les lésions moléculaires font distinguer :- les PHHF sans délétion qui sont souvent associées à des mutations ponctuelles des promoteurs
des gènes γ ; elles sont de beaucoup les plus fréquentes. Leurs mécanismes impliquent des sitesde liaison de facteurs de transcription et de régulation modulant l’expression des gènes γ, parexemple. Elles se limitent à une stimulation de la synthèse des chaînes γ et ne modifient pasl’expression du gène β.
- les PHHF avec délétion étendue. Il s’agit de délétions importantes, incluant les gènes δ et β, dontla longueur et la localisation sont variables.
4-2.2. Aspect clinique
La forme hétérozygote se caractérise par l’absence de signes cliniques et hématologiques.Les cas de PHHF homozygote sont rares et sont bien tolérées puisqu’il existe une augmentationvraie de la synthèse de l’hémoglobine F.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
14/24
4-2.3. Explorations biologiques
Les différentes techniques d’analyse phénotypique (électrophorèses et CLHP de dépistage,focalisation isoélectrique) montrent seulement une augmentation du pourcentage de l’hémoglobineF.L’analyse des gènes γ par la détermination du rapport Aγ / Gγ ainsi que les techniques moléculaires(PCR… et éventuellement séquençage) permettent d’établir un diagnostic de certitude.
4-3. Quelques hémoglobines anormales
4-3.1. La drépanocytose
a/ Répartition géographique
Les patients atteints de syndrome drépanocytaire majeur sont originaires d’Afrique, des Antilles etdes Etats Unis. La coïncidence entre les zones d’endémie drépanocytaire et celles de l’infestationpalustre a fait émettre l’hypothèse d’un avantage sélectif possédé par les sujets drépanocytaireshétérozygotes par rapport aux individus normaux (AA) vis-à-vis du paludisme. A basse pressiond’oxygène, correspondant à la séquestration dans les organes profonds, il y a un retard decroissance du parasite dans les cellules contenant de l’hémoglobine S. Ainsi les sujets AS sontsusceptibles d’infestation palustre mais toute augmentation de la parasitémie se fait aux dépendsde la survie du parasite, ce qui semble confirmer la rareté des parasitoses élevées et souventmortelles chez ces sujets.
En France, 61 % des sujets originaires de pays à haut risque pour les syndromes drépanocytairesmajeurs résident en région parisienne et 39 % en province (d’après D. Lena-Russo, 1992).
b/ Définition moléculaire
La drépanocytose résulte de la mutation faux-sens la plus répandue dans le monde. Sur le gène βde l’hémoglobine, le codon 6 (GAG) codant pour une glutamine est remplacé par un codon (GTG)codant pour une valine. La drépanocytose fut ainsi le premier exemple démontré de maladiemoléculaire ; il s’agit d’une anomalie transmise sur le mode autosomique récessif.Une origine multiple de la mutation drépanocytaire a été mise en évidence par les études depolymorphisme de restriction de l’ADN : il existe trois haplotypes majeurs (béninois, sénégalais etcentrafricain ou Bantu) et plus récemment une quatrième mutation, indienne, a été identifiée.Le taux d’hémoglobine F chez un drépanocytaire joue un rôle important. Il dépend de facteursgénétiques associés, comme les déterminants α-thalassémiques associés et l’haplotype au seinduquel se trouve la mutation βS ; ainsi l’haplotype Sénégal augmente modérément le taux del’hémoglobine F (12 % environ) alors que l’haplotype asiatique l’augmente plus nettement (16 %).
c/ Physiopathologie
c1/ Physiopathologie au niveau moléculaire
La modification structurale de l’hémoglobine S favorise une polymérisation de la formedésoxygénée. La polymérisation est précédée d’une période de latence variable. Le temps delatence dépend de la concentration en désoxyhémoglobine S ; tous les facteurs physico-chimiquesqui stabilisent la structure désoxygénée le raccourcissent comme la température, le pH… Lapériode de latence correspond à la formation de centres de nucléation constitués par l’agrégationd’un petit nombre de tétramères d’hémoglobine. Ensuite seulement apparaissent des polymèresfibreux qui induisent la déformation du globule rouge.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
15/24
Les contacts de la structure cristalline expliquent les interactions entre l’hémoglobine S et d’autreshémoglobines : hémoglobine normale inhibant la polymérisation (comme l’hémoglobine F) ouhémoglobines anormales la facilitant (hémoglobine D Punjab, hémoglobie O Arab…).
c2/ Physiopathologie au niveau cellulaire
Les propriétés de l’hémoglobine S permettent d’expliquer l’ensemble des manifestationspathologiques de la drépanocytose et, en particulier, le fameux « cercle vicieux » auto-entretenantles crises drépanocytaires. Les causes favorisant la désoxygénation (hypoxie, fièvre, acidose,déshydratation…), donc la falciformation, conduisent à des accidents vaso-occlusifs. Cesobstacles à la circulation accentuent encore le trouble initial.
d/ Aspects cliniques
d1/ Drépanocytose homozygote
Les premiers signes cliniques n’apparaissent qu’après l’âge de six mois, période où l’hémoglobineS remplace progressivement l’hémoglobine F.Le tableau clinique comporte trois sortes de situations : les phases stationnaires, les complicationsaiguës et les complications chroniques.
� Les phases stationnaires :A l’état basal, il existe une anémie qui se traduit par une pâleur cutanéo-muqueuse puis par dessignes cardiaques.La splénomégalie est constante chez le nourrisson puis régresse.L’hépatomégalie est inconstante.Il existe un retard staturo-pondéral chez les enfants en zone tropicale et souvent un retard de lapuberté et de la maturation osseuse.
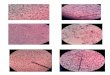
� Les complications aiguës :Les crises douloureuses aiguës, appelées crises drépanocytaires, associent des douleurs à de lafièvre. Elles sont fréquentes chez le petit enfant mais s’espacent au cours de l’adolescence.L’hétérogénéité des symptômes (thoraciques, abdominaux…) génère des problèmesdiagnostiques.Les infections ont une forte incidence dans les premières années de vie et diminuent, sansdisparaître, avec l’âge. Elles sont responsables d’une part importante de la morbidité et de lamortalité de la drépanocytose. Ce sont des méningites, des septicémies, des pneumopathies, desotéomyélites.Des épisodes d’aggravation de l’anémie chronique peuvent résulter d’une situation fébrile, decrises de séquestration splénique (particulières au petit enfant), d’une infection par le parvovirusB19.Les accidents vaso-occlusifs graves sont des complications aiguës qui comportent desmanifestations neurologiques, pulmonaires, des thromboses (thrombose de l’artère centrale de larétine, priapisme…) et des hématuries microscopiques.
� Les complications chroniques :Elles sont fréquentes pendant l’adolescence et la vie adulte. Ce sont des ulcères de jambe, desnécroses osseuses aseptiques (des hanches par exemple), des lésions oculaires (rétinopathieavec hémorragies), des complications pulmonaires (infarctus, infections, insuffisance respiratoirechronique), des atteintes cardiaques, des altérations rénales, des complications hépatobiliaires(lithiase biliaire).
Il a été trouvé des corrélations entre l’évolution clinique et le taux d’hémoglobine F. Les sujetsayant le plus d’hémoglobine F (supérieure à 20 %) ont une forme moins sévère.
Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
16/24
d2/ Drépanocytose hétérozygote
C’est en général un état asymptomatique. Des symptômes évocateurs de syndromedrépanocytaire (hémolyse, douleurs) doivent faire rechercher un facteur aggravant associé(hétérozygotie A/S Antilles).Le trait drépanocytaire expose toutefois à une atteinte rénale (hématuries, défaut du pouvoir deconcentration des urines…), certaines manifestations vaso-occlusives comme l’infarctus splénique,dans des conditions d’hypoxémie profonde par exemple.
d3/ Autres syndromes drépanocytaires
La coexistence d’une ou deux mutations caractéristiques de la drépanocytose et d’une autreanomalie génétique peut être à l’origine de syndromes drépanocytaires majeurs dont la sévéritéest variable (d’après F. GALACTEROS et Y. BEUZARD).
Association Sévérité Fréquence relative
S/S +++ +++
S/S /α thal1 ou α thal2 + à ++ ++
S/β0 thal +++ +
S/β+ thal + à ++ +
S/C + à ++ ++S/D PunjabS/O ArabS/C Harlem
+++ rare
S/PHHF 0 à + rare à +
A/S Antilles + rare
e/ Diagnostic biologique
e1/ Anomalies hématologiques
Les valeurs de l’hémogramme et l’aspect du frottis sanguin varient en fonction du type d’atteintegénétique (d’après J.Y. PELTIER).

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
17/24
Anomalie Numération Aspect sur lame
A/S Hb : NormaleVGM : Normal Normal
S/S Hb : 6-10 g/dlVGM : N ou un peu
drépanocytescellules ciblescorps de Jollyérythroblastes
S/β0 thal Hb : 6-10 g/dlVGM : 65-75 fl
anisocytosepoïkilocytosecellules ciblesdrépanocytescorps de Jolly
S/β+ thal Hb : 7-11 g/dlVGM : 75 fl
anisocytosepoïkilocytosecellules ciblesdrépanocytescorps de Jolly
S/C Hb : 9-12 g/dlVGM : N ou un peu
cellules ciblesdrépanocytes
e2/ Explorations phénotypiques
� Etude électrophorétique- L’électrophorèse à pH alcalin sur un support d’acétate de cellulose dépiste l’hémoglobine Scar la mutation βS modifie le point isoélectrique de la chaîne β et par conséquent celui detétramère α2β2. Cependant de nombreuses mutations ponctuelles induisent un changement decharge analogue à celui provoqué par cette mutation.Il faut donc au moins un autre critère pour pouvoir affirmer la présence de l’hémoglobine S.- L’électrophorèse de l’hémoglobine sur gel de citrate agar à pH acide peut permettre decaractériser l’hémoglobine S.- L’isoélectrofocalisation permet aussi de caractériser l’hémoglobine S. C’est la technique deréférence pour le diagnostic néonatal des hémoglobinopathies ; elle peut alors être réalisée à partirde quelques gouttes de sang recueillies sur un papier buvard.
� Quantification des fractions hémoglobiniques- La chromatographie liquide à haute performance est une étape indispensable pour préciser lediagnostic des syndromes drépanocytaires. Cette technique permet de quantifier précisément lesfractions d’hémoglobines A2, F, A et S.Un patient hétérozygote A/S présente un taux d’hémoglobine S entre 35 et 45 %.Pour un taux d’hémoglobine S inférieur à 35 % (en dehors de toute transfusion sanguine), on peutévoquer l’existence d’une carence martiale ou d’une α-thalassie associée.Quand la concentration d’hémoglobine S dépasse celle de l’hémoglobine A, plusieurs situationssont possibles, dont l’association Sβ+ thalassémie (et éventuellement l’association S hémoglobineinstable).
� Test de solubilitéCe test met en évidence la polymérisation in vitro de l’hémoglobine S.Seules certaines hémoglobines instables donnent une réaction analogue mais sans dépendance àla désoxygénation.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
18/24
e3/ Caractérisation génétique
Elle a deux finalités : la recherche de facteurs pronostiques et le diagnostic prénatal.
� L’étude familiale participe à cette caractérisation génétique. La grande majorité des problèmesdiagnostiques est résolue par cette seule démarche. Cependant elle n’est pas toujours possible ;c’est pour cela que l’on a recours à des techniques de biologie moléculaire.
� Les techniques de biologie moléculaire nécessitent la purification de l’ADN puis utilisent desméthodes de PCR adaptées à chaque problème posé.On peut caractériser les haplotypes de restriction du locus de la famille des gènes de type βglobine. On a tenté de faire des corrélations entre les principaux haplotypes et diversesmanifestations phénotypiques : synthèse d’hémoglobine F, risque vis-à-vis de certainescomplications, sévérité globale de la maladie. Les résultats sont assez contradictoires ; cependantil est possible de retenir que les patients homozygotes pour les haplotypes Sénégal et Arabe-Indien ont une sévérité hématologique atténuée en raison d’une synthèse accrue d’hémoglobine F.Les α-thalassémies délétionnelles (en particulier – 3,7 Kb) sont un élément à prendre en comptedans le polymorphisme phénotypique de la maladie.La technique moléculaire de mise en évidence de mutation est l’outil appliqué au diagnosticprénatal de la forme homozygote. Elle peut être réalisée à partir d’une biopsie de villosité chorialeeffectuée à la 10e ou 11e semaine d’aménorrhée chez les femmes à risque.
4-3.2. L’hémoglobine C
a/ Répartition géographique et définition moléculaire
L’hémoglobine C résulte d’une mutation ponctuelle identifiée : β 6 [A3] Glu → Lys.Par sa fréquence, c’est la seconde des hémoglobines anormales africaines ; elle a son épicentresur le plateau voltaïque.
b/ Physiopathologie
Les hématies contenant l’hémoglobine C sont des cellules partiellement déshydratées, de petitetaille, ayant une charge d’hémoglobine normale. L’augmentation de la concentration enhémoglobine explique la présence des cristaux observés à l’intérieur des hématies. Il existe unperturbation des échanges ioniques transmembranaires.Cette forte concentration hémoglobinique rend compte de la sévérité de l’hétérozygotie composite« hémoglobine S / hémoglobine C ».
c/ Aspects cliniques
� L’hétérozygotie A/C est parfaitement bien tolérée.
� L’homozygote C/C présente un tableau d’anémie hémolytique franc avec splénomégalie.
d/ Diagnostic biologique
d1/ Anomalies hématologiques
� Chez le sujet hétérozygote, le frottis sanguin ne présente pas de particularité et les constantesérythrocytaires sont normales.� Le sujet homozygote C/C présente une discrète anémie microcytaire.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
19/24
Anomalie Numération Aspect sur lame
A/C Hb : NormaleVGM : Normal Normal
C/C Hb : 10-13 g/dlVGM : 65-75 fl Cellules cibles
d2/ Explorations phénotypiques
� Sur acétate de cellulose à pH alcalin, l’hémoglobine C migre plus lentement que l’hémoglobineS, à la même position que l’hémoglobine E et que l’hémoglobine O Arab.
� A l’électrophorèse sur gel d’agar en tampon citrate, l’hémoglobine C est la plus lente detoutes les hémoglobines anormales.
� L’isoélectrofocalisation individualise précisément l’hémoglobine C.
� La chromatographie liquide à haute performance permet d’évaluer le taux de l’hémoglobineC.A l’état hétérozygote, l’hémoglobine C représente 35 à 45 % des hémoglobines ; les hémoglobinesF et A2 ne sont pas quantitativement modifiées. Cette technique permet de suspecter uneassociation avec une α-thalassémie ou l’existence d’une carence martiale quand le taux del’hémoglobine C est inférieur à 35 %.A l’état homozygote, on constate l’absence d’hémoglobine A ; les taux des hémoglobines A2 et Fne sont pratiquement pas modifiés.
4-3.3. L’hémoglobine E
a/ Répartition géographique et définition moléculaire
L’hémoglobine E résulte de la mutation ponctuelle : β 26 [B8] Glu → Lys.Elle est, par le nombre de sujets qui en sont porteurs, la seconde des hémoglobines anormalesaprès la drépanocytose. Elle se rencontre essentiellement dans les populations du Sud-Estasiatique. Cette hémoglobinopathie n’est plus exceptionnelle en France.Dans les populations asiatiques, cette hémoglobine s’associe assez fréquemment avec d’autresanomalies de l’hémoglobine, surtout des α- et des β-thalassémies.
b/ Physiopathologie
La mutation à l’origine de l’hémoglobine E modifie la séquence nucléotidique au voisinage du sitenormal d’épissage et dévoile ainsi un site d’épissage cryptique, normalement non utilisé. Ce sited’épissage caché décale le cadre de lecture et conduit ainsi à rencontrer précocement un signal determinaison. Ainsi l’hémoglobine E est responsable d’un syndrome thalassémique, c’est-à-dired’un déficit de synthèse de l’une des chaînes de globine.
c/ Aspects cliniques
� Les homozygotes E/E présentent un tableau d’anémie hémolytique modérée, bien supportée,voisin d’une thalassémie.
� L’association E/β0-thalassémie correspond à une forme de β-thalassémie intermédiaire desévérité variable.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
20/24
d/ Diagnostic biologique
d1/ Anomalies hématologiques
Selon l’anomalie génétique en cause, on observe :
Anomalie Numération Aspect sur lame
A/E Hb : NormaleVGM : 75 fl Normal
E/E Hb : 10-13 g/dlVGM : 65-75 fl Cellules cibles
E/β0-thalassémie Hb : 2-12 g/dlVGM : 65-75 fl
hypochromiecellules ciblesanisocytosepoïkilocytose
d2/ Explorations phénotypiques
� A l’électrophorèse à pH alcalin, l’hémoglobine E migre à la même position que les hémoglobinesC et O Arab.
� A l’électrophorèse sur gel d’agar, l’hémoglobine E se distingue des précédentes car elle migrecomme l’hémoglobine A, contrairement aux deux autres.
� Sur le tracé d’isoélectrofocalisation, l’hémoglobine E se distingue très discrètement deshémoglobines A2 et C.
� En CLHP, l’hémoglobine E présente le même temps de rétention que l’hémoglobine A2.A l’état hétérozygote, le taux d’hémoglobine E représente seulement 30 à 35 % des hémoglobinesen raison de sa dégradation. Un taux encore plus bas peut faire suspecter une association avecune α-thalassémie ou une carence martiale.A l’état homozygote E/E, on constate l’absence d’hémoglobine A et la présence d’hémoglobines Eet F.Chez les hétérozygotes composites E/β0-thalassémie, seules deux hémoglobines sont visibles :l’hémoglobine E (entre 30 et 95 %) et l’hémoglobine F.
4-3.4. Une hémoglobine instable : l’hémoglobine Djelfa
a/ Définition
Les hémoglobines instables constituent un groupe particulier d’hémoglobines anormalesresponsables d’anémies hémolytiques caractérisées par la présence de corps de Heinz.La première description d’anémie hémolytique par hémoglobine instable a été effectuée enGrande-Bretagne chez un enfant ayant une cyanose associée à une splénomégalie. Il s’agissait del’hémoglobine Köln dont la chaîne β présente une méthionine en position 98 à la place d’une valine(β 98 [FG5] Val → Met).Actuellement une centaine de ce type de mutants de l’hémoglobine est connue.Certaines hémoglobines instables, détruites précocement dans l’érythrocyte, correspondentpratiquement à des thalassémies.Nous avons choisi de décrire ici l’hémoglobine Djelfa que nous avons étudiée au laboratoireMarcel Mérieux.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
21/24
b/ Répartition géographique et définition moléculaire
L’hémoglobine Djelfa est caractérisée par la substitution d’une valine par une alanine en position98 de la chaîne β de la globine, notée : β 98 [FG5] Val → Ala. Cette mutation est située sur l’exon2 du gène β de la globine.
Cette hémoglobine a été mise en évidence pour la première fois par l’équipe de WAJCMAN en1975 chez un Algérien de la région de Djelfa, d’où la dénomination de l’hémoglobine. Cettehémoglobine a été décrite à nouveau en 1989 chez un sujet caucasien en Afrique du Sud.Au laboratoire Marcel Mérieux, nous avons pu étudier ce variant hémoglobinique dans une famillebourguignonne.
c/ Physiopathologie
La valine en position β 98, qui caractérise la mutation de l’hémoglobine Djelfa, est impliquée dansdes contacts avec l’hème et entre sous-unités de globines : contacts α2-β1. La substitution de cetacide aminé est donc responsable d’une perte de l’hème et peut entraîner une modification del’affinité de l’hémoglobine pour l’oxygène.
d/ Aspects cliniques
Le membre de la famille dans laquelle l l’hémoglobine Djelfa a été découverte à l’état hétérozygoteétait une jeune fille de 15 ans qui venait de subir une cholécystectomie suite à des crises decoliques hépatiques en rapport avec une lithiase pigmentaire. L’examen clinique était pauvre ; iln’existait pas de splénomégalie.La mère, la grand-mère maternelle, l’une des trois sœurs de la mère de cette jeune fille étaientégalement porteuses du variant mais elles étaient toutes cliniquement asymptomatiques et neprésentaient pas d’anémie.
e/ Diagnostic biologique
e1/ Anomalies hématologiques
Dans le cas observé, il n’existait pas d’anémie ni d’anomalie morphologique des hématies.La recherche de corps de Heinz n’a pas pu être pratiquée.On retrouvait cependant des signes d’hémolyse : augmentation de réticulocytes, de la bilirubinelibre, de la LDH.
e2/ Explorations biologiques spécifiques
� Les techniques de dépistage ont été au nombre de quatre :� la CLHP automatisée :
La seule anomalie qualitative observée est un petit pic représentant 0,5 à 1,1 % deshémoglobines, avec un temps de rétention de 4,81 à 4,79 minutes, selon les échantillons.Ce constituant hémoglobinique mineur correspond vraisemblablement à une forme dedéshéminisation de l’hémoglobine Djelfa.
� la focalisation isoélectrique :Dans ce système analytique, l’hémoglobine Djelfa n’est pas séparée de l’hémoglobine A : on nemet en évidence aucun pic surnuméraire quantitativement important.
� le test de stabilité à l’isopropanol :L’hémoglobine Djelfa présente une instabilité accrue par rapport à une hémoglobine normale. Aubout de 15 minutes d’incubation, 13 % des hémoglobines du patient hétérozygote pourl’hémoglobine Djelfa précipitent contre 4 % pour le sujet normal. Au bout de 45 minutes, il s’agit de34 % contre 9 %.

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
22/24
� l’électrophorèse des chaînes de globine en gel de polyacrylamide en présence d’uréeet de TritonCette technique permet la séparation de la chaîne β de globine normale et de la chaîne β mutéede l’hémoglobine Djelfa.Ce type de gel peut être scanné permettant ainsi la détermination du pourcentage de la fractionhémoglobinique anormale. La moyenne des pourcentages de l’hémoglobine Djelfa des quatreporteurs hétérozygotes de la famille étudiée est de 33,6 %.
� Une technique complémentaire :� la courbe de fixation de l’oxygène
Une discrète augmentation de l’affinité pour l’oxygène de l’hémoglobine Djelfa avait étéprécédemment décrite mais nous ne l’avons pas retrouvée dans notre étude familiale.
� Des techniques de confirmation :� la CLHP préparative des chaînes de globine
La séparation des chaînes de globine a été réalisée par une technique de chromatographie liquidehaute performance, en phase inverse, sur colonne Vydac C4, selon la technique décrite parRAHBAR. Les différentes fractions sont individualisées et collectées.
� La spectrométrie de masse permet la mesure de la masse moléculaire des chaînes βnormale et mutée, Djelfa. La différence des masses moléculaires des chaînes β normale et βDjelfa est compatible avec la substitution d’une valine par une alanine.
� le séquençageIl nécessite au préalable une amplification des portions codantes du gène β, c’est-à-dire des troisexons. On a ainsi mis en évidence une indétermination sur la deuxième base du codon 98 chez lespatients hétérozygotes pour l’hémoglobine Djelfa. Cette base est soit une thymine qui déterminel’acide aminé valine, soit une cytosine qui permet de coder une alanine.
� l’analyse de restrictionL’analyse de restriction a été réalisé sur des produits de PCR. C’est l’enzyme Cac 8 I qui a étéutilisée car elle fait apparaître un site de restriction au niveau de la mutation. La taille des produitsde PCR et des produits de digestion enzymatique est estimé par rapport à une échelle de poidsmoléculaires, de 100 pb en 100 pb.Le produit de PCR d’un sujet normal, soumis à l’action de la Cac 8 I fait apparaître une seulebande de 866 pb. Le produit de PCR de la patient hétérozygote pour l’hémoglobine Djelfa, soumisà l’action de la Cac 8 I, fait apparaître deux fragments supplémentaires de 698 et 168 pb.
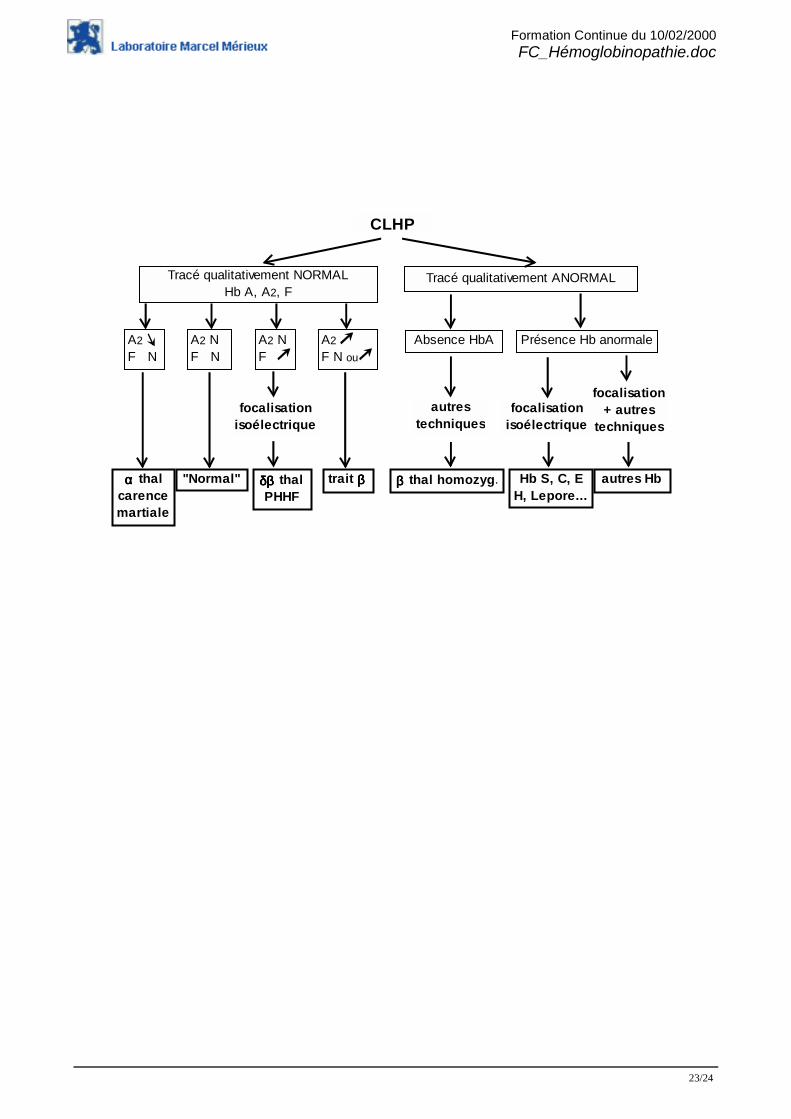
5- En pratique biologique courante
La technique de dépistage la plus informative est actuellement la CLHP ; elle fournit des donnéesqualitatives et aussi quantitatives (pour les hémoglobines A2 et F).La focalisation isoélectrique confirme l’approche chromatographique et permet de porter undiagnostic d’hémoglobinopathie dans la majorité des cas rencontrés.Les anomalies non élucidées par ces deux techniques nécessitent parfois des investigationscomplémentaires complexes et longues.
Notre schéma décisionnel est le suivant :
Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
23/24
autres Hb
focalisation+ autres
techniques
CLHP
Tracé qualitativement NORMALHb A, A2, F
Tracé qualitativement ANORMAL
A2 F N
A2 NF N
A2 F N ou
A2 NF
δβδβδβδβ thalPHHF
αααα thalcarencemartiale
"Normal" trait=
==
=ββββ
Absence HbA
=ββββ thal homozyg.
Présence Hb anormale
Hb S, C, E H, Lepore...
focalisationisoélectrique
autrestechniques
focalisationisoélectrique

Formation Continue du 10/02/2000FC_Hémoglobinopathie.doc
24/24
6- Pour en savoir plus
� Les maladies du globule rouge.H. WAJCMAN, B. LANTZ, R. GIROT.Médecine-Sciences Flammarion, Les éditions Inserm, 1992.
� L’hématologie de Bernard Dreyfus.J. BRETON-GORIUS, F. REYES, H. ROCHANT, J. ROSA, J.P. VERNANT.Médecine-Sciences Flammarion, 1992.
� A syllabus of human hemoglobin variants.T.H. HUISMAN, M.F.H. CARVER, G.D. EFREMOV.The Sickle Cell Anemia Foundation, Augusta, GA, USA, 1996.
� Drépanocytose. Physiopathologie et diagnostic.F. GALACTEROS.Revue de Praticien, 1995, 45, 351-360.
� Epidémiologie des maladies génétiques de l’hémoglobine en France métropolitaine.D. LENA-RUSSO, M.L. NORTH, R. GIROT.Revue de Praticien, 1992, 42, 15, 1867-1872.
� Histoire génétique de la drépanocytose.D. LABIE.Revue de Praticien, 1992, 42, 15,1879-1884.
� Histoire naturelle de la drépanocytose.C. BERCHEL, J.P. DIARA, H. LORET, L. FOUCAN, C. LE TURDU, Y. SAMUEL.Revue de Praticien, 1992, 42, 15,1885-1891.
� Hémoglobinopathies.J.Y. PELTIER.Option/Bio, 1992, 32 et 33.